

Abstract
During cardiac ischemia-reperfusion, excess reactive oxygen species can damage mitochondrial, cellular and organ function. Here we show that cysteine oxidation of the mitochondrial protein Opa1 contributes to mitochondrial damage and cell death caused by oxidative stress. Oxy-proteomics of ischemic-reperfused hearts reveal oxidation of the C-terminal C786 of Opa1 and treatment of perfused mouse hearts, adult cardiomyocytes, and fibroblasts with H2O2 leads to the formation of a reduction-sensitive ∼180 KDa Opa1 complex, distinct from the ∼270 KDa one antagonizing cristae remodeling. This Opa1 oxidation process is curtailed by mutation of C786 and of the other 3 Cys residues of its C-terminal domain (Opa1TetraCys). When reintroduced in Opa1−/− cells, Opa1TetraCys is not efficiently processed into short Opa1TetraCys and hence fails to fuse mitochondria. Unexpectedly, Opa1TetraCys restores mitochondrial ultrastructure in Opa1−/− cells and protects them from H2O2-induced mitochondrial depolarization, cristae remodeling, cytochrome c release and cell death. Thus, preventing the Opa1 oxidation occurring during cardiac ischemia-reperfusion reduces mitochondrial damage and cell death induced by oxidative stress independent of mitochondrial fusion.
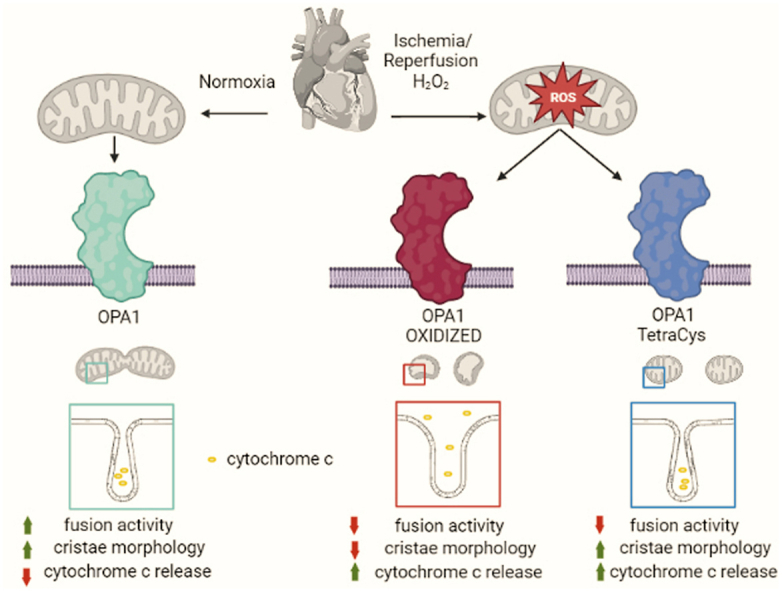
Graphical abstract
Highlights
-
•
The mitochondria-shaping protein Opa1 is specifically oxidized at Cys 786 during cardiac ischemia/reperfusion.
-
•
Oxidative stress leads to Opa1 oxidation and to the formation of a redox sensitive Opa1 oligomer.
-
•
The C-terminal Cys residues Opa1TetraCys mutant is not processed into short Opa1 and does not sustain mitochondrial fusion.
-
•
Opa1TetraCys controls cristae shape and protects mitochondria and cells from hydrogen peroxide.
-
•
Opa1 C-terminal Cys residues dissociate mitochondrial fusion from cristae remodeling in hydrogen peroxide induced cell death.
1. Introduction
Mitochondrial dysfunction is central to cell death and tissue damage in most cardiovascular diseases. Besides converting chemical energy, mitochondria generate potentially damaging side products such as reactive oxygen species (ROS) [1]. Respiratory chain side products, but also monoaminoxidases, p66Shc, cytochrome p450, peroxisomal oxidases and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) are well characterized sources of ROS within the cell [[2], [3], [4], [5]]. On the other side, superoxide dismutase, catalase, glutathione peroxidase/reductase system and the peroxiredoxin/thioredoxin system constitute the antioxidant defense system. Mitochondrial and cytosolic ROS can have both beneficial and detrimental effects depending on the balance between their generation and elimination [6]. In physiological conditions, cardiac ROS signalling regulates heart development, cardiac calcium handling, excitation contraction coupling and vascular tone [7]. Cardiac hypertrophy, heart failure, cardiac ischemia-reperfusion injury and diabetic cardiomyopathy are instead characterized by a common characteristic: excessive ROS production. ROS can damage mitochondria macromolecules such as mitochondrial DNA (mtDNA), lipids and proteins, at or near the site of formation. Therefore, in addition to the role of mitochondria as a source, mitochondria themselves can be damaged by ROS.
Accumulating evidence suggests that mitochondrial redox homeostasis is bidirectionally linked to mitochondrial dynamics. ROS can cause mitochondrial fragmentation and mitochondrial fusion disruption, as well as fission results in ROS accumulation [[8], [9], [10]]. While inhibition of mitochondrial fusion can cause ROS accumulation by the impairment in mitochondrial respiratory chain complexes and supercomplexes assembly, or of ATP synthase oligomerization [11,12], how impaired fission causes ROS accumulation is less clear [13]. Similarly, the molecular mechanisms by which ROS affect the machinery of mitochondrial dynamics are largely unknown. Dynamin related protein1 (Drp1) with its outer mitochondrial membrane (OMM) receptors Fis1, Mid49, Mid51 and MFF control mitochondrial fission [[14], [15], [16], [17], [18]]. Fusion is performed by the coordinated action of mitofusins (Mfn) 1 and 2 in the OMM and Optic atrophy 1 (Opa1), in the inner mitochondrial membrane (IMM) [[19], [20], [21]]. In the heart, five different Opa1 forms are found. Two long Opa1 forms are anchored to the inner mitochondrial membrane (IMM) by its transmembrane domain. Proteolytic cleavage of long Opa1 (L-Opa1) generates three short Opa1 (S-Opa1) forms. In humans, the proteases OMA1 and YME1L1 cleave L-Opa1 at two distinct cleavage sites called S1 and S2. OMA1 is activated by self-cleavage upon mitochondrial membrane depolarization in response to stress, including oxidative stress [[22], [23], [24]]. In addition to its key role in mitochondrial fusion, Opa1 facilitates respiratory chain super complex assembly, impinging on mitochondrial metabolism and blunts cytochrome c release during apoptosis [12,25,26]. A single study addressed molecularly how changes in the cellular redox potential affect the fusion/fission machinery, showing that Mfn2 oxidation promotes its oligomerization and mitochondrial fusion [27]. However, it is not clear whether other mitochondria-shaping proteins including Opa1 are redox-regulated, and whether they are targeted by oxidative damage during ischemia-reperfusion, one of the leading heart conditions characterized by ROS induced cell damage. With this question in mind, we performed oxy-proteomics of Opa1 in hearts undergoing ischemia reperfusion. We find that a C-terminal Cys residue of Opa1 is specifically oxidized and that upon ischemia reperfusion or hydrogen peroxide treatment, Opa1 aggregates in high molecular weight complexes that are sensitive to reducing agents. We determined the molecular identity of the Cys residue responsible for Opa1 oxidation and provide evidence that mutation of this residue and the other three Cys residues in Opa1 C-terminus impairs Opa1 maturation but not its ability to stabilize cristae structure, and more importantly generate an Opa1 version that is resistant to oxidation induced multimerization and protects cells selectively from oxidative stress induced cell death.
