

Abstract

The modularity and synthetic flexibility of metal–organic frameworks (MOFs) have provoked analogies with enzymes, and even the term MOFzymes has been coined. In this review, we focus on molecular catalysis of energy relevance in MOFs, more specifically water oxidation, oxygen and carbon dioxide reduction, as well as hydrogen evolution in context of the MOF–enzyme analogy. Similar to enzymes, catalyst encapsulation in MOFs leads to structural stabilization under turnover conditions, while catalyst motifs that are synthetically out of reach in a homogeneous solution phase may be attainable as secondary building units in MOFs. Exploring the unique synthetic possibilities in MOFs, specific groups in the second and third coordination sphere around the catalytic active site have been incorporated to facilitate catalysis. A key difference between enzymes and MOFs is the fact that active site concentrations in the latter are often considerably higher, leading to charge and mass transport limitations in MOFs that are more severe than those in enzymes. High catalyst concentrations also put a limit on the distance between catalysts, and thus the available space for higher coordination sphere engineering. As transport is important for MOF-borne catalysis, a system perspective is chosen to highlight concepts that address the issue. A detailed section on transport and light-driven reactivity sets the stage for a concise review of the currently available literature on utilizing principles from Nature and system design for the preparation of catalytic MOF-based materials.
1. Introduction
The hydrogen and oxygen evolution reactions (HER and OER, respectively) are crucial for energy storage and at the heart of future energy systems, while the oxygen reduction reaction (ORR) is pivotal to fuel cell technologies. The carbon dioxide reduction reaction (CO2RR) takes a central role in the production of renewable, carbon-based fuels. Moreover, CO2 constitutes a C1 feedstock for value-added commodity chemicals that are essential starting materials for the chemical industry.1,2
The above-mentioned reactions of energy relevance are energetically uphill redox processes and require an energy input either in the form of a chemical reagent, an electrochemical potential, or light (Figure 1) in order to proceed. While the latter energy input mimics principles of natural photosynthesis, and the products are often denoted as “solar fuels”,3 the former means to supply energy are more modular, and are not bound to seasonal variations or day/night cycles.
Figure 1.

Schematic representation of reactions of energy relevance that are addressed in this review.
Irrespective of the kind of energy input, catalysts are required to lower overpotential requirements. Such catalysts are typically distinguished as being either heterogeneous or homogeneous, that is, in different phases or in the same phase. However, this type of differentiation has become increasingly blurred in recent years, and this review differentiates between catalysts which are atomically defined molecular species versus those that are not. The latter category includes, for example, metals or (mixed) metal oxides, alloys, nanoparticles, or other types of nanostructures. Such materials as catalysts of energy relevance impress with high technological readiness and durability, and have their rightful place in the ongoing transformation of our current energy system. At the same time, they fall somewhat short in options to fine-tune reactivity, in particular those of nonprecious metal catalytic sites. This is the stronghold of molecular catalysis, which offers almost limitless possibilities for synthetic modifications to fine-tune electronics and to control distances between specific atoms at the sub-Ångström length scale. In molecular systems, subtle modifications of not only the first but also the second and third coordination sphere around the catalytic metal center can be engineered. Controlling these parameters enables control over reaction outcomes, in particular if multiple products may be feasible at similar energy inputs. This is the case in the CO2RR, which can produce a variety of different products within a narrow potential window. This situation is further complicated by the fact that the HER is a thermodynamically competing reaction,4 putting additional requirements on the catalyst to favor one product over all the others.5
Catalysis of energy relevance is not only crucial for humankind but also in Nature. In fact, the oxygenic atmosphere that enables life on Earth as we know it is a direct result of photosynthetic water oxidation. Nature has evolved enzymes that can activate inert molecules such as water or CO2 using first row transition metals such as Mn, Fe, Co, or Ni in catalytic active sites. These metals are abundant in contrast to noble metals such as platinum or iridium that humankind often uses in this context. Through billions of years of evolution, Nature judiciously optimized environments beyond the first coordination sphere of the metal sites and developed enzymes that rival humankind’s noble metal-based catalysts in terms of overpotential requirements and activity.6
Inspired by Nature’s ingenuity, but also recognizing the necessity of immobilizing molecular catalysts to advance technological readiness, much research has concentrated on the incorporation of molecular catalysts into stabilizing support scaffolds.7 Ideally, such a support should not only structurally stabilize the catalytic site, but also offer synthetic flexibility to tune the chemical environment beyond the first coordination sphere. Examples of this strategy include supramolecular systems such as, among others, cages,8,9 polymers, dendrimers, or zeolites.10−12 Their preparation is often synthetically demanding, and, as in the case of zeolites, may be limited in terms of synthetic flexibility. The support is often conductive, resulting in molecularly functionalized electrodes13 and photoelectrodes. The alternative that is reviewed herein is based on the incorporation of molecularly defined catalytic function into metal–organic frameworks (MOFs), three-dimensional (3D) coordination polymers that consist of metal cations or clusters thereof, so-called secondary building units (SBUs), that are periodically interconnected by organic linker molecules.14 MOFs are highly porous and often exhibit high internal surface areas, both appealing features when designing highly active catalysts. They can also be electrically conducting, either by a hopping mechanism or band-like charge transport.15 In addition, their high degree of modularity combined with ample synthetic functionalization strategies using organic chemistry16 render MOFs an attractive platform for the systematic engineering of outer-sphere interactions.17
1.1. Principles of Enzyme Reactivity as Inspiration for MOF Design
Chemists have long been inspired by the beautiful orchestra that is enzymatic catalysis. Enzymes are the prime example of how outer coordination sphere effects can be leveraged to drive energy efficient and selective catalysis. Coordination spheres are classically divided into the first, second, and third coordination sphere, depending on the distance from the active site metal. The first or “inner” sphere describes those atoms which are bound directly to the metal center. This sphere is comparatively easy to mimic with inorganic coordination chemistry, and, indeed, an incredible array of homogeneous catalysts exists which rely on clever first coordination sphere designs. The second coordination sphere refers to atoms not directly attached to the metal center but near enough to interact directly with the metal center or with a bound substrate. A classic example from the inorganic community is the famous series of [Ni(P2N2)2]2+ catalysts in which a pendant amine not directly attached to the Ni center acts to shuttle protons to and from the active site during proton reduction and hydrogen oxidation, respectively.18 This approach is directly inspired by enzymatic chemistry, as will be discussed below. Last, the third coordination sphere refers to atoms or functional groups that influence catalysis without directly interacting with the active site. In enzymes, this can for example mean long-range ordering of water molecules in such a way as to favor catalysis.19,20 Also channels that regulate substrate and product transport, as for example in photosystem II,21 or redox active cofactors that regulate charge transport22 belong to this category. In this review, we will discuss “outer” coordination sphere effects, which encompasses effects induced by second and third coordination sphere design features. In context of MOFs, structurally discrete interactions between the molecular catalyst unit and the surrounding MOF matrix are an example of second, while measures that control and accelerate transport constitute examples of third coordination sphere effects. In parts of section 3.2 and 3.4, we will go beyond intra-MOF effects and take a broader system perspective on catalytic performance, as governed by the dimension of the MOF-based catalyst and its contacting to the macroscopic world.
To set the stage for the discussion of outer coordination sphere effects in MOFs, we will first highlight some examples of how enzymes utilize outer coordination sphere effects, illustrated on hydrogenase (H2ase) enzymes. The catalytic cycle of the [FeFe] H2ases commences with a mixed-valent FeIIFeI species with one of the CO ligands in a bridging position, exposing a “free” coordination site of Fed, the iron center of the [2Fe2S] subsite that is distal to the [4Fe4S] cluster (Hox in Figure 2).23 Following the cycle in the clockwise H2 oxidation direction, substrate H2 binds to the free coordination site in a η2-fashion and is heterolytically cleaved (Hox → HhydH+). The proton is picked up by a nearby nitrogen base which is conveniently positioned above Fed as part of the azadithiolate (adt) linker. The presence and function of the nitrogen in the adt linkers has long been subject of intense discussions as it could not be unambiguously determined by X-ray crystallography. Spectroscopic work on both enzyme24 and model compounds,25 as well as in situ H2ase enzyme maturations,26 have clearly identified the central atom of the dithiolate bridge to be nitrogen, which functions as a proton relay toward Fed.
Figure 2.

Proposed enzymatic catalytic cycle of [FeFe] H2ases for the oxidation of hydrogen (blue) and the reduction of protons (red).23,27
In the case of some well-studied [FeFe] H2ases,27 the adt linker is locked in a conformation where the nitrogen is on the same side of the [2Fe2S] subsite as Fed. The other conformation of the six-membered Fe-adt metalloheterocycle is sterically inhibited by the surrounding peptide. This outer-sphere effect that forces the proton shuttling base to face the active metal site is absent in the initial synthetic NiP2N2 HER catalyst developed by DuBois and co-workers.28 In this homogeneous mimic, the pendant nitrogen base can undergo inversion, resulting in a protonated conformer where the proton is physically tucked away from the metal site, and can only re-enter the catalytic cycle through a deprotonation pathway.28 Preventing this off-cycle conformation in a subsequent modification produced a considerably faster catalyst that turned over 100 000 times per second, as determined from voltammetric experiments.29 Returning to the catalytic cycle in Figure 2, deprotonation of the HhydH+ state results in a charge shift within the H-cluster that enables a proton transfer from Fed to the adt-N and the formation of HsredH+. Two subsequent one-electron oxidations and N-deprotonation complete the catalytic cycle.
Enzymatic active sites, in particular those that catalyze reactions of energy relevance, are often located in an enzyme’s interior to shield reactive intermediate states from detrimental side reactions. Indeed, decomposition pathways for synthetic molecular species are often bimolecular deactivation or ligand loss.30,31 However, if the active site is shielded by encapsulation, as is the case for proton reduction by [FeFe] H2ases, the active site is protected from such events. In turn, this however means that substrate and products (protons, electrons and hydrogen) need to be transported through the peptide matrix to/from the active site. This transport is highly regulated, and [FeFe] H2ases have dedicated substrate and product channels as well as an energetically highly tuned electron transport chain (Figure 3a).32
Figure 3.

a) Depiction of the proton and electron transfer pathways of FeFe hydrogenase. Adapted with permission from reference (32). Copyright 2020 National Academy of Sciences. b) Active site of FeFe hydrogenase showing how the protonated bridgehead amino group can act, via hydrogen bonding, to stabilize a hydride intermediate. Numbers indicate distances in Å, the red-blue color scheme indicates negative and positive partial charges, respectively. Adapted with permission from reference (33). Copyright 2019 American Chemical Society.
The degree to which outer-sphere effects influence enzymatic performance is difficult to quantify experimentally. Computational work is a valuable tool in this context, and it was used, for example, in the case of [FeFe] H2ases to calculate the energetic effect of the H-bonding interaction between the active site and the surrounding protein matrix. It was shown that the terminal hydride species, which is imperative to the enzyme’s function, is kinetically stabilized by hydrogen bonding interactions (Figure 3b).33 Formation of the thermodynamically more stable μ-H product is prevented by a high energy transition state caused by interactions with the surrounding peptide matrix.34 In other words, catalysis proceeds through a kinetically stabilized state before it converts to the thermodynamically more stable, and thus less reactive, ground state.
Experimentally, the most extreme showcase of the power of outer-sphere effects in molecular design was reported when a synthetic [FeFe]H subunit model was introduced into the apoprotein of an [FeFe] H2ase.26 While [Fe2(adt)(CO)4(CN)]2– is a very limited catalyst by itself,35 its incorporation into a tailor-made environment, that is, the apoprotein, triggers a number of structural rearrangements at the metal cofactor that result in a functional enzyme.36 This work is the most compelling illustration of the effect that delicate interactions between an organometallic active site and a surrounding environment can have on the performance of a catalytic active site.
Electrons are additional “reactants” that need to access the active sites of redox enzymes, a process that may be managed by dedicated electron transport chains. A common electron relay consists of iron–sulfur clusters, prototypically 4Fe4S clusters. In the case of hydrogenases, there are up to six FeS clusters between the surface of the enzyme and the active site.37 These clusters are not all identical, and tuning of their electrochemical potentials by the surrounding peptide matrix gives electron transport a directionality, either to or from the active site, and the enzyme a preference to act as proton reduction, or hydrogen oxidation catalyst. Careful potentiometric titrations coupled with electron paramagnetic resonance (EPR) measurements of the [FeFe] enzyme from C. pasteurianum revealed a redox potential gradient capable of “switching on” electron flow when a sufficiently high amount of reducing equivalents was available (Figure 4).38
Figure 4.

Locations and redox potentials of FeS clusters in the [FeFe] hydrogenase enzyme of C. pasteurianum. Reproduced with permission from reference (38). Copyright 2017 American Chemical Society.
The importance of this carefully tuned long-range electron flow has been demonstrated for hydrogenases. Within the cyanobacterium N. punctiforme is a nickel–iron (NiFe) hydrogenase that principally consumes hydrogen as an electron source. By substituting one of the 4Fe4S clusters with a 3Fe4S cluster, Raleiras et al.39 were able to reverse the preferred direction of electron flow to afford a hydrogen producing enzyme.
The above examples illustrate how the outer coordination sphere of enzymes has evolved to carefully tune product selection (e.g., proton reduction or hydrogen oxidation), manage electron, substrate, and product transport into and out of the enzyme, and stabilize favorable intermediates and transition states. Clearly, management and understanding of outer-sphere effects affords significant opportunities in the design of synthetic catalysts.
1.2. The MOF–Enzyme Analogy: MOFs as Peptide Surrogates to Host Active Sites
MOFs are typically built from metal ions or defined metal clusters (secondary building units, SBUs) that are periodically interconnected by organic linker molecules in two or three dimensions, forming a highly ordered crystal lattice of well-defined chemical topology.40,41 As illustrated by a few examples in Figure 5, the variation of linkers or SBUs can lead to diverse structures, making MOFs highly tunable materials.42 The large number of available SBUs and linkers enables the design of countless MOFs, showcasing unique structures and differing in topology, reactivity, stability, pore size, and internal surface area.43−45 Some MOF materials exhibit internal surface areas as high as 7800 m2 g–1, higher than that of any other material known.46 Consequently, MOFs have attracted large interest for gas storage and separation, a field that is still highly relevant today.47 The large surface area is, however, also relevant for many other applications, and MOFs have been explored as platforms for the incorporation of different functions.48−50 Reviewed herein are catalytic reactions of energy relevance, as large internal surface areas that contain accessible catalytic sites can lead to high activity.43−45 An important aspect to such endeavors is the development of strategies to lend MOFs high hydrolytic stability51 as well as stability under electrocatalysis conditions.52
Figure 5.

Representation of different SBU and linker combinations to afford a selection of MOFs featured in this review. Atom labeling: C, black; O, red; metals, blue polyhedra. H atoms are omitted for clarity. Yellow and orange spheres represent the space in the framework. Reproduced with permission from reference (53). Copyright 2017 Royal Society of Chemistry.
Molecular and nonmolecular species with a desired function (e.g., catalysis) such as nanoparticles or even small enzymes can be integrated into a MOF.54 In general, the functionality can be introduced to the MOF either during its synthesis, often solvothermally, or in a postsynthetic method. The latter includes postsynthetic exchange where linkers or SBU-constituting metals from pristine MOFs are replaced by exogenous counterparts, postsynthetic ligation at a free coordination site of an SBU, postsynthetic metalation, or by using synthetic organic chemistry on linkers with suitable reactive groups.44,55−57
Catalysts of energy relevance have been introduced by most conceivable methodologies. In context of molecularly defined catalysts, the catalysts can in principle be held inside the MOF matrix either by a physical entrapment, nonspecific forces such as hydrophobic–hydrophobic interactions, coordination bonds, including H-bonds, or covalent bonds.44 Of course, the SBU, stabilized by the coordinating linker, can itself be catalytically active,45 as for example seen in many Lewis-acid promoted organic transformations.58,59
1.3. Scope of the Review: Outer Coordination Sphere Effects in MOFs
This review summarizes reports on molecular catalysis of energy relevance in MOFs. Whenever possible, analogies to enzymes are drawn by analyzing interactions between the MOF scaffold and the catalytic site. Such interactions may be in close proximity to the active site, that is in the second coordination sphere, or more remote in the third coordination sphere. The benefits that arise from MOF incorporation may be multiple, and the original research papers in the field have been organized by the most discernible MOF–catalyst interaction. Such interactions may be of simple structural nature, stabilizing molecular integrity of the catalysts under turnover conditions, giving rise to higher turnover numbers (TONs) as compared to those of homogeneous reference compounds (section 3.1).43,44 Given the microporous nature of MOFs, transport to/from catalytic sites that are remote from the MOF crystal surface can easily be limiting the overall efficacy of the material,60,61 and works on facilitating transport issues are addressed in section 3.2.62 The presence of substrate and/or electron transport pathways bears resemblance to corresponding channels in the third coordination sphere of enzymes. In the absence of such engineered intra-MOF transport channels, limitations can be overcome at a system level by simply decreasing the dimension of the catalytic MOF at hand. Such examples will also be reviewed in section 3.2. Zooming closer into the catalytic site and its secondary coordination sphere, specific and atomically well-defined interactions between the catalytic site and the MOF matrix are discussed in section 3.3. Such cooperative effects may, for example, lower the transition state of a rate-determining step of the catalytic cycle, thereby accelerating catalytic turnover.63 The final section 3.4 reviews contributions on different means to interface electrocatalytic MOFs with electrode substrates. This section again addresses catalytic efficiency at a system level, but has its counterpart in the enzyme world where the way by which redox enzymes are anchored on electrode substrates has a great impact on observed current densities.64−66
It is important to point out that different aspects of the present review have been summarized previously.43,44,67−72 The interested reader is referred to excellent reviews on MOF-based biomimicry,45,73−77 MOF-based oxidation reactions,78 in particular OER,79−81 MOF-based HER,82−84 CO2RR,85−87 and ORR,88 often comprising not only molecular but also material catalyst systems.89,90 Drawing parallels between biological systems and catalysis of energy relevance in MOFs, the present review is exclusively focused on molecular catalysts. Catalytic units that are not molecularly defined, as well as entire enzymes that are embedded in MOFs are outside the scope of this review.91−93 The redox equivalents for the reactions may be provided either chemically, electrochemically, or photochemically. The source of redox equivalents may not necessarily be from molecular oxidants/reductants or photosensitizers (PSs) but could also originate from heterogeneous materials such as quantum dots for light harvesting, or conducting substrates in the case of electrochemical processes. Redox equivalents (holes or electrons) and substrates (CO2, water, or O2) come from different sources, and hydrogenation reactions are outside scope of this review.
As higher coordination sphere effects generally aim to accelerate the catalyzed reactions, this review also contains an extensive survey on the kinetics of molecular catalysis in MOFs (section 2). This is important because acceleration of the catalytic reaction may more often than not be masked by other limiting phenomena. In chemically or electrochemically driven catalytic processes, this may be mass or charge transport limitations, while light-driven catalysis comes with the additional complication that sacrificial reductants or oxidants are often present in the MOF only in small amounts. Moreover, productive light-driven charge separations are often reversed by back electron transfer pathways, thereby short-circuiting catalytic mechanisms. All of these phenomena affect the observed rate of product formation of the whole system. In order to benchmark catalytic MOF systems, and to make meaningful and informed improvements, it is thus imperative to discern the limitation under which a specific catalytic MOF system may operate.
2. Kinetics of MOF-Based Molecular Catalysis
A fundamental difference between homogeneous catalysis and catalysis supported in porous materials is the heterogeneous nature of chemical reactions, where mass and charge transport cause substrate, intermediate species, and product concentrations to not only depend on time but possibly space as well. In these situations, concentrations inside the porous support matrix change over the length of the particle or thickness of the film and may differ significantly from bulk concentrations at the boundaries. In the context of biomimicry, introduction of higher coordination sphere effects that may accelerate the intrinsic catalytic rate may not manifest themselves as clear changes in the observed rate of product formation or turnover frequency (TOF). If the substrate and charge transport are relatively slow compared to catalysis, differences in the overall activity as a direct result of outer coordination sphere effects may be difficult or impossible to distinguish. Therefore, to assess the benefit of the pore modifications mentioned above and identify the role of any second coordination sphere interactions, it is necessary to take measurements under conditions where charge and mass transport do not contribute to kinetic control. A detailed kinetic and mechanistic analysis considering both the diffusion of substrate and electron hopping is therefore a prerequisite to determine if molecular catalysis in MOFs can reach enzyme-like properties. In general, this can be accomplished by collecting experimental data over a large range of each operational parameter, as well-demonstrated by recent advances in homogeneous molecular catalysis of electrochemical reactions.94,95
To determine the effect of the framework on the catalytic reaction, it may be required to compare the heterogenized molecular catalyst within the MOF to a homogeneous analogue (if one exists). As has been re-emphasized recently,96,97 varying operational parameters (concentrations, light intensity, or experimental time scales) are used to establish possible mechanistic pathways and kinetic rate laws, while molecular catalysts should be compared based on intrinsic parameters (standard potentials, intrinsic rate constants). Establishment of the TOF expression for a given mechanism by kinetic modeling is consistently required, as it is usually a composite function of several rate constants for different steps in the catalytic cycle, especially where the catalytic mechanism is a network of multiple pathways.
For heterogenized molecular catalysts in porous materials, the transport properties of the framework are also important to consider when comparing catalytic systems. The relevant operational parameter is the geometric length scale (film thickness/particle size) of the finite domain where the reaction-diffusion process is occurring. Varying this will switch the observed catalytic rate between reaction-limited and transport-limited, from which transport properties can be measured (diffusion coefficients). When interpreting kinetic data of experimental systems, a clear distinction between operational and intrinsic parameters as well as between reaction-limited and transport-limited rates is important for drawing mechanistic conclusions, such as the involvement of through-space or second coordination sphere interactions. In the following sections, we summarize several physico-mathematical models that provide a rational basis for kinetic assessment and benchmarking of chemically-, electrochemically-, and photochemically-driven molecular catalysis within MOF particles or films, and point out possibilities and pitfalls. As many reports that will be reviewed herein do not discriminate between the apparent turnover frequencies (TOFapp) that are a composite number and the true TOFtrue that is an intrinsic catalyst metric, we consciously refrain from citing TOFs and TONs. This is to discourage comparisons of performance numbers that should not be compared because they may be influenced by different limiting phenomena. Instead, we provide guidelines that will help the reader to identify these phenomena, and to evaluate whether comparisons of performance parameters of different systems are meaningful.
2.1. Chemically Driven Molecular Catalysis in MOFs
In many examples of MOF-based molecular catalysis, the reaction is driven by a sacrificial chemical reagent, which is added to a suspension of MOF particles. In the case of catalysis of energy relevance, this is a strong oxidant or reductant that delivers electrons or holes to the active sites in the MOF interior, but this situation could apply to thermochemical organic transformations as well. Assuming that catalysis occurs in the MOF interior and not only at exposed surface sites, both the sacrificial reagent and the substrate must diffuse into the MOF pores for the reaction to proceed (Figure 6a). In the case of redox reactions, the sacrificial oxidant/reductant could otherwise initiate a diffusional charge hopping process at the surface of the particles that carries electron or holes to the molecular catalysts within the framework (Figure 6b). Consequently, measuring the intrinsic rate constant of the incorporated molecular catalyst will require accounting for mass transport of the substrate/sacrificial reagent and possibly charge transport of electrons or holes.
Figure 6.

Cases for molecular catalysis in MOF particles. The scenario including a photosensitizer depicted in b) will be discussed further in section 2.3. Reproduced with permission from reference (98). Copyright 2020 Royal Society of Chemistry.
First introduced in the 1930s independently by
Damköhler99 and Thiele,100 the
theoretical basis for catalytic reactions in porous media is well-established
for basic mechanistic schemes. Several simple criteria can be used
to determine the extent of substrate mass transport resistance and
provide the intrinsic rate constant of the catalytic reaction. For
a first-order reaction in a porous catalyst particle at steady-state,
the competition between reaction and diffusion of the substrate is
controlled by the dimensionless parameter ϕ, called the Thiele
modulus (here we will refer to ϕ in eq 1, but the same quantity is also commonly denoted
as the Damköhler number (Da) with  ,
,
| 1 |
where L is the critical length scale of the particle (e.g., radius for spherical particles), DS is the intraparticle diffusion coefficient of the substrate, and kcat is the first-order rate constant of the catalytic reaction. For ϕ ≪ 1, the reaction is kinetically controlled by the intrinsic rate of catalysis. Conversely, when ϕ ≫ 1, there is mixed kinetic control by reaction and diffusion, and the catalytic particle will display significant transport resistance. In this case, when ϕ ≫ 1, this means that the observed rate (v) is proportional to both the diffusion coefficient of the substrate and the (pseudo)first-order catalytic rate constant raised to the one-half power:
| 2 |
Kinetic measurement under these conditions will contain artifacts related to the mass transport of substrate through the particle. Experimentally, these two kinetic regimes can be accessed by systematically varying the size of the particle (given by L) and measured by the internal effectiveness factor, η, which is a metric for the extent of diffusion of the substrate in a catalytic particle. It is defined as the ratio of the observed reaction rate to the reaction rate in the absence of transport effects (when the whole interior of the particle is uniformly exposed to the conditions on the surface). A plot of the effectiveness factor as a function of the Thiele modulus is displayed in Figure 7.
Figure 7.

Plot of the effectiveness factor as a function of the Thiele modulus of spherical particles.
Kinetic measurements taken in the reaction-limited region (η = 1, ϕ ≪ 1) will yield the intrinsic rate constant and should be used for meaningful comparison between heterogenized molecular catalysts. On the other hand, the observed rate in the internal diffusion-limited region (η < 1, ϕ ≫ 1) can be used to calculate important parameters for intra-MOF substrate transport such as diffusion coefficients (DS). We note that transport-limited rates can be useful in obtaining kinetic constants if an independent measurement of both the dimension of the particle (L) and the intra-MOF diffusivity of the substrate (DS) can be made. A physico-mathematical model for the operative mechanism can then be formulated to relate the measured product formation rate to the intrinsic catalytic rate constant(s), with the parameters associated with the diffusion of substrate (L, DS) being known quantities.
2.1.1. Measuring Molecular Diffusion in MOFs and Implications for Chemically Driven Catalysis
As mentioned above, molecular diffusion rates for substrates as well as other coreagents are valuable parameters for analyzing kinetic data, as well as benchmarking transport limitations (via the effectiveness factor). This is quantified by the intracrystalline diffusivity (DS)61 measured under conditions relevant to molecular catalysis, typically with solvent-filled pores. Although the gas-phase self-diffusivity of guest molecules in MOFs has been investigated extensively, solution-phase studies of intracrystalline transport are relatively limited.
In a recent example, Wang et al.101 followed the transport of a luminescent probe (1,3,5,7-tetramethyl-4,4-difluoroboradiazaindacene) through the one-dimensional (1D) pores of NU-1008 using confocal fluorescence microscopy. Measurements conducted in situ on individual crystallites yielded transient concentration profiles, which could be fit to an analytical solution of Fickian diffusion in a finite domain, including transport resistance (“surface barriers”) across the particle/solution interface. Experimentally obtained diffusivities were in the range of 10–8 cm2 s–1, approximately 2 orders of magnitude smaller than that measured in bulk solution of the same solvent. Significant pore confinement and host–guest π–π interactions were suggested as possible reasons for the slower diffusion through the solvent-filled pores of NU-1008. Ideally, microimaging techniques of single crystals, which provide the entire transient concentration profile of guest molecules within the framework, are more accurate at predicting diffusivities. Macroscopic uptake measurements, made by stepping the concentration of the guest molecule in the surrounding bulk solution or atmosphere and recording its change over time, were found to often lead to an incorrect estimation of such transport properties.102
Furthermore, the phenomenon of surface barriers, as mentioned above, frequently has been observed for uptake of solution phase guest molecules into MOF crystallites. This can be described as a finite rate of guest molecule penetration from solution through the outer surface of the MOF.103,104 This kinetic phenomenon was identified as a possible rate-limiting process and subsequently accounted for in models of catalysis within redox-polymer-modified electrodes105 and thus has implications for molecular catalysis in MOFs. The exact physical nature of surface barriers in MOFs is unknown, but reports suggest that defects (possibly pore collapse) at the surface of MOF particles or films formed during solvothermal synthesis slow down mass transfer at the crystal surface (Figure 8).102,106
Figure 8.

Schematic illustration of absorbate molecules encountering “surface barriers” at the crystal surface. In the case of catalytic reactions involving small molecule activation and energy conversion, this phenomenon would occur at the MOF-solution/electrolyte interface. Adapted with permission from reference (106). Copyright 2014 Springer Nature.
In particular, large linear 1D channels may provide advantages for mass transport; however, further research is required in the rational design of MOF structures that are tailored to enhance substrate mass transport for catalysis by considering factors like tortuosity and constriction. Current literature reports have explored a few variations on the framework topology (UiO-6x, NU-1000, PCN, MIL-101), many of which have design properties fitted to other applications (e.g., gas sorption), rather than solution-based catalysis, which requires facile mass transfer through solvent-filled pores.
Several examples exist where the dimensionality of the whole framework has been reduced specifically to enhance mass transport rates. Catalytic two-dimensional (2D) MOFs, sometimes referred to as metal–organic layers (MOLs)107 have been shown to have higher observed rates of product formation than the analogous 3D frameworks with the same catalytic active site. This takes advantage of the fact that a thin slab is the optimal geometric shape to minimize diffusion time of a substrate in a given volume. This strategy has also been employed for electrochemical CO2 reduction mediated by molecular catalysts stabilized within 2D MOF materials.108,109
2.1.2. Relevant Kinetic Models for Chemical Catalysis in Porous Media
While the mathematical analysis of reaction-diffusion in porous particles is well-known for simple, single step reactions,110,111 small molecule activation and solar fuel production in MOFs are inherently multielectron, multistep processes. Explicit treatment of multistep catalytic reactions coupled to mass transport is highly relevant in this regard. Just as in the analysis of homogeneous molecular catalysis of electrochemical reactions,96 a derivation of the analytical rate expressions (approximate or exact when possible) for a given mechanism is important. This is because, for a multistep mechanism, the system of differential equations that needs to be solved is highly coupled and potentially nonlinear, due to intermediates being generated and consumed as well as owing to the presence of multiple reaction pathways. Certainly, each mechanism will have a range of possible solutions for the concentration profiles of substrate and intermediates and the overall observed reaction rate, which depend on limiting values of the intrinsic and operational parameters. Kinetic analysis of molecular catalysis in MOFs could benefit from an update of the single-step Thiele model, tailored for solar fuel reaction mechanisms (OER, HER, ORR, CO2RR, and so forth).
For example, Johnson et al.98 developed a quantitative model for OER under steady reaction-diffusion in spherical MOF particles, catalyzed by a molecular Ru water oxidation catalyst (WOC), and driven by a sacrificial oxidant. It was shown that the diffusion of the chemical oxidant could become significant, where its concentration will vary significantly over the radius of the particle. Two possible OER mechanisms for molecular Ru catalysts (Figure 9a displays the common water-nucleophilic attack mechanism) were analyzed and found to both give rise to nonlinear reaction rates, which were coupled with diffusion in axisymmetric spherical coordinates. All limiting kinetics behaviors can be summarized in a zone diagram (Figure 9b). This analysis allowed for deriving expressions for the observed reaction rate as a function of various operational and intrinsic parameters. Importantly, two definitions for the TOF were proposed: an apparent TOF, which normalized the rate to the total amount of catalyst present and a true TOF, which appropriately considers only the amount of active catalyst in a catalytic particle:
| 3 |
| 4 |
where v is the reaction rate in mol s–1, ntotal is the total moles of catalyst within the MOF particle, and nactive is given by
| 5 |
the integral of the concentration of the catalyst over the volume of the particle V, where r is the radial distance in spherical coordinates. If diffusion is relatively fast, the active catalyst concentration is homogeneous throughout the particle, and TOFapp is equivalent to TOFtrue. However, if diffusion is slow enough compared to the rate of the reaction, the active catalyst concentration will be inhomogeneous across the spatial dimension of the particle (i.e., gradients will develop). When this occurs, it was found that apparent TOFs, as they are most frequently reported, deviate from the true activity of the molecular catalyst and are a composite function of many different parameters (Figure 9c). For example, when diffusional transport of the oxidant is kinetically significant
| 6 |
where R is the radius of the particle, Dox is the diffusivity of the sacrificial oxidant, Cox0 is the bulk oxidant concentration, and Ccat is the total catalyst concentration. The catalytic reaction for the OER can be described by a pseudo-first-order rate constant kcat = kCS0, where CS is the substrate concentration. Here, since water is the substrate and solvent, CS0 remains constant with respect to space and time (for more information on the mechanistic model, relevant assumptions, and derivation of eq 6 see the SI of reference (98)). This predicts that the calculated apparent TOF will not only depend on the size of the particle (R) and the diffusivity of a particular cosubstrate, but will also give a falsified negative one-half reaction order with respect to the catalyst. Significant errors will result if a value such as in eq 6 is inferred to represent only the molecular catalyst’s intrinsic reactivity. Therefore, apparent TOFs calculated using the total amount of catalyst should be viewed as mixed kinetic parameters, unless intra-MOF transport limitations can be exclusively ruled out.
Figure 9.

a) Water-nucleophilic attack mechanism with diffusion of the sacrificial oxidant through MOF particles (approximated as a sphere). The kinetic behavior is completely controlled by two dimensionless governing parameters: λ, a parameter that measures diffusion competing with the catalytic reaction and μ, a competition parameter for the rate-limiting chemical reaction step. b) Zone diagram summarizing all limiting kinetic behaviors with closed-form rate expressions. The upper right-hand compass shows how the variation of each operational and intrinsic parameter translates the system between reaction-limited (bulk reactivity) and transport-limited (surface reactivity) kinetics within the zone diagram. c) TOF expressions for each kinetic zone. Adapted with permission from reference (98). Copyright 2020 Royal Society of Chemistry.
Currently, only a handful of experimental reports of MOFs with applications to heterogeneous catalysis have investigated the relationship between the size of the particles and the observed catalytic activity.112,113 For MOFs, this may be due to difficulties with synthesizing particles of different sizes while keeping all other parameters constant (e.g., defect sites, excess modulator coordinated to nodes). Although, with appropriate reaction-diffusion modeling, it may be possible to interpret kinetic data and identify transport limitations. For example, Wang et al.114 investigated cerium ammonium nitrate (CAN)-driven water oxidation by an Ir complex incorporated into a UiO-69-type framework. The reaction rate was determined to be first order with respect to the CAN concentration through kinetic measurements that followed the consumption of CAN in the bulk solution by UV−vis spectroscopy. Fitting the experimental data to a reaction-diffusion model for the first-order consumption of CAN within the particles revealed that the oxidant only penetrates 11% in depth at steady-state, due to the slow diffusion of the oxidant relative to the fast catalytic reaction.
2.2. Molecular Catalysis of Electrochemical Reactions in MOFs
2.2.1. Conduction Mechanisms
Conduction in MOF materials can occur through several different processes. These can be broadly categorized by the mode through which charge is transported. Band or ohmic conduction operates solely through migration, where an electrostatic potential gradient provides the dominate driving force for charge carrier transport through the MOF material. This requires strong electronic coupling between the linkers or nodes in the MOF, either through-bond or through-space, such that the electronic states are highly delocalized. It is relatively easy to verify this type of conduction when examining the current response of MOFs of this type deposited on electrodes. In the absence of any substrate or coupled chemical reactions, these materials will only display capacitive current as no charge transfer is taking place across the interface. Some MOFs have been reported to exhibit properties similar to semiconductors or even conductors.115−117 A typical example is the Ni3(hexaiminotriphenylene)2 2D MOF reported by Dincǎ and co-workers.118 Upon addition of a substrate into the bulk solution, Faradaic current may be observed as the result of an inner-sphere electrochemical reaction between an active site in the MOF material and a substrate molecule. Electronic structure calculations can be used to indicate whether the MOF should be classified as semiconductor or insulator, where a semiconductor should have a large bandwidth (dispersion). Such calculations have shown that MOFs with carboxylate-based linkers are insulators, as they exhibit very flat and narrow bands and a total dispersion in the valence band of only ∼0.1 eV.119 Optical charge transfer bands that may appear in the absorption spectrum are a result of localized electronic states with little delocalization. This situation applies to a large proportion of MOFs discussed in this review, as many of them are constructed from carboxylate-based linkers.117 It should be pointed out that some authors incorrectly assume that such MOFs are semiconductors with band-like conduction.
Conversely, conduction may occur via electron hopping, formally a diffusion process, where the driving force for charge transport is primarily a chemical potential gradient. This gradient is classically generated through outer-sphere electron transfer at an electrode/MOF interface, but could also be present due to “localized” chemical oxidation or reduction of components of the MOF at the surface of a film or particle. This can occur if the chemical oxidizing/reducing agent is size-excluded from MOF pores and cannot diffuse further into the interior of the MOF particle/film. These MOFs have highly localized electronic states, characterized by a standard potential (E0). Additionally, the current–potential response of MOFs exhibiting this mode of conduction will display Faradaic current in the absence of a substrate, attributed to the outer-sphere reduction or oxidation of the molecular components of the MOF and subsequent diffusional electron hopping through the material.
This distinction in the type of conduction pathway is very important for an accurate interpretation of experimental kinetic data. Outer-sphere electron hopping necessitates stepwise mechanisms (ECEC, EECC; where E represents an electrochemical step and C represents a chemical reaction step). This allows for the concepts and tools developed for homogeneous molecular catalysis of electrochemical reactions120 to be utilized with modification to include diffusion of substrate and diffusional charge transport121−123 for analyzing the current–potential response of this type of MOF. On the other hand, MOFs with band or ohmic conduction will have inner-sphere catalytic mechanisms resembling traditional electrocatalysts.124 Microkinetic models for inner-sphere mechanisms typically rely on Tafel analysis of the current response at high overpotentials.125,126 When the steady-state catalytic current is large, care must be taken to avoid mass transport limitations within the material121 as well as in solution.127 For example, recently Unwin, Dincǎ, and co-workers128 compared data from planar rotating disk electrodes, gas diffusion electrodes, and scanning electrochemical cell microscopy to reveal O2 transport limitations at multiple scales for the ORR by conducting Ni3(HITP)2. It was shown that this MOF’s activity has been underestimated by several orders of magnitude due to mass transport limitations present in previous measurements.
In this review, we mainly focus on MOFs with electron hopping conduction since this type of conductivity also implies discrete molecular active sites, which are more relevant when considering rationally designed bioinspired or biomimetic higher coordination sphere effects. Biological electron transport in and between proteins typically occurs by electron hopping between discrete redox centers, over distances of up to 15 Å for a single step.129,130 It should be noted that some MOF materials may display conduction behavior with characteristics of both mechanisms discussed here (band and hopping), and charge transport can be described as a hybrid process. Recently, this has been shown to be the case for porous metal oxide catalysts for the OER.131,132
2.2.2. Kinetic Analysis of Heterogenized Molecular Catalysis of Electrochemical Reactions
The kinetic analysis of heterogenized molecular catalysis of electrochemical reactions has a long history, starting in the 1980s with redox-polymer-modified electrodes.133 This subject has recently been well-reviewed.134,135 Indeed, these kinetic models can be applied to MOFs as well.60 Rather than presenting these in detail, in this section we will only focus on two specific metrics regularly used to compare the catalytic activity of electroactive MOF film electrodes, namely, the TOF and the onset potential, and briefly discuss their modern-day usage and potential utility.
2.2.2.1. Turnover Frequencies (TOFs)
Many reports that will be discussed in this review present TOFs for catalytic reactions based on comparisons of two or more systems which differ by a certain structural aspect/property. As discussed above, when dealing with catalysis in porous materials the TOF is a function of the intrinsic reaction rate, but potentially also of many other factors, such as diffusion coefficients, particle size, or film thickness. In principle, it is also a transient function, changing dynamically over time and space as the catalytic reaction approaches a steady-state.136
If experimental conditions are controlled, it is possible to measure a steady-state current, which can be then used to compute the TOF. In the ideal case, the catalytic reaction takes place homogeneously throughout the film (the concentration of active catalyst and substrate are at their bulk values over the entire length scale defining the film). Converting the steady-state current density to a TOF, which can be used to compare catalytic systems, then only requires dividing by the total amount of catalyst in the film using Faraday’s constant as a conversion factor.
However, if at the steady-state the concentration of the active
catalyst or the substrate is not spatially homogeneous across the
film, converting the current to a TOF is not as simple. This situation
will arise when the distance that a diffusing species travels into
the film during the time scale of the catalytic reaction is significantly
less than the geometric length scale of the film. Boundary layers
develop where the active catalyst or substrate concentration is depleted
within a small distance from one of the film’s edges. Consequently,
the reaction only takes place in a small reaction layer of size 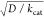 , where D is either the
electron hopping diffusion coefficient or the diffusion coefficient
of the substrate, and kcat is the intrinsic
catalytic rate (s–1). This means that the amount
of active catalyst is much less than the total, and simply dividing
the steady-state current by the total amount of catalyst will result
in an apparent TOF, which underestimates the molecular catalyst’s
true activity (see also discussion around eq 3 and 4 above).
, where D is either the
electron hopping diffusion coefficient or the diffusion coefficient
of the substrate, and kcat is the intrinsic
catalytic rate (s–1). This means that the amount
of active catalyst is much less than the total, and simply dividing
the steady-state current by the total amount of catalyst will result
in an apparent TOF, which underestimates the molecular catalyst’s
true activity (see also discussion around eq 3 and 4 above).
Varying the film thickness can be used as a strategy to diagnose
when transport limitations are present and to position the catalytic
system such that the measured current reflects an intrinsic rate.
If the steady-state current varies linearly with the film thickness,
transport limitations are absent. On the other hand, the catalytic
current remaining constant as the film thickness is increased signals
that the catalytic reaction is faster than diffusion. Again, TOFs
measured under these conditions will underestimate kcat, and will depend on more parameters than just the
catalyst’s intrinsic activity (diffusivities and film thickness).
Generally, using suitably thin films such that 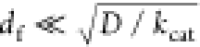 (where df is
the film thickness) remedies these issues.
(where df is
the film thickness) remedies these issues.
2.2.2.2. Onset Potentials
Unless onset potentials are derived from a kinetic model including reaction-diffusion, they should not be used to infer kinetic or mechanistic details. Since the current at the foot of any catalytic wave is an exponential function of the electrode potential,137 it even may be difficult to define the onset without referencing another distinct observable in the current–potential response (e.g., plateau current, catalytic half-wave potential, or standard potential of the catalyst). The point at which the catalytic current emerges from the background (capacitive) current will depend on, for example, noise levels and potential step size of the data.138,139 Other challenges are encountered when the diffusion of substrate is considered. For example, onset potentials appear to change as a function of film thickness in Figure 10 when substrate transport limitations interfere with kinetic control. In fact, they appear to be more and more positive as the film thickness is increased and all other parameters are held constant. One might be tempted to interpret an arbitrary increase in film thickness as a better catalyst, when in fact the opposite is true and the kinetics are under mixed control by substrate diffusion and the catalytic reaction.
Figure 10.

a) Steady-state cyclic voltammograms of catalytic MOF films with different thicknesses (10–500 nm) showing the transition from kinetic control by the catalytic reaction to mixed kinetic control by the catalytic reaction and substrate diffusion when the film thickness is increased (see reference (122) for derivation of the current–potential expression). Under these conditions, the catalytic half-wave potential E1/2 shifts positive as the film thickness increases, which is a result of the influence of substrate diffusion within the film. b) After an optimal film thickness is reached, the current reaches a maximum value and no longer increases if the film is made thicker. The apparent shift in onset potential to more positive values would suggest a more efficient catalyst when an arbitrary increase in the film thickness in fact makes the catalyst less efficient. Other parameters used: kcat = 100 s–1, DS = 10–9 cm2 s–1, catalyst concentration = 0.1 M.
2.3. Photochemically Driven Molecular Catalysis in MOFs
Many examples of light-driven MOF systems for water oxidation or reduction of protons or CO2 can be found in the literature. This is of course coupled to the long-term goal of direct solar energy conversion to a fuel, as an attractive alternative to indirect methods using renewable electricity or biomass as intermediates. From a viewpoint of fundamental understanding, the example of photoactivated enzymes and enzyme complexes shows the power of phototriggered reactions to elucidate complex reactions and identify intermediates. The detailed understanding of photosystems I and II is much greater compared to nonphotochemical energy converting enzymes such as hydrogenases and nitrogenases. Light-activation of enzymes has therefore become a popular strategy, by adding a PS to, for example, ribonucleotide reductase140 or hydrogenases,141,142 following the pioneering work on dye-labeled electron transfer proteins.129 One motivation for covalently linking dyes to proteins is to be able to phototrigger electron transfer reactions with light pulses and follow the photochemical reactions on short time scales with time-resolved spectroscopy. From the viewpoint of solar-driven generation of fuels, there is also the idea that a linked PS may lead to more efficient charge transfer to the enzyme and thus better photocatalytic properties than a bimolecular reaction with a PS in the same solution.
Photoactivation of MOFs has been motivated by similar interests as those above. However, the evaluation and understanding of the performance of these systems are challenging, as the photochemical reaction cycles involve many reaction steps, productive as well as nonproductive ones. Typically, the quantum yield for product formation is less than 10%, which means that other, competing reactions dominate the photochemistry. It is well-recognized that recombination of the photogenerated charges is the most common and general challenge to efficient artificial photosynthesis. Moreover, product formation is also limited by a rather slow photogeneration of redox equivalents, compared to electrochemical or dark chemical redox reactions. Thus, the observed rate is often not limited by the intrinsic ability of the catalyst to turn over. Photochemically driven molecular catalysis can therefore be even more difficult to analyze than electrochemically driven catalysis as discussed above. These points are illustrated and discussed in this section.
2.3.1. Photochemically Driven Catalytic Cycles
Photochemical production of fuels and water oxidation in homogeneous or heterogeneous systems is usually examined by measuring the amount of generated product as a function of time under constant light irradiation. The vast majority of systems examine only one half-reaction, instead of complete artificial photosynthesis, which is water oxidation coupled to reduction of, for example, protons or CO2 to a fuel. Most systems use an alternative sacrificial donor or acceptor instead. For simplicity, we will take photochemical H2 generation as example; see Figure 11 for a simplified reaction scheme of a type commonly used to illustrate the photocatalytic HER in a homogeneous solution or in MOFs.
Figure 11.

A common, simplified reaction scheme for photochemical proton reduction to H2, with a photosensitizer (PS), a catalyst (Cat), and a sacrificial donor (D). The simplicity of the scheme is deceptive, as it only shows a small number of the reaction steps that must be considered.
The TOF is defined as the instantaneous rate of H2 production (in moles per time unit) per active site.136 The TON is usually reported as the maximum amount (mol) of H2 produced per mol catalyst at the time the experiment is stopped, often because the production of H2 has stopped or become very slow. In photochemical reactions, it is common that the reaction stops for other reasons than that the catalyst has degraded. Instead, the sensitizer may have degraded, the sacrificial agent may have been consumed, or the pH may have changed. In these cases, one can demonstrate the longevity of the catalyst by adding more sensitizer or sacrificial agent, or resetting the pH. Many authors therefore also report maximum TONs based on PS.
It is important to realize that the observed TOF in a photocatalytic experiment is not an intrinsic parameter of the catalyst, as was discussed earlier for electrocatalysis. What is evaluated is the entire system and not the catalyst itself. Usually, the observed TOF is limited by the rate of photon absorption and by charge recombination reactions that make reaction quantum yields much lower than unity. This is a consequence of the fact that the photochemical reaction of the OER coupled to the HER or the CO2RR is photosynthetic, which means it uses light energy to drive an uphill reaction, so that charge recombination to reform the starting reactants is a downhill process. This is obviously different from dark reaction methods such as electrocatalysis, which are intrinsically downhill, or photocatalytic conversion of wastewater to H2, which is energy neutral or slightly downhill. For the rest of this review, we will still use the term “photocatalytic” also for photosynthetic systems, although the differences between such systems are important.143 It is furthermore important to point out that even many “sacrificial” donors and acceptors (e.g., ascorbate) will lead to a large degree of recombination, as they are far from completely sacrificial under typical conditions.144 This typically makes the net delivery of redox equivalents to the catalyst rate-limiting, whereas the rate constants of the catalytic steps would often allow for much higher TOFs than those observed. The additional charge separation and recombination reactions make it more complicated to evaluate the potential performance of a catalysts by photochemical experiments.
To demonstrate these points with an example, let us expand the simplified reaction scheme for the photochemical HER in Figure 11 into a more complete, but still minimal scheme in Figure 12. This consists of a PS, a HER catalyst (Cat), and an electron donor (D) in the presence of protons; this could be a MOF system or a homogeneous solution. We assume that the excited *PS is reductively quenched by the D, and the reduced PS– reduces the Cat. The Cat is assumed to undergo an ECEC mechanism, in which the second electron is provided by a second equivalent of PS–. If the D is completely sacrificial, so that every D+ decomposes without interfering with the other reactions, and all other side reactions can be ignored, the quantum yield ΦH2 for the HER reaction is equal to 1:
| 7 |
where nH2 and nphoton are the number of moles of H2 produced and photons absorbed, respectively (as at least two photons are needed to produce one H2, we multiply by two in the numerator). In this case the TOF will simply be equal to the rate of photon absorption, which contains no information on the catalyst rate constants. In practice, however, there are multiple charge recombination reactions and other side reactions, some of which are shown with red dashed arrows in Figure 12. Consequently, the majority of the published MOF studies show ΦH2 ≪ 1, meaning that most of the absorbed photons and charges generated are not productive. Instead, they are involved in a range of energy-wasting charge recombination reactions—often not identified—and even irreversible redox chemistry that cause degradation of the PS, Cat, and/or MOF linkers. The observed TOF is then simply the rate of photon absorption (Ratehv) multiplied by the HER quantum yield:
| 8 |
None of these factors directly reflect the intrinsic reactions of the catalyst.
Figure 12.

