

Abstract
The vast majority of genomic sequences are automatically annotated using various software programs. The accuracy of these annotations depends heavily on the very few manual annotation efforts that combine verified experimental data with genomic sequences from model organisms. Here, we summarize the updated functional annotation of Bacillus subtilis strain 168, a quarter century after its genome sequence was first made public. Since the last such effort 5 years ago, 1168 genetic functions have been updated, allowing the construction of a new metabolic model of this organism of environmental and industrial interest. The emphasis in this review is on new metabolic insights, the role of metals in metabolism and macromolecule biosynthesis, functions involved in biofilm formation, features controlling cell growth, and finally, protein agents that allow class discrimination, thus allowing maintenance management, and accuracy of all cell processes. New ‘genomic objects’ and an extensive updated literature review have been included for the sequence, now available at the International Nucleotide Sequence Database Collaboration (INSDC: AccNum AL009126.4).
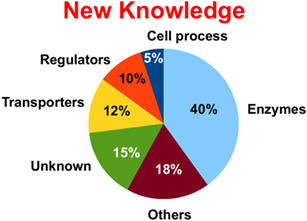
The reference genome of Bacillus subtilis was first sequenced and annotated 25 years ago. At that time, some 42% of the coding sequences could not be assigned a function. In the present annotated update, 15% of the CDS remain functionless. An important contribution of newly identitied functions has been attributed to nucleic acid modifying enzymes and genes acting as class discriminators, controlling growth homeostasis and allowing the cells to display a collective behavior.
INTRODUCTION
The genome sequencing landscape is no longer recognizable in terms of the technology used to sequence model organisms 25 years ago. Sequencing the genome of the first member of the Firmicutes [now Bacillota (Oren & Garrity, 2021)], Bacillus subtilis strain 168, involved more than 30 laboratories worldwide and 151 authors with different cloning and sequencing strategies (Kunst et al., 1997). This multi‐national effort resulted in a patchwork of sequences derived from individual clones of the same strain propagated in the participating laboratories. The corresponding heterogeneity of the sequence quality required subsequent refinement into a final sequence derived from a single clone (Barbe et al., 2009). Our knowledge of B. subtilis in its various environments is improving rapidly and sequences from more than 100 isolates of the species are now routinely available in the International Nucleotide Sequence Database Collaboration (INSDC: DDBJ/ENA‐EBI/GenBank [Arita et al., 2021]). This has been used to define a core and a pan‐genome for the species, allowing minimization of the genome (Michalik et al., 2021; Sutton et al., 2021; Wu, Wang, et al., 2021). Back in 1997 more than half of the genes of the organism had no identifiable role. It was not even certain that they were authentic protein coding sequences (CDSs). The annotation of the genome sequence was therefore partial and incomplete. However, the fact that the sequencing had been carried out by a consortium of scientists from all over the world allowed a particularly rich annotation of the ‘genomic objects’ (i.e. genes and sequences of notable role).
A quarter of a century later, we provide here an updated annotation and a significantly improved metabolic network reconstruction since the previous annotation update (Borriss et al., 2018; Tibocha‐Bonilla et al., 2022). The identification of its relevant biotope is crucial to provide adequate background for annotation of the organism's genomic sequence. It is also important for the further exploitation of this species as both an environmental and industrial organism. Here, we focus on insights into functions that are not part of the core functions of life previously discussed in the annotation of minimal genomes such as that of Mycoplasma mycoides Syn3.0 (Danchin & Fang, 2016; Hutchison et al., 2016).
Just as the power of genome sequencing has dramatically improved our understanding of individual organisms, so too has it revolutionized our understanding of taxonomic relationships among organisms. The genus Bacillus has long been recognized as polyphyletic, being derived from more than one common evolutionary ancestor. Traditionally, the genus has been defined primarily as consisting of Gram‐positive, aerobic and facultatively anaerobic, endospore‐forming bacteria, irrespective of their actual commonality. Until recently, the genus Bacillus consisted of ~280 validly published species, including two important species clades; the ‘Subtilis clade’ (Bacillus subtilis sensu lato) that includes the type strain of the species (B. subtilis Ehrenberg 1835; Cohn 1872; NCBI 3610—often referred to as the Marburg strain), and the ‘Cereus clade’ (B. cereus sensu lato), that includes important human and animal pathogens (e.g. B. anthracis and B. cereus sensu stricto). Because the type strain for the genus is a member of the Subtilis clade, it would normally be expected that the members of the Cereus clade would be transferred to a new genus. However, the International Code of Nomenclature of prokaryotes does not recommend this transfer if renaming confusion could endanger human health. To better understand the phylogeny of the genus Bacillus, the genome sequences of 352 Bacillaceae species have been analysed using multiple independent approaches (Patel & Gupta, 2020). As a result, a plethora of new genera have arisen, with more expected (Gupta et al., 2020).
The Marburg strain, originally named Vibrio subtilis and only later Bacillus subtilis, was isolated from freshly prepared hay infusions in 1872. It was isolated and first characterized by Ferdinand Cohn, who described its life cycle, including the formation of spores and their subsequent germination (Drews, 2000). This species is an epiphyte (Mamphogoro et al., 2020) and sometimes even an endophyte (Kiani et al., 2021), with specific interactions with both the rhizosphere and phylloplane. As such, and in addition to its interest as an industrial workhorse (Harwood, 1992; Su et al., 2020), B. subtilis is recognized for its capabilities to interact with plants (Zuñiga et al., 2020) as well as its importance in protecting plants against various pathogens (Blake et al., 2021). This role implies numerous functions allowing on the one hand a strong interaction with plants, and on the other hand a metabolism generally linked to that of plants. Bacillus subtilis strain 168 was not the original Marburg strain but a tryptophan auxotroph obtained after X‐ray mutagenesis (Zeigler et al., 2008). A vast number of studies have explored this model organism, initially chosen for the importance of sporulation in its life cycle, and not for its biotope. Although not a focus here, the study of sporulation has continued as summarized in recent reviews (Khanna et al., 2020; Stragier, 2022). A significant number of new annotations concern collective movements of the bacterium, biofilm formation and more generally its role in its interaction with dioxygen and leaf adhesion in the phyllosphere (Cámara‐Almirón et al., 2020) as well as root exudates in the rhizosphere (Oppenheimer‐Shaanan et al., 2022). Interaction with plants has also led to a metabolic orientation and preference for a range of metals that differ in importance from the way they are used by the main model of Bacteria, Escherichia coli. This fresh knowledge is summarized here, along with new developments in the metabolism of the bacterium and the identification of several previously unknown functions (Figure 1 and Table S1).
FIGURE 1.
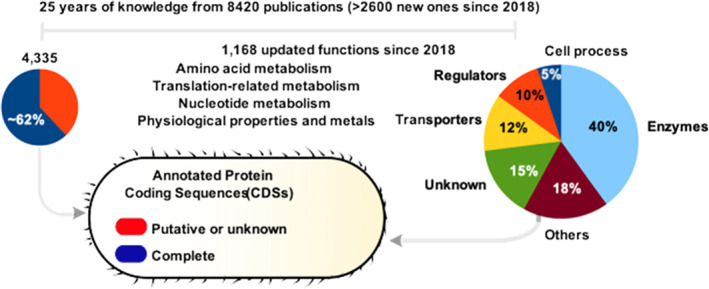
Twenty‐five years of knowledge of the genome sequence of Bacillus subtilis. With these unknowns the B. subtilis model can be an essential partner in our quest to understand the background physical properties of biological agents responsible for the apparent ‘animation’ of chemical biology, a feature essential to proceed with synthetic biology constructs.
KEY ANNOTATION RESOURCES USED TO CREATE THE INTERNATIONAL NUCLEOTIDE SEQUENCE DATABASE (INSDC) UPDATED ENTRY
The reference sequence of strain 168 deposited in the DSMZ collection (https://www.dsmz.de/collection) and in the Institut Pasteur collection (https://www.pasteur.fr/fr/sante‐publique/biobanques‐collections/collection‐institut‐pasteur‐cip) was described in the first sequence and annotation update deposited at the INSDC (DDBJ/ENA‐EBI/GenBank) that ironed out the differences stemming from the variety of clones that had been maintained in individual laboratories around the world (Barbe et al., 2009). It is expected that these isolates of the reference strain 168 did not evolve while staying in the repositories. However, isolates used regularly in the laboratory keep evolving (Richts et al., 2020; Shiwa et al., 2013), and consequently clonal genome sequences should be checked regularly. This is important since the sequencing of individual laboratory isolates often identifies SNP variants that in some cases can reveal additional knowledge (Zhao et al., 2019). This is especially important when discussing the role of specific genes and phenotypes (Gallegos‐Monterrosa et al., 2016). Two related collections, with knock out mutants carrying kanamycin or erythromycin resistant inserts, BKK and BKE, respectively (Koo et al., 2017) are available from the Bacillus Genetic Stock Center (BGSC https://bgsc.org). The American Type Culture Collection (ATCC: https://www.atcc.org) and the Japanese Bioresource Project (https://shigen.nig.ac.jp/bsub/) also keep relevant collections of mutants.
As time goes on, new information on genes and sequences of various strains of the B. subtilis species continues to accumulate (Steinke et al., 2021). The corresponding information is collected in a variety of repositories and databases. Currently, the most popular database, SubtiWiki, follows a simple ‘wiki’ data structure, striving to obtain up‐to‐date basic information on B. subtilis genes from a variety of sources (Pedreira et al., 2022). A counterpart has long been maintained within the MicroScope platform collection (Vallenet et al., 2020). There, annotation is explicitly designed to help users to enter the reference data update deposited at the INSDC whose unique role is to act as the internationally accepted upstream reference data source that can be used by other repositories for value‐added approaches (specialized databases). Importantly, the annotation in the MicroScope platform is substantiated systematically using comparative genomics. The annotation data management system in MicroScope provides the user not only with up‐to‐date annotation but also with access to a variety of tools (see next paragraph) designed to help users verify and extend the annotation (Médigue et al., 2019). The annotation reported here was performed using MicroScope and the presented data refers to that found on 31 December 2022 (https://mage.genoscope.cns.fr/microscope/mage/viewer.php?O_id=7). The metabolic profile reported at the site can also be used to update BsubCyc (https://bsubcyc.org), based on the latest annotations from MicroScope with Gene‐Reaction links using data from the Enzyme Commission, EC (Martínez Cuesta et al., 2015), MetaCyc (Caspi et al., 2020) or Rhea (Bansal et al., 2022): https://mage.genoscope.cns.fr/microscope/search/export.php?format=csv&S_id=843. With reference accession number AccNum AL009126.4, this marks the fourth update of the B. subtilis 168 reference sequence since the debut of the genome sequence in November 1997 (Kunst et al., 1997).
The annotation of genes was initially carried out manually, based on articles reporting experiments performed in vivo (genetics and physiology) and in vitro (biochemistry and structural biology). With the advent of computer science, a new approach, in silico biology, has progressively imposed itself and dominates today. While innovative algorithms and software pipelines provide extremely useful information that, in some cases, can even surpass the quality of in vivo experiments, the main danger of over‐reliance on computational annotations is that errors tend to creep in and lead researchers down the wrong paths (Danchin et al., 2018). When erroneous annotations occur, the gene designation are repeated without verification in subsequent annotations—the so‐called ‘annotation issue’. Nevertheless, the combination of a variety of software and information sources helps users obtain accurate information. The MicroScope platform combines a database reporting annotations on individual genomic objects with a graphical web interface, called MaGe, which provides a set of tools for the user to validate the quality of annotations. For example, in order to benefit from comparative genomics, the display of a genomic sequence with counterparts can be aligned with the large number of genomic sequences present in MicroScope, allowing the visualization of synteny (Vallenet et al., 2013). In parallel, for each gene, the PhyloProfile functionality allows the user to follow a list of genes co‐evolving with the gene of interest (Engelen et al., 2012). The platform also provides direct access to generic databases of proteins: UniProt/SwissProt (https://www.uniprot.org) and Seed/FigFam (http://thefig.info/index.html), as well as protein structures (InterProScan: https://www.ebi.ac.uk/interpro), evolutionary protein genealogy (eggNOG: http://eggnog5.embl.de/#/app/home) and metabolic pathways (available under different flavours in the platform). It is also designed to help users to explore the metabolism of the organism via the establishment of metabolic profiles and pathway curation based on three major metabolism databases: BRENDA (https://www.brenda‐enzymes.org), KEGG (https://www.genome.jp/kegg/) and MetaCyc (https://metacyc.org). Many other resources are also useful to perform expert curation of genome annotation, such as pipelines of the PGAP‐6.3 family (https://github.com/ncbi/pgap/releases). However, accuracy will depend heavily on the quality of manual annotation of reference genomes such as the one described here.
Gene expression begins with transcription, and a variety of RNAs have significance in this process: BSGatlas integrates and unifies multiple existing annotation resources involving ‘non‐coding’ RNAs (ncRNAs), while improving the positional annotation for 70% of the ncRNAs and focusing on precise identification of operons (Geissler et al., 2021). Several other RNA‐centred databases provide further information of specific features of the genome. For example, the MODOMICS database updates RNA modifications annotation (de Crécy‐Lagard et al., 2020). Some but far from all RNA genetic objects (mainly riboswitches and well identified regulatory RNAs) have been introduced in the present annotation. Besides the B. subtilis instantiation in MetaCyc, BsubCyc (https://bsubcyc.org), GapMind is a Web‐based tool for annotating amino acid biosynthesis in the Bacteria and the Archaea clades (http://papers.genomics.lbl.gov/gaps). GapMind incorporates many variant pathways and 130 different reactions, and it analyses a genome in just 15 s. To avoid error‐prone transitive annotations, GapMind relies primarily on a database of experimentally characterized proteins (Price et al., 2020). In the domain of transcription, the ReGPrecise database captures, visualizes and analyses the transcription factor‐dependent regulons as reconstructed by comparative genomics in a variety of prokaryotic genomes, B. subtilis included (Novichkov et al., 2013). Machine learning can be used to identify prophage and horizontally transferred regions (Sirén et al., 2021). Finally, a database collecting metabolites (small molecules but also some macromolecules) that interact with transcription factors can be used to improve annotation by associating a particular transcription factor with its effectors (Koch et al., 2018). The corresponding knowledge has been used here as much as possible.
DEVELOPMENTS IN THE METABOLISM OF BACILLUS SUBTILIS
New information on a large number of B. subtilis metabolic functions has been steadily accumulating over the past 5 years. A list of genes with revised annotation and, in particular, newly assigned or reassigned function is presented in Table S1. As a teaser for further exploration of the re‐annotation of the genome sequence, we outline here an eclectic list of functions that may be of particular interest, grouped into coherent families such as metabolism of the cell's building blocks or nucleic acid‐handling functions. We then discuss how the new findings have allowed us to report new insight in the collective behaviour of B. subtilis communities.
Maintaining the homeostasis of proteinogenic amino acids
Nineteen amino acids and a secondary amine, proline, make the canonical complement of the protein building blocks. The amino acid biosynthesis routes follow similar rules and pathways in most organisms. They invariably involve intermediates that could mimic authentic proteinogenic counterparts, and evolution has had to find ways to prevent these intermediates from entering proteins. This can be achieved spatially, either by channelling, or by maintaining these intermediates at very low concentration. Another common solution is to have the pathway duplicated into a paralogous pathway that uses moonlighting activities of standard or paralogous enzymes (Chan et al., 2014; D'Ari & Casadesús, 1998), channelling chemically tagged intermediates for all the necessary steps until the tag of the labelled precursor is removed and a canonical metabolite is supplied for standard metabolism. To our knowledge, the exact contribution of the tag preference between different acylation groups (essentially, but not solely, acetylation and succinylation) has not yet been introduced in models of metabolism. The difference between authentic proteinogenic amino acids and counterparts that must be prevented from entering translation is well illustrated in methionine metabolism, where homoserine, a non‐proteinogenic amino acid, is acylated (Bastard et al., 2017). In B. subtilis the acylation step is an acetylation, whereas it is a succinylation in Escherichia coli. Bacillus subtilis homoserine O‐acetyltransferase MetAA (formerly MetA) differs from its E. coli succinyltransferase counterpart MetAS by a single amino acid residue (E112 to G112), sufficient to specify the acetylation vs. succinylation reaction, changing the enzyme's specificity (Zubieta et al., 2008). Lysine biosynthesis also involves acylation steps. In B. subtilis, YkuQ (now DapH) catalyses acetyl transfer, not succinyl transfer as does DapD in E. coli. It is a 2,3,4,5‐tetrahydropyridine‐2,6‐dicarboxylate N‐acetyltransferase. To emphasize the fact that the reaction involves a paralogous enzyme (not an authentic orthologue) we named the corresponding gene dapH, not dapD as it is still often named. This protection step is followed by the action of N‐acetyl‐l,l‐diaminopimelate aminotransferase DapX(PatA), the B. subtilis paralogue of E. coli DapC, acting on acetyl‐diaminopimelate instead of succinyl‐diaminopimelate.
Another feature of lysine synthesis has also been updated in the present annotation. It illustrates how, in addition to software‐dependent detection, the correct identification of translation start sites sometimes requires time‐consuming experimental validation and manual annotation (Meydan et al., 2021). For this reason, some genes are missing from databases. The lack of identification of some CDS start sites affected a few entries in the B. subtilis genome annotation. The lysC aspartokinase gene encodes a heterodimeric enzyme composed of two subunits, alpha and beta. The CDS of the alpha subunit (lysCA) begins with an upstream AUG codon, while the CDS of the beta subunit (lysCB) begins with a codon located downstream within the same frame (Kalinowski et al., 1991). A similar situation arises for a second aspartokinase, DapG: two CDSs, dapGA and dapGB code for the two subunits of the protein. Both are now properly annotated in the sequence.
Metabolic accidents are the rule, not the exception (Danchin & Sekowska, 2015; Lerma‐Ortiz et al., 2016) and this constantly generates non‐canonical amino acids in the cell. For example, sulfur‐containing compounds are vulnerable and susceptible to modification by reactive species (Niehaus et al., 2018). Cysteine, in particular, is often subject to accidents. It is also modified by alkylation transfer in normal processes such as DNA repair. During the removal of alkyl groups from DNA by alkyltransferases AdaAB and OgtA, cysteine residues of the enzymes become alkylated. These modified enzymes are subsequently degraded by proteolysis, generating alkylated cysteines. A metabolic pathway has evolved to allow the cells to cope with the presence of these potentially toxic cysteine analogues. Using an acetyl or succinyl tagging group they modify the altered cysteines. This prevents the alkylated cysteines from entering canonical pathways, diverting them instead to degradation or salvage pathways. Then, as discussed above, the metabolite tag is removed, returning a canonical compound to normal metabolism. Combined with inevitably error‐prone editing pathways (Rubio Gomez & Ibba, 2020), this repair pathway further contributes to translation accuracy.
This protection/de‐protection process has been demonstrated for clearance of S‐alkylcysteine metabolites. This involves the snaAtcyJKLMNcmoOIJribRsndAytnM operon (Chan et al., 2014; Hazra et al., 2022). Interestingly, a second pathway has been discovered, that mitigates fumarate‐induced accidents and also involves cysteine adducts. The operon scmKscmLyxeMNOscmPyxeQ encodes (2‐succino)cysteine transport and clearance functions. Notably, as does CmoJ in the previous pathway, ScmK(YxeK, CmoK) catalyses the oxygenation of N‐acetyl‐S‐(2‐succino)cysteine, resulting in its breakdown to oxaloacetate and N‐acetylcysteine (Matthews et al., 2022), which is deacetylated by ScmP(YxeP, SndB) to yield cysteine. The original mechanism of action of ScmK is likely to be similar to that of CmoJ where formation of a cysteine‐sulfoxide intermediate has been demonstrated (Hazra et al., 2022). More in line with chemistry‐motivated expectations, in organisms that do not encounter frequent high levels of dioxygen the corresponding function does not involve oxygen but is replaced by the action of a C‐S lyase (Hillmann et al., 2022). Indeed, the CmoJ/ScmK activity is quite remarkable. It shows that evolution has found a way to use dioxygen in a pathway restoring production of a thiol containing metabolite (in this case cysteine), opening interesting avenues for synthetic biology.
