

Abstract
Recent studies have identified interstitial deletions in the cancer genome as a radiation-related mutational signature, although most of them do not fall on cancer driver genes. Pioneering studies in the field have indicated the presence of loss of heterozygosity (LOH) spanning Apc in a subset of sporadic and radiation-induced intestinal tumors of ApcMin/+ mice, albeit with a substantial subset in which LOH was not detected; whether copy number losses accompany such LOH has also been unclear. Herein, we analyzed intestinal tumors of C3B6F1 ApcMin/+ mice that were either left untreated or irradiated with 2 Gy of γ-rays. We observed intratumor mosaicism with respect to the nuclear/cytoplasmic accumulation of immunohistochemically detectable β-catenin, which is a hallmark of Apc+ allele loss. An immunoguided laser microdissection approach enabled the detection of LOH involving the Apc+ allele in β-catenin-overexpressing cells; in contrast, the LOH was not observed in the non-overexpressing cells. With this improvement, LOH involving Apc+ was detected in all 22 tumors analyzed, in contrast to what has been reported previously. The use of a formalin-free fixative facilitated the LOH and microarray-based DNA copy number analyses, enabling the classification of the aberrations as nondisjunction/mitotic recombination type or interstitial deletion type. Of note, the latter was observed only in radiation-induced tumors (nonirradiated, 0 of 8; irradiated, 11 of 14). Thus, an analysis considering intratumor heterogeneity identifies interstitial deletion involving the Apc+ allele as a causative radiation-related event in intestinal tumors of ApcMin/+ mice, providing an accurate approach for attributing individual tumors to radiation exposure.
Keywords: ApcMin /+ mouse, immunoguided laser microdissection, interstitial deletion, intestinal tumor, radiation signature
INTRODUCTION
Ionizing radiation is a carcinogen that induces cancer by causing DNA double-strand breaks with subsequent mis-rejoining of those breaks. It remains uncertain, however, whether cancer risk can be attributed to low radiation doses (or rates), for which the expected effect is small and below the level of statistical significance given the relatively high frequency of sporadic cancer [1]. To assess whether cancer risk can be attributed to low-level radiation exposure, biomarkers reflecting a key event in carcinogenesis would be of great use [2]. Recent cancer genome resequencing studies have indicated that candidate biomarkers include certain common interstitial deletions and balanced inversions observed in secondary cancers of patients who have received radiation therapy and thyroid cancers diagnosed in people who once resided near the Chernobyl nuclear accident site. Although those biomarkers are concordant with the aforementioned consequences of ionizing radiation, they do not overlap with the tumor suppressor genes that are commonly mutated in various cancers [3, 4]. The existence of such radiation-induced mutations in a causative gene would strongly suggest cancer causation by radiation and thus may be useful for estimating the risk of developing cancer after low dose exposure to radiation.
Epidemiological studies, including those of Japanese atomic bomb survivors, have revealed that radiation exposure increases the risk of developing stomach and colon cancers [5, 6], indicating that the gastrointestinal tract is highly susceptible to radiation-induced cancer. Colon cancer is one of the most common cancers worldwide, and molecular studies have demonstrated the involvement of accumulated mutations in multiple genes, including APC (adenomatous polyposis coli) [7]. APC inactivation leads to nuclear accumulation of β-catenin, which activates canonical Wnt signaling. β-catenin binds to the LEF/TCF family of transcription factors that activate genes promoting cell proliferation and survival, which may lead to adenomas; these precursor lesions oftentimes accumulate further genetic mutations that can lead to colon cancer. Germline mutations in APC cause familial adenomatous polyposis (FAP) [8]. Moreover, Min (multiple intestinal neoplasia) encodes a nonsense mutation of the murine Apc locus. ApcMin/+ mice harbor a germline Apc mutation (g.2549T>A, p.850L>X) associated with multiple intestinal neoplasms mainly in the small intestine, and thus, these mice are one of the most widely used models of FAP [9, 10].
The ApcMin/+ mice, if they have the pure genetic background of strain C57BL/6J (B6), develop large numbers of spontaneous intestinal polyps (mostly adenomas and their precursor lesions); owing to the severe hemorrhage associated with these polyps, B6 ApcMin/+ mice typically die within 20 weeks after birth [11]. Molecular analyses have revealed that the wild-type Apc allele (Apc+) is lost in spontaneous intestinal tumors of B6 ApcMin/+ mice [12]. On the other hand, ApcMin/+ mice on a F1 hybrid background of B6 and C3H strains (C3B6F1) develop much fewer polyps than B6 ApcMin/+ mice and hence live long enough for sufficient observation of progression from adenoma to adenocarcinoma [13]. In addition, with F1 ApcMin/+ mice, it is feasible to study the loss of heterozygosity (LOH) in intestinal tumors using polymorphic markers (e.g. microsatellite loci), and indeed, a radiation-specific LOH pattern involving the Apc locus has been found in tumors that developed after irradiation [14–16]. However, a subset of tumors in these studies retained the Apc+ allele [14–16], advocating the need for further clarification. The copy number variations associated with those LOH patterns also remain unresolved.
We have previously shown that exposure of male C3B6F1 ApcMin/+ mice to ionizing radiation increases the incidence of intestinal tumors [13]. Here, we report a combination of techniques applicable to this animal model, in which LOH and DNA copy number analyses were used to detect radiation-specific causative mutations as a measure for attributing the initiating event in individual tumors to radiation exposure. First, we needed to overcome the issue of intratumor heterogeneity inherent in the intestinal tumors of C3B6F1 ApcMin/+ mice. Second, by isolating β-catenin-overexpressing regions in the tumor, we identified a signature, namely interstitial deletions, that inactivates the Apc+ allele specifically in radiation-induced tumors.
