

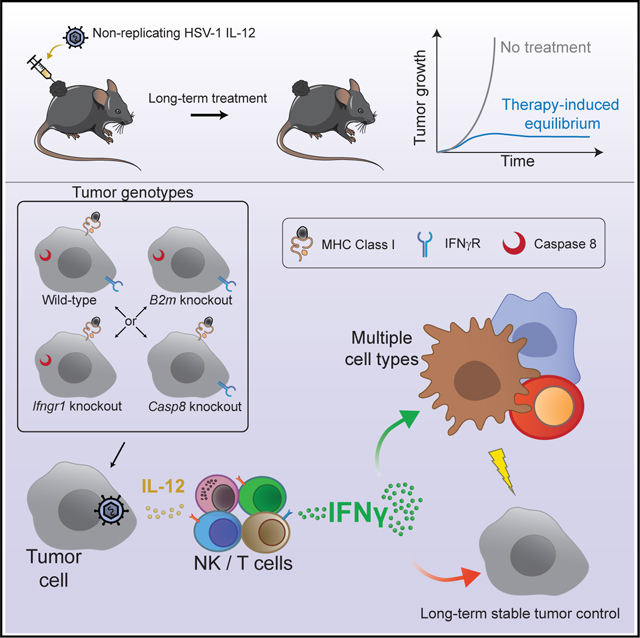
SUMMARY
Tumors in immune equilibrium are held in balance between outgrowth and destruction by the immune system. The equilibrium phase defines the duration of clinical remission and stable disease, and escape from equilibrium remains a major clinical problem. Using a non-replicating HSV-1 vector expressing interleukin-12 (d106S-IL12), we developed a mouse model of therapy-induced immune equilibrium, a phenomenon previously seen only in humans. This immune equilibrium was centrally reliant on interferon-γ (IFNγ). CD8+ T cell direct recognition of MHC class I, perforin/granzyme-mediated cytotoxicity, and extrinsic death receptor signaling such as Fas/FasL were all individually dispensable for equilibrium. IFNγ was critically important and played redundant roles in host and tumor cells such that IFNγ sensing in either compartment was sufficient for immune equilibrium. We propose that these redundant mechanisms of action are integrated by IFNγ to protect from oncogenic or chronic viral threats and establish IFNγ as a central node in therapy-induced immune equilibrium.
In brief
Walsh et al. develop a mouse model of therapy-induced cancer immune equilibrium through injection of a non-replicating interleukin-12-expressing virus. Although interferon-γ (IFNγ) was essential for establishment and maintenance of equilibrium, tumor or host sensing of IFNγ was redundant and tumors were controlled without direct T cell cytotoxicity.
Graphical Abstract
INTRODUCTION
Immunotherapies harness the ability of the immune system to recognize and destroy cancer cells. Cytokine-based therapies, adoptive cell therapies, and antibody blockade of negative regulatory pathways such as cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed death (PD)-1/L1 have demonstrated the effectiveness of immune-stimulating agents for cancer treatment.1 In both humans and mice, the immune system also surveys for nascent cancer development, although the extent to which immune surveillance happens in the absence of therapy is difficult to quantify. Immune equilibrium, or a state of balance between the immune system and clinically undetected or stable tumors, occurs as demonstrated by elegantly designed mouse models and the observation of very long latency periods for certain cancer types such as melanoma.2,3 Many immunotherapies can induce an equilibrium-like state in patients with tumors held in check until immune escape promotes tumor progression.4 Among 105 melanoma patients from the KEYNOTE-001 trial initially classified as having a complete response following anti-PD1 treatment, long-term follow-up revealed that 12% of these complete responders eventually had detectable disease, suggesting their tumors had been held in clinically undetectable equilibrium.5 This equilibrium state in patients is poorly understood and remains a significant clinical barrier to successfully curing patients.
Interferon-γ (IFNγ) signaling is important for the initial response to checkpoint blockade, as highlighted by several CRISPR screens6–8 and evaluation of patients receiving CTLA-4 blockade who displayed primary or acquired resistance.9,10 These studies have found various mutations that inactivate IFNγ sensing in the tumor cells and tend to promote resistance to immunotherapy. In this early response, IFNγ is thought to slow tumor growth and improve CD8+ T cell-directed responses via enhanced antigen presentation.11 IFNγ can also drive regulatory T cell (Treg) fragility in which Foxp3+ Tregs become less suppressive and can secrete IFNγ themselves, allowing for better antitumor responses.12 Additionally, IFNγ can act on the tumor stromal cells, inducing regression of the vasculature supporting tumors, ultimately causing tumor shrinkage.13,14 However, the long-term role of IFNγ in immune equilibrium is unclear. Biopsies cannot be obtained from patients with clinically undetectable disease, and we lack a mouse model of therapy-induced equilibrium. IFNγ has several negative feedback mechanisms, and chronic IFNγ signaling can promote tumor outgrowth.15,16 It is therefore unknown whether long-term exposure to IFNγ is beneficial or deleterious for tumor control.
Viruses can provide an alternative method of immunotherapy. Viral nucleic acids or glycoproteins can serve as pathogen-associated molecular patterns that engage Toll-like receptors (TLRs), such as TLR2 and TLR9,17–19 or other pattern recognition receptors (PRRs),20 to generate an inflammatory response and stimulate maturation of dendritic cells (DCs) to efficiently prime a T cell response against the tumor. Recent advances in genetic engineering of viruses have improved their therapeutic potential, and in 2015 the Food and Drug Administration approved the use of the first oncolytic virus, talimogene laherparepvec (T-VEC), a replication-competent herpes simplex virus 1 (HSV-1) recombinant encoding granulocyte-macrophage colony-stimulating factor (GM-CSF), for the local treatment of melanoma metastases.21
Interleukin-12 (IL-12) is a potent cytokine with multiple functions, including the abilities to stimulate IFNγ production and enhance the growth and cytotoxicity of natural killer (NK), CD8, and CD4 T cells.22 IL-12 also has antiangiogenic properties. Methods of introducing IL-12 into the tumor microenvironment have included recombinant protein injection,23,24 plasmid electroporation,25,26 and microspheres,27 as well as oncolytic virus delivery through both replication-competent28–34 and defective35,36 viral vectors. However, owing to the pleiotropic effects of IL-12, dose-limiting toxicities are a substantial barrier to effective treatment in patients.37 In addition, the standard oncolytic viruses may generate illness in some patients and cannot be given to immunosuppressed patients because they are replication competent, with toxicity increasing further when used as part of a combination therapy.38–40
To improve the safety profile of such oncolytic viruses, we took advantage of the HSV-1 d106S vaccine vector, which unlike T-VEC and most other oncolytic viruses is replication defective.41–43 This virus expresses a GFP viral transgene, which can be replaced with any protein of interest, such as a tumor-specific antigen,44 therapeutic checkpoint inhibitor antibody, or cytokine. We chose to use d106S as a vector to induce a large burst of IL-12 locally within the tumor environment. We discovered that local delivery of IL-12 with the d106S-IL12 vector led to long-term stable equilibrium of B16 melanoma tumors, lasting over 120 days whereby the mice neither clear nor succumb to their tumors. We identify IFNγ as the central factor required for establishment and maintenance of therapy-induced equilibrium and demonstrate redundant mechanisms of action for this critical cytokine.
RESULTS
d106S-IL12 is a model for therapeutic induction of immune equilibrium
Although human cells are readily infectable by HSV-1, B16 murine melanoma cells are non-permissive due to lack of necessary entry receptors (Figure S1A). We therefore transduced B16 melanoma cells with a lentivirus encoding human nectin-1,45 a known HSV-1 entry receptor.46 Whereas B16WT cells could not be infected with d106S-GFP, B16Nectin1 cells were readily infected with the virus; titration of d106S-GFP virus on B16Nectin1 alongside a panel of five human melanoma cell lines showed comparable levels of infection (Figure S1B). Thus, B16Nectin1 cells faithfully model d106S entry into human melanoma cells.
We chose IL-12 as a cargo for local virotherapy given the known role of this cytokine in orchestrating T helper 1 (Th1) responses and the limitations associated with its systemic delivery. We inserted an IL-12 fusion gene cassette in place of GFP to generate the d106S-IL12 virus (Figure S1C) and measured IL-12 (p40) concentration in infected tumor cell supernatants by ELISA. Even at the lowest MOI tested, 0.1 plaque-forming units (PFU)/cell, d106S-IL12 produced abundant levels of cytokine: 9,000 pg per 105 cells over 24 h (Figure S1D). We tested whether d106S-IL12 could robustly induce IL-12 secretion in vivo by inoculating mice with a subcutaneous B16Nectin1 tumor and injecting intratumorally with PBS, d106S, or d106S-IL12 every 3 days starting at day 7. Tumors were collected at day 10 or day 17 post challenge and showed robust levels of IL-12 present (Figure S1E). Our HSV-1 d106S expresses the single-chain version of IL-12, allowing for robust and stable secretion of both subunits, without antagonism from the p40 subunit expression alone.47–49 In addition, the levels of IL-12 secretion that we achieved with d106S-IL12 infection were higher than with previously reported IL-12-expressing oncolytic viruses,28–30,35 demonstrating the robust gene expression capabilities of this vector.
We expected that the viral vector itself would induce an IFN response,50 although this response could be dampened through expression of the viral ICP0 protein.51,52 To examine this, we infected B16Nectin1 cells with d106S or d106S-IL12 virus and co-cultured these cells with murine bone marrow dendritic cells (BMDCs), or separately stimulated BMDCs with virus alone. We measured mRNA expression with a panel of inflammatory genes including IFN-stimulated genes (ISGs) and inflammatory nuclear factor-κB genes. As previously shown in other cell types,51,52 the d106S infection resulted in a minor type I IFN response within the melanoma cells, as shown by IFN-β(IFNb1) expression (Figure S1F). However, co-culture of infected B16 with DCs resulted in a more robust IFN response from DCs (Figures S1F–S1H), which are themselves non-permissive to infection53 but can robustly induce IFN production via innate sensors.
To examine whether the in vitro inflammatory response induced by d106S had an in vivo effect on tumor growth, we challenged mice with B16Nectin1 tumors by subcutaneous injection into the flank. After 5 days of tumor growth, mice were randomized and injected intratumorally with either PBS or d106S (GFP) every 3 days, and tumor growth and survival were monitored. The injection of d106S virus slowed tumor growth (Figure 1A), indicating that some aspects of the viral vector, including the IFN response, were sufficient to delay tumor growth, albeit modestly.
Figure 1. A mouse model of therapy-induced immune equilibrium.

(A) C57BL/6J mice were challenged with 5 × 105 B16Nectin1 cells subcutaneously in the flank. Following 5 days of tumor growth, mice were randomized into treatment groups (N = 6 per group) and received intratumoral injection of 30 μL of either PBS or d106S (1.5 × 107 PFU) every 3 days.
(B and C) Mice were challenged with a primary B16Nectin1 tumor (N = 10 per group) followed by a secondary, contralateral tumor 4 days later. Seven days after initial challenge, only the primary tumor was injected every 3 days with PBS, d106S, or d106S-IL12 for a total of 5 injections.
(D and E) Wild-type C57BL/6J (D) or RAG2−/− mice (E) were inoculated with unilateral B16Nectin1 tumors and injected intratumorally every 3 days with PBS, d106S (wild-type mice only), or d106S-IL12 for a total of 5 injections (N = 5 per group) starting on day 7 for a total of 5 injections. Weight change was normalized to immediately before treatment began. Arrows indicate treatment; Ф indicates blood draw; ⊥ indicates RAG2−/− sacrifice (2 mice) for tumor DNA extraction.
(F) qPCR for viral DNA (ICP8) from DNA extracted from peripheral blood draw (day 22 surviving mice) and tumor (day 25 RAG2−/−). Untreated mice also bore tumors but did not receive treatment. Spike-in control received 8 × 105 PFU of virus into untreated blood. Dotted line indicates limit of detection based on untreated samples.
(G) Wild-type 6694c2 pancreatic cancer cells were infected with varying MOIs of d106S (GFP) and infection measured by flow cytometry (n = 4).
(H) Mice were challenged subcutaneously with 2 × 105 6694c2 cells and treated every 3 days starting on day 7 (N = 5 PBS, N = 5 d106S-IL12).
Values are mean ± SEM. Survival groups were compared using a Bonferroni-corrected log-rank test. *p < 0.05; **p < 0.01.
To test the efficacy of d106S-IL12 therapy, we inoculated mice with a primary tumor and 4 days later inoculated a secondary tumor on the contralateral flank. One week after primary tumor challenge, mice were injected with either PBS, d106S, or d106S-IL12 intratumorally into only the primary tumor. Growth of the primary, treated tumor was reduced slightly with d106S, as seen previously (Figure 1B) and reduced more with d106S-IL12 (Figure 1B). However, d106S failed to reduce growth of the secondary, untreated tumor, while d106S-IL12 partially reduced growth of the distal tumor (Figure 1C), suggesting that d106S acted locally while IL-12 induced a systemic immune response.
Not only did the tumors directly injected with d106S-IL12 have reduced growth compared with d106S but their size remained relatively fixed after several doses, suggesting establishment of equilibrium. However, because the response of contralateral, uninjected tumors was insufficient to stably control tumors, we wished to test whether this equilibrium was due to innate immune activation at the site of injection or induction of adaptive immunity. We inoculated wild-type C57BL/6 and RAG2−/− immunodeficient mice with tumors. In wild-type mice, d106S-IL12 again led to a slowing of tumor growth and prolonged survival with stable tumor masses (Figure 1D). However, in RAG2−/− mice, d106S-IL12 led to only a partial slowing of tumor growth and no equilibrium (Figure 1E), suggesting that adaptive immunity was necessary to stably control tumors. Weight loss was monitored as a proxy for toxicity, as systemic IL-12 in humans and mice is limited by severe toxicity;24,37 no major weight loss was seen in either wild-type or RAG2−/− mice (Figures 1D and 1E). All remaining mice were bled at day 22; DNA was extracted from the peripheral blood and from RAG2−/− tumors at 24 h post viral injection to measure levels of circulating free virus by qPCR for the viral gene ICP8 gene. The level of viral DNA in the blood was at undetectable levels compared with the levels found in the tumor (Figure 1F). In total, these results showed a focused local infection, non-toxicity of the d106S vector even in immunocompromised hosts, and the necessity for adaptive immunity to induce equilibrium in response to d106S-IL12.
