

Abstract
Camptothecin-like compounds are actively employed as anticancer drugs in clinical treatments. The aromathecin family of compounds, which contains the same indazolidine core structure as the camptothecin family of compounds, is also expected to display promising anticancer activity. Therefore, the development of a suitable and scalable synthetic method of aromathecin synthesis is of great research interest. In this study, we report the development of a new synthetic approach for constructing the pentacyclic scaffold of the aromathecin family by forming the indolizidine moiety after synthesizing the isoquinolone moiety. Thermal cyclization of 2-alkynylbenzaldehyde oxime to the isoquinoline N-oxide, followed by a Reissert–Henze-type reaction, forms the key strategy in this isoquinolone synthesis. Under the optimum reaction conditions for the Reissert–Henze-type reaction step, microwave irradiation-assisted heating of the purified N-oxide in acetic anhydride at 50 °C reduced the formation of the 4-acetoxyisoquinoline byproduct to deliver the desired isoquinolone at a 73% yield after just 3.5 h. The eight-step sequence employed afforded rosettacin (simplest member of the aromathecin family) at a 23.8% overall yield. The synthesis of rosettacin analogs was achieved by applying the developed strategy and may be generally applicable to the production of other fused indolizidine compounds.
Keywords: acuminatine; 22-hydroxyacuminatine; benz[6,7]indolizino[1,2-b]quinolin-11(13H)-one; total synthesis; thermal cyclization
1. Introduction
The indolizidine and quinolizidine moieties are important core structures that can be found in several biologically active compounds, such as camptothecin (1) [1], rosettacin (5) [2], and the 8-oxoprotoberberine alkaloids 8–10 [3,4] (Figure 1). The study of indolizidine- or quinolizidine-containing compounds has attracted the interest of many research groups because these compounds exhibit antitumor activity. Among such compounds, camptothecin (1) was isolated by Wani and co-workers in 1966 from the Chinese tree Camptotheca acuminata [1]. This alkaloid was shown to potently inhibit tumor growth by binding to the topoisomerase I enzyme (Top1). Subsequently, drug development studies were conducted with 1 as the lead compound. As a result, irinotecan (2) [5], topotecan (3) [6], and belotecan (4) [7], all compounds bearing substituents on the AB-ring, were developed for use in clinical studies. However, hydrolysis of the E-ring lactone moiety produces hydroxycarboxylates with a high affinity for human serum albumin protein; thus, the E-ring lactone hydrolysis product is responsible for attenuating the activity of 1 derivatives [8]. To address this drawback, the development novel anticancer drugs has focused on the aromathecin family of compounds (benz[6,7]indolizino[1,2-b]quinolin-11(13H)-ones), in which the lactone moiety of 1 is replaced with a benzene ring.
Figure 1.

Structures of natural products containing the indolizinone or quinolizinone scaffold [1,2,3,4,5,6,7,9,10].
To date, three members of the aromathecin family are known: rosettacin (5), 22-hydroxyacuminatine (6) [6], and acuminatine (7) [7]. Together with 1, 6 has been isolated at a very low yield from the seeds of C. accuminata [6]. Moreover, rosettacin, its derivatives, and 6 have been reported to display weak Top1 inhibitory activity [11,12,13,14,15,16]. Therefore, aromathecins can be considered as a new class of Top1 inhibitors which can replace camptothecins as candidates for therapeutic development. To date, many approaches have been reported for the synthesis of aromathecins. These routes primarily focus on a late-stage indolizidine moiety construction by either C- or D-ring formation [15,17,18,19,20,21,22,23,24,25,26]. Indeed, several synthetic procedures have been developed in recent years which enable the facile production of aromathecins. Eycken and co-workers reported the total synthesis of 5 via three synthetic strategies [17,18,19]. The first approach is based on using an intramolecular cascade annulation of O-substituted N-hydroxybenzamides, triggered by Rh(III)-catalyzed sequential C(sp2)-H activation and C(sp3)-H amination, for the synthesis of indolizinones [17]. The next method involves the intermolecular annulation of 2-acetylenic aldehydes with O-substituted N-hydroxybenzamides through Rh(III)-catalyzed C–H activation for the synthesis of indolizinones [18]. The third route comprised the construction of indolizinones by an intramolecular cascade annulation triggered by C–H activation via rhodium hydride intermediate [19]. Huang and co-workers reported the synthesis of 5 via the construction of isoquinolone using carbene-catalyzed aerobic oxidation of isoquinolinium salts, followed by a Pd-catalyzed intramolecular cyclization [20]. Reddy and co-workers developed the synthesis of 5 by a one-pot method for the synthesis of 7-hydroxyisoindolo[2,1-b]isoquinolin-5(7H)-ones from N-(pivaloyloxy)benzamides and 2-alkynyl aldehydes via Rh(III)-catalyzed C–H functionalization [21]. Furthermore, Evano and co-workers developed a method to assemble the indolizinone moiety by copper-catalyzed photoinduced radical domino cyclization of ynamides and applied this method to the synthesis of 5 [22]. Glorius and co-workers reported the synthesis of 5 and its derivatives via the construction of isoquinolones by the intramolecular annulation of a Cp*CoIII-catalyzed C–H activation approach for N-(pent-4-yn-1-yloxy)benzamide production as the key step [23].