MATERIALS AND METHODS
Mice and irradiation protocol
Male B6 ApcMin/+ mice were originally purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained in-house. Female C3H/HeJ mice were obtained from Charles River Laboratories (Kanagawa, Japan). Male B6 ApcMin/+ mice were intercrossed with female C3H/HeJ mice to obtain C3B6F1 ApcMin/+ hybrids, which were identified by genotyping as described [12]. The F1 mice were housed in a conventional clean room under the following conditions: 12-h dark/light cycle, temperature 23 ± 2°C and humidity 50 ± 10%. Animals were irradiated at 2 weeks of age and sacrificed at 30 weeks of age. Irradiation (γ-rays, 0.5 Gy/min) was performed using a Gammacell 40 irradiator (Nordion Inc, Ottawa, Canada) with a 137Cs source. X-ray irradiation (200 kVp, 0.6 Gy/min) was carried out using an X-ray generator (Pantak, East Haven, CT, USA) as described [17]. All animals were fed a laboratory rodent diet and given water ad libitum. All protocols of animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the National Institutes for Quantum Science and Technology (approval number 14–1024 and 19–1005) and were performed in strict accordance with the Institutes of Guide for Care and Use of Laboratory Animals. All efforts were made to minimize suffering.
Tumor isolation and fixation
Mice were sacrificed under isoflurane anesthesia, and the whole gastrointestinal tract was removed. The tract was then separated into stomach, small intestine, caecum and colon, with the small intestine divided further into three segments of equal length. The gut segments were cut along the line of the mesenteric attachment, spread onto paper strips with villi facing upward and fixed in 10% phosphate-buffered formalin for 24 h or in the PAXgene Tissue System fixative (Qiagen, Hilden, Germany) for 3 h, then stored in 70% ethanol. After recording the number and size of tumors larger than 0.5 mm in diameter (Leica Application Suite LAS V4.12; Leica Microsystems, Tokyo, Japan), the tumors were excised and embedded in paraffin.
Histopathological examination and immunohistochemistry
Serial sections were cut at 5 μm thickness and stained with hematoxylin and eosin (H&E). Histological analysis was performed according to the previously reported criteria [18]. After dewaxing and hydration, sections were incubated with an antibody against β-catenin (dilution 1:200; clone 14, BD Biosciences, Franklin Lakes, NJ, USA), a peroxidase-conjugated secondary antibody [Histofine Simple Stain MAX PO (Mouse) kit, Nichirei, Tokyo, Japan], reacted with 3,3′-diaminobenzidine tetrahydrochloride and counterstained with hematoxylin.
DNA extraction from tumor samples
Using a laser microdissection (LMD) system (MMI CellCut, Molecular Machines & Industries GmbH, Eching, Germany), tumor cells were microscopically collected from 50 to 100 sections in 0.5 ml tubes. DNA was subsequently prepared for polymerase chain reaction (PCR) using the QIAamp DNA Micro kit (Qiagen) with minor modifications. Briefly, tumor tissue was suspended in Buffer AL with proteinase K (Qiagen) and incubated at 56°C for 16 h. The DNA samples were heated at 90°C for 1 h, washed and eluted from the spin column prior to use.
LOH analysis
LOH analysis of the Apc locus was performed using HiDi Taq DNA polymerase (myPOLS Biotec GmbH, Konstanz, Germany) and allele-specific primers (Table 1). The PCR cycling program was as follows: 95°C for 2 min and 57°C for 5 min, followed by 42 cycles of 72°C for 30 s, 95°C for 15 s and 57°C for 15 s, and finally 72°C for 2 min. LOH analysis of chromosome 18 microsatellite loci was carried out using AmpliTaq Gold polymerase (Thermo Fisher Scientific, Waltham, MA, USA). The PCR cycling program was 95°C for 9 min and 60°C for 5 min, followed by 20 cycles of 72°C for 15 s, 95°C for 15 s and 60°C for 15 s, 25 cycles of 72°C for 30 s, 95°C for 30 s and 60°C for 30 s, and finally 72°C for 7 min. Primers are listed in Table 1. The PCR products were analyzed by capillary electrophoresis (QIAxcel Advanced System, Qiagen).
Table 1.
Primers for LOH analysis
| Allele or locus | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) |
|---|---|---|
| Apc + allele | [F1]a TTCTGAGAAAGACAGAAGTTT | [R1]a TGTCGTCCTGGGAGGTATGA |
| ApcMin allele | [F2]a TTCTGAGAAAGACAGAAGTTA | [R2]a TTTGGCTATCTGGGCTGCAG |
| D18Mit64b | TCAGATTCACTGCTAAGTCTTTTC | AGCAAGAAAAGCAGGTGAGG |
| D18Mit119b | AGATGCTTGTGAAACATACATATGTG | GAGTGTATAGCGGACTTTTGGG |
| D18Mit120b | ACTGCACTGGTCCCATTTTC | CAATAGTTGGAAATCAGACAGGC |
| D18Mit124b | CCCAAATGGGGTGTCTTTTA | CTGCCACACATTTGTGTGTATG |
| D18Mit184b | CACACATGTGTAGGTAGGTAGGTAGG | CGCACAAGGACTACTGAAACA |
| D18Mit187b | TGCTTGAAGAAAGAGATCCTACG | GACATGCATGCCTGTAACTCC |
aPrimer [names (F, forward; R, reverse)].
bMicrosatellite marker loci.
Array comparative genomic hybridization
DNA samples were amplified using the Sigma-Aldrich GenomePlex WGA2 kit (Merck KGaA, Darmstadt, Germany). Whole-genome amplification (WGA) was carried out with 10–25 ng DNA. The amplified DNA was labeled with either cyanine 3 or 5 using the SureTag Complete DNA Labeling kit (Agilent Technologies, Santa Clara, MA, USA). The array comparative genomic hybridization (aCGH) analysis was performed as described [19, 20]. The microarray data have been deposited in the Gene Expression Omnibus database under accession number GSE228693.
