

Abstract
The spinal dorsal horn contains vesicular glutamate transporter-2 (VGluT2)-expressing excitatory neurons and vesicular GABA transporter (VGAT)-expressing inhibitory neurons, which normally have different roles in nociceptive transmission. Spinal glutamate NMDAR hyperactivity is a crucial mechanism of chronic neuropathic pain. However, it is unclear how NMDARs regulate primary afferent input to spinal excitatory and inhibitory neurons in neuropathic pain. Also, the functional significance of presynaptic NMDARs in neuropathic pain has not been defined explicitly. Here we showed that paclitaxel treatment or spared nerve injury (SNI) similarly increased the NMDAR-mediated mEPSC frequency and dorsal root-evoked EPSCs in VGluT2 dorsal horn neurons in male and female mice. By contrast, neither paclitaxel nor SNI had any effect on mEPSCs or evoked EPSCs in VGAT neurons. In mice with conditional Grin1 (gene encoding GluN1) KO in primary sensory neurons (Grin1-cKO), paclitaxel treatment failed to induce pain hypersensitivity. Unexpectedly, SNI still caused long-lasting pain hypersensitivity in Grin1-cKO mice. SNI increased the amplitude of puff NMDA currents in VGluT2 neurons and caused similar depolarizing shifts in GABA reversal potentials in WT and Grin1-cKO mice. Concordantly, spinal Grin1 knockdown diminished SNI-induced pain hypersensitivity. Thus, presynaptic NMDARs preferentially amplify primary afferent input to spinal excitatory neurons in neuropathic pain. Although presynaptic NMDARs are required for chemotherapy-induced pain hypersensitivity, postsynaptic NMDARs in spinal excitatory neurons play a dominant role in traumatic nerve injury-induced chronic pain. Our findings reveal the divergent synaptic connectivity and functional significance of spinal presynaptic and postsynaptic NMDARs in regulating cell type-specific nociceptive input in neuropathic pain with different etiologies.
SIGNIFICANCE STATEMENT Spinal excitatory neurons relay input from nociceptors, whereas inhibitory neurons repress spinal nociceptive transmission. Chronic nerve pain is associated with aberrant NMDAR activity in the spinal dorsal horn. This study demonstrates, for the first time, that chemotherapy and traumatic nerve injury preferentially enhance the NMDAR activity at primary afferent–excitatory neuron synapses but have no effect on primary afferent input to spinal inhibitory neurons. NMDARs in primary sensory neurons are essential for chemotherapy-induced chronic pain, whereas nerve trauma causes pain hypersensitivity predominantly via postsynaptic NMDARs in spinal excitatory neurons. Thus, presynaptic and postsynaptic NMDARs at primary afferent–excitatory neuron synapses are differentially engaged in chemotherapy- and nerve injury-induced chronic pain and could be targeted respectively for treating these painful conditions.
Keywords: α2δ-1, dorsal root ganglion, spinal cord, K-Cl cotransporter-2 (KCC2), synaptic plasticity, synaptic transmission
Introduction
Chronic neuropathic pain is often caused by peripheral nerve injury and certain cancer therapeutics. Both traumatic nerve injury and chemotherapy-induced neuropathy augment glutamate NMDAR activity in the spinal cord dorsal horn, which serves to amplify nociceptive input from primary sensory neurons (Woolf and Salter, 2000; H. Y. Zhou et al., 2011; Deng et al., 2019a). In addition to conventional postsynaptic NMDARs, presynaptic NMDARs are present in the spinal dorsal horn and are located immediately at the active zone adjacent to the vesicle release site (Liu et al., 1994). However, NMDARs expressed at primary afferent central terminals are normally in a quiescent state; they become tonically activated in opioid-induced hyperalgesia (Zhao et al., 2012; H. Y. Zhou et al., 2012) and many neuropathic pain conditions (Yan et al., 2013; Xie et al., 2016; J. Chen et al., 2018; G. F. Zhang et al., 2021). In this regard, treatment with cancer chemotherapeutic drugs, such as paclitaxel and bortezomib, mainly potentiates presynaptic NMDAR activity in the spinal cord (S. R. Chen et al., 2014c; Xie et al., 2017a, 2017b). By contrast, traumatic injury to peripheral nerves increases the activity of both presynaptic and postsynaptic NMDARs in the spinal dorsal horn (Yan et al., 2013; L. Li et al., 2016; J. Chen et al., 2018). Intrathecal injection of NMDAR antagonists effectively reverses chronic pain caused by both traumatic nerve injury and chemotherapeutic drugs (Yamamoto and Yaksh, 1992; Mao et al., 1993; Chaplan et al., 1997; Xie et al., 2016; Xie et al., 2017b). Nevertheless, the role of presynaptic or postsynaptic NMDARs in neuropathic pain cannot be defined by using NMDAR antagonists administered intrathecally. At present, the functional significance of spinal presynaptic NMDARs in the development of chronic neuropathic pain has not been determined explicitly.
The spinal dorsal horn contains heterogeneous populations of neurons that are molecularly and functionally diverse; these neurons can be broadly divided into excitatory and inhibitory neurons. In the superficial dorsal horn, most glutamatergic excitatory neurons express vesicular glutamate transporter-2 (VGluT2), whereas GABAergic/glycinergic inhibitory neurons express vesicular GABA transporters (VGATs; also known as vesicular inhibitory amino acid transporter) (Koga et al., 2017; Browne et al., 2020; S. R. Chen et al., 2022; Sullivan and Sdrulla, 2022). VGluT2 and VGAT neurons generally have distinct roles in nociceptive transmission at the spinal cord level. In this regard, spinal VGluT2 neurons mediate nociceptive transmission in chronic pain induced by tissue inflammation and nerve injury (Wang et al., 2018), whereas spinal VGAT neurons tonically inhibit nociceptive transmission (Koga et al., 2017). Traumatic nerve injury increases the prevalence of LTP in VGluT2, but not VGAT, neurons in the spinal dorsal horn (Huang et al., 2022). Also, opioid exposure selectively potentiates NMDAR-mediated nociceptive primary afferent input to VGluT2, but not VGAT, dorsal horn neurons (S. R. Chen et al., 2022). However, it is unclear which cell types in the spinal cord are engaged in processing NMDAR-mediated primary afferent input in different neuropathic pain conditions.
To address these gaps in knowledge, we conducted this study to determine how NMDAR-mediated primary afferent input is altered in VGluT2- and VGAT-expressing spinal dorsal horn neurons after treatment with paclitaxel and traumatic nerve injury, the two most commonly used animal models of neuropathic pain. We also determined the functional significance of NMDARs expressed in primary sensory neurons in the development of chronic pain in these two models. Our study showed, for the first time, that in both neuropathic pain conditions, increased NMDAR activity drives primary afferent input preferentially to spinal VGluT2 neurons. However, presynaptic and postsynaptic NMDARs at the spinal cord level are differentially involved in chronic pain induced by paclitaxel treatment and traumatic nerve injury. In addition, changes in presynaptic and postsynaptic NMDAR activity in the spinal cord are not interdependent in neuropathic pain. This new information advances our mechanistic understanding of spinal nociceptive circuits involved in different neuropathic pain conditions.
Materials and Methods
Animals
All animal protocols and experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas M. D. Anderson Cancer Center (approval #1174-RN00) and followed the Guide for the care and use of laboratory animals (National Institutes of Health). C57BL/6 mice (8-14 weeks old, sex- and age-matched) were used in this study. Data were pooled from male and female mice because we observed no sex differences in the electrophysiological and behavioral data during our study. The animals were maintained in a housing facility at 24°C on a 12 h light-dark cycle. Mice were housed at no more than 5 per cage, with free access to food and water.
VGluT2-ires-Cre knock-in mice (#028863), VGAT-ires-Cre knock-in mice (#028862), and tdTomato-floxed mice (#007909) were obtained from The Jackson Laboratory. The genetic background of these mouse strains is C57BL/6J. The VGluT2Cre/+:tdTomatoflox/flox and VGATCre/+:tdTomatoflox/flox mice were obtained by crossing male VGluT2-ires-Cre or VGAT-ires-Cre mice with female tdTomato-floxed mice, respectively (Wang et al., 2018; S. R. Chen et al., 2022). The specificity of VGluT2-tagged excitatory neurons and VGAT-tagged inhibitory neurons in the spinal dorsal horn has been validated previously (Koga et al., 2017; Wang et al., 2018; Browne et al., 2020).
Grin1flox/flox mice were purchased from The Jackson Laboratory (#005246); AvilCre/+ mice were kindly provided by Fan Wang (Massachusetts Institute of Technology), and genotyping was performed as previously described (da Silva et al., 2011). In brief, male AvilCre/+ mice were crossed with female Grin1flox/flox mice to generate male AvilCre/+:Grin1flox/+ mice and then crossed again to female Grin1flox/flox mice to obtain AvilCre/+:Grin1flox/flox mice, which are referred to as Grin1-cKO mice. To generate inducible conditional KO (cKO) of Grin1 in primary sensory neurons, female Grin1flox/flox mice were crossed in a similar way with male AvilCreERT2/+ mice (#032027, The Jackson Laboratory) to produce AvilCerERT2/+:Grin1flox/flox mice. To induce cKO of Grin1 (Grin1-icKO) in adult mice, tamoxifen (#T5648, Millipore Sigma) was dissolved at a concentration of 20 mg/ml in corn oil and intraperitoneally injected into AvilCerERT2/+:Grin1flox/flox mice (75 mg/kg per day for 5 consecutive days). At least 2 weeks were allowed for sufficient GluN1 knockdown before the final experiments (Lau et al., 2011).
Neuropathic pain models
For chemotherapy-induced neuropathic pain, paclitaxel (2 mg/kg; #00703-3213-01, TEVA Pharmaceuticals) was intraperitoneally injected into mice every 2 d, for a total of 4 injections (days 1, 3, 5, and 7; total dose, 8 mg/kg), as described previously (Polomano et al., 2001; S. R. Chen et al., 2014c). Animals were intraperitoneally injected with vehicle (Cremophor EL/ethanol, 1:1) as the vehicle group. The presence of pain hypersensitivity was confirmed 14 d after drug treatment.
Spared nerve injury (SNI) surgery was performed as previously described (Laedermann et al., 2014; Huang et al., 2022). In brief, mice were anesthetized with 2%-3% isoflurane, and an incision was made on the left lateral thigh to locate the sciatic nerve. The tibial and common peroneal nerve branches were ligated with a 6–0 silk suture and sectioned distal to the ligation sites under a surgical microscope, leaving the sural nerve intact. The sham group was subjected to the same surgical procedure without the nerve injury.
Nociceptive behavioral assessment
To assess tactile allodynia, mice were placed in individual chambers on a mesh floor and allowed to acclimate for 30 min. A series of calibrated von Frey filaments (Stoelting) was applied vertically to the plantar surface of the hindpaw with sufficient force to bend the filaments for 6 s; brisk paw withdrawal or flinching was considered a positive response. If a response occurred, the filament of the next lower force was applied. If there was no response, the filament of the next greater force was applied. The tactile stimulus that produced a 50% likelihood of withdrawal was calculated using the “up-down” method, as described previously (Chaplan et al., 1994).
To quantify mechanical nociception, we tested the withdrawal threshold in response to a noxious pressure stimulus (Randall-Selitto test). The Analgesy-Meter device (Stoelting) was pressed to trigger a motor that applied a constantly increasing force to the hindpaw. When the animal displayed pain, either by withdrawing the paw or vocalizing, the device was immediately released and the nociceptive withdrawal threshold was read on the screen (J. Zhang et al., 2018).
Thermal sensitivity was measured using a radiant heat source (IITC Life Sciences), as previously described (Y. Zhang et al., 2016). Mice were placed in a plastic cylinder on a glass plate that had been preheated to 30°C and allowed to acclimate for up to 30 min. A radiant light that generated noxious heat was focused onto the plantar surface of the hindpaw. The withdrawal latency was recorded on a timer when the hindpaw was quickly moved away from the light.
Viral vector injection
We used Cre-expressing AAV8 viral vectors to induce Grin1 cKO in the spinal cord and DRG of male and female Grin1flox/flox mice via intrathecal injection, as we performed previously (L. Li et al., 2016). Cre-expressing vectors (pAAV.CMV.HI.eGFP-Cre.WPRE.SV40; #105545-AAV8) and control vectors (pAAV.CMV.PI.eGFP.WPRE.bGH, #105530-AAV8) were purchased from Addgene. In short, Grin1flox/flox mice were anesthetized with inhalation of 2%-3% isoflurane. The viral vector solution was intrathecally injected at L4-L6 levels (10 μl, titer ≥ 1 × 1013 vg/ml) via lumbar puncture (Sun et al., 2019). Mice were allowed to recover in a warm cage before being returned to the housing facility and used for final experiments 2 weeks after recovery. The dorsal spinal cord and DRG tissues at L4-L6 levels were collected from the viral vector-injected mice after behavioral tests to confirm the GluN1 knockdown.
Spinal cord slice preparation and electrophysiological recording
Mice were anesthetized via inhalation of 3% isoflurane, and the lumbar spinal cords were quickly removed via laminectomy. The spinal cords were sectioned into 400-μm-thick transverse slices using a vibratome and immersed with sucrose-modified ACSF containing the following (in mm): 234 sucrose, 26 NaHCO3, 1.2 NaH2PO4, 3.6 KCl, 2.5 CaCl2, 1.2 MgCl2, and 25 glucose (mixed with 95% O2 and 5% CO2). The slices were immediately preincubated in the 95% O2 and 5% CO2 oxygenated Krebs solution containing the following (in mm): 117 NaCl, 25 NaHCO3, 1.2 NaH2PO4, 3.6 KCl, 2.5 CaCl2, 1.2 MgCl2, and 11 glucose at 34°C for at least 1 h before recording.
