

Abstract
Mitochondrial β-oxidation is the most prominent pathway for fatty acid oxidation but alternative oxidative metabolism exists. Fatty acid ω-oxidation is one of these pathways and forms dicarboxylic acids as products. These dicarboxylic acids are metabolized through peroxisomal β-oxidation representing an alternative pathway, which could potentially limit the toxic effects of fatty acid accumulation. Although dicarboxylic acid metabolism is highly active in liver and kidney, its role in physiology has not been explored in depth. In this review, we summarize the biochemical mechanism of the formation and degradation of dicarboxylic acids through ω- and β-oxidation, respectively. We will discuss the role of dicarboxylic acids in different (patho)physiological states with a particular focus on the role of the intermediates and products generated through the peroxisomal β-oxidation. This review is expected to increase the understanding of dicarboxylic acid metabolism and spark future research.
Introduction
Fatty acid oxidation (FAO) is a metabolic process that provides energy in times of starvation and metabolic stress and in the post-absorptive state. In these physiological situations, circulating lipid levels increase through the generation of free fatty acids via adipose tissue lipolysis and very-low-density lipoprotein particles through hepatic lipogenesis. Two organelles have the capacity to metabolize fatty acids through β-oxidation, the mitochondrion and the peroxisome. The primary pathway for long-chain FAO is mitochondrial [1], while longer-chain fatty acids (>C22) are selectively metabolized in the peroxisome [2]. In both organelles, FAO sequentially removes acetyl-CoA units from the acyl-CoA chain. Acetyl-CoA generated through mitochondrial FAO is a substrate for ketone body synthesis and fuels the tricarboxylic acid (TCA) cycle. Reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) are also formed through mitochondrial FAO and are used in oxidative phosphorylation to generate ATP directly. Ketone bodies serve as an auxiliary fuel to glucose, particularly in the brain. Thus, mitochondrial FAO is crucial in energy homeostasis. The peroxisomal function is crucial for the homeostasis of a variety of carboxylic acids, such as very long-chain fatty acids (VLCFA, >C22) and prevents toxicity due to their accumulation. The importance of mitochondrial and peroxisomal FAO in human physiology is highlighted by patients with inherited disorders caused by defects in these pathways. Patients with mitochondrial FAO defects display a clinical phenotype that may include fasting-induced hypoketotic hypoglycemia, cardiomyopathy, heartbeat disorders, sudden infant death, myopathy and rhabdomyolysis [1]. Defects in peroxisomal FAO also lead to multisystem disease and often involve the nervous system [2–4]. Neonatal screening for mitochondrial FAO disorders allows early intervention and has reduced the number of infants with neurologic damage or sudden death due to hypoglycemia. Unfortunately, some of the symptoms displayed by patients with FAO defects such as myopathy respond poorly to dietary interventions and other treatments [5]. Peroxisomal FAO disorders often lead to severe neurological disease with no curative treatment options available [4]. Therefore, there is a clear need to improve the understanding of the pathophysiology of FAO disorders to develop better therapies.
Mitochondrial β-oxidation is quantitatively the most important pathway for FAO, but alternative metabolic pathways are induced as an auxiliary mechanism when mitochondrial β-oxidation is overloaded or defective. One of these is peroxisomal β-oxidation [6]. While peroxisomes normally oxidize specific carboxylic acids such as VLCFAs, branched-chain fatty acids, bile acid precursors, and fatty dicarboxylic acids (DCAs), they can also contribute to the β-oxidation of medium and long-chain fatty acids that are typically handled by mitochondria [7,8]. Under specific physiological or pathological conditions such as fasting or a mitochondrial FAO defect, respectively, long-chain fatty acids undergo microsomal ω-oxidation, generating DCAs, which are chemically defined by two carboxyl groups, one at each side of the aliphatic chain (α- and ω-carbons). DCAs then undergo peroxisomal β-oxidation [9–14], which generates chain-shortened DCAs and acetyl-CoA units. DCA β-oxidation is particularly prominent when lipid flux is increased and when the mitochondrial FAO is impaired, which is demonstrated by the increased levels of DCAs in urine (dicarboxylic aciduria, see DCAs in human disease). Thus, fatty acid ω-oxidation and subsequent DCA β-oxidation may be an important metabolic pathway under lipid metabolic stress. However, compared with the β-oxidation of monocarboxylic acids, ω-oxidation is a poorly understood metabolic pathway. Isolated defects in DCA metabolism have not been described yet and detailed information on the physiological significance of DCA metabolism is not available. The biochemical mechanism of DCA β-oxidation has only been partially resolved. In this review, we will describe the current knowledge on DCAs, including their metabolism, the compartmentalization of DCA metabolism, the physiological functions of DCAs, and the association of DCAs with human disease. We will also identify gaps in our knowledge and define future directions for research.
DCA generation by ω-oxidation of monocarboxylic acids
Historical perspective
Fatty acid ω-oxidation was first described by Verkade et al. [15]. They administered triundecanoin, a triglyceride made of undecanoic acid (C11 fatty acid), to healthy subjects, including Verkade himself. Unexpectedly, they found a large amount of undecanedioic acid (C11-DCA) excreted in the urine [15]. This was the first observation of ω-oxidation in vivo, but the biochemical pathway was still unknown. In 1961, three independent groups started to elucidate the mechanism of fatty acid ω-oxidation in vitro. Wakabayashi and Shimazono demonstrated the formation of a hydroxylated intermediate in the ω-oxidation of sorbic acid (C6:2) to muconic acid (C6:2-DCA). This reaction was catalyzed by an enzyme that they called a “mixed-function oxidase”, which was present in the microsomal fraction of the guinea pig liver [16]. Robbins reported similar findings for the ω-oxidation of medium-chain fatty acids (C8-C12) by mammalian liver and kidney homogenates to their corresponding DCAs and described in more detail the biochemical reactions [17]. The first reaction is the microsomal conversion of the monocarboxylic acid to the ω-hydroxy acid and requires NADPH (nicotinamide adenine dinucleotide phosphate, reduced) and oxygen (Figure 1). Mitz and Heinrikson isolated an NAD+-dependent alcohol dehydrogenase that catalyzed the second step, the ω-hydroxy acid conversion to an ω-oxo acid (Figure 1) [18]. In the last step, the ω-oxo acid is oxidized to the corresponding DCA in an NAD+-dependent reaction catalyzed by an aldehyde dehydrogenase (Figure 1) [19]. Other cofactors required in these reactions are ATP and Mg2+. Preiss and Bloch reported the first proof of ω-oxidation of physiological long-chain fatty acids in rat liver homogenates in 1964. While investigating the desaturation of stearate and palmitate to oleate and palmitoleate in rat liver preparations, they found that more polar metabolites were produced in smaller amounts. Among these polar metabolites, they identified ω-hydroxy acids, ω−1-hydroxy acids and DCA derivatives of these fatty acids [20]. Combined, these studies provide the basis for a more detailed description of the ω-oxidation pathway that came with the discovery of the role of cytochrome P450 (CYP) enzymes in the first step of ω-oxidation.
Figure 1.
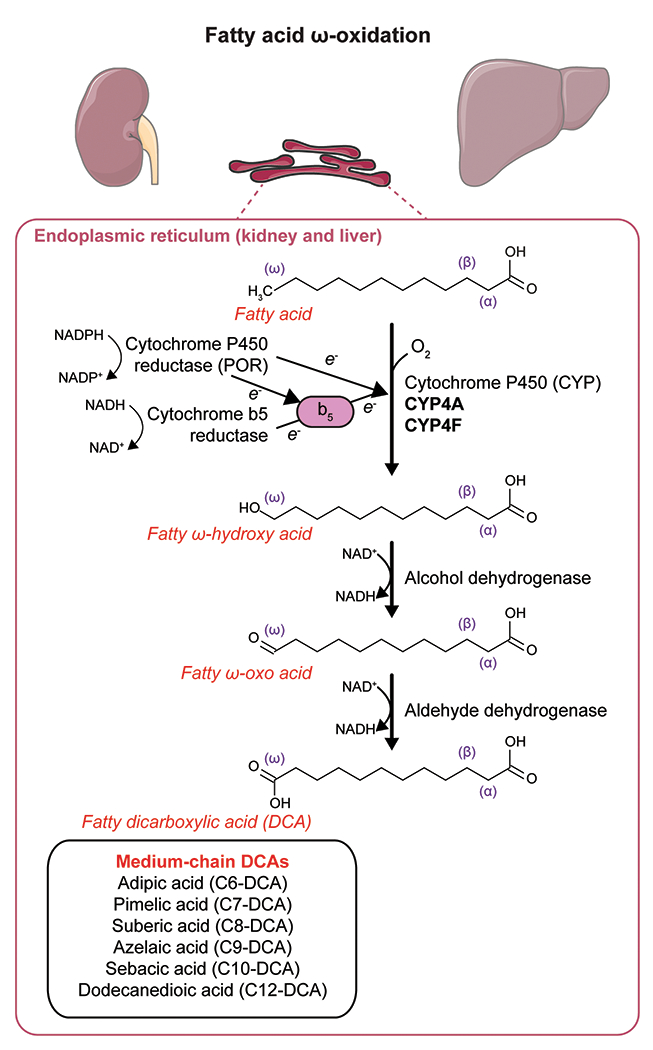
Enzymatic steps of fatty acid ω-oxidation using the example of lauric acid (C12) ω-oxidation to dodecanedioic acid (C12-DCA). This figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
The first reaction in ω-oxidation is the microsomal NADPH-dependent hydroxylation of the monocarboxylic acid to an ω-hydroxy acid (Figure 1). In the early 1960s, it was known that this reaction could take place in microsomes, i.e. artificially fragmented membrane vesicles that originate from the endoplasmic reticulum [21], but the identity of the enzyme was unknown. Two independent groups identified CYP and NADPH-cytochrome p450 oxidoreductase as two enzymes involved in the hydroxylation of lauric acid and stearic acid [22,23]. Later, the third component of the first hydroxylation reaction of ω-oxidation, NADH-cytochrome b5, was discovered [24].
CYP enzymes
CYP enzymes are well known for their role in hepatic drug metabolism and xenobiotic detoxification [25], but they also metabolize endogenous compounds. The CYP enzymes that catalyze the ω-oxidation of fatty acids to DCAs belong to the CYP4A and CYP4F subfamilies [26–28]. CYP4A11 and CYP4A22 enzymes in humans, and Cyp4a10, Cyp4a12a, Cyp4a12b and Cyp4a14 in mice, catalyze the 12-hydroxylation of lauric acid to 12-ω-hydroxy lauric acid [28], the first step in the generation of dodecanedioic acid (C12-DCA, Figure 1). The initial studies suggested that ω-oxidation was restricted to saturated fatty acids of medium-chain length [19,29]. The affinity of the ω-hydroxylation enzymes for saturated monocarboxylic acids is variable depending on the chain length of the fatty acid. Mortensen et al. reported a higher affinity for capric acid (C10, Km = 0.003 mM) and lauric acid (C12, Km = 0.2 mM) than for caprylic acid (C8, Km = 8.2 mM) or caproic acid (C6, Km = 8.8 mM) in microsomes isolated from rat liver [30]. However, CYP enzymes hydroxylate many different fatty acids besides medium-chain saturated fatty acids, and the substrate preference is different for each CYP enzyme.
Most studies have investigated the ω-hydroxylation of lauric acid (C12) by CYP4A enzymes. Besides lauric acid, human CYP4A11 is active toward myristic acid (C14), palmitic acid (C16), stearic acid (C18), oleic acid (C18:1 omega-9), and arachidonic acid (C20:4 omega-6) [31,32]. CYP4A22 is another CYP4A enzyme that is active toward lauric and palmitic acid [33]. CYP4F enzymes also have ω-hydroxylase activity toward fatty acids, including CYP4F2, the major arachidonic acid ω-hydroxylase [34,35]. CYP4F2, together with CYP4FB3, are also active toward very long-chain fatty acids (C>22), a peroxisomal substrate [27,36,37]. Phytanic acid, another peroxisomal substrate, can also be metabolized by ω-oxidation, which became evident from studies in Refsum disease patients [38,39]. In this case, the CYP enzymes that catalyze phytanic acid ω-oxidation in humans are CYP4F3A, CYP4F3B, CYP4F2, and CYP4A11 [40–42].
Regulation of ω-oxidation
The formation of DCAs by ω-oxidation and their subsequent β-oxidation is regulated by similar pathways, as exemplified by the parallel induction of peroxisomal β-oxidation enzymes and CYP-mediated microsomal ω-oxidation by hypolipidemic drugs of the fibrate class [6,43–46]. In this review, we will focus on the regulation of enzymes of the CYP4A and CYP4F families, the major fatty acid ω-hydroxylases. The expression of the most relevant CYP enzymes for fatty acid ω-oxidation (CYP4A11, CYP4A22, CYP4F2, CYP4F3) is particularly high in liver and kidney and, in the case of CYP4F enzymes, also in the small intestine. These organs rely heavily on FAO for energy homeostasis, in line with the supportive role of ω-oxidation. In human liver, the expression of CYP4A11 is significantly higher than CYP4A22 [47]. CYP4A11 is also the major lauric ω-hydroxylase in human liver [26].
Specific physiological and pathophysiological conditions such as fasting, a high-fat diet, or diabetes, induce the expression of CYP4A enzymes. The induction of CYP4A enzymes by fibrates and these different stimuli is mediated by peroxisome proliferator-activated receptor α (PPARα) [48–52]. PPARα is a member of the nuclear receptor family and regulates the expression of genes involved in fatty acid homeostasis. PPARα binds as a heterodimer with the retinoid X receptor (RXR) to peroxisome proliferator response elements (PPREs) in the promoter region of target genes [53]. CYP4A induction by PPARα is coordinated with the transcriptional induction of mitochondrial and peroxisomal β-oxidation enzymes [54], in line with the role of ω-oxidation in the facilitation of FAO. Free fatty acids are activating ligands of PPARα [55], thereby creating an autoregulatory loop in fatty acid homeostasis that regulates the activation of the microsomal, mitochondrial and peroxisomal fatty acid metabolizing pathways.
One interesting aspect of the effects of peroxisome proliferators and the induction of CYP4A enzymes by PPARα is the differential response between rodents and humans. Chronic exposure to peroxisome proliferators leads to hepatomegaly and hepatocellular carcinoma only in rodents and has been associated with a disproportionate increase in peroxisomal hydrogen peroxide production [45,56,57]. Induction of CYP4A11 has been described after overexpression of PPARα and treatment with a peroxisome proliferator in HepG2 cells [47]. However, treatment with peroxisome proliferators only induces CYP4A enzymes robustly in mouse hepatocytes and not in human hepatocytes [58,59], and PPREs have been characterized in rodent CYP4A genes but not in human CYP4A genes [54,60]. These data suggest that PPARα may not mediate CYP4A induction in humans. The development of a mouse model containing a human CYP4A11 transgene allowed the study of human CYP4A11 regulation in vivo [61]. CYP4A11 transgenic mice exhibited an increase in hepatic CYP4A11 mRNA and protein levels after fasting or treatment with fibrates in a PPARα-dependent manner [61]. Thus, the importance of PPARα-mediated regulation of CYP4A enzymes in humans remains to be determined.
