

Significance
Aneuploidy syndromes are collectively common and impact diverse organs—but it remains unclear whether and how aneuploidy effects on gene expression vary across human tissues. Here, we systematically examine the effects of varying chromosome X, Y, and 21 dosages in 3 different cell types: lymphoblastoid cell lines, fibroblasts, and induced pluripotent stem cell-derived excitatory neurons. We detail how—for all three chromosomes—cis effects of aneuploidy (i.e., expression changes on the aneuploidic chromosome) generalize across tissues, but trans effects (expression changes off the chromosome) do not. These findings illuminate the mechanistic bases of gene dosage disorders and the ways in which we may use accessible peripheral tissues to model effects in harder to access organs like brain.
Keywords: sex chromosome aneuploidy, trisomy 21, dosage compensation, X-Y gametologs, X-chromosome inactivation
Abstract
Aneuploidy syndromes impact multiple organ systems but understanding of tissue-specific aneuploidy effects remains limited—especially for the comparison between peripheral tissues and relatively inaccessible tissues like brain. Here, we address this gap in knowledge by studying the transcriptomic effects of chromosome X, Y, and 21 aneuploidies in lymphoblastoid cell lines, fibroblasts and iPSC-derived neuronal cells (LCLs, FCL, and iNs, respectively). We root our analyses in sex chromosome aneuploidies, which offer a uniquely wide karyotype range for dosage effect analysis. We first harness a large LCL RNA-seq dataset from 197 individuals with one of 6 sex chromosome dosages (SCDs: XX, XXX, XY, XXY, XYY, and XXYY) to i) validate theoretical models of SCD sensitivity and ii) define an expanded set of 41 genes that show obligate dosage sensitivity to SCD and are all in cis (i.e., reside on the X or Y chromosome). We then use multiple complementary analyses to show that cis effects of SCD in LCLs are preserved in both FCLs (n = 32) and iNs (n = 24), whereas trans effects (i.e., those on autosomal gene expression) are mostly not preserved. Analysis of additional datasets confirms that the greater cross-cell type reproducibility of cis vs. trans effects is also seen in trisomy 21 cell lines. These findings i) expand our understanding of X, Y, and 21 chromosome dosage effects on human gene expression and ii) suggest that LCLs may provide a good model system for understanding cis effects of aneuploidy in harder-to-access cell types.
Aneuploidies are an important risk factor in diverse fields of medicine (1–3) and also provide naturally occurring probes for basic science research on human genome regulation (4–6). Because aneuploidy syndromes typically impact multiple tissue types (2, 3), and gene regulation varies between tissue types (7, 8), it becomes essential to understand how the same aneuploidy impacts gene expression in different human cell types/tissues. However, to date, very few published studies have examined the transcriptional effects of aneuploidy on different human tissues (9, 10), and there are currently no cross-tissue studies that consider more than one aneuploidy. The current study seeks to address these gaps in knowledge by systematically profiling effects of the three most common chromosomal aneuploidies—those involving chromosomes X, Y, and 21 (11, 12)—on measures of gene expression in 3 different human cell types: lymphoblastoid cell lines (LCLs), fibroblast cell lines (FCLs), and cortical neurons derived from induced pluripotent stem cells (iNs).
Our primary analyses focus on the effects of X and Y chromosome aneuploidy using human RNA-seq data from LCLs, FCLs, and iNs spanning 6 different sex chromosome dosages (SCDs): XX, XXX, XY, XXY, XYY, and XXYY (total n = 253). Several factors motivate our initial focus on sex chromosome aneuploidies. First, they are collectively the most common form of human aneuploidy [occurring in 1:400 births (12)]. Second, their high karyotypic diversity provides an especially powerful test for aneuploidy effects because the same chromosome dosage change can be observed in multiple contexts [facilitating detection of genes showing obligate dosage sensitivity (ODS) where expression levels always vary with SCD]. Expression variation across karyotypically diverse SCD groups also enables agnostic clustering of genes by dosage sensitivity (13). Third, the question of whether transcriptional effects of aneuploidy generalize across tissues is highly relevant for a mechanistic understanding of X and Y chromosome aneuploidy effects across diverse immune, metabolic and neuropsychiatric outcomes (14, 15). Finally, understanding SCD effects has relevance beyond aneuploidy given that euploid males and females also differ in SCD. Specifically, XY males uniquely express Y-linked genes, whereas the process of X chromosome inactivation (XCI) in XX females—from which ~15% of X-linked genes escape (16)—leads to more expressed copies of some X-linked genes in females compared to males (17). In contrast, males and females both express two copies of X-Y gametologs (ancestral sex-chromosome genes that have retained a copy on the Y chromosome) (18, 19) and of genes located in the pseudoautosomal region (PAR).
Here, we adopt a stepwise approach to profiling SCD effects on gene expression in different cell types, first using a large primary LCL dataset to systematically characterize SCD effects on human gene expression across six different SCDs (XX, XXX, XY, XXY, XYY, and XXYY). We apply conjunction and clustering analyses to define core transcriptional effects of SCD variation in LCLs. Next, we test whether observed effects of X and Y chromosome dosage (XCD and YCD, respectively) on gene expression in LCLs align with those in FCLs (n = 32) and iNs (n = 24, 2 technical replicates from each of 2 individuals in each SCD group) samples representing the same 6 SCD groups (XX, XXX, XY, XXY, XYY, and XXYY). In doing so, we separately test cross-cell type reproducibility of statistically significant SCD effects for sex chromosome genes (i.e., cis effects when on the dosage-altered sex chromosome) vs. autosomal genes (i.e., trans effects)—inspired by existent cross-tissue studies of aneuploidy effects on gene expression in maize, which find that genes in cis to an aneuploidy show more stable expression change across tissues than genes in trans (20–22). Inclusion of iNs in this cross-tissue test addresses a particularly pressing need because sex chromosome aneuploidy effects on human neural tissue remain largely uncharacterized despite the prominent impact of these aneuploidies on human brain structure and function (14, 23). This gap in knowledge primarily reflects lack of easy access to human brain tissue, which could potentially be addressed by development of viable in vitro models from patient-derived induced pluripotent stem cells. The current study generates and presents an initial characterization of in vitro neuronal models in sex chromosome aneuploidy.
Finally—having characterized the degree to which statistically significant cis vs. trans effects of aneuploidy generalize across tissues for the X and Y chromosomes—we determine the extent to which a similar pattern is seen for cis vs. trans effects of chromosome 21 aneuploidy (i.e., cis effects on expression of chr21 genes; trans effects on expression beyond chr21) by analyzing cross-tissue measures of gene expression in Down syndrome (DS) (10).
Results
Detailing SCD Effects on Gene Expression in LCLs.
We first used our core LCL dataset derived from 197 individuals spanning 6 SCD groups (Fig. 1) to identify differentially expressed genes (DEGs at FDR < 0.05) in each of 15 unique pairwise SCD group contrasts (Figs. 1 and 2A, Materials and Methods, and SI Appendix, Text S1.1). We reran these analyses in sample-size-matched SCD group contrasts to verify that the rank ordering of contrasts by DEG count was not an artifact of contrast differences in sample size (SI Appendix, Fig. S1 A–D, Pearson’s r = 0.95 for DEG concordance across contrasts, Materials and Methods, SI Appendix, Text S1.2).
Fig. 1.

Sex chromosome dosage (SCD) sample characteristics and group contrasts within this sample. (A) Sample characteristics for each SCD karyotype group in each of 3 cell types: Lymphoblastoid (LCLs, n = 197), Fibroblast (FCLs, n = 32) and ipsc-induced Neuronal (iNs, n = 24) cell lines. *The 4 iN samples for each SCD group consisted of 2 technical replicates (clones) for each of 2 individuals. (B) The 15 unique pairwise contrasts between SCD groups are labeled according to whether each contrast is associated with changes in X chromosome dosage (XCD, n = 11 contrasts), Y chromosome dosage (YCD, n = 12 contrasts) or total sex chromosome dosage (tSCD, i.e., the combined number of X and Y chromosomes, n = 11 contrasts), marked as ΔX, ΔY and ΔtSCD, respectively.
Fig. 2.