Statistical analysis
The data were analyzed using Prism 8.0 software (GraphPad Inc, San Diego, CA, USA). The statistical significance of differences between two sets of independent data was determined using the Student’s or Welch’s t-test based on the results of the F-test. The relative proportions of adenocarcinoma as well as LOH patterns were compared using Fisher’s exact test.
RESULTS
Radiation facilitates intestinal tumorigenesis in C3B6F1ApcMin/+mice
Mice were irradiated with 2 Gy of γ-rays at 2 weeks of age and sacrificed at 30 weeks of age. Mean body weight at the time of sacrifice was significantly lower in the irradiated male mice than the nonirradiated males (Table 2). Tumor incidence in the small intestine of C3B6F1 ApcMin/+ mice of both sexes was 100%; in the colon, irradiation slightly increased the incidence in males, but the difference was not significant (Table 2). Irradiation significantly increased the mean number of tumors in the small intestine of both sexes; in the colon, again, radiation nonsignificantly increased the number of tumors in males. Thus, radiation facilitated tumorigenesis in the small intestine of both male and female C3B6F1 ApcMin/+ mice.
Table 2.
Frequency of intestinal tumors in C3B6F1 ApcMin/+ mice
| Sex | Treatment | Mice | Body weight at sacrifice (g)a | Mice with tumors (%)b | Tumors/mousea | Proportion of ACc (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| SId | Colon | SIe | Colon | SI | Colon | ||||
| Male | 0 Gy | 5 | 49.8 ± 3.2 | 5 (100) | 2 (40) | 15.4 ± 2.5 | 0.4 ± 0.5 | 14/63 (22) | 1/2 (50) |
| 2 Gy | 5 | 41.8 ± 2.6** | 5 (100) | 4 (80) | 26.2 ± 9.1* | 0.8 ± 0.4 | 29/87 (33)* | 3/4 (75) | |
| Female | 0 Gy | 5 | 38.8 ± 2.7 | 5 (100) | 1 (20) | 13.6 ± 3.8 | 0.2 ± 0.4 | 9/58 (16) | 0/1 (0) |
| 2 Gy | 5 | 36.8 ± 3.5 | 5 (100) | 1 (20) | 24.4 ± 4.1** | 0.2 ± 0.4 | 22/73 (30) | 0/1 (0) | |
aData represent the mean ± SD.
bIncludes adenoma and adenocarcinoma (AC).
cOnly tumors >1.5 mm in diameter were subjected to pathology analysis using HE-stained sections.
dSI = small intestine.
eNumber of tumors in each mouse (male, 0 Gy: 15, 19, 16, 15 and 12; male, 2 Gy: 29, 33, 33, 11 and 25; female, 0 Gy: 11, 14, 12, 11 and 20; female, 2 Gy: 23, 19, 29, 28 and 23).
* P < 0.05.
** P < 0.005 vs 0 Gy.
Radiation increases the incidence of adenocarcinoma among intestinal tumors
Histopathologically, around 70% of the small intestinal tumors of C3B6F1 ApcMin/+mice were adenomas without invasion, showing irregular glandular structures, increased nuclear-to-cytoplasmic ratio and cellular atypia compared with normal mucosa (Fig. 1A and B). Adenocarcinomas developed at lower frequencies and were characterized by their invasion into the submucosal layer (Fig. 1C), as reported previously [13]. To quantify the progression from adenoma to adenocarcinoma, tumors larger than 1.5 mm in diameter were randomly selected, sectioned, stained with H&E and analyzed histopathologically. Irradiation with γ-rays increased the proportion of adenocarcinoma in both male (33 vs 22% for nonirradiated, P < 0.05) and female mice (30 vs 16%, P = 0.06) (Table 2). Notably, tumor morphology did not differ between the nonirradiated and irradiated groups.
Fig. 1.
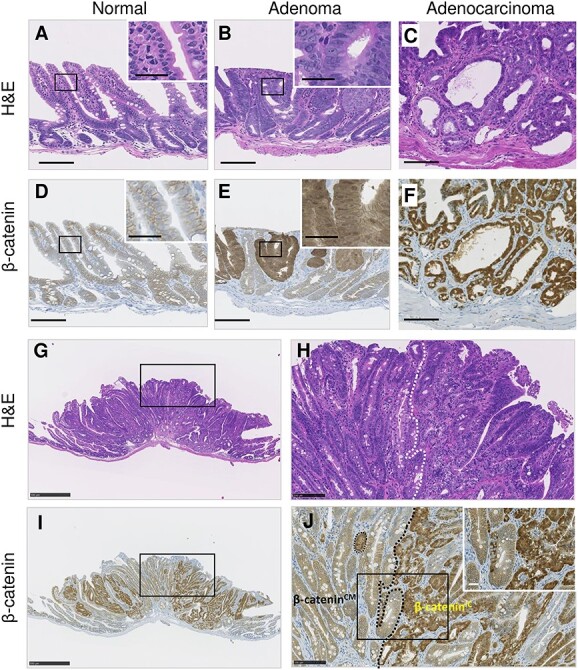
Representative histology and β-catenin immunostaining images of normal and neoplastic intestinal FFPE tissues. (A–C, G and H) H&E staining. (D–F, I and J) β-catenin immunostaining. (G–I) Histology of a representative adenocarcinoma. Insets in A–F are magnified views of the rectangles. (H and J) Enlarged images of rectangles in G and I, respectively. The dotted line indicates the boundary between the β-cateninIC and β-cateninCM regions. Scale bars, 100 μm (A–F, H and J), 25 μm (insets) and 500 μm (G and I).
