

Abstract
The CANTOS and colchicine trials suggest an important role of inflammasomes and their major product IL-1β in human atherosclerotic cardiovascular disease (CVD). Moreover, studies in mouse models indicate a causal role of inflammasomes and IL-1β in atherosclerosis. However, recent studies have led to a more granular view of the role of inflammasomes in atherosclerosis. Studies in hyperlipidemic mouse models suggest that prominent activation of the NLRP3 inflammasome requires a second hit such defective cholesterol efflux, defective DNA repair, clonal hematopoiesis or diabetes. Similarly in humans some mutations promoting clonal hematopoiesis increase coronary artery disease risk in part by promoting inflammasome activation. Recent studies in mice and humans point to a wider role of the AIM2 inflammasome in promoting CVD including in some forms of clonal hematopoiesis and diabetes. These developments suggest a precision medicine approach in which treatments targeting inflammasomes or IL-1β might be best employed in clinical settings involving increased inflammasome activation.
Keywords: cardiovascular disease, clonal hematopoiesis, diabetes, inflammasome, interleukin 1β, macrophage, mouse models
Introduction
Cardiovascular disease (CVD) comprising ischemic heart disease and stroke remains the major cause of death in the US1 and is a leading cause of mortality and disability globally.2 The downward trend in CVD in part reflecting treatment of traditional risk factors has stalled and reversed in recent years, paralleling the rise in obesity, metabolic syndrome and diabetes.3, 4 Even in clinical trials with marked LDL lowering, there is a large burden of residual atherosclerotic CVD, pointing to the need for new treatment and prevention approaches.5 Recent clinical trials suggest that inflammation mediated by inflammasome activation, which results in cellular release of the cytokines interleukin 1β (IL-1β) and IL-18, contributes to residual CVD risk. Thus, anti-inflammatory therapies using IL-1β antibodies6 or colchicine7, 8 have shown amelioration of CVD in subjects on lipid-lowering therapies, confirming the role of inflammation in CVD and suggesting an important role of inflammasomes in residual CVD risk in humans.9 However, IL-1β antibodies caused a small increase in infections, including fatal sepsis, and colchicine doubled the risk of pneumonia. This suggests the need for novel anti-inflammatory treatments that may be less immunosuppressive and for targeting populations at greater inflammatory risk. An improved mechanistic understanding of the role of inflammasomes in athero-thrombotic CVD might help to find new treatments acting on inflammasomes or their downstream products, and to identify patients who would benefit most from such treatments. We will review the role of inflammasomes in atherosclerotic CVD, focusing on NLRP3 (NACHT [nucleotide triphosphatase containing domain], leucine rich repeat [LRR]- and pyrin domain [PYD]-containing protein 3) and AIM2 (Absent In Melanoma 2) inflammasomes and their roles in atherosclerosis and its complications. We will emphasize more recent developments in the field, while referring the reader to several outstanding reviews relevant to this topic.9–12 Inflammasome activation also has an important role in initiation and progression of heart failure as reviewed elsewhere.10, 13–15
NLRP3 and AIM2 inflammasomes
Inflammasomes are cytoplasmic supramolecular complexes that form primarily in innate immune cells in response to exogenous microbial invasion or endogenous damage signals.16 NLRP3 is an inflammasome sensor. NLRP3 inflammasome activation is a two-step process with priming events increasing the expression of inflammasome components and mediators, such as Nlrp3, Casp1 and Il1b, and an activation step involving oligomerization and conformational rearrangement of NLRP3, binding of the PYD domain to the adaptor ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain) and activation of the effector, caspase-1.17 Active caspase-1 mediates cleavage and activation of pro-IL-1β, pro-IL-18 and Gasdermin D (GSDMD);17, 18 cleaved GSDMD N-terminal fragments oligomerize and form membrane pores that permit the secretion of active IL-1β and IL-18 as well as release of pro-inflammatory cell contents such as high mobility group proteins and ATP (Figure 1). GSDMD pore formation can also lead to a programmed type of cell death termed pyroptosis.17 IL-1β promotes inflammasome priming via its receptor IL-1R1 which leads to further NLRP3 inflammasome activation through an autocrine or paracrine positive feedback loop.
Figure 1. Mechanisms of NLRP3 inflammasome activation in lesions of atherosclerosis.
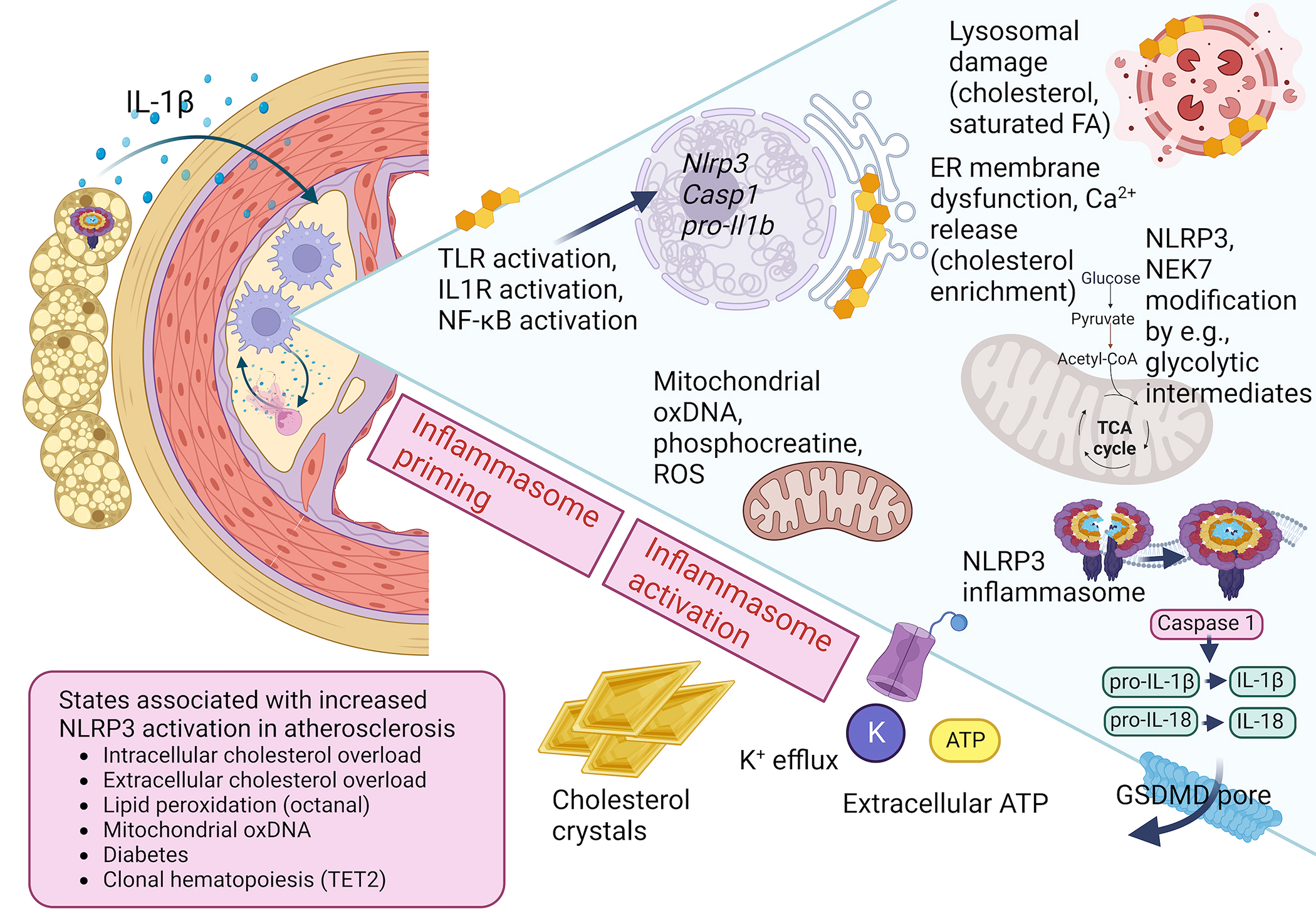
Classical activation of the NLRP3 inflammasome in macrophages requires a priming step, mediated by TLR activation or IL1R activation, NF-κB activation, and subsequent induction of Nlrp3, Il1b and Casp1 gene expression. The NLRP3 inflammasome complex is then assembled and activated by a number of putative stimuli, including mitochondrial products, lysosomal membrane damage, ER membrane dysfunction, extracellular ATP, and K+ efflux. Cholesterol or saturated fatty acid accumulation in intracellular organelles is believed to contribute to NLRP3 activation in lesions of atherosclerosis. Extracellular cholesterol crystals can damage the plasma membrane, leading to NLRP3 activation. Non-vascular tissues, such as PVAT, can contribute to IL-1β release through NLRP3 activation, in turn promoting vascular dysfunction and atherosclerosis. Components of the NLRP3 inflammasome are also influenced by modification by phosphorylation, ubiquitylation, dicarboxypropylation and deglutathionylation; some of these processes are mediated by intermediates of cellular metabolism. The ultimate result of NLRP3 inflammasome activation is the activation of caspase-1 and subsequent cleavage and generation and release of mature IL-1β and IL-18, as well as cleavage of GSDMD, which can result in GSDMD-pore formation and cell death through pyroptosis. States associated with a heightened activation of the NLRP3 inflammasome include CH TET2 mutations and diabetes. The membrane-associated cage structure of the NLRP3 inflammasome is based on the recent study by Andreeva and colleagues.27 Created with BioRender.com
While danger signals are involved in the NLRP3 priming step, acting via toll-like receptors (TLRs) and NF-kB activation, the activation step may be promoted by a variety of different damage-associated molecular patterns (DAMPs) and other factors, including extracellular ATP, bacterial toxins and membrane damage by crystals of uric acid or cholesterol.19 Moreover, increased mitochondrial reactive oxygen species (ROS) formation has often been associated with NLRP3 inflammasome activation. In response to lipopolysaccharide (LPS; a TLR4 ligand) preincubation and NLRP3 activation signals, oxidative damage of newly synthesized mitochondrial DNA may lead to release of oxidized DNA fragments that activate the NLRP3 inflammasome,20 possibly via direct interaction with NLRP3.21 However, a recent study has suggested that mitochondrial generation of creatine phosphate, which may promote formation of cytosolic ATP and activation of NLRP3 via its NACHT domain, mediates activation rather than ROS generated in the mitochondrial electron transport chain.22 Almost all NLRP3 activators converge ultimately on K+ efflux19 (Figure 1).
The NLRP3 inflammasome is tightly regulated at several additional levels. For example, NLRP3 is subject to multiple posttranscriptional modifications including phosphorylation and ubiquitylation. Phosphorylation of specific serine residues leads to binding of the deubiquitylating enzyme BRCC3 (BRCA1/BRCA2-Containing Complex Subunit 3) whose activity controls NLRP3 activation.23, 24 A distinct phosphorylation site inhibits NLRP3 activation in response to prostaglandin E2 and subsequent activation of protein kinase A.25 GSDMD pore formation may also be regulated by multiple processes. Thus, oxidation of C92 in GSDMD by ROS has been shown to promote oligomerization.26 An increased generation of ROS in macrophages might therefore promote IL-1β and IL-18 release and pyroptosis by increasing GSDMD pore formation in addition to activating the NLRP3 inflammasome upstream of GSDMD cleavage.
