

Abstract
Endolysosomal defects in neurons are central to the pathogenesis of prion and other neurodegenerative disorders. In prion disease, prion oligomers traffic through the multivesicular body (MVB) and are routed for degradation in lysosomes or for release in exosomes, yet how prions impact proteostatic pathways is unclear. We found that prion-affected human and mouse brain showed a marked reduction in Hrs and STAM1 (ESCRT-0), which route ubiquitinated membrane proteins from early endosomes into MVBs. To determine how the reduction in ESCRT-0 impacts prion conversion and cellular toxicity in vivo, we prion-challenged conditional knockout mice (male and female) having Hrs deleted from neurons, astrocytes, or microglia. The neuronal, but not astrocytic or microglial, Hrs-depleted mice showed a shortened survival and an acceleration in synaptic derangements, including an accumulation of ubiquitinated proteins, deregulation of phosphorylated AMPA and metabotropic glutamate receptors, and profoundly altered synaptic structure, all of which occurred later in the prion-infected control mice. Finally, we found that neuronal Hrs (nHrs) depletion increased surface levels of the cellular prion protein, PrPC, which may contribute to the rapidly advancing disease through neurotoxic signaling. Taken together, the reduced Hrs in the prion-affected brain hampers ubiquitinated protein clearance at the synapse, exacerbates postsynaptic glutamate receptor deregulation, and accelerates neurodegeneration.
SIGNIFICANCE STATEMENT Prion diseases are rapidly progressive neurodegenerative disorders characterized by prion aggregate spread through the central nervous system. Early disease features include ubiquitinated protein accumulation and synapse loss. Here, we investigate how prion aggregates alter ubiquitinated protein clearance pathways (ESCRT) in mouse and human prion-infected brain, discovering a marked reduction in Hrs. Using a prion-infection mouse model with neuronal Hrs (nHrs) depleted, we show that low neuronal Hrs is detrimental and markedly shortens survival time while accelerating synaptic derangements, including ubiquitinated protein accumulation, indicating that Hrs loss exacerbates prion disease progression. Additionally, Hrs depletion increases the surface distribution of prion protein (PrPC), linked to aggregate-induced neurotoxic signaling, suggesting that Hrs loss in prion disease accelerates disease through enhancing PrPC-mediated neurotoxic signaling.
Keywords: amyloid, ESCRT, neurodegeneration, proteostasis, synapse, ubiquitin
Introduction
Prion diseases are rapidly progressive neurodegenerative disorders, characterized pathologically by aggregated prion protein (PrPSc), reactive astrocytic gliosis, neuronal spongiform degeneration, and synaptic loss in the CNS (Van Everbroeck et al., 2000; Gambetti et al., 2003; Sikorska et al., 2004). PrPC is highly expressed in neurons (Collinge et al., 1994; Fournier et al., 1995; Herms et al., 1999) and binds PrPSc as well as amyloid-β oligomers, triggering synaptic dysfunction (Laurén et al., 2009; Fang et al., 2018). Clinical prion disease is coincident with synapse loss and the formation of large intracellular vacuoles in the soma and neuronal processes (Liberski et al., 1992; Belichenko et al., 2000; Cunningham et al., 2003), yet the pathways triggering synaptic degeneration are unclear.
Pioneering efforts using in vitro models have identified prion conversion sites at the plasma membrane (Veith et al., 2009; Goold et al., 2013) and along the endocytic pathway (Borchelt et al., 1992; Veith et al., 2009), namely, in early recycling endosomes (Godsave et al., 2008; Marijanovic et al., 2009) and in multivesicular bodies (MVBs; Yim et al., 2015). Moreover, detection of PrPSc in endosomes (Borchelt et al., 1992; Magalhães et al., 2005) and exosomes (Fevrier et al., 2004; B.B. Guo et al., 2015) indicates that prions transit through the ESCRT (endosomal sorting complexes required for transport) pathway. Within the ESCRT pathway, endosomal proteins are guided into MVBs for eventual fusion with lysosomes or release in exosomes (Bilodeau et al., 2002; Raiborg et al., 2002; Shih et al., 2002; Bache et al., 2003). The first complex of the pathway, ESCRT-0, is a heterotetramer comprised of two proteins, Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate) and STAM1 (signal transducing adaptor molecule 1) (Asao et al., 1997; Ren et al., 2009), which bind and facilitate the trafficking of ubiquitin-tagged membrane proteins into MVBs. In prion-infected neuroblastoma cells, depleting Hrs disrupts the trafficking of the cellular prion protein, PrPC, into MVBs and sharply reduces prion conversion (Vilette et al., 2015; Yim et al., 2015), suggesting a key role for Hrs in PrPC trafficking and prion propagation.
Multiple lines of evidence suggest that endolysosomal dysregulation may be central to prion disease progression. For example, SORCS1 (VPS10), an endosomal trafficking and sorting protein, and syntaxin-6, a SNARE [soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor] protein involved in retrograde transport, have been identified in genome-wide association studies as risk factors for developing sporadic Creutzfeldt-Jakob disease (sCJD) (C.A. Brown et al., 2014; Jones and Mead, 2020). Additionally, PrPSc deposition sites in the brain correlate with visible endolysosomal defects in neurons, for example, spongiform degeneration (Kovács et al., 2007; Liberski et al., 2010) linked to PIKfyve loss (Lakkaraju et al., 2021). Yet how intraendosomal prion conversion impacts endosomal maturation in neurons and glial cells remains poorly understood, and the relationship between prion replication, intraneuronal vacuolation, and synaptic loss is unclear.
Despite compelling evidence for intracellular prion conversion in vitro, the subcellular sites and functional impact of intracellular prion conversion in vivo have been challenging to investigate. We reasoned that a cell-targeted approach to manipulate the ESCRT pathway provides an opportunity to identify mechanistically how this pathway modulates prion disease progression. We first discovered that prion-infected mice and humans show a profound and specific reduction of Hrs and STAM1, but not other ESCRT proteins. We then used Hrs-depleted neurons and Hrsf/f mice (Tamai et al., 2008) to investigate in a cell-specific manner how Hrs depletion affects prion replication, endolysosomal trafficking, autophagy, and synaptic degeneration. Intriguingly, only mice with neuron-specific Hrs depletion showed a marked disease acceleration and the early development of severe postsynaptic biochemical and structural defects, including the accumulation of ubiquitinated proteins at the synapse, an expanded postsynapse, and features of deregulated glutamate homeostasis. Collectively, these findings provide evidence that reduced neuronal Hrs (nHrs) exacerbates synaptic degeneration and may contribute to the rapid clinical progression characteristic of prion disease. This study also shows marked ESCRT-0 alterations at the synapse in a human neurodegenerative disease, and offers an entry point into understanding the disrupted clearance of ubiquitinated synaptic proteins.
Materials and Methods
Mouse lines and animal care
Hrsf/f mice (C57BL/6J background; Tamai et al., 2008) were bred to Syn1-Cre, GFAP-Cre, or LysM-Cre mice (The Jackson Laboratory). The synapsin-1 promoter drives widespread neuron-specific Cre recombinase expression from approximately E13, however with lower Cre expression in the cerebellum (Zhu et al., 2001). Hrsf/f Cre+ and Cre− littermates were maintained under specific pathogen-free conditions on a 12/12 h light/dark cycle. Mice had access to standard laboratory chow and water ad libitum.
All animal studies were performed following procedures to minimize suffering and were approved by the Institutional Animal Care and Use Committee at University of California, San Diego (UCSD). Protocols were performed in strict accordance with good animal practices, as described in the Guide for the Use and Care of Laboratory Animals published by the National Institutes of Health.
Patients with sporadic Creutzfeldt-Jakob disease and controls
The sCJD brain tissues used in this study were obtained from a prospective study of sCJD patients evaluated between July 2015 and 2018 at University of California, San Francisco (UCSF) Memory and Aging Center clinical research program for rapidly progressive neurologic disease (Extended Data Table 1-1). All patients with sCJD had extensive clinical testing, including brain MRI, CSF analysis for 14-3-3, neuron-specific enolase (NSE), and total tau, and were classified premortem as probable sCJD by UCSF clinical and radiologic diagnostic criteria (Geschwind et al., 2007; Staffaroni et al., 2017). Cases were diagnosed postmortem as sCJD by Western blotting.
sCJD Patient demographic data and clinical features Download Table 1-1, DOCX file (14.4KB, docx) .
Autopsy verified, nonpathologic, aged (control) brain samples were obtained from the Neuropathology Core and brain bank of the Shiley-Marcos Alzheimer's Disease Research Center at University of California, San Diego (UCSD; Extended Data Table 1-2, Table 1-3). These control brains were devoid of significant neurodegenerative disease pathology (postmortem) and disease-related cognitive impairment at the antemortem assessment proximate to death.
Control case demographic data Download Table 1-2, DOCX file (13.4KB, docx) .
Demographic data summary of sCJD cases and controls Download Table 1-3, DOCX file (13.1KB, docx) .
This study was approved by the ethics committee at University of California, San Francisco. Informed consent was received by all patients (IRB Study number 10-04905). All brain tissues used were de-identified samples collected at autopsy (NIH Office of Human Subjects Research Protections, exemption 4).
PRNP genotyping
The open reading frame of the PRNP gene was sequenced from all patient samples to test for mutations in the PrPC sequence and to determine the genotype at polymorphic codon 129 (methionine or valine). There were no mutations discovered in any of the patients with sCJD. The sCJD prion subtype was determined by Western blot analysis at the National Prion Disease Pathology Surveillance Center (NPDPSC) at Case Western Reserve University (Extended Data Fig. 4-1).
Primary neuron culture
Primary cortical neurons from postnatal day (P)0 Hrsf/f mouse pups were cultured in neurobasal media containing 2% B27-Plus supplement and 1× GlutaMAX (all from Invitrogen). In brief, the cerebral cortices were dissected in buffer (1 m MgCl2, 1 m CaCl2,1 m HEPES, and 2.77 m glucose), dissociated with 0.25% trypsin at 37°C for 20 min, treated with DNase, and triturated. Debris was removed by passing the cells through a 70-µm cell strainer. Cells were then centrifuged for 10 min and resuspended in supplemented neurobasal media.
Prion inoculations
Groups of 10–15 male and female Hrsf/f Cre+ and Cre− littermates (6–11 weeks old) were anesthetized with ketamine and xylazine and inoculated into the left parietal cortex with 30 µl of 1% 22L prion-infected brain homogenate from terminally-ill mice in sterile PBS. Strain 22L is a mouse-adapted prion originally derived from sheep scrapie (Kim et al., 1990; a kind gift from Michael Oldstone). Prion-inoculated mice were monitored three times weekly for the development of terminal prion disease, including ataxia, kyphosis, stiff tail, hind leg clasp, and hind leg paresis, and were euthanized at intermediate timepoints or at the onset of terminal disease. The brain was halved, and one hemisphere was immediately fixed in formalin. Specific brain regions were separately collected for certain experiments. Fixed brains were treated for 1 h in 96% formic acid, postfixed in formalin for 2–4 d, cut into 2-mm transverse sections, and paraffin-embedded for histologic analysis. The remaining hemi-brain was immediately frozen for biochemical studies. Survival time was calculated from the day of inoculation to the day of terminal clinical disease. No mice were excluded from the analysis except where noted in the figure legend.
Immunoblot analysis of mouse brains
Brain tissue was homogenized in PBS using a Beadbeater tissue homogenizer. For PrPSc levels, protein levels were quantified using a BCA (bicinchoninic acid) protein assay, and equal amounts of homogenates were lysed in PBS containing 2% sarcosyl and endonuclease (benzonase), and digested with 50 µg/ml proteinase-K (PK) at 37°C for 30 min, with the reaction stopped by boiling samples for 5 min in LDS loading buffer (Invitrogen). Samples were electrophoresed in 10% or 4–12% Bis-Tris gels (Invitrogen) and transferred to a nitrocellulose membrane by wet blotting. Membranes were incubated overnight with monoclonal antibody POM1 (discontinuous epitope at C-terminal domain; Polymenidou et al., 2008) at 1:10,000 in blocking buffer followed by a 1-h incubation with an HRP-conjugated anti-mouse IgG secondary antibody (Jackson ImmunoResearch).
For the experimental measurement of Hrs, STAM1, Tsg101, and CHMP2B levels in prion-infected brain, previously inoculated groups of male and female C57BL/6, VMDK, tga20, or glycosylphosphatidylinositol (GPI)-anchorless PrPC expressing mice (also inoculated into the left parietal cortex) (Aguilar-Calvo et al., 2017, 2018, 2020) were used for Western blotting.
To analyze shed (ADAM10-cleaved) PrPSc levels, PrPSc was concentrated by performing sodium phosphotungstic acid precipitation before Western blotting (Wadsworth et al., 2001). In brief, 10% brain homogenate in an equal volume of 4% Sarkosyl in PBS was digested with benzonase followed by treatment with 50 μg/ml PK at 37°C for 30 min. After addition of 4% sodium phosphotungstic acid in 170 mm MgCl2 and protease inhibitors (Complete TM, Roche), extracts were incubated at 37°C for 30 min and centrifuged at 18,000 × g for 30 min at 25°C. Pellets were resuspended in 2% N-lauryl sarcosine before electrophoresis and immunoblotting. Membranes were incubated with sPrPG228 antibody (Linsenmeier et al., 2018), and developed using a chemiluminescent substrate. After developing, the same membranes were then stripped with Restore Western blot stripping buffer (Thermo Fisher Scientific), and then incubated with anti-prion protein (PrP) POM1 antibody.
For synaptic and endolysosomal protein level analysis, brain homogenates were lysed in PBS containing 2% sarcosyl, benzonase, PhosStop, and Complete Mini protease inhibitor for 30 min on ice, and then centrifuged for 5 min at 2000 × g to remove debris. Equal amounts of brain protein were loaded into gels for Western blot analysis. Actin or GAPDH was used as a loading control. For probing phosphorylated proteins, membranes were stripped twice for 15 min each with Restore Stripping Buffer (Thermo Fisher Scientific) and reprobed for total protein.
Immunoblot analysis of sCJD brain samples
Frontal cortex was homogenized in PBS using a Beadbeater tissue homogenizer. Protein levels were quantified using a BCA protein assay, and 30 µg of protein per sample was lysed in PBS containing 2% sarcosyl, endonuclease (benzonase), phosphatase inhibitors (PhosStop), and protease inhibitors (Complete Mini) for 15 min at 37°C, and then centrifuged for 30 s at 18,000 × g to remove debris. Samples were boiled for 5 min in LDS loading buffer (Invitrogen) and electrophoresed through a 10% Bis-Tris gel (Invitrogen) before transfer to a nitrocellulose membrane.
Antibodies for Western blottings
The following antibodies were used for Western blotting: anti-PrP (1:10,000, mouse anti-PrP POM1; Polymenidou et al., 2008), anti-Hrs (1:5000, Cell Signaling Technology), anti-β actin (1:5000, Genetex), anti-GAPDH (1:5000, Novus), anti-STAM1 (1:1000, Cell Signaling Technology), anti-Tsg101 (1:500, Cell Signaling Technology), anti-CHMP2B (1:1000, Cell Signaling Technology), anti-ubiquitin (1:7000, Dako), anti-Rab11a (1:1000, Cell Signaling Technology), anti-Rab7 (1:1000, Cell Signaling Technology), anti-Rab5 (1:1000, Cell Signaling Technology), anti-GluN1 (1:1000, Cell Signaling Technology), anti-VPS35 (1:10,000, Genetex), anti-LC3-I/II (1:1000, Cell Signaling Technology), anti-p62 (1:4000, Abnova), anti-synapsin-1 (1:10,000, Thermo Fisher Scientific), anti-synaptophysin (1:10,000, Invitrogen), anti-PSD95 (1:5000, Calbiochem), anti-K63 linked ubiquitin (1:1000, Cell Signaling Technology), anti-K48 linked ubiquitin (1:1000, Cell Signaling Technology), anti-VAMP2 (1:10,000, Cell Signaling Technology), anti-SNAP25 (1:10,000, Cell Signaling Technology), anti-mGluR5 (1:2000, Cell Signaling Technology), anti-GluA1 (1:1000, Cell Signaling Technology), anti-phosphorylated GluA1 S831 (1:1000, Cell Signaling Technology), and anti-phosphorylated GluA1 S845 (1:1000, Cell Signaling Technology).
