

Abstract
Although cannabinoid receptors (CB) are recognized as targets for renal fibrosis, the roles of endogenous cannabinoid anandamide (AEA) and its primary hydrolytic enzyme, fatty acid amide hydrolase (FAAH), in renal fibrogenesis remain unclear. The present study used a mouse model of post-ischemia-reperfusion renal injury (PIR) to test the hypothesis that FAAH participates in the renal fibrogenesis. Our results demonstrated that PIR showed upregulated expression of FAAH in renal proximal tubules, accompanied with decreased AEA levels in kidneys. Faah knockout mice recovered the reduced AEA levels and ameliorated PIR-triggered increases in blood urea nitrogen, plasma creatinine as well as renal profibrogenic markers and injuries. Correspondingly, a selective FAAH inhibitor, PF-04457845, inhibited the transforming growth factor-beta 1 (TGF-β1)–induced profibrogenic markers in human proximal tubular cell line (HK-2 cells) and mouse primary cultured tubular cells. Knockdown of FAAH by siRNA in HK-2 cells had similar effects as PF-04457845. Tubular cells isolated from Faah−/− mice further validated the protection against TGF-β1–induced damages. The CB 1 or CB2 receptor antagonist and exogenous FAAH metabolite arachidonic acid failed to reverse the protective effects of FAAH inactivation in HK-2 cells. However, a substrate-selective inhibitor of AEA-cyclooxygenase-2 (COX-2) pathway significantly suppressed the anti-profibrogenic actions of FAAH inhibition. Further, the AEA-COX-2 metabolite, prostamide E2 exerted anti-fibrogenesis effect. These findings suggest that FAAH activation and the consequent reduction of AEA contribute to the renal fibrogenesis, and that FAAH inhibition protects against fibrogenesis in renal cells independently of CB receptors via the AEA-COX-2 pathway by the recovery of reduced AEA.
Keywords: Fatty acid amide hydrolase, Renal fibrogenesis, Endocannabinoid system, Anandamide
1. Introduction
Acute kidney injury (AKI) is an important risk factor for chronic kidney disease (CKD) development and progression [1–3]. Chronic proximal tubular injury with subsequent fibrosis is a key factor in the transition of AKI to CKD [4–6]. Mildly injured proximal tubular epithelial cells (PTECs) initiate a process of adaptive repair in epithelial cells. Severe injured PTECs undergo the change of epithelial phenotype, initiate inflammation and profibrogenic responses and eventually result in renal fibrosis [4–7]. This transition is featured by downregulation of epithelial cell markers, such as E-cadherin or zonula occludins-1 (ZO-1) [8], increased production of profibrogenic mediators IL-1β, TGF-β1 [9,10] and profibrogenic markers α-smooth muscle actin (α-SMA), type I/III collagens [11,12]. Therefore, PTECs play a crucial role in the renal fibrogenesis of progressive CKD. In particular, TGF-β1 is a major mediator to promote profibrogenic effects of PTECs [13–15].
The endogenous cannabinoid (endocannabinoid) system, consisting of two major endocannabinoids, anandamide (AEA) and 2-arachidonoylglycerol (2-AG), that bind and activate the cannabinoid receptors, CB1 and CB2, plays an important physiological function in the kidneys. The cannabinoid receptors have been identified as potential targets in renal fibrosis [16,17]. Lecru et al. showed that inhibition of CB1 reduced renal fibrosis in a unilateral ureter obstruction (UUO) model and attenuated TGF-β1-induced fibrogenesis on renal myofibroblast [18]. The role of CB2 receptor in kidney fibrosis, on the other hand, remains controversial. Studies demonstrated that inhibition of CB2 receptor reduced kidney fibrosis after UUO [19,20], whereas combination of a CB1 receptor antagonist and CB2 receptor agonist prevented diabetic nephropathy [21]. In addition, studies investigating the role of AEA in kidney fibrosis are hampered by its rapid hydrolysis from the primary catabolic enzymes fatty acid amide hydrolase (FAAH) [22]. Thus, FAAH represents a viable target to modulate the endogenous cannabinoid tone, but its role in profibrogenic effects of tubular cells and renal fibrosis remains unspecified.
Growing attention has focused on the development of FAAH inhibitors as potential therapeutic agents for inflammation, and hypertension [23–27]. Notably, the renal cortex possesses a higher level of FAAH expression than renal medulla [28]. Moreover, a selective FAAH inhibitor modulates blood pressure and diuresis after renomedullary infusion [27]. These findings indicate that FAAH is highly associated with renal function. Therefore, the primary focus of the present study was to investigate the in vivo functional contribution of FAAH to post-ischemia-reperfusion (PIR)-induced kidney damage and fibrogenesis in mice. In addition, we explored the mechanism underlying the contribution of the FAAH pathway to the profibrogenic effects of TGF-β1 in renal PTECs.
2. Methods (details in Supplementary Methods)
2.1. Animal
Twelve weeks-old male littermate wild-type (WT, C57BL/6 J) and Faah−/− mice (genetic background C57BL/6 J) were used. All procedures were approved by the Institutional Animal Care and Use Committee of the Virginia Commonwealth University.
2.2. Mouse ischemic AKI to CKD model
Wild-type (WT) and Faah−/− mice were subjected to a renal post-ischemia-reperfusion injury (PIR), which mimics the transition of AKI to CKD [19,29,30]. On day 1, left renal pedicles were clipped by microvascular clamps for 30 min followed by reperfusion. Mice in both the sham and PIR groups underwent right nephrectomy on day 11. Blood samples and the left kidneys were then harvested before euthanasia on day 13. Right nephrectomy would allow the assessment of left renal functions by measuring plasma creatinine and blood urea nitrogen (BUN). Sham-operated control mice were subjected to a similar surgical protocol but did not undergo ischemic injury.
2.3. Renal function analysis
Plasma creatinine was analyzed by LabAssay Creatinine kit (Fujifilm) [31] and BUN was assessed by Urea Nitrogen Reagent Kit (Alfa Wassermann) using Vet Axcel Chemistry Analyzer (Alfa Wassermann) according to manufacturer’s guidance.
2.4. Culture of human proximal tubular epithelial cell line and drug treatment
Human proximal tubular epithelial cells (HK-2) were purchased from AddexBio company and cultured as described [32,33]. Cells were pre-treated with 1 μM PF-04457845 (PF, a selective FAAH inhibitor, Tocris Bioscience) [34], 1 μM Rimonabant (CB1 antagonist, Cayman) [18], 1 μM SR144528 (SR, CB2 antagonist, Cayman) [35], 10 μM arachidonic acid (AA, Cayman) [36], 30 nM LM-4131 (Indomethacin Morpholiny-lamide, substrate-selective AEA-COX-2 inhibitor, Cayman) [37,38] or 100 nM Prostamide E2 (Cayman) [39] 30 min prior to the incubation of 5 ng/ml TGF-β1 (PeproTech) for 48 h [40].
2.5. Primary culture of mouse kidney proximal tubular cells
Isolation and culture of primary proximal tubule cells was performed using collagenase digestion and sieving of renal cortex as previously reported with modifications [41–43]. Tubules were collected and cultured in hormonally defined serum-free DMEM/F12 (Gibco) medium supplemented with epidermal growth factor (EGF, 10 ng/ml), triiodothyronine (T3, 5 × 10–12 M), selenium (50 nM), transferrin (5 μg/ml), insulin (5 μg/ml), hydrocortisone (50 nM) and streptomycin (100μg/ml) /penicillin (100 IU/ml, Gibco). Identification of proximal tubular cells was confirmed by immunostaining of a proximal tubule marker, megalin (1:200, Santa Cruz). Cells were then treated with PF and/or TGF-β1 as described above.
2.6. Western blot
Western blot analysis was performed as previously described [44]. Total protein (15 μg) of cells or renal cortex was loaded into polyacrylamide gel followed by electroblotting onto a PVDF membrane. Primary antibodies used are listed in the Supplementary Methods. Blot signals were amplified by HRP-conjugated secondary antibodies (Cell Signaling Technology) in a concentration of 1:5000 for 1 h and incubated with SuperSignal™ West Pico PLUS Substrate (Thermo Fisher Scientific). Protein bands were detected with Odyssey Fc imaging system and intensity of bands was determined by ImageJ software (NIH).
2.7. Histological examinations
Mouse kidneys were cut longitudinally and fixed in 4 % paraformaldehyde for at least 24 h and then switched to 70 % ethanol. The fixed tissues were embedded in paraffin and cut into 5 μm sections. Paraffin-embedded kidney sections were stained with Periodic-Acid Schiff (PAS, Sigma), picrosirius red (Abcam) [45,46], and Masson’s trichrome (Polysciences) [19,20].
2.8. Immunohistochemical (IHC) staining in kidney tissues
In brief, paraffin-embedded sections were deparaffinized and processed to subsequent antigen retrieval, endogenous peroxidase blocking with 3 % H2O2, and nonspecific sites blocking in 10 % serum. Primary antibodies used are listed in the Supplementary Methods. Primary antibodies were incubated overnight at 4 °C. Rabbit secondary antibody was then incubated for 1 h at room temperature followed by streptavidin HRP incubation, DAB developing, and counterstained with hematoxylin. At least 10 fields for each sample were evaluated for all results. Staining area was calculated by using ImageJ (NIH) [46].
2.9. Immunocytochemical staining in cultured cells
Cells were cultured on glass coverslips, fixed in 4 % paraformaldehyde, incubated with primary antibody at 4°C overnight, and then fluorescence conjugated-secondary antibody for 1 h at room temperature. Positive areas were calculated by ImageJ [47].
2.10. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) for measurement of lipids
AEA of mouse kidney cortex and HK-2 cells were extracted three times with chloroform/methanol (2:1, v/v) and a 0.73 % sodium chloride mixture. The chloroform was collected and pooled. Dried residues were reconstituted in 0.1 ml of chloroform followed by 1 ml of cold acetone (Pharmco, Fisher Scientific). After centrifugation for 5 min at 10000 rpm at 4 °C, the upper layer was collected and dried under nitrogen gas. The extracts were reconstituted with 0.1 ml of methanol and placed in auto-sampler vials for analysis. The LC-MS/MS was performed by the Sciex 6500+ LC-MS/MS as we previously reported [28].
2.11. siRNA transfection
HK-2 cells were grown to 90 % confluence in normal medium before transfection of FAAH or CB2 siRNA (Thermo Fisher) using Lipofect-amine 3000 transfection reagent (Thermo Fisher) according to the manufacturer’s guidance. For a six-well plate, each well was transfected with 75 pmol of siRNA in Opti-MEM medium (Gibco). Following 5 h incubation, cells were then maintained in normal medium overnight and subjected to TGF-β1 stimulation. A negative control siRNA (Silencer, Ambion, Thermo Fisher Scientific) was employed to reflect a baseline for target gene knockdown.
2.12. Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). The significance of differences in mean values within and between multiple groups was evaluated using one-way ANOVA by a Bonferroni’s multiple comparisons test or two-way ANOVA followed by a Tukey’s multiple comparisons test. Student’s t-test was used to evaluate statistical significance of differences between two groups. P < 0.05 was considered statistically significant.
