

Summary
Genetic and in vivo evidence suggests that aberrant recognition of RNA-containing autoantigens by Toll-like receptors (TLRs) 7 and 8 drives autoimmune diseases. Here we report on the preclinical characterization of MHV370, a selective oral TLR7/8 inhibitor. In vitro, MHV370 inhibits TLR7/8-dependent production of cytokines in human and mouse cells, notably interferon-α, a clinically validated driver of autoimmune diseases. Moreover, MHV370 abrogates B cell, plasmacytoid dendritic cell, monocyte, and neutrophil responses downstream of TLR7/8. In vivo, prophylactic or therapeutic administration of MHV370 blocks secretion of TLR7 responses, including cytokine secretion, B cell activation, and gene expression of, e.g., interferon-stimulated genes. In the NZB/W F1 mouse model of lupus, MHV370 halts disease. Unlike hydroxychloroquine, MHV370 potently blocks interferon responses triggered by specific immune complexes from systemic lupus erythematosus patient sera, suggesting differentiation from clinical standard of care. These data support advancement of MHV370 to an ongoing phase 2 clinical trial.
Keywords: Toll-like receptors, lupus, pharmacology, autoimmunity, innate immunity, antagonist
Graphical abstract
Highlights
-
•
MHV370 is a selective, orally bioavailable inhibitor of Toll-like receptors 7 and 8
-
•
MHV370 blocks activation of cell types that drive lupus, including B cells and pDCs
-
•
MHV370 shows efficacy in mouse models of lupus-like disease
-
•
The data support advancement of MHV370 to phase 2 clinical trials
Toll-like receptors 7 and 8 have been implicated in the pathogenesis of autoimmune diseases by genetic and in vivo evidence. Hawtin et al. show in vitro and in vivo efficacy data of MHV370, a selective inhibitor of TLR7 and TLR8, and translational data supporting a phase 2 clinical trial.
Introduction
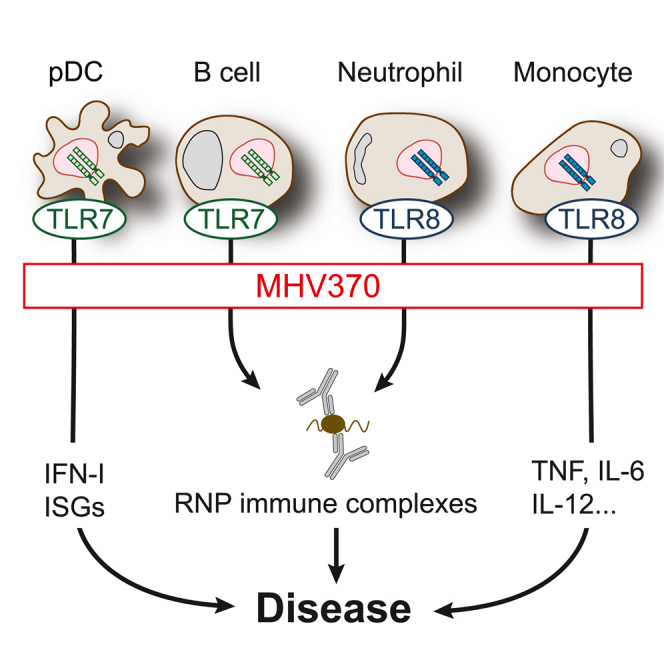
Toll-like receptors TLR7 and TLR8 are endosomal receptors for GU-rich single-stranded RNA (ssRNA). In humans, TLR7 is expressed by B cells and type I interferon (IFN)-producing plasmacytoid dendritic cells (pDCs), and TLR8 is predominantly expressed by monocytes, macrophages, and neutrophils. TLR7/8 activation leads to MyD88/IRAK/TRAF6 signaling, and downstream activation of monocytes/B cells mostly via nuclear factor (NF)-κB, or of pDCs via IRF7.1
TLR7- and TLR8-expressing cells are involved in multiple autoimmune diseases. The genetic link of TLR7 to systemic lupus erythematosus (SLE) is particularly strong.2 For example, elevated TLR7 copy number is a risk factor for juvenile SLE,3 and the strong female bias of SLE has been explained by biallelic expression of X-linked TLR7 in immune cells in women.4 Furthermore, TLR7 is overexpressed in peripheral blood mononuclear cells (PBMCs) of patients suffering from SLE.5,6 On a functional level, a TLR7 gain of function was recently found in a pediatric lupus patient and was sufficient to cause lupus-like disease when introduced in mice.7 Furthermore, TLR7-deficient mice are protected in multiple mouse models of lupus,8,9,10,11 and overexpression of either TLR7 or human TLR8 in mice leads to spontaneous autoimmunity.12,13 In addition, autoantibodies from SLE patients directed against ribonucleoproteins (RNPs) activate pDCs for IFN-α production in a TLR7-dependent fashion.14
Based on these data, TLR7/8 inhibition has been proposed as a therapeutic approach for SLE and other autoimmune diseases. In further support of this hypothesis, oligonucleotide inhibitors of TLR7 and low-molecular-weight (LMW) inhibitors of TLR7/8 showed efficacy in mouse models of lupus.15,16 However, clinical trial data of TLR7/8 antagonists in patients suffering from autoimmune diseases are not yet available. One clinical study was aimed at TLR7/8/9 blockade with an injectable oligonucleotide in psoriasis patients, but it did not show an acceptable risk-benefit profile.17 This was most likely due to the challenge of establishing a robust pharmacokinetic/pharmacodynamic (PK/PD) relationship with this modality, low tolerability, and possibly a disease-promoting potential of TLR9 inhibition.18 Therefore, we chose to develop a selective LMW antagonist against TLR7 and TLR8 to ensure an excellent tolerability profile and a robust PK/PD relationship.
The oral TLR7/8 antagonist MHV370 was identified via phenotypic cellular screening19 and further structure-based optimization. Here we show that MHV370 blocked the production of IFN-α and other pro-inflammatory cytokines in vitro and in vivo. MHV370 was protective in chronic mouse models, as it interfered with the expression of cytokines and interferon-stimulated genes (ISGs) following repeated challenge with the TLR7 agonist R848 or following injection of tetramethylpentadecane (TMPD). It also prevented and halted development of lupus-like disease in the NZB/W F1 model. Furthermore, MHV370 blocked the secretion of pro-inflammatory cytokines triggered by immune complexes derived from SLE patient sera predominantly expressing autoantibodies against RNPs. This opens the possibility that MHV370 will become a safe and effective approach to treat systemic autoimmune diseases, and a clinical phase 2 study is currently under way (ClinicalTrials.gov: NCT04988087).
Results
MHV370 is a selective and specific TLR7/8 antagonist in humans and mice
The cellular activity and TLR selectivity profile of MHV370 (Figures 1A and S1) was initially assessed using an engineered human Ramos NF-κB/AP-1 reporter B cell line. This B cell lymphoma derived cell line expresses TLR3, TLR7, and TLR9, but not TLR8 (Figure S2A). Consistently, this cell line is responsive to the TLR7 ligand CL307 and the TLR9 ligand ODN2006, but not to the TLR8 ligand TL8-506, and the TLR7/8 ligand R848 acts as a TLR7 agonist on these cells (Figure S2B). As shown in Figure 1B, MHV370 potently inhibited CL307- and R848-driven reporter gene activity in these cells with a half-maximal inhibitory concentration (IC50) of 15 ± 10 nM and 7 ± 0.1 nM, respectively (mean ± SD), demonstrating potent TLR7 inhibition. In contrast, MHV370 showed no or very limited inhibitory activity against endosomal TLR9 following ODN2006 stimulation, or against TLR3 following Poly(I:C) stimulation. In addition, MHV370 did not interfere with the activity driven by other TLRs (TLR1/2, TLR2/6), tumor necrosis factor (TNF) receptor, and NOD1, at concentrations up to 10 μM (data not shown, each n = 4). Assays on HEK cells transfected with TLR7 or TLR8 confirmed that MHV370 inhibited both TLR7 and TLR8 responses with IC50 values of 1.1 ± 0.4 nM and 4.5 ± 1.1 nM (mean ± SD), respectively (Figure S2C). The activity of MHV370 on TLR7 and TLR8 was dissected further using human primary cells. As is common for profiling of functional antagonists, we ascertained in profiling assays that MHV370 did not lead to cell toxicity at the tested dose ranges, i.e., at concentrations of up to 10 μM (Figure S2D). Whenever we aimed to observe robust TLR7-dependent IFN-α secretion as readout, the TLR7/8 agonist ssRNA was used instead of R848.20 A fixed dose of MHV370 did not block ODN2216/TLR9-dependent IFN-α secretion from isolated human pDCs, yet it blocked ssRNA-driven IFN-α responses from pDCs (Figure S2E). MHV370 potently suppressed production of multiple cytokines following activation of isolated monocytes with the TLR8-specific agonist TL8-0506, i.e., TNF, interleukin-6 (IL-6), and IL-1β (Figure S2F), at IC50 values that are comparable with values obtained following stimulation with the TLR7/8 agonists ssRNA and R848 (Table S2). As human pDCs express predominantly TLR7 and monocytes express mostly TLR8,13 these data demonstrate that MHV370 is a TLR7/8 antagonist on human primary cells. To further investigate the potency of MHV370 on human primary cells, we studied MHV370 on PBMCs following stimulation with TLR agonists. MHV370 inhibited ssRNA-induced IFN-α and TNF responses on PBMCs with nanomolar potencies (Table 1). Within PBMCs, TLR7-expressing pDCs are the main producers of IFN-α, and TLR8-expressing monocytes are the main producers of TNF. Therefore, these results allowed us to infer the potency of MHV370 on TLR7 and TLR8. The mean IC50 for TLR7 in human PBMCs (4 nM) was similar to its potency on Ramos cells (7 nM) and HEK-TLR7 transfectants (1 nM). Beyond its effect on IFN-α and TNF, MHV370 potently suppressed the production of multiple cytokines following activation of PBMCs with ssRNA, i.e., CXCL10, TNF, IL-6, IL-1β, and IL-12 (Figure 1C), at IC50 values that are comparable with values obtained following stimulation with the TLR7/8 agonist R848 and the TLR8 agonist TL8-506 (Table S3). As observed in Ramos cells, MHV370 was TLR7/8 selective, as it did not significantly interfere with cytokine secretion following PBMC stimulation with agonists of TLR4 (Figure S2G), TLR1/2, TLR5, or the IL-1 receptor (IL-1R), at concentrations up to 10 μM (each tested on n = 5 donors, data not shown). Furthermore, MHV370 showed micromolar potency following TLR9/ODN2216 stimulation of PBMCs (Figure S2G and Table 1). A Schild analysis of PBMCs using the TLR7/8 agonist R848 and IL-6 as readout indicated that MHV370 is a competitive and reversible antagonist (Figures 1D and 1E). TLR7/8 selectivity of MHV370 was also reflected in human blood, as MHV370 potently inhibited cytokine secretion following activation of TLR7 and TLR8 by ssRNA, while it showed micromolar potency for TLR9 and was inactive following TLR4 activation (Figure 1F and Table 1).
Figure 1.

MHV370 inhibits TLR7/8-specific activation of human cells
(A) Structure of MHV370.
(B) Reporter gene activity in Ramos B cells stimulated with agonists for TLR3 (polyIC), TLR7 (CL307), TLR7/8 (R848), or TLR9 (ODN2006), representative of two (TLR7) or four (TLR3, TLR7/8, and TLR9) independent experiments.
(C) ssRNA-mediated pro-inflammatory cytokine release from PBMCs.
(D and E) (D) R848-stimulated IL-6 release from human PBMCs with increasing fixed concentrations of MHV370, one representative of two experiments and (E) Schild plot analysis of (D), means of two experiments ± SD.
(F) IFN-α (TLR7/ssRNA, TLR9/ODN2216-driven) or TNF (TLR4/LPS, TLR8/ssRNA-driven) release from human blood.
(G) R848 (TLR7/8)-, TL8-506 (TLR8)-, or PMA-mediated ROS secretion from neutrophils; means of 8, 6, and 6 donors ± SD, respectively.
(H) B cell activation markers following activation with R848.
(C), (F) and (H) show data from one out of n donors (for n see Tables 1, S3, and S4). Data are standardized to conditions without MHV370, and data points are means of two (B, F, H) or three (C, D) technical replicates ± SD. See also Figure S2.
Table 1.
MHV370 inhibition of TLR7, TLR8, and TLR9 responses
| Cells | TLR7 (ssRNA-IFN-α) | TLR8 (ssRNA-TNF) | TLR9 (ODN-IFN-α) |
|---|---|---|---|
| Human PBMCs | 4.1 ± 0.4 (39) | 70 ± 7.4 (37) | 1,300 ± 170 (20) |
| Human blood | 8.4 ± 1.0 (16) | 31 ± 4.4 (14) | 1,320 ± 150 (12) |
| Mouse blood | 58 ± 10 (7) | 12 ± 4.2 (3)a | >10,000 (4) |
IC50 values (nM) shown are the mean ± SEM of (n) separate experiments or donors.
Murine response driven via TLR7.
In addition, MHV370 potently inhibited both IFN-α and TNF in mouse whole blood, following stimulation with ssRNA (Figure S2H and Table 1). This demonstrated inhibitory activity of MHV370 on murine TLR7, because rodent TLR8 does not recognize ssRNA.21 Of note, the potency of MHV370 on mouse whole blood was about 10-fold lower than on human whole blood, and as observed in human cells. MHV370 did not show functional inhibition of TLR9 or TLR4 at concentrations up to 10 μM (Table 1 and data not shown, n = 4) or any effect on cell viability (Figure S2D). Collectively, these results demonstrate that MHV370 is a potent, selective, and reversible antagonist of TLR7 and TLR8 in humans, and a selective TLR7 antagonist in mice.