Immunohistochemical labeling and histologic analysis of mouse brains
Tissue sections were cut from blocks of formalin-fixed paraffin embedded mouse brain. Four-micron tissue sections were stained on a Ventana Discovery Ultra (Ventana Medical Systems) with antibodies to glial fibrillary acidic protein (GFAP) (1:6000; Dako), ionized calcium-binding adaptor molecule 1 (Iba-1) (1:3000; Wako), ubiquitin (1:3000; Dako), NeuN (1:1000; Millipore Sigma), and lysosomal-associated membrane protein 1 (LAMP1) (Developmental Studies Hybridoma Bank, University of Iowa;1:150). Antigen retrieval was independently optimized for each epitope to yield the maximal signal-to-noise ratio. For PrP, slides were incubated in protease 2 (P2; Ventana) for 20 min followed by antigen retrieval in CC1 (Tris-based; pH 8.5; Ventana) for 64 min at 95°C. For GFAP, only the protease P2 was used (Ventana) for 16 min. For LAMP1, Iba-1, and ubiquitin, all retrieval employed CC1 (Tris-EDTA, pH 8.5) for either 24 (LAMP1) or 40 min (Iba-1, ubiquitin, NeuN). Following retrieval, all antibodies were incubated on the tissue for 32 min at 37°C. The secondary antibody (HRP-coupled goat anti-rabbit; OmniMap system; Ventana) was applied to the sections for 12 min at 37°C. The primary antibody was visualized using the DAB (3′-diaminobenzidine) chromagen followed by a hematoxylin counterstain. Because of the low expression of LAMP1, an additional amplification step was included before the DAB reaction using the Ventana HQ-Amp system for 12 min.
Transmission electron microscopy of mouse brains
Brain sections from uninfected and prion-infected wild-type (WT), Hrsf/fSyn1-Cre+, and Hrsf/fSyn1-Cre− mice were fixed (transcardially perfused or immersion-fixed) in modified Karnovsky's fixative (2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 m sodium cacodylate buffer, pH 7.4) to obtain high resolution ultrastructure of the synapses. Following initial fixation, all brain samples were treated for 1 h in 96% formic acid, washed in 0.15 m sodium cacodylate buffer, and postfixed in modified Karnovsky's fixative.
CA1 hippocampal sections were then immersed in 1% osmium tetroxide in 0.15 m cacodylate buffer for 1 h and stained in 2% uranyl acetate for 1 h. Samples were dehydrated in ethanol, embedded in Durcupan epoxy resin (Sigma-Aldrich), sectioned at 50–60 nm on a Leica UCT ultramicrotome, and placed on Formvar and carbon-coated copper grids. Sections were stained with 2% uranyl acetate for 5 min and Sato's lead stain for 1 min. Grids were viewed using a JEOL 1200EX II (JEOL) transmission electron microscope and photographed using a Gatan digital camera (Gatan).
The electron microscopy images from the CA1 region of the hippocampus were analyzed using ImageJ. N represents number of synapses analyzed across 12 mice. Degree of curvature was determined by measuring the angle of the synapses using three points along the synaptic cleft. Postsynaptic density (PSD) area was quantified by capturing the electron dense region underlying the postsynaptic membrane.
qRT-PCR for hrs transcripts
Brain samples were collected and rapidly frozen on dry ice. To isolate RNA from brain, brains were thawed in RNAice Later (Thermo Fisher Scientific), lysed with PureLink RNA isolation lysis buffer, immersed in 5.3 m guanidine isothiocyanate for 24 h to denature proteins. RNA was isolated using the PureLink RNA isolation kit (Thermo Fisher Scientific). cDNA synthesis was performed using the Promega cDNA synthesis kit, random hexamers, M-MLV RT buffer 5×, dNTP mix (10 mm), and M-MLT RT. qRT-PCR was performed on a StepOnePlus (Applied Biosystems) instrument using cDNA, TaqMan Gene Expression Master MixII, AmpErase Uracil N-Glycosylase (Life Technologies), and FAM-labeled primers for Hgs (assay ID: Mm00468635_m1, 4331182, Thermo Fisher Scientific) and control murine GAPDH (Thermo Fisher Scientific). Three mice per group were analyzed in triplicate.
Lentivirus generation
To generate the recombinant lentivirus, HEK293T cells at 80% confluency were co-transfected with the shuttle vector (Cre-FUWG2, empty-FUWG2, or mCherry-GFP-LC3) and the two helper plasmids, delta8.9 and VSV-G envelope vector, using a CalPhosTM transfection kit (Takara). The media was centrifuged at 2000 × g for 10 min, filtered through a 0.22-μm filter, incubated with PEG-it (System Bioscience) overnight at 4°C, and centrifuged at 1500 × g for 30 min at 4°C. The media was removed from the viral pellet, and the pellet centrifuged again at 1500 × g for 5 min. Pelleted viral particles were resuspended in cold sterile PBS and aliquoted for storage at −80°C.
PrPSc partial purification
22L prion-infected and uninfected 10% brain homogenates were lysed in 2% sarcosyl in PBS containing benzonase and 50 mm MgCl2, incubated for 30 min at 37°C, and centrifuged at 4°C at 18,000 × g for 30 min. The pellets containing PrPSc were resuspended in 100 μl of PBS and heated to 65°C to sterilize samples for cell culture studies.
Quantification of PrPSc levels in neurons
Primary cortical neurons from P0 Hrsf/f mouse pups plated in 12-well plates for 6 d in vitro (DIV) were transduced with either Cre-lentivirus or the vector control lentivirus. Five days after transduction, the cells were infected with partially purified 22L prions or similarly prepared mock brain for 3 d. Neurons were then washed with PBS, lysed on ice in 2% sarcosyl with benzonase for 30 min, and proteins quantified by BCA. Equivalent levels of protein (100 μg) were aliquoted and digested with PK (10 μg/ml final concentration) for 30 min at 37°C. PrPSc in the cell lysates was measured by Western blotting using anti-prion protein (PrP) antibody, POM1 (Polymenidou et al., 2008).
For quantification of additional proteins, equal protein concentrations of prion-infected and uninfected neuronal lysates were immunoblotted. Protein signals were captured and quantified using the Fuji LAS 4000 imager and Multigauge V3.0 software. N = 4 experiments, each with technical duplicates.
Cell surface protein biotinylation and analysis
Primary cortical neurons from P0 Hrsf/f mouse pups plated in 6-well plates were transduced with either Cre-lentivirus or vector control-lentivirus (as described above) after 6 DIV. After one week, cells were washed with ice cold PBS. HEPES (0.1 m) with or without biotin (1 mg/ml; Life Technologies) was added to neurons, and neurons were incubated for 30 min rocking on ice at 4°C. The reaction was quenched twice with 100 mm ice-cold glycine in 0.1 m HEPES buffer for 15 min, washed with PBS, and lysed in IP buffer (10 mm Tris-HCl, 150 mm NaCl, 10 mm EDTA, 0.5% NP40, 0.5% DOC; pH 7.4 with CompleteMini protease inhibitors). The lysate was then incubated on ice for 30 min, and centrifuged at 10,000 × g for 5 min. The supernatant was collected, and the protein concentration was determined by BCA. Protein was saved for assessing the input. Protein samples were incubated with streptavidin-conjugated magnetic beads overnight at 4°C. The magnetic beads were washed with lysis buffer and proteins eluted in lysis buffer with 0.2 m DTT, boiled at 50°C for 5 min, and then at 95°C for 5 min before loading onto 10% Bis-Tris gels for immunoblotting.
Autophagic flux assessment using mCherry-GFP-LC3 tandem reporter
Hippocampal primary neurons from P0 Hrsf/f mouse pups (100,000 cells/well) cultured on 12-mm round coverslips were transduced with the mCherry-GFP-LC3 tandem reporter lentivirus and Cre-lentivirus or the vector-control lentivirus. One week after transduction, neurons were incubated with bafilomycin (50 nm final) or rapamycin (0.25 μm final) for 4 h. To identify the Cre-transduced cells, cells were then fixed in 4% paraformaldehyde (PFA) and stained with anti-Cre-antibody (Cell Signaling Technology) and an Alexa-647 conjugated secondary antibody. Cells were then fixed with 4% paraformaldehyde (PFA; Fisher Scientific). For the fixation, PFA was diluted 1:1 in media (2% final), added directly to the cells, and incubated for 20 min at 37°C. The PFA was removed and 4% PFA was added for an additional 10 min at room temperature. The cells were then permeabilized in 0.5% Triton X-100 in PBS buffer for 5 min, blocked in 3% BSA in 0.1% Triton X-100 in PBS for 1 h, incubated with primary antibody in blocking buffer overnight at 4°C, and then incubated with secondary antibody (1:250, Jackson ImmunoResearch Laboratories) for 1 h followed by DAPI (Sigma-Aldrich). Coverslips were mounted onto a glass slide using ProLong Gold Antifade mounting media (Invitrogen). For neuron imaging analysis of LC3 puncta, N represents the number of cells quantified from three experiments. Only the immunostained neurons were analyzed.
Western blot analysis
To quantify the relative immunoblot signal intensities, protein signals were captured using a chemiluminescent substrate (Supersignal West Dura ECL, Thermo Fisher Scientific) and visualized on a Fuji LAS 4000 imager. The chemiluminescent signals were quantified using the Multigauge V3.0 software.
Quantification of histology
The Iba-1 and GFAP IHC stained slides were scanned on a Zeiss AxioScan.Z1 (Carl Zeiss Microscopy LLC) using a 40× objective to obtain maximal spatial resolution. The AxioScan.Z1 and software Zen2 (Carl Zeiss Microscopy LLC) was used for automatic thresholding and tissue detection. All tissue sections encompassing the entire rostral-caudal extent of the hemi-brain were scanned using the default stitching parameters to combine the individual tiles into a single image. Whole slide images were imported into Definiens Software for quantitative analysis (Definiens). The color contrast of the DAB and hematoxylin counterstain were used to determine the basic region of interest (ROI). Using the hematoxylin as a guide for the nuclei of individual cells, the output values were all cells within the tissue, cells positive for IHC, and the area of tissue stained for either GFAP or Iba-1. Quantification of LAMP1 area was performed using ImageJ. The percent area covered by LAMP1 in the frontal cortex (layers 1–6) was selected using the Shanbhag auto-threshold. Positive nuclei in the NeuN stain were quantified using ImageJ.
Statistical analyses
Log-rank (Mantel–Cox) tests were performed to assess survival differences between groups. A Student's t test (two-tailed, unpaired) was used to determine the statistical significance between two groups. ANOVA testing was performed using one-way or two-way analysis with Tukey's post hoc test to assess group effects. Statistical tests and outlier determination were performed using GraphPad Prism. All data in bar charts and dot plots are shown as mean ± SEM. For all analyses, p < 0.05 was considered significant.
Results
Hrs levels are markedly reduced in mice infected with diverse prion strains
The ESCRT pathway delivers ubiquitinated membrane proteins into MVBs for clearance by lysosomal degradation or release in exosomes (Bilodeau et al., 2002; Raiborg et al., 2002; Erpapazoglou et al., 2012), tightly regulating select receptors (Schmidt and Teis, 2012), including EGFR (Miller et al., 1986; Katzmann et al., 2002). Given the evidence that prions also traffic into MVBs (Fevrier et al., 2004; Yim et al., 2015), we aimed to evaluate how ESCRT proteins are impacted by prion infection, first measuring ESCRT proteins in prion-infected and uninfected mouse brain by immunoblotting. Surprisingly, Hrs and STAM1 levels were markedly reduced (∼80% and 65%, respectively) in terminal prion-infected brain (strain 22L; Fig. 1A–D). In contrast, Tsg101 (ESCRT-I) was modestly increased and CHMP2B (ESCRT III) was unchanged (Fig. 1C,D), suggesting that the reduction in ESCRT-0 was not simply because of neuron loss or a generally downregulated ESCRT pathway. Notably, mice infected with six additional prion strains, including GPI-anchored (subfibrillar) and GPI-anchorless (fibrillar) prions, also showed markedly reduced Hrs levels at terminal disease (Fig. 1A,B; Extended Data Fig. 1-1), indicating that the Hrs reduction is not strain-specific, but a widespread occurrence in the prion-infected brain. To assess whether human patients also show a reduction in Hrs, we analyzed the frontal cortex of sCJD patients (n = 9) and age-matched controls (n = 11). Similar to prion-infected mice, Hrs was reduced in the frontal cortex by ∼50% (Fig. 1E,F). Hrs (also known as Hgs) transcript levels in terminal prion-infected (22L) and control mice were similar (Fig. 1G), indicating Hrs loss occurs post-transcriptionally.
Figure 1.
