

Abstract
Photo‐thermal catalytic CO2 hydrogenation is currently extensively studied as one of the most promising approaches for the conversion of CO2 into value‐added chemicals under mild conditions; however, achieving desirable conversion efficiency and target product selectivity remains challenging. Herein, the fabrication of Ir‐CoO/Al2O3 catalysts derived from Ir/CoAl LDH composites is reported for photo‐thermal CO2 methanation, which consist of Ir‐CoO ensembles as active centers that are evenly anchored on amorphous Al2O3 nanosheets. A CH4 production rate of 128.9 mmol gcat⁻ 1 h⁻1 is achieved at 250 °C under ambient pressure and visible light irradiation, outperforming most reported metal‐based catalysts. Mechanism studies based on density functional theory (DFT) calculations and numerical simulations reveal that the CoO nanoparticles function as photocatalysts to donate electrons for Ir nanoparticles and meanwhile act as “nanoheaters” to effectively elevate the local temperature around Ir active sites, thus promoting the adsorption, activation, and conversion of reactant molecules. In situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) demonstrates that illumination also efficiently boosts the conversion of formate intermediates. The mechanism of dual functions of photothermal semiconductors as photocatalysts for electron donation and as nano‐heaters for local temperature enhancement provides new insight in the exploration for efficient photo‐thermal catalysts.
Keywords: “nanoheaters”, CO2 conversion, CoO carriers, Ir nanoparticles, photo‐thermal catalysis
This work prepares Ir‐CoO/Al2O3 catalysts to realize the highly efficient photo‐thermal catalytic CO2 methanation under mild conditions. The CoO nanoparticles function as photocatalysts to donate electrons for Ir nanoparticles and meanwhile act as “nanoheaters” to effectively elevate the local temperature around Ir active sites, thus promoting the adsorption, activation, and conversion of reactant molecules.
1. Introduction
Conversion of CO2 to useful chemicals offers a renewable approach to ameliorate the greenhouse effect, which emerges as one of the practical strategies to achieve net‐zero emission goals.[ 1 ] Among the reported conversion approaches, reduction of CO2 to CH4 with H2 generated from renewable sources has been intensively explored, as CH4 is a clean fuel with high energy density and can be readily transported via natural gas pipelines.[ 2 ] Compared with the solely light‐driven or the conventional thermal catalytic Sabatier reaction approaches, photo‐thermal catalytic processes where external heat and photo energy are coupled, overcome the low activity in sole solar‐driven catalytic processes and the harsh reaction conditions in thermal catalysis, thereby offering new avenues for efficient CO2 conversion under relatively mild conditions.[ 3 ] In addition to the activation of reactants by external thermal energy, the photo‐excited charge carriers generated on semiconductor materials or the hot electrons produced on the surface of plasmonic metals could be injected into the antibonding orbitals of the reactants to activate the reactant molecules for redox reactions.[ 4 ] It has been demonstrated that hot electrons on surface of Au nanoparticles effectively induced the dissociation of H2, which is a key step in many CO2 reduction reactions.[ 5 ] As such, effective utilization of photo‐induced charge carriers is critical for enhancing the photochemical contribution in photo‐thermal catalytic process.[ 6 ] To date, most reported photo‐thermal catalysts for gas‐phase heterogeneous catalysis are made of metal oxide‐based semiconductors decorated with metal nanoparticles, with inherent limitations including narrow light absorption spectrum and inferior separation efficiency of photo‐induced charge carriers.[ 7 ] To overcome these obstacles, approaches including band gap engineering, introduction of defects, and internal electric field building have been studied to promote charge carrier generation and migration for metal/metal oxide catalysts.[ 8 ]
In addition to the promoted reactants adsorption/activation effect originating from the charge carriers, it is also found that the elevated local temperature around the active sites could dramatically accelerate the reaction kinetics. Recent advances have highlighted that localized photo‐to‐thermal conversion over photothermal materials, triggered via localized surface plasmon resonance (LSPR) or charge carrier relaxation, could dramatically accelerate the catalytic reaction by increasing the temperature of active sites.[ 9 ] Zeng et al. encapsulated Au and Pt nanoparticles into ZIF‐8 to construct Au&Pt@ZIF catalysts, and found that the increased surface temperature of Pt active sites due to the LSPR effect of adjacent Au nanoparticles under light irradiation could enhance the photothermal CO2 hydrogenation for methanol production effectively.[ 10 ] However, most of the investigated photothermal materials are noble metals such as Au and Ag, and their high cost and scarcity present obstacles to industrial application. To this end, the exploitation of inexpensive and naturally‐abundant nanomaterials that could replace noble metals as “nanoheaters” is of great significance. Based on these analyses, we believe that the fabrication of composited catalysts with both high charge carrier separation efficiency and photo‐to‐thermal conversion ability could be a promising approach to achieving efficient photo‐thermal conversion of CO2.
Herein, we report dramatically enhanced photo‐thermal CO2 methanation performances over Ir‐CoO/Al2O3 catalysts derived from Ir/CoAl LDH composites, where CoO carriers play dual critical roles in the hybrid by forming the intimate Ir‐CoO interfaces, expediting photo‐induced charge carrier generation and transportation, and functioning as “nanoheaters” to rapidly elevate the local temperature around the active sites, which benefit the adsorption and activation of CO2 molecules. The Al2O3 nanosheets with high thermal stability could effectively prohibit the agglomeration of tiny Ir‐CoO active centers during the reaction and thus assure the long durability of the composited catalysts. Thanks to the aforementioned merits, the optimized catalysts (0.16%Ir‐CoO/Al2O3) achieved an unprecedented CH4 production rate of 128.9 mmol gcat⁻ 1 h⁻1 with 92% selectivity and exceptional stability. By contrast, Ir/Al2O3 without CoO exhibits much lower catalytic activity under consistent conditions, highlighting the significance of CoO nanoparticles. To reveal the essence of intimate interaction between Ir and CoO, we impregnate Ir nanoparticles on the already prepared CoO/Al2O3 surface, and find that the produced Ir/CoO/Al2O3 catalysts show a CH4 production rate of 32 mmol gcat⁻ 1 h⁻1, which is much lower than that of Ir‐CoO/Al2O3. In situ diffuse reflectance infrared Fourier transform spectroscopy (In situ DRIFTS) measurements reveals that light irradiation brings no changes to the reaction pathways but efficiently boosts the rate‐determining step. To sum up, under light irradiation, the intimate interaction between Ir and CoO brings efficient charge generation and enhanced photothermal effect, which facilitated the adsorption and activation of reactant molecules and intermediate species and resulted in a higher CH4 production rate.