We then tested whether equilibrium could be established in another model using the pancreatic cancer clone 6694c2,54 which is also known to be resistant to checkpoint blockade.54,55 Without genetic perturbation, these 6694c2 cells were naturally permissive to HSV infection, unlike native B16 (Figure S1A), although less well infected than B16Nectin1 (Figures 1G and S1B). Even with lower levels of HSV infection, d106S-IL12 could readily control cc6942 tumors and ultimately establish equilibrium (Figure 1H). This indicated that equilibrium establishment using d106S-IL12 could occur in multiple murine tumor models.
Tumor-intrinsic resistance does not facilitate outgrowth in response to d106S-IL12
To better understand the mechanism of equilibrium, we wanted to determine the requirement for ongoing treatment. We tested whether three total doses or continuous treatment of mice every 3 days could induce stable equilibrium. Despite an initial reduction in B16 tumor size with only three treatments, all mice with treatment cessation failed to clear their tumors; their tumors grew out and they did not survive for as long as mice receiving continuous d106S-IL12 therapy (Figure 2A). However, even with continuous dosing of mice, all mice failed to fully clear their tumors and, instead, the majority entered an equilibrium phase (Figures 2A and 2B). Therefore, we concluded that continuous therapy was necessary to keep tumors in immune equilibrium. However, immune equilibrium was not established in all tumors, as some did not appear to respond to therapy and continued to grow out during the treatment window (Figure 2B), suggesting a means of resistance.
Figure 2. Ongoing treatment with d106S-IL12 generates immune equilibrium but not intrinsic resistance.
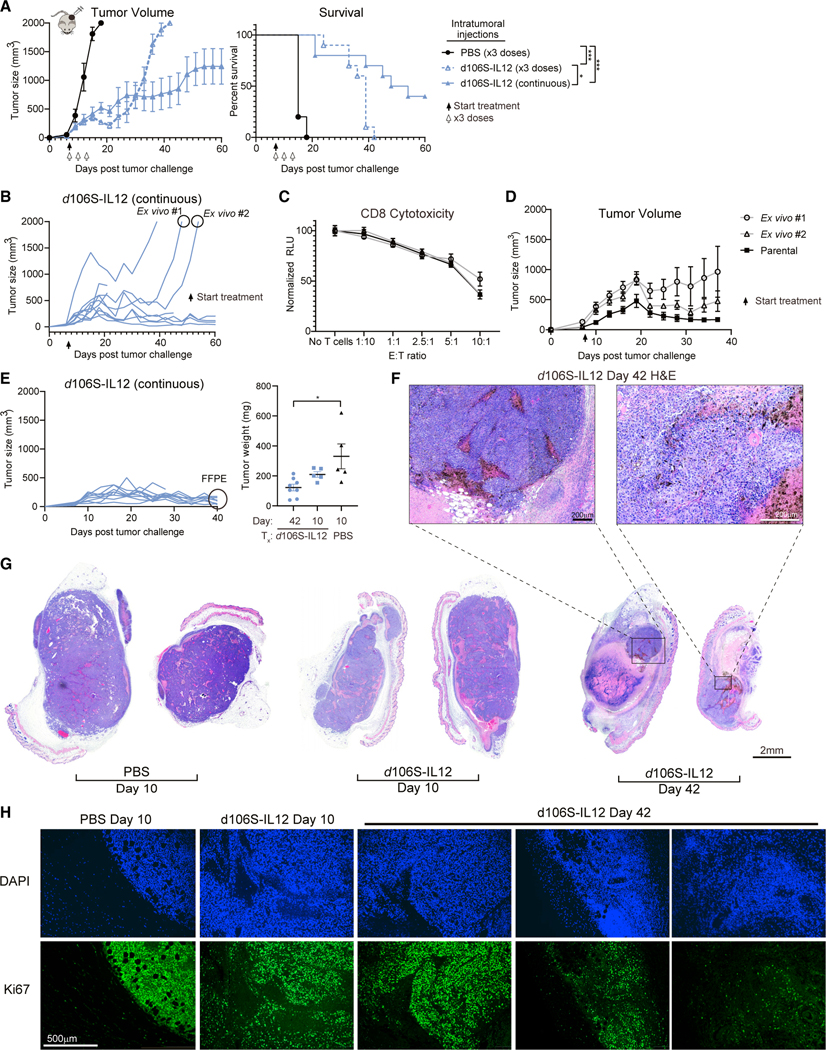
(A) Mice were challenged with unilateral B16Nectin1 tumors (5 × 105 cells) and received either continuous intratumoral dosing every 3 days or only 3 total doses as indicated starting at day 7 (N = 5 mice for PBS, N = 10 mice per d106S-IL12 group). Solid arrow indicates start of continuous dosing; white arrows indicate 3 doses.
(B) Individual growth curves from (A); 2 outgrowing tumors were collected for ex vivo culture.
(C) Activated CD8+ TRP1high cells were co-cultured for 24 h with IFNγ-stimulated ex vivo or parental B16Nectin1 cells, and cytotoxicity was measured by CellTiter-Glo. See also Figure S2. Effector/target (E:T) ratios are shown.
(D) Mice were challenged with 5 × 105 cells from either ex vivo cultures or B16Nectin1 parental line (N = 5 per tumor type). Continuous treatment with d106SIL12 began at day 7, as indicated by the arrow.
(E) B16Nectin1 tumors were treated continuously with d106S-IL12 as before and collected at day 42 post tumor growth (N = 8) or separately treated once with PBS or d106S-IL12 and collected at day 10 (N = 5). Tumor weights are shown. Tumors were formalin fixed and paraffin embedded (FFPE) and stained with hematoxylin and eosin (H&E).
(F and G) Representative H&E of whole tumor slices (G) or zoomed in on day 42 tumors (F).
(H) FFPE sections were also stained with anti-Ki67 and DAPI and imaged by immunofluorescence. See also Figure S3.
Values are mean ± SEM. Tumor weights were compared with a one-way ANOVA and Tukey’s test; survival was compared using a Bonferroni-corrected log-rank test. *p < 0.05.
To determine whether resistance was due to mutations in the tumor permitting immune escape or other mechanisms, we cultured two of these outgrowing tumors ex vivo to generate cell lines (Figure 2B). One possible mechanism of resistance could be loss of nectin-1, the HSV entry receptor, therefore preventing viral transduction and IL-12 production. We determined by flow cytometry that nectin-1 levels were modestly reduced, resulting in a 5%–50% reduction in infectivity (Figures S2A and S2B). However, given the high expression of IL-12 from this vector (Figures S1C and S1D), this reduction in infectivity is unlikely to fully explain the rapid outgrowth of the tumor, so we investigated alternative mechanisms of resistance.
Another possible mechanism of resistance was reduction in major histocompatibility complex (MHC) class I levels, which allows tumors cells to avoid immune surveillance by CD8+ T cells.56 However, we found that the ex vivo tumor cells had higher levels of MHC class I on their surface compared with the parental tumor cell line, in both the absence and presence of IFNγ stimulation (Figure S2C). Additionally, levels of PD-L1 were not enhanced on the surface of these cells (Figure S2D). Co-culture of ex vivo tumor cells with melanocyte-specific TRP1high effector CD8+ T cells57 showed no resistance to T cell cytotoxicity compared with the parental tumor cell line (Figure 2C). These results suggested that the tumor outgrowth seen in vivo was likely tumor cell extrinsic. To test this, we challenged mice with either the parental tumor cell line or the ex vivo tumor cells and began continuous injection with d106S-IL12 virus on day 7. Although the ex vivo lines grew slightly faster, treatment with d106S-IL12 still had a therapeutic benefit, as all tumors responded to therapy (Figure 2D). We therefore conclude that their original rapid outgrowth was due to stochastic loss of immune equilibrium rather than tumor-intrinsic factors.
Cellular characteristics of tumors treated with IL-12 virotherapy
To confirm that continuously treated tumors were indeed live cells held in equilibrium rather than a mass of dead cells, we collected day-42 tumors from mice dosed every 3 days with IL-12 virotherapy and day-10 tumors from mice treated with PBS or d106S-IL12 only once (Figure 2E). From whole sections of this tissue, the tumor was readily identifiable even at day 42, although there was an increase in the portion of necrotic tissue compared with day-10 PBS or d106S-IL12 tumors (Figures 2F and 2G). Ki67 staining revealed a heterogeneous mix of high, medium, and low areas of tumor proliferation at day 42 (Figure 2H). These results indicated that the tumors held in therapy-induced equilibrium were indeed viable and actively proliferating.
These paraffin-embedded tumors were stained for cleaved caspase 3 or Ki67 by immunohistochemistry (Figure S3A), and the ratio of Ki67 to cleaved caspase 3 was determined. There was a significantly higher ratio of Ki67 to cleaved caspase 3 in day-10 PBS tumors compared with day-42 d106S-IL12 tumors, suggesting higher levels of proliferation in these control-treated tumors (Figure S3B). Interestingly, by day 42 this ratio of Ki67 to cleaved caspase 3 had reached 1, suggesting tumors were in equilibrium (Figure S3B). Tumors were also collected from mice on day 17 and stained by immunohistochemistry for CD31 to image the tumor vasculature. We saw no decrease in the quantity of CD31 staining in tumors treated with d106S-IL12 (Figures S3C and S3D).
To profile the immune infiltrate in these tumors treated with IL-12 virotherapy, we analyzed treated tumors by flow cytometry and single-cell RNA sequencing (scRNA-seq). We prepared scRNA-seq libraries from CD45+ live cells and identified 16 distinct clusters (Figure 3A) based on expression of various genes (Figure 3C). We saw differences in immune cell populations (Figure 3B) that were recapitulated by flow cytometry of these same tumors (Figures S4A and S4B). d106S-IL12 induced an early increase in CD8+ T cell infiltration at day 10, which returned to near baseline by day 17, coinciding with an increase in granulocytes and monocytic cells (Figures 3B, S4A, and S4B). We performed gene set enrichment analysis (GSEA) to compare PBS-treated tumors with d106S-IL12-treated tumors. One of the top pathways upregulated by IL-12 virotherapy was IFNγ response (Figure 3D). IFNγ-response genes were most prominently expressed in macrophage/monocyte and neutrophil clusters, although there was a general increase in IFNγ-response genes from all cell types (Figure 3E).
Figure 3. Single-cell RNA sequencing reveals IFNγ response with d106S-IL12 and reduction in Tregs.

Mice with B16Nectin1 tumors were treated 4 times in total and sacrificed on day 16 (PBS) or day 17 (d106S/d106S-IL12). Tumors from each group were pooled (n = 5) and sorted for CD45+ live cells and subjected to single-cell RNA sequencing.
(A) Unsupervised clustering analysis and resulting uniform manifold approximation and projection (UMAP) of all cells.
(B and C) Cell cluster frequency (B) and violin plot (C) of canonical cluster marker genes.
(D) GSEA for hallmark IFNγ-response genes.
(E) IFNγ-response score UMAP based on scaled and summed expression of hallmark IFNγ-response genes for each cell.
(F and G) Subclustering of T cells revealed several unique clusters (F) based on canonical gene expression (G).
(H) Distribution of T cell subclusters across treatment conditions. See also Figure S4.
(I) Foxp3-GFP mice were challenged with B16Nectin1 tumors and injected 4 times every 3 days with PBS, d106S, or d106S-IL12 (N = 5 PBS/d106S, N = 4 d106S-IL12) starting on day 7 post-tumor challenge. On day 17, tumors were collected for flow cytometry and analyzed for presence of Foxp3-GFP+ CD4+ T cells. Representative flow plots are shown.
(J) Extracellular staining for TIM-3 and intracellular staining for T bet was performed on mice from (I).
Groups were compared with one-way ANOVA and Tukey’s multiple comparisons test. NES, normalized enrichment score; MFI, mean fluorescence intensity. *p < 0.05; **p < 0.01; ****p < 0.0001.
Subclustering of the T cell scRNA-seq data further delineated T cell subsets (Figure 3F) which were identified by canonical marker expression (Figure 3G). We discovered an increase in CD4+ T cells cells expressing high levels of IFNγ present in samples treated with d106S-IL12 (Figure 3H). There was also a marked absence of Foxp3+ Tregs in both samples treated with virus (Figure 3H). To confirm these findings, we inoculated Foxp3-GFP mice with B16Nectin1 tumors and treated them as in our scRNA-seq experiment. Flow cytometry revealed a similar decrease in Tregs in both d106S- and d106S-IL12-treated tumors (Figure 3I). Additionally, we measured TIM-3 expression, which is regulated by the Th1 transcriptional regulator, T-bet, and can serve as a marker of Th1 cells.58 We saw an increase in both TIM-3 and T-bet staining by flow cytometry of CD4+ T cells (Figure 3J), consistent with an expansion of Th1 cells.59
IFNγ is a critical node for establishing and maintaining immune equilibrium
Next, we wanted to study the cellular and molecular determinants for establishment and maintenance of equilibrium. Long-term experiments with B16Nectin1 confirmed that continuous dosing of mice with d106S-IL12 every 3 days led to reductions in tumor size and long-term sustained equilibrium, maintaining stable masses for more than 100 days (Figures 4A and 4B). Previous work has implicated IFNγ and T cells as controlling tumors in spontaneous equilibrium,3 although it is unclear what role they play in therapy-induced equilibrium. Because the d106S vector could induce a type I IFN response, we first tested whether blocking this response early in treatment with anti-IFNα/β receptor (TNFΑR) affected establishment of equilibrium. Anti-TNFΑR antibodies were given every 3 days starting with the first dose of d106S-IL12. TNFΑR blockade caused a more rapid initial growth of tumors treated with d106S-IL12, but mice treated with blocking antibodies ultimately stabilized their tumors around days 20–22 and were controlled, though at a higher overall set point than mice not receiving TNFΑR blockade (Figure 4C, left). This indicates that the establishment of equilibrium can occur at a range of tumor sizes and does not necessitate type I IFN.
Figure 4. Continuous d106S-IL12 injections lead to stable tumor control dependent on IFNγ.

(A) Mice treated continuously starting at day 7 (arrow) (N = 5 for PBS; N = 10 for d106S-IL12).
(B) Individual tumor growth from (A).
(C) Mice with B16 tumors began intratumoral treatment with PBS or d106S-IL12 on day 7 and proceeded with treatment every 3 days. Some mice were also treated intraperitoneally (i.p.) with anti-TNFΑR blocking antibodies (100 mg) every 3 days (N = 5 for PBS, N = 10 for d106S-IL12), while the rest of the mice received no blockade (N = 62). At day 40, surviving mice were rerandomized to new treatment groups and injected i.p. with either isotype control, anti-CD4, anti-CD8, anti-IFNγ, or anti-NK1.1 depleting antibodies (100 mg) every 3 days while also continuing to receive d106S-IL12 treatment (curves go down in size as the rerandomized groups contain only remaining live mice, while left-side curves account for endpoint tumors). See also Figure S5.