The NLRP3 inflammasome activity is also regulated by subcellular localization. Recent cryo-electron microscopy (cryo-EM) studies have revealed that inactive NLRP3 forms a double ring cage held together by leucine-rich repeat (LRR) interactions that shield the PYRIN domain and prevent premature activation.27 The centrosomally located, mitotic kinase NEK7 (NIMA-related kinase 7) licenses the assembly and activation of the NLRP3 inflammasome in interphase independent of its catalytic activity;28, 29 given that amounts of NEK7 in the cell are limiting, this makes NLRP3 inflammasome activation and mitosis mutually exclusive processes.29 An integrated model based in part on cryo-EM imaging of NLRP3 bound to ASC and NEK7 in its active state has suggested that following priming NLRP3 is localized both in the cytosol and on the trans-Golgi network in monomeric and cage forms, respectively.27, 30 On stimulation, membrane-bound NLRP3 undergoes conformational changes in the cage form and the trans-Golgi network disperses into vesicles. These NLRP3-containing vesicles are trafficked on microtubules to the microtubule organizing center,31, 32 where centrosomally-localized NEK7 may interact with NLRP3 to open the cage, possibly leading to rearrangement into active NLRP3 oligomers. Recruitment of ASC then helps to complete NLRP3 disc formation and transduce the activation signal. An alternative NLRP3 activation mechanism has been described in human monocytes that involves TLR4-TRIF-RIPK1-FADD-CASP8 signaling and does not require centrosomal localization, NEK7 or ASC speck formation.33 Furthermore, in human induced pluripotent stem cell-derived macrophages an IKKβ-PI4P trans-Golgi dependent mechanism bypasses the need for NEK7 in NLRP3 inflammasome activation.34 Together these studies reveal multiple pathways of NLRP3 inflammasome activation and possible species differences in the requirement for NEK7.
The specific NLRP3 inhibitor MCC95035 that has been widely used in preclinical studies to define the role of NLRP3 in metabolic diseases such as atherosclerosis36 and diabetes37 interacts directly with the NLRP3 NACHT domain, preventing ATP hydrolysis and keeping NLRP3 inactive.38, 39 The cryo-EM structure of MCC950 bound to NLRP3 has revealed a complex, specific interaction, paving the way for rational design of future NLRP3 inhibitors.40 It has been speculated that specific NLRP3 inhibition may be less immunosuppressive than inhibition of downstream components common to all inflammasomes,9 but this remains to be determined.
AIM2 is an interferon-induced sensor of fragments of double stranded DNA (dsDNA) via sequence-independent electrostatic interactions.41–43 dsDNA-induced polymerization of AIM2 leads to inflammasome assembly, ASC binding and caspase-1 activation (Figure 2). AIM2 has an important role in detecting dsDNA derived from bacteria or viruses and plays a key role in host defense.44 AIM2 can also be activated by DNA replication stress and oxidative damage of nuclear DNA during neurodevelopment45 or in response to radiation-induced nuclear DNA damage in enterocytes or bone marrow cells.46 DNA fragments recognized by AIM2 can be derived from mitochondria, chromosomal DNA and potentially from extracellular sources such as circulating DNA fragments or neutrophil extracellular traps (NETs).47, 48
Figure 2. Mechanisms of AIM2 inflammasome activation in lesions of atherosclerosis.
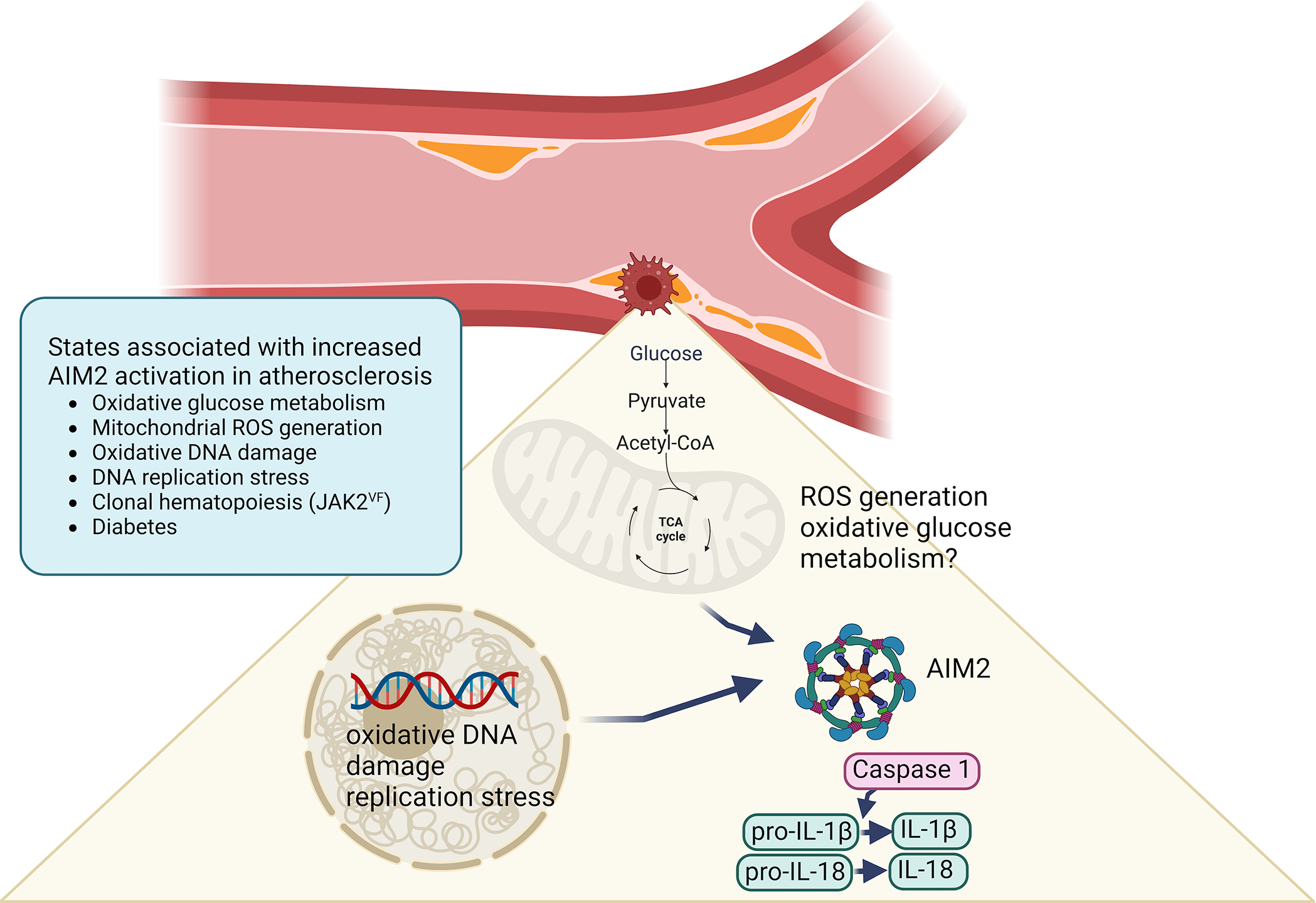
Emerging evidence suggests a causative role of the AIM2 inflammasome in atherosclerosis in the setting of clonal hematopoiesis due to the JAK2VF mutation and in diabetes. The mechanism of AIM2 activation in lesion macrophages has been proposed to be due to increased oxidative glucose metabolism, mitochondrial ROS generation, oxidative DNA damage and DNA replication stress.52 Created with BioRender.com
Role of the NLRP3 inflammasome in murine atherosclerosis
A role of the NLRP3 inflammasome in promoting atherosclerosis was first shown in Western diet-fed LDL receptor-deficient (Ldlr−/−) mice.49 Genetic deletion of the inflammasome components Nlrp3, Asc or Il1b led to significantly reduced early atherosclerosis. While many reports have confirmed these findings (reviewed in11), others have found no significant impact of genetic deletion or inhibition of inflammasomes on atherosclerosis in high cholesterol diet-fed apolipoprotein E-deficient (Apoe−/−) or Ldlr−/− mice.36, 50–54 The reasons for the different results are uncertain but could be related to female-specific effects of NLRP3,55 stage of lesion development, microbiome effects or other experimental variables. Some studies have found no impact of deletion of Nlrp3 or Casp1/11, or inhibition of IL-1β in Western diet-fed female Ldlr−/− mice in either early or late lesions.56 The variable results could be interpreted to suggest that there is only a low level of underlying inflammasome activation in the standard mouse atherosclerosis models. When additional factors are introduced to promote inflammasome activation, by deleting genes mediating cholesterol efflux51 or oxidative DNA damage repair,55 introducing mutations that cause clonal hematopoiesis,52, 57 or inducing diabetes37, 58 there is a more clearcut impact of NLRP3 inflammasomes on atherosclerosis (Figure 1).
Monocytes infiltrating atherosclerotic lesions and lesion macrophages may undergo NLRP3 inflammasome priming in response to TLR signaling induced by oxidized LDL59 or other danger signals.9 Several different factors may be involved in the second step of inflammasome activation in atherosclerosis. NLRP3 activation may result from extracellular ATP released from dying cells, exposure of macrophages to extracellular cholesterol crystals or formation of cholesterol crystals from modified LDL in the endosomal system leading to lysosomal damage and cathepsin release.49, 60 Cholesterol crystals are prominent in advanced atherosclerotic lesions and likely are involved in NLRP3 inflammasome activation. More generally, cholesterol crystal accumulation in tissues has been associated with inflammation and impaired function. Cholesterol crystal accumulation in the skin in ACAT1 (acyl-CoA cholesterol acyltransferase 1) knockout mice has been associated with massive xanthomatosis and inflammatory cell infiltration,61 while accumulation of ageing- or injury-associated myelin debris in the phagocytes in the central nervous system is associated with cholesterol crystal formation and NLRP3 inflammasome activation, with reversal by stimulation of cholesterol efflux by cyclodextrin or LXR activator treatment.62
In contrast to these observation, small refractile crystals observed in early foam cell atherosclerotic lesions49 may represent extracellular cholesteryl ester droplets that are in liquid or liquid crystal form at body temperature but are only formed when samples are cooled below body temperature63, 64 so the role of cholesterol crystals in early lesions remains uncertain. In mice with myeloid cell-deficiency of the cholesterol efflux promoting transporters ABCA1 and ABCG1, macrophage cholesterol accumulation and NLRP3 inflammasome activation occurs without evidence of lysosomal damage.51 While part of the mechanism involves plasma membrane cholesterol accumulation, increased TLR4 signaling and inflammasome priming, the response to activation signals also appears to be increased.51, 65 Moreover, accumulation of cholesterol in the ER may be required for NLRP3 inflammasome activation.66 Thus, it seems that while cholesterol crystals can activate the NLRP3 inflammasome in response to macrophage membrane damage in advanced lesions, cholesterol accumulation in membranes and intracellular organelles is also probably involved in NLRP3 inflammasome priming and activation and involves mechanisms that remain to be clearly defined.
A recent study reported that activation of Olfactory Receptor 2 (OLR2) in vascular macrophages by its ligand octanal promoted atherosclerosis via the NLRP3 inflammasome.67 Injection of octanal, a lipid peroxidation product found in plasma and atherosclerotic lesions, promoted atherosclerosis, while transplantation of Olr2−/− bone marrow into Ldlr−/− mice reduced atherosclerosis and blunted the impact of octanal injection, revealing a novel, therapeutically targetable mechanism of atherogenesis. Octanal increased cAMP, Ca2+ fluxes and ROS production in macrophages and LPS+octanal treatment increased IL-1β and lactate dehydrogenase secretion (a marker of membrane permeability), suggesting that octanal might provide a second signal to activate the NLRP3 inflammasome. However, secretion of IL-1α (which is not a direct product of the NLRP3 inflammasome9, 68), TNF-α and CCL4 were also increased by octanal+LPS treatment suggesting a widespread pro-inflammatory effect of octanol treatment; in vivo evidence that atherogenic properties of octanal were dependent on NLRP3 was not provided. Thus, the precise in vivo pro-atherogenic mechanisms underlying effects of lipid peroxidation products and the in vivo role of inflammasomes in this process are somewhat uncertain.
Together, these studies highlight roles for cellular and lesion overaccumulation of cholesterol and maybe lipid peroxidation products in NLRP3 activation and suggest that the NLRP3 inflammasome plays a much more prominent role in atherogenesis in mouse models when multiple exacerbating factors are involved.