A more complete, but still idealized, reaction scheme for photochemical HER than in Figure 11. The primary reaction of the excited *PS is reductive quenching by the donor, and the catalyst is assumed to follow an ECEC mechanism. Productive reaction steps are shown with black arrows, and charge recombination reactions with red arrows.
In the minimal scheme of Figure 12, the HER quantum yield is given by the products of the yields for photochemical charge generation, electron transfer to catalyst, and catalyst turnover. Each of these processes competes with charge recombination, as shown in red dashed arrows, and the resulting yields are strongly dependent on the competing reactions. Moreover, the catalyst cycles through several intermediate states, each with its own reactivity toward PS– and recombination with D+, which makes the exact kinetics complex. Any change of reaction conditions, or modification of the catalyst, may affect several productive and competing steps. It is therefore not straightforward to determine how the rate constants of the actual catalytic steps have changed. In photocatalytic MOFs, the situation can be even more complicated due to transport limitations and resulting concentration gradients. These may cause the quantum yield ΦH2 to have a spatial dependence within the MOF particle/film. Finally, it should also be pointed out that several MOFs exhibit photochemical instability. Such processes will obscure kinetic information, in particular on longer time scale experiments and under high-energy light irradiation.145
In a large majority of cases, for both homogeneous and MOF-based systems, the observed TOF is not limited by the catalyst’s capacity to turn over. Instead, it is limited by the rate of light absorption and a low quantum yield, and consequently the slow rate of electron delivery to the catalyst.146 An efficient PS is excited only on the order of once every second under full sunlight, and most catalysts can turn over faster than that if electrons and protons are provided. To match the rate of light absorption with that for the intrinsic turnover capacity of the catalyst, multiple PS molecules per catalyst are needed working as a light-harvesting antenna. As one example, Reisner and co-workers147 have used TiO2 particles, each with a large number of attached PS molecules and only few Cat molecules, and found that the photocatalysis rate was independent of light intensity. In most cases in the literature, however, the ratio PS:Cat is not that large. MOF systems on the other hand offer the possibility to implement an antenna function with multiple PS per catalyst, as will be discussed in section 2.3.3.
It is clear from eq 8 that the experimental rate cannot be compared to a dark kinetic experiment when all reactants are added at time t = 0 and the reaction progress is followed over time. In the photochemical experiment, one reactant is generated only slowly, via light absorption, and the reaction can never be faster than the rate of photon absorption. Whether a reaction is light-limited can experimentally be verified through varying the light intensity by, for example, inserting a neutral density filter. If the TOF changes in proportion to the light intensity, the TOF is light-limited. This straightforward test is quite informative and should be routinely reported, yet it is only rarely done. In one case, photochemical water oxidation was compared in homogeneous and heterogeneous systems by Bonnet and co-workers,148 who carefully examined the dependence of the TOF on the concentration of all reactants as well as on the light intensity. With [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) as PS, [Ru(bda)(isoq)2] (H2bda = 2,2′-bipyridine-6,6′-dicarboxylic acid; isoq = isoquinoline) as a catalyst, and persulfate as a donor, the TOF increased linearly with increasing light intensity until ∼1 sun irradiation was reached and the TOF maximized. Even if the reaction was no longer light-limited, the limiting step was still not catalytic turnover. Instead, they concluded that electron transfer from the catalyst to the oxidized PS limited the reaction. Only when using slower catalysts in the homogeneous system could they observe rate limitations from slow catalyst turnover. It should be noted that even in this case, the obtained TOF was lower than the intrinsic rate of the catalyst. The catalyst sluggishness instead reduced the quantum yield ΦH2 because competing, nonproductive reactions had time to occur to a greater extent. Interestingly, when they used a liposomal system instead, in which amphiphilic analogues of their PS and Cat were bound to liposomes, they found that photogeneration of PS+ was limiting instead.149
To illustrate this argument, we can assume that the limiting step of a HER catalyst is the final protonation of the hydride state, that under the experimental conditions has a rate constant of 103 s–1. This catalyst then has the potential to reach up to a TOF = 103 s–1. But if the hydride state at the same time recombines with D+ with a pseudo-first-order constant of 105 s–1 under certain conditions, ΦH2 will be low and a TOF ≪ 103 s–1 will be observed. This may then be interpreted as a slow catalyst, but the problem is rather the rapid recombination reaction. The TOF would only be equal to 103 s–1 under conditions where the rate of photon absorption multiplied by ΦH2 (eq 8) is equal to 103 s–1 per catalyst, which is unlikely for most systems studied to date. The effects of synthetic improvement of the intrinsic catalyst rate may then be masked by effects on the charge recombination.
These reports of homogeneous photoinduced catalysis described above are examples that underline the importance of varying the experimental conditions to elucidate qualitative kinetic features. However, a quantitative relationship between reported initial TOFs or maximal TONs and the parameters of the system (light absorption, catalytic rate constants, back electron transfer, concentration of sacrificial donor and PS, and so forth) is required for benchmarking and reliably comparing photocatalytic systems. Costentin et al.146 recently reported a general analytical kinetic model for such reactions. They derive an analytical expression for the time evolution of the TON that takes the form of a relatively simple third-order polynomial. This is a function of several dimensionless parameters, which represent the competition between various steps in the mechanism. It was identified that in all situations the kinetic information that can be obtained depends on the rate of light absorption. Additionally, this analytical derivation demonstrated that under certain conditions, the rate of the catalytic reaction cannot be retrieved from the time evolution of the TON, as this tends asymptotically to a function that only depends on quenching, back electron transfer, and reduction of the catalyst, in addition to light absorption. An important realization from this analysis was that the TON increases nonlinearly with time due to accumulation of the oxidized donor and the resulting increasing rate of back electron transfer. As a consequence, even within relatively short irradiation times the concept of an initial TOF is no longer meaningful. Finally, by extending this general kinetic model to included deactivation of the catalyst or PS as well as a two-electron stoichiometry, the authors were able to analyze experimental data of light-driven HER by a cobalt tetraazamacrocyclic catalyst in water with ascorbate as the sacrificial donor and either [Ru(bpy)3]2+ or a triazatriangulenium organic dye as the PS. With the addition of the diffusion of the various reaction components, a general kinetic model such as this one that allows for the derivation of the TON versus time expression could be used to understand photoinduced processes in MOFs with molecular catalysts.
It is clear that also MOF-based systems may operate under quite different limiting conditions. A similar analysis as that for liposomes above, with a systematic variation of concentrations and light intensities, has to the best of our knowledge not been performed for MOFs. A few interesting cases exist where the rate of product formation was compared for different catalyst loadings, leading to mechanistic insights, as discussed below (see section 2.3.5 and section 3).
The above discussion may seem discouraging for attempts at gaining mechanistic information on a catalyst through photochemical methods, but it is important to appreciate that a catalyst is always tested as a part of a system. Under electrochemical conditions interfacial electron transfer is ideally assumed to be rapid and the chemical steps limiting. Even in the many cases where this assumption is correct, the same catalyst under dark chemical reduction may be limited by bimolecular electron transfer from the reductant. Moreover, mass transport may limit the overall catalytic process, as discussed in section 2.2.1 for chemical and electrochemical catalysis. As exemplified above, however, more detailed information can be obtained also with photochemical catalysis, provided that necessary kinetic investigations are conducted where the TOF is measured as a function of different reactant concentrations as well as light intensity. In section 2.3.5, we outline some important qualitative differences in photochemical performance as a function of catalyst concentration and light intensity, to illustrate the possibilities to distinguish between cases where different factors are limiting the overall reaction, which is obviously helpful for improved understanding. Moreover, with time-resolved spectroscopic methods, using pulsed excitation light, catalyst intermediates and their reaction steps can be identified and directly followed in a way that may be challenging with other experimental approaches.
2.3.2. Time-Resolved Spectroscopy: Possibilities and Challenges
Time-resolved spectroscopy with pulsed laser excitation is very useful to determine the rates of photochemical reactions. Time-resolved fluorescence can give high-quality kinetic data for the decay of the initial excited state. Often the assignment of the fluorescent state is quite clear, but in more complex systems, such as MOFs, this may not be the case. Typical complications may be electronic interactions between nearby chromophores leading to excimers, exciplexes or larger aggregates. But even in the absence of such complications, the interpretation of the fluorescence decay is not straightforward. In the MOF literature, the quenching of fluorescence lifetime and/or intensity when the dye is introduced in the MOF is usually assumed to reflect the desired electron transfer reaction to or from the dye. However, many other mechanisms besides electron transfer exist that can quench fluorescence, so fluorescence quenching is insufficient evidence for productive charge separation. These mechanisms include energy transfer to either fluorescent or “dark” states, enhanced intersystem-crossing (for example by the heavy-atom effect) to form a nonfluorescent dye triplet state, paramagnetic quenching by radicals or transition metals, and formation of different types of nonfluorescent aggregates. Therefore, while fluorescence quenching is great for the acquisition of high-quality kinetic data, additional evidence is needed to safely assign the kinetics to charge separation.
Transient absorption spectroscopy can be used to detect the electron transfer products. In many cases where this has been used as evidence for charge separation in MOFs, the transient absorption spectra have only revealed the ground state bleach, which decay without formation of other detectable products. Since the species of the proposed charge-separated states in these cases actually have clear absorption characteristics, the data instead prove that productive charge separation did not occur, in contrast to the conclusions of these papers. In some cases, the authors used a so-called state-filling model, as used for semiconductors, to explain why the charge-separated state would not show a signal. Briefly, the model says that the transient ground state bleach is because electrons fill conduction-band states, but that valence-band holes give negligible contribution to the transient signal. However, this is not a valid model for an insulating MOF with local excited states. The absorption and fluorescence bands of the free dyes in solution are still clearly resolved in the MOF in many cases, for example the Soret and Q-bands of porphyrin linkers. This clearly shows the molecular nature of the ground and excited states in the MOF.150,151 Therefore, the charge-separated state in such MOFs, where the porphyrin unit is oxidized, would have shown a strong porphyrin ground state bleach and clear positive signals from the resulting porphyrin radical. The absence of such signals is evidence against—and not for—charge separation.
It is also important to use the proper reference system when assessing MOFs with transient spectroscopy. Insertion of a transition-metal ion in porphyrins or phthalocyanines introduces low-lying charge-transfer and metal-centered states that typically lead to accelerated radiation-less decay to the ground state (<1 ns). This is true for popular catalyst metal ions such as NiII, CoII, and FeIII. That the same thing happens in a MOF should not come as a surprise.150,152,153 The porphyrin excited state, and its fluorescence, will be quenched relative to the free base, but this should not be taken as evidence for productive electron transfer to the putative catalytic metal in the MOF. It is just the intrinsic photophysics of the metal porphyrin.
Even in cases where quenching of the *PS is due to charge separation, covalently linked dye–acceptor or dye–donor dyads typically show rapid recombination of the charge-separated species. Long-lived charge separation usually requires the addition of further acceptor or donor molecules to form triads, tetrads and so forth.154−156 The rate constant for recombination is often faster than that for charge separation, so that no or very little of the charge-separated intermediate is seen. This is a very likely scenario in MOFs, too, and could explain the spectroscopic observations. It is important to realize that if the yield of long-lived charge-separated states is very small, this may not be observed in the transient absorption experiment. If, for example, 1% of the initially separated charges jump to neighboring sites and escape recombination, this can be challenging to detect. Nevertheless, it can still suffice to drive a photocatalytic reaction with ∼1% quantum yield, and many reports show quantum yields in that range or much lower. Thus, the absence of a detectable charge-separated state in a transient absorption experiment is not necessarily in disagreement with detection of photoproducts in the photocatalytic experiments. What the transient experiments then show is that the excited state is quenched and the yield of long-lived charge separation is small.
Some examples where long-lived charge separation in MOFs has been observed by transient spectroscopy include [Ru(bpydc)(bpy)2]2+-decorated (bpydc = 2,2′-bipyridine-5,5′-dicarboxylate) UiO-67.157,158 Santiago-Portillo et al.158 showed relatively long-lived products and by adding electron or hole scavengers they could attribute the signals to a charge-separated state with electrons on the zirconia cluster and holes on the bipyridine ligand. It is interesting that zirconia clusters are widely taken as electron acceptors in the MOF field, whereas bulk zirconia has a very high lying conduction band, and zirconia nanoparticles are used as noninjecting reference substrates in the dye-sensitized solar cell field, with the same type of carboxylate linkers.159 We speculate that there may be different defect sites on the smaller zirconia clusters of MOFs. Lin et al.157 used both the [Ru(bpydc)(bpy)2]2+-sensitizer and a [Ru(terpy)(bpydc)Cl]+ catalyst (terpy = 2,2′:6′,2″-terpyridine) as linkers and could detect rapid (21 ns) generation of oxidized and/or excited catalyst. From the slow decay of their transient, with components up to ca. 280 μs, it was clear that this decay was due to the oxidized catalyst.
Time-resolved laser spectroscopy initiates light reactions with a laser pulse that excites a minor part of the sample and triggers charge-separation reactions. The experiment is typically repeated several times to obtain data of good quality. Thus, the sample has to be regenerated by charge recombination between each laser pulse. If a sacrificial donor or acceptor is used instead, the sample will evolve to form more and more of catalyst in reduced (or oxidized for OER) states with increasing number of flashes, and the transient absorption results will change. With nanosecond flash photolysis one may get useful kinetic data in a single flash and thus track the changes in kinetics as the system evolves, but interpretation becomes increasingly complex. One usually has a mixture of catalyst states, with optical absorption spectra that are often quite indistinct so that their concentrations cannot easily be determined. This has been possible in some cases with some notable examples being TiO2 particles with molecular HER catalysts.160,161 In these cases, the different redox states of the catalyst showed sufficiently distinct absorption spectra for quantification. Another approach to follow later steps in the photoredox cycle is to prereduce (or preoxidize) the sample electrochemically, and do in situ laser spectroscopy, to be able to measure the reactions of a well-defined state. Finally, pump–pump–probe experiments have been performed, where a second laser pulse is applied at a selected time after the first, to achieve accumulative charge separation—i.e., successive photoinduced charge separation after sequential photon absorptions162—on the two-electron level, in nonsacrificial systems.163−168 Accumulative electron transfer has even proven possible in molecular systems under light levels corresponding to solar irradiation.169 These methods have yet to be applied to study solar fuels-forming photocatalytic MOFs.
2.3.3. Coimmobilizing PS and Catalyst in MOFs
Many molecular dyads and larger assemblies, including MOFs, have been prepared and studied where both PS and Cat are immobilized at close distance, thus designing the reactive coordination sphere of the catalysts. One motivation for immobilizing the PS and the catalyst is to perform fundamental studies, where the linkage removes the need for diffusional steps in the reaction that may limit and obscure the kinetics of the PS–catalyst electron transfer itself. A more pragmatic motivation is the desire to increase photocatalytic performance, exerting better control over the electron transfer reactions with linked reactants, and to increase the electron transfer rate. It is true that a linked PS–catalyst system will typically result in faster electron transfer to or from the catalyst than for the corresponding bimolecular reaction, especially when low concentrations of reactants are used. However, this does not necessarily lead to faster overall catalysis (higher TOF), because this electron transfer step may not be rate-limiting. As discussed above, the observed rate of catalysis is given by the rate of photon absorption multiplied by the photoproduct quantum yield. Photon absorption is not improved by linking the reactants, and the product quantum yield is only favored by a fast electron transfer to or from excited *PS if this reaction competes with some other reaction, such as spontaneous decay of the *PS to the ground state. With an initial reductive quenching of *PS by a donor as an example, recombination of PS– with D+ typically leads to large losses of efficiency, unless the sacrificial reaction of the donor is ultrafast. A fast electron transfer from PS– to Cat can then improve the system performance, but only if recombination of D+ with the reduced catalyst is slower than recombination of D+ with PS–. Thus, while MOFs with a relatively short PS–catalyst distance have often been assumed to give a rapid initial electron transfer, this is far from certain to lead to efficient product formation.
If the primary reaction with excited PS* is with the catalyst instead of with a donor—such as in many MOFs where the PS is the linker and the Cat is the node—the electron transfer products (PS+ and Cat–) will typically recombine much faster than in a bimolecular system. This may therefore lead to much lower efficiency for the linked system than for the corresponding bimolecular system. In bimolecular, photoinduced electron transfer, the charge-separated products will have a lifetime >1 ms under one sun irradiation before they recombine by diffusional encounters. In linked systems, the charges typically recombine much faster than that. While photosystem II, with its optimized, well-ordered array of electron transfer components, achieves a charge-separation yield of about 100% and a lifetime of ∼1 s,170 such a performance has proven extraordinary difficult to achieve in dyads, triads, and so forth.154−156 Lifetimes and yields approaching these values have only been reached in heterogeneous molecular materials (e.g., organic and dye-sensitized solar cells). In this context, MOFs provide an interesting possibility for a molecularly designed and well-arranged 3D material to realize efficient charge separation and transport, coupled to catalysis. In section 2.3.4, we will discuss different topological arrangements of PS and Cat in MOFs.
A major advantage with linking the components in MOFs is that a high local concentration of PS around each catalyst can be achieved. A similarly high concentration in homogeneous solution would lead to extreme absorbance values, such that light would not reach far into the solution/suspension. It is also likely to lead to uncontrolled aggregation and precipitation. With a high concentration of PS on a MOF, they may act as antennas for the catalyst, so that the rate of photon absorption can match that of the intrinsic capacity of the catalyst to turn over. Thus, it is an advantage to have a high PS:Cat ratio, so that the catalyst molecules do not compete for photogenerated redox equivalents. An increase in catalyst concentration can lead to not just a lower TOF (per catalyst) but even to a lower rate of product formation, because the slower rate of turnover of each catalyst means a great probability that the redox equivalents are lost in competing reactions.
A high PS density in MOFs can enable efficient PS-to-PS energy transfer, and following a primary quenching with a donor/acceptor also hopping of redox equivalents can be facilitated. Rapid and efficient excitation energy transfer has been demonstrated in MOFs with either porphyrin or transition-metal complex PSs.171,172 The amplified photoluminescence quenching, where a small number of quenchers are sufficient to quench a large part of the MOF’s fluorescence, proves that efficient light-harvesting coupled to charge-separation centers is possible. Even in photocatalytic MOFs, light-harvesting energy transfer coupled to the CO2RR has been reported, and reduced intermediates of the [Re(bpydc)(CO)3Cl] catalyst were detected by in situ FTIR spectroscopy.173
For organic molecules,174 and recently a transition-metal complex,175 symmetry-breaking charge separation has been demonstrated, in which electron transfer between one *PS and one neighboring PS produces one PS+ and one PS–. This effect can be used to obtain efficient charge separation in materials with high density of PS, and principles and design criteria have been discussed in recent reviews.176,177 To the best of our knowledge, this possibility remains to be explored in MOFs.
A high density of PS can also lead to energy-wasting reactions, however, such as exciton–exciton annihilation (at high photon fluxes), and concentration quenching of a single *PS by neighboring ground state PS molecules.178−180 The exact mechanism behind the concentration quenching is often not identified, but molecule–molecule interactions can lead to excitonic states with short lifetimes.
As pointed out above, incorporation of a dye and a catalyst at short distance in the same MOF typically increases the rates of both charge separation and recombination. Moreover, additional difficulties and competing reactions exist when the catalyst has to cycle through different oxidation states, as has been outlined by Hammarström.162 Taking a general donor–photosensitizer–acceptor triad as an example (Figure 13), the initial photoinduced charge separation has to overcome the usual recombination reactions. When the second photon is absorbed, there are not only the corresponding recombination reactions (black, dashed arrows) but there is a large driving force also for reverse electron transfer (red, dashed arrows) where the excited dye sends an electron back to the donor that was oxidized by the first photon, and/or take an electron from the reduced acceptor. This means that the second photon absorption reverses the result of the first photon absorption. In addition, if the original acceptor/donor states are closed shell species, their resulting reduced/oxidized states are typically paramagnetic and have low lying electronically excited states, which may quench the dye in reactions that are not productive for photochemical charge separation and catalysis. These competing reactions are important to consider in the design of a photocatalytic system, especially for complete water splitting or the CO2RR coupled to the OER. In sacrificial half-reactions, the competing reactions of the dye excited state can often be counteracted through a sufficiently rapid initial quenching by the irreversible donor/acceptor, which then blocks all recombination reactions. On the other hand, the ultimate goal should be a complete artificial photosynthetic system, and then one cannot rely on this strategy. The progress on sacrificial half-reactions may therefore hide the difficulties involved in making a complete system.
Figure 13.

Reaction scheme for accumulative charge separation in a donor–photosensitizer–acceptor (DPA) triad undergoing successive absorption of two photons. Solid arrows represent productive reactions after the first (blue) and second (red) photon absorption; dashed arrows represent charge recombination reactions, where the reverse electron transfer reactions are shown in red. Reproduced with permission from reference (162). Copyright 2015 American Chemical Society.
2.3.4. Design of MOFs for Photochemical Charge Separation and Catalysis
The design of photoactive MOFs for solar fuels generation can be based on different topological arrangements, with either the PS or the catalyst (HER is the example in Figure 14) residing in the MOF and the other in solution or on the surface (Figure 14a, b), or with both components inside the MOF (Figure 14c, d).
Figure 14.

Different arrangements of a photosensitizer (PS) and/or a catalyst (Cat) within MOFs. a) Only a Cat within the MOF in combination with a PS and a sacrificial donor (D) outside the matrix. b) Only a PS within the MOF in combination with a D. In a) and b) electron transfer to the catalyst may occur by charge hopping in the MOF or a diffusional redox relay (dashed arrow). c) Both a Cat and a PS within the MOF matrix located but still in combination with a D. d) An arrangement where a PS is combined with two different Cats within a MOF to catalyze water splitting without the use of any sacrificial agents.
With only the catalyst in the MOF (Figure 14a), the PS is often too large to diffuse into the pores. Therefore, only catalysts at the outer MOF layer will react, unless electron transport over the dimension of the MOF is sufficiently rapid to kinetically compete with turnover. This transport can be through electron hopping through the MOF structure, mediated by the MOF backbone and/or through hopping between catalyst sites, or through an electron relay that diffuses through the pores. These transport properties may limit the overall reaction, similar to the dark chemical case discussed above.98 With an external PS and a 100% sacrificial agent, the difference to the dark oxidation case is only that the steady-state concentration of reductant/oxidant is always small, but is continuously generated. This can still lead to transport limitations and a case where mainly the outer catalyst layers are active, but if their intrinsic activity is sufficiently high, one would still only see that the TOF is limited by light generation of oxidizing equivalents. Reports of such systems are discussed in section 3.1.
Systems where only the PS resides in the MOF (Figure 14b) typically contain nonmolecular catalysts on the MOF surface, and these are outside the scope of this review. Their electron transport is limited by analogous limitations as case a) above, but several MOFs show efficient energy transfer between sensitizers that may transport excitons to the MOF interface. Here charge separation and catalysis can occur efficiently with comparatively small transport limitations (see the following for some examples of energy transfer in MOFs171−173,181,182).
Many systems exist with both a PS and a catalyst in the MOF (Figure 14c). They may be introduced at different concentrations in a native MOF structure, as linkers or inside the pores. In other cases, they constitute the entire MOF, often with the linker being the PS and the nodes as the proposed catalyst for either the HER/CO2RR or the OER. Electron transport between the sensitizer and the catalyst may occur via hopping through the MOF or via a diffusional relay. In many cases with a high density of PS and catalyst sites, however, charge separation is believed to occur directly between the sensitizer and a nearby catalyst, either through electron transfer from the excited state or through direct excitation into a ligand-to-metal charge transfer band. Typical examples of the latter are MOFs with meso-4-carboxyphenyl porphyrin ligands and zirconia nodes. Nevertheless, transport of the sacrificial donor/acceptor and the substrate, as well as of the resulting products, are still necessary. This does not exclude that one could run out of donor/acceptor when irradiating for several hours. Even with modest catalyst bulk concentrations and TONs, the quantum yield is often low so that a large amount of donor/acceptor has been consumed.
The short distance between the sensitizer linker and the catalyst node is favorable not only for rapid charge separation but also for rapid charge recombination. It is also important to remember that at least two charge-separation events are needed to complete a catalytic cycle. This design therefore relies on a rapid regeneration of the sensitizer, through electron transfer to or from electron relays or neighboring sites of the MOF, to avoid recombination and also allow for absorption of a second photon. Even if this is achieved, it is important to realize that the catalyst is now in a different redox state than in the dark, and may quench the sensitizer in other ways than by productive charge separation; this was discussed in section 2.3.3.
An ideal system (Figure 14d) includes both the OER and the HER or the CO2RR in the same MOF, which is complete artificial photosynthesis, and is thus independent of sacrificial donors or acceptors. This requires that electron transport in the favorable direction is rapid enough, compared to charge recombination, so that a high product yield is obtained. There are robust guidelines, based on electron transfer theory, how to control electron transfer in terms of energetic factors and electronic coupling (super exchange and hopping transport).183−185 However, from decades of work on artificial photosynthesis, it is clear that this is not easy to realize. Nevertheless, several reports of complete artificial photosynthesis in MOFs have appeared in recent years.150,186−191 The total product formation has in most cases been 100 μmol g–1 or less, which, assuming a reasonable molar mass of 2000 g mol–1, would correspond to 0.2 turnovers per SBU, or, more specifically, per molecular formula. With such low yields, it is particularly important to verify that the products really originate from water splitting and CO2 reduction, and not from degradation of the MOF or other components. Therefore, most authors report isotopically labeled 13CO2 and H218O to prove the origin of the detected products. Even if the yields are low, the fact that some product can be detected is interesting, and by comparison there are no homogeneous (molecular) photochemical systems that have been unambiguously shown to perform complete artificial photosynthesis. Instead, semiconductor systems, colloidal systems as well as electrodes, with or without molecular catalysts, have demonstrated complete artificial photosynthesis, in some cases with quite impressive yields and energy conversion efficiencies.192,193 Classic photoelectrochemical systems with semiconductor/liquid junctions may show quite good performance. The systems with highest efficiencies rely on photoinduced charge separation in well-developed photovoltaic materials, which are isolated from the solution and coupled to catalysts via an ohmic contact layer.194−196 As discussed above, most MOFs used in this field are insulators and not semiconductors, and charge transport would occur by electron hopping or carrier diffusion. It is interesting to speculate why any charges at all escape recombination to instead form products. It is possible that the heterogeneous nature of the system leads to strong kinetic heterogeneity of charge recombination and that some low-probability events can actually accumulate redox equivalents on one catalyst and achieve turnover before recombination occurs. There will always be defects in a MOF that may act as charge traps, which may be favorable for photocatalysis under the right conditions. A further difference to homogeneous systems is that the local concentrations of sensitizers and catalysts are very high in MOFs. When at least one linker per pore is a sensitizer, the local concentration is in the order of 1 M. This is several orders of magnitude larger than in a typical homogeneous experiment and will lead to a higher rate of charge generation per unit volume in the MOF, which should favor multielectron catalysis.
To promote charge separation in MOFs, it would be interesting to introduce redox gradients in MOFs, to retard charge recombination. One particularly interesting report on complete artificial photosynthesis in MOFs used separate MOFs for the HER and the OER, respectively, which were coupled via electron relays.189 This system showed an apparent quantum yield for complete water splitting of 1.5%. It was carefully designed to improve charge separation and counteract recombination by spatially separating them in different regions of a liposome solution.
2.3.5. Analysis of Reactions by Variation of the Catalyst Concentration and Light Intensity
Mechanistic insight may be gained by varying not only the light intensity but also the catalyst concentration in a MOF. This is similar to our point made in section 2.1 in that it is imperative to vary reaction parameters to get viable information in electrocatalytic experiments. Figure 15a illustrates a thought experiment of photochemical H2 production as a function of irradiation time, for four different, hypothetical MOF systems w, x, y, and z. The four MOFs show different initial rates, which means different quantum yields, and it is of interest to understand the origin of this difference. With only the results of Figure 15a at hand, we can only speculate based on our ideas of how the MOFs are designed, but not determine the origin of the behavior with any certainty. Further experiments with variations of conditions could be helpful in that regard, such as variation of catalyst concentration and light intensity.
Figure 15.

Qualitative illustration of the HER for four hypothetical MOF systems that show different TOFs and rate dependencies on [Cat]. a) Rate of H2 production as a function of time at a fixed [Cat], b) maximum (initial) rate of H2 production as a function of [Cat] and c) TOF as a function of [Cat] (data corresponding of panel b, with TOF = Rate/[Cat]). Note that trends shown may hold in a limited range of [Cat] values investigated, but, for example, the rate must go to zero as [Cat] goes to zero.
In Figure 15b and c the maximum rates and the TOFs, respectively, of the same MOFs are plotted as a function of catalyst concentration. The MOFs show qualitatively different behavior. MOF w (blue line) shows a strong increase in rate with increasing [Cat], and even the TOF increases. This may be explained by cooperative catalysis for a case where the actual catalytic steps are rate-limiting, but it may also be caused by increased MOF conductivity by hopping via catalyst sites. For cooperative catalysis, this behavior would disappear at lower light intensities when the catalytic steps are no longer limiting, while limitation by electron hopping would still remain. Thus, a combination of variations of [Cat] and light intensity could offer mechanistic insight. In one example of cooperative catalysis, Choi et al.197 measured photochemical CO2 reduction in UiO-67 with some linkers substituted for the photocatalyst [Re(bpydc)(CO)3Cl]. When varying the photocatalyst loading they found that three Re complexes per unit cell gave the highest amount of CO product, which led the authors to propose cooperative catalysis, although light-intensity variations were not reported. Infrared spectroscopy gave support for interaction between the Re complexes at higher loading levels.
MOF x (yellow line) shows a linear dependence of rate on [Cat] and a constant TOF, which suggests that the catalysts are not competing for redox equivalents. Roy et al.198 studied photochemical H2 production on MIL-101(Cr) with Fe2(1,2-dithiolate)(CO)6 as catalyst linked via amide bonds. They found that the rate of the HER was proportional to the catalyst concentration, which they proposed as evidence that the reaction was not limited by the photogeneration of reducing equivalents. This requires a high steady-state concentration of electrons and/or a slow catalyst, and one would expect the quantum yield to be rather high at high [Cat]. An alternative interpretation is instead that the reaction is light-limited. The increase in rate with [Cat] would then be because of a higher rate of electron transfer from PS– to Cat, which competes more favorably with PS– recombination. Light-intensity variations would be able to distinguish between these two scenarios also in this case.
For MOF y (green line), the rate is independent of [Cat], and different possible scenarios exist. These include the case of a MOF where the reaction is limited by the light generation of reducing equivalents, but where these equivalents are long-lived once they are formed, for example due to an efficient sacrificial donor. Electron transport to the catalysts would in this case not be limiting, and the [Cat] would therefore not be important. The TOF would then be proportional to the light intensity.
MOF z (purple line), in contrast, shows a decrease in rate with increasing [Cat], and a strong decrease in TOF. This is typical for a case where photogeneration of reducing equivalents is slow and the catalysts are competing for electrons. When they do not get the second electron fast enough, it has a higher risk of recombination (or other side reactions). Therefore, a higher [Cat] (beyond a certain optimum) has a negative effect on the overall reaction rate. This situation has been shown in homogeneous systems.199,200 If the catalyst is incorporated in a MOF, its reduction kinetics may be slower because of transport limitations, but also recombination with D+ may be slowed down, so that further losses are avoided. This case has been suggested when comparing a homogeneous Fe2(μ-Cl2bdt)(CO)6 (μ-Cl2bdt = 1,4-dichloro-2,3-benzenedithiolato) catalyst with a hydrogenase enzyme.201 Case z in Figure 15 would definitely show light-dependent rates, as the rate of electron generation is increased, and the dependence may even be stronger than a linear one.
The above scenarios are by no means exhaustive, and other conditions may give rise to a similar behavior as the examples given. Yet, they illustrate what insights a variation of parameters may give, here emphasizing the effects of varying the catalyst concentration and the light intensity. Variations of [PS] or [substrate] and kinetic modeling will provide additional information and insights. Because of the complexity of a complete photochemical reaction scheme, a detailed kinetic model may not be useful. Nevertheless, important insights may be gained by a simplified treatment and identification of the limiting step(s) of photocatalysis.
3. MOF–Catalyst Interactions
MOFs can exert a variety of effects on the electronic properties, reactivities, and catalytic performances of molecular catalysts that they host. For example, the MOF matrix can lead to a structural stabilization of the catalyst, thereby leading to higher TONs as compared to the same catalyst in homogeneous phase. Even though this is a global effect that does not necessarily require specific interactions between the catalyst and the surrounding medium at the atomic level, it is of great technological importance. It also mimics the situation in many enzymes in which active sites are often buried in the interior of the protein matrix, to protect them from bimolecular encounters which could lead to dimerization/oligomerization, competing reactions or even decomposition. Following a concise review of different means of structural stabilization by the MOF matrix in section 3.1, we will subsequently discuss examples in which the MOF plays more specific roles. Working our way from third coordination sphere effects toward the active sites, cases are presented in which the MOF has been engineered to facilitate transport from the crystal surface to the catalyst. Specific pathways for substrate or products are reminiscent of the situation in many enzymes, where substrate access is highly regulated and can even determine the reaction outcome. Finally, sophisticated cooperative effects that are imposed on the catalyst by the surrounding MOF are being discussed. Parallels to second coordination sphere effects as defined in enzymes will be drawn. The chapter also contains examples on how the size/thickness of MOF crystals/thin films may give rise to transport limitations, as well as a system perspective on how MOFs with molecular catalysts are being interfaced with the surrounding media. While these are not effects within the catalyst-containing MOFs, both phenomena will have huge implications to their overall activity. Contacting is also an important topic in enzymes, some of which, for example, are embedded or associated with membranes or possess specific binding sites, for example, for ferredoxin, which connects the enzymes’ electron transport chain to the surrounding.37
3.1. Stabilization of Molecular Integrity
The crystallinity and topological stability of MOFs can contribute to the structural stabilization of incorporated catalysts in various ways. Taking the degree of bonding interactions between the catalyst and the surrounding matrix, may it be a MOF or a peptide, as a basis for classification, catalyst incorporation can be divided into four themes (Figure 16). The first class contains examples in which the active site is merely physically encapsulated by the matrix without any specific atomic interaction (Figure 16a). This strategy is often referred to as the ship-in-a-bottle approach, expressing the fact that the catalyst needs to be bigger than the pore window of the MOF to prevent catalyst leakage. In the second class, matrix and catalyst interact through one specific interaction, for example through a linker that contains a dangling group that interacts with the catalyst through a covalent or coordination bond (Figure 16b). Catalysts that are decorated with a Lewis basic group that coordinates to the SBU belong into this design as well. Similar patterns in biology can, for example, be found in [FeFe] H2ases in which a cysteine-S is an integral part of the H-cluster active site. This cysteine is the only amino acid that provides a ligand to the active site. The two aforementioned approaches differ from the one in Figure 16c in that the catalyst of the latter is an integral part of the MOF, with one of the catalyst ligands being at the same time a MOF linker. In analogy, multiple first coordination sphere interactions can also be found, for example, in [NiFe] hydrogenases, carbon monoxide dehydrogenases or photosystem II. A common structural feature of these enzymes is that the active site metals also contain small inorganic ligands such as CO and CN–, μ-S, or μ-O ligands, respectively. Finally, all permanent ligands at the active site metals may be provided by the surrounding matrix (Figure 16d). In MOFs, this represents a situation in which the SBU exhibits the catalyst function, and all permanent ligands to the active site metals are provided by the MOF linkers. The latter class bears analogy to the nonheme diiron enzymes,202 the entire first coordination sphere of which is composed of amino acid side chains and substrate-derived ligands.
Figure 16.

Schematic illustration of a catalyst moiety (purple sphere) inside a matrix (enzyme or MOF) with increasing degree of matrix/catalyst interaction from a) to d). Catalyst a) entrapped inside the matrix, b) coordinated to one dangling group, c) anchored by two groups, and d) fully fixed into the matrix.
Irrespective of the bonding interactions described in Figure 16, MOF incorporation sterically isolates the molecular catalysts and protects from bimolecular catalyst encounters that, in the case of homogeneous counterparts, are often a major pathway for catalyst deactivation. This holds for reduction catalysts where low-valent metals may engage in unwanted bond forming reactions between catalyst molecules and even more so oxidation catalysts, where the formation of metal oxides is often thermodynamically favored. Another general effect that contributes to catalyst longevity is a tendency to slow down permanent ligand dissociation. This includes also situations where ligands may temporarily decoordinate from the metal, but cannot diffuse away due to the confinement provided by the MOF, thereby enabling recoordination and catalyst reconstitution.
Different techniques and types of experiments are being used in the field to describe and quantify the extent to which the MOF stabilizes the structural integrity of the incorporated catalysts. These include comparisons with homogeneous reference systems, recycling experiments that show that the catalytic material is active in repeated runs, as well as spectroscopic work, using diagnostic markers that are indicative of the structurally intact catalyst. The latter strategy gives perhaps the most information about the fate of the catalyst and can also be used to define the term “stabilization” more clearly. As MOFs are hundreds of nanometers to micrometer size objects, there is the possibility that not all catalysts are exposed to turnover conditions and that catalysis occurs only in a thin shell at the MOF/solution interphase (as schematically shown in Figure 17). This may be the case for MOFs in which transport of, for example, redox equivalents or a substrate, as well as a reductant or an oxidant, is limited, as discussed in section 2. In such cases, a certain proportion of the total catalyst population may be merely spectators and is therefore unchanged after the catalytic activity of the MOF ceases. As this scenario will result in postcatalysis materials with intact catalysts, it may first appear as structural stabilization, and additional experiments may be needed to establish the role of all catalysts within the MOF crystal. Spectator catalysts will also have implications on the apparent TONs and TOFs, as described in section 2. If these metrics are normalized to the total amount of catalysts, they may largely underestimate the actual TOFs and TONs in the environment provided by the MOF.
Figure 17.

Spheres representing MOF crystals, where a) the entire crystal was relatively homogeneously exposed to turnover conditions or b) only the surface layer was sufficiently exposed. The gradient on the left illustrates the exposure to the turnover conditions from low to high.
Prior to discussing specific examples of the four incorporation types in detail, one example of a MOF/catalyst system based on model complexes of the [FeFe] hydrogenase will be presented to illustrate the spectator catalyst topic, while another group of materials based on cobaloxime catalysts will be presented to illustrate the different incorporation designs.
[FeFe] Hydrogenase Active Site Models in MOFs: A Case Study
One of the most obvious biomimetic strategies to design and prepare MOFs for the HER is to take inspiration from [FeFe] H2ase enzymes. Reports of MOFs into which structural analogues of this enzyme active site have been incorporated have entered the scene relatively early. Already in 2013, Pullen et al.203 reported the incorporation of [FeFe](dcbdt)(CO)6 (dcbdt = 1,4-dicarboxylbenzene-2,3-dithiolato), a well-established enzyme active site model complex and proven HER catalyst, into UiO-66. A postsynthetic linker exchange strategy was chosen as the catalyst was not compatible with the harsh solvothermal conditions, and an incorporation yield of 14% was achieved. The resulting UiO-66-[FeFe](dcbdt)(CO)6 (Figure 19a) in combination with [Ru(bpy)3]2+ as a PS and ascorbate as an electron donor under illumination catalyzed the HER with overall higher TONs and even initial rates higher than those of a homogeneous reference system. IR studies after the ceased HER revealed that the vast majority of the [FeFe](dcbdt)(CO)6 was still structurally intact, thus suggesting that MOF integration has a stabilizing effect on the structure of the catalyst. In a follow-up study, it was shown that photocatalytic HER activity from the suspension of UiO-66-[FeFe](dcbdt)(CO)6 in a solution of [Ru(bpy)3]2+ and ascorbate could be recovered after 60 min of stirring without replacing any other component.204 This unexpected phenomenon was proposed to be due to intracrystal linker scrambling that brings structurally intact catalysts from the crystal interior to the surface, where they can then engage in catalysis. This interpretation also implies that the [FeFe](dcbdt)(CO)6 complexes in the inner MOF layers are not being reduced during earlier rounds of photocatalytic HER, and merely lie dormant. This hypothesis is consistent with results from another study that showed that a large proportion of [FeFe](dcbdt)(CO)6 complexes in UiO-66-[FeFe](dcbdt)(CO)6 remains in the initial oxidized state even in the presence of a large excess of cobaltocene,198 a chemical reductant of similar strength as [Ru(bpy)3]+ that is produced photochemically by reductive quenching of the *[Ru(bpy)3]2+ excited state in the earlier works. In contrast, the same [FeFe](dcbdt)(CO)6 complex can almost quantitatively be reduced when incorporated in MIL-101(Cr), which is a MOF with larger pore diameters that allows more facile diffusion of the cobaltocene reductant to the [FeFe](dcbdt)(CO)6 complexes.198
Figure 19.

Examples of MOFs with incorporated model complexes of the [FeFe] H2ase active site. a) Incorporation of the H2ase mimicking complex [FeFe](dcbdt)(CO)6 into UiO-66 by postsynthetic exchange. Reproduced with permission from reference (203). Copyright 2013 American Chemical Society. b) Postsynthetic decoration of ZrPF with [FeFe] H2ase active sites model complexes to form [FeFe]@ZrPF. Reproduced with permission from reference (207). Copyright 2014 Royal Society of Chemistry. c) Azide-functionality-driven click reaction to covalently attach a Fe2 catalyst to the linker units of a UiO-type MOF. Reproduced with permission from reference (208). Copyright 2019 Elsevier.
The accumulated results of these publications are that the mentioned TONs in the first report203 are most likely a significant underestimate, as only a fraction of total catalysts species is active during the initial round of irradiation. This also means that the active catalysts are greatly stabilized compared to the homogeneous reference catalyst. In homogeneous phase, it is well-established that one-electron reduced states of the catalyst are prone to engage in scission of an Fe–S bond to produce a free thiolate that can attack a second Fe2 complex to form tetra-nuclear species as those shown in Figure 18.205,206 This bimolecular decomposition pathway is most likely prevented by incorporating the catalyst in the MOF.
Figure 18.

Dimerization of one-electron reduced model complexes of the [FeFe] H2ase active site that can be prevented owing to incorporation into MOFs.205,206
The structural stabilization becomes more apparent in MOFs into which both a PS as well as an active site model complex have been incorporated. In such systems, it is more likely that all catalysts are exposed to a photochemically produced reductant. Early work by Feng and co-workers207 described such a system based on a robust zirconium-porphyrin-based MOF (ZrPF). Herein, the [FeFe] H2ase active site model complex [((SCH2)2NC(O)C5H4N)Fe2(CO)6] is connected to the ZnTCPP linker through axial coordination by the pyridine moiety at the Fe2 complex (Figure 19b). Under illumination and in the presence of ascorbate, the Fe2-decorated MOF [FeFe]@ZrPF exhibits HER activity that vastly exceeds that of the corresponding homogeneous reference system. In another example, a multifunctional UiO-69 that contained a [Ru(bpy)3]2+-derived metallo-linker and an azide-functionalized tetraphenyl-dicarboxylate was prepared. The latter engages in a click reaction with an acetylene-containing Fe2 complex to form a covalent attachment between the MOF linker and the Fe2 catalyst (UiO-MOF-Fe2S2 in Figure 19c).208 Under illumination and in the presence of ascorbate, this system resulted in a photochemical HER with higher yields than that of the corresponding homogeneous reference system under otherwise identical conditions. IR studies of both MOF systems, the [FeFe]@ZrPF and UiO-MOF-Fe2S2, after ceased photocatalysis showed complete disappearance of the IR frequencies that are attributed to the Fe2 catalyst. As decomposition is a result of catalyst reduction, it can be seen as an indicator that all catalysts have been exposed to turnover conditions. This being the case, both of the systems show higher TONs than the homogeneous references, illustrating the effect of the MOF matrix for catalyst stabilization.
From a biomimetic perspective perhaps the most complete mimic of [FeFe] H2ases was reported in 2021.209 In this paper, not only a model of the active site was incorporated but also redox-active linkers that model the [4Fe4S] electron transport chain of the enzyme. The system was realized by taking advantage of earlier work on PCN-700,210 which possesses two missing linker defect sites of specific size and positions in the crystal lattice. These sites were postsynthetically filled by the [FeFe](dcbdt)(CO)6 active site mimic and a naphthalene diimide-based redox linkers. Rigorous studies on the dual-functionalized MOF by cyclic voltammetry reveal similarities to the natural system but also important limitations in the MOF–enzyme analogy. Most importantly, restrictions apply to the total concentration charge-balancing counter cations that can be accommodated within the MOF. With the capacity of countercation uptake in the MOF being limited, not all linkers can be reduced electrochemically, even if the applied potential is well beyond the standard reduction potential of the isolated linker.209
Cobaloximes in MOFs: A Case Study
Over the years, the same catalyst motif has sometimes been incorporated into MOFs by multiple strategies of the ones outlined in Figure 16. This is nicely illustrated for a series of MOFs with incorporated cobaloximes, one of the most well-studied molecular HER catalysts.211 Cobaloximes contain two bidentate 1,2-diglyoxime ligands that occupy the equatorial coordination sphere of the central Co cation. Axial monodentate ligands such as pyridine, halides, or solvent molecules typically complete the primary coordination sphere of cobaloximes. As proton reduction involves the reduction of the Co center, usually to a CoI level, these axial ligands are prone to decoordination; in fact, it is at these positions that hydride formation occurs. Structural decomposition pathways of the catalyst include reduction and hydrogenation of the glyoxime ligands followed by nanoparticle formation,30,212−214 as well as decoordination of the diglyoxime ligand, as evidenced by increased TONs in photocatalysis experiments (Eosin Y as PS and triethanolamine (TEOA) as donor) to which exogenous diglyoxime ligand was added.215 This effect was rationalized by excess diglyoxime pushing the decoordination equilibrium to the complex side. Incorporation of cobaloximes into heterogeneous matrices such as MOFs as an alternative means to stabilize their molecular structure has been in the center of attention in numerous studies. From an incorporation point of view, most conceivable ways to incorporate cobaloximes have been explored, most of which show the desired effect that the molecular integrity of the catalyst is increased.
For example, a Co-dioxime-diimine has been assembled in the pores of a photoactive NH2-MIL-125(Ti) following a ship-in-a-bottle strategy (Figure 20a).216 The resulting Co@NH2-MIL-125(Ti) could be recycled a couple of cycles over the time scale of days and functioned as a light-driven HER catalyst in the presence of triethylamine (TEA). In related work, the parent cobaloxime was assembled from the diglyoxime ligands and the Co salt in the presence of NH2-MIL-125(Ti) to give a material that photocatalytically produced H2. In these experiments, TEOA was used as a sacrificial donor, and H2 production rates could be increased by additional Eosin Y as a cophotosensitizer.217
Figure 20.

Examples of cobaloxime analogues incorporated in MOFs: a) Ship-in-a-bottle approach, based on the incorporation of CoIIIBr2(LH) into NH2-MIL-125(Ti) to form Co@MOF. Adapted with permission from reference (216). Copyright 2015 Royal Society of Chemistry. b) Cobaloxime complex covalently linked to the MOF matrix, via postsynthetic functionalization on the aromatic rings of MIL-101(Cr) after chlorination and immobilization of the catalyst. Adapted with permission from reference (218). Copyright 2018 Royal Society of Chemistry. c) Cobaloxime analogues can serve as structural linkers, when comprising carboxylate anchors as shown in Co(dcpgH)(dcpgH2)Cl2. Reaction with preassembled Zr-oxo clusters led to the formation of UU-100(Co). Adapted with permission from reference (219). Copyright 2015 American Chemical Society.
Going beyond the ship-in-a-bottle approach, cobaloximes were incorporated by a coordination linkage in the interior of appropriately functionalized MIL-101(Cr) (Figure 20b).218 The resulting material was shown to be an efficient photocatalyst with a high rate for H2 evolution in the presence of TEOA as an electron donor and Eosin Y as a PS. The obtained results are consistent with the hypothesis that cobaloxime species, [CoI(dmgH)2] and [CoIII(dmgH)2(H)] (dmgH = dimethylglyoxime), that are detached from the pyridine groups during the photocatalytic cycle remain in close proximity of the pyridine anchors inside the MOF-pores. Restoration of the Co–pyridine coordination bond during catalysis is therefore more likely to occur inside the MOF-pores compared to the situation in the homogeneous system. Consequently, the heterogeneous MOF–cobaloxime hybrid displayed enhanced HER activity due to the presence of the axial N-ligand. Eventual deactivation of the system likely occurs through diffusion of detached [Co(dmgH)2] units out of the MOF-cages, followed by loss of the glyoxime ligands from cobalt.
Finally, cobaloximes have also been incorporated as structural linkers in a MOF (Figure 20c).219 For this purpose, tetraphenyl-cobaloxime was decorated with four carboxyl-groups for anchorage to Zr6-cluster-based SBUs. The resulting material, UU-100, showed electrocatalytic hydrogen evolution over more than 20 h when grown on glassy carbon substrates. This result illustrates the power of MOF incorporation, as electrochemical reference experiments on the homogeneous linker lead to complete catalyst degradation after a couple of turnovers. In contrast to the homogeneous case where ligand decoordination from the Co center is not restricted, the diglyoxime ligands in UU-100(Co) are held in place by their anchorage to four difference SBUs. Even if transiently decoordinating from the catalytic Co center, the ligand stays in the vicinity, offering the possibility for recoordination.
3.1.1. Catalysts Entrapped within MOF Pores
In the absence of a ligand at the molecular catalyst that could synthetically be modified to allow for MOF incorporation, a ship-in-a-bottle strategy, as illustrated in Figure 16a, can be employed. This strategy is suitable for catalysts that are smaller than the pores of the chosen MOF, but larger than its pore windows. In that way, the catalyst is physically trapped inside the MOF crystal without directly interacting with the framework in a specific, atomically defined manner. In the absence of covalent or coordination bonds, this section also includes examples in which the molecular catalyst is imbedded in the MOF pores by electrostatic or van der Waals interactions. Catalysts can be trapped during the solvothermal synthesis of the MOF, as well as that they can be assembled in the MOF pores in a postsynthetic process.
3.1.1.1. Polyoxometallates (POMs)
Polyoxometalates (POMs) as catalysts for energy-relevant transformations have received significant attention in recent years due to their high catalytic activity, but they have also been criticized for their instability, sometimes being merely precursor species for poorly defined, heterogeneous, yet very active metal oxides.220,221 For example, certain Co4POMs have been found to transform under certain catalytic conditions into CoOx oxide, which itself is a competent WOC.222 Such transformations appear to occur via initial leaching of CoII from the Co4POM into the bulk solution, followed by formation of the CoOx oxide.
POMs have been incorporated into MOFs by the encapsulation approach, wherein the POM is physically trapped inside a MOF’s pores. In 2018, Das and co-workers223 encapsulated both a molecular cobalt complex and a Keggin K6[CoW12O40] POM inside the pores of ZIF-8. The POM@MOF species proved to be a competent WOC at a neutral pH, while the homogeneous POM was not observed to evolve O2. The authors argue that for successful stabilization, the guest must be bigger than the MOF pore windows and yet fit within the cavities. Without meeting these size-match criteria, leaching can occur, with resultant possible formation of heterogeneous metal oxides or hydroxides.
The handful of examples demonstrating water oxidation by POMs encapsulated inside MOFs suggest that POM stability is enhanced compared to that of their homogeneous analogues, however, as discussed above, the probable decomposition pathway to CoOx begins with leaching of CoII from the POM clusters. It is not yet clear why MOF encapsulation guards against this pathway. As recently as 2020, the enhanced stability of POMs in MOFs was purely attributed to preventing leaching of the POM units.224 For a more complete review of stabilization of POMs in MOFs for diverse catalytic applications largely not related to solar fuels, see the recent summary of Dolbecq and co-workers.225
POMs have also been trapped inside UiO-type MOFs to obtain stabilized HER catalysts (Figure 21).226,227 In these materials, the encapsulating MOF also hosted a [Ru(bpy)3]2+ PS as an integral part of the constituting MOF linkers, resulting in a system that was well set up for light-induced electron transfer reactions to the POM-based HER catalyst. Photophysical and electrochemical studies established the oxidative quenching of the excited PS by the POM as the initiating step of HER.
Figure 21.

Example of catalyst and PS coimmobilization within a MOF by a ship-in-a-bottle strategy for light-driven HER. a) structure of a Ru-based PS linker. b) Polyhedral view of the structure of [Ni4(H2O)2(PW9O34)2]10– (Ni4P2). c) Structural model showing unoccupied tetrahedral cavities and the central Ni4P2-loaded octahedral cavity. Adapted with permission from reference (227). Copyright 2016 WILEY-VCH Verlag GmbH & Co.
MOF-encapsulated POMs with high negative charges, when entrapped in MOFs, can also attract additional positively charged guests into the MOF matrix. This strategy has been reported for a MIL-101(Cr) framework into which anionic Wells–Dawson-type POMs had been incorporated during solvothermal MOF synthesis.228 Owing to the high negative charge of the POM, the POM@MOF composite allows for the adsorption of cationic rutheniumtris(bipyridyl) ([RuII(bpy)3]2+) PS, resulting in a POM@PSs@MOF. In the presence of TEOA, this hybrid system catalyzes the light-driven HER with much higher activity than that of the corresponding homogeneous reference system.
3.1.1.2. Polypyridyl Complexes
Polypyridines are popular ligands to transition metals in catalysis of energy relevance. 2,2′-Bipyridine (bpy) and 2,2′:6′,2″-terpyridine (terpy), as well as ligand motifs in which the pyridines are separated by saturated units, in principle, allow for synthetic modifications to introduce anchoring points for SBU coordination and thus MOF production. On the other hand, polypyridine-based catalysts are typically rather large, which qualifies them also for a ship-in-a-bottle incorporation into suitable MOF scaffolds.
One such example is a dinuclear manganese-based WOC, MnTD ([(terpy)Mn(μO)2Mn](terpy)]3+), that has received considerable attention both for its high activity and similarity to the oxygen-evolving complex of photosystem II. Kinetic analysis of MnTD has revealed that catalysis is first order in MnTD, but the major degradation products require multiple MnTD equivalents.229 Significant efforts had focused on bulking up the ligand substituents to improve resistance to bimolecular decomposition until Nepal and Das229 reported the postsynthetic assembly of MnTD inside the pores of MIL-101(Cr) via a stepwise process. The pristine MIL-101(Cr) was first soaked in a solution of terpy ligand for 18 h followed by addition of Mn(OAc)2 and K-oxone to form the MnTD catalyst in the pores of MIL-101(Cr),229 as evidenced by FTIR and EPR spectroscopic studies. Chemical oxidation experiments established that while homogeneous MnTD had a higher initial catalytic rate, the MnTD@MIL-101(Cr) material (Figure 22) maintained catalytic activity for a longer time period as long as fresh oxidant was supplied. To permit comparison between homogeneous and MOF-encapsulated catalysis, the same initial quantity of MnTD was used in both scenarios. The authors were careful to note that this “analytical quantification techniques would not distinguish the catalytically active and inactive forms of Mn” within the MIL-101(Cr) MOF and that therefore the catalytic enhancement as a result of MOF incorporation could be higher than reported.229 Intriguingly, the authors commented that Hatton and co-workers230 found no difference in catalytic rate for Baeyer condensation reactions using an analogous ship-in-a-bottle approach with MIL-101(Cr) for crystallite sizes ranging from ca. 400 nm up to 10 μm, pointing toward catalysis occurring throughout the MIL-101(Cr) crystals. Nepal and Das229 argued that based on these literature results, water oxidation for their system is likely also not transport limited, though no kinetic comparison of different particle sizes was examined. Hansen and Das231 demonstrated that the previously described MnTD@MIL-101(Cr) material was catalytically active for up to seven continuous days of operation. A gradual decrease of catalytic activity was rationalized as possible buildup of insoluble CeIII species originating from the employed sacrificial oxidant CAN, emphasizing the need to avoid sacrificial reagents and instead rely on electrode constructs. An analogous MOF host with pores large enough to host two molecules of MnTD had noticeably decreased catalytic lifetime (7 h versus 7 days), supporting the notion that catalyst site isolation leads to enhanced stability.231 Since their initial report, the group of Das has reported similar success for water oxidation using an encapsulated Co complex as a WOC in a Co framework, Co-WOC-1.232
Figure 22.

