

Abstract
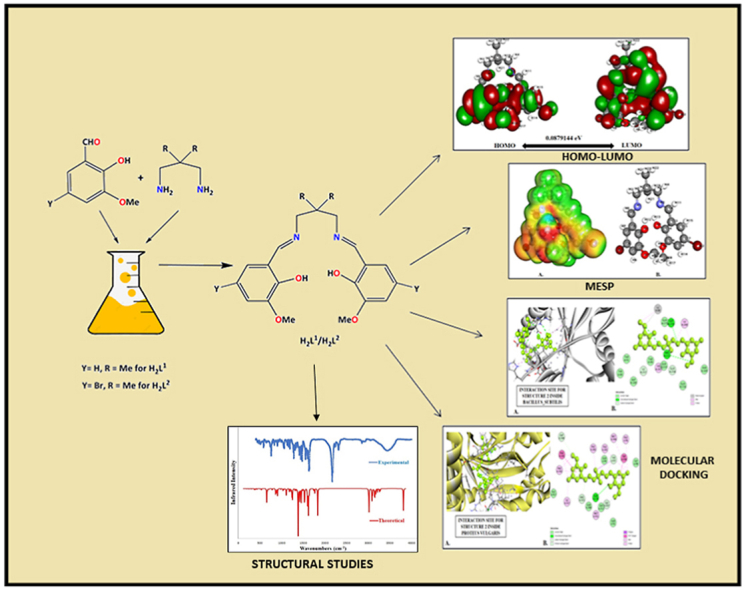
In this article, we have synthesized two contemporary ortho-vanillin-based Salen-type ligands (H2L1/H2L2) characterized by modern spectroscopic tools. EDX analysis supports the elemental composition (C, N, O, and Br). SEM examined the morphology of the synthesized compounds. The molecular geometry was optimized in the gas phase using B3LYP-D3/6-311G (d, p) level. The global reactivity parameters, HOMO-LUMO energy gap (Δ), atomic properties, MESP, and ADME/T, vividly explore the chemical reactivity and toxicity of two Salen-type ligands. The DFT simulated IR/NMR characterized essential structural assignments, and UV-Visible spectra were employed to predict the optical properties. The article demonstrated in silico molecular docking against the Gm + ve Bacillus subtilis (6UF6), and Gm -ve Proteus Vulgaris establishes the ligand binding ability with essential amino acids through conventional H-bonding or other significant interactions. The docking simulation is compared for two compounds better than the control drugs and confirms the antimicrobial activity. The theoretical drug-like properties have been explored in-depth by ADME/T using the SWISSADME database. The analysis estimated the molecule's lipophilicity, the consensus P0/W, and water solubility. Thus, using various pharmaco-logical parameters, toxicity explains where the electron-withdrawing Br group plays a more toxic effect in H2L2 than in H2L1.
Keywords: Salen ligand, DFT, Molecular docking, Molecular dynamics, ADME/T, Toxicity
Graphical abstract
Highlights
-
•
Two contemporary Salen-type ligands were in situ synthesized and characterized.
-
•
Molecular geometry was optimized using the B3LYP-D3/6-311G (d, p) level approach.
-
•
HOMO-LUMO, MESP, atomic properties, and structural IR/NMR/UV-Visible spectra were analyzed.
-
•
In silico MD was performed against the Gm + ve Bacillus subtilis (6UF6) and Gm -ve Proteus Vulgaris.
-
•
ADME/T investigates drug-like properties and toxicity predictions using the Br effect.
1. Introduction
In recent years, the chemistry of Schiff base (SBs) has gained increasing attention due to many incredible discoveries that cater to it with a brand-new look. The privileged ligands are the critical splendours of natural compounds characterized by the imine, CH N functional group, first said by H. Schiff in 1864 (Scheme S1) [1]. The Salen ligands are notable among the SBs, which are unique and exciting given the current research scenario of global researchers. This class of SBs is much more helpful to synthetic chemists because of their synthetic accessibility and various structural features depending on the nature of the substituents. It is used as a photodetector in biological systems and as a photo stabilizer and optical sound recording technology [2]. Interestingly, N/O-donor-containing SBs, always display a wide range of exciting biological activities, including homogeneous and heterogeneous catalysis reactions [3]. The properties of thermal stability [1], NLO [4], and proton transfer [5,6] are of equal interest. Furthermore, synthetic chemists have recently excited Salen-type ligands containing electron-withdrawing substituents in search of novel properties [4]. It is used to manufacture anticancer drugs and in design and synthesis as low-toxic and has applications such as antibacterial and antifungal, anti-inflammatory effects [3], antituberculosis [7], antioxidant [2], and kills parasitic worms [8]. The research is not exhaustive but flourished to Carbon steel corrosion, naked-eye acetate anion detection, and drug-like properties [[9], [10], [11], [12]]. Furthermore, the contemporary Salen-type compounds in synthesizing coordination complexes are the prime theme for crystal complex synthetic purposes [[13], [14], [15], [16], [17]] since this remarkable research opens the concept of supramolecular chemistry associated with tetrel/Chalcogen bonding and σ-hole interaction [18,19]. At the same time, the topological analysis of the electron density distribution and supramolecular chemistry corroboration by employing Hirshfeld surface analysis are also potentially distinctive approaches to studying bonding and crystal packing in various Salen-type ligand crystal complex systems [[20], [21], [22], [23], [24], [25], [26], [27]]. Still, scientists are giving too little concentration of the SBs chemistry concerning DFT-based studies like in silico molecular docking and reactivity by HOMO-LUMO, MESP, and ADME/T for drug-like properties and toxicity predictions. Incredibly, in silico docking, ADME/T, and toxicity prediction analysis are unveiled research in the literature, particularly if we consider the SBs. Therefore, in modern time docking simulation for Schiff base compounds is developing particular interest since it finds antimicrobial or drug-like activities. The analysis has been one of drug discovery's most fundamental and essential strategies [28]. It allows the prediction of molecular interactions that hold a protein and a ligand together in the bound state. Nowadays, ADME/T applicability plays a crucial role in drug discovery and development. ADME/T determined the pharmacokinetic and pharmacodynamic properties of the ligand molecules [29]. Now many in silico models for predicting ADME/T chemical properties are being developed. The simplest form of toxicity prediction is cytotoxicity, where the drug molecule causes severe damage to the cells. It is ADME/T, the online servers to predict toxicity, drug-likeness, and molecular descriptor calculation. ADME/T indicates compounds' mutagenicity and carcinogenicity to avoid toxicity [29].
In this article, our research aims to understand better a few DFT-simulation analyses of the Salen-type compounds (H2L1/H2L2). The global chemical reactivity parameters, energy difference of HOMO-LUMO, and MESP were investigated to ensure the Salen-type ligand's reactivity. Further, comparing the experimental and theoretical FTIR, NMR, and UV-Visible spectra can confirm the ligands' structure. In silico molecular docking examines the antimicrobial potency of the compounds. The theoretical drug-like properties and toxicity predictions were studied by ADME/T, where the electron-withdrawing Br group shows a more toxic effect in H2L2 than in H2L1.
2. Experimental section
2.1. Starting materials
Most of the research chemicals were of analytical grade and used as received without further purification. Ortho vanillin (99%), Bromo-ortho vanillin (97%), and 2,2-dimethyl-1,3-diaminopropane (99%) were purchased directly from Sigma Aldrich Company, USA, and the methanol solvent was a spectroscopic grade.
2.2. Physical measurements
Elemental analyses of the synthesized compounds were performed using a PerkinElmer 2400 CHN Elemental Analyzer. FT-IR spectra were recorded as KBr pellets (4000-400 cm−1) having 16 scans at a wave number resolution of 4 cm−1 on a Perkin–Elmer spectrum RX 1 using detector DTGS (Deuterated triglycine sulphate). A Bruker 400 MHz collected NMR spectra, and a 75.45 MHz NMR spectrometer using TMS as an internal standard in NMR solvent like DMSO‑d6. JEOL Model JSM6390LV analyzed SEM micrographs. The EDX was performed on EDX OXFORD XMX N using W filament. UV-Visible spectra (200–1100 nm) in methanol solvent were determined using the Hitachi model U-3501 spectrophotometer.