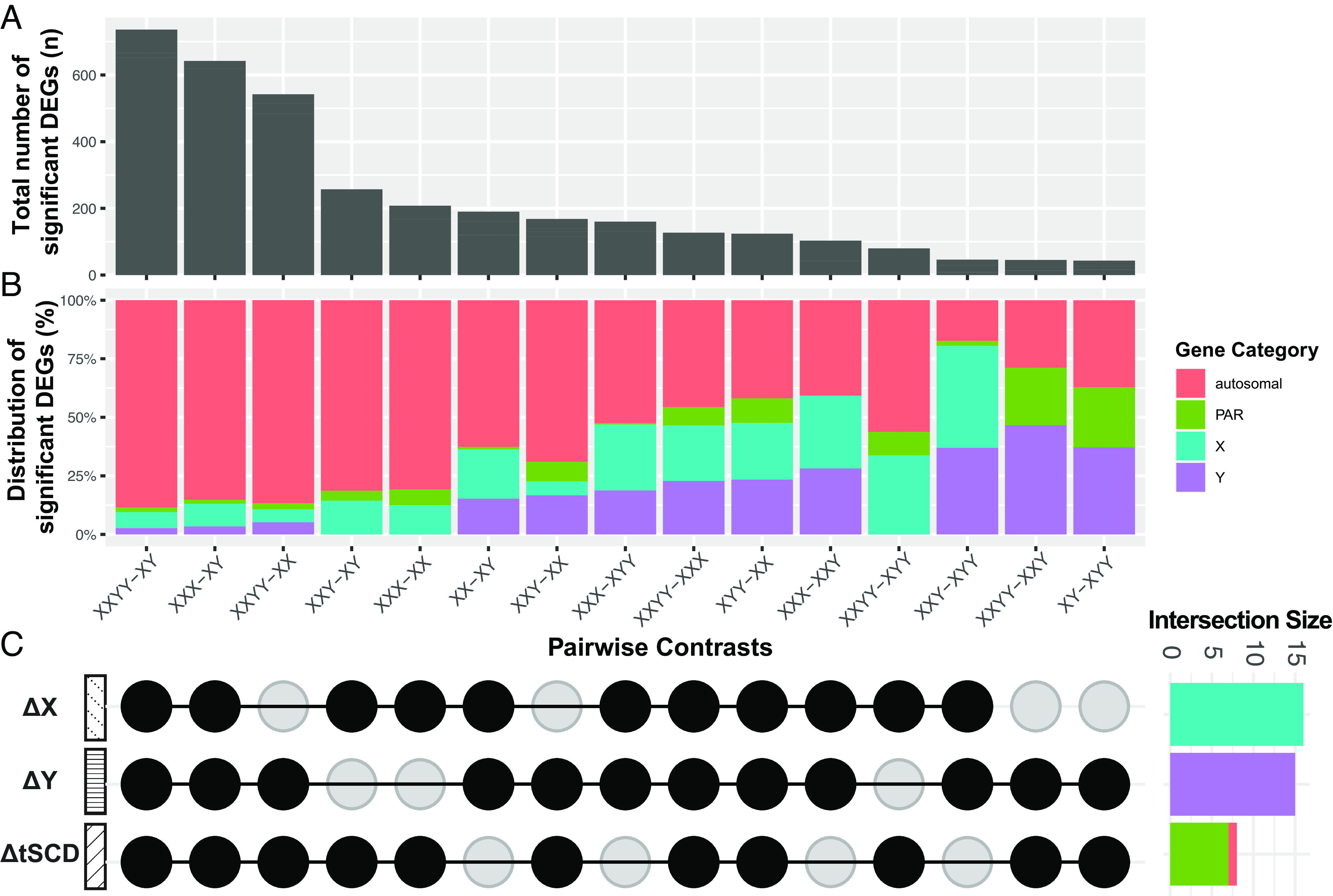
Distribution of differentially expressed genes (DEGs) across pairwise sex chromosome dosage (SCD) group contrasts in lymphoblastoid cell lines (LCLs). (A) Bar chart showing the total number of DEGs in each group contrast (False-discovery rate q < 0.05; full DEG list Dataset S1), (B) Stacked bar chart showing the proportion of DEGs in each contrast by chromosome of origin: autosomal, PAR, X and Y (X- and Y-specific nonPAR). (C) Upset plot showing which SCD group contrasts capture each of the 3 main modes of SCD variation: changes in X chromosome dosage (ΔX), Y chromosome dosage (ΔY), and total SCD (ΔtSCD). Side panel bar chart for the number of DEGs shared across all group contrasts in each mode of SCD variation. The genes specified by these intersection sets therefore show obligate dosage sensitivity (ODS) to each mode of SCD variation (ODS genes listed in Tables 1–3).
Of the 25,075 genes considered in these analyses of SCD effects on LCL gene expression, 1,476 were DEGs in at least one of the 15 unique SCD group contrasts (Dataset S1). Approximately 90% of these 1,476 DEGs were autosomal, although—as expected—the proportion of sex-linked DEGs was significantly enriched relative to background genome proportions (Fisher’s Exact Test: P < 1.7e-12). The chromosomal origin of DEGs was nonrandom across SCD contrasts (chi-square test: P < 2.2e-16, Fig. 2B), and regression models estimating DEG counts from SCD contrast features (Materials and Methods, SI Appendix, Text S1.2) showed that counts of: autosomal DEGs tracked with XCD and tSCD; PAR DEGs with tSCD; X-linked DEGs mostly with XCD; and, Y-linked DEGs with YCD, respectively (SI Appendix, Fig. S1E). Thus, impacts of SCD on autosomal gene expression (i.e., trans effects) are more prominent for variations in X rather than Y chromosome dosage.
Detection of DEGs provides a categorical view of gene expression changes in each SCD contrast variation but loses information on the distribution of expression changes across genes. Therefore, in addition to gene-level fold changes provided in Dataset S1, we took two approaches to examine distributions of expression change in each SCD group contrast. First, we constructed “ratio distribution plots” as pioneered in prior work on aneuploidy (6, 24). In this approach (6, 24), gene-level expression differences between two groups are represented by the ratio between mean expression values in each group (SI Appendix, Text S1.3). The cross-gene distribution of these ratios is tested for normality and significant deviations from normality are taken as evidence of “modulation” (6, 24), interpreted as the aneuploidy altering the distribution of gene expression values. Of note, because ratio plots convey the proportion of expression changes that exist in different ranges of fold change, they complement DEG analyses by better capturing the relative balance between dosage compensation effects as compared to direct dosage effects (positive modulation) or inverse dosage effects (negative modulation). We find that ratio distributions do indeed significantly deviate from normality for cis and trans effects in almost all SCD group contrasts (SI Appendix, Fig. S2). Ratio plots also highlight the prominence of direct dosage effects with YCD changes as compared to the greater degree of dosage compensation with XCD changes [notwithstanding the substantial direct dosage effects of XCD in both ratio plots (SI Appendix, Fig. S2) and DEG analysis (Dataset S1)]. Second, we generated classical volcano plots showing the magnitude and statistical significance of gene-level fold changes in each SCD group contrast (SI Appendix, Text S1.4 and Fig. S3).
We next used conjunction of DEG lists across SCD contrasts to identify genes showing ODS (i.e., recurrent DEGs across contrasts) to each mode of SCD variation in LCLs (Fig. 2C). We identified 18 genes with ODS to XCD changes, which were all X-linked genes known to escape XCI (Table 1) (16, 17), and 15 genes with ODS to YCD changes, which were all Y-linked (Table 2). These results replicate and expand upon the smaller number of ODS genes reported in our past microarray study in a smaller LCL sample size (13)—likely reflecting the benefits of larger sample size and greater dynamic range from RNA-seq in our current study. As in our past study, the expanded sets of ODS genes identified here were also strongly enriched for X-Y gametologs (P < 0.001, Tables 1 and 2). The combined set of 33 sex-linked ODS genes showed statistically significant gene ontology (GO) term enrichment for histone and protein demethylation—reflecting the prominent involvement of several gametolog gene pairs in these processes (e.g., KDM5C-KDM5D and KDM6A-UTY, corrected P < 0.005, Datasets S4 and S5). Genes with ODS to XCD showed GO term enrichment for translational initiation and regulation of translation (capturing known functions of EIF1AX and RPS4X, Dataset S4). Conjunction analyses also identified 8 genes with ODS to tSCD changes (Table 3), comprising 7 genes from the PAR1 region and 1 autosomal gene (HCG11). This observation aligns with prior reports that PAR1 genes escape XCI, whereas PAR2 genes do not (16, 17). Genes with ODS to changes in XCD, YCD, and tSCD in our full dataset were also recovered in control analyses using sample-size matched SCD groups (SI Appendix, Fig. S1C and Dataset S6).
Table 1.