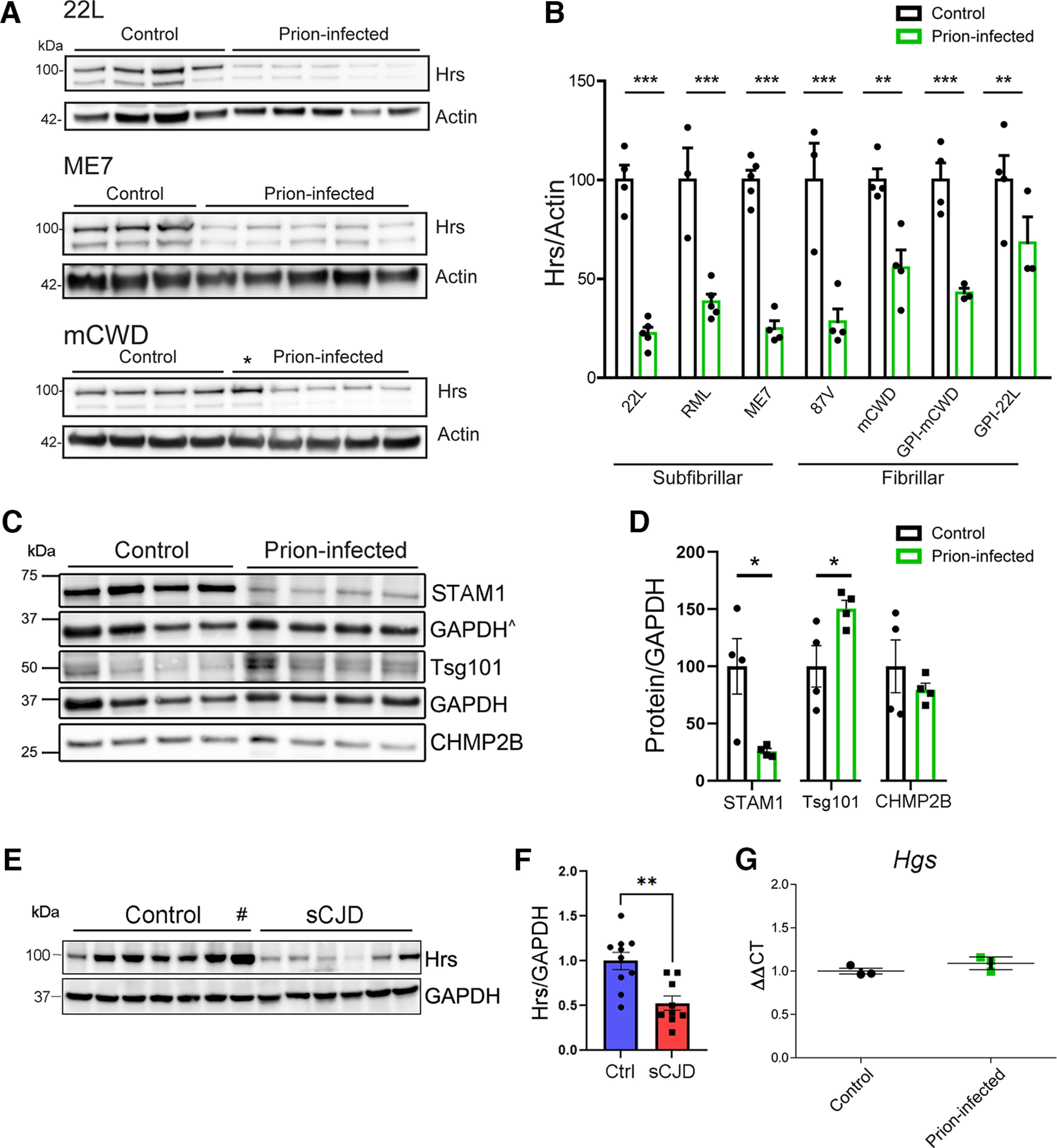
Specific reduction in ESCRT-0 proteins, Hrs and STAM1, in prion-infected mouse and human brain. A, B, Representative Western blottings and quantification of Hrs in whole-brain lysate from uninfected and terminal prion-infected mice challenged with various prion strains (n = 3–5 mice/group). *Mouse was euthanized at an early time point and not quantified (98 dpi, 55% of disease course). C, D, Representative Western blottings and quantification of ESCRT proteins from uninfected control and prion-infected (strain 22L) mice (n = 4 mice/group). ^GAPDH signal used for normalizing CHMP2B (same membrane). E, F, Representative Western blottings and quantification of Hrs in frontal cerebral cortex of unaffected or sCJD affected brain samples (n = 11 controls, n = 9 sCJD). #Outlier identified by Grubbs' test (excluded from graph). G, Hgs transcript levels in uninfected and prion-infected (22L) mouse brains. Relative protein levels or transcripts were normalized to the average of the unaffected mice or humans. Data shown as mean ± SEM; each dot represents an individual mouse or human; unpaired, two-tailed Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001. Data supported by Extended Data Figure 1-1 and Extended Data Tables 1-1, 1-2, and 1-3.
Reduced Hrs in brain of WT and transgenic mice infected with distinct prions. Hrs immunoblots of brain lysates from prion-infected and uninfected control WT and transgenic mice expressing GPI-anchorless PrPC (quantification shown in Fig. 1B). Prion strains include mouse-adapted prions RML, 87V, GPI-anchorless mCWD, and GPI-anchorless 22L. *Mouse euthanized because of intercurrent illness at ∼42% of the expected survival time. Download Figure 1-1, TIF file (7MB, tif) .
Hrs depletion in astrocytes or microglia does not alter prion disease progression
To investigate how Hrs impacts prion disease, we depleted Hrs in a cell-specific manner using an Hrsf/f mouse model, which has loxP sites surrounding exons 2–4 and a frameshift that results in a stop codon in exon 5 (Tamai et al., 2008). Single-cell transcripts from cerebral cortex show higher Hrs transcripts in neurons as compared with astrocytes and microglia (Allen Brain Atlas) (mouse and human; Extended Data Fig. 2-1; Allen Institute for Brain Science, 2020, 2021; Yao et al., 2021a,b). Neuronal Hrs depletion in Hrsf/fSyn1-Cre mice (synapsin-1 promoter) has been linked to growth retardation and focal degeneration of CA3 hippocampal neurons during aging (Tamai et al., 2008). To deplete Hrs in astrocytes, microglia, and neurons, we crossed Hrsf/f mice with GFAP-Cre (Hrsf/fGFAP-Cre+), LysM-Cre (Hrsf/fLysM-Cre+), and synapsin-1-Cre (Hrsf/fSyn1-Cre+) mice, respectively, and inoculated Cre+ and Cre− littermate control mice with 22L prions (Fig. 2A). The 22L prion strain replicates in astrocytes (Carroll et al., 2016); however, we found no differences in the survival times of prion-infected Cre+ and Cre− mice with astrocytic Hrs depletion [prion-infected Hrsf/fGFAP-Cre+: 153 ± 2 vs Cre−: 151 ± 2 d postinoculation (dpi); Fig. 2B]. Additionally, there were no differences in survival times following microglial Hrs depletion (Hrsf/fLysM-Cre+: 143 ± 2 vs Cre−: 146 ± 1 dpi; Fig. 2C). PrPSc levels and distribution, spongiform degeneration, and astrocyte reactivity were indistinguishable in Cre+ and Cre− mice from Hrsf/fGFAP-Cre and Hrsf/fLysM-Cre lines (Fig. 2D–G; Extended Data Fig. 2-2A). Ubiquitinated proteins showed an upward trend in Hrsf/fGFAP-Cre+, but not in the prion-infected Hrsf/fLysM-Cre+ brain (Extended Data Fig. 2-2B,C). To further confirm these results, we repeated the prion transmission experiment in Hrsf/fGFAP-Cre and Hrsf/fLysM-Cre mice and evaluated brains at the 75% time point of disease (115 and 114 dpi), and yet again found no differences in PrPSc levels or brain lesions (Extended Data Figs. 2-2A, 2-3), suggesting that ESCRT-0 in astrocytes and microglia does not significantly impact prion pathogenesis.
Figure 2.
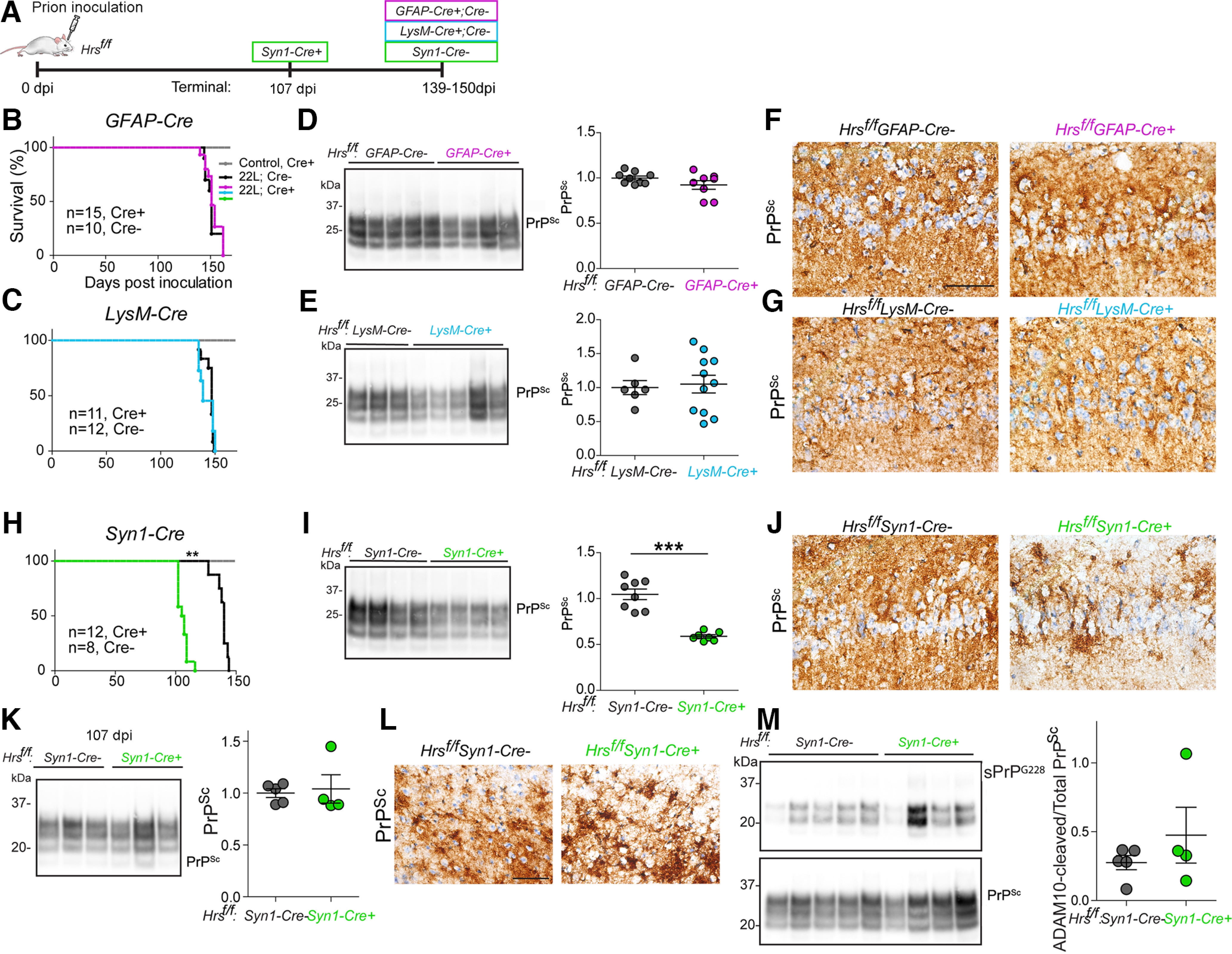
Neuronal, but not astrocytic or microglial, Hrs depletion accelerates prion disease progression. A, Schematic illustrates the timeline of disease progression for prion-inoculated mice. Days to average onset of terminal prion disease are shown. Created with biorender.com. B, C, Survival curves of Hrsf/fGFAP-Cre (B) and Hrsf/fLysM-Cre (C) intracerebrally inoculated with prions (gray line shows uninfected Cre+ control mice). D, E, Representative Western blottings and quantification of PK-resistant PrPSc in brain lysates (n = 8-9 GFAP-Cre mice/group; n = 6–11 LysM-Cre mice/group). F, G, Brain sections immunolabeled for PrP show diffuse PrPSc deposits in the hippocampus (CA1). H–J, Survival curves, representative Western blottings and quantification of PrPSc, and PrP-immunolabeled hippocampal sections from prion-inoculated Hrsf/fSyn1-Cre mice (n = 7–8 Syn1-Cre mice/group). K, Western blottings and quantification of PrPSc in PK-digested brain lysates from Hrsf/fSyn1-Cre− and Cre+ mice collected at 107 dpi (n = 4–5 mice/group). L, Brain sections immunolabeled for PrP show similar diffuse PrPSc deposits in the cerebral cortex. M, Western blottings and quantification of PrPSc in NaPTA-precipitated, PK-digested brain lysates from Hrsf/fSyn1Cre− and Cre+ mice at 107 dpi with antibodies against ADAM-10-cleaved PrP (sPrPG228) or total PrPSc (POM1; n = 4–5 Hrsf/fSyn1-Cre mice). Log-rank (Mantel–Cox) test (panels B, C, H) and unpaired, two-tailed Student's t test (panels D, E, I, K, M), **p < 0.01 and ***p < 0.001. Scale bars = 100 µm. Data supported by Extended Data Figures 2-1, 2-2, 2-3, 2-4, and 2-5.
Mouse and Human Hgs transcripts. Single-cell RNAseq data from the “Allen cell types database – mouse whole cortex and hippocampus [dataset]” (Allen Institute for Brain Science, 2020) and “Allen cell types database – human multiple cortical areas [dataset]” (Allen Institute for Brain Science, 2021) was used to plot the Hgs transcript levels in the mouse or human cortex. A, Heatmap representing the average levels of Hgs in excitatory, inhibitory, and non-neuronal cells (astrocytes and microglia; Class) from mouse cerebral cortex. Neuronal populations were further subdivided (subclass). Hgs represented as copy number per million (CPM) + 1. B, Bar graph of the same data in A showing the distribution and average Hgs expression in excitatory and inhibitory neuronal populations, as well as astrocytes and microglia. Each dot represents an individual cell. C, Bar graph showing the HGS levels in excitatory and inhibitory neurons, as well as astrocytes and microglia cells in human cerebral cortex. Each dot represents the CPM trimmed mean expression per subclass. Subclass was determined using taxonomy of transcriptomic cell types previously published. For B, C, transcriptomic cell type was used to determine cell identity (Yao et al., 2021a). Bar graphs: data shown as mean ± SEM; one-way ANOVA with Tukey's multiple comparisons post hoc test. ***p < 0.001. Download Figure 2-1, TIF file (12.3MB, tif) .
Spongiform degeneration and astrocyte reactivity in prion-infected Hrsf/fGFAPCre+ and Hrsf/fLysCre+ brain were similar to controls. A, Representative hematoxylin and eosin (HE) and GFAP (astrocytes)-immunolabeled brain sections (cerebral cortex) from prion-infected Hrsf/fGFAP-Cre− and Cre+ mice (top) and Hrsf/fLys-Cre− and Cre+ mice (bottom) at both preterminal and terminal disease stages. B, C, Western blottings and quantification of ubiquitinated protein levels in terminal (B) Hrsf/fGFAP-Cre− and Cre+ and (C) Hrsf/fLysM-Cre− and Cre+ brain (n = 4–5/group). Data shown as mean ± SEM; each dot represents an individual mouse; unpaired, two-tailed Student's t test, Scale bar = 100 µm. Download Figure 2-2, TIF file (19MB, tif) .
Neuronal Hrs depletion profoundly accelerates prion disease
Uninfected Hrsf/fSynCre+ mice showed ∼50% reduction in Hrs and STAM1 levels in the rostral brain, which included cerebral cortex, basal ganglia, hippocampus, and thalamus [53 ± 9% (Hrs) and 51 ± 8% (STAM1); Cre expression is low in the cerebellum; Zhu et al., 2001; Extended Data Fig. 2-4A], albeit with no differences in total PrPC levels (Extended Data Fig. 2-4B). Remarkably, Hrs depletion in neurons accelerated prion disease progression by nearly 25% (Cre+: 106 ± 2 vs Cre−: 139 ± 2 dpi; Fig. 2H), with Cre+ mice showing typical terminal signs of prion disease, including kyphosis, ataxia, and inactivity. The short survival time was surprising considering that Hrs depletion in cultured cells reportedly reduces prion conversion (Vilette et al., 2015; Yim et al., 2015), which we reproduced using Hrs-depleted primary neurons infected with prions (Extended Data Fig. 2-5A). We noted that Hrs-depleted neurons showed no change in total PrPC yet show an increase (30%) in biotinylated surface PrPC expression (Extended Data Fig. 2-5B,C), suggesting that reducing Hrs in neurons leads to a retention of cell surface PrPC.
Similar PrPSc levels in prion-infected Hrsf/fGFAP-Cre and Hrsf/fLysM-Cre brains at the 75% time point. Western blotting and quantification of PrPSc levels in PK-digested brain lysates of (A) Hrsf/fGFAP-Cre− and Cre+ (top) and (B) Hrsf/fLysM-Cre− and Cre+ mice (bottom) at 114 and 115 dpi (∼75% of the expected incubation period). There were no differences in PrPSc levels (n = 3/group). Data shown as mean ± SEM; each dot represents an individual mouse; unpaired, two-tailed Student's t test. Download Figure 2-3, TIF file (8.7MB, tif) .
ESCRT-0 proteins, Hrs and STAM1, are reduced in Hrsf/fSyn1-Cre mice. A, Western blotting and quantification of brains of Syn1-Cre+ and Cre− mice show a reduction in Hrs and STAM1 protein levels (n = 4/group). B, Total PrPC levels are unchanged in neuronal Hrs-depleted brain (n = 3–5/group). Data shown as mean ± SEM; each dot represents an individual mouse; unpaired, two-tailed Student's t test, *p < 0.05. Download Figure 2-4, TIF file (9.2MB, tif) .