2.3. Computational methodology
2.3.1. DFT
For the keen interest of diverse medicinal and biological activities, quantum chemical investigations were undertaken, like DFT and surface electrostatic potential (MESP). The program package Gaussian 09 [30,31] and basis sets 6-311G (d, p) is used to carry out DFT calculations. A mixed MCMM/low-mode search with 1000 steps per rotatable bond was used for the conformational search. An energy window of 21kJ/mol−1 was chosen for retention conformers. The DFT calculations were carried out in a water solvent system. The improved geometry resulted in compound energies that may utilize to interpret the biological activity and molecular properties, such as the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO). The HOMO and LUMO are crucial in quantum chemistry because they allow molecule stability and reactivity research. In a standard system, however, the HOMO is an electron donor, whereas the LUMO is an electron acceptor. Using the B3LYP/6-311G (d, p) level, the total energies of HOMO-LUMO and other characteristics of the examined compounds were computed. Global quantum mechanical (QM) reactivity descriptors determined the chemical hardness, chemical softness (S), and electronegativity. Also, the electronic chemical potential and global electrophilicity index. The B3LYP-D/6-311G (d, p) approach generated the salt molecular electrostatic potential (MESP) diagram. It is an ESP representation placed over a generally constant electron density (ED) surface with a colour spectrum ranging from dark red to deep blue. Furthermore, the intriguing electronic properties of the synthesized compounds have been investigated through density functional theory (DFT) calculations after the optimization of the ligands' structure.
2.3.2. Molecular docking: preparation of receptor structure
The gram-positive bacterium, Bacillus subtilis (PDB ID: 6UF6) [32] and Gram-negative bacterium, Protease Vulgaris (PDB ID: 5HXW) [33] Crystal structure receptors were obtained from Protein Data Bank (PDB) [34]. The receptor was built using the Schrödinger suite [35], and prime altered the receptor protein's internal structure and detected the missing side-chain atoms in the protein receptor residues. A hydrogen-bonded network was employed to add hydrogens, allowing the technique to determine the protonation state. PDB format is used to store the protein.
2.3.3. Preparation of ligand structure
The schematic representation of the complexes of particular interest was used to create the ligands. Compounds are identified as H2L1 and H2L2 and Control Drugs in the 3D structure produced on Avogadro software, and their three-dimensional structures were given by LigPrip (LigPrep, Schrödinger, LLC, New York, NY, 2018-3). The arrangements were maintained in MAE (Maestro) format using the default settings of the OPLS3e force field.
2.3.4. Docking calculations
Molecular docking is used to identify the atomic level contact between ligands and proteins. Using Auto dock [36], the chosen complexes, H2L1 and H2L2, were docked onto the binding site of the complete receptor protein of the Gram-positive bacterium, Bacillus subtilis, and Gram-negative bacterium, Protease Vulgaris. The binding site is defined by Auto dock using rectangular grid boxes. The grid box centre can be specified explicitly or implicitly. The grid box centre is then determined using the atoms' mean coordinates and the docking box presented in the window. The size and precise location of the box may also be modified to meet the needs. For the characteristics and form of the receptors, multiple sets of parameters that result in more precise estimates for the ligand poses were applied on a grid [[37], [38], [39]]. It was meant to encompass the grid's critical zone, which qualifies residue as a grid centre. Additional default values [40,41] were utilized. The ideal docked conformation with the lowest docking score value was supplied for each ligand. Intermolecular non-covalent interactions were detected for docked complexes under default settings, including Conventional hydrogen bonding, Carbon hydrogen bond, Van der Waals interactions, Metal-acceptor interactions, Alkyl interactions, pi-alkyl interactions, pi-sigma interactions, pi-pi T-shaped, and pi-donor hydrogen bonding. Auto dock requires receptor and ligand representations in the. pdbqt file format is an enhanced protein data bank format including atomic charges, atom type definitions, and topological information for ligands (rotatable bonds). Auto dock Tools package is used to do these file preparations. Ligands for future docking operations can be provided by supplying a directory holding a library of docked ligands. Docking runs may be performed immediately after defining the binding site and preparing the receptor and ligand.
2.3.5. ADME/T
The structure of ligands (H2L1 and H2L2) is subjected to ADME/T (Adsorption, Distribution, Metabolism, and Excretion) prediction. ADME/T is prime beneficial for assessing the pharmacodynamic properties of a potential medicinal synthesized chemical. SWISS-ADME (https://www.swissadme.ch) [42] is a website that allows users to draw their own ligand or drug molecule or include SMILES data from PubChem and analyze parameters such as lipophilicity (iLOGP, XLOGP3, WLOGP, MLOGP, SILICOS-IT, Log P0/w), water solubility-Log S (ESOL, Ali, SILICOS-IT), drug-likeness rules. H2L1 and H2L2 of the ligands are converted into SMILES and enter the search area.
2.4. Ligand synthesis
Salen-type ligands have been successfully synthesized in situ following the literature-based method (Scheme 1) [43]. The reflux condensation of 3-methoxy-2-hydroxybenzaldehyde (0.152 g, 1 mmol) with 2,2-dimethyl-1,3-diaminopropane (0.0371 g, 0.5 mmol) in (50 mL) methanol at 80 °C for 3 h prepared H2L1. A similar procedure was employed to synthesize H2L2, except here, 5-Bromo-3-methoxy-2-hydroxybenzaldehyde (0.231 g, 1 mmol) instead of 3-methoxy-2-hydroxybenzaldehyde. Finally, the solvent was removed under a rotary vacuum and the yellow powder product separated upon cooling the solution. The yellow product is air-stable and resistant to the atmosphere, and a desiccator and tight air-dried collected it.
Scheme 1.

Synthetic outlines for the contemporary ligands (H2L1/H2L2).
N,N’-bis(3-methoxysalicylidene)-2,2-dimethyl-1,3-propanediamine (H2L1): Yield: 0.174 g Anal. Calc. for C21H26N2O4: C, 68.09; H, 7.07; N, 7.56 Found: C, 68.11; H, 7.10; N, 7.60%. IR (KBr cm−1) selected bands: ν(C꞊N), 1638 vs, ν(C–Ophenolic) 1245 s, ν(O-H) 3436 s, 1H NMR (DMSO‑d6, 400 MHz): δ (ppm): 1.31–1.34 (s, 3H), 4.04 (H), 6.77–7.03 (m, 3H), 8.53 (m, 1H), 13.90 (m,1H), 3.99 (s, 2H), 0.98 (s, 6H), 13C NMR (DMSO‑d6, 75.45 MHz): δ (ppm):15.2 (CH3), 64.2–67.0 (O-CH2),116.3–147.6 (Arom-C),152.2 (C–OH), 167.3 (CH N), 24.0 (C–(CH3)2, 36.1 ((CH3)2-C–(CH2)2), UV-Vis (λmax CH3OH): 226, and 263 nm. N,N’-bis(5-bromo-3-methoxysalicylidenimino)-1,3-diaminopropane (H2L2): Yield: 0.215 g Anal. Calc. for C21H24 Br2N2O4: C, 47.75; H, 4.58; N, 5.30 Found: C, 47.72; H, 4.61; N, 5.27%. IR (KBr cm−1) selected bands: ν(C꞊N), 1648 vs, ν(C–Ophenolic) 1229 s, ν(O-H) 3430 s, 1H NMR (DMSO‑d6, 400 MHz): δ (ppm): 3.75 (s, 3H), 7.06–7.17 (m, 1H), 8.48 (m, 3H5), 13.91 (m, H), 3.04–3.75 (s, 2H), 2.48 (s, 6H), 13C NMR (DMSO‑d6, 75.45 MHz): δ (ppm): 54.94–56.46 (O-CH3),117.23–150.18 (Arom-C),153.71 (C–OH), 165.77 (CH N), 31.40 (-C–(CH2)2), UV-Vis (λmax CH3OH): 229 nm.