(D) Wild-type, TCRα−/−, or IFNγ−/− mice (N = 5/10, 5/8, 5/10 mice for PBS/d106S-IL12) were treated with d106S-IL12 or PBS every 3 days.
(E) Wild-type mice were treated as in (C) and rerandomized at day 40 into isotype control (N = 5) or anti-CD4+ anti-CD8+ anti-NK1.1 (100 μg each; N = 4) treatment every 3 days.
Values are mean ± SEM. Survival was compared using a Bonferroni-corrected log-rank test. *p < 0.05.
We then tested whether perturbing the equilibrium state would allow for tumor outgrowth, as this was the key parameter initially used to define immune equilibrium in mice.3 Stable equilibrium using d106S-IL12 was established in a large cohort of mice, which were rerandomized at day 40 into new groups to receive either anti-CD4, anti-CD8, anti-IFNγ, anti-NK1.1, or isotype control depleting antibodies every 3 days while continuing to receive d106S-IL12. Cell depletions were confirmed by flow cytometry (Figure S5). Interestingly, we found that IFNγ blockade was the only depletion that significantly altered tumor growth and overall survival (Figure 4C). Although there was some stochastic outgrowth of isotype-treated tumors, there was no difference in tumor sizes or survival of these mice compared with mice depleted of CD4+ T cells, CD8+ T cells, or NK cells (Figure 4C).
We confirmed this dependence on IFNγ for stable tumor control using IFNγ-deficient mice. These mice failed to adequately control B16Nectin1 tumors treated with d106S-IL12 (Figure 4D), suggesting that IFNγ was necessary not only for maintenance but also for establishment of equilibrium. TCRa−/− mice, which lack CD4+ and CD8+ T cells, showed an intermediate phenotype in which tumor growth was still slowed significantly over PBS treatment (Figure 4D). This intermediate phenotype argued that αβ T cells play a role in stabilizing tumor growth and establishing equilibrium, but they are not uniquely required for antitumor immunity during d106S-IL12 treatment. Combining CD4, CD8, and NK1.1 depleting antibodies at day 40 led to rapid outgrowth of tumors and poor survival of mice (Figure 4E). This suggested that these cell types played complementary roles, likely through their shared ability to secrete IFNγ, which is consistent with our findings in RAG2−/− mice, which lack not only αβ but also γδ T cells, another potential source of IFNγ.60
Consistent with the importance of IFNγ in our model of equilibrium, transcriptomic data from The Cancer Genome Atlas (TCGA) were analyzed, and a positive association was found between IL-12, IFNγ-stimulated gene expression, and increased survival in melanoma patients (Figures S6A and S6B). Previous analysis has shown that cytolytic activity score (CYT; expression of Gzma and Prf1) is associated with increased survival benefit in cancer patients.61 We observed that, indeed, melanoma patients with higher CYT fared better, as did patients with higher expression of IFNγ-response genes (Figure S6C).
Direct T cell cytotoxicity is not required to control tumors treated with IL-12 virotherapy
Although we had seen that CD8+ T cells were dispensable after equilibrium establishment, we wished to test the role of cell-mediated cytotoxicity in generating tumor control induced by d106S-IL12. We therefore generated B2m CRISPR-Cas9 knockout B16 cells deficient for MHC class I expression, with in vitro growth and HSV infectivity similar to those of wild-type B16Nectin1 cells (Figures S7A and S7D). The β2m−/− cells did not have detectable MHC class I on their cell surface (Figure S7G) and were resistant to antigen-specific CD8+ T cell killing in vitro (Figure 5A). When we implanted B16Nectin1 wild-type or β2m−/− cells into mice, we observed that d106S-IL12 treatment controlled tumors regardless of MHC class I status (Figure 5B), indicating that direct antigen presentation to CD8+ T cells was not required for establishment of equilibrium.
Figure 5. d106S-IL12 therapy controls tumors independent of direct T cell cytotoxicity.

(A) TRP1high effector CD8+ cells were co-cultured for 24–48 h with IFNγ-stimulated, CFSE-labeled B16Nectin1 wild-type or β2m CRISPR-Cas9 knockout cells, and cytotoxicity was measured by fluorescent confluence using a Celigo image cytometer. See also Figure S7.
(B) Mice bearing wild-type or β2m−/− B16Nectin1 tumors were injected every 3 days starting at day 7.
(C) Wild-type or perforin (Prf1)-deficient mice were challenged with B16Nectin1 tumors and treated every 3 days (N = 5 per group for WT; N = 4 per group for Prf1−/−).
(D) Mice bearing wild-type or Caspase8−/− B16Nectin1 tumors were treated every 3 days starting at day 7.
(E) 6694c2WT and 6694c2β2m—/— murine pancreatic cancer cells were cultured for 24 h with or without IFNγ (50 ng/mL), and H2-Kb/Db (MHC-I) expression was measured by flow cytometry. MFI, mean fluorescence intensity.
(F and G) 6694c2WT cells (F) or 6694c2β2m—/— cells (G) were implanted subcutaneously, and treatment began on day 7 and proceeded every 3 days. N = 5 for PBS, 5/10 for d106S-IL12 groups (6694c2/B16, respectively) unless otherwise specified.
Values are mean ± SEM. Survival groups were compared using a log-rank test.
Because we had seen that depletion of multiple cell types led to more tumor outgrowth, we tested whether loss of effector function by multiple cell types elicited a change in tumor control. We challenged wild-type or perforin-deficient (Prf1−/−) mice with wild-type B16Nectin1 tumors and observed equivalent levels of control elicited by IL-12 virotherapy (Figure 5C). Therefore, perforin/granzyme cytotoxicity from CD8+ or CD4+ T cells or NK cells was not required. Cytotoxic effector cells can also kill target cells through tumor necrosis factor α(TNFα) or other TNF-family death ligands, such as FasL. We therefore generated B16 Casp8 knockout cells, which lack caspase 8 (Figures S7B, S7E, and S7H), critical for extrinsic apoptosis, and we observed that caspase-8-deficient tumor cells were still able to be controlled by d106S-IL12 therapy to the same degree as wild-type cells (Figure 5D), suggesting that this cytotoxic pathway was not required. Using previously established β2m−/− 6694c2 pancreatic cancer cells62 (Figure 5E), we confirmed that even in the absence of direct CD8+ T cell recognition, d106S-IL12 could elicit stable tumor control of highly refractive pancreatic cancer (Figures 5F and 5G).
IFNγ-insensitive tumors are resistant to checkpoint blockade, but not d106S-IL12
Based on the importance of IFNγ in establishing and maintaining stable tumor control in response to d106S-IL12 therapy, we hypothesized that large amounts of IFNγ could act directly on the tumor and slow its growth. To this end, we generated IFNγr1 and Stat1 CRISPR-Cas9 knockout B16Nectin1 cells that would be deficient in binding or signaling downstream of IFNγ, respectively. These cells had similar in vitro growth kinetics and HSV infectivity (Figures S7C and S7F). While wild-type cells exhibited slowed growth kinetics in vitro in the presence of IFNγ, IFNγR1−/− (Figure 6A) and STAT1−/− cells did not (Figure S7I). The knockout cells also did not upregulate MHC class I in response to IFNγ (Figures 6B and S7J). RNA sequencing of wild-type cells showed a robust upregulation of hundreds of genes in response to IFNγ stimulation in vitro, while the IFNγ-insensitive cells were transcriptionally unaffected (Figures 6C and S7K).
Figure 6. Tumors lacking IFNγ response are still controlled by d106S-IL12 therapy but not checkpoint blockade.

CRISPR-Cas9 gene editing was used to generate IFNγR1 (and STAT1; see also Figure S7) knockout B16Nectin1 cell lines.
(A) Wild-type or IFNγR1−/− cells were cultured with varying concentrations of IFNγ, and confluence was determined every 24 h by Celigo image cytometer.
(B) Wild-type or IFNγR1−/− cells were cultured in vitro with 50 ng/mL IFNγ and H2-Kb/Db (MHC-I) expression determined by flow cytometry 24 h later. Representative flow plots from multiple independent experiments.
(C) RNA sequencing was performed on wild-type or IFNγR1−/− cells cultured in vitro in the absence or presence (100 ng/mL) of IFNγ for 24 h (n = 3 per group). Differential gene expression was performed comparing IFNγ− versus IFNγ+ samples within the same genotype.
(D) Wild-type mice were challenged with either wildtype or IFNγR1−/− B16Nectin1 tumors and treated with PBS or d106S-IL12 starting at day 7 or anti-PD-1+ anti-CTLA-4 antibodies (150 mg each) i.p. starting at day 4 and proceeding every 3 days (N = 5 mice per group).
(E) Pooled survival across two experiments as in (D) (N = 10 mice per group).
Values are mean ± SEM, N = 3 unless otherwise noted. Confluence was compared by two-way ANOVA with Dunnett’s multiple comparisons test. Survival groups were compared using a log-rank test. *p < 0.05; ***p < 0.001; ****p < 0.0001.
After validating that IFNγR1−/− B16Nectin1 cells were truly insensitive to IFNγ-mediated effects, we challenged wild-type mice with either control or IFNγR1−/− tumor cells, established palpable tumors, and began treatment with dual checkpoint blockade or IL-12 virotherapy. Checkpoint blockade typically fails to protect mice with tumors that are insensitive to IFNγ6,11 and, indeed, dual checkpoint blockade using anti-PD-1 and anti-CTLA-4 antibodies failed to protect mice with IFNγ-insensitive tumors while significantly protecting mice with wild-type B16Nectin1 tumors (Figures 6D and 6E). Surprisingly, all tumors treated with d106S-IL12 were controlled regardless of IFNγ-sensing ability (Figures 6D, 6E, and S7L). This suggested that despite IFNγ being critical for tumor control, this cytokine did not need to act directly on tumor cells to exert an effect.
IFNγ can act directly on tumors or multiple host cell types to generate stable equilibrium
As signaling of IFNγ in the tumor cells was found to be dispensable for equilibrium, we next sought to determine which host cells were responding. As expected based on our previous results, we confirmed that IFNγR1-deficient tumors were still maintained in equilibrium when treated with IL-12 virotherapy (Figure 7A). We hypothesized that IFNγ was acting on host cells to control tumors. Surprisingly, when we challenged IFNγR1−/− mice with wild-type tumors, we discovered that these mice could still largely control their tumors, suggesting that tumor sensing of IFNγ was now controlling tumor growth (Figure 7B).
Figure 7. IFNγ can control tumors through actions on host or tumor cells.

(A) Wild-type mice bearing wild-type or IFNγR1−/− B16Nectin1 tumors were injected every 3 days starting at day 7 (N = 5 for PBS, N = 10 for d106S-IL12 groups).
(B) Wild-type B16Nectin1 tumors were inoculated into wild-type, IFNγR1−/−, or IFNγ−/− mice and treated every 3 days (N = 5/10, 5/7, and 4/4 PBS/d106SIL12 groups, respectively).
(C) Wild-type or IFNγR1−/− mice were challenged with wild-type or IFNγR1−/− tumors and treated every 3 days (N = 5/10 PBS/d106SIL12 for all groups except 5/9 for IFNγR1−/− tumors into IFNγR1−/− mice).
(D) Survival from (C). Wild-type or IFNγR1−/− bone marrow was used to inoculate irradiated wild-type or IFNγR1−/− recipient mice. Mice were rested for several months before B16 IFNγR1−/− tumor challenge and subsequent d106S-IL12 treatment (every 3 days beginning day 7).
(E) Tumor volume and survival of bone marrow chimera mice challenged with B16 IFNγR1−/− tumors. Groups from top to bottom: N = 10, 9, 9, and 5.
Values are mean ± SEM. Survival groups were compared using a Bonferroni-corrected log-rank test. **p < 0.01.
Based on these findings, we concluded that IFNγ could act on the tumor cells directly and/or on host cells to indirectly control tumors via downstream effects of IFNγ. Either pathway could be compensatory when the flow of IFNγ to tumor or host cells was disrupted by IFNγ-sensing defects. To test this multimodal hypothesis, we challenged wild-type or IFNγR1−/− mice with wild-type or IFNγR1−/− tumors. Indeed, wild-type mice bearing wild-type tumors controlled their tumors to a similar extent as wild-type mice bearing IFNγR1−/− tumors or IFNγR1−/− mice bearing wild-type tumors (Figures 7C and 7D). However, when neither tumors nor the host could sense IFNγ, the stable control of tumors was lost and tumors grew progressively (Figures 7C and 7D).
To understand which host cell types were involved in sensing of IFNγ to control IFNγR1−/− tumors, we generated bone marrow chimeric mice of irradiated wild-type or IFNγR1−/− background bearing wild-type or IFNγR1−/− bone marrow. We challenged these mice with IFNγR1−/− tumors to ensure that IFNγ could only act on the host cells. Interestingly, IFNγR1−/− tumors were able to be controlled in all groups except IFNγR1−/− mice with IFNγR1−/− bone marrow (Figure 7E), which phenocopy the total IFNγR1-deficient mice that did not control tumors well (Figures 7C and 7D). This indicates that both hematopoietic and non-hematopoietic cells can respond to IFNγ to control tumors, suggesting a highly compensatory, multimodal mechanism of tumor control during equilibrium in which IFNγ is a key node.
DISCUSSION
In this study, we have generated a highly reproducible model of therapy-induced tumor equilibrium. We discovered that IFNγ is the central player in both establishment and maintenance of equilibrium through a variety of redundant mechanisms. Most current immunotherapies rely on direct CD8+ T cell killing, with the consequence that loss of antigen presentation or defects in the IFNγ-sensing pathway allows tumors to escape CD8+ T cell-mediated immune pressure. Here we found that IL-12 virotherapy was effective across multiple settings in which the tumor cells were not directly recognizable by CD8+ T cells, including loss of β2m. The perforin-granzyme pathway and caspase-8-dependent death receptor pathways were likewise dispensable. This suggests that although CD8+ T cell direct cytotoxicity may play a role in initial tumor control, immune equilibrium is not maintained by CD8+ T cells directly recognizing tumor cells.
Numerous studies have demonstrated the importance of IFNγ for immunosurveillance and control of tumors,3,60,63 although it has been less clear how IFNγ functions in equilibrium. Here we confirm the importance of IFNγ for controlling tumors but demonstrate that its protective effects are not mediated solely through its direct action on tumors to slow their growth and increase antigen presentation. IFNγ can also serve to coordinate an immune response against tumors. IFNγ can enhance the activation of macrophages to express nitric oxide synthase to release reactive nitrogen oxide species and increase their phagocytic function. Nitric oxide species may damage tumor cells64 but also can promote immunosuppression that may dampen tumor control.65,66 Tumor cell control by macrophages can also be due to enhanced phagocytosis. Work from our lab has shown that in the context of cIAP1/2 antagonism, T cells can repolarize macrophages to phagocytose live tumor cells in a manner independent of direct T cell recognition of tumor MHC or IFNγ sensing by the tumor.62 Through scRNA-seq of the tumor microenvironment, we saw a strong IFNγ response generated by many cells, but most notably by macrophages.