Hrs depletion in primary neurons reduces prion conversion and increases PrPC cell surface expression. A, B) Representative Western blotting and quantification of PK-digested (A) or undigested (B) Hrsf/f primary neuron lysates transduced with lentivirus encoding control vector or Cre (Cre− or Cre+) reveals a decrease in PrPSc in Hrs-depleted neurons but no change in PrPC expression. Relative PrPC and PrPSc levels were normalized to the average of the Cre− control lysates (n = 8 from 4 independent experiments, each with technical duplicates, mean and SEM shown). C, Representative Western blottings and quantification of biotin-tagged, streptavidin-precipitated cell surface PrPC from Hrsf/f primary neuron lysates reveal increased levels of surface PrPC in Hrs-depleted neurons. Control with no biotin labeling also shown (n = 4 experimental replicates). Data shown as mean ± SEM. Unpaired, two-tailed Student's t test, *p < 0.05, ***p < 0.001. Download Figure 2-5, TIF file (12.5MB, tif) .
To determine whether the disease acceleration was because of more rapid prion conversion or prion spread, we measured the proteinase-K (PK) resistant PrPSc levels at terminal disease. Notably, there was less PrPSc in the terminal Hrsf/fSyn1-Cre+ brain (45 ± 2% lower than control; Fig. 2I,J). To directly compare PrPSc levels between Hrsf/fSyn1-Cre+ and Hrsf/fSyn1-Cre− mice (hereafter referred to as Cre+ and Cre−) at the same time point, we repeated the experiment and assessed the PrPSc levels and histologic features in prion-infected brain at 107 dpi, when the Cre+, but not the Cre−, mice were showing terminal clinical signs, including kyphosis, weight loss, and ataxia. Here, we observed no differences in the PrPSc levels between the Cre+ and Cre− mice as both were similarly low as compared with terminal Cre− mice (Fig. 2K), suggesting that prion conversion rate was unaffected by Hrs depletion. Additionally, there were no differences in PrPSc morphology or distribution (Fig. 2L) or in ADAM10-cleaved PrPSc levels at 107 dpi (Fig. 2M). Therefore, neuronal Hrs (nHrs) depletion markedly accelerated disease progression in the context of unaltered prion conversion kinetics, revealing an uncoupling of prion aggregate load and neurotoxicity.
We next assessed the possibility that spongiform degeneration or neuroinflammation was driving the early disease onset in the Cre+ mice. However, there was significantly less spongiform change and microglial activation in the terminal Cre+ mice (Fig. 3A–C), reflective of a short incubation period (note: Cre+ and Cre− brains at the same time point showed similar mild spongiform change; Extended Data Fig. 3-1). In stark contrast, astrocyte activation was consistently increased in terminal Cre+ brain (Fig. 3D,E). Thus, terminal Cre+ mice showed PrPSc levels, spongiosis, and microglial activation similar to Cre− mice at 107 dpi, while astrocytic activation was excessive, revealing an unusual uncoupling in the microglial and astrocytic response (astrogliosis and microgliosis typically occur in parallel in prion disease; Ojeda-Juárez et al., 2022). Collectively, these data suggest that neither high PrPSc levels nor spongiform change were driving the acceleration to terminal disease, while the astrocytic reactivity may reflect increased synaptic dysregulation or higher metabolic activity, the latter recently reported in a familial prion disease organoid model (Foliaki et al., 2023).
Figure 3.

Reduced microgliosis with increased astrocytic response in the brains of prion-infected neuronal Hrs-depleted mice. A, Representative hematoxylin and eosin (HE) prion-infected Hrsf/fSyn1-Cre− and Cre+ brain sections (thalamus). Iba1 (microglia)-immunolabeled (B, C) and GFAP (astrocytes)-immunolabeled (D, E) brain sections and quantification of labeled area (thalamus; n = 3–4 Hrsf/fSyn1-Cre mice/group). Scale bar = 100 µm. Data shown as mean ± SEM. Unpaired, two-tailed Student's t test, **p < 0.01. Data supported by Extended Data Figure 3-1.
No change in the developmental timeline of spongiform change or lysosomal accumulation in the cortex of neuronal Hrs-depleted mice. Representative hematoxylin and eosin (HE) and LAMP1 (lysosomes)-immunolabeled brain sections (cerebral cortex) from prion-infected Hrsf/fSyn1-Cre− and Cre+ mice collected at the same time point (107 dpi). Scale bar = 100 µm. Download Figure 3-1, TIF file (10.3MB, tif) .
Hrs depletion impairs ubiquitinated protein clearance at the synapse
Depleting ESCRT proteins reportedly disrupts autophagic flux in cultured cells (Filimonenko et al., 2007; J.A. Lee et al., 2007; Tamai et al., 2007). We depleted Hrs in cultured Hrsf/f neurons using a Cre-lentivirus and found that autophagosomes accumulated when autophagy was induced with rapamycin (Extended Data Fig. 4-1A–D).
Hrs depletion impacts neuronal autophagic flux. A, Schematic of mCherry-GFP-LC3 pH-sensitive sensor. GFP is quenched in the low pH of lysosomes, while mCherry is unaffected (autophagosomes and lysosomes appear yellow or red, respectively). B, Hrsf/f primary neurons transduced with mCherry-GFP-LC3 and control or Cre recombinase lentivirus for one week, and incubated with an inhibitor of autophagosome–lysosome fusion, bafilomycin (Baf), or inducer of autophagosome formation, rapamycin (Rap). C, Quantification of yellow puncta (autophagosomes) and red puncta (lysosomes) in each cell. Graphs indicate percent autophagosomes in each cell (total = autophagosomes + lysosomes) and (D) number of autophagosomes per cell. Data shown as mean ± SEM (C) and (D) were calculated for n = 3 independent experiments, n = 30–50 cells per condition. One-way ANOVA with Tukey's multiple comparisons post hoc test. *p < 0.05. Download Figure 4-1, TIF file (7.8MB, tif) .
We next measured the autophagic protein levels in prion-infected brain. At terminal disease, prion-infected Cre− brain showed an accumulation of p62 and LC3-II/I, suggesting impaired autophagic clearance as previously reported (Boellaard et al., 1991; Liberski et al., 2008; Xu et al., 2012; López-Pérez et al., 2019, 2020; Fig. 4A). However, terminal prion-infected Cre+ brain showed p62 and LC3-II/I levels similar to uninfected brains (Fig. 4A). Histologically, LAMP1 immunostaining in the brain was also lower in the terminal Cre+ brain as compared with terminal Cre− brain (Fig. 4B), however, in mice collected at the same time point, LAMP1 labeling in Cre− brain was similar to preterminal Cre− brain (Extended Data Fig. 3-1), suggesting that there was no detectable difference in the accumulation of autophagosomes or in lysosome formation, excluding dysregulated autophagy as a likely driver of the accelerated disease progression.
Figure 4.
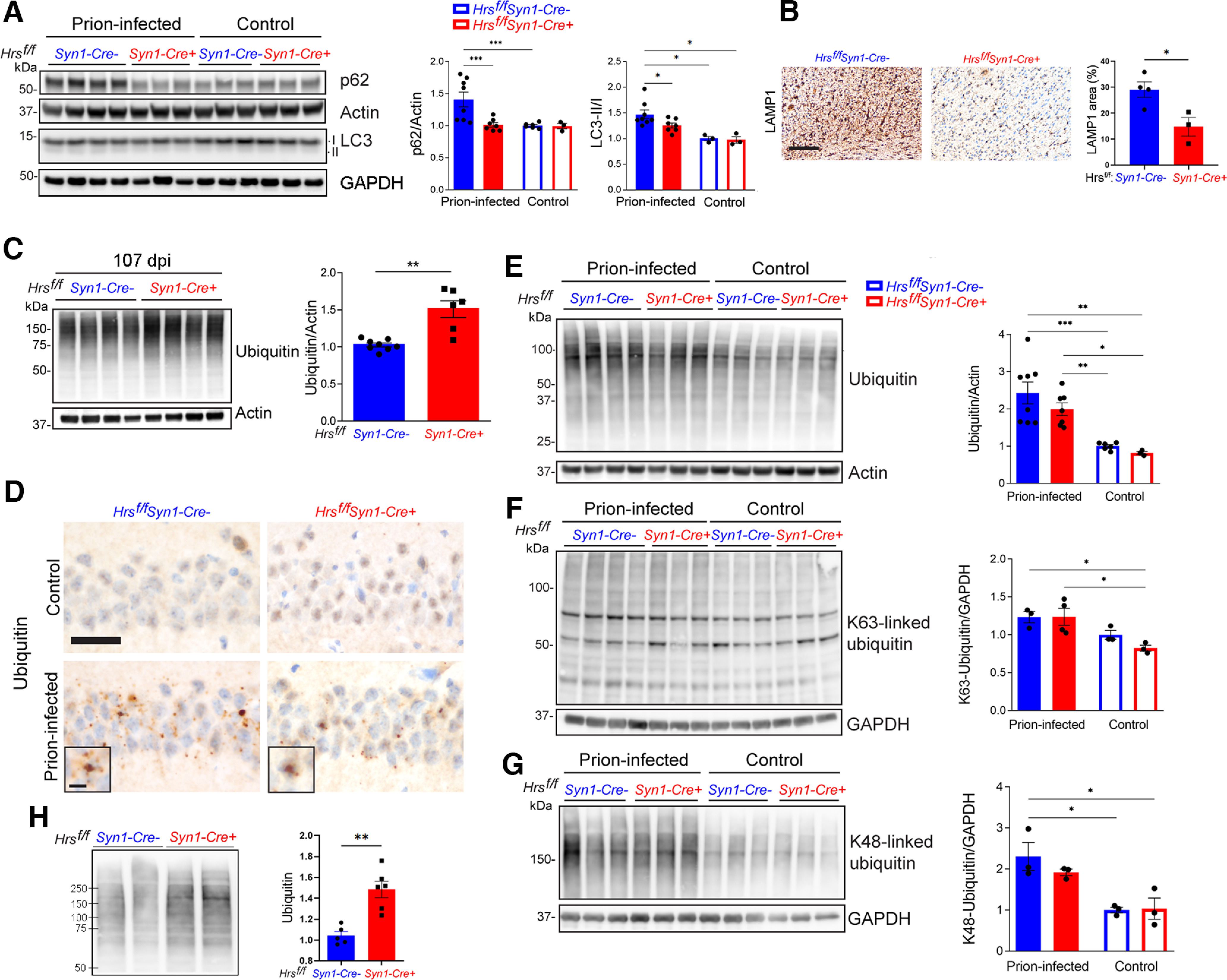
Neuronal Hrs contributes to ubiquitinated protein clearance in prion disease. A, Representative Western blottings and quantification of p62 and LC3 II/I proteins in brain lysates of age-matched uninfected and prion-infected Hrsf/fSyn1-Cre− and Cre+ mice. B, Terminal prion-infected brain sections immunolabeled for LAMP1 (cerebral cortex) and quantification. C, Representative Western blottings and quantification of total ubiquitinated proteins from prion-infected Hrsf/fSyn1-Cre− and Cre+ mice collected at 107 dpi. D, Uninfected or terminal prion-infected brain sections immunolabeled for ubiquitin. E–G, Representative Western blottings and quantification of total, K63-ubiquitinated, and K48-ubiquitinated proteins in uninfected and prion-infected animals at terminal disease (terminal timepoints for Cre− mice: 127, 138, 136, 144 dpi, for Cre+ mice: 102, 102, 102 dpi). H, Representative Western blotting and quantification of ubiquitinated proteins in synaptosomes collected from Hrsf/fSyn1-Cre− and Cre+ brain at 103 dpi. Data shown as mean ± SEM shown. Relative protein levels were normalized to the average of the uninfected mice (n = 3–8 Hrsf/fSyn1-Cre mice/group for immunoblots). Two-way ANOVA with Tukey's multiple comparisons post hoc test (A, E–G), unpaired, two-tailed Student's t test (C, H). *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars = 200 µm (B) and 20 µm (D). Scale bar = 10 µm for ubiquitin inset (D). Data supported by Extended Data Figures 4-1, 4-2, and 4-3.
Considering that ESCRT-0 sorts ubiquitinated membrane proteins within developing MVBs (Chau et al., 1989; Thrower et al., 2000; Lauwers et al., 2009) and that Hrs depletion increases ubiquitinated proteins as observed as a trend here (cortex of young mice; p = 0.13; Extended Data Fig. 4-2) and previously shown by others (hippocampus and cortex, increasing with age; Tamai et al., 2008), we reasoned that ubiquitinated proteins may accumulate at earlier disease stages in the Cre+ mice. By 107 dpi, ubiquitinated proteins were already 50% higher in the prion-infected Cre+ than the Cre− brain, indicating an early marked disruption in the clearance of ubiquitinated proteins (Fig. 4C). Both K63-linked and K48-linked ubiquitinated proteins were higher in Cre+ mice, suggesting that the impairment of both major degradative pathways, lysosomal and proteosomal, was exacerbated by Hrs depletion (Extended Data Fig. 4-3). At terminal disease, ubiquitinated protein accumulation was similarly increased in the Cre− and Cre+ mice (Fig. 4D–G). Collectively, these results indicate that nHrs depletion accelerates ubiquitinated protein accumulation in prion-infected mice.
Ubiquitinated protein levels in the cerebral cortex of uninfected Hrsf/fSyn1-Cre+ and Cre− mice. Western blotting of ubiquitinated proteins shows no significant difference in cerebral cortex from young uninfected Hrsf/fSyn1-Cre+ mice, although there was an upward trend (p = 0.13). All mice were female and ranged between 57 and 73 d old. Data shown as mean ± SEM. Unpaired, two-tailed Student's t test. Download Figure 4-2, TIF file (4.5MB, tif) .
Hrs depletion accelerates ubiquitinated protein accumulation in prion-infected mice. A, B, Representative Western blottings and quantification of the brain lysates reveal an increase in K63-linked and K48-linked ubiquitinated proteins in the prion-infected Hrsf/fSyn1-Cre+ mice at 102 dpi (n = 6–8 mice/group; all mice collected at same time point; 154–178 d old). Data shown as mean ± SEM. Unpaired, two-tailed Student's t test, *p < 0.05, **p < 0.01. Download Figure 4-3, TIF file (12.7MB, tif) .
The ubiquitination of synaptic membrane proteins is critical to maintaining synaptic homeostasis (Bingol and Schuman, 2005; Patrick, 2006; Haas and Broadie, 2008). To determine whether the ubiquitinated protein accumulation at the synapse of nHrs-depleted mice was accelerated, we next isolated synaptosomes from the forebrain of prion-infected Cre+ and Cre− mice at the same time point, when Cre+ mice were terminal. Notably, the synaptosomes showed markedly higher ubiquitinated protein levels in Cre+ mice (42% increase), indicating that nHrs is critical for ubiquitinated protein clearance at the synapse (Fig. 4H).
Loss of nHrs accelerates severe synaptic structural defects during prion infection
To investigate how nHrs depletion and the accumulation of ubiquitinated proteins impact synapse structure, we imaged adjacent synaptic fields from the hippocampal CA1 stratum radiatum and lacunosum moleculare from uninfected and prion-infected mice at ∼84, 107, and 139 dpi, and measured the curvature, postsynaptic density (PSD) area, and PSD length (Fig. 5A,B). By 84 dpi, the synapses of the prion-infected Hrsf/fSynCre+ mice were already highly curved (primarily concave) and the PSD was thick and irregular, and by 107 d, there was an increase in the PSD length as compared with the uninfected mice (unchanged in uninfected Cre+; Fig. 5C–F). By 139 dpi, the terminal Cre− mice also showed highly curved synapses and thick PSDs (Fig. 5C–F), indicating that the Cre+ mice develop a similar synaptic phenotype but on a markedly accelerated timeline. Notably, an increase in PSD length and area has been reported with activity at glutamatergic excitatory synapses (Tao-Cheng, 2019). Together, the numerous highly concave synapses as well as longer and deeper PSDs are consistent with postsynaptic expansion in the terminal prion-infected mice, with the Cre+ mice developing a synaptic phenotype >30 d earlier, suggesting that neuronal Hrs loss is detrimental to synapses.