Genes with obligate dosage sensitivity (ODS) to X chromosome dosage changes in lymphoblastoid cell lines (LCLs)
| Ensembl ID | Symbol | Chr | Gene description | X Escape | Y Gametolog |
|---|---|---|---|---|---|
| ENSG00000005889 | ZFX | X | Regulation of transcription, multicellular organism development | Yes | ZFY |
| ENSG00000006757 | PNPLA4 | X | Lipid metabolic process, triglyceride catabolic process | Yes | |
| ENSG00000069509 | FUNDC1 | X | An activator of hypoxia-induced mitophagy | Yes | |
| ENSG00000072501 | SMC1A | X | Chromosome cohesion during cell cycle and in DNA repair | Yes | |
| ENSG00000086712 | TXLNG | X | Regulation of cell cycle | Yes | TXLNGY |
| ENSG00000126012 | KDM5C | X | Chromatin organization, chromatin remodeling | Yes | KDM5D |
| ENSG00000130021 | PUDP | X | Yes | ||
| ENSG00000130741 | EIF2S3 | X | GTP binding and translation initiation factor activity | Yes | |
| ENSG00000130985 | UBA1 | X | Timely DNA repair and for response to replication stress | Yes | |
| ENSG00000147050 | KDM6A* | X | In utero embryonic development, chromatin organization | Yes | UTY |
| ENSG00000169249 | ZRSR2 | X | Spliceosomal complex assembly, RNA splicing | Yes | |
| ENSG00000173674 | EIF1AX* | X | Translation, translational initiation | Yes | EIF1AY |
| ENSG00000183943 | PRKX | X | Endothelial cell proliferation, signal transduction | Yes | PRKY |
| ENSG00000198034 | RPS4X | X | Multicellular organism development, translational initiation | Yes | RPS4Y1/RPS4Y2 |
| ENSG00000215301 | DDX3X | X | Immune system process, regulation of translation | Yes | DDX3Y |
| ENSG00000225470 | JPX | X | Positive regulation of gene expression | Yes | |
| ENSG00000229807 | XIST* | X | Dosage compensation by inactivation of X chromosome | Yes | |
| ENSG00000285679 | Lnc-VCX-2 | X | Yes |
Table 2.
Genes with obligate dosage sensitivity (ODS) to Y chromosome dosage changes in lymphoblastoid cell lines (LCLs)
| Ensembl ID | Symbol | Chr | Gene description | X Gametolog |
|---|---|---|---|---|
| ENSG00000012817 | KDM5D | Y | Chromatin organization, chromatin remodeling | KDM5C |
| ENSG00000067048 | DDX3Y* | Y | Gamete generation, cell differentiation | DDX3X |
| ENSG00000067646 | ZFY* | Y | Regulation of transcription, DNA-templated | ZFX |
| ENSG00000099725 | PRKY | Y | Protein phosphorylation, signal transduction | PRKX |
| ENSG00000114374 | USP9Y* | Y | Proteolysis, spermatogenesis, cell migration | USP9X |
| ENSG00000129824 | RPS4Y1 | Y | Multicellular organism development, translational initiation | RPS4X |
| ENSG00000131002 | TXLNGY | Y | Syntaxin binding | TXLNG |
| ENSG00000154620 | TMSB4Y* | Y | Actin filament organization, regulation of cell migration | TMSB4X |
| ENSG00000165246 | NLGN4Y | Y | Cell adhesion, chemical synaptic transmission, learning | NLGN4X |
| ENSG00000183878 | UTY* | Y | Chromatin organization, regulation of gene expression | UTX |
| ENSG00000198692 | EIF1AY | Y | Translation, translational initiation | EIF1AX |
| ENSG00000231535 | LINC00278 | Y | ||
| ENSG00000241859 | ANOS2P | Y | A pseudogene, associated with Kallmann Syndrome | |
| ENSG00000260197 | Lnc-KDM5D-1 | Y | ||
| ENSG00000286009 | AC244213.1 | Y |
Table 3.
Genes with obligate dosage sensitivity (ODS) to total sex chromosome dosage changes in lymphoblastoid cell lines (LCLs)
| Ensembl ID | Symbol | Chr | Gene description (associated diseases) | PAR1 | Location |
|---|---|---|---|---|---|
| ENSG00000169084 | DHRSX | X | Cholesterol ester storage disease and partington X-linked mental retardation syndrome | Yes | Xp22.33 and Yp11.2 |
| ENSG00000169093 | ASMTL | X | Melanotic neurilemmoma and chronic tic disorder | Yes | Xp22.3 and Yp11.3 |
| ENSG00000169100 | SLC25A6 | X | Influenza and bubonic plague | Yes | Xp22.32 and Yp11.3 |
| ENSG00000178605 | GTPBP6 | X | Sengers syndrome and mongolian spot | Yes | Xp22.33 and Yp11.32 |
| ENSG00000182162 | P2RY8 | X | Childhood B cell acute lymphoblastic leukemia and B-lymphoblastic leukemia/lymphoma with Iamp21 | Yes | Xp22.33 and Yp11.3 |
| ENSG00000197976 | AKAP17A | X | Hodgkin's lymphoma, mixed cellularity and autism | Yes | Xp22.33 and Yp11.32 |
| ENSG00000214717 | ZBED1 | X | Chronic tic disorder and retinitis pigmentosa | Yes | Xp22.33 and Yp11 |
| ENSG00000228223 | HCG11 | 6 |
All of these 8 ODS genes except HCG11 are on PAR1.
To characterize non-ODS gene expression changes in each SCD group contrast, we applied biological process GO enrichment analyses to significant DEGs in each of the 15 contrasts (Dataset S1) after excluding ODS genes. Six of all 15 SCD group contrasts yielded statistically significant GO term enrichments for non-ODS genes (Dataset S7), with a notable recurrence across the contrasts of GO terms relating to oxidative stress.
Finally, to complement contrast-level DEG analyses, we also examined SCD effects in LCLs through unsupervised clustering of sex chromosome genes by their mean expression profiles across SCD groups (Fig. 3, Dataset S8, and SI Appendix, Text S1.5). Our previously published application of this clustering approach to microarray data in a smaller sample of LCLs (13) had supported a theoretically predicted “Four Class Model”, dividing sex chromosome genes into the following groups with contrasting SCD sensitivity profiles: 1) PAR genes tracking with tSCD count; 2) Y-linked genes tracking with YCD; and X-linked genes aligning with one of two idealized response profiles—3) XCI escape genes (XCIE) tracking with XCD, and 4) genes subject to XCI (XCIS) showing stable expression across any group with one or more X chromosomes. Reapplication of this same clustering method to the current expanded RNA-seq dataset in LCLs provided strong independent support for the Four Class Model, which was further refined by additional differentiation between Y-linked gene sets with variable magnitudes of SCD sensitivity (Fig. 3, Datasets S8 and S9, and SI Appendix, Text S1.5). We established through supplementary analyses that those genes within the Y-linked clusters showing little expression change with YCD (k2 and k4) tended to be Y-linked genes with low levels of expression outside the testes (SI Appendix, Text S1.5 and Fig. S5). Further replicating our prior results (13), we also found that the most SCD-sensitive clusters of sex-linked genes in our current analyses were enriched for gametolog genes [XCIE-cluster (k3, Fig. 3): odds ratio (OR)=57, P = 5.3e-10, Y-cluster (k7, Fig. 3): OR = 1,237, P < 2.2e-16, Dataset S8 and SI Appendix, Text S1.5]. GO enrichment analysis of all clusters identified statistically significant GO terms associated with maintenance of protein location and demethylation in the Y-linked clusters (k2 and k4, respectively), as well as translational initiation and demethylation in the XCIE cluster (k3) (Dataset S10).
Fig. 3.