MHV370 inhibits TLR8-dependent activation of neutrophils and TLR7-dependent activation of B cells
Neutrophils and B cells have been implicated in lupus pathogenesis22 with a potential role of excessive TLR7 and TLR8 activation in these cells.4,23 For this reason, we studied whether MHV370 selectively suppressed neutrophil activation following TLR7/8 or TLR8 stimulation. MHV370 potently blocked reactive oxygen species (ROS) production following stimulation with R848 or TL8-506 with IC50 values of 3.8 ± 0.8 nM and 5.2 ± 1.5 nM, respectively (mean ± SD), but not with phorbol myristate acetate (PMA) (Figure 1G). TL8-506 is a more TLR8-selective derivative of the TLR8 agonist VTX-2337.24 The inhibition was likely via TLR8, as human neutrophils express high levels of TLR8 but not TLR7.13 Moreover, MHV370 interfered with human B cell activation following stimulation with R848, showing potent inhibition of Akt phosphorylation and CD69 expression as well as immunoglobulin M (IgM) and IL-6 secretion (Figure 1H and Table S4). As B cells express TLR7 but not TLR8,13 this result supports TLR7 inhibition. Activation of B cell responses via the B cell receptor was unaffected by MHV370 up to 16 μM (n = 2, data not shown), showing that MHV370 did not cause broad immunosuppression. Together, these results demonstrate that MHV370 can suppress activation of a broad variety of immune cells downstream of TLR7 and TLR8 activation.
In vivo efficacy of MHV370 after single-dose administration
To demonstrate in vivo efficacy and selectivity of MHV370, we analyzed acute cytokine release in plasma of mice following intravenous administration of a TLR7 or TLR9 agonist. In line with our in vitro findings, MHV370 efficiently inhibited TLR7-dependent IFN-α and TNF production in a dose-dependent manner (Figures 2A and S3A), but not TLR9-dependent IFN-α (Figure S3B). A partial reduction of TLR7-dependent IFN-α and TNF was observed at mean blood exposures of 70 nM, which is consistent with the potency of MHV370 on TLR7 in mouse whole blood (IC50 of 58 or 12 nM for IFN-α and TNF, respectively, Table 1). To establish a PK/PD relationship in mice, CD69 expression on B cells was identified as a potential TLR7-dependent ex vivo PD marker. Following administration of a single oral dose of MHV370 to mice (5 mg/kg), CD69 expression was monitored by ex vivo stimulation of blood with R848 at different time points. MHV370 led to a marked inhibition of CD69 expression over a period of 8 h (Figure 2B), with CD69 levels returning to pre-dose levels at later time points, consistent with lower MHV370 blood exposure levels. This suggested that a compound dose of approximately 5 mg/kg orally would need to be administered twice daily to mice to achieve continuous suppression of the TLR7 pathway in long-term models.
Figure 2.

MHV370 suppresses acute TLR7-dependent immune activation in mice
(A) TLR7/ssRNA-induced IFN-α in mouse plasma. MHV370 levels in blood are indicated above the bars (nM ± SD). Pooled data from two independent experiments, means ± SD. Data points represent individual mice. ∗p < 0.05, ∗∗∗∗p < 0.0001, ANOVA with Tukey’s post test.
(B) Expression of CD69 on B cells after ex vivo stimulation of blood with R848 (bars) and MHV370 exposures (gray line) following 5 mg/kg MHV370 orally. Means ± SD of five mice.
See also Figure S3.
Therapeutic administration of MHV370 suppresses chronic TLR7 activation in vivo
The above results encouraged us to investigate whether MHV370 was able to interfere with chronic TLR7 activation in a therapeutic setting. In line with published data,25 daily dosing of R848 for 14 days led to immune activation in mice, including splenomegaly, B cell activation, and elevated cytokine levels in blood (Figures 3A, S3C, and S3D). This was accompanied by increased gene expression of, e.g., ISGs (Figures 3B, S3E, and S3F). All of these parameters were significantly reduced, close to levels observed in naive mice, when MHV370 was administered therapeutically from day 7 onward. Consistently, a gene set enrichment analysis showed strong activation of several inflammatory pathways in mice treated with R848 and vehicle (including ISGs, and TLR and TNF pathways), which were all downregulated in mice treated with R848 and MHV370 (Figures 3C and 3D). This demonstrated that therapeutic dosing of MHV370 can normalize multiple pro-inflammatory parameters in a situation of chronic TLR7 activation and encouraged us to analyze the effect of MHV370 in more complex models of inflammation.
Figure 3.

MHV370 suppresses chronic TLR7-dependent immune activation in mice
(A) Spleen weight, CD69 expression on B cells, and TNF in serum. Data points represent individual mice with means ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001, ANOVA with Tukey’s post test.
(B) Scaled and normalized log counts per million of the most significant genes (adjusted p value <1e−8, absolute fold change >2) differentially expressed in naive mice (green horizontal bar), R848/MHV370-treated (blue), and R848/vehicle-treated (red) mice. Hierarchical clustering of genes and samples after grouping of samples by experimental group.
(C) Anti-correlation of global gene expression changes in mice treated with R848/vehicle compared with naive mice (x axis) vs. mice treated with R848/MHV370 compared with R848/vehicle-treated mice (y axis). Data points represent values for individual genes, line represents linear fit to the data (Pearson correlation coefficient = −0.8, p value <2.2e−16).
(D) Gene set enrichment analysis of the R848/vehicle vs. naive and the R848/MHV370 vs. R848/vehicle comparisons using the Hallmark and C2 Canonical Pathways gene set collections from MSigDB. Red, upregulated pathways; blue, downregulated pathways. The size of each circle is proportional to −log10 of the adjusted p value obtained in the enrichment test (−log10(padj)). Full circles, terms with padj < 0.01; empty circles, terms with padj > 0.01.
See also Figure S3.
MHV370 suppresses ISGs in a TMPD-induced peritonitis model
Elevated expression of an ISG signature is a hallmark of SLE and related rheumatologic diseases.26 Consequently, it was important to demonstrate the ability of MHV370 to suppress ISGs in vivo. Intraperitoneal injection of the mineral oil TMPD leads to TLR7-dependent acute inflammation characterized by influx of inflammatory monocytes into the peritoneum and ISG induction.11 Prophylactic oral treatment of mice with MHV370 led to a dose-dependent reduction of inflammatory monocytes and monocyte chemoattractant CCL2 in the peritoneum on day 7 (Figures 4A and 4B). Of note, MHV370 dose-dependently suppressed a panel of ISGs in peritoneal cells and blood (Figures 4C and 4D). Ex vivo stimulation of the PD marker CD69 was reduced in peripheral blood of mice in a dose-dependent manner (Figure 4E), and its reduction correlated with inhibition of ISGs (Figure 4F). Together, these results demonstrate that MHV370 curbs ISG expression in blood of mice and that a simpler ex vivo PD marker tracked with this readout. These data also suggest that ex vivo inhibition of CD69 on B cells might serve as a TLR7-dependent PD biomarker in clinical studies and may correlate with ISG inhibition.
Figure 4.

In vivo efficacy of MHV370 in the TMPD peritonitis model
(A and B) Frequencies of (A) CD11b+Ly6C+ inflammatory monocytes and (B) CCL2 levels in peritoneal lavages.
(C and D) Expression of ISGs (C) in peritoneal cells and (D) in blood cells. ISG expression was standardized to expression in naive mice with each ISG measured in triplicate.
(E) CD69 expression on B cells from blood following ex vivo stimulation with R848.
(F) Correlation of data from (D) and (E) for individual mice; dotted line, 95% confidence interval. Open symbols represent vehicle-treated mice.
Data points in (A) to (E) represent individual mice, means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001, ANOVA with Dunnett’s post test, comparing MHV370 groups with vehicle group.
MHV370 protects from lupus-like disease in the NZB/W F1 mouse model
The above results encouraged us to analyze the efficacy of MHV370 in the spontaneous, chronically progressive NZB/W F1 model of lupus. We hypothesized that TLR7 inhibition alone was sufficient to interfere with a complex autoimmune pathology in a multifactorial model. Treatment of NZB/W F1 mice with MHV370-laced food (Figures S4A and S4B) strongly impaired progression of lupus-like disease as shown by multiple readout parameters. Proteinuria was effectively prevented (Figure 5A), consistent with moderately reduced blood urea nitrogen (BUN; Figure 5B), which is often used as a systemic marker of impaired kidney function. Inhibition of proteinuria was already achieved at a dose of 0.01% MHV370 in food, where blood exposures periodically dropped below concentrations required for complete inhibition of the TLR7 pathway (IC50 for the ex vivo blood PD marker CD69 is 35 nM, see Figures S4C and S4D). Histological analysis demonstrated that MHV370 treatment protected the kidneys of NZB/W F1 mice from damage: MHV370 reduced glomerulopathy (Figure 5C), IgG deposits in the glomeruli (Figure 5D), and infiltration of kidneys with CD45R+ B cells (Figure 5E) as well as CD138+ plasma cells (Figure S4E). In addition, MHV370 significantly reduced serum levels of CXCL13 (Figure 5F), and serum CXCL13 levels weakly correlated with glomerular IgG deposition (Figure 5G). This shows that serum CXCL13 might become a useful serum biomarker of kidney protection in lupus-like disease following administration of MHV370.
Figure 5.

In vivo efficacy of MHV370 in the NZB/W F1 lupus model
(A) Proteinuria. Lines represent individual mice.
(B) BUN levels at termination. ∗p < 0.05, ANOVA with Dunnett’s post test.
(C) Kidney histopathology and glomerulopathy score.
(D) IgG deposition in kidneys and score.
(E) B cell infiltration in kidneys and score.
(F) CXCL13 protein levels in serum at termination. ∗∗p < 0.01, ∗∗∗p < 0.001, ANOVA with Dunnett’s post test.
(G) Correlation of CXCL13 levels in serum with glomerular IgG deposition; dotted line, 95% confidence interval.
In (B) to (G), data points represent individual mice, in (B) to (F), means ± SD. Open symbols, vehicle-treated mice; open squares, vehicle-treated mice which were terminated prematurely as they reached the humane endpoint of the license. In (C) to (E), ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, Mann-Whitney test. Scale bars, 50 μm.
See also Figures S4–S6.
Since antinuclear antibodies (ANAs) are a clinical hallmark of lupus, it was important to assess whether MHV370 treatment affected their production. IgG autoantibody levels against Smith (Sm), ribosomal P (RiboP), and the N-methyl-D-aspartate receptor (NMDAR) were reduced with MHV370 treatment, and a similar trend was observed for anti-Ro60 antibodies (Figures S5A–S5D). Serum antibodies against double-stranded DNA (dsDNA) and histones were not reduced (Figures S5E and S5F). Therefore, MHV370 had a strong effect on lupus pathology in all mice, yet the effect on ANA types was more selective, affecting mostly specificities against RNA-containing autoantigens and NMDAR. Of note, the latter and anti-RiboP are elevated and potentially even pathogenic in neuropsychiatric SLE.27,28,29 Together, our results from the NZB/W F1 model showed inhibition of multiple disease parameters with MHV370, including complete inhibition of proteinuria and histological protection of kidneys as well as reduction of specific autoantibodies.
The experiment in NZB/W F1 mice was repeated independently at a different research site with slightly different readout parameters (Figure S6). This ensured that in vivo efficacy of MHV370 was independent of mouse housing and handling conditions. When MHV370 twice-daily treatment was started therapeutically, i.e., after detection of robust proteinuria, it reduced proteinuria and increased survival (Figures S6A and S6B). In addition, IgM and IgG deposition in kidneys, as detected by histology, was substantially reduced with MHV370 treatment (Figures S6C and S6D). This confirmed the efficacy of MHV370 in an independent experiment and suggested therapeutic in vivo efficacy.
Together, our results from the NZB/W F1 model suggest that MHV370 may have an impact on several key disease parameters of SLE, in line with a recent study describing TLR7 as a disease driver in this model.30
Inhibition of patient sera immune-complex-mediated IFN-α production and ISGs
Sera from SLE patients contain autoantibodies against nuclear and ribosomal proteins. As immune complexes with necrotic cell extract (NE), these sera can activate cells for the production of IFN-α,31,32 and in some cases through activation of TLR7.14 Since type I IFNs correlate with disease severity in lupus33 and drive ISGs, a hallmark of SLE,26 we analyzed whether MHV370 blocked the interferogenic activity of patient-derived immune complexes in vitro (Figure 6A). SLE patient sera were grouped into three main categories based on their autoantibody profiles: (1) RNP/Sm and/or SS-A only; (2) dsDNA only; or (3) RNP/Sm and/or SS-A plus dsDNA (for full autoantibody profiles of patients, see Table S5). For all three categories of sera, immune complexes with NE specifically stimulated IFN-α secretion (Figure 6B) and ISG expression (Figure S7A) from PBMCs. MHV370 (0.3 μM) completely blocked immune-complex-driven responses derived from patient sera that were positive for RNP/Sm and/or SS-A autoantibodies or for RNP/Sm and/or SS-A plus dsDNA autoantibodies, yet immune-complex-driven responses for patient sera expressing dsDNA antibodies were blocked to a much lesser extent (Figure 6C). Since the majority of SLE patients receive anti-malarials such as hydroxychloroquine (HCQ) as standard-of-care treatment,34 and since these compounds can also influence endosomal TLR responses,35 we analyzed whether immune-complex-driven IFN-α responses were sensitive to HCQ treatment. In marked contrast to MHV370, and when tested at a concentration that matched the steady-state blood exposure level of SLE patients,36 HCQ showed strongest inhibition of IFN-α responses elicited by dsDNA immune complexes (Figure 6C). To examine this difference in more detail, we studied how potently MHV370 or HCQ suppressed immune-complex-induced IFN-α responses in a patient who was autoantibody positive for RNP/Sm plus dsDNA. MHV370 alone potently suppressed IFN-α release in a dose-dependent manner with an IC50 of 1.0 ± 0.3 nM (mean ± SEM, n = 3, Figure 6D, filled circles). HCQ showed an approximately 1,000-fold lower potency than MHV370, with an IC50 of 800 ± 380 nM (mean ± SEM, n = 5, Figure 6D, open circles). Consistent with these data, HCQ was a 3,300-fold less potent antagonist of ISG expression than MHV370 following stimulation of PBMCs with ssRNA (Figure S7B and Table S6).