While macrophages may play some role in controlling tumors in our system, the bone marrow chimera experiment suggests that both hematopoietic and non-hematopoietic cell types can sense and respond to IFNγ to control IFNγ-insensitive tumors. The antiangiogenic properties of IFNγ are well known and may be a possible mechanism for non-hematopoietic cells to control tumors in response to the cytokine.13,14,67 Additionally, tissue-resident macrophages arise from self-renewing populations independent of bone marrow hematopoiesis and are radioresistant.68 Thus, wild-type mice with IFNγR1−/− bone marrow could still have tissue-resident macrophages capable of responding to IFNγ to control IFNγ-insensitive tumors when treated with IL-12 virotherapy. Wild-type mice with IFNγR1−/− bone marrow would also still have stromal vasculature that could respond to IFNγ and reduce blood flow, causing tumors to be better controlled.14 Although we did not examine the vasculature in these chimeric mice, wild-type mice did not have a reduction in CD31+ vascular endothelial cells with d106S-IL12 treatment.
IFN-response genes are induced by TNFα, IFNβ, and IFNγ. A previous study found that PD-1 resistance driven by either IFNγ-sensing defects or β2m−/− tumors could be overcome by co-administration of anti-PD-1 with either a TLR9 agonist or an IL-2 agonist, respectively.11 TLR9 agonism induces type I IFN production that signals through TNFΑR on tumor cells and has overlapping function with IFNγ.11 Although our virotherapy induces a type I IFN response, blockade of TNFΑR in mice only affected the initial set point size of tumors but does not affect establishment of immune equilibrium, despite repeated administration of virotherapy and continual type I IFN production. Outgrowth of tumors in IFNγ−/− and RAG2−/− mice, as well as IFNγR1−/− mice with IFNγR1−/− tumors, was delayed relative to control-treated tumors, which was likely due to type I IFN-mediated effects from the viral vector, although these tumors ultimately did not enter stable equilibrium. We therefore conclude that IFNγ has a central role in establishment and maintenance of equilibrium that is not recapitulated by IFNα or IFNβ.
The concept of equilibrium assumes an equal balance between tumor cell proliferation and tumor cell death. One means of achieving equilibrium is to induce tumor cell quiescence, thereby reducing the proliferation to zero. Tumor cell quiescence has been associated with resistance to CD8 T cell-mediated immunotherapy and can be induced by IFN signaling.69 Here we were surprised to find actively proliferating tumor cells in late-stage equilibrium tumors, suggesting that therapy-induced immune equilibrium is an active state of tumor control. Although the overall levels of proliferating cells were lower, partially due to large areas of necrotic tissue, we observed an equal ratio of proliferating cells to those undergoing apoptosis. The fact that tumors grow progressively upon cessation of virotherapy and the fact that tumor cell loss of IFN sensing is inadequate to prevent establishment of equilibrium support the hypothesis that immune equilibrium can occur by means other than IFNγ-induced tumor cell quiescence.
IFNγ may have evolved as a multimodal cytokine to protect the host against acute and chronic threats from transformed or infected cells. Although the antiviral effects of type I IFNs are best studied, IFNγ also has antiviral effects. In both acute and chronic hepatitis C virus infection, IFNγ levels are correlated with decreased viral load,70 and patients without IFNγ-expressing CD8+ T cells fare worse than those with IFNγ+ CD8+ T cells.71 Hepatitis B virus can be controlled directly by IFNγ acting on hepatocytes to reduce infection in a non-cytolytic manner.72 IFNγ was found to be the dominant IFN expressed in recurrent HSV-2 lesions, causing high levels of ISG expression by epidermal cells, which dampens infection.73 In babies born to human immunodeficiency virus (HIV-1)-positive mothers, IFNγ responses in the breast milk were associated with a 70% reduction in infant HIV acquisition.74 Additionally, IFNγ levels are negatively correlated with HIV-1 viral set point.75 Thus, IFNγ serves as a multipurpose cytokine: capable of initiating and integrating antiviral and antitumor responses through innate and adaptive immune cells as well as being directly antiviral and antitumor against affected cells.
Because the effects of downstream IFNγ are multimodal, involving several cell types that can possibly secrete the cytokine, including CD8+ and CD4+ T cells and NK cells as well as many cell types that can respond to IFNγ to control tumors, escape from equilibrium is more difficult to achieve. However, escape still does occur stochastically in some mice, modeling the clinical situation in humans who experience disease recurrence after long periods of tumor control. Acquired resistance has been linked with the inability of tumors to respond to IFNγ in patients,9,10 suggesting that host cell responsiveness to IFNγ may be impaired within some human tumors due to immune exclusion or other heterogeneities. Escape from equilibrium is a huge unmet clinical problem, and our model provides an opportunity to study these and other questions related to the mechanisms behind therapy-induced equilibrium, with the ultimate goal of understanding how to overcome equilibrium and effect tumor elimination. The seminal study by Koebel et al. demonstrated in mice that immune equilibrium was a distinct phase from elimination and escape, showing that low-dose carcinogen exposure can produce tumors held in check by the immune system for long periods of time.3 Only subsequent depletion of both CD4+ and CD8+ T cells, IFNγ, IL-12, or a combination of cellular and cytokine blockade resulted in escape and the outgrowth of these stable tumors. Because combination blockades were shown to be no more effective at disrupting equilibrium than individual depletions, it can be inferred that these factors control tumors in redundant ways, likely through IFNγ-mediated effects.3 Here, we demonstrate the central role of IFNγ in the establishment and maintenance of equilibrium in the context of immunotherapy. We demonstrate that in the absence of multiple pathways including type I IFN, tumor sensing of IFNγ, CD8+ T cell direct recognition of tumor MHC class I, perforin-granzyme-mediated cytotoxicity, and extrinsic death receptor signaling such as Fas/FasL IFNγ can still ultimately orchestrate long-term durable tumor control.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stephanie Dougan (stephanie_dougan@dfci.harvard.edu)
Materials availability
All unique materials in this study may be made available upon request from the lead contact.
Data and code availability
Bulk and single-cell RNA-sequencing data are available at the Gene Expression Omnibus (GEO) under accession numbers: GSE212829 and GSE222795. Links can be found in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this manuscript is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| InVivoMAb anti-mouse IFNAR-1 | BioXCell | #BE0241; RRID:AB_2687723 |
| InVivoMAb anti-mouse CD4 | BioXCell | #BE0003-1; RRID:AB_1107636 |
| InVivoMAb anti-mouse CD8α | BioXCell | #BE0061; RRID:AB_1125541 |
| InVivoMAb anti-mouse IFNγ | BioXCell | #BE0312; RRID:AB_2736992 |
| InVivoMAb anti-mouse NK1.1 | BioXCell | #BE0036; RRID:AB_1107737 |
| InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin | BioXCell | #BE0090; RRID:AB_1107780 |
| InVivoMAb anti-mouse PD-1 (CD279) | BioXCell | #BE0146; RRID:AB_10949053 |
| InVivoMAb anti-mouse CTLA-4 (CD152) | BioXCell | #BE0164; RRID:AB_10949609 |
| Pacific Blue™ anti-mouse/human CD11b Antibody | Biolegend | 101,224; RRID:AB_755986 |
| PE/Cyanine7 anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody | Biolegend | 108,416; RRID:AB_313381 |
| Brilliant Violet 570™ anti-mouse Ly-6C Antibody | Biolegend | 128,030; RRID:AB_2562617 |
| Brilliant Violet 421™ anti-mouse CD4 Antibody | Biolegend | 100,438; RRID:AB_11203718 |
| Brilliant Violet 785™ anti-mouse CD8a Antibody | Biolegend | 100,750; RRID:AB_2562610 |
| APC anti-mouse CD11c Antibody | Biolegend | 117,310; RRID:AB_313779 |
| Brilliant Violet 421™ anti-T-bet Antibody | Biolegend | 644,816; RRID:AB_10959653 |
| APC anti-mouse CD366 (Tim-3) Antibody | Biolegend | 119,706; RRID:AB_2561656 |
| PE anti-human CD111 (Nectin-1) Antibody | Biolegend | 340,404; RRID:AB_2174152 |
| Brilliant Violet 711™ anti-mouse CD45 Antibody | Biolegend | 103,147; RRID:AB_2564383 |
| APC anti-mouse CD274 (B7-H1, PD-L1) Antibody | Biolegend | 124,312; RRID:AB_10612741 |
| Alexa Fluor ®488 anti-mouse I-A/I-E Antibody | Biolegend | 107,615; RRID:AB_493524 |
| PE anti-mouse H-2Kb/H-2Db Antibody | Biolegend | 114,607; RRID:AB_313598 |
| FITC anti-mouse NK-1.1 Antibody | Biolegend | 108,705; RRID:AB_313392 |
| Anti-GAPDH antibody [6C5] | Abcam | ab8245; RRID:AB_2107448 |
| Caspase-8 Antibody (Mouse Specific) | Cell Signaling | #4927; RRID:AB_2068301 |
| Anti-Ki67 antibody | Abcam | ab15580; RRID:AB_443209 |
| Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | ThermoFisher | ThermoFisher A-11008; RRID:AB_143165 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB ® 5-alpha Competent E. coli (High Efficiency) | NEB | C2987H |
| HSV-1 d106S | Liu et al.41 | N/A |
| HSV-1 d106S-IL12 | This paper | N/A |
|
| ||
| Chemicals peptides and recombinant proteins | ||
|
| ||
| UltraPure™ BSA (50 mg/mL) | ThermoFisher | AM2618 |
| Lipofectamine LTX with Plus Reagent | ThermoFisher | 15,338,100 |
| Lipofectamine™ Stem Transfection Reagent | ThermoFisher | STEM00001 |
| OptiMEM | ThermoFisher | 31,985,062 |
| HBSS, calcium, magnesium, no phenol red | ThermoFisher | 14,025,126 |
| HEPES (1 M) | ThermoFisher | 15,630,080 |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher | 15,140,122 |
| L-Glutamine (200 mM) | ThermoFisher | 25,030,081 |
| Recombinant Murine GM-CSF | Peprotech | 315-03 |
| Recombinant Murine IL-4 | Peprotech | 214-14 |
| Recombinant Murine IFNγ | Peprotech | 315-05 |
| Recombinant Human IL-2 | Peprotech | 200-02 |
| XhoI | NEB | R0146S |
| NotI | NEB | R0189S |
| BbsI | NEB | R0539S |
| SwaI | NEB | R0604S |
| BsmBI-v2 | NEB | R0739S |
| T4 DNA ligase | NEB | M0202S |
| Puromycin Dihydrochloride | ThermoFisher | A1113803 |
| Hygromycin B (50 mg/mL) | ThermoFisher | 10,687,010 |
| Ambion™ DNase I (RNase-free) | ThermoFisher | AM2222 |
| NP-40 Surfact-Amps™ Detergent Solution | ThermoFisher | 85,124 |
| Sodium deoxycholate | ThermoFisher | 89,904 |
| Halt™ Protease and Phosphatase Inhibitor Cocktail (100X) | ThermoFisher | 78,440 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Zombie NIR™ Fixable Viability Kit | Biolegend | 423,105 |
| Dynabeads™ Mouse T-Activator CD3/CD28 for T cell Expansion and Activation | ThermoFisher | 11456D |
| EasySep™ Mouse CD8+ T cell Isolation Kit | Stemcell Technologies | 19,853 |
| Quick Ligation™ Kit | NEB | M2200S |
| ELISA MAX™ Deluxe Set Mouse IL-12/IL-23 (p40) | Biolegend | 431,604 |
| RNeasy Mini Kit | Qiagen | 74,106 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | 4,368,814 |
| SYBR Green Master Mix | ThermoFisher | 4,309,155 |
| Micro BCA™ Protein Assay Kit | ThermoFisher | 23,235 |
| Tumor Dissociation Kit, mouse | Miltenyi | 130-096-730 |
| 10x Chromium Single Cell A Chip Kit | 10x Genomics | PN-1000152 |
| 10x Chromium Single Cell 5′ Library and Gel Bead Kit | 10x Genomics | PN-1000006 |
|
| ||
| Deposited data | ||
|
| ||
| Bulk RNA-sequencing | This paper | NCBI GEO: GSE212829 |
| Single-cell RNA-sequencing | This paper | NCBI GEO: GSE222795 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| B16F10 melanoma | ATCC | CRL-6475 |
| B16F10-nectin1 | This paper | N/A |
| B16F10-nectin1 Stat1-knockout | This paper | N/A |
| B16F10-nectin1 Ifngr1-knockout | This paper | N/A |
| B16F10-nectin1 B2m-knockout | This paper | N/A |
| B16F10-nectin1 Casp8-knockout | This paper | N/A |
| KPC c6694c2 | Li et al.54 | N/A |
| KPC c6694c2 B2m-knockout | Roehle et al.62 | N/A |
| Vero E11 | Samaniego et al.43 | N/A |
| Vero E11 Cas9 | Oh et al.76 | N/A |
| K28; K29; UACC-257 | Patient derived cell lines, Dana-Farber Cancer Institute | N/A |
| A375 | ATCC | CRL-1619 |
| SK-MEL-2 | ATCC | HTB-68 |
| HEK293T | ATCC | CRL-3216 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000,664 |
| Mouse: B6.Cg-Rag2tm1.1Cgn/J | Jackson Laboratory | RRID:IMSR_JAX:008,449 |
| Mouse: B6.129S2-Tcratm1Mom/J | Jackson Laboratory | RRID:IMSR_JAX:002,116 |
| Mouse: B6.129S7-Ifngtm1Ts/J | Jackson Laboratory | RRID:IMSR_JAX:002,287 |
| Mouse: B6.129S7-Ifngr1tm1Agt/J | Jackson Laboratory | RRID:IMSR_JAX:003,288 |
| Mouse: B6.Cg-Rag2tm1.1Cgn TcrbLn5Sdou TcraLn3Sdou/J | Dougan et al.57 | RRID:IMSR_JAX:030,958 |
|
| ||
| Oligonucleotides | ||