3. Results
3.1. Post-ischemia-reperfusion renal injury model causes kidney injury and fibrogenesis, as well as increased FAAH protein expression
To study in vivo renal fibrogenesis, we utilized a mouse model of renal post-ischemia-reperfusion (PIR), which produced a progression of AKI to CKD [19,29,30]. PIR-induced kidney damages were confirmed by the significant increases in biomarkers of kidney injury, neutrophil gelatinase-associated lipocalin (NGAL), and profibrogenic marker, α-SMA in WT mice (Fig. 1A–C). Immunofluorescence double-staining revealed that FAAH was primarily expressed in proximal tubular marker megalin-stained tubules in the cortex (Fig. 1D). Furthermore, PIR resulted in increased FAAH expression, especially in proximal tubules (Fig. 1E, upper panel), which was associated with a concomitant increased expression of PIR-induced α-SMA (Fig. 1C). These findings suggest that FAAH may play a role in the process of renal fibrogenesis. PIR also increased TGF-β1 (Fig. 1E, lower panel), which replicated the findings by Li et al. [29]. Therefore, we used TGF-β1 as a profibrogenic inducer of PTECs in subsequent in vitro studies.
Fig. 1.
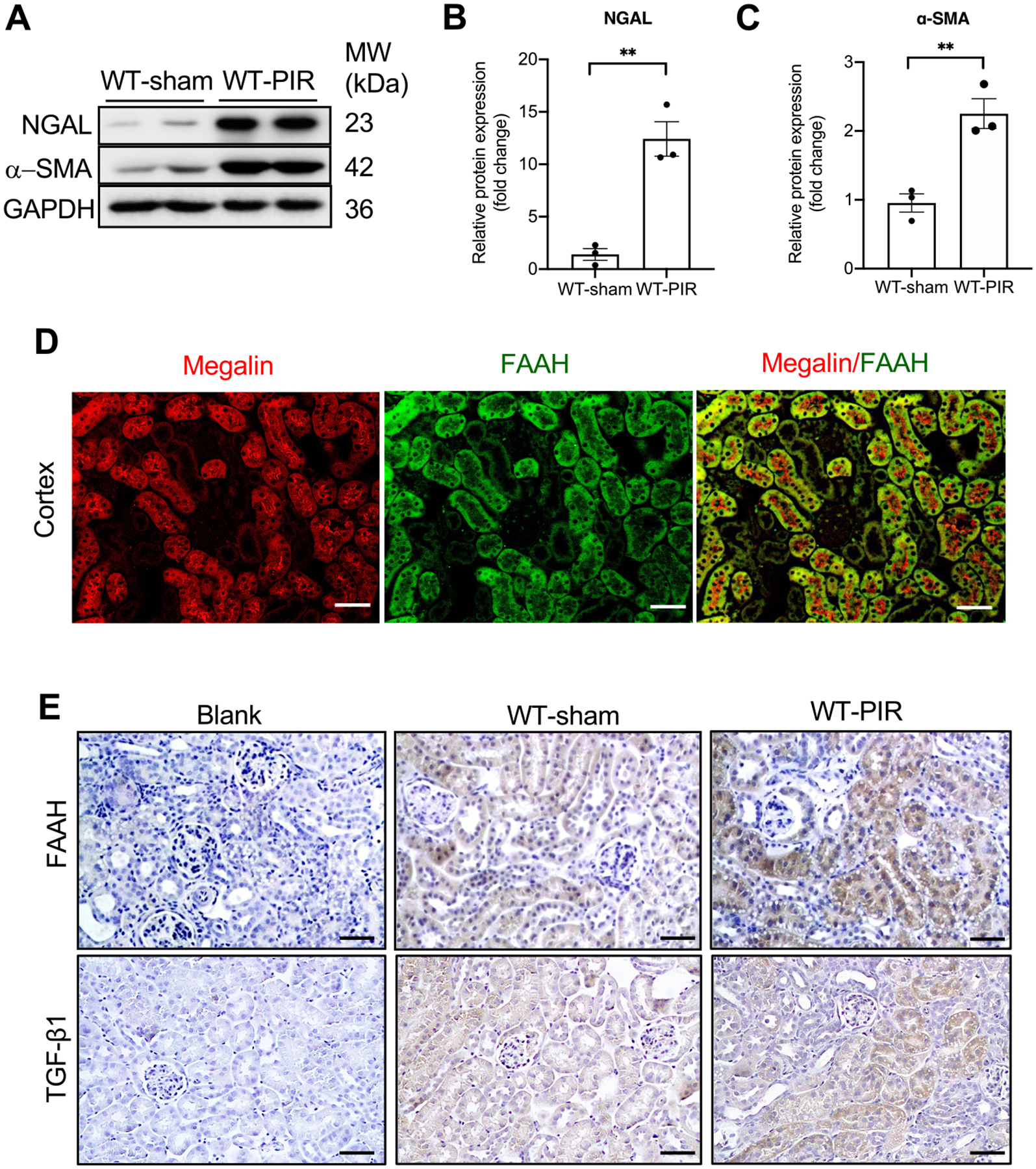
PIR increases levels of NGAL, α-SMA and FAAH in mouse kidneys. (A-C) Representative immunoblots and quantitative data for NGAL and α-SMA by Western blot analysis (n = 3). All values were normalized to the average of WT-sham. **P <0.01 by Student’s t-test. (D) Immunostaining of WT kidney cortex showing megalin (red, a proximal tubule marker), FAAH (green) and the merged image of megalin and FAAH. White bar = 100 μm. (E) Representative immunohistochemical staining showing FAAH and TGF-β1 in mouse kidneys. Blank: absence of anti-FAAH or anti- TGF-β1 antibody. Black bar = 100 μm.
3.2. PIR injury stimulates FAAH activity and concomitantly decreases AEA levels
We further measured the content of AEA, the major substrate of FAAH, in the renal cortex from WT and Faah−/− mice. Firstly, the protein expression of Faah deletion was validated in mice (Fig. 2A). FAAH activity was determined by measuring levels of AEA using LC-MS/MS. As expected, Faah−/− sham mice showed significantly elevated levels of AEA compared with WT-sham (Fig. 2B, C). Surprisingly, PIR led to significant decreases of AEA, which was recovered in Faah−/− mice (Fig. 2B, C). These results demonstrate that profibrotic stimulus in kidneys leads to downregulation of AEA and this endocannabinoid imbalance could be ameliorated by inactivation of FAAH.
Fig. 2.
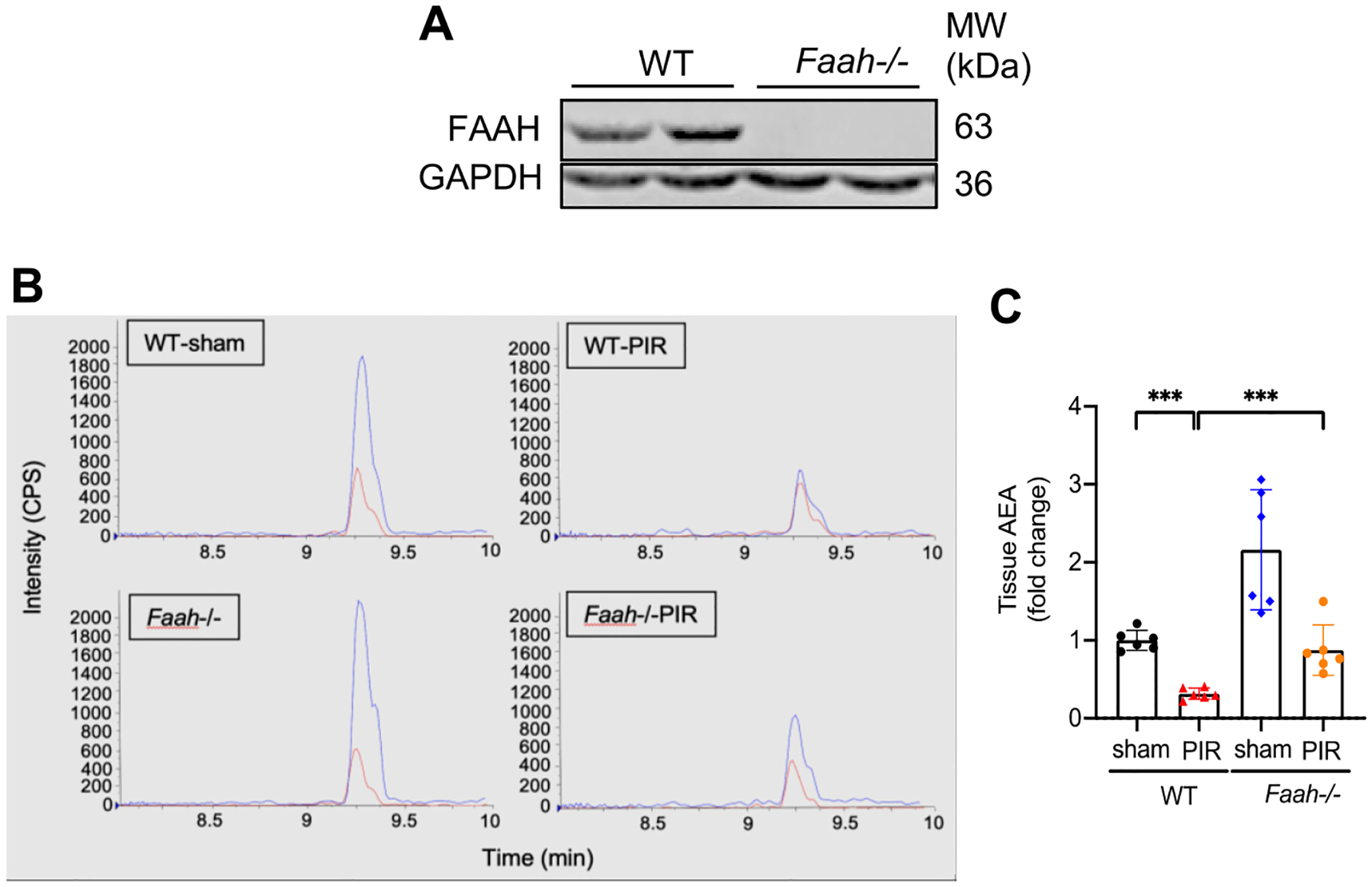
Effects of PIR and FAAH KO on levels of AEA in mouse kidneys. (A) Representative immunoblots validating the Faah deletion in mouse kidneys. (B) Representative liquid chromatography/tandem mass spectrometry (LC-MS/MS) chromatograms of tissue AEA extracted from renal cortex. AEA (blue) & AEA-d 8 (red, internal standard) from renal cortex extracts. Y-axis (intensity of AEA, CPS, counts per second) and X-axis (retention time of AEA). (C) Quantitative data (n = 6) showing the relative fold change of AEA. All values were normalized to tissue weight of mouse cortex. *** P < 0.001 by two-way ANOVA Tukey’s multiple comparisons test.
