

Summary
Ancient DNA preserved in the dental pulp offers the opportunity to characterize the genome of some of the deadliest pathogens in human history. However, while DNA capture technologies help, focus sequencing efforts, and therefore, reduce experimental costs, the recovery of ancient pathogen DNA remains challenging. Here, we tracked the kinetics of ancient Yersinia pestis DNA release in solution during a pre-digestion of the dental pulp. We found that most of the ancient Y. pestis DNA is released within 60 min at 37°C in our experimental conditions. We recommend a simple pre-digestion as an economical procedure to obtain extracts enriched in ancient pathogen DNA, as longer digestion times release other types of templates, including host DNA. Combining this procedure with DNA capture, we characterized the genome sequences of 12 ancient Y. pestis bacteria from France dating to the second pandemic outbreaks of the 17th and 18th centuries Common Era.
Subject areas: Archeology, Biochemistry, Molecular biology
Graphical abstract
Highlights
-
•
Short tooth powder digestion times improve recovery of Yersinia pestis ancient DNA
-
•
Variable Y. pestis detection success across teeth from the same individual
-
•
Sequencing of 12 second plague pandemic genomes from France
-
•
Different strains during the Thirty Years’ War and the Great Plague of Marseille
Archeology; Biochemistry; Molecular biology
Introduction
Ancient DNA preserved within archaeological and paleontological remains provides genetic information about our evolutionary past.1 The increasing throughput of next-generation sequencing instruments has considerably improved our capacity to characterize ancient genomes, despite environmental microbes often producing the dominant fraction of the DNA extracts.2 Specific types of remains, including petrosal bones,3 ossicles,4 and the tooth cementum,5 are known to be generally less prone to environmental DNA contamination. Together with improved decontamination and DNA extraction procedures, involving various pre-digestion treatments of sample powder through bleaching6,7 and/or washing buffers,8,9,10 these biological remains have facilitated the characterization of ancient genomes. The development of DNA capture methodologies targeting million-scale SNP (Single Nucleotide Polymorphism) panels,11,12 whole chromosomes13 or even the entire genome14,15 have provided a further opportunity to focus sequencing efforts on the DNA fraction of interest, therefore, increasing sensitivity and reducing sequencing costs. As a result, there has been an acceleration since the first ancient human genomes have been sequenced in 201016,17,18 to the thousands of ancient human genomes characterized, with recent human paleogenomic studies typically including hundreds of individuals.19,20
Besides host and environmental microbial material, ancient DNA extracts can also preserve molecules originating from pathogens that circulated in the blood system when the host died.21 In cases where the pathogenic load is high and environmental contamination is low, shotgun sequencing has allowed for the successful characterization of the entire genome sequence from ancient pathogens, including viruses (e.g., smallpox,22,23 hepatitis B24,25), bacteria (e.g., Mycobacterium leprae26), and protozoan parasites (e.g., Plasmodium vivax27). Reconstructing ancient pathogen genomes, however, most commonly requires DNA capture technologies, both due to the molecular complexity of ancient DNA extracts and the generally limited genome size of pathogens, relative to their hosts.21,28 Additionally, the type of material considered for ancient pathogen genome characterization is critical, as tissues favoring host DNA preservation, such as petrosal bones, show virtually no bacterial pathogen DNA.29,30 Despite these limitations, more than five hundred ancient pathogen genomes have been sequenced,31 revealing important insights into the epidemiological outbreaks with substantive morbidity and mortality.
Plague represents the best-studied infectious disease from an ancient DNA perspective.32 DNA analyses of human individuals buried during the Justinian plague and the Black Death have confirmed Y. pestis as the etiological agent responsible for massive human loss in the sixth-eighth century Common Era (c. CE)33,34 and the 14th c. CE,35 respectively. Such analyses have, however, revealed that plague already circulated during the late Neolithic and early Bronze Age,36,37,38,39,40,41 and have started reconstructing the evolutionary trajectory of key genetic changes underlying virulence and transmission.38,42 Genetic evidence has also tracked the expansion of the second plague pandemic from a Central Asian homeland43 and the following diversification of several lineages through space and time.44
Despite such advances, the characterization of ancient pathogen genomes remains difficult. Relatively low detection rates and minimal target ancient DNA contents are commonly observed in mass graves, even when the association with known pandemic, such as the second plague pandemic, is documented historically.45 While the performance of various DNA capture systems is well established,46,47,48 no single study has attempted to optimize the molecular toolkit for pathogen DNA extraction and capture. In this study, we investigated the temporal dynamics of plague and host DNA release from dental pulp, using a total of 120 human teeth dated to the 17th and 18th c. CE. These remains originated from five French archaeological sites associated with the second plague pandemic. We found that the vast majority of the pathogen DNA can be recovered following a short pre-digestion wash of the dental pulp, while the recovery of the host DNA benefits from further digestion. Therefore, focusing on the pre-digestion wash improves the ratio of Y. pestis-to-host DNA. Whether such an approach could also enhance the recovery of other ancient pathogens in different preservation contexts and/or tissue types remains to be tested. Combined with targeted capture, pre-digestion washes helped us characterize 12 ancient Y. pestis genomes, including from material showing limited DNA preservation. Our work contributes to ongoing efforts aimed at mapping the genetic diversity of second pandemic plague strains.
Results and discussion
Sample characteristics
We collected 120 teeth from 89 human individuals that were excavated from French mass graves and cemeteries related to the second plague pandemic (Figure 1, Data S1). More specifically, a total of 56 teeth originated from the sites of La Major (1720 CE, Marseille) and Les Rayettes (1721 CE, Martigues), both associated with the last major plague outbreak in France. This epidemic started after the ship Grand Saint Antoine departed from the Levant and landed on the Marseille harbor in 1720 CE. It rapidly decimated half of the population of the city, before it spread to the Provence region, hitting Marseille a second time (causing considerably lower mortality), and finally vanished in 1722 CE.49 Overall, it is estimated that this plague outbreak killed more than a quarter of the population of the Provence region. The remaining 64 teeth were collected from 58 individuals buried during the 17th c. CE, including from the Lariey Puy-Saint-Pierre cemetery (1629–1630 CE), as well as the Maladrerie Saint-Lazare (1630–1670 CE, Beauvais) and the Hôtel-Dieu Saint-Jean-Baptiste (1630–1670 CE, Amiens). While the Lariey cemetery was previously analyzed for Y. pestis and human ancient DNA,45 the latter two sites were genetically investigated here for the first time. These sites correspond to plague mass graves formed at the time of the Thirty Years’ War.
Figure 1.

Sample corpus
(A) Sample location and dates. The archaeological sites analyzed in this study are all from France and are shown in purple, with respect to those previously published, following the coloring scheme from Spyrou and colleagues,43 for consistency. Pictures of the mass graves studied with the exception of the archaeological site of Lariey (12 teeth), which was investigated by Seguin-Orlando and colleagues45 (the number of teeth investigated, N, is indicated on each picture).
(B) La Major (Marseille, 1720 CE).
(C) Les Rayettes (Martigues, 1721 CE).
(D) Hôtel-Dieu Saint-Jean-Baptiste (Amiens, 1630–1670 CE).
(E) Maladrerie Saint-Lazare (Beauvais, 1630–1670 CE). See also Tables S1 and S2.
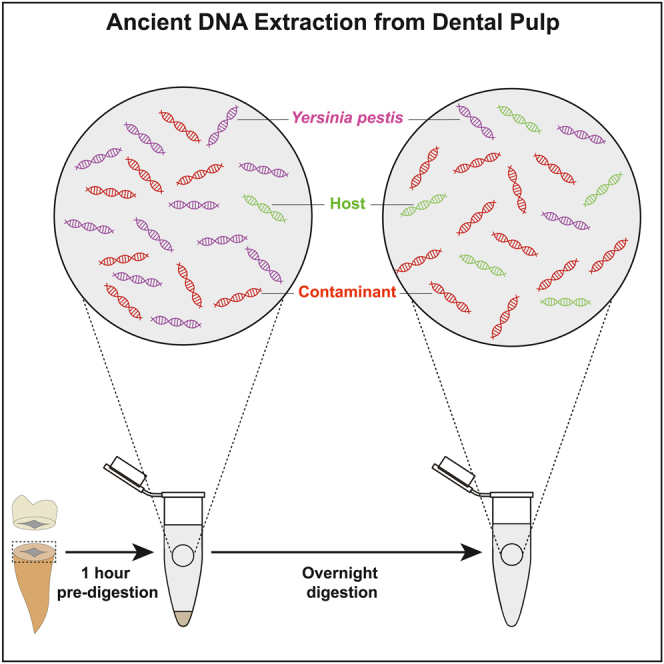
A total of 33 teeth were first subjected to the standard procedure for ancient DNA extraction routinely applied at the Centre for Anthropobiology and Genomics of Toulouse (CAGT) ancient DNA facilities. Briefly, the dental pulp powder is collected by cutting the tooth at the cementum-enamel junction in order to drill the pulp chamber from inside with a diamond ball bit. The powder is first pre-digested in a DNA extraction buffer for 1 h at 37°C. The undigested tooth pellets are then collected and digested until completion in a fresh DNA extraction buffer, following overnight incubation with agitation at 42°C. This procedure was previously optimized for recovering ancient DNA from the host,50 but nonetheless led to the successful characterization of two ancient human and Y. pestis genomes from the Lariey cemetery, following shotgun sequencing on Illumina platforms.45 Stringent alignment of shallow sequencing data against the Y. pestis reference genome34,51 revealed limited, if any, Y. pestis DNA preservation across all the samples investigated (< 0.93%), regardless of the archaeological site considered (Table S1). DNA capture procedures were, thus, implemented to attempt to enrich for Y. pestis and human mtDNA templates, using the synthetic RNA 80-mers probes designed by Wagner and colleagues.33 In this experiment, a total of 5 and 55 amplified DNA libraries were selected from individuals excavated at Lariey and La Major, respectively, as representatives of the entire range of DNA preservation conditions. Bioinformatic demultiplexing and collapsing of capture DNA reads provided between 14,386 and 2,250,565 sequences per individual, of which only those from two individuals from Lariey (LAR8 and LAR11) provided sufficient coverage of the Y. pestis genome (Table S1). Further metagenomics analyses in MetaPhlAn 452 confirmed a greater Y. pestis content in capture versus shotgun collapsed reads for 12 libraries prepared from six individual teeth (Figures 2A and 2B). This supported numbers of classified microbial species dropping with capture, as expected for a procedure aimed at focusing sequencing efforts on Y. pestis only. Accordingly, the fraction of environmental and oral bacteria identified following capture was reduced relative to shotgun experiments (Figure 2B).
Figure 2.

MetaPhlAn 4 microbial taxonomic profiles of the tooth material analyzed
(A and B) Comparison of microbial diversity in shotgun versus capture sequence datasets. These experiments were carried out on the extracts resulting from a 1-h pre-digestion, followed by an overnight digestion of the remaining pellets (1h+ON). (A, B) Relative abundances of microbial species.