The regulation of the CYP4F enzyme family shows some differences compared to the CYP4A family. Unlike members of the CYP4A family, peroxisome proliferators inhibit CYP4F2 gene promoter activity, whereas saturated fatty acids stimulate it in a PPARα-independent regulation [62]. Retinoic acids also regulate CYP4F2 gene activity, with activating and repressive functions mediated by RXRα heterodimers and RARα, respectively [63]. CYP4F2 gene activity is also induced by statins, drugs used for the treatment of hypercholesterolemia, in a process that is mediated by the activation of the sterol regulatory element-binding protein 2 (SREBP2) [64]. This regulatory mechanism by statins may also be shared by CYP4A enzymes [65]. In line with the known role of CYP4F in the metabolism of eicosanoids that act as inflammatory mediators, such as leukotriene B4 [66], CYP4F genes are also regulated by inflammatory cytokines [67].
The basal and inducible expression of CYP4A enzymes shows a marked sexual dimorphism, mainly in rodents. In mice, Cyp4a12 shows male-specific expression and a sex-specific differential response to fibrates, in which Cyp4a12 levels only increase in female mice after fibrate treatment [68,69]. Cyp4a12 expression is inducible by androgens in castrated males and females, especially in the kidney [70]. In the kidney, Cyp4a14 shows a female-specific pattern of expression, whereas Cyp4a10 expression is higher in females than in males varying with the strain of mice [69]. Moreover, Cyp4a14 KO mice present with male-specific hypertension associated with increased plasma androgens and increased Cyp4a12 expression in male KO kidneys [70]. In rats, Cyp4a2 expression in liver and kidney is male-specific, whereas Cyp4a1 and Cyp4a3 are induced to a greater degree in the liver of male rats after clofibrate treatment [71]. Female rats are also less susceptible to fibrate-induced peroxisome proliferation than males, as judged by the degree of liver hepatomegaly and the induction of peroxisomal β-oxidation enzymes [72,73]. How this sexual dimorphism affects DCA metabolism is not known yet. However, our recent studies in mice with a deficiency of EHHADH, an important peroxisomal enzyme for DCA β-oxidation, have identified sex-specific differences in the liver and kidneys [13,74] and sexual dimorphism of peroxisomal β-oxidation enzymes in mouse kidneys [74].
Besides their role in fatty acid catabolism, ω-oxidation catalyzed by CYP4A hydroxylases catalyzes the formation of arachidonic acid metabolites, which have been implicated in diverse biological functions, including the regulation of renal vasculature and ion channels. This aspect of ω-oxidation has been reviewed elsewhere [75] and is not part of this review’s scope. In this review, we focus on the biological roles of DCAs. To answer this question, we need to know how DCAs are further metabolized after their formation by ω-oxidation. Pettersen found in 1972 that palmitic acid (C16), a long-chain monocarboxylic acid, was the precursor of adipic acid (C6-DCA) excreted in the urine of ketotic rats [76]. Thus, DCAs formed by ω-oxidation of long-chain fatty acids are shortened by β-oxidation. The where and how of DCA β-oxidation have been debated for years. In the next section, we provide the current state-of-the-art on the location and mechanisms of DCA β-oxidation.
DCA β-oxidation
Historical perspective
The early studies of Verkade et al. already suggested that DCAs undergo β-oxidation. When they administered the odd-chain triglyceride triundecanoin, besides large amounts of C11-DCA, they also found small amounts of azelaic acid (C9-DCA) and pimelic acid (C7-DCA) in the urine [77]. The same phenomenon was observed for even-chain triglycerides. Administration of tricaprin, composed of C10 chains, led to the excretion of smaller amounts of suberic acid (C8-DCA) and adipic acid in the urine [77]. These medium-chain DCAs adipic and suberic acids are almost absent in normal human urine so the authors concluded that, most likely, longer-chain DCAs undergo β-oxidation to generate chain-shortened DCAs.
DCA β-oxidation was confirmed by several groups using fatty acids with isotope-labeled carbons. Pettersen detected 14C-labeled adipic acid in urine after administering 14C-labeled palmitic acid (C16) to ketotic rats, proving that long-chain monocarboxylic acids are the precursors of medium-chain DCAs [76]. Passi et al. detected, in serum and urine, 3H- or 14C-labeled DCAs that were 2, 4, or 6 carbon atoms shorter than the injected [10,11-3H]dodecanedioic acid or [1,9-14C]azelaic acid, respectively [78]. Cerdan et al. detected 13C-labeled chain-shortened DCAs derived from [1,2,11,12-13C4]dodecanedioic acid in situ in the liver of intact rats by nuclear magnetic resonance spectroscopy [79]. Thus, medium-chain DCAs are formed by ω-oxidation of long-chain fatty acids and subsequent β-oxidation. DCA β-oxidation can start from either carboxyl terminal group, a specific feature of dicarboxylic acids [80,81].
Mitochondrial DCA β-oxidation
The location of DCA β-oxidation has been a matter of debate for years. Different reports have shown that both mitochondria and peroxisomes participate in this process. [9,82–84]. In those first studies, DCA β-oxidation was thought to happen in peroxisomes and mitochondria, the latter being dependent on the carnitine shuttle [30,85]. However, even though DCA β-oxidation was measured in mitochondrial fractions, it was more efficient in the peroxisomal fraction [85], suggesting that the peroxisome was the major organelle for DCA β-oxidation.
Long-chain FAO in the mitochondrion requires activation of the fatty acid into an acyl-CoA and subsequent transport through the carnitine shuttle [1]. Even though mitochondria can β-oxidize long-chain DC-carnitines such as hexadecanedioylcarnitine (C16-DC-carnitine) in vitro under certain conditions, the reaction is 10 times slower than with palmitoylcarnitine (C16-carnitine) [86,87]. Moreover, O2 uptake with C16DC-carnitine is very low and transitory [86]. Vamecq and Draye observed that, in intact mitochondria from rat liver, dodecanedioic acid is β-oxidized only if the mitochondria had been previously permeabilized with digitonin [83]. Kølvraa and Gregersen reported the production of C4-DCA (succinic acid) in the mitochondria from exogenously added dodecanedioic acid and sebacic acid (C10-DCA) but not from suberic or adipic acid [9]. This could be explained because suberic and adipic acid cannot be transported across the mitochondrial membranes or due to the low affinity of the mitochondrial acyl-CoA dehydrogenases for the CoA esters of suberic and adipic acid. In contrast, exogenously added suberic and adipic acid were β-oxidized in the peroxisome due to the higher affinity of the peroxisomal acyl-CoA oxidase [9]. Moreover, the affinity of mitochondrial carnitine palmitoyltransferases toward DCA-CoA and DC-carnitines is much lower than for the corresponding monocarboxylic acids [10,86]. In a recent study, medium-chain acyl-CoA dehydrogenase (MCAD) was demonstrated to have activity with the CoA esters of dodecanedioic and adipic acid in vitro [88]. We recently provided evidence that DCAs are metabolized by mitochondrial fatty acid β-oxidation in mice when their peroxisomal import is not functional [14]. These data suggest that the mitochondrion contributes to DCA β-oxidation, but the extent of that contribution is relatively small. Mitochondrial β-oxidation of DCAs is restricted at the level of transport into the mitochondria and at the dehydrogenation step. It seems evident that mitochondrial DCA β-oxidation is less efficient than peroxisomal β-oxidation and unlikely to be of great physiological significance. However, mitochondria may play role when the peroxisomal machinery for DCA β-oxidation is defective.
Peroxisomal DCA β-oxidation
Lazarow and De Duve discovered that peroxisomes perform β-oxidation in the 70s [6]. They described a system that oxidizes monocarboxylic acids and is insensitive to cyanide, a poison that inhibits mitochondrial respiration [6]. Peroxisomal FAO follows a similar biochemical mechanism as mitochondrial FAO but with specific differences in the substrate specificity, the handling of reducing equivalents, the transport of fatty acids into the organelle, and the enzymes that participate in the process [2]. Most notably, the first step of peroxisomal β-oxidation is catalyzed by acyl-CoA oxidases (ACOX) that reduce oxygen to hydrogen peroxide illustrating the role of this organelle in peroxide metabolism [4,89]. Peroxisomal β-oxidation is induced by clofibrate and other hypolipidemic drugs that induce peroxisomal proliferation in the liver [90,91]. Besides DCAs, peroxisomal β-oxidation handles specific substrates such as VLCFA, bile acid precursors, and branched-chain fatty acids. Defects in peroxisome biogenesis or single peroxisomal enzymes illustrate the essential role of peroxisomes in human physiology. For more details about diseases caused by peroxisomal defects, the reader is referred to excellent reviews on this topic [2–4].
Mortensen and coworkers discovered one of the clues on the subcellular location of DCA β-oxidation. They found that dodecanedioic acid β-oxidation in rat liver was enhanced by clofibrate, an inducer of peroxisomal β-oxidation [92]. Another interesting observation of Mortensen and his team was that cyanide, a mitochondrial respiratory chain inhibitor, not only did not inhibit DCA β-oxidation but enhanced it, both in control and clofibrate-treated rats [92]. Therefore, they pointed to the peroxisome as the place where DCA β-oxidation occurs. When Mortensen and coworkers incubated peroxisomal fractions with dodecanedioic and sebacic acid, they observed an increase in the concentration of chain-shortened adipic and suberic acid, alongside a decrease in dodecanedioic and sebacic acid amounts [30]. Moreover, hypolipidemic drugs concomitantly increased CYP450 enzyme activity, content, and peroxisomal β-oxidation [56,93,94]. Three studies confirmed that peroxisomes are the primary site for DCA β-oxidation. These studies used human fibroblasts from patients with established mitochondrial or peroxisomal FAO defects [11], hepatocytes from mice with peroxisomal gene knockouts [12], and HEK-293 cells in which selected genes encoding for proteins involved in mitochondrial or peroxisomal β-oxidation were knocked out through CRISPR/Cas9 genome editing [13].
Multiple enzymes accommodating the different substrates can catalyze each of the β-oxidation steps in peroxisomes and mitochondria. The pathway for DCA β-oxidation in peroxisomes is composed of ABCD3, ACOX1, the two bifunctional proteins EHHADH and HSD17B4, and the two thiolases SCPx and ACAA1 (Figure 2). Several lines of evidence support the essential role of ABCD3 (also known as PMP70) in long-chain DCA transport into the peroxisome. First, the expression of the human ABCD3 transporter but not that of human ABCD1 or ABCD2 restored C16-DCA (hexadecanedioic acid) β-oxidation in the β-oxidation-deficient pxa1/pxa2Δ yeast mutant [95]. Second, ABCD3 KO HEK-293 cells are deficient in hexadecanedioic acid β-oxidation [13]. Finally, we recently reported the accumulation of long-chain DCAs in the liver of Abcd3 KO mice [14], providing in vivo evidence for the role of ABCD3 in DCA β-oxidation.
Figure 2.
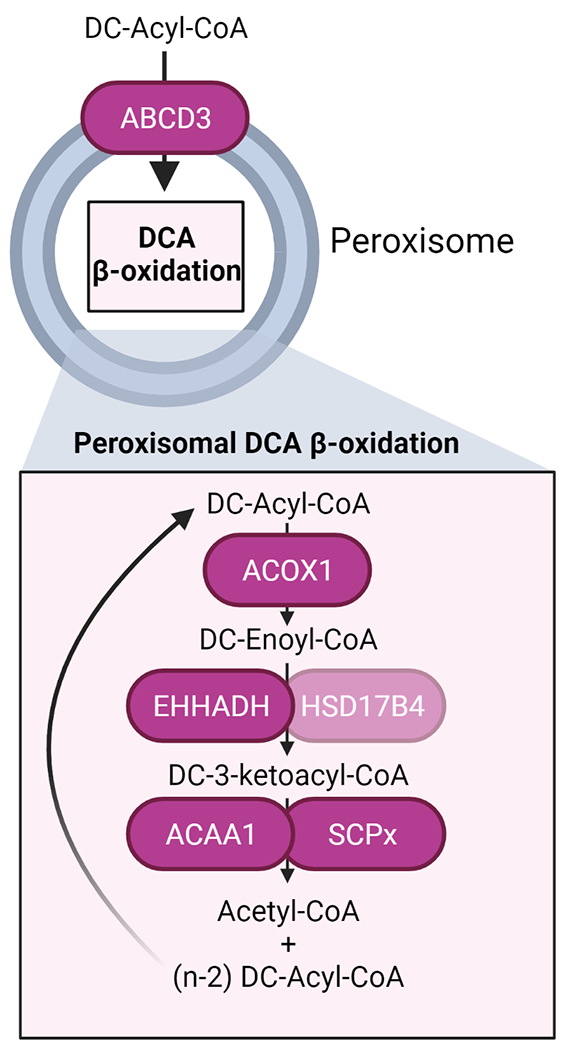
Schema of the peroxisomal steps in DCA β-oxidation with the name of the enzymes involved in each step. Created with BioRender.com
Acyl-CoA oxidases catalyze the first step in peroxisomal β-oxidation. Data from ACOX1 deficient patient fibroblasts and ACOX1 KO HEK-293 cells revealed a defect in hexadecanedioic acid β-oxidation suggesting that ACOX1 is essential for DCA β-oxidation [11,13]. The other two human acyl-CoA oxidases, ACOX2 and the recently identified ACOX3 [96], seem to play a less significant role in DCA β-oxidation. Although ACOX2 is active toward hexadecanedioyl-CoA in rat liver [97], there is no mention of DCA changes in three reports describing patients with ACOX2 deficiency [96,98,99] and in two patients with ACOX3 deficiency [100]. However, in vivo data supporting an essential role of ACOX1 in DCA β-oxidation are also lacking.
L- and D-bifunctional protein (EHHADH and HSD17B4, respectively) catalyze the second and third steps in peroxisomal β-oxidation. Several in vitro and in vivo studies suggest that EHHADH is more important than HSD17B4 in DCA β-oxidation. Fibroblasts from patients with DBP (HSD17B4) deficiency showed normal hexadecanedioic acid β-oxidation activity [11]. Because no patients with EHHADH deficiency have been reported yet, the same authors studied the activities of human EHHADH and HSD17B4 enzymes with enoyl-C16:1DC-CoA in the fox2Δ yeast mutant. The fox2 gene is the yeast ortholog of the human HSD17B4 gene, and these mutants cannot metabolize enoyl-CoAs to the corresponding keto-acyl-CoAs. Both human bifunctional proteins formed 3-keto-C16DC-CoA, but EHHADH had a 3-times lower apparent Km for C16:1DC-CoA than HSD17B4 [11]. We recently showed that EHHADH and HSD17B4 have overlapping functions in DCA β-oxidation in HEK-293 cells and that their contribution may depend mainly on their expression level [13]. The absence of patients with EHHADH deficiency makes animal models essential for studying EHHADH function. Dirkx et al. reported that tetradecanedioic acid (C14-DCA) β-oxidation was reduced in hepatocyte cultures from Ehhadh KO mice but not from Hsd17b4 KO mice [12]. These studies provided the first evidence that EHHADH has a specific role in DCA metabolism.
EHHADH expression is exceptionally inducible, and the highest levels of expression are found in liver and kidney, the same organs with the highest rates of ω-oxidation and peroxisomal β-oxidation. Consistent with this, a mouse gene coexpression subnetwork using Ehhadh as the central node contained other relevant genes for fatty acid ω-oxidation and subsequent DCA β-oxidation, such as Cyp4a10, Cyp4a14, Abcd3, Acaa1b, and Acot4, among others, suggesting these genes function in a coordinated metabolic network [101]. EHHADH was also identified as a regulatory gene in a CYP450 regulatory transcriptional network in human liver [102]. In vivo studies have confirmed the prominent role of EHHADH in DCA β-oxidation. When EHHADH-deficient mice were fed with a coconut oil diet enriched in medium-chain triglycerides or with lauric acid, the generation of DCAs through ω-oxidation with apparently impaired β-oxidation caused severe hepatic toxicity [103]. We recently reported that EHHADH-deficient mice present with an accumulation of medium-chain 3-OH-DCAs and an increased suberic/adipic acid ratio in plasma, liver and urine, indicating an impaired medium-chain DCA β-oxidation [13].