Figure 5.
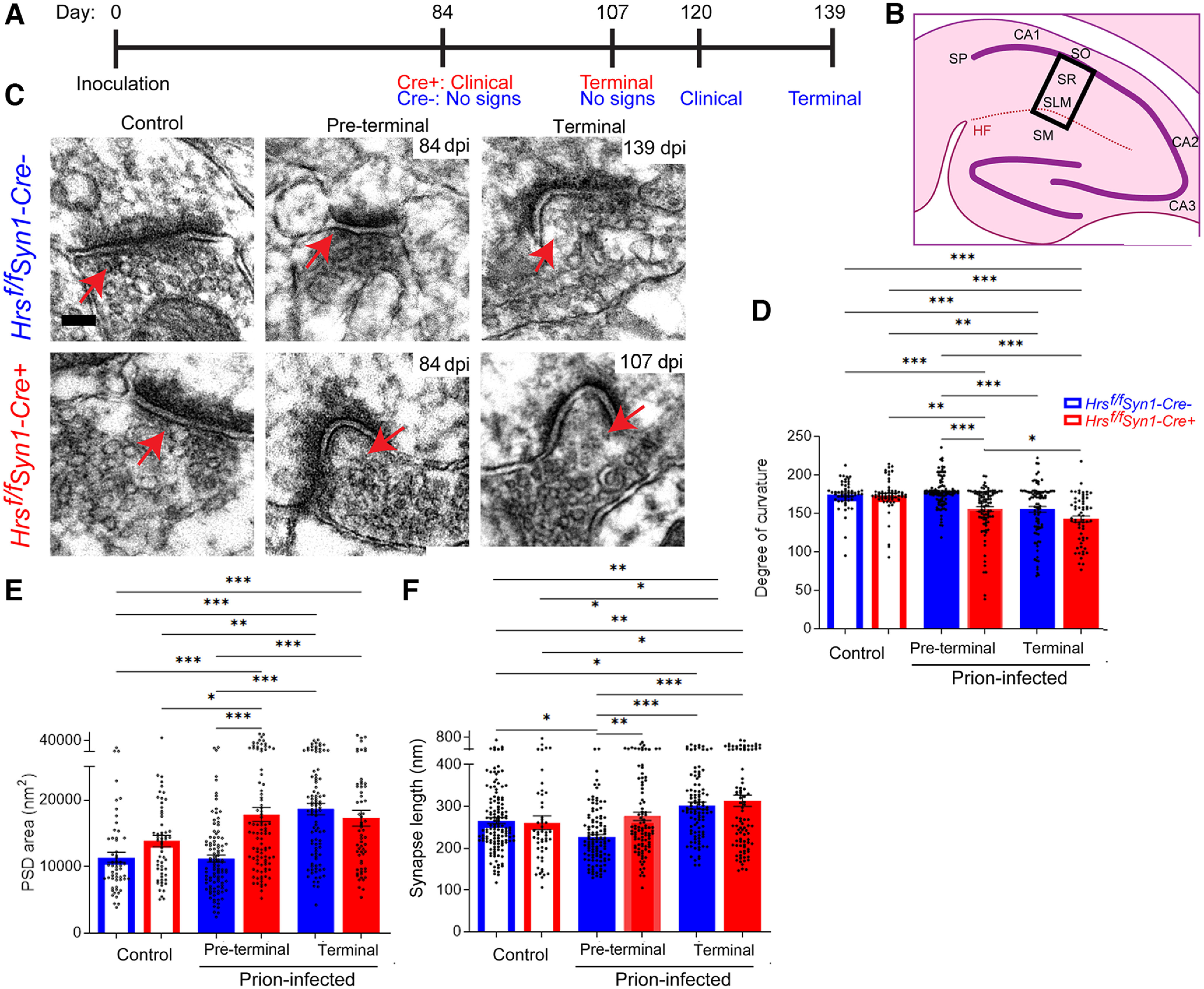
Neuronal Hrs depletion accelerates severe synaptic structural phenotype in prion disease. A, Timeline depicts the clinical status of the prion-infected Hrsf/fSyn1-Cre+ and Hrsf/fSyn1-Cre− mice at four timepoints. B, Schematic shows the CA1 hippocampal region examined by electron microscopy (boxed). SO = stratum oriens; SP = stratum pyramidale; SR = stratum radiatum; SLM = stratum lacunosum-moleculare; SM = stratum moleculare; HF = hippocampal formation. C, Representative transmission electron microscopy images of uninfected and prion-infected Hrsf/fSyn1-Cre− and Cre+ brain at three disease timepoints. D, Quantification of the degree of curvature of the postsynapse. E, Quantification of the postsynaptic density (PSD) area. (F) Quantification of the synapse length. N ≥ 26 synapses per mouse from 15 mice (Uninfected: n = 2 Hrsf/fSyn1-Cre− and Cre+ mice/group; Prion-infected: n = 5 and 6 Hrsf/fSyn1-Cre−/WT and Cre+ mice, respectively). Data shown as mean ± SEM. Scale bar = 100 nm. One way-ANOVA with Tukey's multiple comparisons post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
nHrs depletion accelerates synaptic protein alterations in prion disease
To determine whether the levels of synaptic vesicles (SVs) and synaptic proteins were altered, we next analyzed a panel of presynaptic and postsynaptic proteins at ∼84 d, 107 d (terminal Cre+ mice), and 139 d (terminal Cre− mice). Although Hrs binds SNAP25 on SVs (Sun et al., 2003), neither nHrs depletion nor prion-infection altered the levels of the SV proteins, VAMP2, SNAP25, or synaptophysin, at any time point (Fig. 6A,B; Extended Data Fig. 6-1). However, at 107 dpi, prion-infected Cre+ brain showed less synapsin-1 (syn1), which helps regulate SV number and neurotransmitter release (Patzke et al., 2019; 107 dpi: Cre+: 0.91 ± 0.09, Cre−: 1.35 ± 0.05, p = 0.06; Fig. 6C,D), with levels similarly reduced in Cre− mice by ∼30 d later (Cre+: 0.66 ± 0.05 and Cre−: 0.55 ± 0.05 relative to uninfected brain; Fig. 6A,B). Additionally, both Rab5 and VPS35 were significantly reduced or trending low (p = 0.07), respectively, although were not altered in synaptosomes (Fig. 7A,B,E; Extended Data Fig. 7-1). Other Rab GTPases (Rab7 and Rab11a) were unaltered in the Cre+ mice (note only Rab5 is reduced starting at 84 dpi; Fig. 7A–G), collectively suggesting that Hrs loss during prion-infection impacts proteins involved in early endosomes.
Figure 6.
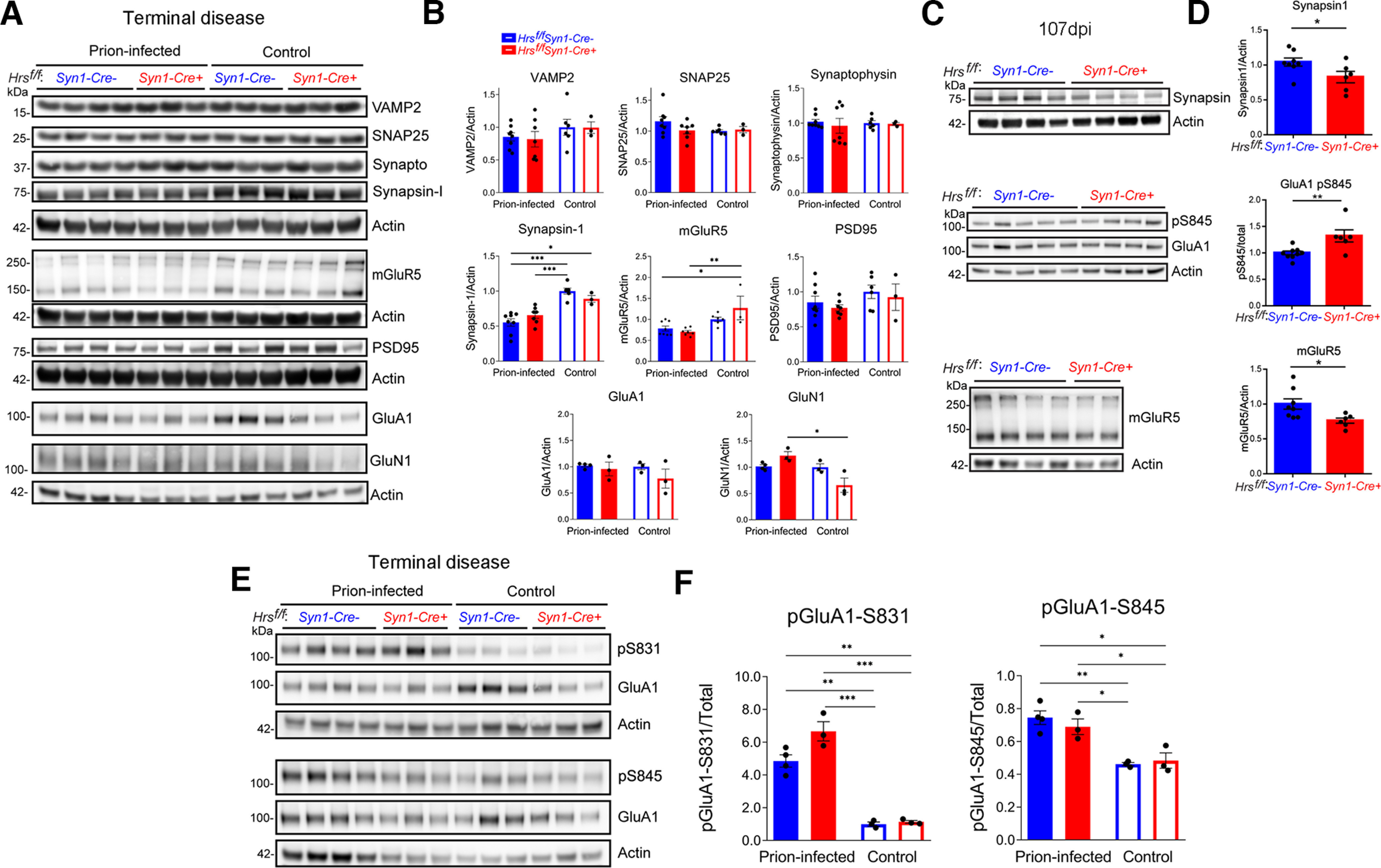
Neuronal Hrs depletion accelerates synaptic protein loss and the accumulation of phosphorylated GluA1 receptors. A–D, Representative Western blottings and quantification of synaptic proteins in brain lysates of age-matched uninfected and prion-infected Hrsf/fSyn1-Cre− and Cre+ mice at terminal disease (A, B) or at 107 dpi (C, D; n = 3–8 mice/group). E, F, Representative Western blottings show phosphorylated GluA1 receptors (S831 and S845) relative to total protein levels (n = 3–4 mice/group). Data shown as mean ± SEM. Relative protein levels were normalized to the average of the uninfected Hrsf/fSyn1-Cre mice. Two-way ANOVA with Tukey's multiple comparisons post hoc test (A, B and E, F), unpaired, two-tailed Student's t test (C, D). *p < 0.05, **p < 0.01, ***p < 0.001. Data supported by Extended Data Figures 6-1, 6-2, and 6-3.
Figure 7.
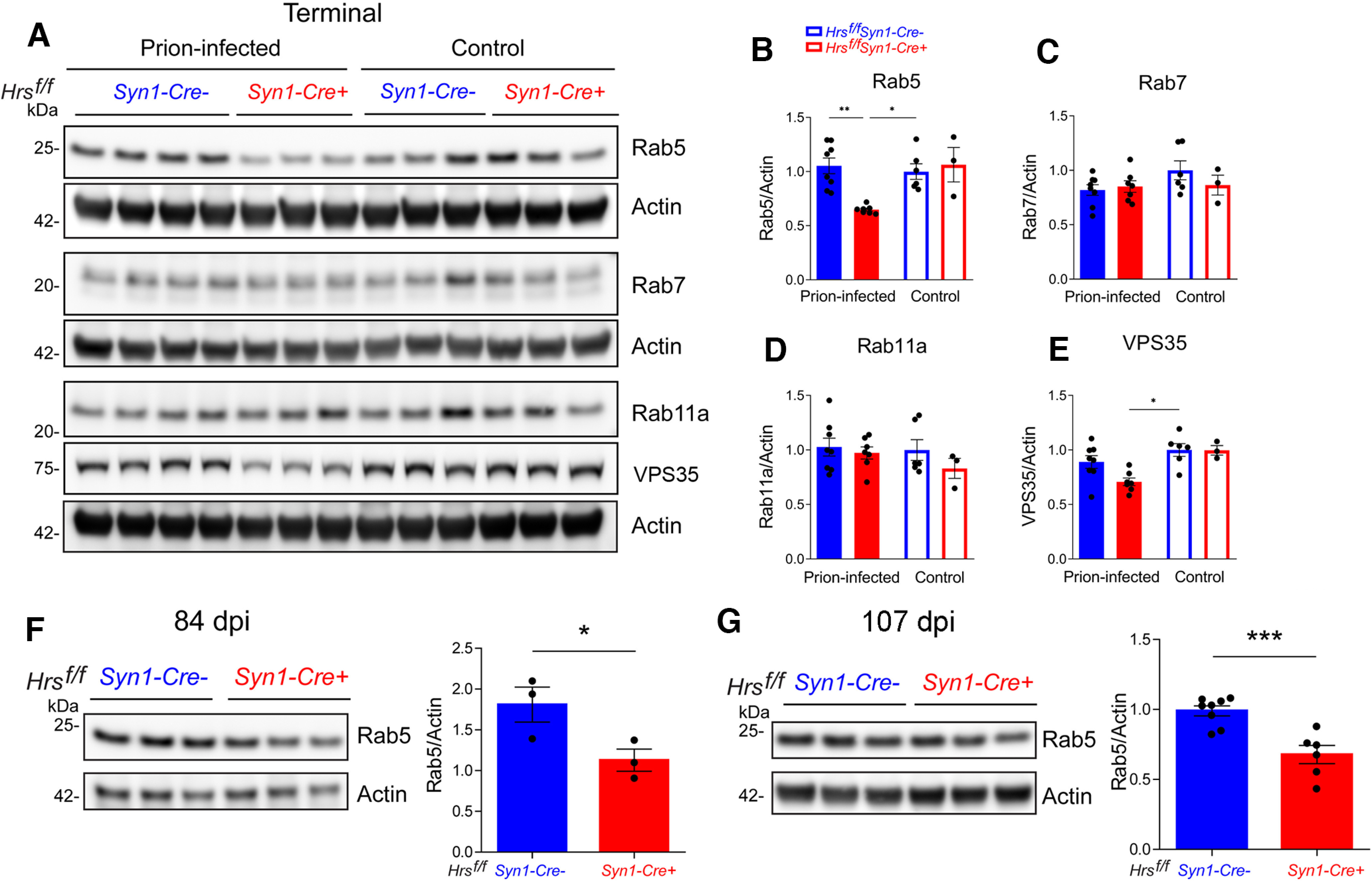
Depleting Hrs in prion-infected mice reduces early endosomal proteins. A–E, Representative Western blottings and quantification of endosomal proteins in brain lysates of uninfected and prion-infected Hrsf/fSyn1-Cre− and Cre+ mice. Relative protein levels were normalized to the average of the uninfected Hrsf/fSyn1-Cre− levels. F, G, Western blottings and quantification of Rab5 in prion-infected Hrsf/fSyn1-Cre− and Cre+ mice at 84 d (F) and 107 d (G) after inoculation. N = 3–8 mice per group. Data shown as mean ± SEM. Two-way ANOVA with Tukey's multiple comparisons post hoc test. B–E, *p < 0.05, **p < 0.01. Unpaired, two-tailed Student's t test (F, G), *p < 0.05, ***p < 0.001.
