

Significance
Hfq is a key posttranscriptional regulator that modulates base-pairing interactions between sRNAs and their target mRNAs. Although there are more than 100 different sRNAs in Pseudomonas aeruginosa, information on their regulatory targets was available for only a few. Here, we identify the targets of 89 Hfq-associated sRNAs in this important opportunistic pathogen. Furthermore, we show that a single sRNA called PhrS dominates the RNA–RNA interaction landscape in this organism by pairing with approximately 800 targets. Our study therefore provides insight into the potential regulatory roles for many sRNAs in P. aeruginosa and identifies an sRNA with an acutely outsized influence on the RNA–RNA interactions occurring on Hfq in this bacterium.
Keywords: small RNA, Hfq, PhrS, posttranscriptional regulator
Abstract
The RNA chaperone Hfq plays important regulatory roles in many bacteria by facilitating the base pairing between small RNAs (sRNAs) and their cognate mRNA targets. In the gram-negative opportunistic pathogen Pseudomonas aeruginosa, over a hundred putative sRNAs have been identified but for most, their regulatory targets remained unknown. Using RIL-seq with Hfq in P. aeruginosa, we identified the mRNA targets for dozens of previously known and unknown sRNAs. Strikingly, hundreds of the RNA–RNA interactions we discovered involved PhrS. This sRNA was thought to mediate its effects by pairing with a single target mRNA and regulating the abundance of the transcription regulator MvfR required for the synthesis of the quorum sensing signal PQS. We present evidence that PhrS controls many transcripts by pairing with them directly and employs a two-tiered mechanism for governing PQS synthesis that involves control of an additional transcription regulator called AntR. Our findings in P. aeruginosa expand the repertoire of targets for previously known sRNAs, reveal potential regulatory targets for previously unknown sRNAs, and suggest that PhrS may be a keystone sRNA with the ability to pair with an unusually large number of transcripts in this organism.
Pseudomonas aeruginosa is an important opportunistic pathogen whose ability to survive and adapt to a variety of environments is thought to depend on the activity of regulatory proteins that tailor the organism’s gene expression profile to fit cellular demands. This regulation occurs at both the transcriptional and posttranscriptional levels. Prominent among the global posttranscriptional regulators in P. aeruginosa is Hfq. This RNA-binding protein has been well studied in a variety of bacteria where it has been shown to play critical roles in stabilizing small RNAs (sRNAs) and modulating their ability to pair with target transcripts (reviewed in refs. 1 and 2). Hfq-mediated pairing of an sRNA with a target mRNA can in turn alter the translation or stability (or both) of the target mRNA species and often occurs in close proximity to the Shine–Dalgarno sequence of the mRNA (reviewed in refs. 1–4). In P. aeruginosa, Hfq is important for growth and for virulence (5, 6) and can work in concert either with sRNAs or with the unusual regulatory protein Crc (7–10). Crc is thought to stabilize the binding of Hfq to target transcripts that carry the so-called ARN motifs that Hfq binds directly to either repress or activate translation (10).
In P. aeruginosa, there are approximately 150 different sRNAs, although some estimates place this number as high as 500 (11–14). However, information on the regulatory targets of these sRNAs is available for only a handful (reviewed in ref. 15). One of the first sRNAs shown to work through pairing with an mRNA in P. aeruginosa is PhrS (16). This sRNA was found to positively control the expression of the gene encoding the transcription regulator MvfR (also known as PqsR) by pairing with the mRNA specifying a small open reading frame (ORF) positioned immediately upstream of the mvfR ORF (16). PhrS was found to stimulate the translation of this small ORF which in turn stimulated the translation or stability of mvfR (16). Because MvfR positively regulates the transcription of the pqs genes involved in the biosynthesis of the quorum-sensing molecules HHQ and PQS, PhrS promotes HHQ and PQS synthesis. Moreover, this sets up a positive feedback loop because both HHQ and PQS appear to bind MvfR directly to promote the activity of this global transcription regulator (17). PhrS is therefore thought to promote the synthesis of quorum-sensing molecules in P. aeruginosa by pairing with a single target transcript that encodes a global transcription regulator.
Here, we use RIL-seq (RNA interaction through ligation and sequencing) to capture sRNAs together with their target transcripts on Hfq in P. aeruginosa. Using this approach, we identified interaction partners for 89 sRNAs in both the exponential and stationary phases of growth. Strikingly, we found that the sRNA PhrS is a major component of the RNA–RNA interaction network in this organism with the capacity to pair with a large number of different target transcripts. In addition, our findings suggest that PhrS functions in a more complex way to control the abundance of quorum-sensing molecules, not only up-regulating the synthesis of MvfR, but also down-regulating the synthesis of a second transcription regulator called AntR whose activity would otherwise limit PQS synthesis.
Results
RIL-Seq Analysis with Hfq in P. aeruginosa.
To identify the targets of Hfq-associated sRNAs in P. aeruginosa, we performed RIL-seq (18) to capture sRNA-target duplexes on Hfq (Fig. 1A). To do this, we used cells of P. aeruginosa strain PAO1 that synthesize Hfq with a vesicular stomatitis virus glycoprotein-G (VSV-G) epitope tag fused to its C terminus (Hfq-V) from the endogenous hfq locus on the chromosome (19). Wild-type PAO1 cells that synthesize Hfq without a VSV-G epitope tag served as a control. Cells were grown in LB medium at 37 °C to the exponential (OD600 of 0.5) and stationary (OD600 of 2.0) phases of growth and exposed to UV to allow cross-linking of RNAs to Hfq. Following cell lysis, Hfq-V together with those RNAs cross-linked to it was immunoprecipitated using antibodies recognizing the VSV-G epitope tag. After trimming with RNase, the ends of duplex RNAs captured on Hfq were ligated to one another and the resulting RNA was subjected to paired-end sequencing. Based on the findings of prior RIL-seq studies with Hfq in other bacteria, chimeric fragments with reads mapping to two distinct genomic locations were expected primarily to correspond to a portion of an sRNA together with a portion of its corresponding target transcript (such as an sRNA–mRNA target pair), whereas fragments with reads that map to a single genomic location were expected primarily to correspond to any Hfq-bound RNA species (18, 20–23). For each growth phase, chimeric reads from each of the triplicate RIL-seq datasets were combined into a single dataset that included only statistically significant chimeras (so-called S-chimeras; ref. 18). In addition, because a small number of chimeric reads were present in the RIL-seq datasets obtained with control cells that did not synthesize epitope-tagged Hfq, we applied an additional filter that removed 90% of the chimeras from the control datasets (24). Thus, the final stringently filtered RIL-seq datasets generated here include only those S-chimeras with at least 32 chimeric reads in cells grown to exponential phase or at least 27 chimeric reads in cells grown to stationary phase (Dataset S1).
Fig. 1.
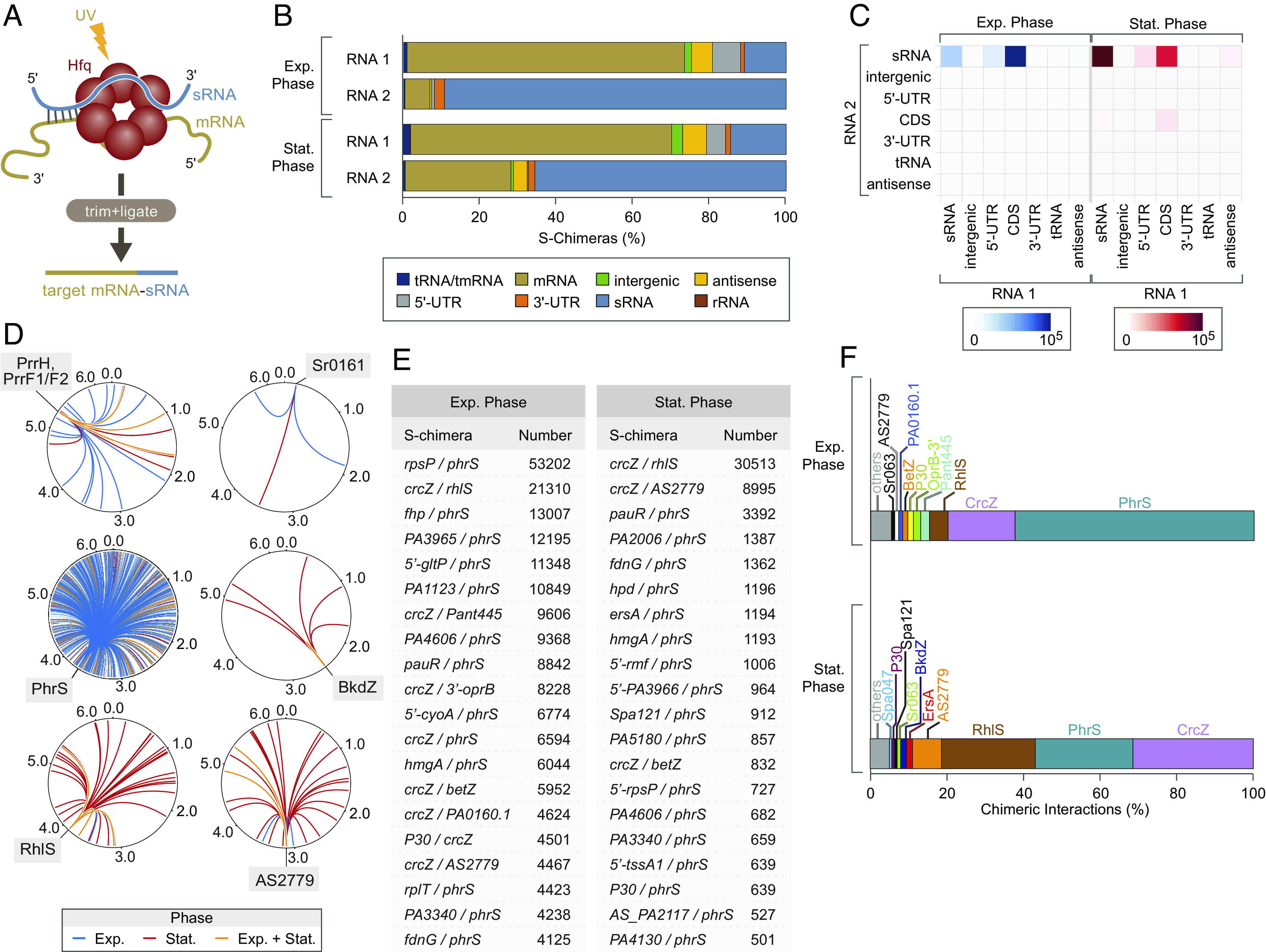