2. Results and Discussion
2.1. Characterizations
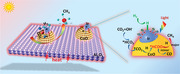
The as‐synthesized Ir nanoparticles (≈1.7 nm) were first supported on the surface of CoAl layered double hydroxide (LDH) to give the Ir/CoAl LDH composites, which were then calcined in a 5 vol.% H2/N2 stream for 2 h to produce the Ir‐CoO/Al2O3 catalysts (Figure 1a). X‐ray diffraction (XRD) patterns confirmed the successful synthesis of Ir/CoAl LDH and the Ir‐CoO/Al2O3 hybrids, and the absence of Ir diffraction peaks in the patterns of composites could be attributed to the low content and high dispersity of the tiny Ir nanoparticles (Figure S2a, Supporting Information).[ 11 ] Transmission electron microscopy (TEM) images manifested that Ir‐CoO/Al2O3 composites inherited the sheet‐like architecture of Ir/CoAl LDH precursors (Figure S4c, Supporting Information) and it should be noticed that the Ir nanoparticles were supported on CoO nanoparticles (≈10 nm) rather than on the porous Al2O3 substrate (Figure 1b,c and Figure S2c, Supporting Information). The corresponding high‐resolution TEM (HRTEM) image showed the lattice fringes with a distance of 0.22 and 0.24 nm which can be indexed to Ir (111) and CoO (111), respectively (Figure 1d). The typical atomic arrangement (ABCABC…) of Ir nanoparticles delineated the face‐centered‐cubic (fcc) structure (inset of Figure 1d). To further corroborate the microstructure, spherical aberration (Cs) corrected high‐angle annular dark‐field scanning TEM (HAADF‐STEM) images and corresponding energy‐dispersive X‐ray (EDX) elemental mappings (Figure 1e–j) were recorded and the results clearly showed that Ir elements were distributed on CoO, which confirmed the close contact between Ir and CoO. The detection of Al and O elements in supports implied that the supports were composed of amorphous Al2O3. In addition, the EDX line scan analysis provided further evidence that the Ir nanoparticles sat on CoO carriers (Figure 1k,l).
Figure 1.
a) Schematic preparation procedures of Ir‐CoO/Al2O3 catalysts. b) TEM image, c) atomic‐resolution image, d) HRTEM image, e) aberration corrected HAADF‐STEM images and f–j) corresponding elemental mappings of 0.16% Ir‐CoO/Al2O3. (l) Compositional line scan profiles of O (red), Al (black), Co (blue) and Ir (green) of 0.16% Ir‐CoO/Al2O3 recorded along the arrow shown in the HAADF‐STEM image (k).
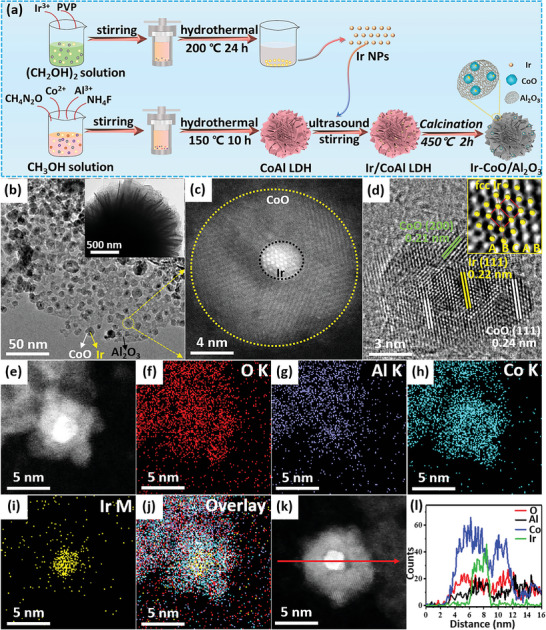
X‐ray photoelectron spectroscopy (XPS) measurements were carried out to probe the surface chemical properties and the interaction between the constituents of catalysts. In the core‐level Ir 4f spectrum of 0.16% Ir/CoAl LDH (Figure 2a), two peaks located at 64.1 eV and 61.1 eV were assigned to Ir 4f5/2 and Ir 4f7/2 orbitals, suggesting that the loaded Ir existed in metallic state.[ 12 ] The Co 2p spectrum could be deconvoluted into four peaks, and the two located at 797.4 eV and 781.3 eV corresponded to Co2+ species, while the other two at 803.5 eV and 786.7 eV could be mainly indexed to the shake‐up satellite peaks of Co2+.[ 11 , 13 ] It is worth noting that the Ir 4f5/2 and Ir 4f7/2 peaks of 0.16%Ir‐CoO/Al2O3 had shifted to lower binding energy regions with respect to 0.16%Ir/CoAl LDH, indicating the higher electron density on Ir species. This phenomenon could be attributed to the electron transfer from CoO to Ir, which agrees well with the fact that the work function of Ir and the electron affinity are CoO of 5.1 eV and 4.43 eV, respectively.[ 14 ] The O 1s peaks of 0.16% Ir/CoAl LDH could be principally deconvoluted into two types of O species, which respectively corresponded to adsorbed oxygen (Oads) at 533.1 eV and hydroxyl species (Ohyd) at 531.8 eV.[ 15 ] Noticeably, two peaks at 530.4 eV and 531.5 eV observed on 0.16% Ir‐CoO/Al2O3 could be ascribed to metal‐oxygen bonds (Co‐O and Al‐O), which was in line with the XRD result and demonstrated the formation of metal oxides.[ 15 ] In addition, XPS spectra under light irradiation (visible irradiation provided with Xenon lamp) were further obtained to study the direction of electron transfer in the Ir‐CoO/Al2O3 composites (Figure S5, Supporting Information). Under light irradiation, the binding energy of Ir 4f shift to lower energy level, while the binding energy of Co 2p shift to higher energy level, suggesting that the photogenerated electrons transfer from CoO to Ir nanoparticles. These results validated the intimate interaction and electron transfer between Ir nanoparticles and CoO in Ir‐CoO/Al2O3 composites.