The extent of intracellular accumulation of β-catenin varies within individual tumors
Previous studies using B6 ApcMin/+ mice revealed that β-catenin accumulates in both the nucleus and cytoplasm of tumors [21]. In C3B6F1 ApcMin/+ mice herein, β-catenin localized to the cell membrane (CM) [herein, named CM localization, β-cateninCM] of the normal intestinal epithelium (Fig. 1A and D) and accumulated in both the nucleus and cytoplasm [intracellular (IC) localization, β-cateninIC] of adenoma and adenocarcinoma (Fig. 1B, C, E and F), as previously reported [21]. Although histologically atypical cells composed the tumors, they were spatially heterogeneous in that the accumulation of β-catenin exhibited a mosaicism with certain regions showing substantial IC localization and other regions displaying mostly CM localization (Fig. 1G–J).
The Apc+ allele is lost in β-cateninIC regions within intestinal tumors
Previous analyses of intestinal tumors from hybrid (AKR × B6)F1 and (CBA × B6)F1 ApcMin/+ mice revealed a radiation-specific LOH pattern on chromosome 18, where Apc resides [14, 16]; these studies, however, used bulk tumor tissues without examining β-catenin expression. Our first analysis of LOH at the Apc locus did not give reproducible results using the bulk tumor samples from C3B6F1 ApcMin/+ mice. Because the IC accumulation of β-catenin is most likely due to the Apc+ allele loss, we hypothesized that the status of the Apc+ allele differs between the β-cateninIC and β-cateninCM regions. Therefore, we examined the presence of the ApcMin and Apc+ alleles in the regions of intestinal tumors that differed with respect to β-catenin IC accumulation (Fig. 2). To isolate those regions, we first performed β-catenin immunostaining on formalin-fixed paraffin-embedded (FFPE) sections of tumors and used LMD to collect (i) whole-tumor tissues, (ii) β-cateninIC regions and (iii) β-cateninCM regions (Fig. 2A). DNA was extracted from each region and subjected to PCR with two forward primers differing in the specificity of the 3′ end (2549) of each of the ApcMin and Apc+ alleles to detect them independently (Fig. 2B and C). As expected, the two alleles were detected in ear-skin DNA of C3B6F1 ApcMin/+ (Fig. 2C, lane 3) and paternal B6 ApcMin/+ mice (Fig. 2C, lane 1), whereas only the Apc+ allele was detected in maternal C3H Apc+/+ mice (Fig. 2C, lane 2). Regarding the five tumor tissue samples isolated as illustrated in Fig. 2A, both the alleles were detected in the whole tumor (Fig. 2C, lane 4), whereas only the ApcMin allele was detected in the β-cateninIC regions (Fig. 2C, lane 5) and both the alleles were detected in the β-cateninCM regions (Fig. 2C, lane 6). These results suggested that tumorigenesis in the β-cateninIC regions is initiated by the loss of the Apc+ allele and that the β-cateninCM regions have distinct origins. Furthermore, an analysis of the whole tumor (i.e. including both the β-cateninIC and β-cateninCM regions, Fig. 2C, lane 4) revealed no evidence for the loss of the Apc+ allele, indicating that the combination of β-catenin immunostaining and LMD is necessary for accurate tissue collection.
Fig. 2.
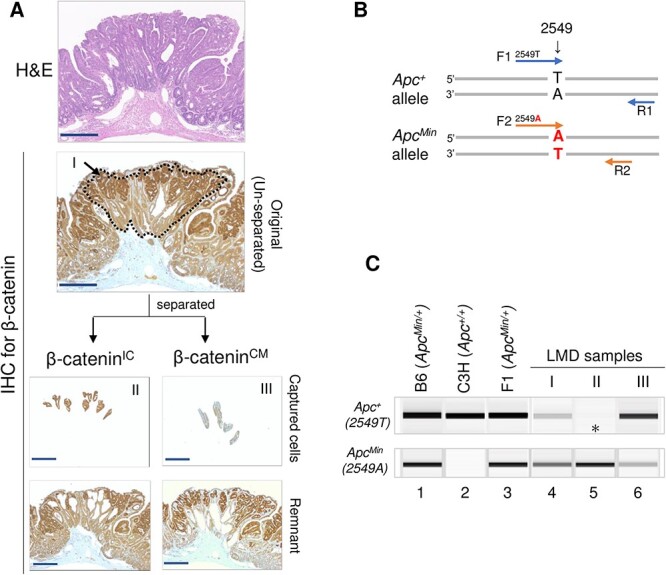
Immunoguided LMD approach for cell collection and analysis of Apc+ allele loss. (A) Guided by the nuclear accumulation of β-catenin, LMD was used to collect cells in the unseparated (I), β-cateninIC (II) and β-cateninCM (III) regions. IHC = immunohistochemical staining. (B) Primer set used to detect Apc+ and ApcMin alleles. (C) LOH results showing loss of Apc+ in the β-cateninIC region (asterisk). Lanes 1–3, ear-skin DNA from B6 ApcMin/+ , C3H Apc+/+ and C3B6F1 ApcMin/+ mice; lanes 4–6, samples corresponding to I, II and III in A. Results are representative of five tumors. Scale bars, 250 μm (A).
Choice of fixative and prestaining affect the subsequent analysis of paraffin-embedded samples
Because our results presented in Fig. 2 implied insensitivity of analyses using the whole tumor, we aimed to examine more precisely the status of chromosome 18 by a DNA copy number analysis combined with a LOH analysis in more homogeneous tumor segments in which we had defined the β-catenin expression in C3B6F1 ApcMin/+ mice. Technically, however, the quality of DNA recovered from the immunohistochemically stained FFPE tissue samples was not sufficient for comprehensive genomic analyses. Because formalin causes DNA crosslinks and thereby decreases the accuracy and reliability of subsequent DNA analyses, we substituted formalin with the PAXgene reagent, which is a formalin-free tissue fixative that preserves nucleic acids better than does formalin [21, 22] (Fig. 3A). Fortunately, the quality of the β-catenin staining in FFPE and PAXgene-fixed paraffin-embedded (PFPE) tissues was comparable (Fig. 3B–G). Because DNA recovery tends to be low in immunostained tissue samples [23], we took an approach in which β-catenin expression was identified by immunohistochemical staining (Fig. 3C and F) and DNA was recovered from the corresponding unstained samples of proximal tissue (Fig. 3D and G). With this approach, both FFPE and PFPE tissues yielded sufficient amounts of DNA for reliable and reproducible PCR analysis (Fig. 3H, ‘PCR’). With FFPE tissues, the copy number changes at the Apc locus were not detectable using WGA and oligonucleotide aCGH technologies; with PFPE tissues, however, a decrease in copy number was indeed detected (Fig. 3H, ‘WGA-aCGH’).