2. Results
2.1. Opa1 is oxidatively modified during heart ischemia-reperfusion
Mitochondrial damage is a key amplifier of cardiac ischemia-reperfusion injury, during which Opa1 undergoes cleavage. Remarkably, increased Opa1 levels protect the heart from ischemia-reperfusion by blunting the cristae remodeling pathway and preventing cell death [28]. However, it is unclear whether and how Opa1 is inactivated by ischemia-reperfusion. We therefore used a validated system of resin-assisted oxidized proteins capturing approach and LC-MS/MS [29] to explore whether oxidized Opa1 peptides were enriched in mouse hearts subjected to ischemia-reperfusion injury in a retrograde Langendorff perfusion system. In this approach, free thiols were blocked by N-ethylmaleimide, and oxidized thiols were then reduced with DTT and captured using a thiol-binding resin. Resin-bound proteins were subjected to trypsin digestion, and the remaining resin-bound peptides were eluted with DTT and identified by LC-MS/MS. Compared to control hearts, the resin captured the Opa1 peptide containing C786 following ischemia-reperfusion (Fig. 1A). Homology modelling of Opa1 showed that this Cys residue at the C-terminal stalk domain of the protein was likely to be exposed to the solvent, like the 3 other C-terminal Cys residues (C853, C856, C874; Fig. 1B–E). C853 corresponds to C812 of the crystallized C. thermophilus Opa1 orthologue Mgm1, whereas Cys 786, 856 and 874 do not appear to be conserved (Fig. S1A). These residues are instead conserved between H. sapiens and M. musculus Opa1 (Fig. S1B). In C. thermophilus Mgm1, C812 is at the margin of the paddle domain, forms a disulfide crossbridge with C821 and is only partially involved in the control of Mgm1 GTPase activity [30]. Thus, Opa1 is specifically oxidized in a residue conserved between human and mouse Opa1 and homology modelling indicates that this C786 and the other Cys residues in Opa1 C-terminus might be exposed to an oxidizing environment.
Fig. 1.

Cysteine 786 of Opa1 is oxidized in hearts undergoing ischemia-reperfusion ex vivo.
(A) MS/MS spectra of the Opa1 peptide containing Cys786 from hearts subjected to ischemia-reperfusion injury bound to the thiopropyl Sepharose resin. Representative MS/MS spectra showing fragmentation of the NRTQEQCVHNETKNELEK peptide. The peptide sequence above the representative spectrum shows theoretical b ion identifications (red, N terminal fragments) and theoretical y ion identifications (blue, C terminal fragments). Peaks in the spectrum that are marked red correspond to matched b ions and peaks that are marked blue correspond to matched y ions. The number paired with each ion identification (i.e., b2, y4, etc.) indicates the number of amino acids present on N terminal fragments for b ions and C terminal fragments for y ions. This peptide identification was observed in hearts subjected to ischemia-reperfusion injury, not in control hearts at baseline.
(B) Predicted structural model of recombinant Opa1 protein, corresponding to Mus musculus isoform 1 (Uniprot: P58281), lacking the mitochondrial targeting sequence (MTS) and the transmembrane domain (TM). C-terminal cysteines are highlighted in red. Inset, close view of the Cys location in the stalk and PH domain of Opa1.
(C–E) Close views of Opa1 C-terminal cysteines and their surrounding residues: Cys786 (C), Cys583 and Cys586 (D), and Cys874 (E). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
We next verified if oxidative stress led to the formation of intra or intermolecular Opa1 disulfide bridges. We induced oxidative stress in whole hearts in a retrograde Langendorff perfusion system and in isolated adult cardiomyocytes. Ischemia-reperfusion of isolated mouse hearts or their perfusion with H2O2 led to the accumulation of an Opa1 immunoreactive band running at approx. 180 kDa, a size compatible with the formation of a crosslink between long (L-) and short (S-) Opa1 that cooperate in Opa1 functions [31,32]. These bands were not detectable in reducing PAGE, suggesting that the Opa1-containing 180 KDa band was disrupted by the strong reducing agent DTT. In H2O2-perfused hearts, we additionally identified a higher MW (∼230 kDa) adduct that was also sensitive to DTT and could include proteins other than Opa1 under these harsh oxidizing conditions (Fig. 2A, arrowhead). We observed the accumulation of the180 KDa, DTT-sensitive Opa1 immunoreactive band also in adult cardiomyocytes undergoing anoxia/reperfusion or treatment with H2O2 (Fig. 2B). Densitometric analysis indicated that while this band was absent in control conditions, it represented ∼9% of the total Opa1 in cardiomyocytes exposed to these oxidizing conditions (Fig. 2C). The formation of these redox adducts was rapid, as they were retrieved as early as after 15 min following treatment of mouse embryonic fibroblasts (MEFs) with H2O2 (Fig. 2D). We finally wished to address whether the Opa1-immunoreactive adducts retrieved in the non-reducing PAGE of H2O2-perfused hearts contained both L- and S-Opa1. We therefore ran reducing and non-reducing PAGE from control and H2O2-perfused hearts, excised the lanes and ran them in a second dimension reducing and non-reducing PAGE. We first determined the position of L- and S-Opa1 in the 2nd dimension non-reducing electrophoresis by a first dimension reducing electrophoresis of control perfused hearts. We visualized two Opa1-immunoreactive bands: a higher MW band corresponding to the position of L-Opa1 in the 1st dimension reducing PAGE and a lower MW band corresponding to S-Opa1 in the 1st dimension gel. Thus, L-and S-Opa1 can be identified in a non-reducing 2nd dimension PAGE. We next ran a 2nd dimension non-reducing PAGE from a 1st dimension non-reducing gel and immunoblotted it for Opa1. We retrieved the same L-and S-Opa1 corresponding to the L-and S-Opa1 identified in the first dimension, plus an Opa1 immunoreactive band at ∼180 kDa that corresponded to the oxidized adduct identified in the first dimension. Finally, we ran a 2nd dimension reducing PAGE from the 1st dimension non-reducing gel. Here, immunoblotting for Opa1 identified L- and S-Opa1 bands at the position corresponding to the oxidized adduct identified in the first dimension (Fig. 2E). Thus, this ∼180 kDa adduct contains both L- and S-Opa1.
Fig. 2.

Opa1 forms redox sensitive oligomers upon H2O2 or ischemia reperfusion in vitro and ex-vivo.
(A) Mouse hearts in a Langendorff system were perfused with oxygenated buffer (Untr), 1 mM H2O2 (H2O2) or underwent ischemia reperfusion (I/R). Tissues were lysed and equal amounts of protein (20 μg) from cardiac tissues were separated by SDS-PAGE in the indicated reducing or non-reducing conditions. Immunoblotting was performed using the indicated antibody. Red P: membranes were stained using Red Ponceau. Asterisks indicate a ∼180 KDa Opa1 immunoreactive band that disappears in reducing SDS-PAGE; arrowhead a ∼240 KDa Opa1 immunoreactive band that is also sensitive to reduction.
(B) Adult cardiac myocytes were perfused with oxygenated buffer (Untr), subjected to I/R or treated with 1 mM H2O2 for 30 min and lysed. Equal amounts of protein (20 μg) were separated by reducing or non-reducing SDS-PAGE and immunoblotted using the indicated antibodies. Asterisks indicate a ∼180 KDa Opa1 immunoreactive band that disappears in reducing SDS-PAGE.
(C) Percentage of oxidized Opa1 calculated by densitometric analysis in non-reducing immunoblots from experiments as in B. N = 5 independent experiments (open dots). Line indicates mean, whiskers SEM. *, p < 0.05 in a one-way ANOVA with Tukey's mean comparison.
(D) MEFs were treated with 1 mM H2O2 for the indicated time and lysed. Equal amounts of protein (15 μg) were separated by reducing or non-reducing SDS-PAGE and immunoblotted using the indicated antibodies. Asterisks indicate the ∼180 KDa Opa1 immunoreactive band that disappears in reducing SDS-PAGE; arrowhead the ∼240 KDa Opa1 immunoreactive band also sensitive to reduction.