RIL-seq identifies RNA–RNA interactions occurring on Hfq in P. aeruginosa. (A) Schematic of RIL-seq approach. Biological triplicates of P. aeruginosa PAO1 wild-type and hfq-V strains were grown in LB at 37 °C to exponential phase (OD600 ≈ 0.5) and stationary phase (OD600 ≈ 2.0) and exposed to ultraviolet radiation, forming cross-links between nucleic acids and proteins. Hfq–RNA complexes were immune precipitated, subjected to limited RNase treatment, and neighboring RNAs were ligated by RNA Ligase. The resulting chimeric RNAs were sequenced using Illumina sequencing technology. (B) Breakdown of types of RNA species in RNA 1 and RNA 2 positions of S-chimeras obtained from cells grown to the exponential (Exp.) or stationary (Stat.) phases of growth. The individual RNAs from each S-chimera were classified according to genomic annotation and the fraction of each annotation type is plotted. (C) Quantification of chimeric species composed of the indicated types of RNA–RNA interactions in cells grown to the exponential (Exp.) or stationary (Stat.) phases of growth. Heatmaps depict the frequency for each type of interaction detected among all S-chimeras. Rows represent the second RNA in the chimera, columns represent the first RNA in the chimera. (D) Circos plot representations of RNA targets for the indicated sRNAs. Interactions that occur in cells grown to exponential phase are in blue, those occurring in cells grown to stationary phase are in red, and those that occur during both growth phases are in orange. Plots were drawn using Circos software version 0.69-9 (http://circos.ca/). (E) Top 20 S-chimeras found in cells grown to exponential (Left) and stationary (Right) phases of growth. Each S-chimera interaction pair is listed as RNA 1/RNA 2. The number column indicates the total number of chimeric fragments detected by RIL-seq for each interaction pair. (F) Relative abundance of sRNAs detected in S-chimeras following RIL-seq with Hfq in P. aeruginosa in cells grown to the exponential (Exp.) or stationary (Stat.) phases of growth. Data represent the relative abundance for each sRNA detected in S-chimeras, which was determined by dividing the number of chimeric fragments involving a particular sRNA by the total number of chimeric fragments containing any sRNAs. Note that some chimeric fragments consist of two sRNAs and those chimeras were counted more than once, with the total number of S-chimeras being updated accordingly.
RIL-seq with Hfq in P. aeruginosa identified 997 S-chimeras in cells grown to exponential phase and 702 S-chimeras in cells grown to stationary phase (Dataset S1). The S-chimeras we found associated with Hfq in exponential phase through RIL-seq more than triple the number of RNA–RNA interactions discovered under similar growth conditions using a different global proximity ligation approach called high GRIL-seq that identifies paired RNA species irrespective of whether their pairing is mediated by an RNA-binding protein (25, 26). The total number of S-chimeras we found associated with Hfq in P. aeruginosa are comparable to those identified through RIL-seq with Hfq in Escherichia coli and Salmonella, as well as Vibrio cholerae, indicative of an extensive Hfq-mediated RNA–RNA interaction network in this organism (18, 20, 21, 23, 24). As observed in RIL-seq studies of Hfq in other bacteria, we found that sRNAs typically constitute the second read of the S-chimeras presumably because the U-rich sequences in their 3′ ends are bound by the proximal surface of Hfq, limiting their availability for ligation (Fig. 1B) (18, 20, 21, 23, 24). Indeed, MEME analyses (27) indicated that a U-rich sequence preceded by G residues was enriched in the RNA 2 sequences of S-chimeras, whereas a sequence motif consisting of 5 ARN repeats that can be bound by the distal surface of Hfq (1) was enriched in the RNA 1 sequences (SI Appendix, Fig. S1). These findings suggest that RNA 1 and RNA 2 of each chimera represent transcripts bound to different surfaces of Hfq. Consistent with previous RIL-seq findings, the most predominant Hfq-associated chimeric RNA species identified in exponentially growing cells comprised sRNAs paired with mRNAs from coding sequences (Fig. 1C). In addition, as with RIL-seq findings with Hfq in Salmonella and Vibrio cholerae, as well as with Hfq CLASH in E. coli (20, 21, 28), sRNA–sRNA interactions were prevalent in our RIL-seq data, especially in cells grown to stationary phase (Fig. 1C). Thus, RIL-seq reveals a plethora of both mRNA and non-mRNA targets for sRNAs in P. aeruginosa. To facilitate access to the RIL-seq datasets, we developed an interactive web-based browser for both the exponential phase (https://genome.ucsc.edu/s/michael-gebhardt/PAO1_RIL-seq_ExpPhase) and the stationary phase datasets (https://genome.ucsc.edu/s/michael-gebhardt/PAO1_RIL-seq_StatPhase).
RIL-Seq with Hfq Identifies Interaction Partners for Previously Known as Well as Previously Unknown sRNAs.
We detected interaction partners for 89 sRNAs in P. aeruginosa using RIL-seq (Dataset S1), 68 of which are either known sRNAs or putative sRNAs identified through RNA-seq studies (11–14). Among our data, we found evidence supporting direct interactions between several known sRNAs and their targets that have been identified previously in P. aeruginosa using high GRIL-seq (25). For example, we identified the exsA mRNA encoding a transcription regulator of the type III secretion system as a target for Sr0161 in our RIL-seq dataset, and we identified many of the known regulatory targets for the well-studied iron-regulated sRNAs PrrF1 and PrrF2 (Dataset S1) (25). These findings suggest that RIL-seq identifies bona fide targets for sRNAs in P. aeruginosa.
We also identified targets for many previously identified sRNAs whose interaction partners were either few or completely unknown such as the sRNA AS2779 (SPA116), whose expression is reduced in multidrug-resistant isolates of P. aeruginosa (29), and the sRNA RhlS (SPA104) that regulates quorum sensing (30). In particular, using RIL-seq, we identified four potential targets for AS2779 in exponential phase and 26 in stationary phase, and we identified 5 and 41 potential targets for RhlS in exponential phase and stationary phase, respectively (Fig. 1D and Dataset S1). Consistent with recent findings obtained using a reverse GRIL-seq approach in which sRNAs are sought that interact with a specific target mRNA (31), our RIL-seq study identified the vfr transcript, specifying the virulence regulator Vfr, as a target for RhlS (Dataset S1). For both AS2779 and RhlS, it is noteworthy that they interact with most of their partner RNAs during the stationary phase of growth, as represented in the Circos plots in Fig. 1D. Consistent with these observations, both sRNAs have been reported to be induced in response to quorum sensing (30), and northern blotting with RNA isolated from wild-type and Δhfq mutant cells establishes for AS2779 and confirms for RhlS that each sRNA is Hfq-dependent and preferentially made during the stationary phase of growth (SI Appendix, Fig. S2) (30). Thus, our RIL-seq studies provide insight into possible regulatory functions of a variety of sRNAs in P. aeruginosa.
Our RIL-seq data revealed an extensive network of potential interactions between sRNAs in P. aeruginosa as well as the apparent rewiring of this network upon the transition from the exponential to stationary phases of growth (Dataset S1 and SI Appendix, Fig. S3). In stationary phase cells, the two most abundant S-chimeras found associated with Hfq are those involving the sRNAs CrcZ and RhlS and those involving CrcZ and AS2779 (Fig. 1E). The large number of chimeras between CrcZ and RhlS in stationary phase strongly influences the observed shift in the prevalence of sRNA–sRNA interactions between mid-log and stationary phase cells (Fig. 1C). The sRNA CrcZ has been well studied and contains multiple ARN motifs that allow it to serve as an RNA sponge for Hfq (7, 8). In P. aeruginosa, CrcZ is highly abundant, is Hfq-dependent (SI Appendix, Fig. S2), is the most abundant sRNA species associated with Hfq in our RIL-seq studies (SI Appendix, Fig. S4), and appears to target many of the sRNAs found among our RIL-seq dataset (Dataset S1). Furthermore, CrcZ has been found to pair with other sRNAs such as PrrF1 in prior GRIL-seq studies (26). Taken together with earlier observations (25, 26, 32), our findings raise the possibility that CrcZ may act as a sponge for both Hfq and certain sRNAs. However, as noted previously (26), it remains to be determined whether any chimeric reads that contain CrcZ reflect a biological activity of this sRNA or represent an artifact due to CrcZ’s abundance and association with Hfq.