Our research group has been interested in the unique structure and pharmacological action of condensed heteroaromatic compounds and we have been searching for highly active compounds based on these naturally occurring compounds and their derivatives. To date, we have achieved the total synthesis of several such compounds using various types of 6π-electron system (hexatriene, 1- and 2-azahexatriene) electrocyclic reactions, such as asiaticumine [27], marinoquinolines [28], trigonoine B [29], girinimbine [30], and karnatakafuran B [31]. We have also published the total synthesis of (R)-(–)-pyridindolols using another synthetic approach that involved thermal cyclization of 3-alkynylindole-2-aldoxime to construct the key β-carboline N-oxide intermediate [32,33].
Recently, we developed a versatile synthetic route to produce 8-oxoprotoberberine alkaloids (alangiumkaloids A (9) and B (10)) through the synthesis of isoquinolone and via B-ring construction [34] (Scheme 1). The key step in this was the synthesis of isoquinolone 13 through the thermal cyclization of 2-alkynylbenzaldehyde oxime 11 to afford isoquinoline N-oxide 12, followed by a Reissert–Henze-type reaction. The lithium aluminum hydride (LiAlH4) reduction of both the obtained isoquinolone 13 and 1-acetoxyisoquinoline 14, followed by quinolizidine ring construction (B-ring formation), completed the first total synthesis of alangiumkaloids 9 and 10. Employing this strategy, we also synthesized a series of alangiumkaloid analogs and are currently conducting exploratory antitumor activity research into these compounds.
Scheme 1.

Key synthetic transformations for the synthesis of alangiumkaloids A and B.
In this paper, we report on the synthesis of rosettacin (5) and its derivatives, a feat achieved by applying the synthetic strategies from our previous work to construction of a benz[6,7]indolizino[1,2-b]quinolin-11(13H)-one scaffold.
2. Results and Discussion
Our retrosynthetic analysis of 5 is presented in Scheme 2. The rosettacin scaffold can be synthesized by late-stage construction of the indolizine ring (CD-ring) moiety through bond formation between C13 and N12. The precursor isoquinolone 15 (DE-ring) can be obtained from a Reissert–Henze-type reaction of isoquinoline N-oxide 16, which can be formed from the thermal cyclization of 2-alkynylbenzaldehyde oxime 17. We envisioned that 17 could be synthesized from the Sonogashira reaction between 2-iodoquinoline 18 and 2-ethynylbenzaldehyde 19. The advantage of this synthetic strategy was that it allowed for easy access to rosettacin analogs by derivatizing both the starting quinolines and 2-ethynylbenzaldehydes.
Scheme 2.

Retrosynthetic analysis of rosettacin (5).
We began the synthesis of three quinolines (AB-ring) in possession of the necessary substitutions, as shown in Scheme 3. The starting materials, 2-chloroquinolines 18a and 18b, were synthesized from acetanilide according to the method of Bhuyan and co-workers [35]. Subsequently, 2-chloroquinolines 18a and 18b were heated with NaI and concentrated HCl in acetonitrile (MeCN) to obtain 2-iodoquinolines 20a and 20b at 92% and 90% yields, respectively [36]. Treatment of 20b with iodine and K2CO3 in methanol (MeOH) provided methyl ester 21 at an 89% yield. Alternatively, the reduction of 20b with NaBH4 followed by hydroxy group methylation provided 3-methoxymethylquinoline 23 at an 80% yield over 2 steps.