3.3. Genetic deletion of FAAH improves kidney function and renal tubulointerstitial fibrosis following post-ischemia-reperfusion renal injury
To study in vivo contribution of FAAH to the pathogenesis of AKI to CKD, we compared renal functional markers including plasma creatinine and blood urea nitrogen (BUN), levels of profibrotic proteins and histological assessments between WT and Faah−/− mice 13 days after PIR. Faah−/− PIR mice showed reduced plasma creatinine and blood urea nitrogen (BUN) compared with WT-PIR mice, whose levels remained high (Fig. 3A, B). Moreover, Faah−/− PIR mice exhibited pronounced reductions in expression of the kidney injury marker NGAL and profibrogenic marker α-SMA compared with WT-PIR mice (Fig. 3C, D). Histological evaluation by PAS staining showed that WT-PIR mice exhibited significant tubular damages in proximal tubules as indicated by tubular atrophy, vacuolar degeneration, loss of brush border and cast formation in proximal tubules compared with WT-sham mice (Fig. 3E, F). Faah deletion protected kidneys from PIR-induced proximal tubular damages, which occurred in WT mice (Fig. 3E, F). The extent of renal fibrosis was measured by picrosirius red and Masson’s trichrome staining, demonstrating significant collagen deposition in tubulointerstitial area of WT-PIR mice, but reduced fibrotic area in Faah−/− PIR cortex (Fig. 3E, G, H). Taken together, these results indicate that FAAH is essential for mediating PIR-induced renal fibrogenesis, and that genetic deletion of FAAH improves kidney function and renal fibrogenesis following PIR.
Fig. 3.
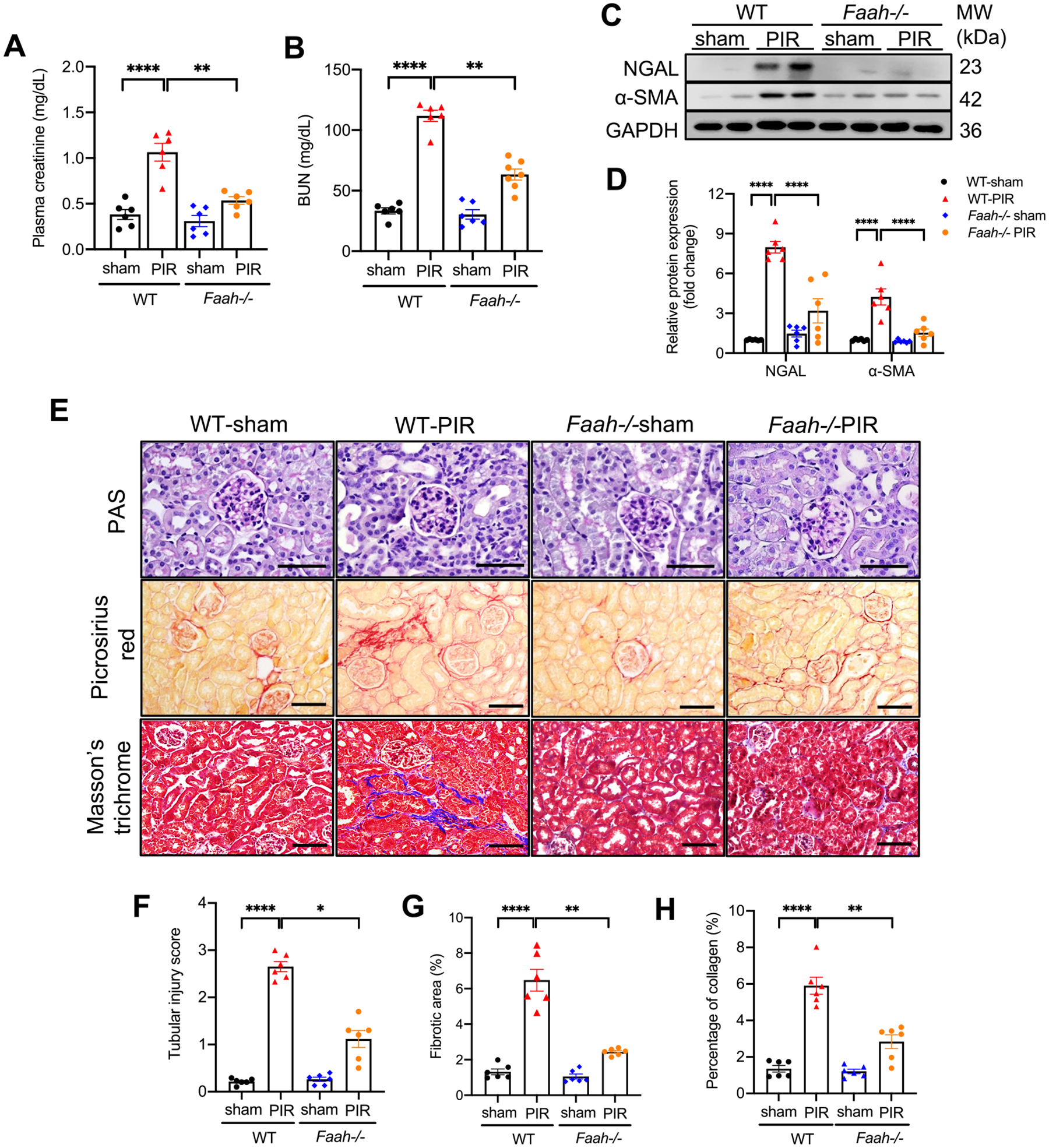
Effect of FAAH KO on renal dysfunction, kidney injury and profibrogenic markers in response to PIR. (A-B) Plasma creatinine and BUN (mg/dL) in WT and Faah−/− mice. (C–D) Gel documents and quantitative data (n = 6) showing levels of NGAL and α-SMA in WT and Faah−/− mice. All values were normalized to WT-sham. (E-H) Representative photomicrographs and quantitative data of Periodic-Acid Schiff (PAS) staining, picrosirius red staining and Masson’s trichrome. (F) Semi-quantification of tubular injury scores of PAS staining. (G) Percentage of positive picrosirius red staining area. (H) Percentage of positive Masson’s trichrome staining area. *P < 0.05, **P < 0.01, **** P < 0.0001 by two-way ANOVA Tukey’s multiple comparisons test. At least 10 fields for each sample were evaluated for all results. Black bar = 100 μm.
3.4. Genetic knockout of Faah also attenuates inflammatory responses after post-ischemia-reperfusion renal injury
Infiltration of inflammatory cells plays important roles in the initiation and pathogenesis of AKI and progression to CKD [48]. A macrophage marker CD68+ and a T-cell marker CD43+ were used to assess immune cell infiltration. Immunohistochemistry showed significant lower renal infiltrations of CD68+ and CD43+ cells (Fig. 4A–C) in Faah−/− mice than WT mice in response to PIR injury. In addition, PIR resulted in a remarkably increased proinflammatory marker, pro-IL1β in WT mice but much lower expression in Faah−/− mice (Fig. 4D, E), suggesting that inactivation of FAAH could also reduce inflammation triggered by PIR injury.
Fig. 4.
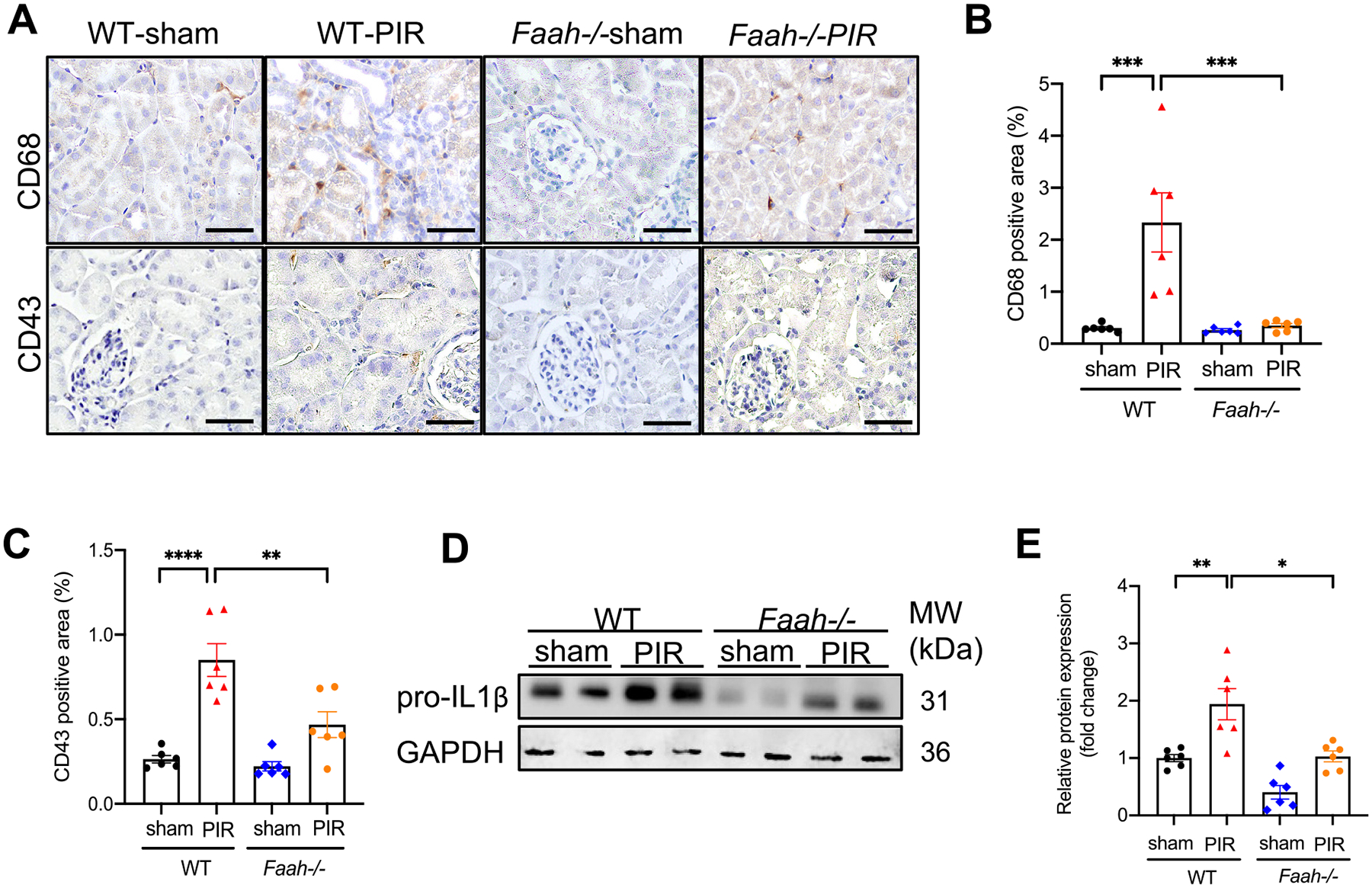
Effect of FAAH KO on PIR-induced inflammatory responses in kidneys. (A) Representative immunohistochemical staining showing CD68 (a macrophage marker) and CD43 (a T-cell marker) in kidney tissues. (B) Percentage of positive area of CD68 and (C) CD43. At least 10 fields for each sample were evaluated for all results. Black bar = 100 μm. (D-E) Representative immunoblots and quantitative data (n = 6) showing the level of proinflammatory marker, pro-IL1β in WT and Faah−/− mice. All values were normalized to WT-sham. *P < 0.05, **P < 0.01, *** P < 0.001, **** P < 0.0001 by two-way ANOVA Tukey’s multiple comparisons test.