Figure 6.

MHV370 inhibits SLE patient sera immune-complex-driven responses and ex vivo blood pathway markers in SLE patients
(A) Schematic illustration of the functional immune-complex assay using SLE patient serum.
(B) IFN-α release from PBMCs stimulated by immune complexes of healthy volunteer (HV) or SLE patient sera with and without necrotic extract (NE). Data points represent individual HV sera (n = 12) or SLE patient sera (n = 20), as means from 2 to 23 PBMC stimulations, and three technical replicates per stimulation. Bars denote means ± SD. Colors indicate autoantibody profiles of individual SLE patients.
(C) Normalized IFN-α release from PBMCs stimulated with SLE immune complexes in presence of medium, MHV370 (0.3 μM) or HCQ (1 μM). Data points are means of individual SLE patient sera (n = 5), with 2–10 PBMC stimulations per serum, and three technical replicates per stimulation. Bars denote means ± SD.
(D) IFN-α release from PBMCs stimulated with immune complexes from an SLE patient (positive for RNP/Sm and dsDNA) in the presence of MHV370 (filled circles), HCQ (open circles), and HCQ in the presence of increasing fixed amounts of MHV370 (gray symbols), as indicated. Means ± SD, one representative of two experiments.
(E) Expression of individual ISGs (performed in triplicate) and a five-gene ISG signature in blood of five individual HV and SLE patients.
(F) Marker expression in blood of individual SLE patients (n = 4 or 5), following ex vivo stimulation with R848 (pAKT, CD69, TNF) or ssRNA (IFN-α), normalized to matched HV blood.
In (B) and (C), data points are means of at least two technical replicates. ns, not significant; ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ANOVA with (B) Tukey’s or (C) Kruskal-Wallis post test. See also Figure S7.
To demonstrate combinatorial efficacy of MHV370 and HCQ in this context, healthy PBMCs were stimulated with SLE immune complexes in the presence of variable concentrations of HCQ combined with defined amounts of MHV370. Addition of low concentrations of MHV370 (1–10 nM) to HCQ already led to almost complete suppression of IFN-α (Figure 6D, gray symbols). Furthermore, MHV370 (0.1 μM) strongly suppressed expression of a five-panel ISG signature by PBMCs stimulated with SLE immune complexes, whereas HCQ (1 μM) was only mildly effective (Figure S7C). Collectively, these results suggest that lupus patients expressing autoantibodies in their serum may respond to MHV370 in the clinic, even when they are treated with anti-malarials.
Consistent with published data,26 SLE patients in this study displayed elevated ISG expression levels compared with healthy controls (Figure 6E). Although ISGs have utility as clinical markers of disease activity,37 they can be induced downstream of several interferogenic nucleic acid sensing pathways. Similarly, elevated IFN-α protein is observed in sera of a fraction of SLE patients,38 although it is not uniquely TLR7 dependent. This prompted us to identify more specific pharmacodynamic biomarkers for TLR7 and TLR8. Blood from SLE patients or healthy volunteers (HVs) was stimulated with the TLR7/8 agonist R848 or ssRNA ex vivo. Expression levels of phospho-Akt (pAkt) on B cells from SLE patients following R848 stimulation were very similar to those from healthy controls (Figure 6F) and, as B cells express TLR7 and not TLR8,13 pAkt represented an attractive proximal pharmacodynamic readout for TLR7. Similarly, TNF release in SLE patient blood following R848 stimulation was comparable with healthy control blood (Figure 6F). Since TNF in peripheral blood was inferred to be mostly derived from TLR8-expressing monocytes, it might serve as a pharmacodynamic biomarker for TLR8 activation. For additional markers of TLR7 activation, SLE patients displayed mostly reduced or variable levels of IFN-α or CD69 expression on B cells compared with healthy controls following ex vivo stimulation of blood with ssRNA and R848, respectively (Figure 6F). Reduced IFN-α levels were not surprising, as SLE patients may have reduced pDC frequencies in peripheral blood.39 In addition, persistent TLR7 activation during chronic inflammation may lead to tachyphylaxis in some cells.40 For example, CD69 is already highly expressed in PBMCs from SLE patients,41 such that further induction ex vivo in patients may be limited. To confirm that MHV370 can block these pharmacodynamic markers in the blood of patients, blood was pretreated with MHV370 and stimulated ex vivo with TLR7/8 agonists. Indeed, MHV370 potently inhibited pAkt and CD69 expression as well as IFN-α and TNF secretion in blood from SLE patients and HVs with the same potency (Table S7). These results suggested that patients and HVs are equally sensitive to MHV370 treatment and that the above pharmacodynamic biomarkers are of potential value in phase 1 or phase 2 clinical studies with MHV370.
Together, the data presented in this paper demonstrate that MHV370 blocks a broad array of TLR7 and TLR8 functional responses on multiple cell types and that it has disease-modifying potential. This lays the foundation for successful use of MHV370 in phase 2 studies that include patients with autoimmune diseases.
Discussion
This study introduces MHV370, a potent and selective, orally bioavailable LMW antagonist of TLR7 and TLR8. It inhibits TLR7/8-dependent secretion of multiple cytokines from monocytes and pDCs, as well as B cell and neutrophil activation. MHV370 shows dose-dependent efficacy in acute and chronic mouse models. It blocks immune activation following single or repeat injection of TLR7/8 agonists and induction of ISGs following injection of TMPD. In the spontaneous NZB/W F1 mouse model of lupus, prophylactic or therapeutic dosing of MHV370 interfered with multiple disease parameters. These results position MHV370 as a potential future treatment option for SLE patients.
Autoimmune diseases are chronic conditions that contribute to significant morbidity and mortality in affected patients. Arguably one of the most complex autoimmune diseases is SLE, with very diverse symptoms and heterogeneous disease progression.34 This has led to only two approvals by the US Food and Drug Administration for new SLE treatments in this disease over the past 60+ years, the antibody drugs belimumab, which interferes with B cell expansion, and anifrolumab, which interferes with type I IFN signaling. In the pivotal BLISS trial, belimumab led to a small 7% and 14% SRI (SLE responder index) response rate above placebo at 1 mg/kg and 10 mg/kg, respectively,42 and anifrolumab failed to reach its predefined endpoint in one of two phase 3 studies.43 The present study gives reason to believe that MHV370 combines inhibitory activity on the pathways targeted by anifrolumab and belimumab, i.e., the type I IFN axis and B cell activation, yet unlike these two other treatments, it does not deplete or paralyze entire arms of the immune system. As such, MHV370 may have an advantageous safety profile. For example, herpes virus infections were increased in patients treated with anifrolumab.44 Protective immunity against herpes viruses appears to rely strongly on TLR3 function,45,46,47 and MHV370 does not block TLR3. Therefore, the preclinical profile of the TLR7/8 antagonist MHV370 strongly suggests that it may show transformational efficacy and superior safety in SLE patients and related indications. Indeed, a phase 2 clinical trial to evaluate safety, tolerability, and efficacy in Sjögren’s syndrome and mixed connective tissue disease is under way (ClinicalTrials.gov: NCT04988087), with no other TLR7/8 antagonist being studied in these indications.
MHV370 has advantages over oligonucleotide-based antagonists such as the TLR7/8/9 antagonist IMO-8400, which has progressed to a small phase 2 study of psoriasis.17 For example, it is challenging to quantify oligonucleotide drugs in blood to establish a robust PK/PD relationship, and oligonucleotides cannot be dosed orally. More recently, preclinical and first-in-man data on an LMW TLR7/8 antagonist, M5049, have been published.16,48 M5049 showed efficacy in an IFN-α accelerated NZB/W F1 model, which expressed very high levels of ISGs. However, since ISG expression is variable in patients suffering from SLE and other autoimmune diseases,49 it was important to ascertain that MHV370 worked in the classical, spontaneous NZB/W F1 model to collect evidence that MHV370 may even show efficacy in patients with lower ISG expression. In addition, efficacy of M5049 in target tissues has not been reported, while MHV370 influences multiple parameters of lupus-related disease in kidneys. Preclinical data on therapeutic efficacy of M5049 are not available either, while we show that therapeutic use of MHV370 for only 7 days in a situation of chronic TLR7 activation normalized multiple TLR7-dependent pro-inflammatory pathways in mice. It is noteworthy that MHV370 ameliorated renal disease in the NZB/W F1 model and reduced autoantibodies against the RNP subunit Smith (Sm) most profoundly, while having no effect on anti-dsDNA autoantibodies. This result is consistent with data from the MRL/lpr model, where TLR7 deficiency improved renal disease and led to significant reduction of anti-Sm and anti-RNP but not anti-dsDNA autoantibodies.8 Together, these data suggest that TLR7 may be a specific driver of autoantibodies against RNPs, possibly in a B cell intrinsic manner.50 It will be informative to study how MHV370 will affect autoantibody subtypes in treated patients. Since autoantibodies against Sm are associated with disease severity51 and with specific pathogenic features,52 it is conceivable that a subset of specific TLR7/8-driven symptoms of lupus will improve most profoundly with MHV370.
By linking cellular and mouse model investigations, our study on MHV370 suggests that interference with multiple pro-inflammatory cell types can result in efficacy in complex autoimmune diseases. Our result also suggests how MHV370 may behave in combination with standard-of-care treatments for SLE. Since TLR7 activation renders pDCs steroid insensitive,53 a TLR7/8 antagonist such as MHV370 may become a steroid-sparing treatment. Moreover, our data using immune complexes from SLE patient sera suggest that MHV370 may show efficacy even in the presence of clinically relevant concentrations of HCQ. This makes MHV370 attractive for combination treatments, implying that MHV370 could change the current pharmacological treatment paradigm in SLE.
Taken together, our study provides broad support for the concept that TLR7/8 antagonism as a potentially transformational treatment for SLE patients. Ongoing clinical trials will investigate how MHV370 provides tangible patient benefit.
Limitations of the study
One obvious limitation of our study is the fact that TLR8 inhibition cannot be studied in mouse models in a straightforward manner. Depending on the mouse model, murine TLR8 may either contribute to lupus-like disease54 or restrain TLR7-dependent pathology.55 Moreover, rodent TLR8 does not recognize the same ssRNA ligand as human TLR8.21 Therefore, our conclusions on TLR8 are based on human in vitro assays. Our data show that MHV370 interferes with the activation of myeloid cells from humans (monocytes, neutrophils), which predominantly express TLR8. Since low-density granulocytes are typically involved in SLE56 and many SLE patients have a granulopoiesis signature,57 it is likely that dual TLR7/8 antagonism will show superior efficacy over single TLR7 or TLR8 antagonists. The pathogenic role of TLR8 in humans is further suggested by chronic inflammatory disease in TLR8 gain-of-function patients58 and by the association of a TLR8 signature to the ISG-high subtype of lupus patients.59
Another limitation of this study is that we studied MHV370 only in preclinical models of SLE. However, the involvement of TLR7 and TLR8 has been described for other systemic autoimmune rheumatic diseases.49,60 This indicates the broad scope of target indications for MHV370, and translational studies with MHV370 are in progress to support such indications.