Scheme 3.

Synthesis of 3-substituted 2-iodoquinolines 20a, 21, and 23.
Next, we prepared the key isoquinolone synthesis precursors: 2-alkynylbenzaldehyde oximes 25 (Scheme 4). The Sonogashira reaction of 2-iodoquinolines 21, 20a, and 23 with 2-ethynylbenzaldehyde 19 in the presence of CuI, triethylamine (Et3N) and bis(triphenylphosphine)palladium(II) chloride (PdCl2(PPh3)2) provided 2-alkynylbenzaldehydes 24a, 24b, and 24c at 44%, 71%, and 84% yields, respectively. The treatment of 24a–c with hydroxylamine produced oximes 25a–c at 68%, 53%, and 86% yields, respectively.
Scheme 4.

Synthesis of key isoquinolone precursor 2-alkynylbenzaldehyde oxime 25.
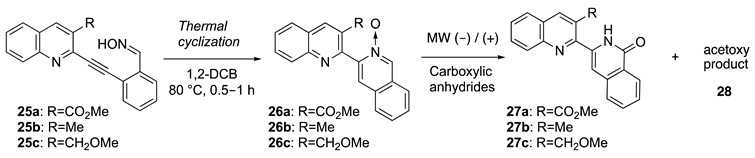
As shown in Table 1, we next investigated the synthesis of isoquinolone 27 via the construction of isoquinoline N-oxide 26 from oxime 25. First, oxime 25a was heated in 1,2-dichlorobenzene (1,2-DCB) at 180 °C until the starting material was no longer detectable by thin-layer chromatography (TLC) analysis. The reaction was quenched by the evaporation of 1,2-DCB in vacuo to obtain N-oxide 26. Subsequently, without further purification, the crude N-oxide 26a was heated in acetic anhydride (Ac2O) at 110 °C. Disappointingly, the desired isoquinolone 27a was not obtained and an unidentifiable mixture of compounds was produced (entry 1). Reducing the cyclization temperature to 80 °C, the thermal cyclization of 25a proceeded smoothly; however, the subsequent heating of the resulting crude 26a in Ac2O at 110 °C also did not afford isoquinoline 27a (entry 2). Therefore, compounds containing an ester functional group were deemed to be unsuitable for the Reissert–Henze-type reaction and studies of oxime 25a ceased.
Table 1.
Optimization studies for the thermal cyclization synthesis of isoquinolone 27 starting from 2-alkynylbenzaldehyde oxime 25.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Starting Material | Method (1) | Carboxylic Anhydride | MW (2) | Temp. (°C) | Time (h) | Yield (%) | |||
| No. | R | No. | 27 | 28 | ||||||
| 1 (3) | 25a | CO2Me | A | Ac2O | − | 110 | 2 | a | − | − |
| 2 | 25a | CO2Me | A | Ac2O | − | 110 | 1.5 | a | − | − |
| 3 | 25b | Me | A | Ac2O | − | 110 | 3 | b | 6 | 33 |
| 4 | 25c | CH2OMe | A | Ac2O | − | 110 | 3 | c | 22 | 40 |
| 5 | 25c | CH2OMe | B | Ac2O | − | 110 | 2 | c | 31 | 43 |
| 6 | 25c | CH2OMe | A | Ac2O | − | 50 | 24 | c | 48 | 27 |
| 7 | 25c | CH2OMe | B | Ac2O | − | 50 | 41 | c | 41 | 33 |
| 8 | 25c | CH2OMe | B | Ac2O | − | rt | 51 | c | 60 | 18 |
| 9 | 25c | CH2OMe | B | Ac2O | + | 110 | 2 | c | 20 | 29 |
| 10 | 25c | CH2OMe | A | Ac2O | + | 50 | 14 | c | 39 | 15 |
| 11 | 25c | CH2OMe | B | Ac2O | + | 50 | 3.5 | c | 73 | 21 |
| 12 | 25c | CH2OMe | A | (CF3CO)2O | − | rt | 1 | c | 14 | − |
| 13 | 25c | CH2OMe | A | Ac2O, PTSA (4) | − | 50 | 39 | c | 28 | 10 |
(1) Solutions of 2-alkynylbenzaldehyde oxime 25 in 1,2-dichlorobenzene (1,2-DCB) were heated at 80 °C until the starting material was no longer detectable by thin-layer chromatography (TLC) analysis. The reaction was quenched by evaporation of the solvent under reduced pressure. Method A: The obtained crude N-oxide 26 was used for the next reaction without further purification. The appropriate carboxylic anhydride was added to crude N-oxide 26 and heated to the desired study temperature. Method B: The obtained N-oxide 26 was purified by recrystallization (73% yield). Recrystallized N-oxide 26 was added to the appropriate carboxylic anhydride and heated to the desired study temperature. (2) MW: microwave; (3) the thermal cyclization of oxime 25a was carried out at 180 °C; (4) PTSA: p-toluenesulfonic acid.