Structure of the MnTD water oxidation catalyst and its catalytic activity as either a homogeneous solution or encapsulated inside MIL-101(Cr). Atom labeling: C, gray; O, red; N, blue; Mn, purple. H atoms are omitted for clarity. Adapted with permission from reference (229). Copyright 2013 WILEY-VCH Verlag GmbH & Co.
In 2017, Park et al.233 reported the functionalization of the anionic zinc-adeninate framework bMOF-100 via postsynthetic cation exchange with [Ru(bpy)3]2+. Thereby, the mesopores of bMOF-100 were spacious enough to host on average 2.43 [Ru(bpy)3]2+ cations. [Ru(bpy)3]2+@bMOF-100 was further immobilized on the surface of a glassy carbon electrode (GCE) to investigate its electrocatalytic and electrochemiluminescence properties. In an aqueous electrolyte solution, the cyclic voltammograms of Ru@bMOF-100 showed an irreversible RuIII/II couple that the authors interpreted as an electrocatalytic OER.
Following the theme of light-driven proton reduction based on a photoresponsive MOF and a molecular HER catalyst, a molecular NiII catalyst, [Ni(dmobpy)(2-mpy)2] (dmobpy = 4,4′-dimethoxy-2,2′-bipyridine, 2-mpy = 2-mercapto-pyridyl) was incorporated in a photoactive MOF, NH2-MIL-125(Ti). Synthetically, the catalyst was assembled in the pores of the MOF by simple soaking of the two ligands and a NiII precursor. 1H NMR spectroscopic evaluation of digested Ni@NH2-MIL-125(Ti) as well as other spectroscopic studies support the formation of the [Ni(dmobpy)(2-mpy)2] catalyst within the composite. In the presence of TEA as electron donor, Ni@NH2-MIL-125(Ti) catalyzed the light-driven HER at appreciable rates for up to 50 h, substantially longer than corresponding reference experiments.234 The same NH2-MIL-125(Ti) MOF was also used to host a CoII-based molecular HER catalyst, [CoII(TPA)CI][CI] (TPA = tris(2-pyridylmethyl)-amine).235 Similar to the previous example, the Co complex was assembled inside the MOF cages by successive exposure of the NH2-MIL-125(Ti) MOF to the ligand, followed by the CoII salt. The molecular integrity of the catalyst was proven by NMR spectroscopy and mass spectrometry of digested samples. In the presence of TEOA as an electron donor, the Co@NH2-MIL-125(Ti) hybrid catalyzed the light-driven HER at a rate that greatly exceeded that of a physical mixture of NH2-MIL-125(Ti) and the same CoII complex in solution, illustrating the necessity for close proximity between the light-responsive MOF and the encapsulated catalyst for appreciable electron transfer.
In terms of biomimicry, also model complexes of the [NiFe] H2ase active site have been incorporated into MOFs. NiFe@PCN-777 was synthesized by simple encapsulation of [NiFe] ([LN2S2NiIIFeIICp(CO)]BF4; LN2S2 = 2,2′-(2,2′-bipyridine-6,6′-diyl)bis(1,1′-diphenylethanethiolate, Cp = cyclopentadienyl) into the Zr-based MOF PCN-777 through soaking. The interactions between the PCN-777 matrix and NiFe are nonspecific, but involve electrostatic interactions of the Zr-cluster surface with the [LN2S2NiIIFeIICp(CO)]+ cation and/or the BF4– anion. Electrochemical reductions associated with the molecular [NiFe] complex could be observed in cyclic voltammograms of NiFe@PCN-777 that had been immobilized on fluorine-doped tin oxide (FTO) by electrophoretic deposition. In the presence of acid, cyclic voltammograms of NiFe@PCN-777 show a current enhancement that is consistent with proton reduction, the magnitude of which is however largely similar to the currents observed for analogous films of the parent PCN-777. The large background current of the latter on FTO makes it difficult to determine the extent to which the molecular catalyst in NiFe@PCN-777 is involved in catalysis, and prevents a more rigorous kinetic treatment of the system.236
Yan et al.237 reported the construction of [NiII(bpet)(H2O)2] (bpet = 1,2-bis((pyridin-2-ylmethyl)thio)ethane) in the pores of the photosensitizing Ru-UiO-67 ([Zr6(μ3-O)4(μ3–OH)4(bpdc)5.7(Ru(bpydc)(bpy)2)0.3]·(OAc)0.6) (bpdc = biphenyl-4,4′-dicarboxylate) framework. The photocatalytically active composite Ni@Ru-UiO-67 was produced in a typical ship-in-the-bottle fashion by entrapment of bpet in Ru-UiO-67, followed by its metalation with Ni(ClO4)2·6H2O. By varying the amount and ratio of bpet and the NiII precursor, three different composites Ni1@Ru-UiO-67, Ni2@Ru-UiO-67, and Ni3@Ru-UiO-67 were prepared, which differed in their NiII loadings of 0.15, 0.23, and 0.31 wt %, respectively. All Ni@Ru-UiO-67 composites displayed photocatalytic activity for the reduction of CO2, with Ni3@Ru-UiO-67 showing 99% selectivity for CO with an appreciable TON.
3.1.2. Catalysts Anchored in MOF Pores
While the previous section reviewed MOF–catalyst composites in which the catalyst was trapped either physically or by nonspecific electrostatic interactions in the MOF pores, examples in the present section rely on more specific catalyst–MOF interactions. This design generally allows more control as to the spatial positions of the constituting components, also resembling the situation in enzymes more closely. The molecular catalysts are either covalently bound to a MOF linker, or encapsulated through defined noncovalent interactions, including hydrogen bonds. The noncovalent interactions may also be between Lewis basic groups that are “dangling” off the MOF linkers and that coordinate to the transition-metal catalyst, or between the Lewis acidic SBUs and catalysts that are equipped with a Lewis base.
3.1.2.1. Catalysts Anchored to Linkers
Zhang et al.238 postsynthetically added terpyridine ligands via covalent bonds to the internal pores of MIL-101(Cr), followed by complexation of manganese to yield a di-μ-oxo dimanganese unit. This material was found to evolve oxygen upon addition of a chemical oxidant and, notably, no leaching of Mn-species was found in the bulk solution after catalysis. The authors also recreated the MnTD@MIL-101(Cr) material of Nepal and Das229 (see section 3.1.1.2) and found that leaching of Mn did occur under similar catalytic conditions. Based on this comparison, it was argued that covalent attachment gives rise to more stable MOF/catalyst hybrids than the ship-in-a-bottle approach.
Using the same parent MIL-101(Cr) MOF, Ott and co-workers239 described the postsynthetic incorporation of the molecular WOC [Ru(bda)(L)2] (bda = 2,2′-bipyridine-6,6′-dicarboxylate; L = pyridine-based axial ligands) into two different MIL-101(Cr)-derived frameworks. The WOCs were incorporated through one of the axial pyridine ligands that in turn were covalently bound to functionalized linkers in the MIL-101(Cr). The benefits of stabilizing the structural integrity of the WOC within the MIL-101(Cr) matrix became evident during water oxidation experiments over a longer time period and in the presence of a higher concentration of the oxidant CAN. After 1 h, one of the MIL-101(Cr)@Ru(bda) materials displayed TONs of 1500, around 10 times higher than that of the homogeneous control experiment. The study also contained attempts to elucidate whether catalysis is surface-confined or a bulk phenomenon, as well as whether catalysis proceeds by a nucleophilic attack or radical coupling mechanism. In a follow-up study, the same group presented a quantitative kinetic model that allowed to distinguish between surface and bulk reactivity in a catalytic MOF (see also discussion around Figure 9 for details).98
Liang et al.240 demonstrated the integration of Ru-based molecular WOCs into the MIL-101(Cr) framework through an amide bond connection to the linkers of the scaffold. The authors described the homogeneous control catalysts [Ru(terpy)(pic)3](PF6)2 (pic = 4-picoline) and [Ru(terpy-Ac)(pic)3](PF6)2 (terpy-Ac = [2,2′:6′,2″-terpyridine]-4′-carboxylic acid), as well as the three novel heterogeneous systems with covalently anchored Ru-based catalysts MIL-101(Cr)-[Ru(terpy-Ac)(pic)2Cl](PF6), MIL-101(Cr)-[Ru(terpy)(isc)Cl2] (isc = isonicotinic acid), and MIL-101(Cr)-[Ru(terpy)(isc)(pic)2](PF6)2. The Ru-based catalysts were built into the MOF structure through amide bond at the terpy-Ac ligand in case of MIL-101(Cr)-[Ru(terpy-Ac)(pic)2Cl](PF6), or the isc ligand in case of the other two materials. The heterogeneous hybrid materials were found to be robust catalysts for the OER, with the CAN-driven OER efficiency of MIL-101(Cr)-[Ru(terpy-Ac)(pic)2Cl](PF6) being 120 times higher compared to that of the nonimmobilized Ru-catalyst. The experimental results revealed that the Ru-aqua species, [Ru(terpy)(pic)2(OH2)]2+, is the actual active species in the OER. The formation of the aqua complex proceeds preferentially by replacement of chloride ligands. In the absence of chloride ligands at the Ru complex as in MIL-101(Cr)-[Ru(terpy)(isc)(pic)2](PF6)2, isc that constitutes the anchor to the MOF matrix is replaced by water, explaining the experimentally found poor stability of this construct during water oxidation experiments.
MOFs can also serve as host matrices for the concomitant incorporation of molecular catalysts and PSs (different arrangements presented in Figure 14). In 2018, Wang et al.241 established such a system by entrapping the molecular catalyst [Cp*Rh(bpy-4,4′-dc)]2+ (Cp* = Cp* = η5-C5Me5) and the PS [Ru(bpy)2(bpy-4,4′-dc)]2+ within NH2-MIL-101(Al). The host framework acts as a protective cage for the captured components which are held in place by H-bonding interactions between the carboxylate-functionalized complexes and the NH2-functionalized MOF matrix. Owing to these interactions, leaching out during photocatalysis is inhibited. Utilization of the resulting Rh–Ru@NH2-MIL-101(Al) in the CO2RR displayed a change in selectivity toward the exclusive production of formate and suppression of H2 production, compared to the homogeneous counterpart. In 2021, Stanley et al.242 followed with a similar demonstration to coimmobilize fac-ReBr(CO)3(bpy-4,4′-dc) and a Ru-based PS within the pores of a NH2-MIL-101(Al) MOF (Figure 23a and b). This system showed selective CO evolution under photocatalytic CO2 reduction conditions from 1.5 to 40 h, indicating improved stability compared to the homogeneous reference. Furthermore, in contrast to typical homogeneous systems, not catalyst but PS degradation was identified as the major performance-limiting factor, corroborating the high potential of MOF platforms to stabilize catalytically active sites.
Figure 23.

a) Noncovalent anchoring of the carboxylic acids of the encapsulated catalyst to the amine functionalities of NH2-MIL-101(Al). b) Photoinduced electron cascade from triethanolamine (TEOA) over the [Ru(bpy)2(bpydc)]Cl2 PS (red) to the ReBr(CO)3(bpy-4,4′-dc) catalyst (orange) for the CO2 reduction reaction in a host–guest environment. Adapted with permission from reference (242). Copyright 2021 American Chemical Society. c) Catalyst (orange) and PS (red) location in the isoreticular UiO (66, 67, 68) host series. Adapted with permission from reference (243). Copyright 2021 WILEY-VCH Verlag GmbH & Co.
In another study, Stanley et al.243 reported the coimmobilization of the molecular CO2 reduction catalyst [ReBr(CO)3(bpy-4,4′-dc)] and the PS [Ru(bpy)2(bpydc)]Cl2 within the isoreticular series of amino-decorated UiO-66, -67, and -68 frameworks (Figure 23c). The series displays a range of maximum pore diameters of 8.0, 13.1, and 17.2 Å, for UiO-66, -67, and -68 respectively, and will thus host the molecular Recatalyst (12 Å) or/and the Ru-PS (15 Å) either on the MOF surface, as in UiO-66, or within the pores within the pores in case of the more spacious analogous. Immobilization of the molecular species is guaranteed through interactions of the carboxy groups at the molecular components with the SBUs and the amine moieties of the linkers. When illuminated and in the presence of TEOA, the UiO-66 assembly with surface confined catalyst and PS showed limited TONs and deactivation after 1.5 h due to the instability of the molecular catalyst on the MOF surface. In the UiO-67 assembly, the smaller catalyst was able to enter the pores, while the larger PS remained on the outside, thereby partially blocking the pores. Consequently, ReRu-67 showed very low activity, most likely due to disabled electron transfer between the surface anchored PS and the entrapped catalyst. In case of UiO-68, the catalyst as well as the PS were able to be coimmobilized within the framework. ReRu-68 constructs with a slight excess of PS displayed TONs of about 10, which is comparable to those of the homogeneous reference system, but higher stability over a longer period of time. Greatly improved TONs were obtained by replacing TEOA with another sacrificial electron donor, namely, BIH (1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole), leading to increased molecular stability and activity. ReRu-66 in combination with BIH reached final TONs of 419 ± 31 until its deactivation within 5 h. While ReRu-67 still showed limited reactivity, ReRu-68 displayed total TONs of 506 ± 29 after two 24 h cycles.
In 2019, Liu et al.244 anchored catalytically active CoII centers postsynthetically into a robust Zr-based MOF (Zr-DMBD), functionalized with dangling thiol groups. The well-defined Co–thiolate units in the resulting Zr-DMBD–Co MOF were capable to convert CO2 into CO in a photocatalytic assay consisting of a Ru-based PS and TEOA as sacrificial reductant. A series of Zr-DMBD–Co with different CoII contents, designated as Zr-DMBD–Co-x (x = wt % of CoII ions), were synthesized to investigate the influence of the CoII loading on the catalytic activity. Of the Zr-DMBD–Co materials that were studied, the one with the lowest Co loading, gave the highest formal TON and TOF and a CO selectivity of 98% over 10 h.
3.1.2.2. Catalysts Anchored to SBUs
In 2018, Paille et al.245 encapsulated the sandwich-type POM [(PW9O34)2Co4(H2O)2]10– (P2W18Co4) inside the pores of MOF-545 (also known as PCN-222) as shown in Figure 24a. Encapsulation was performed by soaking the pristine MOF in an aqueous solution of the P2W18Co4 POM, and relied on strong H-bond interactions between the POM and the Zr6 cluster SBU, as suggested by density functional theory (DFT) calculations. Photocatalysis experiments in the presence of the sacrificial electron acceptor Na2S2O8 demonstrated that the porphyrin linkers of MOF-545 were able to act as PSs for POM oxidation (Figure 24b). Recycling experiments revealed similar initial water oxidation rates and only slightly decreased TONs using recovered POM@MOF. Post mortem X-ray diffraction (XRD), thermogravimetric analysis, energy-dispersive X-ray spectroscopy, and X-ray absorption near edge spectroscopy studies showed no noticeable difference between the material before and after catalysis, indicating that this strategy effectively stabilizes the POM against decomposition.
Figure 24.

a) Structure of MOF-545 with encapsulated P2W18Co4 POM water oxidation catalyst and b) proposed mechanism of water oxidation wherein initial photoexcitation of the MOF porphyrin linker is quenched by a sacrificial electron acceptor, followed by oxidation of the encapsulated POM. Reproduced with permission from reference (245). Copyright 2018 American Chemical Society.
Subsequent work by the same group demonstrated that this POM@MOF material could be prepared as thin films on ITO electrodes, permitting electro- and photocatalysis experiments.246 The method of preparing the thin films had a significant impact on the experimental TONs, as electrophoretic deposition gave thicker films than simple drop casting. The authors stated that lower TONs in thicker films may be due to incomplete illumination of the deposited material, while transport limitation were not considered in the work. Analogous work by Das and co-workers247 showed greatly improved electrocatalytic stability of the K6[CoW12O40] Keggin POM toward water oxidation via encapsulation in the pores of ZIF-8.
In 2019, Hod and co-workers248 anchored the porphyrin-based ORR catalyst Hemin to the SBUs of UiO-66 through solvent-assisted ligand incorporation. This process occurs at missing-linker defect sites at the SBUs that are filled by coordination of the carboxylate-containing Hemin. Accordingly, it is shown that the catalyst incorporation yield is increased upon increased number of such defect sites in the as-prepared UiO-66. The distribution of the Hemin catalysts is however not homogeneous throughout the crystal. Five different UiO-66@Hemin materials that differ in catalyst amount and spatial distribution have been prepared, and are shown to be good catalysts for the ORR. As shown by the same group in 2020, the ORR activity of the system can further be improved by axial coordination of 2-methylimidazole to the central Fe atom of the Hemin moiety.249
FeX (X = Br–, Cl–, AcO–, and BF4–) could be supported on a hexanuclear Zr6O4(OH)4 cluster that constituted the SBU of a mixed-linker MOF, containing nitro-quaterphenyl dicarboxylate and p-phenanthroline dibenzoate (PT).250 The PT-sites were used to assemble a [Cu(PT)(dppe)]+-type PS (dppe = 1,2-bis(diphenylphosphino)ethane) by a series of postsynthetic modifications. The resulting FeX@Zr6-Cu promotes the light-driven HER with TONs of up to 33 700 and TOFs of up to 880 h–1. Photocatalytic H2 evolution activities of FeX@Zr6-Cu were found to correlate with the lability of the counteranions X, consistent with the generation of open coordination sites at the single atom Fe centers by decoordination of labile X groups to facilitate the formation of Fe-hydride intermediates.
Zr6-cluster SBUs also served as the anchoring site for the attachment of transition metals for the HER. In one such report, diverse metal ions (Ni2+, Co2+, Cu2+, Ru3+) were bound to Zr6-oxo clusters of different MOFs in a microwave-assisted method. The introduction of the single metal sites was followed by hydroxylation, sulfidation, or further oxidations. Among the prepared materials, a Ni-loaded UiO-66-NH2, was shown to catalyze the light-driven HER in the presence of TEA.251
Lin and co-workers252 described a novel photosensitizing MOL, Hf12-Ru, consisting of Hf12 SBUs and [Ru(bpy)3]2+-derived dicarboxylate linkers. Hf12-Ru was postsynthetically modified by replacing weakly coordinating trifluoroacetic acid units on the SBUs with MBA (2-(5′-methyl-[2,2′-bipyridin]-5-yl)acetic acid). The resulting Hf12-Ru-MBA was then metalated with [Re(CO)5Cl] or [Mn(CO)5Br] to afford the hybrid systems Hf12-Ru-Re and Hf12-Ru-Mn, respectively, bearing Ru-based PSs and catalysts for the light-driven CO2RR in the presence of electron donors (Figure 25a). From the systems studied, Hf12-Ru-Re exhibited the highest TON of 8613 under artificial visible light irradiation, and of 670 under ambient sunlight. The activity is facilitated by electron transfers from the photogenerated, reduced PS to the catalytic centers that are in close proximity of each other on the MOL skeleton.
Figure 25.

a) Postsynthetic monocarboxylic acid exchange of Hf12-Ru toward the synthesis of Hf12-Ru-M (M = Re, Mn) employed in the sunlight-driven CO2 reduction to CO. Reproduced with permission from reference (252). Copyright 2018 American Chemical Society. b) Synthetic pathway toward Zr-MBA-Ru/Re-MOF via postsynthetic exchange of formates with MBA ligands at the SBUs and the consecutive metalation of their N,N′-chelating sites with [Ru(bpy)2Cl2] and [Re(CO)5Cl], to afford a hybrid system with a Ru-based PS and a molecular Re catalyst for visible light-driven CO2 reduction. Reproduced with permission from reference (191). Copyright 2021 Royal Society of Chemistry. c) Depiction of the PCN-222(Zn)/Re system, enabling electron transport through the MOF to the surface-attached Re active sites for the photocatalytic reduction of CO2. Adapted with permission from reference (173). Copyright 2021 American Chemical Society.
Following a similar postsynthetic sequence, Karmakar et al.191 immobilized not only a CO2 reduction catalyst at the SBU but also a Ru-based PS. The chosen Zr-MOF (=MOF-808) was first postsynthetically modified through a solvent-assistant exchange of formates with MBA ligands at the SBUs. By consecutive postsynthetic metalation with [Ru(bpy)2Cl2] and [Re(CO)5Cl], both functional units were installed at the N,N′-chelating sites of the MBA ligands, to afford Zr-MBA-Ru/Re-MOF (Figure 25b). The photocatalytic CO2RR with the hybrid system was investigated and CO formation was established with a selectivity of >99%. Control experiments with Zr-MBA-Re-MOF and Zr-MBA-Ru-MOF separately employed as a photocatalyst showed only trace amounts of a CO2 reduction product, supporting the hypothesis that the close proximity between the PS and the catalyst within the MOF pores shortens the transport distances of the charge carriers. Interestingly, the photocatalysis experiments were conducted without an exogenous electron donor, implying that water may be the terminal electron donor.
Hu et al.253 described in 2019 the efficient photoreduction of CO2 to HCOOH and CO by 2D light-harvesting MOLs. The parent MOL (Zr-TCBPE-MOL; TCBPE = tetrabenzoatetraphenylethylene) displays 12 available sites on the Zr6O4(OH)4 SBUs whereof only four are connected to TCBPE linkers, leaving the others accessible for other carboxylate ligands. By postsynthetic modification, the H2bpydc-anchored (H2bpydc = 2,2′-bipyridine-5,5′-dicarboxylic acid) catalytic complexes H2bpydc-Re(CO)3Cl and [H2bpydc-IrCp*OH]NO3 can be introduced by coordinating their dicarboxylate moieties to the MOL’s SBUs. The Ir or Re compounds can either bridge two SBUs within one MOL or interconnect two MOLs. Employing Zr-TCBPE-MOL-Ir as a photocatalyst, CO2 was reduced to both formic acid and formaldehyde. In comparison, the homogeneous counterpart under the same reaction conditions generated significantly less product.
Recently, Choi et al.173 described the porphyrinic MOF PCN-22(Zn) onto which the prototypic CO2RR catalyst (bpy-4,4′-dc)ReI(CO)3Cl was installed via carboxylate anchoring groups at SBUs close to the crystal surface. Upon illumination, the synthesized MOF-Re(I) hybrid was able to produce CO over 59 h with BIH (1,3-dimethyl-2-phenyl-1,3-dihydrobenzimidazole) as electron donor, without significant loss in catalytic activity. The observed enhanced catalytic performance of the MOF-Re(I) was ascribed to efficient exciton migration between excited Zn-porphyrins and electron transfer to the Re-based active centers (Figure 25c).
3.1.3. Catalysts as Linkers
Molecular catalysts can be used as integral linkers in MOFs if they contain ligands that can be synthetically modified to introduce multiple anchoring groups. The location of each catalyst in such materials is known more precisely than in the previous section in which the catalysts were hosted in the MOF pores attached to the MOF matrix through some sort of tether. The molecular catalysts in this section can also be seen as metallo-linkers and can either be included in the solvothermal syntheses or introduced into the MOF by postsynthetic strategies. In the former cases, the catalytically active metallo-linker may be the sole linker that the MOF is composed of, or complemented by a second, perhaps sterically less demanding colinker to produce mixed-linker MOFs. The most frequent postsynthetic methods include metalation of MOF-linkers with suitable binding pockets, or postsynthetic linker exchange in which inert linkers from the pristine MOF are exchanged by catalytically active metallo-linkers.
3.1.3.1. 2,2′-Bipyridine (bpy)-Related Work
A significant amount of work in this section is based on UiO-67, as its constituting biphenyldicarboxylate linker can be exchanged by bipyridine dicarboxylate that is identical in length but can host a variety of metal fragments. This strategy is particularly advantageous in context of the OER, as decomposition of, for example, ruthenium-based WOCs has long been attributed to intermolecular pathways.31 Consequently, several groups have targeted the site isolation of ruthenium moieties as metallo-linkers inside MOFs.254−256 Resonance Raman analysis found that while signals for dimeric ruthenium species were observed after chemical oxidation of a homogeneous ruthenium catalysts, no such signals were observed under the same chemically driven catalysis conditions with the analogous MOF-immobilized catalyst.255 Other bpy linker-based WOCs incorporated into MOFs with improved stability include UiO-67 decorated with iridium bipyridine units257 and a UiO-type MOF with expanded linkers using iridium catalytic centers.114 Taken together, these reports demonstrate promising catalytic stability and recyclability of the MOF-based hybrid materials.
Chamber et al.258 realized the first photosensitized rhodium-based MOF for CO2 reduction with a high selectivity for formate formation. In this report, a catalytically active Rh-based half-sandwich complex Cp*Rh(bpydc)Cl2 was introduced into UiO-67 via postsynthetic linker exchange to form Cp*Rh@UiO-67 that was employed in the CO2RR together with homogeneous Ru(bpy)3Cl2 as PS. Even though catalytic activities of the homogeneous and heterogeneous systems were found comparable, the MOF-based system offered the advantage of higher stability and selectivity for formate production, and could also be recycled without loss of activity. Similarly, Liao et al.259 described a MOF platform with incorporated Ru- and Rh-based half sandwich units for CO2 reduction and H2 evolution. The presented MOFs RuCl@UiO, RuOH2@UiO, RhCl@UiO, and RhOH2@UiO were synthesized via postsynthetic linker exchange of the UiO-67 parent system with the complexes [Ru(Cy*)(bpydc)Cl]Cl·H2O (H2RuCl, Cy* = p-cymene), [Ru(Cy*)(bpydc)(OH2)](NO3)2 (H2RuOH2), [Rh(Cp*)(bpydc)Cl]Cl2·H2O (H2RhCl), and [Rh(Cp*)(bpydc)(OH2)](NO3)2·1.5H2O (H2RhOH2), respectively. In combination with Ru(bpy)3Cl2 and the sacrificial electron donors TEOA or N,N-dimethylaniline (DMA), all of the modified MOFs possess the ability to produce H2 and reduce CO2 into CO and HCOO–. Of the systems investigated, RhOH2@UiO displayed the best photocatalytic HER activity while the Ru-based MOF catalysts showed better performance in terms of CO2 reduction selectivity against H2 evolution. A long-term hydrogen evolution lasting for 174 h without significant decrease in efficiency was achieved in the RhOH2@UiO-DMA system, illustrating the power of the MOF matrix to stabilize the structural integrity of molecular catalysts for extended periods of time under turnover conditions. Overall, accommodating the molecular catalysts in the MOF platform showed superior and long-term photocatalytic stability as well as higher TONs than corresponding homogeneous catalysts for both the HER and the CO2RR.
In 2020, Benseghir et al.260 demonstrated the coimmobilization of the Keggin-type POM PW12O403– and the catalytically active complex Cp*Rh(bpydc)Cl2 in UiO-67. The POM was encapsulated inside the pores of the MOF scaffold in a ship-in-the-bottle approach, while the Rh-based catalytic complex was introduced by subsequent postsynthetic linker exchange to derive the composite (PW12,Cp*Rh)@UiO-67. The material was evaluated for photocatalytic CO2 reduction toward formate and hydrogen. In comparison with the POM-free Cp*Rh@UiO-67 the POM-assisted composite showed a doubled formate production yield. Prepared as drop-casted thin films on ITO plates, (PW12,Cp*Rh)@UiO-67 was investigated under photocatalytic conditions to elaborate its recyclability. During the first and second run of irradiation, only slight activity decreases of 8% and 9% were observed, respectively. Compared to analogous experiments of (PW12,Cp*Rh)@UiO-67 as suspension, the ITO immobilized system showed higher TONs of 175 versus 14.6 over 3 h, which was attributed to more efficient illumination of the crystallites in the thin film.
In 2017, Yaghi and co-workers197 described the plasmon-enhanced photocatalytic CO2-to-CO conversion by a Ren-MOF-coated Ag nanocube hybrid system. The ReI(CO)3(bpydc)Cl acts as the photocatalyst and was incorporated into a UiO-67 framework to form a series of Ren-MOF with n = 0 to 24 complexes per unit cell (Figure 26a). Of the materials prepared, Re3-MOF showed the highest photocatalytic activity in the presence of TEA as an electron donor. Further, Re3-MOF was coated onto plasmonic Ag nanoparticles to afford Ag⊂Re3-MOF, which showed a 7-fold improved photocatalytic CO2-to-CO conversion activity compared to Re3-MOF. This activity enhancement was explained by electric fields at the surface of the Ag nanocubes and its influence on the spatially confined catalytically active Re centers.
Figure 26.

a) Combination of Zr6O4(OH)4(−CO2)12 SBUs with bpdc and ReTC linkers to form Ren-MOF. Depicted is the structure of Re3-MOF identified from single-crystal X-ray diffraction. Atom labeling: C, black; O, red; Zr, blue polyhedra; Re, yellow; Cl, green. H atoms are omitted for clarity. Adapted with permission from reference (197). Copyright 2017 American Chemical Society. b) Depiction of the ternary CdS/UiO-bpy/Co composite, obtained through a stepwise modification of a UiO-bpy framework with Cd(CH3COO)2·2H2O in DMSO to form CdS/UiO-bpy, followed by the metalation of the free bpy sites with CoCl2 in THF. Adapted with permission from reference (261). Copyright 2018 Royal Society of Chemistry. c) Example of catalyst and PS coimmobilization within a UiO-67-based framework as metallo-linker for the light-driven HER. Reproduced with permission from reference (263). Copyright 2018 American Chemical Society.
In 2018, Chen et al.261 reported on a MOF composite, CdS/UiO-bpy/Co, with an inorganic CdS semiconductor quantum dot and the redox catalyst Co(bpydc)Cl2. The composite was synthesized by the stepwise postsynthetic modification of UiO-bpy to introduce the functional components (Figure 26b). The ternary system was described to feature good CO2 adsorption properties and facilitate efficient charge separation and diffusion. It was also shown to promote the light-driven reduction of CO2 to CO in the presence of TEOA as an electron donor.
The bpydc linker in UiO-67(bpy) can not only be used as an integral part of a molecular catalyst but also as a ligand of archetypical [Ru(bpy)3]2+-type PSs. Such a strategy places the photosensitizing and catalytic units in close proximity and may promote fast light-induced electron transfer. In this context, we encourage the reader to revisit the section in chapter 2 that also discusses potential drawbacks of such spatial arrangements that can, for example, promote back electron transfers. Examples of systems in which PS and catalysts are incorporated as metallo-linkers include Ru-Pt@UiO-67 (Figure 26c), which contains both a PS and a Pt-based HER catalyst.262 The system was later studied by time-resolved techniques to deduce light-induced charge transfer kinetics and the detection of the catalytically relevant PtI species.263 A related Co-Ru-UiO-67(bpy) system follows the same design principles with the noble metal-based catalyst having been replaced by a Co complex.264 Another bipyridine-embedded UiO-type MOF, namely, Pt-n_Ir_BUiO with an incorporated iridium-based PS was shown to be active in the light-driven HER for weeks.265 The longevity was ascribed to bpydc linkers in close vicinity to the HER catalyst and the PS which are available to recapture metal fragments that may have temporarily decoordinated from their original bpydc linker. As a result, the system has a certain self-healing capacity, and colloid formation that was observed in homogeneous reference experiments was not observed in the Pt-n_Ir_BUiO MOF.
Besides the aforementioned transition-metal complexes comprising Co,264,266 Ru,254−256,259,267 Rh,258,259 Ir,114,257 and Pt262,263,265 active sites, multiple MOF systems with bpy-containing CO2RR catalysts of the general formula M(L)(CO)3X, where M is a transition metal, L a bidentate bipyridine-based linker, and X a halide, have been reported. As one of the most well-documented deactivation pathways for this type of catalysts is the bimolecular dimerization,268,269 site isolation of the catalysts as integral linkers in MOFs is highly motivated. Among other materials, Lin and co-workers257 were first to report a UiO-67 system with an integrated ReI(CO)3(bpydc)Cl catalyst. The resulting hybrid MOF catalyzed the light-driven reduction of CO2 to CO in the presence of TEA with a TON of 10.9, three times higher than that of the homogeneous complex. Similarly, Fei et al.270 reported the synthesis of a mixed linker UiO-67-bpydc MOF with an equimolar ratio of H2bpydc and H2bpdc (biphenyl-4,4′-dicarboxylic acid). Via postsynthetic metalation, the earth-abundant, but thermally unstable, molecular catalyst Mn(bpydc)(CO)3Br was incorporated into the robust MOF platform, to achieve site-isolated catalytically active moieties. Furthermore, the UiO-67 matrix enhanced the stability of the Mn active sites, enabling their reuse for up to three cycles. In combination with a ruthenium-based PS in solution and 1-benzyl-1,4-dihydronicotinamide as a sacrificial reductant, UiO-67-bpy-Mn(bpy)(CO)3Br represents an efficient catalytic system for the selective reduction of CO2 to formate under visible light irradiation, reaching a TON for formate production of 50 over 4 h and a selectivity of 96%. Also the corresponding UiO-67-Re-30% system with an incorporated Re(CO)3(bpydc)Cl catalyst was reported. In this MOF, the Re complex acts as both a PS and a catalyst for the reduction of CO2 to CO in the presence of an electron donor.
In 2018, Deng et al.271 presented the postsynthetic metalation of MOF-253 (Al(OH)(bpydc)) with [Re(CO)5Cl] to produce a photocatalytically active MOF-253-Re(CO)3Cl system. In addition to superior catalytic performance over the homogeneous counterpart, the synthesized MOF was found to selectively reduce CO2 to formate instead of CO. Trying to enhance the light absorption of the formed heterogeneous system, further treatment with [Ru(bpy)2Cl2] afforded Ru-MOF-253-Re. Due to the augmented visible light absorption, Ru-MOF-253-Re showed enhance photocatalytic CO2RR performance. While the TON of the MOF is comparable to Ru–Re supramolecular structures based on covalent bonds, the assembly of a hybrid Ru–Re system within a MOF is synthetically easier and generally more modular.
Going beyond Re and Mn-based bpydc catalyst systems, Sun et al.267 presented the incorporation of a Ru-based catalyst into the bipyridine linkers of MOF-253 to afford MOF-253-Ru(CO)2Cl2. Even though MOF-253-Ru(CO)2Cl2 itself is capable to reduce CO2 when illuminated, its performance can be enhanced by the coimmobilization of a Ru(bpy)2(bpydc)2+ PS, introduced into MOF-253-Ru(CO)2Cl2 by a second postsynthetic metalation strategy, to improve light absorption. The optimum activity was observed when the molar ratio of PS:catalyst was 1:1. By doing so, the amount of formate production was increased by a factor of 12, while the amount of produced CO was unchanged. The TON for formate formation was increased from 0.3 for the homogeneous catalyst Ru(bpydc)(CO)2Cl2, over 2.9 for the nonsensitized MOF, to 35.8 for the sensitized MOF-253-Ru(CO)2Cl2. In 2019, Liu et al.266 described photocatalytic syngas production by the (Co/Ru)n-UiO-67(bpydc) framework, assembled in a two-step postsynthetic metalation procedure. The thereby obtained MOF platform enabled fast electron transfer from the PS to the molecular cobalt catalyst, and exhibited a 29.2-fold increase in syngas production yield compared to the homogeneous reference system.
Even though MOF systems with bpydc as linkers are versatile and suitable to anchor a variety of molecular catalysts, the pores of the MOFs are relatively small, hindering diffusion and high concentrations of sterically demanding active sites. To prevent such performance limitations, elongated linkers can be employed to build isoreticular systems with enlarged pores. In 2016, Lin and co-workers272 described a UiO-type MOF consisting of the elongated metallo-linker Re(CO)3Cl(bpydb) (bpydb = 4,4′-(2,2′-bipyridine-5,5′-diyl)dibenzoic acid). In contrast to the mixed linker systems described by the same group in 2011,257 the elongated linker allowed the synthesis of a MOF that is exclusively formed from the catalytically active metallo-linker. After 6 h of photocatalytic CO2 reduction in acetonitrile, the Re-MOF showed a TON for CO of 6.44, roughly six times higher than that of the corresponding homogeneous complex. Employing phenanthroline instead of bpydb, Lin and co-workers273 further reported the synthesis of two mixed-linker phenanthroline-based (mPT) frameworks, mPT-Cu/Co and mPT-Cu/Re, including cuprous PSs and molecular Co or Re catalysts, suitable for photocatalytic HER and CO2RR, respectively. The catalytically active systems were derived through postsynthetic metalation of the parent mPT-MOF platform, comprising Zr-based SBUs and H2TPHN (2″-nitro-[1,1′:4′,1″:4″,1‴-quaterphenyl]-4,4‴-dicarboxylic acid) and H2PT (4,4′-(1,10-phenanthroline-3,8-diyl)dibenzoic acid) linkers, with Cu(CH3CN)4PF6, dppe (1,2-bis(diphenylphosphino)ethane), and either CoCl2 or Re(CO)5Cl (Figure 27). Simultaneous arrangement of Cu PSs and the molecular Co or Re catalysts led to a multifunctional MOF platform with high photocatalytic activity. The mPT-Cu/Co MOF catalyzed the HER with a TON of 18 700, while mPT-Cu/Re catalyzed the CO2RR with a TON of 1328, nearly 2 orders of magnitude higher than the TONs of their homogeneous counterparts.
Figure 27.

Schematic representation of the framework composition of mPT-Cu/Co and mPT-Cu/Re, utilized in the HER or CO2RR, respectively. Adapted with permission from reference (273). Copyright 2020 American Chemical Society.
3.1.3.2. Porphyrin/Macrocycle-Related Works
MOFs with porphyrin-derived linkers, most prominently H2TCPP (4,4′,4″,4‴-(porphyrin-5,10,15,20-tetrayl)tetrabenzoate)), have been prepared relatively early, and considering the known catalytic activity of homogeneous porphyrins to catalyze reactions of energy relevance, TCPP-containing MOFs have early moved to the center of attention. Al-TCPP, that is, (AlOH)2H2TCPP, in which infinite Al(OH)O4 chains are interconnected by porphyrin linkers into a 3D microporous framework, was platinated at the porphyrin sites.153 The resulting Al-TCPP-Pt is an efficient catalyst for the light-driven HER, with the PtII-porphyrin linkers acting as both PS and HER catalyst in the presence of a reductive quencher. The activity of Al-TCPP-Pt is superior to that of a reference system consisting of Pt nanoparticles that are stabilized by the same sensitizing Al-TCPP. The authors argue that the difference in activity stems from the site isolation of the Pt centers and associated maximized atom utilization for catalysis as compared to the nanoparticle system. Another example of a PtIITCPP HER catalyst that was used as linkers in conjunction with Ti-oxo clusters as metal nodes was reported as the HER catalyst under illumination in the presence of ascorbic acid.274
In 2017, Fateeva and co-workers275 investigated the electrocatalytic ORR performance of Co-Al-PMOF, consisting of Co-porphyrin linkers interconnected by aluminum oxide SBUs. The authors suggested that an increasing porosity to promote oxygen and product transport inside micropores does not always imply more efficient reduction chemistry. The authors stated that the distance between the catalytic centers is more pivotal, and influences whether the reaction proceeds via a 2- or a 4-electron pathway. In related work, PCN-224(Co), a MOF consisting of Zr6 clusters and Co-TCPP linkers, was deposited on carbon substrates as an ink together with surface-activated multiwalled CNTs.276 The composite material was shown to be an active electrocatalyst for both the ORR and OER.
In 2021, Liang et al.277 successfully grafted cobalt porphyrins as the active sites on a MOF surface through ligand exchange. The generated hybrid materials showed improved ORR activity with substantial anodic shift (>70 mV) of the half-wave potential compared to the molecular electrocatalysts without covalent immobilization. The authors described both, ZIF-8, a framework mainly selective for the 2e– ORR toward hydrogen peroxide, and ZIF-67, its analogue, showing higher selectivity for the 4e– ORR reducing H2O2 further to H2O (Figure 28). Control experiments with cobalt-free porphyrins grafted on ZIF-8 revealed that the Co porphyrin linker is the catalytically active site. To demonstrate the practical potential of the prepared porphyrin@MOF hybrids, they were employed as electrode catalysts in Zn–air batteries, exhibiting performances comparable to that of Pt/C.
Figure 28.

Grafting of Co tetra(imidazolyl)porphyrin onto the Zn-based ZIF-8 framework via a ligand exchange. Adapted with permission from reference (277). Copyright 2021 WILEY-VCH Verlag GmbH & Co.
In 2013, Liu et al.278 investigated the CO2 adsorption and activation properties of the unmetalated (SP) and Cu2+-metalated (SCu) version of a porphyrin-based MOF. Compared to SP, SCu showed both enhanced performance in CO2 capture and photocatalytic conversion of CO2 to methanol. Thereby the product evolution rate catalyzed by SCu was about 7 times higher than that of SP. FTIR measurements revealed that the CO2 adsorption and its activation over Cu2+ played a crucial role in the conversion of CO2. Similarly, Zhang et al.279 demonstrated the incorporation of CoII and ZnII cations into the stable scaffold of MOF-525. The authors claimed that the presence of single atoms within the TCPP ligands of MOF-525-Co and MOF-525-Zn enhanced their photoinduced electron–hole pair separation efficiency significantly, achieving long-lived electrons able to be transferred to the catalytically active metal centers for the CO2RR to CO and CH4.
In 2019, Huang et al.280 presented the synthesis of MCF-55, an analogue of MOF-525, both of which are composed of Zr6O4(OH)4 SBUs but differ in their constituting linkers. While the latter is based on TPCC linkers, the former contains H4tactmb (1,4,7,10-tetrazazcyclododecane-N,N′,N″,N‴-tetra-p-methylbenzoic acid) azamacrocycle linkers. Both MOFs were postsynthetically metalated to MCF-55-M and MOF-525-M (M = Co, Ni), and the resulting materials investigated for their catalytic activity for the light-driven CO2RR in the presence of [Ru(phen)3]Cl2 (phen = phenanthroline) and TEOA. An interesting feature of the H4tactmb-M (M = Co, Ni) linker is that its methylene groups create a hydrophobic microenvironment around the metal active sites, resulting in repulsive forces to H2O and enhanced CO2 to CO activity. MCF-55-Co displayed a near-quantitative selectivity for CO and a TOF significantly larger compared to that of the TCPP-based MOF-525-Co(279) that lacks the hydrophobic surroundings. Owing to the stronger CO2 affinity of Ni compared to Co, MCF-55-Ni showed an even higher TOF that was furthermore constant for five consecutive runs of 10 h each.
Hod et al.281 described the postsynthetic metalation of electrophoretically deposited MOF-525 (Zr6O4(OH)4(H2TCPP)3) thin films on conductive FTO surfaces to generate the catalytically active redox-conductive Fe_MOF-525 (Zr6O4(OH)4(FeTCPP)3). This approach afforded a high surface coverage of heterogenized catalyst for the reduction of CO2 to CO, where the metalloporphyrins act as catalysts and redox-hopping channels to distribute charge carrier to active sites, not directly linked to the electrode. Throughout the catalysis, a mixture of CO and H2 in about equal amounts was generated with a Faradaic efficiency (FE) of ∼100%.
Sadeghi et al.282 investigated the electrocatalytic performance of the porphyrin-based framework Zn/PMOF, synthesized from Zn(NO3)2·6H2O and H2TCPP, to afford the metalated ZnTCPP linkers within the framework. Zn/PMOF was employed in the light-driven CO2RR in the presence of H2O vapor as a sacrificial electron donor under UV−vis light. During 4 h of illumination, 10.43 μmol CH4 was produced with 0.3 g of Zn/PMOF, outperforming bare ZnO by 80.6% under the same conditions but still giving much less than one CH4 per pore unit.
In 2020, Zhou et al.283 reported the synthesis of a Cu-based porphyrinic MOF for efficient and selective CO2 reduction toward hydrocarbons. The material was prepared from CuTCPP interconnected by Cu paddle wheel dimers as SBUs. The Cu-based MOF displayed a FE of 73.6% and a current density of 7.5 mA cm–2 at −1.4 V vs RHE (reversible hydrogen electrode) for the production of methane and ethylene. These products are exclusively formed as a function of MOF incorporation of the catalyst, as control experiments with the homogeneous Cu-porphyrin catalyst generated mainly CO as the major product. In the MOF, CO was found to be an intermediate for the generation of hydrocarbons. The porous MOF architecture with abundant copper-based active sites enables not only a high CO2 absorption but also confines the CO intermediates spatially for the selective reduction into hydrocarbons, revealed by finite-element simulations.
In related work on multiple Cu environments, Gu and co-workers284 investigated the electrocatalytic CO2RR with Cu2(COO)4 paddle wheel-based Cu-porphyrinic MOF nanosheets (Cu2(CuTCPP)) and compared their performance to CuO, Cu2O, Cu, CuTCPP, and the CuO/CuTCPP composite. Cu2(CuTCPP) was exposed to potentials between −1.40 V and −1.65 V vs Ag/Ag+, and formate and acetate were found to be the predominant products, accompanied by small amounts of CO and CH4, as well as H2. At an applied potential of −1.40 V, formate and acetate were detected with an FE of 28.1% and 11.6%, respectively, which increased to 61.5% and 12.3%, respectively, at −1.55 V. It was found that the catalyst’s cathodic current density decreased rapidly during the first hour and stabilized thereafter at 4.5 mA cm–2, indicating cathodized restructuring of the catalyst. The structural changes at −1.55 V were observed by XRD, scanning electron microscopy, transmission electron microscopy, X-ray photoelectron spectroscopy (XPS) and FTIR experiments, revealing that the Cu-paddle wheel nodes probably changed to CuO, Cu2O, and Cu4O3 under cathodization conditions. Despite of the restructuring, the material shows a vastly different product selectivity compared to Cu, CuTCPP, CuO, and Cu2O, which all showed predominantly HER activity.
Qin et al.285 described the ionic liquid-induced formation of the 2D Mn-MOF [BMI]2(Mn[Mn(H2O)2-TCPP](H2O)2) (BMI = 1-butyl-3-methylimidazolium), bearing high loading of MnII-metalated porphyrin linkers interconnected by Mn nodes. [BMI]2(Mn[Mn(H2O)2-TCPP](H2O)2) exhibited a high MnTCPP loading of 67 wt % and was synthesized from the metal-free H2TCPP ligand, which was metalated with MnII during the MOF formation in the ionic liquid [BMI]Br. [BMI]2(Mn[Mn(H2O)2-TCPP](H2O)2) was shown to be a photocatalyst for the conversion of CO2 and water vapor to CH4 and CO in the gas–solid interface.
Huang et al.286 demonstrated the fabrication of a core–shell MOF@MOF structure in a one-pot synthesis by taking advantage of differing nucleation kinetics of the two MOFs PCN-222-Ni and UiO-67-NH2. Their difference in nucleation kinetics is due to the fact that linkers with high connectivity show faster nucleation rates than those with low connectivity. Consequently, PCN-222-Ni which consists of tetra-topic linkers nucleated first, while UiO-67-NH2 with ditopic linkers nucleated later, forming a shell around the already formed PCN-222-Ni to afford the P@U composite. The combination of the two MOFs in one composite material resulted in the formation of a Z-scheme heterojunction, where the electrons in UiO-67-NH2 can be excited and then transferred to PCN-222-Ni, which is catalytic for the reduction of CO2 to HCOOH. The P@U composite combines the crystallinity, robustness, and high porosity of the parent MOFs with an optimized band alignment, an enhanced photoresponse and an increased charge separation. Consequently, the P@U composite catalyzed CO2 photoreduction with a rate for formic acid formation that is higher than that of the parent MOFs PCN-222-Ni and UiO-67-NH2. In these experiments, no exogenous electron donor was added, implying that water was the terminal reductant.
In contrast to the aforementioned applications of metalloporphyrins as catalytically active sites, Yaghi and co-workers287 synthesized the catecholate framework MOF-1992, by connecting tetratopic cobalt phthalocyanin-2,3,9,10,16,17,23,24-octaol ligands with Fe3(−C2O2−)6(OH2)2 clusters. Owing to sterically accessible Co active sites and good charge transfer properties, MOF-1992-covered carbon black cathodes reached per-site TONs of 5800 over 6 h for the electrocatalytic reduction of CO2 to CO in water with a FE of 80%.
A series of four structural analogues of conductive 2D MOFs MPc–Cu–X (M = Co, Ni and X = O, NH) comprising metallophthalocyanine (MPc) linkers and Cu nodes was prepared by Meng et al.288 (Figure 29). The authors investigated the influence of the metal center within the MPc and the heteroatom connectivity to the SBU on the electrochemical CO2RR activity. At an applied reductive potential, CoPc–Cu–O exhibited the highest selectivity toward CO formation with a FE of 85% when employed as a composite with carbon black (1:1 m/m) and a FE of 79% without any conductive additive. DFT studies revealed that CoPc-based and O-interconnected MOFs display lower activation energies to form carboxyl intermediates, which accounts for the higher activity and selectivity compared to the NiPc-based and NH-linked analogues.
Figure 29.

Series of MPc–Cu–X (M = Co, Ni, X = O, NH) MOFs containing CoPc and NiPc units connected by Cu bis(diimine) and Cu bis(dioxolene) linkages. Adapted with permission from reference (288). Copyright 2020 American Chemical Society.
In 2021, Qiu et al.289 investigated the 2D-MOF PcCu-Cu-O (Pc = phthalocyanine) with two different Cu environments for the electrochemical CO2RR. The MOF was synthesized from square-planar CuO4 nodes and PcCu-(OH)8 ((2,3,9,10,16,17,23,24-octahydroxyphthalo-cyaninato)copper) ligands. Applied as a powder on a GCE with Nafion binder, PcCu-Cu-O displayed a current density of 7.3 mA cm–2 at a potential of −1.2 V vs RHE in 0.1 M KHCO3, and a FE of 50% for C2H4 production. The FE for C2H4 production was twice of that of the homogeneous CuPc reference, and was attributed to the synergistic effect between the CuO4 and the CuPc units. According to the authors, CO is produced at the CuO4 SBU and then migrates to the C2H4-producing (CuPc) site, thereby lowering the energy barrier for the C–C dimerization. This is based on the facts that CuO4 has a high activity for the reduction of CO2 to CO and a lower *CO adsorption energy (16 kJ mol–1) compared to that of the CuPc (48 kJ mol–1), thereby being able to serve as CO source.
3.1.3.3. Miscellaneous
The use of metal-free, purely organic molecular linkers as active sites for the OER inside MOFs has been claimed. Lu and co-workers290 prepared a lanthanide-based MOF using the linker triphenylene-2,6,10-tricarboxylic acid (H3TTCA) and demonstrated that this MOF was not only stable from pH 1–13 but could photoelectrochemically catalyze water oxidation. A catalytic cycle for the observed OER activity was, however, not reported. The homogeneous parent ligand H3TTCA does not evolve oxygen under similar conditions, which the authors suggest to be due to the instability of such organic catalysts when not site-isolated.
A 3D dithiolene-based MOF, Cu[Ni(pdt)2] (pdt = 2,3-pyrazinedithiolate), was evaluated as an electrocatalyst for the HER.291Cu[Ni(pdt)2] is constructed by nickel bis(pyrazine-2,3-dithiolate) complexes that are linked by 4-coordinated square planar Cu centers through the N atoms of the pyrazine moiety. Cu[Ni(pdt)2] forms a 3D structure with 1D square channels along the c direction. Cu[Ni(pdt)2] catalyzes the electrochemical HER from aqueous electrolyte solution at pH 1.3 with good current density after an initial activation step which involved the cleavage of Cu–N bonds upon applied negative potential in the acidic reaction medium. Computational results suggested that the copper centers were not involved in the HER, whereas the high H affinity of the pyrazine linker suggested a ligand-based HER mechanism.
Dou et al.292 presented the strategy of ligand doping to enhance the activity of MOFs for the electrocatalytic reduction of CO2. In this report, as-synthesized ZIF-8 was first activated to generate open Zn sites which were subsequently coordinated by electron-donating 1–10-phenanthroline units to form ZIF-8-LD (LD = ligand doped). The electron-donating properties of the phenanthroline ligand positively impacts the catalytically active sp2 carbon atoms in the imidazole ligands, facilitating the activation of CO2 and its conversion to CO. Compared to the parent ZIF-8 scaffold, ZIF-8-LD displayed enhanced electrocatalytic activity and a high selectivity for the production of CO with a FECO of 90.6%.
Molecular Ru-based water oxidation catalysts of the general type (tda)Ru(py)2 (tda = 2,2′:6′,2″-terpyridine-6,6′′-dicarboxylic acid) have been decorated with carboxylates at the axial pyridine groups, and used as linkers in Zr6O4(OH)4 SBU-based MOFs. Liseev at al.293 postsynthetically introduced a ditopic linker of this type (with 4-carboxylpyridine ligands) into a UiO-type MOF with ethynyldibenzoic acid linkers. The resulting mixed linker MOF was, however, a rather poor OER catalyst. In subsequent work, the same group reported a mixed linker analogue of NU-1000, prepared from the native H4TBAPy (H4TBAPy = 4,4′,4″,4‴-(pyrene-1,3,6,8-tetrayl)tetrabenzoic acid) linker and a tetratopic analogue of the Ru metallolinker with pyridine-3,5-dibenzoic acid ligands.294 In analogy to the homogeneous reference systems,295 the Ru complexes in the mixed linker MOF could be activated and engaged in electrocatalytic OER, albeit only with a FE of 37%. Interestingly, it was suggested that hole transport to Ru catalysts in the interior of the MOF crystals may be mediated by the TBAPy colinker, which, however, also engaged in irreversible oxidation processes, consistent with the low FE.
3.1.4. Catalysts as SBUs
This section summarizes a class of catalytic materials in which the SBU is the catalytically active site. The SBU, and thus catalyst assembly, is enabled and stabilized by the coordinating linkers, and the overall stability of the MOF. In a way, this scenario resembles the situation in many metalloenzymes, where the active sites are held in place by amino acid side chains that are positioned in space at a defined distance by the surrounding peptide environment. The section is structured by the nature of the metals that the SBU is composed of.
3.1.4.1. Titanium/Zirconium/Iron
Most examples in this section deal with MOFs that are built from high-valent ZrIV or TiIV based SBU, and that engage in light induced electron transfer processes. Frequently, the linker is excited and oxidatively quenched by electron transfer to produce a reduced metal state at the SBU, which then drives the reductive chemistry. For a thorough discussion of possible photophysical processes in such systems, please see section 2.3 of this review. The organic linker that is oxidized in the process needs to be rereduced to enter another absorption/charge-separation cycle. In some examples, this is accomplished by the use of an organic electron donor, but in others, also water oxidation is proposed for this process. The latter is often implied in passing, and mechanistic details as to the water oxidation process are vague.
The oldest example in this section stems from 2012, when Li and co-workers296 described the light-driven reduction of CO2 to formate catalyzed by the Ti-containing MOF, NH2-MIL-125(Ti). This MOF was synthesized by employing 2-aminoterephthalic acid (H2ATA) instead of benzene-1,4-dicarboxylic acid (H2BDC) that was used for the parent framework, MIL-125(Ti). The 2-aminoterephthalate (ATA) linker is important for catalytic function as it shifts the optical absorption of the MOF into the visible region of the electromagnetic spectrum. Upon photoexcitation, Ti4+ is reduced to Ti3+ by the transfer of an electron from the excited ATA linker to the SBU, creating a charge-separated state that is able to reduce CO2. The electrons for this process are provided by a sacrificial electron donor, in the present case TEOA, that reduces that transiently oxidized ATA back to its ground state. The strategy of light-induced charge separation that involve ATA and derivatives thereof has been utilized in numerous subsequent studies, and the thereby generated charge-separated states employed for different transformations.
NH2-MIL-125(Ti) was also investigated for photocatalytic CO2 conversion as an emulsion with CO2/water under high pressures. In the presence of TEOA as electron donor, Luo et al.297 reported an increased efficiency of the CO2/H2O/NH2-MIL-125(Ti) emulsion for the conversion of CO2 to HCOO–, as compared to the same MOF system in conventional organic solvents. The authors identified the large interfacial area and outstanding mass transport under the high-pressure conditions as contributing factors for the improved activity.
Further, Li and co-workers298 conducted photoluminescence studies on NH2-UiO-66(Zr) and suggested a photoinduced electron transfer from the excited ATA linker to the Zr-oxo clusters to form Zr3+ species. The latter species was proposed to drive the reduction of CO2 to formate (Figure 30a). Light absorption of the amino-linkers could be pushed further into the visible spectrum by partial substitution of ATA to DTA (2,5-diaminoterephthalate) linkers, resulting into a mixed NH2-UiO-66(Zr) framework with improved photocatalytic activity. Two years later, the same group prepared the mixed metal Ti-substituted NH2-UiO-66(Zr/Ti) framework by postsynthetic exchange. This material displayed improved photocatalytic performance for CO2RR and HER.299 DFT and electron spin resonance studies proposed that the introduced Ti acts as an electron mediator between the ATA light absorbing linker and the SBU-based ZrIV centers. The interested reader realizes that this thermodynamic ordering is opposite to that of bulk metal oxide nanoparticles where the conduction band of ZrO2 is considerably higher than that of TiO2.300
Figure 30.