Through RIL-seq with Hfq, we identified interaction partners for several putative sRNAs in P. aeruginosa (Dataset S1 and SI Appendix, Table S1). One of these, referred to here as BkdZ, is derived from the 3′ UTR of lpdV, which is part of the bkdA1 operon whose gene products are involved in the assimilation of branched-chain amino acids (33). Northern blotting with RNA isolated from wild-type and Δhfq mutant cells indicates that BkdZ is an Hfq-dependent sRNA that is preferentially made during the stationary phase of growth (SI Appendix, Figs. S2 and S5). Consistent with these findings, a Circos plot representation (34) illustrates that BkdZ pairs with target transcripts preferentially during stationary phase (Fig. 1D). Intriguingly, our RIL-seq findings indicate that BkdZ targets the 5′ UTR of bkdA1 (Dataset S1), which is itself a distinct sRNA (13). The function of any potential interaction between BkdZ and the bkdA1 5′ UTR is unclear. Specifically, such an interaction might influence the translation or abundance of the bkdA1 mRNA. Alternatively, an interaction between BkdZ and the bkdA1 5′ UTR-derived sRNA might serve to alter the activity or abundance of either or both of the participating sRNAs. Nevertheless, our findings with BkdZ demonstrate the utility of RIL-seq to identify previously uncharacterized Hfq-associated sRNAs in P. aeruginosa (35), together with their putative interaction partners, including RNAs that may be generated from the same polycistronic transcript from which the sRNA is derived (36).
PhrS Is the Predominant sRNA in the Hfq RIL-Seq Dataset.
Strikingly, analysis of the S-chimeras associated with Hfq in cells grown to exponential phase reveals that the sRNA interaction network is dominated by a single sRNA called PhrS (Fig. 1F). Indeed, we find that PhrS pairs with 741 different targets in exponential phase cells, which represents 74% of the 997 distinct S-chimeras found associated with Hfq under these conditions (Dataset S1). Furthermore, chimeras that contain PhrS are the most prevalent in the top 20 most abundant S-chimeras found associated with Hfq in cells grown to exponential phase (Fig. 1E). Analysis of the S-chimeras associated with Hfq in cells grown to stationary phase reveals that PhrS is a prominent component of the sRNA interaction network under these growth conditions as well; of the 702 S-chimeras identified in stationary phase, 246 (35%) contained PhrS (Dataset S1). Similar to what we find with cells grown to exponential phase, chimeras containing PhrS are the most prevalent in the top 20 most abundant S-chimeras found associated with Hfq in stationary phase (Fig. 1E). A Circos plot illustrates that there are both common and distinct targets of PhrS at the exponential and stationary phases of growth, likely reflecting, at least in part, whether certain transcripts are found in either one or both of the growth phases analyzed here (Fig. 1D). By combining all of the S-chimeras containing PhrS (that pass the chimera number cutoffs used here), regardless of the growth phase in which they are detected, we see that PhrS has ~800 targets, which is an unusually large target repertoire for an sRNA. PhrS was only known previously to target two RNA species: a polycistronic RNA encoding the transcription regulator MvfR and the CRISPR leader RNA (16, 37). Although PhrS was thought to exert the majority of its regulatory effects by targeting the transcript specifying MvfR (16), our findings raise the possibility that PhrS controls the translation and (or) abundance of many transcripts by pairing with them directly.
PhrS Controls Targets Identified through RIL-Seq Using a Seed Region Close to the Terminator Hairpin.
To determine whether PhrS can exert regulatory effects on those target transcripts identified through RIL-seq, we initially asked whether PhrS could regulate the translation of the mRNA from i) the hmgA gene encoding the enzyme homogentisate 1,2-dioxygenase, and ii) the mRNA from the PA3340 gene, which encodes a homolog of the motility gene fimV. These mRNAs were in the top 20 most abundant chimeras containing PhrS in our RIL-seq dataset obtained with stationary phase cells (the phase of growth where PhrS is most abundant) (Fig. 1E) and neither had previously been shown to be subject to control by PhrS (16). Analysis of the hmgA portion of hmgA-PhrS chimeras as well as the PA3340 portion of PA3340-PhrS chimeras indicated that PhrS targets the 5′ UTR of each mRNA (Fig. 2 A and B). To test whether PhrS could influence the translation of hmgA and PA3340, we made reporter plasmids that contained translational fusions of hmgA and PA3340 to lacZ whose expression was under the control of the corresponding native hmgA or PA3340 promoter. As controls, we made reporter plasmids that contained transcriptional fusions of the hmgA and PA3340 promoters to lacZ. Each of these plasmids was then introduced into wild-type P. aeruginosa strain PAO1, or a previously made ΔphrS mutant derivative (19). Alongside these reporter plasmids, we introduced a vector that contained phrS under the control of an arabinose-inducible promoter (19), or a vector that expressed the unrelated RyhB sRNA from E. coli (38) under the control of an arabinose-inducible promoter, or an empty control vector that did not encode any sRNA. Cells were then grown in the presence of arabinose to an OD600 of ~2.0 in LB and assayed for β-galactosidase activity. Expression of the hmgA translational reporter was slightly higher in ΔphrS mutant cells compared to wild-type cells, and ectopic expression of phrS in ΔphrS mutant cells resulted in ~3-fold repression of reporter gene expression (Fig. 2C). Expression of RyhB did not influence the expression of the hmgA translational reporter (Fig. 2C). Loss or ectopic expression of phrS resulted in minimal effects on expression of the hmgA transcriptional reporter (SI Appendix, Fig. S6). In addition, expression of the PA3340 translational reporter was ~1.5-times higher in ΔphrS mutant cells compared to wild-type cells (Fig. 2D). Furthermore, ectopic expression of phrS resulted in ~4-fold repression of reporter gene expression (Fig. 2D). Loss or ectopic expression of phrS resulted in minimal effects on expression of the PA3340 transcriptional reporter (SI Appendix, Fig. S6). Taken together, these findings suggest that PhrS can repress the translation or stability of the hmgA and PA3340 transcripts, establishing that RIL-seq can identify the regulatory targets of sRNAs in P. aeruginosa.
Fig. 2.
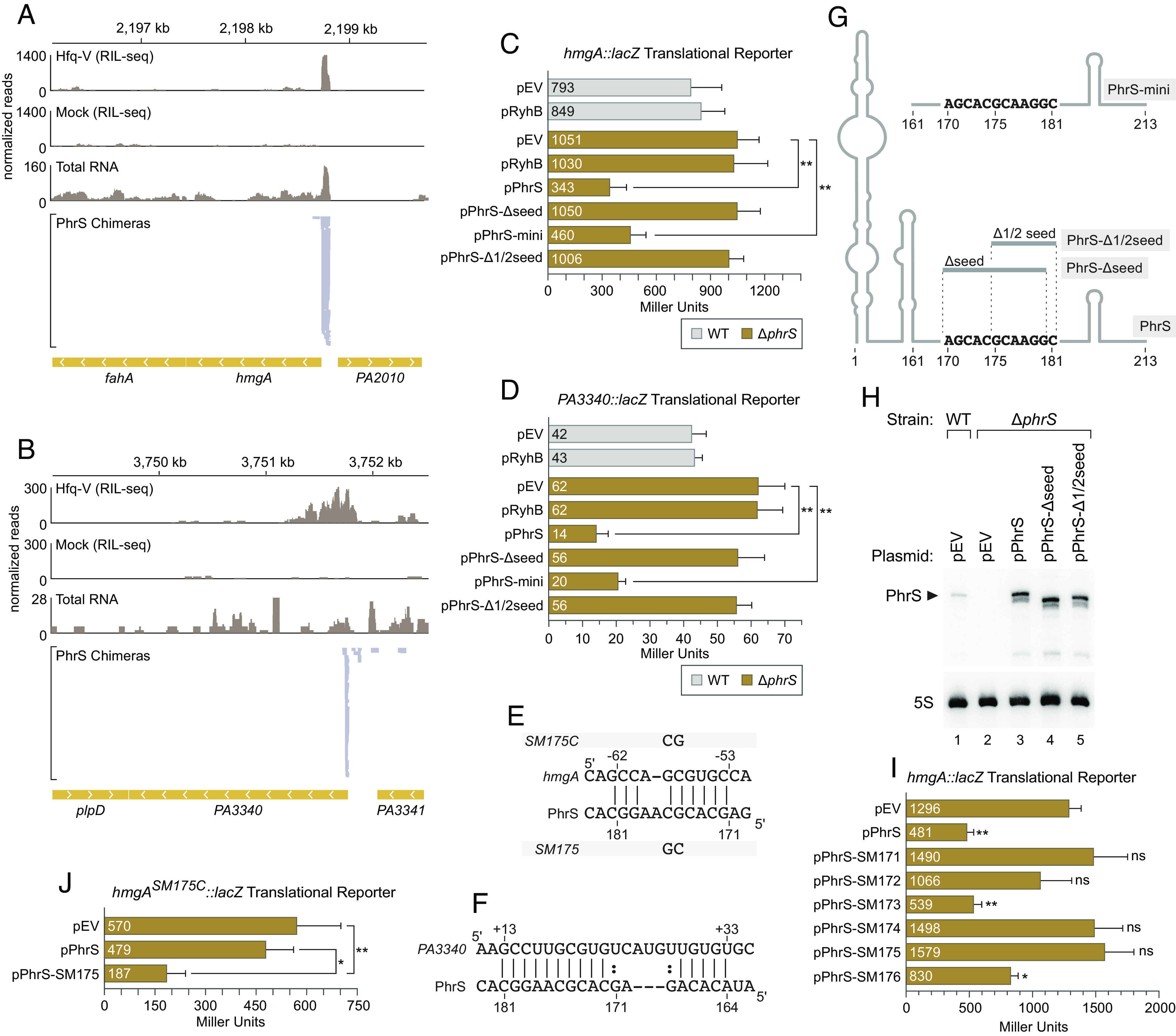
PhrS controls targets hmgA and PA3340 identified through RIL-seq. (A) RIL-seq identifies the hmgA mRNA as a target for PhrS. RIL-seq panels: Reads from RIL-seq with PAO1 Hfq-V cells (Hfq-V) grown to stationary phase compared to RIL-seq with wild-type cells (Mock) that do not synthesize any epitope-tagged Hfq. Total RNA panel: Total RNA-seq reads from PAO1 Hfq-V cells grown to stationary phase. Only reads corresponding to the minus strand are shown. PhrS Chimeras panel: Portions of hmgA mRNA found in hmgA/phrS S-chimeras following RIL-seq with Hfq-V in stationary phase cells. (B) RIL-seq identifies the PA3340 mRNA as a target for PhrS. RIL-seq and total RNA-seq panels are as in A. PhrS Chimeras panel: Portions of PA3340 mRNA found in PA3340/phrS S-chimeras following RIL-seq with Hfq-V in stationary phase cells. In both A and B, genomic location is indicated above data panels and genes are annotated at the bottom. Chevrons within genes indicate direction of transcription. (C) β-galactosidase activity (in Miller Units) of PAO1 WT cells (in gray) or PAO1 ∆phrS mutant cells (in brown) containing an hmgA–lacZ translational fusion as well as the indicated plasmids. Plasmid pEV is the empty vector control, pPhrS encodes PhrS, pRyhB encodes the E. coli sRNA RyhB, pPhrS-∆seed encodes PhrS-∆seed, pPhrS-mini encodes PhrS-mini, and pPhrS-∆1/2seed encodes PhrS-∆1/2seed (see schematic in G). (D) β-galactosidase activity (in Miller Units) of PAO1 WT cells (in gray) or PAO1 ∆phrS mutant cells (in brown) containing a PA3340–lacZ translational fusion as well as the indicated plasmids. Plasmids are as in C. (E) IntaRNA prediction of base-pairing between PhrS and hmgA mRNA. Numbers above indicate nucleotide position relative to the start codon of hmgA and numbers below indicate nucleotide position in PhrS. (F) IntaRNA prediction of base-pairing between PhrS and PA3340 mRNA. Numbers above indicate nucleotide position relative to the start codon of PA3340 and numbers below indicate nucleotide position in PhrS. (G) Schematic of PhrS and indicated mutant derivatives. Sequence of putative seed positioned from 170-181 is shown. (H) Northern blot of PhrS from RNA isolated