|
| ||
| GCGAGTCTCGAGATGTGTCCTCAGAAGCTAACC | IDT | XhoI_IL12_Fow |
| ATAGAAGCGGCCGCTCAGGCGGAGCTCAGATAG | IDT | NotI_IL12_Rev |
| CAGGCGCCCAATACGACCAAATC | Merkl et al.77 | GAPDH fow DNA |
| TTCGACAGTCAGCCGCATCTTCTT | Merkl et al.77 | GAPDH rev DNA |
| CAGGCGCCCAATACGACCAAATC | Merkl et al.77 | ICP8 fow DNA |
| GAGACCGGGGTTGGGGAATGAATC | Merkl et al.77 | ICP8 rev DNA |
| GTAACCCGTTGAACCCCATTCGT | IDT | m18S fow |
| CCATCCAATCGGTAGTAGCGAC | IDT | m18S rev |
| TGAAGCTTGACGCGGTACAT | IDT | mIfit2 fow |
| GCTGCCCTGAGGAGTGTATC | IDT | mIfit2 rev |
| ACCACAGCCCTCTCCATCAA | IDT | mIfnb1 fow |
| TTGAAGTCCGCCCTGTAGGT | IDT | mIfnb1 rev |
| AGCCGATGGGTTGTACCTTG | IDT | mTNF fow |
| ATAGCAAATCGGCTGACGGT | IDT | mTNF rev |
| CACTTCACAAGTCGGAGGCT | IDT | mIl6 fow |
| CTGCAAGTGCATCATCGTTGT | IDT | mIl6 rev |
| TGCAAGCTATGGCTCACTTCA | IDT | mIl1a fow |
| CTTCCCGTTGCTTGACGTTG | IDT | mIl1a rev |
| TCCTCCAAAACACTGCTGACA | IDT | mSting1 fow |
| GAGTCAAGCTCTGAAGGCCC | IDT | mSting1 rev |
| GGAAAGTCGTAAGGGGACCT | IDT | mCgas fow |
| TAGCTTGTCCGGTTCCTTCCT | IDT | mCgas rev |
| CGCTGCAACTGCATCCATATC | IDT | mCxcl10 fow |
| GATAGGCTCGCAGGGATGAT | IDT | mCxcl10 rev |
| GCAAACTTCCGTTGTGCCAT | IDT | mIrf1 fow |
| TCGGCTGGACTTGGACTTTC | IDT | mIrf1 rev |
| ACGCTGCCTATGATGTCTCG | IDT | mStat1 fow |
| AGAAAAGCGGCTGTACTGGT | IDT | mStat1 rev |
| GGTGTACGAACTTAGCCGGG | IDT | mIrf7 fow |
| AATGATCCTGGGGACACACC | IDT | mIrf7 rev |
|
| ||
| Recombinant DNA | ||
|
| ||
| Tandem p40p35 | Addgene | Addgene plasmid #108665 |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Ran et al.78 | Addgene plasmid #62988 |
| lentiCRISPRv2 | Sanjana et al.79 | Addgene plasmid #52961 |
| pVSV-G | Stewart et al.80 | Addgene plasmid #8454 |
| psPAX2 | Addgene | Addgene plasmid #12260 |
| hNectin-1 | Nakashima et al.45 | N/A |
| pd27B HSV-1 shuttle plasmid | Oh et al.76 | N/A |
| pd27-IL12 HSV-1 shuttle plasmid | This paper | N/A |
| Infectious d106S DNA | Watanabe et al.42 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Fiji/ImageJ2 | Schindelin et al.81 | RRID:SCR_002285 |
| cellSens Imaging software | Olympus | RRID:SCR_014551 |
| FlowJo v10.8 | TreeStar | RRID:SCR_008520 |
| Prism v9 | GraphPad | RRID:SCR_002798 |
| Cellranger v6.1.0 | 10x Genomics | http://software.10xgenomics.com/; RRID:SCR_023221 |
| R v4.2.2 | R Project | https://www.r-project.org/; RRID:SCR_001905 |
| RStudio v2022.12.0 | Posit | https://posit.co/downloads/; RRID:SCR_000432 |
| DropletUtils v1.10.3 | Griffiths et al.82 | Bioconductor |
| Seurat v4.0.2 | Stuart et al.83 | https://satijalab.org/seurat/ |
| fgsea v.1.16.0 | https://doi.org/10.1101/060012 | Bioconductor |
| Molecular Signatures Database | Liberzon et al.84 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp; RRID:SCR_016863 |
| Salmon v1.8.0 | Patro et al.85 | https://combine-lab.github.io/salmon/ |
| tximport v1.18.0 | Soneson et al.86 | Bioconductor |
| DESeq2 v1.30.1 | Love et al.87 | Bioconductor |
| Gencode v27 | http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/ | http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/ |
| ggplot2 v3.3.6 | Hadley Wickham | https://ggplot2.tidyverse.org/ |
| cBioPortal | Cerami et al.88; Gao et al.89 | https://www.cbioportal.org/; RRID:SCR_014555 |
| survival v3.2-13 | Terry M Therneau | https://CRAN.R-project.org/package=survival |
| survminer v0.4.9 | Alboukadel Kassambara | https://cran.r-project.org/web/packages/survminer/index.html |
| Gene Ontology Resource | http://geneontology.org/ | http://geneontology.org/; RRID:SCR_002811 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and Nectin-1 generation
B16F10 and HEK293T cells were purchased from American Type Culture Collection. B16 cells were transduced with a previously described lentivirus encoding human nectin-1 (hNectin-1 vector a gift from Dr. Antonio Chiocca.45 Following hygromycin selection (500mg/mL), expression of nectin-1 was validated by infecting B16Nectin1 cells with d106S virus and selecting clones that were GFP-positive by flow cytometry analysis (FACSCalibur). KPC 6694c2 cells were derived from an LSL-KrasG12D;p53+/floxed, Pdx-cre, YFP-floxed mouse as described previously and were a gift from Dr. Benjamin Stanger.54 The Vero-based E11 complementing cell line,43,76 which expresses ICP27 and ICP4, was used to grow d106S virus. K28, K29, A375, UACC-257, and SK-MEL-2 patient melanoma cell lines were a kind gift from Dr. Frank Hodi (Dana-Farber Cancer Institute). B16, E11, and patient melanoma cells were cultured in DMEM with 10% heat-inactivated FBS and 1% PenStrep. Murine bone-marrow derived dendritic cells (BMDCs) were obtained as described previously.57 Briefly, bone marrow aspirates were cultured with complete RPMI media (with 10% heat-inactivated FBS, 2mM glutamine, 1% PenStrep, 1mM sodium pyruvate, 0.1mM β-ME) supplemented with 20 ng/mL each recombinant mGM-CSF and mIL-4 (Peprotech). Fresh media was added every 2 days for 6 days. BMDCs were used for co-culture experiments at day 6–8 of differentiation.
Animals
C57BL/6J mice aged 6 to 8 weeks, RAG2−/− (#008449), TCRα−/− (#002116), IFNγ−/− (#002287), and IFNγR1−/− (#003288) mice were purchased from Jackson Laboratories. TRP1high mice (deposited as jax #030958) were previously generated.57 All mice were housed in the Dana-Farber Cancer Institute Animal Resources Facility. Roughly 80% of mice used were females. All animal experiments were performed in accordance with the DFCI IACUC-approved protocols (#14–019 and #14–037) and are in compliance with the NIH/NCI ethical guidelines for tumor-bearing animals.
METHOD DETAILS
Plasmids
The coding sequence for the murine IL-12 fusion protein was cloned from Tandem p40p35, a gift from Nevil Singh; Addgene plasmid #108665. The sequence was amplified with XhoI_IL12_Fow and NotI_IL12_Rev primers (IDT). The PCR product was cloned into the pd27B shuttle plasmid by XhoI/NotI (NEB) digestion, followed by Quick Ligation (NEB). The correct insertion into the shuttle plasmid was confirmed by Sanger sequencing (Harvard Biopolymers Facility).
CRISPR-Cas9 knockouts
STAT1, IFNγR1, and B2m mutants were generated using pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene #62988).78 Single-guide RNAs (sgRNA) were cloned into this vector using BbsI (NEB) and T4 ligation (NEB). Cells were transfected with sgRNA-containing plasmids using Lipofectamine Stem reagent (ThermoFisher), selected with puromycin (2 μg/mL), FACS-sorted for purity, and absence of protein was confirmed by flow cytometry following addition of murine IFNγ (50 ng/mL) (Peprotech) to the media. Caspase8 mutants were generated using the lentiCRISPRv2 plasmid (Addgene # 52961) and sgRNAs cloned in using BsmBI (NEB) digestion, followed by Quick Ligation (NEB). Lentivirus was generated by transfection of HEK293T cells with lentiCRISPRv2 (Addgene #62961),79 pVSVG (Addgene #8454),80 and psPAX2 (Addgene #12260) using Lipofectamine LTX reagent (ThermoFisher). Supernatants were collected 48 h after transfection, filtered through a 0.45μM pore, and used to infect tumor cells; puromycin was used to select cells. Knockout was confirmed by Western blot as described previously51 using anti-caspase 8 primary antibody (Cell Signaling #4927) and anti-GAPDH (abcam 8245).
Recombinant herpes simplex virus generation
The pd27-IL12 shuttle plasmid was linearized by SwaI (NEB) digestion. E11 complementing cells that express Cas9 and sgRNAs targeting the native HSV genome76 were co-transfected with linear pd27B-IL12 and infectious d106S DNA. Infectious d106S DNA was isolated as described previously.42 The progeny viruses were harvested and fluorescence-negative plaques were isolated and purified three times. Each plaque isolate was analyzed for evidence of IL12 insertion and subsequent lack of GFP by PCR. Viral stocks of both d106S and d106S-IL12 were grown and titered on E11 complementing cells.43 B16Nectin1 cells were infected at varying multiplicities of infection (MOIs) with d106S or d106S-IL12. Twenty-four hours following infection, the supernatant was harvested, and IL-12 production measured by IL12p40 ELISA (BioLegend).
BMDC co-culture and mRNA expression analysis
For mRNA expression analysis of co-culture experiments, B16Nectin1 cells were first infected at MOI 10 with d106S or d106S-IL12. Infected B16 cells were washed multiple times with media before addition of BMDCs for co-culture. Additionally, both B16Nectin1 and BMDCs in isolation were infected with d106S or d106S-IL12 at MOI 10 without co-culture. Total RNA was extracted from single and co-culture infections using the Qiagen RNeasy Kit and DNase treated with the DNA-free kit (Ambion). DNase-treated RNA was reverse-transcribed using the High Capacity cDNA Reverse Transcription kit (ThermoFisher). cDNA was quantified using the Fast SYBR Green Master Mix and StepOne Real-Time PCR System (ThermoFisher). qPCR reactions were carried out in triplicate. Samples were normalized to 18S rRNA.
DNA extraction for virus quantification
Mice bearing B16Nectin1 tumors were bled retroorbitally into a tube containing 0.5M EDTA to prevent clotting. d106S-IL12 virus was spiked into blood from untreated mice bearing B16Nectin1 tumors as a positive control. DNA was extracted from the blood using the Qiagen Blood/Tissue DNA extraction kit. Two RAG2−/− mice near endpoint were sacrificed the day following a final injection of d106S-IL12. Their tumors were placed into DMEM containing 1% FBS, homogenized, freeze-thawed three times, filtered, and centrifuged at 14,000 x g for 10 min to yield a clarified viral lysate. DNA was extracted from this lysate as described above. qPCR reactions were carried out in triplicate to quantify copies of viral ICP8 DNA. Samples were normalized to GAPDH DNA.
Chemokine/cytokine analysis
B16Nectin1 tumors collected on day 10 and day 17 following one or four intratumoral injections, respectively, were snap frozen in liquid nitrogen. Frozen tumors were ground into a powder and tissue homogenate generated by addition of ice-cold RIPA lysis buffer (150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 25mM Tris) and 1:100 protease/phosphatase inhibitor cocktail (ThermoFisher). Homogenate was incubated shaking for 30 min at 4◦C and subsequently centrifuged at 14,000 x g for 15 min to yield clarified lysate. The clarified supernatant was subjected to a 32-plex cytokine/chemokine array (Eve Technologies). The protein concentrate of lysate was determined using the Micro BCA Protein Assay Kit (ThermoFisher) to normalize samples to contain an equal amount of protein.
Tumor inoculations and in vivo experiments
B16Nectin1 and 6694c2 cells were screened prior to in vivo use for murine pathogens, including mycobacteria (Charles River Laboratories). B16Nectin1 or 6694c2 cells were cultured until 80–90% confluent, trypsinized, washed and resuspended in Hank’s balanced salt solution (HBSS) at 2 × 106 cells/mL or 8 × 105 cells/mL, respectively. Mice were shaved and 250μL (5 × 105,2 3 105) cells were injected subcutaneously in the left flank, or right flank for bilateral/rechallenge experiments. Before injections, mice were randomized into treatment groups. Weight change was measured relative to the day before treatment began. For survival experiments, tumor size was measured every three to four days by precision calipers. Mice were euthanized when tumor volume exceeded 2,000 mm3 or tumors developed ulcerations. Intratumoral injections of 30μL PBS, d106S (1.5 × 107 PFU/30μL), or d106S-IL12 (1.5 × 107 PFU/30μL) were performed every three days.
Histopathology, immunohistochemistry, and immunofluorescence
B16Nectin1 tumors and surrounding tissue were excised and submerged in 10% formalin. Tissue was paraffin embedded, sectioned, and stained with hematoxylin and eosin by the Harvard Rodent Histopathology Core. For immunohistochemistry, unstained tissue sections were stained with CD31, cleaved caspase 3, or Ki67, and counterstained for nuclei (hematoxylin) by the Harvard Specialized Histopathology Core. For immunofluorescence, unstained tissue sections were dewaxed and rehydrated; antigen retrieval was performed in 10mM sodium citrate. Following blocking, sections were stained with anti-Ki67 (Abcam 15,580). Secondary staining was performed with goat anti-rabbit AF488 (ThermoFisher A-11008) and samples counterstained with DAPI. All sections were imaged with an Olympus 1X73 inverted fluorescence microscope and Olympus DP74 color camera using Olympus cellSens Imaging software. Stitched images were processed with the Olympus cellSens Imaging software. Color deconvolution of the red chromogen staining was performed with ImageJ/Fiji.81 CD31, Ki67, or cleaved caspase 3 area was determined by ImageJ/Fiji analysis of stitched sections for percent area of red chromogen within the tumor area (including necrotic tissue).