Finally, our study was aimed at a detailed pharmacological characterization of the TLR7/8 antagonist MHV370 in vitro and in vivo. To achieve this, we relied on robust, well-established experimental systems. We expect that these will lay the foundation for more mechanistic insights into TLR7 and TLR8 biology in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Human CD19-APC (clone HIB19) | Biolegend | Cat#302212; RRID: AB_314242 |
| Anti-Human CD69-PE (clone FN50) | Biolegend | Cat#310906; RRID: AB_314841 |
| Anti-Human CD20-Alexa488 (clone H1) | BD Biosciences | Cat#558056; RRID: AB_647080 |
| Anti-Human phospho-Akt-Alexa647 (Ser473) | BD Biosciences | Cat#560343; RRID: AB_1645397 |
| Anti-Mouse CD16/32 (Clone 2.4G2) | BD Biosciences | Cat#553142; RRID: AB_394657 |
| Anti-Mouse Ly6G-APC (Clone 1A8) | BD Biosciences | Cat#560599; RRID: AB_1727560 |
| Anti-Mouse Ly6C-Alexa488 (Clone ER-MP20) | Bio-Rad | Cat#MCA2389A488; RRID: AB_2137342 |
| Anti-Mouse CD11b-PerCP-Cy5.5 (Clone M1/70) | BD Biosciences | Cat#550993; RRID: AB_394002 |
| Anti-Mouse CD19-FITC (Clone 1D3) | BD Biosciences | Cat#553785; RRID: AB_395049 |
| Anti-Mouse CD69-PE (Clone H1.2F3) | BD Biosciences | Cat#553237; RRID: AB_394726 |
| Anti-Mouse IgG-HRPO (Goat polyclonal) | Sigma Aldrich | Cat#A8924; RRID: AB_258426 |
| Anti-human IgM, polyclonal Fc5m fragment specific | Jackson ImmunoResearch | CAT#109-005-129; RRID: AB_2337543 |
| Anti-human IgM-HRPO, polyclonal, Fc5m fragment specific | Jackson ImmunoResearch | CAT#109-035-129; RRID: AB_2337588 |
| Human IgM control, unconjugated | Sigma Aldrich | CAT#I8260; RRID: AB_1163621 |
| Anti-Mouse IgG (Goat polyclonal) | Nordic-MUbio | Cat#GAM/IgG(H + L) |
| Anti-Mouse CD138 (Clone 281-2) | BD Biosciences | Cat#553712; RRID: AB_394998 |
| Anti-Mouse B220/CD45R (Clone RA3-6B2) | Bio-Rad | Cat#MCA1258G; RRID: AB_323211 |
| Anti-Mouse CD3 (Clone SP7) | Thermo Fisher Scientific | Cat#RM-9107-A; RRID: AB_149924 |
| Rabbit-anti-Rat IgG (H + L)-biotin | Vector Laboratories | Cat#BA4001; RRID: AB_10015300 |
| Donkey-anti-Goat IgG (H + L)-biotin | Jackson ImmunoResearch | Cat#705-065-147; RRID: AB_2340397 |
| Goat-anti-Rabbit IgG (H + L)-biotin | Jackson ImmunoResearch | Cat#111-065-144; RRID: AB_2337965 |
| Biotinylated anti-human IgG (Goat) | SouthernBiotech | Cat#2043-08 |
| DELFIA Streptavidin-Eu antibody | PerkinElmer | Cat#1244-360 |
| Biological samples | ||
| Human blood from healthy donors with informed consent | Santémed Gesundheitszentrum AG Basel | N/A |
| Buffy coats from healthy donors | Interregionale Blutspende (IRB) SRK Bern | N/A |
| Sera from SLE patients or healthy donors | University Hospital Basel | Study BASICHR0003 |
| Sera from SLE patients or healthy donors | Synexa Life Sciences | Study PJMR0012114 |
| Sera from SLE patients or healthy donors | Cincinnati Children’s Hospital | Study CCHMC IRB # 01-12-2X |
| Chemicals, peptides, and recombinant proteins | ||
| Pam3CSK4 (TLR1/2 agonist) | InvivoGen | Cat#tlrl-pms |
| FSL-1 (TLR2/6 agonist) | InvivoGen | Cat#tlrl-fsl |
| PolyIC (TLR3 agonist) | InvivoGen | Cat#tlrl-pic |
| LPS-0111:B4 (TLR4 agonist) | InvivoGen | Cat#L4391 |
| CL307 (TLR7 agonist) | InvivoGen | Cat#tlrl-c307 |
| TL8-506 (TLR8 agonist) | InvivoGen | Cat#tlrl-tl8506 |
| ODN1585 (TLR9 agonist) | InvivoGen | Cat#tlrl-1585 |
| ODN2006 (TLR9 agonist) | InvivoGen | Cat#tlrl-2006 |
| ODN2216 (TLR9 agonist) | InvivoGen | Cat#tlrl-2216 |
| Tri-DAP (NOD1 agonist) | InvivoGen | Cat#tlrl-tdap |
| Human recombinant IL-1β and TNF | Novartis | |
| R848 (TLR7/8 agonist) | Enzo life sciences | Cat#ALX-420-038 |
| ssRNA40 (5′-GCCCGUCUGUUGUGUGACUC-3′) with protective phosphorothioate linkages | Microsynth | custom-synthesized |
| R0006 (5′-UUGUUGUUGUUGUUGUUGUU-3′) with protective phosphorothioate linkages | Microsynth | custom-synthesized |
| DOTAP (N-[1-(2,3 Dioleoyloxy) propyl]-N,N,N tri-methyl ammonium methylsulfate) | Sigma-Aldrich (Roche) | Cat#11202375001 |
| MHV370 | This paper | |
| Normocine | InvivoGen | Cat#ant-nr-05 |
| Hydroxychloroquine | Novartis | |
| human IL-3 | PeproTech | Cat#200-03 |
| Peroxidase substrate kit | Bio-Rad | Cat#172-1064 |
| Lymphocyte Separation Medium 1077 | Corning | Cat#25-072-CV |
| Ficoll Plus | Cytiva | Cat#17-1440-03 |
| Phorbol-myristate-acetate (PMA) | Sigma Aldrich | Cat#P1585 |
| Dihydrorhodamine 123 (DHR) | Sigma Aldrich | Cat#D1054 |
| Tetramethylpentadecane (TMPD) | Sigma Aldrich | Cat#P9622 |
| Aqua viability dye solution | Thermo Fisher Scientific | Cat#L34957 |
| Powdered rodent chow | Provimi Kliba | Cat#NAFAG3890 |
| ANAcombi (RNP-70, Sm, RNP/Sm, SS-A, SS-B, Scl-70, Centromer B, Jo-1) | Orgentec | Cat#Org539 |
| Histone from calf thymus | Sigma Aldrich | Cat#H9250 |
| Salmon sperm DNA | Thermo Fisher Scientific | Cat#AM9680 |
| Ribosomal P antigen | Immunovision | Cat#PAG3000 |
| Smith | Immunovision | Cat#SMA3000 |
| DWEYS peptide | Bachem | Cat#4066380 |
| Critical commercial assays | ||
| Bright-Glo assay | Promega | Cat#E2620 |
| QUANTI-Blue | InvivoGen | Cat#rep-qbs |
| RNeasy Mini RNA isolation kit | Qiagen | Cat#74104 |
| Super-Script III First-Strand synthesis Super-Mix | Invitrogen | Cat#11752050 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | Cat#4369016 |
| CXCL10 (AlphaLisa) | PerkinElmer | Cat#AL259 |
| IL-1b(HTRF) | CisBio | Cat#62HIL1B |
| IL-6 (HTRF) | CisBio | Cat#62HIL06 |
| IL-12p40 DuoSet ELISA | R&D systems | Cat#DY1240 |
| CellTiter-Glo assay | Promega | Cat#G7571 |
| ATPlite Luminescence assay | PerkinElmer | Cat#6016943 |
| EasySep Human Plasmacytoid DC Enrichment Kit | Stemcell Technologies | Cat#19062 |
| IFNα (AlphaLisa) | PerkinElmer | Cat#AL297 |
| EasySep Human monocyte Enrichment Kit | Stemcell Technologies | Cat#19059 |
| EasySep human B cell enrichment kit | Stemcell Technologies | Cat#I9054 |
| Human IL-6 DuoSet ELISA | R&D systems | Cat#DY206 |
| Mouse IFNα Platinum ELISA | InvitroGen | Cat#BMS6027TEN |
| Mouse TNF DuoSet ELISA | R&D Systems | Cat#DY410 |
| Mouse CXCL10 ELISA | InvitroGen | Cat#BMS6018 |
| Mouse CCL2 ELISA | Biolegend | Cat#432701 |
| Ion AmpliSeq Transcriptome Mouse Gene Expression Kit | Thermo Fisher Scientific | Cat#A26325 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34957 |
| RiboPure blood RNA isolation kit | Thermo Fisher Scientific | Cat#AM1951 |
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| Protein Assay Kit | Bio-Rad | Cat#5000006 |
| Mouse Anti-SSA/Ro60 Igs (total) ELISA | Alpha Diagnostic | Cat#5710 |
| Mouse CXCL13 Quantikine ELISA kit | R&D systems | Cat#MCX130 |
| S Test Reagent Cartridges BUN | Hitachi Chemical Diagnostics | Cat#10018947 |
| Uristix | Bayer | N/A |
| Streptavidin DABMap kit | Roche | Cat#05266360001 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368813 |
| TaqMan Fast Universal PCR Master Mix | Applied Biosystems | Cat#4352042 |
| DELFIA Enhancement solution | Perkin Elmer | Cat#1244-105 |
| Deposited data | ||
| Data: Gene expression profiling of blood from mice | This paper | https://doi.org/10.5281/zenodo.7573856 |
| Code: Gene expression profiling of blood from mice | This paper | https://doi.org/10.5281/zenodo.7575672 |
| Experimental models: Cell lines | ||
| HEK293-NFkB-luciferase reporter cells | Promega | Cat#E8520 |
| Ramos Blue B reporter cells | InvivoGen | Cat#rms-sp RRID: CVCL:X591 |
| U-937 cells | ATCC | Cat#CRL-1593.2 |
| Experimental models: Organisms/strains | ||
| 129S2/SvPasCrl mice (129Sv) | Charles River | Cat#287 |
| Balb/cByJ (Balb/c) | Charles River | Cat#627 |
| NZBWF1/J | Jackson Labs | Cat#100008 |
| Oligonucleotides | ||
| Primers for qPCR, see Table S1 | This paper | |
| Recombinant DNA | ||
| pUNO1-hTLR07-HA3x | InvivoGen | Cat#puno1hahtlr7 |
| pUNO1-hTLR08-HA3x | InvivoGen | Cat#puno1ha-htlr8 |
| Software and algorithms | ||
| GraphPad Prism (version 9.2.0) | GraphPad | https://www.graphpad.com |
| FlowJo (version 8.8.7) | TreeStar | https://www.flowjo.com |
| Excel XL fit 5.0 with XLfit add-in (version 5.2.0) | Microsoft/IDBS | https://www.idbs.com/xlfit |
| edgeR Bioconductor package | Robinson et al.61 | |
| limma | Ritchie et al.62 | |
| fgsea package | Korotkevich et al.63 | |
| MSigDB | Liberzon et al.64 | |
| Other | ||
| SpectraMax 340PC | Molecular Devices | N/A |
| SpectraMax Plus 384 microplate reader | Molecular Devices | N/A |
| Envision | Perkin Elmer | N/A |
| LSR Fortessa flow cytometer | BD Biosciences | N/A |
| LSRII flow cytometer | BD Biosciences | N/A |
| Liquid chromatography coupled to mass spectrometry (LC-MS/MS). | Novartis | N/A |
| ViiA 7 Real-Time PCR System | Applied Biosystems | N/A |
| HITACHI M40 Clinical Analyzer | AxonLab | N/A |
| XT Discovery | Ventana | N/A |
| HT200 Nanozoomer | Hamamatsu | N/A |
| QuantStudio 12K flex instrument and software | Thermo Fisher Scientific | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tobias Junt (tobias.junt@novartis.com).
Materials availability
Requests for MTAs for MHV370 should be directed to the lead contact.
Experimental model and study participant details
Human subjects
For in vitro studies, fresh human blood was collected from healthy donors, from which no additional details are available, with informed consent (Santémed Gesundheitszentrum AG Basel, Switzerland). Buffy coats were obtained from healthy donors, from which no additional details are available, from Interregionale Blutspende (IRB) SRK Bern.
Sera were obtained from SLE patients or healthy volunteers with informed written consent in non-interventional clinical trials BASICHR0003 (University Hospital Basel, Switzerland), PJMR0012114 (Synexa Life Sciences, Bellville, South Africa), or CCHMC IRB # 01-12-2X (Cincinnati Children’s Hospital, Cincinnati, OH). Samples were collected in an anonymized fashion. All human research was conducted in accordance with the declaration of Helskini, and after approval by the respective institutional Ethic Boards: For BASICHR0003, Ethikkommission Nordwest-und Zentralschweiz; for PJMR0012114: South African Medical Association Research Ethics Committee (SAMAREC); for IRB # 01-12-2X: Cincinnati Children’s Hospital Medical Center Institutional Review Board.
Animals
Female mice were obtained from Charles River (129/Sv, Freiburg, Germany; Balb/c, Saint-Germain-sur-l’Arbresle, France, both 6–8 week old). Female 12-week-old NZB/W F1 mice (NZBWF1/J) were obtained from Jackson Labs (Bar Harbor, ME). All mice were housed under specific pathogen free conditions with a 12/12h light/dark cycle and ad libitum access to water and food. For acute cytokine release, chronic TLR7 activation and TMPD experiments, animals were randomized into age-and sex-matched groups of n = 5 based on weight. For the NZB/W F1 experiment, randomization strategy based on proteinuria in groups of n = 14 is stated below. Cohort sizes of mice to achieve statistically significant results were calculated based on an a priori power analysis. We calculated power and sample sizes required based on pilot experiments. The NZB/W F1 experiment was conducted at two different sites (Basel, Switzerland and La Jolla, CA) to ensure reproducibility. All animal studies were approved by the Veterinary Office Basel Stadt or the Institutional Animal Care and Use Committee of Novartis Pharma, La Jolla, CA. They were performed under strict adherence to Swiss law for animal protection or the Animal Welfare Act, and AAALAC requirements.
Cell lines
TLR-transfected HEK293 reporter cell lines (from female human fetus) were generated and cultured as described previously.19 Briefly, pUNO1-hTLR7-HA3x (InvivoGen, puno1ha-htlr7) and pUNO1-hTLR8-HA3x (InvivoGen, puno1ha-htlr8) were stably expressed in HEK293-NFkB-luciferase reporter cells (Promega). Cells were maintained in DMEM, high glucose, GlutaMAX supplement (Thermo Fisher Scientific) with 10% FBS and 1% Penicillin/Streptomycin (all Thermo Fisher Scientific), in presence of 5 mg/mL puromycin and 5 mg/mL blasticidin (both InvivoGen). Assays were run without antibiotics and in presence of 3% FBS.
Ramos Blue B reporter cells (from white male human 3-year-old donor) were obtained from InvivoGen under license (Toulouse, France) and cultured in IMDM/GlutaMAX (Thermo Fisher) supplemented with 10% FBS, 100 μg/mL Normocine (InvivoGen), 50 U/mL-50 μg/ml Pen-Strep (Thermo Fisher).
Human PBMC isolation
Fresh human blood, collected in S-Monovette Heparin tubes (Sarstedt), was obtained from healthy individuals. PBMCs were prepared by diluting blood 1:1 with phosphate-buffered saline (PBS) and transferred to pre-prepared Leucosep tubes (Greiner Bio-one) containing 15 mL of LSM1077 (Corning). The PBMC layer was carefully removed from the plasma/separation medium interface following centrifugation (800g, 20 min at 22°C without brake). PBMCs were washed with PBS, centrifuged (400g, 10 min at 22°C), and resuspended in growth media (RPMI1640+GlutaMAX-I supplemented with 0.05 mM 2-mercaptoethanol, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 5% v/v FBS).