The AIM2 inflammasome and atherosclerosis
Although much less studied than the NLRP3 inflammasome, emerging evidence in mice and humans suggests a role of the AIM2 inflammasome in atherosclerotic CVD. The first study to implicate the AIM2 inflammasome in murine atherosclerosis was performed in high cholesterol diet-fed Apoe−/− mice.69 Genetic deletion of Aim2 or AIM2 inhibition using synthetic oligonucleotides reduced the levels of both IL-1β and IL-18 in atherosclerotic lesions, decreased necrotic core size and increased fibrous cap thickness, suggesting plaque stabilization. The authors speculated that sources of extracellular DNA such as NETs might be providing DNA fragments for AIM2 activation. The AIM2 inflammasome has also been implicated in atherosclerosis and plaque instability in clonal hematopoiesis in both mice and humans and in plaque area in diabetic mice58 (see below). The underlying mechanisms of AIM2 inflammasome activation seem to involve increase oxidative glucose metabolism, mitochondrial ROS generation, oxidative DNA damage and DNA replication stress52 (Figure 2).
During microbial infections the enzyme cholesterol 25-hydroxylase is induced in response to TLR activation and signaling by Type 1 interferons, playing a role in host defense.70 In mice with cholesterol 25-hydroxylase-deficiency, increased cholesterol synthesis and cholesterol enrichment of the ER and mitochondria results in increased mitochondrial ROS generation, DNA damage and AIM2 inflammasome activation without cholesterol crystal formation.71 Thus, there may also be links between intracellular cholesterol accumulation and AIM2 inflammasome activation.
Emerging roles for NLRP3 and AIM2 inflammasomes in atherosclerosis associated with metabolic dysfunction and diabetes
If there is only a low level of inflammasome activation in the standard mouse atherosclerosis models, it is reasonable to hypothesize that inflammasome inhibition would have a greater beneficial effect on atherosclerosis in mouse models in which there is increased underlying NLRP3 or AIM2 activation. One such condition could be diabetes, which is associated with increased oxidative stress, dyslipidemia and elevated levels of DAMPs, which could act to prime and activate inflammasomes.37 72, 73
Recent research supports a role for inflammasomes as mediators of inflammation and atherosclerosis in response to metabolic dysfunction and diabetes. For example, when Apoe−/− mice rendered diabetic using the β-cell toxin streptozotocin were treated with the small-molecule NLRP3 inhibitor MCC950, they were largely protected from the increased atherosclerosis, increased lesion macrophage content, increased lesion necrotic cores, and increased aortic gene expression of inflammatory mediators observed in untreated diabetic mice.37 Importantly, atherosclerotic lesions were unaltered after MCC950 treatment in nondiabetic mice, suggesting that diabetes sensitizes the NLRP3 inflammasome pathway. The main lesion cell types showing an increase in NLRP3 levels in response to diabetes appeared to be smooth muscle cells and endothelial cells, but macrophages responded much more strikingly to the inhibitor ex vivo. The effect of the NLRP3 inhibitor was not due to significant improvement of blood glucose or plasma lipids. Because this study used systemic inhibition of NLRP3, it did not provide information on which cell type is most important in terms of NLRP3 inhibition in vivo. It is possible that NLRP3 inhibition in several cell types mediated the protective effects. Recent data suggest that NLRP3 activation occurs in hematopoietic cells in the setting of diabetes because NLRP3-deficient hematopoietic chimerism in a high fat diet-fed streptozotocin Ldlr−/− diabetes mouse model resulted in smaller atherosclerotic lesions, as compared with diabetic mice with wildtype bone marrow.58 Together, these results are consistent with the interpretation that diabetes leads to activation of the NLRP3 inflammasome in hematopoietic cells, likely myeloid cells, and that this in turn promotes atherosclerosis.
What could trigger NLRP3 inflammasome activation in myeloid cells in diabetes? Cellular or extracellular overaccumulation of cholesterol is perhaps the most likely culprit. In addition, recent research points to metabolites and enzymes involved in metabolism as possible mediators, although most of this work so far has been done in cultured macrophages. In vivo verification is an important next step. The cellular relationships among inflammasomes and metabolism are likely due to intracellular changes in flux of metabolic pathways mediated by enzymatic activities or altered substrate levels under conditions of metabolic dysfunction.74 Thus, the NLRP3 inflammasome can be activated or inhibited by intracellular changes in glycolytic enzymes, glycolytic flux and metabolites of the TCA cycle.75 As glucose enters the macrophage, it is phosphorylated by hexokinase isoforms as the first step in glycolysis. Inhibition of hexokinase 1 or pyruvate kinase M2, which catalyzes the terminal step in glycolysis, dampens NLRP3 activation, while hexokinase 2 dissociation from the outer mitochondrial membrane promotes NLRP3 assembly. Moreover, itaconate, produced via the TCA cycle, has been shown to inhibit NLRP3 but not AIM2 inflammasome activation through a mechanism involving itaconate-mediated dicarboxypropylation of C548 in NLRP3, potentially impairing the ability of NLRP3 to interact with NEK7.76 NEK7 can also be modified by metabolites: deglutathionylation of NEK7 C253 has been shown to increase in NLRP3 inflammasome activation.77 Yet other studies have shown that itaconate can inhibit the NLRP3 inflammasome by several different mechanisms, reviewed in75. However, most of these studies have employed itaconate tool compounds that are more reactive than itaconate, and a recent study failed to find any effect of itaconate on NLRP3 activation.78 The role of glycolytic metabolites and enzymes in modulating NLRP3 activity in macrophages therefore appears to depend on experimental design. The in vivo relevance of these findings is still unclear because increased glycolytic flux in myeloid cells is not sufficient to induce increased atherosclerosis.79
In addition to glucose and glutamine metabolites, saturated fatty acids can activate the NLRP3 inflammasome in cultured macrophages, perhaps by damaging the lysosomal membrane.80 Conversely, unsaturated fatty acids have a protective effect.81 While the physiological relevance of exposing cultured macrophages to selected fatty acids may be questionable, the role of fatty acid metabolites can be investigated in vivo. Deletion of acyl-CoA synthetase 1, an enzyme that converts free fatty acids to acyl-CoAs for distribution into membrane phospholipids, lipid droplets, beta oxidation and other fates, has been shown to protect macrophages from palmitate-induced NLRP3 inflammasome activation in response to TLR4 activation.82 The finding that myeloid cell-targeted acyl-CoA synthetase 1-deletion protected mice from diabetes-accelerated atherosclerosis could therefore possibly be due in part to reduced inflammasome activation.83
Another possible culprit of NLRP3 inflammasome activation in monocytes has recently been proposed to be apolipoprotein C3 (APOC3). Serum levels of APOC3 predict incident CVD in three distinct cohorts of individuals with type 1 diabetes, indicating that serum APOC3 is a biomarker for increased CVD risk.84 Mechanistic research in a mouse model of type 1 diabetes-accelerated atherosclerosis demonstrated that APOC3 is not only a biomarker, but also a causal mediator of atherosclerosis in diabetic mice.85 The link of APOC3 to NLRP3 inflammasome activation was highlighted by studies demonstrating that delipidated APOC3 activates the NLRP3 inflammasome in human monocytes by an alternative pathway including caspase-8 and dimerization of TLR2 and TRL4.86 This pathway did not require the classical NLRP3 priming step, consistent with the known alternative NLRP3 activation mechanism in human monocytes.33 The reason human monocytes do not adhere to the classical priming and activation steps might be that these cells naturally release ATP upon stimulation with LPS and other pathogen-sensing receptor ligands.87
However, APOC3 is a lipoprotein-bound protein in circulation, and the physiological relevance of delipidated APOC3 as an NLRP3 activator can therefore be questioned. Indeed, Hsu et al recently demonstrated that lipid-bound APOC3 does not share the ability of delipidated APOC3 to induce inflammasome activation in monocytes.88 The reason for the loss of APOC3’s inflammasome activation ability when bound to lipid particles could be due to masking of the TLR-binding when APOC3 is bound to lipid. It is also possible that lipid particles could neutralize very low levels of endotoxin contamination in purified APOC3 preparations. In further support for the lack of endogenous APOC3 to induce NLRP3 inflammasome activation in vivo; silencing of hepatic APOC3 expression in diabetic mice lowered plasma APOC3 levels but did not result in lowered plasma levels of IL-18 or IL-1β, and elevated levels of APOC3 did not increase plasma IL-18 or IL-1β.88 Interestingly, a variant within the NLRP3 gene locus associated with higher NLRP3 inflammasome activation (rs10754555) and CVD showed interaction with plasma APOC3 and triglyceride levels.89 This finding highlights the links among NLRP3, plasma triglycerides, APOC3 and CVD, but does not demonstrate a causative role for APOC3 upstream of NLRP3 activation. Rather, mouse studies suggest that APOC3 might be placed downstream of the NLRP3 inflammasome because diabetic mice with hematopoietic NLRP3-deficiency exhibited reduced serum APOC3 levels.58 While it cannot be ruled out that lipid-free APOC3 exists in lesions of atherosclerosis at high enough levels of activate the NLRP3 inflammasome in newly recruited monocytes, APOC3 most likely acts through its ability to slow the clearance of atherogenic triglyceride-rich lipoproteins and remnant lipoprotein particles, thereby increasing accumulation of these atherogenic particles in the artery wall.84
The NLRP3 inflammasome can also be activated by metabolic alterations in cell types adjacent to the atherosclerotic lesion, inducing IL-1β-mediated effects in lesion cells through paracrine effects. Large arteries are surrounded by perivascular adipose tissue (PVAT) expressing the mitochondrial uncoupling protein 1 (UCP1). A recent study demonstrated that UCP1 expression is downregulated in PVAT in obese rodents, concomitant with an impaired endothelium-dependent vasorelaxation. These phenotypes were mimicked by UCP1-deficiency, and UCP1-deficient Apoe−/− mice exhibited increased atherosclerosis, as compared with Apoe−/− mice without UCP1-deficiency.90 The proposed mechanism, based on a series of co-culture experiments, revealed activation of the NLRP3 inflammasome in PVAT due to downregulation of UCP1 and a subsequent increase in mitochondrial ROS generation, resulting in increased local release of IL-1β from the PVAT, and increased endothelial dysfunction and atherosclerosis as a result (Figure 1). The study went on to demonstrate that expression of UCP1 selectively in adipose tissue prevented the increased coronary atherosclerosis in a streptozotocin pig model of diabetes, and that this effect was associated with a normalized release of IL-1β from PVAT (similar to that in non-diabetic pigs). The effect of adipocyte UCP1 was not due to normalization of plasma glucose or cholesterol, which were both markedly elevated in the two diabetic pig groups as compared with the non-diabetic pigs.90 Although this study nicely demonstrates that forced expression of UCP1 in adipose tissue in pigs results in protection from atherosclerosis associated with diabetes, it also shows that the described UCP1 mechanism is not required for diabetes-accelerated atherosclerosis because pigs are normally deficient in UCP1 and the wildtype diabetic pigs exhibited increased inflammasome activation and coronary atherosclerosis as compared with wildtype non-diabetic pigs.
There is still little understanding on how metabolic dysfunction and diabetes affect the AIM2 inflammasome, and more research is needed in this area. Recent studies from the authors’ laboratories support the conclusion that hematopoietic AIM2-deficiency results in smaller atherosclerotic lesions in fat-fed diabetic Ldlr−/− mice.58 The mechanism of AIM2 activation in hematopoietic cells in the setting of diabetes is unknown.