Next, we investigated the synthesis of isoquinolone 27b from oxime 25b. N-oxide 26b was obtained by heating oxime 25b in 1,2-DCB at 80 °C. Thereafter, crude 26b was heated in Ac2O at 110 °C to afford the desired 27b along with acetoxy 28b at 6% and 33% yields, respectively (entry 3).
Subsequently, the synthesis of isoquinolone 27c from oxime 25c was investigated according to Method A (entry 4). Isoquinolone 27c and the acetoxy product 28c were obtained at 22% and 40% yields, respectively. From these exploratory results, oxime 25c was selected for use in further optimization studies of the Reissert–Henze-type reaction to isoquinolone 27c.
To examine the effect of N-oxide 26c purity on the subsequent reaction, the crude N-oxide 26c obtained by thermal cyclization was purified by crystallization (purification yield 73%). Purified N-oxide 26c was heated in Ac2O at 110 °C (entry 5), resulting in slightly improved yields of 27c (from 22% to 31%). When employing crude 26c, lowering the Reissert–Henze-type reaction temperature to 50 °C further improved the yield of 27c to 48% yield (entry 6). Notably, the production of acetoxy 28c was reduced at the lower reaction temperature. Returning to Method B, the reaction of purified 26c at 50 °C did not improve the yield of 27c (entry 7). However, employing ambient temperature (rt) conditions, stirring purified 26c in Ac2O improved the yield of 27c to 60% and further reduced the production of 28c to 18% (entry 8), although the reaction time required was significantly longer (51 h). From these early optimization studies, it may be asserted that the Reissert–Henze-type synthesis of isoquinolone 27c produced better yields when starting from the purified N-oxide 26c.
Next, the effect of microwave (MW) technology on the Reissert–Henze-type reaction was studied. Unfortunately, the yield of 27c decreased to 20% when purified 26c was heated in Ac2O at 110 °C under MW irradiation (entry 9). To directly compare the effects of MW irradiation (entry 6 vs. entry 10), crude 26c was heated in Ac2O at 50 °C under MW, the reaction time required was greatly reduced from 24 h to 14 h and the production of 28c was further reduced. Gratifyingly, using the same conditions of entry 10, purified 26c was transformed into 27c with a greatly improved yield of 73% in a short 3.5 h reaction time. Briefly, the use of trifluoroacetic anhydride in place of Ac2O and the addition of an acid catalyst were investigated; however, the yield of 27c did not improve further (entries 12 and 13). Therefore, the conditions of entry 11 were deemed optimal for the Reissert–Henze-type synthesis of isoquinolone 27c from 25c.