(E) 2D reducing/non-reducing SDS-PAGE of total lysates (20 μg) of mouse hearts perfused in a Langendorff system with oxygenated buffer (Untr) or 1 mM H2O2 (H2O2) for 15 min, as indicated. Equal amounts of protein (15 μg) were separated by a 1st dimension reducing or non-reducing SDS-PAGE and the lane was excised and mounted on the top of a 2nd dimension reducing or non-reducing SDS-PAGE. In parallel, an identical 1st dimension lane was transferred onto PVDF membranes and immunoblotted using and anti Opa1 antibody for reference. The top images show this anti Opa1 immunoblot on the 1st dimension reference lane. The bottom images show the corresponding anti Opa1 immunoblot of the 2nd dimension SDS-PAGE ran in the indicated conditions, transferred onto PVDF membranes and immunoblotted using and anti Opa1 antibody. The dotted lines indicate the molecular species immunoreactive for Opa1 in the different 1st and 2nd dimension immunoblots.
2.2. C856 and C874 are required for Opa1 cleavage and mitochondrial elongation
To characterize the role of the four C-terminus Cys in Opa1 oxidation, we generated Opa1 mutants where we substituted each Cys with a Ser. We re-expressed these Opa1 mutants in Opa1−/− MEFs and verified their expression, cleavage, and sensitivity to oxidation by monitoring the formation of the 180 KDa DTT-sensitive band characterized in Fig. 2. The single Opa1C786S and Opa1C853S mutants were properly cleaved into S-Opa1. Unexpectedly, mutation of Cys 856 or 874 increased the L-to S-Opa1 ratio, suggesting that these 2 Cys residues are required for basal Opa1 cleavage by the inner mitochondrial membrane proteases Yme1L1 [33]. We therefore generated multiple Cys mutants and verified whether they were properly cleaved. Every mutant containing the C856S and/or the C874S mutation was less efficiently cleaved into the S- form, confirming that these two Cys residues are required for Opa1 maturation (Fig. 3A). We therefore verified if the impaired cleavage was associated with defects in mitochondrial elongation which requires the proper ratio between L- and S-Opa1 [33,34]. While re-expression in Opa1−/− MEFs of Opa1WT as well as of Opa1C786S and of Opa1C853S restored mitochondrial elongation, all the Opa1 mutants containing the C856S and/or C874S substitutions that were not efficiently cleaved were similarly unable to elongate mitochondria (Fig. 3B and C). Thus, Opa1 requires Cys 856 and 874 for efficient maturation into its S- forms and to drive mitochondrial elongation.
Fig. 3.

Single, double, and triple Opa1 C-terminal Cys mutants generate the 180 KDa Opa1 oligomer upon H2O2 treatment.
(A) Equal amounts of proteins (15 μg) from MEFs of the indicated genotypes treated where indicated with 1 mM H2O2 for 30 min were separated by non-reducing SDS-PAGE and immunoblotted using the indicated antibodies. Asterisks indicate the ∼180 KDa Opa1 reduction-sensitive oligomer.
(B) Representative confocal images of mitochondrial morphology in WT and Opa1−/− MEFs cotransfected with mtRFP and the indicated plasmids for 24 h. Scale bar: 20μm.
(C) Quantification of mitochondrial major axis length. Boxes represent 25th-75th percentiles with median values, whiskers 10th-90th percentiles. Values of the individual (4–13, at least 50 cells/experiment) independent experiments performed as in B are plotted as open dots. *. P < 0.05 in a one way ANOVA with Tukey's means comparison between Opa1WT and the indicated Opa1 mutants.
2.3. Mutation of all four Opa1 C-terminal cys residues prevents its oxidation
We next moved to the analysis of the relative role of each Cys residue in Opa1 oxidation. Because the proteomic results (Fig. 1A) pointed to C786 as oxidized during I/R, we first tested whether treatment with H2O2 promoted the formation of the 180 KDa band in Opa1−/− MEFs re-expressing Opa1C786S. Surprisingly, this was the case, suggesting that a different, solvent-exposed Cys might be oxidized in place of C786 when this latter residue was mutated. Similarly, the 180 KDa band was retrieved upon H2O2 treatment in Opa1−/− fibroblasts reconstituted with all the single and multiple Cys mutants, even if Opa1C853S and the other Opa1 mutants containing the C853S substitution appeared less sensitive to oxidation (Fig. 3A).
Because all the mutants characterized so far were still undergoing formation of the 180 KDa adduct upon oxidative stress, we decided to generate a quadruple C→S mutant of all C-terminal Cys residues, Opa1C786-853-856-874S (Opa1TetraCys). First, we inspected cleavage of Opa1TetraCys into S- forms in reducing SDS-PAGE. As expected, this mutant was also not efficiently matured, and by immunoblotting we could retrieve only L-Opa1TetraCys in the reconstituted Opa1−/− MEFs (Fig. 4A). Different from the other Cys mutants generated here, cleavage of Opa1TetraCys appeared completely blocked in basal, unstressed conditions. Upon oxidative stress, Opa1TetraCys was conversely cleaved, like the other Opa1 mutants. Because stress induced Opa1 cleavage depends on Oma1 [24,33], these results indicate that H2O2-induced Opa1 cleavage depends on processes different than the ones responsible for Opa1 maturation and that are independent of the Cys residues analyzed here. Non reducing SDS-PAGE indicated that Opa1TetraCys was not retrieved in the 180 KDa band induced by oxidative stress. Thus, all the C-terminal Opa1 Cys residues can participate in Opa1 oxidative damage, and formation of the 180 kDa Opa1 containing band upon oxidative damage can be abrogated when these Cys residues are mutated into Ser (Fig. 4A).
Fig. 4.

The Opa1TetraCys mutant that is not oxidized upon H2O2 treatment restores cristae shape but not mitochondrial fusion.
(A) Cells of the indicated genotype treated with 1 mM H2O2 for 30 min where indicated were lysed and equal amounts of proteins (15 μg) were separated by reducing or non-reducing SDS-PAGE and immunoblotted using the indicated antibodies. Asterisks indicate the ∼180 KDa Opa1 reduction-sensitive oligomer.
(B) Representative confocal images of mitochondrial fusion experiments. MEFs of the indicated genotype were co-transfected with mtPAGFP and mtRFP. At time 0, mtPAGFP was activated in a region of interest (white circle) and cells were imaged by real time confocal microscopy and the frame acquired at time = 30 min is shown. Bar, 20μm.
(C) Quantification of mitochondrial fusion in experiments as in (B). Data represents mean of ±SEM of 4 independent experiments.
(D) Representative electron micrographs of MEFs of the indicated genotype. The boxed area of the top images is magnified in the bottom images. Scale bars: 1 μm (top) and 500 nm (bottom).
(E) Average ± SEM of morphometric analysis of cristae width in 105 (EV), 309 (Opa1WT) and 287 (Opa1TetraCys) mitochondria of the indicated genotype from 4 independent experiments. *, p < 0.001 in a two-sample t-Test.
We next verified whether this mutant induced mitochondrial elongation. Opa1TetraCys was unable to reconstitute the elongated mitochondrial network when reintroduced in Opa1−/− MEFs (Fig. 3B). This result was expected given that Opa1TetraCys harbors the C856S and C874S substitutions that impair Opa1-driven mitochondrial elongation. Because mitochondrial morphology results from the balance between fusion and fission, we measured the rate of mitochondrial fusion in Opa1−/− MEFs reconstituted with Opa1TetraCys. In a well characterized fusion assay based on the real time measurement of the diffusion of a photoactivable GFP (paGFP) targeted to the mitochondrial matrix [35], fusion of mitochondria expressing Opa1TetraCys was negligible and indistinguishable from that observed in Opa1−/− mitochondria (Fig. 4B and C). Thus, the Opa1TetraCys mutant that is not matured into S- forms is unable to sustain mitochondrial fusion.