Little is known regarding the last step of peroxisomal DCA β-oxidation. Two thiolases catalyze this last step, SCPx and ACAA1. ACAA1 belongs to the so-called classical peroxisomal β-oxidation pathway, together with ACOX1 and EHHADH [101]. This pathway is highly inducible by PPARα-dependent mechanisms. Indeed, Acaa1b, one of the two ACAA1 thiolases present in mice, was present in the gene coexpression subnetwork of mouse Ehhadh, suggesting its involvement in DCA β-oxidation [101]. However, patients with rhizomelic chondrodysplasia punctata type I, which lack the mature form of ACAA1, do not present a defect in long-chain DCA β-oxidation [11], arguing against an essential role for ACAA1 in DCA β-oxidation in humans. We recently reported considerable functional overlap between SCPx and ACAA1 in hexadecanedioic acid β-oxidation in HEK-293 cells, with a more important role for ACAA1 in the β-oxidation of medium-chain DCAs [13].
Similar to monocarboxylic acids, DCAs are activated to an acyl-CoA ester before β-oxidation via a dicarboxylyl-CoA synthetase that was identified in the microsomal fraction of rat liver [104–106] and was not able to activate adipic and suberic acid to the corresponding DC-CoA [104]. However, the molecular identity of the dicarboxylyl-CoA synthetase remains obscure. Other work identified a peroxisomal acyl-CoA synthetase, although not specific for DCAs, on the cytoplasmic side of the peroxisome. The most likely candidates are the long-chain acyl-CoA synthetases ACSL1 and ACSL4 [107,108]. ACSL1 seems a good candidate since it is present in the Ehhadh-centered mouse gene coexpression subnetwork that contained other relevant genes for fatty acid ω-oxidation and subsequent DCA β-oxidation [101]. Interestingly, ACSL1 is present in different proteomics studies of hepatic peroxisomes [109,110], and some of these studies have identified an interaction between ACSL1 and ABCD3 [109,111], supporting the idea that ACSL1 activates DCAs to channel them into the peroxisomes. Another candidate is the long-chain acyl-CoA synthetase FATP2 (also known as ACSVL1, and encoded by the SLC27A2 gene), which has been identified in the endoplasmic reticulum and in the peroxisome [112,113]. However, the role of these acyl-CoA synthetases in peroxisomal DCA metabolism has not been studied yet.
The β-oxidation rates of DCAs decrease as the chain length decreases. Different experiments have shown that suberic and, in particular, adipic acid, are metabolized at much lower rates than longer chain DCAs [10,114–117], which is likely one of the reasons for their appearance in urine. Other reasons for their appearance in urine may be related to their glomerular filtration and tubular reuptake. Fatty acid ω-oxidation and peroxisomal β-oxidation present a very similar specificity pattern, being less active with carbon chains ≤ 8 [118,119]. It has been debated whether fatty acid chain-shortening proceeds beyond C6 in the peroxisome [118,119]. The detection of 3-hydroxy-adipic acid and its derivative 3-hydroxyadipic acid 3,6-lactone in the urine of man and mice [13,120] suggests that peroxisomal DCA β-oxidation proceeds beyond adipic acid, thus generating the anaplerotic substrate succinic acid. We have discussed this possibility in detail in the next section.
The physiological role of DCAs
The physiological relevance of DCA metabolism and ω-oxidation are tightly connected, which is illustrated by the fact that ω-oxidation and urinary DCA excretion are increased under conditions in which the FAO flux is increased, such as fasting, diabetes, high-fat feeding, and after treatment with hypolipidemic drugs of the fibrate class [28,44,121–125]. Under these conditions, ω-oxidation is induced predominantly in metabolically demanding organs such as liver and kidney [19,20,126,127]. The general biological relevance of ω-oxidation has been reviewed by Miura [128]. Here, we will review the physiological roles of fatty DCAs in metabolism, focusing on the role of the intermediates and products generated by the peroxisomal β-oxidation of DCAs.
Energy homeostasis and generation of essential intermediary metabolites
The ω-oxidation of medium-, long-, and very long-chain fatty acids has received considerable interest because this pathway may represent an alternative way to reduce the accumulation of toxic fatty acids in conditions with increased lipid flux, such as fasting, diabetes, and a high-fat diet. However, the physiological role of fatty acid ω-oxidation remains poorly defined. One way to address this problem is to estimate the contribution of ω-oxidation and peroxisomal DCA β-oxidation to total FAO. A first study performed by Antony and Landau in 1968 reported that ω-oxidation contributed 4% to total stearate (C18) oxidation in rat liver slices [129]. However, they did not consider β-oxidation from the original carboxyl group after ω-oxidation of the fatty acid. Thus, the actual contribution of ω-oxidation was not properly calculated. Other studies in liver of fasted or diabetic animals estimated a relative contribution of ω-oxidation that reached up to 20% of total FAO, depending on the length of the fatty acid used and the experimental conditions, being highest with lauric acid in conditions of increased lipid flux such as starvation and diabetes [81,130,131]. In other tissues with high rates of FAO, such as heart or skeletal muscle, the contribution of DCA β-oxidation to total FAO is very low, sometimes even undetectable [86,87,114,132], most likely as a result of the low level of ω-oxidation in these organs. These data suggest that the contribution of ω-oxidation and subsequent DCA β-oxidation to total FAO is relatively minor in normal conditions but becomes physiologically relevant in conditions of metabolic stress caused by an increased lipid flux, especially in liver and kidney.
Anaplerosis
Anaplerosis is the addition of intermediates to the TCA cycle, which compensates for pathways that consume TCA cycle intermediates (cataplerosis), such as gluconeogenesis or lipid synthesis [133]. DCA metabolism received attention because of its potential contribution to anaplerosis. This is in stark contrast to monocarboxylic fatty acids, which are not anaplerotic. It is generally accepted that peroxisomal β-oxidation of DCAs yields adipic and suberic acid as end-products that are lost in the urine. However, if DCA β-oxidation proceeds for one or two more cycles, respectively, the resulting product succinic acid can enter the TCA cycle [134]. However, the generation of succinic acid in peroxisome DCA β-oxidation has remained controversial. Peroxisomal succinic acid production has been detected after administering longer-chain DCAs even though the affinity of peroxisomal acyl-CoA oxidase towards adipic acid is much lower than for longer-chain DCAs [9,10,135,136]. Wada and Usami’s study with 14C-labeled DCAs found a gluconeogenic potential of DCAs that was even higher when the rats were starved or diabetic [81]. Mortensen’s observations further corroborated this hypothesis by showing that medium-chain DCAs, such as adipic acid, also showed gluconeogenic potential [137,138]. Several studies using stable isotope-labeled DCAs were consistent with this hypothesis and detected labeled carbons in TCA intermediates, such as succinate and citrate, after administration of the labeled DCA, supporting the previous findings of the anaplerotic potential of DCAs [80,85,114]. Other studies did not find such a contribution [79,139]. More recently, Jin et al. used mass isotopomer analysis and demonstrated the anaplerotic potential of dodecanedioic acid [140]. We documented the excretion of 3-hydroxyadipic acid in the urine of fasted mice [13]. Together with the low but detectable labeling of TCA cycle intermediates in mouse liver slices after incubation with [U-13C]-dodecanedioic acid [13], these data indicate that adipic acid can indeed undergo another round of peroxisomal β-oxidation. These findings, together with the discovery of ACOT4, a peroxisomal acyl-CoA thioesterase with a specific affinity for succinyl-CoA [141,142], provide substantial evidence to support the notion that peroxisomal DCA β-oxidation can generate succinate and thus provide substrates for anaplerosis. In addition, a recent study has suggested that dodecanedioic acid administration can refill the TCA cycle in fibroblasts from patients with very-long chain acyl-CoA dehydrogenase (VLCAD) deficiency [143].
The anaplerotic potential of DCAs and their potential ease of administration (medium-chain DCAs are water-soluble in contrast to long- and medium-chain triglycerides that have to be administered as emulsions) motivated several studies to investigate their use as nutritional compounds [144]. One study showed that odd- and even-chain DCAs were well tolerated and did not provoke side effects in volunteers [144]. Moreover, infusion of dodecanedioic acid in diabetic patients decreased plasma glucose significantly, suggesting that DCAs may be useful as fuel substrates for diabetes patients. However, the anaplerotic potential of DCA β-oxidation under different physiological and pathophysiological situations remains to be established.
Destination of peroxisomal acetyl-CoA units: Cholesterol and lipid metabolism
Peroxisomal DCA β-oxidation generates acetyl-CoA units, presumably released in the form of acetate or acetylcarnitine [145–147]. Administration of DCAs of different chain lengths increases the extramitochondrial pool of acetyl-CoA [115,135,145,148,149]. The most plausible source of this pool of acetyl-CoA is peroxisomal DCA β-oxidation. One of the suggested destinations of this pool of acetyl-CoA is the biosynthesis of cholesterol [149,150]. Recently we confirmed that disruption of peroxisomal DCA β-oxidation in Ehhadh KO and Abcd3 KO mice induces alterations in hepatic cholesterol synthesis [13,14]. In other tissues, such as the heart, peroxisome-derived acetyl-CoA contributes substantially to the cardiac malonyl-CoA pool, thus participating in the regulation of fatty acid oxidation and lipogenesis [151,152]. Alternatively, acetyl-CoA is converted into acetylcarnitine by the peroxisomal carnitine acetyltransferase (CRAT), with subsequent transport to and metabolism in the mitochondrion [147,153]. Peroxisomal DCA β-oxidation may also participate in protein acetylation and succinylation, important regulatory mechanisms that control metabolic processes [154,155].
DCAs in human disease
Another fact that has attracted much attention to DCA metabolism is the detection of significant amounts of medium-chain or long-chain DCAs in urine under various pathological conditions. This is relevant because DCA levels are usually low in the urine of healthy subjects and animals [13,14,122,156]. Currently, dicarboxylic aciduria is a metabolic feature that is helpful for the diagnosis of several inborn errors of metabolism [156], particularly mitochondrial FAO defects. However, they have also been detected in cases of glycogen storage disorders and HMG-CoA lyase deficiency [156]. Urinary DCAs can also be present in acquired conditions such as Reye-like and Reyés syndrome, Jamaican vomiting disease [157], celiac disease [158], and diabetes [159], or even under non-pathological conditions, such as after medium-chain triglyceride (MCT) administration [160,161], or after prolonged fasting [76,122].
In Reyés syndrome patients, the urinary excretion of DCAs appeared to decrease with increasing chain length, with adipic and suberic acid the most abundant DCAs [162,163]. Strikingly, long-chain DCAs were the most abundant in the serum of comatose patients with Reye syndrome [162], suggesting that the chain length of the accumulated DCA may be associated with the severity of the disease. Accumulating medium-chain DCAs themselves can also be toxic and result in liver failure. This was demonstrated in Ehhadh KO mice fed with lauric acid or a coconut oil diet (rich in lauric acid), which accumulate high levels of dodecanedioic acid [103]. In summary, dicarboxylic aciduria may hint at an underlying biochemical defect behind a clinical presentation, but cannot be used as the only biochemical parameter to diagnose an inherited metabolic disease. Moreover, the effects of DCA accumulation on human health are poorly understood. In this section, we review the role that DCA metabolism plays in different diseases.
DCA metabolism in mitochondrial FAO defects
Abnormal large amounts of medium-chain DCAs are detected in the urine of patients with a mitochondrial FAO defect. Dicarboxylic aciduria is a consistent biochemical feature in cases of systemic carnitine deficiency [164], CACT (carnitine acylcarnitine translocase) deficiency [165], CPT2 (carnitine palmitoyltransferase 2) deficiency [166], MADD (multiple acyl-CoA dehydrogenase deficiency) [167], VLCAD deficiency [168], MCAD deficiency [169], LCHAD or MTP (long-chain 3-hydroxyacyl-CoA dehydrogenase/mitochondrial trifunctional protein) deficiency [170] and some cases of CPT1A (carnitine palmitoyltransferase 1A) deficiency [171]. A recent report found increased levels of hexadecanedioic acid in dried blood spots from five severe VLCAD deficiency patients compared with nine mild cases [172]. The presence of urinary DCAs is also characteristic of mouse models of mitochondrial FAO defects [13,173–176]. The most likely biochemical explanation for the increased DCA excretion in many mitochondrial FAO disorders is an increased compensatory fatty acid ω-oxidation and subsequent peroxisomal DCA β-oxidation [92,177]. Another common feature in some mitochondrial FAO disorders is the excretion of unsaturated DCAs originating from oleic and linoleic acids [175,178–181].
Besides urinary DCAs, medium-chain dicarboxylylcarnitines (DC-carnitines) are present in mitochondrial FAO defects, such as severe cases of CPT2 deficiency [166], CACT deficiency [165] and the LCAD (long-chain acyl-CoA dehydrogenase) KO mouse model [13,182]. C6-C12 DC-carnitines have also been detected in the urine of one Reye syndrome case without an underlying metabolic disorder [183]. The metabolic explanation for the presence of these medium-chain DC-carnitines is still unknown. We hypothesize that these DC-carnitines are generated in the peroxisome given the high rate of DCA β-oxidation due to the mitochondrial defects. Indeed, peroxisomes possess the carnitine acyltransferases CRAT and CROT, which are theoretically capable of generating these metabolites [184]. Although CROT is the most likely origin of the DC-carnitines, this remains to be experimentally tested [147]. Another possibility we cannot exclude is that monocarboxylylcarnitines generated from the mitochondrial FAO defect undergo ω-oxidation in the microsomes to generate the DC-carnitines present in plasma and urine.
Impact of DCA metabolism on mitochondrial function
Mitochondria are not well equipped to metabolize DCAs. Thus, a substrate overload of DCAs may have negative consequences on the function of this organelle. For example, the DCA-rich serum of patients with Reye’s syndrome inhibits mitochondrial respiration and ATP synthesis in isolated mitochondria, acting as a mitochondrial uncoupler [185]. The amount of DCAs in the serum of Reye’s syndrome corresponded directly to the reduction in ATP synthesis. Moreover, substantial serum concentrations of DCAs may not be bound to albumin, increasing the potential toxicity of these metabolites [186]. The antimicrobial action of azelaic acid (C9-DCA), which is used in treating acne and hyperpigmentary disorders [187], is in part mediated by an inhibition of mitochondrial respiration and some mitochondrial respiratory enzymes. This effect was observed in rat liver hepatocytes after adding dodecanedioic acid and other medium-chain DCAs [188]. Sebacic acid induced swelling of respiring mitochondria [189].
Recently, we reported that the mitochondrial FAO machinery metabolizes DCAs in Abcd3 KO mice, a mouse model of defective peroxisomal DCA β-oxidation [14]. Alongside the detection of medium-chain DCAs and dicarboxylylcarnitines in plasma and liver of Abcd3 KO mice, we also observed a decrease in the production of medium-chain dicarboxylylcarnitines in liver slices from Abcd3 KO mice when treated with the CPT2 inhibitor L-aminocarnitine [14]. These results support that DCAs can be metabolized in mitochondria under exceptional circumstances. The mechanisms of DCA import into the mitochondria will need to be established to fully understand the effects of DCAs on mitochondrial function. Besides the low-efficient carnitine shuttle for DCA transport into the mitochondria, other mechanisms have been proposed [189]. Overall, the mechanisms of mitochondrial metabolism of medium- and long-chain DCAs remain largely unknown.