Fig. 3.
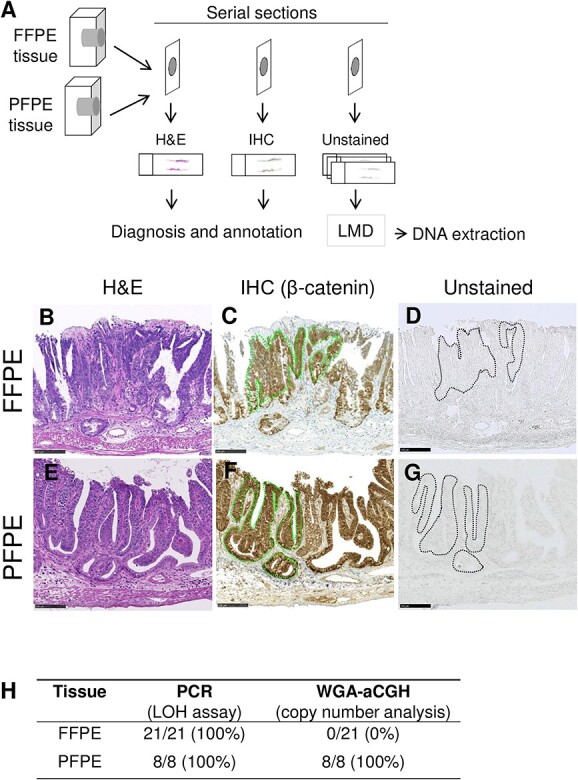
Effect of the formalin-free fixative PAXgene on tissue staining and DNA quality. (A) Staining and DNA extraction strategy using FFPE and PFPE samples. IHC = immunohistochemical staining. (B–G) Serial sections were used for diagnosis on H&E staining (B and E), β-catenin immunostaining (C and F) and DNA isolation without staining (D and G). Dotted lines in C, D, F and G identify the β-cateninIC regions. (H) Summary of detection sensitivity of LOH and copy number variants at the Apc locus. Scale bars, 100 μm (B–G).
Pattern of chromosome 18 LOH in β-cateninIC regions correlates with etiology
The LOH of six microsatellite markers on chromosome 18 was analyzed using DNA obtained from normal ear-skin samples and intestinal tumors (nonirradiated, n = 8; 2-Gy X-irradiated, n = 14). For the intestinal tumor samples, tissues of the β-cateninIC regions were collected from unstained PFPE samples. LOH at the Apc locus was observed in all tumors from the nonirradiated (8 of 8) and irradiated (14 of 14) groups (Fig. 4A and B). The pattern of LOH was dichotomous, including those spanning nearly the entire chromosome and those beginning upstream of Apc and ending in the middle of the chromosome (Fig. 4B). Interestingly, the latter pattern was observed only in the irradiated group (11 of 14, 79%), whereas the former pattern was observed in both nonirradiated (8 of 8, 100%) and irradiated (3 of 14, 21%) groups. These results indicated that the latter and former LOH patterns were characteristics of the radiation-induced and spontaneous tumors, and thus we refer to them as ‘R’ and ‘S’ types, respectively (Fig. 4). In addition, the LOH analysis of tumor tissues exhibiting β-catenin accumulation provided the information for accurate prediction of etiology of the intestinal carcinogenesis.
Fig. 4.

LOH and aCGH analyses using PFPE intestinal tumor samples. (A) LOH analysis of six microsatellite markers and the Apc locus on chromosome 18 in LMD samples from the 0 Gy (n = 8) and 2 Gy X-ray exposure groups (n = 14). N = normal tissue. (B) Mapping of the LOH results. Solid circle, LOH; open circle, no LOH. n, number of cases with the indicated LOH pattern; S = spontaneous type; R = radiation-induced types (R1–R3). (C) Changes in DNA copy number identified by aCGH analysis. Dark gray denotes regions with copy number loss detected by aCGH.
Whether copy number losses associate with such LOH in radiation-induced tumors has not been addressed in previous studies on ApcMin/+ mice [14–16]. To evaluate the pattern of DNA copy number aberrations, tumors displaying representative LOH patterns were chosen from each group for aCGH analysis (i.e. 1 with S-type LOH in the nonirradiated group, 1 with S-type LOH in the irradiated group and 3 with R-type LOH in the irradiated group). Tumors with the S-type LOH pattern exhibited normal copy numbers throughout chromosome 18, indicating that the LOH was caused by chromosomal mis-segregation or mitotic recombination (Fig. 4C). In contrast, a tumor with the R-type LOH pattern harbored copy number losses spanning chromosome 18qB1–D3 (Fig. 4C), indicating that this type of LOH was caused by interstitial deletion. These results suggest that the interstitial (or R-type) LOH pattern is most likely caused by an interstitial chromosomal deletion and hence is a signature for radiation-associated tumors.
