

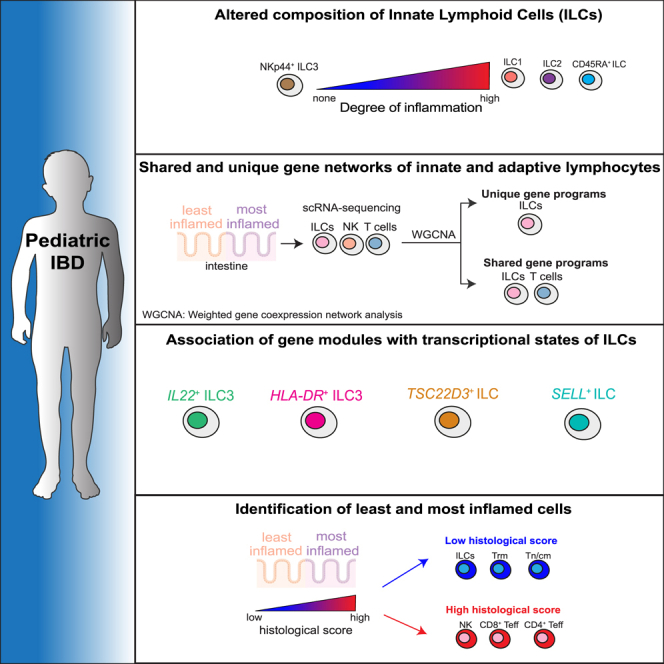
Summary
Innate lymphoid cells (ILCs) are considered innate counterparts of adaptive T cells; however, their common and unique transcriptional signatures in pediatric inflammatory bowel disease (pIBD) are largely unknown. Here, we report a dysregulated colonic ILC composition in pIBD colitis that correlates with inflammatory activity, including accumulation of naive-like CD45RA+CD62L− ILCs. Weighted gene co-expression network analysis (WGCNA) reveals modules of genes that are shared or unique across innate and adaptive lymphocytes. Shared modules include genes associated with activation/tissue residency, naivety/quiescence, and antigen presentation. Lastly, nearest-neighbor-based analysis facilitates the identification of “most inflamed” and “least inflamed” lymphocytes in pIBD colon with unique transcriptional signatures. Our study reveals shared and unique transcriptional signatures of colonic ILCs and T cells in pIBD. We also provide insight into the transcriptional regulation of colonic inflammation, deepening our understanding of the potential mechanisms involved in pIBD.
Keywords: ILC, IBD, pediatric inflammatory bowel disease, gut immunity, single-cell RNA sequencing, tissue-resident T cells
Graphical abstract
Highlights
-
•
Innate lymphoid cell (ILC) balance is altered in pediatric IBD (pIBD)
-
•
Colonic ILCs and T cells have shared and unique transcriptional programs
-
•
PIBD inflammation is characterized by transcriptional changes in both ILCs and T cells
The role of innate lymphoid cells (ILCs) in pediatric IBD (pIBD) remains understudied. Here, Kokkinou et al. show that while some transcriptional features of colonic ILCs in pIBD are unique, some are shared with T cells. pIBD inflammation leads to transcriptional and cellular changes in both ILCs and T cells.
Introduction
Pediatric inflammatory bowel disease (pIBD) includes Crohn’s disease (CD) and ulcerative colitis (UC), which have common symptoms like abdominal pain, frequent bloody diarrhea, and, for children with CD, sometimes also growth retardation.1 Around 25% of patients with IBD have been diagnosed before the age of 18 years, and the overall prevalence of pIBD is increasing.2 PIBD is characterized by more severe symptoms and a more extensive disease at onset than adults, and the CD phenotype shows less isolated ileal manifestations and more colonic inflammation compared with adult patients with CD.2 The etiology and pathogenesis of IBD is considered multifactorial. Host genetic as well as environmental factors may result in a dysregulated immune response and dysbiosis that is believed to drive chronic inflammation.3
T cells are implicated in intestinal inflammation in both mouse models4 and in humans, where conventional (CD4+ and CD8+)5,6,7 as well as unconventional T cells, including innate-like, γδ T cells and mucosal invariant (MAIT)8,9 T cells, show dysregulated composition. In patients with IBD, biologic treatments antagonizing immunomodulating factors that either activate T cells (interleukin [IL]-12/23, ustekinumab, etc.) or are expressed by T cells (α4β7 integrin; velodizumab, tumor necrosis factor [TNF]; infliximab, etc.) are effective in the majority of the patients.10
Innate lymphoid cells (ILCs), including natural killer (NK) cells, are viewed as the counterparts of adaptive T cells, which, in concert, execute specialized effector programs.11 In the inflamed adult IBD colon and ileum, the mucosal ILC composition is altered, resulting in reduced frequencies of IL-22-producing NKp44+ type 3 ILCs (ILC3s) and increased interferon (IFN)-γ-producing ILC1s12,13 as well as IL-17-producing ILC3s.14 However, it remains unknown whether these alterations in ILC frequencies are also present in pIBD.
Single-cell RNA sequencing (scRNA-seq) studies have provided transcriptional signatures of T cells in IBD mucosa of children and adults.5,15,16,17,18,19 However, characterization of intestinal ILCs in parallel to T cells in pediatric patients with IBD has not yet been performed.
Here, we provide a comprehensive characterization of ILCs and T cells in colonic biopsies from newly diagnosed and treatment-naive pediatric patients with IBD colitis and non-IBD controls. Flow cytometry showed inflammation-specific alterations of ILC frequencies, including the accumulation of CD45RA+CD62L− naive-like ILCs, which correlated with disease severity. scRNA-seq revealed gene co-expression networks that were shared between or unique to innate and adaptive lymphocytes. Finally, a nearest-neighbor-based approach provided unbiased means to identify the “most inflamed” or “least inflamed” lymphocytes in our dataset based on the histological inflammation score of the samples. Overall, our study provides a comprehensive immunophenotypic and transcriptional characterization of ILCs and T cells in pediatric patients with IBD colitis. We uncover previously unknown co-regulatory dynamics of innate and adaptive lymphocytes that potentially play a role in disease pathogenesis and may be targeted for therapeutic intervention.
Results
Dysregulated colonic ILC composition correlates with inflammatory activity in pIBD colitis
To characterize ILCs in pediatric patients with newly diagnosed IBD, we collected colonic biopsies from endoscopically inflamed and non-inflamed sites from 19 treatment-naive patients with CD or UC colitis (Table S1). Pediatric patients who underwent colonoscopy because of intestinal symptoms but did not have IBD or other endoscopic inflammation served as the non-IBD control group (n = 10). For the patients with IBD, endoscopic inflammation was evaluated with the SWIBREG20 local endoscopic disease activity score (denoted “endoscopic score”) (Table S2), and local microscopic inflammation was evaluated with a modified version of the D′ Haens et al. histological score (denoted “histo score”; see STAR Methods).21
Human intestinal CD127+ ILCs were analyzed by flow cytometry as previously described13,22 (Figure S1A). The identity of ILC1s, ILC3s, and NK cells was confirmed by staining of lineage-defining transcription factors in both blood and colon samples (Figure S1B). In contrast to CD3+ T cells and CD56+ NK cells, the frequency of CD127+ ILCs among lymphocytes was reduced in endoscopically inflamed IBD compared with non-IBD and non-inflamed IBD colon mucosa (Figure 1A). The majority of ILCs expressed the tissue residency marker CD69 (Figure S1C) but also showed increased expression of markers associated with naive T cells, CD45RA and CD62L (Figure S1C). While HLA-DR expression was elevated in pIBD ILCs, the expression remained unchanged in total CD3+ T cells (Figure S1C). However, elevated HLA-DR expression has been previously detected in CD4+ T cells in IBD.5 Further phenotyping of pIBD T cells and NK cells can be seen in Figure S1C.
Figure 1.

Dysregulated colonic ILC composition correlates to inflammatory activity in pIBD colitis
(A) Bar graphs showing the frequencies of total ILCs, T cells, and NK cells (gated as shown in Figure S1A) in non-IBD (n = 10), endoscopically non-inflamed (n = 12), and endoscopically inflamed (n = 15) pIBD colon samples. A total of 8 samples were derived from paired inflamed and non-inflamed samples. Frequencies shown out of CD45+ lymphocytes.
(B) Bar graphs showing the frequencies of ILC1s, ILC2s, NKp44+ ILC3s, and NKp44− ILCs (gated as shown in Figure S1A) in non-IBD (n = 10), endoscopically non-inflamed (n = 12), and endoscopically inflamed (n = 15) pIBD colon samples. Frequencies shown out of total ILCs.
(C) Correlation plots between frequencies of ILC1s, ILC2s, NKp44+ ILC3s, and NKp44− ILCs and the endoscopic and histological scores determined for each of the endoscopically non-inflamed (n = 12) and endoscopically inflamed (n = 15) pIBD colon samples.
Statistical significances in (B) were detected with Mann-Whitney test. Bar graphs are shown as median ± interquartile range (IQR). Spearman’s rank correlation test was applied for assessing correlations between variables. In (A)–(C), cells from each donor were analyzed in independent experiments.
We detected a skewed ILC composition in endoscopically inflamed IBD versus non-IBD and non-inflamed IBD colon, as illustrated by increased frequencies of NKp44− ILCs and ILC1s, while NKp44+ ILC3 frequencies were reduced (Figure 1B), similar to our previous findings in adult IBD.13 There was a positive correlation between the endoscopic and histo scores and the frequencies of ILC2s and NKp44− ILCs, whereas the frequency of NKp44+ ILC3s was inversely correlated with both endoscopic and histo scores (Figure 1D). Overall, our findings show that the mucosal ILC composition is dysregulated in patients with pIBD, displaying a pattern similar to what we previously reported for adult IBD.