(C and D) Same as A, B except that different pre-digestion times were contrasted from 10 to 60 min and from 0 to 10 min. The analyses were restricted to two teeth from the same sample (AMIENS268t4 and AMIENS268t2).
While targeted DNA capture improved the Y. pestis DNA recovery rate, the vast majority of the sequence data for other individuals consisted of PCR duplicates (37.72–98.03%), which limited the cost-effectiveness as an approach for characterizing the plague genome. The high proportion of PCR duplicates indicated that the total amount of plague DNA templates present in the original extracts was extremely limited. We, thus, decided to identify DNA extraction procedures that could enhance the recovery of ancient plague DNA molecules.
Pre-digestion DNA release
Previous work reported increased proportions of host DNA in libraries constructed following complete digestion, relative to those obtained pre-digestion.8,9 This suggested that pre-digestion could wash away a fraction of the environmental microbial DNA in contrast to the host DNA, which would be preserved deeper into the calcified matrix, and thus would require longer digestion time to be released.53 We reasoned that the Y. pestis DNA would be mostly restricted to the blood circulation and would not penetrate the deeper extracellular matrix. Under this assumption the pathogen DNA would follow release kinetics similar to the environmental microbial DNA. To test this theory, we purified the DNA from 17 1-h pre-digested fractions (1h) to prepare libraries for the capture of Y. pestis and human mtDNA genomes. The DNA contents of the pre-digested samples were compared to those obtained following capture of the DNA libraries prepared after complete pellet digestion (1h+O/N; Figures 3A and 3B). Normalizing for sequencing efforts after Y. pestis whole-genome capture, we found that most of the libraries constructed following 1 h pre-digestion (1h, without overnight digestion) contained larger proportions of Y. pestis sequences. We also observed inverse trends with clonality, which is reduced in 1-h pre-digested samples (Figure S1). Combined, these results support the presence of more Y. pestis DNA molecules in the pre-digested fraction.
Figure 3.

Number of unique high-quality alignments identified following different extraction conditions
(A and B) One hour of pre-digestion wash (1h, dark brown) versus complete pellet digestion overnight (1h+O/N, light brown).
(C and D) 10, 20, 30, 40, 50, and 60 min of pre-digestion wash (from purple to yellow).
(E and F) 0, 0.5, 1, 2.5, 5, 7.5, and 10 min of pre-digestion wash (from blue to green). Analyses are repeated 10 times, following random sampling of 4,557 collapsed reads in each experimental condition for normalizing sequencing efforts. Data are represented as mean ± SEM. Alignments are shown against the Y. pestis CO92 genome (left; A, C, E, pathogenic) and the human mtDNA genome (right; B, D, F, rCRS). See also Figure S1.
Human mtDNA content followed no particular trend, representing lower, similar or greater proportions of the sequence data depending on the specimen considered. This indicated that only some of the remains analyzed still contained a substantial fraction of human DNA in the undigested pellets and that DNA preservation was considerably more limited in others.
Encouraged by the increased proportions of Y. pestis DNA present in the pre-digested fraction, we further investigated the kinetics of DNA release during the 60 min (min) pre-digestion wash. To achieve this, we generated fresh dental pulp powder for four additional teeth from individuals that showed positive plague identification in our previous experiments. The powder was then subjected to six consecutive partial digestion steps at 37°C of 10 min each (10 min, 20 min, 30 min, 40 min, 50 min, and 60 min; Figures 3C and 3D). The remaining undigested pellets were collected after each step, and incubated further in the same volume of a fresh extraction buffer. The resulting six pre-digested fractions were purified and used for library preparation, amplification, DNA capture, and sequencing (Figures 3C and 3D). Normalizing for sequencing efforts after Y. pestis whole-genome capture, we found that most of the Y. pestis DNA was released in the extraction buffer during the first 10 min pre-digestion. The proportion of Y. pestis DNA released during further pre-digestion steps was consistently lower than that released during the previous incubations. This indicated rapid Y. pestis DNA release in the extraction buffer, and diminishing returns with increasing pre-digestion times. Human mtDNA release followed a similar trend for one tooth (MAJ46t9), but exhibited no consistent trend across the four teeth investigated. Interestingly, MetaPhlAn 4 taxonomic profiling of microbial diversity for the single of the four teeth for which sufficient sequencing data were generated (≥ 100,000 collapsed reads; AMIENS268t2), revealed increasingly abundant off-target microbial hits with pre-digestion times (Figures 2C and 2D). This translated into an increasing number of classified microbial species, consistent with capture probes not being saturated by Y. pestis DNA templates only allowing aspecific annealing, including to oral bacterial DNA (e.g., Desulfobulbus oralis; Figure 2D).
We repeated the same experiment for four additional teeth belonging to the same four individuals, except that shorter pre-digestion times were considered (0.5 min, 1 min, 2.5 min, 5 min, 7.5 min, and 10 min; Figures 3E and 3F). The proportions of Y. pestis and human mtDNA present in each resulting pre-digested fraction were compared to those found in the absence of pre-digestion (0 min), which were estimated after suspending the dental pulp powder in the pre-heated extraction buffer, and immediately centrifugating the pellets to collect the supernatant.
Two of the teeth examined were found to contain almost no Y. pestis DNA considering pre-digestion times up to 10 min (MAJ46t11 and SQ8723t4; Figures 3E and 3F). Other teeth from these individuals, however, were tested positive for Y. pestis in previous experimental conditions. This highlights that the success of Y. pestis detection strongly depends on the tooth material considered, possibly due to differences in DNA preservation across tooth microenvironments and/or tooth physical integrity conditioning colonization success from the oral and environmental communities.
The other two of the four teeth examined showed inconsistent kinetics of DNA release, with one sample (AMIENS268t4) releasing roughly twice as much Y. pestis DNA between 2.5 and 5 min than during the following 2.5 min (between 5 and 7.5 min), but the same proportion again between 7.5 and 10 min. For the last tooth (LAR27t4), the proportion of Y. pestis DNA collected in the last experimental condition was 6.43-fold–10.10-fold larger than during the two previous steps, despite lasting for 2.5 min each. It was also equivalent to the proportion of Y. pestis DNA obtained following a 1 min pre-digestion (Figure 3E). The AMIENS268t4 tooth provided sufficient numbers of collapsed reads for metagenomic profiling with MetaPhlAn 4, which revealed the presence of other microbes than Y. pestis in some but not all pre-digestion conditions (Figures 2C and 2D). Combined, these results indicated stochastic effects dominating DNA release of both Y. pestis and other microbes during the first 10 min of pre-digestion. No specific trend could again be observed for the release of human mtDNA.
As the pre-digestion conditions between 10 and 60 min provided consistent results across the four teeth investigated, we decided to further characterize the underlying kinetics of DNA release in the pre-digestion buffer. We found that the proportion of unique stringent alignments characterized decreased exponentially through time (Figures 4A and 4B). This was true for both Y. pestis and human mtDNA, which followed trends that were not significantly different (ANCOVA test, p values ≥ 0.19). As a result, the cumulative proportion of Y. pestis and human mtDNA templates sequenced rapidly reached saturation, leaving only 5% (1%) of unreleased templates after 28.0–38.5 min (43.1–59.2 min) of pre-digestion (Figures 4C, 4D, and S2). This indicated that pre-digesting the dental pulp powder for 30 to 60 min in the experimental conditions tested would be sufficient to release most of the Y. pestis DNA present. We, thus, performed the DNA analysis of the remaining teeth following a conservative pre-digestion for 60 min at 37°C to extract the DNA fraction of the pathogen almost entirely. The supernatant was purified and concentrated for preparing a total of 176 DNA libraries for Y. pestis and human mtDNA enrichment.
Figure 4.

Kinetics of DNA release during pre-digestion wash
(A and B) Percentage of DNA released following pre-digestion wash of incremental time (10 min–1 h). The results are shown for the AMIENS268t2 (A) and MAJ46t9 (B) teeth (see Figure S2 for LAR27t2 and SQ8723t2 results). High-quality sequence alignments are aligned against the pathogen genome (CO92 and plasmids, dark purple, and dark orange) or the human mtDNA genome (Revised Cambridge Reference Sequence rCRS, light purple, and light orange).
(C and D) Prediction for the DNA fraction remaining to be released following incremental pre-digestion wash. Predictions are based on the linear models shown in panels A and B for Y. pestis (dark purple and dark orange) and human mtDNA (light purple and light orange). The blue and dark orange dashed lines indicate the times when respectively 95% and 99% of the DNA has been released. See also Figure S2.
Phylogenetic analyses
The improved DNA extraction procedures developed in this study allowed us to characterize the genome sequence of Y. pestis strains present in 12 ancient individuals. Patterns of nucleotide mis-incorporations and base compositional profiles at the genomic positions preceding and following read termini were characteristic of authentic ancient DNA data generated following USER-treatment of extracts (Figure S3). Specifically, USER enzymes cleave those cytosine residues that are deaminated postmortem, leaving only slightly inflated C→T (G→A) mis-incorporation rates at read starts (ends), as well as an excess of ancient DNA templates starting immediately after cytosine residues.2 Read-to-reference edit distance distributions confirmed a closer genetic proximity to Y. pestis relative to the closest outgroup, Yersinia pseudotuberculosis (Figure S4).
Phylogenetic reconstructions revealed that most of the second pandemic Y. pestis genomes clustered together within a monophyletic group, including strains from the Black Death to the 18th c. CE (Figures 5 and S5, Table S2). Within this cluster, the 12 ancient Y. pestis genomes characterized in this study grouped together with all the other post-Black Death ancient Y. pestis genomes hitherto sequenced. This was true genome-wide (Figure S6), or when considering only a subset of 29,609 parsimony informative sites located outside non-core, repetitive or highly conserved regions, which are reported to be less prone to environmental contaminant mis-mapping (Figures 5 and S5).54 The latter analysis confirmed the 1338–1339 CE genome from KaraDjigach, Kyrgystan, as basal to all subsequent strains sequenced, belonging to the second and third pandemic. Phylogenetic reconstructions also supported all modern branch 1 genomes responsible for the third pandemic descending from a subset of 14th c. CE strains identified in the Netherlands (BergenOpZoom), the United Kingdom (London), and Russia (Bolgar), as previously reported.35,55,56 Additionally, the LAR27 genome from Lariey characterized in this study (1629–1630 CE) appeared closely related to the two genomes previously reported from the same site (LAR8 and LAR11), confirming their genetic proximity with contemporary Y. pestis genomes from San Procolo a Naturno (Italy, 1636 CE).45 The Y. pestis genomes from Beauvais and Amiens (1630–1670 CE) also clustered with this group, suggesting common epidemiological origins. This lineage further spread during or in the aftermath of the Thirty Years’ War (Figures 5 and S5), which is recognized as one of the most destructive conflicts in European history, it affected Central Europe between 1618 and 1648 CE, a time contemporary with the ancient Y. pestis genome from Brandenburg, which is located basally within this group.
Figure 5.

Phylogenetic reconstruction
(A) Maximum Likelihood tree (IQ-TREE), considering 29,609 genome-wide variant positions known to provide reliable phylogenetic signal. This analysis included 185 previously published genomes, but the tree shown is restricted to second and third pandemic genomes, disregarding branch lengths, for clarity (the tree with branch length is shown in Figure S5). Node supports are estimated from SH-aLRT calculations (left) and ultrafast bootstrap approximation (right).