DISCUSSION
In this study, we characterized radiation-induced intestinal tumors, using C3B6F1 ApcMin/+ mice. Our results clarify the mosaicism of β-catenin overexpression in intestinal tumors of ApcMin/+ mice and identify novel differences in genomic variation (presence or absence of the Apc+ allele) indicative of tissue mosaicism, i.e. loss of the Apc+ allele in the β-cateninIC region and retention of the Apc+ allele in the β-cateninCM region. In addition, by devising a sample recovery method, we were also able to show for the first time in F1 ApcMin/+mice that deletion of the Apc site predicted by LOH analysis was confirmed by aCGH analysis. Our study establishes a highly sensitive method for classifying dichotomous LOH types, namely chromosomal mis-segregation/mitotic recombination (S type) and interstitial deletion (R type), and demonstrates that the R-type LOH is a signature of radiation-induced intestinal tumors.
Herein, β-catenin expression was mosaic in the intestinal tumors of C3B6F1 ApcMin/+ mice (Fig. 1). The mosaicism of β-catenin has been reported in human colorectal tumors [24, 25]. The mechanism underlying this heterogeneity is not well understood, although intratumor heterogeneity is sometimes related to genetic [26] rather than epigenetic changes [27]. This heterogeneity has been reported for intestinal tumors of ApcMin/+ mice [28, 29] as well as patients of FAP [30, 31], implying that distinct polyp types from different mammals may have a similar physiological basis. Although previous studies have used F1 ApcMin/+ mice to suggest radiation-specific LOH patterns in bulk intestinal tumors [14, 16], those studies identified a subset of tumors displaying retention of the Apc+ allele, implying substantial contamination of normal cells or heterogeneity of the tumor tissue. In addition, these studies never associated LOH with copy number variation. A major challenge has been the difficulty in obtaining homogenous tumor samples with a sufficient amount and quality of DNA for aCGH experiments. In this study, the immunoguided LMD method was used to collect tumor samples based on the tissue phenotype instead of collecting bulk tumors. Specifically, we established an accurate LOH determination method by collecting and analyzing tumor cells with a common tumorigenesis mechanism using β-catenin accumulation as an indicator, in accordance with the molecular mechanism of multistep tumorigenesis triggered by Apc gene inactivation. We additionally overcome the abovementioned challenge by combining the use of a formalin-free fixative, an LMD approach guided by immunostaining images of proximal sections, and the application of WGA before aCGH. Haines et al. [15] previously collected tumor tissues for LOH analysis using pathological (i.e. H&E-stained) images as a guide. The application of LMD to immunostained tissues has been shown effective in separating cells that are morphologically very similar but have immunohistochemically discernible characteristics [32], although the integrity of the analytes (e.g. DNA) in the processed samples is often compromised by the use of a formalin-containing fixative and the staining procedure itself [23]. We herein combined those techniques to confirm that all tumors of ApcMin/+ mice lacked the Apc+ allele, in contrast to what has been reported in studies using bulk tumors [14, 16]. For the first time, we further associated these LOH patterns with copy number variations. We thus could identify chromosomal mis-segregation/mitotic recombination (S type, i.e. LOH with two copies) and interstitial deletion (R type, i.e. LOH with one copy) events that affect the Apc+ allele, with the latter occurring only in the irradiated group, thus corroborating and extending the results reported for F1 ApcMin/+ mice [14–16]. The method adopted herein offers a promising tool for identifying radiation-induced oncogenic changes in other animal models.
In the irradiated group, we identified not only tumors with the R-type LOH pattern (R-type tumors) but also those with the S-type LOH pattern (S-type tumors) (Fig. 4). This is consistent with the results obtained with Ptch1+/− mice and Tsc2+/− Eker rats [19, 20, 33, 34], each of which is also a model animal with germline heterozygous deletions of a tumor suppressor gene. Therein, the remaining wild-type allele undergoes spontaneous deletion, leading to the development of S-type tumors in nonirradiated animals, whereas both S- and R-type tumors develop in irradiated animals. Especially in Ptch1+/− mice, which develop only one tumor per individual, the incidence of R-type tumors increases, whereas that of S-type tumors decreases in a dose-dependent manner [33]. Therefore, it is possible that competition between cells with S-type and R-type LOH patterns would result in a reduced incidence of S-type tumors. The possibility remains, however, that radiation-induced changes in the intestinal microenvironment may increase the incidence of S-type tumors. To test this possibility, it will be meaningful to analyze the dose dependence of the incidence of S- and R-type tumors in F1 ApcMin/+ mice, which develop multiple intestinal tumors in each individual mouse.
Studies of radiation carcinogenesis in rodent models have shown that chromosomal deletions affect tumor suppressor genes in mouse acute myeloid leukemia [35], lymphoma, sarcoma and mammary carcinoma in Trp53+/− mice [36–38], intestinal tumors in Apc-deficient mice [14–16, 39], medulloblastoma in Ptch1+/− mice [33, 40], renal tumors in Tsc2+/− Eker rats [34] and rat mammary cancer [41–44]. This is a feature not often seen in tumors of nonirradiated animals and is most likely attributable to radiation exposure. ApcMin/+ mice, Ptch1+/− mice and Tsc2+/− Eker rats are special animal models of carcinogenesis owing to a heterozygous germline mutation of a cancer driver gene, and thus it must be considered whether LOH and copy number variations that are a consequence of chromosomal structural instability caused by the absence of one allele can be a general mechanism. To date, only medulloblastoma in Ptch1+/− mice and renal tumors in Tsc2+/− Eker rats have been properly demonstrated (i.e. with evidence of copy number losses) to have an interstitial deletion as a radiation signature in solid tumors [19, 20, 33, 34]. Because these tumors are not of much relevance to human radiation risk, it is significant that we extended these findings to a model of intestinal tumors, one of the diseases relevant to human radiation risk. Furthermore, even in wild-type mice, deletion of any one of the tumor suppressor genes Pax5, Sfpi1, Ikzf1, Pten or Cdkn2a has been reported specifically in irradiated animals in copy number analyses of lymphomas, acute myeloid leukemia and thymic lymphoma [35, 45, 46]. Thus, we speculate that the inactivation of tumor suppressor genes via interstitial deletion is a common mechanism of radiation-induced carcinogenesis. This mechanism could be used for discriminating radiation-induced tumors of ApcMin/+ mice exposed to low levels of radiation from those developed by spontaneous mechanisms, as previously performed with Ptch1+/− mice [19, 20].