In the same way, salvage of sulfur‐containing methionine is important for the cell. MsrC (YtsP) is a widespread reductase experimentally identified in E. coli and other Enterobacteria. It is specific for reduction of the free form of Met‐(R)‐sulfoxide, complementing other methionine sulfoxide reductases (e.g. MsrA and MsrB) that act on proteins or peptides. MsrC belongs to the UPF0067 family and is similar to the Saccharomyces cerevisiae enzyme (Lin et al., 2007). This enzyme may be particularly important to protect B. subtilis against oxygen stress in the phylloplane. Finally, PepA(YuiE) is a leucyl aminopeptidase demonstrated in Pseudomonas aeruginosa and E. coli where its 3D structure is known. It is conserved in M. mycoides Syn3.0. Unexpectedly, the enzyme belongs to a recombination complex in E. coli. This enzyme is also involved in UV tolerance in a cyanobacterium where it exhibits cysteinyl‐glycinase activity. This involves glutathione turnover in E. coli and connects the enzyme to protection against and repair of UV damage (Weiss, Fang, et al., 2022). Because there is no glutathione in B. subtilis the details of an analogous pathway should be further investigated.
Proline and translation‐related metabolism
The 20th protein proteinogenic residue, proline, is an exception. Indeed, this metabolite is not an amino acid but a secondary amine (Pavlov et al., 2009). Its entry into polypeptide chains requires specific steps encoded in several genes that differ in different organisms (Rajkovic & Ibba, 2017). Among the key functions recently identified in B. subtilis is the post‐translational modification of the elongation factor EF‐P, essential for the translation of runs of proline residues and possibly involved in other as yet unknown activities. This factor is not strictly essential in B. subtilis but nevertheless critical for motility (Hummels & Kearns, 2020) and for optimal sporulation (Feaga et al., 2023). In all organisms, EF‐P, which mimics tRNA, is modified on a conserved lysine or arginine residue (lysine 32 in the case of B. subtilis), at a position analogous to the 3'OH‐tRNA aminoacylation site. Here, the modification is 5‐aminopentanol, the synthesis of which is still not fully deciphered (Witzky et al., 2018). Six proteins are involved in this process: EfpB(YnbB), EfpC(GsaB), EfpI(YmfI), EfpO(YaaO), EfpP(YfkA) and EfpG(YwlG). PhyloProfile revealed that the six corresponding genes have co‐evolved, validating this implication. Structural analyses showed that EF‐P can retain unique intermediate modifications upon inactivation of several of these genes. This suggests that the final steps of 5‐aminopentanol synthesis are likely assembled directly on EF‐P. EfpO, EfpP and EfpG are not strictly essential, indicating that they are involved in formation of a substrate that can also be synthesized by paralogous pathways, prior to lysine 32 modification. The expression of EfpB is strongly repressed in the presence of ClO2 in Listeria sp. Predicted to have carbon‐sulfur lyase activity, EfpB probably removes a fatty acid‐related substrate from an acyl carrier protein, forming hydroxypentanone in the first step of the modification. This group is subsequently dehydrated into pentanone. EfpC is a paralogue of HemL, glutamate‐1‐semialdehyde aminomutase that synthesizes 5‐aminolevulinic acid. It is thought to facilitate addition of the final amine group onto pentenone. Finally, EfpI(YmfI) is a NADPH‐dependent reductase that catalyses the reduction of EF‐P‐5 aminopentanone to EF‐P‐5‐aminopentanol (Witzky et al., 2018).
The presence of proline in polypeptides also affects their degradation because proline residues make variants of the canonical peptide bond. In the genome, the peptidase‐encoding papA gene is located in a context compatible with a proline‐related activity since it lies next to gene efpA. This is consistent with a clean‐up activity designed to degrade aborted translation peptides at proline residues. Indeed, the peptidases PapA (YqhT) and PapB (YkvY) are responsible for the hydrolysis of various types of Xaa‐Pro dipeptides and Xaa‐Pro‐Xaa tripeptides. Furthermore, PapA has co‐evolved with EfpA, EfpB, EfpG and EfpI. These co‐evolving functions also include activities characteristic of B. subtilis such as peptide deformylase DefB and nanoRNase NrnA. A counterpart exists as a thermostable enzyme in Pyrococcus furiosus (Ghosh et al., 1998). PapA is conserved in M. mycoides Syn3.0 and in Mycoplasma pneumoniae (Burgos et al., 2020). Among interesting phenotypes of papA, inactivation of the gene leads to very few spores. PapB is involved in osmoprotection as proline, besides its role in polypeptides, is also the major osmoprotectant, requiring efficacious salvage (Zaprasis et al., 2013). Bacillus subtilis uses proline as its major, newly synthesized osmostress‐protectant. Depending on the degree of the imposed osmotic stress, very large quantities of proline need to be produced, which requires a separate, osmotically stimulated proline biosynthetic pathway that is freed from the genetic and biochemical constrains used to control proline production for anabolic purposes (Hoffmann & Bremer, 2017).
Nucleotide metabolism: A new role for the metabolism of cytosine derivatives
Nucleotide metabolism was deciphered during the early days of modern biochemistry, long before genomic sequences were known. It may come as a surprise to see it present here. Previous knowledge highlighted an implicit hierarchy in the way nucleotides were involved in biomass construction and energy supply. In this view, purine nucleotides played the primary role in energy storage and management, with ATP as the key intermediate. Apart from an enigmatic involvement in phospholipid synthesis known for a long time (Jennings & Epand, 2020), a singular role for CTP was not expected. Many energy‐dependent factors are involved in targeting spatial locations in the cell essentially involving ATP or GTP (see details below). Yet CTP was shown to be required in a key process involving chromosome segregation (Osorio‐Valeriano et al., 2019). NocA(YyaA) and ParB(Spo0J) are needed to prevent Z ring assembly over the bacterial nucleoid and help fine tune the assembly of the Z ring at mid‐cell during the cell cycle (Yu et al., 2021). ParB localizes to both poles of the pre‐divisional cell following completion of DNA replication and before asymmetric septation, specifying its orientation and imposing directionality on its subsequent transport through the septum. The protein recognizes a 16‐bp sequence, the centromere region parS found in parB itself and 8–10 times in the origin‐proximal 20% region of the chromosome. CTP‐binding converts ParB dimers to DNA clamps, allowing unidimensional diffusion along the DNA, sliding over large distances from parS centromere sites where ParB is specifically loaded (Guo, Sattler, et al., 2022; Guo, Zhao, et al., 2022; Jalal et al., 2021). Another unexpected role of cytosine derivatives emerged when it was discovered that competence also involves cytosine. The ComGA complex associates to the cell poles, enabling genetic competence (Hahn et al., 2021) and the dCMP deaminase ComEB acts as a dynamic polar localization factor for ComGA within the competence machinery (Burghard‐Schrod et al., 2020).
The revelation that competence and chromosome segregation rely on a CTP‐dependent activity leads to a conjecture where CTP synthesis is the unique node that couples growth, energy and metabolism. This is supported by the observation that CTP synthase (PyrG) plays a key role in growth homeostasis in B. subtilis (Emami et al., 2020), a feature consistent with the conservation of the gene in the synthetic minimal genome of M. mycoides Syn3.0 and in the minimal genomes of cell growing in cytosine derivative‐containing media (Breuer et al., 2019; Danchin & Fang, 2016; Hutchison et al., 2016). Strikingly, this role was corroborated after a study of SARS‐CoV‐2 multiplication aimed at deciphering why the initial evolutionary trend of the virus led to the loss of cytosine residues in its RNA genome (Ou et al., 2020). CTP synthesis is now understood as the universal means for cells to cope with ‘non‐homothetic’ growth, by regulating the supply of metabolites generated in the cytoplasm. In a sphere, growth requires a supply of basic metabolites that grows as r 3 if r is the radius. Membranes grow as r 2 or slightly less but significantly more than r for other cell shapes while the genome grows as r. Therefore, if the supply of substrates is too large, a ‘force’ will manifest itself in various ways, creating wrinkles in the membranes, changing from a sphere to a cylinder, creating appendages, synthesizing intracellular membranes, designing new regulations, etc., and with an even stronger constraint on genome synthesis, a situation that benefits viral multiplication (Ou et al., 2020). The key role of CTP in regulating growth homeostasis was confirmed by the role of the antiviral protein viperin, which, remarkably, synthesizes an inhibitory analogue of CTP (Wein & Sorek, 2022), also identified as an antiphage resource in many bacterial species (Bernheim et al., 2020).
Evolution has shaped CTP synthase for this specific role, with its expression regulated by reiterative transcription at the promoter of the pyrG gene (Shin et al., 2020). A key feature of the enzyme is that, in all organisms where its structure has been explored, it makes filamentous structures, the cytoophidia, with important consequences for the spatial distribution and regulation of enzyme activity (Thangadurai et al., 2022). This parallels the CTP‐dependent overall metabolic activity controlling growth homeostasis which must act simultaneously on cytoplasmic, membrane and genome metabolism. In the cytoplasm, CTP is a precursor of RNA (messenger RNA and stable RNA). It is also required to complete the synthesis and repair of some tRNAs (via the addition of CCA to their 3′OH end). In B. subtilis this applies to 26 tRNA genes lacking a CCA‐3′OH terminus, including the single tRNACys gene (Campos Guillén et al., 2019). In addition to its role in coordinating synthesis of the bacterial proteome driven by ribosome synthesis (You et al., 2013), CTP is involved in membrane synthesis via the formation of CDP‐diglycerides and related metabolites (Centola et al., 2021), and in capsule biosynthesis (Li, Gale, et al., 2021; Litschko et al., 2021). This involvement in growth further extends to sporulation through the control of spore surface growth by glucose‐1‐phosphate cytidylyltransferase (Shuster et al., 2019). Finally, DNA synthesis relies on RNA turnover and phospholipid synthesis because the de novo synthesis of CTP does not include a step involving CDP, an essential precursor to deoxyribonucleotide synthesis (Danchin, 1997). This notable pervasive constraint should allow researchers to check the consistency of metabolic models (in particular, to verify that they are not severely hyperstatic), as the requirement for RNA turnover should be explicitly present in the models for them to function properly.
Several other structural features of the upstream pathway of pyrimidine metabolism underscore the unique role of cytosine. In particular, glutamine‐dependent Carbamoyl phosphate synthetase (PyrAA/PyrAB), Aspartate carbamoyltransferase (PyrB) and Dihydroorotase (PyrC), form a ubiquitous CAD complex. In Bacteria the complex is transient and still poorly characterized (Del Caño‐Ochoa et al., 2019). Expressed from the pyrEFDKpyrAApyrABpyrCBP operon in B. subtilis, CAD integrates nitrogen and energy metabolism. Nucleic acid turnover must also affect the fine‐tuning of non‐homothetic growth homeostasis in addition to de novo pyrimidine synthesis. Far from straightforward, the catabolism and salvage of cytosine‐containing nucleotides is unusual. Instead of using these metabolites, cytosine and cytidine are channelled into uracil and uridine, which must travel the entire anabolic pathway to regenerate CTP. Cytidine deaminase is an example. Remarkably, the translation of the corresponding gene, ccdA, is completed by a second translation with a programmed ribosomal −1 frameshift. It occurs at the frequency of 16% at the sequence CGA AAG, 9 bp upstream of the in‐frame stop codon. The frameshift event is activated by a RBS‐like sequence located 14 bp upstream of the shift site resulting in a gene product extended by 13 amino acid residues (Mejlhede et al., 1999). This second CDS is now included in the current annotation as gene cddB. It presumably encodes a regulatory subunit allowing interaction of the enzyme with protein complexes involved in nucleotide biosynthesis, possibly cytoophidia (Chang et al., 2022).
In parallel, cytidine kinase, which would allow cytidine to re‐enter pathways leading to CTP, is absent in B. subtilis as well as in cells with a streamlined genome (Breuer et al., 2019). Uridine kinase can substitute for this activity but with very poor activity on cytidine. PynN, a promiscuous pyrimidine/pyridine nucleotide nucleotidase contributes to recycling of pyrimidine (deoxy)nucleotide monophosphates (Ulrych et al., 2020). It is conserved in Firmicutes and some Mollicutes. Cytosine phosphoribosyltransferase is another major enzyme that could salvage cytosine, but does it exist? Related paralogous enzymes are widespread. No less than nine phosphoribosyltransferases are encoded in the genome of B. subtilis: for adenine, ATP, hypoxanthine/guanine, quinolinate, nicotinate, orotate, anthranilate, uracil and xanthine, respectively, AptA, HisG, HprT, NadC, PncB, PyrE, TrpD, UppA and XptA. Surprisingly, this is not the case for cytosine phosphoribosyltransferase, which is conspicuous by its absence throughout the tree of life (Ou et al., 2020). Yet it seems clear that from one or more of the existing enzymes, there must be a regular emergence of enzymes with this activity during the inexorable evolution of the genome. The fact that this particular form of phosphoribosyltransferase does not appear suggests that its presence is counter‐selected, reinforcing the idea that global metabolism is poised to force all cytosine metabolism through CTP synthase. With the information gathered here, we can justify this ubiquitous role as a means of coping with non‐homothetic growth.
Finally, a surprising observation may need to be experimentally validated as it may further confirm the growth‐related coordinating role of cytosine derivatives. CcpA is a protein involved in the glucose regulation of many genes, mediating carbon catabolite repression. Remarkably, its structure is similar to that of the CytR repressor from E. coli, that is known to bind cytidine. There is no counterpart of CcpA in E. coli which has another catabolite repression control system that uses the protein Crp bound to 3′,5′‐cyclic AMP. Surprisingly, it has been discovered that the cAMP‐Crp complex is also able to bind cytidine to modulate carbon repression (Lauritsen et al., 2021). We may therefore wonder whether this modulation of catabolite control by pyrimidines is not a general feature that would be implemented in cells via convergent evolution. While B. subtilis has been used to produce pyrimidines, not much is known about the metabolic constraints that limit production (Zhu et al., 2015). CcpA is known to bind a variety of metabolites but the possibility that it would bind pyrimidines has yet to be explored.
A sample of nucleic acid wielding functions
Our revised genome annotation also introduces several new annotations relevant to nucleic acid structure and function. The expression pattern of the frlBONMD operon allowed identification of RulR(YlxR, YmxB) as a key structural determinant of the 3D structure of RNAs (Osipiuk et al., 2001). RulR associates with the RNA K‐turns recognition protein RulQ(YlxQ, YmxC) to modulate gene expression via stabilization of this RNA structure (Ogura et al., 2020). One of the major ‘unknown unknowns’ of M. mycoides Syn3.0 required for the stabilization of 3D RNA structures (Danchin & Fang, 2016), RulR is regulated by glucose‐sensitive arginine phosphorylation/dephosphorylation involving the phosphatase PrpB(YwlE) (Ogura, 2020). RulR controls the bimodal expression of the promoter of operon frlBONMD allowing fructoselysine utilization. Encoded in the frlBONMD operon, the plant‐related enzyme fructosamine‐6‐P deglycase FrlB(YurP) belongs to the catabolic pathway of the glycation product fructose‐ε‐lysine that undergoes ATP‐dependent phosphorylation by a specific kinase (FrlD), followed by the conversion of fructoselysine 6‐phosphate into glucose 6‐phosphate and lysine. Detailed analysis of its activity suggests that it acts on alpha‐glycated amino acids rather than on ε‐glycated lysine (Wiame et al., 2005). Its expression is regulated by CodY and repressed by root exudate. RulS (YbaB, YbxF), also known as alternative ribosomal protein L7A, is another recently discovered specific RNA binding factor that recognizes K‐turns. It is often present in Firmicutes and conserved in Archaea and Eukarya. In B. subtilis the protein stabilizes riboswitches (Oshima et al., 2018; Skeparnias & Zhang, 2021).
Somewhat similar to E. coli ThiI, TrmG is a persulfide ATP pyrophosphatase involved in tRNA 4‐thiouridine modification. This protein is present in M. mycoides but does not belong to the minimal gene set required for independent life (Hutchison et al., 2016). Most, if not all, protein functions involved in the key anticodon nucleotide N34 of tRNA, as well as residue N37 contiguous to the anticodon have been identified since the previous annotation update (Table 1).
TABLE 1.
Updated annotation of the enzyme functions for modification of bases 34 and 37 of tRNAs.
| Label | Gene | Synonyms | Product | EC number |
|---|---|---|---|---|
| BSU22780 | folEA | mtrA | GTP cyclohydrolase I first step of preQ1 biosynthesis | 3.5.4.16 |
| BSU03340 | folEB | yciA, folE2 | alternate GTP cyclohydrolase I first step of preQ1 biosynthesis; active under conditions of severe Zn limitation | 3.5.4.16 |
| BSU27350 | houN | yrrN | enzyme subunit involved in tRNA 5‐methoxyuridine synthesis for tRNA U34 modification | 3.4.‐.‐ |
| BSU27340 | houQ | yrrO | enzyme subunit for synthesis of tRNA 5‐methoxyuridine for tRNA U34 modification | 3.4.‐.‐ |
| BSU27510 | iscSA | yrvO, iscS | cysteine desulfurase involved in U34 tRNA thiolation | 2.8.1.7 |
| BSU17010 | miaB | ymcB, tmtA | tRNA‐2‐methylthio‐N(6)‐dimethylallyladenosine synthase (isopentenyl‐adenosine A37 tRNA methylthiolase) | 2.8.4.3 |
| BSU41020 | mnmE | thdF, trmE | tRNA modification GTPase and tRNA‐U34 5‐formylation enzyme | 3.6.‐.‐ |
| BSU41010 | mnmG | gidA, trmF | tRNA uridine 5‐carboxymethylaminomethyl modification enzyme | _ |
| BSU25430 | mtaB | yqeV, rimO, tmtB | tRNA N(6)‐threonylcarbamoyladenosine (t(6)A) methylthiotransferase | 2.8.4.5 |
| BSU00530 | pthA | spoVC, pth | peptidyl‐tRNA hydrolase | 3.1.1.29 |
| BSU27720 | queA | _ | S‐adenosylmethionine tRNA ribosyltransferase‐isomerase | 2.4.99.17 |
| BSU13720 | queC | ykvJ | 7‐cyano‐7‐deazaguanine (preQ0) synthase | 6.3.4.20 |
| BSU13730 | queD | ykvK | 6‐carboxy‐5,6,7,8‐tetrahydropterin synthase; queuosine biosynthesis | 4.1.2.50 |
| BSU13730 | queE | ykvL | 7‐carboxy‐7‐deazaguanine synthase | 4.3.99.3 |
| BSU13750 | queF | ykvM | NADPH‐dependent 7‐cyano‐7‐deazaguanine reductase (moonlighting hydratase) | 1.7.1.13 |
| BSU08910 | queG | ygaP, yhbA | epoxyqueuosine reductase | 1.17.99.6 |
| BSU15750 | rlmN | yloN | 23S rRNA m2A2503 methyltransferase and tRNA A37 C2 methyltransferase | 2.1.1.192 |
| BSU00180 | tadA | yaaJ | tRNA specific adenosine A34 deaminase | 3.5.4.33 |
| BSU27540 | tcdA | yrvM, csdL | tRNA threonylcarbamoyladenosine dehydratase (t(6)A37 dehydratase) | 6.1.‐.‐ |
| BSU27710 | tgt | _ | tRNA‐guanine transglycosylase | 2.4.2.29 |
| BSU00670 | tilS | yacA, mesJ | tRNA(ile2) lysidine synthetase | 6.3.4.19 |
| BSU15060 | tmcAL | ylbM | N4‐acetylcytidine tRNA C34 acetylase (acetyladenylate synthase) | 2.3.1.193 |
| BSU02330 | trhO | ybfQ | tRNA uridine(34) hydroxylase | 1.14.‐.‐ |
| BSU29900 | trmB | ytmQ | tRNA G46 (guanine‐N(7)‐)‐methyltransferase | 2.1.1.33 |
| BSU16030 | trmD | _ | tRNA(m1G37)methyltransferase | 2.1.1.228 |
| BSU29580 | trmG | ytbJ, thiI, trmI | persulfide ATP pyrophosphatase involved in tRNA 4‐thiouridine modification | 2.8.1.4 |
| BSU08930 | trmL | cspR, ygaR | tRNA (cytidine(34)‐2’‐O)‐methyltransferase; tRNA(Leu) methylation of cmnm5Um | 2.1.1.207 |
| BSU00340 | trmNF | yabB, trmN, trmN6 | tRNA1(Val) (adenine(37)‐N6)‐methyltransferase | 2.1.1.223 |
| BSU27360 | trmR | yrrM | O‐methyltransferase for modification of tRNA U34 into 5‐methoxyuridine (mo5U) | 2.1.1.‐ |
| BSU05920 | tsaB | ydiC | tRNA(NNU) t(6)A37 threonylcarbamoyladenosine modification | _ |
| BSU36950 | tsaC | tamT, ywlC, ipc‐29d, rimN | tRNA(NNU) t(6)A37 threonylcarbamoyladenosine modification; threonine‐dependent ADP‐forming ATPase | 2.7.7.87 |
| BSU05940 | tsaD | gcp, ydiE | tRNA(NNU) t(6)A37 threonylcarbamoyladenosine modification; glycation binding protein | 2.3.1.234 |
| BSU05910 | tsaE | ydiB | tRNA(NNU) t(6)A37 threonylcarbamoyladenosine modification; ADP binding protein | _ |
EtrM(YhaM) is an omnipresent promiscuous 3′‐to‐5′ exonuclease acting on various substrates, but mainly RNAs. It is a nonspecific enzyme that targets the majority of transcript ends generated either by transcription termination or by endonucleolytic cleavage and trims a few nucleotides. This activity is also involved in 23S rRNA maturation (Bechhofer & Deutscher, 2019). Disruption of the gene results in a cold shock phenotype in Streptococcus sp. (Lécrivain et al., 2018). The protein is highly conserved and present in the streamlined genome of M. mycoides Syn3.0 (Danchin & Fang, 2016).