Because Opa1 is also a master cristae structure regulator [[36], [37], [38]], we decided to address if the morphological defects observed in Opa1TetraCys cells were accompanied by changes in cristae shape. Electron microscopy analysis revealed that cristae morphology was normal in Opa1−/− MEFs reconstituted with Opa1TetraCys and cristae lumen was even narrower than in MEFs reconstituted with Opa1WT (Fig. 4D and E). Finally, we tested if reintroduction of Opa1TetraCys was sufficient to ameliorate the mitochondrial dysfunction caused by the lack of Opa1. In permeabilized Opa1−/− MEFs re-expressing Opa1TetraCys mitochondrial respiration was partially restored (Figs. S3A–C) and in real time experiments of mitochondrial membrane potential measurements in intact Opa1−/− MEFs, Opa1TetraCys protected like Opa1WT from depolarization induced by oligomycin, a sensitive assay of latent mitochondrial dysfunction [39] (Fig. S3D). Thus, mutations of the four C-terminal Cys residues impair Opa1 maturation into its S- forms and mitochondrial fusion, but not the ability of Opa1 to control cristae shape and restore mitochondrial function.
2.4. Opa1TetraCys reduces cell death induced by oxidative stress
We finally tested whether Opa1TetraCys protected cells from H2O2-induced cell death [40]. While in response to the intrinsic apoptotic stimuli etoposide and staurosporine, viability of Opa1−/− MEFs reconstituted with Opa1WT or Opa1TetraCys was superimposable, cells expressing Opa1TetraCys were protected from H2O2 induced cell death (Fig. 5A). Mechanistically, we found that Opa1TetraCys protected cells from H2O2-induced loss of mitochondrial membrane potential, as evidenced in real time imaging experiments of the potentiometric probe tetramethyl rhodamine methyl ester (TMRM) (Fig. 5B and C). Opa1TetraCys also protected from cytochrome c release induced by H2O2 (Fig. 5D and E). Because Opa1 forms a 220 KDa oligomer that protects mitochondria from cristae remodeling and cytochrome c release [36], we compared the stability of this oligomer during oxidative stress in Opa1−/− MEFs expressing Opa1WT or Opa1TetraCys. Indeed, H2O2 destabilized the 220 KDa oligomer in Opa1−/− MEFs expressing Opa1WT, whereas it remained stable in MEFs expressing Opa1TetraCys (Fig. 5F). Accordingly, in Opa1TetraCys MEFs treated with H2O2 cristae were narrower whereas BAK oligomerization was similar to that observed in Opa1WT MEFs (Fig. S4), further supporting that Opa1TetraCys protects from cell death by stabilizing mitochondrial ultrastructure without affecting outer mitochondrial membrane permeabilization that allows cytochrome c release egress from mitochondria. Taken together, these data indicate that cristae remodeling, cytochrome c release and cell death induced by H2O2 are blunted when specific C-terminal Cys residues of Opa1 are replaced by non-oxidable Ser residues.
Fig. 5.

Opa1TetraCys MEFs are protected from H2O2 induced cell death.
(A) MEFs of the indicated genotype were treated as indicated, collected, stained with annexin V/PI and analyzed by cytofluorimetry. Boxes represent SEM with mean values, whiskers 10th-90th percentiles. Values of the individual independent experiments are plotted as dots. *, p < 0.05 in a two sample t-test between the indicated groups.
(B) Pseudocolor-coded images of TMRM fluorescence in MEFs of the indicated genotypes treated with 1 mM H2O2. The pseudocolor scale is indicated. Top images represent the initial frame of the real-time sequence, while bottom ones were acquired at time = 40 min. Bar, 40μm.
(C) Quantitative analysis of TMRM fluorescence changes over mitochondrial regions in experiments as in B. Where indicated, 1 mM H2O2 and 2 μM FCCP were added. Data represents mean ± SEM of 4 independent experiments.
(D) Representative confocal images of cytochrome c subcellular distribution. MEFs of the indicated genotype were transfected with mtRFP (red), treated where indicated with 1 mM H2O2 for 30 min, fixed and immunostained for cytochrome c (green). Bar, 15 μm
(E) Localization index of cytochrome c in MEFs of the indicated genotype treated where indicated with 1 mM H2O2 for 30 min. Plots represent mean ± SEM of 9–13 independent experiments performed as in D. Open dots indicate values of individual experiments. *, p < 0.05 in a one way ANOVA with Tukey's mean comparison between the indicated conditions.
(F) Mitochondria (0.5 mg/mL) isolated from MEFs of indicated genotype were incubated with 100 μM H2O2 for the indicated time. Upon crosslinking with 10 mM EDC, equal amounts (15 μg) of mitochondrial proteins were separated by SDS-PAGE and immunoblotted using the anti Opa1 antibody. Asterisks indicate the Opa1 oligomer. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3. Discussion
By combining proteomics, biochemistry, genetics, and functional assays we unveil that oxidation of specific Opa1 Cys residues amplifies oxidative stress-induced mitochondrial dysfunction and cell death.
ROS accumulation and more broadly oxidative stress have been implicated in a plethora of cardiac diseases, including heart failure, ischemia reperfusion, cardiac hypertrophy, and diabetic cardiomyopathy [41]. Mitochondria not only can generate ROS at different sites of the respiratory chain, as well as at the level of the outer mitochondrial membrane MAO A and B [42], but are also main targets of ROS. In fact, ROS generated at different sites of the respiratory chain selectively oxidize different mitochondrial proteins, including Opa1 [43]. Targeting Opa1 might explain the mitochondrial fragmentation observed in cells undergoing oxidative stress. Indeed, mitochondrial fragmentation under these conditions has been ascribed to the rapid Opa1 degradation by the IMM protease Oma1 that is activated upon treatment of mitochondria with H2O2, respiratory chain inhibitors, K+ ionophores and uncouplers [23]. However, whether Oma1 is directly or indirectly affected by oxidative stress remains unexplored. Conversely, our proteomics data indicate that in a model of cardiac ischemia-reperfusion Opa1 Cys 786 is specifically oxidized. Biochemical analyses support that oxidative stress in whole hearts, isolated cardiomyocytes, and fibroblasts rapidly leads to the appearance of a 180 KDa Opa1 oligomer sensitive to strong reducing agents. Size-wise, this oligomer is compatible with a dimer formed by L- and S-Opa1 forms. Conversely, the Opa1-containing oligomer that participates in the regulation of cristae shape has an apparent MW of 270 KDa and is likely composed by one S-Opa1 and two L-Opa1 molecules [36]. In fact, 2D gel electrophoresis confirms that the 180 KDa Opa1 oligomer is formed by L- and S-Opa1, suggesting that oxidative stress leads to the formation of intermolecular disulfide bonds between these L- and S-Opa1 forms. The Cys786 residue is predicted by homology modelling to be exposed to the solvent like the three others Cys residues found in Opa1 C-terminus, C853, C856 and C874, suggesting that all of them could participate in intermolecular disulfide bond formation. Indeed, formation of the Opa1 oxidation 180 KDa product is curtailed only when all C-terminal Cys residues are mutated. Functionally, prevention of Opa1 oxidation by mutagenesis of all four C-terminal Cys residues protects mitochondria from cytochrome c release, mitochondrial depolarization and cell death induced by hydrogen peroxide. Thus, ROS can damage mitochondria by directly targeting Opa1. Because oxidation of Cys residues can result in protein aggregation and degradation [44], the Opa1 oxidation discovered here appeared as a natural prerequisite for its cleavage by Oma1. However, our biochemical studies indicate no difference between cleavage of the Opa1TetraCys mutant generated here and WT Opa1 in response to oxidative stress. This was surprising, because mitochondrial membrane potential was sustained in Opa1TetraCys expressing cells treated with hydrogen peroxide and depolarization is a canonical signal for Oma1 activation [45,46]. Thus, signals other than depolarization might activate Oma1 during oxidative stress to drive Opa1 cleavage.