An important unresolved issue in radiation protection is whether there is a dose-dependent increase or threshold dose of cancer risk in the dose range of several tens of mGy. We anticipate that this approach will help clarify the effects of low dose and low dose rate radiation.
ACKNOWLEDGEMENTS
The authors thank the staff at the Department of Radiation Effects Research and the Laboratory Animal and Genomics Section for technical assistance.
Footnotes
†Presentation at a Conference: The 65th Annual Meeting of the Japanese Radiation Research Society; the 63rd, 64th and 65th Annual Radiation Research Society Meetings and the 56th Annual Meeting on Radioisotopes and Radiation Research.
Contributor Information
Hiromi Yanagihara, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
Takamitsu Morioka, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
Shunsuke Yamazaki, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
Yutaka Yamada, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
Hirotaka Tachibana, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan; Department of Biology, Graduate School of Science, Chiba University, Chiba, Japan.
Kazuhiro Daino, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan; Department of Biology, Graduate School of Science, Chiba University, Chiba, Japan.
Chizuru Tsuruoka, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
Yoshiko Amasaki, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
Mutsumi Kaminishi, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
Tatsuhiko Imaoka, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
Shizuko Kakinuma, Department of Radiation Effects Research, National Institute of Radiological Sciences, National Institutes for Quantum Science and Technology, Chiba, Japan.
CONFLICT OF INTEREST
The authors declare no conflicts of interest associated with this manuscript.
FUNDING
This work was supported by the Japan Society for the Promotion of Science via KAKENHI (grant numbers JP15K00550 to Y.Y., JP18K18197 to H.Y. and JP 21H04932 to S.K.).
DATA AVAILABILITY
The microarray data are available in the Gene Expression Omnibus database under accession number GSE228693.
References
- 1. Laurier D, Ruhm W, Paquet F et al. Areas of research to support the system of radiological protection. Radiat Environ Biophys 2021;60:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Preston RJ, Ruhm W, Azzam EI et al. Adverse outcome pathways, key events, and radiation risk assessment. Int J Radiat Biol 2021;97:804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Behjati S, Gundem G, Wedge DC et al. Mutational signatures of ionizing radiation in second malignancies. Nat Commun 2016;7:12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morton LM, Karyadi DM, Stewart C et al. Radiation-related genomic profile of papillary thyroid carcinoma after the Chernobyl accident. Science 2021;372:eabg2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sugiyama H, Misumi M, Brenner A et al. Radiation risk of incident colorectal cancer by anatomical site among atomic bomb survivors: 1958-2009. Int J Cancer 2020;146:635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakata R, Preston DL, Brenner AV et al. Radiation-related risk of cancers of the upper digestive tract among Japanese atomic bomb survivors. Radiat Res 2019;192:331–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fearon ER, Cho KR, Nigro JM et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 1990;247:49–56. [DOI] [PubMed] [Google Scholar]
- 8. Miyoshi Y, Ando H, Nagase H et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci U S A 1992;89:4452–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Su LK, Kinzler KW, Vogelstein B et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992;256:668–70. [DOI] [PubMed] [Google Scholar]
- 10. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990;247:322–4. [DOI] [PubMed] [Google Scholar]
- 11. Moser AR, Luongo C, Gould KA et al. ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur J Cancer 1995;31a:1061–4. [DOI] [PubMed] [Google Scholar]
- 12. Luongo C, Moser AR, Gledhill S et al. Loss of Apc+ in intestinal adenomas from min mice. Cancer Res 1994;54:5947–52. [PubMed] [Google Scholar]
- 13. Morioka T, Yamazaki S, Yanagihara H et al. Calorie restriction suppresses the progression of radiation-induced intestinal tumours in C3B6F1 Apc (min/+) mice. Anticancer Res 2021;41:1365–75. [DOI] [PubMed] [Google Scholar]
- 14. Luongo C, Dove WF. Somatic genetic events linked to the Apc locus in intestinal adenomas of the min mouse. Genes Chromosom Cancer 1996;17:194–8. [DOI] [PubMed] [Google Scholar]
- 15. Haines J, Dunford R, Moody J et al. Loss of heterozygosity in spontaneous and X-ray-induced intestinal tumors arising in F1 hybrid min mice: evidence for sequential loss of APC(+) and Dpc4 in tumor development. Genes Chromosom Cancer 2000;28:387–94. [DOI] [PubMed] [Google Scholar]
- 16. Ellender M, Harrison JD, Meijne E et al. Intestinal tumours induced in Apc(min/+) mice by X-rays and neutrons. Int J Radiat Biol 2011;87:385–99. [DOI] [PubMed] [Google Scholar]
- 17. Tokairin Y, Kakinuma S, Arai M et al. Accelerated growth of intestinal tumours after radiation exposure in Mlh1-knockout mice: evaluation of the late effect of radiation on a mouse model of HNPCC. Int J Exp Pathol 2006;87:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boivin GP, Washington K, Yang K et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 2003;124:762–77. [DOI] [PubMed] [Google Scholar]
- 19. Tsuruoka C, Blyth BJ, Morioka T et al. Sensitive detection of radiation-induced Medulloblastomas after acute or protracted gamma-ray exposures in Ptch1 heterozygous mice using a radiation-specific molecular signature. Radiat Res 2016;186:407–14. [DOI] [PubMed] [Google Scholar]