from PAO1 wild-type cells (WT) or PAO1 ∆phrS mutant cells (∆phrS) harboring the indicated plasmids. Cultures were grown to stationary phase (OD600 ≈ 2.0) in the presence of 0.2% (w/v) arabinose, after which time RNA was isolated. The RNAs were separated on polyacrylamide gels and subjected to northern blot analysis using radiolabeled oligonucleotides specific to the indicated RNA species on the same membrane. The arrowhead indicates the size of full-length PhrS RNA. (I) β-galactosidase activity (in Miller Units) of PAO1 ∆phrS mutant cells containing an hmgA–lacZ translational fusion as well as the indicated plasmids. Plasmid pEV is the empty vector control, pPhrS encodes PhrS, pPhrS-SM171 through pPhrS-SM176 encodes the indicated SM mutant derivative of PhrS. (J) β-galactosidase activity (in Miller Units) of PAO1 ∆phrS mutant cells containing a version of the hmgA–lacZ translational fusion with mutation SM175C (as indicated in panel E) as well as the indicated plasmids. The SM175 mutation in phrS is predicted to restore base-pairing with the SM175C mutant derivative of the hmgA 5′ UTR. β-galactosidase assays in panels C, D, I, and J were conducted with biological triplicate cultures and repeated independently at least twice. Data are shown from a single representative experiment with error bars representing one SD of the mean. Significance was assessed by one-way ANOVA with Bonferroni post-test correction. Asterisks indicate significant differences with P-value ≤ 0.05 (*), P-value ≤ 0.01 (**); for Panel I, ns indicates a nonsignificant difference compared to pEV.
A characteristic feature of sRNAs is that they employ so-called seed regions to pair with target transcripts (39). Analysis of the predicted pairing between PhrS and the 5′ UTRs of hmgA and PA3340 using IntaRNA (40) implicated a potential seed region in PhrS from position 170-181 as being involved in a direct interaction with each mRNA (Fig. 2 E and F). This contains the portion of PhrS implicated previously in interaction with the mvfR transcript (16). To test whether this putative seed region of PhrS was important for repression of the hmgA and PA3340 translational reporters, we made three mutant versions of PhrS. The first of these (referred to here as PhrS-Δseed) has been described before (16), lacks nucleotides 170 to 180, and thus lacks the region of PhrS predicted to base-pair with hmgA and PA3340 (Fig. 2G). The second mutant, referred to as PhrS-Δ1/2seed, lacks nucleotides 175 to 181 and thus lacks a portion of PhrS predicted to base-pair with hmgA and PA3340 (Fig. 2G). The third mutant, referred to here as PhrS-mini, has been described before (16) and is a truncated version of PhrS consisting of nucleotides 161 to 213 that contain the putative seed region as well as the intrinsic terminator hairpin of PhrS (Fig. 2G). Whereas ectopic expression of wild-type PhrS and PhrS-mini in ΔphrS mutant cells resulted in strong repression of both the hmgA and PA3340 translational reporters, ectopic expression of PhrS mutants Δseed and Δ1/2seed did not (Fig. 2 C and D) despite the fact that they were present in amounts equal to that of wild-type PhrS (Fig. 2H). These findings support the idea that PhrS employs a seed region located around position 170 to 181 to directly repress the translation of hmgA and PA3340.
To test further whether PhrS targeted the hmgA mRNA directly, we first made a series of phrS mutants (SM171-176) that contained dinucleotide substitutions within the region of predicted base-pairing between hmgA and PhrS (from 171 to 177). Expression of each of these mutants and wild-type PhrS in ΔphrS mutant cells revealed that dinucleotide substitutions between 174 and 176 reduced the ability of PhrS to control the expression of the hmgA translational reporter, with the dinucleotide substitution CG at position 175 to 176 (mutant SM175) having a particularly pronounced effect (Fig. 2I). Importantly, although PhrS mutant SM175 failed to repress the expression of the wild-type version of the hmgA translational reporter, it efficiently repressed a mutant version of the reporter in which the 5′ UTR of hmgA was mutated to restore pairing with the SM175 mutant (Fig. 2 E and J). These findings demonstrate that PhrS exerts its regulatory effects on hmgA by pairing with the 5′ UTR of the hmgA mRNA through a seed region located close to its terminator hairpin.
Ectopic Expression of PhrS Results in Widespread Changes in Gene Expression.
To test whether PhrS can influence the abundance of target transcripts on a global scale, we performed a pulse-expression analysis of wild-type PhrS in cells grown to stationary phase—the phase of growth in which PhrS has been shown previously to exert regulatory effects by targeting the transcript encoding the transcription regulator MvfR (16). As a control, we also performed a pulse-expression analysis of PhrS-Δseed, which lacks the putative seed region. RNA-seq analysis indicated that pulse-expression of wild-type PhrS and PhrS-Δseed for 20 min in cells of our ΔphrS mutant strain grown to stationary phase resulted in changes in the expression of 667 and 569 genes, respectively, by a factor of 2 or more (Fig. 3A and Dataset S2). Our RIL-seq analysis indicated that PhrS targets 246 different transcripts under these same conditions and 35 of these (14%) were among those whose abundance changed following pulse-expression of PhrS (Fig. 3B). For an additional 13 predicted PhrS targets, we observed 2-fold or greater changes in the expression of downstream genes, suggesting that interaction between PhrS and the direct target resulted in a regulatory effect on a co-cistronic gene. Pulse-expression therefore suggests that 48 of the 246 direct targets of PhrS identified by RIL-seq may be bona fide regulatory targets. Of these direct targets, 20 were also associated with gene expression changes following expression of PhrS-Δseed (Dataset S2). We infer from this that the seed region of PhrS located from position 170 to 181 is important for the regulatory effects of PhrS associated with 28 direct targets identified by RIL-seq in stationary phase. Thus, as has been observed previously with a variety of sRNAs in E. coli (22, 28), ectopic expression of PhrS in P. aeruginosa results in pronounced changes in the abundance of only a subset of its predicted direct targets and associated genes.
Fig. 3.

RNA-seq analysis of the effects of PhrS pulse-expression on transcript abundance. (A) Pulse-expression of PhrS for 20 min alters the abundance of 677 transcripts. Results of RNA-seq analyses comparing transcript abundance in PAO1 ∆phrS mutant cells containing either plasmid pEV (empty vector control) or plasmid pPhrS (encoding PhrS) following the addition of inducer for 20 min in stationary phase (OD600 ≈ 2.0). (B) Overlap between transcripts whose abundance alters following pulse-expression of PhrS for 20 min in stationary phase and those transcripts that are targeted by PhrS in stationary phase, as indicated by RIL-seq. (C) Overlap between transcripts whose abundance alters following pulse-expression of wild-type PhrS for 20 min in stationary phase and those transcripts whose abundance alters following pulse-expression of PhrS-∆seed for 20 min in stationary phase.
Most transcripts whose abundance changed in response to either wild-type PhrS or PhrS-Δseed are common to both datasets and were not identified as targets for PhrS by RIL-seq (Fig. 3C and Datasets S1 and S2). Ectopic expression of an sRNA can alter the ability of other sRNAs in the cell to compete for the available Hfq (41–43) and this competition may account for some of the gene expression changes we observe. Consistent with this possibility, we find that 13 sRNAs are down-regulated following ectopic expression of both PhrS and PhrS-Δseed (Dataset S2). Although pulse-expression of an sRNA is designed to identify direct targets of the sRNA, some of the genes that are positively regulated following pulse-expression of PhrS (e.g., pqsABCDE, and phnAB; Dataset S2) are known to be positively regulated by MvfR whose translation is in turn directly controlled by PhrS (16, 44). Further, the expression of several genes encoding transcription factors such as rpoH, algU, and algB are strongly up-regulated following ectopic expression of both PhrS and PhrS-Δseed, which could account for the similar widespread effects of these sRNAs on gene expression (Dataset S2). We also note that the relatively high level at which we induce PhrS in our studies (SI Appendix, Fig. S7) may explain, at least in part, the difference between our findings and those of a prior study in which microarray analyses uncovered just over 50 genes whose expression altered following pulse-expression of PhrS (16).