Figure 2.
a) Ir 4f, b) Co 2p, and c) O 1s core‐level XPS spectra of 0.16% Ir/CoAl LDH and 0.16% Ir‐CoO/Al2O3. d) Temperature changes over 0.16% Ir/Al2O3, CoO/Al2O3 and 0.16% Ir‐CoO/Al2O3 catalysts (diluted in quartz sands) under visible light irradiation (420–780 nm, 2 W cm⁻2) and the IR image of 0.16% Ir‐CoO/Al2O3 under light irradiation (inset). (e–g) Induced electric field distributions and (h–j) temperature distributions on CoO, Ir nanoparticles and Ir‐CoO composites under light irradiation (420 nm, 2 W cm⁻2).
As aforementioned, the photothermal effect has been demonstrated to play significant roles in promoting chemical conversion by effectively increasing the local temperature around the active sites. Therefore, the photo‐to‐thermal conversion ability of Ir/Al2O3, CoO/Al2O3, and Ir‐CoO/Al2O3 catalysts was studied (Figure 2d). Under light irradiation (420‐780 nm, 2 W cm⁻2), the surface temperature of the three catalysts (50 mg of the catalysts were diluted into 1.2 g of quartz sands) increased rapidly and reached the plateaus after ≈3 min. The peaked surface temperature of 0.16% Ir/Al2O3 and CoO/Al2O3 were 53 and 75 °C, respectively. Notably, the highest surface temperature of 0.16% Ir‐CoO/Al2O3 reached 90 °C, which was 37 and 15 °C higher than that of 0.16% Ir/Al2O3 and CoO/Al2O3, respectively. To gain insight into the enhancement mechanism of photothermal effect of Ir‐CoO/Al2O3, the induced electric field distributions of CoO, Ir nanoparticles and Ir‐CoO in the presence of 420 nm illumination were simulated by COMSOL Multiphysics based on finite element methods. As shown in Figure 2e, the sole CoO exhibited extremely weak electric field intensity under light irradiation and Ir nanoparticle showed some electric field strength due to the LSPR effect (Figure 2f).[ 16 ] Surprisingly, when the Ir nanoparticle were in close contact with CoO, an extremely intense localized electromagnetic field was produced at the interface, resulting in near‐field enhancement due to the LSPR effect near the semiconductor surface (Figure 2g).[ 17 ] High field intensity also suggested more charge carrier generation and transportation,[ 18 ] which is consistent with the observation that 0.16% Ir‐CoO/Al2O3 exhibited much higher photocurrent response compared with CoO/Al2O3 (Figure S6, Supporting Information) and this benefits the activation of reactant molecules upon acceptance electrons in the antibonding orbitals.[ 19 ] Moreover, the generated heat arising from the decay process of the enhanced localized electromagnetic field would also result in the dramatic temperature increase around the active sites, which would effectively reduce the activation energy of the reactant molecules as well.[ 20 ] To this end, the steady temperature distributions of related samples were also simulated (Figure 2h–j). The result indicates that under light irradiation, the generated heat originating from relaxation of the induced high electric field gives rise to significant temperature increase at the interfaces and the thermal energy is energetically transferred to the Ir nanoparticles due to the higher thermal conductivity of Ir. Therefore, the CoO nanoparticles at the Ir‐CoO interfaces acted as photocatalysts to provide charge carriers and as “nanoheaters” to increase the local temperature rapidly around the Ir active sites, which led to the efficient activation of reactants.
2.2. Photo‐Thermal Catalytic Performance
The photo‐thermal CO2 hydrogenation reaction was used as the probe reaction to evaluate the catalytic performance of Ir/Al2O3, Ir‐CoO, CoO/Al2O3, Ir‐CoO/Al2O3, and Ir/CoO/Al2O3. To avoid the flow choking caused by the agglomeration of nanocatalysts during reaction process, we packed the reactor with mixture of 50 mg catalysts and 1.2 g quartz sands (≈0.42 mm in diameter). As shown in Figure 3a, the composition of the catalysts and the temperature greatly affected the catalytic performances. All catalysts exhibited growing catalytic activities with the increase of reaction temperature and the catalytic behaviors of different catalysts were compared at 250 °C. The CoO/Al2O3 catalyst showed negligible catalytic activity with a CH4 production rate of 6.85 mmol gcat⁻ 1 h⁻1 at 250 °C under light irradiation, indicating the necessity of catalytically active Ir nanoparticles. Compared with CoO/Al2O3, all the Ir‐CoO/Al2O3 catalysts showed significantly enhanced catalytic activity, and the CH4 production rate displayed a volcano‐like tendency with respect to the Ir content. The highest CH4 production rate of 128.9 mmol gcat⁻ 1 h⁻1 (80.6 mol gIr⁻ 1 h⁻1) was obtained at 250 °C under light irradiation (external heat is about 200 °C, Figure S7, Supporting Information) when catalyzed by 0.16% Ir‐CoO/Al2O3 (Figure 3a), significantly outperforming other reported metal‐based catalysts (Figure 3c and Table S2, Supporting Information). Moreover, it was found that the selective production of CH4 was enhanced with the rise of temperatures and the 0.16% Ir‐CoO/Al2O3 catalyst exhibited a prominent selectivity of 92% to CH4, corresponding to a CO2 conversion of 65.5% at 250 °C (Figure 3b). In comparison with Ir‐CoO/Al2O3, the Ir/Al2O3 and Ir‐CoO catalysts showed substantially lower catalytic activity of 4.51 mmol gcat⁻ 1 h⁻1 and 56.9 mmol gcat⁻ 1 h⁻1, respectively, indicating that the Ir and CoO interaction and the stabilizing effect of Al2O3 supports play significant roles in the CO2 hydrogenation process. This assumption could also be verified by the catalytic performance and structure evolution of 0.16% Ir/CoAl LDH catalyst (Figure S8, Supporting Information). In addition, to further reveal the significance of the Ir and CoO interaction on the catalytic performance, we impregnated Ir nanoparticles on CoO/Al2O3 surface to produce the Ir/CoO/Al2O3 catalyst and compared its activity with that of Ir‐CoO/Al2O3. To our surprise, the Ir/CoO/Al2O3 catalyst exhibited only mediocre CH4 production rate of 32 mmol gcat⁻ 1 h⁻1 under the constant condition (≈1/4 of that obtained over Ir‐CoO/Al2O3, Figure 3a), indicating that the efficient fabrication of the Ir‐CoO interface was critical in achieving excellent catalytic performance. Furthermore, we explored the structure and catalytic performance of the composites with different metal nanoparticles (Ru, Rh, Pt and Pd), and the corresponding XRD patterns, TEM images and catalytic activity are shown in Figure S9 (Supporting Information). These results further revealed that the intimate interaction between metal nanoparticles and CoO were critical for the enhanced catalytic performance.