DCA metabolism in peroxisomal disorders
Urinary organic acid analysis is also helpful in the diagnostics of peroxisomal disorders. The central role of peroxisomes in DCA β-oxidation is illustrated by the presence of high levels of long-chain DCAs in urine and long-chain DC-carnitines in plasma (C16- to C22DC-carnitine) in peroxisome biogenesis disorders (PBD) patients [190–192]. We made similar observations in the liver of Abcd3 KO mice [14]. Plasma long-chain DC-carnitines in patients with DBP (HSD17B4) deficiency are comparable to controls [190], indicating that DCA β-oxidation is functionally normal in DBP deficiency, and thus supporting a major role for EHHADH in DCA β-oxidation. Interestingly, long-chain DC-carnitines accumulate in patients with peroxisome biogenesis defects [190], who do not have β-oxidation enzymes in the peroxisomal matrix, suggesting that, under some conditions, CPT1 may handle long-chain DC-CoAs. This is supported by our observation of increased octadecenedioylcarnitine (C18:1-DC) in the liver of Abcd3 KO mice [14].
Patients with PBD also excrete adipic acid, suberic acid, and sebacic acid in the urine [192–195]. The presence of these medium-chain DCAs (C6-C10) in the urine of patients who do not have functional peroxisomes suggests that DCA β-oxidation is not exclusively a peroxisomal process and is consistent with our recent findings in the Abcd3 KO mice, which also accumulate medium-chain DCAs [14]. The pattern of DCA excretion in peroxisomal disorders differs from mitochondrial FAO defects, where adipic acid is the most abundant DCA in urine, followed by suberic acid and sebacic acid. In contrast, PBD patients present a relatively more prominent accumulation of suberic acid and sebacic acid than adipic acid [194], pointing to a block in the conversion of suberic and sebacic acid into adipic acid due to the lack of functional peroxisomes. Data in mouse models of peroxisomal dysfunction are consistent with this impairment in sebacic and suberic acid β-oxidation, as shown by the increase in the suberic/adipic acid ratio in Ehhadh KO mice [13], and the increased levels of suberic and sebacic acid in the urine of Abcd3 KO mice [14].
Patients with peroxisomal disorders also excrete odd-chain (C7-C15) DCAs [192,194]. The origin of these odd-chain DCAs has not been elucidated yet. They may arise from ω-oxidation of odd-chain fatty acids from dietary origin (such as dairy fat), or as a result of increased odd-chain fatty acid synthesis using propionyl-CoA as a primer, or from decarboxylation of 2-hydroxy fatty acids [192,196,197]. Other groups have proposed that medium-chain odd-numbered DCAs are generated by the peroxidation of polyunsaturated fatty acids [198], thus reflecting lipid peroxidation. However, the specific detection of odd-chain DCAs in peroxisomal disorders with chain lengths longer than 9 carbon atoms strongly suggests that these metabolites are generated in a mechanism similar to even-chain DCAs, i.e. through β-oxidation.
Patients with defects in α-oxidation of phytanic acid to pristanic acid (Refsum disease) excrete 3-methyladipic acid in the urine [38,39]. Moreover, the levels of urinary 3-methyladipic acid correlate with the plasma levels of phytanic acid [199], indicating that phytanic acid can undergo ω-oxidation. Therefore, ω-oxidation is an alternative pathway that enables the metabolism of a fraction of the accumulated phytanic acid. In fact, fatty acid ω-oxidation has been proposed as a rescue pathway in mitochondrial and peroxisomal fatty acid oxidation disorders, aiming to reduce the detrimental effects caused by the accumulation of fatty acids in these disorders [200].
DCA and diabetes
Available evidence supports that alterations in FAO are associated with insulin resistance and thus may contribute to the development of diabetes. Diabetic patients display high rates of FAO, reflected by elevated levels of acylcarnitines [201]. Inhibition of mitochondrial FAO induces the accumulation of ectopic lipids in the skeletal muscle, which is associated with an impairment in insulin signaling [201,202], and a compensatory increase in peroxisomal FAO [203,204]. Two recent reports in mice have shown that the expression of genes involved in fatty acid ω-oxidation and peroxisomal β-oxidation is increased in the liver and kidney of streptozocin-induced diabetic mice [205,206], but DCA levels in plasma or urine were not reported. Under these conditions, one would expect an induction of ω-oxidation. Indeed, diabetic rats and humans present high amounts of medium-chain DCAs in urine and DC-carnitines in plasma and a higher rate of DCA β-oxidation [123,138,159,207–209]. Excessive peroxisomal DCA metabolism would lead to an increased production of hydrogen peroxide, which is formed in the first reaction of peroxisomal β-oxidation [89]. It has been proposed that peroxisome-derived hydrogen peroxide is responsible for pancreatic β-cell lipotoxicity [210]. However, the role of DCAs in the pathophysiology of diabetes is not known yet.
DCAs have been proposed as an alternative nutritional substrate for type 2 diabetes patients based on the idea that their anaplerotic potential will improve glucose uptake in skeletal muscle and thus help prevent hyperglycemia. Animal and human studies have shown that administration of sebacic acid reduces postprandial glycemia in type 2 diabetes patients [211], and improves fasting glycemia and glucose tolerance in diabetic db/db mice [212]. Administration of dodecanedioic acid reduced muscle fatigue during exercise in diabetic patients [213]. Along similar lines, the risk of diabetes in a Chinese population was inversely associated with C10DC-carnitine and C12DC-carnitine plasma levels [214]. These findings suggest that DCAs improve energy utilization and metabolic flexibility. However, this possible beneficial effect of DCAs and the mechanisms behind them need to be better understood and deserve further studies.
Cardiovascular disease
Several studies have reported associations between DCA metabolism and cardiovascular disease. Shah et al. found an association between short- and long-chain DC-carnitines plasma levels and adverse cardiovascular events [215]. Elevated plasma tetradecanedioic and hexadecanedioic acid levels are associated with ischemic stroke [216]. Ruiz et al. found increased DC-carnitine levels (C6DC-, C16DC-, and C18DC-carnitine) together with increased circulating C26-carnitine (a metabolite that accumulates due to peroxisomal dysfunction) in two cohorts of patients with heart failure [217]. Furthermore, plasma levels of hexadecanedioic acid are associated with an increased risk of heart failure in two independent cohorts of patients [218], and also with high systolic and diastolic blood pressure levels [219]. These observations are supported by studies in rats indicating that hexadecanedioic acid increases blood pressure levels when added to the diet [219]. Abnormally elevated levels of long-chain DCAs (C14-, C16-, and C18-DCA) are also present in the lungs of patients with pulmonary arterial hypertension [220]. These data suggest that DCA metabolism plays a relevant role in cardiometabolic health. However, fatty acid ω-oxidation rates are low in the heart, suggesting that the generation of DCAs in cardiac tissue is less likely to be the direct cause of these cardiometabolic problems. Despite the low levels of fatty acid ω-oxidation in the heart, it has been shown that peroxisomal FAO can contribute to the acetyl moiety of cardiac malonyl-CoA [151], and that dodecanedioic acid β-oxidation contributes to the acetyl moiety of cardiac malonyl-CoA [152]. These results indicate that DCA β-oxidation may play a role in the cardiovascular system.
The mechanism behind the association between DCA metabolism and blood pressure changes is not clear. 20-HETE (20-hydroxyeicosatetraenoic acid), a major player in blood pressure regulation [221], is formed by ω-oxidation of arachidonic acid by CYP450 enzymes, mainly CYP4F2 and CYP4A11 [35]. A loss-of-function SNP in CYP4A11 is associated with hypertension [222], and mice deficient in Cyp4a10 and Cyp4a14 (CYP4A11 orthologs) are more prone to develop hypertension [70,223]. Given the known association between hexadecanedioic acid and blood pressure [218,219], we speculate that peroxisomal DCA β-oxidation may play a role in the control of blood pressure. The underlying mechanisms have not been elucidated yet, but the availability of animal models defective in DCA β-oxidation provides an excellent opportunity to investigate this association further.
Conclusion
Fatty acid ω-oxidation, defined as the biological oxidation of fatty acids at the ω-carbon, is a well-established pathway in animals and plants. The mechanism of ω-oxidation of long-chain monocarboxylic acids has been elucidated in detail. However, the biological relevance of DCA metabolism remains largely unknown. In particular, there is a lack of understanding of the physiological consequences of alterations in this metabolic pathway. Future studies should aim to fill this knowledge gap. Animal models that are deficient in one of the components of this pathway, such as the Abcd3, Ehhadh, Cyp4a10 and Cyp4a14 KO mice, are a great resource for investigating this aspect of DCA metabolism. A better understanding of fatty DCA metabolism may help the development of therapies for inborn errors of metabolism and other metabolic diseases in which DCAs may play a role.
Funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01 DK113172. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- CPT
carnitine palmitoyltransferase
- CYP
cytochrome P450
- DCA
dicarboxylic acid
- FAD
flavin adenine dinucleotide
- FAO
fatty acid oxidation
- MCAD
medium-chain acyl-CoA dehydrogenase
- MCT
medium-chain triglyceride
- NAD
nicotinamide adenine dinucleotide
- NADP
nicotinamide adenine dinucleotide phosphate
- PBD
peroxisome biogenesis disorder
- TCA
tricarboxylic acid
- VLCAD
very long-chain acyl-CoA dehydrogenase
- VLCFA
very long-chain fatty acids
REFERENCES
- 1.Houten SM, Violante S, Ventura FV and Wanders RJA (2016) The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol 78(1):23–44. doi: 10.1146/annurev-physiol-021115-105045. [DOI] [PubMed] [Google Scholar]
- 2.Wanders RJA and Waterham HR (2006) Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem 75(1):295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- 3.Waterham HR, Ferdinandusse S and Wanders RJA (2016) Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta - Mol. Cell Res 1863(5):922–933. doi: 10.1016/j.bbamcr.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Wanders RJA, Baes M, Ribeiro D, Ferdinandusse S and Waterham HR (2023) The physiological functions of human peroxisomes. Physiol. Rev 103(1):957–1024. doi: 10.1152/physrev.00051.2021. [DOI] [PubMed] [Google Scholar]
- 5.Spiekerkoetter U, Bastin J, Gillingham M, Morris A, Wijburg F and Wilcken B (2010) Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J. Inherit. Metab. Dis 33(5):555–561. doi: 10.1007/s10545-010-9188-1. [DOI] [PubMed] [Google Scholar]
- 6.Lazarow PB and De Duve C (1976) A fatty acyl CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. U. S. A 73(6):2043–2046. doi: 10.1073/pnas.73.6.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Violante S, IJlst L, te Brinke H, Koster J, Tavares de Almeida I, Wanders RJA, Ventura FV, Houten SM, De Almeida IT, … Houten SM (2013) Peroxisomes contribute to the acylcarnitine production when the carnitine shuttle is deficient. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1831(9):1467–1474. doi: 10.1016/j.bbalip.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Violante S, Achetib N, van Roermund CWT, Hagen J, Dodatko T, Vaz FM, Waterham HR, Chen H, Baes M, … Houten SM (2019) Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 33(3):4355–4364. doi: 10.1096/fj.201801498R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kølvraa S and Gregersen N (1986) In vitro studies on the oxidation of medium-chain dicarboxylic acids in rat liver. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 876(3):515–525. doi: 10.1016/0005-2760(86)90039-1. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki H, Yamada J, Watanabe T and Suga T (1989) Compartmentation of dicarboxylic acid β-oxidation in rat liver: importance of peroxisomes in the metabolism of dicarboxylic acids. Biochim. Biophys. Acta - Gen. Subj 990(1):25–30. doi: 10.1016/S0304-4165(89)80007-8. [DOI] [PubMed] [Google Scholar]
- 11.Ferdinandusse S, Denis S, van Roermund CWT, Wanders RJA and Dacremont G (2004) Identification of the peroxisomal β-oxidation enzymes involved in the degradation of long-chain dicarboxylic acids. J. Lipid Res 45(6):1104–1111. doi: 10.1194/jlr.M300512-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Dirkx R, Meyhi E, Asselberghs S, Reddy J, Baes M and Van Veldhoven PP (2007) β-Oxidation in hepatocyte cultures from mice with peroxisomal gene knockouts. Biochem. Biophys. Res. Commun 357(3):718–723. doi: 10.1016/j.bbrc.2007.03.198. [DOI] [PubMed] [Google Scholar]
- 13.Ranea-Robles P, Violante S, Argmann C, Dodatko T, Bhattacharya D, Chen H, Yu C, Friedman SL, Puchowicz M and Houten SM (2021) Murine deficiency of peroxisomal l-bifunctional protein (EHHADH) causes medium-chain 3-hydroxydicarboxylic aciduria and perturbs hepatic cholesterol homeostasis. Cell. Mol. Life Sci 78(14):5631–5646. doi: 10.1007/s00018-021-03869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ranea-Robles P, Chen H, Stauffer B, Yu C, Bhattacharya D, Friedman SL, Puchowicz M and Houten SM (2021) The peroxisomal transporter ABCD3 plays a major role in hepatic dicarboxylic fatty acid metabolism and lipid homeostasis. J. Inherit. Metab. Dis 44(6):1419–1433. doi: 10.1002/jimd.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verkade PE, Elzas M, Van Dee Lee J, Wolff H. H. De, Verkade-sandbergen A and Van Der Sande D (1933) Fat metabolism. 1. Hoppe. Seylers. Z. Physiol. Chem 215225–257. [Google Scholar]