Despite our demonstration that PhrS can specifically control the expression of both an hmgA and a PA3340 translational reporter fusion (Fig. 2), neither hmgA nor PA3340 was among the transcripts whose abundance changed by a factor of 2 or more following pulse-expression of PhrS (Dataset S2). The hmgA transcript was, however, one of the 13 predicted direct PhrS targets with an associated downstream gene(s) whose expression was altered following pulse-expression of PhrS (Dataset S2). Thus, similar to other sRNAs (22, 28), PhrS may influence the translation of target transcripts such as hmgA and PA3340 without appreciably altering their overall abundance.
PhrS Directly Targets the mRNA Encoding AntR, a Regulator of PQS Synthesis.
PhrS is thought to positively control the synthesis of the quorum-sensing signals HHQ and PQS by targeting a single transcript specifying the transcription regulator MvfR (16). Specifically, PhrS was found to positively control the translation of mvfR by pairing directly with the mRNA of a small ORF positioned immediately upstream of mvfR (16). PhrS promotes translation of this small ORF and in so doing promotes the translation of MvfR. In this manner, PhrS results in an increase in the abundance of MvfR with a concomitant increase in the expression of MvfR-regulated genes involved in the synthesis of PQS (16). However, MvfR is not the only transcription regulator known to influence PQS synthesis in P. aeruginosa. In particular, the LysR-type transcription activator AntR negatively controls the abundance of PQS by activating expression of the antABC genes whose products convert anthranilate, the precursor of PQS, to catechol (45). We noticed that the antA and antC genes were among the repressed genes we identified following ectopic expression of PhrS (Fig. 3A and Dataset S2). We also noticed that antR-PhrS chimeras were present in our RIL-seq dataset (Dataset S1), with PhrS appearing to target the 5′ UTR of antR (Fig. 4A) using the seed sequence located between positions 170–181 of PhrS (Fig. 4B). To determine whether native levels of PhrS influenced the expression of antR and the antABC operon, we compared expression of the antR, antA, and antB genes in wild-type and ΔphrS mutant cells of PAO1 using qRT-PCR. The results depicted in Fig. 4C show that expression of antR is ~5-fold higher in ΔphrS mutant cells compared to wild-type cells, whereas expression of antA and antB is ~40-fold and 15-fold higher, respectively, in ΔphrS mutant cells compared to wild-type cells. Moreover, ectopic expression of PhrS complemented the effects of the ΔphrS mutation on the expression of antR, antA, and antB (Fig. 4C). These findings establish that PhrS exerts a strong negative effect on antR expression, as well as the antAB genes (and thus presumably the entire antABC operon), when produced at native levels.
Fig. 4.

PhrS targets the antR transcript. (A) RIL-seq identifies the antR mRNA as a target for PhrS. RIL-seq panels: Reads from RIL-seq with PAO1 Hfq-V cells (Hfq-V) grown to stationary phase compared to RIL-seq with wild-type cells (Mock) that do not synthesize any epitope-tagged Hfq. Total RNA panel: Total RNA-seq reads from PAO1 Hfq-V cells grown to stationary phase. Only reads corresponding to the minus strand are shown. PhrS Chimeras panel: Portions of antR mRNA found in antR/phrS S-chimeras following RIL-seq with Hfq-V in stationary phase cells. Genomic location is indicated above data panels and genes are annotated at the bottom. Chevrons within genes indicate direction of transcription. (B) IntaRNA prediction of base-pairing between PhrS and antR mRNA. Numbers above indicate nucleotide position relative to the start codon of antR and numbers below indicate the nucleotide position in PhrS. The positions and mutations in the PhrS-SM179 mutant and the antR-M2C mutant are shown. (C) Native levels of PhrS influence the abundance of the antR, antA, and antB transcripts. qRT-PCR analyses of antR, antA, and antB relative transcript abundance in PAO1 wild-type cells harboring the empty vector pEV (WT pEV, gray), or in PAO1 ∆phrS mutant cells harboring pEV (∆phrS pEV, blue), or in PAO1 ΔphrS mutant cells harboring the vector pPhrS encoding PhrS (∆phrS pPhrS, pink). Total RNA was extracted from triplicate cultures of the indicated cells that had been grown for 6 h (OD600 ≈ 2.0) in the presence of 0.2% (w/v) arabinose. The resulting RNA was treated with DNAse, converted to cDNA, and qRT-PCR was performed. Data are plotted as the relative transcript abundance of the indicated genes compared to the transcript for clpX using the comparative Ct method (2−∆∆CT). The qRT-PCR experiment was repeated twice independently and data from a representative experiment are shown as the mean transcript abundance. Error bars represent one SD and circles indicate the individual data points. Significance was assessed by one-way ANOVA with Bonferroni post-test correction. P-values indicate the significance levels; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. (D) β-galactosidase activity (in Miller Units) of PAO1 WT cells (in gray) or PAO1 ΔphrS mutant cells (in brown) containing an antR–lacZ translational fusion as well as the indicated plasmids. Plasmid pEV is the empty vector control, pPhrS encodes PhrS, pRyhB encodes RyhB, pPhrS-∆seed encodes PhrS-∆seed, pPhrS-mini encodes PhrS-mini, and pPhrS-∆1/2seed encodes PhrS-∆1/2seed (see schematic in Fig. 2G). (E) β-galactosidase activity (in Miller Units) of PAO1 ∆phrS mutant cells containing an antR–lacZ translational fusion as well as the indicated plasmids. Plasmid pEV is the empty vector control, pPhrS encodes PhrS, pPhrS-SM173 through pPhrS-SM182 encodes the indicated SM mutant derivative of PhrS. (F) Northern blot of PhrS from RNA isolated from PAO1 wild-type cells (WT) or PAO1 ∆phrS mutant cells (∆phrS) harboring the indicated plasmids. Plasmid pEV is the empty vector control, pPhrS encodes PhrS, pPhrS-SM178 through pPhrS-SM181 encodes the indicated SM mutant derivative of PhrS. PAO1 wild-type cells (WT) or PAO1 ∆phrS mutant cells (∆phrS) harboring the indicated plasmids were grown to stationary phase (OD600 ≈ 2.0) in the presence of 0.2% (w/v) arabinose, after which time RNA was isolated. The RNAs were separated on polyacrylamide gels and subjected to northern blot analysis using radiolabeled oligonucleotides specific to the indicated RNA species on the same membrane. The arrowhead indicates the size of full-length PhrS. (G) Western blot with anti-VSV-G antibody to detect AntR-V in cells grown to stationary phase. Whole cell lysates prepared from cells of the indicated strains that had been grown for 18 h were resolved by SDS-PAGE, transferred to PVDF membranes, and subjected to western blot analysis. Lane 1, wild-type PAO1 cells that do not encode AntR-V. Lane 2, PAO1 M2C-AntR-V mutant cells that encode AntR-V together with the M2C mutation altering the 5′ UTR of the antR transcript; these cells encode wild-type PhrS. Lane 3, PAO1 phrS-SM179 M2C-AntR-V mutant cells that encode AntR-V together with the M2C mutation and encode PhrS mutant SM179 from the native chromosomal location. The SM179 mutation in phrS is predicted to restore base-pairing with the M2C mutant derivative of the antR 5′ UTR. The arrowhead indicates the predicted size of AntR-V (approximately 39 kDa). Western blot experiments were completed with biological triplicate cultures and repeated independently at least twice. Data from a single experiment are shown. Size markers on the right indicate the migration of standards from a protein ladder. (H) β-galactosidase activity (in Miller Units) of PAO1 WT cells or PAO1 phrS-SM179 mutant cells containing a version of the antR–lacZ translational fusion plasmids with the M2C mutation altering the 5′ UTR of the antR transcript. For β-galactosidase assays in E, F, and I, the experiments were repeated independently at least twice with biological triplicate cultures. Data from a representative experiment are plotted as the mean and error bars represent one SD of the mean. Significance was assessed by one-way ANOVA with Bonferroni post-test correction. Asterisks indicate significant differences with P-value ≤ 0.05 (*), P-value ≤ 0.01 (**), and P-value ≤ 0.001 (***).
To begin to determine whether PhrS regulates the abundance of the antR mRNA through a direct interaction, we first made a reporter plasmid that contained a translational fusion of antR to lacZ whose expression was under the control of the native antR promoter. As a control, we made a reporter plasmid that contained the antR promoter region positioned upstream of lacZ. Each of these plasmids was then introduced into wild-type or ΔphrS mutant cells of P. aeruginosa strain PAO1 alongside vectors encoding wild-type PhrS, PhrS-Δseed, PhrS-Δ1/2seed, PhrS-mini, RyhB, or an empty control vector. Cells were then grown in the presence of arabinose to an OD600 of ~2.0 in LB and assayed for β-galactosidase activity. Expression of the antR translational reporter was ~3-fold higher in ΔphrS mutant cells compared to wild-type cells, suggesting that native levels of PhrS mediate a strong regulatory effect on antR (Fig. 4D). In addition, we found that ectopic synthesis of PhrS or PhrS-mini in ΔphrS mutant cells resulted in ~16-fold and 20-fold repression of reporter gene expression, respectively, whereas ectopic synthesis of PhrS mutants Δseed or Δ1/2seed did not (Fig. 4D). Loss or ectopic expression of phrS resulted in a modest effect on expression of the antR transcriptional reporter (SI Appendix, Fig. S6). Taken together, these findings suggest that PhrS represses the translation and/or stability of the antR mRNA using a seed region located between nucleotides 170 to 181 of PhrS.