3.5. TGF-β1 enhances the expression and activity of FAAH in cultured proximal tubular epithelial cells
Based on the PIR-induced FAAH upregulation in renal proximal tubular epithelial cells (PTECs), we employed a human PTEC cell line (HK-2) to investigate the molecular mechanism underlying the protective effects of FAAH deletion against PIR. TGF-β1, the prominent mediator in CKD [49–51], caused considerably increased expression of FAAH in HK-2 cells (Fig. 5A, B) in accord with the in vivo upregulation of FAAH in proximal tubules. FAAH activity of HK-2 cells was then inhibited by a FAAH inhibitor (PF-04457845, PF, 1 μM) with or without TGF-β1. As expected, LC-MS/MS assay showed that PF increased levels of AEA in HK-2 cells compared to control cells (Fig. 5C, D). Notably, TGF-β1 led to a significant reduction in cellular AEA and that this effect was counteracted by PF (Fig. 5C, D). These data strongly indicate that TGF-β1 is an important stimulator that facilitates AEA metabolism by FAAH, leading to an endocannabinoid dysregulation.
Fig. 5.
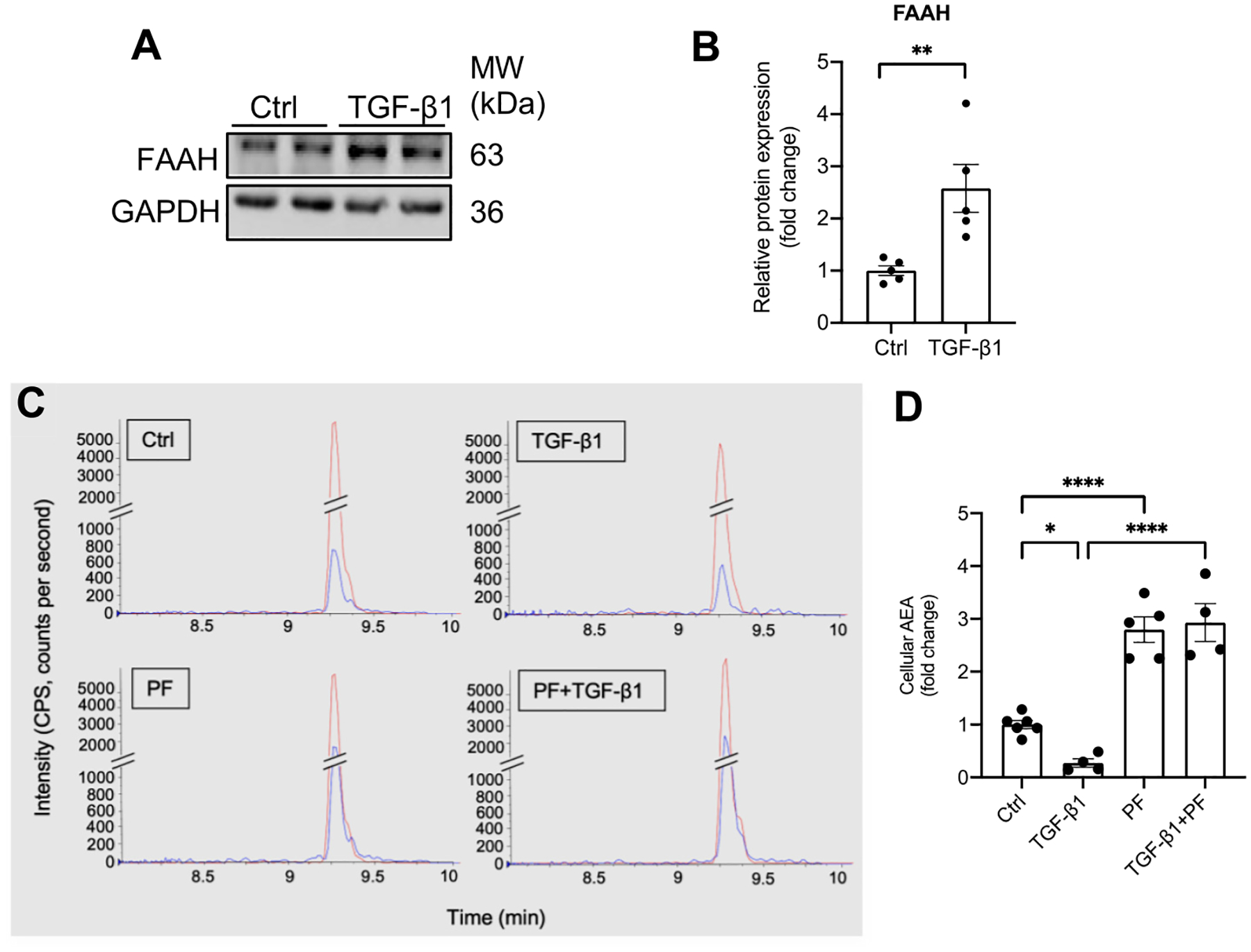
Effect of TGF-β1 on FAAH expression and AEA levels in cultured renal proximal tubular epithelial cells (HK-2 cells). (A-B) Representative immunoblots and quantitative data showing FAAH levels in HK-2 cells by Western blot analysis. Values were normalized to Ctrl (mean ± SEM). **P < 0.01 versus Ctrl group by Student’s t-test. (C) Representative LC-MS/MS chromatograms of cellular AEA extracted from HK-2 cells with different treatments, AEA (blue) and AEA–d 8 (red, internal standard). Y-axis (intensity of AEA, CPS, counts per second) and X-axis (retention time of AEA). (D) Quantitative data showing the relative fold change of AEA. All values were factored to total protein (n = 4–6) and normalized to Ctrl group and shown as mean ± SEM. *P < 0.05, **** P < 0.0001 by two-way ANOVA Tukey’s multiple comparisons test. Ctrl: control. PF: PF-04457845.
3.6. Pharmacological or genetic inhibition of FAAH abrogates TGF-β1–induced expression of profibrogenic proteins in renal PTECs
As the data presented above demonstrate the importance of TGF-β1 in FAAH-AEA regulation, we next sought to clarify the underlying mechanism of the protective effects by FAAH inhibition on in vitro TGF-β1–induced profibrotic effects. Western blot analysis showed that TGF-β1 increased expression of the proinflammatory marker pro-IL1β (Fig. 6A, B), profibrogenic markers α-SMA and collagen I/III (Fig. 6C, D), while PF abolished these effects in HK-2 cells (Fig. 6A–D). PF also attenuated TGF-β1-induced loss of epithelial marker E-cadherin (Fig. 6E, F). Immunocytochemistry revealed that TGF-β1 caused a significant decrease in ZO-1 expression by 40 % and disarrangement of proximal tubular epithelial cells, which were significantly recovered by PF (Fig. 6G, H). In addition, TGF-β1 induced morphology changes from an epithelial phenotype to a more profibrogenic phenotype, a spindle shape in HK-2 cells, whereas cells treated with PF maintained a classical cobblestone morphology even in the presence of TGFβ1 (Fig. 6I). These data suggest that pharmacological inactivation of FAAH effectively abrogates TGF-β1-induced profibrogenic effects in renal proximal epithelial cells.
Fig. 6.
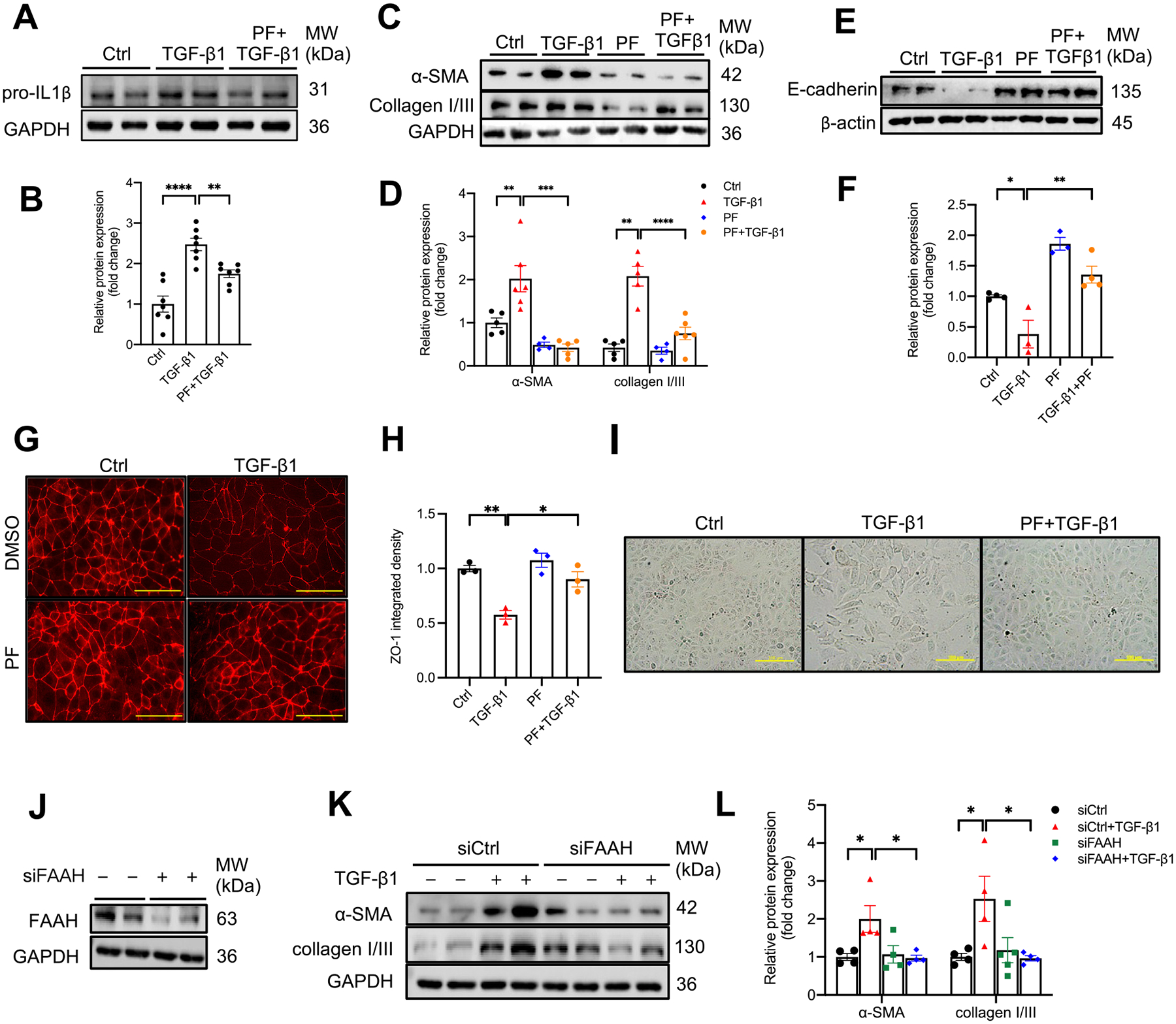
Effect of FAAH inactivation by a selective FAAH inhibitor and siRNA on TGF-β1-induced profibrogenic effects in HK-2 cells. (A-D) Representative immunoblots and summarized data showing pro-IL1β (A & B), α-SMA, collagen I/III (C & D) and E-cadherin expressions (E & F) in TGF-β1-treated HK-2 cells with/without a selective FAAH inhibitor (PF, PF-04457845, 1 μM). (G) Representative immunocytochemistry and (H) summarized integrated intensity of ZO-1 (n = 3). Yellow bar = 75 μm. (I) Representative micrographs of HK-2 cells showing morphological change under different treatments. Yellow bar = 100 μm. (J) Immunoblot verification of siRNA-mediated FAAH knockdown in HK-2 cells. (K-L) Representative immunoblots and summarized data showing the effects of FAAH knockdown on TGF-β1-induced α-SMA and collagen I/III expressions. All values were normalized to Ctrl or siCtrl group and shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 by two-way ANOVA Tukey’s multiple comparisons test. Ctrl: control, PF: PF-04457845.