Method details
Synthesis of MHV370
The synthesis scheme of MHV370 is shown in Figure S1. Synthesis of (4-((tert-butoxycarbonyl)amino)bicyclo[2.2.2]octan-1-yl)methyl 4-(trifluoromethyl)benzenesulfonate (1): To the mixture of tert-butyl (4-(hydroxymethyl)bicyclo[2.2.2]octan-1-yl)carbamate (1.021 g, 4 mmol), 4-(trifluoromethyl)benzene-1-sulfonyl chloride (1.566 g, 6.40 mmol) and DCM (10 mL) was added triethylamine (1.115 mL, 8.00 mmol) and DMAP (0.049 g, 0.400 mmol) at room temperature (rt). The resulting mixture was stirred at rt overnight. LC/MS indicated the reaction was complete: mainly two peaks, product peak with m/z 408 (M + H-56) and intermediate peak (formed from SM2 and DMAP) with m/z 331. Aqueous work up followed by ISCO purification (EtOAc/hexane) to get (4-((tert-butoxycarbonyl)amino)bicyclo[2.2.2]octan-1-yl)methyl 4-(trifluoromethyl)benzenesulfonate (1) as a white solid (1.75g, 94%). 1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 8.4 Hz, 2H), 8.06 (d, J = 8.4 Hz, 2H), 6.39 (s, 1H), 3.74 (s, 2H), 1.75–1.57 (m, 6H), 1.42–1.26 (m, 15H). MS (M + H+-56): 408.0.
Synthesis of tert-butyl (4-((3-methyl-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (2): A suspension of 3-methyl-1H-pyrazolo[4,3-c]pyridine (2.26 g, 16.97 mmol), in DMSO (68 mL) was treated with (4-((tert-butoxycarbonyl)amino)bicyclo[2.2.2]octan-1-yl)methyl 4-(trifluoromethyl)benzenesulfonate (1) (7.87 g, 16.97 mmol) and cesium carbonate (11.06 g, 33.9 mmol). The mixture was heated to 120°C for 18 h to complete, before being cooled to rt and diluted in ethyl acetate and water. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified on a 120 g silica gel column using 0–80% ethyl acetate in hexane and extended to 80% ethyl acetate in hexane to afford 3.63 g (9.79 mmol, yield: 58%) of tert-butyl (4-((3-methyl-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (2). (Further extension of the gradient to 100% ethyl acetate resulted in the elution of 903.4 mg (2.44 mmol, yield: 14%) the regioisomer by-product tert-butyl (4-((3-methyl-2H-pyrazolo[4,3-c]pyridin-2-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate.)
1H NMR (400 MHz, Methanol-d4) δ 9.07 (s, 1H), 8.37 (d, J = 6.1 Hz, 1H), 7.60 (d, J = 6.2 Hz, 1H), 6.18 (broad s, 1H), 4.17 (s, 2H), 2.71 (s, 3H), 1.87 (dd, J = 10.0, 5.9 Hz, 6H), 1.66 (dd, J = 10.0, 5.9 Hz, 6H), 1.47 (s, 9H).). MS (M + H+): 371.2.
Synthesis of tert-butyl (4-((3-methyl-2H-pyrazolo[4,3-c]pyridin-2-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (3): Tert-butyl (4-((3-methyl-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (2) (3.6267 g, 9.79 mmol) was dissolved in EtOH to a concentration of 0.05 M. An H-Cube instrument fitted with a 10% Pd/C, 70mm catcart and equilibrated with EtOH at 1 mL per minute at 10 bar of hydrogen for 10 min. The solution of 2 was then passed through the instrument at 1 mL per minute. LC-MS analysis was consistent with >95% conversion. The reaction solution was concentrated and loaded on a 120 g silica gel column using 0–100% IPA in DCM with 1% ammonia as modifier and then extended to 100% IPA with 1% ammonia as modifier to elute tert-butyl (4-((3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (3) 2.53 g (6.75 mmol, yield: 69%) 1H NMR (400 MHz, CD3OD) δ 3.73 (s, 2H), 3.65 (s, 2H), 3.04 (t, J = 5.8 Hz, 2H), 2.65 (t, J = 5.9 Hz, 2H), 2.12 (s, 3H), 1.87–1.74 (m, 6H), 1.61–1.47 (m, 6H), 1.39 (s, 9H). MS (M + H+): 376.
Synthesis of tert-butyl (4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (4): To a pressure flask containing tert-butyl (4-((3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (3) (0.565 g, 42.5 mmol) was added 4-bromo-1,6-dimethyl-1H-pyrazolo[3,4-b]pyridine (0.94g, 2.5 mmol), cesium carbonate (1.63 g, 5.0 mmol),Pd2(dba)3 (0.057g, 0.062 mmol), RuPhos (0.14 g, 0.3 mmol) and THF (25 mL). The mixture was heated at 80°C for 18 h to complete, then cooled to rt. The mixture was diluted in ethyl acetate and water. After partition, the aqueous layer was extracted with EtOAc once more. Both organic layers were combined, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified via flash chromatography using 0–100% B/A (A = heptane; B = 25% ethanol in ethyl acetate) to elute tert-butyl (4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (4) 1.28 g (2.35 mmol, yield: 94%). 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1H), 6.27 (s, 1H), 4.49 (s, 2H), 4.38–4.25 (m, 1H), 4.11 (s, 3H), 3.94 (t, J = 5.5 Hz, 2H), 3.70 (s, 2H), 2.87 (t, J = 5.6 Hz, 2H), 2.62 (s, 3H), 2.25 (s, 3H), 1.89–1.75 (m, 6H), 1.58 (m, 6H), 1.42 (s, 9H). MS (M + H+): 520.
Synthesis of 4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-amine (5): To a 500 mL round bottom flask containing tert-butyl (4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamate (4) (4.86 g, 9.35 mmol) was added methanol (6 mL), then 4N HCl in dioxane (23.5 mL, 94 mmol). The mixture was stirred at rt for 18 h then concentrated in vacuo. The residue was treated with isopropanol portionwise at 70°C to get all solid dissolved. The solution was cooled naturally to rt and aged for 18 h. Then the solid was filtered and the filtrate was concentrated and the crystallization process repeated. Both batches were combined and dried under vacuum at 40°C for 18 h to afford of 4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-amine 5 as HCl salt: 4.54 g (9.22 mmol, yield: 99%). 1H NMR (400 MHz, CD3OD) δ 8.51 (s, 1H), 6.88 (s, 1H), 5.06–4.92 (m, 2H), 4.24 (s, 2H), 4.11 (s, 3H), 3.97 (s, 2H), 3.10 (t, J = 5.6 Hz, 2H), 2.69 (s, 3H), 2.38 (s, 3H), 1.88–1.59 (m, 12H); MS (M + H+): 420.3. Ambersep 900 OH (17 mL, 0.8 meq/mL, prewashed with 60 mL of MeOH) was added into a solution of 4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-amine as HCl salt (5-HCl) (1.53 g, 2.7 mmol) in MeOH (100 mL). The mixture was stirred at rt for 1 h then filtered, washed with 50 mL of MeOH and concentrated. The crude product was added (by solid loading) to a 12 g silica gel column and was eluted with 2–9% MeOH (containing small amount of ammonia) in DCM. Collected fractions and concentrated to give the product, 4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-amine, as the free base (5): 769 mg (1.80 mmol, yield: 66.5%) 1H NMR (400 MHz, CD3OD) δ 8.15 (s, 1H), 6.45 (s, 1H), 4.54 (s, 2H), 4.00 (s, 3H), 3.97 (t, J = 5.6 Hz, 2H), 3.73 (s, 2H), 2.90 (t, J = 5.6 Hz, 2H), 2.55 (s, 3H), 2.23 (s, 3H), 1.55 (s, 12H); MS (M + H+): 420.3. Elemental Analysis (C, H, N) calcd: 68.70, 7.93, 23.37; found: 68.18, 7.99, 22.94.
Synthesis of (S)-N-(4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)morpholine-3-carboxamide MHV370: To a mixture of 5 (5.00 g, 10.73 mmol), (S)-4-(tert-butoxycarbonyl)morpholine-3-carboxylic acid (2.53 g, 10.94 mmol) and DIEA (2.81 mL, 16.09 mmol) in DCM (Volume: 107 mL) was added HATU (4.24 g, 11.15 mmol) at 0°C then stirred at rt for 60 min. The mixture was washed with saturated NH4Cl, then saturated sodium carbonate. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was loaded on a 40 g silica gel column and eluted using 0–100% ethyl acetate/EtOH (3:1) in hexane to afford tert-butyl (S)-3-((4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)carbamoyl)morpholine-4-carboxylate (7.50 g, 11.85 mmol, 111% yield).
This material was dissolved in 4N HCl in dioxane (53.6 mL, 215 mmol) and methanol (6 mL). The mixture was stirred at rt overnight and then concentrated. The residue was concentrated, redissolved in ethyl acetate and washed with 1N NaOH. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was loaded on a 40 g silica gel column using 0–100% IPA in DCM with 3% NH3 as modifier to elute the product. One pure fraction was selected, concentrated and then suspended in ethyl ether. The solid was filtered and wash twice with ethyl ether. The solid was dried under high vacuum at 90°C for 2 days to afford (S)-N-(4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)morpholine-3-carboxamide (MHV370) (5.77 g, 10.51 mmol, 98% yield); 1H NMR (400 MHz, Methanol-d4) δ 8.52 (s, 1H), 7.92 (s, 1H), 6.88 (s, 1H), 5.06–4.91 (m, 2H), 4.25 (s, 2H), 4.19–4.09 (m, 4H), 4.03–3.89 (m, 4H), 3.78–3.56 (m, 4H), 3.22 (ddd, J = 12.9, 11.2, 3.7 Hz, 1H), 3.17–3.05 (m, 2H), 2.70 (s, 3H), 2.41 (s, 3H), 1.93 (dd, J = 8.9, 5.0 Hz, 6H), 1.72–1.54 (m, 6H). MS (M + H+): 533.4. MHV370 is an active clinical candidate, and it was used at >98% purity.
HEK cell reporter cell line assays
TLR-transfected HEK293 reporter cell lines were treated with increasing concentrations of MHV370 or vehicle control (0.25% v/v DMSO) in culture medium for 60 min at 37°C and 5% CO2. After stimulation with R848 for 6 h (0.3 μM for TLR7 and 4 μM for TLR8), luciferase activity was assessed in supernatants with Bright-Glo assay (Promega) and quantified using an Envision (Perkin Elmer).
Ramos Blue B cell reporter cell line assays
Cells were treated with increasing concentrations of MHV370 or vehicle control (0.25% v/v DMSO) in culture medium for 30 min at 37°C and 5% CO2. Cells were stimulated with Pam3CSK4 (10 μg/mL), FSL-1 (1 μg/mL), PolyIC, (0.3 μg/mL with 25 μg/mL DOTAP), CL307 (9 mM), R848 (3 μM), ODN2006 (0.1 μM), Tri-DAP (10 μg/mL) and TNF (0.1 μg/mL) for 20 h at 37°C and 5% CO2. Secreted alkaline phosphatase was quantified in supernatants using QUANTI-Blue using a SpectraMax Plus 384 microplate reader (Molecular Devices, Sunnyvale, CA, USA). RNA was extracted from Ramos Blue B cells using a RNeasy Mini RNA isolation kit (Qiagen). Isolated RNA was reverse transcribed using Super-Script III First-Strand synthesis Super-Mix (Invitrogen). Quantitative Real Time PCRs (qRT-PCRs) were performed on a ViiA 7 Real-Time PCR System (Applied Biosystems) using 10 ng cDNA, TaqMan Gene Expression Master Mix (Applied Biosystems) and TaqMan probes (see Table S1). The relative expression of each gene was analyzed in duplicates using the 2-ΔΔCt method, with fold change = 2-Δ(ΔCt), where ΔCt = Cttarget – CtrRNA 18S control and Δ(ΔCt) = ΔCtsample – ΔCtcontrol sample and means of duplicates were calculated.
Human PBMC assays
PBMCs were pre-incubated for 30 min with either MHV370 or HCQ, then stimulated with TLR agonists at doses described within65 or with R848 (3 μM), TL8-506 (40 nM). CXCL10 was quantified by AlphaLisa (PerkinElmer), IL-1β and IL-6 by Homogeneous Time Resolved Fluorescence (HTRF, CisBio, Bedford, MA, USA) and IL-12p40 by DuoSet ELISA (R&D systems). Cell viability was not impaired across the tested dose range of MHV370, as assessed by CellTiter-Glo assay (Promega). For a Schild plot analysis, PBMCs were treated with the indicated concentrations of MHV370 for 30 min prior to stimulation with increasing concentrations of R848 for 20 h at 37°C and 5% CO2. The Schild plot was constructed using GraphPad Prism (GraphPad).
Human and murine whole blood assays
Fresh human blood or blood from 129/Sv mice was collected into citrate S-Monovette 9NC tubes (Sarstedt). Blood was diluted 1:1 with RPMI1640 and incubated with increasing concentrations of MHV370 or vehicle control (0.25% v/v DMSO final per well) in RPMI1640 for 30 min at 37°C and 5% CO2. Blood was stimulated with various TLR agonists or with R848 (3 μM) and cytokines quantified as described.65 Cell viability was assessed by ATPlite Luminescence assay (PerkinElmer) and not impaired across the tested dose range of MHV370.
Human pDC assay
pDCs were isolated from PBMCs using the EasySep Human Plasmacytoid DC Enrichment Kit (Stemcell Technologies, Vancouver, Canada). Isolated pDCs were resuspended in complete RPMI1640 containing 10 ng/mL recombinant human IL-3 (PeproTech, London, UK). Cells were incubated with MHV370 or vehicle control (0.25% v/v DMSO) for 30 min prior to stimulation with ssRNA40 (10 μg/mL with 25 μg/mL DOTAP) or ODN2216 (0.3 μM) for 20 h at 37°C and 5% CO2. IFNα was quantified from supernatants by AlphaLisa, and cell viability was not impaired for the entire dose range of MHV370, as assessed by CellTiter-Glo assay.