To test further whether PhrS targeted the antR mRNA directly, we employed a series of ten phrS mutants (SM173-182) that contained dinucleotide substitutions beginning just within the region of predicted base-pairing between antR and PhrS (position 173 to 174) and ending just after it (at position 182 to 183). Expression of each of these mutants alongside wild-type PhrS in ΔphrS mutant cells indicated that dinucleotide substitutions beginning at positions 178 through 181 reduced the ability of PhrS to negatively regulate the expression of the antR translational reporter, with the dinucleotide substitution CC at position 179 to 180 (mutant SM179) having the most pronounced effect (Fig. 4E). Northern blotting indicated that all but one of the four PhrS mutants that displayed a reduced ability to regulate antR expression, including SM179, were at least as abundant as wild-type PhrS (Fig. 4F). These findings support the idea that specific sequences within the PhrS seed region that are predicted to pair with the antR mRNA are required for PhrS to control the translation and/or stability of the antR transcript.
We next asked whether PhrS base-pairs directly with the antR mRNA when produced at native levels. To do this, we first made a mutant version of our PAO1 strain that i) synthesized AntR with a VSV-G epitope tag fused to its C terminus (AntR-V) from the native chromosomal location, and ii) contained a dinucleotide substitution (referred to as M2C) within the 5′ UTR of antR designed to restore the potential base-pairing with PhrS mutant SM179 (creating strain PAO1 M2C-AntR-V) (Fig. 4B). We next made a derivative of PAO1 M2C-AntR-V that synthesized PhrS mutant SM179 with the dinucleotide substitution CC at position 179 to 180 from the native chromosomal location (PAO1 phrS-SM179 M2C-AntR-V). Western blotting with cells isolated from stationary phase revealed that when compared to wild-type PhrS, PhrS mutant SM179 efficiently reduced the abundance of AntR-V in cells containing the complementary M2C mutations that were predicted to restore base-pairing with the PhrS mutant (Fig. 4G). Furthermore, we found that expression of an antR-lacZ translational reporter harboring the M2C mutation was specifically reduced in cells expressing PhrS mutant SM179 when compared to cells expressing wild-type PhrS (Fig. 4H). These findings establish that PhrS exerts its regulatory effects on AntR by base-pairing directly with the antR mRNA.
To determine whether PhrS influences the abundance of PQS through interaction with the antR mRNA, we measured the abundance of PQS in stationary phase culture supernatants isolated from wild-type, ΔphrS, and M2C mutant cells by thin-layer chromatography. The results depicted in SI Appendix, Fig. S8, indicate that cells of the ΔphrS and M2C mutants produce less PQS than wild-type cells and similar amounts of PQS to one another. PQS is thought to influence the production of the redox-active compound pyocyanin by controlling the expression of the pyocyanin biosynthetic genes (17). In support of our observations with PQS, we also found that cells of the ΔphrS and M2C mutant produced less pyocyanin than that of wild-type cells (SI Appendix, Fig. S8). Taken together, these findings suggest that PhrS controls the abundance of PQS through its direct interaction with the antR transcript.
Although ΔphrS and M2C mutant cells produced similar amounts of PQS and pyocyanin to one another (SI Appendix, Fig. S8), our findings do not rule out the possibility that PhrS may need to interact with both the antR transcript and the mvfR transcript to exert effects on the production of PQS and pyocyanin in P. aeruginosa. Thus, in combination with the prior demonstration that PhrS positively regulates the translation of MvfR (16), our findings suggest that PhrS controls PQS through a two-tiered mechanism involving positive control of a transcription regulator (MvfR) that promotes PQS synthesis and negative control of a transcription regulator (AntR) that reduces PQS synthesis (Fig. 5).
Fig. 5.

Model for PhrS-mediated control of PQS synthesis. PhrS promotes the conversion of anthranilate to PQS through a two-tiered mechanism involving targeting the mvfR (pqsR) transcript to enhance the production of the transcription regulator MvfR (PqsR) that activates the expression of genes involved in PQS synthesis. PhrS also serves to direct the flow of anthranilate toward PQS synthesis by directly targeting the antR transcript to inhibit the production of the transcription regulator AntR that would otherwise redirect anthranilate toward the TCA cycle.
Discussion
Using RIL-seq, we have uncovered an extensive network of RNA–RNA interactions occurring on Hfq in cells of P. aeruginosa. These interactions involve eighty-nine sRNAs together with their target transcripts, providing a compendium of targets for a variety of sRNAs in this important opportunistic pathogen. We found that the sRNA PhrS is an especially prominent player that interacts with hundreds of different transcripts on Hfq and provide evidence that PhrS exerts control over a key quorum-sensing molecule through a mechanism that is more complex than previously thought.
PhrS as a Keystone sRNA.
PhrS was one of the first sRNAs to be characterized in P. aeruginosa and only two direct targets had been reported previously (16, 37). A striking finding from our RIL-seq studies with Hfq in P. aeruginosa is that PhrS appears to pair with approximately 800 different target RNAs. Although PhrS is one of the most abundant sRNAs on Hfq in P. aeruginosa (the sixth most abundant in both exponential and stationary phase cells) (SI Appendix, Fig. S4), it pairs with many more target RNAs than other sRNAs of similar or greater abundance. For example, in exponential phase, PrrF1 and PrrF2 are of similar abundance on Hfq to PhrS but pair with 23 targets under these conditions, whereas PhrS pairs with 741 targets (Fig. 1D) (Dataset S1). Thus, compared with other sRNAs in P. aeruginosa, PhrS has an outsized influence on the number of RNA–RNA interactions occurring on Hfq in this organism. By analogy with keystone predators in ecological niches, we therefore consider PhrS a keystone sRNA.
What Enables PhrS to Pair with So Many Targets?
Other sRNAs exist that appear to target 100 or more different transcripts. For some of these sRNAs, such as GcvB in E. coli, which has been reported to pair with more than 150 targets, the presence of multiple seed sequences helps to account for their large target repertoire (18). There are also examples of Hfq-associated sRNAs identified through RIL-seq studies that are thought to use a single seed region to pair with more than 300 targets, such as CyaR and ArcZ from E. coli (18). PhrS uses the same seed region to exert its regulatory effects on MvfR and AntR (16) (Fig. 4). This is the same seed region PhrS uses to control the translation and/or abundance of the hmgA and PA3340 transcripts (Fig. 2), and the same region responsible for changes in the expression of many direct targets following pulse-expression of PhrS (Fig. 3). Indeed, we suspect that PhrS uses the same single seed region to pair with many of its targets, and that this single seed is responsible for most of the interactions we observe using RIL-seq. We speculate that the flexibility of this seed region may be explained by the fact that it comprises an accessible region with a length that can accommodate a set of overlapping GC-rich pairings. We note that the ability to pair with so many different transcripts raises the possibility that the regulatory effects of PhrS will be shaped by the transcriptomic signature of the cell under any given growth condition, with competition between targets (both mRNA and sRNA) for the available pool of PhrS contributing to the overall regulatory effects of this sRNA (46).
A Revised Model for the Control of PQS Synthesis by PhrS.
The quorum-sensing signal PQS and its precursor HHQ are key regulators of virulence gene expression in P. aeruginosa. PhrS was thought to control PQS synthesis by positively regulating the translation of a single transcript encoding the transcription activator MvfR (16). MvfR positively regulates the expression of numerous genes including the phnAB and pqs genes (47, 48). The products of phnAB convert chorismic acid to anthranilate, while the products of the pqsABCD genes convert anthranilate into HHQ, which is subsequently converted into PQS by the enzyme PqsH (44 and references therein) (Fig. 5). Here, we present evidence that PhrS also negatively controls the translation and/or stability of the antR transcript encoding the transcription activator AntR. AntR positively regulates the expression of the antABC genes encoding enzymes that convert anthranilate, a precursor of PQS, into catechol (45), thus diverting it from the PQS pathway. Consistently, we find that the negative control of antR by PhrS enhances the production of both PQS and the redox active molecule pyocyanin, whose synthesis is governed by PQS. These findings with PhrS mirror prior work demonstrating that the iron-responsive sRNAs PrrF1 and PrrF2 influence the production of both HHQ and PQS by modulating the abundance of AntR (49, 50). PhrS therefore likely up-regulates the synthesis of PQS through a two-tiered mechanism—by i) directly targeting the mvfR transcript, PhrS enhances the production of a transcription regulator that serves to direct the flow of anthranilate toward PQS synthesis, and by ii) directly targeting the antR transcript, PhrS limits the production of a second transcription regulator that would otherwise redirect anthranilate toward catechol synthesis, thus increasing the pool of anthranilate available for the synthesis of PQS (Fig. 5).
It is noteworthy that PhrS exerts regulatory effects over PQS synthesis by targeting two distinct transcription regulators. Indeed, the targeting of mRNAs encoding transcription regulators presumably serves to amplify the regulatory effects of PhrS on the abundance of transcripts. Our RIL-seq studies have identified a variety of additional mRNAs specifying transcription regulators as targets for PhrS in P. aeruginosa, including those encoding the key virulence regulators Vfr, AmrZ, and LadS (Dataset S1) (51–54). Thus, although PhrS appears to directly target hundreds of different transcripts, this sRNA may exert a portion of its regulatory effects indirectly by controlling the abundance of transcription regulators.
Insights into Potential Regulatory Targets of sRNAs in P. aeruginosa.