Figure 3.
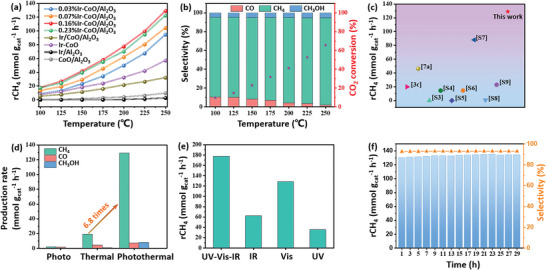
a) Effect of temperature on CH4 yield rate over Ir‐CoO, Ir/Al2O3, CoO/Al2O3, Ir‐CoO/Al2O3 and Ir/CoO/Al2O3 catalysts. b) Product selectivity and CO2 conversion of 0.16%Ir‐CoO/Al2O3 catalyst under light irradiation at different temperatures. c) Comparison of the CH4 production rate of our work with those of representative previous studies (numbers in the figure are their corresponding reference numbers). d) Production rate of CH4, CO and CH3OH of 0.16% Ir‐CoO/Al2O3 catalyst under different conditions. e) CH4 yield rate over 0.16% Ir‐CoO/Al2O3 catalyst under various irradiation conditions with constant light intensity. f) CH4 yield rate and selectivity of 0.16% Ir‐CoO/Al2O3 catalyst during long‐time reaction at 250 °C under light irradiation. Reaction conditions: 100–250 °C, 0.1 MPa, 2 W cm⁻2 in light intensity, GHSV = 24000 cm3 h⁻1 gcat⁻ 1, and H2:CO2 = 4:1.
To investigate the effect of light on catalytic activity, the CH4 production rates under thermal catalytic (in dark), solely light‐driven and photo‐thermal conditions were compared and the results are shown in Figure 3d. At 250 °C in dark, a CH4 production rate of 18.8 mmol gcat⁻ 1 h⁻1 was achieved over 0.16% Ir‐CoO/Al2O3 catalyst and a much lower value of 1.9 mmol gcat⁻ 1 h⁻1 was obtained in the presence of sole light irradiation, suggesting that the photo‐thermal catalytic CO2 hydrogenation was dominantly a thermal‐driven process. Strikingly, ≈sevenfold enhancement of CH4 production rate (128.9 mmol gcat⁻ 1 h⁻1) was observed in photo‐thermal process compared with the thermal catalytic process, demonstrating that light irradiation could significantly enhance the thermal catalytic reaction (Figure 3d and Figure S10a, Supporting Information). The corresponding Arrhenius plot showed one‐stage linear plots with activation energies of 24.77 kJ mol⁻1 in dark and 21.57 kJ mol⁻1 under light irradiation, elucidating that illumination decreased the apparent activation energy (Figure S10b, Supporting Information) and it attributed to the improved activation of CO2 molecules and the formate intermediate. Additionally, to explore the effect of light wavelength ranges on the catalytic activity, a series of tests were performed over 0.16% Ir‐CoO/Al2O3 catalyst at 250 °C under Xe lamp irradiation with different filters (the light intensity was fixed at ≈2W cm⁻2). As shown in Figure 3e, the CH4 production rate under infrared (62.4 mmol gcat⁻ 1 h⁻1) and UV light (35.7 mmol gcat⁻ 1 h⁻1) irradiation was much lower than that under visible light (128.9 mmol gcat⁻ 1 h⁻1) irradiation, indicating that the plasmonic absorption in the visible light region of Ir‐CoO/Al2O3 catalysts greatly contributed to the high activity. To further highlight the significance of light source, the catalytic activity of 0.16% Ir‐CoO/Al2O3 was also evaluated under full spectrum irradiation and a much higher CH4 production rate of 177.9 mmol gcat⁻ 1 h⁻1 was achieved, far exceeding the performance under visible irradiation. This could be attributed to the greatly increased photo‐induced electrons generated due to the strong absorption across the UV‐Vis‐NIR region of Ir‐CoO/Al2O3 catalysts. These results clearly showed that the external heat and light irradiation synergistically enhanced CO2 hydrogenation performance, achieving a much higher CH4 production rate than the sum under sole light irradiation and sole thermal catalytic condition. However, it is still challenging to quantify the individual contribution from light irradiation and external heating process.
To assess the industrial application potential of Ir‐CoO/Al2O3 catalysts, a 30 h long‐term stability test was conducted at 250 °C under light irradiation in the flow reactor. As shown in Figure 3f, no noticeable decrease in CH4 production rate was detected during 30 h of catalytic reactions, indicating that the Ir‐CoO/Al2O3 catalysts were highly stable. Besides, the stability of catalysts in dark (250 °C) was also investigated (Figure S11, Supporting Information). It was found that the production rate and selectivity of CH4 decreased with time, which suggested that the light irradiation benefited the catalyst stability. In addition, XRD pattern, TEM image and XPS spectra of the spent 0.16% Ir‐CoO/Al2O3 were recorded to examine the structure evolution and the results demonstrated the robust stability of the composites in the CO2 hydrogenation reaction process (Figures S12 and S13, Supporting Information). To validate that the product originated from CO2 hydrogenation, we conducted the study with different reaction gases (Figure S14). Obviously, no products were detected in the H2+Ar and CO2+Ar atmosphere, demonstrating that the produced CO, CH4 and CH3OH should be attributed to CO2 hydrogenation process.