During our Reissert–Henze-type reaction optimization studies, because of our work on the synthesis of alangiumkaloids (Scheme 1), we predicted that the obtained acetoxy 28c was the 1-acetoxyisoquinoline product (Scheme 5). Therefore, we attempted to remove the acetyl group of 28c by LiAlH4 reduction. However, the 1H-NMR spectra of the obtained compound 29 did not match that of isoquinolone 27c. Further 2D-NMR studies (heteronuclear multiple-bond correlation (HMBC) and nuclear Overhauser effect spectroscopy (NOESY) measurements) were performed to verify the structure of the obtained 28c. The through-space proton interaction correlations are shown in Figure 2. In particular, 28c was identified as 4-acetoxyisoquinoline because the NOESY correlation of the methyl protons (2.13 ppm) of the acetyl group to C5-H (7.98 ppm) and C8’-H (8.13 ppm) was observed. Consequently, compound 29, obtained from 28c reduction, was confirmed to be 4-hydroxy-3-(3-methoxymethylquinolin-2-yl)isoquinoline and compound 28b was also confirmed to be 4-acetoxyisoquinoline. The formation of both the 4-acetoxyisoquinoline product together with the isoquinolone product has been reported by Robison and co-workers [37]. However, why the Reissert–Henze-type reaction of our N-oxide 26 afforded 4-acetoxyisoquinoline remains to be elucidated. In the future, we will investigate this phenomenon in greater detail.
Scheme 5.

Synthesis of deacetyl compound 29 from predicted acetoxy compound 28c.
Figure 2.

The NOESY and HMBC correlations of acetoxyisoquinoline 28c, and structure of 28b.
To complete the synthesis of rosettacin (5), we applied the conditions reported by Ciufolini and co-workers for C-ring formation [38]. Heating 27c with H2SO4 in ethanol (EtOH) produced 5 at an 88% yield (Scheme 6). Thus, the total synthesis of rosettacin (5) was achieved through an eight-step sequence at a 23.8% overall yield.
Scheme 6.

Synthesis of rosettacin (5) by construction of C-ring.
As described above, we established a method for the synthesis of benz[6,7]indolizino[1,2-b]quinolin-11(13H)-one, which is the core structure of compounds in the aromathecin family. Next, we tried to synthesize 22-hydroxyacuminatin (6) by applying the same methodology (Scheme 7). Sonogashira coupling of 2-iodoquinoline 23 and 2-ethynylbenzaldehyde 30 in the presence of CuI, Et3N and PdCl2(PPh3)2 produced 2-alkynylbenzaldehyde 31 at an 87% yield. The treatment of 31 with hydroxylamine in EtOH produced 2-alkynylbenzaldehyde oxime 32 at an 81% yield. Subsequently, heating 32 in 1,2-DCB at 80 °C did not deliver the isoquinoline N-oxide cyclization product 33. Instead, the treatment of 32 in 1,2-DCB at 180 °C produced 33 at a 36% yield. Next, heating 33 in Ac2O at 50 °C under MW irradiation produced the desired isoquinolone 34 and 4-acetoxyisoquinoline 35 at yields of 54% and 20%, respectively. Finally, when 34 was heated with H2SO4 in EtOH, the desired 22-hydroxyacuminatine (6) was not produced, but acuminatine (7) was isolated at a 79% yield. The formation of 7 is believed to have proceeded with dehydroxylation after the removal of the MOM group along with scaffold formation. The physical and spectroscopic data for our synthetic rosettacin (5) were consistent with previously reported values in all respects [26]. Furthermore, 1H-NMR, 13C-NMR, and mass spectroscopy characterization of all our synthetic compounds supported the identified structures, the details of which can be found in the Supporting Information section.
Scheme 7.

Synthesis of acuminatine (7).
3. Materials and Methods
All non-aqueous reactions were carried out under an atmosphere of nitrogen in dried glassware unless otherwise noted. Solvents were dried and distilled according to standard protocols. Analytical thin-layer chromatography was performed with silica gel 60PF254 (Merck). Silica gel column chromatography was performed with silica gel 60 (70–230 mesh, Kanto Chemical Co. Lit., Nihonbashi, Tokyo, Japan). All melting points were determined on Yanagimoto micromelting point apparatus MP-500D (Yanaco Technical Sciences Co. Lit., Taito-ku, Tokyo, Japan) and are uncorrected. Proton nuclear magnetic resonance (1H-NMR) spectra were recorded on a JEOL JNM-ECZ400S (JEOL Resonance Co. Lit., Akishima, Tokyo, Japan). Chemical shifts were reported relative to Me4Si (δ 0.00). Multiplicity is indicated by one or more of the following: s (singlet); d (doublet); t (triplet); q (quartet); m (multiplet); br (broad). Carbon nuclear magnetic resonance (13C-NMR) spectra were recorded on a JEOL JNM-ECZ400S z at 100 MHz. Chemical shifts were reported relative to CDCl3 (δ 77.0) and DMSO-d6 (δ 39.7). Infrared spectra were recorded with ATR method using a Horiba FT-720 FREEXACT-II spectrophotometer (Horiba Ltd., Kyoto, Japan) and Technologies DuraScop (ST. Japan Inc., Chuo-ku, Tokyo, Japan). Low- and high-resolution mass spectra were recorded on JEOL JMS-700 spectrometers (JEOL Resonance Co.Lit., Akishima, Tokyo, Japan) via the use of a direct inlet system. The microwave-assisted reaction was carried out at 180 W and 2450 MHz with Discover (CEM corporation, Matthews NC, USA).