Human monocyte assay
Monocytes were isolated from PBMCs using the EasySep Human monocyte Enrichment Kit (Stemcell Technologies). They were resuspended in complete RPMI1640 and incubated with MHV370 or vehicle control (0.25% v/v DMSO) for 30 min prior to stimulation with ssRNA40 (1 μg/mL with 10 μg/mL DOTAP), R848 (3 μM) or TL8-506 (40 nM) for 20 h at 37°C and 5% CO2. Cytokines were quantified from supernatants using cytokine specific HTRF kits (Cisbio) and cell viability was not impaired across the tested dose range of MHV370 as assessed by CellTiter-Glo.
Human B cell assay
Human B cells were enriched from PBMCs using the EasySep human B cell enrichment kit (Stemcell Technologies). B cells were incubated with increasing concentrations of MHV370 or vehicle control (0.25% v/v DMSO) in assay medium (RPMI1640, 10% ultralow IgG FCS, 1% Penicillin/Streptomycin, 1% Insulin-Transferrin-Selenium) for 1 h at 37°C and 5% CO2. For stimulation, cells were incubated with R848 (0.5 μM) in assay medium or in medium alone. After 48 h at 37°C and 5% CO2, IL-6 was quantified from supernatants using the human IL-6 DuoSet ELISA (R&D systems). After 72 h, IgM was quantified in the supernatants by custom ELISA as follows: Plates were coated with 2.6 μg/mL goat anti-human IgM (Jackson Immunoresearch) in PBS, washed with wash buffer (PBS supplemented with 0.05% v/v Tween 20), blocked with wash buffer containing 1 mM EDTA for 1h, and washed again. B cell supernatants or human IgM standard (Sigma-Aldrich) were added for 1 h. Plates were washed, 0.8 μg/mL peroxidase-coupled goat anti-human IgM (Jackson Immunoresearch) were added in wash buffer containing EDTA for 1 h. The assay was developed using a peroxidase substrate kit (Bio-Rad, Hercules, CA). Absorbance was measured using a SpectraMax 340PC (Molecular Devices).
For human B cell assays in whole blood, diluted and citrate-anticoagulated blood was incubated with increasing concentrations of MHV370 or vehicle control (0.25% DMSO) for 1 h prior to stimulation with R848 (0.2 μg/mL) for 30 min (pAkt) or overnight (CD69) at 37°C and 5% CO2. For pAkt staining, cultures were incubated with pre-warmed Lyse-Fix buffer for 15 min at 37°C, then with Phosflow Perm/Wash buffer (BD) for 15 min at room temperature, then stained with fluorescent anti-CD20 and anti-pAkt. Cells were washed, resuspended in FACS buffer and analyzed. For CD69 staining, cultures were directly stained with fluorescent anti-CD19 and anti-CD69. Cells were incubated with pre-warmed Lyse-Fix Buffer (BD) for 15 min at 37°C, washed in FACS buffer (PBS w/o Ca2+/Mg2+/2% FBS/5 mM EDTA) and analyzed using an LSRII flow cytometer (BD Biosciences). MFI for CD69 or pAkt on B cells was analyzed using FlowJo (version 8.8.7, TreeStar, Ashland, OR, USA). Background was subtracted and MFI values standardized to mean MFI of R848 stimulation (100%). To study the effect of MHV370 on B cell receptor-mediated activation, a serial 3-fold dilution series of MHV370 (starting at 16 μM) was added to heparinized blood for 1.5 h at 37°C, and cells were activated with 50 μg/mL anti-human IgM for 20 min at 37°C. Blood was incubated with Lyse/Fix then Perm/Wash buffer (BD), then stained with anti-pAkt(Ser437) and anti-CD20, washed in Perm/Wash, resuspended and analyzed on an LSRII flow cytometer (BD).
Human neutrophil assay
Fresh human blood was collected into Vacutainer LH 170 I.U Plus Blood Collection Tubes (BD Biosciences) and neutrophils were isolated by Ficoll (Cytiva) gradient centrifugation. Erythrocytes were lysed using ammonium chloride-potassium buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) and pellets washed in PBS (containing 0.4% w/v BSA and 1 mM EDTA) and resuspended in RPMI1640/GlutaMAX/5% FBS. Cells were incubated with increasing concentrations of MHV370 or vehicle control (0.1% v/v DMSO) for 30 min prior to stimulation with R848 (10 μM), TL8-506 (1 mM) or Phorbol-myristate-acetate (PMA, 50 ng/mL) for 30 min at 37°C and 5% CO2. Cell viability was assessed by CellTiter-Glo. ROS was detected with the addition of 2 μM dihydrorhodamine (DHR) for 15 min at 37°C. Fluorescence was measured using an LSR Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo (TreeStar). Signals were normalized to vehicle treated samples.
Acute cytokine release in vivo assay
MHV370 in MC/Tween (0.5% methylcellulose/0.5% Tween-80) or vehicle alone was administered p.o. to 129/Sv mice (5 per group). At 1 h, mice were injected i.v. with 20 μg R0006 or 20 mg CpG1585, each pre-complexed with 140 μg DOTAP in Hanks Balanced Salt solution (HBSS). At 3 h, blood was withdrawn into EDTA Microvettes (Sarstedt) and plasma TNF and IFNα quantified using a mouse IFNα Platinum ELISA (Invitrogen) and a DuoSet TNF ELISA (R&D Systems), as single technical replicate per mouse. MHV370 serum concentrations were quantified by liquid chromatography coupled to mass spectrometry (LC-MS/MS).
Chronic TLR7 activation in vivo assay
129/Sv mice (5 per group) were treated with daily R848 i.p. (2.5 mg/kg) for 14 days. From day 7 onward, one group received oral doses of 15 mg/kg MHV370 b.i.d., one group vehicle. At day 14, both groups were compared to naive 129/Sv mice. Readouts were spleen weight, FACS for activated B cells (B220. CD69) in blood, serum ELISA for CXCL10 (Invitrogen), CCL2 (Biolegend, San Diego, CA), TNF (R&D Systems), all as single technical replicate per mouse, and transcriptomic profiling of blood via amplicon sequencing (Ion AmpliSeq Transcriptome Mouse Gene Expression Kit, Thermo Fisher Scientific).
Ex vivo PD assay
Citrate-anticoagulated (1:4 with a 4% citrate solution) blood was diluted 1:1 in RPMI1640, incubated for 24 h with 0.04 μg/mL R848 in RPMI1640 or RPMI1640 alone at 37°C and 5% CO2. Erythrocytes were lysed, samples stained for 30 min with Aqua viability dye solution (Thermo Fisher Scientific) in PBS at 4°C, then with anti-CD19 and anti-CD69. MFI for CD69 on CD19+ B cells was analyzed as single technical replicate per mouse using an LSRII flow cytometer (BD Biosciences) and FlowJo. MFI results were expressed as percentages using the following formula: MFI [(x - unstimulated control)/(stimulated control - unstimulated control)] x 100%.
Mouse TMPD peritonitis
Female Balb/c mice (5 per group) were treated p.o. with the indicated doses of MHV370 in 0.5% MC/0.5% Tween80 or vehicle alone b.i.d. starting on day −1. On day 0, mice were injected i.p. with 0.5 mL Tetramethylpentadecane (TMPD). Naive mice were included as controls. At termination, peritoneal cavities were washed with HBSS containing 2% FBS. Cells were pelleted and CCL2 quantified in supernatants by ELISA (BioLegend, San Diego, CA, USA). To quantify peritoneal inflammatory monocytes, an aliquot was stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific) in PBS, then stained with anti-CD11b, anti-Ly6C and anti-Ly6G. Samples were acquired on an LSR Fortessa and frequencies of CD11b+Ly6ChighLy6G-monocytes analyzed using FlowJo v.9.1.2 software (TreeStar). CD69 on CD19+ B cells was analyzed as an ex vivo PD marker (see above).
ISG expression in blood and peritoneal lavages
RNA was extracted from citrate-anticoagulated (1:4) terminal blood or from peritoneal cells using a mouse RiboPure blood RNA isolation kit (Ambion, Thermo Fisher Scientific) and RNeasy Micro Kit (Qiagen), respectively. Isolated RNA was reverse transcribed using Super-Script III First-Strand synthesis Super-Mix (Invitrogen). Quantitative Real Time PCRs (qRT-PCRs) were performed on a ViiA 7 Real-Time PCR System (Applied Biosystems) using 10 ng cDNA, TaqMan Gene Expression Master Mix (Applied Biosystems) and TaqMan probes (see Table S1). The relative expression of each gene was analyzed in triplicates using the 2−ΔΔCt method, with fold change = 2-Δ(ΔCt), where ΔCt = Cttarget – CtHPRT control and Δ(ΔCt) = ΔCtsample – ΔCtcontrol sample and means of triplicates were calculated. The global induction of a panel of ISGs was calculated by averaging 2−ΔΔCt values across all ISG transcripts for each individual naive mouse, averaging ISG expression levels within the naive group and normalized to 100%. Averages of 2−ΔΔCt values across all ISG transcripts from all individual mice were standardized to this value. To correlate ISG expression to an ex vivo PD marker, ISG expression values were plotted against expression of CD69 on B cells following ex vivo stimulation (see above).
Mouse NZB/W F1 model of lupus
Mice were included into the study at 23 weeks of age, i.e. when a majority had detectable proteinuria. Mice were randomized into groups of 14 mice, with average proteinuria of 0.1–0.5 mg/mL. Body weight was recorded twice weekly, proteinuria was recorded weekly using a quantitative Protein Assay (Bio-Rad) with BSA as standard.
Compound treatment
MHV370 was administered via food: MHV370, powdered rodent chow (Provimi Kliba, Kaiseraugst, Switzerland) and water were mixed, and food pellets were dehydrated for 24 h at 37°C. Compound-free control (vehicle) food was prepared in parallel. Groups of mice received 0.01% and 0.1% MHV370 in food or vehicle food, respectively. Throughout the experiment, mice received fresh food every two weeks. Daily food consumption per mouse (calculated by dividing food intake by number of mice per cage) and weight gain of mice were similar for all groups. Administration of MHV370 via food led to near-constant exposure levels in blood, 55 nM for 0.01% and 1424 nM for 0.1%, variable by feeding cycles. Ex vivo blood stimulation and analysis of B220+CD69+ B cells resulted in an ex vivo IC50 of 35 nM (Figure S4D). In mice receiving 0.1% MHV370 in food, blood exposures were always substantially higher, so that complete TLR7 inhibition is assumed throughout the experiment.
Termination criteria
Individual mice had to be terminated once they reached the pre-specified endpoint of the animal license, i.e. once mice had lost 20% of their BW within 10 days, or once a proteinuria level of ≥10 mg/mL was reached and confirmed 24–48 h later. The study was terminated once 50% of animals of the vehicle group had developed clinically relevant proteinuria (≥5 mg/mL).
PK measurements
At the indicated time points, blood was sampled into EDTA-coated tubes (Eppendorf) and terminal blood was collected by cardiac puncture using MITRA devices (Neoteryx LLC, Torrance, CA, USA). MHV370 concentrations in whole blood were determined by LC-MS/MS.
Ex vivo PD marker
Anticoagulated whole blood was diluted 1:1 in RPMI1640. For stimulation, 0.07 μg/mL R848 or medium was added at 37°C and 5% CO2. Blood was analyzed by flow cytometry for the MFI of CD69 on CD19+ B cells as described above.
Antinuclear antibody (ANA) measurements
At the indicated time points, mice were bled and serum isolated using Microtubes Z-gel (Sarstedt). Nunc Maxisorp plates were coated overnight at 4°C using PBS solutions of: 100 μg/mL salmon sperm DNA (Thermo Fisher Scientific), 10 μg/mL calf thymus histone (Sigma Aldrich), 1 U/well ribosomal P (Immunovision, Springdale, AK, USA), 1 U/well Smith antigen (Immunovision) or 20 μg/mL DWEYS peptide (Bachem, Bubendorf, Switzerland). DWEYS peptide coating supported detection of antibodies against NMDAR.
Plates were washed, blocked, prior to the addition of mouse serum dilutions of 1:100 or 1:300 in PBS/1% BSA for 2 h. Washed plates were incubated with HRP-conjugated anti-IgG-specific detection antibodies for 2 h (1:10′000). Washed plates were incubated with 100 μL/well of TMB substrate (BD Biosciences) and the reaction stopped by addition of 1 N HCl. The OD (450 nm) of samples was measured using a Spectramax M5 (Molecular Devices) and data expressed with background subtracted. Ro60 immunoglobulin titers were determined in 1:100 serum dilutions using an ELISA kit (Alpha Diagnostic, San Antonio, TX, USA) and reported as U/ml.
Measurement of CXCL13 in serum
At termination, CXCL13 was quantified from sera using the Quantikine ELISA kit (R&D systems, Minneapolis, MN, USA). Measurements were taken as single technical replicate per mouse.