2.3. Proposed Reaction Mechanisms of CO2 Methanation
To investigate the reduction behavior of catalysts and the interaction between Ir species and support, H2 temperature‐programmed reduction (H2‐TPR) measurements were carried out. As depicted in Figure 4a, the CoO/Al2O3 showed two broad peaks at 594 and 684 °C, which could be attributed to the reduction of CoO to metallic Co and the reduction of cobalt aluminates species, respectively.[ 21 ] For 0.16% Ir‐CoO/Al2O3, the peak at 340 °C was assigned to the reduction of the IrO2 species that strongly interacted with CoO and the other two peaks at high temperature regions corresponded to the reduction of CoO and cobalt aluminates species.[ 22 ] Notably, the addition of Ir led to the decrease in the reduction temperature of CoO and cobalt aluminates species toward 505 and 623 °C, respectively, which could be ascribed to the efficient dissociation and spillover of H2, elucidating that H2 molecules could be easily dissociated on Ir‐CoO/Al2O3 to participate in the hydrogenation reaction. In addition, the CO2 adsorption properties of CoO/Al2O3 and 0.16% Ir‐CoO/Al2O3 catalysts were studied using CO2 temperature‐programmed desorption (CO2‐TPD) and the results are shown in Figure 4b. The desorption peak at around 473 °C on CoO/Al2O3 catalyst was related to the chemically adsorbed CO2 molecules.[ 15a ] The 0.16% Ir‐CoO/Al2O3 catalyst exhibited three desorption peaks at 233, 360, and 518 °C, which could be assigned to the weakly and strongly adsorbed CO2 molecules on the surface of 0.16% Ir‐CoO/Al2O3 catalyst. Apparently, compared with CoO/Al2O3, the 0.16%Ir‐CoO/Al2O3 catalyst showed stronger CO2 adsorption capacity when the temperature was lower than 400 °C, indicating that the Ir species could promote adsorption of CO2 on the catalyst. Generally, valid CO2 adsorption is one of the prerequisites for high CO2 conversion efficiency.[ 2b ] This may be a credit from the formation of Ir‐CoO interface, which promotes the adsorption of CO2.
Figure 4.
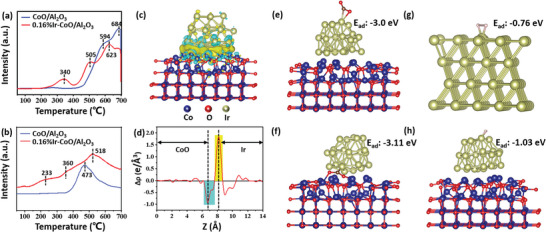
a) H2‐TPR and b) CO2‐TPD profiles of CoO/Al2O3 and 0.16% Ir‐CoO/Al2O3. c) Side view of the charge density difference and d) planar‐averaged electron density difference ∆ρ (z) for Ir‐CoO, and the cyan and yellow areas represent electron depletion and accumulation, respectively. Optimized geometries of CO2 molecules adsorbed on e) Ir surface and at f) Ir‐CoO interface. Optimized geometries of H2 molecules adsorbed on g) Ir and h) Ir‐CoO. The blue, red, yellowish‐green, grey and brown spheres represent Co, O, Ir, H, and C atoms, respectively.
To understand the effect of charge transfer between Ir nanoparticles and CoO on the Sabatier reaction kinetics, density functional theory (DFT) calculations were conducted. As revealed by the charge difference distribution of Ir‐CoO in Figure 4c, electron transfer from CoO to Ir nanoparticles is energetically favored through the intimate interfaces, which is consistent with the XPS results. In addition, the planar‐averaged charge density difference along the Z direction displays the change of charge density, which enables us to observe the surface charge of Ir and CoO directly (Figure 4d). The positive values and negative values represent electron accumulation and depletion, respectively.[ 23 ] This result unveils that the CoO close to the interface is positively charged, whereas the Ir near the interface is negatively charged due to the electron transfer from CoO to Ir.