Suggested mechanisms for the photocatalytic reduction of CO2 by a) NH2-UiO-66(Zr),298 b) UiO-68-R (R = F, CH3, OCH3),301 c) Fe-featured NH2-MIL frameworks,302 and d) NNU-28.303 Adapted with permission from the respective references. Copyright 2013 WILEY-VCH Verlag GmbH & Co, 2019 Royal Society of Chemistry, 2014 American Chemical Society, and 2016 Royal Society of Chemistry, respectively.
In a related work, Gao et al.188 described a Ti/Zr porphyrinic MOF, Ti/Zr-MOF-525, which was able to perform gas-phase CO2 reduction into CH4 and CO under visible light irradiation. No exogenous organic electron donor was added, and the CO2RR was accompanied by water oxidation. The Ti/Zr-MOF-525 was afforded from Zr-MOF-525 in a metal metathesis procedure that replaced Zr with Ti to afford a material with a Ti/Zr atomic ratio of 9:1. The Ti/Zr-MOF-525 yielded CH4 and CO as CO2 reduction produces in yields that were roughly 18 times higher than those of Zr-MOF-525. Overall, catalysis proceeded with less than one turnover per SBU.
In 2019, Wei et al.301 reported the postsynthetic modification of UiO-68-NH2 by a Schiff-base condensation with 3-fluorobenzaldehyde, 3-methylbenzaldehyde, and 3-methoxybenzaldehyde to afford three novel MOFs, UiO-68-F, UiO-68-CH3, and UiO-68-OCH3, respectively (Figure 30b). The MOF with the most electron-rich linker of the series, UiO-68-OCH3, performs best in the light-driven CO2-to-CO reduction in the presence of TEOA. After 6 h, the TONs of UiO-68-F, UiO-68-CH3, and UiO-68-OCH3 per SBU were determined to be 0.44, 2.88, and 4.08, respectively. Recycling experiments for the different frameworks revealed that the CO production does not level off for three consecutive runs over a total of 18 h.
Li and co-workers302 described a set of Fe-MOFs (MIL-101(Fe), MIL-53(Fe), MIL-88B(Fe)). The constituting Fe-oxo cluster SBUs could directly be photoexcited, and after reductive quenching produced FeII species that catalyze the reduction of CO2 to formate. Owing to unsaturated Fe sites, MIL-101(Fe) showed the highest activity among the examined MOFs. The amine-functionalized Fe-MOFs, NH2-MIL-101(Fe), NH2-MIL-53(Fe), and NH2-MIL-88B(Fe), assembled with H2ATA instead of H2BDC, displayed improved photocatalytic performance compared to their unfunctionalized controls (Figure 30c). Photocatalytic CO2-to-formate conversion experiments over the series of Fe-MOFs and their amino-functionalized derivatives showed that NH2-MIL-101(Fe) performed the best. The authors suggest that both ATA excitation and subsequent electron transfer to the SBUs, as well as the direct excitation of the Fe–O clusters contribute to catalytic turnover under irradiation and in the presence of an electron donor.
In contrast to the dual excitation route described above, Chen et al.303 described a dual catalytic mechanism in the light-driven reduction of CO2 to formate, catalyzed by the Zr-MOF NNU-28 ([Zr6O4(OH)4(L)6]·6DMF, H2L = 4,4′-(anthracene-9,10-diylbis(ethyne-2,1-diyl))dibenzoic acid; DMF = dimethylformamide) with an anthracene-based linker (Figure 30d). Photocatalytic experiments and EPR studies revealed that CO2 reduction can occur directly at the photosensitizing anthracene-based organic linker or at the Zr6 oxo clusters after ligand-to-metal charge transfer. Taking advantage of the same principle, Sun et al.304 reported the use of an amino-functionalized photosensitizing MOF Zr-SDCA-NH2 (H2SDCA-NH2 = 2,2′-diamino-4,4′-stilbenedicarboxylic acid), for CO2-to-formate reduction.
Based on the work on NNU-28, SK et al.190 reported an anthracene-based Zr-MOF, of which the activated form was employed as a photoactive catalyst for the CO2RR, accompanied by water oxidation. The Zr-MOF was afforded in a solvothermal synthesis from ZrOCl2·8H2O and the anthracene-based linker. The latter was postulated to be the photoresponsive unit that is oxidatively quenched by the Zr-cluster SBU that catalyzes the CO2RR. As water is proposed as the terminal donor in the process, this implies that the anthracene linker catalyzes water oxidation, a process that is unprecedented in the homogeneous phase. Either way, the system evolved formic acid as the only CO2 reduction product with a TOF of less than one per hour.
Logan et al.305 reported the synthesis of a series of isoreticular NHR-MIL-125 (R = methyl, ethyl, isopropyl, n-butyl, n-heptyl, cyclopentyl, and cyclohexyl) frameworks and connected the experimentally observed reduction of the optical bandgap to an increase in the photocatalytic CO2RR activity. NHCyp-MIL-125 (Cyp = cyclopentyl) displayed a smaller bandgap (Eg = 2.30 eV), a longer-lived excited-state (τ = 68.8 ns) and a larger apparent quantum yield (Φapp = 1.80%) compared to those of the parent NH2-MIL-125 (Eg = 2.56 eV, Φapp = 0.31%, τ = 12.8 ns) and was the most active catalyst for formate production under blue light illumination over 120 h. We note that these MOFs are, however, not expected to be semiconductors (see section 2.3.3), so instead of “bandgap” one should discuss the lowest excited state energy as for a general molecular PS. A general finding of the report was that linkers with secondary amines displayed larger apparent quantum yields and longer excited-state lifetimes compared to their primary amine analogues.
Recently, Chen et al.306 presented a combination of dual excitation and dual catalytic function in a mixed linker MOF, D-TiMOF. This MOF is based on the widely used light-responsive NH2-MIL-125(Ti) (TiMOF) and was developed to further increase its photocatalytic activity. For this reason, photosensitizing Zn-porphyrins, ZnTCPP, were installed at the Ti-oxo SBUs as a second light-harvester in addition to the already present ATA linkers. Photocatalytic CO2 reduction experiments were performed for TiO2, ZnTCPP, TiMOF, D-TiMOF, and the physical mixture of TiMOF and ZnTCPP. TiMOF exhibited higher catalytic activity than TiO2 which was attributed to the CO2 sorption ability of MOFs. The presence of ZnTCPP in D-TiMOF led to an 11-fold enhancement in CO2-to-CO conversion efficiency as compared to that of the parent TiMOF. The significantly enhanced activity is accredited to the synergistic effect between the Ti-oxo clusters and ZnTCPP, not only providing photoelectrons for the Ti-sites but also offering a second reactive site.
In contrast to the benzene-based linkers, the following frameworks consist of either larger π-systems or porphyrins in combination with Zr-oxo clusters. Considering the benefits of incorporating large π-conjugated units into MOFs, Qin et al.307 reported the synthesis of the Zr-based MOF PCN-136, Zr6(μ3-O)4(μ3-OH)4(OH)6(H2O)6(HCHC) (HCHC = hexakis(4-carboxyphenyl)hexabenzocoronene). Owing to the low solubility and the unfavorable conformation of the HCHC ligand, PCN-136 could only be obtained from aromatization-driven postsynthetic annulation of the hexaphenylbenzene unit from the parent pbz-MOF-1 (pbz = polybenzene) scaffold. In comparison with pbz-MOF-1, the combination of isolated photosensitizing hexabenzocoronene moieties and catalytically active Zr-oxo nodes in PCN-136 led to an enhanced photocatalytic activity for the reduction of CO2 to formate.
Based on porphyrinic TCPP linkers, Qiu et al.308 reported the synthesis of two mixed-linker MOFs, [(CH3)2NH2]4[Zn4O]4[ZnTCPP]5[BTB]8/3 (PCN-137) and [Zr6(μ3-O)4(μ3-OH)4][TCPP][TBTB]8/3 (PCN-138) (BTB = 1,3,5-benzene(tris)benzoate and TBTB = 4,4′,4″-(2,4,6-trimethylbenzene-1,3,5-triyl)tribenzoate). Employed as photocatalysts for the CO2RR in the presence of triisopropanolamine as an electron donor, PCN-138 displayed high selectivity for formate production over 12 h in an aqueous medium. The authors postulate that this performance was due to simultaneous immobilization of porphyrinic PS units and active Zr-oxo nodes within the MOF platform, resulting in a material that features a broad absorption in the visible region and a high CO2 sorption performance.
Xu et al.151 demonstrated the selective conversion of CO2 to formate with TEOA as an electron donor and a PCN-222 framework as a catalyst, consisting of H2TCPP light-harvesting porphyrinic linkers and Zr6 clusters as SBUs. Mechanistic investigations revealed that the porphyrinic linkers act as PSs that upon illumination transfer electrons to the Zr-oxo clusters, where CO2 is reduced to formate. Under similar conditions, H2TCPP was also investigated as a homogeneous photocatalyst for the CO2RR, but produced noticeable smaller amounts of product compared to the MOF.
Aziz et al.309 described the change in electronic structure in a series of Fe-doped ZnTCPP-based MOFs (Al-PMOFs) employed for the reduction of CO2 to CH3OH and CH4, by incorporating Fe at the SBUs as well as into the TCPP linkers. While the incorporation of Fe at the porphyrinic sites led to an increase of the highest occupied molecular orbital (HOMO) level, the Fe-doping at the metal nodes lowered the conduction band significantly. Hence, the most promising results were achieved by partially introducing Fe into the Al SBUs (1:1), while keeping the Zn porphyrinic centers unchanged, leading to a nearly ideal bandgap of 1.9 eV and band edge positions suitable for water splitting and CO2 reduction at pH 7. We again note that these MOFs are unlikely to be semiconductors (per section 2.3.3), so one should discuss the lowest excited state energy rather than the “bandgap”.
Zhang and co-workers310 reported a series of Zr-based MOFs with large π-conjugated organic linkers for a near-infrared light-driven CO2RR. The photoinduced electron transfer pathway from the excited π-conjugated linkers to the Zr cluster SBU was investigated by ultrafast spectroscopic studies, XPS, and in situ EPR experiments. In the presence of TEOA, the tetrakis(4-carboxybiphenyl)naphthoporphyrin)-based MOF (TNP-MOF) with the largest π-conjugated linker displayed the highest CO2 reduction rate under NIR light irradiation (λ ≥ 730 nm), as well as the highest apparent quantum efficiencies of up to 2.03 and 1.11% at 760 and 808 nm, respectively.
3.1.4.2. Cobalt
The Co4O4(OAc)4(py)4 cubane is a competent WOC partly due to labile carboxylate ligands, though it is claimed that this comes at the cost of long-term stability. As a homogeneous compound, loss of the carboxylate ligands is proposed to result in cluster aggregation and deactivation.311 Tilley and co-workers311 constructed a MOF containing isolated cubane sites (Figure 31a) and demonstrated that this material remained stable up to pH 14. As noted by Wang and Meyer,312 this advance did result in notable stability improvement, though a significant proportion of the cubane units remained inaccessible due to the insulating nature of the parent MOF. The comparison between the cubane@MOF material and photosystem II was specifically drawn, noting that while photosystem II has an elegant electron transport chain in place, cubane@MOF lacked such a system.312 This points toward the possible future rational design of biomimetic MOFs wherein both catalytic sites and electron transport chains are incorporated.209
Figure 31.

a) Proposed redox-hopping mechanism to achieve a [CoIII2CoIV2] or [CoIII3CoV] intermediate by redox disproportionation of two [CoIII3CoIV] clusters in Co4-TPT. Reprinted with permission from reference.311 Copyright 2019 National Academy of Science. b) Suggested mechanism for the photocatalytic reduction of CO2 by Co-MOF. Reprinted with permission from reference.318 Copyright 2018 American Chemical Society.
The Co centers in ZIF-67, a MOF in which Co-centers are coordinated by four imidazolate linkers, have been utilized for light-driven HER. In order to do so, Ru2+-based PSs, RuN3, were coordinated to ZIF-67 crystals to afford a RuN3/ZIF-67 hybrid system. Following photon absorption, the excited RuN3 dye engages in energy transfer with the ZIF-67, which upon addition of water and a sacrificial electron donor engages in the HER. Catalysis ceases after 10 h due to RuN3 decomposition and can be restored by addition of a fresh PS.313
Qin et al.314 presented the coprecipitation of Co(NO3)2·6H2O and 2-methylimidazole to assemble nanosized ZIF-67 crystals. The latter function as a catalyst for the light-driven HER and CO2-to-CO reduction in cooperation with Ru(bpy)3Cl2 as a PS and TEOA as an electron donor. By varying the excitation wavelength, it was shown that the combined yields of the reaction products correlated with the absorption spectrum of the PS, showing that excitation of the PS is the initial step of the photocatalytic cycle.
The catalytic activity of CoII was utilized in a cobalt-based MOF, Co–MB, prepared from Co(NO3)2, 3,3′-methylenediphthalic acid (H4mda) and 1,4-bis(imidazol-1-ylmethyl)-benzene (bib) in an aqueous sodium hydroxide solution under hydrothermal conditions.315 In Co–MB, two crystallographically independent CoII centers exhibit distorted tetrahedral coordination geometries with two carboxylate oxygen atoms from two different mda4– anions and two nitrogen atoms from two different bib ligands creating the primary coordination sphere. Co–MB catalyzes the light-driven HER in conjunction with various fluorescein-based PSs and TEA as electron donor. The material could be recycled three times without compromising its activity as catalyst for the light-driven HER, illustrating the power of the MOF matrix to stabilize molecularly defined active sites.
In 2014, Wang et al.316 described a robust cobalt imidazolate framework Co-ZIF-9 that serves as a CO2 reduction catalyst in conjunction with a Ru-based PS and TEOA as an electron donor. Even though a homogeneous mixture of the ligand and Co2+ was proven to be able to catalyze CO and H2 evolution, the combined CO and H2 production performances as well as the TON were superior for the MOF system. Furthermore, stability experiments with recovered Co-ZIF-9, redispersed in fresh dye solution, showed no noticeable alteration in the activity of the reused catalyst over 2.5 h. Similarly, Zhao et al.317 presented a pillared layer Co6-MOF that catalyzed the CO2-to-CO reduction, when combined with a Ru-based PS. Compared to the homogeneous control, the MOF system exhibited higher stability, giving rise to higher TONs.
In 2018, Liao et al.318 reported the solvothermal one-pot reaction of Co(NO3)2·6H2O and H4L (2′-amino-[1,1′:4′,1″-terphenyl]-3,3″,5,5″-tetracarboxylic acid), to form Co-MOF ([Co3(HL)2·4DMF·4H2O]) for both photocatalytic hydrogen evolution and CO2 reduction. While the HER was driven by a Ru-based PS under illumination, the internal light absorbing organic linker in combination with the catalytically active Co–O clusters was sufficient to drive the photocatalytic CO2 reduction process. Under visible light irradiation and in the presence of BNAH (1-benzyl-1,4-dihydronicotinamide) as a sacrificial electron donor, excited HL3– transfers electrons to the SBUs, where the trinuclear [CoIICoIICoII] cluster is reduced to a [CoICoIICoII] oxidation state that reduced CO2 to CO or formate (Figure 31b).
3.1.4.3. Copper
An unusual active site has been reported in a mixed linker-mixed metal Na/Cu-MOF.319 The dinuclear Cu2 site is asymmetric, with one of the CuII ions displaying a five-coordinate square-pyramidal geometry while the other adopts a six-coordinate octahedral geometry. In close proximity is an octahedrally coordinated NaI cation. In comparison to a number of reference systems, including a system that lacks the NaI cation, it is shown that Na/Cu-MOF is a superior catalyst for the light-driven HER in the presence of TEA and fluorescein as a PS.
Missing linker defect engineering has been used to turn a catalytically incompetent metal-triazolate (MET) MOF into a defective, but catalytically active material.320 In a defect-free state, each copper center is coordinated by six nitrogen atoms from different 1H-1,2,3-triazole linkers in an octahedral geometry. The coordination sphere of the metal centers is permanently saturated, and as a result, the MET-Cu does not catalyze the HER. However, by decreasing the amount of triazole linkers relative to the Cu salt, as well as variation of other reaction parameters, defective MET-Cu (MET-Cu-D) in which some of the Cu centers lack permanent ligands was prepared. Using Eosin Y as a PS and TEA as a sacrificial electron donor, MET-Cu-D was found to be a competent catalyst for the light-driven HER. Postsynthetic “repair” of MET-Cu-D by addition of a triazole linker decreased the HER activity, providing indirect proof that the partially under-coordinated Cu centers are indeed responsible for the observed HER activity.
In 2012, Mao et al.321 demonstrated the first application of a MOF as an ORR electrocatalyst. The work was based on Cu-BTC (BTC = 1,3,5-benzenetricarboxylate) that, however, revealed its structural instability under ORR conditions in aqueous media. Therefore, the water-stable Cu-bpy-BTC was synthesized and used as an electrocatalyst in form of MOF-modified GCEs. In a phosphate buffer at pH 6.0, cyclic voltammograms of Cu-bpy-BTC showed well-defined reduction peaks and a current enhancement at −0.20 V in the presence of O2, consistent with catalytic turnover. Rotating ring-disk electrode voltammetry of the CuII-based MOF showed an almost 4e– reduction pathway, consistent with reduction of oxygen to water without generation of H2O2 as side product.
In 2012, the electrochemical synthesis of the bulk MOF Cu3(BTC)2, also known as HKUST-1, was described, followed by its application for the electrocatalytic reduction of CO2 to oxalic acid as thin films on GCEs.322 Within the framework, CuI, which is formed at a potential of −0.62 V vs Ag/Ag+ is the catalyst for the CO2RR. The authors demonstrated that the synthesized Cu3(BTC)2 framework is able to form the oxalate anion upon 2-electron reduction and dimerization of CO2.
In 2019, Zhang et al.323 described the zeolite-related boron imidazolate framework BIF-29, which features mononuclear copper in a square-planar coordination mode. In this geometry, the Cu sites are highly active and are able to catalyze the light-driven reduction of CO2 to CO in combination with a Ru-based PS and TEOA as a sacrificial electron donor. Theoretical studies revealed the contribution of the Cu sites with weakly bound water molecules (Cu–O: 2.538 Å) for the absorption and activation of CO2 and the further stabilization of the *COOH intermediate. Furthermore, steady-state and time-resolved fluorescence experiments indicated that the Cu sites engaged in charge separation. Solar-driven catalytic CO2 reduction employing BIF-29 led to CO evolution with a selectivity of 82.6%.
In 2021, Liu et al.324 reported a series of tricycloquinazoline-based 2D MOFs as electrocatalysts for the reduction of CO2 toward methanol. The electron-deficient but nitrogen-rich catechol linker HHTQ (2,3,7,8,12,13-hexahydroxytricycloquinazoline) was employed to coordinate to transition metals (Cu2+ or Ni2+) to afford 2D graphene-like sheets of M3(HHTQ)2 (M = Cu or Ni). Cu3(HHTQ)2 as an electrocatalyst for the CO2RR displayed high selectivity toward CH3OH with a FE of up to 53.6% at −0.4 V vs RHE, exhibiting larger CO2 adsorption energies and higher activities than both, the isostructural Ni3(HHTQ)2 and the archetypical Cu3(HHTP)2 (HHTP = 2,3,6,7,10,11-hexahydroxytriphenylene).
3.1.4.4. Nickel
Mani et al.325 reported Ni-BTB-BPE ([Ni3(BPE)4(BTB)2(H2O)2]·2DMF·2H2O (BTB = 1,3,5-tris(4-carboxyphenyl)benzene and BPE = 1,2-bis(4-pyridyl) ethane), in which the BTB linkers interconnect the Ni ions to form 2D Kagome-type layers, which are then connected through the BPE linker to form the 3D construct. Ni-BTB-BPE showed both ORR and OER activity but only at the surface Ni sites due to the nonporous structure of Ni-BTB-BPE. The catalytic activity of Ni-BTB-BPE for oxygen reduction and evolution was investigated when adsorbed together with Ketjenblack on a conducting substrate. Ni-BTB-BPE/C showed a high selectivity for the four-electron pathway with OH– as the major product.
The 2D layered MOF [Ni2(PymS)4]n (PymSH = pyrimidine-2-thiol) with thiolate-bridged binuclear NiII nodes was reported to catalyze the light-driven HER in the presence of a variety of PSs and sacrificial donors, with fluorescein and TEA being the optimal combination.326 The MOF was formed by the reaction of Ni(OAc)2 and 2-mercapto-pyrimidine in the presence of KOH and was a superior catalyst compared to only the NiII salt of the ligand. The [Ni2(PymS)4]n catalyst possessed a 2D lamellar structure built from binuclear nodes [Ni2(PymS)4], in which the NiII ions are in turn connected by PymS– ligands through S and N atoms.
Another interesting SBU active site and its activity for the HER was recently reported by Budnikova and co-workers.327 Using 4,4′-bipyridine as cross-linking units, Ni and Co redox-active coordination polymers with ferrocene-containing diphosphinate ligands were tethered together to form 3D MOFs. The nickel or cobalt atoms were coordinated to four phosphinic oxygen atoms in the equatorial plane and two nitrogen atoms from the 4,4′-bpy ligand in the axial positions. It is shown that the 4,4′-bpy linkers reduced the overvoltage for HER, resulting in lower Tafel slopes, consistent with more favorable HER kinetics.
Ultrathin 2D nanosheets, produced by exfoliation of a MOF and consisting of bis(4′-carboxy-2,2′:6′,2″-terpyridine) ruthenium linkers and 3d transition-metal (Mn, Co, Ni, and Zn) SBUs, were reported as good photocatalytic HER materials.328 Combined experimental and theoretical insights revealed a reductive quenched pathway of the *Ru2+ excited state by the ascorbate electron donor, followed by electron transfer to the transition-metal nodes. The nodes at the edge of the nanosheets were proposed as the active sites. DFT calculations attributed the different catalytic activities of the nanosheets to different hydrogen atom adsorption free energies at the varying transition-metal nodes. Of the systems investigated, the Ni–Ru nanosheets exhibited the highest HER rates.
In 2020, Fang et al.150 described the synthesis of the framework PCN-601, combining pyrazolyl porphyrinic light harvesters and Ni-based SBU active sites. PCN-601 with a Ni center in the porphyrin linker catalyzed the gas phase reduction of CO2-to-CH4. The reduction was accompanied by the oxidation of H2O vapor to H2O2.
Very recently, Iqbal et al.329 explored the synergistic effect of the incorporation of Ni and Fe in a 2D MOF for the electrochemical CO2RR. Surrounded by four nitrogen atoms of dipyrazino quinoxaline-2,3,6,7,10,11-hexamine, the metal centers are located in a porphyrin-like environment. DFT calculations showed that the simultaneous incorporation of Ni and Fe leads to a system with a high FE of 98.2% for CO evolution and a stability of up to 30 h under an applied potential of −0.5 V vs RHE.
3.1.4.5. Other Metals: Ruthenium, Cerium, Europium, Bismuth, Aluminum, and Zinc
A series of diruthenium-based MOFs [Ru2(BDC)2X]n (X = Cl, Br, and BF4) have been shown to catalyze the light-driven HER in the presence of [Ru(bpy)3]2+, methyl viologen, and EDTA for longer than 4 h.330 While in the early report, the PS was in the solution phase, subsequent work has focused on immobilizing light absorbing PSs as linker units in conjunction with catalysts that are stabilized or supported as SBUs. Ru2 paddle wheel SBUs (Figure 32a) have been combined with TCPP linkers to afford two novel MOFs, Ru-TBP and Ru-TBP-Zn. In these systems, it is shown that the excited porphyrin linkers are oxidatively quenched by the Ru2 SBUs as the initial step of the HER process.331
Figure 32.

a) An example of a molecular catalyst stabilized as secondary building unit in form of a Ru2 paddle wheel in Ru-TBP-Zn.331 b) A molecular catalyst stabilized as secondary building unit, demonstrated with a hexanuclear Ce6 cluster in Ce6-BTB.332 c) Suggested mechanism for the photocatalytic reduction of CO2 by Eu-Ru(phen)3-MOF.333 Adapted with permission from the respective references. Copyright 2018 American Chemical Society, 2020 American Chemical Society, and 2018 Springer Nature, respectively.
Moving away from scarce metal catalysts, a hexanuclear cerium cluster as catalytic site could be stabilized in a Ce6–BTB MOL consisting of the Ce6 clusters as SBUs and BTB linkers (Figure 32b). When postsynthetically modified with molecular Ir- or Ru-based PSs, both MOLs engaged in the photocatalytic HER under visible light irradiation.332
In 2018, Yan et al.333 described a MOF with large pore dimensions, consisting of dinuclear Eu2(μ2-H2O) SBUs with photosensitizing Ru(phen)3-derived metalloligands (phen = phenanthroline). Time-resolved photoluminescence studies in combination with femtosecond transient optical absorption spectroscopy were consistent with a charge transfer from excited Ru-PS to the Eu–O nodes on a time scale of 1–300 ns. The reduced [EuII–H2O–EuII] oxidation state of the SBU is catalytically competent for the selective reduction of CO2 to formate (Figure 32c). The formate production rate of for Eu-Ru(phen)3-MOF was notably higher than the reported values of NH2-MIL-125(Ti),296NH2-UiO-66(Zr),298 and PCN-222(151) under similar reaction conditions.
In 2020, Li et al.334 reported a 2D bismuth framework (Bi-MOF), consisting of Bi–O rods connected with tritopic BTC linkers. This MOF showed permanently accessible pore channels for the efficient electrochemical CO2RR to HCOOH. Bi-MOF displayed a remarkable FE for formic acid formation over a broad potential window, reaching its maximum of 92.2% at −0.9 V vs RHE with a durability of over 30 h. The partial current density for HCOOH around 41.0 mA mgBi–1 was 4 times higher compared to that of commercial Bi2O3 and Bi sheets. Possible explanations are the finding that the Bi3+ state was able to be preserved within the Bi-MOF system and that the channels with accessible Bi active sites favor the formation of *HCOO and suppress the unwanted side-reaction to hydrogen, as suggested by theoretical calculations.
Toma and co-workers335 presented the power of confinement of metal centers based on investigations on the aluminum containing MOF, MIL-53(Al). Aluminum as a foil is not active for the CO2RR but instead shows high selectivity for the HER. Confined in MIL-53(Al), aluminum restrains the HER and enhances the carbon dioxide reduction up to a total FE of 40% for CO and formic acid at a potential of −1.1 V vs RHE.
Bie et al.336 reported the synthesis of Spiro-Zn-MOF, a 3D framework consisting of the organic PS SATC (5,5′-(10H,10′H-9,9′-spirobi[acridine]-10,10′-diyl)diisophthalic acid) and Zn2+-based SBUs. The linker is a donor/acceptor structure that displayed an excited state lifetime up to a microsecond, which was preserved also in the Spiro-Zn-MOF. The long-lived excited state and charge transfer to the Zn nodes allowed for the photocatalytic CO2RR toward CO. Proof for the electron transfer to the Zn2+-based SBU was obtained by EPR analysis of illuminated samples of Spiro-Zn-MOF, which showed a signal that was attributed to Zn+. The authors pointed out that no organic donor was required for catalysis, implying that electrons are provided from water oxidation, which is however not commented on further. Control experiments with pure SATC, zinc nitrate, and a homogeneous mixture of SATC and zinc nitrate did not afford any CO.
3.1.4.6. Bimetallic SBUs
The electrochemical potential that needs to be applied at a catalytic metal site to engage in catalysis can be modulated by including a second metal center, often resulting in a structurally more complex SBU. Examples of such MOFs with tuned redox potentials have been reported. For example, MOFs which have been doped with a second metal cation have been shown to have redox potentials more suitable for fuel-forming reactions. In 2016, Wang and co-workers337 used cathodic electrochemical deposition on nickel foam to prepare a series of such bimetallic MOFs based on the BTC linker with either Ni, Fe, or both Ni and Fe as metals in the SBU (Figure 33). The mixed metal Fe/Ni-BTC MOFs, where the Fe:Ni ratio was 1:12, yielded the lowest overpotential for electrochemical OER catalysis. Based on literature precedent, the authors suggested that the nickel sites in the SBU were the active catalytic sites for the OER. Intriguingly, the Fe was suggested to play a dual role in improving catalysis, by increasing framework conductivity and by shifting the Ni2+/3+ redox potential to milder values. Since then, similar data has been published supporting this cooperative tuning of the electronics of the active metal site by incorporation of a second metal.338−341
Figure 33.

Electrochemical synthesis of Ni-BTC or mixed metal Fe/Ni-BTC MOFs on nickel foam. Adapted with permission from reference.337 Copyright 2016 American Chemical Society.
Yao et al.342 recently reported the synthesis of N-NiFe-MOF that was obtained through a postsynthetic nitridation process from NiFe-MOF, a BDC-based MOF that was obtained from a NiFe layered double hydroxide. During the nitridation, NH3 coordinates to the open metal sites in NiFe-MOF to form N-NiFe-MOF, which is a precatalyst for the OER. Based on DFT calculations and experimental results, the authors suggested that the amino ligands at the open metal sites are oxidized to nitric oxides during the OER, thereby affording NiFe(NO)-MOF, which is a potent OER catalyst in a 1.0 M KOH solution. The transformation from N-NiFe-MOF to NiFe(NO)-MOF introduces an electron deficiency on the Fe atoms and promotes the OER through the stabilization of the intermediate *O species, thereby decreasing the required overpotential for the conversion to *OOH.
Yue et al.343 reported the synthesis of 2D nanosheets of the trimetallic framework NiCoFe–NDA (NDA = 2,6-naphthalenedicarboxylic acid) on nickel foam (NiCoFe–NDA/NF) as a highly active electrocatalyst for the OER. The NiCoFe–NDA/NF electrode displayed a much higher OER activity and stability when compared to the monometallic versions. Thereby, the NiCoFe–NDA suffered topological transformation and underwent an in situ phase transformation, inducing surface-rich metal oxyhydroxides with low-coordination environments, providing a multimetal coupling effect which is beneficial for the water oxidation.
Yoon et al.344 synthesized bimetallic conductive 2D MOFs, CoxNiy-CAT, consisting of HHTP linkers and varied ratios of Co2+ to Ni2+ metal ions, and employed them as catalysts in the ORR. The authors found that compared to monometallic Co2+ or Ni2+ MOFs (Co-CAT or Ni-CAT), the bimetallic MOFs displayed higher ORR activity, possibly due to the combination of abundant Co–O moieties serving as catalytically active sites and the high conductivity within Ni-CAT. The optimized Co0.27Ni0.73-CAT material had a higher electron transfer number of 3.95 (lower H2O2 production) and better durability (87% after 10 000 s) than its monometallic congeners.
Shinde et al.345 reported the synthesis of hexaiminobenzene (HIB)-based MOFs (Mn/Fe-HIB-MOF) as bifunctional ORR and OER electrocatalysts. The Mn/Fe-HIB-MOF was afforded from HAB in ammoniacal solutions of MnII and FeII salts, followed by a thermal treatment. Mn/Fe-HIB-MOF exhibited precisely controlled M2+ species in quintet-shelled hollow spheres, fast electron and mass transfer pathways, and catalytically effective MII–N4 moieties. Being a bidirectional electrocatalyst, Mn/Fe-HIB-MOF was shown to function well as electrode material in rechargeable liquid zinc air batteries. Zinc air batteries made with Mn/Fe-HIB-MOF displayed a remarkable cycling stability for 600 h (3600 cycles@25 mA cm−2) and excellent mechanical robustness.
3.1.4.7. POM
Huang et al.346 presented two structurally analogous POM-based metalloporphyrin coordination frameworks (POMCFs), NNU-13 ([PMoV8MoVI4O35(OH)5Zn4]2[ZnTCPP][2H2O]·xGuest) and NNU-14 ([PMoV8MoVI4O35(OH)5Zn4]2[ZnTCPP][2H2O]·yGuest). The systems were composed of catalytically active Zn-ε-Keggin nodes and photosensitizing ZnTCPP linkers, and catalyzed CO2-to-CH4 conversion in >96% selectivity in an aqueous solution in the presence of TEOA as a sacrificial agent and in the absence of any additional PS. CO that was formed during the reaction was shown to be a reaction intermediate that was further reduced to CH4. Owing to the slight structural deformation, NNU-14 displayed slightly lower CH4 selectivity of 96.2% compared to the 96.6% for NNU-13. Under illumination, the electrons are easily transferred from photoexcited ZnTCPP units to the POM clusters, where coordinated CO2 gets reduced.
In 2019, Li et al.347 revealed the novel POM-based framework TBA5[P2Mo16VMo8VIO71(OH)9Zn8(L)4] (NNU-29; L2– = 4,4′-((((perfluoropropane-2,2-diyl)bis(4,1-phenylene))bis(oxy))bis(methylene))dibenzoate and TBA+ = tetrabutylammonium). This system displayed catalytically active molybdenum POM clusters that , in combination with [Ru(bpy)3]2+ as a PS and TEOA as an electron donor, engaged in CO2-to-formate reduction in an aqueous solution with a selectivity of 97.9% after 16 h. The authors suggested that the competing HER is suppressed by the hydrophobic linkers that shield the catalytically active sites from water. Control experiments with a physical mixture of Zn4-ε-Keggin and H2L revealed a lower selectivity of 78.5% and yield for HCCOH production, demonstrating the benefits of heterogenizing CO2RR catalysts in MOFs.
3.1.4.8. Transition Metal–Heteroatom SBUs
Cobalt dithiolene HER catalysts can be structurally stabilized by incorporation into 2D metal–organic surfaces.348 For this purpose, cobalt dithiolene catalytic sites were assembled in extended architectures, using a tritopic benzenehexathiol linker through a liquid–liquid interfacial process (Figure 34). In this process, an acetonitrile/ethyl acetate solution of [Co(MeCN)6][BF4]2 was layered on top of an aqueous solution of sodium benzenehexathiolate (C6S6Na6). It was demonstrated that integration of cobalt dithiolene catalysts into a metal–organic surface generated highly active electrocatalytic cathode materials for the HER from water. These surfaces display high catalyst loadings and remarkable stability even in acidic aqueous solutions.
Figure 34.

Synthesis of the cobalt dithiolene surfaces, through a liquid–liquid interfacial process. Adapted with permission from reference (348). Copyright 2015 American Chemical Society.
In 2017, Liu et al.349 designed well-defined M–O6 coordination sites within two 2D porous metal-catecholates (M–CATs, M = Ni or Co), consisting of HHTP interconnected with either of the bivalent metal ions, NiII or CoII. The catalytic activity was found to come exclusively from the M–O6 sites and the ORR was predominantly displayed as a four-electron pathway.
A related Ni and Co-based 2D MOF with mixed THT (2,3,6,7,10,11-triphenylenehexa-thiol) and THA (2,3,6,7,10,11-triphenylenehexamine) linkers has also been reported.350 The mixed linker THTA-M 2D MOF (M = Ni or Co) with a metal dithiolene-diamine MS2N2 SBU was evaluated for the electrocatalytic HER, and compared to the activity of the THT-M and THA-M analogues with metal bis(dithiolene) (MS4), and metal bis(diamine) (MN4) sites, respectively. It was found that the electrocatalytic HER activity followed the order of MS2N2 > MN4 > MS4, and that protonation occurred preferentially on the metal atoms. Heterolytic H2 elimination was favored on the M–N units in the MS2N2 active sites.
Recently, Zhou et al.351 systematically investigated a series of 30 MNxO4–x–HTP (M = Fe, Co, Ni, Ru, Rh, and Pd; x = 0–4; HTP = hexatriphenylene) 2D conductive MOFs with the help of DFT calculations as catalysts for the electrocatalytic HER, OER, and ORR. The active centers of these materials are the transition-metal–heteroatom linkages. The strong interaction between the metal and the NxO4–x moiety guaranteed the stability of the MOF structure. Tuning the transition metal and the local coordination number of N/O in the catalysts modulated the catalytic activity. Among the investigated materials, RhN3O1–HTP was found to be an efficient bifunctional catalyst for both the HER and OER, with an OER overpotential of 0.33 V. Besides, RhN1O3–HTP is predicted to be highly promising for the OER and ORR with calculated overpotentials of 0.28 and 0.27 V, respectively.
3.2. Facilitated Transport
Substrate and charge transport within porous materials such as MOFs are important phenomena that can easily limit overall catalytic efficacy. Consequently, very good reviews on the topic have recently appeared.60,61 In this section, articles are reviewed that specifically address transport in context of catalysis. As outlined in section 2, hopping electron/hole transport that is accompanied by diffusion–migration of charge-balancing counterions, mass transport of reactants/products, or in case of photochemical schemes, the diffusion of sacrificial electron donors/acceptors may all limit catalysis. In some instances, structural modifications to the MOF may not selectively alter only one of these potential bottlenecks, and improved catalytic performance, as for example manifested in increased current densities in electrochemical experiments, may in fact be due to several factors.
In general, two approaches have been taken in the literature to overcome transport limitations. In the first set of examples (section 3.2.1), transport limitations are kept to a minimum at a system level by working with ultrathin films or few-nanometer-sized crystallites.81,352 In section 3.2.2, parent MOFs are structurally altered to facilitate one or more of the above-mentioned transport phenomena. From a biomimetic perspective, this strategy bears resemblance to enzymes in which substrate transport is often actively controlled and aided by specific amino acids that form channels to/from the active site.
3.2.1. Minimizing Transport Limitations by Size Reduction
A number of papers have systematically addressed the interplay between catalytic efficacy and the size of MOF particles in case of solution experiments, or thicknesses in case of MOF films on electrodes. It should be pointed out that the examples herein do not have a biomimetic aspect, but address transport limitations at a system level.
Morris and co-workers255 reported a study on Ru-UiO-67, that is, UiO-67 with Ru-based WOCs metallo-linkers. Variation of the MOF particle size between 200 and 1200 nm gave no significant differences in oxygen yield when normalized for total number of active sites, suggesting that catalysis was not limited by charge transport hopping kinetics. Additional work by the group of Morris demonstrated that charge transport can, however, indeed be limited to a surface-confined regime when the Ru-to-Ru distance exceeds a certain percolation threshold, that is, if the distance between the redox active hopping sites is too large, thereby slowing down electron transport significantly. Increasing the amount of redox-active Ru catalysts will move the system into a regime where charge transport into the MOF interior is no longer limiting.256
In another report, Chen et al.353 prepared a series of (Co)PCN222 frameworks with varied particle sizes spanning from 200 to 1000 nm via a coordination control synthesis technique. The ORR activity of these particles has been found to be proportional to their size. The smallest particle sized MOF had the highest mass activity, while the largest one displayed the highest surface area normalized activity of the three sizes tested. In this example, the correlation between catalytic activity and crystal size was suggested to be due to a noticeable decrease in the electrolyte ions’ diffusion distance and an increase in the active material’s specific surface area.
Two systematic reports on the correlation between MOF film thicknesses and catalytic current densities for thin MOF films have been reported. In 2015, Kornienko et al.354 reported the cobalt-porphyrin MOF Al2(OH)2CoTCPP, grown on a conducting substrate as a selective electrocatalyst for the reduction of CO2 to CO in aqueous media. The Al2(OH)2CoTCPP were made by the initial deposition of a known amount of Al2O3 on the electrode surface by ALD, followed by exposure to the H2TCPP linker. By controlling the amount of ALD cycles, and thus the amount of available Al3+ cations, films of different film thicknesses could be prepared. It was found that ultrathin films exhibit relatively small current densities, which increase with increasing film thickness to a maximum value after which current densities start to drop again (Figure 35a). This behavior was interpreted in that the catalytic performance is maximized at a starting layer thickness of 50 ALD cycles, which offers a balance of charge transport, mass transport, and active-site density.
Figure 35.

a) Al2(OH)2CoTCPP MOF and current densities obtained from the electrocatalytic reduction of CO2 from thin films. Maximum activity is obtained from films produced from 50 ALD cycles of the Al2O3 precursor.354 b) Structure of CoBHT and comparison of the current density (mA cm–2) versus thickness (nm) at −0.20 V vs RHE (blue), −0.22 V vs RHE (green), −0.25 V vs RHE (pink), and −0.27 V vs RHE (purple).355 Adapted with permission from the respective references. Copyright 2015 and 2018 American Chemical Society, respectively.
The importance of film thickness on the efficacy of MOF-based catalysts has also been demonstrated on a series of CoBHT-modified GCEs. CoBHT is a 2D-MOF consisting of benzenehexathiolate linkers (BHT) that are interconnected by CoII-cations. These sheets stack to form films, the thickness of which was systematically varied in a study by Downes et al.355 It was found that the initial increase in film thickness from 23 to 244 nm decreased the overpotential to achieve 10 mA cm–2 from 246 to 185 mV (Figure 35b). This finding is consistent with an increase of the number of catalytically active sites, as expected from an increase in film thickness. However, increasing the film thickness further to 1000 nm did not lead to a further increase in the number of electrochemically accessible active sites, and a reduced HER activity was observed, quantified by an increased overpotential of 213 mV. In this regime, charge transport and proton permeation through the thick films limit high electrocatalytic activity. The thickness-dependent HER activity as observed for the CoBHT films is an experimental manifestation of how transport limitations can become dominant in electrodes for electrocatalytic HER.
In addition to the reports above, further example that benefit from decreased dimensions to lessen transport limitations have been reported. Taken advantage of this strategy, ultrathin nanosheet NiFe-MOF arrays, consisting of metal–oxygen-layers (MO6 units; M = Ni or Fe, in a Fe/Ni ratio of 23%) interconnected by 2,6-naphthalenedicarboxylate linkers, have been fabricated on different substrates through a dissolution–crystallization mechanism.356 The metal ions (Ni, Fe) are octahedrally coordinated, and each metal ion is coordinated to two trans monodentate carboxylates and four equatorial water molecules, while each naphthalene dicarboxylate bridges two metal atoms. These materials exhibit intriguing properties for electrocatalysis including highly exposed active molecular metal sites owning to ultrasmall thickness of nanosheets, improved electrical conductivity and a combination of hierarchical porosity. The NiFe-MOF array demonstrated electrocatalytic HER performance with a small overpotential of 134 mV at 10 mA cm–2.356 The same system was also found to be highly active for electrochemical oxygen evolution from a 0.1 M aqueous KOH solution.
Another example of a nanometer sized catalyst layer is a conductive Cu-MOF with HHTP linkers that was prepared as a shell around presynthesized Cu2O nanocube cores. Subsequent removal of the Cu2O core by a redox-etching approach in the presence of FeIII produced a thin Fe(OH)x shell underneath the surface Cu-MOF layer. The resulting Fe(OH)x@Cu-MOF nanoboxes catalyzed the electrochemical HER with low overpotentials, and X-ray absorption spectroscopy and DFT calculations indicated coordinatively unsaturated Cu–O centers as highly active catalytic sites that accelerate the formation of key *H intermediates toward fast HER kinetics.357
In 2020, Kang et al.358 reported the growth of the copper paddle wheel MOF Cu2(L) (H4L = 4,4′,4″,4‴-(1,4-phenylenebis(pyridine-4,2,6-triyl))tetrabenzoic acid) on a Cu-foam electrode by electrosynthesis in the presence of the ionic liquid EmimOAc (1-ethyl-3-methylimidazolium acetate). Owing to the electrosynthesis conditions, Cu2(L)-e/Cu featured structural defects throughout the MOF film, affording undercoordinated CuII centers which show strong interactions with CO2. Cu2(L)-e/Cu was able to reduce carbon dioxide to formic acid with a current density of 65.8 mA cm–2 and a FE of 90.5% for formic acid after 2 h. In comparison, Cu2(L)-t that was synthesized in a conventional solvothermal way and loaded onto carbon paper (CP) to form Cu2(L)-t/CP exhibited a considerably lower current density. Electrochemical impedance spectroscopy measurements revealed that Cu2(L)-e/Cu showed a significantly lower charge-transfer resistance compared to the Cu2(L)-t/CP, reflecting the microscopic morphology of Cu2(L)-e/Cu, that form a compact and thin coating of the Cu-foam electrode, instead of the Cu2(L)-t, which was prepared as larger crystals. In a follow-up paper, the same group further explored the technology to prepare catalytically active MOF materials by electrosynthesis. In that paper, a MFM-300(In)-e/In composite material was prepared by the electrosynthetic growth of MFM-300(In) on an indium foil in the presence of a biphenyl-3,3′,5,5′-tetracarboxylic acid linker.359 Owing to a templating effect by the ionic liquid EmimOAc that was used during electrosynthesis, the MFM-300(In) contained highly active In3+ defect sites. MFM-300(In)-e/In was found to be a good catalyst for the electrochemical CO2RR to formic acid, with a current density of 46.1 mA cm–2 at −2.15 V vs Ag/Ag+ and a FE of 99.1%. The high activity of MFM-300(In)-e/In is supported by a low interfacial charge-transfer resistance of 9.5 Ω cm–2.
Ye et al.360 reported the incorporation of a [Re(bpydc)(CO)3Cl] CO2 reduction catalyst into surface-grafted MOF (SURMOF) thin films on conductive FTO electrodes by a liquid phase epitaxy method. The thereby obtained catalytically active thin films were found to be highly oriented, grown exclusively along the [001] direction. Charge transport by a redox hopping mechanism along the [001] direction is particularly efficient, and consequently, the Re-SURMOF performed well as an electrocatalyst for the reduction of CO2 to CO.
In 2016, Zhao et al.361 developed ultrathin MOF nanosheets (UMOFNs), comprising Ni2+, Co2+, and BDC, for the electrocatalytic OER under alkaline conditions. The NiCo-UMOFNs showed high electrocatalytic activity as well as long-term stability. While installed on GCEs, the NiCo bimetallic UMOFNs required an overpotential of 250 mV to reach a current density of 10 mA cm–2. Loading the catalyst on copper foam enables decreasing the overpotential to 189 mV. At a constant overpotential of 250 mV, a highly stable current density was obtained for at least 200 h, with a high associated FE of 99.3%. Supported by, for example, X-ray spectroscopy and DFT calculations, the authors proposed that the surface atoms in the MOF nanosheets are coordinatively unsaturated and the active catalytic centers. Further was the electronic interplay between the Ni and Co atoms suggested to be crucial for the electrocatalytic activity. To substantiate this, the single-metal-binding motifs, Co-UMOFNs and Ni-UMOFNs, were prepared as well and showed lower activity.
In 2018, Zhu et al.362 employed exfoliated nanosheets from the conductive 2D-MOF Ni3(HITP)2 (HITP = 2,3,6,7,10,11-hexaaminotriphenylene) as an efficient CO2RR catalyst to produce CO in high selectivity of 97%. The reaction was light-driven and performed with [Ru(bpy)3]2+ as a PS and TEOA as an electron donor. The dimension of the Ni3(HITP)2 nanosheets provided high conductivity and highly accessible Ni–N4 active sites, giving rise to an efficient catalytic performance of Ni3(HITP)2. In 2015, Zhang et al.363 described the self-assembly of the flower-like bifunctional Ru-MOF, [Cd2(Ru(bpy-4,4′-dc)3)·12H2O]n, from nanosheets of the same material. The nanoflower Ru-MOF catalyzed the light-driven reduction of CO2 to formate in ca. 150% higher yields than solid-micro crystalline versions of the same Ru-MOF. Moreover, the nanostructure enhanced the stability of the agglomerated nanosheets, which gives rise to higher photostability and recyclability of the photocatalyst.
3.2.2. Engineering Transport Pathways
In the case when the reaction of energy relevance is driven by a chemical reductant or oxidant, these species have to physically diffuse into the MOF matrix to engage all catalysts in the reaction at hand. Similarly, electron donors or acceptors need to diffuse into the MOF in light-driven transformations that are initiated by PSs inside the MOF crystal, or photoactive MOFs. The transport of these sometimes rather large entities will more often than not result in severe transport limitations. Finally, also electrons and holes in electrocatalytic systems have to be transported to the active sites, and strategies to accelerate this process are in high demand. In enzymes, mass and charge transport often occur in well-defined channels364 and between redox-active cofactors,365 respectively. In the case of hydrogenases, the redox potentials of the [4Fe4S] clusters are adjusted to give the enzyme an inherent redox gradient that contributes to bias the enzyme toward either the anodic or cathodic reaction. Such a level of sophistication has not been realized in MOFs in which gradients may, however, develop dynamically under operation.60 The fundamental principles behind transport phenomena in MOFs are outlined in section 2.2 of this review.
In the case an oxidant or a reductant has to physically diffuse through the MOF crystal, ion pairing can lead to pore clogging. This phenomenon was shown on a NH-MIL-101(Cr) into which an [FeFe] hydrogenase active site mimic was incorporated. Electron transfer between the neutral [FeFe] site and neutral cobaltocene afforded the reduced [FeFe]− species and oxidized cobaltocenium ion, which formed ion pairs that were stable on the time scale of hours, as shown by altering IR spectra over these time scales. This effect was shown to be lessened in NH-MIL-101(Cr)-[FeFe] with lower [FeFe] loadings.198
The role of mass transport as a phenomenon potentially limiting the light-assisted HER has been addressed in a Ru-NH2-MIL-125(Ti) series of MOFs.366 Even though the exact molecular structure of the catalyst in these systems is not entirely clear, an interesting transport phenomenon was observed. In order to prepare these materials, pristine NH2-MIL-125(Ti) powder was treated with RuCl3·xH2O in a supercritical CO2/methanol solution. This treatment did not only introduce the Ru catalyst into the MOF but also led to a certain degree of hollowness of the Ru-NH2-MIL-125(Ti), as observed by TEM. The hollowness is attributed to the formation of mesopores in the MOF, a process that is only observed under supercritical fluid conditions in the presence of RuCl3·xH2O. The effect becomes increasingly pronounced as the processing time is increased. The presence of mesopores reduces the Brunauer–Emmett–Teller surface areas of the materials but increases their pore volumes significantly. From a biomimetic perspective, such pore enlargements could be compared to the presence of channels in enzymes. In comparison to NH2-MIL-125(Ti), into which a Ru nanoparticle had been loaded by conventional methods, the Ru-NH2-MIL-125(Ti) catalyst that was prepared under supercritical fluid conditions exhibited a higher light-driven HER activity in the presence of TEOA. While the incorporated Ru centers contribute to the electronic structure of the MOF, the highly mesoporous structure of Ru-NH2-MIL-125(Ti) also contributes to accelerating catalytic reaction by enhancing both the exposure of catalytic active sites and the facilitated transport of the TEOA donor.
In 2019, Zhong et al.367 synthesized a Cu phthalocyanine-based 2D conjugated MOF, PcCu-O8-Co, displaying a layer-stacked structure that featured Co-bis(dihydroxy) complexes (Co–O4) as linkages. When contacted on an electrode with carbon nanotubes (CNTs), PcCu-O8-Co is a decent catalyst for the ORR in alkaline solutions. A possible reason for the excellent performance was the presence of dimensionally controlled micropores (1.5 nm) in combination with mesopores (2–10 nm) in PcCu-O8-Co, both of which are favorable for substrate and electrolyte transport during the ORR.
Identifying factors that govern the diffusional charge hopping transport, as well as identifying means to accelerate this process has been subject of intense research,117 and reports in context of catalysis are reviewed hereafter. First, various reports address the dependency of material crystallinity and crystal orientation relative to the electrode surface as factors that govern diffusional electron hopping charge transport. Also here, parallels can be drawn between the presence and directionality of channels in enzymes, and diffusion pathways in MOFs that are disrupted when the material is poorly crystalline. It has also been shown that partial pore collapse at the crystal surface can lead to surface barriers that impede mass and ion ingress into the MOF,61 thereby slowing down catalysis. In 2020, Park et al.368 presented 2D MOF systems, consisting of M-N4 moieties (M = Ni, Cu) and hexaaminobenzene (HAB) linkers. The authors conducted studies to find correlations between crystallinity and conductivity, and the effect on the materials activities as ORR catalysts. Highly crystalline Ni-HAB-H exhibited high activity, enhanced stability, and better electron conduction compared to its low crystalline analogue (Ni-HAB-L), most likely due to amorphous features of Ni-HAB-L resulting in less exposed active sites and slow mass transport. Additionally, theoretical modeling suggested that along with metal cations as active center, the in-plane linker site can also act as catalytically active site for the ORR.
Meng et al.369 reported a porous Co framework ([Co1.5(tib)(dcpna)]·6H2O), based on 1,3,5-tris(1-imidazolyl)-benzene (tib) and 5-(3′,5′-dicarboxylphenyl)nicotinic acid (H3dcpna), as an OER electrocatalyst. A high OER activity was ascribed to 1D open channels along the b axis of the MOF, which exposed more active sites and facilitates electrolyte penetration. A comparison of the material with a speculative interpenetrated analogue could support this hypothesis.
The fact that charge hopping transport is coupled to diffusion–migration of charge-balancing electrolyte counterions has been found to also affect electrochemical catalysis. Such findings are indicative that charge transport is limiting in such systems. The first report that addressed this topic was as early as in 2011 by Nohra et al.370 The report is based on three MOFs that were prepared from POMs and tridentate 1,3,5-benzene tricarboxylate linkers, capped by Zn2+ ions. The channels of the POM-based MOFs (POMOFs) are occupied with tetrabutylammonium (TBA) counterions. It turned out that carbon paste electrodes of one of the POMOFs showed a remarkably high electrocatalytic activity in 1 M lithium chloride at pH = 1 (HCl). Lithium chloride as a supporting electrolyte was important for high current densities, as a change to sodium and potassium led to a decrease in activity by a factor of 2.8 and 2.5, respectively. The effect was ascribed to the sizes of the hydrated alkali ions as well as their propensity to carry water molecules into the POMOF. The relative hydrated ionic radii for alkali ions are in the order Li+ (340 pm) > Na+ (276 pm) > K+ (232 pm). Compared to the radius of the unhydrated TBA (494 pm) in the as-prepared POMOF, hydrated Li+ appeared as the cation most likely to ensure POMOF stability. In addition, the large and firmly hydrated Li+ ions will carry the largest number of water molecules into the structures, a circumstance that is favorable for HER electrocatalysis.
Another example of introducing electroactive units into a catalytic MOF to increase charge transport was reported by Mukhopadhyay et al.371 In this work, ZIF-8 functions as a host, Fe-salen as the active OER catalyst, and the Keggin SiW12 POM was incorporated to facilitate charge transport, as shown by lower overall electrical resistance of the resulting composite system.
Following a different strategy, Xin et al.372 reported the introduction of electron-rich metallocenes into the pores of MOFs by chemical vapor deposition. Following this strategy, a series of MCp2@MOF-545-Co (MOF-545-Co = Zr6O8(H2O)8(CoTCPP)2, M = Fe, Co, Ni) frameworks were prepared and were shown to be effective CO2RR electrocatalysts. The MCp2 moieties serve as additional electron hopping transport sites, creating electron transport channels within the MOF toward the active metalloporphyrin sites (Figure 36). In addition, the presence of the metallocenes in close proximity to the MOF-linkers leads to a reduced CO2 adsorption energy, as revealed by DFT calculations, and thereby contributes to an enhanced electrocatalytic performance.
Figure 36.