Presynaptic and postsynaptic protein expression in brain of prion-infected Hrsf/fSyn1-Cre mice at 84 dpi. A, B, Representative Western blottings and quantification of synaptic proteins in prion-infected Hrsf/fSyn1-Cre− and Cre+ mice at 84 dpi (n = 3 mice/group). Unpaired, two-tailed Student's t test revealed no significant differences. Data shown as mean ± SEM. Download Figure 6-1, TIF file (12.3MB, tif) .
Postsynaptic proteins in brain of prion-infected Hrsf/fSyn1-Cre mice at 107 dpi. Representative Western blottings (A) and quantification (B) of synaptic proteins in prion-infected Hrsf/fSyn1-Cre− and Cre+ mice at 107 dpi (n = 6–8 mice/group). Unpaired, two-tailed Student's t test revealed no significant differences. Data shown as mean ± SEM. Download Figure 6-2, TIF file (11.9MB, tif) .
Neuronal quantification in uninfected or prion-infected cerebral cortex. Images and quantification of NeuN immunolabeled brain sections (frontal cortex) from Hrsf/fSyn1-Cre− and Cre+ uninfected or prion-infected mice reveal no differences in the numbers of NeuN positive nuclei. Identically-sized areas were assessed (427,193 µm2). Data shown as mean ± SEM. One-way ANOVA with Tukey's multiple comparisons post hoc correction (no significant differences). Scale bar = 100 µm. Download Figure 6-3, TIF file (7.1MB, tif) .
Synaptosomal protein levels from prion-infected Hrsf/fSyn1-Cre+ and Cre− brain. Representative Western blotting and quantification of Rab5, VSP35, Hrs, and synaptophysin proteins in synaptosomes collected from Hrsf/fSyn1-Cre− and Cre+ brain at 103 dpi. For the Rab5 t test, p = 0.175. Data shown as mean ± SEM shown. Relative protein levels were normalized to the average of the uninfected mice (n = 4–6 Hrsf/fSyn1-Cre mice/group). Unpaired, two-tailed Student's t test. **p < 0.01. Download Figure 7-1, TIF file (6.9MB, tif) .
Postsynaptic membrane proteins, including AMPA receptors (AMPARs; Schwarz et al., 2010), are regulated by ubiquitination (Mabb and Ehlers, 2010), therefore may be altered by Hrs depletion. At 107 dpi, phosphorylated AMPA receptors, pGluA1-S845 were increased in the Cre+ mice (-S831 was unaltered), suggesting that nHrs loss exacerbates GluA1 retention in the postsynaptic membrane (Fig. 6C,D; Extended Data Fig. 6-2). By terminal disease, all prion-infected mice showed an increase in pGluA1 at S831 and S845 (Fig. 6E,F), suggesting that prion infection leads to increased GluA1 membrane retention (S845; Roche et al., 1996; Esteban et al., 2003; H.K. Lee et al., 2003) and increased sensitivity to depolarization (S831; Roche et al., 1996; H.K. Lee et al., 2003). Notably, mGluR5 (metabotropic glutamate receptor) levels were reduced in the terminal Cre+ mice at 107 dpi and were similarly low in the Cre− mice, but only at the terminal disease time point (Fig. 6A–D), indicating Hrs depletion is linked to an accelerated loss of mGluR5. The total levels of PSD95, GluA1, and GluN1 were unchanged or slightly elevated (GluN1; Fig. 6A,B; Extended Data Figs. 6-1, 6-2), while NeuN immunolabeled cortical sections showed similar numbers of total neurons in prion-infected Cre+ and Cre− mice (Extended Data Fig. 6-3). Thus, these data show an accelerated accumulation of pGluA1-S845 and loss of mGluR5 in the Cre+ prion-infected mice, suggesting that nHrs facilitates glutamate receptor homeostasis. Collectively, this study defines how a marked ESCRT-0 reduction in prion-affected mouse and sCJD brain alters glutamate receptor homeostasis and accelerates synaptic degeneration in prion disease.
Discussion
Synaptic alterations occur early and correlate with clinical onset in prion disease (Johnston et al., 1997; Belichenko et al., 2000; Jeffrey et al., 2000; D. Brown et al., 2001; Ferrer, 2002; Chiti et al., 2006; Mallucci et al., 2007; Sisková et al., 2009), yet the sequence of events underlying synaptic degeneration is poorly understood. For example, it is unknown how the synapse responds to the impaired clearance of ubiquitinated proteins in the prion-affected brain (Cammarata and Tabaton, 1992; Ironside et al., 1993; McKinnon et al., 2016). Here, we show a profound reduction of ESCRT-0 and a derangement of glutamate receptors, together with ubiquitinated protein accumulation at the synapse in the prion-affected brain. Using a rigorous conditional approach to manipulate Hrs in a cell-specific manner in vivo, we found that nHrs depletion accelerates prion-induced synaptic degeneration and shortens survival, despite unaltered prion conversion kinetics, thus uncoupling PrPSc levels and neurotoxicity. Structural studies show the accelerated development of an expanded and concave postsynapse enveloping the presynapse, concurrent with increased pGluA1-S845 and severe astrogliosis, which was not observed in uninfected Hrs-depleted mice. Our findings reveal Hrs as a pivotal component of the synaptic machinery that is severely altered in prion disease.
ESCRT-0 sorts ubiquitinated membrane cargo, including AMPAR (Ehlers, 2000; Schwarz et al., 2010; Wollert and Hurley, 2010), to MVBs for clearance by autophagy or exosomal release and thus is critical for the rapid regulation of presynaptic and postsynaptic proteins (Colledge et al., 2003; Schwarz et al., 2010; Watson et al., 2015; Sheehan et al., 2016). Here, we find markedly reduced Hrs while ubiquitinated synaptic proteins are increased in prion-infected mice, linking PrPSc to reduced Hrs function. Moreover, depleting nHrs accelerated major biochemical and structural synaptic aberrations during prion disease, including (1) phosphorylated GluA1 receptor (S845) accumulation, (2) reduced synapsin-1 and mGluR5 receptors, and (3) severe enlargement of postsynapses. The heightened pGluA1-S845 levels are indicative of increased synaptic GluA1 containing AMPARs, including homomeric GluA1 Ca2+-permeable AMPARs, which may contribute to Ca2+ influx-triggered excitotoxicity (C. Guo and Ma, 2021). Furthermore, the buildup of ubiquitinated substrates at the synapse may contribute to PSD expansion (Wang et al., 2017) and postsynaptic overgrowth (Wan et al., 2000; DiAntonio et al., 2001; Wang et al., 2017) and is consistent with the aberrant postsynaptic structures others have observed (Sisková et al., 2013), collectively suggestive of activated excitatory synapses in prion disease.
Given that AMPARs are linked to excitotoxicity and are cleared in part by transit through MVBs, a crucial question raised by our study is, how does the reduced Hrs impact AMPAR recycling in prion disease? AMPAR and other PSD proteins are regulated in part by K63 ubiquitination (Widagdo et al., 2015; Ma et al., 2017), and decreased ESCRT-0 engagement of ubiquitinated AMPAR may promote AMPAR recycling to the plasma membrane and membrane retention instead of degradation (Ehlers, 2000), which enhances AMPAR-mediated signaling. In support of this possibility, we found markedly elevated phosphorylated GluA1 (S831 and S845) levels in all terminal prion-infected mice, suggestive of heightened AMPAR activity and synaptic upscaling and consistent with the curved and elongated postsynaptic densities observed here and previously reported following synaptic activation (Desmond and Levy, 1983; Yim et al., 2015; Tao-Cheng, 2019). As we are the first to report increased GluA1 receptor phosphorylation in prion disease (previously in Ojeda-Juárez et al., 2022), determining whether glutamate receptor alterations are common among diverse prion diseases would be an important next step. Our results may have some parallels to Parkinson's disease models, in which α-synuclein oligomers activate NMDA receptors (NMDAR), alter AMPAR composition, and increase AMPAR recruitment to the synapse (Diógenes et al., 2012). Taken together, our studies favor a testable model in which heightened excitatory synaptic activity occurs in conjunction with reduced ESCRT-0, further exacerbating the retention of AMPAR and promoting heightened calcium entry and detrimental downstream signaling responses.
Here, we find that reducing nHrs accelerated the synaptic derangement and markedly accelerated progression to terminal disease. We also report an increase in surface PrPC with Hrs depletion. Given the substantial experimental evidence showing that (1) neuronal PrPC is necessary and sufficient for prion-induced toxicity (Mallucci et al., 2003; Sonati et al., 2013), (2) PrPC expression negatively correlates to survival time (Büeler et al., 1994; Fischer et al., 1996), and (3) mice with increased neuronal surface PrPC expression from depletion of the PrPC sheddase, ADAM10, show accelerated disease progression (Altmeppen et al., 2015), the most parsimonious explanation is that nHrs loss also increases surface retention of PrPC in vivo, potentiating neurotoxic signaling and advancing synaptic impairment in prion disease. In this case, increased surface PrPC in glia would not impact neuronal toxicity or survival time, consistent with the negative findings in astrocyte and microglia Hrs-depleted mice. Further, mice overexpressing PrPC show accelerated disease progression despite less PrPSc in brain than wild-type mice (Fischer et al., 1996; Altmeppen et al., 2015), similar to our results and suggesting that PrPSc levels do not universally correlate with survival time (Fischer et al., 1996). The clinical implication is that as Hrs levels decline, neuronal surface PrPC may rise and accelerate disease progression, underscoring the importance of therapeutic strategies to reduce PrPC expression in prion-affected patients. Future studies to further resolve how Hrs regulates neuronal PrPC levels, PrPSc-induced signaling, and neuronal activity are necessary to provide additional insight into the cascade of synaptic alterations that develops in prion disease.
Our studies also shed light on ESCRT-0 function at the synapse. Previous studies report that Hrs transits in axons on a subset of Rab5+ vesicles (Birdsall et al., 2022) and engages in SV homeostasis, facilitating the activity-dependent clearance of select SV proteins in cultured neurons (Sheehan et al., 2016). Additionally, teetering mice that express mutant Hrs show reduced release of acetylcholine at the neuromuscular junction, potentially contributing to motor deficits (Watson et al., 2015). Here, Hrs depletion was linked to an early rise in pGluA1-S845 levels in prion disease, suggesting that ESCRT-0 has an important role in regulating AMPAR. We speculate that Hrs regulation may be linked to neuronal activity and may underlie the low Hrs levels reported here. Future studies of ESCRT-0 regulation and function at the presynapse and postsynapse and of ESCRT pathway alterations in neurodegenerative disease would be warranted.
In summary, this study demonstrates that (1) Hrs is markedly reduced in the brain in prion disease, regardless of strain, (2) Hrs loss does not detectably impact prion conversion in vivo, and (3) neuronal Hrs depletion slows ubiquitinated protein clearance at the synapse, increases surface PrPC, and accelerates synaptic degeneration, which together may contribute to the rapid clinical deterioration in prion disease. Our findings raise a number of questions with respect to prion disease progression in humans and animals, including, what are the mechanistic underpinnings of the post-transcriptional reduction in Hrs, and how is the reduced Hrs linked to AMPAR activity and the accelerated synaptic derangement? Manipulating Hrs and the ESCRT pathway to facilitate ubiquitinated protein clearance at the synapse provides a new avenue of investigation with therapeutic implications for prion and potentially other neurodegenerative diseases, such as Alzheimer's and Parkinson's disease, in which misfolded oligomers also bind PrPC.
Footnotes
This study was supported by National Institutes of Health Grants NS069566 (to C.J.S.), NS076896 (to C.J.S.), K99AG061251 (to P.A.-C.), NS111978 (to S.R.), R35GM127121 (to J.T.), AG031189 (to M.D.G.), AG062562 (to M.D.G.), AG055619 (to M.D.G.), P30AG06429 (to R.A.R., University of California, San Diego Shiley-Marcos Alzheimer's Disease Research Center), NS047101 (University of California, San Diego Microscopy Core), AG062429 (University of California, San Diego Shiley-Marcos Alzheimer's Disease Research Center), and 1S10OD023527 (Electron Microscopy Facility) and by the Michael J. Homer Family Fund (M.D.G.). J.A.L. was supported by a Ruth L. Kirschstein Institutional National Research Award from the National Institutes of Health (F31NS103588). We thank the National Prion Disease Pathology Surveillance Center (NPDPSC) for prion typing and all patients and their families for participating in research. We thank Dr. Mahsa Pourhamzeh for experimental assistance, Dr. Daniela Boassa for valuable discussion of synapse ultrastructure, Timothy Meerloo and Ying Jones at the University of California, San Diego Electron Microscopy Facility for TEM sample preparation, Jennifer Santini at the University of California, San Diego Light Microscopy Core for technical assistance, Jeffrey Metcalf from the laboratory of Dr. Robert Rissman and the Shiley-Marcos Alzheimer's Disease Research Center at University of California, San Diego for providing human tissues for this study, and the animal care staff at University of California, San Diego for excellent animal care. We also thank Dr. Anna Maria Cuervo for the kind gift of the mCherry-GFP-LC3 plasmid and Drs. Markus Glatzel and Hermann Altmeppen for the kind gift of the sPrPG228 antibody.
The authors declare no competing financial interests.
References
- Aguilar-Calvo P, Xiao X, Bett C, Eraña H, Soldau K, Castilla J, Nilsson KP, Surewicz WK, Sigurdson CJ (2017) Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci Rep 7:43295. 10.1038/srep43295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Calvo P, Bett C, Sevillano AM, Kurt TD, Lawrence J, Soldau K, Hammarstrom P, Nilsson KPR, Sigurdson CJ (2018) Generation of novel neuroinvasive prions following intravenous challenge. Brain Pathol 28:999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Calvo P, Sevillano AM, Bapat J, Soldau K, Sandoval DR, Altmeppen HC, Linsenmeier L, Pizzo DP, Geschwind MD, Sanchez H, Appleby BS, Cohen ML, Safar JG, Edland SD, Glatzel M, Nilsson KPR, Esko JD, Sigurdson CJ (2020) Shortening heparan sulfate chains prolongs survival and reduces parenchymal plaques in prion disease caused by mobile, ADAM10-cleaved prions. Acta Neuropathol 139:527–546. 10.1007/s00401-019-02085-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen Institute for Brain Science (2020) Allen cell types database – mouse whole cortex and hippocampus [dataset]. Available from http://celltypes.brain-map.org/rnaseq.
- Allen Institute for Brain Science (2021) Allen cell types database – human multiple cortical areas [dataset]. Available from http://celltypes.brain-map.org/rnaseq.