Blood urea nitrogen (BUN) measurement
Blood BUN was measured with a HITACHI M40 Clinical Analyzer (AxonLab, Baden, Switzerland) using S Test Reagent Cartridges BUN (Hitachi Chemical, Chiyoda, Japan). BUN levels below detection limit (<0.5 mmol/L) were set to 0.5 mmol/L for graphical display (1/13 mice in 0.01% MHV370 group, 1/12 in 0.1% MHV370 group, 3/11 in 0.1% MHV370 advanced disease group)
Histology
Formalin fixed, paraffin embedded sections (FFPE, 3 μm) of kidneys of vehicle and 0.1% MHV370-treated animals were stained using the periodic acid Schiff (PAS) reaction and examined in a blinded fashion. For immunohistochemistry, sections were deparaffinized in xylene, rehydrated through graded ethanol and loaded on a Ventana Discovery XT stainer (Roche Diagnostics AG, Rotkreuz, Switzerland). After heat induced epitope retrieval of the slides with cell conditioning solution (CC1, Roche Diagnostics AG), slides were incubated with primary antibodies diluted in Ventana Antibody Diluent (Roche Diagnostics AG) and fixed with 0.05% glutaraldehyde in 0.9% NaCl. For detection, slides were incubated with dilutions of biotinylated secondary antibodies in Ventana Antibody Diluent and subsequently developed with streptavidin DABMap Kit (Roche Diagnostics). Primary/secondary antibody pairs and respective dilutions were as follows: (1) For IgG stains: Goat-anti-mouse IgG (Nordic-MUbio, 1:1000)/donkey-anti-goat IgG-biotin (Jackson Immunoresearch, 1:500); (2) for CD138 stains: Rat-anti-mouse CD138 (BD Biosciences, 1:400, 1.25 μg/mL)/rabbit-anti-rat IgG-biotin (Jackson Immunoresearch, 1:200); (3) for B220/CD45R stains: rat-anti-mouse B220/CD45R (Bio-Rad, 1:2000, 0.5 μg/mL)/rabbit-anti-rat IgG-biotin (Jackson Immunoresearch, 1:200); (4) for CD3 stains: rabbit-anti-mouse CD3 (Thermo Fisher Scientific, 1:40, 5 μg/mL)/goat-anti-rabbit IgG-biotin (Jackson Immunoresearch, 1:1000). Slides were counter-stained with Ventana hematoxylin II and bluing reagent (Roche Diagnostics AG), washed, dehydrated and mounted. Digital images of sections were recorded using Aperio ImageScope version 12.3.2.8013 software (Leica Biosystems, Wetzlar, Germany). Severity of glomerulopathy (mesangial proliferation and IgG deposition) and inflammation as well as lymphoid cell infiltration (CD45R + B cells and CD138+ plasma cells separately) were defined on a scale of 0–5 (0 – no pathology, 1 – minimal/low, 2 - slight, 3 - moderate, 4 – marked/high, 5 – severe changes/very high number).
The study in the NZB/W F1 model was repeated at a different site (San Diego). Differences in protocol were: (1) Proteinuria measurement by a dipstick (Uristix, Bayer) rather than on a continuous scale, (2) twice daily dosing of MHV370 instead of continuous food dosing, (3) inclusion at a proteinuria score of 2, which corresponds to 0.3–1 mg/mL, i.e. higher than above. Body weight and proteinuria were measured weekly in NZB/W F1 mice, beginning at 20 weeks of age. Animals were randomized at a proteinuria score of 2 to experimental groups of n = 15. (Score 0: no proteinuria, 1: trace, 2: >0.3 mg/mL, 3: >1 mg/mL, 4: >3 mg/mL, 5: >20 mg/mL). Proteinuria was measured throughout the experiment. Animals were taken out of the experiment once body weight loss reached 20% or an animal had two consecutive scores of 5, and the experiment was terminated when 50% of animals of any group were taken out of the experiment. If an animal was removed from an experiment, the last proteinuria score measured was used for the remainder of the study for data analysis. Animals were treated p.o. with the indicated doses of MHV370 b.i.d. in MC/Tween throughout the experiment. At termination, the left kidney was placed into 10% Neutral buffered formalin (Advantik) for 24 h. Kidneys were washed twice with water and placed into 70% ethanol/water until paraffin embedding. 5 mm serial sections were cut and stained with either hematoxylin/eosin, anti-IgM (Rabbit anti mouse IgM 1:10,000 life technologies) or anti-IgG (Alexa Fluor 488 goat-anti-mouse IgG, 1:100, H&L Jackson Immuno) on an XT Discovery (Ventana). Sections were mounted in immu-mount (Thermo-Fisher); IgM-stained sections were Scanned on an HT200 Nanozoomer (Hamamatsu). For quantification of IgM or IgG staining, the ratios of IgM or IgG positively stained areas to total section area were determined on ten sections per mouse, and the average ratio was recorded.
Stimulation of PBMCs with lupus patient sera
Fresh human PBMCs were incubated with MHV370, HCQ or vehicle (0.1% v/v DMSO) at indicated concentrations in RPMI1640 for 30 min at 37°C and 5% CO2. Cells were stimulated with SLE or HV serum pre-complexed with cellular necrotic extract prepared from U-937 cells as described31 for 20 h at 37°C and 5% CO2. Quantification of IFNα in supernatants was performed by AlphaLisa.
Expression of ISGs from human PBMCs
RNA from stimulated PBMCs were extracted using RNeasy kit (Qiagen). RNA (100 ng) was reverse transcribed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. Quantitative Real Time PCRs (qRT-PCRs) were performed using 10 ng cDNA, TaqMan Fast Universal PCR Master Mix (Applied Biosystems) for a panel of human ISGs (see Table S1) and using QuantStudio 12K flex instrument and software (Thermo Fisher Scientific). The relative expression change of each gene was performed from biological replicates (see legends) using the 2−ΔΔCt method as described with Ramos B cells. The global induction of a panel of ISGs was determined by normalizing the relative induction of each individual ISG to 100%.
Autoantibody titers in lupus patient sera
Detection of autoantibody IgG titers in patient sera was performed using pre-coated plates (ANAcombi: RNP-70, Sm, RNP/Sm, SS-A, SS-B, Scl-70, Centromer B, Jo-1; Orgentec, Mainz, Germany) or Poly-L-lysine pre-coated maxisorp black plates (Thermo Fisher) for dsDNA. Patient or HV sera were pre-diluted (1:250 or 1:50 with serial dilutions for dsDNA including reference controls) with reagent buffer (1% BSA, 0.05% v/v Tween 20 in PBS) in 50 μL and added to plates. After 2 h on an orbital shaker, plates were washed (PBS/0.05% v/v Tween 20) and biotinylated anti-human IgG (1:5000, 50 μL/well) was added for 2 h. Plates were washed four times prior to addition of Streptavidin-Eu antibody (1:1000, 50 μL/well, PerkinElmer) for 1 h. After washing, plates were analyzed using an EnVision Multiplate reader following addition of Enhancement solution (PerkinElmer) for 15 min in the dark. Qualitative titers of each serum were calculated according to the manufacturer’s protocol (Orgentec) with scoring index: negative <1.0, borderline 1.0–1.2 and positive >1.2. For dsDNA IgG titers, quantification based on scoring: negative <20 IU/mL, borderline 21–100 IU/mL and positive >100 IU/mL (IgG).
Quantification and statistical analysis
Cellular profiling data were analyzed using Excel XL fit 5.0 (Microsoft) with XLfit add-in (IDBS; version 5.2.0) or using GraphPad Prism. Specific cytokine concentrations were determined following extrapolation from standard curves of the appropriate reference cytokines supplied with kits. For each agonist, EC90 values were experimentally determined to assess individual IC50 values. IC50 values were calculated by nonlinear regression after fitting of curves to the experimental data. Statistical analysis was performed using GraphPad Prism as indicated on Figure legends.
Analysis of amplicon sequencing data
Raw gene counts obtained from blood of mice subjected to chronic TLR7 activation were first filtered to select genes with at least 10 counts per million in at least 4 samples. These were normalized using the weighted trimmed mean of M-values using the edgeR Bioconductor package61 and used for a differential expression analysis using limma.62 Mice treated with R848 and MHV370 were compared to mice treated with R848 plus vehicle and naive mice. Genes with an adjusted p value (false discovery rate) < 0.01 and an absolute fold change >2 were considered differentially expressed. Gene set enrichment analysis was performed using the fgsea package63 and the Hallmark and C2 Canonical Pathways gene set collections from MSigDB.64 Terms with a false discovery rate adjusted p value (false discovery rate) < 0.01 were considered significant.
Additional resources
The data presented in this manuscript led to the initiation of a clinical Ph2 trial for MHV370 (clinicaltrials.gov identifier NCT04988087).
Acknowledgments
Thanks to Patrick Schmutz, Hélène Marsot, Alice Terrier, and Kathrin Wagner for excellent technical support. This study was funded by Novartis Pharma AG.
Author contributions
Conceptualization, S.H., T.J., P.G., and T.S.; methodology, S.H., C.A., G.C.-Z., T.J., and C.M.-S.; investigation, C.A., G.C.-Z., L.B., D.B., T.B., M.C., E.F., R.F., F.L., C.M.-S., F.R., J.S.R., G.R., T.S., and S.Z.; validation, S.H. and T.J.; formal analysis, E.F., T.J., and S.H.; resources: S.A., B.B., C.B., H.I.B., J.D., D.K., P.M., B.N.-H., and G.W.; writing – original draft and review & editing, visualization, and supervision, S.H. and T.J.; project administration, P.L. and R.S.
Declaration of interests
All authors except S.A., B.B., H.I.B., J.D., D.K., P.M., F.R., and J.S.R. are current employees and shareholders of Novartis Pharma AG. MHV370 is described in patent WO2018047081.
Published: May 16, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101036.
Supplemental information
Data and code availability
-
•
All AmpliSeq data of this study have been deposited at Zenodo and are publicly available as of the date of publication. DOI is listed in the key resources table (https://doi.org/10.5281/zenodo.7573856). Microscopy data reported in this paper will be made available by the lead contact upon request.
-
•
All original code for analysis of AmpliSeq data have been deposited on Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table (https://doi.org/10.5281/zenodo.7575672).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Junt T., Barchet W. Translating nucleic acid-sensing pathways into therapies. Nat. Rev. Immunol. 2015;15:529–544. doi: 10.1038/nri3875. [DOI] [PubMed] [Google Scholar]
- 2.Lee Y.H., Choi S.J., Ji J.D., Song G.G. Association between toll-like receptor polymorphisms and systemic lupus erythematosus: a meta-analysis update. Lupus. 2016;25:593–601. doi: 10.1177/0961203315622823. [DOI] [PubMed] [Google Scholar]
- 3.García-Ortiz H., Velázquez-Cruz R., Espinosa-Rosales F., Jiménez-Morales S., Baca V., Orozco L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann. Rheum. Dis. 2010;69:1861–1865. doi: 10.1136/ard.2009.124313. [DOI] [PubMed] [Google Scholar]
- 4.Souyris M., Cenac C., Azar P., Daviaud D., Canivet A., Grunenwald S., Pienkowski C., Chaumeil J., Mejía J.E., Guéry J.C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018;3:eaap8855. doi: 10.1126/sciimmunol.aap8855. [DOI] [PubMed] [Google Scholar]
- 5.Lyn-Cook B.D., Xie C., Oates J., Treadwell E., Word B., Hammons G., Wiley K. Increased expression of Toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol. Immunol. 2014;61:38–43. doi: 10.1016/j.molimm.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Komatsuda A., Wakui H., Iwamoto K., Ozawa M., Togashi M., Masai R., Maki N., Hatakeyama T., Sawada K. Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2008;152:482–487. doi: 10.1111/j.1365-2249.2008.03646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown G.J., Cañete P.F., Wang H., Medhavy A., Bones J., Roco J.A., He Y., Qin Y., Cappello J., Ellyard J.I., et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605:349–356. doi: 10.1038/s41586-022-04642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen S.R., Shupe J., Nickerson K., Kashgarian M., Flavell R.A., Shlomchik M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Fairhurst A.M., Hwang S.H., Wang A., Tian X.H., Boudreaux C., Zhou X.J., Casco J., Li Q.Z., Connolly J.E., Wakeland E.K. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur. J. Immunol. 2008;38:1971–1978. doi: 10.1002/eji.200838138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savarese E., Steinberg C., Pawar R.D., Reindl W., Akira S., Anders H.J., Krug A. Requirement of Toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. 2008;58:1107–1115. doi: 10.1002/art.23407. [DOI] [PubMed] [Google Scholar]
- 11.Lee P.Y., Kumagai Y., Li Y., Takeuchi O., Yoshida H., Weinstein J., Kellner E.S., Nacionales D., Barker T., Kelly-Scumpia K., et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J. Exp. Med. 2008;205:2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deane J.A., Pisitkun P., Barrett R.S., Feigenbaum L., Town T., Ward J.M., Flavell R.A., Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guiducci C., Gong M., Cepika A.M., Xu Z., Tripodo C., Bennett L., Crain C., Quartier P., Cush J.J., Pascual V., et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 2013;210:2903–2919. doi: 10.1084/jem.20131044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrat F.J., Meeker T., Gregorio J., Chan J.H., Uematsu S., Akira S., Chang B., Duramad O., Coffman R.L. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005;202:1131–1139. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pawar R.D., Ramanjaneyulu A., Kulkarni O.P., Lech M., Segerer S., Anders H.J. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J. Am. Soc. Nephrol. 2007;18:1721–1731. doi: 10.1681/ASN.2006101162. [DOI] [PubMed] [Google Scholar]
- 16.Vlach J., Bender A.T., Przetak M., Pereira A., Deshpande A., Johnson T.L., Reissig S., Tzvetkov E., Musil D., Morse N.T., et al. Discovery of M5049: a novel selective toll-like receptor 7/8 inhibitor for treatment of autoimmunity. J. Pharmacol. Exp. Therapeut. 2021;376:397–409. doi: 10.1124/jpet.120.000275. [DOI] [PubMed] [Google Scholar]