Enhanced electron transfer in MCp2@MOF through the introduction of electron-rich metallocenes, promoting the electrocatalytic CO2 reduction. Adapted with permission from reference (372). Copyright 2020 Elsevier.
While the above papers relied on charge transport by a diffusion-like charge hopping mechanism between discrete redox active units, reminiscent of charge transport between redox active cofactors in enzymes, the following papers rely on charge transport by different mechanisms. In 2018, Dincǎ and co-workers373 reported 2D hexagonal MOFs, Ni3(HITP)2, and Cu3(HHTP)2 along with Co3(HHTP)2 and Ni3(HHTP)2 as trigonal MOFs with high intrinsic band-like conductivity. Such semiconductor-like charge transport is unprecedented in nature, but conceivable in MOFs due to extended conjugation paths. The dependence (nonzero order) of ORR overpotential on [H+] during reduction with the hexagonal MOFs suggested that electron transfer during ORR is proton-coupled while trigonal MOFs lack the electron delocalization in the ab plane and hence disruption of the π-stacking in the c direction was observed.
In 2021, Zhao et al.374 reported a π–d conjugated truxone–Cu MOF that also showed band-like conduction with semiconductor characteristics. The 2D MOF, consisting of 2,3,7,8,12,13-hexahydroxyl truxone linkers interconnected by copper ions, showed typical characteristics of a semiconductor with an intrinsic conductivity of 4.0 mS cm–1 at 30 °C and a small energy gap of 0.24 eV as determined by DFT calculations. Owing to its good electrical conductivity and redox reversibility of both the truxone and the metal center, truxone–Cu MOF-modified electrode was successfully applied to catalyze the ORR.
In 2020, Wang et al.375 reported coordinatively unsaturated Co-BTC-IMI MOFs with high intrinsic activity for both the ORR and OER. The material consisted of BTC linkers that interconnected the CoII cations, aided by imidazoles that are integral to the structure, mainly incorporated by π–π interactions with the benzene rings of the BTC linker. The high activity of Co-BTC-IMI for the electrocatalytic ORR and OER was ascribed to accessible coordination sites at the catalytic Co centers as well as good electric conductivity that is promoted by the through-space π–π interactions.
In 2018, Wang et al.376 presented a series of POM-porphyrinic MOFs, so-called PMOFs, synthesized by a hydrothermal method that links redox active Zn-ε-Keggin clusters (ε-PMoV8MoVI4O40Zn4) as SBUs with metalloporphyrin linkers. The combination of these two structural units in this PMOF leads to outstanding electron transport properties. Of the metalloporphyrin linkers tested, the Co-porphyrin Co-PMOF exhibited the most impressive CO2RR performance with a TOF of 1656 h–1 and a faradaic efficiency for CO of >94%. Remarkably, Co-PMOF catalyzed the electrochemical CO2RR to CO at a constant current density of 17 mA cm–2 for 36 h without any significant drop in stability.
In 2020, Das and co-workers371 described the in situ coencapsulation of the WOC catalyst Fe-salen (i.e., Fe(salen)Cl) and the Keggin POM SiW12 (i.e., H4[SiW12O40]) into the pores of the zeolitic network ZIF-8 to form FSWZ-8 ([Fe(salen)(OH)]+H4[SiW12O40]·HCl)@ZIF-8), an efficient and robust electrocatalyst for the OER at a neutral pH. Incorporation of SiW12 resulted in accelerated formation of FSWZ-8, higher catalyst loadings, and in the lowering of the required overpotential for the electrochemical OER by more than 150 mV. Furthermore, the POMs lower the electrical resistance and hence ease the charge transport. The composite system exhibited high TOFs of around 5 s–1 and FE of overall 94% during electrocatalytic water oxidation.
Catalysis of energy relevance inevitably involves protons. In order to not become limiting, their facile transport through the MOF crystal is of vital importance. In fact, MOFs as proton conductors is a field of research in itself, with some excellent reviews having emerged recently.377,378 One interesting example is that of UiO-66-NH2 that was decorated with sulfonate groups.379 Two materials were prepared that differed by the length of the carbon chain between the −SO3H group and the amino group of the BDC-NH2 linker. It was found that the material with the shorter C2H4 linker exhibited a record proton conductivity of 1.64 × 10–1 S cm–1 at 80 °C, while that of the C3H6 analogue was 4.6 × 10–3 S cm–1. Even though this report does not directly address catalysis, it teaches an important lesson in that modifications that may seem rather insignificant (ethyl versus propyl tether) can hugely impact performance. In the present case, if catalysis had been limited by proton diffusion, choosing the right alkyl sulfonate tether would have increased the material’s TOF.
The effect of proton sources on the electrocatalytic ORR was also investigated by Morris and co-workers in 2017.380 Using PCN-223-Fe, a MOF consisting of Zr6-oxo clusters and FeIII-porphyrin linkers, which was synthesized on a conductive FTO substrate, it could be shown that the ORR activity increased with increasing strength of the acid that was added to the electrolyte solution. Interestingly, the use of the stronger trifluoroacetic acid gave a lower product selectivity than that promoted by the weaker acetic acid. For the latter, the amount of H2O2 was less than 6%, while the former acid gave rise to 34%. The results show the importance to not only supply protons to the catalytic centers at appreciable rates, but also at appropriate activity to warrant good product selectivity.
One example exists wherein two isostructural MOFs that differ in the constituting metal of the SBU (zinc or cobalt) were prepared and directly compared for the OER.381 Notably, the FJU-82-Co had a ca. 300 mV lower overpotential relative to the FJU-82-Zn analogue, yet had only a slightly higher electronic conductivity (1.96 × 10–12 vs 1.67 × 10–12 S cm–1, respectively). Subsequent measurement of the two MOFs by alternative current impedance spectroscopy revealed that FJU-82-Co had a proton conductivity 1 order of magnitude larger than that of the Zn analogue. While Zn has a lower intrinsic OER activity than Co, the authors ventured that since the OER mechanism involved release of a proton, the rate of the OER could be limited by proton transport out of the MOF. FJU-82-Co had a larger unit cell, suggesting that solvated protons would diffuse more quickly than within FJU-82-Zn. While the authors did not measure the diffusion rate of water (the OER substrate) in their two MOFs, it is possible that the water diffusion rate was also larger for FJU-82-Co in addition to having a superior proton conductivity.
Furthermore, hydrogen bonding within MOFs for OER has been observed and postulated. The single crystal structure of the cobalt coordination polymer Co-HL ([(Co2(HL)(H2O)5)3H2O]n), synthesized from H5L (= 5,5′-[(5-carboxy-1,3-phenylene)bis(oxy)]diisophthalic acid), exhibited a clearly visible hydrogen bonding network (Figure 37).382 Comparison of the linear sweep voltammograms of this coordination polymer with its deuterated analogue Co-DL revealed a lower overpotential requirement for Co-HL, strongly suggesting that the hydrogen bond network near the presumed active metal sites was directly involved in OER, thereby being reminiscent to enzymatic chemistry. Related work has found further evidence of proton shuttling and proton binding in MOFs for the OER.383−385
Figure 37.

Crystallographic structure of the hydrogen bonding network of the coordination polymer Co-HL. Atom labeling: Co, green; C, gray; H, white; O (ligand), red; O (water), pink. Adapted with permission from reference (382). Copyright 2020 American Chemical Society.
A metal–organic cage (MOC) [Pd6(RuL3)8]28+ (MOC-16; L = 2-(pyridin-3-yl)-1H-imidazo[4,5-f][1,10]-phenanthroline) consisting of six Pd vertices and eight Ru2+-based photosensitizing units that cover one face of the cage each has been incorporated into the Zn2+/methylimidazole (MeIm)-based ZIF-8.386 The ZIF-8 is constructed around the MOC through coordination-assisted secondary assembly, where MOC-16@ZIF-8 precursors are initially grown from a mixture of MOC-16, Zn2+, and 2-MeIm, which then crystallize to the MOC@MOF. Subsequent partial substitution of MeIm in the ZIF-8 by carbonate ions gives rise to CZIF with molecularly intact MOC-16 in the CZIF scaffold. The transformation of the ZIF-8 to the CZIF is of crucial importance for the photocatalytic HER activity of the MOC. While the ZIF-8 matrix is highly hydrophobic and does not support light-induced HER in the presence of electron donors, the hydrophilicity that is introduces into the CZIF matrix by the constituting CO32– ions guarantees sufficient proton transport to the catalytic sites. In other words, the hydrophilic character of the carbonate ions in the CZIF structure is integral to a H-bonded water network that assists in proton transport to catalytic sites.
3.3. Cooperative Effects
In this section, original reports are reviewed in which the active site interacts with the surrounding MOF matrix in a way that alters the catalytic mechanism as compared to the situation in homogeneous solution phase. Specific catalyst–matrix interactions that, for example, stabilize transition states in the catalytic cycle, offer alternative mechanistic routes, or promote proton-coupled electron transfers pathways are being discussed. Such cooperative effects go beyond “simple” stabilization of the molecular integrity of the catalyst and intermediates as discussed in section 3.1, and are one of the strongholds of enzymatic chemistry. The latter is famous for intricately designed active site pockets, within which amino acid side chains may stabilize transition states or temporarily host substrates like protons. In the context of MOFs, careful design can enable hydrogen bond formation and noncovalent interactions through changes in the microenvironment within the pores to activate substrates, stabilize intermediates, and thereby enhance catalytic efficiencies. The section also includes reports of SBU-based catalysts, the thermodynamics of which are altered by the surrounding MOF matrix, as manifested, for example, by linkers that enforce a coordination geometry on the SBU that is different to the relaxed structure in solution. Finally, this section also contains reports in which the MOF matrix is shown to impart a more global effect on the molecular catalyst, for example by controlling solvation or concentrating substrate, which are effects that resemble outer coordination sphere effects in enzymes.
3.3.1. Microenvironment-Induced Tuning of Catalyst Performance
Historically, MOFs have first been investigated for their gas sorption activities. It is thus not surprising that this effect has also been explored in the context of CO2RR activity. Kajiwara et al.387 demonstrated the synergistic effect between gas adsorption properties and catalytically active sites in a UiO-67-based MOF, modified by the CO2 reduction catalyst [RuII(H2bpydc)(terpy)(CO)]2+ (H2RuCO). The catalytic activity of the obtained Zr-bpdc/RuCO composite was not only comparable to the corresponding homogeneous system, but was in contrast maintained even under low CO2 pressure of a 5% CO2/Ar gas mixture, presenting an important opportunity for the use of low-concentration CO2 streams as C1 sources.
In 2020, Zhang et al.388 described the postsynthetic modification of the 2D MOF nanosheets Zr-BTB by anchoring the cobalt porphyrin-derived catalysts CoTCPP to the Zr6 cluster SBUs. The resulting material, CoTCPP/Zr-BTB, was a good catalyst for the electrochemical reduction of CO2 to CO, significantly outperforming the molecular reference system. The microenvironment around the CoTCPP catalysts was further tuned by postsynthetic coordination of a selection of carboxylate-containing ligands to the SBUs. Of the modified 2D MOFs that were tested, the ones with p-sulfamidobenzoicacid (PSABA) ligands, CoTCPP/Zr-BTB-PSABA, showed the best performance with FECO that increased by about 10% compared to the system that lacked the additional carboxylate ligand. This increase in FECO is accompanied by a decrease of the material’s HER activity, suggesting that HER pathways at the SBUs are shut down by the postsynthetic ligation, thereby improving the utilization of electrons for the CO2RR.
Huang et al.338 reported the synthesis of a series of 3D pillared-layered monometallic (SyA-Co and SyA-Fe) and bimetallic CoFe MOFs with syringic acid (SyA) linkers. It was found that the optimal Co:Fe ratio for the performance of the materials as electrochemical OER catalyst was 2:1 (SyA-Co2Fe). Besides the series of pristine MOFs, an N,S codoped version of the CoFe-MOF was prepared by adding thiourea into the solvothermal mixture. These N,S codoped materials, (SyA-Co2Fe-ST), showed a remarkable increase in the OER performance compared to the pristine MOF SyA-Co2Fe, with low overpotentials and a long-term stability for at least 16 h. The improved electrocatalytic activity of SyA-Co2Fe-ST was mainly attributed to in situ surface modifications that alter the pore structures, tune the electronic and surface structures of the catalysts, while also promoting the formation of new active species and facilitating charge and mass transport for the OER process.
Zhong et al.389 demonstrated a synergetic effect in a series of layer-stacked 2D PcM-O8-M (Pc = phthalocyanine; M = Cu or Zn) frameworks that can be exploited for the electrocatalytic CO2RR to CO. These 2D MOFs consist of zinc/copper phthalocyaninato linkers and zinc/copper-bis(dihydroxy) complex (ZnO4) SBUs. Spectroscopic studies combined with DFT calculation revealed that the ZnO4 complexes of PcCu-O8-Zn exhibit high catalytic activity for CO2-to-CO conversion, while CuN4 complexes in the Pc macrocycles act to promote proton and electron transfer during the reaction process. PcCu-O8-Zn in combination with carbon nanotubes (PcCu-O8-Zn/CNT) exhibited highly selective catalytic activity for CO2-to-CO conversion (88%), high TOFs, and excellent stability. By varying the metal centers (Cu and Zn) of ligand/linkage as well as applied potentials, the H2/CO ratio could be tuned from 1:7 to 4:1. Interestingly, the authors found that the CuN4 linker in PcCu-O8-Zn shows the lowest energy barriers for HER, while also promoting the lowest free energy for the generation of rate-determining *COOH intermediate at the ZnO4 SBU. It is this synergistic effect that is suggested to result in the good CO2RR activity of PcCu-O8-Zn/CNT.
3.3.2. Lattice Effects
Lattice effects have been observed in the photochemical and photoelectrochemical OER using MOFs. In one example, the HOMO–LUMO gap of the triphenylene-2,6,10-tricarboxylic acid (H3TTCA) organic linker decreased upon MOF incorporation (LUMO = lowest unoccupied molecular orbital).290 On its own, H3TTCA is not catalytic for oxygen evolution, but once inside a MOF it could be photoexcited and drive photochemical OER. In a related example, the HOMO of the porphyrin linker within a MOF-545 hybrid material increased in potential relative to that of its homogeneous analogue; the resulting increased HOMO–LUMO gap provided more driving force for the OER.245
Lattice strain has also been utilized to tune the electronics for MOF-based OER and ORR. In 2019, Cheng et al.390 compared the OER and ORR activity of a NiFe MOF to its lattice-strained congeners (Figure 38). Increasing duration of ultraviolet irradiation yielded more strained NiFe MOFs. Analysis of the resulting materials suggested that increased strain upshifted the oxygen 2p level, resulting in a stronger covalent interaction between Ni and oxygen. Comparison of their linear sweep voltammograms revealed that increases in strain directly decreased OER overpotentials, and the most strained NiFe MOF had better durability than the noble metal RuO2 during long-term bulk electrolysis. The lattice-strained NiFe-MOF facilitated fast and efficient 4-electron pathways for both the ORR and OER compared to the low-efficiency 2-electron catalytic kinetics for pristine MOFs. This use of the MOF lattice to tune the catalytic site electronics is an excellent example of how the actual MOF structure can be leveraged to improve catalysis, much as how the protein environment around an enzyme’s active site is tuned to support catalysis.
Figure 38.

Structure of lattice-strained NiFe MOF. Adapted with permission from reference (390). Copyright 2019 Springer Nature.
In 2020, Yang et al.391 demonstrated the influence of lattice strain on the electrocatalytic CO2RR activity of a Zn-porphyrinic MOF. The strain was introduced by a vaper-phase infiltration (VPI) method in the gas phase. First, the pristine porphyrinic framework MOF-p was synthesized starting from a carbon fiber electrode onto which a thin layer of Al2O3 had been deposited by ALD, followed by its treatment with H2TCPP. Subsequently, Zn was introduced into the porphyrinic linkers of pristine MOF-p by VPI with diethyl zinc and H2O (10 cycles) to afford MOF-Zn-inf-10c. To induce lattice strain, additional VPI cycles (x = 20, 40, 60, or 80 cycles) resulted in the formation of additional ZnO clusters within the MOF pores (MOF-Zn-inf-xc). XRD measurements revealed that increasing number of VPI cycles (0–60 cycles) correlated with shifts of the characteristic (201) peak, offering the possibility to fine-tune the internal strain of the resulting MOF materials. The effect of the lattice strain on the intrinsic catalytic activity of the ZnTCPP sites was investigated for the electrochemical CO2RR toward CO in a CO2-saturated solution (DMF/H2O, v/v = 9:1). Out of the afforded infiltration samples, MOF-Zn-inf-60c outperformed the other VPI samples with an overpotential positively shifted by 200 mV and a maximum FE close to 100% at −1.8 V vs SHE (standard hydrogen electrode). Increasing the VPI cycles from 20 to 60 increased selectivity and was argued to be directly linked to the generation of internal strain through gradually filling up of the pores with ZnO clusters.
3.3.3. Discrete Cooperative Effects between Catalyst Linkers and Surrounding MOF Matrix
A discrete cooperative effect between two platinum centers during the HER was reported for Pt-MOF-253.392 The structure of the MOF is such that its Al-based SBUs position bpydc linkers on top of each other. Upon metalation with PtCl2, the Pt(bpydc) metallo-linkers in the resulting Pt-MOF-253 act both as a PS as well as a HER catalyst. Interestingly, the Pt-MOF-253 catalyzed the light-driven HER in the presence of TEOA as electron donor circa five times faster than the corresponding reference system with Pt(bpydc)Cl2 in homogeneous solution phase. While different factors may be at play that explain the difference in activity, one contributing factor was suggested to be a cooperative effect between two Pt centers at adjacent bpydc linkers. It was proposed that upon excitation and reductive quenching of one metallo-linker, a PtIII-hydride intermediate is formed by an oxidative addition pathway. This species interacts with an adjacent PtII metallo-linker to form a mixed-valence hydride-bridged diplatinum(II,III) intermediate. This hypothesis is supported by low temperature luminescence, studies and extended X-ray absorption fine structure data that suggest a short Pt···Pt distance in Pt-MOF-253. The interpretation of the diplatinum intermediate is further supported by the observation that the initial H2 evolution rate exhibits a first order dependence on the catalyst concentration at low catalyst loading, while it deviates from a linear relationship upon higher Pt concentrations. The authors, however, also point out that this effect may be due to inefficient reductive quenching that becomes more severe under high Pt conditions.392
Another example of catalyst linker interations was reported by Cichocka et al.393 in 2020. A new kind of porphyrinic MOF, denoted as PCN-226, was prepared from CoTCPP linkers and Zr-oxide chain structures. The latter give rise to good chemical and redox stability of the MOF, and, unlike other Zr-based PCN MOFs, generate a compact packing structure of the porphyrin linkers. As a result, PCN-226(Co) had a superior ORR performance compared to previously published cluster-based MOFs. According to theoretical calculations, the spatial arrangement in PCN-226(Co) brings porphyrin molecules within a distance of 7 Å, which is favorable for the adsorption of *O, *OH, and *OOH intermediates. The chain structure not only improves the overall stability of PCN-226(Co) but also allows the redox-active centers to engage in high reaction kinetics.
Another cooperative effect was reported by Lin and co-workers108 who described the postsynthetic SBU decoration of 2D TPY-MOL ([Hf6(μ3-O)4(μ3-OH)4(HCO2)6(terpy)2] with cobalt-protoporphyrin (CoPP) units via carboxylate exchange. The thus obtained TPY-MOL-CoPP features basic pyridine sites as part of the terpyridine-based linkers and the CoPP catalyst anchored to the SBU, but otherwise pointing into the MOF pore. In this spatial arrangement, CO2 binding to the CoPP is aided by protonated pyridine moieties (pyH+) to coactivate CO2 by the formation of the [pyH+-–O2C-CoPP] adduct (Figure 39). This interaction facilitates the CO2RR over the HER, hence leading to a CO/H2 selectivity of more than 80%. The control experiment with BTB-MOL-CoPP that lacks the pyridine moieties, and thus the hydrogen bond interactions, resulted in a lower CO/H2 selectivity of 2.7:1, illustrating the effect of the catalytic pocket.
Figure 39.

Near proximity between the pyridinium moiety and the CoPP active site leads to a CO2-activating microenvironment within TPY-MOL-CoPP. Reproduced with permission from reference (108). Copyright 2019 American Chemical Society.
In 2017, Ryu et al.394 reported a series of amino-functionalized Re-containing frameworks, comprising the ReI(CO)3(bpydc)(Cl) metallo-linker and the amino-containing bpdc-(NH2)2 linker within a UiO-67 scaffold. The system was investigated for the light-driven conversion of CO2 to CO, using TEA as an electron donor. Of the systems prepared, the one in which about 30% of the linkers contained the amino substituents, Re-MOF-NH2(33%), showed the highest activity for photocatalytic CO2RR, with a 3-fold increase in comparison with Re-MOF without any amino groups. Supported by DFT calculations, the authors proposed a number of cooperative effects between the Re-based metallo-linker and adjacent amino groups to explain this improved performance (Figure 40a). At the start of the catalytic cycle (Figure 40b), the amino-group can reversibly react with CO2 to form a carbamate, where the OH of the carboxylic group forms a weak hydrogen bond with the chloride of the Re complex. Following light absorption and reductive quenching by the TEA donor, the Re center attacks the electrophilic carbonyl carbon to form the metallo-carboxylate along with the regeneration of the amino group. It is notable that the Re center at this stage is seven-coordinate with the chloride being H-bound to the amino-group. Upon a second light absorption/reductive quenching cycle, the metal-bound COOH is transformed to CO and water as a byproduct.
Figure 40.

a) Structure and cooperative interaction between an amino-functionality of H2bpdc-(NH2)2 and H2ReTC within Re-MOF-(NH2) (X%), employed in the photocatalytic CO2-to-CO conversion. Atom labeling: C, black; O, red; Zr, blue polyhedra; Re, yellow; Cl, green. H atoms are omitted for clarity. b) Proposed mechanism for the amine-assisted conversion of CO2 to CO based on DFT calculations. Adapted with permission from reference (394). Copyright 2017 Springer Nature.
Mao et al.395 reported the synthesis of a UiO-66-derived electrocatalyst into which FeTCPPCl ((5,10,15,20-tetrakis(4-carboxyphenyl)porphyrinato)-FeIII chloride) linkers had been incorporated. The resulting FeTCPP⊂UiO-66 consists of Zr-oxo clusters interconnected by BDC and FeTCPPCl and was demonstrated to catalyze the electrochemical CO2RR in an aqueous solution. The UiO-66 framework provides protons at the Zr-oxo clusters that act to counterbalance negative charge that emerges at the porphyrin-bound carboxylates during electrochemical CO2 reduction, thereby enhancing the CO2 reduction activity of the iron porphyrin linkers. The authors considered a concerted proton-coupled electron transfer mechanism, which contributed to the high FECO of ∼100% at a low overpotential.
In 2016, Dincǎ and co-workers118 reported a 2D MOF, namely, Ni3(HITP)2, with a high intrinsic conductivity of 40 S cm–1. Thin films of Ni3(HITP)2 on glassy carbon rotating disk electrodes were found to catalyze the electrochemical ORR in a 0.1 M KOH solution with decent overpotential, affording predominantly H2O2 (87.5%) in a two-electron pathway. Further systematic theoretical investigations by Sun and Chen396 dealt with the elucidation of the reaction mechanism of Ni3(HITP)2 and the origin of the high electrocatalytic ORR activity. In addition to the expected Ni–N catalytic site, it was found that the hydrogens at the nitrogen heteroatoms of HITP also serve as an active site for the ORR (Figure 41). This site showed a higher adsorption energy for O2 compared to the Ni–N site, while Mulliken population analysis revealed that the oxidation state of the HITP ligand can be altered during the ORR.
Figure 41.

Relative energy diagram for the ORR pathway where oxygen is adsorbed in a bridged configuration between the two protons at the nitrogen centers of HITP. Reprinted with permission from reference (396). Copyright 2017 Elsevier.
In 2019, Li et al.397 presented the solvothermal synthesis of two Co-based MOFs, [Co2(HAD)2(AD)2(BA)]·DMF·2H2O (AD-MOF-1; HAD = adenine and BA = butanedioic acid) and [Co2(HAD)2(AD)2(IA)2]·DMF (AD-MOF-2; IA = isobutyric acid) and investigated their activities for the light-driven reduction of CO2 to formic acid. Under illumination and in the presence of triisopropanolamine as electron donor, AD-MOF-2 displayed a HCOOH production rate in aqueous media more than twice as high as that of AD-MOF-1 in acetonitrile. Experimental and theoretical studies revealed that light-absorption is localized at the adenine, and following reductive quenching, also CO2RR catalysis occurs at this site, rather than at the Co centers. During the catalytic cycle, CO2 binds to the aromatic nitrogen atom of the adenine moieties, assisted by the amino group in ortho-position to the nucleophilic adenine-N.
3.3.4. Discrete Cooperative Effects between SBU-Based Catalysts and Surrounding MOF Matrix
An interesting report describing how MOFs can not only stabilize unusual active sites but also alter their reactivity in the light-driven HER was reported for mixed-node MOF-74(Fe,Co).398 The material consisted of Co/Fe mixed-metal SBUs that are interconnected with 2,5-dihydroxyterephthalic acid linkers. Using in situ X-ray absorption spectroscopy, it was possible to elucidate details of the catalytic mechanism, and the chemistry around the Co and Fe centers in MOF-74(Fe,Co). Under standard catalytic conditions in the presence of [Ru(bpy)3]Cl2 and TEOA, it is shown that both Co and Fe centers in their reduced oxidation states are catalytically active sites. Interestingly, it is shown that the accumulation of these states is responsible for the induction period that is observed before continuous hydrogen evolution. Under steady-state HER conditions, the SBU features a Co center with an elongated Co–O bond distance, while the Fe center adopts a more distorted octahedral geometry compared to the situation in the as-synthesized geometry. According to the authors, it is this cooperative effect of the two metal centers that leads to these structural changes upon reduction, and that is responsible for the high HER activity of the mixed-metal MOF-74(Fe,Co).
Light-driven HER that is catalyzed by the cooperative action of two low-valent Cu ions that represent the SBU of a MOF has also been reported.399 Starting from the as-prepared [Cu2(μ-Cl)2(bbta)] MOF (Cu2–Cl2-bbta or MAF-X29, H2bbta = 1H,5H-benzo(1,2-d:4,5-d′)bistriazole) in which the CuII centers are bridged by charge balancing μ-chloride ions, light-induced reduction leads to decoordination of the chloride ligands, and the formation of the catalytically active dinuclear CuI–CuI site to form Cu2-bbta (Figure 42). The ligand exchange that is triggered by Cu-reduction was followed spectroscopically, and Cu2-bbta is found to be a competent catalyst for the light-driven HER, using [Ru(bpy)3]Cl2 as a PS and TEOA as an electron donor. In accordance with DFT calculation on the catalytic mechanism, the high activity of Cu2-bbta is attributed to a cooperative effect in which the two Cu ions jointly activate water in a pathway with a comparably low-energy potential energy surface, ultimately leading to the Cu hydride intermediate. Further experimental proof for the cooperative effect is obtained from the reference material Cu-btdd (H2btdd = bis(1H-1,2,3-triazolo-[4,5-b],[4′,5′-i])dibenzo-[1,4]-dioxin) with a single Cu catalytic center that exhibits a significantly lower HER activity.
Figure 42.

Cooperative effect between two CuI centers to active water under the formation of a Cu–H state. Reprinted with permission from reference (399). Copyright 2020 Royal Society of Chemistry.
Another example of a MOF with SBUs consisting of a dinuclear Cu-site was reported as a photocatalytic HER material. In this report, Cu-I-bpy, a MOF consisting of discrete Cu2I2 clusters that are interconnected with 4,4′-bipyridine linkers acts as both a light absorber and a catalyst.400 Following light absorption, reductive quenching by the TEA electron donor led to the reduction of the CuI centers. The thereby produced Cu0 state reacts with protons to afford CuI hydride species that are crucial intermediates for HER. Interestingly, the distance between two CuI centers of the Cu2I2 cluster is appropriate for the interaction between two CuI hydrides, offering a pathway for facile hydrogen generation.
Sargent and co-workers401 demonstrated in 2018 that the product selectivity of HKUST-1 as a catalyst material for the CO2RR can be modulated by controlling the coordination number and geometry of the constituting paddle-wheel Cu dimer SBU. The latter was distorted to an asymmetric motif by separating adjacent benzene tricarboxylate moieties using a thermal treatment, thereby altering the local atomic structure, oxidation state, and bonding strain of the Cu dimers. Using EPR and in situ X-ray absorption spectroscopy, Cu clusters with low coordination numbers were observed, giving rise to an unusually high FE of 45% for ethylene production.
A cooperative effect at Co-based SBUs was identified by Wang et al.402 when investigating a series of cobalt-based MOFs (MAF-X27-Cl, MAF-X27l-Cl, MAF-X27-OH, MAF-X27l-OH, MOF-74-Co, and Co-ZIF-9) for their activity in the photocatalytic CO2RR in conjunction with [Ru(bpy)3]Cl2 and TEOA. The MOFs have in common that they contain a catalytically active Co site but differ in the surrounding coordination environment, most importantly in the presence or absence of μ-OH– ligands at the cis-positions neighboring the Co centers. Under 1 atm CO2 and identical photocatalysis conditions, the MOFs with μ-OH– ligands showed the best CO selectivity and TOF. In these MOFs, the TOFs are only marginally reduced when the CO2 partial pressure was decreased to 0.1 atm, clearly pointing to a cooperative role of μ-OH– ligands. Indeed, computational results suggested that the μ-OH– ligands acted as hydrogen-bonding donors to stabilize the Co–CO2 adducts and served as proton sources to promote the C–O bond dissociation (Figure 43).
Figure 43.

Perturbation DFT-derived CO2 binding structure for the reduced forms of MAF-X27-OH, showing the cooperative environment of the μ-OH– ligands, stabilizing the Co–CO2 adducts and promoting the C–O bond dissociation. Adapted with permission from reference (402). Copyright 2018 American Chemical Society.
In 2019, Lian et al.403 reported a study on M3HITP2 (M = Co or/and Ni) conductive coordination polymers with different Co/Ni ratios. The nonpreferred interaction of the HITP linker with Co2+, the reduced crystallinity, and the π–π layering of Co3HITP2 predicted a deformed quadrilateral configuration and a lack of coplanarity due to the unpaired electron in the 3dz2 orbital. This distortion limits charge transport through space, resulting in a significant reduction in electric conductivity. Despite the lower conductivity, Co3HITP2 displayed outstanding electrocatalytic ORR and OER activity, which can be attributed to the more active metal center with unpaired 3d electrons. Importantly, the ORR mechanism was observed to undergo a transition from the two-electron pathway on Ni3HITP2 to the four-electron pathway on Co3HITP2.
3.4. Contacting
While examples in the preceding sections have concentrated on effects within the MOF crystals, this section clearly takes a system perspective on catalytic efficacy and focuses on the importance of the MOF/electrode interface. Reports are reviewed in which this interface has been addressed specifically, and in many cases optimized for improved performance. The section bears certain analogies to protein film electrochemistry,404,405 and the use of enzymes for potential bioelectrocatalysis applications.406 In the latter contexts, it has been shown that the enzyme/substrate interfaces are of vital importance.66 For example, in case of 2D electrode substrates, it is important for electrocatalytic function that the enzyme is immobilized in a way that enables interfacial electron transfer at sufficiently high rates. This entails that enzymes need to reside on the electrode in a way that either the active site itself, or the most outer unit of an electron transport chain is in close spatial proximity to the electrode surface. Consequently, multiple strategies to “wire” enzymes to electrode surfaces have been developed.407 In particular, increasing the physical contact area between enzyme and electrode by nanostructured substrates have the potential to give rise to enhanced current densities.408 Further efforts to enlarge electrode surface areas, often based on carbon nanotubes, have been reported to increase enzyme loadings and current densities.409
Considering the above, it is not surprising that the performance of MOF-based electrocatalytic systems also exhibits a dependence on the immobilization and contacting methods. This was illustrated in a comparative study on a UiO-66 sample into which model complexes of the [FeFe] H2ase active site had been incorporated. Due to the low abundance of the [FeFe] metallo-linker, the resulting UiO-66-[FeFe] was poorly conducting, and only close-to-surface [FeFe] sites could electrochemically be addressed.410 It was shown that 3D electrodes based on multiwalled CNTs inks best engulfed the MOF particles, giving rise to the highest current responses of the investigated substrate anchorage strategies.
3.4.1. MOFs in 3D Electrodes
Carbon nanotubes (CNTs) have been used extensively together with catalytic MOFs as conducting components to produce inks that are then deposited on electrode surfaces for electrocatalysis experiments. In this context, Huang et al.411 reported a systematic study on the optimal ratio between catalytic MOF materials and contacting CNTs. In this contribution, an iron triazolate framework FeTa2 and its performance as composite with varying amounts of conductive Ketjenblack (KB) carbon for the ORR in alkaline electrolyte was investigated. Maximum current densities were observed in FeTa2-KB composites that contained 50 wt % of the FeTa2 catalyst. The authors hypothesized that this composition is the “sweet spot”, balancing maximum catalyst loading and conductivity, provided by FeTa2 and KB, respectively.
A similar observation was reported by Dong et al.412 in a study on the Fe porphyrin-based framework PCN-222(Fe). Mixed in different ratios with carbon black and deposited onto carbon paper through a dip-coating method, the resulting PCN-222(Fe)/C composites were investigated for electrocatalytic CO2-to-CO reduction activity. The composites showed decreased activity the higher the catalyst content was (PCN-222(Fe):C > 1:1), consistent with poor MOF crystallite contacting with low amounts of carbon black. Interestingly, also product selectivity depended on PCN-222(Fe)/C composition, with the materials with better PCN-222(Fe) contacting (PCN-222(Fe):C < 1:1) showing less HER activity.
MOFs are, however, not only coimmobilized with CNTs but also grown directly onto them. In 2016, Fang et al.413 reported such a hybrid material when growing a cobalt-containing zeolitic imidazolate MOF on CNTs to produce Co-MOF@CNTs. The authors showed an efficient “wiring” of the Co-MOF through the CNTs, and that the addition of CNTs can improve the conductivity of the composite. Consequently, the Co-MOF@CNTs showed OER and ORR catalytic activity comparable to that of commercial RuO2 and 20 wt % Pt/C catalysts, respectively, while also exhibiting good stability.
Lin and co-workers414 described a Hf12-CoDBP/CNT composite by growing a MOF that consisted of a Co-porphyrin-type linker (DBP = 5,15-di(p-benzoato)-porphyrin) and a Hf12-based SBU on CNTs. It was shown that the covalent attachment of the MOF nanoplates to conductive CNTs improved interfacial electron transfer from the electrode to the Co-porphyrin active sites, which in turn are involved in diffusional electron hopping charge transport. The Hf12-CoDBP/CNT assembly electrocatalyzed the HER with a TON of 32 000 in 30 min, corresponding to an apparent TOF of 17.7 s–1.
Going beyond the “wiring” function, Ma et al.415 recently illustrated that different conducting substrates can alter product selectivity in the ORR reaction. For this purpose, Co-MOF comprising Al octahedral nodes connected by CoTCPP linkers were grown on CNTs and reduced graphene oxide (rGO). The two different carbon supports promote film growth of Co-MOF stacks with different surface orientations, which in turn caused differences in conductivity normal to the substrate surface. The Co-MOF nanoplates on rGO exhibit a lower overpotential for the ORR, but more importantly also a different product selectivity. By changing from the CNTs to the rGO support, the electrocatalytic mechanism of the ORR is also altered from the two-electron reduction to the four-electron reduction pathway.
3.4.2. 3D Electrodes from 2D Layered Architectures
For long-term durability of future devices, the structural stability of the electrode/MOF interface is of high importance. Already in 2014, Jiang et al.416 conducted a study on this topic by investigating different MOF composites consisting of catalytically active NPC-4 (Cu2(TMBDI)(H2O)2) (TMBDI = 2,3,5,6-tetramethyl-benzene-1,4-di-isophthalate) MOF layers for the electrochemical ORR. When immobilized directly on GCEs, the application of NPC-4 was limited due to its detachment from the electrode surface. To remedy this shortcoming, rGO was immobilized first onto the GCE surface prior to the MOF-immobilization. The extra rGO layer served as a binder and an electron transfer mediator, providing a large specific surface area and more contact points for the adhesion of the MOF than the bare GCE.
An alternative approach for efficient “MOF wiring” is the construction of 3D electrodes, which was already demonstrated in 2012 by Loh and co-workers.417 The authors reported the composite material (G-dye-FeP)n, consisting of pyridine-functionalized rGO nanosheets that were incorporated as a building block in varying ratios into an FeTCPP-based MOF during solvothermal synthesis (Figure 44). Incorporation of the pyridine-functionalized graphene into the MOF produced a composite graphene/MOF material with enhanced catalytic ORR activity. The structure and electrochemical property of the hybrid MOF were investigated as a function of the weight percentage of the functionalized graphene added to the iron porphyrin framework. The results showed that the addition of pyridine-functionalized graphene changes the crystallization process of the iron porphyrin MOF increases its porosity and enhances charge transport, resulting in facile four-electron ORR.
Figure 44.

Schematic representation of a) reduced graphene oxide (rGO), b) G-dye, c) TCPP, d) (Fe–P)n MOF, e) (G-dye-FeP)n MOF, and f) magnified view of layers inside the framework of (G-dye-FeP)n MOF showing how graphene sheets intercalated between porphyrin networks. Reprinted with permission from reference (417). Copyright 2012 American Chemical Society.
The following year, the same group reported a composite material consisting of graphene oxide (GO) and a copper-based MOF for the electrocatalytic HER, the OER, as well as the ORR. The MOF, abbreviated as Cu-MOF, consisted of paddle wheel Cu2(COO)4(ted)2 (ted = triethylenediamine) SBUs that are linked by BDC to form a 2D net parallel to the xy plane that is then further connected by ted to form the 3D Cu-MOF. GO sheets decorated with −OH and epoxy groups on either side of the sheets are analogous to the BDC pillar connectors and used in the solvothermal MOF preparation in amounts ranging from 2 to 10 wt %. The resulting GO-incorporated Cu-MOF composites exhibited smaller overpotentials and higher currents for all three electrocatalytic reactions and showed better stability in acidic media compared to that of the pure MOF. It was postulated that the enhanced electrocatalytic properties and stability in acid of the GO-MOF composite is due to the unique porous scaffold structure, improved charge transport, and synergistic interactions between the GO and the MOF.418
In 2016, Sohrabi et al.419 described the synthesis of the composite PCN-222-G-py, a combination of an iron porphyrin MOF and pyridine-functionalized graphene (G-py). The pyridine groups at the graphene substrates act as axial ligands to the Fe porphyrins the MOF is composed of, altering the electronic properties of the latter. PCN-222-G-py deposited on a GCE catalyzed the ORR in acidic media at a potential that was anodically shifted by 100 mV compared to that of pristine PCN-222. The pyridine functional groups connected the graphene sheets of the MOF facilitated interfacial electron transport and thus the electrocatalytic ORR.
3.4.3. Unusual Electrolytes and Compartmentalization
Apart from the electrode/MOF interface, also the MOF/electrolyte interface can be designed for improved catalytic activity and device durability. One such paper addressed the role of ionic liquids (ILs) as replacements of traditional electrolyte solutions in polar organic solvents or water. ILs possess a wide electrochemical window and exhibit good conductivity. In addition, they are able to interact with CO2 by physical adsorption, thereby promoting the CO2 activation. This effect was utilized by Kang et al.420 who investigated the electrochemical reduction of CO2 to CH4 with a Zn-BTC MOFs that had electrophoretically been deposited on carbon paper (CP). Electrocatalysis was performed in different Bmim-based (Bmim = 1-butyl-3-methylimidazolium) ILs. Out of the investigated ILs (BmimBF4, BmimOTF, BmimPF6, and BmimClO4), the fluorine-containing ILs displayed the highest total current densities, owing to the interaction between fluorine with CO2. The combination of ILs and the Zn-MOF/CP cathode gave rise to the CO2-to-CH4 electroreduction with a FE exceeding 80% at a current density higher than 3 mA cm–2 and an overpotential of 0.25 V. Control experiment with 0.1 M BmimBF4 in acetonitrile showed much lower selectivity for CH4, demonstrating that ILs are responsible for the high selectivity of the CO2RR toward CH4.
MOFs that catalyze the CO2RR and ORR in a light-driven process have also been organized as gas-permeable MOF membrane systems.421 The MOFs that were used for this study were based on highly defective NH2-UiO-66 (A-aUiO) crystals into which Ir and Pd single-atom (SA) active sites had been coordinated to the Zr6O4(OH)4 cluster SBUs. These SA/A-aUiO (SA = Ir or Pd) crystallites were used to fabricate flexible and gas-permeable membranes via layer-by-layer deposition onto porous polytetrafluoroethylene (PTFE) films (Figure 45). The membranes can be run in gas-membrane-gas (GMG) mode in which humidified CO2 gas is fed through the membrane together with isopropanol vapor. The water and isopropanol in the CO2 stream were used as the proton source and sacrificial agent, respectively. Running the Ir/A-aUiO membrane in the GMG mode under illumination, CO2 is converted to HCOOH with a near-unity selectivity and an impressive apparent quantum efficiency of 15.76% at 420 nm. Control experiments with a more traditional gas–liquid–solid reaction mode resulted in significantly lower quantum efficiencies, proving the favorable effect of the GMG operational mode that promotes gas diffusion. Analogous observations have been made for the photocatalytic O2-to-H2O2 reduction, using membranes consisting of Ir/A-aUiO MOFs.
Figure 45.

Humidified gases (e.g., CO2, O2) can be fed through the gas-permeable MOF/PTFE membranes and photocatalytically reduced to value-added chemicals (e.g., HCOOH and H2O2) under visible light irradiation and ambient conditions. Adapted with permission from reference (421). Copyright 2021 Springer Nature.
The final contribution in this section is possibly the most advanced organization of catalytic MOFs as components for a complete light-driven water splitting system.189 In fact, the compartmentalization of the half reactions and their communication promoted by proton and redox shuttles bears strong resemblance to the Z-scheme in natural photosynthesis.422 In this contribution, MOFs for the light-driven HER and OER have been embedded in the lipid bilayer and interior of a liposome, respectively, and were shown to promote water splitting into its constituting elements with an apparent quantum yield of 1.5 ± 1% without the need of any sacrificial agents (Figure 46a). The MOF nanosheets that were used for the light-driven HER half reaction consisted of light-harvesting Zn-porphyrin (ZnTCPP) and catalytic Pt-porphyrin (PtTCPP) linkers, and hexanuclear Hf-based SBUs. For facile incorporation into the hydrophobic lipid bilayer of the liposome, the constituting components of the HER-MOF were functionalized with hydrophobic groups. The MOF for OER consisted of [Ru(bpy)3]2+-based PSs and Ir-bpy catalyst linkers, and Zr-based SBUs, localized in the hydrophilic interior of the liposome. The two half reactions are electrically coupled by means of Fe3+/2+ tetrachlorobenzoquinone-based redox and proton mediators. Careful analyses of the MOF–liposome assembly by electrochemical and transient absorption techniques revealed the sequence of events that led to the overall water splitting process (Figure 46b). Photo-oxidation of the [Ru]2+ linker in the OER-MOF by Fe3+ followed by hole transfer to the catalytic Ir linker, generates high-valent Ir species that oxidize water to O2 with concomitant release of protons. The produced Fe2+ is reoxidized by TCBQ (tetrachlorobenzoquinone) at the lipid/water interface to afford TCBQH (tetrachlorobenzohydrosemiquinone) that is the reducing agent for the HER half reaction. On the cathodic side, photon absorption by the [ZnTCPP]/[PtTCPP] leads to a triplet state, which undergoes charge separation to generate [PtTCPP]− and [ZnTCPP]+. The [ZnTCPP]+ oxidizes the TCBQH back to TCBQ and releases the proton; the [PtTCPP]− is protonated to [H–PtTCPP], which accepts another electron and proton to release H2 and to complete the overall water splitting cycle. The use of two relays and their different hydrophobicity/hydrophilicity allows for more favorable concentration gradients compared to those in a homogeneous system, and is believed to be a key to the performance of this system. The authors identified potential for improvements by better balancing the intrinsic rates of the two photosystems, where currently the HER reaction was slower and allowed reduced forms of the relay to accumulate, which in turn increased recombination with the OER MOF components. This study is a beautiful example of organization on a microscopic and macroscopic level. The MOFs stabilized the light-harvesting and catalytic functional components as linkers in three dimensions, while the OER- and HER-MOF nanosheets themselves are designed to localize in the lipid bilayer or the interior of the liposome, respectively.
Figure 46.

MOFs for photocatalytic HER and OER embedded in lipid bilayer for overall photocatalytic water splitting. a) Systematic representation of the constituting materials and b) energy level diagram of the reaction. Adapted with permission from reference (189). Copyright 2021 Springer Nature.
4. Conclusion
A number of distinct factors differentiate MOFs from other solid supports into which molecular catalysts may be incorporated. The modular nature of MOFs allows for a large variety of SBUs and linkers to be combined in unique topologies. Coordination compounds or metal clusters that may be unstable in a homogeneous solution phase may be constructed as discrete molecular entities as SBUs in MOFs. If such SBUs are catalytically active, as shown for many examples in section 3.1.2, unprecedented catalyst motifs can be designed, synthesized, and investigated. In addition, known catalysts can be incorporated into MOFs by a variety of strategies (sections 3.1.1–3.1.3), thereby boosting structural integrity under turnover conditions.
Furthermore, the arsenal of postsynthetic modification methods that can be employed on existing frameworks is unrivalled by any other incorporation matrix. It is this feature that triggers the imagination of chemists and provokes drawing parallels between MOFs and enzymes. If a MOF pore is regarded as a reaction vessel with linkers and SBUs (more or less) fixed in space, additional functionality can be introduced at specific sites by organic chemistry methods to tailor pore interiors. If these decorations interact favorably with a simultaneously present catalyst, the situation truly resembles second and third coordination sphere effects in enzymes. In the latter, it is a globally (more or less) rigid protein framework that provides defined points in space to which amino acid side chains are attached that interact with catalytic active sites.
The major difference between enzymatic and MOF-borne catalysis lies in the fact that enzymes contain one active site and a protein environment that is optimized to support its function. Under biological conditions, every enzyme, and thus every active site is supplied with electrons/holes and substrate from the surrounding environment to enable its fast and energy efficient operation. Reflecting the importance of transport in biological systems, nature has optimized the third (and fourth) coordination sphere to facilitate charge and mass transport to/from the active sites through carefully designed channels.
It is at this stage that the MOF/enzyme analogy does not hold any longer. In contrast to single entity enzymes with one active site, MOF crystals or thin films exhibit significantly higher concentrations of active sites, sometimes periodically reoccurring in three dimensions within distances as short as a nanometer. Catalyst concentrations in MOFs can approach 1 M, and the importance of transport phenomena cannot be overestimated in such a situation. Consequently, principles concerning charge and mass transport in MOFs are outlined in section 2.1 and 2.2. In an ideal case, high active site concentrations can give rise to significant current densities in electrocatalytic experiments, but only if every catalyst in the MOF is supplied with a sufficient flux of redox equivalents and substrates. In porous materials such as MOFs, this requirement may often not be met, and catalysts in the interior of the bulk MOF may lie dormant. Making things worse, the presence of transport limitations may often remain unnoticed, and transport as a phenomenon that limits efficiency is still underappreciated in the field. As outlined in section 2, it is highly advisable to vary as many parameters as possible (including light intensity, catalyst concentration and particle size), and to match these data to physical models as a basis for understanding kinetic bottlenecks in MOF/molecular catalyst systems.
That such scrutinous studies can be highly rewarding and yield significant enhancements in catalytic efficiency and current density was recently illustrated in a thought experiment on molecular electrocatalysis in MOFs by Johnson et al.60 (Figure 47). Under the assumption of facile substrate transport and a reasonable intrinsic catalyst turnover frequency, an apparent electron diffusion coefficient (Deapp) of 5 × 10–10 cm2 s–1 was shown to sustain a maximum (plateau) current density of 10 mA cm–2 in a 0.1 μm thick MOF film. Utilizing the unique properties of MOFs to tailor specific properties, one could imagine that of De, may be increased to 5 × 10–8 cm2 s–1. As a result of the faster charge transport, the catalytic MOF film can be made thicker (1 μm) without compromising efficiency, reaching a maximum current density of 100 mA cm–2.
Figure 47.