MadA(YerA) is a N6‐methyladenosine deaminase that metabolizes the nucleoside N6‐methyladenosine involved in epigenetic regulation of bacterial metabolism. The gene is located in an island with sporulation‐related genes of poorly identified function (Shi et al., 2020). The protein co‐evolves with BofC, ‘bypass of forespore C’, an intercompartment signalling factor expressed in the forespore. This modification, generating inosine and methylamine, protects RNA and DNA against misincorporation of methyladenine. The enzyme has a binuclear metal centre coordinated by histidines, and residues Phe91 and Gln150 play a crucial role in catalysis (Jiang, Wang, et al., 2021). Finally, present in defective prophage 6 island, AoxN(YobN) is an amine oxidase with demonstrated activity on histamine, tyramine, putrescine and cadaverine. It co‐evolves with a variety of catalytic enzymes including N6‐methyladenosine deaminase MadA and spore coat proteins, but it is not expressed in spores (Pištěková et al., 2022).
A sample of metabolic functions identified since the last annotation update of the B. subtilis genome sequence
In B. subtilis, an organism that does not code for selenocysteine, two non‐canonical menaquinone‐linked formate dehydrogenases, FdhED(YrhED) and ForCE(YjgCE), are similar to selenocysteine‐containing formate dehydrogenases. The transcription of the forCE operon depends on sigma(B). It is upregulated in swarming conditions, in the presence of high salt or ethanol. In contrast, the transcription of fdhED is upregulated in germinating spores and in exponential growth. It is repressed in oxygen‐limited conditions by the ResED two‐component system. The enzymes belong to a new family of enzymes that reversibly catalyse the oxidation of formate to CO2. The ForC subunit hosts the molybdenum/tungsten‐bis‐pyranopterin guanine dinucleotide cofactor at the formate oxidation site as well as five [Fe‐S] clusters in the same polypeptide. ForE is the partner subunit that couples formate oxidation to quinone reduction and the menaquinone‐7 reduction site is likely located at the ForCE interface (Arias‐Cartín et al., 2022).
PadR/PadC constitute a sensor/catabolism system used for the identification and catabolism of a variety of aromatic compounds. PadF(YveF) and PadG(YveG) have a positive impact on catabolism, assisting the release of PadC by PadR repression. The understanding of this system has been used in metabolic engineering. At this point, the experimental data are suggestive of padF and padG belonging to a single gene that has been disrupted by mutation (Jiang, Li, et al., 2021).
Absent from E. coli but present in many bacterial clades, PncA(YueJ) deaminates nicotinamide to nicotinic acid. It is important for the salvage of nicotinamide coenzymes and equilibrating the concentration of the different precursors of NAD. The 3D structure of the enzyme has been established in B. subtilis (Shang et al., 2018).
RbsD is often misannotated as a transporter component. The protein belongs to the mutarotase family, and its activity has been demonstrated in E. coli. The spontaneous interconversion of the beta‐pyran and beta‐furan forms of d‐ribose is slow. As in the case of another catalyst designed to accelerate a spontaneous reaction (e.g. hydrolysis of 6‐phosphogluconolactone catalysed by Pgl in the Entner‐Doudoroff pathway), RbsD is critical for facilitating rapid ribose catabolism (Rogalski et al., 2021). It also catalyses the conversion between beta‐allofuranose and beta‐allopyranose. Excessive expression of the enzyme was shown to result in methylglyoxal accumulation (Kim et al., 2004).
FpsC(YvkC) acts in ATP‐dependent phosphotransferase acting on polyphenolic secondary metabolites found in plants (flavonoids) to generate the corresponding monophosphates, AMP and orthophosphate, suggesting dissipation of energy in a process of discrimination (see below). It is a promiscuous phosphotransferase that efficiently phosphorylates structurally diverse flavonoids, including isoflavones, flavones, flavonols, flavanones and flavonolignans. The phosphorylation mainly occurs on the hydroxyl group at C‐7 of A‐ring or C‐4′ of B‐ring in flavonoid skeleton. Interestingly, for synthetic biology purposes, FpsC is regio‐selective for the ortho‐3′,4′‐dihydroxy moiety of catechol‐containing structures, such as luteolin and quercetin, to produce phosphate conjugates at C‐4′ or C‐3′ of B‐ring (Hsu et al., 2023). Another enzyme metabolizing quercetin, the dioxygenase QdoI(YxaG), cleaves two carbon–carbon bonds, generating carbon monoxide (Bowater et al., 2004), a plant gasotransmitter involved in potassium‐related stress signalling (Lana et al., 2021).
New insights into cell envelope synthesis
The B. subtilis cell envelope contains the cell membrane and a cell wall containing peptidoglycan and teichoic acid polymers (both lipid‐teichoic acid and wall‐teichoic acid). External layers, comprised of extracellular polysaccharides, are important in biofilm formation, as considered further below. The process of peptidoglycan biosynthesis is well understood in B. subtilis and enzymes have been assigned to all major steps in the process. Some of the recently assigned functions include MurJ as the major lipid II flippase with the sigma M‐activated Amj protein as a stress‐inducible flippase (Meeske et al., 2015), RodA and FtsW as transglycosylases (Meeske et al., 2016), and most recently UptA(YngC) as an undecaprenylphosphate retrograde transporter. The UptA protein is not the only mechanism contributing to undecaprenylphosphate recycling since mutants are relatively unaffected in growth (Roney & Rudner, 2022). Redundancy is common in cell envelope synthesis functions, with multiple proteins catalysing many of the key steps including lipid II flippases, transglycosylases and transpeptidasese, lipoteichoic acid synthases, and numerous autolytic enzymes. The recently identified operon controlled by the YclJK two component system gtrA(yngA)angB(yngB)uptA(yngC) is involved in decoration of teichoic acid, a major component of biofilm matrix, by glucose in oxygen‐limited conditions depending on the presence of undecaprenyl phosphate (Wu, Rismondo, et al., 2021).
GlmR(YvcK) has an important role in metabolism originally revealed in mutant screens that displayed mutant morphological defects suggestive of a role in cell wall synthesis. GlmR is essential for growth on Krebs cycle intermediates and substrates of the pentose phosphate pathway (Foulquier et al., 2020). GlmR is now understood to function as a key regulator of GlmS, the branchpoint enzyme that diverts carbon from the central metabolite fructose‐6‐phosphate, which is limiting in cells growing on gluconeogenic carbon sources, into peptidoglycan biosynthesis (Kawai et al., 2019; Patel et al., 2018). The consequences of a GlmR defect can be overcome providing cells with N‐acetyl‐glucosamine (GlcNAc), even under conditions where GlcNAc cannot re‐enter central metabolism and serve as a carbon source for growth. The protein is required for growth on Krebs cycle intermediates and substrates of the pentose phosphate pathway. GlmR functions as a direct activator of GlmS activity, and this function is antagonized by the downstream metabolite UDP‐GlcNAc in a process involving complex formation with YvcJ (Foulquier et al., 2020). In addition, GlmR is phosphorylated on threonine 304 by the protein kinase PrkC and phosphorylated GlmR is dephosphorylated by the cognate phosphatase PrpC, although the function of this modification is not yet understood. In Listeria monocytogenes GlmR is also required for cell wall homeostasis, but functions at a different step, as an accessory N‐acetyl‐glucosamine uridyltransferase (Pensinger et al., 2021).
In addition to these new insights into primary metabolic functions, there is an increasing appreciation that enzymes may have secondary activities due to promiscuous activity with compounds (paralogues) similar to their canonical substrates. This type of ‘underground metabolism’ (aka ‘paralogous metabolism’) has been described for phosphoglucomutase (PgcA), the enzyme that catalyses the interconversion of glucose 6‐phosphate (Glc‐6‐P) and glucose 1‐phosphate (Glc‐1‐P), a precursor of UDP‐glucose (UDP‐Glc). PgcA has a secondary activity as a phosphoglucosamine mutase (Patel et al., 2019). The primary phosphoglucosamine mutase (GlmM) is critical for the synthesis of aminosugars in support of peptidoglycan synthesis. Like GlmM, PgcA can also conver glucosamine 6‐phosphate (GlcN‐6‐P) to glucosamine 1‐phosphate (GlcN‐1‐P). Furthermore, this activity can be enhanced by a gain‐of‐function mutation selected as a suppressor for the loss of GlmR, which activates the upstream enzyme, GlmS, as noted above (Patel et al., 2019).
As a second example, glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) catalyses another paralogous reaction using erythrose 4‐phosphate as a substrate, an intermediate of the pentose phosphate pathway. The resulting 4‐phosphoerythronate (4PE) is toxic and in mammals is degraded by a phosphoglycolate phosphatase (PGP) and in yeast by the Pho13 phosphatase (Collard et al., 2016). Remarkably, in B. subtilis this product of paralogous metabolism is detoxified by CpgA, a GTPase with a canonical role as a late‐stage assembly factor for the 30S ribosomal subunit. In the absence of CpgA, 4PE accumulates and inhibits 6‐phosphogluconate dehydrogenase (GndA). This impedes the pentose phosphate pathway and leads to accumulation of 6‐phosphogluconate, which is in turn a potent inhibitor of phosphoglucose isomerase (Pgi). The resultant shutdown of PPP and glycolysis leads to metabolic gridlock and severe growth impairment in media containing carbon sources that feed into upper glycolysis of the pentose phosphate pathway (Sachla & Helmann, 2019).
Updated manual curation expands the metabolic network of Bacillus subtilis
Reconstruction of genome‐scale metabolic networks highly relies on the quality of available annotation. Metabolic models are knowledge‐based networks that contain all known metabolic reactions and their associated genes of a target organism (Orth et al., 2010). These models enable researchers to predict the growth features of the organism by simulating the metabolic fluxes in the network as well as the contextualization of multi‐omics data (Passi et al., 2021). Model simulations include individual and combinations of environmental and genetic conditions. The improved genome annotation of B. subtilis will facilitate the development of manually curated networks. The first metabolic model for B. subtilis was created in 2007 (Oh et al., 2007). It contained 844 genes, 990 metabolites, and 1250 reactions. Currently, nine metabolic models of B. subtilis are available. Three of these models have been automatically reconstructed and the rest have been manually curated. Despite the exhaustive efforts to keep a metabolic network of B. subtilis updated the most comprehensive model contains only 1162 genes (Machado et al., 2018), limiting their simulation capabilities. Figure 2 shows information about the newly annotated activities in the genome that are contained in the available metabolic networks and metabolism and expression model of B. subtilis.
FIGURE 2.
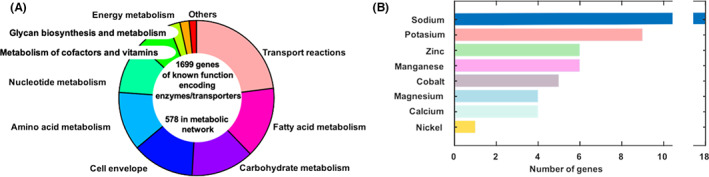
Breakdown of newly annotated metabolic functions in the genome of Bacillus subtilis. (A) Using the available metabolic models we determined the gene pathways associations for 40% of the new functions (total 1699, comprising the genes listed in Table S1, and a further list of 578 genes in metabolic networks characterized from the present annotation). (B) Out of the new annotated function used in metabolic models we found that 53 genes are explicitly associated with metals for transport and metabolism.
COLLECTIVE BEHAVIOUR OF BACILLUS SUBTILIS CELLS: SWIMMING, SWARMING AND BIOFILM FORMATION
Bacillus subtilis cells are motile, involved in swimming and swarming. The biomechanical machinery that allows this behaviour is dependent on flagella, appendages that are present in many bacterial species. The genes responsible for flagella synthesis and activity have been accurately annotated for a long time (77 genes in the present annotation). Recent work has better characterized the single set of flagellar rotor proteins that interact with two distinct stator‐force generators that energize rotation, the H+‐coupled MotAB complex (Sanchez et al., 2022) and the Na+‐coupled MotPS complex (Terahara et al., 2020). The stator elements consisting of (MotA)4(MotB)2 complexes, are anchored to the cell wall, extend through the cell membrane, and interact with FliG in the cytoplasmic C‐ring rotor of the flagellum. A regulator acting as a molecular clutch, MotI(DgrA, YpfA), disengages MotA, binds cyclic di‐GMP and controls swarming (Kunz et al., 2020). The clutch is a simple, rapid and potentially reversible form of motility control. MotP(YtxD) MotS(YtxE) supports Na+‐stimulated motility, chemotaxis on soft agar surfaces and biofilm formation. MotP binds to the peptidoglycan layer through the peptidoglycan‐binding domain of MotS to act as the stator functions efficiently at elevated viscosity in the presence of 200 mM NaCl (Naganawa & Ito, 2020). MotPS also supports motility in soft agar plugs immersed in liquid. MotPS does not, however, support surfactin‐dependent swarming on higher concentration agar surfaces.
SmiA(YvzG), swarming motility inhibitor A, is an adaptor protein for the energy‐dependent LonA discriminator‐mediated degradation of SwrAA, the master activator of flagellar biosynthesis that controls the number of flagella in liquid environments and the assembly of flagella in response to cell contact with solid surfaces (Ermoli et al., 2021). In strain 168 the swrAA gene is affected by a frameshift due to a single insertion of an adenine in the codon for Tyr‐12 is therefore likely to be non‐functional. A functional counterpart of this protein is conserved in wild type B. subtilis strains as well as in a narrow panel of related species such as Bacillus pumilus. The swrAA gene appears to result from a recent horizontal gene transfer or de novo creation. When intact, stoichiometric excess of SmiA causes substrate‐independent inhibition of LonA‐dependent turnover (Olney et al., 2022).
Biofilms are sessile or slowly moving microbial communities enclosed in an extracellular matrix (ECM) of exported biopolymers. In the present annotation, 216 gene entries include ‘biofilm’, showing that the process of forming sessile cultures of B. subtilis is an important feature of its lifestyle (Table S2). The function of several of these genes has been recently uncovered. PdeB(YmdB), a phosphodiesterase that acts against 2′,3′ (and also 3′,5′ cyclic nucleotide monophosphates in vitro) is a bistable switch that controls the decision between swimming and biofilm formation (Ryan‐Payseur & Freitag, 2018; Zhang et al., 2020). A pdeB mutant is unable to form biofilms and inactivation of the gene leads to haemolysis in L. monocytogenes. This defect is suppressed by the deletion of the sinR gene encoding the master regulator of biofilm formation. Deletion of pdeB affects the expression of more than 800 genes. The mutants displayed unordered developmental colony patterns. Reflecting the importance of the phosphodiesterase function beyond biofilms, a counterpart is present in M. mycoides Syn3.0, required for management of degradation products of RNA which are often cyclic phosphodiesters (Danchin & Fang, 2016).
Biofilms attach to many surface types and to the interface formed by other bacteria or fungi. They improve resilience of the cells when compared to cells developing in planktonic conditions. This is due both to the physico‐chemical structure of the biofilm which plays a protective role and to its heterogeneity, such as the division of the biofilm population into functionally distinct cell subgroups. A 15 gene multipartite operon, epsABCDEFGHIJKLMNO, is required for biosynthesis of a matrix exopolysaccharide that binds chains of cells together in bundles (Qin et al., 2022). The operon encodes an inhibitor of motility, EpsE. EpsE arrests flagellar rotation in a manner similar to that of a clutch, by disengaging motor force‐generating elements in cells embedded in the ECM (Guttenplan et al., 2010). Through the action of another protein encoded in the operon, EpsG, the polysaccharide further improves the matrix structure via binding extracellular DNA (Peng et al., 2020). The expression of the operon is regulated by SlrR, which in combination with the anti‐repressor SlrA represses sigma(D)‐dependent flagellar genes and activates the eps and tapA operons. A complex between the SlrR antagonist quorum sensing regulator SinR and its anti‐regulator SinI regulates transcription of operons involved in matrix production (epsA‐O), synthesis of the amyloid‐like protein TasA (Verma et al., 2020). Interestingly the process of biofilm formation is regulated by the growth rate (Chen et al., 2023), and the derepression of the BslA(YuaB) hydrophobin fosters biofilm formation (Charlton et al., 2022; Kim et al., 2021). In this mechanosensitive process an epigenetic switch couples biofilm formation with the inhibition of motility through SlrR, where the SinR/SlrR heterodimer represses autolysin and motility genes. The process further requires the RemA regulator of extracellular matrix genes (Hoffmann et al., 2021).
Besides containing polymers, biofilms involve a variety of metabolites for their construction and turnover. Several transporters affecting biofilms have been identified. They are the target of antibiotics synthesized by competitor or symbiotic species and are involved in the formation of multispecies biofilms. IcaC(YfiQ), an homologue of O‐acetyl transferase WecH(YiaH) of E. coli important for enterobacterial common antigen metabolism (Rai & Mitchell, 2020) and a paralogue of IcaC from Staphylococcus sp., is an acetyl‐glucosamine metabolite exporter acetylase component for poly‐beta‐1,6‐N‐acetyl‐d‐glucosamine metabolism involved in biofilm formation. The similarity with WecH makes IcaC a likely component of the machinery exporting important biofilm metabolites (Pearson et al., 2020). Overexpression of operon ytrBCDEF encoding an ABC transporter involved in transport of an unidentified metabolite also affects biofilms in B. subtilis (Benda et al., 2021). In the same way, permease BifN(YfiN, LnrN) alters biofilm morphology, possibly via export of important biofilm precursors (Stubbendieck & Straight, 2017).
Biofilm management is also influenced by the process of translation. As a case in point, serine starvation causes ribosomes to pause on specific serine codons, leading to a decrease in the translation rate of sinR and ultimately triggers biofilm induction. Remarkably, this process reveals a previously unacknowledged role of the codon usage bias. The level of all five serine tRNA iso‐acceptors is decreased in stationary phase compared with exponential phase but the level of the three iso‐acceptors recognizing UCN serine codons is reduced to a much greater extent than the two that recognize AGC and AGU serine codons (Greenwich et al., 2019). Another specific involvement of the translation process is revealed by inactivation of the rpsU gene, which affects motility and biofilm formation (Takada et al., 2014). In line with a role of codon usage, RpsU (ribosomal protein S21) co‐evolves with functions affecting modification of the N34 and N37 positions of the anticodon in tRNAs. Counterparts of the protein were recruited and evolved by bacteriophages to interfere with the host translation machinery (Al‐Shayeb et al., 2020). Finally, RpsU is a modulator of translation initiation promoting plant growth in B. velezensis and its synthesis stimulated by root exudate (Clarke et al., 2022).
Biofilms often allow interactions between different microbial species as well as a commensal or symbiotic host. Metabolites produced by the host play an important role in their formation, development and stability. L‐lactate is produced in roots as an electron sink for the plant during hypoxia. It is also made in leaves in response to wounding and it may be important in plant defence against pathogens (Maurino & Engqvist, 2015). The operon lutABC encodes three iron–sulfur‐containing subunits of an oxidase required for l‐lactate utilization and biofilm formation (Chai et al., 2009). Its expression is activated by the sulfur‐sensitive regulator CymR and regulated by FbpB and FsrA (Pi & Helmann, 2017). The lactate utilization regulator LutR acts not only on the lactate oxidase operon lutABC, but also on the lactate permease gene lutP. In E. coli, the YkgEFG complex is homologous to the B. subtilis LutABC oxidative lactate catabolism electron chain (Augustiniene & Malys, 2022). However, E. coli does not appear to metabolize lactate and its expression is regulated in this organism by redox conditions and regulator HxpR(YieP). It responds to chlorine and other oxidants and is possibly involved in the metabolism of 3‐hydroxypropionate (Nguyen‐Vo et al., 2020). This suggests that the exact function of operon lutABC should be revisited in B. subtlis.