3. Results and discussion
3.1. Synthetic perception
Two contemporary Salen-type ligands (H2L1/H2L2) were obtained following the literature-based in situ method appended in the references (Scheme 1) [43]. Although we tried to synthesize them as crystal products after repeating the experimental procedure, we were unsuccessful. The yield of the products derived from such practices is satisfactory, with deep yellow amorphous powder. The synthesized compounds are readily soluble in methanol, characterized by elemental and various spectroscopic techniques. The synthesized ligands are structurally compartmental compared to other trendy ligands. Carefully overlooking the ligand structure consists of imines (C N), O-CH3, C–O, C–Br, C–H, C C, and free phenolic –OH group (see Scheme 1). Additionally, H2L2 is structurally linked to electron-withdrawing bromine substituent. Besides, ligands contain aromatic rings with C–H and C C bonds (Scheme 1) where carbon atoms are sp2 hybridized. The ligand structure has been confirmed using an experimental IR/1H/13C NMR and UV-Visible spectral study after corroboration with DFT-calculated theoretical spectra. The theoretical IR and NMR study conclusively support the presence of aromatic ring carbon, azomethine carbon, and other significant carbon atoms, including the OH group and azomethine (CH N) linkage. In addition, these ligands, after deprotonation, generate an N2O2-imine chelating position (Scheme S2-S3) [17,18], which vividly explores coordination chemistry by mimicking [17,18,44]. The literature on contemporary ligands, DFT, ADME/T toxicity predictions, and molecular dynamics-based research is unveiled. For the first time, we are divulging such trendy ligands DFT, ADME/T toxicity prediction, and molecular dynamics experiments. Notably, in this research work, we aim to investigate the influence of the electron-withdrawing bromine group and whether there is a striking difference in properties between the two ligands based on molecular dynamics simulations. Besides, remarkable global reactivity parameters, energy difference of HOMO-LUMO, atomic properties, and MESP studies are added, which are welcome research potential.
3.2. Spectroscopic characterization
3.2.1. Experimental and theoretical IR spectra
Generally, FTIR Spectroscopy, known as Fourier-transform infrared spectroscopy, deals with the vibration of molecules commonly used to establish a newly synthesized compound's functional group and, thereby, the predictions of the probable structures. Each active group has discrete vibrational energy to identify a particular molecule by combining all the functional groups. Our synthesized Salen-type ligands structural frameworks consist of imines (C N), O-CH3, C–O, C–Br, C–H, C C, and free phenolic –OH group (see Scheme 1). Besides, ligands contain aromatic rings with C–H and C C bonds (Scheme 1). The aromatic ring and imine atoms are sp2 hybridized, whereas oxygen in the free –OH group is sp3 hybridized. The C C bonds in the aromatic ring are planer and sp2 hybridized. Herein the structure of the ligands has been investigated using an experimental IR study after corroboration with DFT-calculated theoretical FTIR spectra. The theoretical IR calculations (cm−1) are done using the B3LYP/6-311G (d, p) level of theory in gas phase, and the essential IR bands in the ligand's structural skeleton are assigned in Fig. 1, Fig. 2 and Table 1. The observed and theoretical assigned IR peaks are C N, C–O, O-H, O-CH3, C–Br, and C–H. Except for C N, all other theoretical IR peaks correlate well with the experimental values. Aside from that, for C–H vibrations, generally, three types of bands are noticed: ῀1480 (rocking), ῀2790 (symmetric stretching), and ῀2860 (asymmetric stretching) for both ligands. Here, the calculated and observed values slightly differ. In both compounds, experimental IR stretching vibration of the azomethine group (CH N) was observed in 1638–1648 cm−1 (Fig. S1) [45], while the theoretical value ranges from 1720 to 1721 cm−1. The experimental free-OH stretching was observed at 3430-3436 cm−1 but theoretically fits well within the 3000-3011 cm−1. The experimental (C–Ophenolic) vibration was identified at 1245 and 1229 cm−1, while the theoretical range was 1258–1259 cm−1. Here the calculated and observed band values are in nearly good agreement. Finally, the experimental and theoretical IR comparison can confirm the Salen-type ligand's structure.
Fig. 1.

Experimental (top) and DFT computed (bottom) IR plot of H2L1.
Fig. 2.

Experimental (top) and DFT computed (bottom) IR plot of H2L2.
Table 1.
Comparison of observed and calculated wavenumbers (cm−1), and assignments for Salen compounds in gas phase using B3LYP/6-311G (d, p) level theory (Assignments refer to Scheme 1).
| H2L1 |
B3LYP/6-311G (d, p) level |
Assignments |
|---|---|---|
| Experimental | Calculated | |
| 1638 | 1721 | ν(C N) |
| 1245 | 1259 | ν(C–Ophenolic) |
| 3436 | 3000 | ν(O-H) |
| 1043,1061 | 1047,1084 | ν(C–O) |
| 1026 | 1023 | ν(O-CH3) |
| 675 | 678 | ν(C–Br) |
| 1475 (rock) | 1479, 2939 | ν(C–H) |
| 2792 (sym stretch) | ||
| 2856 (asym stretch) | ||
| H2L2 | ||
| 1648 | 1720 | ν(C N) |
| 1229 | 1258 | ν(C–Ophenolic) |
| 3430 | 3011 | ν(O-H) |
| 1037,1064 | 1049,1086 | ν(C–O) |
| 1025 | 1021 | ν(O-CH3) |
| 1490 (rock) | 1482, 2945 | ν(C–H) |
| 2796 (sym stretch) | ||
| 2861 (asym stretch) | ||
3.2.2. UV-visible spectrum
The experimental UV-Vis absorption spectra of the ligands were analyzed using spectroscopic grade methanol solvent, ensuring the synthesized compounds exhibited electronic transitions. Two ligands vividly show the UV-Visible bands value near 226 nm, 263 nm, and 229 nm, respectively (Fig. S2A). These values confirm that synthesized compounds exhibit π→π*/n→π* type optical shifts. These spectral data mentioned above are identical to the previously reported ligands [46,47]. The experimental UV-Visible spectral bands are compared with theoretical values to support the trendy ligand compound's structure. DFT computed UV-Visible spectra (TDDFT) were analyzed in water solvent using the B3LYP/6-311G (d, p) level of theory (see correlation Figs. S2B–S2C). The Salen compounds theoretical UV-Visible band values were observed near 215.69 nm, 258.30 nm, and 224.9 nm, respectively, which are π→π*/n→π* (C C and C O) type electronic transitions. According to TDDFT (HOMO, LUMO, Absorption Bands and oscillator strength values), the calculated details in Table S1A explores the electronic shifts predominantly H-1→L+2, and H-3→L+1 with Oscillator Strength (f) 0.8872 and 0.3219, respectively, for H2L1. In the case of H2L2, the electronic transition belongs to H-1→L+4 with Oscillator Strength (f) 1.2044. The slight deviation of the UV spectrum values is due to changing the solvent's polarity and the positions of those UV maxima. Hence, reasonable concordances are observed between experimental and theoretical UV-Visible spectra. The correlation of calculated and theoretical UV-Visible band values characterized the trendy ligand's optical properties.