Single-cell transcriptional profiling of colonic ILCs, NK cells, and T cells in pIBD colitis
To characterize the transcriptional signatures of both innate and adaptive lymphocytes in pIBD, we performed 3′ 10× scRNA-seq of biopsies from six treatment-naive pediatric patients with newly diagnosed CD-colitis (total n = 12 samples) (Figure 2A; Table S3). To assess the transcriptional signatures of inflammation within the same patient, biopsies were obtained from the endoscopically least and most inflamed area of the colon and subsequently fluorescence-activated cell sorted (FACS) for Lin−CD127+CD161+ (ILCs), Lin+CD127−CD56+ (NK cells), and CD3+ (T cells) (Figure S2A). After quality control, normalization, and integration of the datasets, 44,717 lymphocytes were obtained for analysis. Louvain clustering analysis allowed for the separation of the pre-sorted cell populations into 12 clusters, which were represented by all patients and samples (Figures 2B and S2B). Clusters were annotated based on signature genes (Figure 2C), where one cluster represented ILCs expressing IL7R, ID2, and IL22, two clusters represented different activation stages of NK cells expressing ID2, ZBTB16, PRF1, GZMA, and IFNG, and nine clusters represented subsets of CD4+ and CD8+ T cells. These included naive/central memory CD8+ and CD4+ T cells (CD8+ and CD4+ Tn/cm cells; CD8, CD4, CCR7, SELL, TCF7, and CD27); CD8+ T effector cells (CD8+ Teff cells; CD8B, PRF1, GZMA, and IFNG); CD8+ T effector memory cells (CD8+ Tem cells; CD8B, KLRC2, and ZNF683); CD8+ T resident memory cells (CD8+ Trm cells; CD69, ITGA1, and ITGAE); CD4+ Teff cells (CD4, IFNG, and GZMA); CD4+ Trm cells (CD4, CD69, and ICOS); T follicular helper cells (Tfh cells; CD4, CXCR5, PDCD1, and FAS); and activated CD4+T/regulatory T cells (Activ. CD4+T/Tregs; CD4, CTLA4, ICOS, FOXP3, and IL10) (Figures 2B and 2C).
Figure 2.

Single-cell transcriptional profiling of colonic ILCs, NK cells, and T cells in pIBD colitis
(A) Schematic illustration of the scRNA-seq workflow of the total 12 paired colonic biopsy samples obtained from the endoscopically least and most inflamed areas of the 6 pediatric patients with CD-colitis (n = 6 donors).
(B) UMAP visualization of the scRNA-seq data showing the unbiased clustering of total 44 717 ILCs, NK cells, and T cells.
(C) Dotplot displaying expression of lineage-identity genes for the annotation of the clusters in (B).
In (A)–(C), cells from each donor were analyzed in independent experiments.
Altogether, scRNA-seq revealed an overall diverse landscape of innate and adaptive lymphocytes in the pediatric colonic mucosa.
Gene module analyses of colonic innate and adaptive lymphocytes reveal subset-specific and shared programs of co-expressed genes
Complex biological processes like inflammation are orchestrated by co-expressed gene networks. In addition, ILCs and T cells largely overlap in terms of phenotype and functions, suggesting coordinated gene regulation. To identify such patterns of gene expression and regulation dynamics, we performed weighted gene co-expression network analysis (WGCNA),23,24 where we used the top 4,000 variable genes to discern 50 modules of co-regulated genes among the lymphocytes (Figure 3B). With this approach, we identified gene networks that were either confined to a specific cell type or shared across cell types. More specifically, we identified three modules (1, 11, and 43) preferentially expressed by the ILC cluster (Figures 3C and S3A). Intriguingly, module 1 showed coordinated expression between key ILC cytokine genes IL22, CSF2 (encoding GM-CSF), the co-stimulatory molecule CD83, and IL4I1, the latter encoding a secreted L-amino acid oxidase reported to promote aryl hydrocarbon receptor (AHR) activation while limiting Th17 proliferation.25,26 The expression of SOX4, IL4R, and CYSLTR1 (encoding the cysteinyl leukotriene receptor) in module 11 showed that although a distinct cluster of ILC2s could not be identified in our data, ILCs expressing transcripts with known regulatory function in ILC2s/Th2 cells are present in the pIBD colon.
Figure 3.

Gene module analyses of colonic innate and adaptive lymphocytes reveal subset-specific and shared programs of co-expressed genes
(A) Summary flow chart used for the analysis of scRNA-seq data and subsequent gene module and nearest-neighbor analysis.
(B) Circular dendrogram illustrating the number and composition of each module and the contribution of each module to discriminating cell clusters and donors. Connecting lines represent modules with highly correlated expressions (kNN [k nearest neighbor]). Modules selected for analysis and described in the text are marked with an asterisk.
(C–I) A selection of genes in each module marked with an asterisk in (B) illustrated by dotplots. The expression of the modules across the annotated clusters is shown in Figure S3.
Two gene modules were shared between the two innate lymphocyte clusters (ILCs and NK cells; modules 8 and 19), including the cytotoxicity-associated adapter proteins TYROBP and FCER1G, as well as the effector molecule AREG that was expressed together with BHLHE40, encoding a transcriptional regulator previously reported to control cytokine production in T cells27,28 (Figures 3D and S3B). Highlighting the shared features of ILCs and T cells, module 26 was coordinately expressed by ILCs and Trm cells, including innate-like genes (ID2 and KLRB1) and genes associated with tissue residency (ZFP36 and NR4A2) (Figures 3E and S3C). Module 7 was shared by ILCs and CD4+ Teff cells, Trm cells, and Activ. T cells and included genes typically expressed by Th17 cells (RBPJ, ICOS, CCL20, and CCR6) (Figures 3F and S3D). In line with previous reports showing antigen-presenting features of ILCs,29,30,31,32 module 20 contained a set of transcripts associated with antigen presentation (HLA-DRA and HLA-DPA1) that were shared by ILCs and Teff cells (Figures 3G and S3E). Additional shared signatures between ILCs and T cells were related to genes that regulate tissue residency (FOS and NR4A1; module 18) as well as quiescence (TSC22D3, DUSP1, ZFP36L2, and KLF6; module 22) (Figures 3H and S3F), the latter recently shown to be expressed by plastic skin ILCs in a mouse model of psoriasis.33 Lastly, module 30 showed coordinated expression of KLF2 and TCF7, genes typically expressed by naive T cells. However, module 30 was highly expressed by ILCs and naive T cells, indicating the presence of ILCs with naive features as previously described in the adult gut24,34 (Figures 3I and S3G).
Overall, these findings reveal subset-specific and common gene signatures that may regulate ILCs and T cells.
Gene modules associate with ILC subclusters and identify quiescent/naive-like ILCs
To better understand the expression pattern of unique and shared gene modules among ILCs, we focused our analysis only on the ILCs. Reclustering of ILCs revealed 7 transcriptionally distinct clusters that were annotated by differential expression (DE) analysis (Figures 4A, 4B, S4A, and S4B). Interestingly, the pIBD ILC subclusters displayed a similar degree of heterogeneity with ILCs from adult IBD colon, while tonsillar ILCs were less heterogeneous (Figures S4C and S4D). Intriguingly, the module 1 genes CD83, IL4I1, IL22, and CSF2 were confined to ILC subcluster 3 (Figures 4C and S4E), but these genes associated poorly with expression of the key ILC3 genes NCR2 (Figure 4C) and RORC (Figure S4F), which were widely expressed among the ILC clusters. We further identified that module 20 genes (HLA-DPA1, HLA-DRA, and HLA-DQB1) were uniquely expressed (Figures 4B, 4D, and S4E) by subcluster 7, suggesting that they represent antigen-presenting ILCs. Furthermore, subcluster 1 expressed many genes in modules 18 and 22, including TSC22D3, DUSP1, and CD69 (Figures 4B, 4E, and S4E), previously associated with quiescent and tissue-resident ILCs.33 Interestingly, in line with our previous report,34 we identified a SELL-expressing ILC subcluster 4 (Figures 4B and 4F), with low expression of NCR2, HLA-DR, and RORC transcripts (Figures 4B, 4D, and S4F), likely representing naive-like ILCs enriched in adult IBD. Genes in the “naivety” module 30 (Figure 3I) were not solely expressed by subcluster 4. In fact, TCF7 was homogenously expressed across all ILCs, and KLF2 was also expressed by subcluster 1 (Figures 4B and 4G). These data indicated the presence of at least two subclusters of ILCs with features of naive T cells. To determine the developmental relationships between the ILC subclusters, we performed trajectory analysis with Monocle 3 using subcluster 4 (Figure 4H) or 1 (Figure S4G) as the root, respectively. We found that regardless of the root, subclusters 3 (IL22-expressing effector ILCs), 4 (naive ILCs), 1 (tissue resident/quiescent ILCs), and 6–8 (ILC1-like ILCs, HLA-DR+ ILCs, SOX4+ ILCs) branch separately from each other, with cluster 0 as the branching point.
Figure 4.

Gene modules associate with ILC subclusters and identify quiescent/naive-like ILCs
(A) UMAP visualization of ILC subclusters after reclustering main cluster 1 shown in Figure 2B.
(B) Dotplot showing selected ILC-module genes (Figures 3D–3I). The expression is shown within the annotated subclusters of ILCs depicted in (A).
(C–G) UMAP visualization of selected ILC-module genes in the ILC subclusters. Dotted lines mark the subcluster where the depicted gene is highly and differentially expressed. A list of differentially expressed genes (DEGs) per ILC subcluster is shown in Figure S4A.
(H) Developmental trajectories of ILC subclusters performed with Monocle 3. Subcluster 4 (dotted circle) was used as the root.
(I) Frequencies of CD45RA+ ILCs among ILCs expressing combinations of HLA-DR and NKp44 in inflamed pIBD (n = 12–14) and non-IBD (n = 8) colon biopsies (bar graphs are shown as median). The samples were analyzed in independent experiments.