- 20. Tsuruoka C, Kaminishi M, Shinagawa M et al. High relative biological effectiveness of 2 MeV fast neutrons for induction of Medulloblastoma in Ptch1+/− mice with radiation-specific deletion on chromosome 13. Radiat Res 2021;196:225–34. [DOI] [PubMed] [Google Scholar]
- 21. Mathieson W, Marcon N, Antunes L et al. A critical evaluation of the PAXgene tissue fixation system: morphology, immunohistochemistry, molecular biology, and proteomics. Am J Clin Pathol 2016;146:25–40. [DOI] [PubMed] [Google Scholar]
- 22. Southwood M, Krenz T, Cant N et al. Systematic evaluation of PAXgene(R) tissue fixation for the histopathological and molecular study of lung cancer. J Pathol Clin Res 2020;6:40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tangrea MA, Mukherjee S, Gao B et al. Effect of immunohistochemistry on molecular analysis of tissue samples: implications for microdissection technologies. J Histochem Cytochem 2011;59:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang S, Wang Z, Shan J et al. Nuclear expression and/or reduced membranous expression of beta-catenin correlate with poor prognosis in colorectal carcinoma: a meta-analysis. Medicine (Baltimore) 2016;95:e5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takayama T, Shiozaki H, Shibamoto S et al. Beta-catenin expression in human cancers. Am J Pathol 1996;148:39–46. [PMC free article] [PubMed] [Google Scholar]
- 26. Mamlouk S, Childs LH, Aust D et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat Commun 2017;8:14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ryser MD, Yu M, Grady W et al. Epigenetic heterogeneity in human colorectal Tumors reveals preferential conservation and evidence of immune surveillance. Sci Rep 2018;8:17292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thliveris AT, Halberg RB, Clipson L et al. Polyclonality of familial murine adenomas: analyses of mouse chimeras with low tumor multiplicity suggest short-range interactions. Proc Natl Acad Sci U S A 2005;102:6960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zahm CD, Szulczewski JM, Leystra AA et al. Advanced intestinal cancers often maintain a multi-ancestral architecture. PLoS One 2016;11:e0150170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Novelli MR, Williamson JA, Tomlinson IP et al. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science 1996;272:1187–90. [DOI] [PubMed] [Google Scholar]
- 31. Thirlwell C, Will OC, Domingo E et al. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology 2010;138:1441–54, 1454.e1-7. [DOI] [PubMed] [Google Scholar]
- 32. Domazet B, Maclennan GT, Lopez-Beltran A et al. Laser capture microdissection in the genomic and proteomic era: targeting the genetic basis of cancer. Int J Clin Exp Pathol 2008;1:475–88. [PMC free article] [PubMed] [Google Scholar]
- 33. Ishida Y, Takabatake T, Kakinuma S et al. Genomic and gene expression signatures of radiation in medulloblastomas after low-dose irradiation in Ptch1 heterozygous mice. Carcinogenesis 2010;31:1694–701. [DOI] [PubMed] [Google Scholar]
- 34. Inoue T, Kokubo T, Daino K et al. Interstitial chromosomal deletion of the tuberous sclerosis complex 2 locus is a signature for radiation-associated renal tumors in Eker rats. Cancer Sci 2020;111:840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook WD, McCaw BJ, Herring C et al. PU.1 is a suppressor of myeloid leukemia, inactivated in mice by gene deletion and mutation of its DNA binding domain. Blood 2004;104:3437–44. [DOI] [PubMed] [Google Scholar]
- 36. Umesako S, Fujisawa K, Iiga S et al. Atm heterozygous deficiency enhances development of mammary carcinomas in p53 heterozygous knockout mice. Breast Cancer Res 2005;7:R164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kemp CJ, Wheldon T, Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet 1994;8:66–9. [DOI] [PubMed] [Google Scholar]
- 38. Bouffler SD, Kemp CJ, Balmain A et al. Spontaneous and ionizing radiation-induced chromosomal abnormalities in p53-deficient mice. Cancer Res 1995;55:3883–9. [PubMed] [Google Scholar]
- 39. van der Houven van Oordt CW, Smits R, Williamson SL et al. Intestinal and extra-intestinal tumor multiplicities in the Apc1638N mouse model after exposure to X-rays. Carcinogenesis 1997;18:2197–203. [DOI] [PubMed] [Google Scholar]
- 40. Pazzaglia S, Tanori M, Mancuso M et al. Two-hit model for progression of medulloblastoma preneoplasia in patched heterozygous mice. Oncogene 2006;25:5575–80. [DOI] [PubMed] [Google Scholar]
- 41. Iizuka D, Imaoka T, Takabatake T et al. DNA copy number aberrations and disruption of the p16Ink4a/Rb pathway in radiation-induced and spontaneous rat mammary carcinomas. Radiat Res 2010;174:206–15. [DOI] [PubMed] [Google Scholar]
- 42. Showler K, Nishimura M, Daino K et al. Analysis of genes involved in the PI3K/Akt pathway in radiation- and MNU-induced rat mammary carcinomas. J Radiat Res 2017;58:183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moriyama H, Daino K, Ishikawa A et al. Exome of radiation-induced rat mammary carcinoma shows copy-number losses and mutations in human-relevant cancer genes. Anticancer Res 2021;41:55–70. [DOI] [PubMed] [Google Scholar]
- 44. Nishimura M, Daino K, Fukuda M et al. Development of mammary cancer in γ-irradiated F1 hybrids of susceptible Sprague-Dawley and resistant Copenhagen rats, with copy-number losses that pinpoint potential tumor suppressors. PLoS One 2021;16:e0255968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tachibana H, Daino K, Ishikawa A et al. Genomic profile of radiation-induced early-onset mouse B-cell lymphoma recapitulates features of Philadelphia chromosome-like acute lymphoblastic leukemia in humans. Carcinogenesis 2022;43:693–703. [DOI] [PubMed] [Google Scholar]
- 46. Nakayama T, Sunaoshi M, Shang Y et al. Calorie restriction alters the mechanisms of radiation-induced mouse thymic lymphomagenesis. PLoS One 2023;18:e0280560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The microarray data are available in the Gene Expression Omnibus database under accession number GSE228693.