3.2.3. 1H/13C NMR studies
The NMR spectroscopic study (1H/13C NMR) successfully investigated the newly synthesized compound's structure. The 1H NMR spectroscopic study successfully diagnoses the nature of the proton's nuclei, while 13C characterizes the carbon atoms in the synthesized compounds (see ligands structure). The experimental 1H/13C NMR chemical shifts of two Salen-type compounds taken in DMSO‑d6 solvent are given in Table 2 referencing TMS. The experimental NMR values are compared with the theoretical NMR predicted by the GIAO method (gauge-including atomic orbital) with the hybrid B3LYP/6–311 G (d, p) method in gas phase and employing the approximate RMSD values. Herein the experimental and DFT calculated NMR comparison has been done according to the ligand structure (see Scheme 1). The NMR essential assignments can be considered based on structural studies of two Salen-type compounds and given in Fig. 3, Fig. 4. The significant theoretical 1H NMR assignments are restricted to the aromatic proton, methyl/methoxy proton, azomethine proton (CH N), and phenolic proton (OH). Similarly, compared with experimental ones, theoretical 13C NMR analysis identifies aromatic ring carbon, azomethine carbon, and other significant carbon atoms. The ligands possess free phenolic proton (OH) peaks identified the chemical shift 13.90–13.91 ppm (Fig. S3-Fig. S4), while theoretically, this chemical shift recognized 13.41 ppm. The azomethine linkage formation (CH N) supports the successful Schiff base synthesis, and such azomethine proton chemical shifts were observed at 8.48–8.53 ppm. In contrast, theoretically, for azomethine proton, a good correlation was observed near 8.30–8.50 ppm for both ligands. The aromatic ring protons resonate at 6.77–7.17 ppm; theoretically, the range covers 6.56–7.92 ppm. In addition, the experimental NMR peak at 2.48 ppm is assigned to the CH3 group protons, while for DFT calculated value is 2.65 ppm. In addition, experimental 13C NMR characterizes the structural carbon atoms in the prepared ligands (Fig. S5-Fig. S6), presented in the experimental section. Further, compared with experimental ones, theoretical 13C NMR analysis identifies aromatic ring carbon, azomethine carbon, and other carbon atoms. The experimental 13C NMR azomethine carbon resonates at 165.77–167.30 ppm. In addition, theoretically, such carbon shifts were observed at 159.58–160.47 ppm. The experimental aromatic carbon shifts were identified near 116.30–150.18 ppm, concerning the theoretical value of 109.82–152.78 ppm. We observed that excellent correlations were obtained for the 1H nucleus than the 13C atoms since the DFT calculations were performed using the 6-311G (d, p) basis set, producing better results having approximate RMSD values. Therefore, experimental, and theoretical NMR corroboration confirm the structure of the ligands.
Table 2.
Comparison of observed and calculated 1H and13C chemical shifts (δ in ppm) for assignments of Salen compounds in gas phase using B3LYP/6-311G (d, p) level theory (Assignments refer to Scheme 1).
| H2L1 | B3LYP/6-311G (d, p) level | Experimental |
|---|---|---|
| s, 3H | 1.15 | 1.31–1.34 |
| H | 5.02 | 4.04 |
| m, 3H | 6.56–7.92 | 6.77–7.03 |
| m, 1H | 8.50 | 8.53 |
| m, 1H | 13.41 | 13.90 |
| s,2H | 3.98 | 3.99 |
| s,6H | 0.68 | 0.98 |
| RMSD | 0.9031 | - |
| 13C NMR | ||
| CH3 | 24.80 | 15.2 |
| O-CH2 | 74.65 | 64.2–67.0 |
| Arom-C | 109.82–152.78 | 116.3–147.6 |
| C–OH | 152.78 | 152.2 |
| CH=N | 160.47 | 167.3 |
| C–(CH3)2 | 41.98 | 24.0 |
| (CH3)2-C–(CH3)2 | 54.49 | 36.1 |
| RMSD | 3.2913 | |
| H2L2 | ||
| s, 3H | 3.48 | 3.75 |
| m, 1H | 6.81 | 7.06–7.17 |
| m, 3H | 8.30 | 8.48 |
| m, H | 12.94 | 13.91 |
| s, 2H | 3.47 | 3.94 |
| s, 6H | 2.65 | 2.48 |
| RMSD | 0.9110 | - |
| 13C NMR | ||
| O-CH3 | 54.82 | 54.94–56.46 |
| Arom-C | 113.50–151.43 | 117.23–150.18 |
| C–OH | 151.37 | 153.71 |
| CH=N | 159.58 | 165.77 |
| C–(CH3)2 | 24.66 | 31.40 |
| RMSD | 3.3192 | - |
Fig. 3.

Theoretical 1H NMR (top) and 13C NMR (bottom) curve for H2L1.
Fig. 4.

Theoretical 1H NMR (top) and 13C NMR (bottom) curve for H2L2.
3.2.4. EDX-SEM analysis
Today, EDX analysis is gaining importance in coordination chemistry, as it accurately reveals the elemental composition of synthesized compounds. Using the EDX tool, we recognize the chemical composition, such as C, N, Br, and O, as the main elements present in the two compounds (Table S1B/Figs. S7A–S7B). In addition, the SEM technique is used to study the morphological features of the contemporary Salen ligands. Today, SEM is widely used in materials chemistry to determine surface morphology. Therefore, SEM is used for the morphological characteristics of the newly synthesized compounds. We performed an SEM experiment on our amorphous Salen-type powder compounds (ESI2/Figs. 8–S9). The SEM micrographs showed that the morphology is distributed in overlapping sheets almost identical to the other Schiff base compounds.
4. HOMO-LUMO
At first, we compared the reactivity based on the HOMO-LUMO energy gap computed by DFT. The values and dispersion of the FMO are critical predictors of reactivity. HOMO reflects the molecule's capacity to donate electrons, whereas LUMO represents the molecule's ability to accept electrons [48]. The HOMO-LUMO energy gap [49] indicates the kinetic stability of the molecule. The optimized molecular structure calculates the HOMO-LUMO energies and corresponding energy gaps. Fig. 5 depicts the 2D structure of energy gaps H2L1 between HOMO and LUMO. In contrast, Fig. 6 illustrates the energy gap between HOMO and LUMO of H2L2. The energy gap between HOMO-LUMO H2L1 is 0.0918 eV while H2L2 that an energy gap of 0.0879 eV H2L2 projected favourable features since the discovered tiny energy gap reflects higher polarizability and reactivity. The H2L2 is a suitable candidate for use as a chelating agent due to the little energy gap (0.082 eV), significant softness (22.74 eV), and small chemical hardness (0.043 eV). Additionally, the electronegativity of H2L1 is somewhat greater, indicating that it is slightly more stable than H2L2. Thus, H2L2 is more reactive than H2L1. Table 3 explores the calculated ionization potential, dipole moment, chemical hardness, and other parameters. Generally, a significant energy gap suggests a hard and stable molecule, whereas a small energy gap indicates a soft and reactive molecule. H2L2 has an energy gap of 0.087 eV as a result, the molecule is delicate and reactive. In the case of HOMO, charges are dispersed across the. However, when comparing HOMO with LUMO, the negative and positive charges are not distributed evenly. HOMO is packed up in the bromine area (Fig. 6), indicating that practically all the atoms in the bromine region have both positive and negative charges. Charges are concentrated on all carbon atoms in LUMO. However, negative charges are focused on the oxygen atom. Table 3 shows Dipole moment calculations, energy gap, ionization energy, electronegativity, electron affinity, and hardness. Besides, electrophilicity and electronic potential for two compound calculations are also shown. Aside from reactivity comparison, we have divulged bond lengths and bond order profiles (Table S2) in the synthesized compounds using DFT. Notably, the DFT calculation supports the single and double-bond nature of the compounds. Further, DFT computed bond lengths value is nearly comparable with the experimental values.
Fig. 5.

HOMO-LUMO map with energy gap for H2L.1.
Fig. 6.

HOMO-LUMO map with energy gap for H2L2.
Table 3.