Electrophysiological recordings of spinal dorsal horn neurons were performed as previously described (Pan and Pan, 2004; S. R. Chen et al., 2014a; G. F. Zhang et al., 2021). In brief, spinal cord slices were fixed in a glass-bottomed recording chamber and continuously perfused by oxygenated Krebs solution at 34°C at a speed of 3 ml/min. The tdTomato-labeled neurons in spinal lamina II were identified using a combination of epifluorescence illumination and infrared and differential interference contrast optics on an upright microscope (S. R. Chen et al., 2022; Huang et al., 2022). Whole-cell voltage-clamp recordings were performed using 5-8 mΩ glass electrodes filled with the internal solution containing the following (in mm): 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, 0.5 Na2-GTP, 5 ATP-Mg, and 10 lidocaine N-ethyl bromide (QX314) (280-300 mOsm, pH 7.3). QX314 was used to block voltage-gated Na+ channels and suppress action potentials. EPSCs were recorded at a holding potential of –60 mV. Miniature EPSCs (mEPSCs) were recorded in the presence of 0.5 μm TTX (#HB1035, Hello Bio). Because postsynaptic NMDARs are blocked by extracellular Mg2+ at negative holding potentials, mEPSCs are mediated by AMPARs (D. P. Li et al., 2002; Pan and Pan, 2004). To evoke glutamate release from primary afferents, a tungsten bipolar electrode was used to electrically stimulate (0.6 mA, 0.5 ms, 0.1 Hz) the ipsilateral dorsal root. Monosynaptic EPSCs were identified on the basis of the constant latency and the absence of conduction failure in response to 20 Hz stimulation, as we described previously (H. Y. Zhou et al., 2010). Two EPSCs were evoked by a pair of stimuli at 50 ms intervals to calculate the paired-pulse ratio (PPR), which was the amplitude of the second synaptic response divided by the amplitude of the first synaptic response (Xie et al., 2016; Deng et al., 2019b).
In some experiments, we recorded postsynaptic NMDAR currents elicited by puff application of 100 μm NMDA (#M3262, Millipore Sigma) directly onto lamina II neurons at 150 μm distance using a positive pressure system (Toohey). The internal solution contained the following (in mm): 110 Cs2SO4, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 10 HEPES, 0.3 Na2-GTP, 2 Mg-ATP, and 10 QX314 (280-300 mOsm, pH 7.3). MgCl2 was replaced with CaCl2 in the extracellular solution to eliminate the Mg2+ block of NMDARs at negative holding potentials (S. R. Chen et al., 2014a; J. Chen et al., 2018). To record GABA reversal potential of lamina II neurons, we used the chloride-impermeable, gramicidin-based perforated patch-clamp method (H. Y. Zhou et al., 2012; S. R. Chen et al., 2014c). The internal solution contained the following (in mm): 130 potassium acetate, 15 KCl, 5 NaCl, 1 MgCl2, and 10 HEPES (280-300 mOsm, pH 7.3). Gramicidin was freshly dissolved in DMSO and then diluted to a final concentration of 50 μg/ml in the internal solution. We recorded currents induced by puff application of 100 μm GABA (#A2129, Millipore Sigma) at various holding potentials, ranging from –90 to –30 mV in 10 mV steps. Linear regression was performed for the voltage dependence of chloride-mediated currents, and the intercept with the abscissa was taken as the GABA reversal potential (H. Y. Zhou et al., 2012). All signals were filtered at 1-2 kHz and digitized at 10 kHz using a DigiData 1320A and a MultiClamp 700B amplifier (Molecular Devices). 2-Amino-5-phosphonopentanoate (AP5) was purchased from Hello Bio (#HB0252). All drugs were freshly prepared in artificial cerebrospinal fluid before the recording and delivered at their final concentrations using syringe pumps.
Western immunoblotting
The lumbar dorsal spinal cord and DRG tissues were removed from mice anesthetized with 3% isoflurane and then dissected and homogenized in ice-cold RIPA buffer (#89901, Fisher Scientific) containing cocktails of protease and phosphatase inhibitors (#78442, Thermo Scientific). The homogenate was centrifuged at 12,000 × g for 20 min at 4°C, and the supernatant was collected as total protein. The 40 μg samples were subjected to 4%-15% SDS-polyacrylamide gel (#NP0336BOX, Fisher Scientific) and transferred to PVDF membrane (#IPVH00010, Millipore Sigma). The blots were probed with rabbit anti-GluN1 (1:1000; #G8913, Millipore Sigma) or rabbit anti-GAPDH antibody (1:7000; #5174, Cell Signaling Technology). The specificity of the anti-GluN1 antibody has been validated in previous studies (Huang et al., 2020; G. F. Zhang et al., 2021). An ECL kit (#34 580, Fisher Scientific) was used to visualize the protein bands. For protein quantification, the protein band intensity of GluN1 was normalized to that of GAPDH in the same blot. The mean values of GluN1 proteins in samples from WT mice were considered to be 1.
Study design and statistical analysis
Data are mean ± SEM. The investigators conducting the experiments were blinded to the mouse genotype and treatment. The sample sizes of behavioral and electrophysiological experiments were similar to those we published previously (H. Y. Zhou et al., 2010; S. R. Chen et al., 2014a; Xie et al., 2017a; Huang et al., 2022). Only one neuron was recorded from each spinal cord slice, and at least 3 animals were used for each recording protocol. The input resistance was monitored, and the recording was abandoned if it changed >15%. The amplitude of evoked EPSCs and puff NMDA currents was quantified by averaging 6 consecutive traces using Clampfit 11 software (Molecular Devices). The frequency and amplitude of mEPSCs were analyzed using the MiniAnalysis program (Synaptosoft), and the Kolmogorov-Smirnov test was used to assess the normality of data distribution. We used two-tailed Student's t tests to compare the differences between two groups and one-way or Brown-Forsythe and Welch ANOVA to compare the data between more than two groups. Two-way ANOVA, followed by Tukey's or Dunnett's post hoc test, was used to determine the differences between more than two groups for behavioral and electrorheological data. All statistical analyses were performed using Prism 9 software (GraphPad Software). A p value <0.05 was considered statistically significant.
Results
Paclitaxel treatment increases presynaptic NMDAR activity in spinal VGluT2 dorsal horn neurons
VGluT2- and VGAT-expressing dorsal horn neurons have a distinct role in processing and integrating nociception (Koga et al., 2017; Wang et al., 2018). Both nerve ligation injury and paclitaxel treatment increase presynaptic NMDAR activity in the spinal cord (Yan et al., 2013; L. Li et al., 2016; Xie et al., 2016; Y. Chen et al., 2019). We first determined whether paclitaxel treatment-induced presynaptic NMDAR activity in the spinal cord is cell type-specific. VGluT2Cre/+:tdTomatoflox/flox and VGATCre/+:tdTomatoflox/flox mice were treated systemically with paclitaxel or vehicle, and spinal cord slices were obtained 2 weeks after treatment. We then conducted whole-cell recording in tdTomato-tagged VGluT2 or VGAT neurons in lamina II in spinal cord slices. The baseline frequency, but not the amplitude, of mEPSCs in VGluT2 neurons was much higher in paclitaxel-treated (n = 16 neurons) than in vehicle-treated mice (n = 14 neurons) (5.897 ± 0.769 Hz vs 2.712 ± 0.496 Hz, p = 0.0055, F(1,28) = 8.961; Fig. 1A,B). Bath application of 50 μm AP5, a specific NMDAR antagonist, for 6 min led to a rapid reduction in the frequency of mEPSCs of VGluT2 neurons in paclitaxel-treated mice (5.897 ± 0.769 Hz vs 4.380 ± 0.560 Hz, p = 0.0003, F(1.598,44.73) = 10.40; n = 16 neurons) but had no effect in vehicle-treated mice (Fig. 1A,B). These results indicate that presynaptic NMDARs in VGluT2 neurons in the spinal dorsal horn are tonically activated by paclitaxel treatment.
Figure 1.
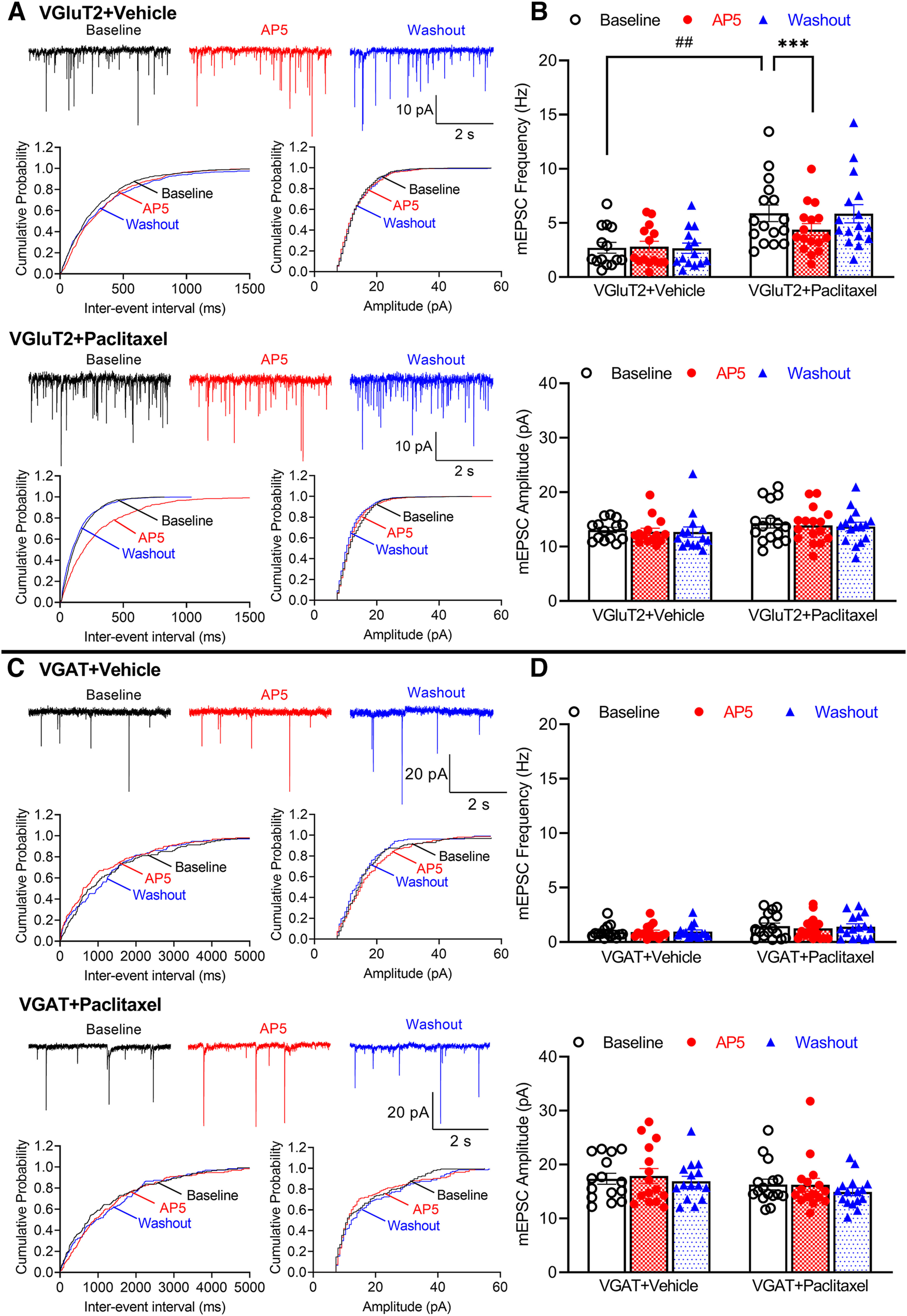
Paclitaxel treatment augments presynaptic NMDAR activity in spinal VGluT2, but not VGAT, neurons. A, Representative recording traces and cumulative probability plots show the effect of bath application of 50 μm AP5 on mEPSCs of spinal lamina II VGluT2 neurons from vehicle-treated and paclitaxel-treated mice. B, Mean data show the effect of AP5 on the frequency and amplitude of mEPSCs of lamina II VGluT2 neurons from vehicle- (n = 14 neurons from 3 mice) and paclitaxel-treated mice (n = 16 neurons from 3 mice). Two-way ANOVA showed that there was a significant main effect for paclitaxel treatment (p = 0.0055, F(1,28) = 8.961) and AP5 treatment (p = 0.0003, F(1.598,44.73) = 10.40) as well as a significant interaction between the paclitaxel and AP5 treatment (p < 0.0001, F(2,56) = 14.35). C, Original recording traces and cumulative probability plots show the effect of bath application of 50 μm AP5 on mEPSCs of lamina II VGAT neurons from vehicle-treated and paclitaxel-treated mice. D, Summary data show the effect of AP5 on the frequency and amplitude of mEPSCs of lamina II VGAT neurons from vehicle-treated (n = 15 neurons from 3 mice) and paclitaxel-treated mice (n = 16 neurons from 3 mice). Data are mean ± SEM. ##p < 0.01; ***p < 0.001; two-way ANOVA followed by Tukey's post hoc test.
By contrast, neither the frequency nor amplitude of baseline mEPSCs of VGAT neurons differed significantly between paclitaxel-treated (n = 16 neurons) and vehicle-treated mice (n = 15 neurons, Fig. 1C,D). In vehicle-treated mice, the baseline frequency of mEPSCs was much higher in VGluT2 than in VGAT neurons (2.712 ± 0.496 Hz vs 0.915 ± 0.153 Hz, p = 0.0166, W(5,36.36) = 6.869, Brown-Forsythe and Welch ANOVA), whereas the baseline amplitude of mEPSCs was significantly smaller in VGluT2 than in VGAT neurons (13.04 ± 0.502 pA vs 17.36 ± 1.011 pA, p = 0.0051, W(5,37.26) = 6.954, Brown-Forsythe and Welch ANOVA). Furthermore, bath application of 50 μm AP5 had no effect on the frequency or amplitude of mEPSCs in VGAT neurons in vehicle- and paclitaxel-treated mice (Fig. 1C,D). These data suggest that paclitaxel treatment induces tonic activation of presynaptic NMDARs, which predominantly drives glutamatergic input to VGluT2-expressing excitatory neurons in the spinal dorsal horn.
Treatment with paclitaxel increases NMDAR activity at primary afferent central terminals that form synapses with spinal VGluT2 neurons
NMDARs are expressed at primary afferent terminals in the spinal superficial dorsal horn (Liu et al., 1994). In chemotherapy-induced neuropathic pain, presynaptic NMDARs at primary afferent central terminals are tonically activated (Xie et al., 2016, 2017b). To determine specifically whether the paclitaxel-induced increase in NMDAR activity occurs at primary afferents that synapse with VGluT2 or VGAT neurons in the spinal dorsal horn, we recorded EPSCs of tdTomato-tagged neurons in lamina II that were monosynaptically evoked from the dorsal root, which reflect glutamate release elicited from primary afferent terminals. The baseline amplitude of evoked EPSCs was significantly larger in VGluT2 neurons in paclitaxel-treated (n = 15 neurons) than in vehicle-treated mice (n = 14 neurons) (536.6 ± 26.87 pA vs 366.2 ± 14.56 pA, p < 0.0001, F(1,27) = 32.41; Fig. 2A). Bath application of 50 μm AP5 for 6 min rapidly reversed the increased amplitude of monosynaptic EPSCs of VGluT2 neurons in paclitaxel-treated mice but had no such effect in vehicle-treated mice (Fig. 2A).