(J) Representative flow cytometry plots of CD45RA versus CD62L (marked in red is the CD45RA+ ILC subset), gated out of NKp44−HLA-DR− ILCs, in inflamed pIBD and non-IBD colon biopsies.
(K) Frequency of CD45RA+ ILCs out of CD161+ ILCs in non-ΙBD (n = 8), endoscopically non-inflamed (n = 10), and endoscopically inflamed pIBD (n = 14) colon biopsies. Mann-Whitney test (p < 0.05), mean with SD. The samples were analyzed in independent experiments.
(L) Correlation plots (Spearman, p < 0.05) between the frequency of CD45RA+ ILCs and the endoscopic and histological scores determined for each of the endoscopically non-inflamed (n = 8) and endoscopically inflamed (n = 14) pIBD colon samples.
To better characterize the naive/quiescent ILCs in pIBD colon, we stained ILCs for markers of activation/differentiation. We have recently described that the less-differentiated and less-activated (NKp44−HLA-DR−) ILCs in human tissues, including adult IBD gut, form a heterogeneous population that includes naive-like/quiescent ILCs marked by the presence of surface CD45RA expression.34 Confirming and extending our previous findings, CD45RA also marked such cells in pIBD (Figure 4I). As we observed in the adult IBD gut, surface CD62L was not expressed on this population despite the presence of SELL transcripts (Figure 4J). Similarly to adult IBD, the frequency of these CD45RA+CD62L− ILCs (referred to as CD45RA+ ILCs) was increased in the endoscopically inflamed pIBD gut samples, and their frequency was positively correlated with the local endoscopic as well as histo score (Figures 4K and 4L).
In summary, our analyses reveal gene networks confined to certain ILC subsets, including a naive-like module. Using previously described surface markers that identify naive-like/quiescent ILCs in tissues, we observed an accumulation of CD45RA+ ILCs in the inflamed gut of pediatric patients with IBD that correlated with the degree of inflammation.
Nearest-neighbor-smoothed histo scores reveal the most and least inflammatory cells
Next, we set out to explore inflammation-related transcriptional signatures of innate and adaptive lymphocytes. During endoscopy, the grade of inflammation is evaluated based on the presence of visual signs.35 In contrast, histological findings more precisely assess both the acute and chronic cellular changes occurring in the intestinal epithelium and lamina propria.36 We were therefore interested in defining the transcriptional signatures of these cellular changes with scRNA-seq (Figures 5A and 5B). Indeed, when plotting the histo scores on the uniform manifold approximation and projection (UMAP), we could observe overrepresentation of lymphocytes from samples with high histo scores in certain clusters (Figure 5C). However, this crude representation did not consider donor representability. Therefore, we repurposed a nearest-neighbor-based smoothing method deployed elsewhere.37,38,39 Here, each cell was assigned the average histo scores of the 100 nearest-neighbor cells in the Euclidean space, which was created by a 100-dimensional principal-component analysis based on the 4,000 most variable genes (Figure 5D). To balance out the number of neighbors for each histological group (defined below), we conducted subsampling of the data so that all three groups consisted of the same number of neighbors (Figure S5A). Distribution of lymphocytes across tissues with different histo scores allowed for categorization of the cells into three groups: least inflamed (histo score ≤ 4), “intermediate” (histo score = 5–8), and most inflamed lymphocytes (histo score ≥ 9) (Figure 5E). Visualization of the smoothed histo scores revealed enrichment of the most inflamed and least inflamed lymphocytes in certain clusters (Figures 5F and S5B). The most inflamed samples were mostly derived from 4 samples (Figure S5C). However, similar enrichment was predicted when batch correction was applied (Figure S5D) and also when we performed differential abundance analysis with DAseq (Figure S5E).38 The most inflamed lymphocytes were predominantly NK cells (cluster 2), CD8+ Teff cells (cluster 5), CD4+ Tef cells (cluster 9), and Activ. T cells/Tregs (cluster 12) (Figure 5F). Conversely, the least inflamed cells were enriched in ILCs (cluster 1), CD8+ Tn/cm cells (cluster 4), CD4+ Tn/cm cells (cluster 8), CD8+ Trm cells (cluster 7), and CD4+ Trm cells (cluster 10).
Figure 5.

Nearest-neighbor-smoothed histological scores reveal the most and least inflammatory cells
(A) Schematic illustration showing the histological scores from the endoscopically least and most inflamed paired colonic biopsies used for scRNA-seq.
(B) Pie chart showing the total number of cells from areas of different histological scores from all 12 pIBD colon biopsy samples.
(C) The raw histological score of each cell derived from each sample depicted onto the main UMAP from Figure 2B.
(D) Schematic illustration of the Euclidean neighbor smoothing. Each cell was given the average histological scores of its 100 transcriptionally closest neighbors. These smoothed histological scores were used for the subsequent analysis.
(E) Histogram showing the cell type distribution across the histological scores derived from the nearest-neighbor analysis. Cells were categorized into 3 groups: ≤4 (least inflamed), 5–8 (intermediate), and ≥9 (most inflamed).
(F) Visualization of cells from the nearest-neighbor smoothing on the main UMAP with cells derived from the ≥9 histological (histo) score (most inflamed) and ≤4 histo score (least inflamed) groups, which also have neighbors from at least 5 samples.
(G and H) Violin plots showing the expression level of selected DEGs (log2 fold change [FC] > 0.10 and adjusted p value [adj. p value] < 0.05) upregulated in the most (G) and least (H) inflamed cells versus the rest of the cells in each cluster.
(I) Frequencies of CD4+CD69+, CD4+CD161+CD69+, CD8+CD69+, and CD8+CD161+CD19+ cells in colonic adult non-IBD (n = 8) and IBD samples (n = 8). Mann-Whitney test (p < 0.05), mean with SD. The samples were analyzed in two independent experiments.
To uncover transcriptional signatures of the most inflamed lymphocytes, we performed DE analysis in the most inflamed cells (shown as MOST) versus the rest of the cells (shown as REST) within each cluster. A total of 96 genes were upregulated in the most inflamed cells with 35 genes being shared among the clusters (Table S4). The most inflamed NK cells showed elevated expression of the transcript for the antimicrobial molecule granulysin (GNLY) and genes involved in immunoregulatory interactions (CD81 and CLEC2B), while the most inflamed CD8+ T cells expressed the selenoprotein K (SELK) and the cAMP regulator CREM (Figure 5G). Interestingly, the most inflamed CD4+ Teff cells displayed a cytotoxic signature with expression of the granzymes GZMK and GZMH (Figure 5G), while the most inflamed Activ. CD4+ T/Tregs upregulated genes involved in redox balance (SELK, PRDX1, PRDX2, and SOD1), the caspase inhibitor CARD16, and the inhibitory molecules LAIR2 and TNFRSF18 (GITR) (Figure 5G). Similarly, to transcriptionally characterize the least inflamed lymphocytes, we performed DE analysis in the least inflamed cells (shown as LEAST) versus the rest of the cells (shown as REST) (Figure 5H). The least inflamed lymphocytes demonstrated larger transcriptional differences compared with the most inflamed lymphocytes with an upregulation of 226 genes, of which 70 were shared among the clusters (Table S4). Transcripts related to tissue residency such as NR4A2 and JUND as well as PTGER4 and ICOS were commonly expressed by the least inflamed cells across several clusters (Figure 5H). Additionally, the least inflamed ILCs revealed expression of CD83, involved in dendritic cell maturation as well as lymphocyte activation.40 The least inflamed CD8+ and CD4+ Tn/cm cells expressed molecules related to their identity (SELL, LEF1, CCR7, TCF7) like the least inflamed Trm cells that additionally showed overexpression of genes associated with tissue residency (ICOS, JUND, NR4A2, and PTGER4) (Figure 5H). The least inflamed CD8+ Trm cells additionally upregulated genes related to “innateness” (RORA, KLRB1) and effector functions (IFNG, TNF, CCL4) (Figure 5H). To validate these findings, we performed flow cytometry using intestinal specimens of adult patients with and without IBD (Table S5). We showed that, indeed, CD4+ Trm and CD8+ Trm cells (defined as CD69+ and/or CD161+) were lower in frequency in IBD, as predicted by the nearest-neighbor analysis (Figure 5I). The prediction of ILCs being mostly represented in the least inflamed landscape was additionally confirmed by our flow cytometry analysis of total ILCs (Figure 1A).
Collectively, based on nearest-neighbor analysis of transcriptional profiles and histo scores, we identified most inflamed and least inflamed lymphocytes with potential roles in colonic inflammation. The genes expressed by these cells deserve further functional exploration to assess their usefulness as targets for immunotherapy in IBD.
Discussion
In our study, we combined high-dimensional single-cell transcriptional and immunophenotypic analyses to comprehensively characterize innate and adaptive lymphocytes from colonic biopsies obtained from treatment-naive pediatric patients with newly diagnosed IBD colitis. We focused on pIBD as it generally shows a more aggressive phenotype and allows for studies of disease mechanisms that are less influenced by diagnostic delay,41 age-related environmental exposures, for example smoking, and co-morbidities compared with adults. A number of studies have investigated IBD-associated patterns of T cells and ILCs in human tissues at the single-cell level.5,15,16,18,19 However, these studies describe discrete cell populations of either ILCs or T cells without directly comparing these two cell types. Therefore, we complemented our protein analysis of ILCs with in-parallel single-cell transcriptional profiling of ILCs, NK cells, and T cells to uncover transcriptional networks that govern both shared and unique properties. Through our approach, we provide insights into ILC and T cell biology in pIBD. Understanding the regulation of these transcriptional networks could possibly lead to the development of therapies for IBD.