Single-cell RNA-sequencing processing
Tumors were collected and processed as described previously.90 Briefly, the tumor was excised, manually dissociated with scissors, and incubated in RPMI with tumor digestion enzymes (Miltenyi tumor dissociation kit, Cat. No. 130–096-730) for 30 min at 37◦C. Digested tumors were filtered through a 40μM strainer and cells were collected and resuspended in FACS buffer (PBS with 2% FBS). Cells were counted with a hemacytometer and equal portions of cells from each mouse in a sample group were pooled before staining with anti-CD45 (Biolegend clone 30-F11) and ZombieNIR (Biolegend #423105) and sorted for ZombieNIR− CD45+ events into collection buffer (RPMI 1640, 25mM HEPES, 10% FBS) on a BD FACS Aria II at the Dana Farber Flow Sorting Core. Sorted cells were washed one time with 5mL PBS +0.05% ultrapure BSA, counted via hemacytometer, and resuspended at a concentration of 1000 cell/μL. Cell suspensions were loaded onto a 10x Chromium instrument with the Single Cell A Chip per manufacturer’s protocol with a targeted recovery of 5000 cells/sample. Library preparation was performed with the Chromium Single Cell 5′ Library and Gel Bead Kit (#1000006), and samples were sequenced on an Illumina HighSeq instrument with 2 × 150bp sequencing.
Single-cell RNA-sequencing analysis
The 10x Cellranger pipeline (v6.1.0) was used to align reads to the Mm10 reference genome and generate a single-cell feature count matrix for each library using default parameters. The count matrices were imported for downstream analysis into R using the Seurat (v4.0.2) package. The DropletUtils (v1.10.3) package82 was used to exclude empty droplets with an FDR <0.01. Genes expressed in fewer than 3 cells were discarded from further analysis. Barcodes were classified as cells if they satisfied the following criteria: minimum of 500 genes per cell and percentage of mitochondrial reads less than 3 MADs from the median. Counts from all samples were merged into one matrix, split by sample, then log-normalized individually. 2000 variable features were identified for each sample, from which 2000 integration anchors were identified and used to integrate the datasets together to minimize batch effects.83 The integrated dataset was then scaled and subjected to dimensionality reduction using Principal Component Analysis (PCA) of the integration anchors. Uniform Manifold Approximation and Projection (UMAP) embedding was generated from the top 14 dimensions of the PCA. Clusters were identified first by constructing a Shared Nearest Neighbor (SNN) graph based on each cell’s 20-nearest neighbors and then applying modularity refinement with the Louvain algorithm. Markers for each cluster were identified by comparing expressions using the ‘FindAllMarkers’ function.
The average foldchange between gene expression in the PBS vs. d106S-IL12 samples was calculated with the ‘FoldChange’ function and used for Gene Set Enrichment Analysis (GSEA). GSEA was performed with the fgsea package (v.1.16.0), using the Molecular Signature (v7.2) Hallmark gene signatures.84 Genes from the “Hallmark Interferon Gamma Response” were scaled and summed for each cell to visualize expression across samples. T cells were subclustered based on expression of at least two CD3 chains (Cd3d, Cd3e, CD3g), but no expression of Csf1r, Cd79a, Lgmn, or Lyz2. The top 2000 variable genes from these T cells were used for PCA and the top 12 PCA dimensions were used for UMAP generation. The R packages Seurat or ggplot2 (v3.3.6) were used for visualization of scRNA-seq data.
Antibody blockade in vivo
For type I IFN blockade mice were injected i.p. with 200mL (100mg) of anti-TNFΑR1 (BioXCell Clone MAR1–5A3) starting on day seven and continuing every three days until day 34. For equilibrium disruption experiments: at day 40 surviving mice treated with d106S-IL12 were rerandomized and injected with 200μL (100μg) of either: anti-CD4 (BioXCell Clone GK1.5), anti-CD8α (BioXCell Clone 2.43), anti-IFNγ (BioXCell Clone H22), anti-NK1.1 (BioXCell Clone PK136), isotype antibody control (BioXCell Clone LTF-2) or anti-CD4 + anti-CD8α + anti-NK1.1 (100μg each) starting on day 40 every three days for a total of seven injections while continuing to receive d106S-IL12 treatment. For checkpoint blockade experiments: at day four post tumor challenge mice received 150μL (150μg) each of anti-PD-1 (BioXCell Clone RMP1–14) and anti-CTLA-4 (BioXCell Clone 9D9) every three days for five total injections.
Bone marrow chimeras
Wild-type or IFNγR1−/− recipient mice were irradiated with 4.5 Gy using a 137Cs source, rested for 5 h and irradiated with another 4.5 Gy. Irradiated mice were placed on Baytril-water for 14 days. Wild-type or IFNγR1−/− mice donor mice were sacrificed, and bone marrow collected. Bone marrow with matching genotypes was pooled and evenly distributed among irradiated recipients by tail vein injection 8 h following second irradiation. Bone marrow chimeras were housed for several months prior to tumor challenge to ensure survival following reconstitution.
Ex vivo cultures
Two mice bearing endpoint tumors not responding to d106S-IL12 therapy were sacrificed. Their tumors were excised, manually dissociated with sterile scissors in a biosafety cabinet, and filtered through a 40mM strainer into DMEM containing 10% heat-inactivated FBS and 1% PenStrep. Each tumor was diluted successive times to generate a non-confluent monolayer. Cells were washed and media changed every one-two days; confluent wells were trypsinized and replated until a homogeneous monolayer was apparent (about 1–2 weeks). Cells were used in downstream infection and flow cytometry experiments after >1 week of culture.
CD8 cytotoxicity
CD8 cytotoxicity assays were performed as previously described.91 Briefly, CD8 T cells from TRP1high Mice57 were isolated with EasySep Mouse CD8+ T cell Isolation Kit (StemCell), resuspended in complete RPMI +100U/mL hIL2 (Peprotech #200–02) and stimulated with CD3/CD28 beads Dynabeads (Thermofisher #11456D) for 48 h. Target B16Nectin1 or ex vivo cultures were plated in complete DMEM with 50 ng/mL IFNγ (Peprotech #315–05) on the day before co-culture. On day seven following T cell isolation, CD8 T cells were plated at various E:T ratios with or without the presence of tumor cells. 24 h following co-incubation of T cells with tumor cells, media was carefully removed and cell viability was assessed using CellTiterGlo (Promega). Relative luminescence was normalized to wells containing the same number of T cells without tumor cells present. For T cell cytotoxicity comparing wildtype B16Necti1 to β2m−/− B16Nectin1, tumor cells were stained with 5μM CFSE prior to plating with 50 ng/mL IFNγ; T cell cytotoxicity was measured as fluorescent confluence loss comparing T cell containing wells to wells with tumor cells only using a Celigo image cytometer (Nexcelom 200-BFFL-5c).
Cell growth
B16Nectin1+ knockout cell line growth was compared to B16Nectin1+ wildtype cells using a Celigo image cytometer. To compare IFNγ-mediated growth delay, wild-type, STAT1−/−, or IFNγR1−/− B16Nectin1 cells were plated in 96-well plates with varying amounts of IFNγ present during plating. Confluence measurements were made every 24 h after plating in 96-well plates.
Flow cytometry
Tumors were processed for flow cytometry as above for scRNA-seq: manual dissociation, enzyme digest, filtering, followed by staining with flow cytometry antibodies as below. Peripheral blood or spleens were collected and ACK-lysed prior to staining with flow cytometry antibodies for 20 min at 4◦C. Cells were washed once with PBS and fixed with 1% formalin in PBS before analysis, which was performed on a Sony Biotechnology SP6800 Spectral Analyzer and analyzed with the Sony Biotechnology SP6800 Software and FlowJo (Tree Star, Ashland, OR). Flow cytometry antibodies used in this study were purchased from Biolegend:, anti-CD11b (clone M1/70), anti-CD11c (clone N418), anti-CD4 (clone GK1.5), anti-CD45 (clone 30-F11), anti-CD8 (clone 53–6.7), anti-Gr1 (clone RB6–8C5), anti-Ly6C (clone HK1.4), anti-Tbet (clone 4B10), anti-TIM3 (clone RMT3–23), anti-CD111(Nectin-1) (clone R1.302), anti-CD274 (PD-L1) (clone 10F.9G2), anti-IA/IE (clone M5/114.15.2), anti-Kb/Db (clone 28–8-6), anti-NK1.1 (clone PK136), and ZombieNIR (#423105). Intracellular staining for T-bet was performed with eBioscience Foxp3/Transcription Factor Staining Buffer Set (Invitrogen 00–5523-00).
Bulk RNA-sequencing
Wild-type, STAT1−/−, and IFNγR1−/− B16Nectin1 cells were cultured in vitro for 24 h with 0 or 100 ng/mL IFNγ (Peprotech). Total RNA was extracted from cells of biological triplicates using the Qiagen RNeasy Plus Kit. Library construction and Illumina sequencing was performed by Genewiz. Quantification of transcripts from reads was performed using Salmon (v1.8.0)85 with the mouse GRCm39 reference genome and Gencode transcript release v27. The R package tximport (v1.18.0)86 was used to quantify transcript abundance and DESeq2 (v1.30.1)92 was used to identify differentially expressed genes. Volcano plots were generated using ggplot2 (v3.3.6).
TCGA analysis
TCGA transcriptomic and clinical data were obtained from cBioPortal website88,89 for melanoma patients. For each gene, patients were stratified into expression above or below the median and the hazard ratio and p value based on the log rank test was calculated using the survival (v3.2–13) and survminer (v0.4.9) R packages. survminer was also used to plot TCGA survival curves. To adjust for false discovery, p values were corrected with the Benjamini-Hochberg procedure. CYT status was calculated as above/below the median of the geometric mean of GZMA and PRF1 genes.61 IFNγ-response was calculated as above/below the median of scaled and summed expression of genes in the Gene Ontology database terms GO0034341, GO0071346, and GO0060335 (response to type II interferon, cellular response to type II interferon and positive regulation of type II interferon-mediated signaling pathway, respectively).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
All data were analyzed with GraphPad Prism or R. All data were presented as mean with S.E.M. errors bars. Kaplan-Meier survival curves of mice were analyzed using the log rank test with a Bonferroni correction to adjust for multiple comparisons. Data were considered significant when p < 0.05; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Supplementary Material
Highlights.
Model of cancer immune equilibrium using a non-replicating virus expressing IL-12
IFNγ is critical for establishing and maintaining equilibrium
Multiple mechanisms of T cell cytotoxicity are redundant for stable tumor control
Tumors deficient for MHC class I or IFNγ sensing can be stably controlled
ACKNOWLEDGMENTS
M.J.W. was funded by NIH predoctoral fellowship F31 CA235898-01. This research was funded by a grant from the Melanoma Research Alliance to D.M.K. and S.K.D.; NIH grant P01 AI098681-07 to D.M.K.; NIH grant 1R01 AI158488-01; the Ludwig Center at Harvard; a Technology Impact award from the Cancer Research Institute to S.K.D.; and S.K.D. is a Pew-Stewart Scholar in Cancer Research. M.D. is funded by an NIH Mentored Clinical Scientist Development award 1K08DK114563-01 and an American Gastroenterological Association Research Scholars award. We thank F. Stephen Hodi and the DFCI Center for Immuno-Oncology for human melanoma cell lines and Antonio Chiocca for the hNectin-1 plasmid. We thank Jeho Shin and Daniel Heid for technical assistance.
DECLARATION OF INTERESTS
M.D. receives research funding from Novartis and is on the Scientific Advisory Board for Neoleukin Therapeutics. S.K.D. receives research funding from Novartis, Bristol-Meyers Squibb, and Eli Lilly and is co-founder and on the Scientific Advisory Board of Kojin. M.J.W., D.M.K., and S.K.D. together have a patent application “Immunotherapeutic Virus for Treatment of Cancer” pending for using d106S as an immunomodulatory agent.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112219.