Our RIL-seq study provides evidence for the existence of a large number of RNA–RNA interactions that occur on Hfq in P. aeruginosa at two different points in the growth curve. The S-chimeras we found associated with Hfq in mid-log phase through RIL-seq more than triple the number of RNA–RNA interactions discovered under similar growth conditions using a different global proximity ligation approach called high GRIL-seq that identifies paired RNA species irrespective of whether their pairing is mediated by an RNA-binding protein (26). Because sRNAs can mediate regulatory effects by base-pairing with target RNA species, our study provides a compendium of potential regulatory targets for as many as 66 sRNAs during mid-log growth and 62 sRNAs during stationary phase in cells grown in LB. Moreover, because some of these sRNAs appear to be newly identified in our study, such as BkdZ derived from the 3′ UTR of lpdV and a putative sRNA derived from the 3′ UTR of oprB (referred to here as OprB-3′), our study identifies both previously unknown sRNAs and candidate regulatory targets thereof, providing insight into their potential regulatory roles.
The regulatory effects of sRNAs extend beyond the targeting of mRNA species. Indeed, it is becoming increasingly apparent that sRNAs can exert regulatory effects by targeting other sRNAs, serving as so-called molecular sponges to alter the abundance or activity of their target sRNAs (reviewed in refs. 55 and 56). Our RIL-seq analyses in P. aeruginosa reveal an extensive network of sRNA–sRNA interactions in this organism, as well as the rewiring of this network in stationary phase cells (SI Appendix, Fig. S3), in part reflecting the interactions of sRNAs that are made in response to quorum sensing. It will be important to investigate the potential regulatory roles of the large number of sRNA–sRNA interactions our work has uncovered.
Materials and Methods
Bacterial Strains and Plasmids.
The experiments described herein were conducted with P. aeruginosa PAO1 and its derivatives. All strains, plasmids, and oligonucleotides are listed in SI Appendix, Tables S2–S5. All plasmid constructs were confirmed via sequencing. Mutant strains were confirmed by PCR and/or sequencing prior to use.
RIL-Seq.
RIL-seq experimental procedure.
RIL-seq was performed essentially as described previously (57); a detailed protocol is included in SI Appendix. Briefly, triplicate overnight cultures of PAO1 and PAO1 Hfq-V were back-diluted to an initial OD600 of 0.01 and grown until exponential phase (OD600 ≈ 0.5) and early stationary phase (OD600 ≈ 2) at which times, samples were collected and processed for RIL-seq as described in SI Appendix. The RIL-seq computational analyses were carried out as described by Melamed et al. (57) with modifications for the PAO1 genome (SI Appendix). Importantly, the three biological replicates for each time point (i.e., exponential phase and stationary phase) were tested for and found to be reproducible (57) and were thus unified into a single library prior to identification of S-chimeras (SI Appendix, Fig. S9). Visualization of RIL-seq data was performed with IGV (58).
RNA-Seq.
Triplicate overnight cultures of the PAO1 ∆phrS strain harboring an empty vector (pKH6; herein referred to as pEV), pPhrS (pKH6 + PhrS), or pPhrS-∆seed (pKH6 + PhrS-∆seed) were back-diluted to an OD600 of 0.01 in fresh LB + 30 µg/mL gentamicin and grown to early stationary phase (OD600 ≈ 1.75 to 2.0) at which time sRNA expression from pKH6 or its derivatives was induced with arabinose (0.2% w/v) for 20 min. RNA was extracted from 2 mL of culture using Tri-Reagent (Sigma-Aldrich). RNA-sequencing was performed by SeqCenter (Pittsburg, PA). Reads were assessed for quality control and adaptor trimming with bcl2fastq and mapped with bowtie2 (version 2.4.5); read quantification was performed with htseq (59) and differential gene expression analysis was conducted with DESeq2 (60). The DESeq2 datasets can be found in Dataset S2. The volcano plot in Fig. 3A was created in the R package, EnhancedVolcano (61).
Northern Blot Analysis.
Overnight cultures of the indicated strains were refreshed into LB medium containing gentamicin (30 µg/mL) and arabinose (0.2% w/v) and grown for 6 h, at which time RNA was extracted from 2 mL of culture. Following RNA extraction, 5 µg RNA was fractionated on 8% polyacrylamide urea gels containing 6 M urea (UreaGel-8, National Diagnostics) and transferred to a Zeta-Probe GT membrane (Bio-Rad). Following transfer, RNA was cross-linked to the membrane via UV irradiation. Molecular size markers were indicated (RNA low-range ladder, New England Biolabs), and the membranes were hybridized with 32P-end labeled oligonucleotide probes. Northern blots were imaged using Carestream BioMax MR film (Sigma-Aldrich) or captured on phospho-storage screens and imaged with an Azure Sapphire PhosphorImager. The membranes were stripped and reprobed to detect additional RNAs as needed. Northern blots were repeated with two biological replicates, and results from a single representative experiment are shown.
Beta-Galactosidase Assays.
Beta-galactosidase assays were conducted essentially as described previously (62). Cells were permeabilized with SDS and chloroform and assayed for β-galactosidase activity using 2-nitro-phenyl β-D-galactopyranoside (ONPG). Values reported are the average of biological triplicate cultures, with error bars representing one SD. Statistical analyses were performed using one-way ANOVA followed by Bonferroni correction. All β-gal experiments were independently repeated at least twice and results depict a single representative experiment.
qRT-PCR.
qRT-PCR was performed using cDNA derived from RNA collected from the indicated strains. Briefly, overnight cultures were back-diluted to an OD600 of 0.01 in fresh medium (LB + 30 µg/mL gentamicin and 0.2% w/v arabinose) and grown for 6 h, at which point RNA was collected from 2 mL culture using Tri-Reagent. 10 ng cDNA (synthesized using SuperScript IV, Thermo Fisher) was used as template for qRT-PCR reactions using iTaq Universal SYBR Green SuperMix (Bio-Rad) on an ABI QuantStudio 3 instrument (Applied Biosystems). Relative transcript abundance was measured using clpX as a reference transcript and was calculated via the comparative threshold cycle (CT) method (2−∆∆CT) (63). The qRT-PCR experiment was completed with biological triplicate cultures twice on independent samples; data from a single representative experiment are shown. Error bars represent one SD of the mean. Results were analyzed using ANOVA with Bonferroni correction to assess significance.
Western Blotting.
Whole-cell lysates of the indicated strains were resolved by SDS-PAGE on 4 to 12% Bis-Tris NuPAGE gels in MOPS running Buffer (Thermo Fisher) and transferred to PVDF membranes using an XCell-II Blot Module (Thermo Fisher). Membranes were blocked in a 1:5 dilution of Odyssey Blocking Buffer (LI-COR) for 1 h or overnight prior to being probed with anti-VSV-G antibodies (Sigma-Aldrich, cat. no. V4888). The membranes were subsequently washed, blocked, and incubated with donkey anti-rabbit secondary antibodies conjugated with the near-infrared dye 800CW (LI-COR) and imaged with an Azure C600 imaging system (Azure Biosciences). Data from a single, representative replicate are shown, and the experiment was performed independently at least two times with biological triplicates.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Acknowledgments
We thank Stephen Lory for plasmids, Renate Hellmiss for artwork, and Ann Hochschild for discussions and comments on the manuscript. Bioanalyzer and TapeStation analyses were performed in the Boston Children’s Hospital IDDRC Molecular Genetics Core that is supported by NIH award NIH-P30-HD 18655. RNA-seq libraries were constructed and sequenced at SeqCenter. Sequencing of the RIL-seq libraries was performed at the Biopolymers Facility at Harvard Medical School. This work was supported by NIH grant AI143771 (to S.L.D.). M.J.G. was supported by a Postdoctoral Research Fellowship from the Cystic Fibrosis Foundation.
Author contributions
M.J.G., S.M., and S.L.D. designed research; M.J.G., E.A.F., P.B., and K.M. performed research; M.J.G. contributed new reagents/analytic tools; M.J.G., E.A.F., P.B., K.M., S.M., and S.L.D. analyzed data; and M.J.G. and S.L.D. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Michael J. Gebhardt, Email: michael-gebhardt@uiowa.edu.
Simon L. Dove, Email: simon.dove@childrens.harvard.edu.
Data, Materials, and Software Availability
Sequencing data have been deposited in the Gene Expression Omnibus (GEO) at NCBI under accession number GSE216135 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE216135) (64). All data generated or analyzed during this study are included in the manuscript and supporting information. The RIL-seq data generated herein are also available online via an interactive UCSC genome browser. The different tracks are simultaneously displayed through the UCSC Track Hub functionality. The datasets can be accessed using the following links: Experiment 1: Exponential Phase RIL-seq: https://genome.ucsc.edu/s/michael-gebhardt/PAO1_RIL-seq_ExpPhase. Experiment 2: Stationary Phase RIL-seq: https://genome.ucsc.edu/s/michael-gebhardt/PAO1_RIL-seq_StatPhase.