A second surprising finding of our study was that the Opa1TetraCys mutant is not efficiently cleaved into S-Opa1 at steady state, a process that is mediated by the IMM protease Yme1l1 [33]. Opa1TetraCys is also unable to restore mitochondrial elongation when reintroduced in Opa1 deficient MEFs. By analyzing the multiple mutants generated here, we could pinpoint a crucial role for Cys 856 and/or 874 in Opa1 cleavage. Because the GTPase activity of purified recombinant Opa1TetraCys is comparable to that of Opa1WT, we postulate that Opa1TetraCys is unable to restore mitochondrial fusion in Opa1−/− MEFs because of its impaired cleavage. On the other hand, Opa1TetraCys can fully correct mitochondrial cristae shape when expressed in Opa1−/− MEFs. This mutant therefore functionally dissociates the mitochondrial fusion and cristae shaping activity of Opa1 and might represent a useful tool to genetically dissect the multiple facets of Opa1 mitochondrial and cellular biology. These findings are consistent with results obtained in assays of Opa1-mediated liposome fusion. In this reconstituted assay, L-Opa1 can bind cardiolipin in trans to drive membrane fusion, but the fusion reaction is greatly stimulated by the presence of S-Opa1. Conversely, interaction in trans between two L-Opa1 molecules is sufficient for membrane tethering, a therefore for sustaining cristae shape [47].
The evolutionary conserved C-terminal domain of Opa1 is predicted to mediate Opa1 oligomerization [48,49]. Crystals of the C. thermophilus Opa1 orthologue Mgm1 substantiate this prediction and identify that at least one of the Cys residues studied here (C. thermophilus C812, corresponding to Opa1 C853) in the stalk region crucial for the formation of Mgm1 dimers [30]. In Mgm1, C812 forms a disulfide bridge with C821 that is however not conserved in Opa1. In Opa1, C856 is proximal enough to C853 to establish a disulfide bridge, but the orientation of the thiols of these two Cys residues in the predicted Opa1 structure suggests that a conformational change is required to accommodate the bridge. How oxidative stress affects these Cys residues, potentially favoring intermolecular disulfide bridges, remains to be addressed. Structural disulfide bridges are common in the mitochondrial intermembrane space and are retrieved for example in Atp23 [50], MICU1 and MICU2 [51], and anamorsin [52]. Our analysis extends this list to include Opa1: its oxidation represents an important mechanism of mitochondrial damage, cytochrome c release and cell death upon oxidative stress like during heart ischemia-reperfusion.
4. Methods
4.1. Cell biology
4.1.1. Cell culture
SV40 transformed Opa1−/− mouse embryonic fibroblasts (MEFs) were cultured as described in Ref. [36]. Cells were grown in Dulbecco's Modified Medium (DMEM, Invitrogen) containing 4.5 mg/mL glucose and supplemented with 10% fetal bovine serum (FBS, Invitrogen), 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/mL streptomycin (Invitrogen), 100 μM non-essential amino acids (Invitrogen) at 37 °C and 5% CO2 atmosphere.
4.1.2. Transfection, virus production and transduction
Opa1−/− MEFs and HEK293 cells were cultured as previously described [35]. Cells were transfected using Lipofectamine (Invitrogen) following manufacturer's instructions. HEK293 were used as packaging cell line and co transfected with the packaging vector pIK and the appropriate pMSCV as described in Ref. [53]. Viral supernatant was collected and used to transduce Opa1−/− MEFs in the presence of 4 μg/mL Hexadimethrine Bromide (Sigma). Following an overnight transduction, medium was supplemented with Puromycin 5 (μg/ml, Sigma) to select for transduced cells.
4.1.3. Plasmids and molecular biology
Mito-dsRED (mtRFP) was described previously [54]. mt-PAGFP was a gift from M. Karbowski (University of Maryland, USA). pMSCV, pMSCV-Opa1 were previously described in Ref. [55]. pMSCV-Opa1C786S, pMSCV-Opa1C853S, pMSCV-Opa1C856S, pMSCV-Opa1C874S, pMSCV-Opa1C786–853S, pMSCV-Opa1C786–856S, pMSCV-Opa1C786–874S,pMSCV-Opa1C853–856S, pMSCV-Opa1C853–874S, pMSCV-Opa1C856–874S, pMSCV-Opa1C786-853−856S, pMSCV-Opa1C786-853−874S, pMSCV-Opa1C786-856−874S, pMSCV-Opa1C853-856−874S, pMSCV-Opa1C786-853-856-874 (named Opa1TetraCys) were obtained by Site directed mutagenesis kit (Invitrogen) following manufacturer's instructions and using the primer sequences reported in Supplementary Table 1.
4.2. In vivo procedures
4.2.1. Animal handling
Animals were handled by specialized personnel under the control of inspectors of the Veterinary Service of the Local Sanitary Service (ULSS 6-Padova), the local officers of the Ministry of Health. The use of animals and the experimental protocol was approved by the animal welfare coordinator of the OPBA, University of Padova. Organ explant procedures are authorized by the Italian Health Ministry, Office VI (564/2020-PR) and were conducted in accordance with relevant codes of practice for the care and use of animals for scientific purposes.
4.2.2. Cardiac ischemia reperfusion (I/R)
A Langendorff model was used to perform I/R on ex vivo murine hearts [56]. After heparinization, 5 months male mice were cervical dislocated and their hearts were rapidly perfused retrogradely for 5 min with 5 mL/min bicarbonate buffer (118.5 mM NaCl, 3.1 mM KCl, 1.18 mM KH2PO4), 25 mM NaHCO3, 1 mM MgCl2, 1.4 mM CaCl2, and 5.6 mM glucose) insufflated with 95%CO2–5%O2 at 37 °C. Hearts were subjected to 45 min ischemia, followed by 15 min of reperfusion, or to perfusion with bicarbonate buffer containing 1 mM H2O2 for 15 min. At the end of the procedure, hearts were immediately snap frozen in liquid nitrogen and homogenized for Western blot analysis.
4.2.3. Adult mouse cardiomyocytes
Adult murine cardiomyocytes were isolated from 12-weel-old C57BL6/J mice as described [57]. Collagenase digested cardiac myocytes were incubated in buffer with increasing concentration of Ca2+, achieving a concentration of 1.2 mM Ca2+ as in Dulbecco's modified Eagle's medium (DMEM) plating media. Cells were seeded at a density of 6 × 104/ml in a laminin coated 6 well plate. Cardiac myocytes were maintained in DMEM supplemented with 10 mM 2,3-butanedione monoxime (Sigma) for 12 h at 37 °C in presence of 5% CO2. Medium was replaced with HBSS supplemented with 5 mM Hepes (Sigma) and the indicated concentrations of H2O2.