Figure 2.
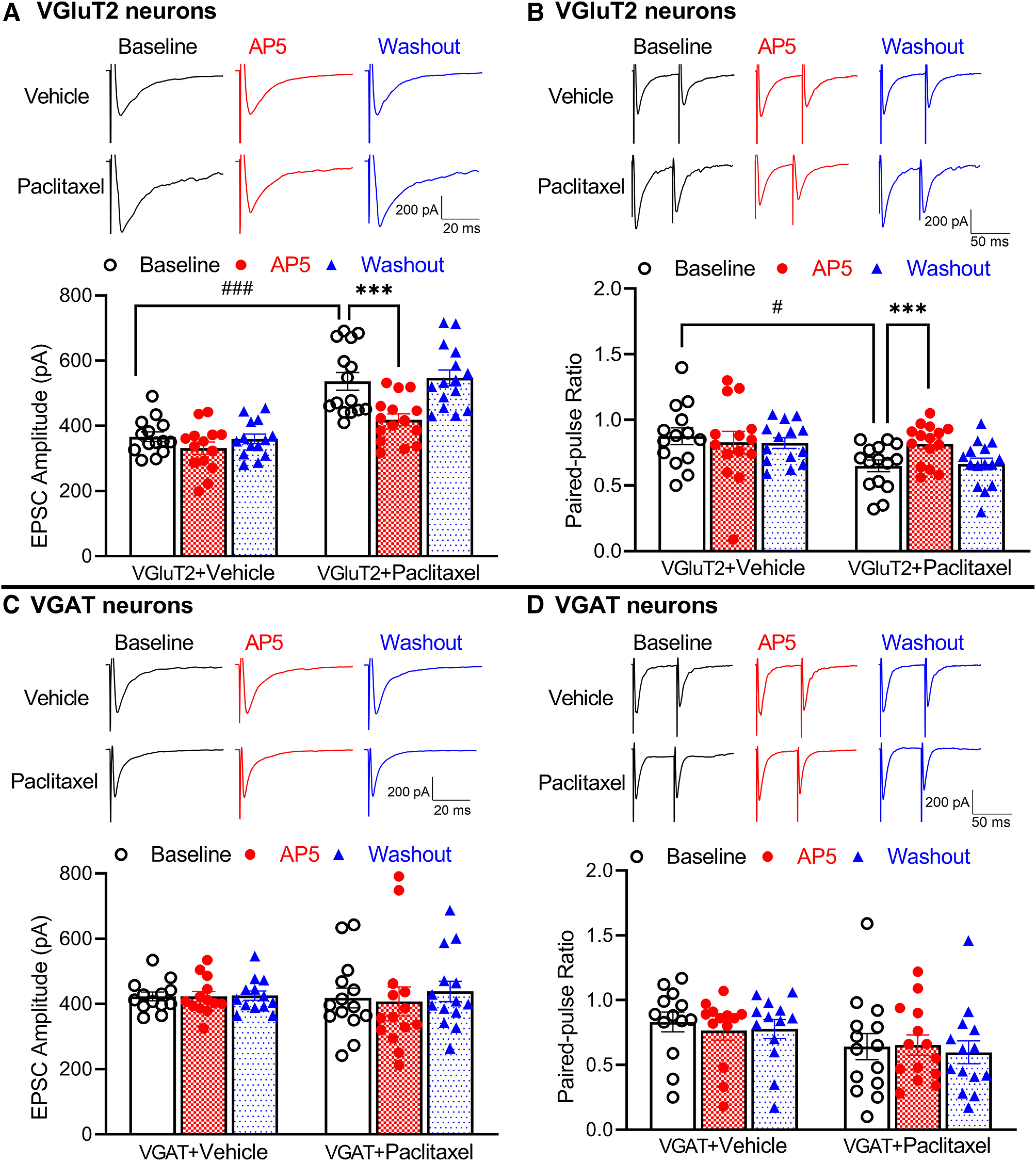
Paclitaxel treatment increases activity of presynaptic NMDARs at primary afferent central terminals that form synapses with VGluT2 neurons. A, B, Representative recording traces and summary data show the effect of 50 μm AP5 on the amplitude of evoked monosynaptic EPSCs (A) and PPR (B) in spinal dorsal horn VGluT2 neurons from vehicle-treated (n = 14 neurons from 3 mice) and paclitaxel-treated mice (n = 15 neurons from 3 mice). Two-way ANOVA showed that there was a significant main effect for paclitaxel treatment (p < 0.0001, F(1,27) = 32.41 in A, p = 0.0228, F(1,27) = 3.977 in B) and AP5 treatment (p < 0.0001, F(1.549,41.82) = 33.74 in A, p < 0.0001, F(1.637,44.2) = 3.081 in B) as well as a significant interaction between the paclitaxel and AP5 treatment (p < 0.0001, F(2,54) = 12.12 in A, p = 0.0076, F(2,54) = 5.355 in B). C, D, Original recording traces and mean data show the effect of 50 μm AP5 on the amplitude of evoked monosynaptic EPSCs (C) and PPR of evoked EPSCs (D) in spinal dorsal horn VGAT neurons from vehicle-treated (n = 13 neurons from 3 mice) and paclitaxel-treated mice (n = 14 neurons from 3 mice). Data are mean ± SEM. #p < 0.05; ###p < 0.001; ***p < 0.001; two-way ANOVA followed by Tukey's post hoc test.
In addition, we recorded the PPR of monosynaptically evoked EPSCs, a measure of the probability of glutamate release from presynaptic terminals, in spinal lamina II neurons. The baseline PPR of evoked EPSCs was significantly lower in VGluT2 neurons in paclitaxel-treated (n = 15 neurons) than in vehicle-treated mice (n = 14 neurons) (0.649 ± 0.044 vs 0.875 ± 0.064, p = 0.0228, F(1,27) = 3.977; Fig. 2B). In VGluT2 neurons from paclitaxel-treated mice (n = 15 neurons), bath application of 50 μm AP5 for 6 min significantly inhibited the first evoked EPSCs more than the second evoked EPSCs, resulting in an increase in the PPR of evoked EPSCs (0.649 ± 0.044 vs 0.817 ± 0.039, p < 0.0001, F(1.637,44.2) = 3.081; Fig. 2B). However, AP5 application had no effect on the PPR of evoked EPSCs in VGluT2 neurons from vehicle-treated mice (n = 14 neurons; Fig. 2B). These data suggest that paclitaxel treatment augments presynaptic NMDAR activity at primary afferent terminals that form synapses with spinal VGluT2 neurons.
By contrast, the baseline amplitude of evoked EPSCs in VGAT neurons did not differ significantly between paclitaxel-treated (n = 14 neurons) and vehicle-treated mice (n = 13 neurons, Fig. 2C). Bath application of AP5 had no effect on the amplitude of evoked EPSCs in these VGAT neurons in both vehicle- and paclitaxel-treated mice (Fig. 2C). The baseline amplitude of evoked EPSCs was much higher in VGAT neurons than in VGluT2 neurons (423.2 ± 13.24 pA vs 366.2 ± 14.56 pA, p = 0.0372, W(5,34.94) = 6.132, Brown-Forsythe and Welch ANOVA) in vehicle-treated animals. In addition, treatment with AP5 had no effect on the PPR of VGAT neurons from either vehicle-treated (n = 13 neurons) or paclitaxel-treated mice (n = 14 neurons; Fig. 2D). Together, these results suggest that paclitaxel treatment potentiates the activity of presynaptic NMDARs at the primary afferent–VGluT2 neuron synapses in the spinal dorsal horn.
Traumatic nerve injury potentiates presynaptic NMDAR activity in spinal VGluT2 dorsal horn neurons
Traumatic nerve injury increases presynaptic NMDARs in the spinal cord (Yan et al., 2013; L. Li et al., 2016; J. Chen et al., 2018). We next determined whether traumatic nerve injury-augmented presynaptic NMDAR activity occurs in a cell type-specific manner in the spinal dorsal horn. VGluT2Cre/+:tdTomatoflox/flox and VGATCre/+:tdTomatoflox/flox were subjected to SNI or sham surgery, and spinal cord slices were obtained from these mice 2 weeks after surgery. Whole-cell recordings were performed in tdTomato-tagged VGluT2 or VGAT neurons in lamina II in spinal cord slices. In mice subjected to SNI, the baseline frequency of mEPSCs (n = 14 neurons) was significantly larger than that of the sham control (n = 16 neurons) in VGluT2 neurons (7.503 ± 1.242 Hz vs 3.328 ± 0.516 Hz, p = 0.0187, F(1,28) = 6.774; Fig. 3A,B). Bath application of 50 μm AP5 for 6 min had no effect on either the frequency or amplitude of mEPSCs of VGluT2 neurons from sham control mice (n = 16 neurons) but decreased the augmented mEPSC frequency in VGluT2 neurons from SNI mice (7.503 ± 1.242 Hz vs 5.281 ± 0.985 Hz, p = 0.0094, F(1.877,25.16) = 10.22, n = 14 neurons; Fig. 3A,B).
Figure 3.
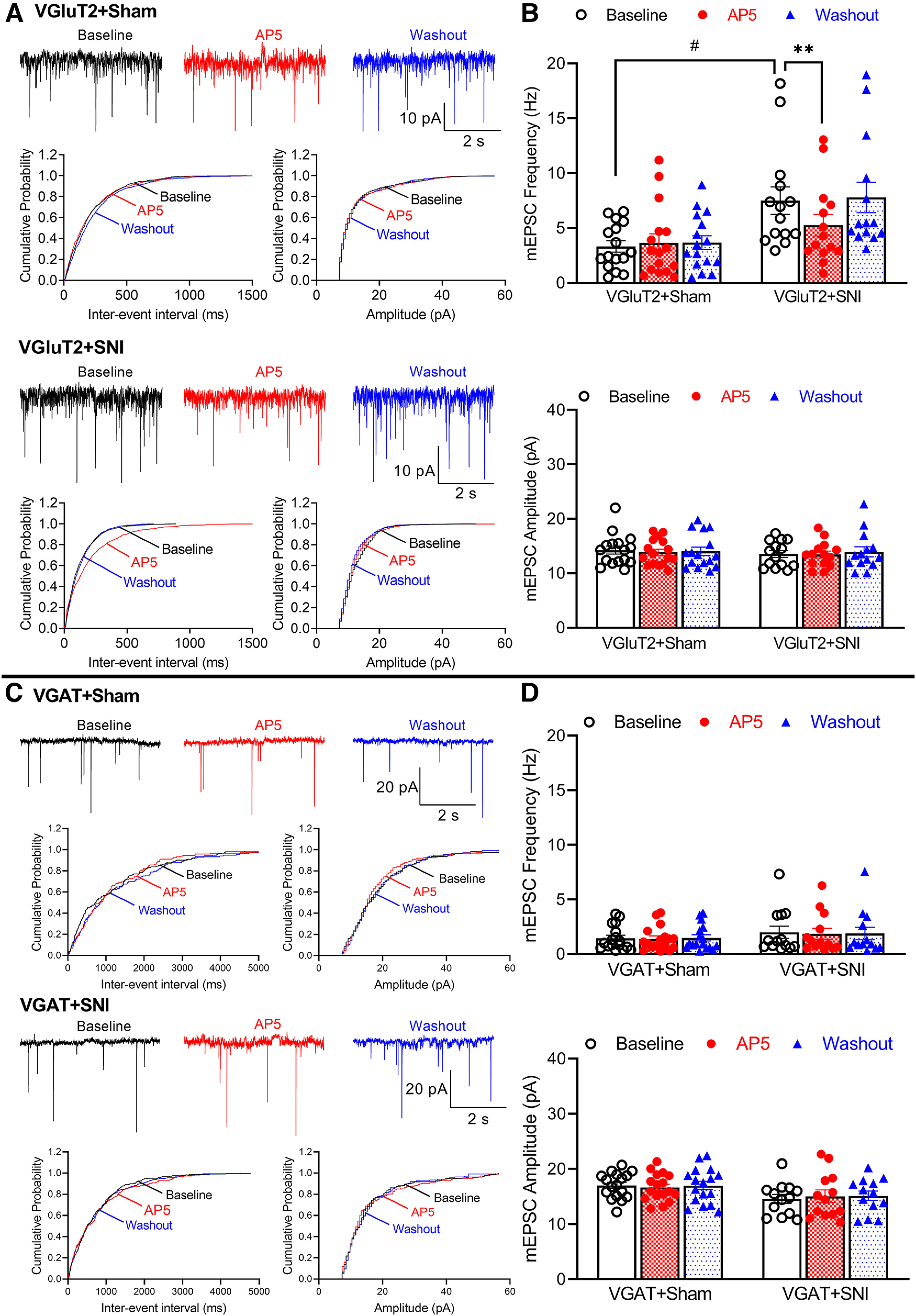
Traumatic nerve injury augments the activity of presynaptic NMDARs in spinal VGluT2, but not VGAT, neurons. A, Original recording traces and cumulative probability plots show the effect of bath application of 50 μm AP5 on mEPSCs of lamina II VGluT2 neurons from sham control and nerve-injured mice. B, Summary data show the effect of AP5 on the frequency and amplitude of mEPSCs of lamina II VGluT2 neurons from sham control (n = 16 neurons from 3 mice) and nerve-injured mice (n = 14 neurons from 3 mice). Two-way ANOVA showed that there was a significant main effect for nerve injury (p = 0.0187, F(1,28) = 6.774) and AP5 treatment (p = 0.0094, F(1.877,25.16) = 10.22) as well as a significant interaction between the injury and AP5 treatment (p = 0.0013, F(2,56) = 7.507). C, Representative recording traces and cumulative probability plots show the effect of bath application of 50 μm AP5 on mEPSCs of lamina II VGAT neurons from sham control and nerve-injured mice. D, Mean data show the effect of AP5 on the frequency and amplitude of mEPSCs of lamina II VGAT neurons from sham control (n = 17 neurons from 3 mice) and nerve-injured mice (n = 13 neurons from 3 mice). Data are mean ± SEM. #p < 0.05; **p < 0.01; two-way ANOVA followed by Tukey's post hoc test.