Remote induction of biofilm generation is triggered by volatiles. Twenty‐six strains of B. subtilis isolated from different habitats were found to produce more than 200 volatile secondary metabolites between them: alcohols, aldehydes, aromatics, esters, hydrocarbons, ketones, nitrogen‐ and sulfur‐containing compounds (Kai, 2020). Strain 168 uses acetic acid as a volatile signal to coordinate the timing of biofilm formation within physically separated cells. VbfBA(YwcBA) is a Na+‐dependent acetate symporter involved in the management of this volatile signal. It has been proposed that the transcription factor CidR binds acetic acid and activates the murein hydrolase holin‐antiholin factors CidA‐CidB (Sadykov et al., 2019), possibly releasing DNA as a component of the biofilm matrix after cell death (Chen et al., 2015).
In Enterobacteriaceae and some Firmicutes, cyclic di‐GMP is involved in biofilm management (Randall et al., 2022). However, in B. subtilis the equivalent role of this signalling molecule remains elusive (Bange & Bedrunka, 2020). In contrast, the role of cyclic di‐AMP is well established. The organism secretes this molecule and its transporters impact biofilm formation and plant root colonization. Several putative exporters of cyclic di‐AMP, including CdaE(YcnB) and YhcA have been characterized. CdaE is highly induced when translation slows down. A double deletion mutant of genes cdaE and yhcA displays a significant decrease in the level of secreted cyclic di‐AMP and this impacts biofilm formation and plant root colonization. Only cyclic di‐AMP secretion (and not its synthesis) is impacted in this strain, indicating that these transporters contribute to export of the molecule (Townsley et al., 2018). Additional studies are needed to determine if these or other, yet‐to‐be‐identified receptors are important for connecting cyclic di‐AMP signalling to the biofilm regulatory network in B. subtilis. A further role of the molecule will be discussed in the section investigating new annotations involving ions.
The development of biofilms often involves microbes of different species. Leu‐Xaa‐Gly (LXG) toxins (proteins with N‐terminal LXG domains), together with their type VII secretion system (T7SS) play a role in competition between bacterial species (Klein et al., 2022; Spencer & Doran, 2022). The B. subtilis 168 genome hosts six toxins of this family, DtxF(YeeF), DtxL(YobL), DtxI(YokI), DtxG(YqcG), RtxN(YwqJ), RtxD(YxiD). Their respective genes are associated with downstream genes encoding putative antitoxins, which, in contrast to the toxins do not share significant similarity (Brantl & Müller, 2019). Three of the corresponding genes, dtxL(yobL), dtxI(yokI) and dtxG(yqcG), are located in the prophage‐like element 6, the SPβ prophage, and the phage‐derived sigK‐intervening element (skin), respectively. Gene dtxF belongs to a horizontal gene transfer island. Induction of DtxF, DtxL, DtxI, DtxG and RtxD caused a large decrease in chromosomal DNA (Kobayashi, 2021) suggesting that these toxins could be DNases acting on the DNA molecules exported in the biofilm matrix. However, they are also found to be RNases when expressed in E. coli. Finally, beside a RNase‐like domain, with an activity which has not been established, RtxN has an unknown deaminase‐like motif which may act on small molecules. These toxins are exported using a T7SS encoded in the operon tgsEDCBA(yukEDCBA)‐tgsFG(yueBC). TgsE(YukE) apparently binds to relevant LXG toxins and promotes their export through the T7SS via contacts with the TgsC(YukC) pseudokinase subunit (Tassinari et al., 2022 ).
THE CRUCIAL ROLE OF METALS
In cells, monovalent ions play a specific role that differs from that of divalent ions. The general background where B. subtilis thrives is highly variable, with alternating episodes of dry and wet conditions. This is reflected by a highly variable concentration of sodium and potassium, with often conditions of high salinity. Divalent metals are likely to be linked to the plant hosts as well as to the nature of the soil surrounding roots. This is reflected in the metal preferences displayed by B. subtilis as compared to other bacterial species. Figure 2B shows the breakdown of functions associated with eight different metals in the metabolic models constructed from the present annotation.
Sodium and potassium
A number of functions encoded in the genome are sensitive to the presence of sodium (73 genes with ‘salinity’ or ‘sodium’ in the annotation, Table S3). This is not unexpected as this ion is omnipresent in the environment. While B. subtilis is not known to require sodium for growth, absence of this ion can impair function of the Tat export system contributing the oxidative stress (Prajapati et al., 2021). Conversely, B. subtilis can accommodate a significant level of the cation in the environment without having its growth impaired (Nguyen & Kumar, 2022), at least as long its major osmostress response systems, the synthesis of the compatible solute proline or the uptake of osmostress‐protectants (e.g. glycine betaine) are intact (Hoffmann & Bremer, 2017). Bacillus subtilis keeps its internal sodium concentration very low and uses several sodium extrusion systems to remove excess sodium of this cytotoxic ion (Górecki et al., 2014). In contrast to sodium, potassium is essential for life (Danchin & Nikel, 2019). Several novel features of the genome involving potassium have been uncovered since our last annotation release at the INSDC. This is particularly well illustrated by the strict requirement for the presence of this cation to allow translation to proceed. The activity of two essential factors required for proper folding and assembly of the ribosome, EngD and RbgA are strictly dependent on the presence of potassium (Seffouh et al., 2019), and potassium is also thought to be a major counterion for ribosome assembly (Rozov et al., 2019).
Open questions related to potassium and cyclic di‐AMP have now been answered. Protein YdaO (recently renamed KimA) acts as a potassium transporter that binds cyclic di‐AMP (Gundlach et al., 2019). This property is also shared by the K+/H+ antiporter KhtT, the potassium exporter CpaA (YjbQ), the osmoprotectant transporter subunit OpuCA, the primary Mg2+ importer MgtE, and DarB(YkuL), a cyclic di‐AMP receptor and pyruvate carboxylase regulator that controls (p)ppGpp synthesis under conditions of potassium starvation (Krüger et al., 2021, 2022). KimA(YdaO) is conserved in M. pneumoniae and co‐evolves with RNA binding and RNA metabolism proteins. A further involvement of cyclic di‐AMP in the control of potassium availability is seen in the presence of the swdA cyclic di‐AMP‐binding riboswitch upstream of operon ktrAB, coding for a high affinity proton‐driven potassium pump (He et al., 2020). KtrB forms a dimer in the membrane, and the soluble regulatory subunit KtrA attaches to the cytoplasmic side of the dimer as an octameric ring conferring Na+ and ATP dependency to the system. Unlike most K+ channels, KtrB lacks the highly conserved T(X)GYG selectivity filter sequence. Only a single glycine residue is found in each pore loop, which raises the question of how selective the ion channel is (Mikušević et al., 2019).
Magnesium and calcium
The cyclic di‐AMP concentration fluctuates in coordination with both the Mg2+ and K+ levels, consistent with the proposal that this messenger may contribute to the cellular response to osmotic stress. Import of K+ upon osmotic upshift is correlated with Mg2+ efflux, and Mg2+ reimport is critical for adaptation. The transient growth inhibition resulting from hyperosmotic stress is coincident with loss of Mg2+ and a decrease in protein translation. Conversely, the reimport of Mg2+ is a limiting factor during resumption of growth (Wendel et al., 2022). This places magnesium transport at the crux of living processes. The expression of the main magnesium transporter, MgtE, which discriminates efficiently against calcium (Teng et al., 2022), is controlled by a magnesium‐dependent riboswitch. The magnesium cation has a critical role in many cell processes, not only as a cofactor for enzyme catalysis and macromolecular biosyntheses but also in coordinating growth. For example, cell length decreases proportionally with increasing Mg2+ from 0.2 to 4.0 mM, with little or no detectable change observed in intracellular Mg2+ (Guo & Herman, 2022). Disruption of the MpfA(YhdP) efflux pump alleviates some of the consequences of ribosomal protein L34 deletion with restoration of a high intracellular magnesium concentration. It displays a similar effect on lack of ribosomal proteins L1, L23, L36, and S6. Expression of mpfA is induced by Mg2+, and induction is independent of the Mer family regulator YhdQ encoded by the neighbouring gene, which is still of unknown function. MpfA is a homologue of the putative Mg2+ efflux transporter CorC in E. coli and MpfA in Staphylococcus aureus. Manganese and cobalt in their octahedral binding structure are practically isosteric with magnesium. Thus, these ions may compete with Mg2+, particularly when the latter is limiting. This likely explains why loss of MpfA, which leads to increased cytosolic magnesium levels, alleviates Mn2+ and Co2+ intoxication (Pi et al., 2020).
In contrast to E. coli, where the role of calcium remains elusive (Luder et al., 2021), this divalent ion is important for B. subtilis in a variety of processes, biofilm formation and in particular sporulation. It is also important for the structural integrity and activity of several extracellular proteins. The ECM preferentially binds calcium ions over other metal ions, including magnesium or the divalent ions used in cells at a lower concentration zinc, manganese and iron. These ions apparently circulate in the water channels generated by the macroscopic wrinkles of the biofilm. They may thus behave as propagated signals triggering vegetative in situ cell multiplication or sporulation (Azulay et al., 2022). CalJ(YetJ) is the B. subtilis bacterial homologue of human Bax‐inhibitor 1 protein (hBI‐1) involved in calcium leak control between the endoplasmic reticulum, organelles and cytoplasm. AceP is the counterpart present in E. coli, identified as an acetate transporter, belonging to the UPF0005 family. Structural characterization of the protein reveals different conformations depending on the pH. It forms a closed compact structure at pH 8 and an open conformation at pH 6, forming a pore through the lipid bilayer. Overexpression of CatJ in E. coli and with CatJ proteoliposomes reveals a pH‐dependent calcium‐leak activity. The ion flux rate is higher at pH 7 where the pore is in equilibrium than at pH 6 where it is open. In both conformations, the structure of the protein is composed by 7 trans‐membrane helices similar to the helix boundaries of the entire family of TMBIM proteins (PF01027) to which hBI‐1 belongs (Li et al., 2020).
Zinc and transition metals
Zinc is essential in all living organisms. It is a cofactor for many enzymes as well as for proteins interacting with nucleic acids. In B. subtilis, zinc homeostasis is tightly regulated by the Zur transcription factor which controls the successive induction of adaptive mechanisms as zinc becomes limiting (Chandrangsu et al., 2017; Shin & Helmann, 2016). Conversely, excess zinc leads to induction of specific efflux systems, and in their absence zinc toxicity ensues due to the mismetallation of another metalloregulator, PerR (Chandrangsu & Helmann, 2016). Intracellular zinc is stored by association with several ribosomal proteins (L31 and L33), and upon deprivation paralogues are induced to displace the Zn‐containing proteins to mobilize zinc (Akanuma et al., 2021). These paralogues include an L31 paralogue and an L33 paralogue, although in strain 168, the rpmGC gene (coding for L33*) is interrupted by a frameshift and is not functional. Zinc also regulates an alternative S14 subunit (S14*) that is derepressed to allow continued ribosome biogenesis when zinc is limited. Zinc is also used in a variety of activities that integrate one‐carbon metabolism into a coherent enzyme system based on a concerted set of zinc‐sensitive steps (Danchin, 2020). Folate biosynthesis depends on the FolEA / FolEB enzymes that catalyse dihydroneopterin triphosphate + formic acid formation from guanosine triphosphate. Whereas FolEA is dependent on zinc, FolEB is independent of zinc and is induced by zinc starvation. However, prior to induction of FolEB, delivery of zinc to FolEA is prioritized by the ZagA zinc metallochaperone. ZagA is representative of a family of GTP‐dependent metallochaperones that function with zinc, with homologues identified in other bacteria (Nairn et al., 2016) and recently in mammals (Weiss, Murdoch, et al., 2022) and yeast (Pasquini et al., 2022) where this protein is defined as ZNG1. While canonically defined as a GTPase, B. subtilis ZagA also responds to ZTP (5′‐aminoimidazole‐4‐carboxamide‐1‐β‐D‐ribofuranosyl‐5′‐triphosphate, known as AICAR triphosphate or Z nucleoside triphosphate), a signal of 10‐formyl‐tetrahydrofolate deficiency in bacteria derived from the purine biosynthetic pathway. ZTP accumulates transiently as FolE begins to fail. This stimulates the interaction between ZagA(YciC), the ZTP activated GTPase A metallochaperone; and FolE, helping to sustain folate synthesis despite declining zinc availability (Chandrangsu et al., 2019). It is unknown whether ZNG1 or any other members of this family might also be activated by ZTP instead of, or in addition to, GTP. Zinc is also critical for the homeostasis of iron–sulfur cluster synthesis. Zinc‐containing SufU is the protein that transfers sulfur from SufS to SufB, and that the SufBCD complex is the site of Fe‐S cluster assembly. A zinc‐ligand exchange reaction upon SufS‐SufU complexation provides a free thiol from Cys41 of SufU as a sulfur acceptor (Yokoyama et al., 2018).
Manganese (Mn) is also an essential nutrient metal ion for B. subtilis, and MntR is the key transcription factor regulating both import and export functions. MntR activates both MneP (primary) and MneS (secondary) manganese exporters in response to elevated manganese. Mutant strains lacking mneP and mneS are Mn2+ sensitive and accumulate elevated levels of Mn2+ (Sachla et al., 2021). The major essential function for manganese in B. subtilis is thought to be as a cofactor for the essential ribonucleotide reductase (RNR). OxdD(YoaN) has been identified as an oxalate decarboxylase with an interesting catalytic mechanism that depends on the presence of two manganese ions, with the N‐terminal ion in the Mn(III) state and the C‐terminal ion acting as an electron hole (Pastore et al., 2021). The gene belongs to the phage 6 region and has been recruited by the sporulation process with its expression dependent on sigma(K). OxD is a spore coat inner layers constituent (Costa et al., 2004), identified in crystalline form in B. pumilus (Garcia‐Ramon et al., 2018).
Bacillus subtilis also utilizes copper, which plays a key role in cytochrome c oxidase (caa3) and thereby contributes to aerobic respiration. Understanding of copper metallobiology is still in its early stages but involves two Cu‐sensing transcription factors. CsoR is a repressor of the copZA operon for copper export and its activity is antagonized by Cu(I). In contrast, YcnK is a repressor of an operon that encoded copper import functions (Damle et al., 2021). As far as is known, the only cytosolic role of copper in B. subtilis is as a signal of copper status and as an intermediate in the pathway that likely contributes to metalation of cytochrome c oxidase.
The fate of cobalt in B. subtilis is enigmatic. Indeed, while this organism does not have a coenzyme B12‐dependent methionine synthase, it possesses an import system for coenzyme B12 controlled by the cobalamin riboswitch srbL that modulates the activity of operon yrvABbtuC(yrvC)pduO(yvqK) (Chan & Mondragón, 2020). The only known enzyme of the bacterium that appears to require cobalamin for its function is in the queuosine biosynthesis pathway, which depends on B12 availability in a variety of bacteria (Romine et al., 2017). Epoxyqueuosine reductase QueG is B12‐dependent in B. subtilis but replaced by a counterpart of a different descent in B12‐independent contexts (Li, Zallot, et al., 2021). The fact that queuosine metabolism is related to cobalt availability is further substantiated by the observation that mutants of the NADPH‐dependent 7‐cyano‐7‐deazaguanine reductase (with moonlighting hydratase activity) QueF can suppress cobalt sensitivity (Pi et al., 2020). The epoxide group in pre‐queuosine is derived from ribose in de novo synthesis, rather than from the oxidation of the dihydroxyclyclopentene ring carbon double bond. However, this double bond is likely to be reactive and some ROSs, in the cellular context, will probably epoxidize it. This potentially provides a new role to QueG, as a buffering/repair system that can use tRNAs as a first line of defence against ROSs. This may have implications for ROS resistance in media containing cobalamin, although this has not been investigated. Another possible reason for bacteria to import cobalamin could be to obtain the Co2+ ion, which under some conditions of metal limitation might help sustain activity of some metal‐dependent enzymes. In this model, cobalamins in the environment would be recognized as an easily scavenged source of cobalt ion, but it is not clear if or how the ion might be released from imported cobalamin.
Nickel is also an ion of apparently little importance to B. subtilis. Nickel can sometimes substitute for other divalent transition ions, but it has been proposed to be critical for the activity of three enzymes: the long known UreCBA heterotrimeric urease, that contains two nickel ions essential for activity, the EgsA(AraM, YseB) sn‐glycerol‐1‐phosphate dehydrogenase (Guldan et al., 2008) and the FarJ(YugJ) furan aldehyde reductase (Cho et al., 2022). However, the role of nickel in the B. subtilis urease has never been established, and this organism lacks both an obvious Ni import system and the Ni metallochaperone often associated with Ni‐dependent ureases. This raises the possibility that this enzyme may use a different cation. The EgsA enzyme is most active in vitro with Ni2+, but no phenotype has been reported for nickel deficient cells. Thus, the role of nickel in B. subtilis biology appears to be limited to a small number of specialized functions. The BstA(YfiT) bacillithiol S‐transferase binds either nickel or zinc (Perera et al., 2018). Nickel has also a role in the synthesis of the gaseous mediator carbon monoxide when it is present at a higher concentration than iron, diverting the activity of acireductone dioxygenase MtnD from its normal role in the methionine salvage pathway (Sekowska et al., 2019).
ENERGY‐DEPENDENT MAXWELL'S DISCRIMINATORS
The need for energy‐dissipating discrimination between classes of entities or processes is omnipresent. Agents with this role enable the cell to cope with variety, including controlling export of artificial compounds that did not yet exist on our planet (Boel et al., 2019). This is also the case for discrimination of spatial locations in the cell. Finally, the maintenance of the cell by allowing either the repair or degradation of inevitable aged or damaged entities must discriminate between these and canonical compounds. Energy is dissipated when these agents return to their ground state where they can once again perform their function (Danchin, 2021; Johnson, 1970; Landauer, 1961). Table S4 lists 150 genes encoding functions involving ATP hydrolysis, 17 genes involving GTP hydrolysis and one gene involving CTP hydrolysis (parB) that may be involved in discrimination processes. Relevant examples are illustrated below. Most (except for transporters which tend to vary as a function of the environment of the organisms of interest) are conserved in the M. mycoides Syn3.0 minimal genome (Danchin & Fang, 2016).
Discrimination in sensing and transport
Transport in and out of the cell is critical for life. Many transporters are permeases that are highly specific for a narrow class or even unique substrates. However, one important feature of transporters is in self/non‐self‐discrimination. They allow cells to discriminate between specific families of inputs and the infinite variety of possible substrates present in the environment, or to export potentially toxic metabolites while simultaneously preventing canonical metabolites from escaping the cell. In addition to constraints that involve the electrical potential of the membrane and affect electrically charged metabolites outside or inside the cell and are therefore energy consuming, discrimination between classes of substrates dissipates energy. This is reflected in the large number of ATP‐driven transporters, as shown in Table S4 (77 genes). The proportion of these transporters with no identified function remains large (26 ‘y’ genes) because the function of membrane proteins is often more difficult to identify than that of their cytoplasmic counterpart. However, several of them have been characterized since the previous annotation (Table 2).
TABLE 2.