- Altmeppen HC, Prox J, Krasemann S, Puig B, Kruszewski K, Dohler F, Bernreuther C, Hoxha A, Linsenmeier L, Sikorska B, Liberski PP, Bartsch U, Saftig P, Glatzel M (2015) The sheddase ADAM10 is a potent modulator of prion disease. Elife 4:e04260. 10.7554/eLife.04260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asao H, Sasaki Y, Arita T, Tanaka N, Endo K, Kasai H, Takeshita T, Endo Y, Fujita T, Sugamura K (1997) Hrs is associated with STAM, a signal-transducing adaptor molecule. Its suppressive effect on cytokine-induced cell growth. J Biol Chem 272:32785–32791. 10.1074/jbc.272.52.32785 [DOI] [PubMed] [Google Scholar]
- Bache KG, Brech A, Mehlum A, Stenmark H (2003) Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol 162:435–442. 10.1083/jcb.200302131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Brown D, Jeffrey M, Fraser JR (2000) Dendritic and synaptic alterations of hippocampal pyramidal neurones in scrapie-infected mice. Neuropathol Appl Neurobiol 26:143–149. 10.1046/j.1365-2990.2000.026002143.x [DOI] [PubMed] [Google Scholar]
- Bilodeau PS, Urbanowski JL, Winistorfer SC, Piper RC (2002) The Vps27p Hse1p complex binds ubiquitin and mediates endosomal protein sorting. Nat Cell Biol 4:534–539. 10.1038/ncb815 [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM (2005) Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol 15:536–541. 10.1016/j.conb.2005.08.016 [DOI] [PubMed] [Google Scholar]
- Birdsall V, Kirwan K, Zhu M, Imoto Y, Wilson SM, Watanabe S, Waites CL (2022) Axonal transport of Hrs is activity dependent and facilitates synaptic vesicle protein degradation. Life Sci Alliance 5:e202000745. 10.26508/lsa.202000745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boellaard JW, Kao M, Schlote W, Diringer H (1991) Neuronal autophagy in experimental scrapie. Acta Neuropathol 82:225–228. 10.1007/BF00294449 [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Taraboulos A, Prusiner SB (1992) Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem 267:16188–16199. 10.1016/S0021-9258(18)41985-0 [DOI] [PubMed] [Google Scholar]
- Brown CA, Schmidt C, Poulter M, Hummerich H, Klöhn PC, Jat P, Mead S, Collinge J, Lloyd SE (2014) In vitro screen of prion disease susceptibility genes using the scrapie cell assay. Hum Mol Genet 23:5102–5108. 10.1093/hmg/ddu233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, Belichenko P, Sales J, Jeffrey M, Fraser JR (2001) Early loss of dendritic spines in murine scrapie revealed by confocal analysis. Neuroreport 12:179–183. 10.1097/00001756-200101220-00043 [DOI] [PubMed] [Google Scholar]
- Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C (1994) High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1:19–30. 10.1007/BF03403528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarata S, Tabaton M (1992) Ubiquitin-reactive axons have a widespread distribution and are unrelated to prion protein plaques in Creutzfeldt-Jakob disease. J Neurol Sci 110:32–36. 10.1016/0022-510x(92)90006-7 [DOI] [PubMed] [Google Scholar]
- Carroll JA, Striebel JF, Rangel A, Woods T, Phillips K, Peterson KE, Race B, Chesebro B (2016) Prion strain differences in accumulation of PrPSc on neurons and glia are associated with similar expression profiles of neuroinflammatory genes: comparison of three prion strains. PLoS Pathog 12:e1005551. 10.1371/journal.ppat.1005551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A (1989) A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243:1576–1583. 10.1126/science.2538923 [DOI] [PubMed] [Google Scholar]
- Chiti Z, Knutsen OM, Betmouni S, Greene JR (2006) An integrated, temporal study of the behavioural, electrophysiological and neuropathological consequences of murine prion disease. Neurobiol Dis 22:363–373. 10.1016/j.nbd.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD (2003) Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40:595–607. 10.1016/s0896-6273(03)00687-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG (1994) Prion protein is necessary for normal synaptic function. Nature 370:295–297. 10.1038/370295a0 [DOI] [PubMed] [Google Scholar]
- Cunningham C, Deacon R, Wells H, Boche D, Waters S, Diniz CP, Scott H, Rawlins JN, Perry VH (2003) Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur J Neurosci 17:2147–2155. 10.1046/j.1460-9568.2003.02662.x [DOI] [PubMed] [Google Scholar]
- Desmond NL, Levy WB (1983) Synaptic correlates of associative potentiation/depression: an ultrastructural study in the hippocampus. Brain Res 265:21–30. 10.1016/0006-8993(83)91329-x [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS (2001) Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412:449–452. 10.1038/35086595 [DOI] [PubMed] [Google Scholar]
- Diógenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, Näsström T, Franquelim HG, Oliveira LM, Castanho MA, Lannfelt L, Bergström J, Ingelsson M, Quintas A, Sebastião AM, Lopes LV, Outeiro TF (2012) Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci 32:11750–11762. 10.1523/JNEUROSCI.0234-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28:511–525. 10.1016/s0896-6273(00)00129-x [DOI] [PubMed] [Google Scholar]
- Erpapazoglou Z, Dhaoui M, Pantazopoulou M, Giordano F, Mari M, Léon S, Raposo G, Reggiori F, Haguenauer-Tsapis R (2012) A dual role for K63-linked ubiquitin chains in multivesicular body biogenesis and cargo sorting. Mol Biol Cell 23:2170–2183. 10.1091/mbc.E11-10-0891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6:136–143. 10.1038/nn997 [DOI] [PubMed] [Google Scholar]
- Fang C, Wu B, Le NTT, Imberdis T, Mercer RCC, Harris DA (2018) Prions activate a p38 MAPK synaptotoxic signaling pathway. PLoS Pathog 14:e1007283. 10.1371/journal.ppat.1007283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I (2002) Synaptic pathology and cell death in the cerebellum in Creutzfeldt-Jakob disease. Cerebellum 1:213–222. 10.1080/14734220260418448 [DOI] [PubMed] [Google Scholar]
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G (2004) Cells release prions in association with exosomes. Proc Natl Acad Sci U S A 101:9683–9688. 10.1073/pnas.0308413101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179:485–500. 10.1083/jcb.200702115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15:1255–1264. 10.1002/j.1460-2075.1996.tb00467.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foliaki ST, Smith A, Schwarz B, Bohrnsen E, Bosio CM, Williams K, Orrú CD, Lachenauer H, Groveman BR, Haigh CL (2023) Altered energy metabolism in Fatal Familial Insomnia cerebral organoids is associated with astrogliosis and neuronal dysfunction. PLoS Genet 19:e1010565. 10.1371/journal.pgen.1010565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier JG, Escaig Haye F, Billette de Villemeur T, Robain O (1995) Ultrastructural localization of cellular prion protein (PrPc) in synaptic boutons of normal hamster hippocampus. C R Acad Sci III 318:339–344. [PubMed] [Google Scholar]
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003) Sporadic and familial CJD: classification and characterisation. Br Med Bull 66:213–239. 10.1093/bmb/66.1.213 [DOI] [PubMed] [Google Scholar]
- Geschwind MD, Josephs KA, Parisi JE, Keegan BM (2007) A 54-year-old man with slowness of movement and confusion. Neurology 69:1881–1887. 10.1212/01.wnl.0000290370.14036.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godsave SF, Wille H, Kujala P, Latawiec D, DeArmond SJ, Serban A, Prusiner SB, Peters PJ (2008) Cryo-immunogold electron microscopy for prions: toward identification of a conversion site. J Neurosci 28:12489–12499. 10.1523/JNEUROSCI.4474-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, Tabrizi SJ (2013) Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J Cell Sci 126:3552–3562. 10.1242/jcs.120477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo BB, Bellingham SA, Hill AF (2015) The neutral sphingomyelinase pathway regulates packaging of the prion protein into exosomes. J Biol Chem 290:3455–3467. 10.1074/jbc.M114.605253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Ma YY (2021) Calcium permeable-AMPA receptors and excitotoxicity in neurological disorders. Front Neural Circuits 15:711564. 10.3389/fncir.2021.711564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas KF, Broadie K (2008) Roles of ubiquitination at the synapse. Biochim Biophys Acta 1779:495–506. 10.1016/j.bbagrm.2007.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schürmann P, Windl O, Brose N, Kretzschmar H (1999) Evidence of presynaptic location and function of the prion protein. J Neurosci 19:8866–8875. 10.1523/JNEUROSCI.19-20-08866.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ironside JW, McCardle L, Hayward PA, Bell JE (1993) Ubiquitin immunocytochemistry in human spongiform encephalopathies. Neuropathol Appl Neurobiol 19:134–140. 10.1111/j.1365-2990.1993.tb00418.x [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Halliday WG, Bell J, Johnston AR, MacLeod NK, Ingham C, Sayers AR, Brown DA, Fraser JR (2000) Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol Appl Neurobiol 26:41–54. 10.1046/j.1365-2990.2000.00216.x [DOI] [PubMed] [Google Scholar]
- Johnston AR, Black C, Fraser J, MacLeod N (1997) Scrapie infection alters the membrane and synaptic properties of mouse hippocampal CA1 pyramidal neurones. J Physiol 500:1–15. 10.1113/jphysiol.1997.sp021994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones E, Mead S (2020) Genetic risk factors for Creutzfeldt-Jakob disease. Neurobiol Dis 142:104973. 10.1016/j.nbd.2020.104973 [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002) Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol 3:893–905. 10.1038/nrm973 [DOI] [PubMed] [Google Scholar]
- Kim YS, Carp RI, Callahan SM, Wisniewski HM (1990) Pathogenesis and pathology of scrapie after stereotactic injection of strain 22L in intact and bisected cerebella. J Neuropathol Exp Neurol 49:114–121. 10.1097/00005072-199003000-00004 [DOI] [PubMed] [Google Scholar]
- Kovács GG, Gelpi E, Ströbel T, Ricken G, Nyengaard JR, Bernheimer H, Budka H (2007) Involvement of the endosomal-lysosomal system correlates with regional pathology in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol 66:628–636. 10.1097/nen.0b013e318093ecc7 [DOI] [PubMed] [Google Scholar]
- Lakkaraju AKK, Frontzek K, Lemes E, Herrmann U, Losa M, Marpakwar R, Aguzzi A (2021) Loss of PIKfyve drives the spongiform degeneration in prion diseases. EMBO Mol Med 13:e14714. 10.15252/emmm.202114714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457:1128–1132. 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauwers E, Jacob C, André B (2009) K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J Cell Biol 185:493–502. 10.1083/jcb.200810114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112:631–643. 10.1016/s0092-8674(03)00122-3 [DOI] [PubMed] [Google Scholar]
- Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB (2007) ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol 17:1561–1567. 10.1016/j.cub.2007.07.029 [DOI] [PubMed] [Google Scholar]
- Liberski PP, Yanagihara R, Wells GA, Gibbs CJ Jr, Gajdusek DC (1992) Comparative ultrastructural neuropathology of naturally occurring bovine spongiform encephalopathy and experimentally induced scrapie and Creutzfeldt-Jakob disease. J Comp Pathol 106:361–381. 10.1016/0021-9975(92)90022-m [DOI] [PubMed] [Google Scholar]
- Liberski PP, Brown DR, Sikorska B, Caughey B, Brown P (2008) Cell death and autophagy in prion diseases (transmissible spongiform encephalopathies). Folia Neuropathol 46:1–25. [PubMed] [Google Scholar]
- Liberski PP, Sikorska B, Hauw JJ, Kopp N, Streichenberger N, Giraud P, Boellaard J, Budka H, Kovacs GG, Ironside J, Brown P (2010) Ultrastructural characteristics (or evaluation) of Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies or prion diseases. Ultrastruct Pathol 34:351–361. 10.3109/01913123.2010.491175 [DOI] [PubMed] [Google Scholar]
- Linsenmeier L, Mohammadi B, Wetzel S, Puig B, Jackson WS, Hartmann A, Uchiyama K, Sakaguchi S, Endres K, Tatzelt J, Saftig P, Glatzel M, Altmeppen HC (2018) Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein. Mol Neurodegener 13:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Pérez Ó, Otero A, Filali H, Sanz-Rubio D, Toivonen JM, Zaragoza P, Badiola JJ, Bolea R, Martín-Burriel I (2019) Dysregulation of autophagy in the central nervous system of sheep naturally infected with classical scrapie. Sci Rep 9:1911. 10.1038/s41598-019-38500-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Pérez Ó, Toivonen JM, Otero A, Solanas L, Zaragoza P, Badiola JJ, Osta R, Bolea R, Martín-Burriel I (2020) Impairment of autophagy in scrapie-infected transgenic mice at the clinical stage. Lab Invest 100:52–63. 10.1038/s41374-019-0312-z [DOI] [PubMed] [Google Scholar]
- Ma Q, Ruan H, Peng L, Zhang M, Gack MU, Yao WD (2017) Proteasome-independent polyubiquitin linkage regulates synapse scaffolding, efficacy, and plasticity. Proc Natl Acad Sci U S A 114:E8760–E8769. 10.1073/pnas.1620153114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Ehlers MD (2010) Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol 26:179–210. 10.1146/annurev-cellbio-100109-104129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhães AC, Baron GS, Lee KS, Steele-Mortimer O, Dorward D, Prado MA, Caughey B (2005) Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J Neurosci 25:5207–5216. 10.1523/JNEUROSCI.0653-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J (2003) Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302:871–874. 10.1126/science.1090187 [DOI] [PubMed] [Google Scholar]
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, Brandner S, Jefferys JG, Collinge J (2007) Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 53:325–335. 10.1016/j.neuron.2007.01.005 [DOI] [PubMed] [Google Scholar]
- Marijanovic Z, Caputo A, Campana V, Zurzolo C (2009) Identification of an intracellular site of prion conversion. PLoS Pathog 5:e1000426. 10.1371/journal.ppat.1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon C, Goold R, Andre R, Devoy A, Ortega Z, Moonga J, Linehan JM, Brandner S, Lucas JJ, Collinge J, Tabrizi SJ (2016) Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin-proteasome system. Acta Neuropathol 131:411–425. 10.1007/s00401-015-1508-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller K, Beardmore J, Kanety H, Schlessinger J, Hopkins CR (1986) Localization of the epidermal growth factor (EGF) receptor within the endosome of EGF-stimulated epidermoid carcinoma (A431) cells. J Cell Biol 102:500–509. 10.1083/jcb.102.2.500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda-Juárez D, Lawrence JA, Soldau K, Pizzo DP, Wheeler E, Aguilar-Calvo P, Khuu H, Chen J, Malik A, Funk G, Nam P, Sanchez H, Geschwind MD, Wu C, Yeo GW, Chen X, Patrick GN, Sigurdson CJ (2022) Prions induce an early Arc response and a subsequent reduction in mGluR5 in the hippocampus. Neurobiol Dis 172:105834. 10.1016/j.nbd.2022.105834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN (2006) Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr Opin Neurobiol 16:90–94. 10.1016/j.conb.2006.01.007 [DOI] [PubMed] [Google Scholar]
- Patzke C, Brockmann MM, Dai J, Gan KJ, Grauel MK, Fenske P, Liu Y, Acuna C, Rosenmund C, Südhof TC (2019) Neuromodulator signaling bidirectionally controls vesicle numbers in human synapses. Cell 179:498–513.e22. 10.1016/j.cell.2019.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B, Hafner-Bratkovic I, Jerala R, Hornemann S, Wuthrich K, Bellon A, Vey M, Garen G, James MN, Kav N, Aguzzi A (2008) The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One 3:e3872. 10.1371/journal.pone.0003872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H (2002) Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol 4:394–398. 10.1038/ncb791 [DOI] [PubMed] [Google Scholar]