Options for optimizing MOF-based molecular catalysis either by adjustment of the film thickness or by increasing a diffusion coefficients. Plotted are current density versus the film thickness (df), under the assumption of facile substrate transport and a reasonable intrinsic catalyst TOF. The shaded region below each curve signifies film thicknesses where the efficiency is less than unity and the amount of active catalyst is less than the amount of total catalyst. The curves are for Deapp = 5 × 10–10 cm2 s–1 (green curve) and 5 × 10–8 cm2 s–1 (red curve). Adapted with permission from reference (60). Copyright 2020 American Chemical Society.
Transport limitations in photochemical schemes in which the light absorber and the catalyst are coimmobilized in close proximity and engage in a fast light-induced electron transfer process may at first glance be less of an issue. This view is, however, highly deceiving, as sacrificial agents that are the final oxidant/reductant need to diffuse into the MOF to provide the holes/electrons for the reaction at hand. As described in section 2.3, this process needs to be fast in order to compete with nonproductive pathways such as charge recombination. Even in a scenario when the oxidant/reductant is too large to diffuse into the MOF and provides holes/electrons to the MOF at the MOF/solution interface, charge transport between discrete redox active sites may limit the overall process. The vast majority of MOFs discussed in this review are not semiconducting, as is often assumed, but instead insulating materials. Therefore, charge transport proceeds through a hopping mechanism which is diffusional in nature. Moreover, it is coupled to the diffusion/migration of charge balancing counterions.
The present review clearly illustrates that the field has realized the effect of MOFs to stabilize structural integrity of a catalyst, which is very important. The inclusion of higher coordination sphere effects, and their successful experimental manifestation is, however, still in its infancy. Papers in which transport, which is third coordination sphere effects, is specifically investigated as discussed in section 3.2 are scarce in general and in context of catalysis even more so. An alternative strategy to lessen the impact of transport limitations is the use of nanometer-sized platelets and nanosheets, which however, comes at the cost of low current densities. With transport not being limiting, it is in such systems that synthetic modifications that lead to accelerated intrinsic catalyst rates are easiest demonstrated. Examples of such strategies and their experimental manifestation as described in section 3.3 are still scarce in the field. On the other hand, all original papers that were discussed herein originate from the past decade, and with the knowledge that has been accumulated, it is certain that MOFs as materials to host molecular catalysts will continue to evolve. For example, the manifestation of potential gradients for long-range electric fields is such an open quest. Such a system would be particularly important for full artificial photosynthesis, where both catalytic reduction and oxidation occurs by light-induced charge separation. Another interesting challenge would be to utilize and even control the structural dynamics of the MOFs,423,424 and couple such processes to transport and catalysis. Extrapolating the current knowledge increase in MOF-based catalysis into the future, our mechanistic understanding will continue to grow, and its translation into exciting new catalysis systems can be expected. One such example on the rational design of MOF-based artificial enzymes was reported very recently by Lin and co-workers.425 The study was based on the integration of active sites for the CO2RR and OER as well as proximal amino acids and other cofactors into tunable MOF monolayers. The MOF layers were first separately optimized for the CO2RR and OER by a diversification, selection and optimization strategy, and then coupled into a complete artificial photosynthesis scheme by the addition of a Co(bpy)32+ redox mediator. The optimized CO2RR MOF featured pendant urea groups in the second coordination sphere of the Hemin active site, resulting in highly active and selective photocatalytic CO2 reduction with a 27-fold increase in activity over the homogeneous control. The most active photocatalytic OER system contained pendant (p-chloro-phenyl)amide groups in the second coordination sphere of an Ir-based molecular catalyst. The coupling of the two optimized MOFs with the redox mediator resulted in a complete artificial photosynthesis system that catalyzed light-driven CO2RR and OER, according to (1 + n)CO2 + 2H2O → CH4 + nCO + (2 + n/2)O2. The turnover frequency of the process was close to 100 h–1, thus over 1 order of magnitude faster than previously reported photocatalysts. This paper is a glimpse into what the future in MOF-based catalysis with orchestrated higher coordination sphere effects may hold.
Acknowledgments
Financial support from the European Research Council through a Consolidator grant to S.O. (ERC-CoG2015-681895_MOFcat), the Swedish Energy Agency (grant number: 42029-2), and the Knut & Wallenberg Foundation (KAW 2019.0071) is gratefully acknowledged.
Biographies
Nina F. Suremann was born in 1996 in Switzerland. She received her Bachelor’s and Master’s degree in Chemistry from the University of Zurich in 2018 and 2020, respectively. In 2019, she completed her Master’s project in the group of Prof. Roger Alberto at the University of Zurich. Her work focused on the development of a mixed-aromatic ring sandwich complex library, starting from a fulvene-benzene-type Rhenium complex and a variety of nucleophiles. In June 2020 she moved to Uppsala, Sweden, where she joined the group of Prof. Sascha Ott as a Ph.D. student. Her current research concerns the investigation of photoelectrocatalytic applications of porphyrin-based MOFs on semiconductors for the reduction of carbon dioxide.
Brian D. McCarthy was born in 1989 in Hillsboro, Oregon, USA, and was inspired to study renewable energy-driven chemistry while working with Prof. Carl Wamser at Portland State University. He earned his Bachelor’s degree in Chemistry at the Massachusetts Institute of Technology while working with Prof. Daniel Nocera and Prof. Mircea Dincǎ. As a DOE Office of Science Graduate Fellow, he studied electrochemically triggered proton-coupled electron transfer with Prof. Jillian Dempsey at the University of North Carolina at Chapel Hill, earning a Ph.D. in 2016. Brian worked as a postdoctoral researcher in the group of Prof. Sascha Ott before joining the battery startup EC Power in 2021.
Wanja Gschwind joined Prof. Sascha Ott’s lab in 2020 to pursue his Ph.D. In his research he investigates the distribution of postsynthetically introduced metal catalysts in MOF single crystals and the potential of redox-active MOFs as electrocatalytic systems for organic transformation. Previous to that he conducted his Master’s thesis at the University of Basel, Switzerland, in the field of antibiotics research. There he explored the metal-dependent substrate promiscuity of the potential antibiotic target DapE.
Amol Kumar is currently a Ph.D. candidate in Prof. Sascha Ott’s lab. His doctoral research focuses on developing redox-active MOF thin films and exploring new paths in organic catalysis. Prior to joining the Ott lab, Amol obtained his Bachelor’s and Master’s degree in Chemistry from IISER Kolkata, India, where he did his Master’s project under the supervision of Dr. Biplab Maji on “Manganese catalyzed selective Alkylation exploring the transfer hydrogenation”.
Ben A. Johnson completed his Ph.D. on transport phenomena in metal–organic frameworks in the group of Prof. Sascha Ott at Uppsala University in 2020. For his Ph.D. work, he was awarded the Anna Sundström Award 2021 from the Swedish Chemical Society for the best Ph.D. thesis in inorganic chemistry in Sweden 2020. In 2021, he moved to TU Munich as a Marie Skłodowska-Curie postdoctoral fellow to continue his career in the group of Prof. Dr. Nicolas Plumeré.
Leif Hammarström is professor of Chemical Physics at Uppsala University, and chairperson of the Swedish Consortium for Artificial Photosynthesis. His research focuses on mechanisms of artificial photosynthesis, which includes photochemistry and photophysics, proton-coupled electron transfer and catalyst mechanisms.
Sascha Ott (born in Germany, 1973) studied chemistry as an undergraduate in Germany and Australia, and obtained his PhD in Chemistry at University College London, U.K., in 2002. Following postdoctoral work with Profs. Björn Åkermark and Licheng Sun at Stockholm University, Sweden, he moved to Uppsala University in 2004, and became Professor in Synthetic Molecular Chemistry at the Department of Chemistry – Ångström Laboratory in 2017. He is the recipient of a Consolidator Grant from the European Research Council (2015), the Göran Gustafsson Prize for younger researchers (2010), and the Lilly och Sven Thuréus Prize from the Royal Society of Sciences Uppsala (2017). Research in the group is centered on different topics at the interface of organic chemistry and molecular inorganic chemistry. These include works on low-coordinate and low-valent phosphorus in an organic chemistry context, and the development of molecular redox catalysts of energy relevance, more recently in the confinements of MOFs.
Abbreviations
The following list of abbreviations contains commonly known abbreviations as well as abbreviations often used throughout this review article. Further abbreviations were used in certain sections, which are then directly introduced and used throughout smaller sections and are therefore not listed here.
For naming conventions of MOFs and certain linkers, we tried to make it as consistent as possible throughout this review. This includes that we changed some “set” names from certain publications to fit into this consistency. For example, the adjustment whenever 2,2′-bipyridine-5,5′-dicarboxylate was mentioned to bpydc, even though the original publication wrote dcbpy or a deviation from it.
| 1D | one-dimensional |
| 2D | two-dimensional |
| Adt | three-dimensional |
| ALD | azadithiolate |
| ATA | 2-aminoterephthalate |
| H2ATA | 2-aminoterephthalic acid |
| BDC | benzene-1,4-dicarboxylate |
| H2BDC | benzene-1,4-dicarboxylic acid |
| bpdc | biphenyl-4,4′-dicarboxylate |
| H2bpdc | biphenyl-4,4′-dicarboxylic acid |
| bpy | 2,2′-bipyridine |
| bpydc | 2,2′-bipyridine-5,5′-dicarboxylate |
| H2bpydc | 2,2′-bipyridine-5,5′-dicarboxylic acid |
| BTB | 1,3,5-benzene(tris)benzoate |
| H3BTB | 1,3,5-benzene(tris)benzoic acid |
| BTC | 1,3,5-benzenetricarboxylate |
| H3BTC | 1,3,5-benzenetricarboxylic acid |
| CAN | carbon nanotubes |
| Cat | catalyst (in relation with figures or equations) |
| CNTs | carbon nanotubes |
| CO2RR | carbon dioxide reduction reaction |
| Cp | cyclopentadienyl |
| Cp* | η5-C5Me5/1,2,3,4,5-pentamethylcyclopentadiene |
| D | electron donor (in relation with figures or equations) |
| dcbdt | 1,4-dicarboxylbenzene-2,3-dithiolato |
| DFT | density functional theory |
| DMF | dimethylformamide |
| EPR | electron paramagnetic resonance |
| FE | Faradaic efficiency |
| FTO | fluorine-doped tin oxide |
| GCE | glassy carbon electrode |
| GO | graphene oxide |
| H2ase | hydrogenase |
| H2TCPP | 4,4′,4″,4‴-(porphyrin-5,10,15,20-tetrayl)tetrabenzoate |
| HER | hydrogen evolution reaction |
| HHTP | 2,3,6,7,10,11-hexahydroxytriphenylene |
| HITP | 2,3,6,7,10,11-hexaaminotriphenylene |
| MIL | Matériaux de l′Institut Lavoisier |
| MOF | metal–organic framework |
| MOL | metal–organic layer |
| OER | oxygen evolution reaction |
| ORR | oxygen reduction reaction |
| PCN | porous coordination network |
| POM | polyoxometalate |
| PS | photosensitizer |
| rGO | reduced graphene oxide |
| RHE | reversible hydrogen electrode |
| SBU | secondary building unit |
| TEA | triethylamine |
| TEOA | triethanolamine |
| terpy | 2,2′:6′,2″-terpyridine |
| TOF | turnover frequency |
| TON | turnover number |
| UiO | Universitetet i Oslo |
| WOC | water oxidation catalyst |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
| ZIF | zeolitic imidazolate framework |
List of Mathematical Symbols in Section 2
The following list summarizes all mathematical symbols used throughout the formulas in section 2 and their respective meaning.
| ϕ | Thiele modulus |
| Da | Damköhler number |
| L | Damköhler number |
| kcat | first-order rate constant of the catalytic reaction |
| (in s–1) | |
| Ds | intraparticle diffusion coefficient of the substrate |
| v | observed reaction rate (in mol s–1) |
| η | internal effectiveness factor |
| TOFapp | apparent TOF; rate normalized to the total |
| amount of catalyst present | |
| TOFtrue | only active catalyst considered in catalytic particle |
| ntotal | total moles of catalyst within the MOF particle |
| nactive | active moles of catalyst within the MOF particle |
| V | volume of the particle |
| Ccat | concentration of the catalyst |
| r | radial distance in spherical coordinates |
| R | radius of the particle |
| Dox | diffusivity of the sacrificial oxidant |
| Cox0 | bulk oxidant concentration |
| Ccat0 | total catalyst concentration |
| CS0 | substrate concentration |
| D | electron hopping coefficient OR diffusion |
| coefficient of the substrate | |
| df | film thickness |
| dfopt | optimal film thickness |
| ΦH2 | HER quantum yield |
| nH2 | number of moles of H2 produced |
| nphoton | number of moles of photons absorbed |
| Ratehv | rate of photon absorption |
Special Issue
This paper is an additional review for Chem. Rev. 2022, volume 122, issue (14), , “Catalysis beyond the First Coordination Sphere”.
Author Contributions
CRediT: Nina F. Suremann visualization, writing – original draft, writing – review & editing. Brian D. McCarthy writing – original draft. Wanja Gschwind visualization, writing – original draft. Amol Kumar writing – original draft. Ben A. Johnson conceptualization, visualization, writing – original draft, writing – review & editing. Leif Hammarström conceptualization, writing – original draft, writing – review & editing, funding acquisition. Sascha Ott conceptualization, writing – original draft, writing – review & editing, funding acquisition.
The authors declare no competing financial interest.
References
- Montoya J. H.; Seitz L. C.; Chakthranont P.; Vojvodic A.; Jaramillo T. F.; Norskov J. K. Materials for Solar Fuels and Chemicals. Nat. Mater. 2017, 16, 70–81. 10.1038/nmat4778. [DOI] [PubMed] [Google Scholar]
- Appel A. M.; Bercaw J. E.; Bocarsly A. B.; Dobbek H.; DuBois D. L.; Dupuis M.; Ferry J. G.; Fujita E.; Hille R.; Kenis P. J.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis N. S.; Nocera D. G. Powering the Planet: Chemical Challenges in Solar Energy Utilization. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15729–15735. 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson E. E.; Kubiak C. P.; Sathrum A. J.; Smieja J. M. Electrocatalytic and Homogeneous Approaches to Conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 2009, 38, 89–99. 10.1039/B804323J. [DOI] [PubMed] [Google Scholar]
- Li X.; Yu J.; Jaroniec M.; Chen X. Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels. Chem. Rev. 2019, 119, 3962–4179. 10.1021/acs.chemrev.8b00400. [DOI] [PubMed] [Google Scholar]
- Jones A. K.; Sillery E.; Albracht S. P.; Armstrong F. A. Direct Comparison of the Electrocatalytic Oxidation of Hydrogen by an Enzyme and a Platinum Catalyst. Chem. Commun. 2002, 866–867. 10.1039/b201337a. [DOI] [PubMed] [Google Scholar]
- Sun L.; Reddu V.; Fisher A. C.; Wang X. Electrocatalytic Reduction of Carbon Dioxide: Opportunities with Heterogeneous Molecular Catalysts. Energy Environ. Sci. 2020, 13, 374–403. 10.1039/C9EE03660A. [DOI] [Google Scholar]
- Fang Y.; Powell J. A.; Li E.; Wang Q.; Perry Z.; Kirchon A.; Yang X.; Xiao Z.; Zhu C.; Zhang L.; et al. Catalytic Reactions within the Cavity of Coordination Cages. Chem. Soc. Rev. 2019, 48, 4707–4730. 10.1039/C9CS00091G. [DOI] [PubMed] [Google Scholar]
- Tan C.; Chu D.; Tang X.; Liu Y.; Xuan W.; Cui Y. Supramolecular Coordination Cages for Asymmetric Catalysis. Chem. Eur. J. 2019, 25, 662–672. 10.1002/chem.201802817. [DOI] [PubMed] [Google Scholar]
- Corma A.; Garcia H. Supramolecular Host-Guest Systems in Zeolites Prepared by Ship-in-a-Bottle Synthesis. Eur. J. Inorg. Chem. 2004, 2004, 1143–1164. 10.1002/ejic.200300831. [DOI] [Google Scholar]
- Kosinov N.; Liu C.; Hensen E. J. M.; Pidko E. A. Engineering of Transition Metal Catalysts Confined in Zeolites. Chem. Mater. 2018, 30, 3177–3198. 10.1021/acs.chemmater.8b01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C.; Zhang A.; Song C.; Guo X. Advances in the Synthesis and Catalysis of Solid and Hollow Zeolite-Encapsulated Metal Catalysts. Adv. Catal. 2018, 63, 75–115. 10.1016/bs.acat.2018.10.002. [DOI] [Google Scholar]
- Murray R. W.Introduction to Molecularly Designed Electrode Surfaces. In Molecular Design of Electrode Surfaces, 1st ed.; John Wiley & Sons, Inc.: New York, 1992; pp 1–48. [Google Scholar]
- Gascon J.; Corma A.; Kapteijn F.; Llabrés i Xamena F. X. Metal Organic Framework Catalysis: Quo Vadis?. ACS Catal. 2014, 4, 361–378. 10.1021/cs400959k. [DOI] [Google Scholar]
- Xie L. S.; Skorupskii G.; Dincǎ M. Electrically Conductive Metal-Organic Frameworks. Chem. Rev. 2020, 120, 8536–8580. 10.1021/acs.chemrev.9b00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaj M.; Cohen S. M. Postsynthetic Modification: An Enabling Technology for the Advancement of Metal-Organic Frameworks. ACS Cent. Sci. 2020, 6, 1046–1057. 10.1021/acscentsci.0c00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayder T. M.; Bensalah A. T.; Li B.; Byers J. A.; Tsung C. K. Engineering Second Sphere Interactions in a Host-Guest Multicomponent Catalyst System for the Hydrogenation of Carbon Dioxide to Methanol. J. Am. Chem. Soc. 2021, 143, 1630–1640. 10.1021/jacs.0c08957. [DOI] [PubMed] [Google Scholar]
- Rakowski DuBois M.; DuBois D. L. Development of Molecular Electrocatalysts for CO2 Reduction and H2 Production/Oxidation. Acc. Chem. Res. 2009, 42, 1974–1982. 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]
- Ball P. Water as an Active Constituent in Cell Biology. Chem. Rev. 2008, 108, 74–108. 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- Rebilly J. N.; Colasson B.; Bistri O.; Over D.; Reinaud O. Biomimetic Cavity-Based Metal Complexes. Chem. Soc. Rev. 2015, 44, 467–489. 10.1039/C4CS00211C. [DOI] [PubMed] [Google Scholar]
- Hussein R.; Ibrahim M.; Bhowmick A.; Simon P. S.; Chatterjee R.; Lassalle L.; Doyle M.; Bogacz I.; Kim I. S.; Cheah M. H.; et al. Structural Dynamics in the Water and Proton Channels of Photosystem II During the S2 to S3 Transition. Nat. Commun. 2021, 12, 6531. 10.1038/s41467-021-26781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeuken L. J.; Jones A. K.; Chapman S. K.; Cecchini G.; Armstrong F. A. Electron-Transfer Mechanisms through Biological Redox Chains in Multicenter Enzymes. J. Am. Chem. Soc. 2002, 124, 5702–5713. 10.1021/ja012638w. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E.; Tye J. W.; Hall M. B. Computational Studies of [NiFe] and [FeFe] Hydrogenases. Chem. Rev. 2007, 107, 4414–4435. 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- Silakov A.; Wenk B.; Reijerse E.; Lubitz W. 14NHYSCORE Investigation of the H-Cluster of [FeFe] Hydrogenase: Evidence for a Nitrogen in the Dithiol Bridge. Phys. Chem. Chem. Phys. 2009, 11, 6592–6599. 10.1039/b905841a. [DOI] [PubMed] [Google Scholar]
- Erdem Ö. F.; Schwartz L.; Stein M.; Silakov A.; Kaur-Ghumaan S.; Huang P.; Ott S.; Reijerse E. J.; Lubitz W. A Model of the [FeFe] Hydrogenase Active Site with a Biologically Relevant Azadithiolate Bridge: A Spectroscopic and Theoretical Investigation. Angew. Chem., Int. Ed. 2011, 50, 1439–1443. 10.1002/anie.201006244. [DOI] [PubMed] [Google Scholar]
- Berggren G.; Adamska A.; Lambertz C.; Simmons T. R.; Esselborn J.; Atta M.; Gambarelli S.; Mouesca J. M.; Reijerse E.; Lubitz W.; et al. Biomimetic Assembly and Activation of [FeFe]-Hydrogenases. Nature 2013, 499, 66–69. 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi M.; Berggren G.. [FeFe] Hydrogenases and Their Functional Models. In Comprehensive Coordination Chemistry III, 3rd ed.; Elsevier Ltd.: Amsterdam, 2021; pp 731–756. [Google Scholar]
- Ho M.-H.; Rousseau R.; Roberts J. A. S.; Wiedner E. S.; Dupuis M.; DuBois D. L.; Bullock R. M.; Raugei S. Ab Initio-Based Kinetic Modeling for the Design of Molecular Catalysts: The Case of H2 Production Electrocatalysts. ACS Catal. 2015, 5, 5436–5452. 10.1021/acscatal.5b01152. [DOI] [Google Scholar]
- Helm M. L.; Stewart M. P.; Bullock R. M.; DuBois M. R.; DuBois D. L. A Synthetic Nickel Electrocatalyst with a Turnover Frequency above 100,000 S–1 for H2 Production. Science 2011, 333, 863–866. 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- Anxolabéhère-Mallart E.; Costentin C.; Fournier M.; Nowak S.; Robert M.; Savéant J. M. Boron-Capped Tris(Glyoximato) Cobalt Clathrochelate as a Precursor for the Electrodeposition of Nanoparticles Catalyzing H2 Evolution in Water. J. Am. Chem. Soc. 2012, 134, 6104–6107. 10.1021/ja301134e. [DOI] [PubMed] [Google Scholar]
- Nagoshi K.; Yamashita S.; Yagi M.; Kaneko M. Catalytic Activity of [(bpy)2(H2O)Ru-O-Ru(H2O)(bpy)2]4+ for Four-Electron Water Oxidation. J. Mol. Catal. A: Chem. 1999, 144, 71–76. 10.1016/S1381-1169(98)00334-3. [DOI] [Google Scholar]
- Lampret O.; Duan J.; Hofmann E.; Winkler M.; Armstrong F. A.; Happe T. The Roles of Long-Range Proton-Coupled Electron Transfer in the Directionality and Efficiency of [FeFe]-Hydrogenases. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 20520–20529. 10.1073/pnas.2007090117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J.; Mebs S.; Laun K.; Wittkamp F.; Heberle J.; Happe T.; Hofmann E.; Apfel U.-P.; Winkler M.; Senger M.; et al. Geometry of the Catalytic Active Site in [FeFe]-Hydrogenase Is Determined by Hydrogen Bonding and Proton Transfer. ACS Catal. 2019, 9, 9140–9149. 10.1021/acscatal.9b02203. [DOI] [Google Scholar]
- Finkelmann A. R.; Stiebritz M. T.; Reiher M. Inaccessibility of the Μ-Hydride Species in [FeFe] Hydrogenases. Chem. Sci. 2014, 5, 215–221. 10.1039/C3SC51700D. [DOI] [Google Scholar]
- Li H.; Rauchfuss T. B. Iron Carbonyl Sulfides, Formaldehyde, and Amines Condense to Give the Proposed Azadithiolate Cofactor of the Fe-Only Hydrogenases. J. Am. Chem. Soc. 2002, 124, 726–727. 10.1021/ja016964n. [DOI] [PubMed] [Google Scholar]
- Esselborn J.; Lambertz C.; Adamska-Venkatesh A.; Simmons T.; Berggren G.; Noth J.; Siebel J.; Hemschemeier A.; Artero V.; Reijerse E.; et al. Spontaneous Activation of [FeFe]-Hydrogenases by an Inorganic [2Fe] Active Site Mimic. Nat. Chem. Biol. 2013, 9, 607–609. 10.1038/nchembio.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubitz W.; Ogata H.; Rudiger O.; Reijerse E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- Artz J. H.; Mulder D. W.; Ratzloff M. W.; Lubner C. E.; Zadvornyy O. A.; LeVan A. X.; Williams S. G.; Adams M. W. W.; Jones A. K.; King P. W.; et al. Reduction Potentials of [FeFe]-Hydrogenase Accessory Iron-Sulfur Clusters Provide Insights into the Energetics of Proton Reduction Catalysis. J. Am. Chem. Soc. 2017, 139, 9544–9550. 10.1021/jacs.7b02099. [DOI] [PubMed] [Google Scholar]
- Raleiras P.; Khanna N.; Miranda H.; Mészáros L. S.; Krassen H.; Ho F.; Battchikova N.; Aro E.-M.; Magnuson A.; Lindblad P.; et al. Turning around the Electron Flow in an Uptake Hydrogenase. EPR Spectroscopy and in Vivo Activity of a Designed Mutant in HupSL from Nostoc Punctiforme. Energy Environ. Sci. 2016, 9, 581–594. 10.1039/C5EE02694F. [DOI] [Google Scholar]
- O’Keeffe M.; Yaghi O. M. Deconstructing the Crystal Structures of Metal-Organic Frameworks and Related Materials into Their Underlying Nets. Chem. Rev. 2012, 112, 675–702. 10.1021/cr200205j. [DOI] [PubMed] [Google Scholar]
- Batten S. R.; Champness N. R.; Chen X.-M.; Garcia-Martinez J.; Kitagawa S.; Öhrström L.; O’Keeffe M.; Paik Suh M.; Reedijk J. Terminology of Metal-Organic Frameworks and Coordination Polymers (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1715–1724. 10.1351/PAC-REC-12-11-20. [DOI] [Google Scholar]
- Betard A.; Fischer R. A. Metal-Organic Framework Thin Films: From Fundamentals to Applications. Chem. Rev. 2012, 112, 1055–1083. 10.1021/cr200167v. [DOI] [PubMed] [Google Scholar]
- Xu C.; Fang R.; Luque R.; Chen L.; Li Y. Functional Metal-Organic Frameworks for Catalytic Applications. Coord. Chem. Rev. 2019, 388, 268–292. 10.1016/j.ccr.2019.03.005. [DOI] [Google Scholar]
- Majewski M. B.; Peters A. W.; Wasielewski M. R.; Hupp J. T.; Farha O. K. Metal-Organic Frameworks as Platform Materials for Solar Fuels Catalysis. ACS Energy Lett. 2018, 3, 598–611. 10.1021/acsenergylett.8b00010. [DOI] [Google Scholar]
- Qin J. S.; Yuan S.; Lollar C.; Pang J.; Alsalme A.; Zhou H. C. Stable Metal-Organic Frameworks as a Host Platform for Catalysis and Biomimetics. Chem. Commun. 2018, 54, 4231–4249. 10.1039/C7CC09173G. [DOI] [PubMed] [Google Scholar]
- Honicke I. M.; Senkovska I.; Bon V.; Baburin I. A.; Bonisch N.; Raschke S.; Evans J. D.; Kaskel S. Balancing Mechanical Stability and Ultrahigh Porosity in Crystalline Framework Materials. Angew. Chem., Int. Ed. 2018, 57, 13780–13783. 10.1002/anie.201808240. [DOI] [PubMed] [Google Scholar]
- Li H.; Wang K.; Sun Y.; Lollar C. T.; Li J.; Zhou H.-C. Recent Advances in Gas Storage and Separation Using Metal-Organic Frameworks. Mater. Today 2018, 21, 108–121. 10.1016/j.mattod.2017.07.006. [DOI] [Google Scholar]
- Getman R. B.; Bae Y. S.; Wilmer C. E.; Snurr R. Q. Review and Analysis of Molecular Simulations of Methane, Hydrogen, and Acetylene Storage in Metal-Organic Frameworks. Chem. Rev. 2012, 112, 703–723. 10.1021/cr200217c. [DOI] [PubMed] [Google Scholar]
- Suh M. P.; Park H. J.; Prasad T. K.; Lim D. W. Hydrogen Storage in Metal-Organic Frameworks. Chem. Rev. 2012, 112, 782–835. 10.1021/cr200274s. [DOI] [PubMed] [Google Scholar]
- Kreno L. E.; Leong K.; Farha O. K.; Allendorf M.; Van Duyne R. P.; Hupp J. T. Metal-Organic Framework Materials as Chemical Sensors. Chem. Rev. 2012, 112, 1105–1125. 10.1021/cr200324t. [DOI] [PubMed] [Google Scholar]
- Burtch N. C.; Jasuja H.; Walton K. S. Water Stability and Adsorption in Metal-Organic Frameworks. Chem. Rev. 2014, 114, 10575–10612. 10.1021/cr5002589. [DOI] [PubMed] [Google Scholar]
- McCarthy B. D.; Beiler A. M.; Johnson B. A.; Liseev T.; Castner A. T.; Ott S. Analysis of Electrocatalytic Metal-Organic Frameworks. Coord. Chem. Rev. 2020, 406, 213137. 10.1016/j.ccr.2019.213137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungtaweevoranit B.; Diercks C. S.; Kalmutzki M. J.; Yaghi O. M. Spiers Memorial Lecture:. Progress and Prospects of Reticular Chemistry. Faraday Discuss. 2017, 201, 9–45. 10.1039/C7FD00160F. [DOI] [PubMed] [Google Scholar]
- Wang X.; Lan P. C.; Ma S. Metal-Organic Frameworks for Enzyme Immobilization: Beyond Host Matrix Materials. ACS Cent. Sci. 2020, 6, 1497–1506. 10.1021/acscentsci.0c00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. M. The Postsynthetic Renaissance in Porous Solids. J. Am. Chem. Soc. 2017, 139, 2855–2863. 10.1021/jacs.6b11259. [DOI] [PubMed] [Google Scholar]
- Cohen S. M. Postsynthetic Methods for the Functionalization of Metal-Organic Frameworks. Chem. Rev. 2012, 112, 970–1000. 10.1021/cr200179u. [DOI] [PubMed] [Google Scholar]
- Deria P.; Mondloch J. E.; Karagiaridi O.; Bury W.; Hupp J. T.; Farha O. K. Beyond Post-Synthesis Modification: Evolution of Metal-Organic Frameworks via Building Block Replacement. Chem. Soc. Rev. 2014, 43, 5896–912. 10.1039/C4CS00067F. [DOI] [PubMed] [Google Scholar]
- Mondloch J. E.; Katz M. J.; Isley W. C. III; Ghosh P.; Liao P.; Bury W.; Wagner G. W.; Hall M. G.; DeCoste J. B.; Peterson G. W.; et al. Destruction of Chemical Warfare Agents Using Metal-Organic Frameworks. Nat. Mater. 2015, 14, 512–516. 10.1038/nmat4238. [DOI] [PubMed] [Google Scholar]
- Yadav A.; Kanoo P. Metal-Organic Frameworks as Platform for Lewis-Acid-Catalyzed Organic Transformations. Chem. Asian J. 2019, 14, 3531–3551. 10.1002/asia.201900876. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Beiler A. M.; McCarthy B. D.; Ott S. Transport Phenomena: Challenges and Opportunities for Molecular Catalysis in Metal-Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 11941–11956. 10.1021/jacs.0c02899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp C. H.; Bukowski B. C.; Li H.; Johnson E. M.; Ilic S.; Morris A. J.; Gersappe D.; Snurr R. Q.; Morris J. R. Nanoconfinement and Mass Transport in Metal-Organic Frameworks. Chem. Soc. Rev. 2021, 50, 11530–11558. 10.1039/D1CS00558H. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Huang Y.; Peng Y.; Huang Z.; Ma Q.; Zhang H. Two-Dimensional Metal-Organic Framework Nanosheets: Synthesis and Applications. Chem. Soc. Rev. 2018, 47, 6267–6295. 10.1039/C8CS00268A. [DOI] [PubMed] [Google Scholar]
- Huang Y. B.; Liang J.; Wang X. S.; Cao R. Multifunctional Metal-Organic Framework Catalysts: Synergistic Catalysis and Tandem Reactions. Chem. Soc. Rev. 2017, 46, 126–157. 10.1039/C6CS00250A. [DOI] [PubMed] [Google Scholar]
- Leger C.; Bertrand P. Direct Electrochemistry of Redox Enzymes as a Tool for Mechanistic Studies. Chem. Rev. 2008, 108, 2379–438. 10.1021/cr0680742. [DOI] [PubMed] [Google Scholar]
- Noll T.; Noll G. Strategies for ″Wiring″ Redox-Active Proteins to Electrodes and Applications in Biosensors, Biofuel Cells, and Nanotechnology. Chem. Soc. Rev. 2011, 40, 3564–3576. 10.1039/c1cs15030h. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Patolsky F.; Katz E.; Hainfeld J. F.; Willner I. ″es″: Nanowiring of Redox Enzymes by a Gold Nanoparticle. Science 2003, 299, 1877–1881. 10.1126/science.1080664. [DOI] [PubMed] [Google Scholar]
- Wang W.; Xu X.; Zhou W.; Shao Z. Recent Progress in Metal-Organic Frameworks for Applications in Electrocatalytic and Photocatalytic Water Splitting. Adv. Sci. 2017, 4, 1600371. 10.1002/advs.201600371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Astruc D. State of the Art and Prospects in Metal-Organic Framework (MOF)-Based and MOF-Derived Nanocatalysis. Chem. Rev. 2020, 120, 1438–1511. 10.1021/acs.chemrev.9b00223. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Ma Y.; Zheng M.; Yang P.; Asiri A. M.; Wang X. Metal-Organic Frameworks for Solar Energy Conversion by Photoredox Catalysis. Coord. Chem. Rev. 2018, 373, 83–115. 10.1016/j.ccr.2017.09.013. [DOI] [Google Scholar]
- Banerjee S.; Anayah R. I.; Gerke C. S.; Thoi V. S. From Molecules to Porous Materials: Integrating Discrete Electrocatalytic Active Sites into Extended Frameworks. ACS Cent. Sci. 2020, 6, 1671–1684. 10.1021/acscentsci.0c01088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu T. N.; Nguyen M. V.; Nguyen H. L.; Yuliarto B.; Cordova K. E.; Demir S. Designing bipyridine-Functionalized Zirconium Metal-Organic Frameworks as a Platform for Clean Energy and Other Emerging Applications. Coord. Chem. Rev. 2018, 364, 33–50. 10.1016/j.ccr.2018.03.014. [DOI] [Google Scholar]
- Dhakshinamoorthy A.; Li Z.; Garcia H. Catalysis and Photocatalysis by Metal Organic Frameworks. Chem. Soc. Rev. 2018, 47, 8134–8172. 10.1039/C8CS00256H. [DOI] [PubMed] [Google Scholar]
- Cohen S. M.; Zhang Z.; Boissonnault J. A. Toward ″metalloMOFzymes″: Metal-Organic Frameworks with Single-Site Metal Catalysts for Small-Molecule Transformations. Inorg. Chem. 2016, 55, 7281–7290. 10.1021/acs.inorgchem.6b00828. [DOI] [PubMed] [Google Scholar]
- Chen K.; Wu C.-D. Designed Fabrication of Biomimetic Metal-Organic Frameworks for Catalytic Applications. Coord. Chem. Rev. 2019, 378, 445–465. 10.1016/j.ccr.2018.01.016. [DOI] [Google Scholar]
- Nath I.; Chakraborty J.; Verpoort F. Metal Organic Frameworks Mimicking Natural Enzymes: A Structural and Functional Analogy. Chem. Soc. Rev. 2016, 45, 4127–4170. 10.1039/C6CS00047A. [DOI] [PubMed] [Google Scholar]
- Ma L.; Jiang F.; Fan X.; Wang L.; He C.; Zhou M.; Li S.; Luo H.; Cheng C.; Qiu L. Metal-Organic-Framework-Engineered Enzyme-Mimetic Catalysts. Adv. Mater. 2020, 32, e2003065 10.1002/adma.202003065. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Ou S.; Wu C. D. Porous Metal-Organic Frameworks for Heterogeneous Biomimetic Catalysis. Acc. Chem. Res. 2014, 47, 1199–1207. 10.1021/ar400265x. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy A.; Asiri A. M.; Garcia H. Metal-Organic Frameworks as Catalysts for Oxidation Reactions. Chem. Eur. J. 2016, 22, 8012–8024. 10.1002/chem.201505141. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S.; Basu O.; Nasani R.; Das S. K. Evolution of Metal Organic Frameworks as Electrocatalysts for Water Oxidation. Chem. Commun. 2020, 56, 11735–11748. 10.1039/D0CC03659E. [DOI] [PubMed] [Google Scholar]
- Shao Q.; Yang J.; Huang X. The Design of Water Oxidation Electrocatalysts from Nanoscale Metal-Organic Frameworks. Chem. Eur. J. 2018, 24, 15143–15155. 10.1002/chem.201801572. [DOI] [PubMed] [Google Scholar]
- Du J.; Li F.; Sun L. Metal-Organic Frameworks and Their Derivatives as Electrocatalysts for the Oxygen Evolution Reaction. Chem. Soc. Rev. 2021, 50, 2663–2695. 10.1039/D0CS01191F. [DOI] [PubMed] [Google Scholar]
- Zhu B.; Zou R.; Xu Q. Metal-Organic Framework Based Catalysts for Hydrogen Evolution. Adv. Energy Mater. 2018, 8, 1801193. 10.1002/aenm.201801193. [DOI] [Google Scholar]
- Reddy D. A.; Kim Y.; Gopannagari M.; Kumar D. P.; Kim T. K. Recent Advances in Metal-Organic Framework-Based Photocatalysts for Hydrogen Production. Sustainable Energy Fuels 2021, 5, 1597–1618. 10.1039/C9SE00749K. [DOI] [Google Scholar]
- Budnikova Y. H. Recent Advances in Metal-Organic Frameworks for Electrocatalytic Hydrogen Evolution and Overall Water Splitting Reactions. Dalton Trans. 2020, 49, 12483–12502. 10.1039/D0DT01741H. [DOI] [PubMed] [Google Scholar]
- Alkhatib I. I.; Garlisi C.; Pagliaro M.; Al-Ali K.; Palmisano G. Metal-Organic Frameworks for Photocatalytic CO2 Reduction under Visible Radiation: A Review of Strategies and Applications. Catal. Today 2020, 340, 209–224. 10.1016/j.cattod.2018.09.032. [DOI] [Google Scholar]
- Chen Y.; Wang D.; Deng X.; Li Z. Metal-Organic Frameworks (MOFs) for Photocatalytic CO2 Reduction. Catal. Sci. Technol. 2017, 7, 4893–4904. 10.1039/C7CY01653K. [DOI] [Google Scholar]
- Li X.; Zhu Q.-L. MOF-Based Materials for Photo- and Electrocatalytic CO2 Reduction. EnergyChem. 2020, 2, 100033. 10.1016/j.enchem.2020.100033. [DOI] [Google Scholar]
- Lu X. F.; Xia B. Y.; Zang S. Q.; Lou X. W. D. Metal-Organic Frameworks Based Electrocatalysts for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2020, 59, 4634–4650. 10.1002/anie.201910309. [DOI] [PubMed] [Google Scholar]
- Chen L.; Luque R.; Li Y. Controllable Design of Tunable Nanostructures inside Metal-Organic Frameworks. Chem. Soc. Rev. 2017, 46, 4614–4630. 10.1039/C6CS00537C. [DOI] [PubMed] [Google Scholar]
- Ifraemov R.; Mukhopadhyay S.; Rozenberg I.; Hod I. Metal-Organic-Framework-Based Photo-Electrochemical Cells for Solar Fuel Generation. J. Phys. Chem. C 2022, 126, 5079–5091. 10.1021/acs.jpcc.2c00671. [DOI] [Google Scholar]
- Du Y.; Jia X.; Zhong L.; Jiao Y.; Zhang Z.; Wang Z.; Feng Y.; Bilal M.; Cui J.; Jia S. Metal-Organic Frameworks with Different Dimensionalities: An Ideal Host Platform for Enzyme@MOF Composites. Coord. Chem. Rev. 2022, 454, 214327. 10.1016/j.ccr.2021.214327. [DOI] [Google Scholar]
- Wang C.; Liao K. Recent Advances in Emerging Metal- and Covalent-Organic Frameworks for Enzyme Encapsulation. ACS Appl. Mater. Interfaces 2021, 13, 56752–56776. 10.1021/acsami.1c13408. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy A.; Asiri A. M.; Garcia H. Integration of Metal Organic Frameworks with Enzymes as Multifunctional Solids for Cascade Catalysis. Dalton Trans. 2020, 49, 11059–11072. 10.1039/D0DT02045A. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Savéant J.-M.. Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, 2019; pp 285–379. [Google Scholar]
- Costentin C.; Robert M.; Savéant J.-M. Molecular Catalysis of Electrochemical Reactions. Curr. Opin. Electrochem. 2017, 2, 26–31. 10.1016/j.coelec.2017.02.006. [DOI] [Google Scholar]
- Costentin C.; Savéant J.-M. Homogeneous Molecular Catalysis of Electrochemical Reactions: Manipulating Intrinsic and Operational Factors for Catalyst Improvement. J. Am. Chem. Soc. 2018, 140, 16669–16675. 10.1021/jacs.8b09154. [DOI] [PubMed] [Google Scholar]
- Costentin C. Molecular Catalysis of Electrochemical Reactions. Overpotential and Turnover Frequency: Unidirectional and Bidirectional Systems. ACS Catal. 2021, 11, 5678–5687. 10.1021/acscatal.1c00744. [DOI] [Google Scholar]
- Johnson B. A.; Ott S. Diagnosing Surface Versus Bulk Reactivity for Molecular Catalysis within Metal-Organic Frameworks Using a Quantitative Kinetic Model. Chem. Sci. 2020, 11, 7468–7478. 10.1039/D0SC02601H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damköhler G. Einflüsse der Strömung, Diffusion und des Wärmeüberganges auf die Leistung von Reaktionsöfen.: I. Allgemeine Gesichtspunkte für die Übertragung eines Chemischen Prozesses aus dem Kleinen ins Große. Z. Elektrochem. Angew. Phys. Chem. 1936, 42, 846–862. [Google Scholar]
- Thiele E. W. Relation between Catalytic Activity and Size of Particle. Ind. Eng. Chem. 1939, 31, 916–920. 10.1021/ie50355a027. [DOI] [Google Scholar]
- Wang R.; Bukowski B. C.; Duan J.; Sheridan T. R.; Atilgan A.; Zhang K.; Snurr R. Q.; Hupp J. T. Investigating the Process and Mechanism of Molecular Transport within a Representative Solvent-Filled Metal-Organic Framework. Langmuir 2020, 36, 10853–10859. 10.1021/acs.langmuir.0c01999. [DOI] [PubMed] [Google Scholar]
- Kärger J.; Binder T.; Chmelik C.; Hibbe F.; Krautscheid H.; Krishna R.; Weitkamp J. Microimaging of Transient Guest Profiles to Monitor Mass Transfer in Nanoporous Materials. Nat. Mater. 2014, 13, 333–343. 10.1038/nmat3917. [DOI] [PubMed] [Google Scholar]
- Titze T.; Lauerer A.; Heinke L.; Chmelik C.; Zimmermann N. E. R.; Keil F. J.; Ruthven D. M.; Kärger J. Transport in Nanoporous Materials Including MOFs: The Applicability of Fick’s Laws. Angew. Chem., Int. Ed. 2015, 54, 14580–14583. 10.1002/anie.201506954. [DOI] [PubMed] [Google Scholar]
- Han S.; Hermans T. M.; Fuller P. E.; Wei Y.; Grzybowski B. A. Transport into Metal-Organic Frameworks from Solution Is Not Purely Diffusive. Angew. Chem., Int. Ed. 2012, 51, 2662–2666. 10.1002/anie.201108492. [DOI] [PubMed] [Google Scholar]
- Leddy J.; Bard A. J.; Maloy J. T.; Savéant J. M. Kinetics of Film-Coated Electrodes: Effect of a Finite Mass Transfer Rate of Substrate across the Film—Solution Interface at Steady State. J. Electroanal. Chem. Interfacial Electrochem. 1985, 187, 205–227. 10.1016/0368-1874(85)85779-8. [DOI] [Google Scholar]
- Heinke L.; Gu Z.; Woll C. The Surface Barrier Phenomenon at the Loading of Metal-Organic Frameworks. Nat. Commun. 2014, 5, 4562. 10.1038/ncomms5562. [DOI] [PubMed] [Google Scholar]
- Feng X.; Song Y.; Lin W. Dimensional Reduction of Lewis Acidic Metal-Organic Frameworks for Multicomponent Reactions. J. Am. Chem. Soc. 2021, 143, 8184–8192. 10.1021/jacs.1c03561. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Shi W.; Yang H.; He Q.; Zeng Z.; Ye J. Y.; He X.; Huang R.; Wang C.; Lin W. Cooperative Stabilization of the [Pyridinium-CO2-Co] Adduct on a Metal-Organic Layer Enhances Electrocatalytic CO2 Reduction. J. Am. Chem. Soc. 2019, 141, 17875–17883. 10.1021/jacs.9b09227. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Wang Y.; Shen Y.; Cai Z.; Li Z.; Liu J.; Chen J.; Xiao C.; Liu H.; Lin W.; et al. Tunable Cobalt-Polypyridyl Catalysts Supported on Metal-Organic Layers for Electrochemical CO2 Reduction at Low Overpotentials. J. Am. Chem. Soc. 2020, 142, 21493–21501. 10.1021/jacs.0c10719. [DOI] [PubMed] [Google Scholar]
- Koros R. M.; Nowak E. J. A Diagnostic Test of the Kinetic Regime in a Packed Bed Reactor. Chem. Eng. Sci. 1967, 22, 470. 10.1016/0009-2509(67)80134-9. [DOI] [Google Scholar]
- Madon R. J.; Boudart M. Experimental Criterion for the Absence of Artifacts in the Measurement of Rates of Heterogeneous Catalytic Reactions. Ind. Eng. Chem. Fundam. 1982, 21, 438–447. 10.1021/i100008a022. [DOI] [Google Scholar]
- Semrau A. L.; Stanley P. M.; Urstoeger A.; Schuster M.; Cokoja M.; Fischer R. A. Substantial Turnover Frequency Enhancement of MOF Catalysts by Crystallite Downsizing Combined with Surface Anchoring. ACS Catal. 2020, 10, 3203–3211. 10.1021/acscatal.0c00550. [DOI] [Google Scholar]
- Stanley P. M.; Parkulab M.; Rieger B.; Warnan J.; Fischer R. A. Understanding Entrapped Molecular Photosystem and Metal-Organic Framework Synergy for Improved Solar Fuel Production. Faraday Discuss. 2021, 231, 281–297. 10.1039/D1FD00009H. [DOI] [PubMed] [Google Scholar]
- Wang C.; Wang J. L.; Lin W. Elucidating Molecular Iridium Water Oxidation Catalysts Using Metal-Organic Frameworks: A Comprehensive Structural, Catalytic, Spectroscopic, and Kinetic Study. J. Am. Chem. Soc. 2012, 134, 19895–19908. 10.1021/ja310074j. [DOI] [PubMed] [Google Scholar]
- Dou J.-H.; Sun L.; Ge Y.; Li W.; Hendon C. H.; Li J.; Gul S.; Yano J.; Stach E. A.; Dincǎ M. Signature of Metallic Behavior in the Metal-Organic Frameworks M3(hexaiminobenzene)2 (M = Ni, Cu). J. Am. Chem. Soc. 2017, 139, 13608–13611. 10.1021/jacs.7b07234. [DOI] [PubMed] [Google Scholar]
- Clough A. J.; Orchanian N. M.; Skelton J. M.; Neer A. J.; Howard S. A.; Downes C. A.; Piper L. F. J.; Walsh A.; Melot B. C.; Marinescu S. C. Room Temperature Metallic Conductivity in a Metal-Organic Framework Induced by Oxidation. J. Am. Chem. Soc. 2019, 141, 16323–16330. 10.1021/jacs.9b06898. [DOI] [PubMed] [Google Scholar]
- Sun L.; Campbell M. G.; Dincǎ M. Electrically Conductive Porous Metal-Organic Frameworks. Angew. Chem., Int. Ed. 2016, 55, 3566–3579. 10.1002/anie.201506219. [DOI] [PubMed] [Google Scholar]