Supporting Information
References
- 1.Vogel J., Luisi B. F., Hfq and its constellation of RNA. Nat. Rev. Microbiol. 9, 578–589 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Updegrove T. B., Zhang A., Storz G., Hfq: The flexible RNA matchmaker. Curr. Opin. Microbiol. 30, 133–138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kavita K., de Mets F., Gottesman S., New aspects of RNA-based regulation by Hfq and its partner sRNAs. Curr. Opin. Microbiol. 42, 53–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hör J., Matera G., Vogel J., Gottesman S., Storz G., Trans-acting small RNAs and their effects on gene expression in Escherichia coli and Salmonella enterica. EcoSal Plus 9 (2020), 10.1128/ecosalplus.ESP-0030-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill I. T., Tallo T., Dorman M. J., Dove S. L., Loss of RNA chaperone Hfq unveils a toxic pathway in Pseudomonas aeruginosa. J. Bacteriol. 201, e00232-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonnleitner E., et al. , Reduced virulence of a hfq mutant of Pseudomonas aeruginosa O1. Microb. Pathog. 35, 217–228 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Sonnleitner E., Abdou L., Haas D., Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 106, 21866–21871 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonnleitner E., Bläsi U., Regulation of Hfq by the RNA CrcZ in Pseudomonas aeruginosa carbon catabolite repression. PLoS Genet. 10, e1004440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonnleitner E., et al. , Interplay between the catabolite repression control protein Crc, Hfq and RNA in Hfq-dependent translational regulation in Pseudomonas aeruginosa. Nucleic Acids Res. 46, 1470–1485 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malecka E. M., et al. , Stabilization of Hfq-mediated translational repression by the co-repressor Crc in Pseudomonas aeruginosa. Nucleic Acids Res. 49, 7075–7087 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livny J., Brencic A., Lory S., Waldor M. K., Identification of 17 Pseudomonas aeruginosa sRNAs and prediction of sRNA-encoding genes in 10 diverse pathogens using the bioinformatic tool sRNAPredict2. Nucleic Acids Res. 34, 3484–3493 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wurtzel O., et al. , The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog. 8, e1002945 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrara S., et al. , Comparative profiling of Pseudomonas aeruginosa strains reveals differential expression of novel unique and conserved small RNAs. PLoS One 7, e36553 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grmez-Lozano M., Marvig R. L., Molin S., Long K. S., Genome-wide identification of novel small RNAs in Pseudomonas aeruginosa. Environ. Microbiol. 14, 2006–2016 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Pusic P., Sonnleitner E., Bläsi U., Specific and global RNA regulators in Pseudomonas aeruginosa. Int. J. Mol. Sci. 22, 8632 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sonnleitner E., et al. , The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol. 80, 868–885 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Xiao G., et al. , MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol. Microbiol. 62, 1689–1699 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Melamed S., et al. , Global mapping of small RNA-target interactions in bacteria. Mol. Cell 63, 884–897 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kambara T. K., Ramsey K. M., Dove S. L., PErvasive targeting of nascent transcripts by Hfq. Cell Rep. 23, 1543–1552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huber M., et al. , An RNA sponge controls quorum sensing dynamics and biofilm formation in Vibrio cholerae. Nat. Commun. 13, 7585 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matera G., et al. , Global RNA interactome of Salmonella discovers a 5’ UTR sponge for the MicF small RNA that connects membrane permeability to transport capacity. Mol. Cell 82, 629–644.e4 (2022). [DOI] [PubMed] [Google Scholar]
- 22.Faigenbaum-Romm R., et al. , Hierarchy in Hfq chaperon occupancy of small RNA targets plays a major role in their regulation. Cell Rep. 30, 3127–3138.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearl Mizrahi S., et al. , The impact of Hfq-mediated sRNA-mRNA interactome on the virulence of enteropathogenic Escherichia coli. Sci. Adv. 7, eabi8228 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melamed S., Adams P. P., Zhang A., Zhang H., Storz G., RNA-RNA interactomes of ProQ and Hfq reveal overlapping and competing roles. Mol. Cell 77, 411–425.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y. F., et al. , Probing the sRNA regulatory landscape of P. aeruginosa: Post-transcriptional control of determinants of pathogenicity and antibiotic susceptibility. Mol. Microbiol. 106, 919–937 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han K., Tjaden B., Lory S., GRIL-seq provides a method for identifying direct targets of bacterial small regulatory RNA by in vivo proximity ligation. Nat. Microbiol. 2, 16239 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey T. L., et al. , MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iosub I. A., et al. , Hfq CLASH uncovers sRNA-target interaction networks linked to nutrient availability adaptation. Elife 9, e54655 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Law C. O. K., et al. , A small RNA transforms the multidrug resistance of Pseudomonas aeruginosa to drug susceptibility. Mol. Ther. Nucleic Acids 16, 218–228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomason M. K., et al. , A rhlI 5’ UTR-derived sRNA regulates RhlR-dependent quorum sensing in Pseudomonas aeruginosa. mBio 10, e02253-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trouillon J., Han K., Attrée I., Lory S., The core and accessory Hfq interactomes across Pseudomonas aeruginosa lineages. Nat. Commun. 13, 1258 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han K., Lory S., Toward a comprehensive analysis of posttranscriptional regulatory networks: A new tool for the identification of small RNA regulators of specific mRNAs. mBio 12, e03608-20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sykes P. J., Burns G., Menard J., Hatter K., Sokatch J. R., Molecular cloning of genes encoding branched-chain keto acid dehydrogenase of Pseudomonas putida. J. Bacteriol. 169, 1619–1625 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krzywinski M. I., et al. , Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009), 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bar A., Argaman L., Altuvia Y., Margalit H., Prediction of novel bacterial small RNAs from RIL-Seq RNA-RNA interaction data. Front. Microbiol. 12, 635070 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoyos M., Huber M., Förstner K. U., Papenfort K., Gene autoregulation by 3’ UTR-derived bacterial small RNAs. Elife 9, e58836 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin P., et al. , High-throughput screen reveals sRNAs regulating crRNA biogenesis by targeting CRISPR leader to repress Rho termination. Nat. Commun. 10, 3728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massé E., Gottesman S., A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 99, 4620–4625 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papenfort K., Bouvier M., Mika F., Sharma C. M., Vogel J., Evidence for an autonomous 5’ target recognition domain in an Hfq-associated small RNA. Proc. Natl. Acad. Sci. U.S.A. 107, 20435–20440 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mann M., Wright P. R., Backofen R., IntaRNA 2.0: Enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 45, W435–W439 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hussein R., Lim H. N., Disruption of small RNA signaling caused by competition for Hfq. Proc. Natl. Acad. Sci. U.S.A. 108, 1110–1115 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang A., et al. , The OxyS regulatory RNA represses rpoS translation and binds the Hfq (HF-I) protein. EMBO J. 17, 6061–6068 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papenfort K., et al. , Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol. Microbiol. 74, 139–158 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Déziel E., et al. , Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc. Natl. Acad. Sci. U.S.A. 101, 1339–1344 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oglesby A. G., et al. , The influence of iron on Pseudomonas aeruginosa physiology: A regulatory link between iron and quorum sensing. J. Biol. Chem. 283, 15558–15567 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P. P., A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 146, 353–358 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallagher L. A., McKnight S. L., Kuznetsova M. S., Pesci E. C., Manoil C., Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J. Bacteriol. 184, 6472–6480 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wade D. S., et al. , Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J. Bacteriol. 187, 4372–4380 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Djapgne L., et al. , The Pseudomonas aeruginosa PrrF1 and PrrF2 small regulatory RNAs promote 2-alkyl-4-quinolone production through redundant regulation of the antR mRNA. J. Bacteriol. 200, e00704-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sonnleitner E., Prindl K., Blasi U., The Pseudomonas aeruginosa CrcZ RNA interferes with Hfq-mediated riboregulation. PLoS One 12, e0180887 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.West S. E., Sample A. K., Runyen-Janecky L. J., The vfr gene product, required for Pseudomonas aeruginosa exotoxin A and protease production, belongs to the cyclic AMP receptor protein family. J. Bacteriol. 176, 7532–7542 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolfgang M. C., Lee V. T., Gilmore M. E., Lory S., Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev. Cell 4, 253–263 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Ventre I., et al. , Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc. Natl. Acad. Sci. U.S.A. 103, 171–176 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones C. J., et al. , ChIP-Seq and RNA-Seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by Pseudomonas aeruginosa. PLoS Pathog. 10, e1003984 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Azam M. S., Vanderpool C. K., Talk among yourselves: RNA sponges mediate cross talk between functionally related messenger RNAs. EMBO J. 34, 1436–1438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denham E. L., The Sponge RNAs of bacteria–How to find them and their role in regulating the post-transcriptional network. Biochim. Biophys. Acta Gene Regul. Mech. 1863, 194565 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Melamed S., et al. , Mapping the small RNA interactome in bacteria using RIL-seq. Nat. Protoc. 13, 1–33 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Thorvaldsdóttir H., Robinson J. T., Mesirov J. P., Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anders S., Pyl P. T., Huber W., HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blighe K., Rana S., Lewis M., EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling (2022). https://github.com/kevinblighe/EnhancedVolcano. Accessed 2 November 2022.
- 62.Gebhardt M. J., Kambara T. K., Ramsey K. M., Dove S. L., Widespread targeting of nascent transcripts by RsmA in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 117, 10520–10529 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Livak K. J., Schmittgen T. D., Analysis of relative gene expression data using real-time quantitative PCR and the 2-∆∆CT Method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 64.Gebhardt M. J., Melamed S., Dove S. L., PAO1 Hfq-V RIL-seq and RNA-seq. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE216135. Deposited 19 October 2022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Data Availability Statement
Sequencing data have been deposited in the Gene Expression Omnibus (GEO) at NCBI under accession number GSE216135 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE216135) (64). All data generated or analyzed during this study are included in the manuscript and supporting information. The RIL-seq data generated herein are also available online via an interactive UCSC genome browser. The different tracks are simultaneously displayed through the UCSC Track Hub functionality. The datasets can be accessed using the following links: Experiment 1: Exponential Phase RIL-seq: https://genome.ucsc.edu/s/michael-gebhardt/PAO1_RIL-seq_ExpPhase. Experiment 2: Stationary Phase RIL-seq: https://genome.ucsc.edu/s/michael-gebhardt/PAO1_RIL-seq_StatPhase.