4.3. Oxidation site identification with resin-assisted capture
4.3.1. Mass spectrometry
Hearts were Langendorff-perfused with Krebs-Henseleit buffer as described [58], and subjected to control perfusion (60 min perfusion period) or an ischemia-reperfusion protocol (60 min equilibration period, 20 min ischemic period, 5 min reperfusion period); hearts were snap frozen in liquid nitrogen immediately following the treatment protocol. Hearts were then powdered on liquid nitrogen with a mortar and pestle and resuspended in 1.5 mL of homogenization buffer containing (in mmol/L): sucrose (Sigma), HEPES-NaOH 7.7 (Life Technologies), EDTA (Sigma), and Neocuproine (Sigma). An EDTA-free protease inhibitor tablet (Roche Diagnostics Corporation, Indianapolis, IN) was introduced just before use. Samples were then homogenized via Dounce glass homogenization on ice and centrifuged at 1000 g for 2 min. The supernatant was recovered as total crude homogenate. Protein concentration was determined using the Bradford protein assay. Cysteine oxidation sites for Opa1 were then identified using an unbiased oxidation-resin-assisted capture (Ox-RAC) enrichment approach as previously described [58]. Samples (1 mg) were diluted in HEN buffer containing (in mmol/L): HEPES-NaOH 7.7 (250), EDTA (1), and Neocuproine (0.1) with 2.5% SDS and an EDTA-free protease inhibitor tablet (Roche Diagnostics Corporation). All buffers were de-gassed prior to use to prevent oxidation of the resin. Homogenates were then incubated with 20 mmol/L ascorbate for 45 min at room temperature to remove S-nitrosylation. Samples were then incubated with 50 mmol/L N-ethylmaleimide (NEM; Sigma) for 20 min at 50 °C to block non-modified (i.e., free) and ascorbate-reduced thiol groups from modification; ascorbate and NEM were removed via acetone precipitation. Samples were then resuspended in HENS and oxidized thiols were then reduced with 10 mmol/L DTT for 10 min at room temperature; DTT was removed via acetone precipitation. Samples were then resuspended in HEN with 1% SDS (HENS). Thiopropyl sepharose (GE Healthcare, Piscataway, NJ) was rehydrated for 25 min in water. Following rehydration, 25 mL of the resin slurry was added to a Handee Mini Spin Column (Pierce) and washed with 5 × 0.5 mL water, followed by 10 × 0.5 mL HEN buffer. Blocked samples were then added to the thiopropyl Sepharose-containing spin column and rotated for 4 h in the dark at room temperature. Resin-bound proteins were then washed with 8 × 0.5 mL HENS buffer, followed by 4 × 0.5 mL HENS buffer diluted 1:10. Samples were then subjected to trypsin digestion (sequencing grade modified; Promega, Madison, WI) overnight at 37 °C with rotation in buffer containing (in mmol/L): NH4HCO3 (50) and EDTA (1). Resin-bound peptides were then washed with 5 × 0.5 mL HENS buffer diluted 1:10, 5 × 0.5 mL 2 mol/L NaCl, 5 × 0.5 mL 80% acetonitrile/0.1% trifluoroacetic acid, and 5 × 0.5 mL HEN buffer diluted 1:10. Peptides were eluted for 30 min at room temperature in elution buffer containing (in mmol/L): DTT (20), NH4CO3 (10), and 50% methanol. The resin was then washed with an additional volume of elution buffer, followed by 2 vol of water. All fractions were combined and concentrated via SpeedVac. Samples were then resuspended in 0.1% formic acid and cleaned with a C18 column (ZipTip; Millipore, Billerica, MA). Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was then performed using an Eksigent nano-LC 1D plus system (Dublin, CA) coupled to an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using CID fragmentation. Peptides were first loaded onto a Zorbax 300SB-C18 trap column (Agilent, Palo Alto, CA) at a flow rate of 5 μL/min for 10 min, and then separated on a reversed-phase PicoFrit analytical column (New Objective, Woburn, MA) using a 40-min linear gradient of 2–40% acetonitrile in 0.1% formic acid at a flow rate of 300 nL/min. LTQ Orbitrap XL settings were as follows: spray voltage 1.5 kV, and full MS mass range: m/z 200 to 2000. The LTQ Orbitrap XL was operated in a data-dependent mode (i.e., one MS1 high resolution [30,000] scan for precursor ions followed by six data-dependent MS2 scans for precursor ions above a threshold ion count of 2000 with collision energy of 35%). The raw file generated from the LTQ Orbitrap XL was analyzed using Proteome Discoverer v1.1 software (Thermo Fisher Scientific) with the NIH six-processor MASCOT cluster search engine (http://biospec.nih.gov, version 2.3). The following search criteria were used: database, Swiss-Prot (Swiss Institute of Bioinformatics); taxonomy, Mus musculus (mouse); enzyme, trypsin; miscleavages, 3; variable modifications, oxidation (M), N-methylmaleimide (C), deamidation (NQ); MS peptide tolerance 25 ppm; MS/MS tolerance as 0.8 Da. Peptides were filtered at a false discovery rate (FDR) of 5%, as determined by a targeted decoy database search with a significance threshold of 0.03.
4.4. Molecular modelling of Opa1 and mutation analysis
4.4.1. Homology modelling
Recombinant Opa1 structure model was built with Phyre2 using an intensive model generation approach that created a complete full-length model of the protein through a combination of multiple template modelling and simplified ab initio folding simulation, as described in Ref. [58]. The top six protein templates selected to model rOpa1 structure based on heuristics were: (i), GTPase HflX from Escherichia coli (PDB: c5ady6), (ii) Mgm1 from Chaetomium thermophilus (PDB: c6ql4B), (iii) Dynamin-1 from Rattus norvegicus (PDB: c3zvrA), (iv) Dynamin 3 from Homo Sapiens (PDB: c5a3fD), (v) Dynamin-1 from Homo sapiens (PDB: c3snhA), (vi) Mgm1 from Saccaharomyces cerevisiae (PDB: c6jsjB). The 93% of the final rOpa1 model was modelled at >90% confidence, 7% was modelled ab initio. Residue scanning wizard of Bioluminate 1.0 in Schrodinger suite (Schrödinger, LLC), was used to study the effects of Cys mutations on Opa1 model. The difference in the stability of mutants compared to wild-type protein was calculated for all the mutations. Molecular graphics images were produced using the Chimera package (Computer Graphics Laboratory, University of California, San Francisco).
4.5. Biochemistry
4.5.1. Immunoblotting and bidimensional redox electrophoresis
Snap frozen heart tissue and harvested cells were homogenized in RIPA buffer (1% Triton X-100, 150 mM NaCl, 50 mM Tris, pH7.4) in the presence of complete protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktail 1 (Sigma) with the addiction of 1 mM N-Ethylmaleimide (NEM, Sigma) to prevent oxidation. All the procedures were performed at 4 °C. Protein concentration was determined by BCA assay (Pierce, Rockford, IL). Upon centrifugation, supernatant was diluted in Laemmli buffer 4X (Invitrogen) supplemented with 2.5% β-mercaptoethanol for reducing gels. Fifteen μg of tissue homogenate or 20 μg of cell homogenate were separated by 3–8% Tris-acetate (NuPage, Invitrogen) or 4–12% Bis-Tris (BioRad) and then transferred onto polyvinylidene difluoride (PVDF, BioRad) membranes. The following antibody were used: Monoclonal anti-Opa1 (1:1000 BD), rabbit polyclonal anti-GRP75 (1:1000 Santa Cruz Biotechnology), monoclonal anti-Vinculin (1:10,000 Sigma). Membranes were probed using isotype-matched secondary antibodies conjugated to horseradish peroxidase. Signal was detected with ECL (Amersham).
4.5.2. Protein crosslinking
For protein crosslinking, mitochondria from the different cell lines were treated as indicated with 10 mM BMH or 1 mM EDC for 30 min as described in Ref. [36], before separation in a 3–8% Tris-acetate gel (NuPAGE, Invitrogen).
4.6. Imaging and transmission electron microscopy
4.6.1. Immunofluorescence
2 × 105 MEFs were seeded onto 24-mm round coverslips. After 24 h, cells were treated as indicated and fixed for 10 min at room temperature with 3.7% (w/v) formaldehyde (Sigma). Cells were permeabilized using 0.1% Triton-X-100 (Sigma) for 10 min, blocked for 1 h with BSA 1% and incubated with primary antibodies. The following antibodies were used: anti-Tom20 (1:500, Santa Cruz Biotechnology), anti-cytochrome c (1:200, BD Pharmingen). Staining was revealed with a goat anti-rabbit or anti-mouse IgG conjugated to FITC or TRITC (Jackson Labs). For confocal imaging, 1 × 104 cells seeded onto 24-mm round glass coverslips were transfected with PA-GFP. After 24 h, cells were placed on the stage of a laser scanning microscope (TCS SP5, Leica). Using the LasAF software (Leica), RFP and YFP were excited using 488 nm or the 543 nm line of the HeNe and Argon with a 63x, 1.4 NA objective (Leica). Confocal images of mtRFP and YFP fluorescence were acquired using the Concolve and VolumeJ plugins of ImageJ (NIH, Bethesda). Length of mitochondria was quantified using the Multimeasure plug-in of ImageJ. All the mitochondria in each cell were considered for the analysis.