The baseline frequency of mEPSCs was much greater in VGluT2 neurons than in VGAT neurons (3.328 ± 0.516 Hz vs 1.455 ± 1.114 Hz, p = 0.0186, W(5,42.18) = 5.301, Brown-Forsythe and Welch ANOVA), whereas the mEPSC amplitude in VGluT2 neurons was significantly smaller than that in VGAT neurons (14.25 ± 0.726 pA vs 17.02 ± 0.568 pA, p = 0.0186, W(5,43.19) = 5.404, Brown-Forsythe and Welch ANOVA) from sham-treated animals. However, in VGAT-expressing lamina II neurons, the baseline frequency and amplitude of mEPSCs did not differ between sham control (n = 17 neurons) and SNI (n = 13 neurons) mice (Fig. 3C,D). In addition, bath application of AP5 had no effect on the frequency or amplitude of mEPSCs in VGAT neurons in both groups of mice (Fig. 3C,D). These findings suggest that traumatic nerve injury preferentially potentiates presynaptic NMDAR activity in VGluT2-expressing excitatory neurons in the spinal dorsal horn.
Traumatic nerve injury augments NMDAR activity at primary afferent central terminals that form synapses with spinal VGluT2 neurons
To determine specifically whether nerve trauma-induced presynaptic NMDAR activity occurs at primary afferent central terminals that synapse with VGluT2 or VGAT neurons in the spinal dorsal horn, we recorded EPSCs in tdTomato-tagged VGluT2 or VGAT neurons in lamina II monosynaptically evoked from the dorsal root. The baseline amplitude of evoked EPSCs in VGluT2 neurons was much larger in SNI mice (n = 14 neurons) than in sham control mice (n = 16 neurons) (463.9 ± 23.44 pA vs 358.5 ± 17.49 pA, p = 0.0041, F(1,28) = 4.322; Fig. 4A). Bath application of 50 μm AP5 for 6 min completely reversed the increased amplitude of evoked EPSCs in VGluT2 neurons from SNI mice (Fig. 4A).
Figure 4.
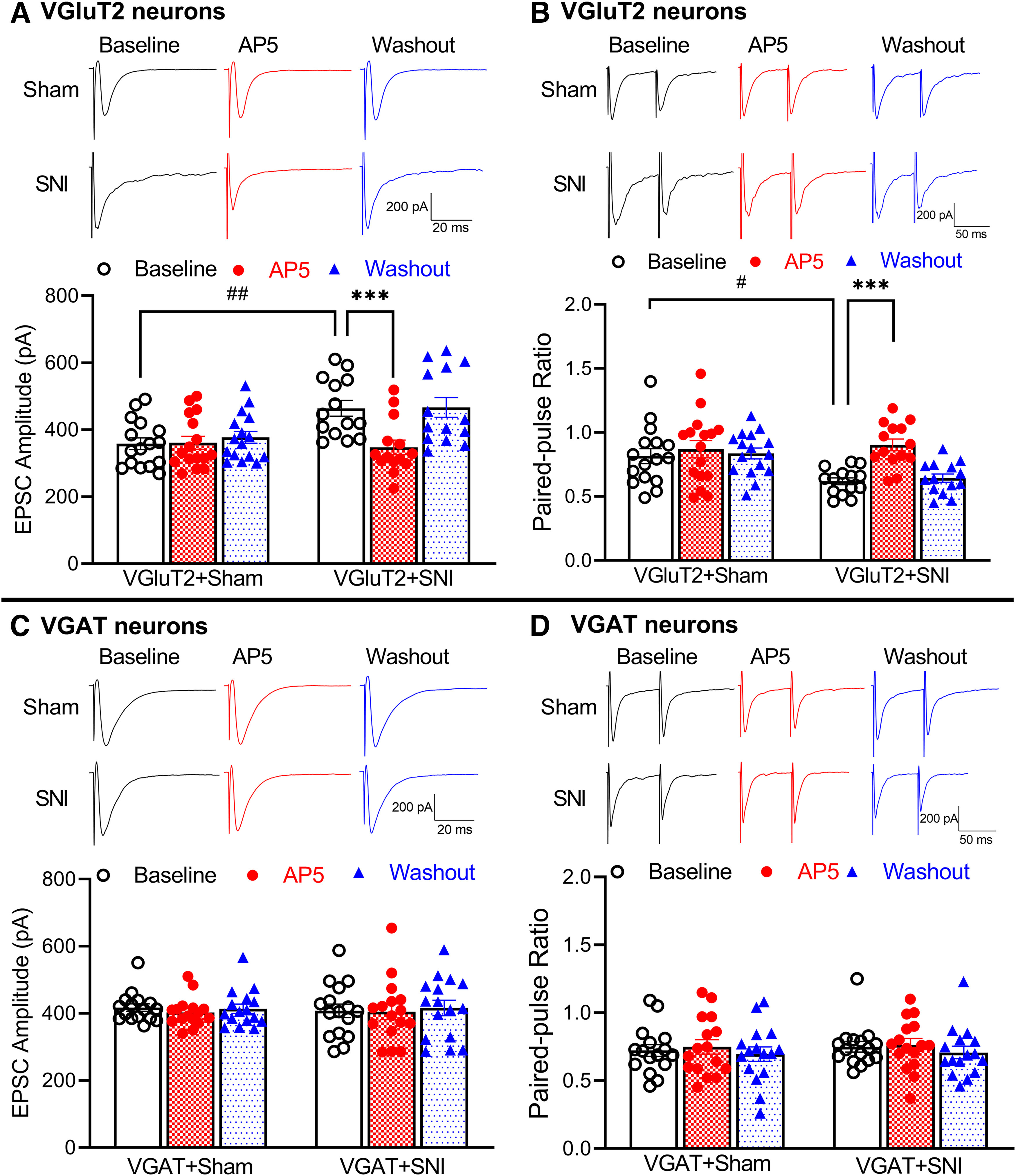
Traumatic nerve injury increases NMDAR activity at primary afferent central terminals that form synapses with spinal VGluT2 neurons. A, B, Original recording traces and summary data show the effect of 50 μm AP5 on the amplitude of evoked monosynaptic EPSCs (A) and PPR of evoked EPSCs (B) in spinal lamina II VGluT2 neurons from sham control (n = 16 neurons from 3 mice) and nerve-injured mice (n = 14 neurons from 3 mice). Two-way ANOVA showed that there was a significant main effect for nerve injury (p = 0.0041, F(1,28) = 4.322 in A, p = 0.0174, F(1,28) = 3.534 in B) and AP5 treatment (p < 0.0001, F(1.805,50.53) = 43.73 in A, p < 0.0001, F(1.925,53.91) = 25.49 in B) as well as a significant interaction between the injury and AP5 treatment (p < 0.0001, F(2,56) = 34.30 in A, p < 0.0001, F(2,56) = 13.11 in B). C, D, Representative recording traces and mean data show the effect of 50 μm AP5 on the amplitude of evoked monosynaptic EPSCs (C) and PPR (D) in spinal lamina II VGAT neurons from sham control (n = 16 neurons from 3 mice) and nerve-injured mice (n = 16 neurons from 3 mice). Data are mean ± SEM. #p < 0.05; ##p < 0.01; ***p < 0.001; two-way ANOVA followed by Tukey's post hoc test.
In VGluT2 neurons, SNI significantly increased the first evoked EPSCs more than the second evoked EPSCs, resulting in the reduced baseline PPR (n = 14 neurons in SNI mice, n = 16 neurons in sham control mice; 0.619 ± 0.027 vs 0.816 ± 0.058, p = 0.0174, F(1,28) = 3.534; Fig. 4B). Moreover, bath application of AP5 significantly increased the PPR of evoked EPSCs in VGluT2 neurons from SNI mice (n = 14 neurons; 0.619 ± 0.027 vs 0.903 ± 0.046, p < 0.0001, F(1.925,53.91) = 25.49; Fig. 4B). However, AP5 application had no effect on the PPR of evoked EPSCs in VGluT2 neurons from sham control mice (n = 16 neurons). These results suggest that traumatic nerve injury potentiates presynaptic NMDAR activity at the primary afferent–VGluT2 neuron synapses in the spinal dorsal horn.
In sham-treated animals, the baseline amplitude of evoked monosynaptic EPSCs was significantly higher in VGAT neurons than in VGluT2 neurons (417.2 ± 11.16 pA vs 358.5 ± 17.49 pA, p = 0.0434, W(5,41.66) = 2.762, Brown-Forsythe and Welch ANOVA). However, the baseline amplitude of evoked EPSCs in VGAT neurons did not differ significantly between SNI (n = 16 neurons) and sham control mice (n = 16 neurons, Fig. 4C). Furthermore, bath application of AP5 had no effect on the amplitude of evoked EPSCs in VGAT neurons in sham control or SNI mice (Fig. 4C). In addition, AP5 had no effect on the PPR of evoked EPSCs in VGAT neurons from sham control mice (n = 16 neurons) or SNI mice (n = 16 neurons, Fig. 4D). Together, these data indicate that traumatic nerve injury selectively increases the activity of NMDARs at primary afferent terminals that form synapses with VGluT2-expressing dorsal horn neurons.
NMDARs in primary sensory neurons are required for chemotherapy-induced chronic pain
Paclitaxel treatment increases presynaptic, but not postsynaptic, NMDAR activity in the spinal dorsal horn (S. R. Chen et al., 2014c; Xie et al., 2016; Y. Chen et al., 2019). However, the role of presynaptic NMDARs expressed in primary sensory neurons in the development of chemotherapy-induced chronic pain has not been explicitly demonstrated. To this end, we generated Grin1-cKO mice that genetically ablated GluN1, an obligatory subunit of NMDARs, from primary sensory neurons by crossing Grin1flox/flox mice to AvilCre/+ mice, as described previously (Huang et al., 2020). Baseline tactile, mechanical, or thermal paw withdrawal thresholds did not differ significantly between the Grin1-cKO and WT mice, which are consistent with previous studies (Huang et al., 2020; G. F. Zhang et al., 2021). Repeated treatment with paclitaxel in WT mice resulted in a large reduction in tactile, pressure, and heat withdrawal thresholds, which lasted for at least 2 weeks after the last drug treatment (n = 8 mice, Fig. 5A). In Grin1-cKO mice, however, treatment with paclitaxel failed to significantly reduce tactile, pressure, or thermal withdrawal thresholds (n = 8 mice, Fig. 5A). Treatment with vehicle had no effect on tactile, pressure, or thermal thresholds in WT and Grin1-cKO mice (n = 8 mice per group, Fig. 5A). These findings indicate that NMDARs in primary sensory neurons are essential for the development of paclitaxel-induced chronic pain.
Figure 5.
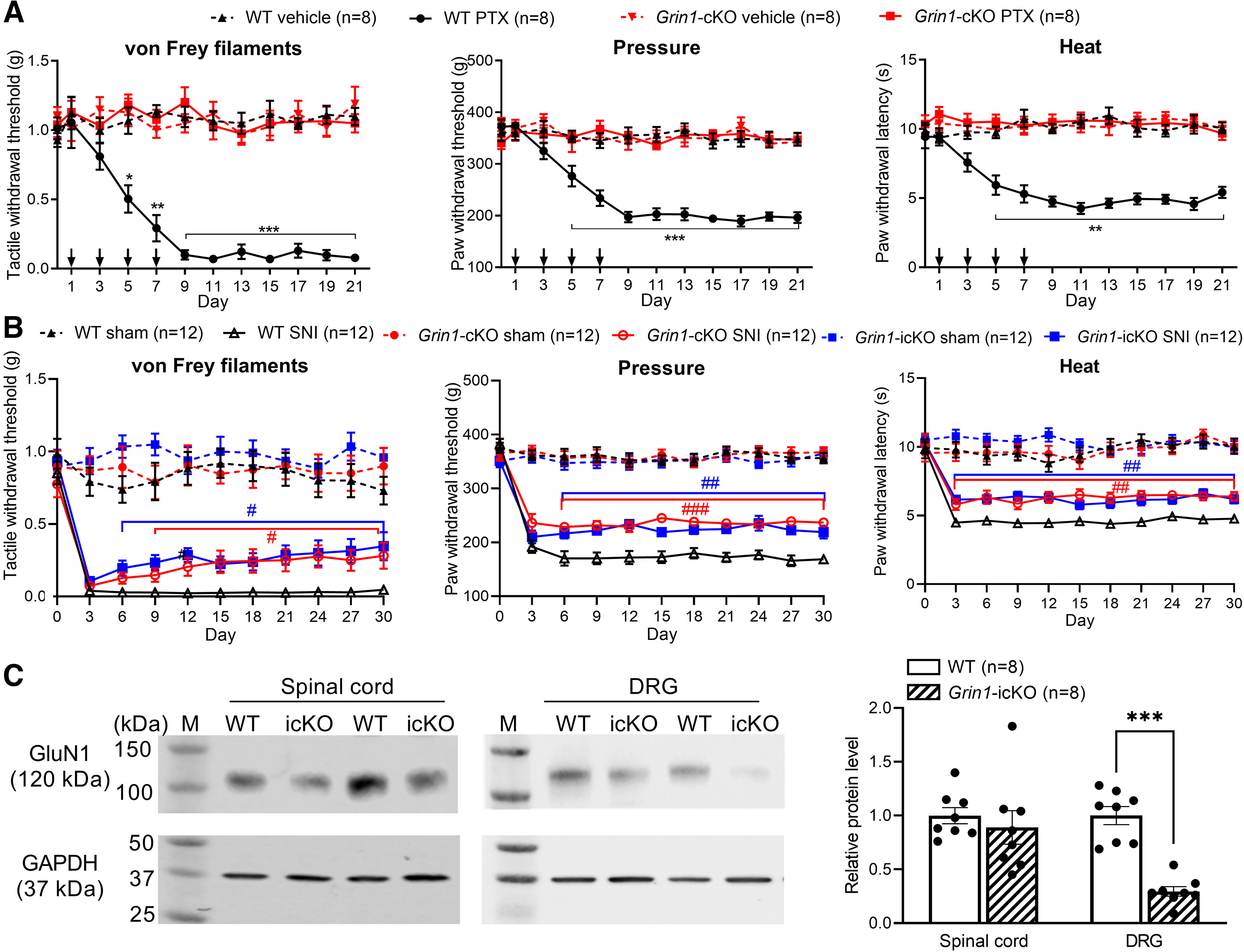
NMDARs in primary sensory neurons are essential for chronic pain induced by paclitaxel treatment, but not by traumatic nerve injury. A, Time course of changes in withdrawal thresholds in response to von Frey filaments, pressure, and noxious heat stimuli in Grin1-cKO mice and their WT littermates (n = 8 mice per group) after systemic treatment with vehicle or paclitaxel (PTX, down arrows indicate drug treatments). Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the baseline (day 0) within the same group (two-way ANOVA followed by Dunnett's post hoc test). Two-way ANOVA showed that there was a significant main effect for genotype (p < 0.0001, F(3,28) = 133.6 in von Frey filaments; p < 0.0001, F(3,28) = 57.63 in pressure; p < 0.0001, F(3,28) = 76.11 in noxious heat) and paclitaxel treatment (p < 0.0001, F(7.368, 206.3) = 5.831 in von Frey filaments; p < 0.0001, F(7.066,197.8) = 13.84 in pressure; p = 0.0022, F(7.524, 210.7) = 3.228 in noxious heat) as well as a significant interaction between the genotype and paclitaxel treatment (p < 0.0001, F(33, 308) = 6.465 in von Frey filaments; p < 0.0001, F(33, 308) = 8.363 in pressure; p < 0.0001, F(33, 308) = 4.505 in noxious heat). B, Time course of changes in withdrawal thresholds in response to von Frey filaments, pressure, and noxious heat stimuli in Grin1-cKO mice, tamoxifen-induced Grin1 cKO (Grin1-icKO) mice, and their WT littermates (n = 12 mice per group) after sham or SNI. Data are mean ± SEM. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the WT group at the same time point (two-way ANOVA followed by Tukey's post hoc test). Two-way ANOVA showed that there was a significant main effect for genotype (p < 0.0001, F(5,66) = 30.12 in von Frey filaments; p < 0.0001, F(5,66) = 117.0 in pressure; p < 0.0001, F(5,66) = 55.67 in noxious heat) and nerve injury (p < 0.0001, F(7.726, 509.9) = 27.80 in von Frey filaments; p < 0.0001, F(7.636, 504.0) = 54.24 in pressure; p < 0.0001, F(6.942, 458.1) = 23.14 in noxious heat) as well as a significant interaction between the genotype and nerve injury (p < 0.0001, F(50, 660) = 5.625 in von Frey filaments; p < 0.0001, F(50, 660) = 10.71 in pressure; p < 0.0001, F(50, 660) = 5.228 in noxious heat). C, Representative blotting images and quantification of the GluN1 protein levels in the dorsal spinal cord and DRG tissues from WT and Grin1-icKO mice (n = 8 mice per group). Data are mean ± SEM. GAPDH was used as a loading control. M, Molecular weight marker. ***p < 0.001 (two-tailed Student's t test).