To further clarify the CO2 adsorption sites on the catalysts surface, the CO2 adsorption behavior on Ir surface and at Ir‐CoO interface is investigated by DFT calculations. The optimized geometries of CO2 adsorbed on Ir surface and at Ir‐CoO interface are exhibited in Figure 4e,f. On the Ir surface, the C‐O bond lengths are increased from 1.16 Å to 1.22 Å and 1.33 Å, and the bond angle decrease from 180° to 142.4°; while at the Ir‐CoO interface, the C‐O bond lengths increase to 1.28 Å and 1.29 Å, and the bond angle decreases to 127.2°, indicating easier dissociation of CO2 molecules at the Ir‐CoO interface.[ 24 ] Besides, the adsorption energy (Eads) of CO2 on the Ir surface and at Ir‐CoO surface are ‐3.0 and ‐3.11 eV, respectively, suggesting that the CO2 is more easily adsorbed at the Ir‐CoO interface. Therefore, the Ir‐CoO interface is considered as the most active adsorption site of CO2 molecules. In addition, to shed light on the effect of the photo‐induced electrons on the adsorption of reactants, the adsorption behavior of H2 molecules on the surface of Ir nanoparticles was also explored by DFT calculations. We simulated the neutral and negatively charged Ir surface by constructing the geometries of Ir(111) and Ir(111)‐CoO, denoted as [Ir(111)] and [Ir(111)]–, respectively. Figure 4g,h shows the optimized geometries of H2 adsorbed [Ir(111)] and [Ir(111)]– surfaces, and the corresponding bond lengths and adsorption energy are also simulated (Table S3, Supporting Information), and again the results indicated that H2 is more easily adsorbed on the [Ir(111)]– surface. The σ*antibonding orbitals of H2 molecules could then accept electrons from the negatively charged Ir surface, and this process would enable the dissociation of adsorbed H2 into H species, which would readily react with the adsorbed CO2 molecules to initiate the hydrogenation process.[ 25 ]
To track the reaction intermediates during the photo‐thermal CO2 methanation process over Ir‐CoO/Al2O3, in situ DRIFTS measurements were carried out. As shown in Figure S15 (Supporting Information), when CO2 and H2 were introduced into the reactor at room temperature, several vibrational peaks could be observed at 1385, 1530 and 1628 cm⁻1. The bands at 1385/1628 and 1529 cm⁻1 were ascribed to the formation of bicarbonate (*HCO3) and carbonates (*CO3) species, respectively,[ 26 ] indicating that CO2 was first adsorbed and existed in the form of *HCO3 and *CO3. Noticeably, these bands of *HCO3 disappeared as the temperature reached 200 °C, whereas vibrations peaks at 1362 and 1583 cm⁻1 corresponding to the formate species (*HCOO) appeared,[ 26 , 27 ] suggesting that *HCO3 was transformed into *HCOO. As the temperature further increased to 300 °C, two new peaks were observed at 1341 and 1427 cm⁻1, which were ascribed to *HCO3 species,[ 28 ] hinting that high temperature was favorable for CO2 activation (Figure 5a). Notably, compared to the spectra at 200 °C, the band at 1583 cm⁻1 of *HCOO exhibited a small shift to the higher frequency region by 15 cm⁻1 at 300 °C. The increase in vibrational frequency indicates that the adsorption of *HCOO species on catalyst surface was weakened.[ 27b ] The band at 2001 cm⁻1 was ascribed to the linear CO adsorption on Ir0,[ 29 ] indicating that *CO species was formed in the process of CO2 methanation. Additionally, a sharp band at 3016 cm⁻1 was observed corresponding to the C‐H stretching vibrations of CH4.[ 30 ] The intensity of these peaks increased over time and peaked after 10 min. Upon light irradiation, the peak intensity of *HCOO species gradually decreased while the CH4 signal gradually increased, indicating that the *HCOO species was the crucial intermediate in the conversion of CO2 to CH4 (Figure 5b). This result revealed that light irradiation could promote the conversion of *HCOO intermediate to CH4, leading to a decreased coverage of *HCOO species on the catalyst surface. In addition, the *CO amount presented a remarkably increasing trend over time, indicating that *CO made no contribution to the generation of CH4. Therefore, the in situ DRIFTS results demonstrated that CO2 methanation on the Ir‐CoO/Al2O3 catalyst followed the formate pathway, which is in line with previous works.[ 26b ]
Figure 5.
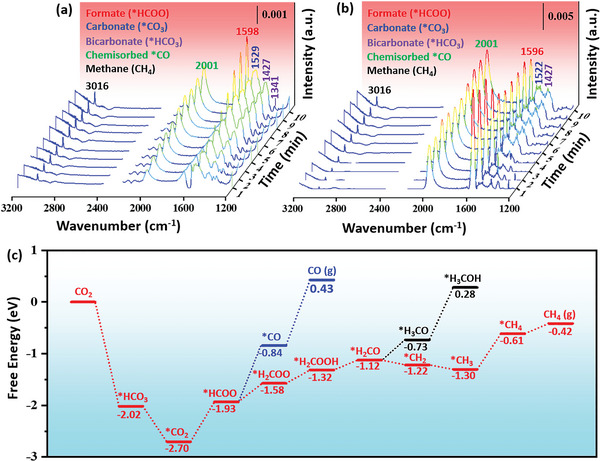
In situ DRIFTS spectra of 0.16% Ir‐CoO/Al2O3 catalyst in dark (a) and under light irradiation (b) at 300 °C for 10 min. c) Relative free energy changes in potential pathways for CO2 hydrogenation to CO, CH4, and CH3OH.
Furthermore, DFT calculations were carried out based on the models of Ir supported on the CoO (200) slabs to illustrate the reaction pathway, and the change of free energy was indicated for each elementary step (Figure 5c). The optimized configurations of the involved intermediates are shown in Figure S16 (Supporting Information). As shown in Figure 5c, the formed *CO2 can be hydrogenated to *HCOO with an energy change of 0.77 eV. The further hydrogenation of *HCOO to *H2COO (energy change: 0.35 eV) is thermodynamically more favorable than the dissociation of *HCOO to *CO (energy change: 1.09 eV), which is consistent with the fact that the selectivity of CO is relatively low. The generated *H2COO is further hydrogenated to *H2COOH, with an energy change of 0.26 eV. Afterward, the *H2COOH dissociates into *H2CO and *OH via the direct C‐O bond cleavage pathway. Notably, the further dissociation of *H2CO to *CH2 is evidently more favorable than the hydrogenation of *H2CO to *H3CO, and therefore, *CH2 is hydrogenated to CH4. Namely, the *H2CO species favor dissociation energetically rather than hydrogenation, resulting in lower CH3OH selectivity and higher selectivity for the targeted CH4.
Based on the detected intermediate species, the possible reaction mechanisms of photo‐thermal CO2 methanation on Ir‐CoO/Al2O3 catalysts are proposed (Figure 6 ). First, adsorbed CO2 is converted into *HCO3 species, which then transform into the CO2*.[ 26b ] Subsequently, *CO2 accepts the H atoms generated via the dissociation of H2 on Ir nanoparticles to form *HCOO intermediates. According to the results of DFT calculations, the *HCOO species could be hydrogenated to *HCOOH and further hydrogenated to *H2COOH. Then *H2COOH undergoes consecutive decomposition to produce *CH2, and *CH2 was eventually hydrogenated to CH4. In addition, a small portion of the *HCOO intermediates disassociate into *CO and H2O, and the *CO is not converted to CH4, but desorbed to produce CO gas. Furthermore, light irradiation does not change the reaction pathway, but promotes the adsorption activation and conversion of *HCOO intermediates, which is the rate‐determining step for the CO2 hydrogenation to CH4, thus effectively facilitating the CH4 formation. Combined with the aforementioned analysis based on DFT calculations and finite element methods based numerical simulations, in the thermal‐driven CO2 hydrogenation process, light irradiation results in the improved adsorption and activation of the reactants molecules by inducing the efficient utilization of charge carriers and enhanced photothermal effect, and simultaneously promotes the conversion of *HCOO intermediates to CH4, thus synergistically boosting the overall CO2 methanation efficiency in the global heating reaction system.