- 16.Wakabayashi K and Shimazono N (1961) Studies in vitro on the mechanism of ω-oxidation of fatty acids. BBA - Biochim. Biophys. Acta 48(3):615–617. doi: 10.1016/0006-3002(61)90069-5. [DOI] [PubMed] [Google Scholar]
- 17.Robbins KC (1961) Enzymatic omega oxidation of fatty acids. Fed. Proc 20(272):. [Google Scholar]
- 18.Mitz MA and Heinrikson RL (1961) Omega hydroxy fatty acid dehydrogenase. BBA - Biochim. Biophys. Acta 46(1):45–50. doi: 10.1016/0006-3002(61)90644-8. [DOI] [PubMed] [Google Scholar]
- 19.Robbins KC (1968) In vitro enzymic omega oxidation of medium-chain fatty acids in mammalian tissue. Arch. Biochem. Biophys 123(3):531–538. doi: 10.1016/0003-9861(68)90174-4. [DOI] [PubMed] [Google Scholar]
- 20.PREISS B and BLOCH K (1964) Omega-oxidation of long chain fatty acids in rat liver. J. Biol. Chem 23985–88. doi: 10.1016/s0021-9258(18)51750-6. [DOI] [PubMed] [Google Scholar]
- 21.Palade GE and Siekevitz P (1956) Liver microsomes; an integrated morphological and biochemical study. J. Biophys. Biochem. Cytol 2(2):171–200. doi: 10.1083/jcb.2.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wada F, Hirata K, Shibata H, Higashi K and Sakamoto Y (1967) Involvement of P-450 in Several Reactions of Lipid Metabolism in Liver Microsomes. J. Biochem 62(1):134–136. doi: 10.1093/oxfordjournals.jbchem.a128627. [DOI] [PubMed] [Google Scholar]
- 23.Lu AY and Coon MJ (1968) Role of hemoprotein P-450 in fatty acid omega-hydroxylation in a soluble enzyme system from liver microsomes. J. Biol. Chem 243(6):1331–1332. doi: 10.1016/s0021-9258(19)56992-7. [DOI] [PubMed] [Google Scholar]
- 24.Hildebrandt A and Estabrook RW (1971) Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions. Arch. Biochem. Biophys 143(1):66–79. doi: 10.1016/0003-9861(71)90186-x. [DOI] [PubMed] [Google Scholar]
- 25.Cooper DY, Levin S, Narasimhulu S, Rosenthal O and Estabrook RW (1965) Photochemical action spectrum of the terminal oxidase of mixed function oxidase systems. Science (80-.) 147(3656):400–402. doi: 10.1126/science.147.3656.400. [DOI] [PubMed] [Google Scholar]
- 26.Powell PK, Wolf I and Lasker JM (1996) Identification of CYP4A11 as the major lauric acid ω-hydroxylase in human liver microsomes. Arch. Biochem. Biophys 335(1):219–226. doi: 10.1006/abbi.1996.0501. [DOI] [PubMed] [Google Scholar]
- 27.Sanders RJ, Ofman R, Duran M, Kemp S and Wanders RJA (2006) ω-oxidation of very long-chain fatty acids in human liver microsomes: Implications for X-linked adrenoleukodystrophy. J. Biol. Chem 281(19):13180–13187. doi: 10.1074/jbc.M513481200. [DOI] [PubMed] [Google Scholar]
- 28.Hardwick JP, Song BJ, Huberman E and Gonzalez FJ (1987) Isolation, complementary DNA sequence, and regulation of rat hepatic lauric acid ω-hydroxylase (cytochrome P-450(LAω)). Identification of a new cytochrome P-450 gene family. J. Biol. Chem 262(2):801–810. doi: 10.1016/s0021-9258(19)75857-8. [DOI] [PubMed] [Google Scholar]
- 29.Kosuke I, Emi K and Masamichi K (1969) Some properties and distribution of the ω-hydroxylation system of medium-chain fatty acids. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 176(4):704–712. doi: 10.1016/0005-2760(69)90251-3. [DOI] [PubMed] [Google Scholar]
- 30.Mortensen PB (1992) Formation and degradation of dicarboxylic acids in relation to alterations in fatty acid oxidation in rats. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 1124(1):71–79. doi: 10.1016/0005-2760(92)90128-I. [DOI] [PubMed] [Google Scholar]
- 31.Palmer CN, Richardson TH, Griffin KJ, Hsu MH, Muerhoff AS, Clark JE and Johnson EF (1993) Characterization of a cDNA encoding a human kidney, cytochrome P-450 4A fatty acid omega-hydroxylase and the cognate enzyme expressed in Escherichia coli. Biochim. Biophys. Acta 1172(1–2):161–6. doi: 10.1016/0167-4781(93)90285-1. [DOI] [PubMed] [Google Scholar]
- 32.Hoch U, Zhang Z, Kroetz DL and Ortiz De Montellano PR (2000) Structural determination of the substrate specificities and regioselectivities of the rat and human fatty acid ω-hydroxylases. Arch. Biochem. Biophys 373(1):63–71. doi: 10.1006/abbi.1999.1504. [DOI] [PubMed] [Google Scholar]
- 33.Kawashima H, Naganuma T, Kusunose E, Kono T, Yasumoto R, Sugimura K and Kishimoto T (2000) Human fatty acid ω-hydroxylase, CYP4A11: Determination of complete genomic sequence and characterization of purified recombinant protein. Arch. Biochem. Biophys 378(2):333–339. doi: 10.1006/abbi.2000.1831. [DOI] [PubMed] [Google Scholar]
- 34.Lasker JM, Chen WB, Wolf I, Bloswick BP, Wilson PD and Powell PK (2000) Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of CYP4F2 and CYP4A11. J. Biol. Chem 275(6):4118–4126. doi: 10.1074/jbc.275.6.4118. [DOI] [PubMed] [Google Scholar]
- 35.Powell PK, Wolf I, Jin R and Lasker JM (1998) Metabolism of arachidonic acid to 20-hydroxy-5,8,11,14-eicosatetraenoic acid by P450 enzymes in human liver: Involvement of CYP4F2 and CYP4A11. J. Pharmacol. Exp. Ther 285(3):1327–1336. [PubMed] [Google Scholar]
- 36.Sanders RJ, Ofman R, Valianpour F, Kemp S and Wanders RJA (2005) Evidence for two enzymatic pathways for ω-oxidation of docosanoic acid in rat liver microsomes. J. Lipid Res 46(5):1001–1008. doi: 10.1194/jlr.M400510-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Sanders RJ, Ofman R, Dacremont G, Wanders RJA and Kemp S (2008) Characterization of the human ω-oxidation pathway for ω-hydroxy-very-long-chain fatty acids. FASEB J. 22(6):2064–2071. doi: 10.1096/fj.07-099150. [DOI] [PubMed] [Google Scholar]
- 38.Brenton DP and Krywawych S (1982) 3-methyladipate excretion in Refsum’s disease. Lancet 319(8272):624. doi: 10.1016/S0140-6736(82)91780-9. [DOI] [PubMed] [Google Scholar]
- 39.Billimoria JD, Gibberd FB, Clemens ME and Whitelaw MN (1982) Metabolism of phytanic acid in Refsum’s disease. Lancet 319(8265):194–196. doi: 10.1016/S0140-6736(82)90760-7. [DOI] [PubMed] [Google Scholar]
- 40.Komen JC, Duran M and Wanders RJA (2004) ω-hydroxylation of phytanic acid in rat liver microsomes: Implications for Refsum disease. J. Lipid Res 45(7):1341–1346. doi: 10.1194/jlr.M400064-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Komen JC, Duran M and Wanders RJA (2005) Characterization of phytanic acid ω-hydroxylation in human liver microsomes. Mol. Genet. Metab 85(3):190–195. doi: 10.1016/j.ymgme.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Komen JC and Wanders RJA (2006) Identification of the cytochrome P450 enzymes responsible for the ω-hydroxylation of phytanic acid. FEBS Lett 580(16):3794–3798. doi: 10.1016/j.febslet.2006.05.069. [DOI] [PubMed] [Google Scholar]
- 43.Gibson GG, Orton TC and Tamburini PP (1982) Cytochrome P-450 induction by clofibrate. Purification and properties of a hepatic cytochrome P-450 relatively specific for the 12- and 11-hydroxylation of dodecanoic acid (lauric acid). Biochem. J 203(1):161–168. doi: 10.1042/bj2030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orton TC and Parker GL (1982) The effect of hypolipidemic agents on the hepatic microsomal drug-metabolizing enzyme system of the rat. Induction of cytochrome(s) P-450 with specificity toward terminal hydroxylation of lauric acid. Drug Metab. Dispos 10(2):110–115. [PubMed] [Google Scholar]
- 45.Reddy JK, Azarnoff DL and Hignite CE (1980) Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature 283(5745):397–398. doi: 10.1038/283397a0. [DOI] [PubMed] [Google Scholar]
- 46.Mannaerts GP, Debeer LJ, Thomas J and De Schepper PJ (1979) Mitochondrial and peroxisomal fatty acid oxidation in liver homogenates and isolated hepatocytes from control and clofibrate-treated rats. J. Biol. Chem 254(11):4585–4595. doi: 10.1016/s0021-9258(17)30051-0. [DOI] [PubMed] [Google Scholar]
- 47.Savas Ü, Hsu MH and Johnson EF (2003) Differential regulation of human CYP4A genes by peroxisome proliferators and dexamethasone. Arch. Biochem. Biophys 409(1):212–220. doi: 10.1016/S0003-9861(02)00499-X. [DOI] [PubMed] [Google Scholar]
- 48.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H and Gonzalez FJ (1995) Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol 15(6):3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kroetz DL, Yook P, Costet P, Bianchi P and Pineau T (1998) Peroxisome proliferator-activated receptor controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J. Biol. Chem 273(47):31581–31589. doi: 10.1074/jbc.273.47.31581. [DOI] [PubMed] [Google Scholar]
- 50.Leone TC, Weinheimer CJ and Kelly DP (1999) A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: The PPARα-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. U. S. A 96(13):7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patsouris D, Reddy JK, Müller M and Kersten S (2006) Peroxisome proliferator-activated receptor α mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 147(3):1508–1516. doi: 10.1210/en.2005-1132. [DOI] [PubMed] [Google Scholar]
- 52.Issemann I and Green S (1990) Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347(6294):645–650. doi: 10.1038/347645A0. [DOI] [PubMed] [Google Scholar]
- 53.Johnson EF, Hsu MH, Savas U and Griffin KJ (2002) Regulation of P450 4A expression by peroxisome proliferator activated receptors. Toxicology 181-182203-206. doi: 10.1016/S0300-483X(02)00282-2. [DOI] [PubMed] [Google Scholar]
- 54.Rakhshandehroo M, Knoch B, Müller B and Kersten S (2010) Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010612089. doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Göttlicher M, Widmark E, Li Q and Gustafsson JÅ (1992) Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. U. S. A 89(10):4653–4657. doi: 10.1073/pnas.89.10.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hess R, Stäubli W and Riess W (1965) Nature of the hepatomegalic effect produced by ethyl-chlorophenoxy- isobutyrate in the rat. Nature 208(5013):856–858. doi: 10.1038/208856a0. [DOI] [PubMed] [Google Scholar]
- 57.Makowska JM, Gibson GG and Banner FW (1992) Species differences in ciprofibrate induction of hepatic cytochrome p450 4A1 and peroxisome proliferation. J. Biochem. Toxicol 7(3):183–191. doi: 10.1002/jbt.2570070308. [DOI] [PubMed] [Google Scholar]
- 58.Rakhshandehroo M, Hooiveld G, Müller M and Kersten S (2009) Comparative analysis of gene regulation by the transcription factor PPARα between mouse and human. PLoS One 4(8):. doi: 10.1371/journal.pone.0006796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Richert L, Lamboley C, Viollon-Abadie C, Grass P, Hartmann N, Laurent S, Heyd B, Mantion G, Chibout SD and Staedtler F (2003) Effects of clofibric acid on mRNA expression profiles in primary cultures of rat, mouse and human hepatocytes. Toxicol. Appl. Pharmacol 191(2):130–146. doi: 10.1016/S0041-008X(03)00231-X. [DOI] [PubMed] [Google Scholar]
- 60.Aldridge TC, Tugwood JD and Green S (1995) Identification and characterization of DNA elements implicated in the regulation of CYP4A1 transcription. Biochem. J 306(2):473–479. doi: 10.1042/bj3060473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Savas Ü, Machemer DEW, Hsu MH, Gaynor P, Lasker JM, Tukey RH and Johnson EF (2009) Opposing roles of peroxisome proliferator-activated receptor α and growth hormone in the regulation of CYP4A11 expression in a transgenic mouse model. J. Biol. Chem 284(24):16541–16552. doi: 10.1074/jbc.M902074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Chen L and Hardwick JP (2000) Promoter activity and regulation of the CYP4F2 leukotriene B4 ω- hydroxylase gene by peroxisomal proliferators and retinoic acid in HepG2 cells. Arch. Biochem. Biophys 378(2):364–376. doi: 10.1006/abbi.2000.1836. [DOI] [PubMed] [Google Scholar]
- 63.Zhang X and Hardwick JP (2000) Regulation of CYP4F2 leukotriene B4 ω-hydroxylase by retinoic acids in HepG2 cells. Biochem. Biophys. Res. Commun 279(3):864–871. doi: 10.1006/bbrc.2000.4020. [DOI] [PubMed] [Google Scholar]
- 64.Hsu MH, Savas Ü, Griffin KJ and Johnson EF (2007) Regulation of human cytochrome P450 4F2 expression by sterol regulatory element-binding protein and lovastatin. J. Biol. Chem 282(8):5225–5236. doi: 10.1074/jbc.M608176200. [DOI] [PubMed] [Google Scholar]
- 65.Kocarek TA and Reddy AB (1996) Regulation of cytochrome P450 expression by inhibitors of hydroxymethylglutaryl-coenzyme A reductase in primary cultured rat hepatocytes and in rat liver. Drug Metab. Dispos 24(11):1197–1204. [PubMed] [Google Scholar]
- 66.Kalsotra A and Strobel HW (2006) Cytochrome P450 4F subfamily: At the crossroads of eicosanoid and drug metabolism. Pharmacol. Ther 112(3):589–611. doi: 10.1016/j.pharmthera.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 67.Kalsotra A, Anakk S, Brommer CL, Kikuta Y, Morgan ET and Strobel HW (2007) Catalytic characterization and cytokine mediated regulation of cytochrome P450 4Fs in rat hepatocytes. Arch. Biochem. Biophys 461(1):104–112. doi: 10.1016/j.abb.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heng YM, Kuo CWS, Jones PS, Savory R, Schulz RM, Tomlinson SR, Gray TJB and Bell DR (1997) A novel murine P-450 gene, Cyp4a14, is part of a cluster of Cyp4a and Cyp4b, but not of CYP4F, genes in mouse and humans. Biochem. J 325(3):741–749. doi: 10.1042/bj3250741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muller DN, Schmidt C, Barbosa-Sicard E, Wellner M, Gross V, Hercule H, Markovic M, Honeck H, Luft FC and Schunck W-H (2007) Mouse Cyp4a isoforms: enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem. J 403(1):109–18. doi: 10.1042/BJ20061328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holla VR, Adas F, Imig JD, Zhao X, Price E, Olsen N, Kovacs WJ, Magnuson MA, Keeney DS, … Capdevila JH (2001) Alterations in the regulation of androgen-sensitive Cyp 4a monooxygenases cause hypertension. Proc. Natl. Acad. Sci. U. S. A 98(9):5211–5216. doi: 10.1073/pnas.081627898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sundseth SS and Waxman DJ (1992) Sex-dependent expression and clofibrate inducibility of cytochrome P450 4A fatty acid ω-hydroxylases. Male specificity of liver and kidney CYP4A2 mRNA and tissue-specific regulation by growth hormone and testosterone. J. Biol. Chem 267(6):3915–3921. doi: 10.1016/s0021-9258(19)50613-5. [DOI] [PubMed] [Google Scholar]