(B) Sample dates CE, with error margins representing the lower and upper boundaries provided by archaeological contexts.
(C and D) represent the coverage and the logarithm of the average sequencing depth-of-coverage achieved for the different CO92 genomes considered. The colors illustrated follow the coloring scheme from Spyrou et al., 2022,43 and the tip shapes indicate the historical context of archaeological samples investigated with respect to Figure 1. See also Figures S5 and S6.
The Y. pestis genome sequences retrieved from the samples of La Major (Marseille, 1720 CE) and Les Rayettes (Martigues, 1721 CE) formed a monophyletic group together with the previously reported genomes from Observance (Marseille, 1722 CE).57 This supports historical sources that describe an initial introduction of the plague in Marseille by the ship Grand Saint Antoine on May 1720 CE,49 and a subsequent rapid spread to the entire Provence region. Interestingly, our phylogenetic reconstructions indicated that the ancient Y. pestis genomes from Observance do not directly descend from those entering Marseille two years earlier and present at La Major. Taken at face value, this may have provided support for the introduction of two closely related plague strains. However, we caution that due to extreme DNA degradation, the genomes from La Major and Les Rayettes could only be characterized with limited coverage (between 1.19-fold and 5.25-fold), despite the improved methodology described in this study. Furthermore, the majority of ancient strains presented in our phylogenetic analysis appear to have excessively long terminal branches, even when compared to modern genetic diversity (Figures S5 and S6). The combination of low-genome coverage, remnant sequencing errors due to DNA degradation, and residual contamination from environmental microbes, can result in excessive terminal branch lengths in ancient genomes. The presence of such long branches can introduce significant biases in phylogenetic placement,58,59 possibly impacting fine-scale evolutionary predictions. Additionally, we note that the Observance genomes were characterized using a different DNA capture system,57 introducing subtle technical batch effects, also potentially impacting phylogenetic reconstruction. This can be illustrated by the single Lariey genome characterized in this study, following DNA capture, which appears phylogenetic close, but does not form a monophyletic clade with those previously characterized by shotgun sequencing in Seguin-Orlando and colleagues.45 In light of these caveats, we consider our phylogenetic reconstructions consistent with the abundance of historical sources supporting a unique introduction of the plague in Marseille in 1720 CE.49 Determining whether the underlying strain first emerged in the Levant, or derived from a European source re-entering Europe through the Grand Saint-Antoine, requires further work. For example, mapping the genetic structure of Y. pestis strains at a finer-grained resolution during the 17th and the 18th c. CE may clarify the geographic sources and spread of related plague outbreaks. Integrating historical evidence will however be essential, given that the mutation rate of the Y. pestis genome provides genetic resolution at the 10-year timescale,60 which is considerably slower than the pace of human circulation and trade.
Genome analyses
We further tested whether the ancient Y. pestis strains sequenced in this study showed specific genetic characteristics compared to their closest phylogenetic relatives. The average depth-of-coverage of the 12 individuals sequenced here ranged between 0.16 and 56.16-fold for the circular chromosome CO92 (median = 3.97-fold, average = 11.67-fold), while it ranged between 0.79 and to221.45-fold, 0.20– to73.46-fold and 5.71– to1092.67-fold for the pCD1, pMT1, and pPCP1 plasmids, respectively. Interestingly, the average proportion of high-quality alignments against the CO92 chromosome was lower-than-expected on the basis of the fraction of CO92 probes used for capture. The average proportions of high-quality alignment against the three different plasmids were, conversely, larger-than-expected on the basis of the probe distribution (Figures 6A–6D). This is in line with bacterial cells containing multiple copies of plasmids. The excess of plasmid DNA relative to probe proportions was, however, significantly more limited in the specimens dated from the 17th c. CE than in those from the 18th c. CE (Figures 6B–6D). This suggests that the latter contained a greater number of plasmid copies per cell than the former. Interestingly, the plasmid copy number can have an impact on virulence,61 in particular the pCD1 plasmid, which carries the T3SS genes responsible for delivering Yersinia outer membrane proteins into the host cells.62 The number of plasmids carried by the 17th and 18th c. CE strains may, thus, have driven differences in their virulence. Whether this genetic feature played a role in the excessive mortality observed during the Great Plague of Marseille, which decimated half the city’s population in only a few months,49 remains to be tested. Whether it also had equally dramatic consequences on the populations of rodent natural hosts, thus, precipitating disease disappearance is also unknown.
Figure 6.

Plasmid copy numbers per bacterial cell
(A–D)Average proportions of sequences mapping against the CO92 circular chromosome (A) and the plasmids (B, C, and D) for the different archaeological sites from the 17th c. CE (orange, Amiens, Beauvais, and Lariey) and the 18th c. CE (light brown, La Major, and Les Rayettes). Probe proportions targeting the CO92 genome and the three plasmids are indicated by the brown bars. Data are represented as mean ± SEM. Statistically significant values (Wilcoxon test) for inter- and intra-century comparisons are indicated. Only those libraries prepared on the DNA extracted following 1-h pre-digestion were considered to avoid technical batch effects, disregarding those for which an insufficient number of high-quality read alignments could be identified (i.e. < 500).
Furthermore, a 49-kb region from the CO92 chromosome was found to be largely absent in the strains from La Major and Les Rayettes (Figures 7A–7E). The cspE gene (cspE_1883773–1883985), which is involved in the survival and the multiplication at low temperature,63 returned spurious non-null coverage, due to sporadic alignments resulting from environmental Yersinia bacteria. The 49-kb region spans the mgtA (mgtA_1888533–1891232, previously named mgtB) and mgtC (YPORS09280_1887434–1888132) genes involved in the survival of the bacteria under condition with low concentrations of Mg2+.64 It was previously reported to also be missing from the genomes of their closest phylogenetic relatives, including 18th c. CE strains from France (Observance, Marseille), Russia (Maist), and Sweden (Pestbacken), as well as mid-16th/mid-17th c. CE strains from the United Kingdom (London).51 Moreover, a 47-kb region overlapping 11 virulence and pathogenicity factors (inv_2040296–2042350, fyuA_2140840–2142861, ybtE_2142992–2144569, ybtT_2144573–2145376, ybtU_2145373–2146473, irp1_2146470–2155961, irp2_2156049–2162156, ybtA_2162347–2163306, ybtP_2163563–2165275, ybtQ_2165262–2167064, ybtX_2167057–2168337) was largely under-covered in all the genomes from La Major, Beauvais, Lariey and Amiens reported here (Figure 7A). They were, however, covered in two genomes from Lariey that were previously characterized using shotgun sequencing. We, thus, consider that the limited coverage observed most likely reflects under-performing probe-template annealing in those regions than genuine genomic rearrangements.
Figure 7.

Genome coverage and %GC variation
(A) CO92 Y. pestis reference genome.
(B) pCD1 plasmid.
(C) pMT1 plasmid.
(D) pPCP1 plasmid. A total of 13 samples sequenced in this study are shown (purple) together with two close relatives from the 17th c. CE (yellow) and the 18th c. CE (green) for comparison.
(E) Fraction of the gene covered at least once for 235 virulence and pathogenicity loci. Second pandemic Y. pestis genomes were considered as long as they were sequenced to a minimum 2-fold average depth-of-coverage. The red box highlights the 49-kb deletion identified, with “X” indicating those virulence and pathogenicity loci showing best Blast hits against environmental Yersinia species, and, thus, likely reflecting false positive alignments against Y. pestis. See also Figures S3, S4, and S7 and Table S3.
We next used snpToolkit56 to characterize the single nucleotide mutations found among the new 17th and 18th c. CE strains sequenced here, and their closest phylogenetic relatives. This analysis revealed a total of 375 SNPs, comprising 148 non-synonymous, 97 synonymous, and 130 intergenic mutations (Figure S7, Table S3). Interestingly, the iron ABC transporter encoded by the YbtP gene, which is essential for the bacterial physiology65 and can deplete Y. pestis avirulence in mice,66 was found to be polymorphic in both strains from the 17th and 18th c. CE. No mutations were otherwise found in the 235 virulence and pathogenicity loci considered in the analyses above. The genomes from Amiens and Beauvais Y. pestis strains (1630–1670 CE) showed remarkable genetic similarity with those from Lariey (1629–1630 CE), as they shared only eight non-synonymous variants not found in the latter (Table S3). One such variant affected the autotransporter YapC gene, which mediates adhesion to human cells and, thus, may affect host innate immune response.67 In contrast, the number of non-synonymous differences found between the 1720–1722 CE genomes from La Major, Les Rayettes, and Observance was larger. Whether this is indicative of currently unidentified minor mis-mapping from other bacteria than Y. pestis, or faster mutation rates in this specific lineage requires further clarification through additional sequencing of complete genomes from Y. pestis strains responsible for the plague epidemic that spreads to the Provence region in the early 18th c. CE.
Conclusion
In this study, we report that ancient Y. pestis DNA is almost entirely released during a 60 min pre-digestion wash of the dental pulp. This finding might have important methodological implications for the workflow of ancient pathogen studies, as DNA extracts obtained following pre-digestion appear to maximize the ratio of ancient pathogen DNA to human DNA. This contrasts to the extracts obtained following complete pellet digestion, which are generally used for the study of ancient plagues, despite containing a much greater ratio of human to pathogen DNA. In the experimental conditions described herein, a pre-digestion of the dental pulp powder for 60 min at 37°C appears sufficient to release at least 99% of the ancient Y. pestis DNA generally preserved. Applying targeted capture to these DNA extracts allowed us to characterize 12 ancient Y. pestis genomes from five French archaeological sites dated to the 17th and 18th c. CE. We find that the number of plasmid copies within Y. pestis bacteria from the 17th c. CE was lower than in those from the 18th c. CE. The Thirty Years’ War likely facilitated the spread of a plague lineage throughout Germany, Italy, and France, all of which were eventually dead-end emergences. The last plague outbreak in France appears to follow a single emergence in Marseille, before spreading to the Provence region.