Flow cytometry revealed a dysregulated composition of mucosal ILCs in pIBD that correlated with the inflammatory activity. More specifically, we observed a reduction of NKp44+ ILC3s and an increase of NKp44− ILCs and ILC1, the latter which could, at least to some extent, be explained by ILC3-ILC1 plasticity as described for adult IBD.12 However, it is currently unclear how well the FACS-based definition of human ILCs correlates with transcriptional and functional features of ILCs in the gut mucosa. Similar to our FACS definition of ILC1, scRNA-seq analysis revealed an ILC1-like cluster expressing TBX21 (encoding T-bet) but low KIT (encoding CD117) and RORC. These cells likely represent the previously described CD127+ ILC1s12 and not the intraepithelial CD103+ ILC1s, as those lack CD127 expression.42 Expression of NCR2, encoding NKp44, was seen in several ILC subclusters, indicating transcriptional heterogeneity among NKp44+ ILC3s. It has been well described that human NKp44+ ILC3s harbor IL-22-producing ILCs43 and that ILC3-derived IL-22 is crucial for gut homeostasis in mice.44 However, here we show that IL22 transcription is confined to a specific ILC subcluster with relatively low expression of RORC. This is biologically possible as RORC-deficient patients are still capable of IL-22 production.45 Of note, IL22 was co-transcribed with other effector molecules such as CSF2, CD83, and IL4I1 in a gene module that was preferentially expressed in ILCs compared with T cells, potentially reflecting the more rapid response of ILCs compared with T cells. Notably, IL4I1 can activate AHR ligands in tumor cells via tryptophan metabolism,25 which in turn supports IL-22 production in ILCs.46 Thus, IL4I1 may promote AHR in ILC3s. Additional studies are needed to investigate how IL4I1 or other co-expressed molecules regulate IL-22 production by intestinal human ILCs.
Although ILC2s represent a large population in the mouse intestine47 and human fetal intestine,48 the healthy human adult intestine is largely devoid of conventional CD127+ ILC2s,48,49 similar to what we also observed in our flow cytometric and scRNA-seq analysis of pIBD colon tissue. Although it is possible that intestinal ILC2s show an unconventional profile that we excluded with our current sorting gating strategy, we identified an ILC-restricted gene module containing genes associated with the regulation of ILC2s. This included positive regulators such as IL4R and CYSTL1 but also SOX4, which can suppress Th2 differentiation.50 Thus, we speculate that the absence of a transcriptionally distinct subset of CD127+ ILC2s in the human intestine could be due to gut environmental factors that suppress ILC2s with a conventional phenotype. This topic requires further investigation.
One of the most striking overlaps between innate and adaptive lymphocytes in our analysis was the expression of a gene module containing genes associated with tissue residency including CD69, CXCR4, and KLF6. This gene module was particularly highly expressed in ILCs and Trm cells, indicating similar programs for tissue residency in innate and adaptive lymphocytes, a topic that deserves further investigation. However, we also noted that this module included genes associated with cellular quiescence, TSC22D3, DUSP1, and ZFP36L2.33 These genes were highly transcribed in a SELL-expressing ILC subcluster (ZFP36L2) and in another ILC subcluster (TSC22D3 and DUSP1), which co-transcribed the transcription factor KLF2, typically expressed by naive T cells. The SELL-expressing ILC subcluster lacked features of mature ILCs including RORC, NCR2, TBX21, or EOMES, reminiscent of the CD62L+ naive-like ILCs we recently described in tonsils.34 The second naive/quiescent cluster, which lacked expression of TBX21 and EOMES, likely represented the quiescent/naive-like ILCs that we previously reported in human tonsil and adult gut,34 where we showed that these less-differentiated quiescent ILCs are marked by the presence of CD45RA expression and, despite expression of SELL transcripts, the absence of CD62L.34 The frequency of this subset was increased in the inflamed gut of adult patients with IBD, and they could differentiate to ILC1s and IL-22-producing ILC3s.34 In the present study, we extended this finding to show that CD45RA+ ILCs accumulate in the inflamed gut of pediatric patients with IBD as well. These cells could be ready for trans-differentiation, as previously shown in a mouse model of psoriasis where ILC3s accumulated by trans-differentiation of ILC2s via a quiescent-like state of ILCs.33
We further identified a gene module that was commonly expressed by a distinct subcluster of ILCs and T cells and that contained transcripts of major histocompatibility complex (MHC) class II molecules involved in antigen presentation. Indeed, we and others have previously shown that ILCs can acquire antigen-presenting properties that in turn can be suppressed by the human tumor microenvironment29 and the gut microenvironment in mice.31 However, the antigen-presenting capacity of ILCs in the context of human IBD, in the presence of a more complex microbiota compared with transgenic mice, remains underexplored. Hence, it is possible that ILCs could orchestrate CD4+ T cell responses during gut inflammation in humans, similarly to what was described in a mouse model of acute colitis.30
To unravel inflammation-driven transcriptional changes in innate and adaptive lymphocytes, we made use of the histological inflammation score of our samples rather than grouping them in two endoscopically defined groups with variable histo scores. A nearest-neighbor-based smoothing of histo scores identified so-called least inflamed cells that were mostly defined as ILCs, CD4+ and CD8+ Tn/cm cells, and innate-like CD8+ Trm cells. The least inflamed colon areas of patients with pIBD may represent tissue that was previously inflamed and now healed or tissue that is yet unaffected by inflammation. Regardless, the transcriptomes of least inflamed cells represent a footprint of innate and adaptive lymphocyte homeostasis in the colon. This includes genes associated with the lineage identity of ILCs, CD4+/CD8+ Tn/cm cells, CD4+ Trm cells, and innate-like CD8+ Trm cells, suggesting that maintained function of these cells is critical for immune homeostasis in the gut and that loss of these cells is associated with gut inflammation. In contrast, the most inflamed cells were dominated by NK cells, cytotoxic CD4+ T cells, Activ. CD4+ T cells/Tregs, and CD8+ Teff cells.
It is possible that the compositional differences in least and most inflamed cells is due to recruitment of lymphocytes from the blood to inflamed areas of the colon. However, it could also be due to plasticity and/or differentiation of cells. Plasticity of ILCs and T cells is well described, and for ILCs, conversion of non-cytotoxic human ILCs to NK cells has been demonstrated in vitro.51 Most inflamed NK cells upregulated transcripts of the antimicrobial protein GNLY and CLEC2B, the latter mediating NK cell activation via ligation to NKp80.52 GNLY has been shown to be released in response to infections and to facilitate antigen presentation in dendritic cells (DCs) via Toll-like receptor 4 (TLR4).53 Interestingly, GNLY-expressing CD94+CD127+ ILCs are expanded in patients with CD,54 illustrating the fluidity in ILC-NK cell functions and the possibly that ILCs are reduced at the expense of NK cells during gut inflammation. Similarly, the underrepresentation of CD4+ Tn/cm and CD4+ Trm cells among the most inflamed cells could be due to differentiation to cytotoxic CD4+ T cells and/or Activ. T cells/Tregs. Several studies have described the presence of cytotoxic CD4+ T cells (CD4+ CLT cells) in IBD,55,56 but their origin remains unknown. We identified a CD4+ T cell subset expressing GZMH and GZMK in inflamed IBD. Interestingly, this is in line with a recent report describing that CD4+ CTL cells exhibit a diverse granzyme expression in nasal polyp tissue, including a subset that expressed these non-conventional granzymes.57 GZMK and GZMH expression is seen in CD8+ T cells with limited TCR repertoire and expression of exhaustion markers in aging58 and autoimmunity,59 suggesting that the CD4+ T cell subset that we detect herein might be a highly differentiated/exhausted subset. Future studies should be aimed to address the origin and function of these CD4+ CLT cells and Activ. T cells/Tregs in the context of gut mucosal inflammation in IBD.
We also noted accumulation of CD8+ Teff cells among the most inflamed cells. While it is tempting to speculate that these cells could be derived from the CD8+ Tn/cm and/or CD8+ Trm cells among the least inflamed cells, further work is required to address this. The role for the CD8+ T cell compartment is conflicting in IBD. A single-cell atlas study of CD8+ T cells in UC described innate features of CD8+IL26+ T cells,18 while another study reported reduced frequencies of cytotoxic IFN-γ+ CD8+ T effector memory cells re-expressing CD45RA (TEMRA) cells in UC mucosa.5 Similarly, we found increased expression of IFNG, TNF, and innate-like transcripts in the least inflamed IBD CD8+ Trm cells, suggesting that these cells have important homeostatic functions in the gut.
In summary, we report that pediatric patients with IBD display dysregulated ILC frequencies that are similar to those observed in adult patients with IBD, suggesting a common pathogenic program in this aspect. Using a combination of high-dimensional single-cell analysis on the transcriptional and protein levels, we provide a roadmap of ILCs and T cells in pIBD, including transcriptional signatures associated with inflammation. Further mechanistic studies are necessary to understand whether and how these findings contribute to pathogenesis and how they could facilitate the design of immunotherapeutic targets.