Newly annotated transport‐related ATP‐binding proteins.
| ID | Gene | Synonym | Function |
|---|---|---|---|
| BSU15650 | atcL | yloB, tcaB | P‐type calcium transport ATPase (sporulation) |
| BSU30380 | bceA | ytsC | bacitracin‐related ABC efflux transporter (ATP‐binding protein) |
| BSU08310 | bifL | yfiL | ABC transporter (ATP‐binding protein) biofilm formation |
| BSU34820 | bmrA | yvcC | efflux transporter (ATP‐binding and permease protein) |
| BSU09710 | bmrC | yheI | efflux ABC transporter subunit (ATP‐binding protein) |
| BSU09720 | bmrD | yheH | efflux ABC transporter (ATP‐binding subunit) |
| BSU33490 | cadA | yvgW | Cd(II), Zn(II) and Co(II) exporter (ATPase) |
| BSU33500 | copA | yvgX | copper [Cu(I)] transporter ATPase |
| BSU40150 | epeA | yydI, liaL | ABC transporter for regulatory peptide EpeX/LiaD* (ATP‐binding protein) |
| BSU16240 | fliI | _ | flagellar‐specific ATPase subunit of export apparatus |
| BSU16250 | fliJ | cheF | flagellar synthesis rod subunit of export ATPase |
| BSU32550 | frlP | yurJ | fructose‐amino acid ABC transporter (ATP‐binding subunit) |
| BSU31100 | ktrB | yubG | potassium transporter ATPase |
| BSU14510 | ktrC | ylxV, yzaC, ykqB | potassium uptake protein; regulator of potassium conductance; ATP‐dependent gating channel |
| BSU13500 | ktrD | ykrM | K + ‐transporting ATPase (glumamate controlled) |
| BSU30760 | mntB | ytgB | manganese ABC transporter (ATP‐binding protein) |
| BSU38810 | msmX | yxkG | multiple sugar (maltodextrins, galactans) transporter ATP‐binding protein |
| BSU33730 | opuBA | proV | choline ABC transporter (ATP‐binding protein) |
| BSU33830 | opuCA | yvbE | glycine betaine/carnitine/choline/choline ABC transporter (ATP‐binding protein) |
| BSU35700 | tagH | _ | ATP‐binding teichoic acid precursor transporter component |
| BSU13220 | thiW | ykoD | thiamine/HMP ABC transporter (ATP‐binding subunit) |
| BSU03140 | tmrB | _ | ATP‐binding tunicamycin resistance protein |
| BSU02860 | znuC | ycdI, adcC | Zn(II) transporter (ATP‐binding protein) |
The following are some examples of recently characterized transporters. Contrary to initial views, the BceAB system does not inactivate or import the antibiotic bacitracin but acts by transiently freeing lipid II cycle intermediates from the inhibitory grip of antimicrobial peptides and thus provides resistance through targeted protection of cell wall synthesis (Kobras et al., 2020). EpeAB is encoded in operon epeXEPAB that encodes a 49‐amino acid peptide EpeX(YydF, LiaD), modified by EpeE(YydG) an AdoMet radical peptide epimerase, a membrane‐embedded protease EpeP(YydH, LiaK) and exported by an ATP‐binding cassette (ABC) transporter. The modified peptide EpeX* elicits cell envelope stress sensed by the LiaRS two‐component system (Popp et al., 2021). MsmX is the energy‐dissipating subunit of transporter maltodextrin‐specific MdxEFG essential for pectin mobilization (Leisico et al., 2020). Finally, the important role of discrimination between various metabolites allowing the cell to adapt its osmotic pressure to the environment, a situation widely met by B. subtilis, is also visible in the various proline and osmoprotectants transporters. OpuB exhibits a rather restricted substrate profile. It imports choline and arsenocholine with high affinity. In contrast, OpuC imports a broad range of osmoprotectants mostly with high affinity, including choline, arsenocholine, glycine betaine and arsenobetaine (Hoffmann & Bremer, 2017; Warmbold et al., 2020). The actual hierarchy in balancing import of these various osmostress‐protectants is not yet known, nor are its links with the amount of energy dissipated in the process.
Discrimination for spatial targeting
Besides ParB, discussed in a previous section, several localization factors such as DnaA (ATP‐dependent), which identifies the correct origin of replication, are encoded in the genome. The Smc/ScpAB complex is critical for correctly folding the nucleoid while avoiding collisions during replication (Anchimiuk et al., 2021; Brandão et al., 2021; Nomidis et al., 2022) and RarA and SbcE localize to their specific sites of action critical for recombination (Romero et al., 2020). DynA, which localizes to the cell division septum, exhibits strictly auto‐regulated GTP hydrolysis. It can tether membranes and mediate nucleotide‐independent membrane fusion in vitro. Deletion of the dynA gene results in defects in cell growth and shape maintenance. This factor plays an important role in a membrane surveillance system that counteracts membrane pore formation provoked by antibiotics and phages. Upon damage, DynA localizes into large and static assemblies, where it acts locally to counteract stress‐induced pores, presumably by inducing lipid bilayer fusion and sealing membrane gaps (Guo, Sattler, et al., 2022; Guo, Zhao, et al., 2022). The role of GTP might be to identify the proper targets of the protein. In some cases, the function involves a mechanical function, and it can be expected that beside one quantum of energy used for discrimination, one or several further energy quanta are also involved in the biomechanical process.
Discrimination in shaping, maintenance and degradation
The accuracy of ribosome biogenesis is mainly controlled using GTP hydrolysis as the energy‐dissipating store. The need for discrimination is expected to facilitate correct RNA folding and formation of RNA‐protein complexes. Indeed, once transcribed, ribosomal RNA alone cannot escape engaging in wrong folding pathways. These incorrect folding pathways must be discriminated against as they would be highly toxic for the cell. Factors that are conserved in the minimal genome of M. mycoides Syn3.0, DeaD helicase (ATP‐dependent), EngD (we have seen that it is dependent on the presence of potassium), HflX (GTP, ATPase) BipA, CpgA, EngA, EngB, EraA, ObgE, RbgA and RgpH are all essential for functional ribosome folding and assembly (Stoll et al., 2022). These factors are not only important for shape discrimination but also for the correct timing of the process, as for example illustrated by RbgA (Seffouh et al., 2022). The details of the way energy is dissipated in the process are expected to be unravelled in the next few years, in particular in the way they interfere with the function of the alarmones (p)ppGpp. This signalling system operates not only via its overlap with GTP binding sites but also via modulators of translation of the ABCF ATP‐binding factors. In a further connection to the translation process, B. subtilis possesses several counterparts of the E. coli EttA factor, EttM(YfmM), EttP(Uup, YfmR), YdiF, VmlR(ExpZ), the functions of which are still under investigation (Takada et al., 2022). Discrimination during maintenance is closely associated with a general process the understanding of which has witnessed considerable improvement during the past 5 years. It constitutes the proteostasis network that we now consider.
THE PROTEOSTASIS NETWORK
Bacteria divide by binary fission and therefore do not have distinguishable somatic and germ‐line cells. Nevertheless, individual cells in a bacterial populations age due to damage to vital cell components such as DNA and proteins. DNA damage can often be repaired using precise or error‐prone DNA repair mechanisms; however, protein damage is more complex and the proteostasis network (Figure 3) constantly monitors the proteome to help maintain protein homeostasis (Balchin et al., 2016; Matavacas & von Wachenfeldt, 2022). In addition to damage, many proteins have a functional ‘shelf life’, and specific degradation signals are incorporated into proteins whose activities are required to fulfil a specific function during a prescribed period of time (e.g. cell cycle, differentiation process, stress response). Proteins that are irreparably damaged or that have come to the end of their functional lifespan are removed by quality control proteases (Harwood & Kikuchi, 2022).
FIGURE 3.
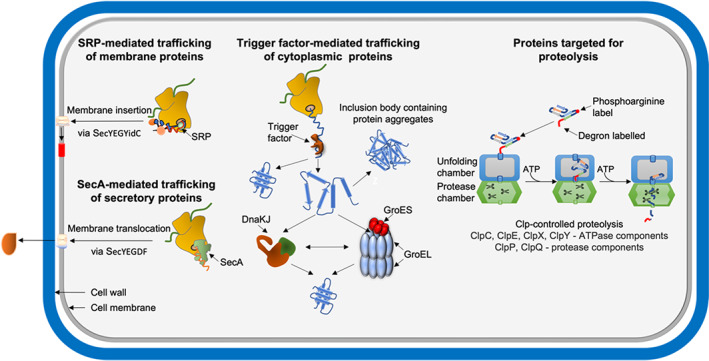
Bacillus subtilis proteostasis network Nascent polypeptide chains (NPCs) emerging from the ribosome are screened to determine their final locations using poorly understood signals within their N‐terminal regions. SRP and SecA targets proteins, respectively, to membrane or extracytoplasmic locations. Trigger factor interacts with cytoplasmic proteins as an intermediate in free folding and chaperone mediated (DnaKJ, GroEL/ES) folding pathways. Misfolded proteins, and proteins with post‐translational modifications or degrons are targeted for degradation by Hsp100 family proteases such as Clp or Lon. High concentrations of misfolded proteins form aggregate due to illicit inter‐ and intra‐molecular interactions.
The folding pathways of newly born proteins are dependent on their final locations. As a growing nascent peptide chain emerges from the exit port of the ribosome, they are scanned by proteins/complexes (e.g. Signal Recognition Particle (SRP), SecA or trigger factor (TF) that are responsible for trafficking them to their final cellular location. Trigger factor guides cytoplasmic proteins to chaperone systems (in B. subtilis DnaJK and GroESGroEL) that promote protein folding by partitioning non‐native states (Balchin et al., 2016). Membrane and secreted proteins are directed to their secretion pathways, respectively by the SRP or SecA (Huber et al., 2017; Schibich et al., 2016). Their folding is mediated by specific folding factors such as propeptides, thiol‐disulfide oxidoreductases (DbdA,B,C,D), peptidylproline cis/trans isomerase (PrsA) and metal ions (Sarvas et al., 2004).
Protein folding is a multistep process that is highly sensitive to the local environment. Proteins also tend to misfold or depart from their properly folded structure as they age. Molecular chaperones generally use energy twice in the process of maintenance and repair: they use a first quantum of energy (phosphate hydrolysis) to perform the discriminating step, allowing them to identify misfolded candidates, and they use at least a second quantum of energy to unfold and refold that candidate (Boel et al., 2019). Most chaperones, such as GroESGroEL and DnaJK, have counterparts in the three domains of life. At least one member of this family is present in the smallest genomes where they fold or re‐fold polypeptides (Danchin & Fang, 2016). If the misfolded or damaged proteins cannot be refolded or disposed of as localized aggregates, they have to be degraded. To facilitate degradation, energy‐dissipating agents target aggregates to specific locations and several energy‐dependent proteases encoded in the genome shred proteins into fragments further processed by the general metabolism (Harwood & Kikuchi, 2022). The ClpX, ClpE and ClpC subunits form ATPase complexes that couple to the ClpP peptidase which acts as a discriminator that allows degradation of foreign, damaged or misfolded proteins (Kim et al., 2022). In some cases, proteins may be stabilized by anti‐adaptors, such as the YirB protein that antagonizes proteolysis of the unstable Spx transcription factor (Rojas‐Tapias & Helmann, 2018).
Selective phosphorylation of arginine residues by MscB plays an important regulatory role in controlling proteostasis (Lilge et al., 2020). During rapid physicochemical transitions (collectively called stress conditions), McsB phosphorylates and inactivates the transcriptional repressors CtsR and HrcA. This triggers the expression of heat shock genes, including McsB itself, which begins by forming dimers. The mass action, or perhaps unknown chemical signals, associated with the increased level of McsB leads to the formation of octamers which form a closed structure. The self‐phosphorylated McsB octamers are the active kinase species responsible for labelling misfolded or altered proteins for degradation. They form a chamber within which the active site of the kinase is accessible to unfolded polypeptides via a narrow entry port. This structure distinguishes potential substrates from functionally active proteins. Proteins that have been phosphorylated are then targeted by proteases (Hajdusits et al., 2021).
Proteostasis is also critical in the case of defective translation. Ribosomes read and decode the genetic information until a stop codon is reached. Stop codons not only signal the end of the protein‐coding sequence but also serve as the binding sites for factors that promote the release of the nascent polypeptide. Messenger RNA molecules that lack appropriate termination signals, due to premature transcription termination, transcription errors or the presence of rare codons, are unable to bind release factors, leading to a loss of translational capacity and the presence of aberrant proteins. Consequently, bacteria have evolved two mechanisms for resolving stalled ribosomes: a trans‐translation system, mediated by transfer messenger RNA (tmRNA) and SmpB, and the ribosome‐associated protein quality control (RQC) pathway mediated by RqcP(YabO)/RqcH (Nadler et al., 2022; Takada et al., 2021). While the SmpB/tmRNA system is unique to eubacteria, the ubiquitous RQC pathway provides an alternative pathway in which stalled ribosomes are split, producing 50S subunits that remain obstructed with peptidyl‐tRNA (Kurita & Himeno, 2022). Recent work on B. subtilis has shown that RqcH and RqcP sense the incomplete peptide, allowing charged tRNAAla to tag the nascent‐chains with an untemplated C‐terminal Ala tail (Lytvynenko et al., 2019). RqcP binds to peptidyl‐tRNA at the P‐site, freeing the A‐site for RqcH to deliver Ala‐tRNAAla. Cycles of RqcP binding and dissociation drives the processivity of alanine‐tailing and the resulting homopolymeric Ala peptide functions as a degron tag, targeting the peptide to the ClpXP protease (Crowe‐McAuliffe et al., 2021).
Finally, another omnipresent energy‐dissipating protease, LonA, uses ATP but also polyphosphate as its energy source (Ropelewska et al., 2020). It degrades protein substrates in a processive fashion (Li, Hsieh, et al., 2021). As stated previously, it uses SmiA as an adaptator for SwrA degradation (Hughes et al., 2018; Olney et al., 2022). Unexpectedly, PrkA, which for a long time has been wrongly annotated as a protein kinase, is able to hydrolyze an exogenous substrate of Lon proteases, alpha‐casein, in an ATP‐dependent manner. This protease activity is essential for PrkA function in sporulation via sigma(K) modulation of activity. Transcriptional analysis demonstrated that deletion of prkA significantly reduced the expression of the transcriptional factor sigma(K) and its downstream genes (Zhang et al., 2022). Mutation in its ATP‐binding motif led to a sporulation defect. PrkA protease activity is tightly regulated by phosphorylation events involving one of the Ser/Thr protein kinase, PrkC. when PrkA is inhibited by 9‐alpha‐D‐arabinofuranosyladenine.
CONCLUSION: TOWARDS A RATIONAL MINIMUM BACILLUS SUBTILIS GENOME
Here we have proposed an eclectic representation of the annotation progress that has been associated to the reference genome of B. subtilis 168 deposited at the INSDC under accession number AL009126.4. Approximately one‐third of the genes of unknown function are clustered in 25 genomic regions containing at least 5 genes. They correspond mainly to prophage regions. Their contribution may be important for industrial uses of this organism, and we can expect a better understanding to develop in the coming years. The region encoding ICEBs1 is important for the management of interactions between B. subtilis and other organisms, plants in particular. Understanding the details of the behaviour of this element, particularly during biofilm formation, is greatly affected by the ability of the donor and recipient to form a biofilm. Conjugative transfer appears to be favoured by the biophysical context of biofilms. Indeed, extracellular matrix production, particularly from recipient cells, is essential for biofilms to promote ICEBs1 transfer (Bean et al., 2022; Lécuyer et al., 2018). Progress has also been made in annotating the six phage regions of the strain 168 genome, but these are the regions that are still poorly understood. They are not present in Mollicutes that have undergone reductive evolution. Most of the corresponding genes are not directly useful to the cell and have been deleted in the construction of simplified genomes. A single phage, SPβ, develops the full life cycle of a temperate phage. Once in the host genome, SPβ phages make complex lysis‐lysogeny decisions and enter a lytic cycle or integrate as a dormant prophage. As a prophage, SPβ confers additional properties to its host. The best known of the synthesis of lantibiotics such as sublancin 168, which is used as a molecular weapon by the host and maintains the prophage (Kohm & Hertel, 2021). The arbitrium quorum‐sensing peptide controls the exit from lysogeny by secreting and sensing the AimP‐derived arbitrium signal, thereby avoiding lysis when they are likely to encounter established lysogens rather than permissive uninfected hosts (Aframian et al., 2022). In this process the prophage master repressor MrpR(YopR) plays a central role (Kohm et al., 2022). Knowledge of this exquisite regulation may be of interest for the generation of adjustable metabolic engineering. Future biotechnological applications are likely to combine in vivo, in vitro and in silico approaches (Danchin, 2022). Membrane vesicle (MV) formation an interesting process for the engineering transition between in vivo and in vitro conditions. Bacillus subtilis releases membrane vesicles during the SOS response, which is associated with cell lysis triggered by the PBSX prophage‐encoded cell‐lytic enzymes XhlAB and XlyA deletions of xhlAB and xlyA had no effect on autolysis‐triggered MV biogenesis, indicating that autolysis is a novel and prophage‐independent pathway for MV production. Interestingly, the cell lysis induced by the surfactant treatment was effectively neutralized by the addition of exogenous purified MVs (Abe et al., 2021).
Eight thousand new references related to B. subtilis have been included in PubMed since the last annotation of the genome (Borriss et al., 2018), many of which are consistent with the identification of gene functions and to our vision of the organism as an industrial workhorse not only for plant protection and yield enhancement but also for metabolic engineering (Kovács, 2019). Here, we have improved the overall identification of gene functions, while focusing on creating a better picture of the organism's intermediary metabolism, a feature that will be essential for industrial applications relating to large‐scale enzyme and metabolite production, as well as the rapidly developing field of plant and animal pre‐ and probiotics. We plan to continue this work over the next few years with the goal of obtaining a complete picture of this model organism that could be used as a useful reference for improved automatic annotation of bacterial genomes.
AUTHOR CONTRIBUTIONS
Erhard Bremer: Investigation (equal); validation (equal); writing – review and editing (equal). Alexandra Calteau: Data curation (equal); resources (equal); software (equal); visualization (equal); writing – review and editing (equal). Antoine Danchin: Conceptualization (lead); data curation (lead); supervision (lead); writing – original draft (lead). Colin Robert Harwood: Data curation (equal); validation (equal); visualization (equal); writing – original draft (equal). John D. Helmann: Data curation (equal); validation (equal); writing – review and editing (equal). Claudine Médigue: Data curation (equal); methodology (equal); resources (equal); software (equal); supervision (equal); writing – review and editing (equal). Bernhard O Palsson: Validation (equal); writing – review and editing (equal). Agnieszka Sekowska: Investigation (equal); resources (equal); validation (equal); writing – review and editing (equal). David Vallenet: Conceptualization (equal); data curation (equal); methodology (equal); project administration (equal); resources (lead); software (lead); supervision (lead); validation (equal); writing – review and editing (equal). Abril Zuñiga: Investigation (equal); validation (equal). Cristal Zuñiga: Data curation (equal); supervision (equal); validation (equal); writing – review and editing (equal).
FUNDING INFORMATION
This work was supported by a grant from the National Institutes of Health (R35GM122461) to J.D.H. and grants from the Agence Nationale pour la Recherche (ANR‐11‐INBS‐0013 and ANR‐21‐ESRE‐0048) to C.M. and D.V.
CONFLICT OF INTEREST STATEMENT
None of the authors revealed a conflict of interest in this work devoted to a model organism.
Supporting information
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGEMENTS
The French Bioinformatics Institute national infrastructures (funded as part of Investissement d'Avenir program managed by Agence Nationale pour la Recherche, contracts ANR‐11‐INBS‐0013 and ANR‐21‐ESRE‐0048) is acknowledged for support within the MicroScope annotation platform. The manuscript style has been reviewed by Marc Abrams, publication coordinator of B.O.P laboratory. Note that because we consider accurate genome sequence annotation as critical for the future of biology we did not prioritize the role of each of the authors of this paper and named them in alphabetic order.