Density Function Theory calculations with other descriptors.
| Name | H2L1 | H2L2 |
|---|---|---|
| Total energy | −1215.67 | −6355.88 |
| Binding energy | −10.2342 | −10.0751 |
| Dipole moment | 1.00852 | 1.74034 |
| HOMO energy eV | −0.163293 | −0.175512 |
| LUMO energy eV | −0.0714509 | −0.0875975 |
| Band Gap Energy eV | 0.0918418 | 0.0879144 |
| Hardness | 0.0459209 | 0.0439572 |
| Softness | 21.77657667859297 | 22.74940169073553 |
| Electronegativity | −0.11737195 | −0.13155475 |
| Electrophilicity | 0.1499989617668915 | 2.239200767460565 |
4.1. MESP analysis
The MESP forecasts a species' relative reactivity locations for nucleophilic and electrophilic assault. The MESP surface analysis of the synthesized compound was obtained utilizing the optimized structure (Fig. 7A–7B) and the B3LYP/6-31G (d, p) basis set. The MESP Iso-values and color Scale shown in Table 4 and Fig. 7, Fig. 8 explores the MESP of the ligands. The compound's colour code ranges from 4.618e3 to 4.618e3. The colours red and blue in the MESP structure indicate more electron-rich and electron-deficient regions, respectively. The chemical exhibits the polarization effect. The MESP has negative potential zones over the electronegative atoms (oxygen and nitrogen) and positive potential over the hydrogen atoms. However, the compound's bromine atom has a lower negative potential site than the other electronegative atoms. As a result, the locations with higher negative electronegative and positive electrostatic potential are more attractive toward synthesized compounds. The interesting is due to nucleophilic and electrophilic species activity. Significantly, we have investigated DFT calculated significant properties such as hybridization, Oxidation, and ESP charge, Mulliken and Hirshfeld charges for two Salen compounds (Table S3).
Fig. 7.

MESP profile (A) and optimized structure (B) of H2L1.
Table 4.
MESP Iso-values and colour Scale.
| Compounds | Iso-value | Spectrum (Color range) |
Histogram Scale | |
|---|---|---|---|---|
| Min | Max | |||
| H2L1 | 3.000000e-02 | 0.0000 | 618.9843 | 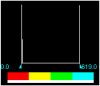 |
| H2L2 | 3.000000e-02 | 0.0000 | 712.4078 | 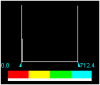 |
Fig. 8.

MESP profile (A) and optimized structure (B) of H2L2.
4.2. Molecular docking
We have performed a docking simulation of our synthesized compounds to inspect whether the compounds act as bacterial agents. Table 5 shows the essential interaction and binding manner of hits with the binding site of amino acids of the Gram-positive bacterium, Bacillus subtilis, and Gram-negative bacterium, Proteus Vulgaris. The chemical's unique linkages enable it to create receptor-ligand solid interactions. Furthermore, the interactions between the chemical and the receptor illustrated in Table 5 make the complex more stable.
Table 5.
The binding energies and detailed docking interactions.
| Important Parameters |
The Gram-positive bacterium, Bacillus subtilis (6UF6) |
The gram-negative bacterium, Proteus Vulgaris (5HXW) |
||||
|---|---|---|---|---|---|---|
| H2L1 | H2L2 | Penicillin G (Control) | H2L1 | H2L2 | Ceftriaxone (Control) | |
| Binding Energy (Kcal/Mol) (Delta G) | −7.7 | −7.0 | −8.9 | −8.7 | −8.2 | −8.1 |
| Carbon hydrogen bonds | Gln219, His271. | Ile257, Gln258, | Asn253 | – | Arg315, Asp340, Gly437, | Gln278 |
| Conventional Hydrogen Bond | Asp256, | Arg215(2), Ser259, | Thr250 | Gln278 | Gln278 | Gln92, Gln99, Arg315 |
| Covalent bond | Thr99 | – | -- | Gln278, Ile413 | Gln278, Ile317 | |
| Alkyl Interaction | Val69, Val92, Ile257. | Ile88, Ile268, | Ala226 | Leu274, Ile317, Ala319, Val343, Ile345, Ile413, | Ile317, Pro342, Met440, Met411, | Met411 |
| Pi- Alkyl Interaction | Ile88, | Ile268, His267, | Val241, Phe251, Trp255 | Ala319, Phe341(2), Val343 | Phe96, Tyr97, Phe201, Phe301, Phe341, Pro342 | Pro342, Trp438 |
| Pi-sigma interaction | -- | -- | -- | Ile317, Val343 | Trp97 | -- |
| Pi-pi T- Shaped | -- | -- | -- | -- | Phe341, Trp438, | -- |
| Metal Acceptor Interaction | Val92, Thr99. | Ile88 | – | Ile317, Val343 | – | – |
| Unfavourable Donor-Donor | -- | -- | -- | -- | -- | Gly439 |
4.2.1. Gram-positive bacterium, Bacillus subtilis (6UF6)
With binding energy of −7.7 kcal/mol, H2L1 nearly filled the binding site. H2L1 forms strong hydrogen bonds with amino acids such as Gln219 and His271. H2L1 has created a solid covalent connection with essential amino acids, including Thr99, at the active site of the Gram-positive bacterium Bacillus subtilis. (Table 5 and Fig. S8). The optimum binding postures were chosen based on binding affinity ratings and orientation inside the active site. With predicted binding energy of −7.0 kcal/mol, H2L2 produces hydrogen bond interactions with amino acids such as Ile257 and Gln258, as well as cluster residues such as Arg215 (2) and Ser259, which have conventional bonds. The penicillin G (a known famous drug) was used as control validation again Bacillus subtilis, having almost a similar range of docking energy −8.9 kcal/mol with some crucial Hydrogen bond interactions at Thr250, which are needed for their stability inside the protein active site (Fig. S10A). Other interactions contribute to the stability of these complexes (Table 5 and Fig. S9).
4.2.2. The gram-negative bacterium, Proteus Vulgaris (5HXW)
H2L1 virtually filled the binding site with an energy of −8.7 kcal/mol, generating common hydrogen bonds with amino acids like Gln278. H2L1 formed two solid covalent bonds with essential amino acids such as Gln278 and Ile413 at the Gram-negative bacterium Proteus Vulgaris' active site (Table 5 and Fig. S10). The best binding postures were determined based on binding affinity ratings and active site orientation. H2L2 creates hydrogen bond interactions with amino acids such as Arg315, Asp340, and Gly437, as well as cluster residues such as Gln278 that have conventional bonds with an estimated binding energy of −8.2 kcal/mol. Ceftriaxone (a known famous drug) was used as control validation again Proteus Vulgaris having almost the equal range of docking energy −8.1 kcal/mol with some crucial, crucial Hydrogen and hydrophobic bond interactions at Gln278. and an additional unfavourable donor-donor interaction was found at Gln439, which is needed for their stability inside the protein active site (Fig. S11A). Other interactions also contribute to the stability of these complexes (Table 5 and Fig. S11).
Furthermore, to validate our docking experiment methods, we have to represent the figure of the compound before and after docking to represent RMSD. When docking, the RMSD value is used to compare the docked conformation to the reference conformation, or another docked conformation. Therefore, the most common method to assess the correctness of the docking geometry is to measure the ligand's root mean square deviation (RMSD) from its reference position in the response complex after the optimal superposition of the receptor molecules. In the below-mentioned Fig. 9, we have submitted docking with RMSD calculations of our two trendy Salen-type ligands.
Fig. 9.