We used siRNA as a complementary approach to inhibit FAAH (Fig. 6J) and showed that silencing of FAAH significantly mitigated TGFβ1-induced upregulation of profibrogenic markers α-SMA and collagen I/III (Fig. 6K, L). These findings demonstrate that genetic knockdown of FAAH elicits comparable anti-profibrogenic effects as pharmacological inactivation of FAAH.
3.7. FAAH inhibition also abrogates TGF-β1–induced profibrogenic responses in mouse primary PTECs
We next isolated mouse primary proximal tubular epithelial cells (PPTECs) to validate the findings in immortalized HK-2 cells. Immunostaining for megalin, a PTEC marker demonstrated the identification of PPTECs (Fig. 7A, right). M1 cells (mouse collecting duct cells) were used as negative control (Fig. 7A, left). Similar to HK-2 cells, PPTECs exposed to TGF-β1 had significantly increased expression of FAAH (Fig. 7B, C). TGF-β1 also increased expressions of pro-IL1β, α-SMA (Fig. 7D, E), whereas PF inhibited these effects. Immunocytochemistry showed that compared with control cells, TGF-β1 upregulated α-SMA (green) expression in megalin-positive stained PPTECs (red), which also presented a profibrogenic phenotype (Fig. 7F). These outcomes were reversed by PF treatment.
Fig. 7.
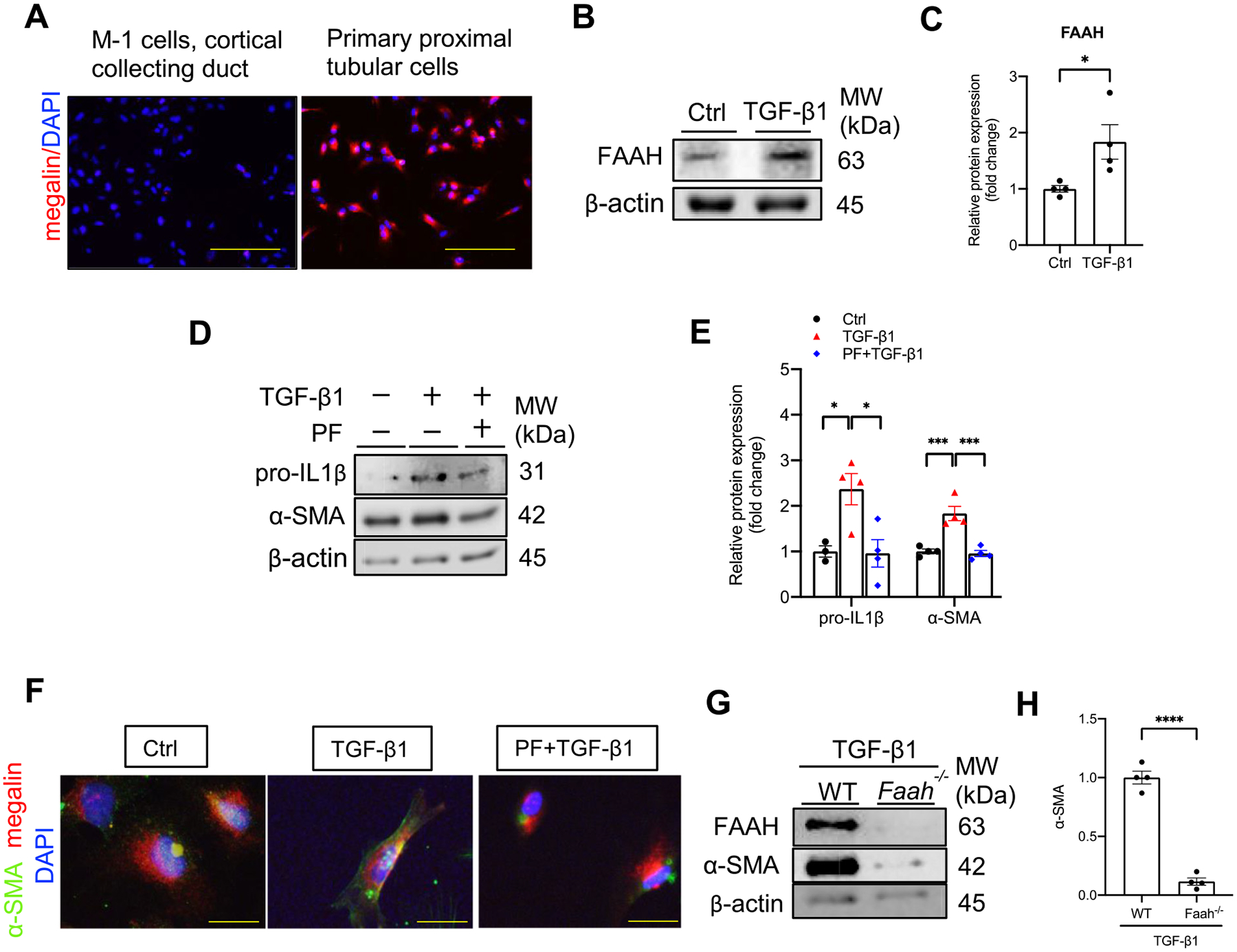
Effect of FAAH inactivation on TGF-β1-induced profibrogenic effects in primary cultured mouse proximal tubular epithelial cells (PPTECs). (A) Immunocytochemistry of mouse primary culture for proximal tubular maker megalin (red) with DAPI nuclear staining (blue). (Left) negative control: M-1 cells, mouse cortical collecting duct. (Right) primary mouse renal proximal tubular cells. Yellow bar =100 μm. (BE–) Representative immunoblots and quantitative data showing FAAH expression (B–C), the effects of PF (FAAH inhibitor, PF-04457845, 1 μM) on proIL-1β and α-SMA expressions (D-E) in the presence of TGF-β1 (5 ng/ml, 48 h). All values were normalized to Ctrl average and shown as mean ± SEM. *P < 0.05, ***P < 0.001 by one-way ANOVA Bonferroni’s multiple comparisons test. (F) Immunocytochemistry showing staining of α-SMA (green) and megalin (red) with DAPI nuclear staining (blue) in PPTECs under different treatments. Yellow bar = 20 μm. (G-H) Representative immunoblots and quantitative data showing α-SMA expression in isolated primary cultures from WT and Faah−/− mice in the presence of TGF-β1. All values were normalized to WT-TGF-β1 average and shown as mean ± SEM. **** P < 0.0001 by Student’s t-test. Ctrl: control, PF: PF-04457845.
We further isolated PPTECs from Faah−/− mice and confirmed their effect against TGF-β1. Western blot verified the genetic knockout of Faah and showed that Faah−/− PPTECs had a significant reduction in α-SMA compared with WT PPTECs after exposure to TGF-β1 (Fig. 7G, H). These data suggest that the PF exerts comparable protections in both PPTECs and HK-2 cell line in response to TGF-β1. In addition, PPTECs isolated from Faah−/− mice further validated the protection against TGF-β1-induced profibrogenesis.
3.8. The protective effects of FAAH inhibition on TGF-β1 induced-profibrogenesis are independent of CB1 and CB2 receptors in PTECs
Because FAAH inhibition increases its substrate AEA, which would possibly activate CB1 and CB2, we next investigated whether cannabinoid receptors mediate the protective effects of FAAH inhibition against TGF-β1 induced-profibrogenesis. Although CB1 receptor has been shown to mediate renal damages [16,18,52,53], its role in the current experimental setting needs to be validated. Immunoblots revealed that CB1 antagonist (Rimonabant, 1 μM) did not revert the protection of PF against TGF-β1-induced α-SMA and collagen I/III (Fig. 8A–C), indicating that the protective effects of PF in PTECs were not via CB1 receptor. Because of controversies on the role of CB2 receptor in kidney diseases, we used both CB2 antagonist (SR, SR144528, 1 μM) and CB2 siRNA to test the role of CB2 receptor in FAAH activation. Immunoblots showed that upon TGF-β1 stimulation, co-treatment of PF and the SR had no significant changes in α-SMA and collagen I/III compared with PF treated-cells (Fig. 8D, E). In addition to the pharmacological method, simultaneous genetic knockdown of both CB2 and FAAH in HK-2 cells was conducted. Silencing of CB2 (Fig. 8F) did not reverse the anti-profibrogenic effects of FAAH siRNA (Fig. 8G, H), which was consistent with the lack of an effect by the CB2 receptor antagonist (Fig. 8D, E). These data indicate that the protective effects of FAAH inhibition are independent of the CB1 and CB2 receptors in HK-2 cells.
Fig. 8.