Clonal hematopoiesis, inflammasomes and CVD
Clonal hematopoiesis (CH) arises from somatic mutations that enhance the fitness of hematopoietic stem cells (HSCs) and the outgrowth of clones of blood cells. CH mutations occur in genes that are involved in epigenetic modifications (TET2, ASXL1, DNMT3A), hematopoietic cytokine signaling (JAK2V617F) and DNA damage repair (PPM1D, TP53). CH involving the more common variants has emerged as a major independent risk factor for CVD.91, 92 CH increases in frequency with aging and may partly explain why aging is a potent CVD risk factor.93 CH increases the risk of both myeloid malignancy and CVD but the latter has a much broader impact on human health.94 The NLRP3 inflammasome promotes accelerated atherosclerosis in chimeric mice modeling TET2 CH,57 while the AIM2 inflammasome aggravates atherosclerosis in JAK2VF CH.52 The JAK2VF mutation although less common than several other CH variants disproportionately increased CVD risk including coronary artery disease (CAD) and thrombotic risk.92 Macrophage-specific expression of Jak2VF or Jak2VF expression in chimeric mice modeling CH increased atherosclerosis and features of plaque instability.52 Single cell RNA-sequencing analysis of plaque immune cells in Jak2VF CH revealed an increase in inflammatory macrophages and a relative depletion of less inflammatory Trem2Hi macrophages. Genetic suppression of inflammasomes or IL-1β inhibition improved features of plaque instability. IL-1β inhibition has also recently been shown to improve features of myeloproliferative neoplasm in Jak2VF mice95, 96 suggesting that this therapy could have multiple benefits in this setting. In addition, Jak2VF CH may increase aortic aneurysm formation in mice and possibly humans, reflecting increased inflammatory resident-like macrophages in the aortic adventitia.97, 98
The association between CH and CVD may involve both forward and reverse mechanisms.99 Forward mechanisms include expansion of HSPCs, increased myelopoiesis, macrophage inflammation and inflammasome activation, while reverse mechanisms include effects of inflammatory cytokines such as IL-1β or IL-6 on hematopoietic stem and multi-potential progenitor cell proliferation and myelopoiesis.99 A recent study in the UK Biobank (UKB) population did not find evidence for a causal association between CH and CVD.100 However, risk estimates in UKB are attenuated by the much healthier status of UKB subjects than the general UK population101 and likely in this study100 by insufficient depth of sequencing and mutation mis-calling.101, 102 Moreover, the Mendelian randomization analysis mainly used SNPs near the DNMT3A gene that were not associated with CVD risk and thus represent a weak instrument as acknowledged by the authors.100
The role of the inflammasome products IL-1β and IL-18 in atherosclerosis
Early studies of IL-1 or IL-1β inhibition in Apoe−/− mice showed an adverse negative effect on outward remodeling of plaques, cap thickness and cap macrophage density.103, 104 These findings seem at odds with the positive outcome of the Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS; NCT01327846).6 However, they also suggest complexity and that there could be both beneficial and adverse effects of IL-1β on plaque development and remodeling with the adverse effects usually predominating. A more recent study in Ldlr−/− mice showed a modest benefit of IL-1β inhibition on plaque area in advanced but not early lesions and no negative impact on outward remodeling.105 In other studies, IL-1β antibodies and NLRP3 inhibition using MCC950 reduced early atherosclerosis in high cholesterol diet fed Apoe−/− mice in association with reduced HSPC proliferation and myelopoiesis as well as decreased entry of leukocytes into plaques.106 Reduced leukocyte entry was related to diminished expression of cell adhesion molecules and leukocyte chemoattractants by the endothelium. In another study involving more advanced lesions, there was no impact of IL-1β antagonism on plaque area or features of stability in control Ldlr−/− mice.52 Overall, the variable results in different studies are consistent with a modest, somewhat variable role of inflammasomes in plaque development in standard mouse atherosclerosis models as suggested above. Imaging studies of carotid plaques in humans showed no impact of anti-IL-1β therapy on plaque volume,107 suggesting that the benefit of this treatment might rather relate to plaque stabilization. In Jak2VF CH mice IL-1 antagonism with anakinra (a decoy receptor that blocks both IL-1α and IL-1β signaling through IL1R1) or blocking IL-1β antibodies did not reduce lesion area but improved features of plaque stability with increased cap thickness, decreased plaque necrosis and decreased macrophage proliferation and burden.52 The mechanisms responsible for these changes, which in humans correlate with lower CAD risk108–111 are largely unknown and warrant further investigation.
In contrast to IL-1β, IL-1α is present on the surface of monocytes and macrophages and other vascular cells in a membrane-bound, active form and is considered an “alarmin.”68 It is released in response to necrotic cell death including pyroptosis and promotes the release of chemokines leading to neutrophil then monocyte infiltration.112 IL-1α, by interacting with IL1R1, can also promote NLRP3 inflammasome priming.9 Inhibition of IL-1α reduced early atherosclerosis in Ldlr−/− mice and slightly impaired outward remodeling of the aorta.105 IL-1α has also been shown to promote thrombus formation in a mouse model of atherosclerotic plaque erosion.113 IL-1α is increased on the surface of monocytes following myocardial infarction or in chronic kidney disease and enhances leukocyte-endothelial cell adhesion.114 Together, these findings suggest that IL-1α has the ability to prime the NLRP3 inflammasome, but also has many other effects that can promote atherosclerosis and athero-thrombosis.
IL-18 although much less studied than IL-1β also has a pro-atherogenic role in mice and likely humans.115–117 IL-18 increases interferon-γ production by NK and T cells118 and its vascular effects are mediated through increased production of interferon-γ,116 which is highly atherogenic.119
Human atherosclerotic plaques with increased necrotic cores and thin fibrous caps are more prone to rupture. Pyroptosis downstream of inflammasome activation is a form of programmed cell necrosis. Does macrophage pyroptosis contribute to necrotic core formation and expansion in lesions of atherosclerosis? Recent studies suggest that macrophage GSDMD expression is not a major contributor to necrotic core size in advanced lesions in the standard models. Thus, deletion of GSDMD in bone marrow cells did not result in smaller necrotic cores in Ldlr−/− mice.52, 58 However, GSDMD has been shown to promote lesion development and necrotic core formation in some mouse models. Whole-body Gsdmd−/− LDLR-deficient mice and Gsdmd−/− Apoe−/− mice exhibited smaller lesions with reduced necrotic cores compared to controls,120, 121 while two other studies found no difference in lesion size in Ldlr−/− mice with hematopoietic GSDMD-deficiency52, 58. Conversely, in the presence of diabetes, hematopoietic GSDMD-deficiency resulted in smaller lesions.58 It is possible that in the standard mouse atherosclerosis models the pro-atherogenic role of GSDMD depends partly on cell types other than macrophages. These findings are consistent with the notion that increased inflammasome activation is needed in order for inhibition of these pathways to show athero-protective effects. Moreover, in Jak2VF CH mice hematopoietic GSDMD-deficiency was associated with no change in lesion area but a decrease in necrotic core and an increase in fibrous cap formation indicating a clear adverse effect of GSDMD in the setting of exaggerated plaque inflammation in CH.52 The frequency of the mutant allele in blood leukocytes was increased by GSDMD-deficiency indicating the preservation of mutant clones. As shown by scRNA-sequencing, deficiency of GSDMD led to increased monocytes, decreased macrophage populations overall but an increase in a small population of pro-inflammatory pro-thrombotic perhaps “zombie” macrophages that failed to undergo pyroptosis.
In addition to pyroptosis, other types of macrophage death, such as ferroptosis, necroptosis and post-apoptotic necrosis are likely to be relevant to necrotic core formation in atherosclerotic lesions.11 An impaired ability of macrophages to clear dead and dying cells through efferocytosis also contributes to necrotic core expansion.122 Thus, while decreasing plaque necrosis seems like a laudable therapeutic goal, it is not clear that therapeutic inhibition of GSDMD would have a beneficial effect on CVD.
Inflammasomes and neutrophil extracellular traps
Neutrophil extracellular traps (NETs), which were first described as having anti-microbial actions123 also have a role in sterile inflammation and promote atherosclerotic plaque vulnerability and athero-thrombosis.124 Westerterp et al showed that myeloid cell deficiency of the cholesterol efflux promoting genes Abca1 and Abcg1 while inducing NLRP3 inflammasome activation also promoted NETosis in atherosclerotic plaques; deficiency of Nlrp3 in bone marrow cells in these mice virtually abolished NETosis in plaques, placing NET formation downstream of inflammasome activation51 (Figure 3). Recent studies have shown that activation of NLRP3 and non-canonical inflammasomes in neutrophils can also induce NETosis.125 To distinguish whether NETosis was due to cholesterol transporter deficiency in macrophages or neutrophils, Yalcinkaya et al generated mice with neutrophil or macrophage-specific deficiency of ABCA1 and ABCG1 and transplanted their bone marrow into Ldlr−/− mice.126 Macrophage ABCA1/ABCG1-deficiency activated inflammasomes in macrophages and neutrophils, and induced NETosis in plaques, while neutrophil ABCA1/ABCG1 deficiency had no impact on NETs. NETosis was suppressed by administering an IL-1β neutralizing antibody. The extent of NETosis in plaques correlated strongly with the degree of neutrophil accumulation, irrespective of blood neutrophil counts, and neutrophil accumulation was decreased by IL-1β antagonism. IL-1β or media transferred from ABCA1/ABCG1 deficient macrophages increased NETosis in both control and ABCA1/ABCG1 deficient neutrophils. This cell-extrinsic effect of IL-1β on NETosis was blocked by the NLRP3 inhibitor MCC950. These studies establish a link between inflammasome mediated IL-1β production in macrophages and NETosis in atherosclerotic plaques. Macrophage-derived IL-1β appears to increase NETosis both by increasing neutrophil recruitment to plaques and by promoting neutrophil NLRP3 inflammasome activation.126
Figure 3. Interactions between the NLRP3 inflammasome and NETosis in atherosclerosis and athero-thrombosis.
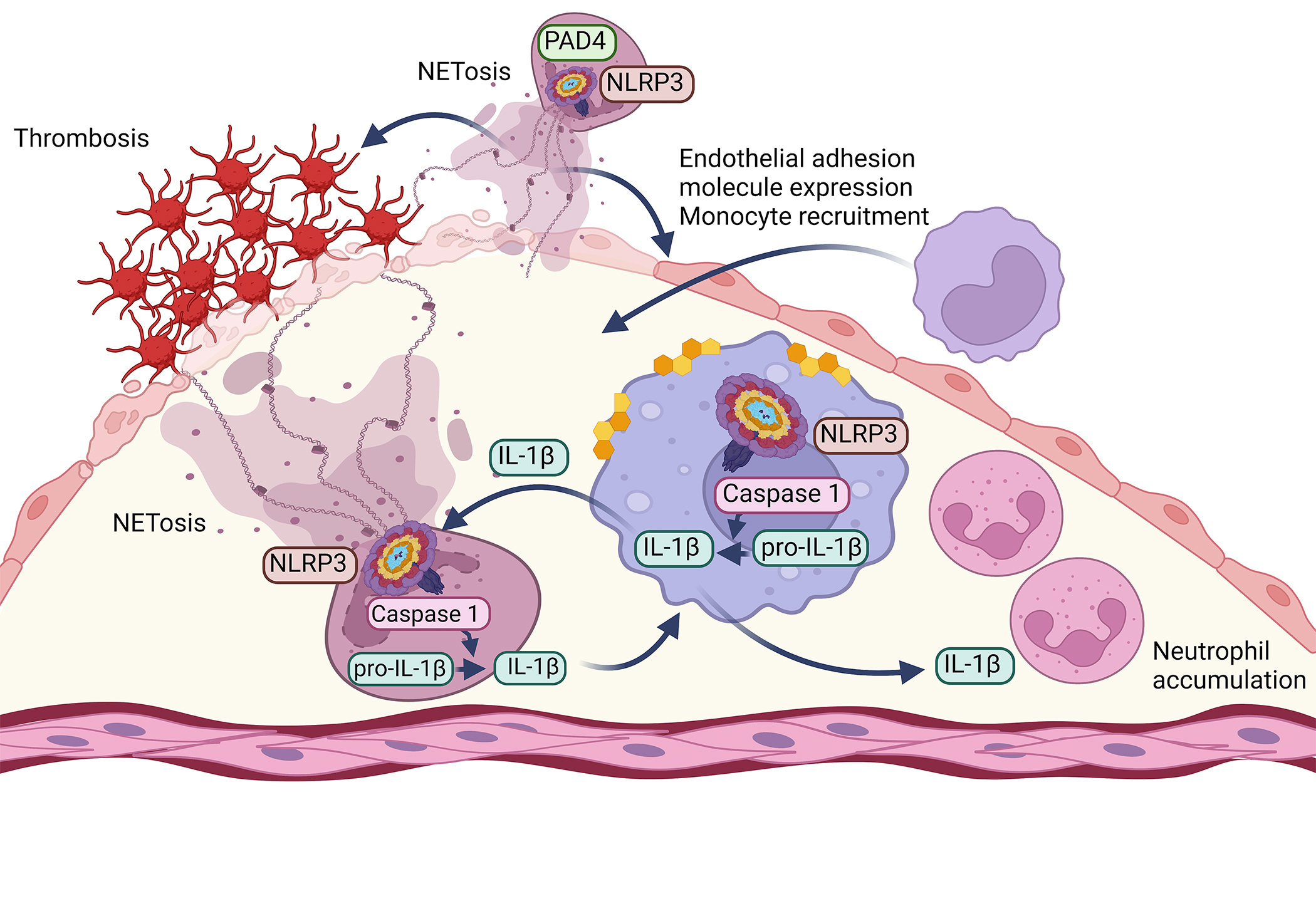
Overaccumulation of cholesterol in macrophages not only induces NLRP3 inflammasome activation in these cells, as shown in Figure 1, but also promotes neutrophil NETosis through a paracrine mechanism. The IL-1β released from macrophages stimulates NLRP3 activation in neutrophils and subsequent NETosis, as well as accumulation of neutrophils in lesions of atherosclerosis. IL-1β released from neutrophils may in turn further activate the NLRP3 pathway in lesion macrophages. NETosis has also been shown to promote lesion erosion and thrombosis, endothelial expression of adhesion molecules, and likely further monocyte recruitment. In neutrophils, the enzyme PAD4, which is essential for NETosis, acts upstream of optimal NLRP3 activation. Created with BioRender.com
There may be a bi-directional relationship between inflammasome activation, IL-1 production and NETs (Figure 3). Warnatsch et al have shown that NETs promote inflammasome priming in macrophages of Apoe−/− mice127 and consistently an inhibitor of PAD4 (peptidyl arginine deiminase 4), an essential enzyme in NET formation, decreased atherosclerosis and thrombosis in Western diet-fed Apoe−/− mice.128 Other studies show that PAD4 is needed for optimal NLRP3 inflammasome/ASC speck assembly.129 However, in Western diet-fed Ldlr−/− mice, deficiency of PAD4 did not affect atherogenesis,130 perhaps consistent with minimal inflammasome activation in this model. In contrast, NETs appear to have a prominent pro-thrombotic role in a model of atherosclerotic plaque erosion.113, 130 Together the studies suggest that while macrophage inflammasome activation and active IL-1β production can initiate NETosis in plaques, there may also be a positive feedback loop from neutrophils to macrophages, providing a feed-forward mechanism. NETosis and inflammasome activation in neutrophils in close proximity to the endothelium may also promote plaque erosion and athero-thrombosis (Figure 3).