Bremer, E. , Calteau, A. , Danchin, A. , Harwood, C. , Helmann, J.D. , Médigue, C. et al. (2023) A model industrial workhorse: Bacillus subtilis strain 168 and its genome after a quarter of a century. Microbial Biotechnology, 16, 1203–1231. Available from: 10.1111/1751-7915.14257
REFERENCES
- Abe, K. , Toyofuku, M. , Nomura, N. & Obana, N. (2021) Autolysis‐mediated membrane vesicle formation in Bacillus subtilis . Environmental Microbiology, 23, 2632–2647. [DOI] [PubMed] [Google Scholar]
- Aframian, N. , Omer Bendori, S. , Kabel, S. , Guler, P. , Stokar‐Avihail, A. , Manor, E. et al. (2022) Dormant phages communicate via arbitrium to control exit from lysogeny. Nature Microbiology, 7, 145–153. [DOI] [PubMed] [Google Scholar]
- Akanuma, G. , Kawamura, F. , Watanabe, S. , Watanabe, M. , Okawa, F. , Natori, Y. et al. (2021) Evolution of ribosomal protein S14 demonstrated by the reconstruction of chimeric ribosomes in Bacillus subtilis . Journal of Bacteriology, 203, e00599–e00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Shayeb, B. , Sachdeva, R. , Chen, L.‐X. , Ward, F. , Munk, P. , Devoto, A. et al. (2020) Clades of huge phages from across Earth's ecosystems. Nature, 578, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anchimiuk, A. , Lioy, V.S. , Bock, F.P. , Minnen, A. , Boccard, F. & Gruber, S. (2021) A low Smc flux avoids collisions and facilitates chromosome organization in Bacillus subtilis . eLife, 10, e65467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias‐Cartín, R. , Uzel, A. , Seduk, F. , Gerbaud, G. , Pierrel, F. , Broc, M. et al. (2022) Identification and characterization of a noncanonical menaquinone‐linked formate dehydrogenase. The Journal of Biological Chemistry, 298, 101384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita, M. , Karsch‐Mizrachi, I. & Cochrane, G. (2021) The international nucleotide sequence database collaboration. Nucleic Acids Research, 49, D121–D124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustiniene, E. & Malys, N. (2022) Identification and characterization of L‐ and D‐lactate‐inducible systems from Escherichia coli MG1655, Cupriavidus necator H16 and pseudomonas species. Scientific Reports, 12, 2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azulay, D.N. , Spaeker, O. , Ghrayeb, M. , Wilsch‐Bräuninger, M. , Scoppola, E. , Burghammer, M. et al. (2022) Multiscale X‐ray study of Bacillus subtilis biofilms reveals interlinked structural hierarchy and elemental heterogeneity. Proceedings of the National Academy of Sciences of the United States of America, 119, e2118107119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin, D. , Hayer‐Hartl, M. & Hartl, F.U. (2016) In vivo aspects of protein folding and quality control. Science, 353, aac4354. [DOI] [PubMed] [Google Scholar]
- Bange, G. & Bedrunka, P. (2020) Physiology of guanosine‐based second messenger signaling in Bacillus subtilis . Biological Chemistry, 401, 1307–1322. [DOI] [PubMed] [Google Scholar]
- Bansal, P. , Morgat, A. , Axelsen, K.B. , Muthukrishnan, V. , Coudert, E. , Aimo, L. et al. (2022) Rhea, the reaction knowledgebase in 2022. Nucleic Acids Research, 50, D693–D700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbe, V. , Cruveiller, S. , Kunst, F. , Lenoble, P. , Meurice, G. , Sekowska, A. et al. (2009) From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology (Reading, England), 155, 1758–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastard, K. , Perret, A. , Mariage, A. , Bessonnet, T. , Pinet‐Turpault, A. , Petit, J.L. et al. (2017) Parallel evolution of non‐homologous isofunctional enzymes in methionine biosynthesis. Nature Chemical Biology, 13, 858–866. [DOI] [PubMed] [Google Scholar]
- Bean, E.L. , McLellan, L.K. & Grossman, A.D. (2022) Activation of the integrative and conjugative element Tn916 causes growth arrest and death of host bacteria. PLoS Genetics, 18, e1010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechhofer, D.H. & Deutscher, M.P. (2019) Bacterial ribonucleases and their roles in RNA metabolism. Critical Reviews in Biochemistry and Molecular Biology, 54, 242–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benda, M. , Schulz, L.M. , Stülke, J. & Rismondo, J. (2021) Influence of the ABC transporter YtrBCDEF of Bacillus subtilis on competence, biofilm formation and cell wall thickness. Frontiers in Microbiology, 12, 587035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim, A. , Millman, A. , Ofir, G. , Meitav, G. , Avraham, C. , Shomar, H. et al. (2020) Prokaryotic viperins produce diverse antiviral molecules. Nature, 589, 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake, C. , Christensen, M.N. & Kovács, Á.T. (2021) Molecular aspects of plant growth promotion and protection by Bacillus subtilis . Molecular Plant‐Microbe Interactions, 34, 15–25. [DOI] [PubMed] [Google Scholar]
- Boel, G. , Danot, O. , de Lorenzo, V. & Danchin, A. (2019) Omnipresent Maxwell's demons orchestrate information management in living cells. Microbial Biotechnology, 12, 210–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borriss, R. , Danchin, A. , Harwood, C.R. , Medigue, C. , Rocha, E.P.C. , Sekowska, A. et al. (2018) Bacillus subtilis, the model gram‐positive bacterium: 20 years of annotation refinement. Microbial Biotechnology, 11, 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowater, L. , Fairhurst, S.A. , Just, V.J. & Bornemann, S. (2004) Bacillus subtilis YxaG is a novel Fe‐containing quercetin 2,3‐dioxygenase. FEBS Letters, 557, 45–48. [DOI] [PubMed] [Google Scholar]
- Brandão, H.B. , Ren, Z. , Karaboja, X. , Mirny, L.A. & Wang, X. (2021) DNA‐loop‐extruding SMC complexes can traverse one another in vivo. Nature Structural & Molecular Biology, 28, 642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantl, S. & Müller, P. (2019) Toxin−antitoxin systems in Bacillus subtilis . Toxins (Basel), 11, E262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer, M. , Earnest, T.M. , Merryman, C. , Wise, K.S. , Sun, L. , Lynott, M.R. et al. (2019) Essential metabolism for a minimal cell. eLife, 8, e36842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghard‐Schrod, M. , Altenburger, S. & Graumann, P.L. (2020) The Bacillus subtilis dCMP deaminase ComEB acts as a dynamic polar localization factor for ComGA within the competence machinery. Molecular Microbiology, 113, 906–922. [DOI] [PubMed] [Google Scholar]
- Burgos, R. , Weber, M. , Martinez, S. , Lluch‐Senar, M. & Serrano, L. (2020) Protein quality control and regulated proteolysis in the genome‐reduced organism mycoplasma pneumoniae . Molecular Systems Biology, 16, e9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cámara‐Almirón, J. , Navarro, Y. , Díaz‐Martínez, L. , Magno‐Pérez‐Bryan, M.C. , Molina‐Santiago, C. , Pearson, J.R. et al. (2020) Dual functionality of the amyloid protein TasA in Bacillus physiology and fitness on the phylloplane. Nature Communications, 11, 1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos Guillén, J. , Arvizu Gómez, J.L. , Jones, G.H. , Hernández Flores, J.L. , Ramos López, M.A. , Cruz Hernández, A. et al. (2019) Analysis of tRNACys processing in the absence of CCAase in Bacillus subtilis . Brazilian Journal of Microbiology, 50, 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi, R. , Billington, R. , Keseler, I.M. , Kothari, A. , Krummenacker, M. , Midford, P.E. et al. (2020) The MetaCyc database of metabolic pathways and enzymes ‐ a 2019 update. Nucleic Acids Research, 48, D445–D453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centola, M. , van Pee, K. , Betz, H. & Yildiz, Ö. (2021) Crystal structures of phosphatidyl serine synthase PSS reveal the catalytic mechanism of CDP‐DAG alcohol O‐phosphatidyl transferases. Nature Communications, 12, 6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai, Y. , Kolter, R. & Losick, R. (2009) A widely conserved gene cluster required for lactate utilization in Bacillus subtilis and its involvement in biofilm formation. Journal of Bacteriology, 191, 2423–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, C.M. , Danchin, A. , Marliere, P. & Sekowska, A. (2014) Paralogous metabolism: S‐alkyl‐cysteine degradation in Bacillus subtilis . Environmental Microbiology, 16, 101–117. [DOI] [PubMed] [Google Scholar]
- Chan, C.W. & Mondragón, A. (2020) Crystal structure of an atypical cobalamin riboswitch reveals RNA structural adaptability as basis for promiscuous ligand binding. Nucleic Acids Research, 48, 7569–7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrangsu, P. & Helmann, J.D. (2016) Intracellular Zn(II) intoxication leads to dysregulation of the PerR regulon resulting in heme toxicity in Bacillus subtilis . PLoS Genetics, 12, e1006515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrangsu, P. , Huang, X. , Gaballa, A. & Helmann, J.D. (2019) Bacillus subtilis FolE is sustained by the ZagA zinc metallochaperone and the alarmone ZTP under conditions of zinc deficiency. Molecular Microbiology, 112, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrangsu, P. , Rensing, C. & Helmann, J.D. (2017) Metal homeostasis and resistance in bacteria. Nature Reviews. Microbiology, 15, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C.‐C. , Peng, M. , Zhong, J. , Zhang, Z. , Keppeke, G.D. , Sung, L.‐Y. et al. (2022) Molecular crowding facilitates bundling of IMPDH polymers and cytoophidium formation. Cellular and Molecular Life Sciences, 79, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton, S.G.V. , Kurz, D.L. , Geisel, S. , Jimenez‐Martinez, J. & Secchi, E. (2022) The role of biofilm matrix composition in controlling colony expansion and morphology. Interface Focus, 12, 20220035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Gozzi, K. & Chai, Y. (2015) A bacterial volatile signal for biofilm formation. Microbial Cell, 2, 406–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Zarazúa‐Osorio, B. , Srivastava, P. , Fujita, M. & Igoshin, O.A. (2023) The slowdown of growth rate controls the single‐cell distribution of biofilm matrix production via an SinI‐SinR‐SlrR network. mSystems, e00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, H.Y. , Nam, M.S. , Hong, H.J. , Song, W.S. & Yoon, S.‐I. (2022) Structural and biochemical analysis of the furan aldehyde reductase YugJ from Bacillus subtilis . International Journal of Molecular Sciences, 23, 1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, J. , Grogan, H. , Fitzpatrick, D. & Kavanagh, K. (2022) Analysis of the effect of Bacillus velezensis culture filtrate on the growth and proteome of Cladobotryum mycophilum . Fungal Biology, 126, 11–19. [DOI] [PubMed] [Google Scholar]
- Collard, F. , Baldin, F. , Gerin, I. , Bolsée, J. , Noël, G. , Graff, J. et al. (2016) A conserved phosphatase destroys toxic glycolytic side products in mammals and yeast. Nature Chemical Biology, 12, 601–607. [DOI] [PubMed] [Google Scholar]
- Costa, T. , Steil, L. , Martins, L.O. , Völker, U. & Henriques, A.O. (2004) Assembly of an oxalate decarboxylase produced under sigmaK control into the Bacillus subtilis spore coat. Journal of Bacteriology, 186, 1462–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe‐McAuliffe, C. , Takada, H. , Murina, V. , Polte, C. , Kasvandik, S. , Tenson, T. et al. (2021) Structural basis for bacterial ribosome‐associated quality control by RqcH and RqcP. Molecular Cell, 81, 115–126.e7. [DOI] [PubMed] [Google Scholar]
- Damle, M.S. , Singh, A.N. , Peters, S.C. , Szalai, V.A. & Fisher, O.S. (2021) The YcnI protein from Bacillus subtilis contains a copper‐binding domain. The Journal of Biological Chemistry, 297, 101078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. (1997) Comparison between the Escherichia coli and Bacillus subtilis genomes suggests that a major function of polynucleotide phosphorylase is to synthesize CDP. DNA Research, 4, 9–18. [DOI] [PubMed] [Google Scholar]
- Danchin, A. (2020) Zinc, an unexpected integrator of metabolism? Microbial Biotechnology, 13, 895–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. (2021) Three overlooked key functional classes for building up minimal synthetic cells. Synthetic Biology, 6, ysab010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. (2022) In vivo, in vitro and in silico: an open space for the development of microbe‐based applications of synthetic biology. Microbial Biotechnology, 15, 42–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. & Fang, G. (2016) Unknown unknowns: essential genes in quest for function. Microbial Biotechnology, 9, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. & Nikel, P.I. (2019) Why nature chose potassium. Journal of Molecular Evolution, 87, 271–288. [DOI] [PubMed] [Google Scholar]
- Danchin, A. , Ouzounis, C. , Tokuyasu, T. & Zucker, J.‐D. (2018) No wisdom in the crowd: genome annotation in the era of big data ‐ current status and future prospects. Microbial Biotechnology, 11, 588–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, A. & Sekowska, A. (2015) The logic of metabolism. Perspectives in Science, 6, 15–26. [Google Scholar]
- D'Ari, R. & Casadesús, J. (1998) Underground metabolism. BioEssays, 20, 181–186. [DOI] [PubMed] [Google Scholar]
- de Crécy‐Lagard, V. , Ross, R.L. , Jaroch, M. , Marchand, V. , Eisenhart, C. , Brégeon, D. et al. (2020) Survey and validation of tRNA modifications and their corresponding genes in Bacillus subtilis sp subtilis strain 168. Biomolecules, 10, E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Caño‐Ochoa, F. , Moreno‐Morcillo, M. & Ramón‐Maiques, S. (2019) CAD, a multienzymatic protein at the head of de novo pyrimidine biosynthesis. Sub‐Cellular Biochemistry, 93, 505–538. [DOI] [PubMed] [Google Scholar]
- Drews, G. (2000) The roots of microbiology and the influence of Ferdinand Cohn on microbiology of the 19th century. FEMS Microbiology Reviews, 24, 225–249. [DOI] [PubMed] [Google Scholar]
- Emami, K. , Wu, L.J. & Errington, J. (2020) A small molecule inhibitor of CTP synthetase identified by differential activity on a Bacillus subtilis mutant deficient in class a penicillin‐binding proteins. Frontiers in Microbiology, 11, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen, S. , Vallenet, D. , Medigue, C. & Danchin, A. (2012) Distinct co‐evolution patterns of genes associated to DNA polymerase III DnaE and PolC. BMC Genomics, 13, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermoli, F. , Bontà, V. , Vitali, G. & Calvio, C. (2021) SwrA as global modulator of the two‐component system DegSU in Bacillus subtilis . Research in Microbiology, 172, 103877. [DOI] [PubMed] [Google Scholar]
- Feaga, H.A. , Hong, H.‐R. , Prince, C.R. , Rankin, A. , Buskirk, A.R. & Dworkin, J. (2023) Elongation factor P is important for sporulation initiation. Journal of Bacteriology, 205, e0037022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulquier, E. , Pompeo, F. , Byrne, D. , Fierobe, H.‐P. & Galinier, A. (2020) Uridine diphosphate N‐acetylglucosamine orchestrates the interaction of GlmR with either YvcJ or GlmS in Bacillus subtilis . Scientific Reports, 10, 15938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos‐Monterrosa, R. , Mhatre, E. & Kovács, Á.T. (2016) Specific Bacillus subtilis 168 variants form biofilms on nutrient‐rich medium. Microbiology (Reading), 162, 1922–1932. [DOI] [PubMed] [Google Scholar]
- Garcia‐Ramon, D.C. , Berry, C. , Tse, C. , Fernández‐Fernández, A. , Osuna, A. & Vílchez, S. (2018) The parasporal crystals of Bacillus pumilus strain 15.1: a potential virulence factor? Microbial Biotechnology, 11, 302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler, A.S. , Anthon, C. , Alkan, F. , González‐Tortuero, E. , Poulsen, L.D. , Kallehauge, T.B. et al. (2021) BSGatlas: a unified Bacillus subtilis genome and transcriptome annotation atlas with enhanced information access. Microbial Genomics, 7, 000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, M. , Grunden, A.M. , Dunn, D.M. , Weiss, R. & Adams, M.W. (1998) Characterization of native and recombinant forms of an unusual cobalt‐dependent proline dipeptidase (prolidase) from the hyperthermophilic archaeon Pyrococcus furiosus. Journal of Bacteriology, 180, 4781–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Górecki, K. , Hägerhäll, C. & Drakenberg, T. (2014) The Na+ transport in gram‐positive bacteria defect in the Mrp antiporter complex measured with 23Na nuclear magnetic resonance. Analytical Biochemistry, 445, 80–86. [DOI] [PubMed] [Google Scholar]
- Greenwich, J. , Reverdy, A. , Gozzi, K. , Di Cecco, G. , Tashjian, T. , Godoy‐Carter, V. et al. (2019) A decrease in serine levels during growth transition triggers biofilm formation in Bacillus subtilis . Journal of Bacteriology, 201, e00155‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldan, H. , Sterner, R. & Babinger, P. (2008) Identification and characterization of a bacterial glycerol‐1‐phosphate dehydrogenase: Ni(2+)‐dependent AraM from Bacillus subtilis . Biochemistry, 47, 7376–7384. [DOI] [PubMed] [Google Scholar]
- Gundlach, J. , Krüger, L. , Herzberg, C. , Turdiev, A. , Poehlein, A. , Tascón, I. et al. (2019) Sustained sensing in potassium homeostasis: cyclic di‐AMP controls potassium uptake by KimA at the levels of expression and activity. The Journal of Biological Chemistry, 294, 9605–9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, L. , Zhao, Y. , Zhang, Q. , Feng, Y. , Bi, L. , Zhang, X. et al. (2022) Stochastically multimerized ParB orchestrates DNA assembly as unveiled by single‐molecule analysis. Nucleic Acids Research, 50, 9294–9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, L. , Sattler, L. , Shafqat, S. , Graumann, P.L. & Bramkamp, M. (2022) A bacterial dynamin‐like protein confers a novel phage resistance strategy on the population level in Bacillus subtilis . mBio, 13, e0375321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, T. & Herman, J.K. (2022) Magnesium modulates Bacillus subtilis cell division frequency. Journal of Bacteriology, 205, e0037522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, R.S. , Patel, S. , Saini, N. & Chen, S. (2020) Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the subtilis and cereus clades of species. International Journal of Systematic and Evolutionary Microbiology, 70, 5753–5798. [DOI] [PubMed] [Google Scholar]
- Guttenplan, S.B. , Blair, K.M. & Kearns, D.B. (2010) The EpsE flagellar clutch is bifunctional and synergizes with EPS biosynthesis to promote Bacillus subtilis biofilm formation. PLoS Genetics, 6, e1001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, J. , DeSantis, M. & Dubnau, D. (2021) Mechanisms of transforming DNA uptake to the periplasm of Bacillus subtilis . MBio, 12, e0106121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdusits, B. , Suskiewicz, M.J. , Hundt, N. , Meinhart, A. , Kurzbauer, R. , Leodolter, J. et al. (2021) McsB forms a gated kinase chamber to mark aberrant bacterial proteins for degradation. eLife, 10, e63505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood, C.R. (1992) Bacillus subtilis and its relatives: molecular biological and industrial workhorses. Trends in Biotechnology, 10, 247–256. [DOI] [PubMed] [Google Scholar]
- Harwood, C.R. & Kikuchi, Y. (2022) The ins and outs of Bacillus proteases: activities, functions and commercial significance. FEMS Microbiology Reviews, 46, fuab046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra, S. , Bhandari, D.M. , Krishnamoorthy, K. , Sekowska, A. , Danchin, A. & Begley, T.P. (2022) Cysteine dealkylation in Bacillus subtilis by a novel flavin‐dependent monooxygenase. Biochemistry, 61, 952–955. [DOI] [PubMed] [Google Scholar]
- He, J. , Yin, W. , Galperin, M.Y. & Chou, S.‐H. (2020) Cyclic di‐AMP, a second messenger of primary importance: tertiary structures and binding mechanisms. Nucleic Acids Research, 48, 2807–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmann, K.B. , Goethel, M.E. , Erickson, N.A. & Niehaus, T.D. (2022) Identification of a S‐(2‐succino)cysteine breakdown pathway that uses a novel S‐(2‐succino) lyase. The Journal of Biological Chemistry, 298, 102639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, T. & Bremer, E. (2017) Guardians in a stressful world: the Opu family of compatible solute transporters from Bacillus subtilis . Biological Chemistry, 398, 193–214. [DOI] [PubMed] [Google Scholar]
- Hoffmann, T. , Mrusek, D. , Bedrunka, P. , Burchert, F. , Mais, C.‐N. , Kearns, D.B. et al. (2021) Structural and functional characterization of the bacterial biofilm activator RemA. Nature Communications, 12, 5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, C. , Tsai, H.‐Y. , Chang, C.‐F. , Yang, C.‐C. & Su, N.‐W. (2023) Discovery of a novel phosphotransferase from Bacillus subtilis that phosphorylates a broad spectrum of flavonoids. Food Chemistry, 400, 134001. [DOI] [PubMed] [Google Scholar]