NMDARs in primary sensory neurons play a minor role in traumatic nerve injury-induced mechanical and thermal hypersensitivity
Traumatic nerve injury increases the activity of both presynaptic and postsynaptic NMDARs at the spinal cord level (Yan et al., 2013; L. Li et al., 2016; J. Chen et al., 2018). We used Grin1-cKO mice to determine the relative importance of NMDARs in primary sensory neurons in nerve injury-induced mechanical and thermal hypersensitivity. SNI in WT mice showed markedly lower paw withdrawal thresholds in response to tactile, pressure, and thermal stimuli, lasting for at least 30 d (n = 12 mice per group, Fig. 5B). Strikingly, in Grin1-cKO mice, SNI still caused a large reduction in tactile, pressure, and thermal withdrawal thresholds. Compared with WT mice, the reduction in tactile, pressure, and thermal thresholds was only slightly attenuated in Grin1-cKO mice (n = 12 mice per group, Fig. 5B).
Traumatic nerve injury, but not paclitaxel treatment, recapitulates developmental reprogramming of the DRG at the neonatal stage (Garriga et al., 2018). Also, constitutively ablating Grin1 may affect the development of DRG neurons. Therefore, we generated AvilCerERT2/+:Grin1flox/flox mice and used tamoxifen to induce cKO of GluN1 in primary sensory neurons in adult mice, referred to as Grin1-icKO mice, to validate the nerve injury effect on mechanical and thermal hypersensitivity observed in Grin1-cKO mice. The baseline tactile, pressure, and thermal withdrawal thresholds did not differ significantly between littermate WT control and Grin1-icKO mice after tamoxifen treatment (n = 12 mice per group, Fig. 5B). We performed SNI or sham surgery in Grin1-icKO mice 3 weeks after tamoxifen injection. However, similar to Grin1-cKO mice subjected to SNI, Grin1-icKO mice also showed a large and persistent reduction in tactile, pressure, and thermal withdrawal thresholds after SNI (n = 12 mice, Fig. 5B). Western blotting analysis showed that the GluN1 protein level in the DRG, but not dorsal spinal cord, was largely reduced in Grin1-icKO mice compared with that in WT mice (p < 0.0001, t(14) = 7.431, n = 8 mice per group; Fig. 5C). Cre expression driven by the Avil promoter occurs in 84%-87% of DRG neurons in AvilCreERT2/+ mice and AvilCre/+ mice (Zappia et al., 2017), which explains the incomplete elimination of GluN1 proteins in the DRG in Grin1-icKO mice. Together, these data indicate that NMDARs in primary sensory neurons do not play a major role in mechanical and thermal hypersensitivity induced by traumatic nerve injury.
Traumatic nerve injury potentiates postsynaptic NMDAR activity in spinal VGluT2 neurons independently of NMDARs in primary sensory neurons
Intrathecal injection of NMDAR antagonists rapidly reverses pain hypersensitivity induced by traumatic nerve injury (Yamamoto and Yaksh, 1992; Chaplan et al., 1997; H. Y. Zhou et al., 2012). Because presynaptic NMDARs expressed in DRG neurons play only a minor role in nerve trauma-induced mechanical and thermal hypersensitivity, we determined how traumatic nerve injury affects the activity of spinal postsynaptic NMDARs in VGluT2 and VGAT neurons in the spinal dorsal horn. We recorded NMDAR currents elicited by puff application of 100 μm NMDA to tdTomato-tagged VGluT2 and VGAT neurons in lamina II. The amplitude of puff NMDAR currents in VGluT2 neurons was much greater in SNI (n = 15 neurons) than in sham-control mice (n = 14 neurons) (376.3 ± 32.01 pA vs 135.4 ± 15.61 pA, p < 0.0001, F(1,55) = 14.33; Fig. 6A). By contrast, the amplitude of puff NMDAR currents in VGAT neurons did not differ significantly between SNI and sham control mice (n = 15 neurons per group, Fig. 6A). These results suggest that traumatic nerve injury preferentially augments postsynaptic NMDAR activity in VGluT2-expressing excitatory dorsal horn neurons.
Figure 6.
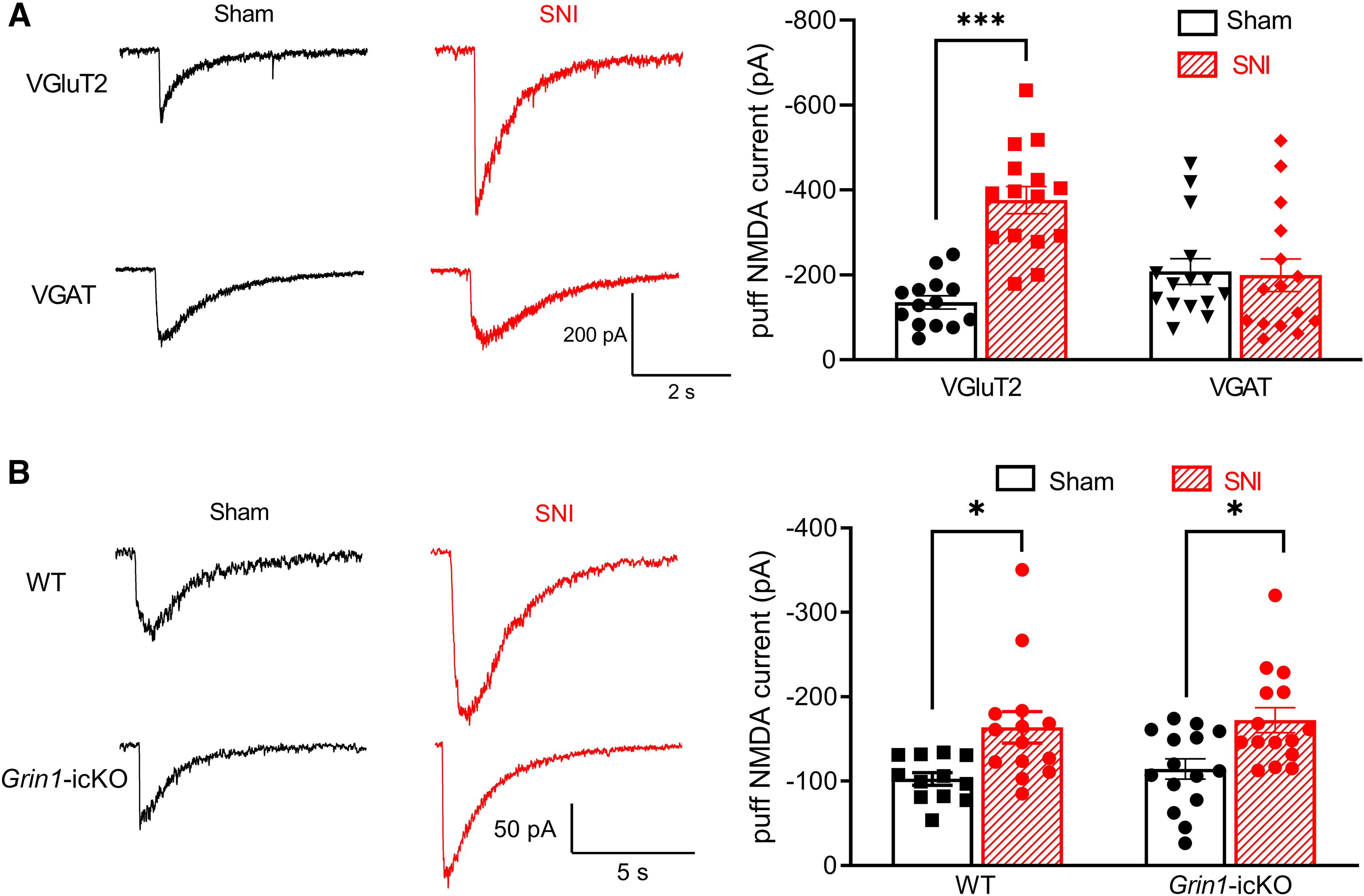
Traumatic nerve injury increases the activity of postsynaptic NMDARs in spinal VGluT2 neurons independently of NMDARs in primary sensory neurons. A, Representative recording traces and mean data show currents elicited by puff application of 100 μm NMDA in lamina II VGluT2 neurons from sham control (n = 14 neurons from 3 mice) and nerve-injured mice (n = 15 neurons from 4 mice) or lamina II VGAT neurons from sham control (n = 15 neurons from 3 mice) and nerve-injured mice (n = 15 neurons from 4 mice). B, Original recording traces and mean data show currents elicited by puff application of 100 μm NMDA in lamina II neurons from sham control (n = 12 neurons from 3 mice) and nerve-injured (n = 14 neurons from 3 mice) WT mice or from sham control (n = 15 neurons from 3 mice) and nerve-injured Grin1-icKO mice (n = 15 neurons from 3 mice). Data are mean ± SEM. *p < 0.05; ***p < 0.001; two-way ANOVA followed by Tukey's post hoc test.
To determine whether postsynaptic NMDAR activity in spinal dorsal horn neurons potentiated by nerve injury is dependent of presynaptic NMDARs in primary sensory neurons, we measured puff NMDAR currents in lamina II neurons in WT and Grin1-icKO mice subjected to SNI. In WT mice, SNI significantly increased the amplitude of puff NMDAR currents in lamina II neurons (n = 14 neurons, 163.8 ± 18.72 pA vs 102.7 ± 7.51 pA, p = 0.0141, F(1,52) = 18.00), compared with that in sham-treated mice (n = 12 neurons; Fig. 6B). Furthermore, in Grin1-icKO mice, SNI caused a similar increase in the amplitude of puff NMDAR currents in lamina II neurons (n = 15 neurons in both sham and SNI groups; 165.7 ± 10.94 pA vs 114.4 ± 12.00 pA, p = 0.0311, F(1,52) = 18.00; Fig. 6B). These data suggest that traumatic nerve injury augments postsynaptic NMDAR activity predominantly in spinal excitatory neurons; this effect is independent of presynaptic NMDARs in primary sensory neurons.
Traumatic nerve injury impairs GABAergic synaptic inhibition of spinal dorsal horn neurons independently of NMDARs in primary sensory neurons
In contrast to chemotherapy-induced neuropathic pain (S. R. Chen et al., 2014c; Xie et al., 2016, 2017b), traumatic nerve injury promotes NMDAR- and calpain-mediated cleavage of K+-Cl− cotransporter-2 (KCC2) proteins to impair normal synaptic inhibition in the spinal dorsal horn (H. Y. Zhou et al., 2012; L. Li et al., 2016). We next determined whether nerve injury-induced diminishment of GABAergic synaptic inhibition results from postsynaptic NMDAR hyperactivity in spinal dorsal horn neurons. In lamina II neurons recorded from sham-treated WT mice, the GABA reversal potential was –63.8 ± 2.15 mV (n = 18 neurons, Fig. 7A–C). SNI caused a significant depolarizing shift in the GABA reversal potential of lamina II neurons in WT mice (–52.6 ± 2.78 mV, n = 17 neurons; p = 0.0297, F(1,67) = 25.44; Fig. 7A–C). Furthermore, in Grin1-icKO mice, SNI caused a similar depolarizing shift in the GABA reversal potential in lamina II neurons (–65.2 ± 2.46 mV in the sham group vs –48.4 ± 3.52 mV in the SNI group, n = 18 neurons per group; p = 0.0003, F(1,67) = 25.44; Fig. 7A–C). These findings suggest that traumatic nerve injury diminishes GABAergic synaptic inhibition in the spinal dorsal horn independently of presynaptic NMDARs produced in primary sensory neurons.
Figure 7.