RMSD Calculations A. For H2L1 (Before docking pose) (Grey Ball Stick structure) superimpose over H2L1 (After docking pose) (Pink Ball Stick structure) with Gram-positive bacterium, Bacillus subtilis. B. H2L1 (Before docking pose) (Grey Ball Stick structure) superimpose over H2L1 (After docking pose) (Blue Ball Stick structure) with Gram-negative bacterium, Proteus Vulgaris. RMSD Calculations A. For H2L2 (Before docking pose) (Grey Ball Stick structure) superimpose over H2L2 (After docking pose) (Violet Ball Stick structure) with Gram-positive bacterium, Bacillus subtilis. B. H2L2 (Before docking pose) (Grey Ball Stick structure) superimpose over H2L2 (After docking pose) (Brown Ball Stick structure) with Gram-negative bacterium, Proteus Vulgaris. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4.3. In-silico ADME/T and toxicity predictions
The ADME/T prediction using the SWISSADME database generated the following results for the two compounds. Following the submission of both structures, the ADME/T prediction using the SWISSADME database produced the findings below. The following results were obtained from the ADME/T forecast utilizing the SWISSADME database for the synthesized compounds. After submitting both structures of the compounds, the ADME/T prediction using the SWISSADME database produced the findings below. The molecule's lipophilicity and the Consensus P0/W. Water Solubility characteristics were estimated. The expected pharmacokinetic data was found to be of high GI absorption, not blood-brain barrier permeant, functions as a P-gp substrate, and does not inhibit CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 cytochromes. Drug similarity variables were discovered to be of a drug-like molecule that violates Lipinski's Rules, as well as drug-likeness score principles such as Veber, Egan, and Muegge, and with a 0.55 Bioavailability score. Medicinal chemistry factors revealed that there was no PAINS alarm and that it violated Brenk's rules with two alerts of being an isolated alkene, one Michael acceptor, no lead likeness, and a molecular weight of more than 350. Additional ADME and toxicity descriptors elements are discussed in (Table 6). Here we are also interested in explaining the Bromine group's function in the compound H2L2. Notably, it is electron-withdrawing, so have influenced biological activity. A compound like H2L2 with the Br group works directly, irritating the skin, mucous membranes, and tissues. The seriousness of poisoning caused by Br depends on the amount, exact route, and length of time of exposure. Apart from that, it is also essential to the age and pre-existing medical condition of the person exposed.
-
•
MW- Molecular Weight.
-
•
Fraction Csp3-the ratio of sp3 hybridizes carbons over the total carbon count of the molecule.
-
•
TPSA-topological polar surface area.
-
•
log Po/w - partition coefficient between n-octanol and water.
-
•
XLOGP3, an atomistic method including corrective factors and a knowledge-based library [28].
-
•
WLOGP, our own implementation of a purely atomistic method based on the fragmental system of Wildman and Crippen.
-
•
MLOGP, an archetype of a topological method relying on a linear relationship with 13 molecular descriptors implemented from refs.
-
•
SILICOS-IT, a hybrid method relying on 27 fragments and 7 topological descriptors.
-
•
BBB -the blood-brain barrier.
-
•
P-gp - permeability glycoprotein.
-
•
CYP - cytochromes P450.
Table 6.
ADME/T toxicity predicted descriptors.
| Parameters |
H2L1 |
H2L2 |
|---|---|---|
| PHYSICO-CHEMICAL PROPERTY | ||
| Formula | C21H26N2O4 | C21H24Br2N2O4 |
| MW (G/Mol) | 370.44 g/mol | 528.23 g/mol |
| Num. Heavy Atoms | 27 | 29 |
| Fraction Csp3 | 0.33 | 0.33 |
| Num. Of Rotatable Bonds | 8 | 8 |
| Num. H-Bond Acceptors | 6 | 6 |
| Num. H-Bond Donors | 2 | 2 |
| Molar Refractivity | 108.88 | 124.28 |
| TPSA (A2) | 83.64 | 83.64 |
| LIPOPHILICITY | ||
| Log Po/W (Ilogp) | 4.06 | 4.74 |
| Log Po/W (XLOGP3) | 3.19 | 4.57 |
| Log Po/W (WLOGP) | 3.68 | 5.20 |
| Log Po/W (MLOGP) | 1.62 | 2.80 |
| Log Po/W (SILICOS-IT) | 5.00 | 6.39 |
| Consensus Log Po/W | 3.51 | 4.74 |
| WATER SOLUBILITY | ||
| Log S (ESOL) Solubility Class |
−3.95 Soluble |
−5.77 moderately Soluble |
| Log S (ALI) Solubility Class |
−4.62 moderately Soluble |
−6.05 Poorly soluble |
| Log S (SILICOS-IT) Solubility Class |
−5.79 moderately soluble |
−7.33 Poorly soluble |
| PHARMACOKINETICS | ||
| GI Absorption | High | High |
| BBB Permeation | No | No |
| P-Gp Substrate | No | No |
| CYP1A2 Inhibitor | No | No |
| CYP2C19 Inhibitor | No | Yes |
| CYP2C9 Inhibitor | No | Yes |
| CYP2D6 Inhibitor | Yes | No |
| CYP3A4 Inhibitor | Yes | Yes |
| Log Kp (Skin Permeation) | −6.29 cm/s | −6.28 cm/s |
| DRUG LIKENESS | ||
| Violation Of Lipinski's Rule Of Five | Yes | Yes |
| Ghose | Yes | No |
| Violation Of Veber Rule | Yes | Yes |
| Egan | Yes | Yes |
| Muegge | Yes | Yes |
| Bioavailability Score | 0.55 | 0.55 |
| MEDICINAL CHEMISTRY | ||
| Leadlikeness | NO | NO |
| Synthetic Accessibility | 3.18 | 3.43 |
| TOXICITY | ||
| ADMET-EXT-CYP2D6 | −2.12503 | −3.7762 |
| ADMET-EXT-CYP2D6#prediction | False | False |
| ADMET-EXT-CYP2D6-Applicability | Within Expected Range | Within Expected Range |
| ADMET-EXT-CYP2D6-Applicability#MD | 11.3664 | 11.566 |
| ADMET-EXT-CYP2D6-Applicability#MDpvalue | 0.0109541 | 0.00733465 |
| ADMET-EXT- Hepatotoxic | −0.131477 | 1.51172 |
| ADMET-EXT- Hepatotoxic #prediction | True | True |
| ADMET-EXT- Hepatotoxic Applicability | Within Expected Range | Within Expected Range |
| ADMET-EXT- Hepatotoxic Applicability #MD | 11.374 | 11.4464 |
| ADMET-EXT- Hepatotoxic Applicability #MDpvalue | 0.00176447 | 0.00136254 |
| ADMET-EXT-PPB | 1.79107 | 0.993801 |
| ADMET-EXT-PPB #Prediction | True | True |
| ADMET-EXT-PPB- Applicability | Within Expected Range | Within Expected Range |
| ADMET-EXT-PPB- Applicability #MD | 11.6676 | 11.1673 |
| ADMET-EXT-PPB- Applicability #MDpvalue | 0.185727 | 0.400759 |
5. Conclusions
In this research, we synthesized two contemporary Salen-type compounds with satisfactory yields. The products were yellow powders characterized by modern spectroscopic tools and EDX-SEM. The DFT studies in gas phase B3LYP-D3/6-311G (d, p) level optimized the trendy ligand's molecular geometry. The global reactivity, HOMO-LUMO parameters, atomic properties, and MESP support the chemical reactivity. DFT-simulated IR/NMR and UV-visible spectral results corroborated the experimental values. Notably, good correlations were obtained when the predicted 1H/13C NMR spectra were compared with the corresponding experimental spectral result values. In addition, the experimental and theoretical spectral results ensure ligand structure. This article in depth demonstrated in silico molecular docking against the Gm + ve Bacillus subtilis (6UF6) and Gm -ve Proteus Vulgaris. The RMSD calculations further validate the docking study. The docking study explains the ligand's binding ability with essential amino acids through conventional hydrogen bonding or other significant interactions. The drug-like properties of ADME/T using the SWISSADME database successfully found the molecule's lipophilicity and the consensus P0/W. The water solubility characteristics were also estimated. The expected pharmacokinetic data was found to be of high GI absorption, not blood-brain barrier permeant, and function as a P-gp substrate. The Br group showed a more toxic effect in H2L2 than in H2L1. These include irritating the skin, mucous membranes, and tissues. The severity of Br poisoning depends on the amount, exact route, and exposure, and it also depends on the age and previous illness of the exposed person. The manuscript is fascinating in respect of future research perspectives. The article's findings ensure that two Salen-type compounds will be used as promising drugs and antimicrobial agents in a flash. Moreover, the presented article's topics gained intriguing interest from molecular dynamic simulation community researchers.
Author contribution statement
Dhrubajyoti Majumdar: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper. Amit Dubey, Aisha Tufail: Performed the experiments; Analyzed and interpreted the data; analysis tools or data, Wrote the paper.