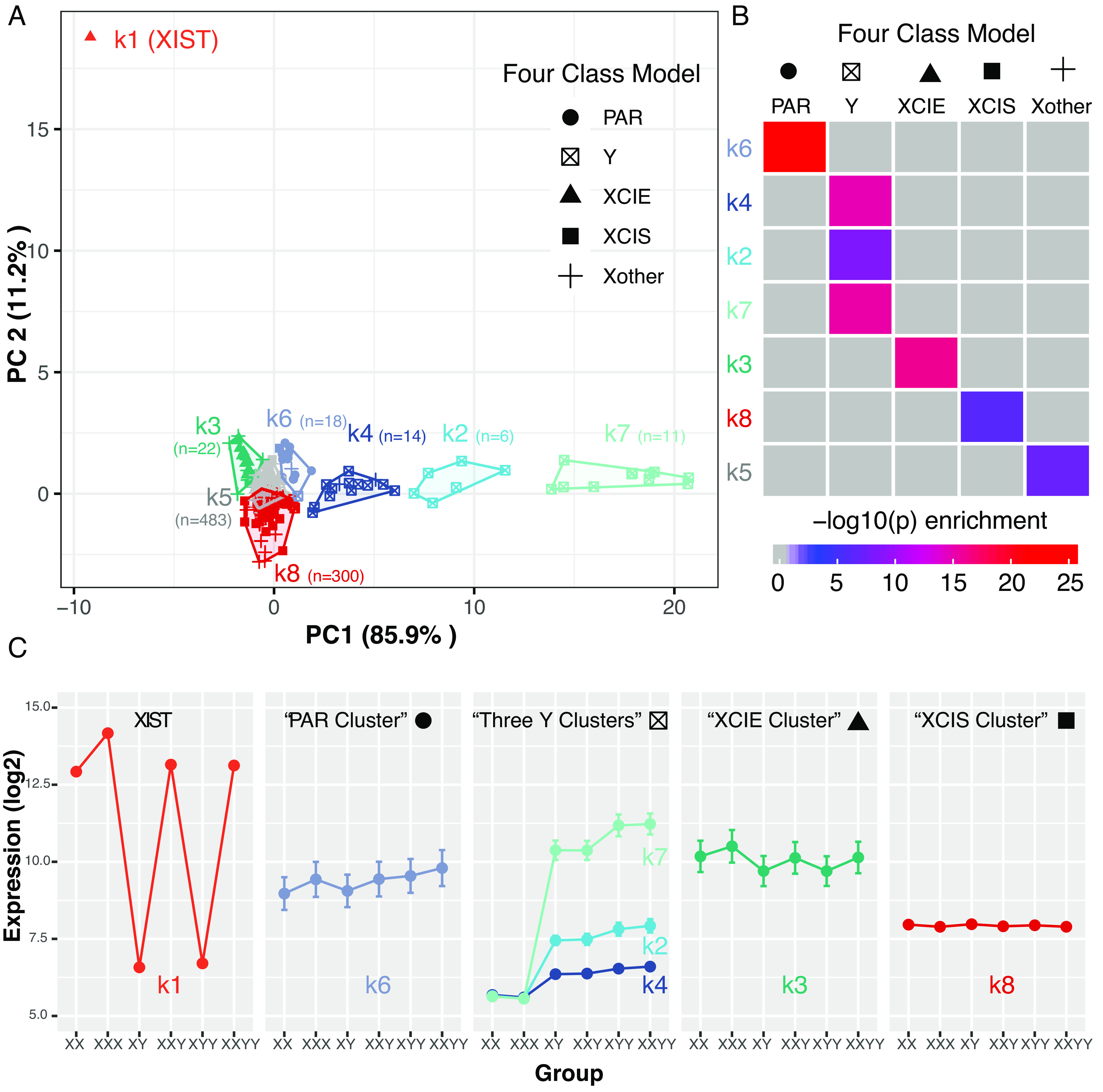
Clustering sex-chromosome genes by sex chromosome dosage (SCD) sensitivity in lymphoblastoid cell lines (LCLs). (A) A total of 855 sex chromosome genes (points) were k-means clustered based on their profiles of mean expression across 6 SCD groups (k = 8 selection by scree plot in SI Appendix, Fig. S4). Genes are plotted by their scores on the first 2 principal components (PCs) of the gene*group mean expression matrix. Cluster designation is encoded by the color of points and cluster boundaries. Point shape encodes assigned gene class according to the theoretical Four Class Model of SCD sensitivity [Pseudoautosomal Region (PAR); Y-linked; X-linked genes that escape XCI (XCIE), are subject to XCI (XCIS), and have no clear XCI status (Xother)]. (B) Heatmap of pairwise Fisher’s exact tests for overlaps between gene sets in k-means cluster and Four Class Model categories. All P-values shown in color survive Bonferroni correction. (C) Average expression of genes in each k-means cluster across SCD groups.
Thus, both ODS identification through DEG conjunction (Fig. 2) and cluster-based analyses (Fig. 3) find that gametologs are core to the proximal, cis-effect of SCD variation on gene expression—reinforcing the already strong candidacy of these genes as likely drivers of downstream SCD effects on biological structure and function (18, 19).
Generalizability of SCD Effects in LCLs to Other Cell Types.
To probe cross-cell type effects, we applied the same analytic pipeline used in LCLs to estimate SCD effects on gene expression in fibroblasts (FCLs) and iPSC-derived neuronal cells (iNs) (DEGs for these two cell types with log2FC values listed in Datasets S11 and S12). The detailed protocol for reprogramming of iPSCs from skin biopsy fibroblasts and differentiation of iPSCs to iNs is described in SI Appendix, Texts S2.1 and S2.2, as are the procedures for characterization of iPSCs and verification of excitatory neuronal status of iNs (SI Appendix, Text S2.3 and Fig. S6 and Dataset S13). Given the substantially smaller sample size of FCLs and iNs datasets relative to LCLs, we assessed cross-cell type stability of SCD effects on gene expression using three complementary approaches (Methods and Materials, SI Appendix, Text S3.1).
We first asked whether ODS gene sets defined in LCLs (Tables 1–3) occupied significantly extreme positions in log2FC ranked gene lists for each SCD contrast in FCLs and iNs. For almost all pairwise SCD group contrasts, the ODS genes identified in LCLs showed a statistically significantly extreme median rank in the log2FC ranked gene lists from FCLs and iNs. This statement held true for genes with ODS to each of the three different modes of SCD variation: XCD, YCD (Methods and Materials, Fig. 4 A–D, Upper) and tSCD (SI Appendix, Fig. S7, Upper). Thus, genes showing consistently altered expression with SCD variation in LCLs—which almost all reside on the sex-chromosomes and are highly enriched for gametologs—also showed elevated sensitivity to SCD in FCLs and iNs.
Fig. 4.
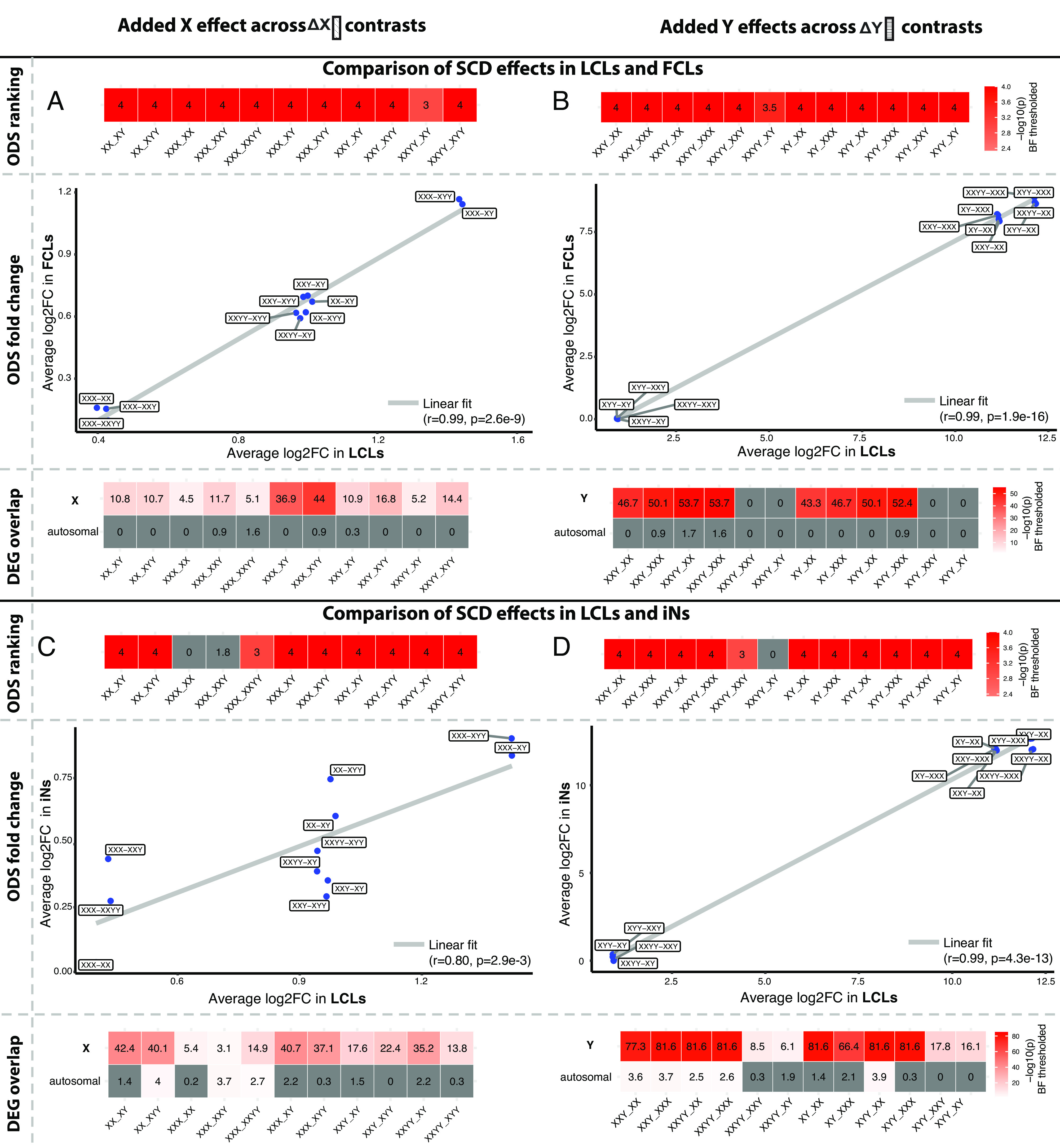
Generalizability of sex chromosome dosage (SCD) effects on gene expression in lymphoblastoid cell lines (LCLs) to fibroblasts (FCLs) and induced neurons (iNs). Results are shown separately for the LCL-FCL comparison (A and B) and the LCL-iN comparison (C and D), and separately for X chromosome dosage (XCD, A and C) and Y chromosome dosage (YCD, B and D) effects. In each panel: The upper 1-row heatmap shows contrast-specific P-values for a rank-based permutation test that asks whether ODS genes for that contrast in LCLs show extreme log2FC values in the non-LCL tissue type; the middle scatterplot correlates the mean log2FC of ODS genes for each contrast in LCLs vs. the mean log2FC of these genes from the equivalent contrast in the non-LCL tissue type; and, the lower heatmap shows the result of Fisher’s exact tests comparing differentially expressed genes (DEGs) for each SCD contrast in LCLs to those in the non-LCL tissue type—stratified by whether or not the DEGs reside on a sex-chromosome. All P-values shown in color survive Bonferroni correction. Of note, the consistency of cis effects between LCLs and iNs shown in XCD contrasts of panel C was also recovered in a sensitivity analysis that excluded 2 iN samples with potential markers of XCI erosion (SI Appendix, Text S3.2 and Fig. S9).