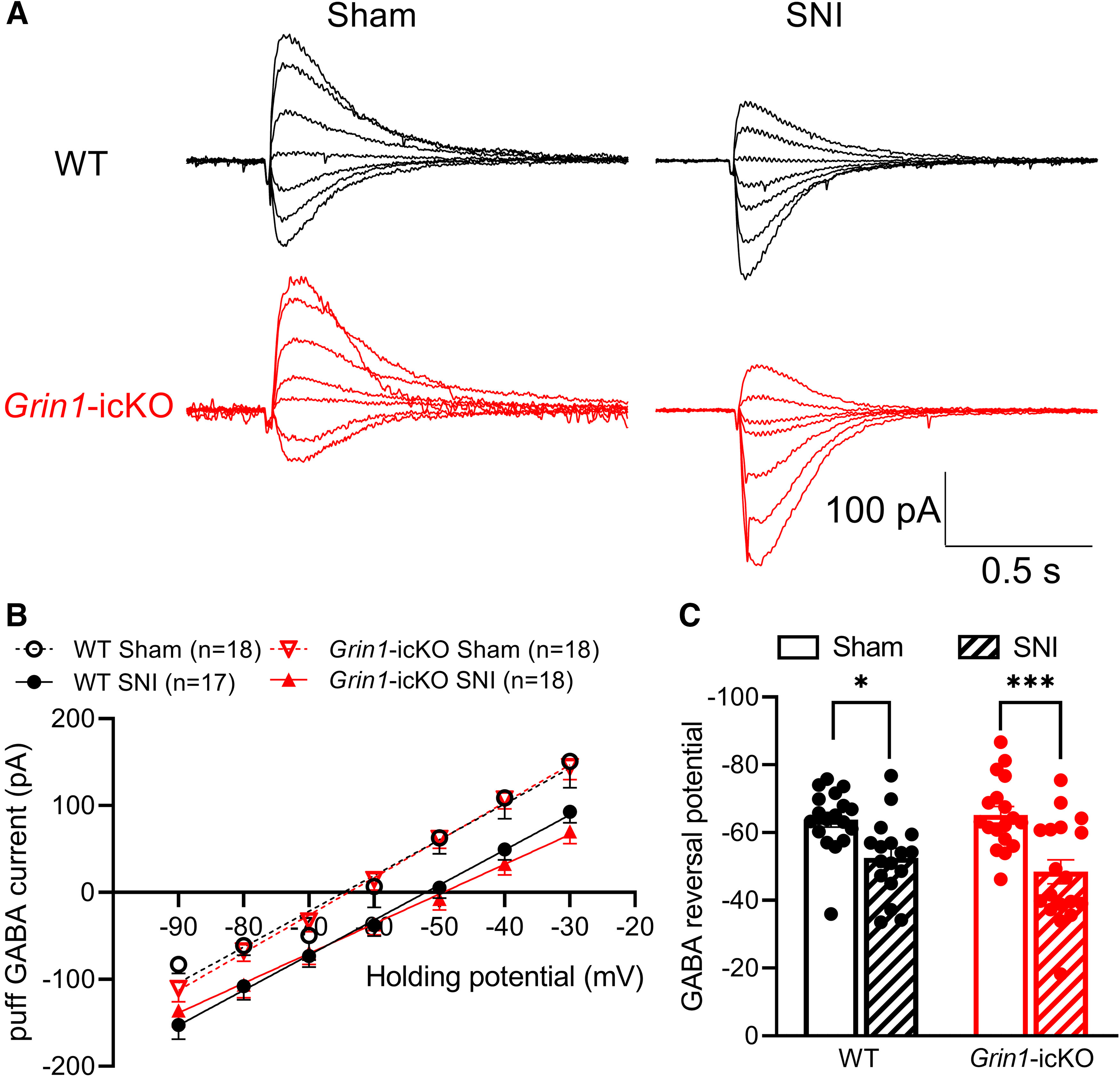
Nerve injury induces a depolarizing shift in spinal GABA reversal potentials independently of NMDARs expressed in primary sensory neurons. A–C, Representative traces (A), mean current–voltage plot (B), and summary data (C) show the puff application of 100 μm GABA-elicited currents recorded at different holding potentials in lamina II neurons from sham control (n = 18 neurons from 6 mice) and nerve-injured (n = 17 neurons from 5 mice) WT mice or from sham control (n = 18 neurons from 5 mice) and nerve-injured Grin1-icKO mice (n = 18 neurons from 5 mice). Data are mean ± SEM. *p < 0.05; ***p < 0.001; two-way ANOVA followed by Tukey's post hoc test.
Knockdown of NMDARs at the spinal cord level diminishes mechanical and thermal hypersensitivity caused by traumatic nerve injury
Because traumatic nerve injury augments postsynaptic NMDAR activity in the spinal cord independently of presynaptic NMDARs, we reasoned that development of mechanical and thermal hypersensitivity after traumatic nerve injury requires both presynaptic and postsynaptic NMDARs at the spinal cord level. To test this hypothesis, we intrathecally injected Cre-expressing AAV8 vectors or GFP-expressing AAV8 control vectors in Grin1flox/flox mice to knock down GluN1 in both the spinal cord and DRG. In mice injected with control vectors, SNI induced a profound and sustained reduction in tactile, pressure, and heat withdrawal thresholds (n = 8 mice, Fig. 8A). By contrast, GluN1 knockdown at the spinal cord level by intrathecal injection of Cre-expressing vectors diminished the reduction in tactile, pressure, and heat withdrawal thresholds induced by SNI (n = 11 mice, Fig. 8A). Immunoblotting analysis showed that the GluN1 protein level in the dorsal spinal cord was much lower in mice injected with Cre-expressing vectors than in mice injected with control vectors (p < 0.0001, t(20) = 5.514, n = 11 mice per group; Fig. 8B). In addition, the GluN1 protein level in the DRG was also largely knocked down in mice injected with Cre-expressing vectors compared with mice treated with control vectors (p < 0.0001, t(20) = 6.221, n = 11 mice per group; Fig. 8B). These data suggest that both presynaptic and postsynaptic NMDARs at the spinal cord level are essential for the full manifestation of traumatic nerve injury-induced mechanical and thermal hypersensitivity.
Figure 8.
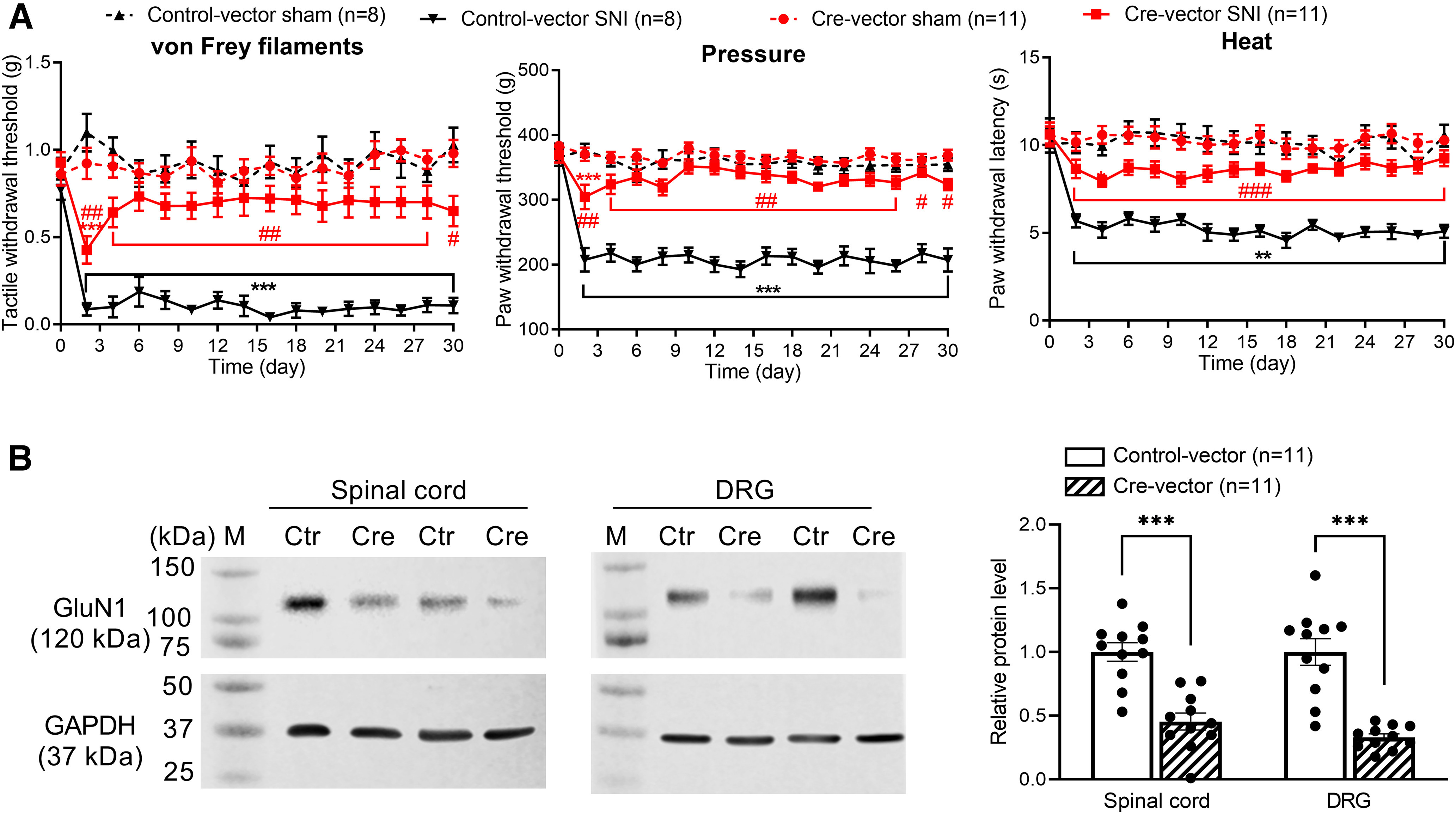
NMDARs at the spinal level mediate traumatic nerve injury-induced chronic pain. A, Time course of changes in withdrawal thresholds in response to von Frey filaments, pressure, and noxious heat stimuli in Grin1flox/flox mice subjected to sham or SNI surgery after intrathecal injection of control vectors (n = 8 mice) or Cre-expressing vectors (n = 11 mice). **p < 0.01, ***p < 0.001 compared with the baseline (day 0) within the same group; ##p < 0.01, ###p < 0.001 compared with the control vector group at the same time point (two-way ANOVA followed by Tukey's post hoc test). Two-way ANOVA showed that there was a significant main effect for the Cre-expressing vector (p < 0.0001, F(3,34) = 41.90 in von Frey filaments; p < 0.0001, F(3,34) = 76.47 in pressure; p < 0.0001, F(3,34) = 91.20 in noxious heat) and nerve injury (p = 0.0007, F(7.157, 243.3) = 3.717 in von Frey filaments; p < 0.0001, F(8.232, 279.9) = 12.26 in pressure; p < 0.0001, F(8.582, 291.8) = 5.668 in noxious heat) as well as a significant interaction between the Cre-expressing vector and nerve injury (p < 0.0001, F(45, 510) = 3.312 in von Frey filaments; p < 0.0001, F(45, 510) = 5.128 in pressure; p < 0.0001, F(45, 510) = 2.260 in noxious heat). B, Original gel images and quantification of GluN1 and GAPDH protein levels in the dorsal spinal cord and DRGs from Grin1flox/flox mice injected with control vectors or Cre-expressing vectors (n = 11 mice per group). Data are mean ± SEM. GAPDH was used as a loading control. M, Molecular weight marker; Ctr, control vectors; Cre, Cre-expressing vectors. ***p < 0.001 (two-tailed Student's t test).
Discussion
Our study provides new evidence that both chemotherapy- and nerve injury-induced chronic pain are associated with increased presynaptic NMDAR activity, predominantly at synapses formed by primary afferent terminals and excitatory dorsal horn neurons. In the present study, we demonstrated that both paclitaxel treatment and traumatic nerve injury augmented presynaptic NMDAR activity in VGluT2, but not VGAT, neurons in the spinal dorsal horn. Increased presynaptic NMDAR activity potentiates neurotransmitter release from primary afferent central terminals in both chemotherapy- and nerve injury-induced neuropathic pain (Yan et al., 2013; Xie et al., 2016). VGluT2 neurons in the spinal dorsal horn play a crucial role in the relay of nociceptive information from primary afferent nerves in painful conditions (Wang et al., 2018; Huang et al., 2022), whereas reducing the excitability of VGAT dorsal horn neurons leads to pain hypersensitivity (Koga et al., 2017). Sciatic nerve injury substantially increases the number of FosB-immunoreactive cells that do not express Pax2, a specific marker of inhibitory neurons, in spinal laminae I-III (Motojima et al., 2021), suggesting that traumatic nerve injury predominantly activates excitatory neurons in the spinal dorsal horn. Spinal GlyT2-expressing inhibitory neurons receive monosynaptic input mainly from DRG neurons expressing NF200 (presumably non-nociceptive neurons) (Foster et al., 2015). On the other hand, little is known about the subtypes of DRG neurons that are monosynaptically connected to VGluT2 dorsal horn neurons. Further studies are needed to determine what subtypes of DRG neurons provide monosynaptic input to spinal excitatory and inhibitory neurons in neuropathic pain conditions.
The observed difference in the baseline frequency of mEPSCs between VGluT2- and VGAT-expressing dorsal horn neurons suggests that a greater number or release probability of glutamatergic afferent input in VGluT2 neurons than in VGAT neurons. Furthermore, the difference in the baseline amplitudes of mEPSCs and evoked EPSCs between the two types of dorsal horn neurons suggests that postsynaptic AMPAR activity is greater in VGAT neurons than in VGluT2 neurons. Recent studies reveal that α2δ-1, known previously as a subunit of voltage-gated Ca2+ channels, is essential for synaptic trafficking and activity of NMDARs in the brain and spinal cord (J. Chen et al., 2018; Ma et al., 2018; Y. Chen et al., 2019; M. H. Zhou et al., 2021; Jin et al., 2023). Traumatic nerve injury induces aberrant sprouting of α2δ-1–expressing primary afferent terminals in the spinal lamina I/II (Yamanaka et al., 2021), which is densely occupied by VGluT2 neurons (S. R. Chen et al., 2022). Thus, it is likely that VGluT2 dorsal horn neurons preferentially form synaptic contacts with α2δ-1–expressing primary afferent terminals and that the NMDAR-mediated sensory input from α2δ-1–expressing DRG neurons to spinal excitatory neurons is selectively potentiated by both chemotherapy-induced peripheral neuropathy and traumatic nerve injury.