Dipankar Sutradhar, Sourav Roy: Performed experiments; Analyzed and interpreted the data.
Data availability statement
The data that has been used is confidential.
Declaration of competing interest
The authors declare no competing interests or personal relationships that could have appeared to impact the work reported in this paper.
Acknowledgments
The current research has not received specific funding from any public, commercial, or not-for-profit sector funding agency. All authors in the manuscript carefully read and approved the final version before submission.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2023.e16057.
Contributor Information
Dhrubajyoti Majumdar, Email: dmajumdar30@gmail.com.
Sourav Roy, Email: souravscott@gmail.com.
Appendix B. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Schiff H. The syntheses and characterization of Schiff base. Ann. Chem. Suppl. 1864;3:343. [Google Scholar]
- 2.Aboul-Fadl T., Mohammed F.A.-H., Hassan E.A.-S. Synthesis, antitubercular activity and pharmacokinetic studies of some Schiff bases derived from 1-alkylisatin and isonicotinic acid hydrazide (INH) Arch Pharm. Res. (Seoul) 2003;3:778–784. doi: 10.1007/BF02980020. [DOI] [PubMed] [Google Scholar]
- 3.Renner C., Asperger A., Seyffarth A., Meixensberger J., Gebhardt R., Gaunitz F. Carnosine inhibits ATP production in cells from malignant glioma. Neurol. Res. 2010;32:101–105. doi: 10.1179/016164109X12518779082237. [DOI] [PubMed] [Google Scholar]
- 4.Soleymanabadi H., Rastegar S.F. J Theoretical investigation on the selective detection of SO2 molecule by AlN nanosheets. J. Mol. Model. 2014;20:2439–2445. doi: 10.1007/s00894-014-2439-6. [DOI] [PubMed] [Google Scholar]
- 5.Soleymanabadi H., Rastegar S.F. DFT studies of acrolein molecule adsorption on pristine and Al-doped graphenes. J. Mol. Model. 2013;19:3733–3740. doi: 10.1007/s00894-013-1898-5. [DOI] [PubMed] [Google Scholar]
- 6.Deshpande N.S., Mahendra G.S., Aggarwal N.N., Gatphoh B.F.D., Siddappa B.C.R. Insilico design, ADMET screening, MM-GBSA binding free energy of novel 1,3,4 oxadiazoles linked Schiff bases as PARP-1 inhibitors targeting breast cancer, Fut. J. Pharmaceut. Sci. 2021;7:174. [Google Scholar]
- 7.Sathe B.S., Jayachandran E., Jagtap V.A., Sreenivasa G.M. Synthesis characterization and anti-inflammatory evaluation of new fluorobenzothiazole Schiff's bases. J. Chem. Pharmaceut. Sci. 2010;3:164–169. [Google Scholar]
- 8.Avaji P.G., Kumar C.H.V., Patil S.A., Shivananda K.N., Nagaraju C. Synthesis, spectral characterization, in-vitro microbiological evaluation and cytotoxic activities of novel macrocyclic bis hydrazone. Eur. J. Med. Chem. 2009;44:3552–3559. doi: 10.1016/j.ejmech.2009.03.032. [DOI] [PubMed] [Google Scholar]
- 9.Oganov A.R., editor. Modern Methods of Crystal Structure Prediction. Wiley-VCH Verlag GmbH Co. KGaA; Weinheim, Germany: 2011. [Google Scholar]
- 10.Abdulridha A.H., Allah M.A.A.H., Makki S.Q., Sert Y., Salman H.I., Balakit A.A. Corrosion inhibition of carbon steel in 1 M H2SO4 using new Azo Schiff compound: electrochemical, gravimetric, adsorption, surface and DFT studies. J. Mol. Liq. 2020;315 [Google Scholar]
- 11.A. A. Balakit, S. Q. Makki, Y. Sert, F.Ucun, M. B.Alshammari, P. Thordason, G. A. EL-Hilti, Synthesis, spectrophotometric and DFT studies of new Triazole Schiff bases as selective naked-eye sensors for acetate anion, Supramol. Chem., 32 (20–20) 519-526.
- 12.Dege N., Gokce H., Dogan O.E., Alpaslan G., Agar T., Muthu S., Sert Y. Quantum computational, spectroscopic investigations on N-(2-((2-chloro-4,5-dicyanophenyl)amino)ethyl)-4-methylbenzenesulfonamide by DFT/TD-DFT with different solvents, molecular docking and drug-likeness researches. J. Colloids Surf. 2022;638 [Google Scholar]
- 13.Majumdar D.J., Tüzün B., Pal T.K., Saini Reena V., Bankura K., Mishra D. Structurally diverse heterobimetallic Pb(II)-Salen complexes mechanistic notion of cytotoxic activity against neuroblastoma cancer cell: synthesis, characterization, protein–ligand interaction profiler, and intuitions from DFT. Polyhedron. 2021;210 [Google Scholar]
- 14.Majumdar D.J., Agrawal Y., Thomas R., Ullah Z., Santra M.K., Das S., Pal T.K., Bankura K., Mishra D. Syntheses, characterizations, crystal structures, DFT/TD-DFT, luminescence behaviors and cytotoxic effect of bicompartmental Zn (II)-dicyanamide Schiff base coordination polymers: an approach to apoptosis, autophagy and necrosis type classical cell death. Appl. Organomet. Chem. 2019;34:e5269. [Google Scholar]
- 15.Majumdar D.J., Dey Swapan, Sreejith S.S., Biswas J.K., Mondal M., Shukla P., Das S., Pal T.K., Das D., Bankura K., Mishra D. Syntheses, crystal structures and photo physical aspects of azido-bridged tetranuclear cadmium (II) complexes: DFT/TD-DFT, thermal, antibacterial and anti-biofilm properties. J. Mol. Struct. 2019;1179:694–708. [Google Scholar]
- 16.Majumdar D.J., Das D., Nag S., Bhattacharyya M., Singh D.K., Parai D., Bankura K., Mishra D. A rare hetero-bimetallic Zn(II)/Ca(II) Schiff base complex: synthesis, crystal structure, DFT, molecular docking and unveiling antimicrobial activity. J. Mol. Struct. 2020;1222 [Google Scholar]
- 17.Majumdar D.J., Dey S., Kumari A., Pal T.K., Bankura K., Mishra D. Dicyanamide-intertwined assembly of two new Zn complexes based on N2O4-type pro-ligand: synthesis, crystal networks, spectroscopic insights, and selective nitroaromatic turn-off fluorescence sensing. Spectrochim. Acta. 2021;254 doi: 10.1016/j.saa.2021.119612. [DOI] [PubMed] [Google Scholar]
- 18.Majumdar D.J., Tüzün B., Pal T.K., Das S. Kalipada bankura, architectural view of flexible aliphatic –OH group coordinated hemi-directed Pb(II)-Salen coordination polymer: synthesis, crystal structure, spectroscopic insights, supramolecular topographies, and DFT perspective. J. Inorg. Organomet. Polym. 2022;32:1159–1176. [Google Scholar]
- 19.Majumdar D.J., Frontera A., Gomila Rosa M., Das S., Bankura K.P. Synthesis, spectroscopic findings and crystal engineering of Pb(ii)–Salen coordination polymers, and supramolecular architectures engineered by σ-hole/spodium/tetrel bonds: a combined experimental and theoretical investigation. RSC Adv. 2022;12:6352–6363. doi: 10.1039/d1ra09346k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eliseeva A.A., Ivanov D.M., Novikov A.S., Rozhkov A.V., Kornyakov I.V., Dubovtsev A Yu. V. Yu. Kukushkin, Hexaiododiplatinate(ii) as a useful supramolecular synthon for halogen bond involving crystal engineering. Dalton Trans. 2020;49:356–367. doi: 10.1039/c9dt04221k. [DOI] [PubMed] [Google Scholar]
- 21.Mikherdov A.S., Novikov A.S., Kinzhalov M.A., Zolotarev A.A., Boyarskiy V.P. Intra-/Intermolecular bifurcated chalcogen bonding in crystal structure of thiazole/thiadiazole derived binuclear (diaminocarbene)PdII complexes. Crystals. 2018;8(3):112. [Google Scholar]