- Ren X, Kloer DP, Kim YC, Ghirlando R, Saidi LF, Hummer G, Hurley JH (2009) Hybrid structural model of the complete human ESCRT-0 complex. Structure 17:406–416. 10.1016/j.str.2009.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL (1996) Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16:1179–1188. 10.1016/s0896-6273(00)80144-0 [DOI] [PubMed] [Google Scholar]
- Schmidt O, Teis D (2012) The ESCRT machinery. Curr Biol 22:R116–R120. 10.1016/j.cub.2012.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Hall BJ, Patrick GN (2010) Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci 30:16718–16729. 10.1523/JNEUROSCI.3686-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan P, Zhu M, Beskow A, Vollmer C, Waites CL (2016) Activity-dependent degradation of synaptic vesicle proteins requires Rab35 and the ESCRT pathway. J Neurosci 36:8668–8686. 10.1523/JNEUROSCI.0725-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih SC, Katzmann DJ, Schnell JD, Sutanto M, Emr SD, Hicke L (2002) Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat Cell Biol 4:389–393. 10.1038/ncb790 [DOI] [PubMed] [Google Scholar]
- Sikorska B, Liberski PP, Giraud P, Kopp N, Brown P (2004) Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt-Jakob disease: a brain biopsy study. Int J Biochem Cell Biol 36:2563–2573. 10.1016/j.biocel.2004.04.014 [DOI] [PubMed] [Google Scholar]
- Sisková Z, Page A, O'Connor V, Perry VH (2009) Degenerating synaptic boutons in prion disease: microglia activation without synaptic stripping. Am J Pathol 175:1610–1621. 10.2353/ajpath.2009.090372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisková Z, Reynolds RA, O'Connor V, Perry VH (2013) Brain region specific pre-synaptic and post-synaptic degeneration are early components of neuropathology in prion disease. PLoS One 8:e55004. 10.1371/journal.pone.0055004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, Swayampakula M, Rahman MH, Das D, Kav N, Riek R, Liberski PP, James MN, Aguzzi A (2013) The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501:102–106. 10.1038/nature12402 [DOI] [PubMed] [Google Scholar]
- Staffaroni AM, Elahi FM, McDermott D, Marton K, Karageorgiou E, Sacco S, Paoletti M, Caverzasi E, Hess CP, Rosen HJ, Geschwind MD (2017) Neuroimaging in dementia. Semin Neurol 37:510–537. 10.1055/s-0037-1608808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Yan Q, Vida TA, Bean AJ (2003) Hrs regulates early endosome fusion by inhibiting formation of an endosomal SNARE complex. J Cell Biol 162:125–137. 10.1083/jcb.200302083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K, Tanaka N, Nara A, Yamamoto A, Nakagawa I, Yoshimori T, Ueno Y, Shimosegawa T, Sugamura K (2007) Role of Hrs in maturation of autophagosomes in mammalian cells. Biochem Biophys Res Commun 360:721–727. 10.1016/j.bbrc.2007.06.105 [DOI] [PubMed] [Google Scholar]
- Tamai K, Toyoshima M, Tanaka N, Yamamoto N, Owada Y, Kiyonari H, Murata K, Ueno Y, Ono M, Shimosegawa T, Yaegashi N, Watanabe M, Sugamura K (2008) Loss of hrs in the central nervous system causes accumulation of ubiquitinated proteins and neurodegeneration. Am J Pathol 173:1806–1817. 10.2353/ajpath.2008.080684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng JH (2019) Stimulation induces gradual increases in the thickness and curvature of postsynaptic density of hippocampal CA1 neurons in slice cultures. Mol Brain 12:44. 10.1186/s13041-019-0468-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J 19:94–102. 10.1093/emboj/19.1.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Everbroeck B, Pals P, Dziedzic T, Dom R, Godfraind C, Sciot R, Brucher JM, Martin JJ, Cras P (2000) Retrospective study of Creutzfeldt-Jakob disease in Belgium: neuropathological findings. Acta Neuropathol 99:358–364. 10.1007/s004010051136 [DOI] [PubMed] [Google Scholar]
- Veith NM, Plattner H, Stuermer CA, Schulz-Schaeffer WJ, Bürkle A (2009) Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur J Cell Biol 88:45–63. 10.1016/j.ejcb.2008.08.001 [DOI] [PubMed] [Google Scholar]
- Vilette D, Laulagnier K, Huor A, Alais S, Simoes S, Maryse R, Provansal M, Lehmann S, Andreoletti O, Schaeffer L, Raposo G, Leblanc P (2015) Efficient inhibition of infectious prions multiplication and release by targeting the exosomal pathway. Cell Mol Life Sci 72:4409–4427. 10.1007/s00018-015-1945-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth JDF, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J (2001) Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immuno-blotting assay. Lancet 358:171–180. 10.1016/s0140-6736(01)05403-4 [DOI] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS (2000) Highwire regulates synaptic growth in Drosophila. Neuron 26:313–329. 10.1016/s0896-6273(00)81166-6 [DOI] [PubMed] [Google Scholar]
- Wang CH, Huang YC, Chen PY, Cheng YJ, Kao HH, Pi H, Chien CT (2017) USP5/Leon deubiquitinase confines postsynaptic growth by maintaining ubiquitin homeostasis through Ubiquilin. Elife 6:e26886. 10.7554/eLife.26886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JA, Bhattacharyya BJ, Vaden JH, Wilson JA, Icyuz M, Howard AD, Phillips E, DeSilva TM, Siegal GP, Bean AJ, King GD, Phillips SE, Miller RJ, Wilson SM (2015) Motor and sensory deficits in the teetering mice result from mutation of the ESCRT component HGS. PLoS Genet 11:e1005290. 10.1371/journal.pgen.1005290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, Anggono V (2015) Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep 10:783–795. 10.1016/j.celrep.2015.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert T, Hurley JH (2010) Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 464:864–869. 10.1038/nature08849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Tian C, Wang SB, Xie WL, Guo Y, Zhang J, Shi Q, Chen C, Dong XP (2012) Activation of the macroautophagic system in scrapie-infected experimental animals and human genetic prion diseases. Autophagy 8:1604–1620. 10.4161/auto.21482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, et al. (2021a) A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184:3222–3241.e26. 10.1016/j.cell.2021.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, et al. (2021b) A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature 598:103–110. 10.1038/s41586-021-03705-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim YI, Park BC, Yadavalli R, Zhao X, Eisenberg E, Greene LE (2015) The multivesicular body is the major internal site of prion conversion. J Cell Sci 128:1434–1443. 10.1242/jcs.165472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF (2001) Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev 15:859–876. 10.1101/gad.862101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
sCJD Patient demographic data and clinical features Download Table 1-1, DOCX file (14.4KB, docx) .
Control case demographic data Download Table 1-2, DOCX file (13.4KB, docx) .
Demographic data summary of sCJD cases and controls Download Table 1-3, DOCX file (13.1KB, docx) .
Reduced Hrs in brain of WT and transgenic mice infected with distinct prions. Hrs immunoblots of brain lysates from prion-infected and uninfected control WT and transgenic mice expressing GPI-anchorless PrPC (quantification shown in Fig. 1B). Prion strains include mouse-adapted prions RML, 87V, GPI-anchorless mCWD, and GPI-anchorless 22L. *Mouse euthanized because of intercurrent illness at ∼42% of the expected survival time. Download Figure 1-1, TIF file (7MB, tif) .
Mouse and Human Hgs transcripts. Single-cell RNAseq data from the “Allen cell types database – mouse whole cortex and hippocampus [dataset]” (Allen Institute for Brain Science, 2020) and “Allen cell types database – human multiple cortical areas [dataset]” (Allen Institute for Brain Science, 2021) was used to plot the Hgs transcript levels in the mouse or human cortex. A, Heatmap representing the average levels of Hgs in excitatory, inhibitory, and non-neuronal cells (astrocytes and microglia; Class) from mouse cerebral cortex. Neuronal populations were further subdivided (subclass). Hgs represented as copy number per million (CPM) + 1. B, Bar graph of the same data in A showing the distribution and average Hgs expression in excitatory and inhibitory neuronal populations, as well as astrocytes and microglia. Each dot represents an individual cell. C, Bar graph showing the HGS levels in excitatory and inhibitory neurons, as well as astrocytes and microglia cells in human cerebral cortex. Each dot represents the CPM trimmed mean expression per subclass. Subclass was determined using taxonomy of transcriptomic cell types previously published. For B, C, transcriptomic cell type was used to determine cell identity (Yao et al., 2021a). Bar graphs: data shown as mean ± SEM; one-way ANOVA with Tukey's multiple comparisons post hoc test. ***p < 0.001. Download Figure 2-1, TIF file (12.3MB, tif) .
Spongiform degeneration and astrocyte reactivity in prion-infected Hrsf/fGFAPCre+ and Hrsf/fLysCre+ brain were similar to controls. A, Representative hematoxylin and eosin (HE) and GFAP (astrocytes)-immunolabeled brain sections (cerebral cortex) from prion-infected Hrsf/fGFAP-Cre− and Cre+ mice (top) and Hrsf/fLys-Cre− and Cre+ mice (bottom) at both preterminal and terminal disease stages. B, C, Western blottings and quantification of ubiquitinated protein levels in terminal (B) Hrsf/fGFAP-Cre− and Cre+ and (C) Hrsf/fLysM-Cre− and Cre+ brain (n = 4–5/group). Data shown as mean ± SEM; each dot represents an individual mouse; unpaired, two-tailed Student's t test, Scale bar = 100 µm. Download Figure 2-2, TIF file (19MB, tif) .
Similar PrPSc levels in prion-infected Hrsf/fGFAP-Cre and Hrsf/fLysM-Cre brains at the 75% time point. Western blotting and quantification of PrPSc levels in PK-digested brain lysates of (A) Hrsf/fGFAP-Cre− and Cre+ (top) and (B) Hrsf/fLysM-Cre− and Cre+ mice (bottom) at 114 and 115 dpi (∼75% of the expected incubation period). There were no differences in PrPSc levels (n = 3/group). Data shown as mean ± SEM; each dot represents an individual mouse; unpaired, two-tailed Student's t test. Download Figure 2-3, TIF file (8.7MB, tif) .
ESCRT-0 proteins, Hrs and STAM1, are reduced in Hrsf/fSyn1-Cre mice. A, Western blotting and quantification of brains of Syn1-Cre+ and Cre− mice show a reduction in Hrs and STAM1 protein levels (n = 4/group). B, Total PrPC levels are unchanged in neuronal Hrs-depleted brain (n = 3–5/group). Data shown as mean ± SEM; each dot represents an individual mouse; unpaired, two-tailed Student's t test, *p < 0.05. Download Figure 2-4, TIF file (9.2MB, tif) .
Hrs depletion in primary neurons reduces prion conversion and increases PrPC cell surface expression. A, B) Representative Western blotting and quantification of PK-digested (A) or undigested (B) Hrsf/f primary neuron lysates transduced with lentivirus encoding control vector or Cre (Cre− or Cre+) reveals a decrease in PrPSc in Hrs-depleted neurons but no change in PrPC expression. Relative PrPC and PrPSc levels were normalized to the average of the Cre− control lysates (n = 8 from 4 independent experiments, each with technical duplicates, mean and SEM shown). C, Representative Western blottings and quantification of biotin-tagged, streptavidin-precipitated cell surface PrPC from Hrsf/f primary neuron lysates reveal increased levels of surface PrPC in Hrs-depleted neurons. Control with no biotin labeling also shown (n = 4 experimental replicates). Data shown as mean ± SEM. Unpaired, two-tailed Student's t test, *p < 0.05, ***p < 0.001. Download Figure 2-5, TIF file (12.5MB, tif) .
No change in the developmental timeline of spongiform change or lysosomal accumulation in the cortex of neuronal Hrs-depleted mice. Representative hematoxylin and eosin (HE) and LAMP1 (lysosomes)-immunolabeled brain sections (cerebral cortex) from prion-infected Hrsf/fSyn1-Cre− and Cre+ mice collected at the same time point (107 dpi). Scale bar = 100 µm. Download Figure 3-1, TIF file (10.3MB, tif) .
Hrs depletion impacts neuronal autophagic flux. A, Schematic of mCherry-GFP-LC3 pH-sensitive sensor. GFP is quenched in the low pH of lysosomes, while mCherry is unaffected (autophagosomes and lysosomes appear yellow or red, respectively). B, Hrsf/f primary neurons transduced with mCherry-GFP-LC3 and control or Cre recombinase lentivirus for one week, and incubated with an inhibitor of autophagosome–lysosome fusion, bafilomycin (Baf), or inducer of autophagosome formation, rapamycin (Rap). C, Quantification of yellow puncta (autophagosomes) and red puncta (lysosomes) in each cell. Graphs indicate percent autophagosomes in each cell (total = autophagosomes + lysosomes) and (D) number of autophagosomes per cell. Data shown as mean ± SEM (C) and (D) were calculated for n = 3 independent experiments, n = 30–50 cells per condition. One-way ANOVA with Tukey's multiple comparisons post hoc test. *p < 0.05. Download Figure 4-1, TIF file (7.8MB, tif) .
Ubiquitinated protein levels in the cerebral cortex of uninfected Hrsf/fSyn1-Cre+ and Cre− mice. Western blotting of ubiquitinated proteins shows no significant difference in cerebral cortex from young uninfected Hrsf/fSyn1-Cre+ mice, although there was an upward trend (p = 0.13). All mice were female and ranged between 57 and 73 d old. Data shown as mean ± SEM. Unpaired, two-tailed Student's t test. Download Figure 4-2, TIF file (4.5MB, tif) .
Hrs depletion accelerates ubiquitinated protein accumulation in prion-infected mice. A, B, Representative Western blottings and quantification of the brain lysates reveal an increase in K63-linked and K48-linked ubiquitinated proteins in the prion-infected Hrsf/fSyn1-Cre+ mice at 102 dpi (n = 6–8 mice/group; all mice collected at same time point; 154–178 d old). Data shown as mean ± SEM. Unpaired, two-tailed Student's t test, *p < 0.05, **p < 0.01. Download Figure 4-3, TIF file (12.7MB, tif) .
Presynaptic and postsynaptic protein expression in brain of prion-infected Hrsf/fSyn1-Cre mice at 84 dpi. A, B, Representative Western blottings and quantification of synaptic proteins in prion-infected Hrsf/fSyn1-Cre− and Cre+ mice at 84 dpi (n = 3 mice/group). Unpaired, two-tailed Student's t test revealed no significant differences. Data shown as mean ± SEM. Download Figure 6-1, TIF file (12.3MB, tif) .
Postsynaptic proteins in brain of prion-infected Hrsf/fSyn1-Cre mice at 107 dpi. Representative Western blottings (A) and quantification (B) of synaptic proteins in prion-infected Hrsf/fSyn1-Cre− and Cre+ mice at 107 dpi (n = 6–8 mice/group). Unpaired, two-tailed Student's t test revealed no significant differences. Data shown as mean ± SEM. Download Figure 6-2, TIF file (11.9MB, tif) .
Neuronal quantification in uninfected or prion-infected cerebral cortex. Images and quantification of NeuN immunolabeled brain sections (frontal cortex) from Hrsf/fSyn1-Cre− and Cre+ uninfected or prion-infected mice reveal no differences in the numbers of NeuN positive nuclei. Identically-sized areas were assessed (427,193 µm2). Data shown as mean ± SEM. One-way ANOVA with Tukey's multiple comparisons post hoc correction (no significant differences). Scale bar = 100 µm. Download Figure 6-3, TIF file (7.1MB, tif) .
Synaptosomal protein levels from prion-infected Hrsf/fSyn1-Cre+ and Cre− brain. Representative Western blotting and quantification of Rab5, VSP35, Hrs, and synaptophysin proteins in synaptosomes collected from Hrsf/fSyn1-Cre− and Cre+ brain at 103 dpi. For the Rab5 t test, p = 0.175. Data shown as mean ± SEM shown. Relative protein levels were normalized to the average of the uninfected mice (n = 4–6 Hrsf/fSyn1-Cre mice/group). Unpaired, two-tailed Student's t test. **p < 0.01. Download Figure 7-1, TIF file (6.9MB, tif) .