REFERENCES
- 1.Dougan M, Dranoff G, and Dougan SK (2019). Cancer immunotherapy: beyond checkpoint blockade. Annu. Rev. Cell Biol 3, 55–75. 10.1146/annurev-cancerbio-030518-055552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunn GP, Old LJ, and Schreiber RD (2004). The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21, 137–148. 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 3.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, and Schreiber RD (2007). Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907. 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 4.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. (2013). Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med 369, 122–133. 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert C, Ribas A, Hamid O, Daud A, Wolchok JD, Joshua AM, Hwu WJ, Weber JS, Gangadhar TC, Joseph RW, et al. (2018). Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J. Clin. Oncol 36, 1668–1674. 10.1200/JCO.2017.75.6270. [DOI] [PubMed] [Google Scholar]
- 6.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. (2017). Identification of essential genes for cancer immunotherapy. Nature 548, 537–542. 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med 375, 819–829. 10.1056/nejmoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-g pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167, 397–404.e9. 10.1016/J.CELL.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torrejon DY, Abril-Rodriguez G, Champhekar AS, Tsoi J, Campbell KM, Kalbasi A, Parisi G, Zaretsky JM, Garcia-Diaz A, Puig-Saus C, et al. (2020). Overcoming genetically based resistance mechanisms to PD-1 blockade. Cancer Discov. 10, 1140–1157. 10.1158/2159-8290.CD-19-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, et al. (2017). Interferon-γ drives Treg fragility to promote anti-tumor immunity. Cell 169, 1130–1141.e11. 10.1016/J.CELL.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B, Karrison T, Rowley DA, and Schreiber H. (2008). IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J. Clin. Invest 118, 1398–1404. 10.1172/JCI33522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, Ivanov A, Szymborska A, Patone G, Kunz S, et al. (2017). Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature 545, 98–102. 10.1038/NATURE22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. (2016). Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554.e12. 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, et al. (2019). Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 178, 933–948.e14. 10.1016/J.CELL.2019.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato A, Linehan MM, and Iwasaki A. (2006). Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. USA 103, 17343–17348. 10.1073/pnas.0605102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lund J, Sato A, Akira S, Medzhitov R, and Iwasaki A. (2003). Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med 198, 513–520. 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurt-jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, and Finberg RW (2004). Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 101, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasmussen SB, Sørensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, and Paludan SR (2007). Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J. Virol 81, 13315–13324. 10.1128/JVI.01167-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. (2015). Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol 33, 2780–2788. 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 22.Zeh HJ, Hurd S, Storkus WJ, and Lotze MT (1993). Interleukin-12 promotes the proliferation and cytolytic maturation of immune effectors: implications for the immunotherapy of cancer. J. Immunother. Emphas. Tumor Immunol 14, 155–161 10.1097/00002371-199308000-00012. [DOI] [PubMed] [Google Scholar]
- 23.Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, and Gately MK (1993). Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J. Exp. Med 178, 1223–1230. 10.1084/jem.178.4.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Momin N, Mehta NK, Bennett NR, Ma L, Palmeri JR, Chinn MM, Lutz EA, Kang B, Irvine DJ, Spranger S, and Wittrup KD (2019). Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci. Transl. Med 11, eaaw2614. 10.1126/scitranslmed.aaw2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay A, Wright J, Shirley S, David AC, Christoph B, Richard JC, Jean SC, and Robert HP (2019). Characterization of abscopal effects of intratumoral electroporation-mediated IL-12 gene therapy. Gene Ther. 26, 1–15. 10.1038/s41434-018-0044-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisenring M, vom Berg J, Kristiansen G, Saller E, and Becher B. (2010). IL-12 initiates tumor rejection via lymphoid tissue–inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol 11, 1030–1038. 10.1038/ni.1947. [DOI] [PubMed] [Google Scholar]
- 27.Mills BN, Connolly KA, Ye J, Murphy JD, Uccello TP, Han BJ, Zhao T, Drage MG, Murthy A, Qiu H, et al. (2019). Stereotactic body radiation and interleukin-12 combination therapy eradicates pancreatic tumors by repolarizing the immune microenvironment. Cell Rep. 29, 406–421.e5. 10.1016/j.celrep.2019.08.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leoni V, Vannini A, Gatta V, Rambaldi J, Sanapo M, Barboni C, Zaghini A, Nanni P, Lollini P-L, Casiraghi C, Campadelli-Fiume G, et al. (2018). A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. Hutt-Fletcher L, ed. 14, e1007209. 10.1371/journal.ppat.1007209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang P, Li X, Wang J, Gao D, Li Y, Li H, Chu Y, Zhang Z, Liu H, Jiang G, et al. (2017). Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun 8, 1395. 10.1038/s41467-017-01385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, and Markert JM (2000). Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 97, 2208–2213. 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bramson JL, Hitt M, Addison CL, Muller WJ, Gauldie J, and Graham FL (1996). Direct intratumoral injection of an adenovirus expressing interleukin-12 induces regression and long-lasting immunity that is associated with highly localized expression of interleukin-12. Hum. Gene Ther 7, 1995–2002. 10.1089/hum.1996.7.16-1995. [DOI] [PubMed] [Google Scholar]
- 32.Quetglas JI, Dubrot J, Bezunartea J, Sanmamed MF, Hervas-Stubbs S, Smerdou C, and Melero I. (2012). Immunotherapeutic synergy between anti-CD137 mAb and intratumoral administration of a cytopathic Semliki Forest virus encoding IL-12. Mol. Ther 20, 1664–1675. 10.1038/mt.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saha D, Wakimoto H, Peters CW, Antoszczyk SJ, Rabkin SD, and Martuza RL (2018). Combinatorial effects of VEGFR kinase inhibitor axitinib and oncolytic virotherapy in mouse and human glioblastoma stem-like cell models. Clin. Cancer Res 24, 3409–3422. 10.1158/1078-0432.CCR-17-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasu Y, Bangma CH, Hull GW, Lee HM, Hu J, Wang J, McCurdy MA, Shimura S, Yang G, Timme TL, and Thompson TC (1999). Adenovirus-mediated interleukin-12 gene therapy for prostate cancer: suppression of orthotopic tumor growth and pre-established lung metastases in an orthotopic model. Gene Ther. 6, 338–349. 10.1038/sj.gt.3300834. [DOI] [PubMed] [Google Scholar]
- 35.Toda M, Martuza R, Kojima H, and Rabkin SD (1998). In situ cancer vaccination: an IL-12 defective vector/replication-competent herpes simplex virus combination induces local and systemic antitumor activity. J. Immunol 160, 4457–4464. [PubMed] [Google Scholar]
- 36.Cao L, Zeng Q, Xu C, Shi S, Zhang Z, and Sun X. (2013). Enhanced antitumor response mediated by the codelivery of paclitaxel and adenoviral vector expressing IL-12. Mol. Pharm 10, 1804–1814. 10.1021/mp300602j. [DOI] [PubMed] [Google Scholar]
- 37.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, and Ryan JL (1997). Effects of single-dose interleukin-12 exposure on interleukin-12 – associated toxicity and interferon-γ production. Blood 90, 2541–2548. [PubMed] [Google Scholar]
- 38.Puzanov I, Diab A, Abdallah K, Bingham CO 3rd, Brogdon C, Dadu R, Hamad L, Kim S, Lacouture ME, LeBoeuf NR, et al. (2017). Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the society for immunotherapy of cancer (SITC) toxicity management working group. J. Immunother. Cancer 5, 95. 10.1186/s40425-017-0300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pauken KE, Dougan M, Rose NR, Lichtman AH, and Sharpe AH (2019). Adverse events following cancer immunotherapy: obstacles and opportunities. Trends Immunol. 40, 511–523. 10.1016/J.IT.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walsh MJ, and Dougan M. (2021). Checkpoint blockade toxicities: insights into autoimmunity and treatment. Semin. Immunol 52, 101473. 10.1016/j.smim.2021.101473. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Broberg E, Watanabe D, Dudek T, Deluca N, and Knipe DM (2009). Genetic engineering of a modified herpes simplex virus 1 vaccine vector. Vaccine 27, 2760–2767. 10.1016/j.vaccine.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe D, Brockman MA, Ndung’u T, Mathews L, Lucas WT, Murphy CG, Felber BK, Pavlakis GN, Deluca NA, and Knipe DM (2007). Properties of a herpes simplex virus multiple immediate-early gene-deleted recombinant as a vaccine vector. Virology 357, 186–198. 10.1016/j.virol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 43.Samaniego LA, Neiderhiser L, and DeLuca NA (1998). Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol 72, 3307–3320. 10.1128/JVI.72.4.3307-3320.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boscheinen JB, Thomann S, Knipe DM, DeLuca N, Schuler-Thurner B, Gross S, Dö rrie J, Schaft N, Bach C, Rohrhofer A, et al. (2019). Generation of an oncolytic herpes simplex virus 1 expressing human MelanA. Front. Immunol 10, 2. 10.3389/fimmu.2019.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakashima H, Nguyen T, Kasai K, Passaro C, Ito H, Goins WF, Shaikh I, Erdelyi R, Nishihara R, Nakano I, et al. (2018). Toxicity and efficacy of a novel GADD34-expressing oncolytic HSV-1 for the treatment of experimental glioblastoma. Clin. Cancer Res 24, 2574–2584. 10.1158/1078-0432.CCR-17-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller CG, Krummenacher C, Eisenberg RJ, Cohen GH, and Fraser NW (2001). Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther 3, 160–168. 10.1006/mthe.2000.0240. [DOI] [PubMed] [Google Scholar]
- 47.Gillessen S, Carvajal D, Ling P, Podlaski FJ, Stremlo DL, Familletti PC, Gubler U, Presky DH, Stern AS, and Gately MK (1995). Mouse interleukin-12 (IL-12) p40 homodimer: a potent IL-12 antagonist. Eur. J. Immunol 25, 200–206. 10.1002/eji.1830250133. [DOI] [PubMed] [Google Scholar]
- 48.Lieschke GJ, Rao PK, Gately MK, and Mulligan RC (1997). Bioactive murine and human interleukin-12 fusion proteins which retain anti-tumor activity in vivo. Nat. Biotechnol 15, 35–40. 10.1038/nbt0197-35. [DOI] [PubMed] [Google Scholar]
- 49.Ling P, Gately MK, Gubler U, Stern AS, Lin P, Hollfelder K, Su C, Pan YC, and Hakimi J. (1995). Human IL-12 p40 homodimer binds to the IL-12 receptor but does not mediate biologic activity. J. Immunol 154, 116–127. [PubMed] [Google Scholar]
- 50.Thomann S, Boscheinen JB, Vogel K, Knipe DM, DeLuca N, Gross S, Schuler-Thurner B, Schuster P, and Schmidt B. (2015). Combined cytotoxic activity of an infectious , but non-replicative herpes simplex virus type 1 and plasmacytoid dendritic cells against tumour cells. Immunology. Immunology 146, 327–338. 10.1111/imm.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orzalli MH, DeLuca NA, and Knipe DM (2012). Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 109, E3008–E3017. 10.1073/PNAS.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eidson KM, Hobbs WE, Manning BJ, Carlson P, and DeLuca NA (2002). Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol 76, 2180–2191. 10.1128/jvi.76.5.2180-2191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salio M, Cella M, Suter M, and Lanzavecchia A. (1999). Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol 29, 3245–3253. 10.1002/(SICI)1521-4141. [DOI] [PubMed] [Google Scholar]
- 54.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. (2018). Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49, 178–193.e7. 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stump CT, Roehle K, Manjarrez Orduno N, and Dougan SK (2021). Radiation combines with immune checkpoint blockade to enhance T cell priming in a murine model of poorly immunogenic pancreatic cancer. Open Biol. 11, 210245. 10.1098/RSOB.210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A. (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. 10.1016/J.CELL.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dougan SK, Dougan M, Kim J, Turner JA, Ogata S, Cho HI, Jaenisch R, Celis E, and Ploegh HL (2013). Transnuclear TRP1-specific CD8 T cells with high or low affinity TCRs show equivalent antitumor activity. Cancer Immunol. Res 1, 99–111. 10.1158/2326-6066.CIR-13-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson AC, Lord GM, Dardalhon V, Lee DH, Sabatos-Peyton CA, Glimcher LH, and Kuchroo VK (2010). T-bet, a Th1 transcription factor regulates the expression of Tim-3. Eur. J. Immunol 40, 859–866. 10.1002/eji.200939842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mayer KD, Mohrs K, Crowe SR, Johnson LL, Rhyne P, Woodland DL, and Mohrs M. (2005). The functional heterogeneity of type 1 effector T cells in response to infection is related to the potential for IFN-gamma production. J. Immunol 174, 7732–7739. 10.4049/JIMMUNOL.174.12.7732. [DOI] [PubMed] [Google Scholar]
- 60.Gao Y, Yang W, Pan M, Scully E, Girardi M, Augenlicht LH, Craft J, and Yin Z. (2003). γδ T cells provide an early source of interferon g in tumor immunity. J. Exp. Med 198, 433–442. 10.1084/jem.20030584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N. (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. 10.1016/J.CELL.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roehle K, Qiang L, Ventre KS, Heid D, Ali LR, Lenehan P, Heckler M, Crowley SJ, Stump CT, Ro G, et al. (2021). cIAP1/2 antagonism eliminates MHC class I-negative tumors through T cell-dependent reprogramming of mononuclear phagocytes. Sci. Transl. Med 13, 5058. 10.1126/scitranslmed.abf5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Teng MWL, Vesely MD, Duret H, McLaughlin N, Towne JE, Schreiber RD, and Smyth MJ (2012). Opposing roles for IL-23 and IL-12 in maintaining occult cancer in an equilibrium state. Cancer Res. 72, 3987–3996. 10.1158/0008-5472.CAN-12-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhat MY, Solanki HS, Advani J, Khan AA, Keshava Prasad TS, Gowda H, Thiyagarajan S, and Chatterjee A. (2018). Comprehensive network map of interferon gamma signaling. J. Cell Commun. Signal 12, 745–751. 10.1007/s12079-018-0486-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Cicco P, Ercolano G, and Ianaro A. (2020). The new era of cancer immunotherapy: targeting myeloid-derived suppressor cells to overcome immune evasion. Front. Immunol 11, 1680. 10.3389/fimmu.2020.01680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mittal D, Gubin MM, Schreiber RD, and Smyth MJ (2014). New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr. Opin. Immunol 27, 16–25. 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Angiolillo AL, Sgadari C, and Tosato G. (1996). A role for the interferon-inducible protein 10 in inhibition of angiogenesis by interleukin-12. Ann. N. Y. Acad. Sci 795, 158–167. 10.1111/j.1749-6632.1996.tb52664.x. [DOI] [PubMed] [Google Scholar]