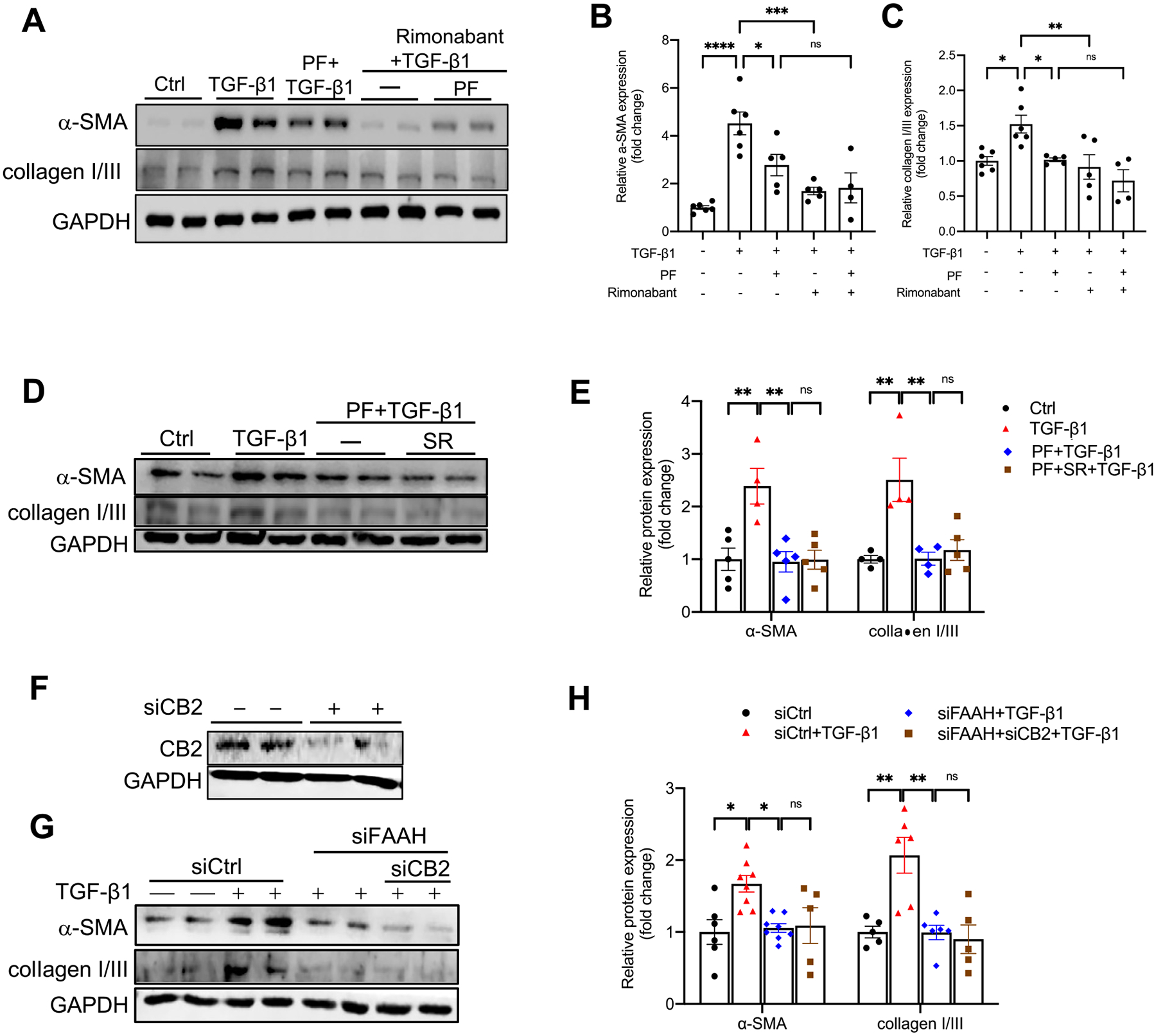
Effect of CB1 or CB2 receptor inactivation on FAAH inhibition-mediated repression of TGF-β1-induced α-SMA and collagen I/III expression in HK-2 cells. (A-E) Representative immunoblots and quantitative data showing α-SMA and collagen I/III expression in HK-2 cells with different combinations of treatments with TGF-β1, FAAH inhibitor (PF, PF-04457845, 1 μM), CB1 receptor antagonist (Rimonabant, 1 μM) (A-C) or CB2 receptor antagonist (SR; SR144528, 1 μM) (D-E). (F) Immunoblots verification of siRNA-mediated CB2 knockdown in HK-2 cells. (G-H) Representative immunoblots and quantitative data showing the effects of simultaneous siRNA silencing of CB2 and/or FAAH on α-SMA and collagen I/III expressions in TGF-β1-treated HK-2 cells. All values were normalized to Ctrl or siCtrl and shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 by one-way ANOVA Bonferroni’s multiple comparisons test. Ctrl: control; siCtrl: control siRNA. ns: not significant.
3.9. The AEA-COX-2 pathway mediates the anti-profibrogenic effects of FAAH inactivation on TGF-β1-treated HK-2 cells
Arachidonic acid (AA) is one of the major metabolites of AEA produced by FAAH. To ascertain whether potentially reduced AA contributed to the protection by FAAH inhibition, we tested if supplement of exogenous AA would eliminate the protection by FAAH inactivation. As seen in Fig. 9A–B, HK-2 cells treated with exogenous AA (10 μM) did not reverse the protective outcome of PF on TGF-β1-induced α-SMA expression (Fig. 9A, B). Conversely, the other leg of AEA metabolism (i.e., the AEA-COX-2-pathway) may mediate the protective effects of FAAH inhibition. We discovered that the substrate-selective COX-2 inhibitor (LM-4131, 30 nM), which inhibits AEA-COX-2 but not AA-COX-2 pathway [37,38], profoundly diminished the anti-profibrogenic effects of PF under TGF-β1 stimulation (Fig. 9C, D). Specifically, LM-4131 annihilated the attenuation of TGF-β1-induced α-SMA and collagen I/III expression by PF. Furthermore, prostamide E2, the major metabolite of AEA-COX-2, mitigated TGF-β1-induced α-SMA expression (Fig. 9E, F), which mimicked the effect of FAAH inhibition. Overall, these results suggest that the protective effects of FAAH inhibition on TGF-β1-induced profibrogenic responses are not mediated by reduction of AA levels and can be realized through enhanced AEA-COX-2 pathway in PTECs.
Fig. 9.
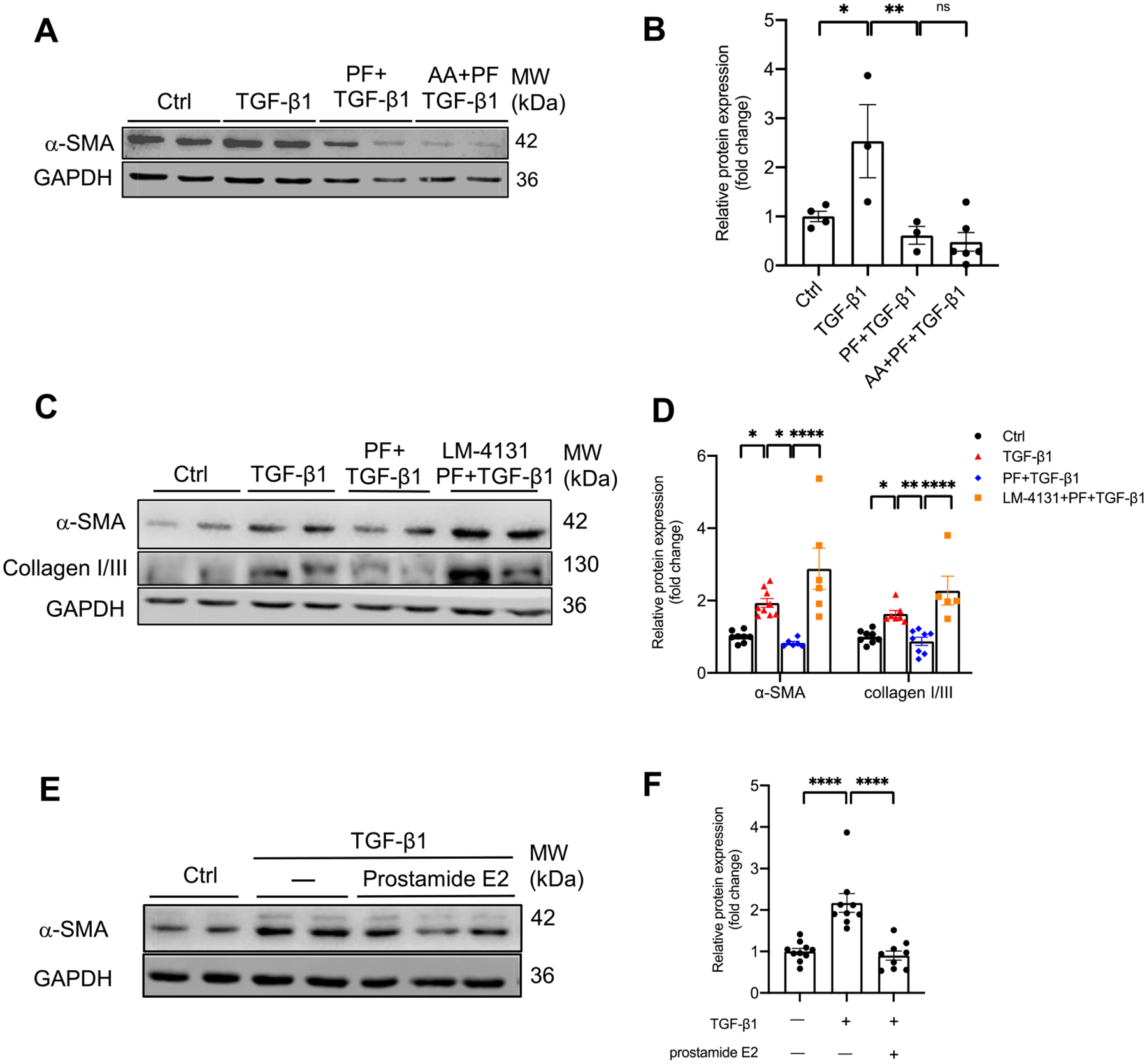
Effects of arachidonic acid (AA) or AEA-COX-2 substrate-selective inhibitor on protective effects of FAAH inactivation on TGF-β1 induced-fibrogenesis in HK-2 cells. (A-B) Representative immunoblots and quantitative data of α-SMA in HK-2 cells with different combinations of treatments with TGF-β1, arachidonic acid (AA, 10 μM) and FAAH inhibitor PF-04457845 (PF, 1 μM). (C–D) Representative immunoblots and quantitative data of α-SMA and collagen I/III expressions in HK-2 cells with different combinations of treatments with TGF-β1, AEA-COX-2 pathway substrate-selective inhibitor (LM-4131, 30 nM), and PF. (E-F) Representative immunoblots and quantitative data of α-SMA expression in HK-2 cells with prostamide E2 (100nM) and TGF-β1. All values were normalized to Ctrl group and shown as mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001 by one-way ANOVA Bonferroni’s multiple comparisons test. Ctrl: control. ns: not significant.
4. Discussion
The present study demonstrates that inactivating FAAH, the rate-limiting enzyme for the degradation of AEA [22], offers protection from PIR-induced renal fibrogenesis and mitigates TGF-β1-induced profibrogenic responses in renal proximal tubular epithelial cells (PTECs). These protective effects are associated with recovery of PIR-induced reductions in AEA levels but not due to either CB receptors activation or decreased metabolites of AEA caused by FAAH inhibition. Instead, the results of the present study suggest that the mechanism underlying this protection may result from increased metabolites of AEA through the AEA-COX-2 pathway when FAAH is inactivated. These results reveal a novel mechanism contributing to the PIR-induced renal fibrogenesis.
The present study used a clinically relevant mouse ischemic AKI to CKD model, PIR, to test the role of FAAH in renal fibrogenesis. Based on Human Protein Atlas database [54] and one report [55], human kidney samples show the highest expression levels of FAAH in proximal tubules but lower levels of FAAH in other segments, which are consistent with that in mouse kidneys in our findings (Fig. 1D). Furthermore, PIR-induced expression of FAAH in proximal tubules (Fig. 1E) was accompanied with notable decreased AEA, suggesting that FAAH may participate in regulating the pathophysiology of proximal tubules during the transition of AKI to CKD. It is worth noting that literature reports different changes in AEA and/or 2-AG in different kidney disease models as well as in acute and chronic phases of kidney injury. For instance, AEA was not altered in 24 h of the renal post-ischemia reperfusion injury (IRI)-induced AKI model [56]. On the other hand, the UUO model decreased AEA levels but increased 2-AG levels [18]. Notably, we did not observe renal protection of Faah−/− mice in the acute phase of 48 h IRI-AKI and the UUO model (data not shown). The above evidence indicates the diverse actions of endocannabinoids in different kidney diseases and that FAAH plays a key role in the progression of ischemic AKI to CKD.