Inflammasomes in human coronary artery disease
The findings that IL-1β antibodies6 and colchicine7, 8 reduced atherosclerotic CVD in humans suggest that inflammasomes have an important role in human atherosclerosis, including in those with diabetes. While active IL-1β is a major product of all inflammasomes, colchicine inhibits the microtubule-dependent assembly of the NLRP3 inflammasome and IL-1β secretion.131 Colchicine is avidly taken up by leucocytes, and its ability to bind to tubulin and interfere with microtubular function affects the expression of cytokines and interleukins, and the ability of neutrophils to marginate, ingress, aggregate, express superoxide and release NETs.132 As described above many of these effects may be secondary to inhibition of the macrophage NLRP3 inflammasome and IL-1β mediated cross-talk to neutrophils and endothelial cells.126 However, colchicine has a variety of other anti-inflammatory effects133 and active IL-1β can be produced by non-inflammasome mediated mechanisms such as cleavage of pro-IL1β by neutrophil elastase or proteinase-3.9 Thus, the positive outcomes of the CANTOS and low dose colchicine (Lo-Do-Co) trials while indicating a beneficial anti-inflammatory effect, do not provide definitive evidence for involvement of inflammasomes in human CVD.
Genetic studies also implicate the NLRP3 inflammasome in human CVD. SNPs in NLRP3 that alter its expression associate with atherosclerotic CVD. Genetic analyses showed that the highly prevalent intronic NLRP3 variant rs10754555 affects NLRP3 gene expression.134 Carriers of the G allele displayed higher NLRP3 inflammasome activation in isolated monocytes and showed significantly higher plasma levels of C-reactive protein, a marker of inflammation. In carriers of the rs10754555 variant, the prevalence of CAD was significantly increased with a significant interaction between rs10754555 and age perhaps suggesting a possible link to CH. In addition, active IL-1β promotes the expression of IL-6.135, 136 In CANTOS, achieved IL-6 levels below the median were associated with benefit of IL-1β antibody treatment, while levels above the median were not.115 Moreover, genetic variants that reduce direct signaling of the IL6 receptor markedly ameliorated the CAD risk of TET2 and DNMT3A CH.137
Relevant to individuals with diabetes, recent findings suggest that sodium glucose co-transporter-2 (SGLT2) inhibitors suppress NLRP3 inflammasome activation. SGLT2 inhibitors are used to lower blood glucose in patients with type 2 diabetes. This class of drugs lowers blood glucose by increasing urinary glucose excretion and has been shown to have a marked beneficial effect on CVD risk, primarily heart failure.138 A recent study suggested that the treatment of individuals with type 2 diabetes with the SGLT2 inhibitor empagliflozin resulted in reduced NLRP3 inflammasome activation in macrophages derived from their peripheral blood mononuclear cells, using ATP or palmitate to trigger NLRP3 activation.139 The mechanism is unclear, but was proposed to be mediated by reduced glucose, insulin, and uric acid, or perhaps by the small increase in ketones in the empagliflozin-treated subjects through a mechanism that was maintained through the 7-day macrophage maturation in vitro. SGLT2 inhibition has been associated with stabilization of atherosclerotic plaques in a mouse model.140
Mutations in Pyrin (gene name MEFV) underlie familial Mediterranean fever and lead to activation of caspase-1 and IL-1β production and to intermittent systemic inflammatory flares.141 Paradoxically, familial Mediterranean fever was reported to be associated with reduced prevalence of ischemic heart disease.142 However, many patients were being treated with colchicine that may have blunted any impact on CVD. A more recent study based on electronic health records suggested a moderate increase in ischemic heart disease risk in familial Mediterranean fever subjects.143 Nonetheless, it remains a mystery why familial Mediterranean fever or cryopyrin-associated periodic syndrome patients (who have activating mutations in NLRP3144) are not more obviously susceptible to CVD. Perhaps chronic low-grade inflammatory processes are more important in the promotion of atherosclerosis than intermittent inflammatory flares.
Analysis of gene expression indicates enrichment of inflammasome components in human carotid plaques versus normal artery.145, 146 However, this can probably be explained by the relative abundance of macrophages in atherosclerotic lesions compared to normal arteries. In some145, 146 but not other147 studies inflammasome components were increased in carotid plaques of symptomatic versus asymptomatic patients. However, symptomatic plaques may rapidly become less inflammatory prior to surgical removal148 blurring differences in plaque characteristics. Advanced human plaques are often fibrous which may limit and bias the recovery of the cells used in single cell analysis. This stresses the importance of immunolocalization or spatial omics studies in sections of intact tissue. Moreover, there is currently an unmet need for the development of antibodies that specifically recognize the activated (cleaved) form of IL-1β in plaques.
Overall, these studies indicate a likely role of inflammasome activation in human atherosclerotic CVD. That inflammasome activation does not appear to have a major role in atherogenesis in standard mouse atherosclerosis models could indicate important species differences. For example, mice do not readily rupture atherosclerotic plaques and may be of limited use in predicting athero-thrombosis. A human-specific mechanism of NLRP3 inflammasome activation in monocytes has been described in which direct activation of TLR4 by PAMPs or DAMPs in plaques could lead to NLRP3 inflammasome activation shortly after monocytes are recruited into the arterial intima.33 Another more likely explanation is that aggravating factors (“second hits”) like reduced cholesterol efflux, inflammasome-promoting CH mutations or diabetes are highly prevalent in humans and have an important role in inflammasome driven CVD.
Emerging evidence suggests an expanded role of inflammasomes in human clonal hematopoiesis and CVD
Very recent studies in patients with CH have provided further evidence for a role of inflammasomes in human athero-thrombotic disease. A post hoc analysis of a subset of patients in CANTOS suggested that IL-1β inhibition benefited patients with TET2 clonal hematopoiesis more than subjects with other forms of CH or patients without CH.149 While consistent with mouse studies on Tet2 CH, the population was too small to make firm conclusions concerning less common CH variants. Strong support for the role of AIM2 in human JAK2VF CH and ASXL1 CH associated atherosclerosis has been obtained in a study using data from ~425,00 subjects in the UK Biobank (UKB) published in preprint.150 The CAD risk of subjects with JAK2VF and ASXL1 CH was increased in those with higher predicted expression of AIM2 based on expression quantitative trait loci (eQTLs) around the AIM2 gene; those without JAK2VF or ASXL1 CH with higher AIM2 expression scores did not have altered risk and thus the interaction of genotype with predicted AIM2 expression was significant. Moreover, predicted increased expression of IFNGR that mediates interferon-γ signaling increased CAD risk in JAK2VF CH, consistent with mouse studies showing that interferon-γ increased AIM2 expression in macrophages.52 Studies in bone marrow-derived macrophages from Asxl1 CH mice showed increased AIM2 but not NLRP3 inflammasome activation compared to controls.150 CH subjects collectively who had higher predicted expression of the IL1 receptor associated protein (IL1RAP) that has an essential role in IL-1 signaling,151 also showed significantly increased CAD risk. These studies point to the broad significance of inflammasome activation in CH-associated CVD risk but suggest distinctive roles for AIM2 and NLRP3 in different forms of CH. This has important implications for the discovery and design of potential novel therapeutic strategies targeting inflammasomes or their downstream products.
Summary and Perspective
The mechanisms underlying NLRP3 and AIM2 inflammasome activation in atherosclerosis and diabetes remain poorly understood. Although widely accepted, the role of cholesterol crystals as the main trigger of NLRP3 inflammasome activation is speculative and the role of cholesterol accumulation in cellular organelles such as ER and mitochondria needs to be considered as an alternative. Mitochondrial ROS production has often been linked to NRLP3 inflammasome activation but its causal role has been questioned and is unclear. AIM2 inflammasome activation has been attributed to both mitochondrial oxidative processes and to oxidative changes and replication stress in nuclear DNA. How these processes are interconnected if at all remains uncertain. An improved mechanistic understanding of the role of inflammasomes in atherosclerosis is needed to foster to the design of more rational treatments.
Diabetes is probably the single most important emerging CVD risk factor acting both via dyslipidemia and vascular inflammation to increase atherosclerosis and complications. Early evidence suggests a role of inflammasomes in diabetes risk. Much more needs to be done to increase the mechanistic understanding of diabetes and inflammatory risk. APOC3 is increased in diabetes and likely increases CVD via remnant accumulation and vascular effects; however, NLRP3 inflammasome activation is not specifically increased by APOC3 in lipoproteins. SGLT2 inhibitors have proven effective treatments for diabetes and found to decrease CVD risk; however, this appears to be primarily a benefit for heart failure rather than atherosclerosis or CAD. Potential links between diabetes, diabetes treatments and inflammasome activation need to be further explored.
Precision medicine approaches targeting mechanism-based treatments to patients who most need those treatments has led to major improvements in survival for multiple types of cancer. In contrast, precision approaches have not yet been adopted in the treatment of atherosclerotic CVD. A thesis developed in this review is that anti-inflammatory treatments should be directed to specific patient groups based on genetic risk such as CH variants or metabolically determined risk such as diabetes. This may lead to an improved benefit/risk ratio with greater benefit and less immunosuppression and infectious complications. Emerging evidence suggests an important role of both NLRP3 and AIM2 inflammasome activation in promoting atherosclerosis and its complications in diabetes and CH. For CH a variety of potential therapies may be considered, but those targeting common downstream factors, such as IL-1β or IL-6, and CH-mutation-specific therapies appear to be most promising.13 A range of new treatments targeting NLRP3 are under development. Although initial clinical trials with MCC950 were stopped due to hepatotoxicity152 other NLRP3 inhibitors with different chemical structures have been developed and are under evaluation.39 In addition, drugs modifying posttranscriptional modifications of NLRP3 that may only cause partial inhibition of inflammasome activation by inhibiting its deubiquitylation are under development153 but may also be less specific. AIM2 inhibitors could also potentially be developed to treat different forms of CH associated with AIM2 activation. Future clinical trials enriched for individuals with CH and high atherosclerotic risk will be required to establish the efficacy and safety of such approaches.