- Miner E. M.; Fukushima T.; Sheberla D.; Sun L.; Surendranath Y.; Dincǎ M. Electrochemical Oxygen Reduction Catalysed by Ni3(hexaiminotriphenylene)2. Nat. Commun. 2016, 7, 10942. 10.1038/ncomms10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassen I.; Burtch N.; Talin A.; Falcaro P.; Allendorf M.; Ameloot R. An Updated Roadmap for the Integration of Metal-Organic Frameworks with Electronic Devices and Chemical Sensors. Chem. Soc. Rev. 2017, 46, 3185–3241. 10.1039/C7CS00122C. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Drouet S.; Robert M.; Savéant J.-M. Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. 10.1021/ja303560c. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Savéant J.-M. Cyclic Voltammetry of Fast Conducting Electrocatalytic Films. Phys. Chem. Chem. Phys. 2015, 17, 19350–19359. 10.1039/C5CP02825F. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Savéant J.-M. Cyclic Voltammetry of Electrocatalytic Films: Fast Catalysis Regimes. ChemElectroChem. 2015, 2, 1774–1784. 10.1002/celc.201500217. [DOI] [Google Scholar]
- Costentin C.; Savéant J.-M. Cyclic Voltammetry Analysis of Electrocatalytic Films. J. Phys. Chem. C 2015, 119, 12174–12182. 10.1021/acs.jpcc.5b02376. [DOI] [PubMed] [Google Scholar]
- Bard A. J. Inner-Sphere Heterogeneous Electrode Reactions. Electrocatalysis and Photocatalysis: The Challenge. J. Am. Chem. Soc. 2010, 132, 7559–7567. 10.1021/ja101578m. [DOI] [PubMed] [Google Scholar]
- Limaye A. M.; Zeng J. S.; Willard A. P.; Manthiram K. Bayesian Data Analysis Reveals No Preference for Cardinal Tafel Slopes in CO2 Reduction Electrocatalysis. Nat. Commun. 2021, 12, 703. 10.1038/s41467-021-20924-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinagawa T.; Garcia-Esparza A. T.; Takanabe K. Insight on Tafel Slopes from a Microkinetic Analysis of Aqueous Electrocatalysis for Energy Conversion. Sci. Rep. 2015, 5, 13801. 10.1038/srep13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J. N.; Prats H.; Toudahl K. K.; Mørch Secher N.; Chan K.; Kibsgaard J.; Chorkendorff I. Is There Anything Better Than Pt for HER?. ACS Energy Lett. 2021, 6, 1175–1180. 10.1021/acsenergylett.1c00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariano R. G.; Wahab O. J.; Rabinowitz J. A.; Oppenheim J.; Chen T.; Unwin P. R.; Dincǎ M. Thousand-Fold Increase in O2 Electroreduction Rates with Conductive MOFs. ACS Cent. Sci. 2022, 8, 975–982. 10.1021/acscentsci.2c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Electron Tunneling through Proteins. Q. Rev. Biophys. 2003, 36, 341–372. 10.1017/S0033583503003913. [DOI] [PubMed] [Google Scholar]
- Page C. C.; Moser C. C.; Chen X.; Dutton P. L. Natural Engineering Principles of Electron Tunnelling in Biological Oxidation-Reduction. Nature 1999, 402, 47–52. 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- Brodsky C. N.; Bediako D. K.; Shi C.; Keane T. P.; Costentin C.; Billinge S. J. L.; Nocera D. G. Proton-Electron Conductivity in Thin Films of a Cobalt-Oxygen Evolving Catalyst. ACS Appl. Energy Mater. 2019, 2, 3–12. 10.1021/acsaem.8b00785. [DOI] [Google Scholar]
- Costentin C.; Nocera D. G. Dual-Phase Molecular-Like Charge Transport in Nanoporous Transition Metal Oxides. J. Phys. Chem. C 2019, 123, 1966–1973. 10.1021/acs.jpcc.8b10948. [DOI] [Google Scholar]
- Andrieux C. P.; Savéant J. M.. Catalysis at Redox Polymer Coated Electrodes; Murray R. W., Ed.; John Wiley & Sons, Inc,: Hoboken, 1992; pp 207–270. [Google Scholar]
- Costentin C.; Savéant J.-M. Molecular Approach to Catalysis of Electrochemical Reaction in Porous Films. Curr. Opin. Electrochem. 2019, 15, 58–65. 10.1016/j.coelec.2019.03.014. [DOI] [Google Scholar]
- Savéant J.-M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. 10.1021/cr068079z. [DOI] [PubMed] [Google Scholar]
- Kozuch S.; Martin J. M. L. Turning over” Definitions in Catalytic Cycles. ACS Catal. 2012, 2, 2787–2794. 10.1021/cs3005264. [DOI] [Google Scholar]
- Tafel J. Über die Polarisation bei Kathodischer Wasserstoffentwicklung. Z. Phys. Chem. 1905, 50U, 641–712. 10.1515/zpch-1905-5043. [DOI] [Google Scholar]
- Batchelor-McAuley C.; Katelhon E.; Barnes E. O.; Compton R. G.; Laborda E.; Molina A. Recent Advances in Voltammetry. ChemistryOpen 2015, 4, 224–260. 10.1002/open.201500042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn A. W.; Levey K. J.; O’Hare D.; Macpherson J. V. Lifting the Lid on the Potentiostat: A Beginner’s Guide to Understanding Electrochemical Circuitry and Practical Operation. Phys. Chem. Chem. Phys. 2021, 23, 8100–8117. 10.1039/D1CP00661D. [DOI] [PubMed] [Google Scholar]
- Chang M. C.; Yee C. S.; Stubbe J.; Nocera D. G. Turning on Ribonucleotide Reductase by Light-Initiated Amino Acid Radical Generation. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 6882–6887. 10.1073/pnas.0401718101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisner E.; Powell D. J.; Cavazza C.; Fontecilla-Camps J. C.; Armstrong F. A. Visible Light-Driven H2 Production by Hydrogenases Attached to Dye-Sensitized TiO2 Nanoparticles. J. Am. Chem. Soc. 2009, 131, 18457–18466. 10.1021/ja907923r. [DOI] [PubMed] [Google Scholar]
- Lubner C. E.; Knorzer P.; Silva P. J.; Vincent K. A.; Happe T.; Bryant D. A.; Golbeck J. H. Wiring an [FeFe]-Hydrogenase with Photosystem I for Light-Induced Hydrogen Production. Biochemistry 2010, 49, 10264–10266. 10.1021/bi1016167. [DOI] [PubMed] [Google Scholar]
- Osterloh F. E. Photocatalysis Versus Photosynthesis: A Sensitivity Analysis of Devices for Solar Energy Conversion and Chemical Transformations. ACS Energy Lett. 2017, 2, 445–453. 10.1021/acsenergylett.6b00665. [DOI] [Google Scholar]
- Neshvad G.; Hoffman M. Z. Reductive Quenching of the Luminescent Excited State of Tris(2,2’-bipyrazine)ruthenium(2+) Ion in Aqueous Solution. J. Phys. Chem. 1989, 93, 2445–2452. 10.1021/j100343a044. [DOI] [Google Scholar]
- Mateo D.; Santiago-Portillo A.; Albero J.; Navalon S.; Alvaro M.; Garcia H. Long-Term Photostability in Terephthalate Metal-Organic Frameworks. Angew. Chem., Int. Ed. 2019, 58, 17843–17848. 10.1002/anie.201911600. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Camara F.; Fortage J.; Collomb M.-N. Photoinduced Catalysis of Redox Reactions. Turnover Numbers, Turnover Frequency, and Limiting Processes: Kinetic Analysis and Application to Light-Driven Hydrogen Production. ACS Catal. 2022, 6246–6254. 10.1021/acscatal.2c01289. [DOI] [Google Scholar]
- Lakadamyali F.; Kato M.; Reisner E. Colloidal Metal Oxide Particles Loaded with Synthetic Catalysts for Solar H2 Production. Faraday Discuss. 2012, 155, 191–205. 10.1039/C1FD00077B. [DOI] [PubMed] [Google Scholar]
- Limburg B.; Bouwman E.; Bonnet S. Rate and Stability of Photocatalytic Water Oxidation Using [Ru(bpy)3]2+ as Photosensitizer. ACS Catal. 2016, 6, 5273–5284. 10.1021/acscatal.6b00107. [DOI] [Google Scholar]
- Limburg B.; Wermink J.; van Nielen S. S.; Kortlever R.; Koper M. T. M.; Bouwman E.; Bonnet S. Kinetics of Photocatalytic Water Oxidation at Liposomes: Membrane Anchoring Stabilizes the Photosensitizer. ACS Catal. 2016, 6, 5968–5977. 10.1021/acscatal.6b00151. [DOI] [Google Scholar]
- Fang Z. B.; Liu T. T.; Liu J.; Jin S.; Wu X. P.; Gong X. Q.; Wang K.; Yin Q.; Liu T. F.; Cao R.; et al. Boosting Interfacial Charge-Transfer Kinetics for Efficient Overall CO2 Photoreduction via Rational Design of Coordination Spheres on Metal-Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 12515–12523. 10.1021/jacs.0c05530. [DOI] [PubMed] [Google Scholar]
- Xu H. Q.; Hu J.; Wang D.; Li Z.; Zhang Q.; Luo Y.; Yu S. H.; Jiang H. L. Visible-Light Photoreduction of CO2 in a Metal-Organic Framework: Boosting Electron-Hole Separation via Electron Trap States. J. Am. Chem. Soc. 2015, 137, 13440–13443. 10.1021/jacs.5b08773. [DOI] [PubMed] [Google Scholar]
- Wu L. Y.; Mu Y. F.; Guo X. X.; Zhang W.; Zhang Z. M.; Zhang M.; Lu T. B. Encapsulating Perovskite Quantum Dots in Iron-Based Metal-Organic Frameworks (MOFs) for Efficient Photocatalytic CO2 Reduction. Angew. Chem., Int. Ed. 2019, 58, 9491–9495. 10.1002/anie.201904537. [DOI] [PubMed] [Google Scholar]
- Fang X.; Shang Q.; Wang Y.; Jiao L.; Yao T.; Li Y.; Zhang Q.; Luo Y.; Jiang H. L. Single Pt Atoms Confined into a Metal-Organic Framework for Efficient Photocatalysis. Adv. Mater. 2018, 30, 1705112. 10.1002/adma.201705112. [DOI] [PubMed] [Google Scholar]
- Wasielewski M. R. Photoinduced Electron Transfer in Supramolecular Systems for Artificial Photosynthesis. Chem. Rev. 1992, 92, 435–461. 10.1021/cr00011a005. [DOI] [Google Scholar]
- Gust D.; Moore T. A.; Moore A. L.; Lee S. J.; Bittersmann E.; Luttrull D. K.; Rehms A. A.; Degraziano J. M.; Ma X. C.; Gao F.; et al. Efficient Multistep Photoinitiated Electron Transfer in a Molecular Pentad. Science 1990, 248, 199–201. 10.1126/science.248.4952.199. [DOI] [PubMed] [Google Scholar]
- Imahori H.; Guldi D. M.; Tamaki K.; Yoshida Y.; Luo C.; Sakata Y.; Fukuzumi S. Charge Separation in a Novel Artificial Photosynthetic Reaction Center Lives 380 ms. J. Am. Chem. Soc. 2001, 123, 6617–6628. 10.1021/ja004123v. [DOI] [PubMed] [Google Scholar]
- Lin S.; Cairnie D. R.; Davis D.; Chakraborty A.; Cai M.; Morris A. J. Photoelectrochemical Alcohol Oxidation by Mixed-Linker Metal-Organic Frameworks. Faraday Discuss. 2021, 225, 371–383. 10.1039/D0FD00021C. [DOI] [PubMed] [Google Scholar]
- Santiago-Portillo A.; Baldoví H. G.; Carbonell E.; Navalón S.; Álvaro M.; García H.; Ferrer B. Ruthenium(II) Tris(2,2′-bipyridyl) Complex Incorporated in UiO-67 as Photoredox Catalyst. J. Phys. Chem. C 2018, 122, 29190–29199. 10.1021/acs.jpcc.8b07204. [DOI] [Google Scholar]
- Tachibana Y.; Moser J. E.; Grätzel M.; Klug D. R.; Durrant J. R. Subpicosecond Interfacial Charge Separation in Dye-Sensitized Nanocrystalline Titanium Dioxide Films. J. Phys. Chem. 1996, 100, 20056–20062. 10.1021/jp962227f. [DOI] [Google Scholar]
- Reynal A.; Lakadamyali F.; Gross M. A.; Reisner E.; Durrant J. R. Parameters Affecting Electron Transfer Dynamics from Semiconductors to Molecular Catalysts for the Photochemical Reduction of Protons. Energy Environ. Sci. 2013, 6, 3291. 10.1039/c3ee40961a. [DOI] [Google Scholar]
- Bozal-Ginesta C.; Mesa C. A.; Eisenschmidt A.; Francas L.; Shankar R. B.; Anton-Garcia D.; Warnan J.; Willkomm J.; Reynal A.; Reisner E.; et al. Charge Accumulation Kinetics in Multi-Redox Molecular Catalysts Immobilised on TiO2. Chem. Sci. 2021, 12, 946–959. 10.1039/D0SC04344C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström L. Accumulative Charge Separation for Solar Fuels Production: Coupling Light-Induced Single Electron Transfer to Multielectron Catalysis. Acc. Chem. Res. 2015, 48, 840–850. 10.1021/ar500386x. [DOI] [PubMed] [Google Scholar]
- Karlsson S.; Boixel J.; Pellegrin Y.; Blart E.; Becker H. C.; Odobel F.; Hammarström L. Accumulative Charge Separation Inspired by Photosynthesis. J. Am. Chem. Soc. 2010, 132, 17977–17979. 10.1021/ja104809x. [DOI] [PubMed] [Google Scholar]
- Karlsson S.; Boixel J.; Pellegrin Y.; Blart E.; Becker H. C.; Odobel F.; Hammarström L. Accumulative Electron Transfer: Multiple Charge Separation in Artificial Photosynthesis. Faraday Discuss. 2012, 155, 233–52. 10.1039/C1FD00089F. [DOI] [PubMed] [Google Scholar]
- Kuss-Petermann M.; Orazietti M.; Neuburger M.; Hamm P.; Wenger O. S. Intramolecular Light-Driven Accumulation of Reduction Equivalents by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2017, 139, 5225–5232. 10.1021/jacs.7b01605. [DOI] [PubMed] [Google Scholar]
- Nomrowski J.; Wenger O. S. Exploiting Potential Inversion for Photoinduced Multielectron Transfer and Accumulation of Redox Equivalents in a Molecular Heptad. J. Am. Chem. Soc. 2018, 140, 5343–5346. 10.1021/jacs.8b02443. [DOI] [PubMed] [Google Scholar]
- Pannwitz A.; Wenger O. S. Proton-Coupled Multi-Electron Transfer and Its Relevance for Artificial Photosynthesis and Photoredox Catalysis. Chem. Commun. 2019, 55, 4004–4014. 10.1039/C9CC00821G. [DOI] [PubMed] [Google Scholar]
- Tran T.-T.; Pino T.; Ha-Thi M.-H. Watching Intermolecular Light-Induced Charge Accumulation on Naphthalene Diimide by Tris(bipyridyl)ruthenium(II) Photosensitizer. J. Phys. Chem. C 2019, 123, 28651–28658. 10.1021/acs.jpcc.9b09556. [DOI] [Google Scholar]
- Chen H. Y.; Ardo S. Direct Observation of Sequential Oxidations of a Titania-Bound Molecular Proxy Catalyst Generated through Illumination of Molecular Sensitizers. Nat. Chem. 2018, 10, 17–23. 10.1038/nchem.2892. [DOI] [PubMed] [Google Scholar]
- Cardona T.; Sedoud A.; Cox N.; Rutherford A. W. Charge Separation in Photosystem II: A Comparative and Evolutionary Overview. Biochim. Biophys. Acta 2012, 1817, 26–43. 10.1016/j.bbabio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Kent C. A.; Liu D.; Meyer T. J.; Lin W. Amplified Luminescence Quenching of Phosphorescent Metal-Organic Frameworks. J. Am. Chem. Soc. 2012, 134, 3991–3994. 10.1021/ja211271m. [DOI] [PubMed] [Google Scholar]
- Son H. J.; Jin S.; Patwardhan S.; Wezenberg S. J.; Jeong N. C.; So M.; Wilmer C. E.; Sarjeant A. A.; Schatz G. C.; Snurr R. Q.; et al. Light-Harvesting and Ultrafast Energy Migration in Porphyrin-Based Metal-Organic Frameworks. J. Am. Chem. Soc. 2013, 135, 862–869. 10.1021/ja310596a. [DOI] [PubMed] [Google Scholar]
- Choi S.; Jung W. J.; Park K.; Kim S. Y.; Baeg J. O.; Kim C. H.; Son H. J.; Pac C.; Kang S. O. Rapid Exciton Migration and Amplified Funneling Effects of Multi-Porphyrin Arrays in a Re(I)/Porphyrinic MOF Hybrid for Photocatalytic CO2 Reduction. ACS Appl. Mater. Interfaces 2021, 13, 2710–2722. 10.1021/acsami.0c19856. [DOI] [PubMed] [Google Scholar]
- Markovic V.; Villamaina D.; Barabanov I.; Daku L. M.; Vauthey E. Photoinduced Symmetry-Breaking Charge Separation: The Direction of the Charge Transfer. Angew. Chem., Int. Ed. 2011, 50, 7596–7598. 10.1002/anie.201102601. [DOI] [PubMed] [Google Scholar]
- Kaul N.; Lomoth R. The Carbene Cannibal: Photoinduced Symmetry-Breaking Charge Separation in an Fe(III) N-Heterocyclic Carbene. J. Am. Chem. Soc. 2021, 143, 10816–10821. 10.1021/jacs.1c03770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. M.; Wasielewski M. R. Mixed Electronic States in Molecular Dimers: Connecting Singlet Fission, Excimer Formation, and Symmetry-Breaking Charge Transfer. Acc. Chem. Res. 2020, 53, 1957–1968. 10.1021/acs.accounts.0c00397. [DOI] [PubMed] [Google Scholar]
- Sebastian E.; Hariharan M. Symmetry-Breaking Charge Separation in Molecular Constructs for Efficient Light Energy Conversion. ACS Energy Lett. 2022, 7, 696–711. 10.1021/acsenergylett.1c02412. [DOI] [Google Scholar]
- Baxendale J. H.; Rodgers M. A. J. Abnormal Decay Kinetics of the Excited State of Ruthenium Trisbipyridyl Ions in Surfactant Solutions. Chem. Phys. Lett. 1980, 72, 424–426. 10.1016/0009-2614(80)80322-8. [DOI] [Google Scholar]
- Baxendale J. H.; Rodgers M. A. J. Fluorescence of Tris(2,2’-bipyridyl)ruthenium(II) in Sodium Dodecyl Sulfate Solutions Below the Critical Micelle Concentration. J. Phys. Chem. 1982, 86, 4906–4909. 10.1021/j100222a014. [DOI] [Google Scholar]
- Ghosh P. K.; Bard A. J. Photochemistry of Tris(2,2’-bipyridyl)(ruthenium(II) in Colloidal Clay Suspensions. J. Phys. Chem. 1984, 88, 5519–5526. 10.1021/j150667a012. [DOI] [Google Scholar]
- Cao W.; Tang Y.; Cui Y.; Qian G. Energy Transfer in Metal-Organic Frameworks and Its Applications. Small Struct. 2020, 1, 2000019. 10.1002/sstr.202000019. [DOI] [Google Scholar]
- Shaikh S. M.; Ilic S.; Gibbons B. J.; Yang X.; Jakubikova E.; Morris A. J. Role of a 3D Structure in Energy Transfer in Mixed-Ligand Metal-Organic Frameworks. J. Phys. Chem. C 2021, 125, 22998–23010. 10.1021/acs.jpcc.1c06427. [DOI] [Google Scholar]
- Marcus R. A.; Sutin N. Electron Transfers in Chemistry and Biology. Biochim. Biophys. Acta, Rev. Bioenerg. 1985, 811, 265–322. 10.1016/0304-4173(85)90014-X. [DOI] [Google Scholar]
- Closs G. L.; Miller J. R. Intramolecular Long-Distance Electron Transfer in Organic Molecules. Science 1988, 240, 440–447. 10.1126/science.240.4851.440. [DOI] [PubMed] [Google Scholar]
- Weiss E. A.; Wasielewski M. R.; Ratner M. A. Molecules as Wires: Molecule-Assisted Movement of Charge and Energy. Top. Curr. Chem. 2005, 257, 103–133. 10.1007/b136068. [DOI] [PubMed] [Google Scholar]
- An Y.; Liu Y.; An P.; Dong J.; Xu B.; Dai Y.; Qin X.; Zhang X.; Whangbo M. H.; Huang B. Ni(II) Coordination to an Al-Based Metal-Organic Framework Made from 2-Aminoterephthalate for Photocatalytic Overall Water Splitting. Angew. Chem., Int. Ed. 2017, 56, 3036–3040. 10.1002/anie.201612423. [DOI] [PubMed] [Google Scholar]
- Dong L. Z.; Zhang L.; Liu J.; Huang Q.; Lu M.; Ji W. X.; Lan Y. Q. Stable Heterometallic Cluster-Based Organic Framework Catalysts for Artificial Photosynthesis. Angew. Chem., Int. Ed. 2020, 59, 2659–2663. 10.1002/anie.201913284. [DOI] [PubMed] [Google Scholar]
- Gao W. Y.; Ngo H. T.; Niu Z.; Zhang W.; Pan Y.; Yang Z.; Bhethanabotla V. R.; Joseph B.; Aguila B.; Ma S. A Mixed-Metal Porphyrinic Framework Promoting Gas-Phase CO2 Photoreduction without Organic Sacrificial Agents. ChemSusChem 2020, 13, 6273–6277. 10.1002/cssc.202002414. [DOI] [PubMed] [Google Scholar]
- Hu H.; Wang Z.; Cao L.; Zeng L.; Zhang C.; Lin W.; Wang C. Metal-Organic Frameworks Embedded in a Liposome Facilitate Overall Photocatalytic Water Splitting. Nat. Chem. 2021, 13, 358–366. 10.1038/s41557-020-00635-5. [DOI] [PubMed] [Google Scholar]
- SK M.; Barman S.; Paul S.; De R.; Sreejith S. S.; Reinsch H.; Grzywa M.; Stock N.; Volkmer D.; Biswas S.; et al. An Anthracene-Based Metal-Organic Framework for Selective Photo-Reduction of Carbon Dioxide to Formic Acid Coupled with Water Oxidation. Chem. Eur. J. 2021, 27, 4098–4107. 10.1002/chem.202004596. [DOI] [PubMed] [Google Scholar]
- Karmakar S.; Barman S.; Rahimi F. A.; Maji T. K. Covalent Grafting of Molecular Photosensitizer and Catalyst on MOF-808: Effect of Pore Confinement toward Visible Light-Driven CO2 Reduction in Water. Energy Environ. Sci. 2021, 14, 2429–2440. 10.1039/D0EE03643A. [DOI] [Google Scholar]
- Ager J. W.; Shaner M. R.; Walczak K. A.; Sharp I. D.; Ardo S. Experimental Demonstrations of Spontaneous, Solar-Driven Photoelectrochemical Water Splitting. Energy Environ. Sci. 2015, 8, 2811–2824. 10.1039/C5EE00457H. [DOI] [Google Scholar]
- Fabian D. M.; Hu S.; Singh N.; Houle F. A.; Hisatomi T.; Domen K.; Osterloh F. E.; Ardo S. Particle Suspension Reactors and Materials for Solar-Driven Water Splitting. Energy Environ. Sci. 2015, 8, 2825–2850. 10.1039/C5EE01434D. [DOI] [Google Scholar]
- Cheng W.-H.; Richter M. H.; Sullivan I.; Larson D. M.; Xiang C.; Brunschwig B. S.; Atwater H. A. CO2 Reduction to CO with 19% Efficiency in a Solar-Driven Gas Diffusion Electrode Flow Cell under Outdoor Solar Illumination. ACS Energy Lett. 2020, 5, 470–476. 10.1021/acsenergylett.9b02576. [DOI] [Google Scholar]
- Cheng W.-H.; Richter M. H.; May M. M.; Ohlmann J.; Lackner D.; Dimroth F.; Hannappel T.; Atwater H. A.; Lewerenz H.-J. Monolithic Photoelectrochemical Device for Direct Water Splitting with 19% Efficiency. ACS Energy Lett. 2018, 3, 1795–1800. 10.1021/acsenergylett.8b00920. [DOI] [Google Scholar]
- Cox C. R.; Lee J. Z.; Nocera D. G.; Buonassisi T. Ten-Percent Solar-to-Fuel Conversion with Nonprecious Materials. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 14057–14061. 10.1073/pnas.1414290111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K. M.; Kim D.; Rungtaweevoranit B.; Trickett C. A.; Barmanbek J. T.; Alshammari A. S.; Yang P.; Yaghi O. M. Plasmon-Enhanced Photocatalytic CO2 Conversion within Metal-Organic Frameworks under Visible Light. J. Am. Chem. Soc. 2017, 139, 356–362. 10.1021/jacs.6b11027. [DOI] [PubMed] [Google Scholar]
- Roy S.; Pascanu V.; Pullen S.; Miera G. G.; Martin-Matute B.; Ott S. Catalyst Accessibility to Chemical Reductants in Metal-Organic Frameworks. Chem. Commun. 2017, 53, 3257–3260. 10.1039/C7CC00022G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynal A.; Pastor E.; Gross M. A.; Selim S.; Reisner E.; Durrant J. R. Unravelling the pH-Dependence of a Molecular Photocatalytic System for Hydrogen Production. Chem. Sci. 2015, 6, 4855–4859. 10.1039/C5SC01349F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streich D.; Astuti Y.; Orlandi M.; Schwartz L.; Lomoth R.; Hammarström L.; Ott S. High-Turnover Photochemical Hydrogen Production Catalyzed by a Model Complex of the [FeFe]-Hydrogenase Active Site. Chem. Eur. J. 2010, 16, 60–63. 10.1002/chem.200902489. [DOI] [PubMed] [Google Scholar]
- Streich D.Stepping into Catalysis: Kinetic and Mechanistic Investigations of Photo- and Electrocatalytic Hydrogen Production with Natural and Synthetic Molecular Catalysts. Ph.D. Thesis, University of Uppsala, Sweden, 2013; pp 56–59. [Google Scholar]
- Jasniewski A. J.; Que L. Jr. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev. 2018, 118, 2554–2592. 10.1021/acs.chemrev.7b00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen S.; Fei H. H.; Orthaber A.; Cohen S. M.; Ott S. Enhanced Photochemical Hydrogen Production by a Molecular Diiron Catalyst Incorporated into a Metal-Organic Framework. J. Am. Chem. Soc. 2013, 135, 16997–17003. 10.1021/ja407176p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozal-Ginesta C.; Pullen S.; Ott S.; Hammarström L. Self-Recovery of Photochemical H2 Evolution with a Molecular Diiron Catalyst Incorporated in a UiO-66 Metal-Organic Framework. Chemphotochem 2020, 4, 287–290. 10.1002/cptc.201900273. [DOI] [Google Scholar]
- Singh P. S.; Rudbeck H. C.; Huang P.; Ezzaher S.; Eriksson L.; Stein M.; Ott S.; Lomoth R. (I,0) Mixed-Valence State of a Diiron Complex with Pertinence to the [FeFe]-Hydrogenase Active Site: An IR, EPR, and Computational Study. Inorg. Chem. 2009, 48, 10883–10885. 10.1021/ic9016454. [DOI] [PubMed] [Google Scholar]
- Aguirre de Carcer I.; DiPasquale A.; Rheingold A. L.; Heinekey D. M. Active-Site Models for Iron Hydrogenases: Reduction Chemistry of Dinuclear Iron Complexes. Inorg. Chem. 2006, 45, 8000–8002. 10.1021/ic0610381. [DOI] [PubMed] [Google Scholar]
- Sasan K.; Lin Q. P.; Mao C. Y.; Feng P. Y. Incorporation of Iron Hydrogenase Active Sites into a Highly Stable Metal-Organic Framework for Photocatalytic Hydrogen Generation. Chem. Commun. 2014, 50, 10390–10393. 10.1039/C4CC03946G. [DOI] [PubMed] [Google Scholar]
- Wang W. J.; Song X. W.; Hong Z. X.; Li B. B.; Si Y. N.; Ji C. Q.; Su K. Z.; Tan Y. X.; Ju Z. F.; Huang Y. Y.; et al. Incorporation of Iron Hydrogenase Active Sites into a Stable Photosensitizing Metal-Organic Framework for Enhanced Hydrogen Production. Appl. Catal., B 2019, 258, 117979. 10.1016/j.apcatb.2019.117979. [DOI] [Google Scholar]
- Castner A. T.; Johnson B. A.; Cohen S. M.; Ott S. Mimicking the Electron Transport Chain and Active Site of [FeFe] Hydrogenases in One Metal-Organic Framework: Factors That Influence Charge Transport. J. Am. Chem. Soc. 2021, 143, 7991–7999. 10.1021/jacs.1c01361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S.; Lu W.; Chen Y. P.; Zhang Q.; Liu T. F.; Feng D.; Wang X.; Qin J.; Zhou H. C. Sequential Linker Installation: Precise Placement of Functional Groups in Multivariate Metal-Organic Frameworks. J. Am. Chem. Soc. 2015, 137, 3177–3180. 10.1021/ja512762r. [DOI] [PubMed] [Google Scholar]
- Dempsey J. L.; Brunschwig B. S.; Winkler J. R.; Gray H. B. Hydrogen Evolution Catalyzed by Cobaloximes. Acc. Chem. Res. 2009, 42, 1995–2004. 10.1021/ar900253e. [DOI] [PubMed] [Google Scholar]
- El Ghachtouli S.; Fournier M.; Cherdo S.; Guillot R.; Charlot M.-F.; Anxolabéhère-Mallart E.; Robert M.; Aukauloo A. Monometallic Cobalt-Trisglyoximato Complexes as Precatalysts for Catalytic H2 Evolution in Water. J. Phys. Chem. C 2013, 117, 17073–17077. 10.1021/jp405134a. [DOI] [Google Scholar]
- Anxolabéhère-Mallart E.; Costentin C.; Fournier M.; Robert M. Cobalt-Bisglyoximato Diphenyl Complex as a Precatalyst for Electrocatalytic H2 Evolution. J. Phys. Chem. C 2014, 118, 13377–13381. 10.1021/jp500813r. [DOI] [Google Scholar]
- Kaeffer N.; Morozan A.; Fize J.; Martinez E.; Guetaz L.; Artero V. The Dark Side of Molecular Catalysis: Diimine-Dioxime Cobalt Complexes Are Not the Actual Hydrogen Evolution Electrocatalyst in Acidic Aqueous Solutions. ACS Catal. 2016, 6, 3727–3737. 10.1021/acscatal.6b00378. [DOI] [Google Scholar]
- Lazarides T.; McCormick T.; Du P.; Luo G.; Lindley B.; Eisenberg R. Making Hydrogen from Water Using a Homogeneous System without Noble Metals. J. Am. Chem. Soc. 2009, 131, 9192–9194. 10.1021/ja903044n. [DOI] [PubMed] [Google Scholar]
- Nasalevich M. A.; Becker R.; Ramos-Fernandez E. V.; Castellanos S.; Veber S. L.; Fedin M. V.; Kapteijn F.; Reek J. N. H.; van der Vlugt J. I.; Gascon J. Co@NH2-MIL-125(Ti): Cobaloxime-Derived Metal-Organic Framework-Based Composite for Light-Driven H2 Production. Energy Environ. Sci. 2015, 8, 364–375. 10.1039/C4EE02853H. [DOI] [Google Scholar]
- Luo S.; Liu X. Y.; Wei X. J.; Fu M.; Lu P.; Li X.; Jia Y. M.; Ren Q.; He Y. Z. Noble-Metal-Free Cobaloxime Coupled with Metal-Organic Frameworks NH2-MIL-125: A Novel Bifunctional Photocatalyst for Photocatalytic NO Removal and H2 Evolution under Visible Light Irradiation. J. Hazard. Mater. 2020, 399, 122824. 10.1016/j.jhazmat.2020.122824. [DOI] [PubMed] [Google Scholar]
- Roy S.; Bhunia A.; Schuth N.; Haumann M.; Ott S. Light-Driven Hydrogen Evolution Catalyzed by a Cobaloxime Catalyst Incorporated in a MIL-101(Cr) Metal-Organic Framework. Sustainable Energy Fuels 2018, 2, 1148–1152. 10.1039/C8SE00072G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S.; Huang Z. H.; Bhunia A.; Castner A.; Gupta A. K.; Zou X. D.; Ott S. Electrocatalytic Hydrogen Evolution from a Cobaloxime-Based Metal-Organic Framework Thin Film. J. Am. Chem. Soc. 2019, 141, 15942–15950. 10.1021/jacs.9b07084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracke J. J.; Finke R. G. Distinguishing Homogeneous from Heterogeneous Water Oxidation Catalysis When Beginning with Polyoxometalates. ACS Catal. 2014, 4, 909–933. 10.1021/cs4011716. [DOI] [Google Scholar]
- Widegren J. A.; Finke R. G. A Review of the Problem of Distinguishing True Homogeneous Catalysis from Soluble or Other Metal-Particle Heterogeneous Catalysis under Reducing Conditions. J. Mol. Catal. A: Chem. 2003, 198, 317–341. 10.1016/S1381-1169(02)00728-8. [DOI] [Google Scholar]
- Stracke J. J.; Finke R. G. Electrocatalytic Water Oxidation Beginning with the Cobalt Polyoxometalate [Co4(H2O)2(PW9O34)2]10-: Identification of Heterogeneous CoOx as the Dominant Catalyst. J. Am. Chem. Soc. 2011, 133, 14872–5. 10.1021/ja205569j. [DOI] [PubMed] [Google Scholar]
- Basu O.; Mukhopadhyay S.; Das S. K. Cobalt Based Functional Inorganic Materials: Electrocatalytic Water Oxidation. J. Chem. Sci. 2018, 130, 93. 10.1007/s12039-018-1494-4. [DOI] [Google Scholar]
- Buru C. T.; Farha O. K. Strategies for Incorporating Catalytically Active Polyoxometalates in Metal-Organic Frameworks for Organic Transformations. ACS Appl. Mater. Interfaces 2020, 12, 5345–5360. 10.1021/acsami.9b19785. [DOI] [PubMed] [Google Scholar]
- Mialane P.; Mellot-Draznieks C.; Gairola P.; Duguet M.; Benseghir Y.; Oms O.; Dolbecq A. Heterogenisation of Polyoxometalates and Other Metal-Based Complexes in Metal-Organic Frameworks: From Synthesis to Characterisation and Applications in Catalysis. Chem. Soc. Rev. 2021, 50, 6152–6220. 10.1039/D0CS00323A. [DOI] [PubMed] [Google Scholar]
- Zhang Z. M.; Zhang T.; Wang C.; Lin Z.; Long L. S.; Lin W. Photosensitizing Metal-Organic Framework Enabling Visible-Light-Driven Proton Reduction by a Wells-Dawson-Type Polyoxometalate. J. Am. Chem. Soc. 2015, 137, 3197–3200. 10.1021/jacs.5b00075. [DOI] [PubMed] [Google Scholar]
- Kong X. J.; Lin Z. K.; Zhang Z. M.; Zhang T.; Lin W. B. Hierarchical Integration of Photosensitizing Metal-Organic Frameworks and Nickel-Containing Polyoxometalates for Efficient Visible-Light-Driven Hydrogen Evolution. Angew. Chem., Int. Ed. 2016, 55, 6411–6416. 10.1002/anie.201600431. [DOI] [PubMed] [Google Scholar]
- Li H.; Yao S.; Wu H. L.; Qu J. Y.; Zhang Z. M.; Lu T. B.; Lin W. B.; Wang E. B. Charge-Regulated Sequential Adsorption of Anionic Catalysts and Cationic Photosensitizers into Metal-Organic Frameworks Enhances Photocatalytic Proton Reduction. Appl. Catal., B 2018, 224, 46–52. 10.1016/j.apcatb.2017.10.031. [DOI] [Google Scholar]
- Nepal B.; Das S. Sustained Water Oxidation by a Catalyst Cage-Isolated in a Metal-Organic Framework. Angew. Chem., Int. Ed. 2013, 52, 7224–7227. 10.1002/anie.201301327. [DOI] [PubMed] [Google Scholar]
- Bromberg L.; Diao Y.; Wu H.; Speakman S. A.; Hatton T. A. Chromium(III) Terephthalate Metal Organic Framework (MIL-101): Hf-Free Synthesis, Structure, Polyoxometalate Composites, and Catalytic Properties. Chem. Mater. 2012, 24, 1664–1675. 10.1021/cm2034382. [DOI] [Google Scholar]
- Hansen R. E.; Das S. Biomimetic Di-Manganese Catalyst Cage-Isolated in a MOF: Robust Catalyst for Water Oxidation with Ce(IV), a Non-O-Donating Oxidant. Energy Environ. Sci. 2014, 7, 317–322. 10.1039/C3EE43040E. [DOI] [Google Scholar]
- Manna P.; Debgupta J.; Bose S.; Das S. K. A Mononuclear Co(II) Coordination Complex Locked in a Confined Space and Acting as an Electrochemical Water-Oxidation Catalyst: A ″Ship-in-a-Bottle″ Approach. Angew. Chem., Int. Ed. 2016, 55, 2425–2430. 10.1002/anie.201509643. [DOI] [PubMed] [Google Scholar]
- Park J.-E.; Oh H.; An J.; Shin I.-S. Post-Synthetic Modification of Mesoporous Zinc-Adeninate Framework with Tris(2,2′-bipyridine) Ruthenium(II) Complex and Its Electrochemiluminescence. Bull. Korean Chem. Soc. 2017, 38, 471–476. 10.1002/bkcs.11115. [DOI] [Google Scholar]
- Meyer K.; Bashir S.; Llorca J.; Idriss H.; Ranocchiari M.; van Bokhoven J. A. Photocatalyzed Hydrogen Evolution from Water by a Composite Catalyst of NH2-MIL-125(Ti) and Surface Nickel(II) Species. Chem. Eur. J. 2016, 22, 13894–13899. 10.1002/chem.201601988. [DOI] [PubMed] [Google Scholar]
- Li Z.; Xiao J. D.; Jiang H. L. Encapsulating a Co(II) Molecular Photocatalyst in Metal-Organic Framework for Visible-Light-Driven H2 Production: Boosting Catalytic Efficiency via Spatial Charge Separation. ACS Catal. 2016, 6, 5359–5365. 10.1021/acscatal.6b01293. [DOI] [Google Scholar]
- Balestri D.; Roux Y.; Mattarozzi M.; Mucchino C.; Heux L.; Brazzolotto D.; Artero V.; Duboc C.; Pelagatti P.; Marchio L.; et al. Heterogenization of a [NiFe] Hydrogenase Mimic through Simple and Efficient Encapsulation into a Mesoporous MOF. Inorg. Chem. 2017, 56, 14801–14808. 10.1021/acs.inorgchem.7b01824. [DOI] [PubMed] [Google Scholar]
- Yan Z.-H.; Ma B.; Li S.-R.; Liu J.; Chen R.; Du M.-H.; Jin S.; Zhuang G.-L.; Long L.-S.; Kong X.-J.; et al. Encapsulating a Ni(II) Molecular Catalyst in Photoactive Metal-Organic Framework for Highly Efficient Photoreduction of CO2. Sci. Bull. 2019, 64, 976–985. 10.1016/j.scib.2019.05.014. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Chen J.; Fan T.; Shen K.; Jiang M.; Li Y. A High-Valent Di-μ-Oxo Dimanganese Complex Covalently Anchored in a Metal-Organic Framework as a Highly Efficient and Recoverable Water Oxidation Catalyst. Chem. Commun. 2018, 54, 4188–4191. 10.1039/C8CC00258D. [DOI] [PubMed] [Google Scholar]
- Bhunia A.; Johnson B. A.; Czapla-Masztafiak J.; Sa J.; Ott S. Formal Water Oxidation Turnover Frequencies from MIL-101(Cr) Anchored Ru(bda) Depend on Oxidant Concentration. Chem. Commun. 2018, 54, 7770–7773. 10.1039/C8CC02300J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; Yang S.; Yang J.; Sun W.; Li X.; Ma B.; Huang J.; Zhang J.; Duan L.; Ding Y. Covalent Immobilization of Molecular Complexes on Metal-Organic Frameworks Towards Robust and Highly Efficient Heterogeneous Water Oxidation Catalysts. Appl. Catal., B 2021, 291, 120070. 10.1016/j.apcatb.2021.120070. [DOI] [Google Scholar]
- Wang X.; Wisser F. M.; Canivet J.; Fontecave M.; Mellot-Draznieks C. Immobilization of a Full Photosystem in the Large-Pore MIL-101 Metal-Organic Framework for CO2 Reduction. ChemSusChem 2018, 11, 3315–3322. 10.1002/cssc.201801066. [DOI] [PubMed] [Google Scholar]
- Stanley P. M.; Thomas C.; Thyrhaug E.; Urstoeger A.; Schuster M.; Hauer J.; Rieger B.; Warnan J.; Fischer R. A. Entrapped Molecular Photocatalyst and Photosensitizer in Metal-Organic Framework Nanoreactors for Enhanced Solar CO2 Reduction. ACS Catal. 2021, 11, 871–882. 10.1021/acscatal.0c04673. [DOI] [Google Scholar]
- Stanley P. M.; Haimerl J.; Thomas C.; Urstoeger A.; Schuster M.; Shustova N. B.; Casini A.; Rieger B.; Warnan J.; Fischer R. A. Host-Guest Interactions in a Metal-Organic Framework Isoreticular Series for Molecular Photocatalytic CO2 Reduction. Angew. Chem., Int. Ed. 2021, 60, 17854–17860. 10.1002/anie.202102729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. C.; Ouyang T.; Xiao R.; Liu W. J.; Zhong D. C.; Xu Z.; Lu T. B. Anchoring Co(II) Ions into a Thiol-Laced Metal-Organic Framework for Efficient Visible-Light-Driven Conversion of CO2 into CO. ChemSusChem 2019, 12, 2166–2170. 10.1002/cssc.201900338. [DOI] [PubMed] [Google Scholar]
- Paille G.; Gomez-Mingot M.; Roch-Marchal C.; Lassalle-Kaiser B.; Mialane P.; Fontecave M.; Mellot-Draznieks C.; Dolbecq A. A Fully Noble Metal-Free Photosystem Based on Cobalt-Polyoxometalates Immobilized in a Porphyrinic Metal-Organic Framework for Water Oxidation. J. Am. Chem. Soc. 2018, 140, 3613–3618. 10.1021/jacs.7b11788. [DOI] [PubMed] [Google Scholar]
- Paille G.; Gomez-Mingot M.; Roch-Marchal C.; Haouas M.; Benseghir Y.; Pino T.; Ha-Thi M. H.; Landrot G.; Mialane P.; Fontecave M.; et al. Thin Films of Fully Noble Metal-Free POM@MOF for Photocatalytic Water Oxidation. ACS Appl. Mater. Interfaces 2019, 11, 47837–47845. 10.1021/acsami.9b13121. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S.; Debgupta J.; Singh C.; Kar A.; Das S. K. A Keggin Polyoxometalate Shows Water Oxidation Activity at Neutral pH: POM@ZIF-8, an Efficient and Robust Electrocatalyst. Angew. Chem., Int. Ed. 2018, 57, 1918–1923. 10.1002/anie.201711920. [DOI] [PubMed] [Google Scholar]
- Shimoni R.; He W.; Liberman I.; Hod I. Tuning of Redox Conductivity and Electrocatalytic Activity in Metal-Organic Framework Films via Control of Defect Site Density. J. Phys. Chem. C 2019, 123, 5531–5539. 10.1021/acs.jpcc.8b12392. [DOI] [Google Scholar]
- Liberman I.; Shimoni R.; Ifraemov R.; Rozenberg I.; Singh C.; Hod I. Active-Site Modulation in an Fe-Porphyrin-Based Metal-Organic Framework through Ligand Axial Coordination: Accelerating Electrocatalysis and Charge-Transport Kinetics. J. Am. Chem. Soc. 2020, 142, 1933–1940. 10.1021/jacs.9b11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi Y. H.; Feng X. Y.; Song Y.; Xu Z. W.; Li Z.; Lin W. B. Metal-Organic Frameworks Integrate Cu Photosensitizers and Secondary Building Unit-Supported Fe Catalysts for Photocatalytic Hydrogen Evolution. J. Am. Chem. Soc. 2020, 142, 10302–10307. 10.1021/jacs.0c03906. [DOI] [PubMed] [Google Scholar]
- Ma X.; Liu H.; Yang W.; Mao G.; Zheng L.; Jiang H. L. Modulating Coordination Environment of Single-Atom Catalysts and Their Proximity to Photosensitive Units for Boosting MOF Photocatalysis. J. Am. Chem. Soc. 2021, 143, 12220–12229. 10.1021/jacs.1c05032. [DOI] [PubMed] [Google Scholar]
- Lan G.; Li Z.; Veroneau S. S.; Zhu Y. Y.; Xu Z.; Wang C.; Lin W. Photosensitizing Metal-Organic Layers for Efficient Sunlight-Driven Carbon Dioxide Reduction. J. Am. Chem. Soc. 2018, 140, 12369–12373. 10.1021/jacs.8b08357. [DOI] [PubMed] [Google Scholar]
- Hu X.; Chen P.; Zhang C.; Wang Z.; Wang C. Energy Transfer on a Two-Dimensional Antenna Enhances the Photocatalytic Activity of CO2 Reduction by Metal-Organic Layers. Chem. Commun. 2019, 55, 9657–9660. 10.1039/C9CC04594E. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Bhunia A.; Ott S. Electrocatalytic Water Oxidation by a Molecular Catalyst Incorporated into a Metal-Organic Framework Thin Film. Dalton Trans. 2017, 46, 1382–1388. 10.1039/C6DT03718F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Ravari A. K.; Zhu J.; Usov P. M.; Cai M.; Ahrenholtz S. R.; Pushkar Y.; Morris A. J. Insight into Metal-Organic Framework Reactivity: Chemical Water Oxidation Catalyzed by a [Ru(tpy)(dcbpy)(OH2)]2+-Modified UiO-67. ChemSusChem 2018, 11, 464–471. 10.1002/cssc.201701644. [DOI] [PubMed] [Google Scholar]
- Lin S.; Pineda-Galvan Y.; Maza W. A.; Epley C. C.; Zhu J.; Kessinger M. C.; Pushkar Y.; Morris A. J. Electrochemical Water Oxidation by a Catalyst-Modified Metal-Organic Framework Thin Film. ChemSusChem 2017, 10, 514–522. 10.1002/cssc.201601181. [DOI] [PubMed] [Google Scholar]
- Wang C.; Xie Z.; deKrafft K. E.; Lin W. Doping Metal-Organic Frameworks for Water Oxidation, Carbon Dioxide Reduction, and Organic Photocatalysis. J. Am. Chem. Soc. 2011, 133, 13445–13454. 10.1021/ja203564w. [DOI] [PubMed] [Google Scholar]
- Chambers M. B.; Wang X.; Elgrishi N.; Hendon C. H.; Walsh A.; Bonnefoy J.; Canivet J.; Quadrelli E. A.; Farrusseng D.; Mellot-Draznieks C.; et al. Photocatalytic Carbon Dioxide Reduction with Rhodium-Based Catalysts in Solution and Heterogenized within Metal-Organic Frameworks. ChemSusChem 2015, 8, 603–608. 10.1002/cssc.201403345. [DOI] [PubMed] [Google Scholar]
- Liao W.-M.; Zhang J.-H.; Wang Z.; Yin S.-Y.; Pan M.; Wang H.-P.; Su C.-Y. Post-Synthetic Exchange (PSE) of UiO-67 Frameworks with Ru/Rh Half-Sandwich Units for Visible-Light-Driven H2 Evolution and CO2 Reduction. J. Mater. Chem. A 2018, 6, 11337–11345. 10.1039/C8TA02962H. [DOI] [Google Scholar]
- Benseghir Y.; Lemarchand A.; Duguet M.; Mialane P.; Gomez-Mingot M.; Roch-Marchal C.; Pino T.; Ha-Thi M. H.; Haouas M.; Fontecave M.; et al. Co-Immobilization of a Rh Catalyst and a Keggin Polyoxometalate in the UiO-67 Zr-Based Metal-Organic Framework: In Depth Structural Characterization and Photocatalytic Properties for CO2 Reduction. J. Am. Chem. Soc. 2020, 142, 9428–9438. 10.1021/jacs.0c02425. [DOI] [PubMed] [Google Scholar]
- Chen C.; Wu T.; Wu H.; Liu H.; Qian Q.; Liu Z.; Yang G.; Han B. Highly Effective Photoreduction of CO2 to CO Promoted by Integration of Cds with Molecular Redox Catalysts through Metal-Organic Frameworks. Chem. Sci. 2018, 9, 8890–8894. 10.1039/C8SC02809E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C. C.; Li T. T.; Cao S.; Chen Y.; Fu W. F. Incorporation of a [Ru(dcbpy)(bpy)2]2+ Photosensitizer and a Pt(dcbpy)Cl2 Catalyst into Metal-Organic Frameworks for Photocatalytic Hydrogen Evolution from Aqueous Solution. J. Mater. Chem. A 2015, 3, 10386–10394. 10.1039/C5TA01135C. [DOI] [Google Scholar]
- Yang S. Z.; Fan D. H.; Hu W. H.; Pattengale B.; Liu C. M.; Zhang X. Y.; Huang J. E. Elucidating Charge Separation Dynamics in a Hybrid Metal-Organic Framework Photocatalyst for Light-Driven H2 Evolution. J. Phys. Chem. C 2018, 122, 3305–3311. 10.1021/acs.jpcc.8b00471. [DOI] [Google Scholar]
- Yang S. Z.; Pattengale B.; Lee S.; Huang J. Real-Time Visualization of Active Species in a Single-Site Metal-Organic Framework Photocatalyst. ACS Energy Lett. 2018, 3, 532–539. 10.1021/acsenergylett.8b00062. [DOI] [Google Scholar]
- Kim D.; Whang D. R.; Park S. Y. Self-Healing of Molecular Catalyst and Photosensitizer on Metal-Organic Framework: Robust Molecular System for Photocatalytic H2 Evolution from Water. J. Am. Chem. Soc. 2016, 138, 8698–8701. 10.1021/jacs.6b04552. [DOI] [PubMed] [Google Scholar]
- Liu M.; Mu Y.-F.; Yao S.; Guo S.; Guo X.-W.; Zhang Z.-M.; Lu T.-B. Photosensitizing Single-Site Metal-Organic Framework Enabling Visible-Light-Driven CO2 Reduction for Syngas Production. Appl. Catal., B 2019, 245, 496–501. 10.1016/j.apcatb.2019.01.014. [DOI] [Google Scholar]
- Sun D.; Gao Y.; Fu J.; Zeng X.; Chen Z.; Li Z. Construction of a Supported Ru Complex on Bifunctional MOF-253 for Photocatalytic CO2 Reduction under Visible Light. Chem. Commun. 2015, 51, 2645–2648. 10.1039/C4CC09797A. [DOI] [PubMed] [Google Scholar]
- Johnson F. P. A.; George M. W.; Hartl F.; Turner J. J. Electrocatalytic Reduction of CO2 Using the Complexes [Re(bpy)(CO)3L]n (n = + 1, L = P(OEt)3, CH3CN; n = 0, L = Cl–, Otf–; bpy = 2,2’-Bipyridine; Otf–= CF3SO3) as Catalyst Precursors: Infrared Spectroelectrochemical Investigation. Organometallics 1996, 15, 3374–3387. 10.1021/om960044+. [DOI] [Google Scholar]
- Benson E. E.; Kubiak C. P. Structural Investigations into the Deactivation Pathway of the CO2 Reduction Electrocatalyst Re(bpy)(CO)3Cl. Chem. Commun. 2012, 48, 7374–7376. 10.1039/c2cc32617e. [DOI] [PubMed] [Google Scholar]
- Fei H.; Sampson M. D.; Lee Y.; Kubiak C. P.; Cohen S. M. Photocatalytic CO2 Reduction to Formate Using a Mn(I) Molecular Catalyst in a Robust Metal-Organic Framework. Inorg. Chem. 2015, 54, 6821–6828. 10.1021/acs.inorgchem.5b00752. [DOI] [PubMed] [Google Scholar]
- Deng X.; Albero J.; Xu L.; Garcia H.; Li Z. Construction of a Stable Ru-Re Hybrid System Based on Multifunctional MOF-253 for Efficient Photocatalytic CO2 Reduction. Inorg. Chem. 2018, 57, 8276–8286. 10.1021/acs.inorgchem.8b00896. [DOI] [PubMed] [Google Scholar]
- Huang R.; Peng Y.; Wang C.; Shi Z.; Lin W. A Rhenium-Functionalized Metal-Organic Framework as a Single-Site Catalyst for Photochemical Reduction of Carbon Dioxide. Eur. J. Inorg. Chem. 2016, 2016, 4358–4362. 10.1002/ejic.201600064. [DOI] [Google Scholar]
- Feng X.; Pi Y.; Song Y.; Brzezinski C.; Xu Z.; Li Z.; Lin W. Metal-Organic Frameworks Significantly Enhance Photocatalytic Hydrogen Evolution and CO2 Reduction with Earth-Abundant Copper Photosensitizers. J. Am. Chem. Soc. 2020, 142, 690–695. 10.1021/jacs.9b12229. [DOI] [PubMed] [Google Scholar]
- Feng H.; Li H.; Liu X.; Huang Y.; Pan Q.; Peng R.; Du R.; Zheng X.; Yin Z.; Li S.; et al. Porphyrin-Based Ti-MOFs Conferred with Single-Atom Pt for Enhanced Photocatalytic Hydrogen Evolution and NO Removal. Chem. Eng. J. 2022, 428, 132045. 10.1016/j.cej.2021.132045. [DOI] [Google Scholar]
- Lions M.; Tommasino J. B.; Chattot R.; Abeykoon B.; Guillou N.; Devic T.; Demessence A.; Cardenas L.; Maillard F.; Fateeva A. Insights into the Mechanism of Electrocatalysis of the Oxygen Reduction Reaction by a Porphyrinic Metal Organic Framework. Chem. Commun. 2017, 53, 6496–6499. 10.1039/C7CC02113E. [DOI] [PubMed] [Google Scholar]
- Sohrabi S.; Dehghanpour S.; Ghalkhani M. A Cobalt Porphyrin-Based Metal Organic Framework/Multi-Walled Carbon Nanotube Composite Electrocatalyst for Oxygen Reduction and Evolution Reactions. J. Mater. Sci. 2018, 53, 3624–3639. 10.1007/s10853-017-1768-0. [DOI] [Google Scholar]