The TGF-β1 signaling pathway has been shown to mediate renal tubular damage, particularly in proximal tubules [49]. Injured tubular epithelial cells per se are also described to initiate the release of TGF-β1, which further triggers the progression of acute tubular injury toward tubular atrophy and is associated with the progression to CKD [49–51]. Here we demonstrate that PIR increases the expression of TGF-β1 in proximal tubules (Fig. 1E), which is consistent with a previous report that also shows in vivo upregulation of TGF-β1 in the PIR model [29]. TGF-β1 is a key mediator that may hinder the repair process of damaged tubules after AKI and can induce profibrotic epithelial phenotype of tubules [50]. These results suggest that PIR-induced TGF-β1 expression plays a role in fibrogenesis in the in vivo model and allow for the utilization of TGF-β1 to investigate mechanisms in in vitro study.
In the present study, Faah−/− mice exerted protection against PIR-induced kidney damage, inflammation and fibrogenesis. These anti-inflammatory and anti-fibrogenic effects by enhancing the AEA in the current PIR model are supported by the literature showing antioxidant effects of FAAH inhibition and/or AEA, given that the oxidative stress critically contributes to the inflammatory processes and fibrogenesis. For example, inhibition of FAAH has been shown to activate Nrf2/antioxidant signaling [57], reduce the activity of reactive oxygen species (ROS)-generating enzymes [58], and prevent the increase of lipid peroxidation in the membranes of erythrocytes from hypertensive rats [59]. The above evidence suggests that anti-inflammatory properties of enhanced AEA by FAAH inhibition [24–26] may be closely related to the antioxidant effects.
Consistent with in vivo data, FAAH inhibition corrected the TGF-β1-downregulated AEA tone in HK-2 cells. However, there were differences in the recovery of AEA levels by FAAH inhibition in the kidneys and cultured cells. The decrease in AEA levels in vivo could result from reduced production and/or enhanced degradation in WT-PIR mice. The AEA levels were only partially recovered in Faah KO-PIR mice compared with the Faah KO-sham mice, indicating that PIR might also trigger the inhibition of AEA synthesis in addition to the enhancement of AEA degradation. Interestingly, in vitro data showed that FAAH inhibitor totally recovered the decrease in AEA levels when cells were exposed to TGF-β1 (Fig. 5C, D). These data indicate that TGF-β1 is an important regulator for FAAH expression and activity as well as the AEA metabolism in renal proximal tubules, whereas in the in vivo setting PIR injury might not only activate TGF-β1 pathway, but also trigger other profibrotic factors and inflammatory factors to reduce AEA biosynthesis. The role of reduced AEA biosynthesis in PIR injury requires further investigation in the future.
The protective effects of FAAH blockade against TGF-β1-induced profibrotic markers were impervious to the abolishment of CB1 or CB2 receptor activation in HK-2 cells in the current study (Fig. 8). Indeed, several studies have reported that CB1 and CB2 receptors contribute to renal fibrosis and CKD [16,18–20,52,53,60]. The lack of involvement of CB receptors in the effects of FAAH inhibition could be explained by the findings that pharmacological effects of AEA mediated by CB receptors are of short duration [61] and that unlike THC or stable analogues of AEA, the pharmacological effects of AEA are preserved in CB1−/− or CB1 antagonist-treated mice or CB2−/− mice [62–64]. The above information indicates that unknown receptors for AEA or AEA metabolites, may elicit effects of AEA that are distinct from the effects of CB receptors. We then explored the possible roles of AEA metabolites in TGF-β1-induced fibrogenesis.
Our observations that exogenous AA failed to reverse the protection argue AA in playing a relevant role in facilitating profibrogenic responses of PTECs by FAAH activation. Consistently, AA-derived mediators have been shown to play protective roles in kidney pathology [65]. Specifically, the eicosanoid metabolites of AA from the cPLA2-α/P450/EET pathway were shown to prevent the renal profibrotic responses caused by UUO and display positive regulatory effects on renal proximal tubules [66,67]. Interestingly, we demonstrated that a substrate-selective AEA-COX-2 inhibitor [37,38,68], which inhibits AEA-COX-2 but not AA-COX-2 pathway, blocked the protection by FAAH inhibition (Fig. 9C, D), revealing that AEA-COX-2 pathway mediates the anti-fibrogenesis effects of FAAH inhibition, which leads to an enhancement of endogenous AEA tone and augmented alternative AEA-COX-2 metabolites. Indeed, several reports have demonstrated the biological effects of AEA-COX-2 pathway [27,28,39,69]. For example, AEA-COX-2 pathway and its major metabolite, prostamide E2 are involved in mitigating renal-associated hypertension [27,28] and homocysteine-induced podocyte injury [39]. Therefore, it is likely that transient high intracellular AEA concentrations might be sufficient to stimulate AEA-COX-2 activity when FAAH is inactivated and that active metabolites produced from the AEA-COX-2 consequently exert physiological actions [70,71].
While our findings suggest that the renal protection of FAAH inhibition is through AEA-COX-2 mediated metabolites, we cannot rule out the possibility that other mechanisms might be involved. Exogenous AEA and its lipoxygenases (LOX) metabolites have been found to activate transient receptor potential vanilloid type I channels (TRPV1) [72]. However, we speculate that TRPV1 may not be involved in the current study given that much higher concentrations of AEA are needed to activate TRPV1 [72] and that COX-2 metabolites of AEA-prostamides have very low potency for TRPV1 receptor [73]. It has also been suggested that prostamides have their unique receptors called prostamide receptors which have different actions from those prostanoid receptors for prostaglandins [70]. It is, therefore, important to examine the effects of the prostamide receptors when FAAH is inactivated in models of renal fibrogenesis in future studies.
Our findings suggest that FAAH is a downstream effector of TGF-β1 but not a feedback regulator of TGF-β1, because the TGF-β1-induced FAAH levels can lead to the downregulation of AEA, a substrate of FAAH, and thereafter the deficiency of AEA-COX-2-mediated anti-inflammatory and antiprofibrogenic actions. AEA and its metabolites have been reported to inhibit nuclear factor-kappaB (NF-kB) activation [74,75]. Moreover, AEA can influence IL-1β synthesis and IL-1 system gene expressions during lipopolysaccharide (LPS)-induced inflammation [76]. The above studies indicate that inhibitions of signaling pathways downstream of TGF-β1 are the possible mechanisms underlying the protection of increased AEA and its metabolites in FAAH KO mice against PIR injury.
In summary, the present study demonstrates that FAAH inactivation, which recovers the reduced levels of AEA, protects against PIR injury or TGF-β1–induced profibrogenic effects through the AEA-COX-2 metabolism pathway. The key findings of the present study include the following: (i) FAAH is a key metabolizing enzyme that participates in the pathogenesis of AKI to CKD; (ii) Decreases in endogenous AEA induced by PIR and TGF-β1 plays an important role in renal fibrogenesis, and (iii) inhibition of FAAH offsets the dysregulation of AEA tone and prevents renal fibrogenesis via the AEA-COX-2 pathway, which represents a novel protective pathway against fibrogenesis in renal cells.
Supplementary Material
Acknowledgments
National Institutes of Health (HL145163, DK107991, DK102539, P30DA033934).
Footnotes
CRediT authorship contribution statement
Chaoling Chen: Conceptualization, Investigation, Methodology, Validation, Data Curation, Writing - Original Draft, Writing - Review & Editing.
Weili Wang: Investigation, Validation, Methodology.
Justin L. Poklis: Investigation, Data Curation, Methodology, Writing - Review & Editing.
Aron H. Lichtman: Conceptualization, Review & Editing, Supervision, Funding acquisition.
Joseph K. Ritter: Conceptualization, Review & Editing, Supervision.
Gaizun Hu: Methodology, Investigation.
Dengpiao Xie: Methodology, Investigation.
Ningjun Li: Conceptualization, Writing-Review & Editing, Supervision, Project administration, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbadis.2022.166456.