Limitations of the study
Although paired non-inflamed biopsies were used to infer inflammatory changes in the inflamed samples, the main limitation of the study is the lack of a non-symptomatic, healthy non-IBD control group in the single-cell analysis. Furthermore, one of the main transcriptional findings, on the reduced presence of Trm cells in colon inflammation, was validated on the protein level using samples from adult patients with IBD. However, we were not able to validate this, or other interesting transcriptional findings, in samples from patients with pIBD. Finally, since we performed our studies in samples from patients with ongoing inflammation, we cannot determine if the observed differences are a cause or consequence of IBD and what the role of these differences are in driving inflammation. In summary, the limitations of this study represent important aspects to address in future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human CD1a FITC (Clone HI149) | Biolegend | Cat#300104 |
| anti-human CD14 FITC (Clone Tuk4) | LifeTech | Cat#MHCD1401-4 |
| anti-human CD19 FITC (Clone 4G7) | BD | Cat#345776 |
| anti-human CD123 FITC (Clone 6H6) | Biolegend | Cat#306014 |
| anti-human BDCA2 FITC (Clone AC144) | Miltenyi | Cat#130-113-192 |
| anti-human FcER1a FITC (Clone AER-27) | Biolegend | Cat#334608 |
| anti-human CD34 FITC (Clone 581) | Biolegend | Cat#343504 |
| anti-human CD94 FITC (Clone DX22) | Biolegend | Cat#305504 |
| anti-human TCRab FITC (Clone IP26) | Biolegend | Cat#306706 |
| anti-human TCRgd FITC (Clone B1) | Biolegend | Cat#331208 |
| anti-human Eomes FITC (Clone WD1928) | eBioscience | Cat#11-4877-42 |
| anti-human NKG2A (CD159a) APC (Clone Z199) | Beckman Coulter | Cat#60797 |
| anti-human Eomes efluor660 (Clone WD1928) | eBioscience | Cat#50-4877-41 |
| anti-human CD69 AF700 (Clone FN50) | Biolegend | Cat#310992 |
| anti-human CD3 AF700 (Clone UCHT1) | BD | Cat#557943 |
| anti-human CD62L APC-Cy7 (Clone DREG-56) | Biolegend | Cat#304814 |
| anti-human CRTH2 V450 (Clone BM16) | BD | Cat#561661 |
| anti-human CD45 V500 (Clone HI30) | BD | Cat#560777 |
| anti-human CD1a biotin (Clone HI149) | Biolegend | Cat#300112 |
| anti-human CD14 biotin (Clone HCD14) | Biolegend | Cat#325624 |
| anti-human CD19 biotin (Clone HIB19) | Biolegend | Cat#302204 |
| anti-human CD123 biotin (Clone 6H6) | Biolegend | Cat#306004 |
| anti-human BDCA2 biotin (Clone GoH3) | Miltenyi | Cat#130-097-923 |
| anti-human FcER1a biotin (Clone AER-37) | eBioscience | Cat#13-5899-82 |
| anti-human CD34 biotin (Clone 581) | Biolegend | Cat#343524 |
| anti-human CD94 biotin (Clone REA113) | Miltenyi | Cat#130-098-967 |
| anti-human TCRab biotin (Clone IP26) | Biolegend | Cat#306704 |
| anti-human TCRgd biotin (Clone B1) | Biolegend | Cat#331206 |
| Streptavidin BV570 | Biolegend | Cat#405227 |
| anti-human CD90 BV605 (Clone 5E10) | Biolegend | Cat#328128 |
| anti-human CD161 BV605 (Clone HP-3G10) | Biolegend | Cat#339915 |
| anti-human CD45 BV650 (Clone HI30) | Biolegend | Cat#304043 |
| anti-human CD56 BV650 (Clone HCD56) | Biolegend | Cat#318343 |
| anti-human CD56 BV711 (Clone HCD56) | Biolegend | Cat#318336 |
| anti-human CD3 BV785 (Clone OKT3) | Biolegend | Cat#317329 |
| anti-human NKG2A (CD159a) PE (Clone 131411) | R&D | Cat#FAB1059P |
| anti-human ROR-gt PE (Clone AFKJS-9) | eBioscience | Cat#12-6988-82 |
| anti-human HLA-DR PE (Clone L243) | eBioscience | Cat#12-9952-41 |
| anti-human CRTH2 PE-CF594 (Clone BM16) | BD | Cat#563501 |
| anti-human CD161 PE-CF594 (Clone HP-3G10) | Biolegend | Cat#339939 |
| anti-human T-bet PE-CF594 (Clone O4-46) | BD | Cat#562467 |
| anti-human NKp44 PE-Cy5 (Clone Z231) | Beckman Coulter | Cat#A66903 |
| anti-human CD117 PE-Cy5.5 (Clone 95C3) | Beckman Coulter | Cat#A66333 |
| anti-human CD127 PE-Cy7 (Clone R34.34) | Beckman Coulter | Cat#A64618 |
| anti-human CD45 BUV395 (Clone HI30) | BD | Cat#563791 |
| anti-human CD45RA BUV737 (Clone HI100) | BD | Cat#564442 |
| anti-human CD3 BV570 (Clone UTCH1) | Biolegend | Cat#300436 |
| anti-human CD69 BV650 (Clone FN50) | Biolegend | Cat#310934 |
| anti-human CD4 BUV496 (Clone SK3) | BD | Cat#564652 |
| anti-human CD8 BUV805 (Clone SK1) | BD | Cat#612889 |
| Biological samples | ||
| Intestinal biopsies from pediatric IBD and non-IBD patients | Karolinska University Hospital or Sachs’ Children and Youth Hospital | N/A |
| Intestinal specimens from adult IBD and non-IBD patients | Karolinska University Hospital | N/A |
| Peripheral blood from pediatric IBD patients | Karolinska University Hospital | N/A |
| Deposited data | ||
| Count matrix from cell ranger | This paper | GEO Accession Number: GSE169136 |
| R scripts | This paper |
https://github.com/ramvinay/Kokkinou_pIBD https://github.com/jtheorell/Kokkinou_gut_neighbors/ |
| Software and algorithms | ||
| R v.4.1.0 | R Core Team | https://cran.r-project.org/ |
| Cell Ranger v.2.1.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software |
| Seurat v.4.1.1 | Butler et al., 201960 | https://github.com/satijalab/seurat/ |
| Harmony v.0.1 | Korsunsky et al., 201961 | https://github.com/immunogenomics/harmony |
| sva v.3.32.1 | Leek et al., 202262 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| Slingshot v.2.2.1 | Street et al., 201863 | https://bioconductor.org/packages/release/bioc/html/slingshot.html |
| WGCNA v.1.70.3 | Langfelder et al., 200823 | https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/ |
| SingleCellExperiment | Amezquita et al., 202064 | https://www.bioconductor.org/packages/release/bioc/html/SingleCellExperiment.html |
| DASeq v1.0.0 | Zhao et al., 202138 | https://github.com/KlugerLab/DAseq |
| DepecheR v.1.14.0 | Theorell et al., 201965 | https://www.bioconductor.org/packages/release/bioc/html/DepecheR.html |
| Other | ||
| LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit | Invitrogen | Cat#L34975 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Invitrogen | Cat#L34957 |
| LIVE/DEAD™ Fixable Green Dead Cell Stain Kit | Invitrogen | Cat#L34970 |
| Collagenase II | Sigma-Aldrich | Cat#C6885 |
| DNAse | Sigma-Aldrich | Cat#10104159001 |
| IMDM | Gibco | Cat#12440-053 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| Chromium™ Single Cell 3' Library & Gel Bead Kit v2, 16 rxns | 10x Genomics | Cat#PN-120237 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Jenny Mjösberg (jenny.mjosberg@ki.se).
Materials availability
The study did not generate new reagents.
Experimental model and subject details
Human patients and study design
Pediatric patients at the Karolinska University Hospital or Sachs’ Children and Youth Hospital were included. All patients were treatment-naive and included upon diagnostic colonoscopy. IBD colitis diagnosis was endoscopically and histologically verified.
For flow cytometry analysis, 16 biopsies were obtained from two areas in the colon of pediatric patients with IBD (n = 19): the endoscopically most affected area (inflamed) and the endoscopically least-affected area (non-inflamed) (Table S1). Some patients had pancolitis or restricted ileal or upper GI-tract involvement, and thus, samples were taken only from the endoscopically inflamed or non-inflamed colonic mucosa, respectively. Non-IBD controls (n = 10), displayed unspecific gastrointestinal symptoms and colonoscopy was performed to rule out IBD diagnosis. All non-IBD controls had endoscopically and histologically normal gut mucosal findings with no signs of active or chronic inflammation.
For the scRNA-seq, treatment-naïve pediatric patients with CD-colitis were included (n = 6). Biopsies were sampled from endoscopically most inflamed and adjacent least inflamed areas of the colon and histologically evaluated (Table S3).
Colonic intestinal specimens from adult patients (Table S5), include non-IBD controls (non-affected intestine from colorectal cancer patients) (n = 8) as well as IBD patients that underwent surgery (n = 8, 2 UC, 6 CD).
Ethical approval
This study was approved by the Swedish Ethical Review Authority (approval numbers 2010/32-31/4, 2018/323-31/1 and 2020-00507). Written informed consents were obtained from all patients and their caregivers.
Characterization and inflammation activity scoring of patients with pIBD
Disease activity was evaluated with endoscopic and histopathological scorings. To evaluate the disease activity endoscopically, we used the endoscopic part of SWIBREG (Swedish Inflammatory Bowel Disease Register) scoring system (Table S2).20 For pediatric CD patients, SES-CD endoscopic score and Paris phenotypic classification were additionally assessed to characterize the patient group. For histopathological scoring, we used a modification of colonic global histologic disease activity score of diagnostic samples collected in close proximity to the research biopsies.21 The evaluation was based on the written statement of an IBD-experienced pathologist. Established IBD biomarkers (shown in Tables S1 and S5), were taken as routine diagnostic examination: blood 0-36 (median 3) and fecal samples 0-60 (median 18) days prior to biopsy sampling and are listed for characterization of the patient groups.
Method details
Cell isolation from intestinal samples and blood
Cell isolation from colonic biopsies was performed as described in Mazzurana et al 2019, where the detailed protocols are provided.66 Briefly, colonic biopsies were collected in wash buffer containing HBSS supplemented with antibiotics. For subsequent enzymatic digestion, wash buffer was removed and replaced with 1mL IMDM (supplemented with antibiotics), collagenase II 0.25 mg/mL; Sigma) and DNase (0.25 mg/mL; Roche). Magnetic stirring was performed at 37°C, 450rpm.
For 3 pIBD patients (Table S1), peripheral blood mononuclear cells (PBMC) were isolated as previously described29,66 using Lymphoprep™ (Stemcell Technologies) with gradient density centrifugation at 2000 rpm for 20 min, room temperature (RT). Cells were collected, washed, counted and subjected to cell staining.