- 68.Hoeffel G, and Ginhoux F. (2015). Ontogeny of tissue-resident macrophages. Front. Immunol 6, 486. 10.3389/FIMMU.2015.00486/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baldominos P, Barbera-Mourelle A, Barreiro O, Huang Y, Wight A, Cho JW, Zhao X, Estivill G, Adam I, Sanchez X, et al. (2022). Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell 185, 1694–1708.e19. 10.1016/j.cell.2022.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Flynn JK, Dore GJ, Hellard M, Yeung B, Rawlinson WD, White PA, Kaldor JM, Lloyd AR, and Ffrench RA; ATAHC Study Group (2011). Early IL-10 predominant responses are associated with progression to chronic hepatitis C virus infection in injecting drug users. J. Viral Hepat 18, 549–561. 10.1111/J.1365-2893.2010.01335.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thimme R, Oldach D, Chang K-M, Steiger C, Ray SC, and Chisari FV (2001). Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med 194, 1395–1406. 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, and Chisari FV (1996). Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4, 25–36. 10.1016/S1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 73.Peng T, Phasouk K, Sodroski CN, Sun S, Hwangbo Y, Layton ED, Jin L, Klock A, Diem K, Magaret AS, et al. (2021). Tissue-resident-memory CD8+ T cells bridge innate immune responses in neighboring epithelial cells to control human genital herpes. Front. Immunol 12, 735643. 10.3389/FIMMU.2021.735643/FULL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lohman-Payne B, Slyker JA, Moore S, Maleche-Obimbo E, Wamalwa DC, Richardson BA, Rowland-Jones S, Mbori-Ngacha D, Farquhar C, Overbaugh J, and John-Stewart G. (2012). Breast milk cellular HIV-specific interferon g responses are associated with protection from peripartum HIV transmission. AIDS 26, 2007–2016. 10.1097/QAD.0B013E328359B7E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roberts L, Passmore JAS, Williamson C, Little F, Bebell LM, Mlisana K, Burgers WA, van Loggerenberg F, Walzl G, Djoba Siawaya JF, et al. (2010). Plasma cytokine levels during acute HIV-1 infection predict HIV disease progression. AIDS 24, 819–831. 10.1097/QAD.0B013E3283367836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oh HS, Diaz FM, Zhou C, Carpenter N, and Knipe DM (2022). CRISPR-Cas9 expressed in stably transduced cell lines promotes recombination and selects for herpes simplex virus recombinants. Curr. Res. Virol. Sci 3, 100023. 10.1016/J.CRVIRO.2022.100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Merkl PE, Orzalli MH, and Knipe DM (2018). Mechanisms of host IFI16, PML, and daxx protein restriction of herpes simplex virus 1 replication. J. Virol 92, e00057–18. 10.1128/jvi.00057-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanjana NE, Shalem O, and Zhang F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/NMETH.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. 10.1261/RNA.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Griffiths JA, Richard AC, Bach K, Lun ATL, and Marioni JC (2018). Detection and removal of barcode swapping in single-cell RNA-seq data. Nat Commun. 9, 2667. 10.1038/s41467-018-05083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R. (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. 10.1016/J.CELL.2019.05.031/ATTACHMENT/2F8B9EBE-54E6-43EB-9EF2-949B6BDA8BA2/MMC3.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liberzon A, Birger C, Thorvaldsdó ttir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The molecular signatures database hallmark gene set collection. Cell Syst. 1, 417–425. 10.1016/J.CELS.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521. 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. 10.1126/SCISIGNAL.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clancy-Thompson E, Devlin CA, Tyler PM, Servos MM, Ali LR, Ventre KS, Bhuiyan MA, Bruck PT, Birnbaum ME, and Dougan SK (2018). Altered binding of tumor antigenic peptides to MHC class I affects CD8+ T cell-effector responses. Cancer Immunol. Res 6, 1524–1536. 10.1158/2326-6066.CIR-18-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dougan M, Ingram JR, Jeong H-J, Mosaheb MM, Bruck PT, Ali L, Pishesha N, Blomberg O, Tyler PM, Servos MM, et al. (2018). Targeting cytokine therapy to the pancreatic tumor microenvironment using PD-L1 specific VHHs. Cancer Immunol. Res 6, 389–401, Published online February 2018:canimm. 10.1158/2326-6066.CIR-17-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk and single-cell RNA-sequencing data are available at the Gene Expression Omnibus (GEO) under accession numbers: GSE212829 and GSE222795. Links can be found in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this manuscript is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| InVivoMAb anti-mouse IFNAR-1 | BioXCell | #BE0241; RRID:AB_2687723 |
| InVivoMAb anti-mouse CD4 | BioXCell | #BE0003-1; RRID:AB_1107636 |
| InVivoMAb anti-mouse CD8α | BioXCell | #BE0061; RRID:AB_1125541 |
| InVivoMAb anti-mouse IFNγ | BioXCell | #BE0312; RRID:AB_2736992 |
| InVivoMAb anti-mouse NK1.1 | BioXCell | #BE0036; RRID:AB_1107737 |
| InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin | BioXCell | #BE0090; RRID:AB_1107780 |
| InVivoMAb anti-mouse PD-1 (CD279) | BioXCell | #BE0146; RRID:AB_10949053 |
| InVivoMAb anti-mouse CTLA-4 (CD152) | BioXCell | #BE0164; RRID:AB_10949609 |
| Pacific Blue™ anti-mouse/human CD11b Antibody | Biolegend | 101,224; RRID:AB_755986 |
| PE/Cyanine7 anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody | Biolegend | 108,416; RRID:AB_313381 |
| Brilliant Violet 570™ anti-mouse Ly-6C Antibody | Biolegend | 128,030; RRID:AB_2562617 |
| Brilliant Violet 421™ anti-mouse CD4 Antibody | Biolegend | 100,438; RRID:AB_11203718 |
| Brilliant Violet 785™ anti-mouse CD8a Antibody | Biolegend | 100,750; RRID:AB_2562610 |
| APC anti-mouse CD11c Antibody | Biolegend | 117,310; RRID:AB_313779 |
| Brilliant Violet 421™ anti-T-bet Antibody | Biolegend | 644,816; RRID:AB_10959653 |
| APC anti-mouse CD366 (Tim-3) Antibody | Biolegend | 119,706; RRID:AB_2561656 |
| PE anti-human CD111 (Nectin-1) Antibody | Biolegend | 340,404; RRID:AB_2174152 |
| Brilliant Violet 711™ anti-mouse CD45 Antibody | Biolegend | 103,147; RRID:AB_2564383 |
| APC anti-mouse CD274 (B7-H1, PD-L1) Antibody | Biolegend | 124,312; RRID:AB_10612741 |
| Alexa Fluor ®488 anti-mouse I-A/I-E Antibody | Biolegend | 107,615; RRID:AB_493524 |
| PE anti-mouse H-2Kb/H-2Db Antibody | Biolegend | 114,607; RRID:AB_313598 |
| FITC anti-mouse NK-1.1 Antibody | Biolegend | 108,705; RRID:AB_313392 |
| Anti-GAPDH antibody [6C5] | Abcam | ab8245; RRID:AB_2107448 |
| Caspase-8 Antibody (Mouse Specific) | Cell Signaling | #4927; RRID:AB_2068301 |
| Anti-Ki67 antibody | Abcam | ab15580; RRID:AB_443209 |
| Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | ThermoFisher | ThermoFisher A-11008; RRID:AB_143165 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB ® 5-alpha Competent E. coli (High Efficiency) | NEB | C2987H |
| HSV-1 d106S | Liu et al.41 | N/A |
| HSV-1 d106S-IL12 | This paper | N/A |
|
| ||
| Chemicals peptides and recombinant proteins | ||
|
| ||
| UltraPure™ BSA (50 mg/mL) | ThermoFisher | AM2618 |
| Lipofectamine LTX with Plus Reagent | ThermoFisher | 15,338,100 |
| Lipofectamine™ Stem Transfection Reagent | ThermoFisher | STEM00001 |
| OptiMEM | ThermoFisher | 31,985,062 |
| HBSS, calcium, magnesium, no phenol red | ThermoFisher | 14,025,126 |
| HEPES (1 M) | ThermoFisher | 15,630,080 |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher | 15,140,122 |
| L-Glutamine (200 mM) | ThermoFisher | 25,030,081 |
| Recombinant Murine GM-CSF | Peprotech | 315-03 |
| Recombinant Murine IL-4 | Peprotech | 214-14 |
| Recombinant Murine IFNγ | Peprotech | 315-05 |
| Recombinant Human IL-2 | Peprotech | 200-02 |
| XhoI | NEB | R0146S |
| NotI | NEB | R0189S |
| BbsI | NEB | R0539S |
| SwaI | NEB | R0604S |
| BsmBI-v2 | NEB | R0739S |
| T4 DNA ligase | NEB | M0202S |
| Puromycin Dihydrochloride | ThermoFisher | A1113803 |
| Hygromycin B (50 mg/mL) | ThermoFisher | 10,687,010 |
| Ambion™ DNase I (RNase-free) | ThermoFisher | AM2222 |
| NP-40 Surfact-Amps™ Detergent Solution | ThermoFisher | 85,124 |
| Sodium deoxycholate | ThermoFisher | 89,904 |
| Halt™ Protease and Phosphatase Inhibitor Cocktail (100X) | ThermoFisher | 78,440 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Zombie NIR™ Fixable Viability Kit | Biolegend | 423,105 |
| Dynabeads™ Mouse T-Activator CD3/CD28 for T cell Expansion and Activation | ThermoFisher | 11456D |
| EasySep™ Mouse CD8+ T cell Isolation Kit | Stemcell Technologies | 19,853 |
| Quick Ligation™ Kit | NEB | M2200S |
| ELISA MAX™ Deluxe Set Mouse IL-12/IL-23 (p40) | Biolegend | 431,604 |
| RNeasy Mini Kit | Qiagen | 74,106 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | 4,368,814 |
| SYBR Green Master Mix | ThermoFisher | 4,309,155 |
| Micro BCA™ Protein Assay Kit | ThermoFisher | 23,235 |
| Tumor Dissociation Kit, mouse | Miltenyi | 130-096-730 |
| 10x Chromium Single Cell A Chip Kit | 10x Genomics | PN-1000152 |
| 10x Chromium Single Cell 5′ Library and Gel Bead Kit | 10x Genomics | PN-1000006 |
|
| ||
| Deposited data | ||
|
| ||
| Bulk RNA-sequencing | This paper | NCBI GEO: GSE212829 |
| Single-cell RNA-sequencing | This paper | NCBI GEO: GSE222795 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| B16F10 melanoma | ATCC | CRL-6475 |
| B16F10-nectin1 | This paper | N/A |
| B16F10-nectin1 Stat1-knockout | This paper | N/A |
| B16F10-nectin1 Ifngr1-knockout | This paper | N/A |
| B16F10-nectin1 B2m-knockout | This paper | N/A |
| B16F10-nectin1 Casp8-knockout | This paper | N/A |
| KPC c6694c2 | Li et al.54 | N/A |
| KPC c6694c2 B2m-knockout | Roehle et al.62 | N/A |
| Vero E11 | Samaniego et al.43 | N/A |
| Vero E11 Cas9 | Oh et al.76 | N/A |
| K28; K29; UACC-257 | Patient derived cell lines, Dana-Farber Cancer Institute | N/A |
| A375 | ATCC | CRL-1619 |
| SK-MEL-2 | ATCC | HTB-68 |
| HEK293T | ATCC | CRL-3216 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000,664 |
| Mouse: B6.Cg-Rag2tm1.1Cgn/J | Jackson Laboratory | RRID:IMSR_JAX:008,449 |
| Mouse: B6.129S2-Tcratm1Mom/J | Jackson Laboratory | RRID:IMSR_JAX:002,116 |
| Mouse: B6.129S7-Ifngtm1Ts/J | Jackson Laboratory | RRID:IMSR_JAX:002,287 |
| Mouse: B6.129S7-Ifngr1tm1Agt/J | Jackson Laboratory | RRID:IMSR_JAX:003,288 |
| Mouse: B6.Cg-Rag2tm1.1Cgn TcrbLn5Sdou TcraLn3Sdou/J | Dougan et al.57 | RRID:IMSR_JAX:030,958 |
|
| ||
| Oligonucleotides | ||
|
| ||
| GCGAGTCTCGAGATGTGTCCTCAGAAGCTAACC | IDT | XhoI_IL12_Fow |
| ATAGAAGCGGCCGCTCAGGCGGAGCTCAGATAG | IDT | NotI_IL12_Rev |
| CAGGCGCCCAATACGACCAAATC | Merkl et al.77 | GAPDH fow DNA |
| TTCGACAGTCAGCCGCATCTTCTT | Merkl et al.77 | GAPDH rev DNA |
| CAGGCGCCCAATACGACCAAATC | Merkl et al.77 | ICP8 fow DNA |
| GAGACCGGGGTTGGGGAATGAATC | Merkl et al.77 | ICP8 rev DNA |
| GTAACCCGTTGAACCCCATTCGT | IDT | m18S fow |
| CCATCCAATCGGTAGTAGCGAC | IDT | m18S rev |
| TGAAGCTTGACGCGGTACAT | IDT | mIfit2 fow |
| GCTGCCCTGAGGAGTGTATC | IDT | mIfit2 rev |
| ACCACAGCCCTCTCCATCAA | IDT | mIfnb1 fow |
| TTGAAGTCCGCCCTGTAGGT | IDT | mIfnb1 rev |
| AGCCGATGGGTTGTACCTTG | IDT | mTNF fow |
| ATAGCAAATCGGCTGACGGT | IDT | mTNF rev |
| CACTTCACAAGTCGGAGGCT | IDT | mIl6 fow |
| CTGCAAGTGCATCATCGTTGT | IDT | mIl6 rev |
| TGCAAGCTATGGCTCACTTCA | IDT | mIl1a fow |
| CTTCCCGTTGCTTGACGTTG | IDT | mIl1a rev |
| TCCTCCAAAACACTGCTGACA | IDT | mSting1 fow |
| GAGTCAAGCTCTGAAGGCCC | IDT | mSting1 rev |
| GGAAAGTCGTAAGGGGACCT | IDT | mCgas fow |
| TAGCTTGTCCGGTTCCTTCCT | IDT | mCgas rev |
| CGCTGCAACTGCATCCATATC | IDT | mCxcl10 fow |
| GATAGGCTCGCAGGGATGAT | IDT | mCxcl10 rev |
| GCAAACTTCCGTTGTGCCAT | IDT | mIrf1 fow |
| TCGGCTGGACTTGGACTTTC | IDT | mIrf1 rev |
| ACGCTGCCTATGATGTCTCG | IDT | mStat1 fow |
| AGAAAAGCGGCTGTACTGGT | IDT | mStat1 rev |
| GGTGTACGAACTTAGCCGGG | IDT | mIrf7 fow |
| AATGATCCTGGGGACACACC | IDT | mIrf7 rev |
|
| ||
| Recombinant DNA | ||
|
| ||
| Tandem p40p35 | Addgene | Addgene plasmid #108665 |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Ran et al.78 | Addgene plasmid #62988 |
| lentiCRISPRv2 | Sanjana et al.79 | Addgene plasmid #52961 |
| pVSV-G | Stewart et al.80 | Addgene plasmid #8454 |
| psPAX2 | Addgene | Addgene plasmid #12260 |
| hNectin-1 | Nakashima et al.45 | N/A |
| pd27B HSV-1 shuttle plasmid | Oh et al.76 | N/A |
| pd27-IL12 HSV-1 shuttle plasmid | This paper | N/A |
| Infectious d106S DNA | Watanabe et al.42 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Fiji/ImageJ2 | Schindelin et al.81 | RRID:SCR_002285 |
| cellSens Imaging software | Olympus | RRID:SCR_014551 |
| FlowJo v10.8 | TreeStar | RRID:SCR_008520 |
| Prism v9 | GraphPad | RRID:SCR_002798 |
| Cellranger v6.1.0 | 10x Genomics | http://software.10xgenomics.com/; RRID:SCR_023221 |
| R v4.2.2 | R Project | https://www.r-project.org/; RRID:SCR_001905 |
| RStudio v2022.12.0 | Posit | https://posit.co/downloads/; RRID:SCR_000432 |
| DropletUtils v1.10.3 | Griffiths et al.82 | Bioconductor |
| Seurat v4.0.2 | Stuart et al.83 | https://satijalab.org/seurat/ |
| fgsea v.1.16.0 | https://doi.org/10.1101/060012 | Bioconductor |
| Molecular Signatures Database | Liberzon et al.84 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp; RRID:SCR_016863 |
| Salmon v1.8.0 | Patro et al.85 | https://combine-lab.github.io/salmon/ |
| tximport v1.18.0 | Soneson et al.86 | Bioconductor |
| DESeq2 v1.30.1 | Love et al.87 | Bioconductor |
| Gencode v27 | http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/ | http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/ |
| ggplot2 v3.3.6 | Hadley Wickham | https://ggplot2.tidyverse.org/ |
| cBioPortal | Cerami et al.88; Gao et al.89 | https://www.cbioportal.org/; RRID:SCR_014555 |
| survival v3.2-13 | Terry M Therneau | https://CRAN.R-project.org/package=survival |
| survminer v0.4.9 | Alboukadel Kassambara | https://cran.r-project.org/web/packages/survminer/index.html |
| Gene Ontology Resource | http://geneontology.org/ | http://geneontology.org/; RRID:SCR_002811 |