References
- [1].Guzzi F, Cirillo L, Roperto RM, Romagnani P, Lazzeri E, Molecular mechanisms of the acute kidney injury to chronic kidney disease transition: an updated view, Int. J. Mol. Sci 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kurzhagen JT, Dellepiane S, Cantaluppi V, Rabb H, AKI: an increasingly recognized risk factor for CKD development and progression, J. Nephrol (2020), 10.1007/s40620-020-00793-2. [DOI] [PubMed] [Google Scholar]
- [3].Selewski DT, Hyatt DM, Bennett KM, Charlton JR, Is acute kidney injury a harbinger for chronic kidney disease? Curr. Opin. Pediatr 30 (2018) 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tian YC, Fraser D, Attisano L, Phillips AO, TGF-beta1-mediated alterations of renal proximal tubular epithelial cell phenotype, Am J Physiol Renal Physiol 285 (2003) F130–F142. [DOI] [PubMed] [Google Scholar]
- [5].Gewin LS, Renal fibrosis: primacy of the proximal tubule, Matrix Biol. 68–69 (2018) 248–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Qi R, Yang C, Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury, Cell Death Dis. 9 (2018) 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lovisa S, Zeisberg M, Kalluri R, Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis, Trends Endocrinol. Metab 27 (2016) 681–695. [DOI] [PubMed] [Google Scholar]
- [8].Liu Y, Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention, J Am Soc Nephrol 15 (2004) 1–12. [DOI] [PubMed] [Google Scholar]
- [9].Burns KD, Interleukin-1beta as a mediator of tubulointerstitial fibrosis, Kidney Int. 62 (2002) 346–347. [DOI] [PubMed] [Google Scholar]
- [10].Jones LK, et al. , IL-1RI deficiency ameliorates early experimental renal interstitial fibrosis, Nephrol. Dial. Transplant 24 (2009) 3024–3032. [DOI] [PubMed] [Google Scholar]
- [11].Fragiadaki M, Mason RM, Epithelial-mesenchymal transition in renal fibrosis - evidence for and against, Int. J. Exp. Pathol 92 (2011) 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rastaldi MP, et al. , Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies, Kidney Int. 62 (2002) 137–146. [DOI] [PubMed] [Google Scholar]
- [13].Yang J, Liu Y, Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis, Am. J. Pathol 159 (2001) 1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fan JM, et al. , Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro, Kidney Int. 56 (1999) 1455–1467. [DOI] [PubMed] [Google Scholar]
- [15].Meng XM, Tang PM, Li J, Lan HY, TGF-beta/Smad signaling in renal fibrosis, Front. Physiol 6 (2015) 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Udi S, et al. , Proximal tubular Cannabinoid-1 receptor regulates obesity-induced CKD, J Am Soc Nephrol 28 (2017) 3518–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Francois H, Lecru L, The role of cannabinoid receptors in renal diseases, Curr. Med. Chem 25 (2018) 793–801. [DOI] [PubMed] [Google Scholar]
- [18].Lecru L, et al. , Cannabinoid receptor 1 is a major mediator of renal fibrosis, Kidney Int. 88 (2015) 72–84. [DOI] [PubMed] [Google Scholar]
- [19].Zhou L, et al. , Targeted inhibition of the type 2 cannabinoid receptor is a novel approach to reduce renal fibrosis, Kidney Int. 94 (2018) 756–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou S, et al. , Cannabinoid receptor type 2 promotes kidney fibrosis through orchestrating beta-catenin signaling, Kidney Int. 99 (2021) 364–381. [DOI] [PubMed] [Google Scholar]
- [21].Barutta F, et al. , Dual therapy targeting the endocannabinoid system prevents experimental diabetic nephropathy, Nephrol. Dial. Transplant 32 (2017) 1655–1665. [DOI] [PubMed] [Google Scholar]
- [22].Cravatt BF, et al. , Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 9371–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tripathi RKP, A perspective review on fatty acid amide hydrolase (FAAH) inhibitors as potential therapeutic agents, Eur. J. Med. Chem 188 (2020), 111953. [DOI] [PubMed] [Google Scholar]
- [24].Tham CS, Whitaker J, Luo L, Webb M, Inhibition of microglial fatty acid amide hydrolase modulates LPS stimulated release of inflammatory mediators, FEBS Lett. 581 (2007) 2899–2904. [DOI] [PubMed] [Google Scholar]
- [25].Schlosburg JE, Kinsey SG, Lichtman AH, Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation, AAPS J. 11 (2009) 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].McDougall JJ, Muley MM, Philpott HT, Reid A, Krustev E, Early blockade of joint inflammation with a fatty acid amide hydrolase inhibitor decreases end-stage osteoarthritis pain and peripheral neuropathy in mice, Arthritis Res. Ther 19 (2017) 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ahmad A, et al. , Modulation of mean arterial pressure and diuresis by renomedullary infusion of a selective inhibitor of fatty acid amide hydrolase, Am J Physiol Renal Physiol 315 (2018) F967–F976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ritter JK, et al. , Production and actions of the anandamide metabolite prostamide E2 in the renal medulla, J. Pharmacol. Exp. Ther 342 (2012) 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li L, et al. , Fibrillin-1-enriched microenvironment drives endothelial injury and vascular rarefaction in chronic kidney disease, Sci. Adv 7 (2021), eabc7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Skrypnyk NI, Harris RC, de Caestecker MP, Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice, J. Vis. Exp (2013), 10.3791/50495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xie D, et al. , Kidney-targeted delivery of prolyl hydroxylase domain protein 2 small interfering RNA with nanoparticles alleviated renal Ischemia/Reperfusion injury, J. Pharmacol. Exp. Ther 378 (2021) 235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ryan MJ, et al. , HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney, Kidney Int. 45 (1994) 48–57. [DOI] [PubMed] [Google Scholar]
- [33].Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV, Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury, Nat. Med 16 (2010) 535–543, 531p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ahn K, et al. , Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain, J. Pharmacol. Exp. Ther 338 (2011) 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tang M, et al. , Celastrol alleviates renal fibrosis by upregulating cannabinoid receptor 2 expression, Cell Death Dis. 9 (2018) 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Brash AR, Arachidonic acid as a bioactive molecule, J. Clin. Invest 107 (2001) 1339–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hermanson DJ, Gamble-George JC, Marnett LJ, Patel S, Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation, Trends Pharmacol. Sci 35 (2014) 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hermanson DJ, et al. , Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation, Nat. Neurosci 16 (2013) 1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li G, et al. , Protective action of anandamide and its COX-2 metabolite against l-homocysteine-induced NLRP3 inflammasome activation and injury in podocytes, J. Pharmacol. Exp. Ther 358 (2016) 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Simic P, et al. , SIRT1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis, Cell Rep. 3 (2013) 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Terryn S, et al. , A primary culture of mouse proximal tubular cells, established on collagen-coated membranes, Am J Physiol Renal Physiol 293 (2007) F476–F485. [DOI] [PubMed] [Google Scholar]
- [42].Kamiyama M, Garner MK, Farragut KM, Kobori H, The establishment of a primary culture system of proximal tubule segments using specific markers from normal mouse kidneys, Int. J. Mol. Sci 13 (2012) 5098–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chung SD, Alavi N, Livingston D, Hiller S, Taub M, Characterization of primary rabbit kidney cultures that express proximal tubule functions in a hormonally defined medium, J. Cell Biol 95 (1982) 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hu G, et al. , Collecting duct-specific knockout of sphingosine-1-phosphate receptor 1 aggravates DOCA-salt hypertension in mice, J. Hypertens 39 (2021) 1559–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lyu B, Wang W, Ji XY, Ritter JK, Li N, Detrimental role of sphingosine kinase 1 in kidney damage in DOCA-salt hypertensive model: evidence from knockout mice, BMC Nephrol. 21 (2020) 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang Z, et al. , Silencing of hypoxia-inducible factor-1alpha gene attenuates chronic ischemic renal injury in two-kidney, one-clip rats, Am J Physiol Renal Physiol 306 (2014) F1236–F1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Boini KM, et al. , Implication of CD38 gene in podocyte epithelial-to-mesenchymal transition and glomerular sclerosis, J. Cell. Mol. Med 16 (2012) 1674–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sato Y, Yanagita M, Immune cells and inflammation in AKI to CKD progression, Am J Physiol Renal Physiol 315 (2018) F1501–F1512. [DOI] [PubMed] [Google Scholar]
- [49].Gewin LS, Transforming growth factor-beta in the acute kidney injury to chronic kidney disease transition, Nephron 143 (2019) 154–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gewin L, et al. , Deleting the TGF-beta receptor attenuates acute proximal tubule injury, J Am Soc Nephrol 23 (2012) 2001–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gentle ME, et al. , Epithelial cell TGFbeta signaling induces acute tubular injury and interstitial inflammation, J Am Soc Nephrol 24 (2013) 787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jourdan T, et al. , Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy, Proc. Natl. Acad. Sci. U. S. A 111 (2014) E5420–E5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dao M, et al. , The cannabinoid receptor 1 is involved in renal fibrosis during chronic allograft dysfunction: proof of concept, J. Cell. Mol. Med 23 (2019) 7279–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Uhlen M, et al. , Proteomics. Tissue-based map of the human proteome, Science 347 (2015) 1260419. [DOI] [PubMed] [Google Scholar]
- [55].Schlosser M, Löser H, Siegmund S, Montesinos-Rongen M, Bindila L, Lutz B, Barrett D, Sarmad S, Ortori C, Grau V, von Brandenstein M, Fries J, The endocannabinoid, anandamide, induces cannabinoid receptor-independent cell death in renal proximal tubule cells, Cell Bio 6 (2017) 33–55. [Google Scholar]
- [56].Anonymous, Increased renal 2-arachidonoylglycerol level is associated with improved renal function in a mouse model of acute kidney injury, Cannabis Cannabinoid Res. 1 (2016) 218–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Li H, et al. , Inhibition of fatty acid amide hydrolase activates Nrf2 signalling and induces heme oxygenase 1 transcription in breast cancer cells, Br. J. Pharmacol 170 (2013) 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Biernacki M, et al. , Redox system and phospholipid metabolism in the kidney of hypertensive rats after FAAH inhibitor URB597 administration, Redox Biol. 15 (2018) 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Dobrzyńska I, Szachowicz-Petelska B, Pędzińska-Betiuk A, Figaszewski ZA, Skrzydlewska E, Effects of hypertension and FAAH inhibitor treatment of rats with primary and secondary hypertension considering the physicochemical properties of erythrocytes, Toxicol. Mech. Methods 30 (2020) 297–305. [DOI] [PubMed] [Google Scholar]
- [60].Chua JT, et al. , Endocannabinoid system and the kidneys: from renal physiology to injury and disease, Cannabis Cannabinoid Res 4 (2019) 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Willoughby KA, Moore SF, Martin BR, Ellis EF, The biodisposition and metabolism of anandamide in mice, J. Pharmacol. Exp. Ther 282 (1997) 243–247. [PubMed] [Google Scholar]
- [62].Adams IB, Compton DR, Martin BR, Assessment of anandamide interaction with the cannabinoid brain receptor: SR 141716A antagonism studies in mice and autoradiographic analysis of receptor binding in rat brain, J. Pharmacol. Exp. Ther 284 (1998) 1209–1217. [PubMed] [Google Scholar]
- [63].Di Marzo V, et al. , Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: evidence for non-CB (1), non-CB(2) receptor-mediated actions of anandamide in mouse brain, J. Neurochem 75 (2000) 2434–2444. [DOI] [PubMed] [Google Scholar]
- [64].Jarai Z, et al. , Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors, Proc. Natl. Acad. Sci. U. S. A 96 (1999) 14136–14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang T, et al. , Arachidonic acid metabolism and kidney inflammation, Int. J. Mol. Sci 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Skibba M, et al. , Epoxyeicosatrienoic acid analog decreases renal fibrosis by reducing epithelial-to-mesenchymal transition, Front. Pharmacol 8 (2017) 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fan F, Roman RJ, Effect of cytochrome P450 metabolites of arachidonic acid in nephrology, J. Am. Soc. Nephrol 28 (2017) 2845–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gamble-George JC, et al. , Cyclooxygenase-2 inhibition reduces stress-induced affective pathology, elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Alhouayek M, Muccioli GG, COX-2-derived endocannabinoid metabolites as novel inflammatory mediators, Trends Pharmacol. Sci 35 (2014) 284–292. [DOI] [PubMed] [Google Scholar]
- [70].Ritter JK, Li G, Xia M, Boini K, Anandamide and its metabolites: what are their roles in the kidney? Front. Biosci. (Schol. Ed.) 8 (2016) 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chen P, et al. , Induction of cyclooxygenase-2 by anandamide in cerebral microvascular endothelium, Microvasc. Res 69 (2005) 28–35. [DOI] [PubMed] [Google Scholar]
- [72].Ross RA, Anandamide and vanilloid TRPV1 receptors, Br. J. Pharmacol 140 (2003) 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Matias I, et al. , Prostaglandin ethanolamides (Prostamides). In vitro pharmacology and metabolism, J. Pharmacol. Exp. Ther 309 (2004) 745–757. [DOI] [PubMed] [Google Scholar]
- [74].Sancho R, Calzado MA, Di Marzo V, Appendino G, Munoz E, Anandamide inhibits nuclear factor-kappaB activation through a cannabinoid receptor-independent pathway, Mol. Pharmacol 63 (2003) 429–438. [DOI] [PubMed] [Google Scholar]
- [75].Nakajima Y, et al. , Endocannabinoid, anandamide in gingival tissue regulates the periodontal inflammation through NF-kappaB pathway inhibition, FEBS Lett. 580 (2006) 613–619. [DOI] [PubMed] [Google Scholar]
- [76].Tomczyk M, Tomaszewska-Zaremba D, Bochenek J, Herman A, Herman AP, Anandamide influences interleukin-1β synthesis and IL-1 system gene expressions in the ovine hypothalamus during Endo-toxin-induced inflammation, Animals (Basel) 11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.