Flow cytometry and cell sorting
For flow cytometry, colon cells or PBMC were stained with dead cell marker (DCM) and antibodies targeting surface antigens for 20-30 min at RT, followed by washing with FACS buffer (PBS containing 1% FCS). The cells were fixed with either 2% PFA for 10 min at RT, or in case of intracellular staining, with Foxp3 Fix/Perm solution kit (eBioscience) for 30 minutes at RT. For the latter, cells were subsequently washed with permeabilization buffer (eBioscience) and stained with antibodies targeting intracellular antigens for 30 min at RT. Samples were immediately acquired on a BD LSR Fortessa or BD Symphony A5 flow cytometer. Analysis of flow cytometry data was performed using FlowJo versions 9.9.6, 10.4.2 and 10.6.2 (TreeStar). A detailed list of the antibodies are shown in the key resources table.
For cell sorting, colonic ILCs, T cells and NK cells were surface-stained with lineage specific antibodies (all antibodies are indicated in the key resources table). Cells were sort-purified in cold RMPI using the BD FACSAria Fusion™. Gating strategy for sorted ILCs, T cells and NK cells is shown in Figure S1A. After sorting cells were immediately subjected to RNA library preparation.
10x genomics single-cell RNA sequencing
Chromium Single-Cell v2 3ʹ Reagents Kit (10x Genomics) was used to generate single-cell RNA Sequencing libraries. In brief, cell suspension was spun down and resuspended in 17.4μl PBS with 0.04% BSA at a final concentration of 1000 cells/μl (target recovery of 10 000 cells). Accordingly, Master Mix was prepared at the adjusted volume of 16.4μl Nuclease-Free water. Cells were loaded on the Chromium Controller Single Cell A Chip to generate Gel Bead Emulsions (GEMs). GEMs were proceeded to GEM-RT incubation, cDNA amplification and library construction following the Chromium™ Single Cell 3′ library Kit v2 User Guide (10x Genomics). Quality controls for cDNA amplification and final barcoded libraries were performed using High Sensitivity DNA Assays from 2100 Agilent Bioanalyzer to assess the quantity and fragment size. Sequencing libraries were loaded at 2.1pM loading concentration on an HiSeq4000 with custom sequencing metrics (single-indexed sequencing run, 28/8/0/98 cycles for R1/i7/i5/R2) (Illumina, San Diego, CA).
Quantification and statistical analysis
Single-cell RNA sequencing data analysis
10x Cell Ranger toolkit (v2.1.1) was used for filtering and aligning reads onto human genome hg19 and counting barcodes and unique molecular identifiers (UMI). Seurat (v4.1.1)60 was used for downstream 10x scRNA-seq data analysis. Briefly, cells with a percentage of mitochondria genes > 5% were considered as dead cells and were removed. Cells with < 200 genes and > 2500 detected genes were excluded. Genes detected in < 3 cells were excluded. Raw UMI counts were scaled and normalized through log transformation. We used Harmony for performing the integration of samples.61 The top 4000 variable genes were selected for PCA and the first 40 dimensions were selected for UMAP and clustering analysis with resolutions 0.6. Seurat function FindAllMarkers (logFC threshold = 0.25) were used to find cluster- and sample-specific marker genes. Further, UMAP clusters were annotated by well-known marker genes and marker genes were detected by the FindAllMarkers function. The functions FeaturePlot, VlnPlot and DotPlot were used for feature plots, violins plots, heatmaps and dot plots, respectively, with default parameters. For subcluster analysis, we used the ComBat function from sva (v3.32.1)62 R package to remove known batches (donors). Wilcoxon test was used for the DE analysis. Trajectory analysis on ILCs was performed using the Slingshot version 2.2.163 R package with Seurat object. We used UMAP coordinates and ILC subclusters 4 and 1 as the starting point (root).
Weighted gene co-expression network analysis (WGCNA)
We used the weighted co-expression network analysis (WGCNA)23 algorithm to identify gene co-expression networks. Specifically, 4000 variable genes were used for calculation of gene-to-gene Pearson correlations. The adjacency matrix was directly used for creation of the topological overlap matrix (TOM). Gene module identification was done with agglomerative hierarchical clustering on the TOM using Ward’s squared linkage (Ward.D2) with a fixed number of 50 modules. Gene counts belonging to the same module were normalized between 0–1 individually and then averaged per cell for calculation of each gene module expression pattern. Modules were prioritized by fitting a general linear model (GLM) using each gene module expression pattern as the effect and the sample metadata (Louvain cluster, Donor) as the explaining variables. Each module and metadata parameter was tested independently. The output of the GLM fit results in the residual metric of “deviance” (devmodel) after fitting the model and “null deviance” (devnull) when considering a NULL model. The percent difference in these two deviances (Δdev) was used as a measure of how much a particular metadata factor explains the expression pattern of a gene module:
This way, modules with higherΔdevwere prioritized for further exploration.
Euclidean neighbor-based smoothing of histology inflammation score
Since samples that are endoscopically scored as non-inflamed or inflamed are composed of samples with variable histological scores we needed a method to better define “inflammatory” and “non-inflammatory” cells to potentially discover genes involved in pIBD inflammation. This method builds on the concept of Euclidean neighbor-based smoothing, which has previously been used both in cytometry and scRNAseq analysis.37,38 Its core assumption is that Euclidean neighbors in the transcriptomic space have similar functions.
The processing of the data was done using the neighSmooth function in the BioConductor package DepecheR, in a similar fashion as in Hart et al37. To even out differences in cell numbers between donors and conditions, the samples were divided into least inflamed (histo score 0-4), intermediate (histo score 7) and most inflamed (histo score 9-12). Then, random subsets of cells were chosen, so that each sample within these overarching inflammation groups had the same number of cells, and simultaneously that the least, intermediate, and most inflamed sample groups had the same number of total cells. In the end, a total number of 22065 neighbor cells were identified, ranging between 1461 and 3681 cells for each sample. After this, the 100 closest Euclidean neighbors were identified from this neighbor cell subset for all cells in the dataset, and each cell was given the average histo score for these 100 nearest neighbors. Each cell was further assessed for the number of samples represented among its 100 nearest neighbors. An arbitrary cutoff of five samples was used to filter out cell populations strongly overrepresented in outlier samples. For this more representative group of cells, aided by a histogram, cutoff values of ≤ 4 for least inflamed cells and ≥ 9 for most inflamed cells were introduced.
To confirm the results of the nearest neighbor-smoothing analysis of the histological scores, DAseq was used.38 As this is a discriminant analysis tool, a few adaptions of the data was necessary. First, all samples with score 7 were excluded. Second, if both samples from one individual had low (<4) or high (≥9) histological scores, the least extreme sample was removed. Subsequently, all remaining cells in the low or the high inflammation group were given these discriminant labels. This was followed by the standard DASeq pipeline. As a prediction threshold, the value of 0.5 was used.
Statistical analyses
Statistical analysis and graphing of the flow cytometry data was performed using Prism 9 software (GraphPad Software, San Diego, US). The sample size for each experiment, the replicate number as well as the precision measures are included in the figure legends.
Acknowledgments
We would like to thank all the patients, and for the pediatric patients, also their parents, for their generous participation. Additionally, we thank all nurses, research nurses, and physicians that contributed to the study and the Flow Cytometry Core Facility, Department of Medicine Huddinge, the Bioinformatics and Expression Analysis core facility at Karolinska Institutet, and the VIB Nucleomics Core, Leuven, Belgium, for their essential help. Support by NBIS (National Bioinformatics Infrastructure Sweden) is gratefully acknowledged. P.C. is financially supported by the Knut and Alice Wallenberg Foundation as part of the National Bioinformatics Infrastructure Sweden at SciLifeLab. This work was supported by the European Crohn's and Colitis Organisation (ECCO) (no grant number) (to A.v.A.); the Swedish Research Council 2018-00384 and 521-2013-2791 (to J.M.); Cancerfonden 170082, 160664, and 190163 (to J.M.); the Foundation for Strategic Research FFL15-0120 (to J.M.); the Knut and Alice Wallenberg Foundation 4-1198/2016 (to J.M.); Karolinska Institutet 2-1536/202 (to J.M.); the European Research Council (ERC) under the European Union Horizon 2020 research and innovation program (grant agreement no. 850963, GUT-SEQ) (to J.M.); the Sigrid Jusélius Foundation (no grant number) (to T.S.); and the Finnish Pediatric Research Foundation (no grant number) (to T.S.).
Author contributions
Conceptualization, J.M., H.R., E. Kokkinou, and A.v.A.; methodology, E. Kokkinou, T.S., R.V.P., A.v.A., J.T., P.C., N.V., and C.S.; investigation, E. Kokkinou, T.S., R.V.P., A.v.A., J.T., P.C., and N.V.; writing – original draft, J.M., H.R., E. Kokkinou, T.S., and R.V.P.; writing – review & editing; P.C., E. Kvedaraite, N.V., M.L., C.S., W.W., A.C., C.A.T., H.S., U.L., C.N., M.L., M.I., M.S., J.-I.H., E.J.V., and Y.T.B.; funding acquisition, J.M. and A.v.A.; resources, E. Kvedaraite, U.L., C.N., M.L., M.I., M.S., J.-I.H., E.J.V., H.R., and J.M.; supervision, J.M.
Declaration of interests
The authors declare no competing interests.
Published: May 8, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101038.
Supplemental information
Data and code availability
-
•
ScRNA-seq data (count matrix) are available at GEO under the accession number GSE169136.