Limitations of the study
In this study, genome variation among ancient Y. pestis strains was only investigated from sequence alignments against one single reference genome. The full extent of structural variation and their possible consequences on transmission, pathogenicity and virulence remains to be characterized. Moreover, the quality of the data generated for most of the ancient genomes presented was limited, which precluded the examination of fine-scale strain evolution and divergence dates. Only a small number of samples were subjected to pre-digestion tests, they were also confined to a single geographic area and, only encompassed preservation conditions from a relatively recent period. Even if our results highlight a strong pattern for the DNA release of Y. pestis, experiments should be extended to various areas, periods and epidemics. Finally, whether the recommended methodology shows similar performance on other types of non-dental tissues, and will equally apply to the entire range of pathogenic bacteria that can circulate in the blood, remains untested.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Osteological remain | This study | MAJ6t |
| Osteological remain | This study | MAJ17t |
| Osteological remain | This study | MAJ19t |
| Osteological remain | This study | MAJ33t |
| Osteological remain | This study | MAJ36t |
| Osteological remain | This study | MAJ38t |
| Osteological remain | This study | MAJ38t2 |
| Osteological remain | This study | MAJ38t3 |
| Osteological remain | This study | MAJ38t4 |
| Osteological remain | This study | MAJ38t5 |
| Osteological remain | This study | MAJ38t6 |
| Osteological remain | This study | MAJ43t |
| Osteological remain | This study | MAJ44t |
| Osteological remain | This study | MAJ46t |
| Osteological remain | This study | MAJ46t2 |
| Osteological remain | This study | MAJ46t3 |
| Osteological remain | This study | MAJ46t4 |
| Osteological remain | This study | MAJ46t5 |
| Osteological remain | This study | MAJ46t6 |
| Osteological remain | This study | MAJ46t7 |
| Osteological remain | This study | MAJ46t8 |
| Osteological remain | This study | MAJ46t9 |
| Osteological remain | This study | MAJ46t11 |
| Osteological remain | This study | MAJ48t |
| Osteological remain | This study | MAJ55t2 |
| Osteological remain | This study | MAJ70t |
| Osteological remain | This study | MAJ72t |
| Osteological remain | This study | MAJ75t |
| Osteological remain | This study | MAJ75t3 |
| Osteological remain | This study | MAJ75t4 |
| Osteological remain | This study | MAJ75t5 |
| Osteological remain | This study | MAJ75t6 |
| Osteological remain | This study | MAJ75t7 |
| Osteological remain | This study | MAJ75t8 |
| Osteological remain | This study | MAJ75t9 |
| Osteological remain | This study | MAJ86t |
| Osteological remain | This study | MAJ87t |
| Osteological remain | This study | MAJ89t |
| Osteological remain | This study | MAJ104t |
| Osteological remain | This study | MAJ1001t |
| Osteological remain | This study | RAY1002t |
| Osteological remain | This study | RAY1005t |
| Osteological remain | This study | RAY1009t |
| Osteological remain | This study | RAY1015t |
| Osteological remain | This study | RAY1015t2 |
| Osteological remain | This study | RAY1015t3 |
| Osteological remain | This study | RAY1028t |
| Osteological remain | This study | RAY1028t2 |
| Osteological remain | This study | RAY3009t |
| Osteological remain | This study | RAY3010t |
| Osteological remain | This study | RAY3015t |
| Osteological remain | This study | RAY3023t |
| Osteological remain | This study | RAY3024t |
| Osteological remain | This study | RAY3027t |
| Osteological remain | This study | RAY3030t |
| Osteological remain | This study | RAY3030t2 |
| Osteological remain | This study | LAR8t |
| Osteological remain | This study | LAR9t |
| Osteological remain | This study | LAR11t |
| Osteological remain | This study | LAR14t |
| Osteological remain | This study | LAR23t |
| Osteological remain | This study | LAR24t |
| Osteological remain | This study | LAR26t |
| Osteological remain | This study | LAR27t |
| Osteological remain | This study | LAR27t2 |
| Osteological remain | This study | LAR27t4 |
| Osteological remain | This study | LAR30t |
| Osteological remain | This study | LAR31t |
| Osteological remain | This study | SQ7044t |
| Osteological remain | This study | SQ8504t |
| Osteological remain | This study | SQ8579t |
| Osteological remain | This study | SQ8833t |
| Osteological remain | This study | SQ8844t |
| Osteological remain | This study | SQ8950t |
| Osteological remain | This study | SQ8519t |
| Osteological remain | This study | SQ8637t |
| Osteological remain | This study | SQ8670t |
| Osteological remain | This study | SQ8772t |
| Osteological remain | This study | SQ8893t |
| Osteological remain | This study | SQ8918t |
| Osteological remain | This study | SQ8962t |
| Osteological remain | This study | SQ8723t |
| Osteological remain | This study | SQ8723t2 |
| Osteological remain | This study | SQ8723t4 |
| Osteological remain | This study | AMIENS109t |
| Osteological remain | This study | AMIENS121t |
| Osteological remain | This study | AMIENS122t |
| Osteological remain | This study | AMIENS133t |
| Osteological remain | This study | AMIENS134t |
| Osteological remain | This study | AMIENS146t |
| Osteological remain | This study | AMIENS148t |
| Osteological remain | This study | AMIENS177t |
| Osteological remain | This study | AMIENS183t |
| Osteological remain | This study | AMIENS209t |
| Osteological remain | This study | AMIENS222t |
| Osteological remain | This study | AMIENS255t |
| Osteological remain | This study | AMIENS260t |
| Osteological remain | This study | AMIENS266t |
| Osteological remain | This study | AMIENS268t |
| Osteological remain | This study | AMIENS268t2 |
| Osteological remain | This study | AMIENS268t4 |
| Osteological remain | This study | AMIENS32t |
| Osteological remain | This study | AMIENS41t |
| Osteological remain | This study | AMIENS46t |
| Osteological remain | This study | AMIENS54t |
| Osteological remain | This study | AMIENS55t |
| Osteological remain | This study | AMIENS63t |
| Osteological remain | This study | AMIENS69t |
| Osteological remain | This study | AMIENS74t |
| Osteological remain | This study | AMIENS75t |
| Osteological remain | This study | AMIENS78t |
| Osteological remain | This study | AMIENS81t |
| Osteological remain | This study | AMIENS82t |
| Osteological remain | This study | AMIENS85t |
| Osteological remain | This study | AMIENS86t |
| Osteological remain | This study | AMIENS91t |
| Osteological remain | This study | AMIENS93t |
| Osteological remain | This study | AMIENS94t |
| Osteological remain | This study | AMIENS97t |
| Osteological remain | This study | AMIENS99t |
| Deposited data | ||
| Sequencing dataset | This study | ENA: PRJEB59233 |
| Chemicals, peptides, and recombinant proteins | ||
| N-Lauroylsarcosine solution 30% 500ml | Dutscher | Cat#348533 |
| Proteinase K 10MG | Thermo Fisher Scientific | Cat#AM2542 |
| H2O, Molecular Biology Grade, Fisher BioReagents | Thermo Fisher Scientific | Cat#10977015 |
| Tween 20 100ML | Thermo Fisher Scientific | Cat#13464259 |
| Ethanol, Absolute, Mol Biology Grade | Thermo Fisher Scientific | Cat#16606002 |
| USER Enzyme | New England Biolabs | Cat#M5505L |
| NEBNext End Repair Module | New England Biolabs | Cat#E6050L |
| Bst DNA Polymerase | New England Biolabs | Cat#M0275L |
| NEBNext Quick Ligation Module | New England Biolabs | Cat#E6056L |
| BSA Molecular Biology Grade | New England Biolabs | Cat#B9000S |
| ACCUPRIME PFX DNA POLYMERASE 100mL | Thermo Fisher Scientific | Cat#10472482 |
| Agencourt AMPure XP - 60ml | Beckman Coulter | Cat#A63881 |
| Buffer PE | QIAGEN | Cat#19065 |
| Buffer PB | QIAGEN | Cat#19066 |
| Buffer EB | QIAGEN | Cat#19086 |
| dNTP Set 100mM 100mL | Thermo Fisher Scientific | Cat#10297018 |
| EDTA 0.5M pH 8.0 Fisher Bioreagents 500ML | Thermo Fisher Scientific | Cat#AM9261 |
| Critical commercial assays | ||
| MinElute PCR Purification kit | QIAGEN | Cat#28006 |
| Tapestation screenTape D1000 HS | Agilent | Cat#5067-5584 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q32854 |
| MiniSeq High Output Reagent Kit (150-cycles) | Illumina | Cat#FC-420-1002 |
| myBaits Custom DNA-Seq | Arbor Biosciences | Cat#300596.v5 |
| Deposited data | ||
| Raw and analyzed data | This study | ENA: PRJEB59233 |
| Software and algorithms | ||
| mapDamage2 | Jónsson et al.,68 | https://ginolhac.github.io/mapDamage |
| PALEOMIX | Schubert et al.69 | https://github.com/MikkelSchubert/paleomix |
| AdapterRemoval | Schubert et al.70 | https://github.com/MikkelSchubert/adapterremoval |
| Samtools | Li and Durbin,71 | https://github.com/samtools/samtools |
| snpToolkit v2.3.0 | Namouchi et al.56 | https://github.com/Amine-Namouchi/snpToolkit |
| IQ-Tree v1.6.12 | Nguyen et al.72 | https://github.com/iqtree/iqtree1 |
| Bowtie2 | Langmead and Salzberg,73 | https://github.com/BenLangmead/bowtie2 |
| BWA | Li and Durbin,71 | https://github.com/lh3/bwa |
| seqtk | - | https://github.com/lh3/seqtk |
| MetaPhlAn 4 | Blanco-Miguez et al.52 | https://github.com/biobakery/MetaPhlAn |
Resource availability
Lead contact
Further information and requests for resources and materials may be directed to lead contact, Prof. Ludovic Orlando (ludovic.orlando@univ-tlse3.fr).
Materials availability
This study did not generate new unique reagents or materials.
Experimental model and subject details
Samples and archaeological sites
The 120 teeth analyzed in this study originate from 89 individual skeletons buried in five archaeological sites (Figure 1, Table S1; see Data S1 for further description of the archaeological contexts). Prior to destructive sub-sampling, precise virtual 3D models were constructed for the majority of teeth investigated (94/120 teeth), using the ArtecMicro© surface scanner (Artec 3D, 1 mm voxel size). Samples and laboratory procedures were registered in the Laboratory Information Management System CASCADE.74
The plague cemetery of Lariey Puy-Saint-Pierre dating to 1629-1630 CE is located in the French Alps, and was excavated in 2002 CE (Minimal number of individuals, MNI = 34). Two out of 10 individuals (LAR8 and LAR11) investigated in this study were previously sequenced by Seguin-Orlando and colleagues in 2021,45 who demonstrated that they were infected with a Y. pestis strain genetically close to those infecting 1636 CE individuals from San Procolo a Naturno, Italy (1636 CE),75 in the context of the Thirty Year’s War.
The excavations at the Hôtel-Dieu Saint-Jean-Baptiste of Amiens, France, were carried out in 2017 CE by the Service archéologie préventive d'Amiens Métropole, under the supervision of Richard Jonvel, who provided access to the city’s ancient plague cemetery (MNI = 259). The 34 individuals studied here come from multiple graves dating to 1630-1670 CE, when Amiens experienced notable military and epidemic turmoil.
The site of the Maladrerie Saint-Lazare (Beauvais, France) was excavated between 2002 and 2014 CE by the Service archéologique municipal de Beauvais (MNI = 490). The discovery of a plague cemetery established since 1623 CE allowed the analysis of 14 individuals coming from the mass grave labelled 8559.
The parish cemetery of La Major cathedral (La Major), excavated in 2008 CE76 was created at the beginning of September 1720 CE during the last great wave of plague in Marseille (1720-1722 CE). A total of 19 individuals were DNA tested, but showed poor macroscopic preservation, consistent with that observed across the many other individuals buried at the same site (MNI = 106).