- 22.Anisimova T.B., Kinzhalov M.A., Guedes da Silva M. Fátima C., Novikov A.S., Kukushkin V. Yu, Pombeiro A.J.L., Luzyanin K.V. Addition of N-nucleophiles to gold(III)-bound isocyanides leading to short-lived gold(III) acyclic diaminocarbene complexes. New J. Chem. 2017;41:3246–3250. [Google Scholar]
- 23.Adonin S.A., Usoltsev A.N., Novikov A.S., Kolesov B.A., Fedin V.P., Sokolov M.N. One- and two-dimensional iodine-rich iodobismuthate(III) complexes: structure, optical properties, and features of halogen bonding in the solid state. Inorg. Chem. 2020;59:3290–3296. doi: 10.1021/acs.inorgchem.9b03734. [DOI] [PubMed] [Google Scholar]
- 24.Kashina M.V., Kinzhalov M.A., Smirnov A.S., Ivanov D.M., Novikov A.S., Kukushkin V. Yu. Dihalomethanes as bent bifunctional XB/XB‐Donating building blocks for construction of metal‐involving halogen bonded hexagons. Chem.--Asian J. 2019;14(21):3915–3920. doi: 10.1002/asia.201901127. [DOI] [PubMed] [Google Scholar]
- 25.Adonin S.A., Gorokh I.D., Novikov A.S., Samsonenko D.G., Korolkov I.V., Sokolov M.N., Fedin V.P. Bromobismuthates: cation-induced structural diversity and Hirshfeld surface analysis of cation–anion contacts. Polyhedron. 2018;139:282–288. [Google Scholar]
- 26.Kulish K.I., Novikov A.S., Tolstoy P.M., Bolotin D.S., Bokach N.A., Zolotarev A.A., Kukushkin V Yu. Solid state and dynamic solution structures of O-carbamidine amidoximes gives further insight into the mechanism of zinc(II)-mediated generation of 1,2,4-oxadiazoles. J. Mol. Struct. 2016;1111:142–150. [Google Scholar]
- 27.Novikov A.S., Kuznetsov M.L. Theoretical study of Re(IV) and Ru(II) bis-isocyanide complexes and their reactivity in cycloaddition reactions with nitrones. Inorg. Chim. Acta. 2012;380:78–79. [Google Scholar]
- 28.Silakari O., Sing P.K. Vol. 1. 2021. Concepts and Experimental Protocols of Modeling, and Informatics in Drug Design; pp. 131–135. [Google Scholar]
- 29.Guan L., Yang H., Cai Y., Sun L., Di P., Li W., Liu G., Tang Y. ADMET-score-a comprehensive scoring function for evaluation of chemical drug-likeness. Med. Chem. Commun. 2019;10:148–157. doi: 10.1039/c8md00472b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., et al. Gaussian Inc.; Wallingford, CT, USA: 2016. Gaussian 09, Revision E.01. [Google Scholar]
- 31.Dennington Roy, Keith Todd, Millam John. Semichem Inc.; Shawnee Mission, KS: 2019. Gauss View, Version 6.1.1. [Google Scholar]
- 32.Li F.K.K., Rosell F.I., Gale R.T., Simorre J.P., Brown E.D., Strynadka N.C.J. Crystallographic analysis of Staphylococcus aureus LcpA, the primary wall teichoic acid ligase. J. Biol. Chem. 2020;295(9):2629–2639. doi: 10.1074/jbc.RA119.011469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ju Y., Tong S., Gao Y., Zhao W., Liu Q., Gu Q., Xu J., Niu L., Teng M., Zhou H. Crystal structure of a membrane-bound l-amino acid deaminase from Proteus vulgaris. J. Struct. Biol. 2016;195(3):306–315. doi: 10.1016/j.jsb.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 34.Rose P.W., Prlic A., Bi C., Bluhm W.F., Christie C.H., Dutta S., Green R.K., Goodsell D.S., Westbrook J.D. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. J. Woo, Nucl. Acids Res. 2015;43:D345–D356. doi: 10.1093/nar/gku1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schrödinger Release 2018-3: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, (2018); Impact, Schrödinger, LLC, New York, NY; Prime. Schrödinger, LLC; New York, NY: 2018. [Google Scholar]
- 36.Morris G.M., Goodsell D.S., Halliday D.S., Huey R., Hart W.E., Belew R., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 37.Dubey A., Marabotti A., Ramteke P.W., Facchiano A. In silico approach to find chymase inhibitors among biogenic compounds. Future Med. Chem. 2016;8:841–851. doi: 10.4155/fmc-2016-0030. [DOI] [PubMed] [Google Scholar]
- 38.Bharadwaj S., Dubey A., Kamboj N.K., Sahoo A.K., Kang S.G., Yadava U. Drug repurposing for ligand-induced rearrangement of Sirt2 active site-based inhibitors via molecular modeling and quantum mechanics calculations. Sci. Rep. 2021;11(1) doi: 10.1038/s41598-021-89627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dubey A., Dotolo S., Ramteke P.W., Facchiano A., Marabotti A. Searching for chymase inhibitors among chamomile compounds using a computational-based approach. Biomolecules. 2018;9(1):5. doi: 10.3390/biom9010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huey R., Morris G.M., Olson A.J., Goodsell D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 41.Trott O., Olson A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7 doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das L.K., Biswas A., Frontera A., Ghosh A. Polymorphism in hetero-metallic tri-nuclear CuII2CdIIcomplexes of salicylaldimine ligand: structural analysis and theoretical study. Polyhedron. 2013;52:1416–1424. [Google Scholar]
- 44.Dolai M., Mistri T., Panja A., Ali M. Diversity in supramolecular self-assembly through hydrogen-bonding interactions of non-coordinated aliphatic –OH group in a series of heterodinuclear CuIIM (M = NaI, ZnII, HgII, SmIII, BiIII, PbII and CdII) Inorg. Chim. Acta. 2013;399:95–104. [Google Scholar]
- 45.Maiti M., Thakurta S., Sadhukhan D., Pilet G., Rosair G.M., Nonat A., Charbonniere L.J., Mitra S. Thermally stable luminescent zinc–Schiff base complexes: a thiocyanato bridged 1D coordination polymer and a supramolecular 1D polymer. Polyhedron. 2013;65:6–15. [Google Scholar]
- 46.Sadhukhan D., Ray A., Rosair G., Charbonnière L., Mitra S. A two-dimensional zinc(II)–Schiff base coordination polymer formed by six-membered metallacyclic repeating motif: structural aspects, thermal and photophysical properties. BCSJ. 2011;84:211–217. [Google Scholar]
- 47.Amirnasr M., Schenk K.J., Salavati M., Dehghanpour S., Taeb A., Tadjarodi A. Synthesis and Characterization of Cobalt(II), Nickel(II), and Zinc(II) Complexes with N,N ′-bis(Trans- Cinnamaldehyde)-1,2-Diiminoethane Ligand, (ca2en): crystal and Molecular Structures of Co(ca2en)Cl2 , Co(ca2en)Br2 and Ni(ca2en)Br2. J. Coord. Chem. 2003;56:231–243. [Google Scholar]
- 48.Karimi S., Rezaeivala M., Sayin K., Tuzun B. Experimental and computational investigation of 3,5-di-tert-butyl-2-(((3-((2-morpholinoethyl)(pyridin-2-ylmethyl) amino)propyl)imino) methyl)phenol and related reduced form as an inhibitor for C–steel. Mater. Chem. Phys. 2022;287 [Google Scholar]
- 49.Rezaeivala M., Karimi S., Sayin K., Tüzün B. Experimental and theoretical investigation of corrosion inhibition effect of two piperazine-based ligands on carbon steel in acidic media. Colloids Surf. A Physicochem. Eng. Asp. 2022;641 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that has been used is confidential.