-
•
All codes (R scripts) have been deposited at: https://github.com/ramvinay/Kokkinou_pIBD and https://github.com/jtheorell/Kokkinou_gut_neighbors/
-
•
Software and packages used for this analysis are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Rosen M.J., Dhawan A., Saeed S.A. Inflammatory bowel disease in children and adolescents. JAMA Pediatr. 2015;169:1053–1060. doi: 10.1001/JAMAPEDIATRICS.2015.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Limbergen J., Russell R.K., Drummond H.E., Aldhous M.C., Round N.K., Nimmo E.R., Smith L., Gillett P.M., McGrogan P., Weaver L.T., et al. Definition of phenotypic characteristics of childhood-onset inflammatory bowel disease. Gastroenterology. 2008;135:1114–1122. doi: 10.1053/J.GASTRO.2008.06.081. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y.Z., Li Y.Y. Inflammatory bowel disease: pathogenesis. World J. Gastroenterol. 2014;20:91–99. doi: 10.3748/WJG.V20.I1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powrie F., Leach M.W., Mauze S., Caddle L.B., Coffman R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993;5:1461–1471. doi: 10.1093/INTIMM/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 5.Mitsialis V., Wall S., Liu P., Ordovas-Montanes J., Parmet T., Vukovic M., Spencer D., Field M., McCourt C., Toothaker J., et al. Single-cell analyses of colon and blood reveal distinct immune cell signatures of ulcerative colitis and Crohn’s disease. Gastroenterology. 2020;159:591–608.e10. doi: 10.1053/J.GASTRO.2020.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smids C., Horjus Talabur Horje C.S., Drylewicz J., Roosenboom B., Groenen M.J.M., van Koolwijk E., Wahab P.J., van Lochem E.G., Wahab P.J. Intestinal T cell profiling in inflammatory bowel disease: linking T cell subsets to disease activity and disease course. J. Crohns Colitis. 2018;12:465–475. doi: 10.1093/ECCO-JCC/JJX160. [DOI] [PubMed] [Google Scholar]
- 7.Maxwell J.R., Zhang Y., Brown W.A., Smith C.L., Byrne F.R., Fiorino M., Stevens E., Bigler J., Davis J.A., Rottman J.B., et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity. 2015;43:739–750. doi: 10.1016/J.IMMUNI.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 8.Trejdosiewicz L.K., Calabrese A., Smart C.J., Oakes D.J., Howdle P.D., Crabtree J.E., Losowsky M.S., Lancaster F., Boylston A.W. Gamma delta T cell receptor-positive cells of the human gastrointestinal mucosa: occurrence and V region gene expression in Heliobacter pylori-associated gastritis, coeliac disease and inflammatory bowel disease - PubMed. Clin. Exp. Immunol. 1991;84:440–444. [PMC free article] [PubMed] [Google Scholar]
- 9.Serriari N.E., Eoche M., Lamotte L., Lion J., Fumery M., Marcelo P., Chatelain D., Barre A., Nguyen-Khac E., Lantz O., et al. Innate mucosal-associated invariant T (MAIT) cells are activated in inflammatory bowel diseases. Clin. Exp. Immunol. 2014;176:266–274. doi: 10.1111/CEI.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Amico F., Peyrin-Biroulet L., Danese S., Fiorino G. New drugs in the pipeline for the treatment of inflammatory bowel diseases: what is coming? Curr. Opin. Pharmacol. 2020;55:141–150. doi: 10.1016/J.COPH.2020.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Colonna M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity. 2018;48:1104–1117. doi: 10.1016/J.IMMUNI.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernink J.H., Peters C.P., Munneke M., Te Velde A.A., Meijer S.L., Weijer K., Hreggvidsdottir H.S., Heinsbroek S.E., Legrand N., Buskens C.J., et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013;14:221–229. doi: 10.1038/NI.2534. [DOI] [PubMed] [Google Scholar]
- 13.Forkel M., VanTol S., Höög C., Michaëlsson J., Almer S., Mjösberg J. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established Crohn’s disease and ulcerative colitis. J. Crohns Colitis. 2019;13:67–78. doi: 10.1093/ECCO-JCC/JJY119. [DOI] [PubMed] [Google Scholar]
- 14.Geremia A., Arancibia-Cárcamo C.V., Fleming M.P.P., Rust N., Singh B., Mortensen N.J., Travis S.P.L., Powrie F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011;208:1127–1133. doi: 10.1084/JEM.20101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin J.C., Chang C., Boschetti G., Ungaro R., Giri M., Grout J.A., Gettler K., Chuang L.S., Nayar S., Greenstein A.J., et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell. 2019;178:1493–1508.e20. doi: 10.1016/J.CELL.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uniken Venema W.T., Voskuil M.D., Vila A.V., van der Vries G., Jansen B.H., Jabri B., Faber K.N., Dijkstra G., Xavier R.J., Wijmenga C., et al. Single-cell RNA sequencing of blood and ileal T cells from patients with Crohn’s disease reveals tissue-specific characteristics and drug targets. Gastroenterology. 2019;156:812–815.e22. doi: 10.1053/J.GASTRO.2018.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang B., Chen Z., Geng L., Wang J., Liang H., Cao Y., Chen H., Huang W., Su M., Wang H., et al. Mucosal profiling of pediatric-onset colitis and IBD reveals common pathogenics and therapeutic pathways. Cell. 2019;179:1160–1176.e24. doi: 10.1016/J.CELL.2019.10.027. [DOI] [PubMed] [Google Scholar]
- 18.Corridoni D., Antanaviciute A., Gupta T., Fawkner-Corbett D., Aulicino A., Jagielowicz M., Parikh K., Repapi E., Taylor S., Ishikawa D., et al. Single-cell atlas of colonic CD8+ T cells in ulcerative colitis. Nat. Med. 2020;26:1480–1490. doi: 10.1038/s41591-020-1003-4. [DOI] [PubMed] [Google Scholar]
- 19.Jaeger N., Gamini R., Cella M., Schettini J.L., Bugatti M., Zhao S., Rosadini C.V., Esaulova E., Di Luccia B., Kinnett B., et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat. Commun. 2021;121:1921–2012. doi: 10.1038/s41467-021-22164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludvigsson J.F., Andersson M., Bengtsson J., Eberhardson M., Fagerberg U.L., Grip O., Halfvarson J., Hjortswang H., Jäghult S., Karling P., et al. Swedish inflammatory bowel disease register (SWIBREG) - a nationwide quality register. Scand. J. Gastroenterol. 2019;54:1089–1101. doi: 10.1080/00365521.2019.1660799. [DOI] [PubMed] [Google Scholar]
- 21.D’Haens G.R., Geboes K., Peeters M., Baert F., Penninckx F., Rutgeerts P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114:262–267. doi: 10.1016/S0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 22.Yudanin N.A., Schmitz F., Flamar A.L., Thome J.J.C., Tait Wojno E., Moeller J.B., Schirmer M., Latorre I.J., Xavier R.J., Farber D.L., et al. Spatial and temporal mapping of human innate lymphoid cells reveals elements of tissue specificity. Immunity. 2019;50:505–519.e4. doi: 10.1016/J.IMMUNI.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559–613. doi: 10.1186/1471-2105-9-559/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mazzurana L., Czarnewski P., Jonsson V., Wigge L., Ringnér M., Williams T.C., Ravindran A., Björklund Å.K., Säfholm J., Nilsson G., et al. Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res. 2021;31:554–568. doi: 10.1038/S41422-020-00445-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadik A., Somarribas Patterson L.F., Öztürk S., Mohapatra S.R., Panitz V., Secker P.F., Pfänder P., Loth S., Salem H., Prentzell M.T., et al. IL4I1 is a metabolic immune checkpoint that activates the AHR and promotes tumor progression. Cell. 2020;182:1252–1270.e34. doi: 10.1016/J.CELL.2020.07.038. [DOI] [PubMed] [Google Scholar]
- 26.Santarlasci V., Maggi L., Mazzoni A., Capone M., Querci V., Rossi M.C., Beltrame L., Cavalieri D., De Palma R., Liotta F., et al. IL-4-induced gene 1 maintains high Tob1 expression that contributes to TCR unresponsiveness in human T helper 17 cells. Eur. J. Immunol. 2014;44:654–661. doi: 10.1002/EJI.201344047. [DOI] [PubMed] [Google Scholar]
- 27.Lin C.C., Bradstreet T.R., Schwarzkopf E.A., Sim J., Carrero J.A., Chou C., Cook L.E., Egawa T., Taneja R., Murphy T.L., et al. Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nat. Commun. 2014;5:3551. doi: 10.1038/NCOMMS4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanda M., Yamanaka H., Kojo S., Usui Y., Honda H., Sotomaru Y., Harada M., Taniguchi M., Suzuki N., Atsumi T., et al. Transcriptional regulator Bhlhe40 works as a cofactor of T-bet in the regulation of IFN-γ production in iNKT cells. Proc. Natl. Acad. Sci. USA. 2016;113:E3394–E3402. doi: 10.1073/PNAS.1604178113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao A., Strauss O., Kokkinou E., Bruchard M., Tripathi K.P., Schlums H., Carrasco A., Mazzurana L., Konya V., Villablanca E.J., et al. Cytokines regulate the antigen-presenting characteristics of human circulating and tissue-resident intestinal ILCs. Nat. Commun. 2020;11:2049. doi: 10.1038/S41467-020-15695-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hepworth M.R., Fung T.C., Masur S.H., Kelsen J.R., McConnell F.M., Dubrot J., Withers D.R., Hugues S., Farrar M.A., Reith W., et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science. 2015;348:1031–1035. doi: 10.1126/SCIENCE.AAA4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehmann F.M., von Burg N., Ivanek R., Teufel C., Horvath E., Peter A., Turchinovich G., Staehli D., Eichlisberger T., Gomez de Agüero M., et al. Microbiota-induced tissue signals regulate ILC3-mediated antigen presentation. Nat. Commun. 2020;11:1794. doi: 10.1038/S41467-020-15612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Von Burg N., Chappaz S., Baerenwaldt A., Horvath E., Bose Dasgupta S., Ashok D., Pieters J., Tacchini-Cottier F., Rolink A., Acha-Orbea H., Finke D. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc. Natl. Acad. Sci. USA. 2014;111:12835–12840. doi: 10.1073/PNAS.1406908111/SUPPL_FILE/PNAS.201406908SI.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bielecki P., Riesenfeld S.J., Hütter J.C., Torlai Triglia E., Kowalczyk M.S., Ricardo-Gonzalez R.R., Lian M., Amezcua Vesely M.C., Kroehling L., Xu H., et al. Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature. 2021;592:128–132. doi: 10.1038/S41586-021-03188-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kokkinou E., Pandey R.V., Mazzurana L., Gutierrez-Perez I., Tibbitt C.A., Weigel W., Soini T., Carrasco A., Rao A., Nagasawa M., et al. CD45RA+CD62L− ILCs in human tissues represent a quiescent local reservoir for the generation of differentiated ILCs. Sci. Immunol. 2022;7:eabj8301. doi: 10.1126/SCIIMMUNOL.ABJ8301. [DOI] [PubMed] [Google Scholar]