Second, we asked whether the average magnitude of expression changes for ODS genes in each LCL SCD group contrast was correlated with the magnitude of ODS gene expression changes in corresponding SCD group contrasts for FCLs and iNs. We found robust evidence in support of such cross-cell type reproducibility for XCD and YCD effects (cross-contrast r values for mean log2FC all >0.8, Methods and Materials, Fig. 4 A–D, Middle) as well as tSCD effects (SI Appendix, Fig. S7, Middle). For XCD effects, this high concordance of expression changes across SCD contrasts captures the fact that the SCD contrasts in our study can be grouped into subsets with differing integer changes in XCD (i.e., addition of 0, 1, or 2 X chromosomes, Fig. 4 A–C). For YCD effects, this high concordance captures the extremely large fold changes in Y chromosome OSD genes between groups with one or more Y chromosomes vs. groups with no Y chromosomes (Fig. 4 B–D). Nevertheless, we observe high intertissue concordance in the magnitude of expression changes for ODS genes across the SCD group contrasts included in our study.
Third, we identified DEGs for each individual SCD group contrast in FCLs (Dataset S11) and iNs (Dataset S12), and—for each contrast in each cell type—we tested if the observed DEG set showed statistically significant overlap with DEGs (Dataset S1) for the corresponding contrast in LCLs (Methods and Materials, SI Appendix, Text S3.1, Fig. 4 A–D, Lower). We ran this test separately for sex-linked and autosomal DEGs to differentiate between cis vs. trans effects of SCD on gene expression. These analyses revealed that cis effects of SCD on gene expression in LCLs almost always showed statistically significant overlap with cis effects in FCLs and iNs, whereas trans effects did not. This statement held true for XCD and YCD effects (lower panels Fig. 4 A–D) as well as tSCD effects (SI Appendix, Fig. S7, Lower). Notably however, some trans effects of SCD variation in LCLs did appear to generalize to iNs, although the specific SCD group contrasts that showed this differed for XCD, YCD, and tSCD effects (Fig. 4 C and D, Lower and SI Appendix, Fig. S7B). We provide complete lists of overlapping DEGs between each pair of cell types per SCD group contrast in Datasets S14 (LCLs and FCLs) and S15 (LCLs and iNs), across all three cell types in Dataset S16.
We conducted two sets of sensitivity analyses to verify robustness of the above observations across several key methodological considerations (Methods and Materials, SI Appendix, Text S3.2). First, given the exceptionally large log2FC changes in XIST expression with variations in XCD, we verified that both rank- and correlation-based tests (Fig. 4 A–C, Top and Middle, respectively) for generalizability of SCD cis effects in LCLs to FCLs and iNs remained high—albeit lowered (r = 0.55)—after exclusion of XIST from analyses (SI Appendix, Fig. S8). Thus, there is concordance in mean log2FC expression change for ODS genes across tissue types even after exclusion of the very large log2FC values for XIST, which otherwise amplify the level of concordance. Second, given past reports of XCI erosion in iPSC models and derived cell types (25–27), we characterized XIST expression and XCI status of iPSC-derived iN lines, and verified that the observed generalizability of cis effects of SCD on gene expression from LCLs to iNs (Fig. 4 C and D) was retained after a strict filtering of iN lines on potential markers for XCI erosion (SI Appendix, Text S3.2 and Fig. S9).
Finally, to give a full view of gene expression changes in each SCD group contrast for FCLs and iNs—above and beyond the question of their convergence with changes in LCLs—we generated ratio (FCLs: SI Appendix, Fig. S10, iNs: SI Appendix, Fig. S11) and volcano plots (FCLs: SI Appendix, Fig. S12, iNs: SI Appendix, Fig. S13) for these two cell types as previously introduced for the LCL analyses above. As for LCLs, ratio plots for almost all SCD group contrasts in FCLs and iNs significantly deviated from normality SI Appendix, Figs. S10 and S11)—which has been taken as evidence for a modulated distribution of gene expression values (6, 24). Of note, although the specific trans DEGs differed between LCLs, FCLs and iNs—comparison of ratio plots for these tissues (SI Appendix, Figs. S2, S10, and S11) showed that the same main modes of trans dosage effect were manifested in all three tissues —with some trans genes showing direct dosage effects (positive modulation), some inverse dosage effects (negative modulation) and some, no effect.
Collectively, the above results across 3 cell types reveal that cis effects of SCD on gene expression in human LCLs are also preserved in human FCLs and iNs. Thus, genes showing ODS to XCD, YCD, and tSCD are strong candidate drivers of SCD effects across multiple cell types. In contrast, SCD effects on autosomal gene expression (i.e., trans effects) appear to be much more variable across cell types—although there is some evidence that for certain SCD group contrasts, trans effects in LCLs are conserved in iNs.
Generalizability of Sex Chromosome Aneuploidy Findings to Trisomy 21.
The above findings regarding cross-cell type generalizability of cis and trans effects of aneuploidy on gene expression (Fig. 4) could theoretically be unique to the X and Y chromosomes rather than a general property of aneuploidy syndromes. To assess this possibility, we harnessed a large-scale cross-tissue dataset compiled for meta-analysis of gene expression alterations in trisomy 21 (10). This published dataset from Toma et al. (10) includes measures of gene expression fold change and lists of DEGs from a total of 67 studies representing human and mouse tissues with trisomy 21.
We first establish that Toma et al.’s (10) meta-analytically derived list of the top 500 genes with recurrent dosage sensitivity to trisomy 21 (defined as being DEGs in at least four independent studies) shows strong and specific enrichment for chr21 genes (P = 3.0e-77, Fig. 5A). This observation suggests that cis effects of trisomy 21 are more likely to recur across different tissues than trans-effects. To achieve a more direct test of this hypothesis akin to that achieved in sex chromosome aneuploidy, we filtered the meta-analytic dataset to retain 9 studies that included human cell lines (LCLs, FCLs or iNs) with trisomy 21 for comparison with control groups (Dataset S17). Concatenating DEGs across studies within each cell type enabled an analysis in trisomy 21 (Fig. 5B) analogous to that applied to sex chromosome aneuploidy (Fig. 4). Specifically, we stratified these DEGs into those residing on chr21 (cis) vs. those on other chromosomes (trans), and—for each class of genes—we computed the degree of DEG overlap between each pair of cell types (Fig. 5B). These analyses established that cis DEGs in trisomy 21 show strong and statistically significant overlaps between all cell types (odds ratio, OR > 379, (−log10(p)>15), whereas trans DEGs only showed a significant overlap between LCLs and FCLs which was much weaker than the LCL-FCL overlap for cis genes (OR = 3.5, (−log10(p) = 4.5). Thus—as observed for aneuploidies of the X- and Y chromosomes in humans (Fig. 4)—altered gene expression in aneuploidy of chr21 also shows more cross-cell type stability for cis than trans effects.