Sources of Funding
This study was supported by NIH grants R01HL107653, R01HL13766 and by the Leducq Foundation (A.R.T.) and R35HL150754, P01HL151328 and R01HL161829 (K.E.B.).
Non-standard abbreviations and acronyms:
- AIM2
Absent In Melanoma 2
- APOC3
apolipoprotein C3
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- CAD
coronary artery disease
- CANTOS
Canakinumab Antiinflammatory Thrombosis Outcome Study
- CH
clonal hematopoiesis
- CVD
cardiovascular disease
- GSDMD
gasdermin D
- HSC
hematopoietic stem cells
- IL-1β
interleukin 1β
- IL-18
interleukin 18
- LPS
lipopolysaccharide
- NEK7
NIMA-related kinase 7
- NET
neutrophil extracellular traps
- NLRP3
NACHT (nucleotide triphosphatase containing domain), leucine rich repeat (LRR)- and pyrin domain (PYD)-containing protein 3
- PVAT
perivascular adipose tissue
- ROS
reactive oxygen species
- SGLT2
sodium glucose co-transporter-2
- TLR
Toll-Like Receptor
- UCP1
uncoupling protein 1
Footnotes
Disclosures
K.E.B. serves on the Scientific Advisory Board of Esperion Therapeutics. A.R.T. is a consultant or Scientific Advisory Board member of Staten Biotechnology, Tensixteen Bio, Commonwealth Serum Laboratories and Beren Pharmaceuticals.
References
- 1.Ahmad FB and Anderson RN. The Leading Causes of Death in the US for 2020. JAMA. 2021;325:1829–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ, Benziger CP, Bonny A, Brauer M, Brodmann M, Cahill TJ, Carapetis J, Catapano AL, Chugh SS, Cooper LT, Coresh J, Criqui M, DeCleene N, Eagle KA, Emmons-Bell S, Feigin VL, Fernandez-Sola J, Fowkes G, Gakidou E, Grundy SM, He FJ, Howard G, Hu F, Inker L, Karthikeyan G, Kassebaum N, Koroshetz W, Lavie C, Lloyd-Jones D, Lu HS, Mirijello A, Temesgen AM, Mokdad A, Moran AE, Muntner P, Narula J, Neal B, Ntsekhe M, Moraes de Oliveira G, Otto C, Owolabi M, Pratt M, Rajagopalan S, Reitsma M, Ribeiro ALP, Rigotti N, Rodgers A, Sable C, Shakil S, Sliwa-Hahnle K, Stark B, Sundstrom J, Timpel P, Tleyjeh IM, Valgimigli M, Vos T, Whelton PK, Yacoub M, Zuhlke L, Murray C, Fuster V and Group G-N-JGBoCDW. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J Am Coll Cardiol. 2020;76:2982–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, Elkind MSV, Evenson KR, Eze-Nliam C, Ferguson JF, Generoso G, Ho JE, Kalani R, Khan SS, Kissela BM, Knutson KL, Levine DA, Lewis TT, Liu J, Loop MS, Ma J, Mussolino ME, Navaneethan SD, Perak AM, Poudel R, Rezk-Hanna M, Roth GA, Schroeder EB, Shah SH, Thacker EL, VanWagner LB, Virani SS, Voecks JH, Wang NY, Yaffe K and Martin SS. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation. 2022;145:e153–e639. [DOI] [PubMed] [Google Scholar]
- 4.Moore JX, Chaudhary N and Akinyemiju T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev Chronic Dis. 2017;14:E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tall AR, Thomas DG, Gonzalez-Cabodevilla AG and Goldberg IJ. Addressing dyslipidemic risk beyond LDL-cholesterol. J Clin Invest. 2022;132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ and Group CT. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 7.Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, The SHK, Xu XF, Ireland MA, Lenderink T, Latchem D, Hoogslag P, Jerzewski A, Nierop P, Whelan A, Hendriks R, Swart H, Schaap J, Kuijper AFM, van Hessen MWJ, Saklani P, Tan I, Thompson AG, Morton A, Judkins C, Bax WA, Dirksen M, Alings M, Hankey GJ, Budgeon CA, Tijssen JGP, Cornel JH, Thompson PL and LoDoCo2 Trial I. Colchicine in Patients with Chronic Coronary Disease. N Engl J Med. 2020;383:1838–1847. [DOI] [PubMed] [Google Scholar]
- 8.Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, Lopez-Sendon J, Ostadal P, Koenig W, Angoulvant D, Gregoire JC, Lavoie MA, Dube MP, Rhainds D, Provencher M, Blondeau L, Orfanos A, L’Allier PL, Guertin MC and Roubille F. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381:2497–2505. [DOI] [PubMed] [Google Scholar]
- 9.Grebe A, Hoss F and Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res. 2018;122:1722–1740. [DOI] [PubMed] [Google Scholar]
- 10.Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW and Dinarello CA. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ Res. 2020;126:1260–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puylaert P, Zurek M, Rayner KJ, De Meyer GRY and Martinet W. Regulated Necrosis in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2022;42:1283–1306. [DOI] [PubMed] [Google Scholar]
- 12.Libby P Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J Am Coll Cardiol. 2017;70:2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tall AR and Fuster JJ. Clonal hematopoiesis in cardiovascular disease and therapeutic implications. Nat Cardiovasc Res. 2022;1:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chavkin NW, Min KD and Walsh K. Importance of clonal hematopoiesis in heart failure. Trends Cardiovasc Med. 2022;32:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans MA and Walsh K. Clonal hematopoiesis, somatic mosaicism, and age-associated disease. Physiol Rev. 2023;103:649–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamkanfi M and Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22. [DOI] [PubMed] [Google Scholar]
- 17.Lamkanfi M and Dixit VM. In Retrospect: The inflammasome turns 15. Nature. 2017;548:534–535. [DOI] [PubMed] [Google Scholar]
- 18.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F and Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. [DOI] [PubMed] [Google Scholar]
- 19.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM and Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T and Karin M. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang YS, Fitzgerald KA, Underhill DM, Town T and Arditi M. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity. 2012;36:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M, Jacobs HT, Reczek CR, Rashidi A, Zhang P, Miska J and Chandel NS. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol. 2022;23:692–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng L, Wang J and Chen J. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J Biol Chem. 2010;285:30982–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren G, Zhang X, Xiao Y, Zhang W, Wang Y, Ma W, Wang X, Song P, Lai L, Chen H, Zhan Y, Zhang J, Yu M, Ge C, Li C, Yin R and Yang X. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortimer L, Moreau F, MacDonald JA and Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol. 2016;17:1176–86. [DOI] [PubMed] [Google Scholar]
- 26.Devant P, Borsic E, Ngwa EM, Xiao H, Chouchani ET, Thiagarajah JR, Hafner-Bratkovic I, Evavold CL and Kagan JC. Gasdermin D pore-forming activity is redox-sensitive. Cell Rep. 2023;42:112008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P and Wu H. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell. 2021;184:6299–6312 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He Y, Zeng MY, Yang D, Motro B and Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, Lyon S, Scott L, Quan J, Sun Q, Russell J, Arnett S, Jurek P, Chen D, Kravchenko VV, Mathison JC, Moresco EM, Monson NL, Ulevitch RJ and Beutler B. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17:250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao L, Magupalli VG and Wu H. Cryo-EM structures of the active NLRP3 inflammasome disc. Nature. 2023;613:595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, Masters L, James JR, Shi Y, Meng G and Mallat Z. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat Commun. 2017;8:15986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, Sharif H, Hu JJ, Evavold CL, Kagan JC, Schmidt FI, Fitzgerald KA, Kirchhausen T, Li Y and Wu H. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020;369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T and Hornung V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44:833–46. [DOI] [PubMed] [Google Scholar]
- 34.Schmacke NA, O’Duill F, Gaidt MM, Szymanska I, Kamper JM, Schmid-Burgk JL, Madler SC, Mackens-Kiani T, Kozaki T, Chauhan D, Nagl D, Stafford CA, Harz H, Frohlich AL, Pinci F, Ginhoux F, Beckmann R, Mann M, Leonhardt H and Hornung V. IKKbeta primes inflammasome formation by recruiting NLRP3 to the trans-Golgi network. Immunity. 2022;55:2271–2284 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Nunez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA and O’Neill LA. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA and Walsh K. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma A, Choi JSY, Stefanovic N, Al-Sharea A, Simpson DS, Mukhamedova N, Jandeleit-Dahm K, Murphy AJ, Sviridov D, Vince JE, Ritchie RH and de Haan JB. Specific NLRP3 Inhibition Protects Against Diabetes-Associated Atherosclerosis. Diabetes. 2021;70:772–787. [DOI] [PubMed] [Google Scholar]
- 38.Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, Chitty JL, Fraser JA, Jennings MP, Robertson AAB and Schroder K. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15:556–559. [DOI] [PubMed] [Google Scholar]
- 39.Coll RC, Schroder K and Pelegrin P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43:653–668. [DOI] [PubMed] [Google Scholar]
- 40.Hochheiser IV, Pilsl M, Hagelueken G, Moecking J, Marleaux M, Brinkschulte R, Latz E, Engel C and Geyer M. Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature. 2022;604:184–189. [DOI] [PubMed] [Google Scholar]
- 41.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E and Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernandes-Alnemri T, Yu JW, Datta P, Wu J and Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Man SM, Karki R and Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol. 2016;46:269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E and Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, Hurt ME, Paysour MJ, Ennerfelt HE and Lukens JR. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature. 2020;580:647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM, Zhu S, Strowig T, Lam FC, Zhao C, Henao-Mejia J, Yilmaz O, Fitzgerald KA, Eisenbarth SC, Elinav E and Flavell RA. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354:765–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antiochos B, Trejo-Zambrano D, Fenaroli P, Rosenberg A, Baer A, Garg A, Sohn J, Li J, Petri M, Goldman DW, Mecoli C, Casciola-Rosen L and Rosen A. The DNA sensors AIM2 and IFI16 are SLE autoantigens that bind neutrophil extracellular traps. Elife. 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soehnlein O and Tall AR. AIMing 2 treat atherosclerosis. Nat Rev Cardiol. 2022;19:567–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V and Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L and Tschopp J. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011;2:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide-van Gemert S, Wang N, Welch CL, Reilly MP, Stroes ES, Moore KJ and Tall AR. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation. 2018;138:898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, Silvestre-Roig C, Kotini AG, Luchsinger LL, Wei Y, Westerterp M, Snoeck HW, Papapetrou EP, Schulz C, Massberg S, Soehnlein O, Ebert B, Levine RL, Reilly MP, Libby P, Wang N and Tall AR. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tumurkhuu G, Shimada K, Dagvadorj J, Crother TR, Zhang W, Luthringer D, Gottlieb RA, Chen S and Arditi M. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ Res. 2016;119:e76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X, McDonald JG, Aryal B, Canfran-Duque A, Goldberg EL, Araldi E, Ding W, Fan Y, Thompson BM, Singh AK, Li Q, Tellides G, Ordovas-Montanes J, Garcia Milian R, Dixit VD, Ikonen E, Suarez Y and Fernandez-Hernando C. Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen S, Markman JL, Shimada K, Crother TR, Lane M, Abolhesn A, Shah PK and Arditi M. Sex-Specific Effects of the Nlrp3 Inflammasome on Atherogenesis in LDL Receptor-Deficient Mice. JACC Basic Transl Sci. 2020;5:582–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, Chew SHS, See P, Chakarov S, Wang XN, Lim LH, Johnson LA, Lum J, Fong CY, Bongso A, Biswas A, Goh C, Evrard M, Yeo KP, Basu R, Wang JK, Tan Y, Jain R, Tikoo S, Choong C, Weninger W, Poidinger M, Stanley ER, Collin M, Tan NS, Ng LG, Jackson DG, Ginhoux F and Angeli V. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity. 2018;49:1191. [DOI] [PubMed] [Google Scholar]