Our study also reveals that traumatic nerve injury increases postsynaptic NMDAR activity in spinal excitatory dorsal horn neurons independently of presynaptic NMDARs. Traumatic nerve injury potentiates both presynaptic and postsynaptic NMDAR activity via α2δ-1 in the spinal dorsal horn. Protein kinase C (PKC) is important for increased presynaptic NMDAR activity in the spinal cord after nerve injury (Yan et al., 2013). PKC-mediated NMDAR phosphorylation, including GluN2A Ser929 and GluN2B Ser1413, promotes the α2δ-1–NMDAR interaction and synaptic trafficking (M. H. Zhou et al., 2021). Furthermore, casein kinase-2 plays a major role in nerve injury-induced potentiation of postsynaptic NMDAR activity in the spinal dorsal horn (S. R. Chen et al., 2014b). Additionally, sciatic nerve injury decreases the expression of calcineurin, a Ca2+/calmodulin-dependent serine-threonine phosphatase, in the DRG and spinal cord (Miletic et al., 2011), and reduced calcineurin activity could also increase the phosphorylation and activity of NMDARs in the spinal dorsal horn (Huang et al., 2020). Of note, the expression level of α2δ-1 in the DRG and spinal cord is upregulated after nerve ligation (Luo et al., 2001; Zhang et al., 2022), and α2δ-1–bound NMDARs are required for the tonic activation of NMDARs in nerve injury-induced neuropathic pain (J. Chen et al., 2018). Thus, the increased activity of presynaptic and postsynaptic NMDARs by nerve injury likely results from elevated NMDAR phosphorylation levels and their coupling to the α2δ-1 protein. Because α2δ-1 is mainly expressed in excitatory neurons (Cole et al., 2005), this may explain our finding that nerve injury preferentially augments postsynaptic NMDAR activity in VGluT2-expressing excitatory neurons in the spinal dorsal horn. In addition to chemotherapy-induced neuropathy, which selectively increases presynaptic NMDAR activity in the spinal cord (Xie et al., 2016, 2017b), we found in the present study that traumatic nerve injury potentiated postsynaptic NMDAR activity in mice in which NMDARs were ablated from DRG neurons. Together, these findings indicate that changes in presynaptic and postsynaptic NMDAR activity are not interdependent in neuropathic pain conditions.
Another major finding of our study is that presynaptic NMDARs have a divergent role in the development of pain hypersensitivity caused by traumatic nerve injury and chemotherapy. In mice that lacked GluN1 in primary sensory neurons, paclitaxel treatment largely failed to result in pain hypersensitivity, indicating that NMDARs produced by primary sensory neurons have a pivotal role in chemotherapy-induced neuropathic pain. Chemotherapy-induced pain hypersensitivity is associated with abnormal afferent nerve discharges (Xiao and Bennett, 2008), which may augment PKC activity and subsequent NMDAR phosphorylation in the DRG (Xie et al., 2016, 2017b). Furthermore, paclitaxel treatment increases presynaptic mGluR5 activity in the spinal cord, which is an upstream signaling of PKC (Xie et al., 2017b). In addition, because calcineurin constitutively restrains phosphorylation and activity of NMDARs (Tong et al., 1995; J. J. Zhou et al., 2022), diminished calcineurin activity in the DRG by paclitaxel treatment may further potentiate NMDAR activity (Nie et al., 2018). α2δ-1 is a phospho-binding protein and preferentially interacts with phosphorylated NMDARs (M. H. Zhou et al., 2021). The C-terminus of α2δ-1 physically interacts with NMDARs to promote the synaptic trafficking of α2δ-1–bound NMDARs in the spinal cord (J. Chen et al., 2018; Y. Chen et al., 2019). Importantly, paclitaxel treatment increases the expression of α2δ-1 in the DRG and the α2δ-1–NMDAR interaction to potentiate presynaptic NMDAR activity (Y. Chen et al., 2019). By contrast, treatment with paclitaxel or bortezomib does not affect postsynaptic NMDAR activity in the spinal dorsal horn (S. R. Chen et al., 2014c; Xie et al., 2017b). Collectively, these findings support the notion that NMDARs in primary sensory neurons are essential for the development of chemotherapy-induced chronic pain.
We found unexpectedly that, although traumatic nerve injury similarly increased presynaptic NMDAR activity in spinal VGluT2 neurons, it still induced profound and persistent pain hypersensitivity in both Grin1-cKO and Grin1-icKO mice in which GluN1 in primary sensory neurons was genetically ablated. We showed that nerve trauma augmented postsynaptic NMDAR activity in VGluT2, but not VGAT, dorsal horn neurons. Thus, in addition to presynaptic NMDARs, postsynaptic NMDAR hyperactivity in excitatory dorsal horn neurons might play a dominant role in chronic pain caused by traumatic nerve injury. Consistent with this hypothesis, we found that knockdown of NMDARs at the spinal cord level via intrathecal injection of Cre-expressing viral vectors in Grin1-floxed mice diminished SNI-induced pain hypersensitivity. Therefore, both NMDARs at primary afferent central terminals and in excitatory dorsal horn neurons are required for the full manifestation of chronic pain after nerve trauma.
Additionally, we showed in this study that traumatic nerve injury impaired GABAergic synaptic inhibition in the spinal dorsal horn independently of presynaptic NMDARs. Nerve injury-induced NMDAR hyperactivity diminishes KCC2-dependent GABAergic/glycinergic synaptic inhibition via Ca2+ and calpain-mediated proteolysis at the spinal cord level (H. Y. Zhou et al., 2012; L. Li et al., 2016). By contrast, treatment with paclitaxel does not affect the KCC2 protein levels in the spinal dorsal horn (S. R. Chen et al., 2014c). We found that the GABA reversal potential of dorsal horn neurons shifted by nerve injury is similar in WT and Grin1-cKO mice, suggesting that increased postsynaptic NMDAR activity impairs normal synaptic inhibition independently of NMDARs produced by primary sensory neurons. Voltage-gated Na+ channels at nerve axons are important for the ectopic bursting discharge after traumatic nerve injury (Matzner and Devor, 1994; Omana-Zapata et al., 1997). In the absence of presynaptic NMDARs, traumatic nerve injury-induced high-frequency, bursting discharges (Pan et al., 1999; S. R. Chen et al., 2002; Devor, 2009) may induce a neuronal activity-dependent increase in casein kinase-2 activity and potentiate NMDAR phosphorylation and its interaction with α2δ-1 to augment postsynaptic NMDAR activity in spinal excitatory neurons. In addition, traumatic nerve injury reduces glutamate transporter activity (Sung et al., 2003; Napier et al., 2012), which may increase ambient glutamate concentrations to cause persistent stimulation of postsynaptic NMDARs in the spinal cord. Therefore, postsynaptic NMDAR hyperactivity and the ensuing synaptic disinhibition in spinal excitatory neurons likely play a dominant role in the development of chronic pain after traumatic nerve injury.
In conclusion, our study demonstrates, for the first time, that chemotherapy- and traumatic nerve injury-induced presynaptic NMDAR hyperactivity occurs predominantly in VGluT2-expressing excitatory neurons in the spinal dorsal horn. However, although presynaptic NMDARs are essential for chemotherapy-induced chronic pain, increased postsynaptic NMDAR activity, via diminishing synaptic inhibition, in VGluT2 dorsal horn neurons likely plays a major role in the development of chronic pain after traumatic nerve injury (Fig. 9). This new information advances our mechanistic understanding of the molecular and cellular basis of central sensitization, illustrating how presynaptic and postsynaptic NMDARs are engaged in the spinal nociceptive circuitry in different neuropathic pain conditions. Our findings also suggest that NMDARs expressed in primary sensory neurons and spinal excitatory neurons could be targeted, respectively, for managing chemotherapy- and traumatic nerve injury-induced neuropathic pain.
Figure 9.
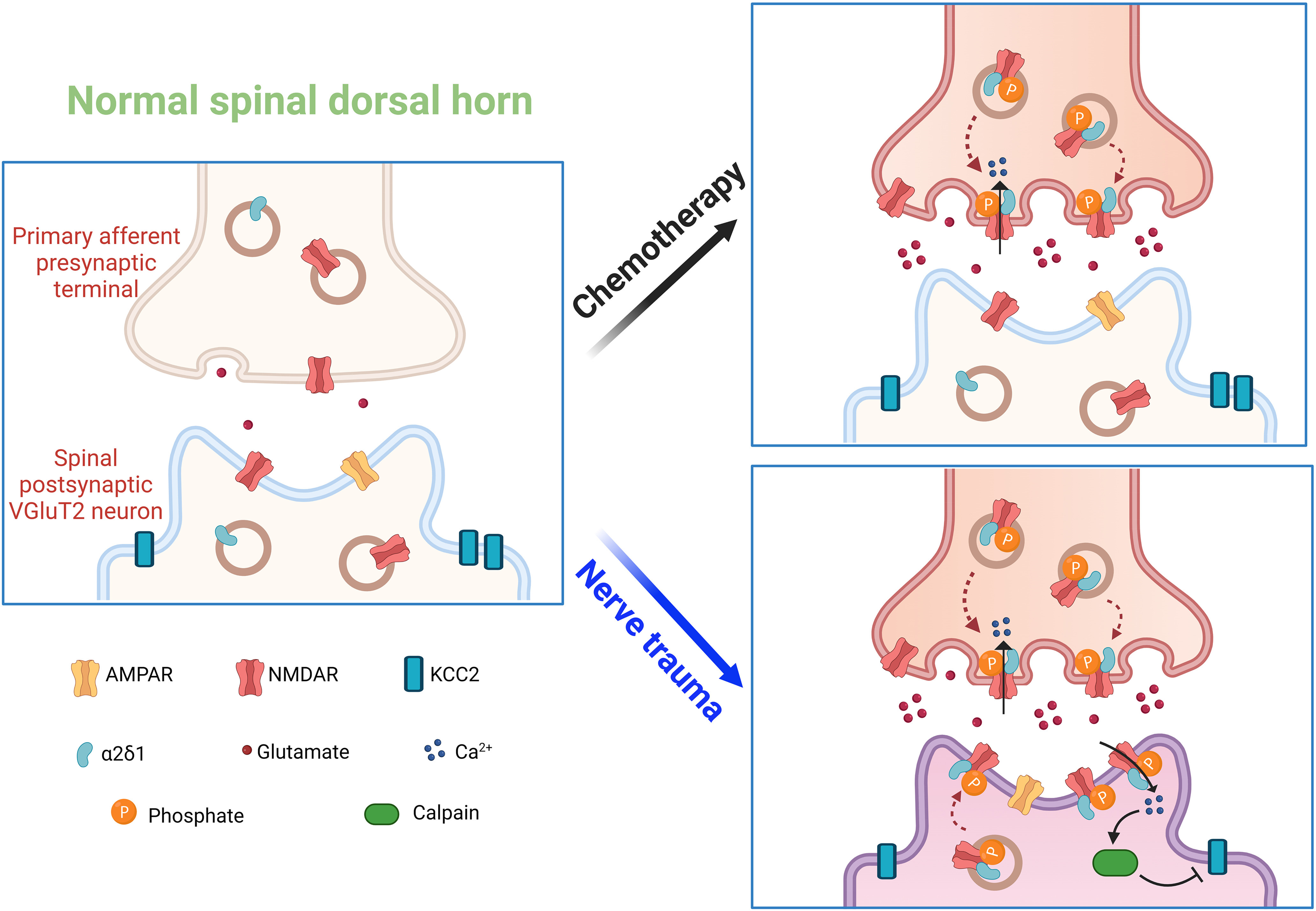
Schematic shows the potential roles of presynaptic and postsynaptic NMDARs in synaptic plasticity induced by chemotherapy and traumatic nerve injury. Under normal conditions, NMDARs are minimally active at presynaptic and postsynaptic sites. Paclitaxel treatment induces NMDAR phosphorylation and its physical interaction with α2δ-1 at the primary afferent central terminals, leading to increased synaptic trafficking and activity of α2δ-1–bound NMDARs and presynaptic glutamate release onto VGluT2-expressing excitatory neurons in the spinal dorsal horn. By comparison, traumatic nerve injury increases the phosphorylation of NMDARs at both primary afferent central terminals and VGluT2 neurons in the spinal dorsal horn. These phosphorylated NMDARs interact with α2δ-1 and traffic to both presynaptic and postsynaptic sites in the spinal cord. Traumatic nerve injury also causes NMDAR- and calpain-mediated KCC2 proteolysis to impair synaptic inhibition in spinal VGluT2 neurons.
Footnotes
This work was supported by National Institutes of Health Grants NS101880 and DA041711 and by the Pamela and Wayne Garrison Distinguished Chair Endowment. We thank Ann Sutton at MD Anderson Cancer Center for proofreading the manuscript.
The authors declare no competing financial interests.