- 35.Mitsuishi T. Correlation between histological findings and endoscopic findings in patients with ulcerative colitis: Basal plasmacytosis is an important finding suggesting active inflammation. JGH Open. 2019;3:100–104. doi: 10.1002/JGH3.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geboes K., Riddell R., Öst A., Jensfelt B., Persson T., Löfberg R. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47:404–409. doi: 10.1136/GUT.47.3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hart G.T., Tran T.M., Theorell J., Schlums H., Arora G., Rajagopalan S., Sangala A.D.J., Welsh K.J., Traore B., Pierce S.K., et al. Adaptive NK cells in people exposed to Plasmodium falciparum correlate with protection from malaria. J. Exp. Med. 2019;216:1280–1290. doi: 10.1084/JEM.20181681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao J., Jaffe A., Li H., Lindenbaum O., Sefik E., Jackson R., Cheng X., Flavell R.A., Kluger Y. Detection of differentially abundant cell subpopulations in scrna-seq data. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/PNAS.2100293118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huhn O., Ivarsson M.A., Gardner L., Hollinshead M., Stinchcombe J.C., Chen P., Shreeve N., Chazara O., Farrell L.E., Theorell J., et al. Distinctive phenotypes and functions of innate lymphoid cells in human decidua during early pregnancy. Nat. Commun. 2020;11:381. doi: 10.1038/S41467-019-14123-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breloer M., Fleischer B. CD83 regulates lymphocyte maturation, activation and homeostasis. Trends Immunol. 2008;29:186–194. doi: 10.1016/J.IT.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 41.Schoepfer A., Santos J., Fournier N., Schibli S., Spalinger J., Vavricka S., Safroneeva E., Aslan N., Rogler G., Braegger C., Nydegger A. Systematic analysis of the impact of diagnostic delay on bowel damage in paediatric versus adult onset Crohn’s disease. J. Crohns Colitis. 2019;13:1334–1342. doi: 10.1093/ECCO-JCC/JJZ065. [DOI] [PubMed] [Google Scholar]
- 42.Fuchs A., Vermi W., Lee J.S., Lonardi S., Gilfillan S., Newberry R.D., Cella M., Colonna M. Intraepithelial type 1 innate lymphoid cells are a unique subset of cytokine responsive interferon-γ-producing cells. Immunity. 2013;38:769–781. doi: 10.1016/J.IMMUNI.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoorweg K., Peters C.P., Cornelissen F., Aparicio-Domingo P., Papazian N., Kazemier G., Mjösberg J.M., Spits H., Cupedo T. Functional differences between human NKp44- and NKp44+ RORC+ innate lymphoid cells. Front. Immunol. 2012;3:72. doi: 10.3389/FIMMU.2012.00072/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sonnenberg G.F., Monticelli L.A., Elloso M.M., Fouser L.A., Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/J.IMMUNI.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim A.I., Li Y., Lopez-Lastra S., Stadhouders R., Paul F., Casrouge A., Serafini N., Puel A., Bustamante J., Surace L., et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell. 2017;168:1086–1100.e10. doi: 10.1016/j.cell.2017.02.021. [DOI] [PubMed] [Google Scholar]
- 46.Lee J.S., Cella M., McDonald K.G., Garlanda C., Kennedy G.D., Nukaya M., Mantovani A., Kopan R., Bradfield C.A., Newberry R.D., Colonna M. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2011;13:144–151. doi: 10.1038/NI.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klose C.S.N., Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016;177:765–774. doi: 10.1038/ni.3489. [DOI] [PubMed] [Google Scholar]
- 48.Mjösberg J.M., Trifari S., Crellin N.K., Peters C.P., Van Drunen C.M., Piet B., Fokkens W.J., Cupedo T., Spits H. Human IL-25-and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011;12:1055–1062. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 49.Simoni Y., Fehlings M., Kløverpris H.N., McGovern N., Koo S.L., Loh C.Y., Lim S., Kurioka A., Fergusson J.R., Tang C.L., et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity. 2017;46:148–161. doi: 10.1016/J.IMMUNI.2016.11.005/ATTACHMENT/38FF1BFA-4E9D-4421-A0AC-4B3B45F9B9AA/MMC1.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuwahara M., Yamashita M., Shinoda K., Tofukuji S., Onodera A., Shinnakasu R., Motohashi S., Hosokawa H., Tumes D., Iwamura C., et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-β and suppresses TH2 differentiation. Nat. Immunol. 2012;13:778–786. doi: 10.1038/NI.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hughes T., Briercheck E.L., Freud A.G., Trotta R., McClory S., Scoville S.D., Keller K., Deng Y., Cole J., Harrison N., et al. AHR prevents human IL-1R1hi ILC3 differentiation to natural killer cells. Cell Rep. 2014;8:150–162. doi: 10.1016/J.CELREP.2014.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Welte S., Kuttruff S., Waldhauer I., Steinle A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat. Immunol. 2006;7:1334–1342. doi: 10.1038/NI1402. [DOI] [PubMed] [Google Scholar]
- 53.Tewary P., Yang D., De La Rosa G., Li Y., Finn M.W., Krensky A.M., Clayberger C., Oppenheim J.J. Granulysin activates antigen-presenting cells through TLR4 and acts as an immune alarmin. Blood. 2010;116:3465–3474. doi: 10.1182/BLOOD-2010-03-273953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krabbendam L., Heesters B.A., Kradolfer C.M.A., Haverkate N.J.E., Becker M.A.J., Buskens C.J., Bemelman W.A., Bernink J.H., Spits H. CD127+ CD94+ innate lymphoid cells expressing granulysin and perforin are expanded in patients with Crohn’s disease. Nat. Commun. 2021;12:5841. doi: 10.1038/S41467-021-26187-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allez M., Tieng V., Nakazawa A., Treton X., Pacault V., Dulphy N., Caillat-Zucman S., Paul P., Gornet J.M., Douay C., et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology. 2007;132:2346–2358. doi: 10.1053/J.GASTRO.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 56.Chen L., He Z., Reis B.S., Gelles J.D., Chipuk J.E., Ting A.T., Spicer J.A., Trapani J.A., Furtado G.C., Lira S.A. IFN-γ + cytotoxic CD4 + T lymphocytes are involved in the pathogenesis of colitis induced by IL-23 and the food colorant Red 40. Cell. Mol. Immunol. 2022;19:777–790. doi: 10.1038/S41423-022-00864-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma J., Tibbitt C.A., Georen S.K., Christian M., Murrell B., Cardell L.O., Bachert C., Coquet J.M. Single-cell analysis pinpoints distinct populations of cytotoxic CD4+T cells and an IL-10+CD109+TH2 cell population in nasal polyps. Sci. Immunol. 2021;6:6356. doi: 10.1126/SCIIMMUNOL.ABG6356/SUPPL_FILE/SCIIMMUNOL.ABG6356_TABLES_S1_TO_S7.ZIP. [DOI] [PubMed] [Google Scholar]
- 58.Mogilenko D.A., Shpynov O., Andhey P.S., Arthur L., Swain A., Esaulova E., Brioschi S., Shchukina I., Kerndl M., Bambouskova M., et al. Comprehensive profiling of an aging immune system reveals clonal GZMK + CD8 + T cells as conserved hallmark of inflammaging. Immunity. 2021;54:99–115.e12. doi: 10.1016/J.IMMUNI.2020.11.005. [DOI] [PubMed] [Google Scholar]
- 59.Perez R.K., Gordon M.G., Subramaniam M., Kim M.C., Hartoularos G.C., Targ S., Sun Y., Ogorodnikov A., Bueno R., Lu A., et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science. 2022;376:eabf1970. doi: 10.1126/SCIENCE.ABF1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/NBT.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/S41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/BIOINFORMATICS/BTS034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018;19:477. doi: 10.1186/S12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amezquita R.A., Lun A.T.L., Becht E., Carey V.J., Carpp L.N., Geistlinger L., Marini F., Rue-Albrecht K., Risso D., Soneson C., et al. Orchestrating single-cell analysis with Bioconductor. Nat. Methods. 2019;172:137–145. doi: 10.1038/s41592-019-0654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Theorell A., Bryceson Y.T., Theorell J. Determination of essential phenotypic elements of clusters in high-dimensional entities—depeche. PLoS One. 2019;14 doi: 10.1371/JOURNAL.PONE.0203247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mazzurana L., Forkel M., Rao A., Van Acker A., Kokkinou E., Ichiya T., Almer S., Höög C., Friberg D., Mjösberg J. Suppression of Aiolos and Ikaros expression by lenalidomide reduces human ILC3-ILC1/NK cell transdifferentiation. Eur. J. Immunol. 2019;49:1344–1355. doi: 10.1002/EJI.201848075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
ScRNA-seq data (count matrix) are available at GEO under the accession number GSE169136.
-
•
All codes (R scripts) have been deposited at: https://github.com/ramvinay/Kokkinou_pIBD and https://github.com/jtheorell/Kokkinou_gut_neighbors/
-
•
Software and packages used for this analysis are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.