- 72.Svoboda D, Grady H and Azarnoff D (1967) Microbodies in experimentally altered cells. J. Cell Biol 35(1):127–152. doi: 10.1083/jcb.35.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kawashima Y, Uy-Yu N and Kozuka H (1989) Sex-related difference in the inductions by perfluoro-octanoic acid of peroxisomal beta-oxidation, microsomal 1-acylglycerophosphocholine acyltransferase and cytosolic long-chain acyl-CoA hydrolase in rat liver. Biochem. J 261(2):595–600. doi: 10.1042/bj2610595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ranea-Robles P, Portman K, Bender A, Lee K, He JC, Mulholland DJ, Argmann C and Houten SM (2021) Peroxisomal L-bifunctional Protein Deficiency Causes Male-specific Kidney Hypertrophy and Proximal Tubular Injury in Mice. Kidney360 2(9):1441–1454. doi: 10.34067/kid.0003772021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Capdevila JH and Falck JR (2018) The arachidonic acid monooxygenase: From biochemical curiosity to physiological/pathophysiological significance. J. Lipid Res 59(11):2047–2062. doi: 10.1194/jlr.R087882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pettersen JE (1972) Formation of n-hexanedioic acid from hexadecanoic acid by an initial ω oxidation in ketotic rats. Clin. Chim. Acta 41(C):231–237. doi: 10.1016/0009-8981(72)90516-5. [DOI] [PubMed] [Google Scholar]
- 77.Verkade PE and Van Der Lee J (1934) Researches of fat metabolism. IV. Two-sided b-oxidation of the dicarboxylic acids formed by w-oxidation of saturated fatty acids. Proc. R. Acad. Amsterdam 37(7):460–647. [Google Scholar]
- 78.Passi S, Nazzaro-Porro M, Picardo M, Mingrone G and Fasella P (1983) Metabolism of straight saturated medium chain length (C9 to C12) dicarboxylic acids. J. Lipid Res 24(9):1140–1147. doi: 10.1016/s0022-2275(20)37897-4. [DOI] [PubMed] [Google Scholar]
- 79.Cerdan S, Kunnecke B, Dolle A and Seelig J (1988) In situ metabolism of 1,ω medium chain dicarboxylic acids in the liver of intact rats as detected by 13C and 1H NMR. J. Biol. Chem 263(24):11664–11674. doi: 10.1016/s0021-9258(18)37836-0. [DOI] [PubMed] [Google Scholar]
- 80.Tserng KY and Jin SJ (1991) Metabolic conversion of dicarboxylic acids to succinate in rat liver homogenates: A stable isotope tracer study. J. Biol. Chem 266(5):2924–2929. doi: 10.1016/s0021-9258(18)49936-x. [DOI] [PubMed] [Google Scholar]
- 81.Wada F and Usami M (1977) Studies on fatty acid omega-oxidation. Antiketogenic effect and gluconeogenicity of dicarboxylic acids. Biochim. Biophys. Acta 487(2):361–368. doi: 10.1016/0005-2760(77)90002-9. [DOI] [PubMed] [Google Scholar]
- 82.Mortensen PB, Gregersen N, Rasmussen K and Kølvraa S (1983) The β-oxidation of dicarboxylic acids in isolated mitochondria and peroxisomes. J. Inherit. Metab. Dis 6(S2):123–124. doi: 10.1007/BF01810358.6422143 [DOI] [Google Scholar]
- 83.Vamecq J and Draye J-P (1989) Peroxisomal and Mitochondrial β-Oxidation of Monocarboxylyl-CoA, ω-Hydroxymonocarboxylyl-CoA and Dicarboxylyl-CoA Esters in Tissues from Untreated and Clofibrate-Treated Rats. J. Biochem 106(2):216–222. doi: 10.1093/oxfordjournals.jbchem.a122835. [DOI] [PubMed] [Google Scholar]
- 84.Draye J-P, Veitch K, Vamecq J. and Van Hoof F. (1988) Comparison of the metabolism of dodecanedioic acid in vivo in control, riboflavin-deficient and clofibrate-treated rats. Eur. J. Biochem 178(1):183–189. doi: 10.1111/j.1432-1033.1988.tb14442.x. [DOI] [PubMed] [Google Scholar]
- 85.Pourfarzam M and Bartlett K (1991) Products and intermediates of the β-oxidation of [U-14C]hexadecanedionoyl-mono-CoA by rat liver peroxisomes and mitochondria. Biochem. J 273(1):205–210. doi: 10.1042/bj2730205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pettersen JE (1973) In vitro studies on the metabolism of hexadecanedioic acid and its mono-l-carnitine- ester. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 306(1):1–14. doi: 10.1016/0005-2760(73)90201-4. [DOI] [PubMed] [Google Scholar]
- 87.Pourfarzam M and Bartlett K (1993) Skeletal muscle mitochondrial beta-oxidation of dicarboxylates. Biochim. Biophys. Acta 1141(1):81–9. doi: 10.1016/0005-2728(93)90192-i. [DOI] [PubMed] [Google Scholar]
- 88.Bharathi SS, Zhang Y, Gong Z, Muzumdar R and Goetzman ES (2020) Role of mitochondrial acyl-CoA dehydrogenases in the metabolism of dicarboxylic fatty acids. Biochem. Biophys. Res. Commun 527(1):162–166. doi: 10.1016/j.bbrc.2020.04.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fransen M, Nordgren M, Wang B and Apanasets O (2012) Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim. Biophys. Acta 1822(9):1363–1373. doi: 10.1016/J.BBADIS.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 90.Lazarow PB (1977) Three hypolipidemic drugs increase hepatic palmitoyl-coenzyme A oxidation in the rat. Science (80-.). 197(4303):580–581. doi: 10.1126/science.195342. [DOI] [PubMed] [Google Scholar]
- 91.Reddy J and Krishnakantha T (1975) Hepatic peroxisome proliferation: induction by two novel compounds structurally unrelated to clofibrate. Science (80-. ) 190(4216):787–789. doi: 10.1126/SCIENCE.1198095. [DOI] [PubMed] [Google Scholar]
- 92.Mortensen PB, Kølvraa S, Gregersen N and Rasmussen K (1982) Cyanide-insensitive and clofibrate enhanced β -oxidation of dodecanedioic acid in rat liver an indication of peroxisomal β-oxidation of N-dicarboxylic acids. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 713(2):393–397. doi: 10.1016/0005-2760(82)90258-2. [DOI] [PubMed] [Google Scholar]
- 93.Sharma R, Lake BG and Gibson GG (1988) Co-induction of microsomal cytochrome P-452 and the peroxisomal fatty acid beta-oxidation pathway in the rat by clofibrate and di-(2-ethylhexyl)phthalate. Dose-response studies. Biochem. Pharmacol 37(7):1203–6. doi: 10.1016/0006-2952(88)90771-x. [DOI] [PubMed] [Google Scholar]
- 94.Lake BG, Gray TJB, Pels Rijcken WR, Beamand JA and Gangolli SD (1984) The effect of hypolipidaemic agents on peroxisomal oxidation and mixed-function oxidase activities in primary cultures of rat hepatocytes. Relationship between induction of palmitoyl-coa oxidation and lauric acid hydroxylation. Xenobiotica 14(3):269–276. doi: 10.3109/00498258409151411. [DOI] [PubMed] [Google Scholar]
- 95.van Roermund CWT, IJlst L, Wagemans T, Wanders RJA and Waterham HR (2014) A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1841(4):563–568. doi: 10.1016/j.bbalip.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 96.Ferdinandusse S, Denis S, van Roermund CWT, Preece MA, Koster J, Ebberink MS, Waterham HR and Wanders RJA (2018) A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta - Mol. Basis Dis 1864(3):952–958. doi: 10.1016/J.BBADIS.2017.12.032. [DOI] [PubMed] [Google Scholar]
- 97.Van Veldhoven PP, Vanhove G, Assselberghs S, Eyssen HJ and Mannaerts GP (1992) Substrate specificities of rat liver peroxisomal acyl-CoA oxidases: Palmitoyl-CoA oxidase (inducible acyl-CoA oxidase), pristanoyl-CoA oxidase (non-inducible acyl-CoA oxidase), and trihydroxycoprostanoyl-CoA oxidase. J. Biol. Chem 267(28):20065–20074. doi: 10.1016/s0021-9258(19)88666-0. [DOI] [PubMed] [Google Scholar]
- 98.Monte MJ, Alonso-Peña M, Briz O, Herraez E, Berasain C, Argemi J, Prieto J and Marin JJG (2017) ACOX2 deficiency: An inborn error of bile acid synthesis identified in an adolescent with persistent hypertransaminasemia. J. Hepatol 66(3):581–588. doi: 10.1016/j.jhep.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 99.Vilarinho S, Sari S, Mazzacuva F, Bilgüvar K, Esendagli-Yilmaz G, Jain D, Akyol G, Dalgiç B, Günel M, … Lifton RP (2016) ACOX2 deficiency: A disorder of bile acid synthesis with transaminase elevation, liver fibrosis, ataxia, and cognitive impairment. Proc. Natl. Acad. Sci 113(40):11289–11293. doi: 10.1073/PNAS.1613228113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim JT, Won SY, Kang KW, Kim SH, Park MS, Choi KH, Nam TS, Denis SW, Ferdinandusse S, … Kim MK (2020) ACOX3 Dysfunction as a Potential Cause of Recurrent Spontaneous Vasospasm of Internal Carotid Artery. Transl. Stroke Res 11(5):1041–1051. doi: 10.1007/s12975-020-00779-z. [DOI] [PubMed] [Google Scholar]
- 101.Houten SM, Denis S, Argmann CA, Jia Y, Ferdinandusse S, Reddy JK and Wanders RJA (2012) Peroxisomal L-bifunctional enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids. J. Lipid Res 53(7):1296–1303. doi: 10.1194/jlr.M024463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang X, Zhang B, Molony C, Chudin E, Hao K, Zhu J, Gaedigk A, Suver C, Zhong H, … Lum PY (2010) Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res 20(8):1020–1036. doi: 10.1101/gr.103341.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ding J, Loizides-Mangold U, Rando G, Zoete V, Michielin O, Reddy JK, Wahli W, Riezman H and Thorens B (2013) The Peroxisomal Enzyme L-PBE Is Required to Prevent the Dietary Toxicity of Medium-Chain Fatty Acids. Cell Rep. 5(1):248–258. doi: 10.1016/j.celrep.2013.08.032. [DOI] [PubMed] [Google Scholar]
- 104.Pettersen JE and Aas M (1973) ATP-dependent activation of dicarboxylic acids in rat liver. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 326(3):305–313. doi: 10.1016/0005-2760(73)90132-X. [DOI] [PubMed] [Google Scholar]
- 105.Adachi H, Mitsuhashi O and Imai Y (1974) Biosynthesis of a mono-coa ester from octadecane-1, 18-dioic acid in rat liver. J. Biochem 76(6):1281–1286. doi: 10.1093/oxfordjournals.jbchem.a130681. [DOI] [PubMed] [Google Scholar]
- 106.Vamecq J, De Hoffmann E and Van Hoof F (1985) The microsomal dicarboxylyl-CoA synthetase. Biochem. J 230(3):683–693. doi: 10.1042/bj2300683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Watkins PA, Howard AE, Gould SJ, Avigan J and Mihalik SJ (1996) Phytanic acid activation in rat liver peroxisomes is catalyzed by long-chain acyl-CoA synthetase. J. Lipid Res 37(11):2288–2295. doi: 10.1016/s0022-2275(20)37477-0. [DOI] [PubMed] [Google Scholar]
- 108.Watkins PA and Ellis JM (2012) Peroxisomal acyl-CoA synthetases. Biochim. Biophys. Acta - Mol. Basis Dis 1822(9):1411–1420. doi: 10.1016/j.bbadis.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kikuchi M, Hatano N, Yokota S, Shimozawa N, Imanaka T and Taniguchi H (2004) Proteomic analysis of rat liver peroxisome. Presence of peroxisome-specific isozyme of Lon protease. J. Biol. Chem 279(1):421–428. doi: 10.1074/jbc.M305623200. [DOI] [PubMed] [Google Scholar]
- 110.Islinger M, Lüers GH, Ka WL, Loos M and Völkl A (2007) Rat liver peroxisomes after fibrate treatment: A survey using quantitative mass spectrometry. J. Biol. Chem 282(32):23055–23069. doi: 10.1074/jbc.M610910200. [DOI] [PubMed] [Google Scholar]
- 111.Young PA, Senkal CE, Suchanek AL, Grevengoed TJ, Lin DD, Zhao L, Crunk AE, Klett EL, Füllekrug J, … Coleman RA (2018) Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J. Biol. Chem 293(43):16724–16740. doi: 10.1074/jbc.RA118.004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Smith BT, Sengupta TK and Singh I (2000) Intraperoxisomal localization of very-long-chain fatty acyl-CoA synthetase: implication in X-adrenoleukodystrophy. Exp. Cell Res 254(2):309–320. doi: 10.1006/EXCR.1999.4757. [DOI] [PubMed] [Google Scholar]
- 113.Steinberg SJ, Wang SJ, Kim DG, Mihalik SJ and Watkins PA (1999) Human very-long-chain acyl-CoA synthetase: cloning, topography, and relevance to branched-chain fatty acid metabolism. Biochem. Biophys. Res. Commun 257(2):615–621. doi: 10.1006/BBRC.1999.0510. [DOI] [PubMed] [Google Scholar]
- 114.Bergseth S, Hokland BM and Bremer J (1988) Metabolism of dicarboxylic acids in vivo and in the perfused kidney of the rat. Biochim. Biophys. Acta 961(1):103–9. doi: 10.1016/0005-2760(88)90135-x. [DOI] [PubMed] [Google Scholar]
- 115.Bergseth S, Poisson JP and Bremer J (1990) Metabolism of dicarboxylic acids in rat hepatocytes. Biochim. Biophys. Acta 1042(2):182–7. doi: 10.1016/0005-2760(90)90005-i. [DOI] [PubMed] [Google Scholar]
- 116.Vamecq J, Draye JP and Brison J (1989) Rat liver metabolism of dicarboxylic acids. Am. J. Physiol. Liver Physiol 256(4):G680–G688. doi: 10.1152/ajpgi.1989.256.4.G680. [DOI] [PubMed] [Google Scholar]
- 117.Poosch MS and Yamazaki RK (1989) The oxidation of dicarboxylic acid CoA esters via peroxisomal fatty acyl-CoA oxidase. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 1006(3):291–298. doi: 10.1016/0005-2760(89)90016-7. [DOI] [PubMed] [Google Scholar]
- 118.Lazarow PB (1978) Rat liver peroxisomes catalyze the β oxidation of fatty acids. J. Biol. Chem 253(5):1522–1528. doi: 10.1016/s0021-9258(17)34897-4. [DOI] [PubMed] [Google Scholar]
- 119.Osumi T and Hashimoto T (1978) Acyl-CoA oxidase of rat liver: a new enzyme for fatty acid oxidation. Biochem. Biophys. Res. Commun 83(2):479–85. doi: 10.1016/0006-291x(78)91015-x. [DOI] [PubMed] [Google Scholar]
- 120.Tserng KY, Jin SJ, Hoppel CL, Kerr DS and Genuth SM (1989) Urinary 3-hydroxyadipic acid 3,6-lactone: structural identification and effect of fasting in adults and children. Metabolism. 38(7):655–61. doi: 10.1016/0026-0495(89)90103-0. [DOI] [PubMed] [Google Scholar]
- 121.Mortensen PB and Gregersen N (1981) The biological origin of ketotic dicarboxylic aciduria. In vivo and in vitro investigations of the ω-oxidation of C6-C16-monocarboxylic acids in unstarved, starved and diabetic rats. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 666(3):394–404. doi: 10.1016/0005-2760(81)90298-8. [DOI] [PubMed] [Google Scholar]