The site of Les Rayettes (Capucins de Ferrières trenches, Martigues, France, MNI = 208), was excavated in 2002 CE. It is located less than 40 kilometers away from La Major. The 12 individuals investigated here were found in multiple mass graves dated to 1720-1721 CE and characteristic of the epidemic ongoing locally at the time.
Method details
Ancient DNA extraction
All molecular work, from dental pulp sampling to library construction and PCR mix preparation, was carried out in the ancient DNA facilities of the CAGT (CNRS UMR 5288/Université Paul Sabatier), following strict experimental standards to avoid and monitor contamination, including use of disposable personal protection equipment, positive air pressure, bleach/UV surface and instrument decontamination, and negative control blanks. PCR amplification, purification, quantitation as well as DNA capture and sequencing were carried out in post-PCR laboratories that are located in a building physically isolated from the ancient DNA facilities.
Between 15 and 80 mg of dental pulp were collected for each of the 120 teeth analyzed (incisors, canines, premolars and molars), following the method developed by Neumann and colleagues,77 which is routinely used at CAGT.45 The extraction was performed following the protocol described by Seguin-Orlando and colleagues,45 which corresponds to a modified version of the Y2 protocol described by Gamba and colleagues.50 The standard protocol makes use of 963 μL of lysis buffer (0.45M EDTA, 0.25 mg/mL proteinase K and 0.5% sodium lauroyl sarcosinate) to carry out a first pre-digestion step of 1 hour at 37°C. The supernatant is then collected following centrifugation at 12,000 rotation per minute (rpm) for 30 seconds and a 963 μL volume of a fresh, pre-heated, lysis buffer is added to proceed to full digestion of the remaining pellets at 42°C overnight. After centrifugation at 12,000 rpm for 30 seconds, 200 μL of the lysis buffer is purified through QIAgen© Minelute columns and concentrated in a final elution buffer consisting of 23 μL EB supplemented with 0.05 % Tween 20 (later referred to as EB-Tween buffer). A total of 22.8 μL purified extract was treated with 7 μL of USER® enzyme (NEB®) for 3 hours at 37°C, following,78 to reduce spurious nucleotide mis-incorporation at those deaminated cytosine residues formed by post-mortem degradation. The pre-digestion supernatant was also purified and processed in parallel to the overnight digestion fraction, to contrast the DNA content following shotgun DNA sequencing and/or DNA capture.
The standard extraction protocol was modified to assess the kinetics of DNA release in the lysis buffer. This was achieved through two main experiments, each focusing on two different teeth from four individuals that were previously tested positive for Y. pestis DNA (AMIENS268, tooth 2 (t2) and t4; LAR27 t2 and t4; SQ8723 t2 and t4, and; MAJ46 t9 and t11). The first experiment followed the standard extraction protocol, with the exception that the whole supernatant was collected every 10 min and replaced by 963 μL of a fresh lysis buffer. Supernatant collection was carried every 10 min until 1 hour of pre-digestion was completed. The second experiment followed the same procedure as the first, except that the supernatants were collected directly after suspending the dental pulp powder in the lysis buffer (0 min), and 0.5, 1, 2.5, 5, 7.5 and 10 min after. Each collected supernatant was further processed following the methodology described above to obtain 29.8 μL of USER-treated DNA extract.
Library construction and PCR amplification
Triple-indexed double-stranded DNA libraries compatible with Illumina sequencing were prepared following the protocol originally described by Rohland et al., 2015,79 as modified by Fages and colleagues.80 This protocol includes a unique combination of two barcodes, each located 3’ of the P5 and P7 library adapters. Due to their location immediately downstream of the sequencing primers, these “internal” barcodes form the first seven nucleotide positions within each sequencing read. Each library was amplified in eight parallel PCRs for eight cycles in the same conditions as described by Seguin-Orlando and colleagues45: in a total reaction volume of 25 μL, using 0.4 μL of AccuPrime™ Pfx DNA polymerase (one unit), 2.5 μL 10X Accuprime™ Pfx reaction mix, 2 μL of DNA library, 1 μL of BSA (20 mg/mL), 17.9 μL of H2O, 0.2 μL of inPE1 primer (25 μM) and 1 μL indexing primer (5 μM) containing a unique 6-bp “external” barcode. The latter corresponds to a third barcode located within the PCR primer.81 The same external index was used in each of the eight parallel amplifications carried out on the same original library. Libraries were then amplified through a second additional PCR round to obtain sufficient DNA mass as input for capture enrichment with a custom myBaits® hybridization capture kit (Daicel Arbor Biosciences). To achieve this, two amplification products for a given library were merged and purified together using Agencourt Ampure XP beads (1:1.2 or 1:1.4 as DNA:beads ratio), and eluted in 11 μL of EB-Tween buffer. This provided four purified amplification products per library, which were further split and processed through two parallel PCR amplifications, using 1 μL of the eluate for each PCR reaction, for 10 cycles to obtain sufficient DNA material for DNA capture. These amplifications formed the second PCR round and were carried out in 25 μL reaction volumes, following the same conditions as for the first amplification round, except that IS5 and IS6 were used as PCR primers.82 The resulting eight PCR products were co-purified on a single MinElute column (QIAGEN©), and eluted in 12 μL of EB-Tween buffer. Library concentration and/or size distribution were measured on a Tapestation 4200 instrument using a High sensitivity D1000 ScreenTapeAssay (Agilent technologies) as well as on a Qubit HS dsDNA assay (Invitrogen).
Capture and sequencing
A total of four to eight libraries from different extracts with unique internal and external barcodes were pooled and concentrated together on a MinElute column (QIAGEN©). This resulted in pools of 7 μL, containing between 2-12 μg of DNA, as recommended in the myBaits® High Sensitivity Protocol Version 5.00. DNA capture was performed according to the aforementioned protocol, consisting of two rounds of 16 to 24 hour hybridization with the probes described by Wagner and colleagues.33 Captured libraries were sequenced using the Paired-End mode on an Illumina MiniSeq instrument (2x81 cycles) at CAGT. A similar procedure was followed for shotgun sequencing, except that only one amplification round of 8-12 PCR cycles was performed on each library, before purification, quantification, pooling and DNA sequencing.
Quantification and statistical analysis
Read demultiplexing and collapsing, as well as adapter and poor-quality end trimming were performed using AdapterRemoval2 version 2.3.0,70 tolerating at most one mismatch in each internal barcode (--barcode-mm-r[12] 1 --minadapteroverlap 3 --mm 5). Read mapping against the human reference genome (hg19, GRCh37) and the revised Cambridge Reference Sequence (rCRS, Accession Number = NC_012920.1) were carried out using Bowtie2 version 2.3.4.1,73 and the recommended parameters from Poullet and Orlando, 2020.82 Read mapping against the Y. pseutotuberculosis reference genome (Accession Number NC_006155.1), the Y. pestis reference genome (strain CO92, Accession Number = NC_003143.1)83 and the three plasmids pCD1 (Accession Number NC_003131.1), pMT1 (Accession Number NC_003134.1) and pPCP1 (Accession Number AL109969.1) were performed using BWA version 0.7.17 “backtrack” mode,71 and the “stringent” parameters defined by Spyrou and colleagues.51 Read (re-)alignment, PCR duplicate removal, quality filtering and mapDamage profiling68 were carried out using the automated PALEOMIX pipeline v1.2.13.2.69 The same procedures were followed using previously published read sequence data (Table S2) to obtain the comparative panel of Y. pestis genomes and plasmids used for phylogenetic reconstructions and/or for assessing the presence/absence of specific virulence genes and lineage-specific mutations, which was summarized using snpToolkit v2.356 (see below).
Basic alignment statistics for each sample, DNA library and experimental conditions are provided in Table S1. When comparing the respective performance of each experimental condition, Y. pestis and human DNA content of each library were estimated for normalized sequencing efforts. This was achieved by considering the fraction of collapsed reads only, and down-sampling the sequencing data 10 times for each library to the number collected for the least sequenced library.
The investigation of pathogen and mitochondrial unique reads released through time was performed on the first set of extraction tests on four teeth, which were digested in lysis buffer during one hour, sampling and replacing the supernatant with fresh lysis buffer every 10 min. Results were normalized by down-sampling the number of reads for each digestion time by the lowest number of reads sequenced across the different digestion times for each sample. As the percentage of unique reads mapping against the pathogen genomes (CO92, pCD1, pMT1 and pPCP1) over digestion time approximatively fit an inverse exponential function, data have been log-linearized. Linear models of the logarithm of the percentage of unique reads mapping against pathogen and mitochondrial DNA (mtDNA) were performed using the R lm command. The remaining Y. pestis and human mtDNA to be released at a given time could be predicted using predict command in R, according to these models.
The proportions of unique reads mapping against the circular chromosome and the different plasmids of the pathogen were calculated for each site to compare the plasmid load between 17th and 18th c. CE strains. These were contrasted to the proportion of probes targeting these different genetic materials. Calculations were based on libraries subjected to a one-hour digestion, as long as at least 500 high-quality alignments against the CO92 genome were found. Inter and intra-century differences in these proportions were compared for 17th and 18th c. CE samples using non-parametric Wilcoxon and Dunn tests in R.
The read-to-reference edit distance distributions (Figure S2) were generated for each of the new genomes characterized in this study from the NM:I field obtained while running the samtools view command, considering either the Y. pestis (CO92, NC_003143.1), or the Y. pseudotuberculosis (IP 32953, NC_006155.1) as a reference. Depth-of-coverage variation was calculated using the PALEOMIX69 coverage command and non-overlapping sliding windows of 1,000 bp for the CO92 chromosome, or 100 bp for the pCD1, pMT1 and pPCP1 plasmids. GC content was estimated for the same windows using seqtk (https://github.com/lh3/seqtk), and visualized together with coverage estimates through circular plots produced using the R circlize library.84 The fraction of the positions that are covered at least once within a given gene was also calculated using the PALEOMIX depth command to assess the presence or absence of 235 plague virulence and pathogenicity loci, 190 of which located on the CO92 chromosome, and 45 on the three bacterial plasmids.43,45,54 Gene coverage plots for virulence genes were generated using the heatmap function from the R ggplot2 library (https://cran.r-project.org/web/packages/ggplot2/index.html). A 49-kb region of CO92 was found absent in most of the phylogenetically-close Y. pestis strains from New Churchyard (BED038, BED034, BED028, BED024, BED030), Azov (Azov38), CHE1, Rostov2033, Observance (OBS107, OBS116, OBS137, OBS110, OBS124), and La Major (MAJ75 and MAJ46). However, the coverage of some genes within this region was sporadically greater than zero in a subset of strains, suggesting the possible presence of spurious alignments, despite stringent alignment parameters. This was further confirmed using BlastN85 against the nucleotide collection (nt), showing best hits against environmental Yersinia strains than Y. pestis. Those cases are highlighted on the heatmap presented on Figure 7E by crossing the cell presenting the corresponding coverage estimate. We used snpToolkit v2.356 and the individual vcf files generated as described above to annotate the new Y. pestis genome and plasmid sequences characterized in this study for synonymous and non-synonymous sequence polymorphisms present in at least 90% of the reads. The minimal depth threshold was set to 3 (-d 3) for bases showing Phred scores above or equal to 30. The samples considered for this analysis were merged by archaeological site using samtools to provide sufficient coverage for the detection of strain-specific SNPs.