- Huber, D. , Jamshad, M. , Hanmer, R. , Schibich, D. , Döring, K. , Marcomini, I. et al. (2017) SecA cotranslationally interacts with nascent substrate proteins in vivo. Journal of Bacteriology, 199, e00622‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A.C. , Subramanian, S. , Dann, C.E. & Kearns, D.B. (2018) The C‐terminal region of Bacillus subtilis SwrA is required for activity and adaptor‐dependent LonA proteolysis. Journal of Bacteriology, 200, e00659‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummels, K.R. & Kearns, D.B. (2020) Translation elongation factor P (EF‐P). FEMS Microbiology Reviews, 44, 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison, C.A. , Chuang, R.‐Y. , Noskov, V.N. , Assad‐Garcia, N. , Deerinck, T.J. , Ellisman, M.H. et al. (2016) Design and synthesis of a minimal bacterial genome. Science, 351, aad6253. [DOI] [PubMed] [Google Scholar]
- Jalal, A.S.B. , Tran, N.T. , Wu, L.J. , Ramakrishnan, K. , Rejzek, M. , Gobbato, G. et al. (2021) CTP regulates membrane‐binding activity of the nucleoid occlusion protein Noc. Molecular Cell, 81, 3623–3636.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, W. & Epand, R.M. (2020) CDP‐diacylglycerol, a critical intermediate in lipid metabolism. Chemistry and Physics of Lipids, 230, 104914. [DOI] [PubMed] [Google Scholar]
- Jiang, T. , Li, C. & Yan, Y. (2021) Optimization of a p‐coumaric acid biosensor system for versatile dynamic performance. ACS Synthetic Biology, 10, 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Z. , Wang, C. , Wu, Z. , Chen, K. , Yang, W. , Deng, H. et al. (2021) Enzymatic deamination of the epigenetic nucleoside N6‐methyladenosine regulates gene expression. Nucleic Acids Research, 49, 12048–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, H.A. (1970) Information theory in biology after 18 years. Science, 168, 1545–1550. [DOI] [PubMed] [Google Scholar]
- Kai, M. (2020) Diversity and distribution of volatile secondary metabolites throughout Bacillus subtilis isolates. Frontiers in Microbiology, 11, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski, J. , Cremer, J. , Bachmann, B. , Eggeling, L. , Sahm, H. & Puhler, A. (1991) Genetic and biochemical analysis of the aspartokinase from Corynebacterium glutamicum . Molecular Microbiology, 5, 1197–1204. [DOI] [PubMed] [Google Scholar]
- Kawai, Y. , Mercier, R. , Mickiewicz, K. , Serafini, A. , Sório de Carvalho, L.P. & Errington, J. (2019) Crucial role for central carbon metabolism in the bacterial L‐form switch and killing by β‐lactam antibiotics. Nature Microbiology, 4, 1716–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna, K. , Lopez‐Garrido, J. & Pogliano, K. (2020) Shaping an endospore: architectural transformations during Bacillus subtilis sporulation. Annual Review of Microbiology, 74, 361–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani, T. , Mehboob, F. , Hyder, M.Z. , Zainy, Z. , Xu, L. , Huang, L. et al. (2021) Control of stripe rust of wheat using indigenous endophytic bacteria at seedling and adult plant stage. Scientific Reports, 11, 14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Kim, W. & Kim, J. (2021) New bacterial surface display system development and application based on Bacillus subtilis YuaB biofilm component as an anchoring motif. Biotechnology and Bioprocess Engineering, 26, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, I. , Kim, E. , Yoo, S. , Shin, D. , Min, B. , Song, J. et al. (2004) Ribose utilization with an excess of mutarotase causes cell death due to accumulation of methylglyoxal. Journal of Bacteriology, 186, 7229–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, L. , Lee, B.‐G. , Kim, M. , Kim, M.K. , Kwon, D.H. , Kim, H. et al. (2022) Structural insights into ClpP protease side exit pore‐opening by a pH drop coupled with substrate hydrolysis. The EMBO Journal, 41, e109755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, T.A. , Grebenc, D.W. , Shah, P.Y. , McArthur, O.D. , Dickson, B.H. , Surette, M.G. et al. (2022) Dual targeting factors are required for LXG toxin export by the bacterial type VIIb secretion system. MBio, 13, e0213722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, K. (2021) Diverse LXG toxin and antitoxin systems specifically mediate intraspecies competition in Bacillus subtilis biofilms. PLoS Genetics, 17, e1009682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobras, C.M. , Piepenbreier, H. , Emenegger, J. , Sim, A. , Fritz, G. & Gebhard, S. (2020) BceAB‐type antibiotic resistance transporters appear to act by target protection of cell wall synthesis. Antimicrobial Agents and Chemotherapy, 64, e02241‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, M. , Pandi, A. , Delépine, B. & Faulon, J.‐L. (2018) A dataset of small molecules triggering transcriptional and translational cellular responses. Data in Brief, 17, 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohm, K. , Floccari, V.A. , Lutz, V.T. , Nordmann, B. , Mittelstädt, C. , Poehlein, A. et al. (2022) The Bacillus phage SPβ and its relatives: a temperate phage model system reveals new strains, species, prophage integration loci, conserved proteins and lysogeny management components. Environmental Microbiology, 24, 2098–2118. [DOI] [PubMed] [Google Scholar]
- Kohm, K. & Hertel, R. (2021) The life cycle of SPβ and related phages. Archives of Virology, 166, 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo, B.‐M. , Kritikos, G. , Farelli, J.D. , Todor, H. , Tong, K. , Kimsey, H. et al. (2017) Construction and analysis of two genome‐scale deletion libraries for Bacillus subtilis . Cell Systems, 4, 291–305.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács, Á.T. (2019) Bacillus subtilis . Trends in Microbiology, 27, 724–725. [DOI] [PubMed] [Google Scholar]
- Krüger, L. , Herzberg, C. , Wicke, D. , Bähre, H. , Heidemann, J.L. , Dickmanns, A. et al. (2021) A meet‐up of two second messengers: the c‐di‐AMP receptor DarB controls (p)ppGpp synthesis in Bacillus subtilis . Nature Communications, 12, 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger, L. , Herzberg, C. , Wicke, D. , Scholz, P. , Schmitt, K. , Turdiev, A. et al. (2022) Sustained control of pyruvate carboxylase by the essential second messenger cyclic di‐AMP in Bacillus subtilis . mBio, 13, e0360221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst, F. , Ogasawara, N. , Moszer, I. , Albertini, A.M. , Alloni, G. , Azevedo, V. et al. (1997) The complete genome sequence of the gram‐positive bacterium Bacillus subtilis . Nature, 390, 249–256. [DOI] [PubMed] [Google Scholar]
- Kunz, S. , Tribensky, A. , Steinchen, W. , Oviedo‐Bocanegra, L. , Bedrunka, P. & Graumann, P.L. (2020) Cyclic di‐GMP signaling in Bacillus subtilis is governed by direct interactions of diguanylate cyclases and cognate receptors. MBio, 11, e03122‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita, D. & Himeno, H. (2022) Bacterial ribosome rescue systems. Microorganisms, 10, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana, L.G. , de Araújo, L.M. , Silva, T.F. & Modolo, L.V. (2021) Interplay between gasotransmitters and potassium is a K+ey factor during plant response to abiotic stress. Plant Physiology and Biochemistry, 169, 322–332. [DOI] [PubMed] [Google Scholar]
- Landauer, R. (1961) Irreversibility and heat generation in the computing process. IBM Journal of Research and Development, 3, 184–191. [Google Scholar]
- Lauritsen, I. , Frendorf, P.O. , Capucci, S. , Heyde, S.A.H. , Blomquist, S.D. , Wendel, S. et al. (2021) Temporal evolution of master regulator Crp identifies pyrimidines as catabolite modulator factors. Nature Communications, 12, 5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lécrivain, A.‐L. , Le Rhun, A. , Renault, T.T. , Ahmed‐Begrich, R. , Hahnke, K. & Charpentier, E. (2018) In vivo 3′‐to‐5′ exoribonuclease targetomes of streptococcus pyogenes . Proceedings of the National Academy of Sciences of the United States of America, 115, 11814–11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lécuyer, F. , Bourassa, J.‐S. , Gélinas, M. , Charron‐Lamoureux, V. , Burrus, V. & Beauregard, P.B. (2018) Biofilm formation drives transfer of the conjugative element ICEBs1 in Bacillus subtilis . mSphere, 3, e00473‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisico, F. , Godinho, L.M. , Gonçalves, I.C. , Silva, S.P. , Carneiro, B. , Romão, M.J. et al. (2020) Multitask ATPases (NBDs) of bacterial ABC importers type I and their interspecies exchangeability. Scientific Reports, 10, 19564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma‐Ortiz, C. , Jeffryes, J.G. , Cooper, A.J.L. , Niehaus, T.D. , Thamm, A.M.K. , Frelin, O. et al. (2016) “Nothing of chemistry disappears in biology”: the top 30 damage‐prone endogenous metabolites. Biochemical Society Transactions, 44, 961–971. [DOI] [PubMed] [Google Scholar]
- Li, C.‐C. , Kao, T.‐Y. , Cheng, C.‐C. & Chiang, Y.‐W. (2020) Structure and regulation of the BsYetJ calcium channel in lipid nanodiscs. Proceedings of the National Academy of Sciences of the United States of America, 117, 30126–30134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F.K.K. , Gale, R.T. , Petrotchenko, E.V. , Borchers, C.H. , Brown, E.D. & Strynadka, N.C.J. (2021) Crystallographic analysis of TarI and TarJ, a cytidylyltransferase and reductase pair for CDP‐ribitol synthesis in Staphylococcus aureus wall teichoic acid biogenesis. Journal of Structural Biology, 213, 107733. [DOI] [PubMed] [Google Scholar]
- Li, Q. , Zallot, R. , MacTavish, B.S. , Montoya, A. , Payan, D.J. , Hu, Y. et al. (2021) Epoxyqueuosine reductase QueH in the biosynthetic pathway to tRNA queuosine is a unique metalloenzyme. Biochemistry, 60, 3152–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Hsieh, K.‐Y. , Kuo, C.‐I. , Su, S.‐C. , Huang, K.‐F. , Zhang, K. et al. (2021) Processive cleavage of substrate at individual proteolytic active sites of the Lon protease complex. Science Advances, 7, eabj9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilge, L. , Reder, A. , Tippmann, F. , Morgenroth, F. , Grohmann, J. , Becher, D. et al. (2020) The involvement of the McsB arginine kinase in Clp‐dependent degradation of the MgsR regulator in Bacillus subtilis . Frontiers in Microbiology, 11, 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Z. , Johnson, L.C. , Weissbach, H. , Brot, N. , Lively, M.O. & Lowther, W.T. (2007) Free methionine‐(R)‐sulfoxide reductase from Escherichia coli reveals a new GAF domain function. Proceedings of the National Academy of Sciences of the United States of America, 104, 9597–9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litschko, C. , Budde, I. , Berger, M. , Bethe, A. , Schulze, J. , Alcala Orozco, E.A. et al. (2021) Mix‐and‐match system for the enzymatic synthesis of enantiopure glycerol‐3‐phosphate‐containing capsule polymer backbones from Actinobacillus pleuropneumoniae, Neisseria Meningitidis, and Bibersteinia Trehalosi . mBio, 12, e0089721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luder, R. , Bruni, G.N. & Kralj, J.M. (2021) Genome‐wide functional screen for calcium transients in Escherichia coli identifies increased membrane potential adaptation to persistent DNA damage. Journal of Bacteriology, 203, e00509–e00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytvynenko, I. , Paternoga, H. , Thrun, A. , Balke, A. , Müller, T.A. , Chiang, C.H. et al. (2019) Alanine tails signal proteolysis in bacterial ribosome‐associated quality control. Cell, 178, 76–90.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado, D. , Andrejev, S. , Tramontano, M. & Patil, K.R. (2018) Fast automated reconstruction of genome‐scale metabolic models for microbial species and communities. Nucleic Acids Research, 46, 7542–7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamphogoro, T.P. , Babalola, O.O. & Aiyegoro, O.A. (2020) Sustainable management strategies for bacterial wilt of sweet peppers (Capsicum annuum) and other Solanaceous crops. Journal of Applied Microbiology, 129, 496–508. [DOI] [PubMed] [Google Scholar]
- Martínez Cuesta, S. , Rahman, S.A. , Furnham, N. & Thornton, J.M. (2015) The classification and evolution of enzyme function. Biophysical Journal, 109, 1082–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matavacas, J. & von Wachenfeldt, C. (2022) Update on the protein homeostasis network in Bacillus subtilis . Frontiers in Microbiology, 13, 865141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, A. , Schönfelder, J. , Lagies, S. , Schleicher, E. , Kammerer, B. , Ellis, H.R. et al. (2022) Bacterial flavoprotein monooxygenase YxeK salvages toxic S‐(2‐succino)‐adducts via oxygenolytic C‐S bond cleavage. The FEBS Journal, 289, 787–807. [DOI] [PubMed] [Google Scholar]
- Maurino, V.G. & Engqvist, M.K.M. (2015) 2‐hydroxy acids in plant metabolism. Arabidopsis Book, 13, e0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Médigue, C. , Calteau, A. , Cruveiller, S. , Gachet, M. , Gautreau, G. , Josso, A. et al. (2019) MicroScope‐an integrated resource for community expertise of gene functions and comparative analysis of microbial genomic and metabolic data. Briefings in Bioinformatics, 20, 1071–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske, A.J. , Riley, E.P. , Robins, W.P. , Uehara, T. , Mekalanos, J.J. , Kahne, D. et al. (2016) SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature, 537, 634–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske, A.J. , Sham, L.‐T. , Kimsey, H. , Koo, B.‐M. , Gross, C.A. , Bernhardt, T.G. et al. (2015) MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis . Proceedings of the National Academy of Sciences of the United States of America, 112, 6437–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejlhede, N. , Atkins, J.F. & Neuhard, J. (1999) Ribosomal −1 frameshifting during decoding of Bacillus subtilis cdd occurs at the sequence CGA AAG. Journal of Bacteriology, 181, 2930–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meydan, S. , Klepacki, D. , Mankin, A.S. & Vázquez‐Laslop, N. (2021) Identification of translation start sites in bacterial genomes. Methods in Molecular Biology, 2252, 27–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik, S. , Reder, A. , Richts, B. , Faßhauer, P. , Mäder, U. , Pedreira, T. et al. (2021) The Bacillus subtilis minimal genome compendium. ACS Synthetic Biology, 10, 2767–2771. [DOI] [PubMed] [Google Scholar]
- Mikušević, V. , Schrecker, M. , Kolesova, N. , Patiño‐Ruiz, M. , Fendler, K. & Hänelt, I. (2019) A channel profile report of the unusual K+ channel KtrB. The Journal of General Physiology, 151, 1357–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler, F. , Lavdovskaia, E. & Richter‐Dennerlein, R. (2022) Maintaining mitochondrial ribosome function: the role of ribosome rescue and recycling factors. RNA Biology, 19, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganawa, S. & Ito, M. (2020) MotP subunit is critical for ion selectivity and evolution of a K+‐coupled flagellar motor. Biomolecules, 10, E691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairn, B.L. , Lonergan, Z.R. , Wang, J. , Braymer, J.J. , Zhang, Y. , Calcutt, M.W. et al. (2016) The response of Acinetobacter baumannii to zinc starvation. Cell Host & Microbe, 19, 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, K. & Kumar, P. (2022) Morphological phenotypes, cell division, and gene expression of Escherichia coli under high concentration of sodium sulfate. Microorganisms, 10, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen‐Vo, T.P. , Ko, S. , Ryu, H. , Kim, J.R. , Kim, D. & Park, S. (2020) Systems evaluation reveals novel transporter YohJK renders 3‐hydroxypropionate tolerance in Escherichia coli . Scientific Reports, 10, 19064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus, T.D. , Folz, J. , McCarty, D.R. , Cooper, A.J.L. , Moraga Amador, D. , Fiehn, O. et al. (2018) Identification of a metabolic disposal route for the oncometabolite S‐(2‐succino)cysteine in Bacillus subtilis . The Journal of Biological Chemistry, 293, 8255–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomidis, S.K. , Carlon, E. , Gruber, S. & Marko, J.F. (2022) DNA tension‐modulated translocation and loop extrusion by SMC complexes revealed by molecular dynamics simulations. Nucleic Acids Research, 50, 4974–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novichkov, P.S. , Kazakov, A.E. , Ravcheev, D.A. , Leyn, S.A. , Kovaleva, G.Y. , Sutormin, R.A. et al. (2013) RegPrecise 3.0 – A resource for genome‐scale exploration of transcriptional regulation in bacteria. BMC Genomics, 14, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura, M. (2020) Glucose‐mediated protein arginine phosphorylation/dephosphorylation regulates ylxR encoding nucleoid‐associated protein and cell growth in Bacillus subtilis . Frontiers in Microbiology, 11, 590828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura, M. , Shindo, K. & Kanesaki, Y. (2020) Bacillus subtilis nucleoid‐associated protein YlxR is involved in bimodal expression of the fructoselysine utilization operon (frlBONMD‐yurJ) promoter. Frontiers in Microbiology, 11, 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, Y.‐K. , Palsson, B.O. , Park, S.M. , Schilling, C.H. & Mahadevan, R. (2007) Genome‐scale reconstruction of metabolic network in Bacillus subtilis based on high‐throughput phenotyping and gene essentiality data. The Journal of Biological Chemistry, 282, 28791–28799. [DOI] [PubMed] [Google Scholar]
- Olney, S.G. , Chien, P. & Kearns, D.B. (2022) SmiA is a hybrid priming/scaffolding adaptor for the LonA protease in Bacillus subtilis . The Journal of Biological Chemistry, 298, 102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer‐Shaanan, Y. , Jakoby, G. , Starr, M.L. , Karliner, R. , Eilon, G. , Itkin, M. et al. (2022) A dynamic rhizosphere interplay between tree roots and soil bacteria under drought stress. eLife, 11, e79679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren, A. & Garrity, G.M. (2021) Valid publication of the names of forty‐two phyla of prokaryotes. International Journal of Systematic and Evolutionary Microbiology, 71, 005056. [DOI] [PubMed] [Google Scholar]
- Orth, J.D. , Thiele, I. & Palsson, B.Ø. (2010) What is flux balance analysis? Nature Biotechnology, 28, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima, K. , Gao, X. , Hayashi, S. , Ueda, T. , Nakashima, T. & Kimura, M. (2018) Crystal structures of the archaeal RNase P protein Rpp38 in complex with RNA fragments containing a K‐turn motif. Acta Crystallographica. Section F, Structural Biology Communications, 74, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk, J. , Górnicki, P. , Maj, L. , Dementieva, I. , Laskowski, R. & Joachimiak, A. (2001) Streptococcus pneumonia YlxR at 1.35 a shows a putative new fold. Acta Crystallographica. Section D, Biological Crystallography, 57, 1747–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio‐Valeriano, M. , Altegoer, F. , Steinchen, W. , Urban, S. , Liu, Y. , Bange, G. et al. (2019) ParB‐type DNA segregation proteins are CTP‐dependent molecular switches. Cell, 179, 1512–1524.e15. [DOI] [PubMed] [Google Scholar]
- Ou, Z. , Ouzounis, C. , Wang, D. , Sun, W. , Li, J. , Chen, W. et al. (2020) A path towards SARS‐CoV‐2 attenuation: metabolic pressure on CTP synthesis rules the virus evolution. Genome Biology and Evolution, 12, 2467–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquini, M. , Grosjean, N. , Hixson, K.K. , Nicora, C.D. , Yee, E.F. , Lipton, M. et al. (2022) Zng1 is a GTP‐dependent zinc transferase needed for activation of methionine aminopeptidase. Cell Reports, 39, 110834. [DOI] [PubMed] [Google Scholar]
- Passi, A. , Tibocha‐Bonilla, J.D. , Kumar, M. , Tec‐Campos, D. , Zengler, K. & Zuniga, C. (2021) Genome‐scale metabolic modeling enables in‐depth understanding of big data. Metabolites, 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore, A.J. , Teo, R.D. , Montoya, A. , Burg, M.J. , Twahir, U.T. , Bruner, S.D. et al. (2021) Oxalate decarboxylase uses electron hole hopping for catalysis. The Journal of Biological Chemistry, 297, 100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, S. & Gupta, R.S. (2020) A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: proposal for six new genera of Bacillus species, Peribacillus gen. Nov., Cytobacillus gen. Nov., Mesobacillus gen. Nov., Neobacillus gen. Nov., Metabacillus gen. Nov. and Alkalihalobacillus gen. Nov. International Journal of Systematic and Evolutionary Microbiology, 70, 406–438. [DOI] [PubMed] [Google Scholar]