Figure 6.
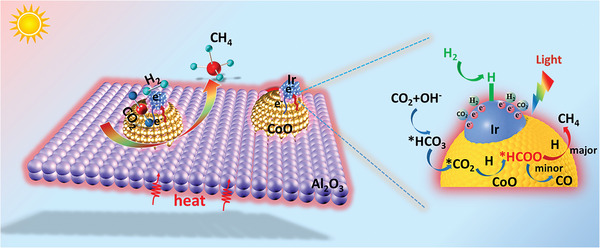
Proposed reaction mechanism for photo‐thermal catalytic Sabatier reaction process over Ir‐CoO/Al2O3.
3. Conclusion
In summary, we have prepared Ir‐CoO/Al2O3 catalysts to realize the highly efficient photo‐thermal catalytic CO2 methanation under mild conditions. Our comprehensive investigation demonstrated that the significantly enhanced catalytic performance was attributed to the intimate interaction between Ir and CoO and the stabilizing effect of the Al2O3 supports. The formation of Ir‐CoO interfaces resulted in strong localized electric field to accelerate charge carrier generation and migration under illumination. DFT calculations revealed that the electron transfer from CoO to Ir nanoparticles favored the H2 dissociation. In addition, CoO also acted as “nanoheaters” to elevate the local temperature of active sites to accelerate the reaction kinetics, which further enhanced the catalytic performance. In situ DRIFTS confirmed that the CO2 hydrogenation on Ir‐CoO/Al2O3 catalysts followed the formate pathway and light irradiation efficiently boosted the activation and conversion of *HCOO intermediates. Owing to these contributions from light irradiation, a high CH4 production rate of 128.9 mmol gcat⁻ 1 h⁻1 (80.6 mol gIr⁻ 1 h⁻1) was achieved over 0.16% Ir‐CoO/Al2O3 catalyst at 250 °C under ambient pressure, outperforming most of the reported metal‐based catalysts. This work provides in‐depth understanding on the synergistic effects in photo‐thermal catalytic process and proposes a novel catalyst for the efficient conversion of CO2 under mild conditions.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 21902085 and 52172213) and Natural Science and Development Foundation of Shenzhen (JCYJ20190807093205660).
Tang Y., Zhao T., Han H., Yang Z., Liu J., Wen X., Wang F., Ir‐CoO Active Centers Supported on Porous Al2O3 Nanosheets as Efficient and Durable Photo‐Thermal Catalysts for CO2 Conversion. Adv. Sci. 2023, 10, 2300122. 10.1002/advs.202300122
Contributor Information
Xiaodong Wen, Email: wxd@sxicc.ac.cn.
Fenglong Wang, Email: fenglong.wang@sdu.edu.cn.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.a) Bushuyev O. S., De Luna P., Dinh C. T., Tao L., Saur G., van de Lagemaat J., Kelley S. O., Sargent E. H., Joule 2018, 2, 825; [Google Scholar]; b) Ye R.‐P., Ding J., Gong W., Argyle M. D., Zhong Q., Wang Y., Russell C. K., Xu Z., Russell A. G., Li Q., Fan M., Yao Y.‐G., Nat. Commun. 2019, 10, 5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Yan X., Sun W., Fan L., Duchesne P. N., Wang W., Kübel C., Wang D., Kumar S. G. H., Li Y. F., Tavasoli A., Wood T. E., Hung D. L. H., Wan L., Wang L., Song R., Guo J., Gourevich I., Jelle A. A., Lu J., Li R., Hatton B. D., Ozin G. A., Nat. Commun. 2019, 10, 2608; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Y., Zhang Y., Fan G., Song L., Jia G., Huang H., Ouyang S., Ye J., Li Z., Zou Z., Joule 2021, 5, 3235. [Google Scholar]
- 3.a) Yang Z., Qi Y., Wang F., Han Z., Jiang Y., Han H., Liu J., Zhang X., Ong W. J., J. Mater. Chem. A 2020, 8, 24868; [Google Scholar]; b) Iglesias‐Juez A., Coronado J. M., Chem 2018, 4, 1490; [Google Scholar]; c) Tang Y., Yang Z., Guo C., Han H., Jiang Y., Wang Z., Liu J., Wu L., Wang F., J. Mater. Chem. A 2022, 10, 12157. [Google Scholar]
- 4. Fang S., Hu Y. H., Chem. Soc. Rev. 2022, 51, 3609. [DOI] [PubMed] [Google Scholar]
- 5. Mukherjee S., Libisch F., Large N., Neumann O., Brown L. V., Cheng J., Lassiter J. B., Carter E. A., Nordlander P., Halas N. J., Nano Lett. 2013, 13, 240. [DOI] [PubMed] [Google Scholar]
- 6. Ghoussoub M., Xia M., Duchesne P. N., Segal D., Ozin G., Energy Environ. Sci. 2019, 12, 1122. [Google Scholar]
- 7.a) Wang C., Fang S., Xie S., Zheng Y., Hu Y. H., J. Mater. Chem. A 2020, 8, 7390; [Google Scholar]; b) Quan F., Zhan G., Mao C., Ai Z., Jia F., Zhang L., Gu H., Liu S., Catal. Sci. Technol. 2018, 8, 6503. [Google Scholar]
- 8.a) Li Q., Ouyang Y., Li H., Wang L., Zeng J., Angew. Chem., Int. Ed. 2022, 61, e202108069; [DOI] [PubMed] [Google Scholar]; b) Meng L., Chen Z., Ma Z., He S., Hou Y., Li H.‐H., Yuan R., Huang X.‐H., Wang X., Wang X., Long J., Energy Environ. Sci. 2018, 11, 294. [Google Scholar]