Microbial taxonomic profiles were obtained using MetaPhlAn 452 on collapsed sequencing unique reads after filtering for those mapping against the human nuclear and mitochondrial genomes following the procedure presented above. To accommodate for variable sequencing efforts across DNA libraries, we random-sampled a number of 100,000 reads. This procedure was repeated 10 times to assess the robustness of the taxonomic assignments, given the limited of reads considered. Reads were mapped against the MetaPhlAn 4 biomarker database using Bowtie2 v.2.3.4.173 with default parameters, and further filtered for minimal mapping quality of 25, and PCR duplicates using the view and rmdup modules from samtools v1.11,86 respectively. Those species showing relative abundances <1% in a given dataset were defined as “low abundance”.
Maximum Likelihood phylogenetic trees were constructed using IQ-TREE v1.6.1272 and a panel of Y. pestis genomes, including 124 modern accessions and 61 second pandemic samples, 12 of which were sequenced here for the first time. Individual base calling was carried out using BCFtools87 (--ploidy 1), requiring minimum base, mapping and genotype Phred quality scores of 30, and filtering those variants located within 10 bp of indels. The best mutational model was selected based on AICc and BIC values with the Model Finder Plus module (MFP), as part of IQ-TREE. Node support was estimated from a total of 1,000 pseudo-replicates using the ultrafast bootstrap (UFBoot) approximation,88 optimized by nearest neighbor interchange (NNI), as well as the SH-like approximate likelihood ratio (aLRT) test.89 The final sequence alignments corresponded, on the first hand, to the entire CO92 chromosome, considering polymorphic sites only, or, on the other hand, were restricted to 29,609 genome-wide variant positions previously characterized to provide robust phylogenetic signal.43,54 Two sequence alignments were considered, requiring a conservative minimum 3-fold average depth-of-coverage per sample. Accessions for the sequence data underlying the comparative panel of 185 Yersinia pestis and 1 Yersinia pseudotuberculosis genomes are provided in Table S2.
Acknowledgments
We thank all the members of the AGES group at CAGT for fruitful discussion and Alice Rabasse for proofreading the manuscript. This research was funded by the CNRS MITI (Mission pour les Initiatives Transverses et Interdisciplinaires) IndigenousHealth program; the ANR LifeChange; the ANR GenIn; the Simone and Cino Del Duca Foundation (Subventions scientifiques 2020, HealthTime Travel) and the European Research Council (ERC), under the European Union’s Horizon 2020 research and innovation program (grant agreement 681605).
Author contributions
L.O. conceived the project and designed the research. N.T., B.C., R.J., S.T., L.B., J.M.F., M.S., and C.C. provided samples and information about historical and archaeological context. P.C., C.D.S., C.T., C.G., and A.S.O. carried out ancient DNA laboratory work with input from L.C., G.T., S.S., and L.C.T., who managed the sequencing runs. P.C., L.L., C.D.S., and L.O. carried out computational analyses, with input from A.S.O. and L.O. provided reagents and material. J.K. and H.P. provided the probe design. P.C., C.D.S., A.S.O., and L.O. wrote the paper with input from J.K., H.P., and M.A.S., and all the coauthors.
Declaration of interests
The authors declare no competing interests.
Published: May 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106787.
Supplemental information
LIMS IDs refer to the collection numbers as indicated in the internal sample database of CAGT. Dates are provided in years CE. The column “Digestion steps” refers to the experimental conditions underlying DNA extraction, with 1h indicating a one-hour pre-digestion wash, 1h+O/N indicating the complete pellet digestion following overnight incubation, and 0.5–60 min referring to the time period after which pre-digestion wash supernatants were collected. The column “Total Unique” reports the number of high-quality alignments against the considered chromosomal (CO92) or plasmid genome sequence (pCD1, pMT1 and pPCP1), or the human mtDNA revised CRS sequence (rCRS) or the human nuclear genome (hg19), after removal of duplicates. The column “Unique Length” reports the average size of the high-quality alignments characterized after removal of duplicates. Global = sequence results for CO92, the three plasmids and rCRS considered altogether.
The column “USER” indicates whether (+) or not (-) the DNA extracts were digested with the USER enzymatic mix prior to DNA library construction.
For Amiens, the AMIENS268 genome sequence was considered as it was characterized with sufficient coverage (56.16-fold). The three sequences obtained for individuals SQ8962, SQ8833 and SQ8723 at Beauvais were merged together to carry out the sequence comparison, as they were only characterized at limited coverage (2.47-fold, 1.30-fold and 8.66-fold, respectively). The three genome sequences obtained for individuals buried at Les Rayettes (RAY1015, RAY1028 and RAY3030) and at La Major (MAJ38, MAJ46 and MAJ75) were also merged as their genome could only be sequenced at limited coverage ≤ 5.25-fold). The three individuals for which CO92 and plasmid genomes could be characterized at Lariey (LAR8, LAR11 and LAR27) were merged to provide comparative sequence data of the closest phylogenetic relatives of those 17th c. CE strains found at Amiens and Beauvais. Similarly, the five individuals previously analyzed from Observance provided the comparative sequence data of the closest phylogenetic relatives of those 18th c. CE strains found at Les Rayettes and La Major. 1: the SNP (i.e. non-reference) allele was identified (0 otherwise). Those non-synonymous sites showing different alleles in Amiens and Beauvais relative to Lariey, but not polymorphic in Les Rayettes, La Major and Observance are highlighted in blue. Conversely, those non-synonymous sites showing different alleles in Les Rayettes and La Major relative to Observance, but not polymorphic in Amiens, Beauvais, and Lariey are highlighted in yellow.
Overview of the 5 archaeological sites investigated in this study. See also Figure 1 and Table S1.
Data and code availability
-
•
Sequencing data can be found on the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home) and are publicly available as of the date of publication under project accession number ENA: PRJEB59233.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Llamas B., Willerslev E., Orlando L. Human evolution: a tale from ancient genomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372:20150484. doi: 10.1098/rstb.2015.0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orlando L., Allaby R., Skoglund P., Der Sarkissian C., Stockhammer P.W., Ávila-Arcos M.C., Fu Q., Krause J., Willerslev E., Stone A.C., Warinner C. Ancient DNA analysis. Nat. Rev. Methods Primers. 2021;1:14–26. doi: 10.1038/s43586-020-00011-0. [DOI] [Google Scholar]
- 3.Pinhasi R., Fernandes D., Sirak K., Novak M., Connell S., Alpaslan-Roodenberg S., Gerritsen F., Moiseyev V., Gromov A., Raczky P., et al. Optimal ancient DNA yields from the inner ear part of the human petrous bone. PLoS One. 2015;10:e0129102. doi: 10.1371/journal.pone.0129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sirak K., Fernandes D., Cheronet O., Harney E., Mah M., Mallick S., Rohland N., Adamski N., Broomandkhoshbacht N., Callan K., et al. Human auditory ossicles as an alternative optimal source of ancient DNA. Genome Res. 2020;30:427–436. doi: 10.1101/gr.260141.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harney É., Cheronet O., Fernandes D.M., Sirak K., Mah M., Bernardos R., Adamski N., Broomandkhoshbacht N., Callan K., Lawson A.M., et al. A minimally destructive protocol for DNA extraction from ancient teeth. Genome Res. 2021;31:472–483. doi: 10.1101/gr.267534.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korlević P., Gerber T., Gansauge M.-T., Hajdinjak M., Nagel S., Aximu-Petri A., Meyer M. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques. 2015;59:87–93. doi: 10.2144/000114320. [DOI] [PubMed] [Google Scholar]
- 7.Boessenkool S., Hanghøj K., Nistelberger H.M., Der Sarkissian C., Gondek A.T., Orlando L., Barrett J.H., Star B. Combining bleach and mild predigestion improves ancient DNA recovery from bones. Mol. Ecol. Resour. 2017;17:742–751. doi: 10.1111/1755-0998.12623. [DOI] [PubMed] [Google Scholar]
- 8.Ginolhac A., Vilstrup J., Stenderup J., Rasmussen M., Stiller M., Shapiro B., Zazula G., Froese D., Steinmann K.E., Thompson J.F., et al. Improving the performance of true single molecule sequencing for ancient DNA. BMC Genom. 2012;13:177. doi: 10.1186/1471-2164-13-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Damgaard P.B., Margaryan A., Schroeder H., Orlando L., Willerslev E., Allentoft M.E. Improving access to endogenous DNA in ancient bones and teeth. Sci. Rep. 2015;5:11184. doi: 10.1038/srep11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Essel E., Korlević P., Meyer M. A method for the temperature-controlled extraction of DNA from ancient bones. Biotechniques. 2021;71:382–386. doi: 10.2144/btn-2021-0025. [DOI] [PubMed] [Google Scholar]
- 11.Mathieson I., Lazaridis I., Rohland N., Mallick S., Patterson N., Roodenberg S.A., Harney E., Stewardson K., Fernandes D., Novak M., et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohland N., Mallick S., Mah M., Maier R., Patterson N., Reich D. Three assays for in-solution enrichment of ancient human DNA at more than a million SNPs. Genome Res. 2022;32:2068–2078. doi: 10.1101/gr.276728.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Q., Meyer M., Gao X., Stenzel U., Burbano H.A., Kelso J., Pääbo S. DNA analysis of an early modern human from Tianyuan Cave, China. Proc. Natl. Acad. Sci. USA. 2013;110:2223–2227. doi: 10.1073/pnas.1221359110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carpenter M.L., Buenrostro J.D., Valdiosera C., Schroeder H., Allentoft M.E., Sikora M., Rasmussen M., Gravel S., Guillén S., Nekhrizov G., et al. Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am. J. Hum. Genet. 2013;93:852–864. doi: 10.1016/j.ajhg.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Enk J.M., Devault A.M., Kuch M., Murgha Y.E., Rouillard J.-M., Poinar H.N. Ancient whole genome enrichment using baits built from modern DNA. Mol. Biol. Evol. 2014;31:1292–1294. doi: 10.1093/molbev/msu074. [DOI] [PubMed] [Google Scholar]