Fig. 5.

Cross-cell type generalizability of trisomy 21 effects on gene expression. (A) Bar graph shows the statistical significance, −log10(p), of enrichment tests in each of 22 autosomes and the X chromosome for a preidentified set of 500 genes that show consistently DE in comparisons of euploid vs. trisomy 21 samples. Only human chromosome 21 (chr21) genes survive Bonferroni correction. (B) Heat map shows the result of Fisher’s exact tests of overlaps of chr21(Upper row) and non-chr21 (Lower) DEGs between a pair of tissues. All −log10(p) values shown in color survive Bonferroni correction (Materials and Methods).
Discussion
Our study advances understanding of aneuploidy effects on human gene expression in several key directions, with a focus on aneuploidies of the X and Y chromosomes, and extension of findings to aneuploidy of chr21.
First, we use the largest single-tissue RNA-seq dataset of human gene expression across multiple SCD variations to date—197 LCLs spanning 6 different SCD groups—to substantially expand the list of genes showing ODS to changes in XCD, YCD, or tSCD. The criterion of ODS provides a strict definition of dosage sensitivity that complements alternative rankings of sex-linked genes by dosage sensitivity (9), and both analytic approaches find an enrichment of dosage sensitivity among X-Y gametolog genes pairs. Moreover, our replication and refinement of the canonical Four Class Model for SCD sensitivity further spotlights X-Y gametologs as showing highly dynamic expression changes in sex chromosome aneuploidy. X-Y gametologs are responsible for a broad range of fundamental functions such as histone demethylation, translational initiation, and regulation of translation, that are critical for regulation of gene expression (28). These observations—together with the striking evolutionary retention of gametologs amidst the otherwise wholesale loss of ancestral protosex chromosome genes from the Y chromosome (18, 19)—strongly prioritize gametologs as genes that are core to the proximal cis-effects of SCD and potential drivers of downstream SCD effects in both euploid and aneuploid contexts. Furthermore, given the high-sequence homology of X-Y gametologs, their shared ODS status also provides a potential basis for the well-established, but poorly understood capacity of X and Y chromosome aneuploidies to exert highly convergent profiles of phenotypic change in behavior (14) and brain anatomy (23, 29).
Second, we use the above findings in LCLs as a solid foundation from which to assess the cross-cell type stability of SCD effects on gene expression. This question provides a window into the fundamental biology of tissue-specific genome regulation (8, 30), and is also important for mechanistic understanding of tissue-specific clinical effects in sex chromosome aneuploidy. Indeed, while all types of sex chromosome aneuploidy have the capacity to impact neurodevelopment (14, 23, 29), they vary greatly in their impact upon nonbrain tissues including the reproductive, immune, and metabolic systems (14, 15). Understanding these tissue-specific clinical effects hinges on understanding tissue- and cell type-specific effects of SCD on gene expression. Our study builds on emerging evidence that cis effects of X chromosome dosage are highly reproducible between human LCLs and FCLs (9) by probing cross-cell type cis and trans effects of both X and Y chromosome dosage on LCLs, FCLs, and iNs. Inclusion of iNs is particularly relevant given that there are robust effects of sex chromosome aneuploidies on brain development (14, 23, 29), but a paucity of postmortem brain samples with which to probe the potential transcriptional basis for these effects.
We find that cis effects of SCD on gene expression (which are most consistent and large for gametologs) are reproducible between LCLs, FCL, and iNs, whereas trans effects of SCD variation – which have been well-described in humans (9, 13, 31) and diverse model organisms (32, 33) – show limited cross-cell type generalizability. Our parallel analyses of trisomy 21 indicate that this observation is not specific to the X and Y chromosomes. The resulting implication—that accessible peripheral tissues in aneuploidy patients may provide a reasonably good model for cis effects of interest in harder-to-access tissues like brain—lowers barriers for i) mechanistic research on the cis-transcriptional effects of aneuploidy and ii) testing if peripheral measures of cis effects severity could provide a potentially useful prognostic marker of interindividual variation in brain-related outcomes of aneuploidy. In further support of this latter idea, we recently showed that interindividual variation in expression of dosage-sensitive sex-linked genes among LCLs from individuals with sex chromosome aneuploidy could predict cooccurring variation in the severity of their neuroanatomical changes (34). Taken together these results encourage greater use of peripheral measures of cis effects in gene dosage disorders as a model and marker for less accessible tissues.
Conversely, we find that trans effects of aneuploidy on gene expression–which are substantial in nature and can impact a far larger number of genes than cis effects (Fig. 2)–show limited generalizability across LCLs, FCLs, and iNs. Thus, the clinical observation that tissues vary in their sensitivity to aneuploidies of chromosomes X, Y, and 21 may be more driven by tissue-specific trans as opposed to tissue-specific cis effects on gene expression. A major priority for future research will be to better-understand the mechanistic basis for tissue-specific trans effects, which could potentially operate across multiple interwoven aspects of genome regulation spanning 3-dimensional chromosome conformation (35), chromatin accessibility (36), the wiring of transcription factor networks (37), DNA methylation (38), and posttranscriptional processing (39). Characterizing the interactions between aneuploidy and these diverse aspects of genome organization in a tissue-specific manner will be a crucial step toward identifying pathways through which chromosome dosage may influence phenotypic variation in euploidy (i.e., between XY males and XX females) and aneuploidy. Future cross-cell type and cross-tissue comparisons will not only need to consider how cis effects on gene expression can influence the wider genome, but to also examine potentially nongenic effects of chromosome dosage on genome function including altered dosage of noncoding regulatory regions (40) and nuclear heterochromatin content (41).
Our findings must be considered in light of several caveats and limitations. First, a focus on gene expression misses the additional translation and posttranslational processes that must necessarily convey aneuploidy effects to protein biology. Second, with respect to sex chromosomes, the current study considers euploid and supernumerary SCD variations, and therefore does not shed light on the cross-tissue nature of sex chromosome haploinsufficiencies such as X-monosomy (the dominant form of Turner syndrome). Third, our cross-tissue analyses are based on only three cell types given the rarity of tissue samples from aneuploidy syndromes. In the future however, improved access to diversity of tissue types and karyotypes in postmortem human tissue would enable more systematic and comprehensive analysis of cross-tissue aneuploidy effects. Another path to this goal would be the creation of iPSC-derived tissue banks in aneuploidy. Such resources remain to be developed, but our current study takes a first step in this direction by generating and characterizing a small set of iNs from sex chromosome aneuploidy patients. Although our iN findings are limited by a small sample size, and variation in some aspects of XCI across lines, we find that these in vitro models for human forebrain excitatory neurons do indeed show SCD cis effects that converge with those in our large LCL dataset. Future work using in vitro models would benefit from retaining the diversity of SCD karyotypes studies here, while increasing the number of available biological replicates per karyotype. It will also be important to develop benchmarked assays for XCI status and fidelity of differentiation in iPSC-derived models as both these factors may influence observed SCD effects.
Notwithstanding the caveats above, our study harnesses RNA-seq data from cell lines representing different aneuploidies and tissue types to refine and extend our understanding of aneuploidy effects on human genome function. By using the largest single-tissue (LCL) resource to date, we i) substantially expand the inventory of genes showing ODS to sex chromosome dosage variation in both euploid and aneuploid contexts, and ii) produce a classification of all sex-linked genes by their sensitivity to SCD. Both of these results indicate that altered expression of gametologs is core to the cis-effects of SCD variation. We then expand analysis of SCD effects to other cell types and conduct parallel analyses in trisomy 21 to show that aneuploidy cis effects on gene expression generalize across tissue types, whereas trans effects do not. Collectively, these results shed light on the broader issue of tissue-specific aneuploidy effects on gene expression, which has so far undergone limited direct research in humans despite being crucial for our understanding of disease processes.