- 9.a) Shao T., Wang X., Dong H., Liu S., Duan D., Li Y., Song P., Jiang H., Hou Z., Gao C., Xiong Y., Adv. Mater. 2022, 34, 2202367; [DOI] [PubMed] [Google Scholar]; b) Luo S., Ren X., Lin H., Song H., Ye J., Chem. Sci. 2021, 12, 5701; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang X., Fan Y., You E., Li Z., Dong Y., Chen L., Yang Y., Xie Z., Kuang Q., Zheng L., Nano Energy 2021, 84, 105950. [Google Scholar]
- 10. Zhang W., Wang L., Wang K., Khan M. U., Wang M., Li H., Zeng J., Small 2017, 13, 1602583. [DOI] [PubMed] [Google Scholar]
- 11. Liu Y., Yu C., Che H., Guo Z., Mu J., Zhang X., Liu A., J. Colloid Interface Sci. 2021, 581, 485. [DOI] [PubMed] [Google Scholar]
- 12.a) Zhang T., Li S.‐C., Zhu W., Ke J., Yu J.‐W., Zhang Z.‐P., Dai L.‐X., Gu J., Zhang Y.‐W., Surf. Sci. 2016, 648, 319; [Google Scholar]; b) Liu Q., Liu Q., Chen Y., Li Y., Su H., Liu Q., Li G., Chin. Chem. Lett. 2022, 33, 374. [Google Scholar]
- 13. Ge B., Tang S., Zhang H., Li W., Wang M., Ren G., Zhang Z., J. Mater. Chem. A 2021, 9, 7967. [Google Scholar]
- 14.a) Lai Y., Zhang Z., Zhang Z., Tan Y., Yu L., Wu W., Wang Z., Jiang T., Gao S., Cheng N., Chem. Eng. J. 2022, 435, 135102; [Google Scholar]; b) Lomocso T. L., Baranova E. A., Electrochim. Acta 2011, 56, 8551; [Google Scholar]; c) Guo P., Xiong Z., Yuan S., Xie K., Wang H., Gao Y., Chem. Eng. J. 2021, 420, 130372. [Google Scholar]
- 15.a) Lian Y., Fang T., Zhang Y., Liu B., Li J., J. Catal. 2019, 379, 46; [Google Scholar]; b) Jing C., Liu X., Yao H., Yan P., Zhao G., Bai X., Dong B., Dong F., Li S., Zhang Y., CrystEngComm 2019, 21, 4934; [Google Scholar]; c) Li F.‐T., Zhao Y., Wang Q., Wang X.‐J., Hao Y.‐J., Liu R.‐H., Zhao D., J. Hazard. Mater. 2015, 283, 371; [DOI] [PubMed] [Google Scholar]; d) Okla M. K., Kokilavani S., Mohebaldin A., Thomas A. M., Soufan W., Abdel‐Maksoud M. A., Abdelgawad H., Raju L. L., khan S. S., Colloid. Surf., A 2022, 640, 128318. [Google Scholar]
- 16. Han H., Xu X., Kan H., Tang Y., Liu C., Wen H., Wu L., Jiang Y., Wang Z., Liu J., Wang F., J. Colloid Interface Sci. 2022, 616, 304. [DOI] [PubMed] [Google Scholar]
- 17. Yuan J., Yi X., Tang Y., Liu M., Liu C., Adv. Funct. Mater. 2020, 30, 1906983. [Google Scholar]
- 18. Zou N., Chen G., Mao X., Shen H., Choudhary E., Zhou X., Chen P., ACS Nano 2018, 12, 5570. [DOI] [PubMed] [Google Scholar]
- 19. Ling L.‐L., Yang W., Yan P., Wang M., Jiang H.‐L., Angew. Chem., Int. Ed. 2022, 61, e202116396. [DOI] [PubMed] [Google Scholar]
- 20. Gao L., Cui X., Wang Z., Sewell C. D., Li Z., Liang S., Zhang M., Li J., Hu Y., Lin Z., Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2023421118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tavasoli A., Abbaslou R. M. M., Trepanier M., Dalai A. K., Appl. Catal., A 2008, 345, 134. [Google Scholar]
- 22. Lin W., Cheng H., He L., Yu Y., Zhao F., J. Catal. 2013, 303, 110. [Google Scholar]
- 23. Liu J., J. Phys. Chem. C 2015, 119, 28417. [Google Scholar]
- 24. Zhang J., Yang Y., Liu J., Xiong B., Appl. Surf. Sci. 2021, 558, 149866. [Google Scholar]
- 25. Brien P. G. O., Ghuman K. K., Jelle A. A., Sandhel A., Wood T. E., Loh J. Y. Y., Jia J., Perovic D., Singh C. V., Kherani N. P., Mims C. A., Ozin G. A., Environ Sci 2018, 11, 3443. [Google Scholar]
- 26.a) Sheng Q., Ye R.‐P., Gong W., Shi X., Xu B., Argyle M., Adidharma H., Fan M., J. Environ. Sci. 2020, 92, 106; [DOI] [PubMed] [Google Scholar]; b) Xu X., Tong Y., Huang J., Zhu J., Fang X., Xu J., Wang X., Fuel 2021, 283, 118867. [Google Scholar]
- 27.a) Zhu J., Zhang G., Li W., Zhang X., Ding F., Song C., Guo X., ACS Catal. 2020, 10, 7424; [Google Scholar]; b) Ren P., Tu W., Wang C., Cheng S., Liu W., Zhang Z., Tian Y., Han Y.‐F., Appl. Catal., B 2022, 305, 121016. [Google Scholar]
- 28. Zhang G., Fan G., Yang L., Li F., Appl. Catal. A 2020, 605, 117805. [Google Scholar]
- 29.a) Ziaei Azad H., Semagina N., ChemCatChem 2014, 6, 885; [Google Scholar]; b) Cheng D., Wang M., Tang L., Gao Z., Qin X., Gao Y., Xiao D., Zhou W., Ma D., Angew. Chem., Int. Ed. 2022, 61, e202202654. [DOI] [PubMed] [Google Scholar]
- 30. Yang C., Liu S., Wang Y., Song J., Wang G., Wang S., Zhao Z.‐J., Mu R., Gong J., Angew. Chem., Int. Ed. 2019, 58, 11242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.