- 17.Balak D.M.W., van Doorn M.B.A., Arbeit R.D., Rijneveld R., Klaassen E., Sullivan T., Brevard J., Thio H.B., Prens E.P., Burggraaf J., Rissmann R. IMO-8400, a toll-like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo-controlled trial in patients with moderate-to-severe plaque psoriasis. Clin. Immunol. 2017;174:63–72. doi: 10.1016/j.clim.2016.09.015. [DOI] [PubMed] [Google Scholar]
- 18.Tilstra J.S., John S., Gordon R.A., Leibler C., Kashgarian M., Bastacky S., Nickerson K.M., Shlomchik M.J. B cell-intrinsic TLR9 expression is protective in murine lupus. J. Clin. Invest. 2020;130:3172–3187. doi: 10.1172/JCI132328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alper P.B., Deane J., Betschart C., Buffet D., Collignon Zipfel G., Gordon P., Hampton J., Hawtin S., Ibanez M., Jiang T., et al. Discovery of potent, orally bioavailable in vivo efficacious antagonists of the TLR7/8 pathway. Bioorg. Med. Chem. Lett. 2020;30:127366. doi: 10.1016/j.bmcl.2020.127366. [DOI] [PubMed] [Google Scholar]
- 20.Rajagopal D., Paturel C., Morel Y., Uematsu S., Akira S., Diebold S.S. Plasmacytoid dendritic cell-derived type I interferon is crucial for the adjuvant activity of Toll-like receptor 7 agonists. Blood. 2010;115:1949–1957. doi: 10.1182/blood-2009-08-238543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forsbach A., Nemorin J.G., Montino C., Müller C., Samulowitz U., Vicari A.P., Jurk M., Mutwiri G.K., Krieg A.M., Lipford G.B., Vollmer J. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. JImmunol. 2008;180:3729–3738. doi: 10.4049/jimmunol.180.6.3729. [DOI] [PubMed] [Google Scholar]
- 22.Gestermann N., Di Domizio J., Lande R., Demaria O., Frasca L., Feldmeyer L., Di Lucca J., Gilliet M. Netting neutrophils activate autoreactive B cells in lupus. J. Immunol. 2018;200:3364–3371. doi: 10.4049/jimmunol.1700778. [DOI] [PubMed] [Google Scholar]
- 23.Zimmermann M., Arruda-Silva F., Bianchetto-Aguilera F., Finotti G., Calzetti F., Scapini P., Lunardi C., Cassatella M.A., Tamassia N. IFNalpha enhances the production of IL-6 by human neutrophils activated via TLR8. Sci. Rep. 2016;6:19674. doi: 10.1038/srep19674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu H., Dietsch G.N., Matthews M.A.H., Yang Y., Ghanekar S., Inokuma M., Suni M., Maino V.C., Henderson K.E., Howbert J.J., et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin. Cancer Res. 2012;18:499–509. doi: 10.1158/1078-0432.CCR-11-1625. [DOI] [PubMed] [Google Scholar]
- 25.Baenziger S., Heikenwalder M., Johansen P., Schlaepfer E., Hofer U., Miller R.C., Diemand S., Honda K., Kundig T.M., Aguzzi A., Speck R.F. Triggering TLR7 in mice induces immune activation and lymphoid system disruption, resembling HIV-mediated pathology. Blood. 2009;113:377–388. doi: 10.1182/blood-2008-04-151712. [DOI] [PubMed] [Google Scholar]
- 26.Banchereau R., Hong S., Cantarel B., Baldwin N., Baisch J., Edens M., Cepika A.M., Acs P., Turner J., Anguiano E., et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell. 2016;165:1548–1550. doi: 10.1016/j.cell.2016.05.057. [DOI] [PubMed] [Google Scholar]
- 27.Omdal R., Brokstad K., Waterloo K., Koldingsnes W., Jonsson R., Mellgren S.I. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur. J. Neurol. 2005;12:392–398. doi: 10.1111/j.1468-1331.2004.00976.x. [DOI] [PubMed] [Google Scholar]
- 28.Kowal C., Degiorgio L.A., Lee J.Y., Edgar M.A., Huerta P.T., Volpe B.T., Diamond B. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc. Natl. Acad. Sci. USA. 2006;103:19854–19859. doi: 10.1073/pnas.0608397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bravo-Zehnder M., Toledo E.M., Segovia-Miranda F., Serrano F.G., Benito M.J., Metz C., Retamal C., Álvarez A., Massardo L., Inestrosa N.C., González A. Anti-ribosomal P protein autoantibodies from patients with neuropsychiatric lupus impair memory in mice. Arthritis Rheumatol. 2015;67:204–214. doi: 10.1002/art.38900. [DOI] [PubMed] [Google Scholar]
- 30.Murakami Y., Fukui R., Tanaka R., Motoi Y., Kanno A., Sato R., Yamaguchi K., Amano H., Furukawa Y., Suzuki H., et al. Anti-TLR7 antibody protects against lupus nephritis in NZBWF1 mice by targeting B cells and patrolling monocytes. Front. Immunol. 2021;12:777197. doi: 10.3389/fimmu.2021.777197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lövgren T., Eloranta M.L., Båve U., Alm G.V., Rönnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 32.Eloranta M.L., Lövgren T., Finke D., Mathsson L., Rönnelid J., Kastner B., Alm G.V., Rönnblom L. Regulation of the interferon-alpha production induced by RNA-containing immune complexes in plasmacytoid dendritic cells. Arthritis Rheum. 2009;60:2418–2427. doi: 10.1002/art.24686. [DOI] [PubMed] [Google Scholar]
- 33.Dall'era M.C., Cardarelli P.M., Preston B.T., Witte A., Davis J.C., Jr. Type I interferon correlates with serological and clinical manifestations of SLE. Ann. Rheum. Dis. 2005;64:1692–1697. doi: 10.1136/ard.2004.033753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaul A., Gordon C., Crow M.K., Touma Z., Urowitz M.B., van Vollenhoven R., Ruiz-Irastorza G., Hughes G. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2016;2:16039. doi: 10.1038/nrdp.2016.39. [DOI] [PubMed] [Google Scholar]
- 35.Kuznik A., Bencina M., Svajger U., Jeras M., Rozman B., Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 2011;186:4794–4804. doi: 10.4049/jimmunol.1000702. [DOI] [PubMed] [Google Scholar]
- 36.Durcan L., Clarke W.A., Magder L.S., Petri M. Hydroxychloroquine blood levels in systemic lupus erythematosus: clarifying dosing controversies and improving adherence. J. Rheumatol. 2015;42:2092–2097. doi: 10.3899/jrheum.150379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirou K.A., Lee C., George S., Louca K., Peterson M.G.E., Crow M.K. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52:1491–1503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]
- 38.Rodero M.P., Decalf J., Bondet V., Hunt D., Rice G.I., Werneke S., McGlasson S.L., Alyanakian M.A., Bader-Meunier B., Barnerias C., et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J. Exp. Med. 2017;214:1547–1555. doi: 10.1084/jem.20161451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Migita K., Miyashita T., Maeda Y., Kimura H., Nakamura M., Yatsuhashi H., Ishibashi H., Eguchi K. Reduced blood BDCA-2+ (lymphoid) and CD11c+ (myeloid) dendritic cells in systemic lupus erythematosus. Clin. Exp. Immunol. 2005;142:84–91. doi: 10.1111/j.1365-2249.2005.02897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michaelis K.A., Norgard M.A., Levasseur P.R., Olson B., Burfeind K.G., Buenafe A.C., Zhu X., Jeng S., McWeeney S.K., Marks D.L. Persistent Toll-like receptor 7 stimulation induces behavioral and molecular innate immune tolerance. Brain Behav. Immun. 2019;82:338–353. doi: 10.1016/j.bbi.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baechler E.C., Batliwalla F.M., Karypis G., Gaffney P.M., Ortmann W.A., Espe K.J., Shark K.B., Grande W.J., Hughes K.M., Kapur V., et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Navarra S.V., Guzmán R.M., Gallacher A.E., Hall S., Levy R.A., Jimenez R.E., Li E.K.M., Thomas M., Kim H.Y., León M.G., et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 43.Morand E.F., Furie R., Tanaka Y., Bruce I.N., Askanase A.D., Richez C., Bae S.C., Brohawn P.Z., Pineda L., Berglind A., et al. Trial of anifrolumab in active systemic lupus erythematosus. N. Engl. J. Med. 2020;382:211–221. doi: 10.1056/NEJMoa1912196. [DOI] [PubMed] [Google Scholar]
- 44.Tummala R., Abreu G., Pineda L., Michaels M.A., Kalyani R.N., Furie R.A., Morand E.F. Safety profile of anifrolumab in patients with active SLE: an integrated analysis of phase II and III trials. Lupus Sci. Med. 2021;8:e000464. doi: 10.1136/lupus-2020-000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang F., Glans H., Enoksson S.L., Kolios A.G.A., Loré K., Nilsson J. Recurrent herpes zoster ophthalmicus in a patient with a novel toll-like receptor 3 variant linked to compromised activation capacity in fibroblasts. J. Infect. Dis. 2020;221:1295–1303. doi: 10.1093/infdis/jiz229. [DOI] [PubMed] [Google Scholar]
- 46.Sironi M., Peri A.M., Cagliani R., Forni D., Riva S., Biasin M., Clerici M., Gori A. TLR3 mutations in adult patients with herpes simplex virus and varicella-zoster virus encephalitis. J. Infect. Dis. 2017;215:1430–1434. doi: 10.1093/infdis/jix166. [DOI] [PubMed] [Google Scholar]
- 47.Zhang S.Y., Jouanguy E., Ugolini S., Smahi A., Elain G., Romero P., Segal D., Sancho-Shimizu V., Lorenzo L., Puel A., et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 48.Port A., Shaw J.V., Klopp-Schulze L., Bytyqi A., Vetter C., Hussey E., Mammasse N., Ona V., Bachmann A., Strugala D., et al. Phase 1 study in healthy participants of the safety, pharmacokinetics, and pharmacodynamics of enpatoran (M5049), a dual antagonist of toll-like receptors 7 and 8. Pharmacol. Res. Perspect. 2021;9:e00842. doi: 10.1002/prp2.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reynolds J.A., Briggs T.A., Rice G.I., Darmalinggam S., Bondet V., Bruce E., Khan M., Haque S., Chinoy H., Herrick A.L., et al. Type I interferon in patients with systemic autoimmune rheumatic disease is associated with haematological abnormalities and specific autoantibody profiles. Arthritis Res. Ther. 2019;21:147. doi: 10.1186/s13075-019-1929-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lau C.M., Broughton C., Tabor A.S., Akira S., Flavell R.A., Mamula M.J., Christensen S.R., Shlomchik M.J., Viglianti G.A., Rifkin I.R., Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn S.S., Jung S.M., Yoo J., Lee S.W., Song J.J., Park Y.B. Anti-Smith antibody is associated with disease activity in patients with new-onset systemic lupus erythematosus. Rheumatol. Int. 2019;39:1937–1944. doi: 10.1007/s00296-019-04445-y. [DOI] [PubMed] [Google Scholar]
- 52.Arroyo-Ávila M., Santiago-Casas Y., McGwin G., Jr., Cantor R.S., Petri M., Ramsey-Goldman R., Reveille J.D., Kimberly R.P., Alarcón G.S., Vilá L.M., Brown E.E. Clinical associations of anti-Smith antibodies in PROFILE: a multi-ethnic lupus cohort. Clin. Rheumatol. 2015;34:1217–1223. doi: 10.1007/s10067-015-2941-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guiducci C., Gong M., Xu Z., Gill M., Chaussabel D., Meeker T., Chan J.H., Wright T., Punaro M., Bolland S., et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature. 2010;465:937–941. doi: 10.1038/nature09102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Umiker B.R., Andersson S., Fernandez L., Korgaokar P., Larbi A., Pilichowska M., Weinkauf C.C., Wortis H.H., Kearney J.F., Imanishi-Kari T. Dosage of X-linked Toll-like receptor 8 determines gender differences in the development of systemic lupus erythematosus. Eur. J. Immunol. 2014;44:1503–1516. doi: 10.1002/eji.201344283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demaria O., Pagni P.P., Traub S., de Gassart A., Branzk N., Murphy A.J., Valenzuela D.M., Yancopoulos G.D., Flavell R.A., Alexopoulou L. TLR8 deficiency leads to autoimmunity in mice. J. Clin. Invest. 2010;120:3651–3662. doi: 10.1172/JCI42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carmona-Rivera C., Kaplan M.J. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013;35:455–463. doi: 10.1007/s00281-013-0375-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bennett L., Palucka A.K., Arce E., Cantrell V., Borvak J., Banchereau J., Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aluri J., Bach A., Kaviany S., Chiquetto Paracatu L., Kitcharoensakkul M., Walkiewicz M.A., Putnam C.D., Shinawi M., Saucier N., Rizzi E.M., et al. Immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain of function. Blood. 2021;137:2450–2462. doi: 10.1182/blood.2020009620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brohawn P.Z., Streicher K., Higgs B.W., Morehouse C., Liu H., Illei G., Ranade K. Type I interferon gene signature test-low and -high patients with systemic lupus erythematosus have distinct gene expression signatures. Lupus. 2019;28:1524–1533. doi: 10.1177/0961203319885447. [DOI] [PubMed] [Google Scholar]
- 60.Ah Kioon M.D., Tripodo C., Fernandez D., Kirou K.A., Spiera R.F., Crow M.K., Gordon J.K., Barrat F.J. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med. 2018;10:eaam8458. doi: 10.1126/scitranslmed.aam8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korotkevich GS V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2016 doi: 10.1101/060012v32021. Preprint at. [DOI] [Google Scholar]
- 64.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knoepfel T., Nimsgern P., Jacquier S., Bourrel M., Vangrevelinghe E., Glatthar R., Behnke D., Alper P.B., Michellys P.Y., Deane J., et al. Target-based identification and optimization of 5-Indazol-5-yl pyridones as toll-like receptor 7 and 8 antagonists using a biochemical TLR8 antagonist competition assay. J. Med. Chem. 2020;63:8276–8295. doi: 10.1021/acs.jmedchem.0c00130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All AmpliSeq data of this study have been deposited at Zenodo and are publicly available as of the date of publication. DOI is listed in the key resources table (https://doi.org/10.5281/zenodo.7573856). Microscopy data reported in this paper will be made available by the lead contact upon request.
-
•
All original code for analysis of AmpliSeq data have been deposited on Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table (https://doi.org/10.5281/zenodo.7575672).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.