Materials and Methods
Acquisition and Preparation of SCD Variation Biosamples.
Euploid and aneuploid participants with varying SCD were recruited through research protocol at the NIMH Intramural Research Program and the University of Oxford. RNA was extracted using standard methods from LCLs (197 individuals), FCLs (32 individuals), and iN (24 samples; 2 vials for each of 12 individuals) (Fig. 1A). All participants with X/Y aneuploidy were nonmosaic based on visualization of at least 50 metaphase spreads in peripheral blood. Stability of karyotype across LCL derivation was confirmed by chromosome FISH in all members of a randomly selected subset of nine LCL samples representing each of the four supernumerary SCA groups.
The SCA LCL, FCL, and iN samples were fully independent from each other (excepting five participants who provided samples for both FCL and iN cell lines). All tissue types included individuals from the same set of 6 SCDs: XX, XXX, XY, XXY, XYY, and XXYY (Fig. 1A). The human iPSC lines used for this study (Dataset S13) were derived from participant skin biopsy fibroblasts using CytoTune® (Life Technologies), and underwent quality control including confirmation of genomic stability through SNP array karyotyping and FACS based pluripotency marker expression (42) (SI Appendix, Text S2.1). iPSC lines were differentiated to cortical neurons using the rapid single-step induction protocol published by Zhang et al. (43) (SI Appendix, Text S2.2). We verified the enrichment of neuronal marker gene sets in each iN sample using gene set enrichment analysis (GSEA) provided by Clusterprofiler (44) (SI Appendix, Text S2.3 and Fig. S6). The research protocol was approved by the institutional review board at the NIMH and South Central-Oxford A Research Ethics Committee (IRAS no 104383). Informed consent or assent was obtained from all children who participated in the study, as well as consent from their parents if the child was under the legal age of majority.
RNA-Sequencing and Differential Expression Analysis in SCD Variation Samples.
LCL and FCL samples were pooled using the Illumina TruSeq Stranded Total RNA Kit RS-122-2201 and randomly distributed across libraries. The pooled libraries were paired end sequenced with read length of 150 bp on the Illumina HiSeq 4000. For each iN sample, an RNA sequencing library was made with the NEB Next Directional RNA kit and paired end sequenced with read length of 75 bp over six lanes on the Illumina HiSeq 4000 to increase the power to detect low-expressed genes.
RNA-seq data from these three cell types were separately submitted to the following workflow (SI Appendix, Text S1.1): i) FastQC (45), MultQC (46), and Trimmomatic (47) for QC and trimming, ii) Salmon (48) for transcript quantification with the Y chromosome masked reference transcriptome when mapping female samples and with the YPAR-gene masked one when mapping male samples to reduce misaligning between the X and Y chromosomes (49), iii) DESeq2 (50) to compare gene expression values across samples with different SCD while adjusting for measured covariates of batch and age, and surrogate variables (SVs) determined by sva package (51). SVs were estimated to exclude gene expression variation linearly attributable to SCD or to the measured covariates. We excluded genes with low expression (<10 read counts in at least three samples) and those that did not converge in the differential expression analysis, resulting in a total of 25,075, 18,761, 25,905 in LCLs, FCLs and iNs. The threshold of false-discovery rate (FDR) smaller than 5% was used to determine statistical significance. Variance stabilizing transformations was applied to counts, producing gene expression on the log2 scale, which has been normalized with respect to library size for downstream clustering analyses. DESeq2 and the rest of analyses were conducted in R version 4.1.0.
Detailing SCD Effects on Gene Expression in LCLs.
We identified significantly (FDR < 0.05) DEGs in each of 15 pairwise independent SCD group contrasts separately in the three cell types (Datasets S1, S11, and S12). Each of these 15 contrasts was labeled based on whether there was a disparity in X-, Y-, and total sex-chromosome dosage between two SCD groups that were compared in the contrast (Fig. 1B). In the core LCL dataset, we identified 1,476 genes (Dataset S1 and Fig. 2) that showed significantly differential expression in at least one of all 15 pairwise contrasts. Sensitivity analyses in a reduced and fully balanced (i.e., sample-size matched) LCL dataset (n = 23 for each of the 6 SCD groups following random removal of samples) demonstrated that the distribution of numbers of significant DEGs across 15 contrasts was not driven by sample size variation across SCD groups (SI Appendix, Text S1.2 and Fig. S1 and Datasets S2, S3, and S6). In the complete LCL dataset, we screened for genes with ODS to each of the three modes of SCD variation—defined as always showing significant differential expression across all SCD group contrasts featuring differences in X chromosome dosage (XCD, n = 11 contrasts), Y chromosome dosage (YCD, n = 12 contrasts) and total sex chromosome dosage (tSCD, n = 11 contrasts) (Fig. 2 and Tables 1–3). Fisher's Exact Tests were used to assess enrichment of gametologs in genes with ODS to changes in XCD and YCD (Tables 1 and 2). GO enrichment analysis was applied in these ODS genes using enrichGO function of clusterprofileR (52) with the default background of all genes in the database (Datasets S4 and S5). Identical GO enrichment analyses were also run for the non-ODS DEGs (Dataset S1) in each SCD group contrast (Dataset S7). Log2 fold changes (log2FC) were adjusted using lfcShrink function (53) and used for downstream analyses. Clustering of sex chromosome genes by the profile of their mean expression in each SCD group was conducted as previously outlined, and as detailed further in SI Appendix, Text S1.5.
Comparing SCD Effects in LCLs to Those in FCLs and iNs.
Using three methods, we examined whether SCD effects on gene expression in LCLs are generalizable to other human tissues, FCLs and iNs (SI Appendix, Text S3.1). First, using rank-based permutation, we tested whether the genes with ODS to changes in XCD, YCD, tSCD, identified in LCLs (Fig. 2 and Tables 1–3), had significantly higher log2FCs than chance in the corresponding XCD, YCD, and tSCD contrasts (Fig. 1B) in FCLs and iNs (Fig. 4 A–D, Upper and SI Appendix, Fig. S7 A and B). This test asks whether genes with ODS in LCLs tend to show high log2FC with the corresponding SCD variation in FCLs and iN. Second, to compare the magnitude of SCD effects on expression between tissues, we calculated the average log2FC for each ODS gene set in each SCD group contrast in LCLs, and we correlated these values across contrasts with their corresponding values in FCLs and iNs (Fig. 4 A–D, Middle and SI Appendix, Fig. S7 A and B). This test assesses whether differences across SCD contrasts in the magnitude of expression change they can induce in LCLs are preserved in FCLs and iNs. We ran sensitivity analyses to recompute these correlations after excluding XIST from analyses given the extreme expression changes in this gene with XCD variation (SI Appendix, Text S3.2 and Fig. S8). Last, we divided the significant DEGs for each SCD contrast in LCLs into X-, Y-linked, PAR and autosomal groups and separately tested whether there was a significant overlap in these DEGs with those from the same contrasts in FCLs and iNs (using Fisher’s Test, Fig. 4 A–D, Lower and SI Appendix, Fig. S7 A and B). This test directly assesses DEG overlaps between tissues for each SCD contrast and does so separately for genes that are in cis vs. trans with the SCD variation.
Given prior reports of XCI erosion in human iPSCs and derived cell types (26, 27), we ran several supplementary analyses to characterize XCI in iN samples within our dataset (SI Appendix, Text S3.2 and Fig. S9 A and B), and we verified the robustness of our main findings regarding generalizability of SCD-induced DEGs between LCLs and iNs to filtering of iN samples by XCI status (SI Appendix, Text S3.2 and Fig. S9C).
Comparing Cross-Tissue Effects of Trisomy 21 on Gene Expression.
We harnessed a meta-analysis of 67 transcriptional studies in trisomy 21 (10) and using two methods, tested whether cross-cell type effects of trisomy 21 on gene expression showed the same dissociable patterns between cis and trans effects as seen for aneuploidies of the X and Y chromosome. First, we downloaded a set of 500 most consistently DE genes that were identified by Toma et al., Table S1 (10), and tested if this gene set was enriched in genes on any of 22 autosomes and X chromosome (no Y-linked genes in this set, Fig. 5A). In this analysis, specific enrichment of DEGs on chr21 would be suggestive of conserved cis effects of trisomy 21 on gene expression across the diverse tissues represented in the DS metanalysis. Second, to match analytical methods in trisomy 21 to those applied to study SCD effects, we filtered the 67 studies in Toma et al. to retain only those including human LCL (n = 3), FCL (n=4), iN (n=2) (Dataset S17), and we downloaded the list of DEGs in each of these selected studies (DEGs defined by Toma et al. as those with adjusted P < 0.05 and absolute fold change >1.5, https://github.com/Ilarius/DS_meta_analysis). We concatenated DEG lists across studies within each cell type to arrive at a single DEG list per cell type capturing significant changes in gene expression induced by trisomy 21. Fisher’s Tests were used to test for significant DEG overlaps between tissues—stratifying for cis (chr21) vs. trans (non-chr21) effects (Fig. 5B) as was done for analyses of SCD variation effects (Fig. 4).