- 57.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA and Walsh K. Clonal hematopoiesis associated with Tet2 deficiency accelerates atherosclerosis development in mice. Science. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsu CC FT, Kanter JE, Kothari V, Kramer F, Tang J, Tall AR, Bornfeldt KE. Hematopoietic NLRP3 and AIM2 inflammasomes promote diabetes-accelerated atherosclerosis, but increased necrosis is independent of pyroptosis. Diabetes 2023;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT and Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA and Moore KJ. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Accad M, Smith SJ, Newland DL, Sanan DA, King LE Jr., Linton MF, Fazio S and Farese RV Jr. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J Clin Invest. 2000;105:711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil MT, Su M, Sen P, Ruhwedel T, Mitkovski M, Trendelenburg G, Lutjohann D, Mobius W and Simons M. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science. 2018;359:684–688. [DOI] [PubMed] [Google Scholar]
- 63.Tall AR and Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res. 2019;60:721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Small DM and Shipley GG. Physical-chemical basis of lipid deposition in atherosclerosis. Science. 1974;185:222–9. [DOI] [PubMed] [Google Scholar]
- 65.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N and Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de la Roche M, Hamilton C, Mortensen R, Jeyaprakash AA, Ghosh S and Anand PK. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J Cell Biol. 2018;217:3560–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Orecchioni M, Kobiyama K, Winkels H, Ghosheh Y, McArdle S, Mikulski Z, Kiosses WB, Fan Z, Wen L, Jung Y, Roy P, Ali AJ, Miyamoto Y, Mangan M, Makings J, Wang Z, Denn A, Vallejo J, Owens M, Durant CP, Braumann S, Mader N, Li L, Matsunami H, Eckmann L, Latz E, Wang Z, Hazen SL and Ley K. Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science. 2022;375:214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281:8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paulin N, Viola JR, Maas SL, de Jong R, Fernandes-Alnemri T, Weber C, Drechsler M, Doring Y and Soehnlein O. Double-Strand DNA Sensing Aim2 Inflammasome Regulates Atherosclerotic Plaque Vulnerability. Circulation. 2018;138:321–323. [DOI] [PubMed] [Google Scholar]
- 70.Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, Freiberg AN, Su L, Lee B and Cheng G. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity. 2013;38:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dang EV, McDonald JG, Russell DW and Cyster JG. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell. 2017;171:1057–1071 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan SF, Ramasamy R and Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eckel RH, Bornfeldt KE and Goldberg IJ. Cardiovascular disease in diabetes, beyond glucose. Cell Metab. 2021;33:1519–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hughes MM and O’Neill LAJ. Metabolic regulation of NLRP3. Immunol Rev. 2018;281:88–98. [DOI] [PubMed] [Google Scholar]
- 75.Olona A, Leishman S and Anand PK. The NLRP3 inflammasome: regulation by metabolic signals. Trends Immunol. 2022;43:978–989. [DOI] [PubMed] [Google Scholar]
- 76.Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, Ruzek MC, Slivka PF, McGettrick AF, Banahan K, Hughes MM, Irvine AD, Fischer R and O’Neill LAJ. The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab. 2020;32:468–478 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hughes MM, Hooftman A, Angiari S, Tummala P, Zaslona Z, Runtsch MC, McGettrick AF, Sutton CE, Diskin C, Rooke M, Takahashi S, Sundararaj S, Casarotto MG, Dahlstrom JE, Palsson-McDermott EM, Corr SC, Mills KHG, Preston RJS, Neamati N, Xie Y, Baell JB, Board PG and O’Neill LAJ. Glutathione Transferase Omega-1 Regulates NLRP3 Inflammasome Activation through NEK7 Deglutathionylation. Cell Rep. 2019;29:151–161 e5. [DOI] [PubMed] [Google Scholar]
- 78.He W, Henne A, Lauterbach M, Geissmar E, Nikolka F, Kho C, Heinz A, Dostert C, Grusdat M, Cordes T, Harm J, Goldmann O, Ewen A, Verschueren C, Blay-Cadanet J, Geffers R, Garritsen H, Kneiling M, Holm CK, Metallo CM, Medina E, Abdullah Z, Latz E, Brenner D and Hiller K. Mesaconate is synthesized from itaconate and exerts immunomodulatory effects in macrophages. Nat Metab. 2022;4:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishizawa T, Kanter JE, Kramer F, Barnhart S, Shen X, Vivekanandan-Giri A, Wall VZ, Kowitz J, Devaraj S, O’Brien KD, Pennathur S, Tang J, Miyaoka RS, Raines EW and Bornfeldt KE. Testing the role of myeloid cell glucose flux in inflammation and atherosclerosis. Cell Rep. 2014;7:356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karasawa T, Kawashima A, Usui-Kawanishi F, Watanabe S, Kimura H, Kamata R, Shirasuna K, Koyama Y, Sato-Tomita A, Matsuzaka T, Tomoda H, Park SY, Shibayama N, Shimano H, Kasahara T and Takahashi M. Saturated Fatty Acids Undergo Intracellular Crystallization and Activate the NLRP3 Inflammasome in Macrophages. Arterioscler Thromb Vasc Biol. 2018;38:744–756. [DOI] [PubMed] [Google Scholar]
- 81.L’Homme L, Esser N, Riva L, Scheen A, Paquot N, Piette J and Legrand-Poels S. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J Lipid Res. 2013;54:2998–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalugotla G, He L, Weber KJ, Daemen S, Reller A, Razani B and Schilling JD. Frontline Science: Acyl-CoA synthetase 1 exacerbates lipotoxic inflammasome activation in primary macrophages. J Leukoc Biol. 2019;106:803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, Li LO, Becker L, Yuan W, Chait A, Braun KR, Potter-Perigo S, Sanda S, Wight TN, Pennathur S, Serhan CN, Heinecke JW, Coleman RA and Bornfeldt KE. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc Natl Acad Sci U S A. 2012;109:E715–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bornfeldt KE. The Remnant Lipoprotein Hypothesis of Diabetes-Associated Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2022;42:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanter JE, Shao B, Kramer F, Barnhart S, Shimizu-Albergine M, Vaisar T, Graham MJ, Crooke RM, Manuel CR, Haeusler RA, Mar D, Bomsztyk K, Hokanson JE, Kinney GL, Snell-Bergeon JK, Heinecke JW and Bornfeldt KE. Increased apolipoprotein C3 drives cardiovascular risk in type 1 diabetes. J Clin Invest. 2019;129:4165–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, Klug M, Schunk SJ, Schmit D, Kramann R, Korbel C, Ampofo E, Laschke MW, Selejan SR, Paschen A, Herter T, Schuster S, Silbernagel G, Sester M, Sester U, Assmann G, Bals R, Kostner G, Jahnen-Dechent W, Menger MD, Rohrer L, Marz W, Bohm M, Jankowski J, Kopf M, Latz E, Niemeyer BA, Fliser D, Laufs U and Speer T. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. 2020;21:30–41. [DOI] [PubMed] [Google Scholar]
- 87.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G and Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A. 2008;105:8067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hsu CC, Shao B, Kanter JE, He Y, Vaisar T, Witztum JL, Snell-Bergeon J, McInnes G, Bruse S, Gottesman O, Mullick AE and Bornfeldt KE. Apolipoprotein C3 induces inflammasome activation only in its delipidated form. Nat Immunol. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, Klug M, Schunk SJ, Schmit D, Kramann R, Korbel C, Ampofo E, Laschke MW, Selejan SR, Paschen A, Herter T, Gaul S, Silbernagel G, Sester M, Sester U, Assmann G, Bals R, Kostner G, Jahnen-Dechent W, Menger MD, Rohrer L, Marz W, Bohm M, Jankowski J, Kopf M, Latz E, Niemeyer BA, Fliser D, Laufs U and Speer T. Reply to: Apolipoprotein C3 induces inflammasome activation only in its delipidated form. Nat Immunol. 2023. [DOI] [PubMed] [Google Scholar]
- 90.Gu P, Hui X, Zheng Q, Gao Y, Jin L, Jiang W, Zhou C, Liu T, Huang Y, Liu Q, Nie T, Wang Y, Wang Y, Zhao J and Xu A. Mitochondrial uncoupling protein 1 antagonizes atherosclerosis by blocking NLRP3 inflammasome-dependent interleukin-1beta production. Sci Adv. 2021;7:eabl4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D and Ebert BL. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S and Ebert BL. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Natarajan P 2022. Hoeg Jeffrey M. Award Lecture: Genomic Aging, Clonal Hematopoiesis, and Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jaiswal S and Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rahman MF, Yang Y, Le BT, Dutta A, Posyniak J, Faughnan P, Sayem MA, Aguilera NS and Mohi G. Interleukin-1 contributes to clonal expansion and progression of bone marrow fibrosis in JAK2V617F-induced myeloproliferative neoplasm. Nat Commun. 2022;13:5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rai S, Grockowiak E, Hansen N, Luque Paz D, Stoll CB, Hao-Shen H, Mild-Schneider G, Dirnhofer S, Farady CJ, Mendez-Ferrer S and Skoda RC. Inhibition of interleukin-1beta reduces myelofibrosis and osteosclerosis in mice with JAK2-V617F driven myeloproliferative neoplasm. Nat Commun. 2022;13:5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Al-Rifai R, Vandestienne M, Lavillegrand JR, Mirault T, Cornebise J, Poisson J, Laurans L, Esposito B, James C, Mansier O, Hirsch P, Favale F, Braik R, Knosp C, Vilar J, Rizzo G, Zernecke A, Saliba AE, Tedgui A, Lacroix M, Arrive L, Mallat Z, Taleb S, Diedisheim M, Cochain C, Rautou PE and Ait-Oufella H. JAK2V617F mutation drives vascular resident macrophages toward a pathogenic phenotype and promotes dissecting aortic aneurysm. Nat Commun. 2022;13:6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yokokawa T, Misaka T, Kimishima Y, Wada K, Minakawa K, Sugimoto K, Ishida T, Morishita S, Komatsu N, Ikeda K and Takeishi Y. Crucial role of hematopoietic JAK2 V617F in the development of aortic aneurysms. Haematologica. 2021;106:1910–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heyde A, Rohde D, McAlpine CS, Zhang S, Hoyer FF, Gerold JM, Cheek D, Iwamoto Y, Schloss MJ, Vandoorne K, Iborra-Egea O, Munoz-Guijosa C, Bayes-Genis A, Reiter JG, Craig M, Swirski FK, Nahrendorf M, Nowak MA and Naxerova K. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell. 2021;184:1348–1361 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kessler MD, Damask A, O’Keeffe S, Banerjee N, Li D, Watanabe K, Marketta A, Van Meter M, Semrau S, Horowitz J, Tang J, Kosmicki JA, Rajagopal VM, Zou Y, Houvras Y, Ghosh A, Gillies C, Mbatchou J, White RR, Verweij N, Bovijn J, Parikshak NN, LeBlanc MG, Jones M, Regeneron Genetics C, Collaboration G-RD, Glass DJ, Lotta LA, Cantor MN, Atwal GS, Locke AE, Ferreira MAR, Deering R, Paulding C, Shuldiner AR, Thurston G, Ferrando AA, Salerno W, Reid JG, Overton JD, Marchini J, Kang HM, Baras A, Abecasis GR and Jorgenson E. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature. 2022;612:301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Scott Beeler AGBKLB J. Genetic causes and cardiovascular consequences of clonal hematopoiesis in the UK Biobank. Nature Cardiovascular Research. 2022; 10.1038/s44161-022-00198-3. [DOI] [PubMed] [Google Scholar]