- 122.Pettersen JE, Jeelum E and Eedjarn L (1972) The occurrence of adipic and suberic acid in urine from ketotic patients. Clin. Chim. Acta 38(1):17–24. doi: 10.1016/0009-8981(72)90202-1. [DOI] [PubMed] [Google Scholar]
- 123.Pettersen JE (1974) Urinary excretion of n hexanedioic and n octanedioic acid in juvenile diabetics with ketonuria. Diabetes 23(1):16–20. doi: 10.2337/diab.23.1.16. [DOI] [PubMed] [Google Scholar]
- 124.Shimojo N, Ishizaki T, Imaoka S, Funae Y, Fuji S and Okuda K (1993) Changes in amounts of cytochrome P450 isozymes and levels of catalytic activities in hepatic and renal microsomes of rats with streptozocin-induced diabetes. Biochem. Pharmacol 46(4):621–627. doi: 10.1016/0006-2952(93)90547-A. [DOI] [PubMed] [Google Scholar]
- 125.Zhang X, Gao T, Deng S, Shang L, Chen X, Chen K, Li P, Cui X and Zeng J (2021) Fasting induces hepatic lipid accumulation by stimulating peroxisomal dicarboxylic acid oxidation. J. Biol. Chem 296100622. doi: 10.1016/j.jbc.2021.100622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mortensen PB (1986) C6-C10-dicarboxylic acids in liver and kidney tissue in normal, diabetic ketotic and clofibrate-treated rats. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 878(1):14–19. doi: 10.1016/0005-2760(86)90338-3. [DOI] [PubMed] [Google Scholar]
- 127.Wada F, Shibata H, Goto M and Sakamoto Y (1968) Participation of the microsomal electron transport system involving cytochrome P-450 in ω-oxidation of fatty acids. BBA - Bioenerg 162(4):518–524. doi: 10.1016/0005-2728(68)90058-3. [DOI] [PubMed] [Google Scholar]
- 128.Miura Y (2013) The biological significance of ω-oxidation of fatty acids. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci 89(8):370–82. doi: 10.2183/PJAB.89.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Antony GJ and Landau BR (1968) Relative contributions of alpha-, beta-, and omega-oxidative pathways to in vitro fatty acid oxidation in rat liver. J. Lipid Res 9(2):267–269. doi: 10.1016/S0022-2275(20)43128-1. [DOI] [PubMed] [Google Scholar]
- 130.Bjorkhem I (1978) On the quantitative importance of omega-oxidation of fatty acids. J. Lipid Res 19(5):585–590. doi: 10.1016/s0022-2275(20)41290-8. [DOI] [PubMed] [Google Scholar]
- 131.Kam W, Kumaran K and Landau BR (1978) Contribution of omega-oxidation to fatty acid oxidation by liver of rat and monkey. J. Lipid Res 19(5):591–600. doi: 10.1016/s0022-2275(20)41291-x. [DOI] [PubMed] [Google Scholar]
- 132.Vamecq J and Draye JP (1987) Interactions between the ω- and β-oxidations of fatty acids. J. Biochem 102(1):225–234. doi: 10.1093/oxfordjournals.jbchem.a122035. [DOI] [PubMed] [Google Scholar]
- 133.Owen OE, Kalhan SC and Hanson RW (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem 277(34):30409–12. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- 134.Weitzel G (1947) Die Bernsteinsäureausscheidung bei Stoffwechselbelastung mit höheren n-Dicarbonsäuren. Hoppe. Seylers. Z. Physiol. Chem 282(5–6):185–191. doi: 10.1515/bchm2.1947.282.5-6.185. [DOI] [PubMed] [Google Scholar]
- 135.Rusoff II, Baldwin RR, Domingues FJ, Monder C, Ohan WJ and Thiessen R (1960) Intermediary metabolism of adipic acid. Toxicol. Appl. Pharmacol 2(3):316–330. doi: 10.1016/0041-008X(60)90060-0. [DOI] [PubMed] [Google Scholar]
- 136.Pettersen JE (1975) In vivo studies on the metabolism of hexanedioic acid. Clin. Chim. Acta 58(1):43–50. doi: 10.1016/0009-8981(75)90483-0. [DOI] [PubMed] [Google Scholar]
- 137.Mortensen PB (1980) The possible antiketogenic and gluconeogenic effect of the ω-oxidation of fatty acids in rats. Biochim. Biophys. Acta - Lipids Lipid Metab 620(2):177–185. doi: 10.1016/0005-2760(80)90199-X. [DOI] [PubMed] [Google Scholar]
- 138.Mortensen PB (1981) Urinary excretion of C4-C10-dicarboxylic acids and antiketogenic properties of adipic acid in ketogenic-stimulated rats due to diabetes, long-chain and short-chain monocarboxylic acids. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 664(2):335–348. doi: 10.1016/0005-2760(81)90056-4. [DOI] [PubMed] [Google Scholar]
- 139.POURFARZAM M and BARTLETT K (1992) Intermediates of peroxisomal β-oxidation of [U-14C]hexadecanedionoate: A study of the acyl-CoA esters which accumulate during peroxisomal β-oxidation of [U-14C]hexadecanedionoate and [U-14C]hexadecanedionoyl-mono-CoA. Eur. J. Biochem 208(2):301–307. doi: 10.1111/j.1432-1033.1992.tb17187.x. [DOI] [PubMed] [Google Scholar]
- 140.Jin Z, Bian F, Tomcik K, Kelleher JK, Zhang GF and Brunengraber H (2015) Compartmentation of metabolism of the C12-, C9-, and C5 n-dicarboxylates in rat liver, investigated by mass isotopomer analysis: Anaplerosis from dodecanedioate. J. Biol. Chem 290(30):18671–18677. doi: 10.1074/jbc.M115.651737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Westin MAK, Hunt MC and Alexson SEH (2005) The Identification of a Succinyl-CoA Thioesterase Suggests a Novel Pathway for Succinate Production in Peroxisomes. J. Biol. Chem 280(46):38125–38132. doi: 10.1074/jbc.M508479200. [DOI] [PubMed] [Google Scholar]
- 142.Hunt MC, Rautanen A, Westin MAK, Svensson LT and Alexson SEH (2006) Analysis of the mouse and human acyl-CoA thioesterase (ACOT) gene clusters shows that convergent, functional evolution results in a reduced number of human peroxisomal ACOTs 1. FASEB J. 20(11):1855–1864. doi: 10.1096/fj.06-6042com. [DOI] [PubMed] [Google Scholar]
- 143.Radzikh I, Fatica E, Kodger J, Shah R, Pearce R and Sandlers YI (2021) Metabolic outcomes of anaplerotic dodecanedioic acid supplementation in very long chain acyl-coa dehydrogenase (Vlcad) deficient fibroblasts. Metabolites 11(8):. doi: 10.3390/metabo11080538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Grego AV and Mingrone G (1995) Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: an update. Clin. Nutr 14(3):143–8. doi: 10.1016/s0261-5614(95)80011-5. [DOI] [PubMed] [Google Scholar]
- 145.Leighton F, Bergseth S, Rortveit T, Christiansen EN and Bremer J (1989) Free acetate production by rat hepatocytes during peroxisomal fatty acid and dicarboxylic acid oxidation. J. Biol. Chem 264(18):10347–10350. doi: 10.1016/s0021-9258(18)81625-8. [DOI] [PubMed] [Google Scholar]
- 146.Kasumov T, Adams JE, Bian F, David F, Thomas KR, Jobbins KA, Minkler PE, Hoppel CL and Brunengraber H (2005) Probing peroxisomal β-oxidation and the labelling of acetyl-CoA proxies with [1–13C]octanoate and [3–13C]octanoate in the perfused rat liver. Biochem. J 389(2):397–401. doi: 10.1042/BJ20050144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Houten SM, Wanders RJA and Ranea-Robles P (2020) Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta - Mol. Basis Dis 1866(5):165720. doi: 10.1016/j.bbadis.2020.165720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hemmelgarn E, Kumaran K and Landau BR (1977) Role of ω oxidation of fatty acids in formation of the acetyl unit for acetylation. J. Biol. Chem 252(12):4379–4383. doi: 10.1016/s0021-9258(17)40275-4. [DOI] [PubMed] [Google Scholar]
- 149.Kondrup J and Lazarow PB (1985) Flux of palmitate through the peroxisomal and mitochondrial β-oxidation systems in isolated rat hepatocytes. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 835(1):147–153. doi: 10.1016/0005-2760(85)90041-4. [DOI] [PubMed] [Google Scholar]
- 150.Dietschy JM and McGarry JD (1974) Limitations of acetate as a substrate for measuring cholesterol synthesis in liver. J. Biol. Chem 249(1):52–58. doi: 10.1016/s0021-9258(19)43089-5. [DOI] [PubMed] [Google Scholar]
- 151.Reszko AE, Kasumov T, David F, Jobbins KA, Thomas KR, Hoppel CL, Brunengraber H and Des Rosiers C (2004) Peroxisomal Fatty Acid Oxidation Is a Substantial Source of the Acetyl Moiety of Malonyl-CoA in Rat Heart. J. Biol. Chem 279(19):19574–19579. doi: 10.1074/jbc.M400162200. [DOI] [PubMed] [Google Scholar]
- 152.Bian F, Kasumov T, Thomas KK, Jobbins KA, David F, Minkler PE, Hoppel CL and Brunengraber H (2005) Peroxisomal and mitochondrial oxidation of fatty acids in the heart, assessed from the 13C labeling of malonyl-CoA and the acetyl moiety of citrate. J. Biol. Chem 280(10):9265–9271. doi: 10.1074/jbc.M412850200. [DOI] [PubMed] [Google Scholar]
- 153.Violante S, Ijlst L, Ruiter J, Koster J, van Lenthe H, Duran M, de Almeida IT, Wanders RJA, Houten SM and Ventura FV (2013) Substrate specificity of human carnitine acetyltransferase: Implications for fatty acid and branched-chain amino acid metabolism. Biochim. Biophys. Acta - Mol. Basis Dis 1832(6):773–779. doi: 10.1016/j.bbadis.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 154.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR and Thompson CB (2009) ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science (80-. ). 324(5930):1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Trefely S, Lovell CD, Snyder NW and Wellen KE (2020) Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab 100941. doi: 10.1016/j.molmet.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kumps A, Duez P and Mardens Y (2002) Metabolic, nutritional, iatrogenic, and artifactual sources of urinary organic acids: A comprehensive table. Clin. Chem 48(5):708–717. doi: 10.1093/clinchem/48.5.708. [DOI] [PubMed] [Google Scholar]
- 157.Tanaka K, Kean EA and Johnson B (1976) Jamaican Vomiting Sickness: Biochemical Investigation of Two Cases. N. Engl. J. Med 295(9):461–467. doi: 10.1056/NEJM197608262950901. [DOI] [PubMed] [Google Scholar]
- 158.Costa CG, Verhoeven NM, Kneepkens CMF, Douwes AC, Wanders RJA, De Almeida IT, Duran M and Jakobs C (1996) Organic acid profiles resembling a β-oxidation defect in two patients with coeliac disease. In Journal of Inherited Metabolic Disease, pp 177–180. doi: 10.1007/BF01799423. [DOI] [PubMed] [Google Scholar]
- 159.Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, Hwang DH, Newman JW and Garvey WT (2009) Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid β-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr 139(6):1073–1081. doi: 10.3945/jn.108.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mortensen PB and Gregersen N (1980) Medium-chain triglyceride medication as a pitfall in the diagnosis of non-ketotic C6-C10-dicarboxylic acidurias. Clin. Chim. Acta 103(1):33–37. doi: 10.1016/0009-8981(80)90227-2. [DOI] [PubMed] [Google Scholar]
- 161.Dias VC, Fung E, Snyder FF, Carter RJ and Parsons HG (1990) Effects of medium-chain triglyceride feeding on energy balance in adult humans. Metabolism. 39(9):887–891. doi: 10.1016/0026-0495(90)90295-N. [DOI] [PubMed] [Google Scholar]
- 162.Tonsgard JH (1986) Serum dicarboxylic acids in patients with Reye syndrome. J. Pediatr 109(3):440–445. doi: 10.1016/S0022-3476(86)80114-7. [DOI] [PubMed] [Google Scholar]
- 163.Gregersen N, Wintzensen H, Kølvraa S, Christensen E, Christensen MF, Brandt NJ and Rasmussen K (1982) C6-c10-dicarboxylic aciduria: Investigations of a patient with riboflavin responsive multiple acyl- CoA dehydrogenation defects. Pediatr. Res 16(10):861–868. doi: 10.1203/00006450-198210000-00012. [DOI] [PubMed] [Google Scholar]
- 164.Karpati G, Carpenter S, Engel AG, Watters G, Allen J, Rothman S, Klassen G and Mamer OA (1975) The syndrome of systemic carnitine deficiency: Clinical, morphologic, biochemical, and pathophysiologic features. Neurology 25(1):16–24. doi: 10.1212/wnl.25.1.16. [DOI] [PubMed] [Google Scholar]
- 165.Röschinger W, Muntau AC, Duran M, Dorland L, IJlst L, Wanders RJA and Roscher AA (2000) Carnitine-acylcarnitine translocase deficiency: Metabolic consequences of an impaired mitochondrial carnitine cycle. Clin. Chim. Acta 298(1–2):55–68. doi: 10.1016/S0009-8981(00)00268-0. [DOI] [PubMed] [Google Scholar]
- 166.Fontaine M, Briand G, Largillière C, Degand P, Divry P, Vianey-Saban C, Mousson B and Vamecq J (1998) Metabolic studies in a patient with severe carnitine palmitoyltransferase type II deficiency. Clin. Chim. Acta 273(2):161–170. doi: 10.1016/S0009-8981(98)00041-2. [DOI] [PubMed] [Google Scholar]
- 167.Przyrembel H, Wendel U, Becker K, Bremer HJ, Bruinvis L, Ketting D and Wadman SK (1976) Glutaric aciduria type II: Report on a previously undescribed metabolic disorder. Clin. Chim. Acta 66(2):227–239. doi: 10.1016/0009-8981(76)90060-7. [DOI] [PubMed] [Google Scholar]
- 168.Vianey-Saban C, Divry P, Brivet M, Nada M, Zabot MT, Mathieu M and Roe C (1998) Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: Clinical characteristics and diagnostic considerations in 30 patients. Clin. Chim. Acta 269(1):43–62. doi: 10.1016/S0009-8981(97)00185-X. [DOI] [PubMed] [Google Scholar]
- 169.Gregersen N, Lauritzen R and Rasmussen K (1976) Suberylglycine excretion in the urine from a patient with dicarboxylic aciduria. Clin. Chim. Acta 70(3):417–425. doi: 10.1016/0009-8981(76)90355-7. [DOI] [PubMed] [Google Scholar]
- 170.Wanders RJA, Ijlst L, Poggi F, Bonnefont JP, Munnich A, Brivet M, Rabier D and Saudubray JM (1992) Human trifunctional protein deficiency: A new disorder of mitochondrial fatty acid β-oridation. Biochem. Biophys. Res. Commun 188(3):1139–1145. doi: 10.1016/0006-291X(92)91350-Y. [DOI] [PubMed] [Google Scholar]
- 171.Korman SH, Waterham HR, Gutman A, Jakobs C and Wanders RJA (2005) Novel metabolic and molecular findings in hepatic carnitine palmitoyltransferase I deficiency. Mol. Genet. Metab 86(3):337–343. doi: 10.1016/j.ymgme.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 172.Knottnerus SJG, Pras-Raves ML, van der Ham M, Ferdinandusse S, Houtkooper RH, Schielen PCJI, Visser G, Wijburg FA and de Sain-van der Velden MGM. (2020) Prediction of VLCAD deficiency phenotype by a metabolic fingerprint in newborn screening bloodspots. Biochim. Biophys. Acta - Mol. Basis Dis 1866(6):. doi: 10.1016/j.bbadis.2020.165725. [DOI] [PubMed] [Google Scholar]