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Dataset S14 (XLSX)
Dataset S15 (XLSX)
Dataset S16 (XLSX)
Dataset S17 (XLSX)
Acknowledgments
This study was supported by the intramural research program of the National Institute of Mental Health (NIMH) (NIH Annual Report Number: ZIAMH002949; Protocol: 89-M-0006; ClinicalTrials.gov Number: NCT00001246) and in part by NIH grants GM131745 and AG073918 (C.M.D.). We thank the patients and their families for participating in this study, as well as the Association for X and Y Chromosome Variations (https://genetic.org) for their assistance with recruitment.
Author contributions
F.J.M., C.M.D., and A.R. designed research; S.L., P.K.R., J.R., E.T., L.S.C., J.B., F.L., F.S., M.Z.C., and A.R. performed research; S.L. and N.A. analyzed data; and S.L., F.J.M., F.S., C.M.D., M.Z.C., and A.R. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
RNA-seq data have been deposited in the Database of Genotypes and Phenotypes (dbGaP) with accession number phs003278.v1.p1 (54).
Supporting Information
References
- 1.Siegel J. J., Amon A., New insights into the troubles of aneuploidy. Annu. Rev. Cell Dev. Biol. 28, 189–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonarakis S. E., et al. , Down syndrome. Nat. Rev. Dis. Primers 6, 1–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bojesen A., Gravholt C. H., Klinefelter syndrome in clinical practice. Nat. Rev. Urol. 4, 192–204 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Veitia R. A., Potier M. C., Gene dosage imbalances: Action, reaction, and models. Trends Biochem. Sci. 40, 309–317 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Kojima S., Cimini D., Aneuploidy and gene expression: Is there dosage compensation? Epigenomics 11, 1827–1837 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hou J., et al. , Global impacts of chromosomal imbalance on gene expression in Arabidopsis and other taxa. Proc. Natl. Acad. Sci. U.S.A. 115, E11321–E11330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierson E., et al. , Sharing and specificity of co-expression networks across 35 human tissues. PLoS Comput. Biol. 11, e1004220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.GTEx Consortium, The GTEx consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.San Roman A. K., et al. , The human inactive X chromosome modulates expression of the active X chromosome. Cell Genom. 3, 100259 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toma I. D., Sierra C., Dierssen M., Meta-analysis of transcriptomic data reveals clusters of consistently deregulated gene and disease ontologies in down syndrome. PLoS Comput. Biol. 17, e1009317 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes-McCormack L. A., et al. , Birth incidence, deaths and hospitalisations of children and young people with Down syndrome, 1990–2015: Birth cohort study. BMJ Open 10, e033770 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen J., Wohlert M., Chromosome abnormalities found among 34,910 newborn children: Results from a 13-year incidence study in Arhus, Denmark. Hum. Genet. 87, 81–83 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Raznahan A., et al. , Sex-chromosome dosage effects on gene expression in humans. Proc. Natl. Acad. Sci. U.S.A. 115, 7398–7403 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong D. S., Reiss A. L., Cognitive and neurological aspects of sex chromosome aneuploidies. Lancet Neurol. 13, 306–318 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Seminog O., Seminog A., Yeates D., Goldacre M., Associations between Klinefelter’s syndrome and autoimmune diseases: English national record linkage studies. Autoimmunity 48, 125–128 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Balaton B. P., Cotton A. M., Brown C. J., Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex Differ. 6, 35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tukiainen T., et al. , Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skaletsky H., et al. , The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Godfrey A. K., et al. , Quantitative analysis of Y-Chromosome gene expression across 36 human tissues. Genome Res. 30, 860–873 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo M., Birchler J. A., Trans-acting dosage effects on the expression of model gene systems in maize aneuploids. Science 266, 1999–2002 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Cooper J. L., Birchler J. A., Developmental impact on trans-acting dosage effects in maize aneuploids. Genesis 31, 64–71 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Makarevitch I., Harris C., Aneuploidy causes tissue-specific qualitative changes in global gene expression patterns in Maize. Plant Physiol. 152, 927–938 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raznahan A., et al. , Globally divergent but locally convergent X- and Y-chromosome influences on cortical development. Cereb. Cortex 26, 70–79 (2016), 10.1093/cercor/bhu174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi X., et al. , “The gene balance hypothesis: Epigenetics and dosage effects in plants” in Plant Epigenetics and Epigenomics: Methods and Protocols, Spillane C., McKeown P., Eds. (Methods in Molecular Biology, Springer, US, 2020), pp. 161–171. [DOI] [PubMed] [Google Scholar]
- 25.Brenes A. J., et al. , Erosion of human X chromosome inactivation causes major remodeling of the iPSC proteome. Cell Rep. 35, 109032 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mekhoubad S., et al. , Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 10, 595–609 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dandulakis M. G., Meganathan K., Kroll K. L., Bonni A., Constantino J. N., Complexities of X chromosome inactivation status in female human induced pluripotent stem cells—a brief review and scientific update for autism research. J. Neurodev. Disord. 8, 22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bellott D. W., et al. , Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508, 494–499 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong D. S., et al. , Influence of the X-chromosome on neuroanatomy: Evidence from Turner and Klinefelter syndromes. J. Neurosci. 34, 3509–16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguet F., et al. , Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X., et al. , Integrated functional genomic analyses of Klinefelter and Turner syndromes reveal global network effects of altered X chromosome dosage. Proc. Natl. Acad. Sci. U.S.A. 117, 4864–4873 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birchler J. A., Hiebert J. C., Krietzman M., Gene expression in adult metafemales of drosophila melanogaster. Genetics 122, 869–879 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun L., et al. , Dosage compensation and inverse effects in triple X metafemales of Drosophila. Proc. Natl. Acad. Sci. U.S.A. 110, 7383–7388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidlitz J., et al. , Transcriptomic and cellular decoding of regional brain vulnerability to neurogenetic disorders. Nat. Commun. 11, 3358 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schoenfelder S., Fraser P., Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet. 20, 437–455 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Tsompana M., Buck M. J., Chromatin accessibility: A window into the genome. Epigenetics Chromatin 7, 33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert S. A., et al. , The human transcription factors. Cell 172, 650–665 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Lea A. J., et al. , Genome-wide quantification of the effects of DNA methylation on human gene regulation. Elife 7, e37513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen M., Manley J. L., Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 10, 741–754 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tosh J., Tybulewicz V., Fisher E. M. C., Mouse models of aneuploidy to understand chromosome disorders. Mamm. Genome 33, 157–168 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Francisco F. O., Lemos B., How do y-chromosomes modulate genome-wide epigenetic States: Genome folding, chromatin sinks, and gene expression. J. Genomics 2, 94–103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volpato V., et al. , Reproducibility of molecular phenotypes after long-term differentiation to human iPSC-derived neurons: A multi-site omics study. Stem Cell Rep. 11, 897–911 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y., et al. , Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu G., Wang L.-G., Han Y., He Q.-Y., clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andrews S., FASTQC. A quality control tool for high throughput sequence data (2010). Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 46.Ewels P., Magnusson M., Lundin S., Käller M., MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C., Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olney K. C., Brotman S. M., Andrews J. P., Valverde-Vesling V. A., Wilson M. A., Reference genome and transcriptome informed by the sex chromosome complement of the sample increase ability to detect sex differences in gene expression from RNA-seq data. Biol. Sex Differ. 11, 42 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leek J. T., Johnson W. E., Parker H. S., Jaffe A. E., Storey J. D., The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu T., et al. , clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stephens M., False discovery rates: A new deal. Biostatistics 18, 275–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raznahan A., Brain imaging of childhood onset psychiatric disorders, endocrine disorders and healthy volunteers. NCBI dGaP. https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003278. Deposited 4 May 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Dataset S14 (XLSX)
Dataset S15 (XLSX)
Dataset S16 (XLSX)
Dataset S17 (XLSX)
Data Availability Statement
RNA-seq data have been deposited in the Database of Genotypes and Phenotypes (dbGaP) with accession number phs003278.v1.p1 (54).