- 102.Caitlyn Vlasschaert1 TM, Heimlich3 J. Brett, Niroula4 Abhishek, Mesbah Uddin4, Weinstock8 Joshua, Sharber2 Brian, Silver9 Alexander J., Xu10 Yaomin, Savona9 Michael, Lanktree17 Matthew B., Rauh19 Michael J., Ebert4 Benjamin L., Pradeep and Natarajan4, Jaiswal22 Siddhartha, Bick2 Alexander G.. A practical approach to curate clonal hematopoiesis of indeterminate potential in human genetic datasets. BioRxiv preprint. 2022; 10.1101/2022.10.21.22281368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL and Owens GK. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest. 2012;122:70–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gomez D, Baylis RA, Durgin BG, Newman AAC, Alencar GF, Mahan S, St Hilaire C, Muller W, Waisman A, Francis SE, Pinteaux E, Randolph GJ, Gram H and Owens GK. Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med. 2018;24:1418–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vromman A, Ruvkun V, Shvartz E, Wojtkiewicz G, Santos Masson G, Tesmenitsky Y, Folco E, Gram H, Nahrendorf M, Swirski FK, Sukhova GK and Libby P. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur Heart J. 2019;40:2482–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hettwer J, Hinterdobler J, Miritsch B, Deutsch MA, Li X, Mauersberger C, Moggio A, Braster Q, Gram H, Robertson AAB, Cooper MA, Gross O, Krane M, Weber C, Koenig W, Soehnlein O, Adamstein NH, Ridker P, Schunkert H, Libby P, Kessler T and Sager HB. Interleukin-1beta suppression dampens inflammatory leucocyte production and uptake in atherosclerosis. Cardiovasc Res. 2022;118:2778–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Choudhury RP, Birks JS, Mani V, Biasiolli L, Robson MD, L’Allier PL, Gingras MA, Alie N, McLaughlin MA, Basson CT, Schecter AD, Svensson EC, Zhang Y, Yates D, Tardif JC and Fayad ZA. Arterial Effects of Canakinumab in Patients With Atherosclerosis and Type 2 Diabetes or Glucose Intolerance. J Am Coll Cardiol. 2016;68:1769–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hansson GK, Libby P and Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.van der Wal AC and Becker AE. Atherosclerotic plaque rupture--pathologic basis of plaque stability and instability. Cardiovasc Res. 1999;41:334–44. [DOI] [PubMed] [Google Scholar]
- 110.Silvestre-Roig C, de Winther MP, Weber C, Daemen MJ, Lutgens E and Soehnlein O. Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ Res. 2014;114:214–26. [DOI] [PubMed] [Google Scholar]
- 111.Libby P The changing landscape of atherosclerosis. Nature. 2021;592:524–533. [DOI] [PubMed] [Google Scholar]
- 112.Chen CJ, Kono H, Golenbock D, Reed G, Akira S and Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6. [DOI] [PubMed] [Google Scholar]
- 113.Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, Nakamura M, Newton G, Luscinskas FW and Libby P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1alpha and Cathepsin G. Arterioscler Thromb Vasc Biol. 2018;38:1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schunk SJ, Triem S, Schmit D, Zewinger S, Sarakpi T, Becker E, Hutter G, Wrublewsky S, Kuting F, Hohl M, Alansary D, Prates Roma L, Lipp P, Mollmann J, Lehrke M, Laschke MW, Menger MD, Kramann R, Boor P, Jahnen-Dechent W, Marz W, Bohm M, Laufs U, Niemeyer BA, Fliser D, Ampofo E and Speer T. Interleukin-1alpha Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation. 2021;144:893–908. [DOI] [PubMed] [Google Scholar]
- 115.Ridker PM, MacFadyen JG, Thuren T and Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. 2020;41:2153–2163. [DOI] [PubMed] [Google Scholar]
- 116.Whitman SC, Ravisankar P and Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(−/−) mice through release of interferon-gamma. Circ Res. 2002;90:E34–8. [DOI] [PubMed] [Google Scholar]
- 117.Whitman SC, Ravisankar P, Elam H and Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. Am J Pathol. 2000;157:1819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dinarello CA, Novick D, Kim S and Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gupta S, Pablo AM, Jiang X, Wang N, Tall AR and Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Opoku E, Traughber CA, Zhang D, Iacano AJ, Khan M, Han J, Smith JD and Gulshan K. Gasdermin D Mediates Inflammation-Induced Defects in Reverse Cholesterol Transport and Promotes Atherosclerosis. Front Cell Dev Biol. 2021;9:715211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Puylaert P, Van Praet M, Vaes F, Neutel CHG, Roth L, Guns PJ, De Meyer GRY and Martinet W. Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in ApoE Knock-Out Mice. Biomedicines. 2022;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yurdagul A Jr., Doran AC, Cai B, Fredman G and Tabas IA. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front Cardiovasc Med. 2017;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brinkmann V and Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. 2012;198:773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Doring Y, Libby P and Soehnlein O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ Res. 2020;126:1228–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, Kruger R, Herzig A and Zychlinsky A. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3. [DOI] [PubMed] [Google Scholar]
- 126.Yalcinkaya M, Fotakis P, Liu W, Endo-Umeda K, Dou H, Abramowicz S, Xiao T, Libby P, Wang N, Tall AR and Westerterp M. Cholesterol accumulation in macrophages drives NETosis in atherosclerotic plaques via IL-1beta secretion. Cardiovasc Res. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Warnatsch A, Ioannou M, Wang Q and Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT and Kaplan MJ. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. 2014;114:947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Munzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, Cherpokova D, Gutch S, Sorvillo N, Shi L, Magupalli VG, Weber ANR, Scharf RE, Waterman CM, Wu H and Wagner DD. NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front Immunol. 2021;12:683803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Franck G, Mawson TL, Folco EJ, Molinaro R, Ruvkun V, Engelbertsen D, Liu X, Tesmenitsky Y, Shvartz E, Sukhova GK, Michel JB, Nicoletti A, Lichtman A, Wagner D, Croce KJ and Libby P. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ Res. 2018;123:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T and Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–60. [DOI] [PubMed] [Google Scholar]
- 132.Imazio M and Nidorf M. Colchicine and the heart. Eur Heart J. 2021;42:2745–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dalbeth N, Choi HK, Joosten LAB, Khanna PP, Matsuo H, Perez-Ruiz F and Stamp LK. Gout. Nat Rev Dis Primers. 2019;5:69. [DOI] [PubMed] [Google Scholar]
- 134.Schunk SJ, Kleber ME, Marz W, Pang S, Zewinger S, Triem S, Ege P, Reichert MC, Krawczyk M, Weber SN, Jaumann I, Schmit D, Sarakpi T, Wagenpfeil S, Kramann R, Boerwinkle E, Ballantyne CM, Grove ML, Tragante V, Pilbrow AP, Richards AM, Cameron VA, Doughty RN, Dube MP, Tardif JC, Feroz-Zada Y, Sun M, Liu C, Ko YA, Quyyumi AA, Hartiala JA, Tang WHW, Hazen SL, Allayee H, McDonough CW, Gong Y, Cooper-DeHoff RM, Johnson JA, Scholz M, Teren A, Burkhardt R, Martinsson A, Smith JG, Wallentin L, James SK, Eriksson N, White H, Held C, Waterworth D, Trompet S, Jukema JW, Ford I, Stott DJ, Sattar N, Cresci S, Spertus JA, Campbell H, Tierling S, Walter J, Ampofo E, Niemeyer BA, Lipp P, Schunkert H, Bohm M, Koenig W, Fliser D, Laufs U, Speer T, e Qc and consortium B. Genetically determined NLRP3 inflammasome activation associates with systemic inflammation and cardiovascular mortality. Eur Heart J. 2021;42:1742–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ridker PM and Rane M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ Res. 2021;128:1728–1746. [DOI] [PubMed] [Google Scholar]
- 136.Ridker PM, MacFadyen JG, Thuren T and Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. 2019. [DOI] [PubMed] [Google Scholar]
- 137.Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, Libby P, Kathiresan S and Natarajan P. Genetic IL-6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE and Investigators E-RO. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373:2117–28. [DOI] [PubMed] [Google Scholar]
- 139.Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, Rim JH, Hwang I, Lee CJ, Lee M, Oh CM, Jeon JY, Gee HY, Kim JH, Lee BW, Kang ES, Cha BS, Lee MS, Yu JW, Cho JW, Kim JS and Lee YH. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. 2020;11:2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen YC, Jandeleit-Dahm K and Peter K. Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Dapagliflozin Stabilizes Diabetes-Induced Atherosclerotic Plaque Instability. J Am Heart Assoc. 2022;11:e022761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Levy R, Gerard L, Kuemmerle-Deschner J, Lachmann HJ, Kone-Paut I, Cantarini L, Woo P, Naselli A, Bader-Meunier B, Insalaco A, Al-Mayouf SM, Ozen S, Hofer M, Frenkel J, Modesto C, Nikishina I, Schwarz T, Martino S, Meini A, Quartier P, Martini A, Ruperto N, Neven B, Gattorno M, for P and Eurofever. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis. 2015;74:2043–9. [DOI] [PubMed] [Google Scholar]
- 142.Langevitz P, Livneh A, Neumann L, Buskila D, Shemer J, Amolsky D and Pras M. Prevalence of ischemic heart disease in patients with familial Mediterranean fever. Isr Med Assoc J. 2001;3:9–12. [PubMed] [Google Scholar]
- 143.Gendelman O, Shapira R, Tiosano S, Pras E, Comaneshter D, Cohen A and Amital H. Familial Mediterranean fever is associated with increased risk for ischaemic heart disease and mortality-Perspective derived from a large database. Int J Clin Pract. 2020;74:e13473. [DOI] [PubMed] [Google Scholar]
- 144.Booshehri LM and Hoffman HM. CAPS and NLRP3. J Clin Immunol. 2019;39:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Paramel Varghese G, Folkersen L, Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, Krohg-Sorensen K, Skjelland M, Espevik T, Aukrust P, Lengquist M, Hedin U, Jansson JH, Fransen K, Hansson GK, Eriksson P and Sirsjo A. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J Am Heart Assoc. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shi X, Xie WL, Kong WW, Chen D and Qu P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J Stroke Cerebrovasc Dis. 2015;24:2455–66. [DOI] [PubMed] [Google Scholar]
- 147.Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, Khan NS, Wong CK, Shamailova R, Hill CA, Wang Z, Remark R, Li JR, Pina C, Faries C, Awad AJ, Moss N, Bjorkegren JLM, Kim-Schulze S, Gnjatic S, Ma’ayan A, Mocco J, Faries P, Merad M and Giannarelli C. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Redgrave JN, Lovett JK, Gallagher PJ and Rothwell PM. Histological assessment of 526 symptomatic carotid plaques in relation to the nature and timing of ischemic symptoms: the Oxford plaque study. Circulation. 2006;113:2320–8. [DOI] [PubMed] [Google Scholar]
- 149.Svensson EC, Madar A, Campbell CD, He Y, Sultan M, Healey ML, Xu H, D’Aco K, Fernandez A, Wache-Mainier C, Libby P, Ridker PM, Beste MT and Basson CT. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab: An Exploratory Analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. 2022;7:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhi Yu TPF, Ruan Yunfeng, Vlasschaert Caitlyn, Nakao Tetsushi, Uddin Md Mesbah, Mack Taralynn, Niroula Abhishek, Brett J and Heimlich SMZ, Gibson Christopher J., Griffin Gabriel K., Wang Yuxuan, Peloso Gina M., Heard-Costa Nancy, Levy Daniel, Vasan Ramachandran S., Aguet François, Ardlie Kristin, Taylor Kent D., Rich Stephen S., Rotter Jerome I., Libby Peter, Jaiswal Siddhartha, Ebert Benjamin L., Bick Alexander G., Tall Alan R., Pradeep Natarajan Genetic modification of inflammation and clonal hematopoiesis-a 1 ssociated coronary artery disease. BioRxiv preprint. 2022; 10.1101/2022.12.08.22283237. [DOI] [Google Scholar]
- 151.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. [DOI] [PubMed] [Google Scholar]
- 152.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD and Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:688. [DOI] [PubMed] [Google Scholar]
- 153.Ren GM, Li J, Zhang XC, Wang Y, Xiao Y, Zhang XY, Liu X, Zhang W, Ma WB, Zhang J, Li YT, Tao SS, Wang T, Liu K, Chen H, Zhan YQ, Yu M, Li CY, Ge CH, Tian BX, Dou GF, Yang XM and Yin RH. Pharmacological targeting of NLRP3 deubiquitination for treatment of NLRP3-associated inflammatory diseases. Sci Immunol. 2021;6. [DOI] [PubMed] [Google Scholar]