- 16.Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H.-Y., et al. A draft sequence of the neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen M., Li Y., Lindgreen S., Pedersen J.S., Albrechtsen A., Moltke I., Metspalu M., Metspalu E., Kivisild T., Gupta R., et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463:757–762. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reich D., Green R.E., Kircher M., Krause J., Patterson N., Durand E.Y., Viola B., Briggs A.W., Stenzel U., Johnson P.L.F., et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allentoft M.E., Sikora M., Refoyo-Martínez A., Irving-Pease E.K., Fischer A., Barrie W., Ingason A., Stenderup J., Sjögren K.-G., Pearson A., et al. Population genomics of stone Age eurasia. bioRxiv 2022. 2022 doi: 10.1101/2022.05.04.490594. Preprint at. [DOI] [Google Scholar]
- 20.Lazaridis I., Alpaslan-Roodenberg S., Acar A., Açıkkol A., Agelarakis A., Aghikyan L., Akyüz U., Andreeva D., Andrijašević G., Antonović D., et al. The genetic history of the southern arc: a bridge between west asia and Europe. Science. 2022;377:eabm4247. doi: 10.1126/science.abm4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spyrou M.A., Bos K.I., Herbig A., Krause J. Ancient pathogen genomics as an emerging tool for infectious disease research. Nat. Rev. Genet. 2019;20:323–340. doi: 10.1038/s41576-019-0119-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pajer P., Dresler J., Kabíckova H., Písa L., Aganov P., Fucik K., Elleder D., Hron T., Kuzelka V., Velemínsky P., et al. Characterization of two historic smallpox specimens from a Czech museum. Viruses. 2017;9:E200. doi: 10.3390/v9080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrari G., Neukamm J., Baalsrud H.T., Breidenstein A.M., Ravinet M., Phillips C., Rühli F., Bouwman A., Schuenemann V.J. Variola virus genome sequenced from an eighteenth-century museum specimen supports the recent origin of smallpox. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020;375:20190572. doi: 10.1098/rstb.2019.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kahila Bar-Gal G., Kim M.J., Klein A., Shin D.H., Oh C.S., Kim J.W., Kim T.-H., Kim S.B., Grant P.R., Pappo O., et al. Tracing hepatitis B virus to the 16th century in a Korean mummy. Hepatology. 2012;56:1671–1680. doi: 10.1002/hep.25852. [DOI] [PubMed] [Google Scholar]
- 25.Krause-Kyora B., Susat J., Key F.M., Kühnert D., Bosse E., Immel A., Rinne C., Kornell S.-C., Yepes D., Franzenburg S., et al. Neolithic and medieval virus genomes reveal complex evolution of hepatitis B. Elife. 2018;7:e36666. doi: 10.7554/eLife.36666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuenemann V.J., Singh P., Mendum T.A., Krause-Kyora B., Jäger G., Bos K.I., Herbig A., Economou C., Benjak A., Busso P., et al. Genome-wide comparison of medieval and modern Mycobacterium leprae. Science. 2013;341:179–183. doi: 10.1126/science.1238286. [DOI] [PubMed] [Google Scholar]
- 27.van Dorp L., Gelabert P., Rieux A., de Manuel M., de-Dios T., Gopalakrishnan S., Carøe C., Sandoval-Velasco M., Fregel R., Olalde I., et al. Plasmodium vivax malaria viewed through the lens of an eradicated European strain. Mol. Biol. Evol. 2020;37:773–785. doi: 10.1093/molbev/msz264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marciniak S., Poinar H.N. In: Paleogenomics: Genome-Scale Analysis of Ancient DNA Population Genomics. Lindqvist C., Rajora O.P., editors. Springer International Publishing; 2019. Ancient pathogens through human history: a paleogenomic perspective; pp. 115–138. [DOI] [Google Scholar]
- 29.Margaryan A., Hansen H.B., Rasmussen S., Sikora M., Moiseyev V., Khoklov A., Epimakhov A., Yepiskoposyan L., Kriiska A., Varul L., et al. Ancient pathogen DNA in human teeth and petrous bones. Ecol. Evol. 2018;8:3534–3542. doi: 10.1002/ece3.3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duchêne S., Ho S.Y.W., Carmichael A.G., Holmes E.C., Poinar H. The recovery, interpretation and use of ancient pathogen genomes. Curr. Biol. 2020;30:R1215–R1231. doi: 10.1016/j.cub.2020.08.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fellows Yates J.A., Andrades Valtueña A., Vågene Å.J., Cribdon B., Velsko I.M., Borry M., Bravo-Lopez M.J., Fernandez-Guerra A., Green E.J., Ramachandran S.L., et al. Community-curated and standardised metadata of published ancient metagenomic samples with AncientMetagenomeDir. Sci. Data. 2021;8:31. doi: 10.1038/s41597-021-00816-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malyarchuk A.B., Andreeva T.V., Kuznetsova I.L., Kunizheva S.S., Protasova M.S., Uralsky L.I., Tyazhelova T.V., Gusev F.E., Manakhov A.D., Rogaev E.I. Genomics of ancient pathogens: first advances and prospects. Biochemistry. 2022;87:242–258. doi: 10.1134/S0006297922030051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner D.M., Klunk J., Harbeck M., Devault A., Waglechner N., Sahl J.W., Enk J., Birdsell D.N., Kuch M., Lumibao C., et al. Yersinia pestis and the plague of Justinian 541-543 AD: a genomic analysis. Lancet Infect. Dis. 2014;14:319–326. doi: 10.1016/S1473-3099(13)70323-2. [DOI] [PubMed] [Google Scholar]
- 34.Keller M., Spyrou M.A., Scheib C.L., Neumann G.U., Kröpelin A., Haas-Gebhard B., Päffgen B., Haberstroh J., Ribera i Lacomba A., Raynaud C., et al. Ancient Yersinia pestis genomes from across western Europe reveal early diversification during the first pandemic (541–750) Proc. Natl. Acad. Sci. USA. 2019;116:12363–12372. doi: 10.1073/pnas.1820447116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bos K.I., Schuenemann V.J., Golding G.B., Burbano H.A., Waglechner N., Coombes B.K., McPhee J.B., DeWitte S.N., Meyer M., Schmedes S., et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature. 2011;478:506–510. doi: 10.1038/nature10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasmussen S., Allentoft M.E., Nielsen K., Orlando L., Sikora M., Sjögren K.G., Pedersen A.G., Schubert M., Van Dam A., Kapel C.M.O., et al. Early divergent strains of Yersinia pestis in eurasia 5,000 Years ago. Cell. 2015;163:571–582. doi: 10.1016/j.cell.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andrades Valtueña A., Mittnik A., Key F.M., Haak W., Allmäe R., Belinskij A., Daubaras M., Feldman M., Jankauskas R., Janković I., et al. The stone Age plague and its persistence in eurasia. Curr. Biol. 2017;27:3683–3691.e8. doi: 10.1016/j.cub.2017.10.025. [DOI] [PubMed] [Google Scholar]
- 38.Spyrou M.A., Tukhbatova R.I., Wang C.-C., Valtueña A.A., Lankapalli A.K., Kondrashin V.V., Tsybin V.A., Khokhlov A., Kühnert D., Herbig A., et al. Analysis of 3800-year-old Yersinia pestis genomes suggests Bronze Age origin for bubonic plague. Nat. Commun. 2018;9:2234. doi: 10.1038/s41467-018-04550-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rascovan N., Sjögren K.G., Kristiansen K., Nielsen R., Willerslev E., Desnues C., Rasmussen S. Emergence and spread of basal lineages of Yersinia pestis during the neolithic decline. Cell. 2019;176:295–305.e10. doi: 10.1016/j.cell.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 40.Yu H., Spyrou M.A., Karapetian M., Shnaider S., Radzevičiūtė R., Nägele K., Neumann G.U., Penske S., Zech J., Lucas M., et al. Paleolithic to Bronze Age siberians reveal connections with first Americans and across eurasia. Cell. 2020;181:1232–1245.e20. doi: 10.1016/j.cell.2020.04.037. [DOI] [PubMed] [Google Scholar]
- 41.Susat J., Lübke H., Immel A., Brinker U., Macāne A., Meadows J., Steer B., Tholey A., Zagorska I., Gerhards G., et al. A 5,000-year-old hunter-gatherer already plagued by Yersinia pestis. Cell Rep. 2021;35:109278. doi: 10.1016/j.celrep.2021.109278. [DOI] [PubMed] [Google Scholar]
- 42.Barbieri R., Signoli M., Chevé D., Costedoat C., Tzortzis S., Aboudharam G., Raoult D., Drancourt M. Yersinia pestis: the natural history of plague. Clin. Microbiol. Rev. 2020;34 doi: 10.1128/CMR.00044-19. e000444-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spyrou M.A., Musralina L., Gnecchi Ruscone G.A., Kocher A., Borbone P.-G., Khartanovich V.I., Buzhilova A., Djansugurova L., Bos K.I., Kühnert D., et al. The source of the Black Death in fourteenth-century central Eurasia. Nature. 2022;606:718–724. doi: 10.1038/s41586-022-04800-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui Y., Yu C., Yan Y., Li D., Li Y., Jombart T., Weinert L.A., Wang Z., Guo Z., Xu L., et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc. Natl. Acad. Sci. USA. 2013;110:577–582. doi: 10.1073/pnas.1205750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seguin-Orlando A., Costedoat C., Der Sarkissian C., Tzortzis S., Kamel C., Telmon N., Dalén L., Thèves C., Signoli M., Orlando L. No particular genomic features underpin the dramatic economic consequences of 17th century plague epidemics in Italy. iScience. 2021;24:102383. doi: 10.1016/j.isci.2021.102383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cruz-Dávalos D.I., Llamas B., Gaunitz C., Fages A., Gamba C., Soubrier J., Librado P., Seguin-Orlando A., Pruvost M., Alfarhan A.H., et al. Experimental conditions improving in-solution target enrichment for ancient DNA. Mol Ecol Res. 2017;17:508–522. doi: 10.1111/1755-0998.12595. [DOI] [PubMed] [Google Scholar]
- 47.Ziesemer K.A., Ramos-Madrigal J., Mann A.E., Brandt B.W., Sankaranarayanan K., Ozga A.T., Hoogland M., Hofman C.A., Salazar-García D.C., Frohlich B., et al. The efficacy of whole human genome capture on ancient dental calculus and dentin. Am. J. Phys. Anthropol. 2019;168:496–509. doi: 10.1002/ajpa.23763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suchan T., Kusliy M.A., Khan N., Chauvey L., Tonasso-Calvière L., Schiavinato S., Southon J., Keller M., Kitagawa K., Krause J., et al. Performance and automation of ancient DNA capture with RNA hyRAD probes. Mol Ecol Res. 2022;22:891–907. doi: 10.1111/1755-0998.13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Signoli M. History of the plague of 1720-1722, in Marseille. Presse Med. 2022;51:104138. doi: 10.1016/j.lpm.2022.104138. [DOI] [PubMed] [Google Scholar]
- 50.Gamba C., Hanghøj K., Gaunitz C., Alfarhan A.H., Alquraishi S.A., Al-Rasheid K.A.S., Bradley D.G., Orlando L. Comparing the performance of three ancient DNA extraction methods for high-throughput sequencing. Mol Ecol Res. 2016;16:459–469. doi: 10.1111/1755-0998.12470. [DOI] [PubMed] [Google Scholar]
- 51.Spyrou M.A., Keller M., Tukhbatova R.I., Scheib C.L., Nelson E.A., Andrades Valtueña A., Neumann G.U., Walker D., Alterauge A., Carty N., et al. Phylogeography of the second plague pandemic revealed through analysis of historical Yersinia pestis genomes. Nat. Commun. 2019;10:4470. doi: 10.1038/s41467-019-12154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blanco-Miguez A., Beghini F., Cumbo F., Mclver L.J., Thompson K.N., Zolfo M., Manghi P., Dubois L., Huang K.D., Maltez Thomas A., et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species with MetaPhlAn 4. bioRxiv. 2022 doi: 10.1101/2022.08.22.504593. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Der Sarkissian C., Ermini L., Jónsson H., Alekseev A.N., Crubezy E., Shapiro B., Orlando L. Shotgun microbial profiling of fossil remains. Mol. Ecol. 2014;23:1780–1798. doi: 10.1111/mec.12690. [DOI] [PubMed] [Google Scholar]