- Patel, V. , Black, K.A. , Rhee, K.Y. & Helmann, J.D. (2019) Bacillus subtilis PgcA moonlights as a phosphoglucosamine mutase in support of peptidoglycan synthesis. PLoS Genetics, 15, e1008434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, V. , Wu, Q. , Chandrangsu, P. & Helmann, J.D. (2018) A metabolic checkpoint protein GlmR is important for diverting carbon into peptidoglycan biosynthesis in Bacillus subtilis . PLoS Genetics, 14, e1007689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov, M.Y. , Watts, R.E. , Tan, Z. , Cornish, V.W. , Ehrenberg, M. & Forster, A.C. (2009) Slow peptide bond formation by proline and other N ‐alkylamino acids in translation. Proceedings of the National Academy of Sciences of the United States of America, 106, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, C.R. , Tindall, S.N. , Herman, R. , Jenkins, H.T. , Bateman, A. , Thomas, G.H. et al. (2020) Acetylation of surface carbohydrates in bacterial pathogens requires coordinated action of a two‐domain membrane‐bound acyltransferase. MBio, 11, e01364–e01320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedreira, T. , Elfmann, C. & Stülke, J. (2022) The current state of SubtiWiki, the database for the model organism Bacillus subtilis . Nucleic Acids Research, 50, D875–D882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, N. , Cai, P. , Mortimer, M. , Wu, Y. , Gao, C. & Huang, Q. (2020) The exopolysaccharide‐eDNA interaction modulates 3D architecture of Bacillus subtilis biofilm. BMC Microbiology, 20, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensinger, D.A. , Gutierrez, K.V. , Smith, H.B. , Vincent, W.J.B. , Stevenson, D.S. , Black, K.A. et al. (2021) Listeria monocytogenes GlmR is an accessory uridyltransferase essential for cytosolic survival and virulence. mBio, 20, e0007323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera, V.R. , Lapek, J.D. , Newton, G.L. , Gonzalez, D.J. & Pogliano, K. (2018) Identification of the S‐transferase like superfamily bacillithiol transferases encoded by Bacillus subtilis . PLoS One, 13, e0192977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi, H. & Helmann, J.D. (2017) Sequential induction of fur‐regulated genes in response to iron limitation in Bacillus subtilis . Proceedings of the National Academy of Sciences of the United States of America, 114, 12785–12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi, H. , Wendel, B.M. & Helmann, J.D. (2020) Dysregulation of magnesium transport protects Bacillus subtilis against manganese and cobalt intoxication. Journal of Bacteriology, 202, e00711‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pištěková, H. , Jančová, P. , Buňková, L. , Šopík, T. , Maršálková, K. , Berčíková, L. et al. (2022) Detection and relative quantification of amine oxidase gene (yobN) in Bacillus subtilis: application of real‐time quantitative PCR. Journal of Food Science and Technology, 59, 909–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp, P.F. , Friebel, L. , Benjdia, A. , Guillot, A. , Berteau, O. & Mascher, T. (2021) The epipeptide biosynthesis locus epeXEPAB is widely distributed in Firmicutes and triggers intrinsic cell envelope stress. Microbial Physiology, 31, 306–318. [DOI] [PubMed] [Google Scholar]
- Prajapati, B. , Bernal‐Cabas, M. , López‐Álvarez, M. , Schaffer, M. , Bartel, J. , Rath, H. et al. (2021) Double trouble: Bacillus depends on a functional tat machinery to avoid severe oxidative stress and starvation upon entry into a NaCl‐depleted environment. Biochimica et Biophysica Acta ‐ Molecular Cell Research, 1868, 118914. [DOI] [PubMed] [Google Scholar]
- Price, M.N. , Deutschbauer, A.M. & Arkin, A.P. (2020) GapMind: automated annotation of amino acid biosynthesis. mSystems, 5, e00291–e00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Y. , Angelini, L.L. & Chai, Y. (2022) Bacillus subtilis cell differentiation, biofilm formation and environmental prevalence. Microorganisms, 10, 1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai, A.K. & Mitchell, A.M. (2020) Enterobacterial common antigen: synthesis and function of an enigmatic molecule. MBio, 11, e01914‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkovic, A. & Ibba, M. (2017) Elongation factor P and the control of translation elongation. Annual Review of Microbiology, 71, 117–131. [DOI] [PubMed] [Google Scholar]
- Randall, T.E. , Eckartt, K. , Kakumanu, S. , Price‐Whelan, A. , Dietrich, L.E.P. & Harrison, J.J. (2022) Sensory perception in bacterial cyclic diguanylate signal transduction. Journal of Bacteriology, 204, e0043321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richts, B. , Hertel, R. , Potot, S. , Poehlein, A. , Daniel, R. , Schyns, G. et al. (2020) Complete genome sequence of the prototrophic Bacillus subtilis subsp. subtilis strain SP1. Microbiology Resource Announcements, 9, e00825–e00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski, E. , Ehrmann, M.A. & Vogel, R.F. (2021) Intraspecies diversity and genome‐phenotype‐associations in Fructilactobacillus sanfranciscensis . Microbiological Research, 243, 126625. [DOI] [PubMed] [Google Scholar]
- Rojas‐Tapias, D.F. & Helmann, J.D. (2018) Stabilization of Bacillus subtilis Spx under cell wall stress requires the anti‐adaptor protein YirB. PLoS Genetics, 14, e1007531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, H. , Serrano, E. , Hernández‐Tamayo, R. , Carrasco, B. , Cárdenas, P.P. , Ayora, S. et al. (2020) Bacillus subtilis RarA acts as a positive RecA accessory protein. Frontiers in Microbiology, 11, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romine, M.F. , Rodionov, D.A. , Maezato, Y. , Anderson, L.N. , Nandhikonda, P. , Rodionova, I.A. et al. (2017) Elucidation of roles for vitamin B12 in regulation of folate, ubiquinone, and methionine metabolism. Proceedings of the National Academy of Sciences of the United States of America, 114, E1205–E1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roney, I.J. & Rudner, D.Z. (2022) Two broadly conserved families of polyprenyl‐phosphate transporters. Nature, 613, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropelewska, M. , Gross, M.H. & Konieczny, I. (2020) DNA and polyphosphate in directed proteolysis for DNA replication control. Frontiers in Microbiology, 11, 585717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov, A. , Khusainov, I. , El Omari, K. , Duman, R. , Mykhaylyk, V. , Yusupov, M. et al. (2019) Importance of potassium ions for ribosome structure and function revealed by long‐wavelength X‐ray diffraction. Nature Communications, 10, 2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio Gomez, M.A. & Ibba, M. (2020) Aminoacyl‐tRNA synthetases. RNA, 26, 910–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan‐Payseur, B.K. & Freitag, N.E. (2018) Bacillus subtilis biofilms: a matter of individual choice. MBio, 9, e02339–e02318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachla, A.J. & Helmann, J.D. (2019) A bacterial checkpoint protein for ribosome assembly moonlights as an essential metabolite‐proofreading enzyme. Nature Communications, 10, 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachla, A.J. , Luo, Y. & Helmann, J.D. (2021) Manganese impairs the QoxABCD terminal oxidase leading to respiration‐associated toxicity. Molecular Microbiology, 116, 729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadykov, M.R. , Windham, I.H. , Widhelm, T.J. , Yajjala, V.K. , Watson, S.M. , Endres, J.L. et al. (2019) CidR and CcpA synergistically regulate Staphylococcus aureus cidABC expression. Journal of Bacteriology, 201, e00371‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, S. , Snider, E.V. , Wang, X. & Kearns, D.B. (2022) Identification of genes required for swarming motility in Bacillus subtilis using transposon mutagenesis and high‐throughput sequencing (TnSeq). Journal of Bacteriology, 204, e0008922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarvas, M. , Harwood, C.R. , Bron, S. & van Dijl, J.M. (2004) Post‐translocational folding of secretory proteins in gram‐positive bacteria. Biochimica et Biophysica Acta, 1694, 311–327. [DOI] [PubMed] [Google Scholar]
- Schibich, D. , Gloge, F. , Pöhner, I. , Björkholm, P. , Wade, R.C. , von Heijne, G. et al. (2016) Global profiling of SRP interaction with nascent polypeptides. Nature, 536, 219–223. [DOI] [PubMed] [Google Scholar]
- Seffouh, A. , Jain, N. , Jahagirdar, D. , Basu, K. , Razi, A. , Ni, X. et al. (2019) Structural consequences of the interaction of RbgA with a 50S ribosomal subunit assembly intermediate. Nucleic Acids Research, 47, 10414–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seffouh, A. , Trahan, C. , Wasi, T. , Jain, N. , Basu, K. , Britton, R.A. et al. (2022) RbgA ensures the correct timing in the maturation of the 50S subunits functional sites. Nucleic Acids Research, 50, 10801–10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekowska, A. , Ashida, H. & Danchin, A. (2019) Revisiting the methionine salvage pathway and its paralogues. Microbial Biotechnology, 12, 77–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, F. , Chen, J. , Wang, L. , Jin, L. , Zou, L. , Bu, T. et al. (2018) Crystal structure of the nicotinamidase/pyrazinamidase PncA from Bacillus subtilis . Biochemical and Biophysical Research Communications, 503, 2906–2911. [DOI] [PubMed] [Google Scholar]
- Shi, L. , Derouiche, A. , Pandit, S. , Rahimi, S. , Kalantari, A. , Futo, M. et al. (2020) Evolutionary analysis of the Bacillus subtilis genome reveals new genes involved in sporulation. Molecular Biology and Evolution, 37, 1667–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, J.‐H. & Helmann, J.D. (2016) Molecular logic of the Zur‐regulated zinc deprivation response in Bacillus subtilis . Nature Communications, 7, 12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, Y. , Hedglin, M. & Murakami, K.S. (2020) Structural basis of reiterative transcription from the pyrG and pyrBI promoters by bacterial RNA polymerase. Nucleic Acids Research, 48, 2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiwa, Y. , Matsumoto, T. & Yoshikawa, H. (2013) Identification of laboratory‐specific variations of Bacillus subtilis strains used in Japan. Bioscience, Biotechnology, and Biochemistry, 77, 2073–2076. [DOI] [PubMed] [Google Scholar]
- Shuster, B. , Khemmani, M. , Nakaya, Y. , Holland, G. , Iwamoto, K. , Abe, K. et al. (2019) Expansion of the spore surface polysaccharide layer in Bacillus subtilis by deletion of genes encoding glycosyltransferases and glucose modification enzymes. Journal of Bacteriology, 201, e00321‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirén, K. , Millard, A. , Petersen, B. , Gilbert, M.T.P. , Clokie, M.R.J. & Sicheritz‐Pontén, T. (2021) Rapid discovery of novel prophages using biological feature engineering and machine learning. NAR Genomics and Bioinformatics, 3, lqaa109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeparnias, I. & Zhang, J. (2021) Cooperativity and interdependency between RNA structure and RNA‐RNA interactions. Noncoding RNA, 7, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, B.L. & Doran, K.S. (2022) Evolving understanding of the type VII secretion system in gram‐positive bacteria. PLoS Pathogens, 18, e1010680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinke, K. , Mohite, O.S. , Weber, T. & Kovács, Á.T. (2021) Phylogenetic distribution of secondary metabolites in the Bacillus subtilis species complex. mSystems, 6, e00057‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll, J. , Zegarra, V. , Bange, G. & Graumann, P.L. (2022) Single‐molecule dynamics suggest that ribosomes assemble at sites of translation in Bacillus subtilis . Frontiers in Microbiology, 13, 999176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stragier, P. (2022) To feed or to stick? Genomic analysis offers clues for the role of a molecular machine in endospore formers. Journal of Bacteriology, 204, e0018722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbendieck, R.M. & Straight, P.D. (2017) Linearmycins activate a two‐component signaling system involved in bacterial competition and biofilm morphology. Journal of Bacteriology, 199, e00186‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, Y. , Liu, C. , Fang, H. & Zhang, D. (2020) Bacillus subtilis: a universal cell factory for industry, agriculture, biomaterials and medicine. Microbial Cell Factories, 19, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, G. , Fogel, G.B. , Abramson, B. , Brinkac, L. , Michael, T. , Liu, E.S. et al. (2021) A pan‐genome method to determine core regions of the Bacillus subtilis and Escherichia coli genomes. F1000Research, 10, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada, H. , Crowe‐McAuliffe, C. , Polte, C. , Sidorova, Z.Y. , Murina, V. , Atkinson, G.C. et al. (2021) RqcH and RqcP catalyze processive poly‐alanine synthesis in a reconstituted ribosome‐associated quality control system. Nucleic Acids Research, 49, 8355–8369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada, H. , Mandell, Z.F. , Yakhnin, H. , Glazyrina, A. , Chiba, S. , Kurata, T. et al. (2022) Expression of Bacillus subtilis ABCF antibiotic resistance factor VmlR is regulated by RNA polymerase pausing, transcription attenuation, translation attenuation and (p)ppGpp. Nucleic Acids Research, 50, 6174–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada, H. , Morita, M. , Shiwa, Y. , Sugimoto, R. , Suzuki, S. , Kawamura, F. et al. (2014) Cell motility and biofilm formation in Bacillus subtilis are affected by the ribosomal proteins, S11 and S21. Bioscience, Biotechnology, and Biochemistry, 78, 898–907. [DOI] [PubMed] [Google Scholar]
- Tassinari, M. , Doan, T. , Bellinzoni, M. , Chabalier, M. , Ben‐Assaya, M. , Martinez, M. et al. (2022) The antibacterial type VII secretion system of Bacillus subtilis: structure and interactions of the pseudokinase YukC/EssB. MBio, 13, e0013422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng, X. , Sheng, D. , Wang, J. , Yu, Y. & Hattori, M. (2022) Ion selectivity mechanism of the MgtE channel for Mg2+ over Ca2. iScience, 25, 105565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terahara, N. , Namba, K. & Minamino, T. (2020) Dynamic exchange of two types of stator units in Bacillus subtilis flagellar motor in response to environmental changes. Computational and Structural Biotechnology Journal, 18, 2897–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangadurai, S. , Bajgiran, M. , Manickam, S. , Mohana‐Kumaran, N. & Azzam, G. (2022) CTP synthase: the hissing of the cellular serpent. Histochemistry and Cell Biology, 158, 517–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibocha‐Bonilla, J.D. , Zuñiga, C. , Lekbua, A. , Lloyd, C. , Rychel, K. , Short, K. et al. (2022) Predicting stress response and improved protein overproduction in Bacillus subtilis . NPJ Systems Biology and Applications, 8, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley, L. , Yannarell, S.M. , Huynh, T.N. , Woodward, J.J. & Shank, E.A. (2018) Cyclic di‐AMP acts as an extracellular signal that impacts Bacillus subtilis biofilm formation and plant attachment. MBio, 9, e00341‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrych, A. , Petráčková, D. , Goldová, J. , Buriánková, K. , Doubravová, L. & Branny, P. (2020) PynA is a pyrimidine 5′‐nucleotidase that functions as an antimutator protein in Streptococcus pneumoniae . The FEBS Journal, 287, 267–283. [DOI] [PubMed] [Google Scholar]
- Vallenet, D. , Belda, E. , Calteau, A. , Cruveiller, S. , Engelen, S. , Lajus, A. et al. (2013) MicroScope–an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Research, 41, D636–D647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallenet, D. , Calteau, A. , Dubois, M. , Amours, P. , Bazin, A. , Beuvin, M. et al. (2020) MicroScope: an integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucleic Acids Research, 48, D579–D589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, N. , Srivastava, S. , Malik, R. , Yadav, J.K. , Goyal, P. & Pandey, J. (2020) Computational investigation for modeling the protein‐protein interaction of TasA(28‐261)‐TapA(33‐253): a decisive process in biofilm formation by Bacillus subtilis . Journal of Molecular Modeling, 26, 226. [DOI] [PubMed] [Google Scholar]
- Warmbold, B. , Ronzheimer, S. , Freibert, S.‐A. , Seubert, A. , Hoffmann, T. & Bremer, E. (2020) Two MarR‐type repressors balance precursor uptake and glycine betaine synthesis in Bacillus subtilis to provide cytoprotection against sustained osmotic stress. Frontiers in Microbiology, 11, 1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wein, T. & Sorek, R. (2022) Bacterial origins of human cell‐autonomous innate immune mechanisms. Nature Reviews. Immunology, 22, 629–638. [DOI] [PubMed] [Google Scholar]
- Weiss, A. , Murdoch, C.C. , Edmonds, K.A. , Jordan, M.R. , Monteith, A.J. , Perera, Y.R. et al. (2022) Zn‐regulated GTPase metalloprotein activator 1 modulates vertebrate zinc homeostasis. Cell, 185, 2148–2163.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, E.L. , Fang, M. , Taton, A. , Szubin, R. , Palsson, B.Ø. , Mitchell, B.G. et al. (2022) An unexpected role for leucyl aminopeptidase in UV tolerance revealed by a genome‐wide fitness assessment in a model cyanobacterium. Proceedings of the National Academy of Sciences of the United States of America, 119, e2211789119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, B.M. , Pi, H. , Krüger, L. , Herzberg, C. , Stülke, J. & Helmann, J.D. (2022) A central role for magnesium homeostasis during adaptation to osmotic stress. MBio, 13, e0009222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiame, E. , Lamosa, P. , Santos, H. & Van Schaftingen, E. (2005) Identification of glucoselysine‐6‐phosphate deglycase, an enzyme involved in the metabolism of the fructation product glucoselysine. The Biochemical Journal, 392, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzky, A. , Hummels, K.R. , Tollerson, R. , Rajkovic, A. , Jones, L.A. , Kearns, D.B. et al. (2018) EF‐P posttranslational modification has variable impact on polyproline translation in Bacillus subtilis . MBio, 9, e00306–e00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C.‐H. , Rismondo, J. , Morgan, R.M.L. , Shen, Y. , Loessner, M.J. , Larrouy‐Maumus, G. et al. (2021) Bacillus subtilis YngB contributes to wall teichoic acid glucosylation and glycolipid formation during anaerobic growth. The Journal of Biological Chemistry, 296, 100384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H. , Wang, D. & Gao, F. (2021) Toward a high‐quality pan‐genome landscape of Bacillus subtilis by removal of confounding strains. Briefings in Bioinformatics, 22, 1951–1971. [DOI] [PubMed] [Google Scholar]
- Yokoyama, N. , Nonaka, C. , Ohashi, Y. , Shioda, M. , Terahata, T. , Chen, W. et al. (2018) Distinct roles for U‐type proteins in iron‐sulfur cluster biosynthesis revealed by genetic analysis of the Bacillus subtilis sufCDSUB operon. Molecular Microbiology, 107, 688–703. [DOI] [PubMed] [Google Scholar]
- You, C. , Okano, H. , Hui, S. , Zhang, Z. , Kim, M. , Gunderson, C.W. et al. (2013) Coordination of bacterial proteome with metabolism by cyclic AMP signalling. Nature, 500, 301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. , Zhou, J. , Gueiros‐Filho, F.J. , Kearns, D.B. & Jacobson, S.C. (2021) Noc corrals migration of FtsZ protofilaments during cytokinesis in Bacillus subtilis . MBio, 12, e02964‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaprasis, A. , Brill, J. , Thüring, M. , Wünsche, G. , Heun, M. , Barzantny, H. et al. (2013) Osmoprotection of Bacillus subtilis through import and proteolysis of proline‐containing peptides. Applied and Environmental Microbiology, 79, 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigler, D.R. , Prágai, Z. , Rodriguez, S. , Chevreux, B. , Muffler, A. , Albert, T. et al. (2008) The origins of 168, W23, and other Bacillus subtilis legacy strains. Journal of Bacteriology, 190, 6983–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, A. , Lebrun, R. , Espinosa, L. , Galinier, A. & Pompeo, F. (2022) PrkA is an ATP‐dependent protease that regulates sporulation in Bacillus subtilis . The Journal of Biological Chemistry, 298, 102436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Wang, H. , Xie, T. , Huang, Q. , Xiong, X. , Liu, Q. et al. (2020) The YmdB protein regulates biofilm formation dependent on the repressor SinR in Bacillus cereus 0‐9. World Journal of Microbiology and Biotechnology, 36, 165. [DOI] [PubMed] [Google Scholar]
- Zhao, H. , Sachla, A.J. & Helmann, J.D. (2019) Mutations of the Bacillus subtilis YidC1 (SpoIIIJ) insertase alleviate stress associated with σM‐dependent membrane protein overproduction. PLoS Genetics, 15, e1008263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H. , Yang, S.‐M. , Yuan, Z.‐M. & Ban, R. (2015) Metabolic and genetic factors affecting the productivity of pyrimidine nucleoside in Bacillus subtilis . Microbial Cell Factories, 14, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta, C. , Arkus, K.A.J. , Cahoon, R.E. & Jez, J.M. (2008) A single amino acid change is responsible for evolution of acyltransferase specificity in bacterial methionine biosynthesis. The Journal of Biological Chemistry, 283, 7561–7567. [DOI] [PubMed] [Google Scholar]
- Zuñiga, C. , Li, T. , Guarnieri, M.T. , Jenkins, J.P. , Li, C.‐T. , Bingol, K. et al. (2020) Synthetic microbial communities of heterotrophs and phototrophs facilitate sustainable growth. Nature Communications, 11, 3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4