- 173.Ibdah JA, Paul H, Zhao Y, Binford S, Salleng K, Cline M, Matern D, Bennett MJ, Rinaldo P and Strauss AW (2001) Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Invest 107(11):1403–1409. doi: 10.1172/JCI12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cox KB, Hamm DA, Millington DS, Matern D, Vockley J, Rinaldo P, Pinkert CA, Rhead WJ, Lindsey JR and Wood PA (2001) Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum. Mol. Genet 10(19):2069–2077. doi: 10.1093/hmg/10.19.2069. [DOI] [PubMed] [Google Scholar]
- 175.Miinalainen IJ, Schmitz W, Huotari A, Autio KJ, Soininen R, Van Themaat EVL, Baes M, Herzig KH, Conzelmann E and Hiltunen JK (2009) Mitochondrial 2,4-dienoyl-CoA reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. PLoS Genet. 5(7):. doi: 10.1371/journal.pgen.1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Janssen U and Stoffel W (2002) Disruption of Mitochondrial β-Oxidation of Unsaturated Fatty Acids in the 3,2- trans -Enoyl-CoA Isomerase-deficient Mouse. J. Biol. Chem 277(22):19579–19584. doi: 10.1074/jbc.M110993200. [DOI] [PubMed] [Google Scholar]
- 177.Veitch K, Draye JP, Vamecq J, Causey AG, Bartlett K, Sherratt HSA and Van Hoof F (1989) Altered acyl-CoA metabolism in riboflavin deficiency. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 1006(3):335–343. doi: 10.1016/0005-2760(89)90021-0. [DOI] [PubMed] [Google Scholar]
- 178.Tserng KY, Jin SJ, Kerr DS and Hoppel CL (1990) Abnormal urinary excretion of unsaturated dicarboxylic acids in patients with medium-chain acyl-CoA dehydrogenase deficiency. J. Lipid Res 31(5):763–771. doi: 10.1016/s0022-2275(20)42775-0. [DOI] [PubMed] [Google Scholar]
- 179.Jin SJ and Tserng KY (1990) Metabolic origins of urinary unsaturated dicarboxylic acids. Biochemistry 29(37):8540–7. doi: 10.1021/bi00489a006. [DOI] [PubMed] [Google Scholar]
- 180.Lindstedt S, Norberg K, Steen G and Wahl E (1976) Structure of some aliphatic dicarboxylic acids found in the urine of an infant with congenital lactic acidosis. Clin. Chem doi: 10.1093/clinchem/22.8.1330. [DOI] [PubMed] [Google Scholar]
- 181.Duran M, De Klerk JBC, Wadman SK, Bruinvis L and Ketting D (1984) The differential diagnosis of dicarboxylic aciduria. J. Inherit. Metab. Dis 7(1 Supplement):48–51. doi: 10.1007/BF03047374. [DOI] [PubMed] [Google Scholar]
- 182.Houten SM, Herrema H, te Brinke H, Denis S, Ruiter JPN, van Dijk TH, Argmann CA, Ottenhoff R, Müller M, … Wanders RJA (2013) Impaired amino acid metabolism contributes to fasting-induced hypoglycemia in fatty acid oxidation defects. Hum. Mol. Genet 22(25):5249–5261. doi: 10.1093/hmg/ddt382. [DOI] [PubMed] [Google Scholar]
- 183.Tracey BM, Chalmers RA, Mehta A, English N, Purkiss P, Valman HB and Stacey TE (1987) Studies on abnormal metabolic function in Reye’s syndrome. J. Inherit. Metab. Dis 10(S2):263–265. doi: 10.1007/BF01811421. [DOI] [Google Scholar]
- 184.Westin MAK, Hunt MC and Alexson SEH (2008) Short- and medium-chain carnitine acyltransferases and acyl-CoA thioesterases in mouse provide complementary systems for transport of β-oxidation products out of peroxisomes. Cell. Mol. Life Sci 65(6):982–990. doi: 10.1007/s00018-008-7576-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tonsgard JH and Getz GS (1985) Effect of Reye’s syndrome serum on isolated chinchilla liver mitochondria. J. Clin. Invest 76(2):816–825. doi: 10.1172/JCI112039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tonsgard JH, Mendelson SA and Meredith SC (1988) Binding of straight-chain saturated dicarboxylic acids to albumin. J. Clin. Invest 82(5):1567–1573. doi: 10.1172/JCI113767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nguyen QH and Bui TP (1995) Azelaic acid: pharmacokinetic and pharmacodynamic properties and its therapeutic role in hyperpigmentary disorders and acne. Int. J. Dermatol 34(2):75–84. doi: 10.1111/j.1365-4362.1995.tb03583.x. [DOI] [PubMed] [Google Scholar]
- 188.Passi S, Picardo M, Nazzaro-Porro M, Breathnach A, Confaloni AM and Serlupi-Crescenzi G (1984) Antimitochondrial effect of saturated medium chain length (C8-C13) dicarboxylic acids. Biochem. Pharmacol 33(1):103–108. doi: 10.1016/0006-2952(84)90376-9. [DOI] [PubMed] [Google Scholar]
- 189.Liu G, Hinch B and Beavis AD (1996) Mechanisms for the transport of α,ω-dicarboxylates through the mitochondrial inner membrane. J. Biol. Chem 271(41):25338–25344. doi: 10.1074/jbc.271.41.25338. [DOI] [PubMed] [Google Scholar]
- 190.Rizzo C, Boenzi S, Wanders RJA, Duran M, Caruso U and Dionisi-Vici C (2003) Characteristic acylcarnitine profiles in inherited defects of peroxisome biogenesis: A novel tool for screening diagnosis using tandem mass spectrometry. Pediatr. Res 53(6):1013–1018. doi: 10.1203/01.PDR.0000064902.59052.0F. [DOI] [PubMed] [Google Scholar]
- 191.Wangler MF, Hubert L, Donti TR, Ventura MJ, Miller MJ, Braverman N, Gawron K, Bose M, Moser AB, … Elsea SH (2018) A metabolomic map of Zellweger spectrum disorders reveals novel disease biomarkers. Genet. Med 20(10):1274–1283. doi: 10.1038/gim.2017.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rocchiccioli F, Aubourg P and Bougneres PF (1986) Medium and long-chain dicarboxylic aciduria in patients with zellweger syndrome and neonatal adrenoleukodystrophy. Pediatr. Res 20(1):62–66. doi: 10.1203/00006450-198601000-00018. [DOI] [PubMed] [Google Scholar]
- 193.Björkhem I, Blomstrand S, Hågå P, Kase BF, Palonek E, Pedersen JI, Strandvik B and Wikström SA (1984) Urinary excretion of dicarboxylic acids from patients with the Zellweger syndrome. Importance of peroxisomes in beta-oxidation of dicarboxylic acids. Biochim. Biophys. Acta 795(1):15–9. doi: 10.1016/0005-2760(84)90099-7. [DOI] [PubMed] [Google Scholar]
- 194.Shimizu N, Yamaguchi S and Orii T (1994) A study of urinary metabolites in patients with dicarboxylic aciduria for differential diagnosis. Pediatr. Int 36(2):139–145. doi: 10.1111/j.1442-200X.1994.tb03149.x. [DOI] [PubMed] [Google Scholar]
- 195.Korman SH, Mandel H and Gutman A (2000) Characteristic urine organic acid profile in peroxisomal biogenesis disorders. In Journal of Inherited Metabolic Disease, pp 425–428. doi: 10.1023/A:1005624523611. [DOI] [PubMed] [Google Scholar]
- 196.Jansen GA and Wanders RJA (2006) Alpha-Oxidation. Biochim. Biophys. Acta - Mol. Cell Res 1763(12):1403–1412. doi: 10.1016/j.bbamcr.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 197.Foulon V, Sniekers M, Huysmans E, Asselberghs S, Mahieu V, Mannaerts GP, Van Veldhoven PP and Casteels M (2005) Breakdown of 2-hydroxylated straight chain fatty acids via peroxisomal 2-hydroxyphytanoyl-CoA lyase: a revised pathway for the alpha-oxidation of straight chain fatty acids. J. Biol. Chem 280(11):9802–9812. doi: 10.1074/JBC.M413362200. [DOI] [PubMed] [Google Scholar]
- 198.Passi S, Picardo M, De Luca C, Nazzaro-Porro M, Rossi L and Rotilio G (1993) Saturated dicarboxylic acids as products of unsaturated fatty acid oxidation. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 1168(2):190–198. doi: 10.1016/0005-2760(93)90124-R. [DOI] [PubMed] [Google Scholar]
- 199.Wierzbicki AS, Mayne PD, Lloyd MD, Burston D, Mei G, Sidey MC, Feher MD and Gibberd FB (2003) Metabolism of phytanic acid and 3-methyl-adipic acid excretion in patients with adult Refsum disease. J. Lipid Res 44(8):1481–1488. doi: 10.1194/jlr.M300121-JLR200. [DOI] [PubMed] [Google Scholar]
- 200.Wanders RJA, Komen J and Kemp S (2011) Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 278(2):182–194. doi: 10.1111/j.1742-4658.2010.07947.x. [DOI] [PubMed] [Google Scholar]
- 201.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JRB, … Muoio DM (2008) Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 7(1):45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 202.Muoio DM and Neufer PD (2012) Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab. 15(5):595–605. doi: 10.1016/j.cmet.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ghosh S, Wicks SE, Vandanmagsar B, Mendoza TM, Bayless DS, Salbaum JM, Dearth SP, Campagna SR, Mynatt RL and Noland RC (2019) Extensive metabolic remodeling after limiting mitochondrial lipid burden is consistent with an improved metabolic health profile. J. Biol. Chem 294(33):12313–12327. doi: 10.1074/jbc.RA118.006074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wicks SE, Vandanmagsar B, Haynie KR, Fuller SE, Warfel JD, Stephens JM, Wang M, Han X, Zhang J, … Mynatt RL (2015) Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc. Natl. Acad. Sci. U. S. A 112(25):E3300–E3309. doi: 10.1073/pnas.1418560112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wang Y, Zhang X, Yao H, Chen X, Shang L, Li P, Cui X and Zeng J (2022) Peroxisome-generated succinate induces lipid accumulation and oxidative stress in the kidneys of diabetic mice. J. Biol. Chem 101660. doi: 10.1016/j.jbc.2022.101660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhang X, Wang Y, Yao H, Deng S, Gao T, Shang L, Chen X, Cui X and Zeng J (2022) Peroxisomal β-oxidation stimulates cholesterol biosynthesis in the liver in diabetic mice. J. Biol. Chem 298(2):101572. doi: 10.1016/j.jbc.2022.101572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Björkhem I (1973) ω-Oxidation of Stearic Acid in the Normal, Starved and Diabetic Rat Liver. Eur. J. Biochem 40(2):415–422. doi: 10.1111/j.1432-1033.1973.tb03210.x. [DOI] [PubMed] [Google Scholar]
- 208.Mortensen PB (1981) C6-C10-dicarboxylic aciduria in starved, fat-fed and diabetic rats receiving decanoic acid or medium-chain triacylglycerol an in vivo measure of the rate of β-oxidation of fatty acids. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 664(2):349–355. doi: 10.1016/0005-2760(81)90057-6. [DOI] [PubMed] [Google Scholar]
- 209.Mortensen PB and Gregersen N (1982) The biological origin of ketotic dicarboxylic aciduria. II. In vivo and in vitro investigations of the β-oxidation of C8-C16-dicarboxylic acids in unstarved, starved and diabetic rats. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab 710(3):477–484. doi: 10.1016/0005-2760(82)90132-1. [DOI] [PubMed] [Google Scholar]
- 210.Elsner M, Gehrmann W and Lenzen S (2011) Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 60(1):200–208. doi: 10.2337/DB09-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Iaconelli A, Gastaldelli A, Chiellini C, Gniuli D, Favuzzi A, Binnert C, Macé K and Mingrone G (2010) Effect of oral sebacic acid on postprandial glycemia, insulinemia, and glucose rate of appearance in type 2 diabetes. Diabetes Care 33(11):2327–2332. doi: 10.2337/dc10-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Membrez M, Chou CJ, Raymond F, Mansourian R, Moser M, Monnard I, Ammon-Zufferey C, Mace K, Mingrone G and Binnert C (2010) Six weeks’ sebacic acid supplementation improves fasting plasma glucose, HbA1c and glucose tolerance in db/db mice. Diabetes, Obes. Metab 12(12):1120–1126. doi: 10.1111/j.1463-1326.2010.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Salinari S, Bertuzzi A, Gandolfi A, Greco AV, Scarfone A, Manco M and Mingrone G (2006) Dodecanedioic acid overcomes metabolic inflexibility in type 2 diabetic subjects. Am. J. Physiol. - Endocrinol. Metab 291(5):E1051–E1058. doi: 10.1152/ajpendo.00631.2005. [DOI] [PubMed] [Google Scholar]
- 214.Sun L, Liang L, Gao X, Zhang H, Yao P, Hu Y, Ma Y, Wang F, Jin Q, … Wu J (2016) Early prediction of developing type 2 diabetes by plasma acylcarnitines: A population-based study. Diabetes Care 39(9):1563–1570. doi: 10.2337/dc16-0232. [DOI] [PubMed] [Google Scholar]
- 215.Shah SH, Sun JL, Stevens RD, Bain JR, Muehlbauer MJ, Pieper KS, Haynes C, Hauser ER, Kraus WE, … Newby LK (2012) Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am. Heart J 163(5):844–850.e1. doi: 10.1016/j.ahj.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 216.Sun D, Tiedt S, Yu B, Jian X, Gottesman RF, Mosley TH, Boerwinkle E, Dichgans M and Fornage M (2019) A prospective study of serum metabolites and risk of ischemic stroke. Neurology 92(16):E1890–E1898. doi: 10.1212/WNL.0000000000007279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ruiz M, Labarthe F, Fortier A, Bouchard B, Legault JT, Bolduc V, Rigal O, Chen J, Ducharme A, … Des Rosiers C (2017) Circulating acylcarnitine profile in human heart failure: A surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. - Hear. Circ. Physiol 313(4):768–781. doi: 10.1152/ajpheart.00820.2016. [DOI] [PubMed] [Google Scholar]
- 218.Yu B, Li AH, Metcalf GA, Muzny DM, Morrison AC, White S, Mosley TH, Gibbs RA and Boerwinkle E (2016) Loss-of-function variants influence the human serum metabolome. Sci. Adv 2(8):e1600800. doi: 10.1126/sciadv.1600800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Menni C, Graham D, Kastenmüller G, Alharbi NHJ, Alsanosi SM, McBride M, Mangino M, Titcombe P, Shin S-Y, … Valdes AM (2015) Metabolomic Identification of a Novel Pathway of Blood Pressure Regulation Involving Hexadecanedioate. Hypertension 66(2):422–429. doi: 10.1161/HYPERTENSIONAHA.115.05544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhao Y, Peng J, Lu C, Hsin M, Mura M, Wu L, Chu L, Zamel R, Machuca T, … De Perrot M (2014) Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS One 9(2):. doi: 10.1371/journal.pone.0088727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kroetz DL and Xu F (2005) REGULATION AND INHIBITION OF ARACHIDONIC ACID ω-HYDROXYLASES AND 20-HETE FORMATION. Annu. Rev. Pharmacol. Toxicol 45(1):413–438. doi: 10.1146/annurev.pharmtox.45.120403.100045. [DOI] [PubMed] [Google Scholar]
- 222.Gainer JV, Bellamine A, Dawson EP, Womble KE, Grant SW, Wang Y, Cupples LA, Guo CY, Demissie S, … Capdevila JH (2005) Functional variant of CYP4A11 20-hydroxyeicosatetraenoic acid synthase is associated with essential hypertension. Circulation 111(1):63–69. doi: 10.1161/01.CIR.0000151309.82473.59. [DOI] [PubMed] [Google Scholar]
- 223.Nakagawa K, Holla VR, Wei Y, Wang W-H, Gatica A, Wei S, Mei S, Miller CM, Cha DR, … Capdevila JH (2006) Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J. Clin. Invest 116(6):1696–1702. doi: 10.1172/JCI27546. [DOI] [PMC free article] [PubMed] [Google Scholar]