References
- Browne TJ, Gradwell MA, Iredale JA, Maden JF, Callister RJ, Hughes DI, Dayas CV, Graham BA (2020) Transgenic cross-referencing of inhibitory and excitatory interneuron populations to dissect neuronal heterogeneity in the dorsal horn. Front Mol Neurosci 13:32. 10.3389/fnmol.2020.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Malmberg AB, Yaksh TL (1997) Efficacy of spinal NMDA receptor antagonism in formalin hyperalgesia and nerve injury evoked allodynia in the rat. J Pharmacol Exp Ther 280:829–838. [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RE, MacLean DM, Zhang Y, Zhou MH, Jayaraman V, Pan HL (2018) The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep 22:2307–2321. 10.1016/j.celrep.2018.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Wozniak KM, Slusher BS, Pan HL (2002) Effect of 2-(phosphono-methyl)-pentanedioic acid on allodynia and afferent ectopic discharges in a rat model of neuropathic pain. J Pharmacol Exp Ther 300:662–667. 10.1124/jpet.300.2.662 [DOI] [PubMed] [Google Scholar]
- Chen SR, Hu YM, Chen H, Pan HL (2014a) Calcineurin inhibitor induces pain hypersensitivity by potentiating pre- and postsynaptic NMDA receptor activity in spinal cords. J Physiol 592:215–227. 10.1113/jphysiol.2013.263814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Zhou HY, Byun HS, Chen H, Pan HL (2014b) Casein kinase II regulates N-methyl-D-aspartate receptor activity in spinal cords and pain hypersensitivity induced by nerve injury. J Pharmacol Exp Ther 350:301–312. 10.1124/jpet.114.215855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Zhu L, Chen H, Wen L, Laumet G, Pan HL (2014c) Increased spinal cord Na(+)-K(+)-2Cl(–) cotransporter-1 (NKCC1) activity contributes to impairment of synaptic inhibition in paclitaxel-induced neuropathic pain. J Biol Chem 289:31111–31120. 10.1074/jbc.M114.600320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Chen H, Jin D, Pan HL (2022) Brief opioid exposure paradoxically augments primary afferent input to spinal excitatory neurons via α2δ-1-dependent presynaptic NMDA receptors. J Neurosci 42:9315–9329. 10.1523/JNEUROSCI.1704-22.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen SR, Chen H, Zhang J, Pan HL (2019) Increased alpha2delta-1-NMDA receptor coupling potentiates glutamatergic input to spinal dorsal horn neurons in chemotherapy-induced neuropathic pain. J Neurochem 148:252–274. 10.1111/jnc.14627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA, Gu G (2005) Differential distribution of voltage-gated calcium channel alpha-2 delta (α2δ) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491:246–269. 10.1002/cne.20693 [DOI] [PubMed] [Google Scholar]
- da Silva S, Hasegawa H, Scott A, Zhou X, Wagner AK, Han BX, Wang F (2011) Proper formation of whisker barrelettes requires periphery-derived Smad4-dependent TGF-beta signaling. Proc Natl Acad Sci USA 108:3395–3400. 10.1073/pnas.1014411108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Chen SR, Pan HL (2019a) Presynaptic NMDA receptors control nociceptive transmission at the spinal cord level in neuropathic pain. Cell Mol Life Sci 76:1889–1899. 10.1007/s00018-019-03047-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Chen SR, Chen H, Luo Y, Dong Y, Pan HL (2019b) Mitogen-activated protein kinase signaling mediates opioid-induced presynaptic NMDA receptor activation and analgesic tolerance. J Neurochem 148:275–290. 10.1111/jnc.14628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M (2009) Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res 196:115–128. 10.1007/s00221-009-1724-6 [DOI] [PubMed] [Google Scholar]
- Foster E, Wildner H, Tudeau L, Haueter S, Ralvenius WT, Jegen M, Johannssen H, Hosli L, Haenraets K, Ghanem A, Conzelmann KK, Bosl M, Zeilhofer HU (2015) Targeted ablation, silencing, and activation establish glycinergic dorsal horn neurons as key components of a spinal gate for pain and itch. Neuron 85:1289–1304. 10.1016/j.neuron.2015.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga J, Laumet G, Chen SR, Zhang Y, Madzo J, Issa JJ, Pan HL, Jelinek J (2018) Nerve injury-induced chronic pain is associated with persistent DNA methylation reprogramming in dorsal root ganglion. J Neurosci 38:6090–6101. 10.1523/JNEUROSCI.2616-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Chen SR, Chen H, Luo Y, Pan HL (2020) Calcineurin inhibition causes α2δ-1-mediated tonic activation of synaptic NMDA receptors and pain hypersensitivity. J Neurosci 40:3707–3719. 10.1523/JNEUROSCI.0282-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Chen SR, Chen H, Zhou JJ, Jin D, Pan HL (2022) Theta-burst stimulation of primary afferents drives long-term potentiation in the spinal cord and persistent pain via α2δ-1-bound NMDA receptors. J Neurosci 42:513–527. 10.1523/JNEUROSCI.1968-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Chen H, Chen SR, Pan HL (2023) α2δ-1 protein drives opioid-induced conditioned reward and synaptic NMDA receptor hyperactivity in the nucleus accumbens. J Neurochem 164:143–157. 10.1111/jnc.15706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K, Kanehisa K, Kohro Y, Shiratori-Hayashi M, Tozaki-Saitoh H, Inoue K, Furue H, Tsuda M (2017) Chemogenetic silencing of GABAergic dorsal horn interneurons induces morphine-resistant spontaneous nocifensive behaviours. Sci Rep 7:4739. 10.1038/s41598-017-04972-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laedermann CJ, Pertin M, Suter MR, Decosterd I (2014) Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol Pain 10:19. 10.1186/1744-8069-10-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau J, Minett MS, Zhao J, Dennehy U, Wang F, Wood JN, Bogdanov YD (2011) Temporal control of gene deletion in sensory ganglia using a tamoxifen-inducible Advillin-Cre-ERT2 recombinase mouse. Mol Pain 7:100. 10.1186/1744-8069-7-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan YZ, Levey AI, Pan HL (2002) Role of presynaptic muscarinic and GABA(B) receptors in spinal glutamate release and cholinergic analgesia in rats. J Physiol 543:807–818. 10.1113/jphysiol.2002.020644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chen SR, Chen H, Wen L, Hittelman WN, Xie JD, Pan HL (2016) Chloride homeostasis critically regulates synaptic NMDA receptor activity in neuropathic pain. Cell Rep 15:1376–1383. 10.1016/j.celrep.2016.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang H, Sheng M, Jan LY, Jan YN, Basbaum AI (1994) Evidence for presynaptic N-methyl-D-aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci USA 91:8383–8387. 10.1073/pnas.91.18.8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL (2001) Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci 21:1868–1875. 10.1523/JNEUROSCI.21-06-01868.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Chen SR, Chen H, Zhou JJ, Li DP, Pan HL (2018) α2δ-1 couples to NMDA receptors in the hypothalamus to sustain sympathetic vasomotor activity in hypertension. J Physiol 596:4269–4283. 10.1113/JP276394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Hayes RL, Lu J, Mayer DJ, Frenk H (1993) Intrathecal treatment with dextrorphan or ketamine potently reduces pain-related behaviors in a rat model of peripheral mononeuropathy. Brain Res 605:164–168. 10.1016/0006-8993(93)91368-3 [DOI] [PubMed] [Google Scholar]
- Matzner O, Devor M (1994) Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J Neurophysiol 72:349–359. 10.1152/jn.1994.72.1.349 [DOI] [PubMed] [Google Scholar]
- Miletic G, Sullivan KM, Dodson AM, Lippitt JA, Schneider JA, Miletic V (2011) Changes in calcineurin message, enzyme activity and protein content in the spinal dorsal horn are associated with chronic constriction injury of the rat sciatic nerve. Neuroscience 188:142–147. 10.1016/j.neuroscience.2011.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motojima Y, Ueta Y, Sakai A (2021) Analysis of the proportion and neuronal activity of excitatory and inhibitory neurons in the rat dorsal spinal cord after peripheral nerve injury. Neurosci Lett 749:135707. 10.1016/j.neulet.2021.135707 [DOI] [PubMed] [Google Scholar]
- Napier IA, Mohammadi SA, Christie MJ (2012) Glutamate transporter dysfunction associated with nerve injury-induced pain in mice. J Neurophysiol 107:649–657. 10.1152/jn.00763.2011 [DOI] [PubMed] [Google Scholar]
- Nie B, Liu C, Bai X, Chen X, Wu S, Zhang S, Huang Z, Xie M, Xu T, Xin W, Zeng W, Ouyang H (2018) AKAP150 involved in paclitaxel-induced neuropathic pain via inhibiting CN/NFAT2 pathway and downregulating IL-4. Brain Behav Immun 68:158–168. 10.1016/j.bbi.2017.10.015 [DOI] [PubMed] [Google Scholar]
- Omana-Zapata I, Khabbaz MA, Hunter JC, Clarke DE, Bley KR (1997) Tetrodotoxin inhibits neuropathic ectopic activity in neuromas, dorsal root ganglia and dorsal horn neurons. Pain 72:41–49. 10.1016/s0304-3959(97)00012-2 [DOI] [PubMed] [Google Scholar]
- Pan HL, Eisenach JC, Chen SR (1999) Gabapentin suppresses ectopic nerve discharges and reverses allodynia in neuropathic rats. J Pharmacol Exp Ther 288:1026–1030. [PubMed] [Google Scholar]
- Pan YZ, Pan HL (2004) Primary afferent stimulation differentially potentiates excitatory and inhibitory inputs to spinal lamina II outer and inner neurons. J Neurophysiol 91:2413–2421. 10.1152/jn.01242.2003 [DOI] [PubMed] [Google Scholar]
- Polomano RC, Mannes AJ, Clark US, Bennett GJ (2001) A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 94:293–304. 10.1016/S0304-3959(01)00363-3 [DOI] [PubMed] [Google Scholar]
- Sullivan SJ, Sdrulla AD (2022) Excitatory and inhibitory neurons of the spinal cord superficial dorsal horn diverge in their somatosensory responses and plasticity in vivo. J Neurosci 42:1958–1973. 10.1523/JNEUROSCI.1860-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Chen SR, Chen H, Pan HL (2019) μ-Opioid receptors in primary sensory neurons are essential for opioid analgesic effect on acute and inflammatory pain and opioid-induced hyperalgesia. J Physiol 597:1661–1675. 10.1113/JP277428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J (2003) Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci 23:2899–2910. 10.1523/JNEUROSCI.23-07-02899.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Shepherd D, Jahr CE (1995) Synaptic desensitization of NMDA receptors by calcineurin. Science 267:1510–1512. 10.1126/science.7878472 [DOI] [PubMed] [Google Scholar]
- Wang L, Chen SR, Ma H, Chen H, Hittelman WN, Pan HL (2018) Regulating nociceptive transmission by VGluT2-expressing spinal dorsal horn neurons. J Neurochem 147:526–540. 10.1111/jnc.14588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW (2000) Neuronal plasticity: increasing the gain in pain. Science 288:1765–1769. 10.1126/science.288.5472.1765 [DOI] [PubMed] [Google Scholar]
- Xiao WH, Bennett GJ (2008) Chemotherapy-evoked neuropathic pain: abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitine. Pain 135:262–270. 10.1016/j.pain.2007.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JD, Chen SR, Chen H, Zeng WA, Pan HL (2016) Presynaptic N-methyl-D-aspartate (NMDA) receptor activity is increased through protein kinase C in paclitaxel-induced neuropathic pain. J Biol Chem 291:19364–19373. 10.1074/jbc.M116.732347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JD, Chen SR, Pan HL (2017a) Presynaptic mGluR5 receptor controls glutamatergic input through protein kinase C-NMDA receptors in paclitaxel-induced neuropathic pain. J Biol Chem 292:20644–20654. 10.1074/jbc.M117.818476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JD, Chen SR, Chen H, Pan HL (2017b) Bortezomib induces neuropathic pain through protein kinase C-mediated activation of presynaptic NMDA receptors in the spinal cord. Neuropharmacology 123:477–487. 10.1016/j.neuropharm.2017.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Yaksh TL (1992) Spinal pharmacology of thermal hyperesthesia induced by constriction injury of sciatic nerve: excitatory amino acid antagonists. Pain 49:121–128. 10.1016/0304-3959(92)90198-K [DOI] [PubMed] [Google Scholar]
- Yamanaka H, Okubo M, Kobayashi K, Noguchi K (2021) Aberrant axo-axonic synaptic reorganization in the phosphorylated L1-cam/calcium channel subunit α2δ-1-containing central terminals of injured c-fibers in the spinal cord of a neuropathic pain model. eNeuro 8:ENEURO.0499-20.2021. 10.1523/ENEURO.0499-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Jiang E, Gao M, Weng HR (2013) Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. J Physiol 591:2001–2019. 10.1113/jphysiol.2012.250522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappia KJ, O'Hara CL, Moehring F, Kwan KY, Stucky CL (2017) Sensory neuron-specific deletion of TRPA1 results in mechanical cutaneous sensory deficits. eNeuro 4:ENEURO.0069-16.2017. 10.1523/ENEURO.0069-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GF, Chen SR, Jin D, Huang Y, Chen H, Pan HL (2021) α2δ-1 upregulation in primary sensory neurons promotes NMDA receptor-mediated glutamatergic input in resiniferatoxin-induced neuropathy. J Neurosci 41:5963–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen SR, Chen H, Pan HL (2018) RE1-silencing transcription factor controls the acute-to-chronic neuropathic pain transition and Chrm2 receptor gene expression in primary sensory neurons. J Biol Chem 293:19078–19091. 10.1074/jbc.RA118.005846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen SR, Zhou MH, Jin D, Chen H, Wang L, DePinho RA, Pan HL (2022) HDAC2 constitutively restrains chronic pain by repressing α2δ-1 expression and associated NMDA receptor activity. J Neurosci 42:8918–8935. 10.1074/jbc.RA118.005846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen SR, Laumet G, Chen H, Pan HL (2016) Nerve injury diminishes opioid analgesia through lysine methyltransferase-mediated transcriptional repression of mu-opioid receptors in primary sensory neurons. J Biol Chem 291:8475–8485. 10.1074/jbc.M115.711812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YL, Chen SR, Chen H, Pan HL (2012) Chronic opioid potentiates presynaptic but impairs postsynaptic N-methyl-D-aspartic acid receptor activity in spinal cords: implications for opioid hyperalgesia and tolerance. J Biol Chem 287:25073–25085. 10.1074/jbc.M112.378737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL (2010) Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci 30:4460–4466. 10.1523/JNEUROSCI.5857-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Pan HL (2011) Targeting N-methyl-D-aspartate receptors for treatment of neuropathic pain. Expert Rev Clin Pharmacol 4:379–388. 10.1586/ecp.11.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Byun HS, Chen H, Li L, Han HD, Lopez-Berestein G, Sood AK, Pan HL (2012) N-methyl-D-aspartate receptor- and calpain-mediated proteolytic cleavage of K+-Cl- cotransporter-2 impairs spinal chloride homeostasis in neuropathic pain. J Biol Chem 287:33853–33864. 10.1074/jbc.M112.395830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JJ, Shao JY, Chen SR, Pan HL (2022) Calcineurin controls hypothalamic NMDA receptor activity and sympathetic outflow. Circ Res 131:345–360. 10.1161/CIRCRESAHA.122.320976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou MH, Chen SR, Wang L, Huang Y, Deng M, Zhang J, Zhang J, Chen H, Yan J, Pan HL (2021) Protein kinase C-mediated phosphorylation and α2δ-1 interdependently regulate NMDA receptor trafficking and activity. J Neurosci 41:6415–6429. 10.1523/JNEUROSCI.0757-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]