- Liang Z.; Guo H.; Zhou G.; Guo K.; Wang B.; Lei H.; Zhang W.; Zheng H.; Apfel U.-P.; Cao R. Metal-Organic-Framework-Supported Molecular Electrocatalysis for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2021, 60, 8472–8476. 10.1002/anie.202016024. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Yang Y.; Sun Q.; Wang Z.; Huang B.; Dai Y.; Qin X.; Zhang X. Chemical Adsorption Enhanced CO2 Capture and Photoreduction over a Copper Porphyrin Based Metal Organic Framework. ACS Appl. Mater. Interfaces 2013, 5, 7654–7658. 10.1021/am4019675. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Wei J.; Dong J.; Liu G.; Shi L.; An P.; Zhao G.; Kong J.; Wang X.; Meng X.; et al. Efficient Visible-Light-Driven Carbon Dioxide Reduction by a Single-Atom Implanted Metal-Organic Framework. Angew. Chem., Int. Ed. 2016, 55, 14310–14314. 10.1002/anie.201608597. [DOI] [PubMed] [Google Scholar]
- Huang N. Y.; Zhang X. W.; Xu Y. Z.; Liao P. Q.; Chen X. M. A Local Hydrophobic Environment in a Metal-Organic Framework for Boosting Photocatalytic CO2 Reduction in the Presence of Water. Chem. Commun. 2019, 55, 14781–14784. 10.1039/C9CC08094E. [DOI] [PubMed] [Google Scholar]
- Hod I.; Sampson M. D.; Deria P.; Kubiak C. P.; Farha O. K.; Hupp J. T. Fe-Porphyrin-Based Metal-Organic Framework Films as High-Surface Concentration, Heterogeneous Catalysts for Electrochemical Reduction of CO2. ACS Catal. 2015, 5, 6302–6309. 10.1021/acscatal.5b01767. [DOI] [Google Scholar]
- Sadeghi N.; Sharifnia S.; Sheikh Arabi M. A Porphyrin-Based Metal Organic Framework for High Rate Photoreduction of CO2 to CH4 in Gas Phase. J. CO2 Util. 2016, 16, 450–457. 10.1016/j.jcou.2016.10.006. [DOI] [Google Scholar]
- Zhou Y.; Chen S.; Xi S.; Wang Z.; Deng P.; Yang F.; Han Y.; Pang Y.; Xia B. Y. Spatial Confinement in Copper-Porphyrin Frameworks Enhances Carbon Dioxide Reduction to Hydrocarbons. Cell Rep. Phys. Sci. 2020, 1, 100182. 10.1016/j.xcrp.2020.100182. [DOI] [Google Scholar]
- Wu J. X.; Hou S. Z.; Zhang X. D.; Xu M.; Yang H. F.; Cao P. S.; Gu Z. Y. Cathodized Copper Porphyrin Metal-Organic Framework Nanosheets for Selective Formate and Acetate Production from CO2 Electroreduction. Chem. Sci. 2019, 10, 2199–2205. 10.1039/C8SC04344B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J. H.; Xu P.; Huang Y. D.; Xiao L. Y.; Lu W.; Yang X. G.; Ma L. F.; Zang S. Q. High Loading of Mn(II)-Metalated Porphyrin in a MOF for Photocatalytic CO2 Reduction in Gas-Solid Conditions. Chem. Commun. 2021, 57, 8468–8471. 10.1039/D1CC02847B. [DOI] [PubMed] [Google Scholar]
- Huang H. B.; Fang Z. B.; Wang R.; Li L.; Khanpour M.; Liu T. F.; Cao R. Engineering Hierarchical Architecture of Metal-Organic Frameworks for Highly Efficient Overall CO2 Photoreduction. Small 2022, 18, e2200407 10.1002/smll.202200407. [DOI] [PubMed] [Google Scholar]
- Matheu R.; Gutierrez-Puebla E.; Monge M. A.; Diercks C. S.; Kang J.; Prevot M. S.; Pei X.; Hanikel N.; Zhang B.; Yang P.; et al. Three-Dimensional Phthalocyanine Metal-Catecholates for High Electrochemical Carbon Dioxide Reduction. J. Am. Chem. Soc. 2019, 141, 17081–17085. 10.1021/jacs.9b09298. [DOI] [PubMed] [Google Scholar]
- Meng Z.; Luo J.; Li W.; Mirica K. A. Hierarchical Tuning of the Performance of Electrochemical Carbon Dioxide Reduction Using Conductive Two-Dimensional Metallophthalocyanine Based Metal-Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 21656–21669. 10.1021/jacs.0c07041. [DOI] [PubMed] [Google Scholar]
- Qiu X. F.; Zhu H. L.; Huang J. R.; Liao P. Q.; Chen X. M. Highly Selective CO2 Electroreduction to C2H4 Using a Metal-Organic Framework with Dual Active Sites. J. Am. Chem. Soc. 2021, 143, 7242–7246. 10.1021/jacs.1c01466. [DOI] [PubMed] [Google Scholar]
- Gong Y. N.; Ouyang T.; He C. T.; Lu T. B. Photoinduced Water Oxidation by an Organic Ligand Incorporated into the Framework of a Stable Metal-Organic Framework. Chem. Sci. 2016, 7, 1070–1075. 10.1039/C5SC02679B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.; Ray D.; Ziebel M. E.; Gaggioli C. A.; Gagliardi L.; Marinescu S. C. Cu[Ni(2,3-pyrazinedithiolate)2] Metal-Organic Framework for Electrocatalytic Hydrogen Evolution. ACS Appl. Mater. Interfaces 2021, 13, 34419–34427. 10.1021/acsami.1c08998. [DOI] [PubMed] [Google Scholar]
- Dou S.; Song J.; Xi S.; Du Y.; Wang J.; Huang Z. F.; Xu Z. J.; Wang X. Boosting Electrochemical CO2 Reduction on Metal-Organic Frameworks via Ligand Doping. Angew. Chem., Int. Ed. 2019, 58, 4041–4045. 10.1002/anie.201814711. [DOI] [PubMed] [Google Scholar]
- Liseev T.; Howe A.; Hoque M. A.; Gimbert-Surinach C.; Llobet A.; Ott S. Synthetic Strategies to Incorporate Ru-Terpyridyl Water Oxidation Catalysts into MOFs: Direct Synthesis vs. Post-Synthetic Approach. Dalton Trans. 2020, 49, 13753–13759. 10.1039/D0DT01890B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe A.; Liseev T.; Gil-Sepulcre M.; Gimbert-Surinach C.; Benet-Buchholz J.; Llobet A.; Ott S. Electrocatalytic Water Oxidation from a Mixed Linker MOF Based on NU-1000 with an Integrated Ruthenium-Based Metallo-Linker. Mater. Adv. 2022, 3, 4227–4234. 10.1039/D2MA00128D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheu R.; Ertem M. Z.; Benet-Buchholz J.; Coronado E.; Batista V. S.; Sala X.; Llobet A. Intramolecular Proton Transfer Boosts Water Oxidation Catalyzed by a Ru Complex. J. Am. Chem. Soc. 2015, 137, 10786–10795. 10.1021/jacs.5b06541. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Sun D.; Chen Y.; Huang R.; Ding Z.; Fu X.; Li Z. An Amine-Functionalized Titanium Metal-Organic Framework Photocatalyst with Visible-Light-Induced Activity for CO2 Reduction. Angew. Chem., Int. Ed. 2012, 51, 3364–3367. 10.1002/anie.201108357. [DOI] [PubMed] [Google Scholar]
- Luo T.; Zhang J.; Li W.; He Z.; Sun X.; Shi J.; Shao D.; Zhang B.; Tan X.; Han B. Metal-Organic Framework-Stabilized CO2/Water Interfacial Route for Photocatalytic CO2 Conversion. ACS Appl. Mater. Interfaces 2017, 9, 41594–41598. 10.1021/acsami.7b13900. [DOI] [PubMed] [Google Scholar]
- Sun D.; Fu Y.; Liu W.; Ye L.; Wang D.; Yang L.; Fu X.; Li Z. Studies on Photocatalytic CO2 Reduction over NH2-UiO-66(Zr) and Its Derivatives: Towards a Better Understanding of Photocatalysis on Metal-Organic Frameworks. Chem. Eur. J. 2013, 19, 14279–14285. 10.1002/chem.201301728. [DOI] [PubMed] [Google Scholar]
- Sun D.; Liu W.; Qiu M.; Zhang Y.; Li Z. Introduction of a Mediator for Enhancing Photocatalytic Performance via Post-Synthetic Metal Exchange in Metal-Organic Frameworks (MOFs). Chem. Commun. 2015, 51, 2056–2059. 10.1039/C4CC09407G. [DOI] [PubMed] [Google Scholar]
- Kay A.; Grätzel M. Dye-Sensitized Core-Shell Nanocrystals: Improved Efficiency of Mesoporous Tin Oxide Electrodes Coated with a Thin Layer of an Insulating Oxide. Chem. Mater. 2002, 14, 2930–2935. 10.1021/cm0115968. [DOI] [Google Scholar]
- Wei Y. P.; Liu Y.; Guo F.; Dao X. Y.; Sun W. Y. Different Functional Group Modified Zirconium Frameworks for the Photocatalytic Reduction of Carbon Dioxide. Dalton Trans. 2019, 48, 8221–8226. 10.1039/C9DT01767D. [DOI] [PubMed] [Google Scholar]
- Wang D.; Huang R.; Liu W.; Sun D.; Li Z. Fe-Based MOFs for Photocatalytic CO2 Reduction: Role of Coordination Unsaturated Sites and Dual Excitation Pathways. ACS Catal. 2014, 4, 4254–4260. 10.1021/cs501169t. [DOI] [Google Scholar]
- Chen D.; Xing H.; Wang C.; Su Z. Highly Efficient Visible-Light-Driven CO2 Reduction to Formate by a New Anthracene-Based Zirconium MOF via Dual Catalytic Routes. J. Mater. Chem. A 2016, 4, 2657–2662. 10.1039/C6TA00429F. [DOI] [Google Scholar]
- Sun M.; Yan S.; Sun Y.; Yang X.; Guo Z.; Du J.; Chen D.; Chen P.; Xing H. Enhancement of Visible-Light-Driven CO2 Reduction Performance Using an Amine-Functionalized Zirconium Metal-Organic Framework. Dalton Trans. 2018, 47, 909–915. 10.1039/C7DT04062H. [DOI] [PubMed] [Google Scholar]
- Logan M. W.; Ayad S.; Adamson J. D.; Dilbeck T.; Hanson K.; Uribe-Romo F. J. Systematic Variation of the Optical Bandgap in Titanium Based Isoreticular Metal-Organic Frameworks for Photocatalytic Reduction of CO2 under Blue Light. J. Mater. Chem. A 2017, 5, 11854–11863. 10.1039/C7TA00437K. [DOI] [Google Scholar]
- Chen S.; Yang F.; Gao H.; Wang J.; Chen X.; Zhang X.; Li J.; Li A. Construction of Dual Ligand Ti-Based MOFs with Enhanced Photocatalytic CO2 Reduction Performance. J. CO2 Util. 2021, 48, 101528. 10.1016/j.jcou.2021.101528. [DOI] [Google Scholar]
- Qin J. S.; Yuan S.; Zhang L.; Li B.; Du D. Y.; Huang N.; Guan W.; Drake H. F.; Pang J.; Lan Y. Q.; et al. Creating Well-Defined Hexabenzocoronene in Zirconium Metal-Organic Framework by Postsynthetic Annulation. J. Am. Chem. Soc. 2019, 141, 2054–2060. 10.1021/jacs.8b11042. [DOI] [PubMed] [Google Scholar]
- Qiu Y. C.; Yuan S.; Li X. X.; Du D. Y.; Wang C.; Qin J. S.; Drake H. F.; Lan Y. Q.; Jiang L.; Zhou H. C. Face-Sharing Archimedean Solids Stacking for the Construction of Mixed-Ligand Metal-Organic Frameworks. J. Am. Chem. Soc. 2019, 141, 13841–13848. 10.1021/jacs.9b05580. [DOI] [PubMed] [Google Scholar]
- Aziz A.; Ruiz-Salvador A. R.; Hernandez N. C.; Calero S.; Hamad S.; Grau-Crespo R. Porphyrin-Based Metal-Organic Frameworks for Solar Fuel Synthesis Photocatalysis: Band Gap Tuning via Iron Substitutions. J. Mater. Chem. A 2017, 5, 11894–11904. 10.1039/C7TA01278K. [DOI] [Google Scholar]
- Zeng J. Y.; Wang X. S.; Xie B. R.; Li Q. R.; Zhang X. Z. Large Pi-Conjugated Metal-Organic Frameworks for Infrared-Light-Driven CO2 Reduction. J. Am. Chem. Soc. 2022, 144, 1218–1231. 10.1021/jacs.1c10110. [DOI] [PubMed] [Google Scholar]
- Nguyen A. I.; Van Allsburg K. M.; Terban M. W.; Bajdich M.; Oktawiec J.; Amtawong J.; Ziegler M. S.; Dombrowski J. P.; Lakshmi K. V.; Drisdell W. S.; et al. Stabilization of Reactive Co4O4 Cubane Oxygen-Evolution Catalysts within Porous Frameworks. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 11630–11639. 10.1073/pnas.1815013116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Meyer T. J. A Strategy for Stabilizing the Catalyst Co4O4 in a Metal-Organic Framework. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 13719–13720. 10.1073/pnas.1909543116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. Z.; Pattengale B.; Kovrigin E. L.; Huang J. Photoactive Zeolitic Imidazolate Framework as Intrinsic Heterogeneous Catalysts for Light-Driven Hydrogen Generation. ACS Energy Lett. 2017, 2, 75–80. 10.1021/acsenergylett.6b00540. [DOI] [Google Scholar]
- Qin J.; Wang S.; Wang X. Visible-Light Reduction CO2 with Dodecahedral Zeolitic Imidazolate Framework ZIF-67 as an Efficient Co-Catalyst. Appl. Catal., B 2017, 209, 476–482. 10.1016/j.apcatb.2017.03.018. [DOI] [Google Scholar]
- Liu Y. H.; Zhang F. J.; Wu P. Y.; Deng C. F.; Yang Q. M.; Xue J. J.; Shi Y. H.; Wang J. Cobalt(II)-Based Metal-Organic Framework as Bifunctional Materials for Ag(I) Detection and Proton Reduction Catalysis for Hydrogen Production. Inorg. Chem. 2019, 58, 924–931. 10.1021/acs.inorgchem.8b03046. [DOI] [PubMed] [Google Scholar]
- Wang S.; Yao W.; Lin J.; Ding Z.; Wang X. Cobalt Imidazolate Metal-Organic Frameworks Photosplit CO2 under Mild Reaction Conditions. Angew. Chem., Int. Ed. 2014, 53, 1034–1038. 10.1002/anie.201309426. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Wang Q.; Sun C.; Zheng T.; Yan L.; Li M.; Shao K.; Wang X.; Su Z. A Hexanuclear Cobalt Metal-Organic Framework for Efficient CO2 Reduction under Visible Light. J. Mater. Chem. A 2017, 5, 12498–12505. 10.1039/C7TA02611K. [DOI] [Google Scholar]
- Liao W.-M.; Zhang J.-H.; Wang Z.; Lu Y.-L.; Yin S.-Y.; Wang H.-P.; Fan Y.-N.; Pan M.; Su C.-Y. Semiconductive Amine-Functionalized Co(II)-MOF for Visible-Light-Driven Hydrogen Evolution and CO2 Reduction. Inorg. Chem. 2018, 57, 11436–11442. 10.1021/acs.inorgchem.8b01265. [DOI] [PubMed] [Google Scholar]
- Shi D. Y.; Cui C. J.; Hu M.; Ren A. H.; Song L. B.; Liu C. S.; Du M. A Microporous Mixed-Metal (Na/Cu) Mixed-Ligand (Flexible/Rigid) Metal-Organic Framework for Photocatalytic H2 Generation. J. Mater. Chem. C 2019, 7, 10211–10217. 10.1039/C9TC03342D. [DOI] [Google Scholar]
- Wang Z. D.; Zang Y.; Liu Z. J.; Peng P.; Wang R.; Zang S. Q. Opening Catalytic Sites in the Copper-Triazoles Framework via Defect Chemistry for Switching on the Proton Reduction. Appl. Catal., B 2021, 288, 119941. 10.1016/j.apcatb.2021.119941. [DOI] [Google Scholar]
- Mao J.; Yang L.; Yu P.; Wei X.; Mao L. Electrocatalytic Four-Electron Reduction of Oxygen with Copper (II)-Based Metal-Organic Frameworks. Electrochem. Commun. 2012, 19, 29–31. 10.1016/j.elecom.2012.02.025. [DOI] [Google Scholar]
- Senthil Kumar R.; Senthil Kumar S.; Anbu Kulandainathan M. Highly Selective Electrochemical Reduction of Carbon Dioxide Using Cu Based Metal Organic Framework as an Electrocatalyst. Electrochem. Commun. 2012, 25, 70–73. 10.1016/j.elecom.2012.09.018. [DOI] [Google Scholar]
- Zhang H. X.; Hong Q. L.; Li J.; Wang F.; Huang X.; Chen S.; Tu W.; Yu D.; Xu R.; Zhou T.; et al. Isolated Square-Planar Copper Center in Boron Imidazolate Nanocages for Photocatalytic Reduction of CO2 to CO. Angew. Chem., Int. Ed. 2019, 58, 11752–11756. 10.1002/anie.201905869. [DOI] [PubMed] [Google Scholar]
- Liu J.; Yang D.; Zhou Y.; Zhang G.; Xing G.; Liu Y.; Ma Y.; Terasaki O.; Yang S.; Chen L. Tricycloquinazoline-Based 2D Conductive Metal-Organic Frameworks as Promising Electrocatalysts for CO2 Reduction. Angew. Chem., Int. Ed. 2021, 60, 14473–14479. 10.1002/anie.202103398. [DOI] [PubMed] [Google Scholar]
- Mani P.; Sheelam A.; Karthik P. E.; Sankar R.; Ramanujam K.; Mandal S. Nickel-Based Hybrid Material for Electrochemical Oxygen Redox Reactions in an Alkaline Medium. ACS Appl. Energy Mater. 2020, 3, 6408–6415. 10.1021/acsaem.0c00615. [DOI] [Google Scholar]
- Feng Y. A.; Chen C.; Liu Z. G.; Fei B. J.; Lin P.; Li Q. P.; Sun S. G.; Du S. W. Application of a Ni Mercaptopyrimidine MOF as Highly Efficient Catalyst for Sunlight-Driven Hydrogen Generation. J. Mater. Chem. A 2015, 3, 7163–7169. 10.1039/C5TA00136F. [DOI] [Google Scholar]
- Khrizanforova V.; Shekurov R.; Miluykov V.; Khrizanforov M.; Bon V.; Kaskel S.; Gubaidullin A.; Sinyashin O.; Budnikova Y. 3D Ni and Co Redox-Active Metal-Organic Frameworks Based on Ferrocenyl Diphosphinate and 4,4’-bipyridine Ligands as Efficient Electrocatalysts for the Hydrogen Evolution Reaction. Dalton Trans. 2020, 49, 2794–2802. 10.1039/C9DT04834K. [DOI] [PubMed] [Google Scholar]
- Huo D. B.; Lin F. F.; Chen S.; Ni Y. R.; Wang R. H.; Chen H.; Duan L. L.; Ji Y. F.; Zhou A. J.; Tong L. P. Ruthenium Complex-Incorporated Two-Dimensional Metal-Organic Frameworks for Cocatalyst-Free Photocatalytic Proton Reduction from Water. Inorg. Chem. 2020, 59, 2379–2386. 10.1021/acs.inorgchem.9b03250. [DOI] [PubMed] [Google Scholar]
- Iqbal R.; Akbar M. B.; Ahmad A.; Hussain A.; Altaf N.; Ibraheem S.; Yasin G.; Khan M. A.; Tabish M.; Kumar A.; et al. Exploring the Synergistic Effect of Novel Ni-Fe in 2D Bimetallic Metal-Organic Frameworks for Enhanced Electrochemical Reduction of CO2. Adv. Mater. Interfaces 2022, 9, 2101505. 10.1002/admi.202101505. [DOI] [Google Scholar]
- Kataoka Y.; Miyazaki Y.; Sato K.; Saito T.; Nakanishi Y.; Kiatagwa Y.; Kawakami T.; Okumura M.; Yamaguchi K.; Mori W. Modification of MOF Catalysts by Manipulation of Counter-Ions: Experimental and Theoretical Studies of Photochemical Hydrogen Production from Water over Microporous Diruthenium (II, III) Coordination Polymers. Supramol. Chem. 2011, 23, 287–296. 10.1080/10610278.2010.527976. [DOI] [Google Scholar]
- Lan G. X.; Zhu Y. Y.; Veroneau S. S.; Xu Z. W.; Micheroni D.; Lin W. B. Electron Injection from Photoexcited Metal-Organic Framework Ligands to Ru2 Secondary Building Units for Visible-Light-Driven Hydrogen Evolution. J. Am. Chem. Soc. 2018, 140, 5326–5329. 10.1021/jacs.8b01601. [DOI] [PubMed] [Google Scholar]
- Song Y.; Pi Y. H.; Feng X. Y.; Ni K. Y.; Xu Z. W.; Chen J. S.; Li Z.; Lin W. B. Cerium-Based Metal-Organic Layers Catalyze Hydrogen Evolution Reaction through Dual Photoexcitation. J. Am. Chem. Soc. 2020, 142, 6866–6871. 10.1021/jacs.0c00679. [DOI] [PubMed] [Google Scholar]
- Yan Z. H.; Du M. H.; Liu J.; Jin S.; Wang C.; Zhuang G. L.; Kong X. J.; Long L. S.; Zheng L. S. Photo-Generated Dinuclear {Eu(II)}2 Active Sites for Selective CO2 Reduction in a Photosensitizing Metal-Organic Framework. Nat. Commun. 2018, 9, 3353. 10.1038/s41467-018-05659-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Gu G. H.; Choi C.; Kolla P.; Hong S.; Wu T.-S.; Soo Y.-L.; Masa J.; Mukerjee S.; Jung Y.; et al. Highly Stable Two-Dimensional Bismuth Metal-Organic Frameworks for Efficient Electrochemical Reduction of CO2. Appl. Catal., B 2020, 277, 119241. 10.1016/j.apcatb.2020.119241. [DOI] [Google Scholar]
- Lee M.; De Riccardis A.; Kazantsev R. V.; Cooper J. K.; Buckley A. K.; Burroughs P. W. W.; Larson D. M.; Mele G.; Toma F. M. Aluminum Metal-Organic Framework Triggers Carbon Dioxide Reduction Activity. ACS Appl. Energy Mater. 2020, 3, 1286–1291. 10.1021/acsaem.9b02210. [DOI] [Google Scholar]
- Bie B.; Jiang Z.; Zhang J.; Deng H.; Yang C. Long Excited State Lifetime of Thermally Activated Delayed Fluorescent Photosensitizer Integrated into Metal-Organic Framework Enables Efficient CO2 Photoreduction. Chem. Eng. J. 2022, 431, 133897. 10.1016/j.cej.2021.133897. [DOI] [Google Scholar]
- Wang L.; Wu Y.; Cao R.; Ren L.; Chen M.; Feng X.; Zhou J.; Wang B. Fe/Ni Metal-Organic Frameworks and Their Binder-Free Thin Films for Efficient Oxygen Evolution with Low Overpotential. ACS Appl. Mater. Interfaces 2016, 8, 16736–16743. 10.1021/acsami.6b05375. [DOI] [PubMed] [Google Scholar]
- Huang Z. Q.; Wang B.; Pan D. S.; Zhou L. L.; Guo Z. H.; Song J. L. Rational Design of a N,S Co-Doped Supermicroporous CoFe-Organic Framework Platform for Water Oxidation. ChemSusChem 2020, 13, 2564–2570. 10.1002/cssc.202000376. [DOI] [PubMed] [Google Scholar]
- Wu C.; Zhang X.; Li H.; Xia Z.; Yu S.; Wang S.; Sun G. Iron-Based Binary Metal-Organic Framework Nanorods as an Efficient Catalyst for the Oxygen Evolution Reaction. Chin. J. Catal. 2021, 42, 637–647. 10.1016/S1872-2067(20)63686-5. [DOI] [Google Scholar]
- Li F. L.; Wang P.; Huang X.; Young D. J.; Wang H. F.; Braunstein P.; Lang J. P. Large-Scale, Bottom-up Synthesis of Binary Metal-Organic Framework Nanosheets for Efficient Water Oxidation. Angew. Chem., Int. Ed. 2019, 58, 7051–7056. 10.1002/anie.201902588. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Liu F.; Wei C.; Li D.; Guo W.; Zhao Q. High Efficiency FeNi-Metal-Organic Framework Grown in-Situ on Nickel Foam for Electrocatalytic Oxygen Evolution. ChemistrySelect 2019, 4, 5988–5994. 10.1002/slct.201901709. [DOI] [Google Scholar]
- Yao N.; Jia H.; Fan Z.; Bai L.; Xie W.; Cong H.; Chen S.; Luo W. Nitridation-Induced Metal-Organic Framework Nanosheet for Enhanced Water Oxidation Electrocatalysis. J. Energy Chem. 2022, 64, 531–537. 10.1016/j.jechem.2021.05.024. [DOI] [Google Scholar]
- Yue K.; Liu J.; Zhu Y.; Xia C.; Wang P.; Zhang J.; Kong Y.; Wang X.; Yan Y.; Xia B. Y. In Situ Ion-Exchange Preparation and Topological Transformation of Trimetal-Organic Frameworks for Efficient Electrocatalytic Water Oxidation. Energy Environ. Sci. 2021, 14, 6546–6553. 10.1039/D1EE02606B. [DOI] [Google Scholar]
- Yoon H.; Lee S.; Oh S.; Park H.; Choi S.; Oh M. Synthesis of Bimetallic Conductive 2D Metal-Organic Framework (CoxNiy-CAT) and Its Mass Production: Enhanced Electrochemical Oxygen Reduction Activity. Small 2019, 15, e1805232 10.1002/smll.201805232. [DOI] [PubMed] [Google Scholar]
- Shinde S. S.; Lee C. H.; Jung J.-Y.; Wagh N. K.; Kim S.-H.; Kim D.-H.; Lin C.; Lee S. U.; Lee J.-H. Unveiling Dual-Linkage 3D hexaiminobenzene Metal-Organic Frameworks Towards Long-Lasting Advanced Reversible Zn-Air Batteries. Energy Environ. Sci. 2019, 12, 727–738. 10.1039/C8EE02679C. [DOI] [Google Scholar]
- Huang Q.; Liu J.; Feng L.; Wang Q.; Guan W.; Dong L.-Z.; Zhang L.; Yan L.-K.; Lan Y.-Q.; Zhou H.-C. Multielectron Transportation of Polyoxometalate-Grafted Metalloporphyrin Coordination Frameworks for Selective CO2-to-CH4 Photoconversion. Natl. Sci. Rev. 2020, 7, 53–63. 10.1093/nsr/nwz096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. X.; Liu J.; Zhang L.; Dong L. Z.; Xin Z. F.; Li S. L.; Huang-Fu X. Q.; Huang K.; Lan Y. Q. Hydrophobic Polyoxometalate-Based Metal-Organic Framework for Efficient CO2 Photoconversion. ACS Appl. Mater. Interfaces 2019, 11, 25790–25795. 10.1021/acsami.9b03861. [DOI] [PubMed] [Google Scholar]
- Clough A. J.; Yoo J. W.; Mecklenburg M. H.; Marinescu S. C. Two-Dimensional Metal-Organic Surfaces for Efficient Hydrogen Evolution from Water. J. Am. Chem. Soc. 2015, 137, 118–121. 10.1021/ja5116937. [DOI] [PubMed] [Google Scholar]
- Liu X.-H.; Hu W.-L.; Jiang W.-J.; Yang Y.-W.; Niu S.; Sun B.; Wu J.; Hu J.-S. Well-Defined Metal-O6 in Metal-Catecholates as a Novel Active Site for Oxygen Electroreduction. ACS Appl. Mater. Interfaces 2017, 9, 28473–28477. 10.1021/acsami.7b07410. [DOI] [PubMed] [Google Scholar]
- Dong R. H.; Zheng Z. K.; Tranca D. C.; Zhang J.; Chandrasekhar N.; Liu S. H.; Zhuang X. D.; Seifert G.; Feng X. L. Immobilizing Molecular Metal Dithiolene-Diamine Complexes on 2D Metal-Organic Frameworks for Electrocatalytic H2 Production. Chem. Eur. J. 2017, 23, 2255–2260. 10.1002/chem.201605337. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Sheng L.; Luo Q.; Zhang W.; Yang J. Improving the Activity of Electrocatalysts toward the Hydrogen Evolution Reaction, the Oxygen Evolution Reaction, and the Oxygen Reduction Reaction via Modification of Metal and Ligand of Conductive Two-Dimensional Metal-Organic Frameworks. J. Phys. Chem. Lett. 2021, 12, 11652–11658. 10.1021/acs.jpclett.1c03452. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Qiao M.; Liu J.; Tao T.; Guo C. Engineering Pristine 2D Metal-Organic Framework Nanosheets for Electrocatalysis. J. Mater. Chem. A 2020, 8, 8143–8170. 10.1039/D0TA03138K. [DOI] [Google Scholar]
- Chen G.; Stevens M. B.; Liu Y.; King L. A.; Park J.; Kim T. R.; Sinclair R.; Jaramillo T. F.; Bao Z. Nanosized Zirconium Porphyrinic Metal-Organic Frameworks That Catalyze the Oxygen Reduction Reaction in Acid. Small Methods 2020, 4, 2000085. 10.1002/smtd.202000085. [DOI] [Google Scholar]
- Kornienko N.; Zhao Y.; Kley C. S.; Zhu C.; Kim D.; Lin S.; Chang C. J.; Yaghi O. M.; Yang P. Metal-Organic Frameworks for Electrocatalytic Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2015, 137, 14129–14135. 10.1021/jacs.5b08212. [DOI] [PubMed] [Google Scholar]
- Downes C. A.; Clough A. J.; Chen K. Y.; Yoo J. W.; Marinescu S. C. Evaluation of the H2 Evolving Activity of Benzenehexathiolate Coordination Frameworks and the Effect of Film Thickness on H2 Production. ACS Appl. Mater. Interfaces 2018, 10, 1719–1727. 10.1021/acsami.7b15969. [DOI] [PubMed] [Google Scholar]
- Duan J.; Chen S.; Zhao C. Ultrathin Metal-Organic Framework Array for Efficient Electrocatalytic Water Splitting. Nat. Commun. 2017, 8, 15341. 10.1038/ncomms15341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W.; Zhang H.; Luan D.; Lou X. W. D. Exposing Unsaturated Cu1-O2 Sites in Nanoscale Cu-MOF for Efficient Electrocatalytic Hydrogen Evolution. Sci. Adv. 2021, 7, eabg2580 10.1126/sciadv.abg2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X.; Li L.; Sheveleva A.; Han X.; Li J.; Liu L.; Tuna F.; McInnes E. J. L.; Han B.; Yang S.; et al. Electro-Reduction of Carbon Dioxide at Low over-Potential at a Metal-Organic Framework Decorated Cathode. Nat. Commun. 2020, 11, 5464. 10.1038/s41467-020-19236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X.; Wang B.; Hu K.; Lyu K.; Han X.; Spencer B. F.; Frogley M. D.; Tuna F.; McInnes E. J. L.; Dryfe R. A. W.; et al. Quantitative Electro-Reduction of CO2 to Liquid Fuel over Electro-Synthesized Metal-Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 17384–17392. 10.1021/jacs.0c05913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L.; Liu J.; Gao Y.; Gong C.; Addicoat M.; Heine T.; Wöll C.; Sun L. Highly Oriented MOF Thin Film-Based Electrocatalytic Device for the Reduction of CO2 to CO Exhibiting High Faradaic Efficiency. J. Mater. Chem. A 2016, 4, 15320–15326. 10.1039/C6TA04801C. [DOI] [Google Scholar]
- Zhao S.; Wang Y.; Dong J.; He C.-T.; Yin H.; An P.; Zhao K.; Zhang X.; Gao C.; Zhang L.; et al. Ultrathin Metal-Organic Framework Nanosheets for Electrocatalytic Oxygen Evolution. Nat. Energy 2016, 1, 16184. 10.1038/nenergy.2016.184. [DOI] [Google Scholar]
- Zhu W.; Zhang C.; Li Q.; Xiong L.; Chen R.; Wan X.; Wang Z.; Chen W.; Deng Z.; Peng Y. Selective Reduction of CO2 by Conductive MOF Nanosheets as an Efficient Co-Catalyst under Visible Light Illumination. Appl. Catal., B 2018, 238, 339–345. 10.1016/j.apcatb.2018.07.024. [DOI] [Google Scholar]
- Zhang S.; Li L.; Zhao S.; Sun Z.; Hong M.; Luo J. Hierarchical Metal-Organic Framework Nanoflowers for Effective CO2 Transformation Driven by Visible Light. J. Mater. Chem. A 2015, 3, 15764–15768. 10.1039/C5TA03322E. [DOI] [Google Scholar]
- Huang X.; Holden H. M.; Raushel F. M. Channeling of Substrates and Intermediates in Enzyme-Catalyzed Reactions. Annu. Rev. Biochem. 2001, 70, 149–180. 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- Munro A. W.; McLean K. J.. Electron Transfer Cofactors. In Encyclopedia of Biophysics; Roberts G. C. K., Ed.; Springer: Heidelberg, 2013; pp 601–606. [Google Scholar]
- Zhang F. Y.; Zhang B. X.; Feng J. Q.; Tan X. N.; Liu L.; Liu L. F.; Han B. X.; Zheng L. R.; Zhang J.; Tai J.; et al. Highly Mesoporous Ru-MIL-125-NH2 Produced by Supercritical Fluid for Efficient Photocatalytic Hydrogen Production. ACS Appl. Energy Mater. 2019, 2, 4964–4970. 10.1021/acsaem.9b00649. [DOI] [Google Scholar]
- Zhong H.; Ly K. H.; Wang M.; Krupskaya Y.; Han X.; Zhang J.; Zhang J.; Kataev V.; Büchner B.; Weidinger I. M.; et al. A Phthalocyanine-Based Layered Two-Dimensional Conjugated Metal-Organic Framework as a Highly Efficient Electrocatalyst for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2019, 58, 10677–10682. 10.1002/anie.201907002. [DOI] [PubMed] [Google Scholar]
- Park J.; Chen Z.; Flores R. A.; Wallnerström G.; Kulkarni A.; Nørskov J. K.; Jaramillo T. F.; Bao Z. Two-Dimensional Conductive Ni-Hab as a Catalyst for the Electrochemical Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2020, 12, 39074–39081. 10.1021/acsami.0c09323. [DOI] [PubMed] [Google Scholar]
- Meng Q.; Yang J.; Ma S.; Zhai M.; Lu J. A Porous Cobalt (II) Metal-Organic Framework with Highly Efficient Electrocatalytic Activity for the Oxygen Evolution Reaction. Polymers 2017, 9, 676. 10.3390/polym9120676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohra B.; El Moll H.; Albelo L. M. R.; Mialane P.; Marrot J.; Mellot-Draznieks C.; O’Keeffe M.; Biboum R. N.; Lemaire J.; Keita B.; et al. Polyoxometalate-Based Metal Organic Frameworks (POMOFs): Structural Trends, Energetics, and High Electrocatalytic Efficiency for Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2011, 133, 13363–13374. 10.1021/ja201165c. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S.; Basu O.; Kar A.; Das S. K. Efficient Electrocatalytic Water Oxidation by Fe(Salen)-MOF Composite: Effect of Modified Microenvironment. Inorg. Chem. 2020, 59, 472–483. 10.1021/acs.inorgchem.9b02745. [DOI] [PubMed] [Google Scholar]
- Xin Z.; Wang Y.-R.; Chen Y.; Li W.-L.; Dong L.-Z.; Lan Y.-Q. Metallocene Implanted Metalloporphyrin Organic Framework for Highly Selective CO2 Electroreduction. Nano Energy 2020, 67, 104233. 10.1016/j.nanoen.2019.104233. [DOI] [Google Scholar]
- Miner E. M.; Wang L.; Dincǎ M. Modular O2 Electroreduction Activity in Triphenylene-Based Metal-Organic Frameworks. Chem. Sci. 2018, 9, 6286–6291. 10.1039/C8SC02049C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Jiang J.; Zhao W.; Li S.-H.; Mi W.; Zhang C. Truxone-Based Conductive Metal-Organic Frameworks for the Oxygen Reductive Reaction. J. Phys. Chem. C 2021, 125, 12690–12698. 10.1021/acs.jpcc.1c03418. [DOI] [Google Scholar]
- Wang H.; Zhang X.; Yin F.; Chu W.; Chen B. Coordinately Unsaturated Metal-Organic Framework as an Unpyrolyzed Bifunctional Electrocatalyst for Oxygen Reduction and Evolution Reactions. J. Mater. Chem. A 2020, 8, 22111–22123. 10.1039/D0TA04331A. [DOI] [Google Scholar]
- Wang Y. R.; Huang Q.; He C. T.; Chen Y.; Liu J.; Shen F. C.; Lan Y. Q. Oriented Electron Transmission in Polyoxometalate-Metalloporphyrin Organic Framework for Highly Selective Electroreduction of CO2. Nat. Commun. 2018, 9, 4466. 10.1038/s41467-018-06938-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Li G. Proton Conductive Zr-Based MOFs. Inorg. Chem. Front. 2020, 7, 3765–3784. 10.1039/D0QI00883D. [DOI] [Google Scholar]
- Lim D.-W.; Kitagawa H. Proton Transport in Metal-Organic Frameworks. Chem. Rev. 2020, 120, 8416–8467. 10.1021/acs.chemrev.9b00842. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S.; Debgupta J.; Singh C.; Sarkar R.; Basu O.; Das S. K. Designing UiO-66-Based Superprotonic Conductor with the Highest Metal-Organic Framework Based Proton Conductivity. ACS Appl. Mater. Interfaces 2019, 11, 13423–13432. 10.1021/acsami.9b01121. [DOI] [PubMed] [Google Scholar]
- Usov P. M.; Huffman B.; Epley C. C.; Kessinger M. C.; Zhu J.; Maza W. A.; Morris A. J. Study of Electrocatalytic Properties of Metal-Organic Framework PCN-223 for the Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2017, 9, 33539–33543. 10.1021/acsami.7b01547. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Lin Q.; Wu W.; Ye Y.; Yao Z.; Ma X.; Xiang S.; Zhang Z. Isostructural MOFs with Higher Proton Conductivity for Improved Oxygen Evolution Reaction Performance. ACS Appl. Mater. Interfaces 2020, 12, 16367–16375. 10.1021/acsami.9b23356. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhang H.; Li B.; Yang J.; Li X.; Zhang T.; He C.; Duan C.; Wang L. Bioinspired Carboxylate-Water Coordination Polymers with Hydrogen-Bond Clusters and Local Coordination Flexibility for Electrochemical Water Splitting. ACS Appl. Energy Mater. 2020, 3, 10515–10524. 10.1021/acsaem.0c01552. [DOI] [Google Scholar]
- Li W.-H.; Lv J.; Li Q.; Xie J.; Ogiwara N.; Huang Y.; Jiang H.; Kitagawa H.; Xu G.; Wang Y. Conductive Metal-Organic Framework Nanowire Arrays for Electrocatalytic Oxygen Evolution. J. Mater. Chem. A 2019, 7, 10431–10438. 10.1039/C9TA02169H. [DOI] [Google Scholar]
- Usov P. M.; Ahrenholtz S. R.; Maza W. A.; Stratakes B.; Epley C. C.; Kessinger M. C.; Zhu J.; Morris A. J. Cooperative Electrochemical Water Oxidation by Zr Nodes and Ni-Porphyrin Linkers of a PCN-224 MOF Thin Film. J. Mater. Chem. A 2016, 4, 16818–16823. 10.1039/C6TA05877A. [DOI] [Google Scholar]
- Xu Q.; Li H.; Yue F.; Chi L.; Wang J. Nanoscale Cobalt Metal-Organic Framework as a Catalyst for Visible Light-Driven and Electrocatalytic Water Oxidation. New J. Chem. 2016, 40, 3032–3035. 10.1039/C5NJ03113C. [DOI] [Google Scholar]
- Luo Y. C.; Chu K. L.; Shi J. Y.; Wu D. J.; Wang X. D.; Mayor M.; Su C. Y. Heterogenization of Photochemical Molecular Devices: Embedding a Metal-Organic Cage into a ZIF-8-Derived Matrix to Promote Proton and Electron Transfer. J. Am. Chem. Soc. 2019, 141, 13057–13065. 10.1021/jacs.9b03981. [DOI] [PubMed] [Google Scholar]
- Kajiwara T.; Fujii M.; Tsujimoto M.; Kobayashi K.; Higuchi M.; Tanaka K.; Kitagawa S. Photochemical Reduction of Low Concentrations of CO2 in a Porous Coordination Polymer with a Ruthenium(II)-CO Complex. Angew. Chem., Int. Ed. 2016, 55, 2697–2700. 10.1002/anie.201508941. [DOI] [PubMed] [Google Scholar]
- Zhang X. D.; Hou S. Z.; Wu J. X.; Gu Z. Y. Two-Dimensional Metal-Organic Framework Nanosheets with Cobalt-Porphyrins for High-Performance CO2 Electroreduction. Chem. Eur. J. 2020, 26, 1604–1611. 10.1002/chem.201904072. [DOI] [PubMed] [Google Scholar]
- Zhong H.; Ghorbani-Asl M.; Ly K. H.; Zhang J.; Ge J.; Wang M.; Liao Z.; Makarov D.; Zschech E.; Brunner E.; et al. Synergistic Electroreduction of Carbon Dioxide to Carbon Monoxide on Bimetallic Layered Conjugated Metal-Organic Frameworks. Nat. Commun. 2020, 11, 1409. 10.1038/s41467-020-15141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W.; Zhao X.; Su H.; Tang F.; Che W.; Zhang H.; Liu Q. Lattice-Strained Metal-Organic-Framework Arrays for Bifunctional Oxygen Electrocatalysis. Nat. Energy 2019, 4, 115–122. 10.1038/s41560-018-0308-8. [DOI] [Google Scholar]
- Yang F.; Hu W.; Yang C.; Patrick M.; Cooksy A. L.; Zhang J.; Aguiar J. A.; Fang C.; Zhou Y.; Meng Y. S.; et al. Tuning Internal Strain in Metal-Organic Frameworks via Vapor Phase Infiltration for CO2 Reduction. Angew. Chem., Int. Ed. 2020, 59, 4572–4580. 10.1002/anie.202000022. [DOI] [PubMed] [Google Scholar]
- Zhou T.; Du Y.; Borgna A.; Hong J.; Wang Y.; Han J.; Zhang W.; Xu R. Post-Synthesis Modification of a Metal-Organic Framework to Construct a Bifunctional Photocatalyst for Hydrogen Production. Energy Environ. Sci. 2013, 6, 3229–3234. 10.1039/c3ee41548a. [DOI] [Google Scholar]
- Cichocka M. O.; Liang Z.; Feng D.; Back S.; Siahrostami S.; Wang X.; Samperisi L.; Sun Y.; Xu H.; Hedin N.; et al. A Porphyrinic Zirconium Metal-Organic Framework for Oxygen Reduction Reaction: Tailoring the Spacing between Active-Sites through Chain-Based Inorganic Building Units. J. Am. Chem. Soc. 2020, 142, 15386–15395. 10.1021/jacs.0c06329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu U. J.; Kim S. J.; Lim H. K.; Kim H.; Choi K. M.; Kang J. K. Synergistic Interaction of Re Complex and Amine Functionalized Multiple Ligands in Metal-Organic Frameworks for Conversion of Carbon Dioxide. Sci. Rep. 2017, 7, 612. 10.1038/s41598-017-00574-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao F.; Jin Y.-H.; Liu P. F.; Yang P.; Zhang L.; Chen L.; Cao X.-M.; Gu J.; Yang H. G. Accelerated Proton Transmission in Metal-Organic Frameworks for the Efficient Reduction of CO2 in Aqueous Solutions. J. Mater. Chem. A 2019, 7, 23055–23063. 10.1039/C9TA07967J. [DOI] [Google Scholar]
- Sun F.; Chen X. Oxygen Reduction Reaction on Ni3(HITP)2: A Catalytic Site That Leads to High Activity. Electrochem. Commun. 2017, 82, 89–92. 10.1016/j.elecom.2017.07.028. [DOI] [Google Scholar]
- Li N.; Liu J.; Liu J. J.; Dong L. Z.; Xin Z. F.; Teng Y. L.; Lan Y. Q. Adenine Components in Biomimetic Metal-Organic Frameworks for Efficient CO2 Photoconversion. Angew. Chem., Int. Ed. 2019, 58, 5226–5231. 10.1002/anie.201814729. [DOI] [PubMed] [Google Scholar]
- Zhou Y. X.; Hu W. H.; Yang S. Z.; Zhang Y. B.; Nyakuchena J.; Duisenova K.; Lee S.; Fan D. H.; Huang J. Site-Selective Probes of Mixed-Node Metal Organic Frameworks for Photocatalytic Hydrogen Generation. J. Phys. Chem. C 2020, 124, 1405–1412. 10.1021/acs.jpcc.9b09634. [DOI] [Google Scholar]
- Huang N. Y.; He H.; Li H.; Liao P. Q.; Chen X. M. A Metal-Organic Framework within Situgenerated Low-Coordinate Binuclear Cu(I) Units as a Highly Effective Catalyst for Photodriven Hydrogen Production. Chem. Commun. 2020, 56, 6700–6703. 10.1039/C9CC09589F. [DOI] [PubMed] [Google Scholar]
- Shi D. Y.; Zheng R.; Sun M. J.; Cao X. R.; Sun C. X.; Cui C. J.; Liu C. S.; Zhao J. W.; Du M. Semiconductive Copper(I)-Organic Frameworks for Efficient Light-Driven Hydrogen Generation without Additional Photosensitizers and Cocatalysts. Angew. Chem., Int. Ed. 2017, 56, 14637–14641. 10.1002/anie.201709869. [DOI] [PubMed] [Google Scholar]
- Nam D. H.; Bushuyev O. S.; Li J.; De Luna P.; Seifitokaldani A.; Dinh C. T.; Garcia de Arquer F. P.; Wang Y.; Liang Z.; Proppe A. H.; et al. Metal-Organic Frameworks Mediate Cu Coordination for Selective CO2 Electroreduction. J. Am. Chem. Soc. 2018, 140, 11378–11386. 10.1021/jacs.8b06407. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Huang N. Y.; Shen J. Q.; Liao P. Q.; Chen X. M.; Zhang J. P. Hydroxide Ligands Cooperate with Catalytic Centers in Metal-Organic Frameworks for Efficient Photocatalytic CO2 Reduction. J. Am. Chem. Soc. 2018, 140, 38–41. 10.1021/jacs.7b10107. [DOI] [PubMed] [Google Scholar]
- Lian Y.; Yang W.; Zhang C.; Sun H.; Deng Z.; Xu W.; Song L.; Ouyang Z.; Wang Z.; Guo J.; et al. Unpaired 3d Electrons on Atomically Dispersed Cobalt Centres in Coordination Polymers Regulate Both Oxygen Reduction Reaction (ORR) Activity and Selectivity for Use in Zinc-Air Batteries. Angew. Chem., Int. Ed. 2020, 59, 286–294. 10.1002/anie.201910879. [DOI] [PubMed] [Google Scholar]
- Leger C.; Elliott S. J.; Hoke K. R.; Jeuken L. J.; Jones A. K.; Armstrong F. A. Enzyme Electrokinetics: Using Protein Film Voltammetry to Investigate Redox Enzymes and Their Mechanisms. Biochemistry 2003, 42, 8653–8662. 10.1021/bi034789c. [DOI] [PubMed] [Google Scholar]
- Hirst J. Elucidating the Mechanisms of Coupled Electron Transfer and Catalytic Reactions by Protein Film Voltammetry. Biochim. Biophys. Acta 2006, 1757, 225–239. 10.1016/j.bbabio.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Chen H.; Simoska O.; Lim K.; Grattieri M.; Yuan M.; Dong F.; Lee Y. S.; Beaver K.; Weliwatte S.; Gaffney E. M.; et al. Fundamentals, Applications, and Future Directions of Bioelectrocatalysis. Chem. Rev. 2020, 120, 12903–12993. 10.1021/acs.chemrev.0c00472. [DOI] [PubMed] [Google Scholar]
- Yates N. D. J.; Fascione M. A.; Parkin A. Methodologies for ″Wiring″ Redox Proteins/Enzymes to Electrode Surfaces. Chem. Eur. J. 2018, 24, 12164–12182. 10.1002/chem.201800750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olloqui-Sariego J. L.; Calvente J. J.; Andreu R. Immobilizing Redox Enzymes at Mesoporous and Nanostructured Electrodes. Curr. Opin. Electrochem. 2021, 26, 100658. 10.1016/j.coelec.2020.100658. [DOI] [Google Scholar]
- Mazurenko I.; Monsalve K.; Infossi P.; Giudici-Orticoni M.-T.; Topin F.; Mano N.; Lojou E. Impact of Substrate Diffusion and Enzyme Distribution in 3D-Porous Electrodes: A Combined Electrochemical and Modelling Study of a Thermostable H2/O2 Enzymatic Fuel Cell. Energy Environ. Sci. 2017, 10, 1966–1982. 10.1039/C7EE01830D. [DOI] [Google Scholar]
- Mijangos E.; Roy S.; Pullen S.; Lomoth R.; Ott S. Evaluation of Two- and Three-Dimensional Electrode Platforms for the Electrochemical Characterization of Organometallic Catalysts Incorporated in Non-Conducting Metal-Organic Frameworks. Dalton Trans. 2017, 46, 4907–4911. 10.1039/C7DT00578D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z. H.; Xie N. H.; Zhang M.; Xu B. Q. Nonpyrolyzed Fe-N Coordination-Based Iron Triazolate Framework: An Efficient and Stable Electrocatalyst for Oxygen Reduction Reaction. ChemSusChem 2019, 12, 200–207. 10.1002/cssc.201801886. [DOI] [PubMed] [Google Scholar]
- Dong B.-X.; Qian S.-L.; Bu F.-Y.; Wu Y.-C.; Feng L.-G.; Teng Y.-L.; Liu W.-L.; Li Z.-W. Electrochemical Reduction of CO2 to CO by a Heterogeneous Catalyst of Fe-Porphyrin-Based Metal-Organic Framework. ACS Appl. Energy Mater. 2018, 1, 4662–4669. 10.1021/acsaem.8b00797. [DOI] [Google Scholar]
- Fang Y.; Li X.; Li F.; Lin X.; Tian M.; Long X.; An X.; Fu Y.; Jin J.; Ma J. Self-Assembly of Cobalt-Centered Metal Organic Framework and Multiwalled Carbon Nanotubes Hybrids as a Highly Active and Corrosion-Resistant Bifunctional Oxygen Catalyst. J. Power Sources 2016, 326, 50–59. 10.1016/j.jpowsour.2016.06.114. [DOI] [Google Scholar]
- Micheroni D.; Lan G. X.; Lin W. B. Efficient Electrocatalytic Proton Reduction with Carbon Nanotube-Supported Metal-Organic Frameworks. J. Am. Chem. Soc. 2018, 140, 15591–15595. 10.1021/jacs.8b09521. [DOI] [PubMed] [Google Scholar]
- Ma W.; Wu F.; Yu P.; Mao L. Carbon Support Tuned Electrocatalytic Activity of a Single-Site Metal-Organic Framework toward the Oxygen Reduction Reaction. Chem. Sci. 2021, 12, 7908–7917. 10.1039/D1SC00997D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M.; Li L.; Zhu D.; Zhang H.; Zhao X. Oxygen Reduction in the Nanocage of Metal-Organic Frameworks with an Electron Transfer Mediator. J. Mater. Chem. A 2014, 2, 5323–5329. 10.1039/C3TA15319C. [DOI] [Google Scholar]
- Jahan M.; Bao Q.; Loh K. P. Electrocatalytically Active Graphene-Porphyrin MOF Composite for Oxygen Reduction Reaction. J. Am. Chem. Soc. 2012, 134, 6707–6713. 10.1021/ja211433h. [DOI] [PubMed] [Google Scholar]
- Jahan M.; Liu Z. L.; Loh K. P. A Graphene Oxide and Copper-Centered Metal Organic Framework Composite as a Tri-Functional Catalyst for HER, OER, and ORR. Adv. Funct. Mater. 2013, 23, 5363–5372. 10.1002/adfm.201300510. [DOI] [Google Scholar]
- Sohrabi S.; Dehghanpour S.; Ghalkhani M. Three-Dimensional Metal-Organic Framework Graphene Nanocomposite as a Highly Efficient and Stable Electrocatalyst for the Oxygen Reduction Reaction in Acidic Media. ChemCatChem. 2016, 8, 2356–2366. 10.1002/cctc.201600298. [DOI] [Google Scholar]
- Kang X.; Zhu Q.; Sun X.; Hu J.; Zhang J.; Liu Z.; Han B. Highly Efficient Electrochemical Reduction of CO2 to CH4 in an Ionic Liquid Using a Metal-Organic Framework Cathode. Chem. Sci. 2016, 7, 266–273. 10.1039/C5SC03291A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y. C.; Chen L. W.; Li J.; Guo Y.; Su X.; Shu M.; Zhang Q.; Gao W. Y.; Li S.; Yu Z. L.; et al. Metal-Organic Framework Membranes with Single-Atomic Centers for Photocatalytic CO2 and O2 Reduction. Nat. Commun. 2021, 12, 2682. 10.1038/s41467-021-22991-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Suzuki H.; Xie J.; Tomita O.; Martin D. J.; Higashi M.; Kong D.; Abe R.; Tang J. Mimicking Natural Photosynthesis: Solar to Renewable H2 Fuel Synthesis by Z-Scheme Water Splitting Systems. Chem. Rev. 2018, 118, 5201–5241. 10.1021/acs.chemrev.7b00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida J.; Tamimi A.; Fei H.; Pullen S.; Ott S.; Cohen S. M.; Fayer M. D. Structural Dynamics inside a Functionalized Metal-Organic Framework Probed by Ultrafast 2D IR Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18442–18447. 10.1073/pnas.1422194112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P.; Fang H.; Mukhopadhyay S.; Li A.; Rudic S.; McPherson I. J.; Tang C. C.; Fairen-Jimenez D.; Tsang S. C. E.; Redfern S. A. T. Structural Dynamics of a Metal-Organic Framework Induced by CO2 Migration in Its Non-Uniform Porous Structure. Nat. Commun. 2019, 10, 999. 10.1038/s41467-019-08939-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan G.; Fan Y.; Shi W.; You E.; Veroneau S. S.; Lin W. Biomimetic Active Sites on Monolayered Metal-Organic Frameworks for Artificial Photosynthesis. Nat. Catal. 2022, 5, 1006–1018. 10.1038/s41929-022-00865-5. [DOI] [Google Scholar]