4.6.2. PA-GFP fusion assay
Fusion assay was performed by seeding 2 × 105 cells onto 24 mm round glass coverslips. Cells were co-transfected with mtRFP and mito-pAGFP for 24 h and imaged using a laser scanning microscope (TCS SP5, Leica). Using LasAF software (Leica), one circular region of interest (ROI) of the same size for each cell was photoactivated by using 100% of the power of the 413 nm laser with a 63X, 1.4NA objective. Frames were acquired each min using the 488 nm and the 563 nm laser lines for a total time of 30 min. Standard deviation of the green fluorescent in the whole cell was measured and normalized for the intensity of the mtRFP fluorescence using the Multi Measure plug-in of ImageJ (NIH, Bethesda).
4.6.3. Transmission electron microscopy
MEFs of the indicated genotypes were treated as indicated and fixed with 1.25% (v:v) glutaraldehyde in 0.1 M Na-cacodylate pH 7.2 for 1 h at 37 °C in a 5%CO2 atmosphere. Samples were left at 4 °C in 0.1 M sodium cacodylate buffer until postfixation. Thin sections were imaged using Tecnai-20 electron microscope (Philips-FEI) as described in Ref. [12].
4.6.4. Assay of mitochondrial membrane potential
MEFs plated on a 24 mm round coverslip were incubated in HBSS-Hepes and stained with 5 nM TMRM (Molecular Probes) in the presence of 1 μM cyclosporine H (a P-glycoprotein inhibitor) for 30 min at 37 °C. Cells were observed using an Olympus IX81 inverted microscope (Melville, NY) equipped with a CellR imaging system. Sequential images of TMRM were acquired every 60 s with a 40× objective (Olympus). Where indicated, H2O2 (1 mM, Sigma), oligomycin (5 nM, Sigma) or the uncoupler carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP, 200 nM, Sigma) were added. TMRM fluorescence analysis over the mitochondrial regions of interest was performed using ImageJ and normalized to the intensity of the initial frame of the real time imaging experiment.
4.7. Analysis of cell death
4.7.1. Flow cytometry
For cell death analysis, 4 × 104 MEFs of the indicated genotype grown in a 6 well plate were treated as indicated. At the indicated time points, cells were harvested and stained with propidium iodide (PI) and annexin-V-FLUOS (BenderMedSystems) according to the manufacturer's protocol. Cell viability was measured by flow cytometry (FACSCalibur, BD Biosciences) as a percentage of annexin-V and PI negative events.
4.8. Statistical analysis
The sample size was not predetermined using statistical methods. No animals were excluded from the analysis. The experiments were not randomized. N of independent experiments, n of cells or mitochondria examined for each condition are indicated. In box plots, center line represents mean, bounds of boxes SEM, whiskers the 10th-90th percentiles; each dot represents an individual measurement. Origin 2019b (OriginLab) was used for statistical analyses. In time course graphs, average ± SEM is plotted. Statistical significance was calculated by one-way ANOVA test with Tukey's mean comparison between indicated samples. A p < 0.05 was considered statistically significant.
Author contributions
MS, FdL and LS conceptualized the project and acquired funds. MS and LS wrote the manuscript. MS performed and analyzed most of the experiments and prepared figures. MJK performed LC-MS/MS experiments; CQ performed GTPase assays; RM performed perfusion experiments; PA performed cardiomyocyte isolation; LA performed oxygraphy experiments; AP performed modelling experiments; EM, FdL and LS supervised the project. All authors edited the manuscript.
Declaration of competing interest
The Authors declare no competing interests.
Acknowledgments
We thank Drs. F. Caicci and F. Boldrin (BioImaging Facility, Department of Biology, University of Padova) for EM samples preparation; Prof. Christian Frezza (U. of Cologne, Germany) and Dr. Andrea Carpi (University of Padova) for helpful discussions. This work was supported by Fondation Leducq TNE15004, by Ministry of Education, University and Research (MIUR) FIRB RBAP11Z3YA_005 and PRIN 2017BF3PXZ, and by Ministry of University and Research (MUR) 2020PKLEPN grants (to LS). LA is the recipient of the MSCA_0000019, MUR Concession Decree No. n. 564 of December 13, 2022, CUP C93C22007550006 funded under the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.2, MUR Call for tender n. 367 of October 7, 2022 funded by the European Union – NextGenerationEU.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2023.102755.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
Data availability
Data will be made available on request.
References
- 1.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Lisa F., Canton M., Carpi A., Kaludercic N., Menabò R., Menazza S., Semenzato M. Mitochondrial injury and protection in ischemic pre- and postconditioning. Antioxidants Redox Signal. 2011;14:881–891. doi: 10.1089/ars.2010.3375. [DOI] [PubMed] [Google Scholar]
- 3.Lenaz G. In: Advances in Mitochondrial Medicine. Scatena R., Bottoni P., Giardina B., editors. Springer Netherlands; Dordrecht: 2012. Mitochondria and reactive oxygen species. Which role in physiology and pathology? pp. 93–136. [DOI] [PubMed] [Google Scholar]
- 4.Matsushima S., Sadoshima J. Yin and yang of NADPH oxidases in myocardial ischemia-reperfusion. Antioxidants. 2022;11:1069. doi: 10.3390/antiox11061069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pei J.F., Li X.K., Li W.Q., Gao Q., Zhang Y., Wang X.M., Fu J.Q., Cui S.S., Qu J.H., Zhao X., Hao D.L., Ju D., Liu N., Carroll K.S., Yang J., Zhang E.E., Cao J.M., Chen H.Z., Liu D.P. Diurnal oscillations of endogenous H(2)O(2) sustained by p66(Shc) regulate circadian clocks. Nat. Cell Biol. 2019;21:1553–1564. doi: 10.1038/s41556-019-0420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sies H., Jones D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020;21:363–383. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 7.Burgoyne J.R., Mongue-Din H., Eaton P., Shah A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012;111:1091–1106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 8.Yu T., Sheu S.S., Robotham J.L., Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008;79:341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu T., Robotham J.L., Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorn G.W., 2nd, Clark C.F., Eschenbacher W.H., Kang M.Y., Engelhard J.T., Warner S.J., Matkovich S.J., Jowdy C.C. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ. Res. 2011;108:12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quintana-Cabrera R., Manjarrés-Raza I., Vicente-Gutiérrez C., Corrado M., Bolaños J.P., Scorrano L. Opa1 relies on cristae preservation and ATP synthase to curtail reactive oxygen species accumulation in mitochondria. Redox Biol. 2021;41 doi: 10.1016/j.redox.2021.101944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cogliati S., Frezza C., Soriano M.E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L.C., Perales-Clemente E., Salviati L., Fernandez-Silva P., Enriquez J.A., Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon Y.S., Yoon D.S., Lim I.K., Yoon S.H., Chung H.Y., Rojo M., Malka F., Jou M.J., Martinou J.C., Yoon G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006;209:468–480. doi: 10.1002/jcp.20753. [DOI] [PubMed] [Google Scholar]
- 14.James D.I., Parone P.A., Mattenberger Y., Martinou J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 15.Smirnova E., Shurland D.-L., Ryazantsev S.N., van der Bliek A.M. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 1998;143 doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otera H., Wang C., Cleland M.M., Setoguchi K., Yokota S., Youle R.J., Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gandre-Babbe S., van der Bliek A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmer C.S., Osellame L.D., Laine D., Koutsopoulos O.S., Frazier A.E., Ryan M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Legros F., Lombes A., Frachon P., Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cipolat S., Martins de Brito O., Dal Zilio B., Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griparic L., Kanazawa T., van der Bliek A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker M.J., Lampe P.A., Stojanovski D., Korwitz A., Anand R., Tatsuta T., Langer T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014;33:578–593. doi: 10.1002/embj.201386474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee H., Smith S.B., Sheu S.S., Yoon Y. The short variant of optic atrophy 1 (OPA1) improves cell survival under oxidative stress. J. Biol. Chem. 2020;295:6543–6560. doi: 10.1074/jbc.RA119.010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bean C., Audano M., Varanita T., Favaretto F., Medaglia M., Gerdol M., Pernas L., Stasi F., Giacomello M., Herkenne S., Muniandy M., Heinonen S., Cazaly E., Ollikainen M., Milan G., Pallavicini A., Pietiläinen K.H., Vettor R., Mitro N., Scorrano L. The mitochondrial protein Opa1 promotes adipocyte browning that is dependent on urea cycle metabolites. Nat.Metabol. 2021;3:1633–1647. doi: 10.1038/s42255-021-00497-2. [DOI] [PubMed] [Google Scholar]