- 54.Andrades Valtueña A., Neumann G.U., Spyrou M.A., Musralina L., Aron F., Beisenov A., Belinskiy A.B., Bos K.I., Buzhilova A., Conrad M., et al. Stone Age Yersinia pestis genomes shed light on the early evolution, diversity, and ecology of plague. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2116722119. e2116722119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spyrou M.A., Tukhbatova R.I., Feldman M., Drath J., Kacki S., Beltrán de Heredia J., Arnold S., Sitdikov A.G., Castex D., Wahl J., et al. Historical Y. pestis genomes reveal the European Black Death as the source of ancient and modern plague pandemics. Cell Host Microbe. 2016;19:874–881. doi: 10.1016/j.chom.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Namouchi A., Guellil M., Kersten O., Hänsch S., Ottoni C., Schmid B.V., Pacciani E., Quaglia L., Vermunt M., Bauer E.L., et al. Integrative approach using Yersinia pestis genomes to revisit the historical landscape of plague during the Medieval Period. Proc. Natl. Acad. Sci. USA. 2018;115:E11790–E11797. doi: 10.1073/pnas.1812865115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bos K.I., Herbig A., Sahl J., Waglechner N., Fourment M., Forrest S.A., Klunk J., Schuenemann V.J., Poinar D., Kuch M., et al. Eighteenth century Yersinia pestis genomes reveal the long-term persistence of an historical plague focus. Elife. 2016;5:e12994. doi: 10.7554/eLife.12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bos K.I., Kühnert D., Herbig A., Esquivel-Gomez L.R., Andrades Valtueña A., Barquera R., Giffin K., Kumar Lankapalli A., Nelson E.A., Sabin S., et al. Paleomicrobiology: diagnosis and evolution of ancient pathogens. Annu. Rev. Microbiol. 2019;73:639–666. doi: 10.1146/annurev-micro-090817-062436. [DOI] [PubMed] [Google Scholar]
- 59.Susko E., Roger A.J. Long branch attraction biases in phylogenetics. Syst. Biol. 2021;70:838–843. doi: 10.1093/sysbio/syab001. [DOI] [PubMed] [Google Scholar]
- 60.Eaton K., Featherstone L., Duchene S., Carmichael A.G., Varlık N., Golding G.B., Holmes E.C., Poinar H.N. Plagued by a cryptic clock: insight and issues from the global phylogeny of Yersinia pestis. Commun. Biol. 2023;6:23. doi: 10.1038/s42003-022-04394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H., Avican K., Fahlgren A., Erttmann S.F., Nuss A.M., Dersch P., Fallman M., Edgren T., Wolf-Watz H. Increased plasmid copy number is essential for Yersinia T3SS function and virulence. Science. 2016;353:492–495. doi: 10.1126/science.aaf7501. [DOI] [PubMed] [Google Scholar]
- 62.Demeure C.E., Dussurget O., Mas Fiol G., Le Guern A.-S., Savin C., Pizarro-Cerdá J. Yersinia pestis and plague: an updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics. Gene Immun. 2019;20:357–370. doi: 10.1038/s41435-019-0065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu T., Keto-Timonen R., Jiang X., Virtanen J.-P., Korkeala H. Insights into the phylogeny and evolution of cold shock proteins: from enteropathogenic yersinia and escherichia coli to eubacteria. Int. J. Mol. Sci. 2019;20:4059. doi: 10.3390/ijms20164059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Groisman E.A., Hollands K., Kriner M.A., Lee E.-J., Park S.-Y., Pontes M.H. Bacterial Mg2+ homeostasis, transport, and virulence. Annu. Rev. Genet. 2013;47:625–646. doi: 10.1146/annurev-genet-051313-051025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Delepelaire P. Bacterial ABC transporters of iron containing compounds. Res. Microbiol. 2019;170:345–357. doi: 10.1016/j.resmic.2019.10.008. [DOI] [PubMed] [Google Scholar]
- 66.Fetherston J.D., Bertolino V.J., Perry R.D. YbtP and YbtQ: two ABC transporters required for iron uptake in Yersinia pestis. Mol. Microbiol. 1999;32:289–299. doi: 10.1046/j.1365-2958.1999.01348.x. [DOI] [PubMed] [Google Scholar]
- 67.Felek S., Lawrenz M.B., Krukonis E.S. The Yersinia pestis autotransporter YapC mediates host cell binding, autoaggregation and biofilm formation. Microbiology. 2008;154:1802–1812. doi: 10.1099/mic.0.2007/010918-0. [DOI] [PubMed] [Google Scholar]
- 68.Jónsson H., Ginolhac A., Schubert M., Johnson P.L.F., Orlando L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 2013;29:1682–1684. doi: 10.1093/bioinformatics/btt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schubert M., Ermini L., Der Sarkissian C., Jónsson H., Ginolhac A., Schaefer R., Martin M.D., Fernández R., Kircher M., McCue M., et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014;9:1056–1082. doi: 10.1038/nprot.2014.063. [DOI] [PubMed] [Google Scholar]
- 70.Schubert M., Lindgreen S., Orlando L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes. 2016;9:88. doi: 10.1186/s13104-016-1900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dolle D., Fages A., Mata X., Schiavinato S., Tonasso-Calvière L., Chauvey L., Wagner S., Der Sarkissian C., Fromentier A., Seguin-Orlando A., Orlando L. CASCADE: a custom-made archiving system for the conservation of ancient DNA experimental data. Front. Ecol. Evol. 2020;8 doi: 10.3389/fevo.2020.00185. [DOI] [Google Scholar]
- 75.Guellil M., Kersten O., Namouchi A., Luciani S., Marota I., Arcini C.A., Iregren E., Lindemann R.A., Warfvinge G., Bakanidze L., et al. A genomic and historical synthesis of plague in 18th century Eurasia. Proc. Natl. Acad. Sci. USA. 2020;117:28328–28335. doi: 10.1073/pnas.2009677117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tzortzis S., Signoli M. Characterization of the funeral groups associated with plague epidemics. Microbiol. Spectr. 2016;4 doi: 10.1128/microbiolspec.PoH-0011-2015. [DOI] [PubMed] [Google Scholar]
- 77.Neumann G.U., Andrades Valtueña A., Fellows Yates J.A., Stahl R., Brandt G. 2020. Tooth Sampling from the Inner Pulp Chamber for Ancient DNA Extraction V1 (protocols.io.Bakqicvw) [DOI] [Google Scholar]
- 78.Gaunitz C., Fages A., Hanghøj K., Albrechtsen A., Khan N., Schubert M., Seguin-Orlando A., Owens I.J., Felkel S., Bignon-Lau O., et al. Ancient genomes revisit the ancestry of domestic and Przewalski’s horses. Science. 2018;360:111–114. doi: 10.1126/science.aao3297. [DOI] [PubMed] [Google Scholar]
- 79.Rohland N., Harney E., Mallick S., Nordenfelt S., Reich D. Partial uracil-DNA-glycosylase treatment for screening of ancient DNA. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20130624. doi: 10.1098/rstb.2013.0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fages A., Hanghøj K., Khan N., Gaunitz C., Seguin-Orlando A., Leonardi M., McCrory Constantz C., Gamba C., Al-Rasheid K.A.S., Albizuri S., et al. Tracking five millennia of horse management with extensive ancient genome time series. Cell. 2019;177:1419–1435.e31. doi: 10.1016/j.cell.2019.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meyer M., Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.prot5448. pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 82.Poullet M., Orlando L. Assessing DNA sequence alignment methods for characterizing ancient genomes and methylomes. Front. Ecol. Evol. 2020;8 [Google Scholar]
- 83.Parkhill J., Wren B.W., Thomson N.R., Titball R.W., Holden M.T., Prentice M.B., Sebaihia M., James K.D., Churcher C., Mungall K.L., et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- 84.Gu Z., Gu L., Eils R., Schlesner M., Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 85.Zhang Z., Schwartz S., Wagner L., Miller W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 86.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup Genome project data processing subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hoang D.T., Chernomor O., von Haeseler A., Minh B.Q., Vinh L.S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018;35:518–522. doi: 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LIMS IDs refer to the collection numbers as indicated in the internal sample database of CAGT. Dates are provided in years CE. The column “Digestion steps” refers to the experimental conditions underlying DNA extraction, with 1h indicating a one-hour pre-digestion wash, 1h+O/N indicating the complete pellet digestion following overnight incubation, and 0.5–60 min referring to the time period after which pre-digestion wash supernatants were collected. The column “Total Unique” reports the number of high-quality alignments against the considered chromosomal (CO92) or plasmid genome sequence (pCD1, pMT1 and pPCP1), or the human mtDNA revised CRS sequence (rCRS) or the human nuclear genome (hg19), after removal of duplicates. The column “Unique Length” reports the average size of the high-quality alignments characterized after removal of duplicates. Global = sequence results for CO92, the three plasmids and rCRS considered altogether.
The column “USER” indicates whether (+) or not (-) the DNA extracts were digested with the USER enzymatic mix prior to DNA library construction.
For Amiens, the AMIENS268 genome sequence was considered as it was characterized with sufficient coverage (56.16-fold). The three sequences obtained for individuals SQ8962, SQ8833 and SQ8723 at Beauvais were merged together to carry out the sequence comparison, as they were only characterized at limited coverage (2.47-fold, 1.30-fold and 8.66-fold, respectively). The three genome sequences obtained for individuals buried at Les Rayettes (RAY1015, RAY1028 and RAY3030) and at La Major (MAJ38, MAJ46 and MAJ75) were also merged as their genome could only be sequenced at limited coverage ≤ 5.25-fold). The three individuals for which CO92 and plasmid genomes could be characterized at Lariey (LAR8, LAR11 and LAR27) were merged to provide comparative sequence data of the closest phylogenetic relatives of those 17th c. CE strains found at Amiens and Beauvais. Similarly, the five individuals previously analyzed from Observance provided the comparative sequence data of the closest phylogenetic relatives of those 18th c. CE strains found at Les Rayettes and La Major. 1: the SNP (i.e. non-reference) allele was identified (0 otherwise). Those non-synonymous sites showing different alleles in Amiens and Beauvais relative to Lariey, but not polymorphic in Les Rayettes, La Major and Observance are highlighted in blue. Conversely, those non-synonymous sites showing different alleles in Les Rayettes and La Major relative to Observance, but not polymorphic in Amiens, Beauvais, and Lariey are highlighted in yellow.
Overview of the 5 archaeological sites investigated in this study. See also Figure 1 and Table S1.
Data Availability Statement
-
•
Sequencing data can be found on the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home) and are publicly available as of the date of publication under project accession number ENA: PRJEB59233.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.