- 26.Frezza C., Cipolat S., Martins dB., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper B., Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 27.Shutt T., Geoffrion M., Milne R., McBride H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012;13:909–915. doi: 10.1038/embor.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varanita T., Soriano M.E., Romanello V., Zaglia T., Quintana-Cabrera R., Semenzato M., Menabo R., Costa V., Civiletto G., Pesce P., Viscomi C., Zeviani M., Di Lisa F., Mongillo M., Sandri M., Scorrano L. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metabol. 2015;21:834–844. doi: 10.1016/j.cmet.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohr M.J., Sun J., Aponte A., Wang G., Gucek M., Murphy E., Steenbergen C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ. Res. 2011;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faelber K., Dietrich L., Noel J.K., Wollweber F., Pfitzner A.K., Muhleip A., Sanchez R., Kudryashev M., Chiaruttini N., Lilie H., Schlegel J., Rosenbaum E., Hessenberger M., Matthaeus C., Kunz S., von der Malsburg A., Noe F., Roux A., van der Laan M., Kuhlbrandt W., Daumke O. Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1. Nature. 2019;571:429–433. doi: 10.1038/s41586-019-1372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akepati V.R., Muller E.C., Otto A., Strauss H.M., Portwich M., Alexander C. Characterization of OPA1 isoforms isolated from mouse tissues. J. Neurochem. 2008;106:372–383. doi: 10.1111/j.1471-4159.2008.05401.x. [DOI] [PubMed] [Google Scholar]
- 32.Song Z., Chen H., Fiket M., Alexander C., Chan D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anand R., Wai T., Baker M.J., Kladt N., Schauss A.C., Rugarli E., Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeVay R.M., Dominguez-Ramirez L., Lackner L.L., Hoppins S., Stahlberg H., Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomes L.C., Di Benedetto G., Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper B., Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 37.Glytsou C., Calvo E., Cogliati S., Mehrotra A., Anastasia I., Rigoni G., Raimondi A., Shintani N., Loureiro M., Vazquez J., Pellegrini L., Enriquez J.A., Scorrano L., Soriano M.E. Optic atrophy 1 is epistatic to the core MICOS component MIC60 in mitochondrial cristae shape control. Cell Rep. 2016;17:3024–3034. doi: 10.1016/j.celrep.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quintana-Cabrera R., Quirin C., Glytsou C., Corrado M., Urbani A., Pellattiero A., Calvo E., Vazquez J., Enriquez J.A., Gerle C., Soriano M.E., Bernardi P., Scorrano L. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 2018;9:3399. doi: 10.1038/s41467-018-05655-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irwin W.A., Bergamin N., Sabatelli P., Reggiani C., Megighian A., Merlini L., Braghetta P., Columbaro M., Volpin D., Bressan G.M., Bernardi P., Bonaldo P. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 2003;35:367–371. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- 40.Scorrano L., Ashiya M., Buttle K., Weiler S., Oakes S.A., Mannella C.A., Korsmeyer S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 41.Murphy E., Ardehali H., Balaban R.S., DiLisa F., Dorn G.W., 2nd, Kitsis R.N., Otsu K., Ping P., Rizzuto R., Sack M.N., Wallace D., Youle R.J. Mitochondrial function, Biology, and role in disease: a scientific statement from the American heart association. Circ. Res. 2016;118:1960–1991. doi: 10.1161/RES.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaludercic N., Mialet-Perez J., Paolocci N., Parini A., Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014;73:34–42. doi: 10.1016/j.yjmcc.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bleier L., Wittig I., Heide H., Steger M., Brandt U., Dröse S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015;78:1–10. doi: 10.1016/j.freeradbiomed.2014.10.511. [DOI] [PubMed] [Google Scholar]
- 44.Pajares M., Jiménez-Moreno N., Dias I.H.K., Debelec B., Vucetic M., Fladmark K.E., Basaga H., Ribaric S., Milisav I., Cuadrado A. Redox control of protein degradation. Redox Biol. 2015;6:409–420. doi: 10.1016/j.redox.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baker M.J., Lampe P.A., Stojanovski D., Korwitz A., Anand R., Tatsuta T., Langer T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014;33:578–593. doi: 10.1002/embj.201386474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang K., Li H., Song Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014;15:576–585. doi: 10.1002/embr.201338240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ban T., Ishihara T., Kohno H., Saita S., Ichimura A., Maenaka K., Oka T., Mihara K., Ishihara N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017;19:856–863. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- 48.Li D., Wang J., Jin Z., Zhang Z. Structural and evolutionary characteristics of dynamin-related GTPase OPA1. PeerJ. 2019;7 doi: 10.7717/peerj.7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu C., Zhao J., Yan L., Qi Y., Guo X., Lou Z., Hu J., Rao Z. Structural insights into G domain dimerization and pathogenic mutation of OPA1. J. Cell Biol. 2020:219. doi: 10.1083/jcb.201907098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weckbecker D., Longen S., Riemer J., Herrmann J.M. Atp23 biogenesis reveals a chaperone-like folding activity of Mia40 in the IMS of mitochondria. EMBO J. 2012;31:4348–4358. doi: 10.1038/emboj.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petrungaro C., Zimmermann K.M., Küttner V., Fischer M., Dengjel J., Bogeski I., Riemer J. The Ca(2+)-dependent release of the mia40-induced MICU1-MICU2 dimer from MCU regulates mitochondrial Ca(2+) uptake. Cell Metabol. 2015;22:721–733. doi: 10.1016/j.cmet.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 52.Banci L., Bertini I., Ciofi-Baffoni S., Boscaro F., Chatzi A., Mikolajczyk M., Tokatlidis K., Winkelmann J. Anamorsin is a [2Fe-2S] cluster-containing substrate of the Mia40-dependent mitochondrial protein trapping machinery. Chem. Biol. 2011;18:794–804. doi: 10.1016/j.chembiol.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 53.Cheng E.H.Y.A., Wei M.C., Weiler S., Flavell R.A., Mak T.W., Lindsten T., Korsmeyer S.J. BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 54.Cereghetti G.M., Stangherlin A., Martins de Brito O., Chang C.R., Blackstone C., Bernardi P., Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaninello M., Palikaras K., Naon D., Iwata K., Herkenne S., Quintana-Cabrera R., Semenzato M., Grespi F., Ross-Cisneros F.N., Carelli V., Sadun A.A., Tavernarakis N., Scorrano L. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat. Commun. 2020;11:4029. doi: 10.1038/s41467-020-17821-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barbato R., Menabò R., Dainese P., Carafoli E., Schiaffino S., Di Lisa F. Binding of cytosolic proteins to myofibrils in ischemic rat hearts. Circ. Res. 1996;78:821–828. doi: 10.1161/01.res.78.5.821. [DOI] [PubMed] [Google Scholar]
- 57.Kaludercic N., Takimoto E., Nagayama T., Feng N., Lai E.W., Bedja D., Chen K., Gabrielson K.L., Blakely R.D., Shih J.C., Pacak K., Kass D.A., Di Lisa F., Paolocci N. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 2010;106:193–202. doi: 10.1161/CIRCRESAHA.109.198366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.