3.1. 2-Iodo-3-methylquinoline (20a)
We added dropwise conc. HCl (0.12 mL) to a solution of 2-chloroquinoline 18a (685 mg, 3.86 mol) and NaI (1.74 × 103 mg, 11.61 mmol) in MeCN (16 mL); then, these were stirred at 85 °C for 16 h. After cooling at ambient temperature, we added H2O (15 mL) and saturated Na2S2O3 (15 mL) to the reaction mixture. The resulting precipitate was filtrated in vacuo. The filtrate was extracted with EtOAc. The organic layer was washed with H2O and brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:4 v/v) to give 2-iodoquinoline 20a (958 mg, 92%) as a yellow solid. mp 181–183 °C (EtOAc-hexane). 1H-NMR (400 MHz, DMSO-d6) δ 2.46 (s, 3H), 7.61 (t, J = 8.2 Hz, 1H), 7.71 (t, J = 8.2 Hz, 1H), 7.89–7.93 (m, 2H), 8.17 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 25.8, 127.2, 127.3, 127.5, 127.6, 128.2, 129.6, 135.11, 135.14, 147.1. MS m/z: 269 (M+). HRMS (EI): calcd for C10H8NI 268.9701; found 268.9711.
3.2. 2-Iodoquinoline-3-carbaldehyde (20b)
The same procedure as above was carried out with 2-chloroquinoline 18b (1.0 × 104 mg, 52.35 mmol) to give 2-iodoquinoline 20b (13.3 g, 90%) as a white solid. mp 150–151 °C (EtOH). IR (ATR) ν = 1685 cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.67 (t, J = 8.2 Hz, 1H), 7.88 (t, J = 8.2 Hz, 1H), 7.97 (d, J = 8.2 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 8.56 (s, 1H), 10.29 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 120.5, 126.5, 128.3, 128.7, 128.9, 129.7, 133.4, 138.7, 151.6, 194.7. MS m/z: 283 (M+). HRMS (EI): calcd for C10H6NOI 282.9494; found 282.9485.
3.3. Methyl 2-Iodoquinoline-3-carboxylate (21)
A suspension of quinoline-3-carbaldehyde 20b (200 mg 0.71 mmol), I2 (683 mg, 2.69 mmol) and K2CO3 (345 mg, 2.50 mmol) in MeOH (6 mL) was stirred at rt for 35 min. After quenching with H2O (5 mL) and saturated Na2S2O3 (5 mL), the resulting precipitate was filtered in vacuo to give methyl ester 21 (198 mg, 89%) as a yellow solid. mp 93–94 °C (EtOH). IR (ATR) ν = 1716 cm−1. 1H-NMR (400 MHz, CDCl3) δ 4.02 (s, 3H), 7.64 (t, J = 8.2 Hz, 1H), 7.82 (t, J = 8.2 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 8.10 (d, J = 8.2 Hz, 1H), 8.49 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 52.9, 115.7, 125.6, 128.0, 128.5, 128.8, 129.0, 132.3, 138.9, 149.8, 166.0. MS m/z: 313 (M+). HRMS (EI): calcd for C11H8NO2I 312.9600; found 312.9605.
3.4. 2-Iodoquinolin-3-ylmethanol (22)
A solution of quinoline-3-carbaldehyde 20b (900 mg, 3.18 mmol) in MeOH (20 mL) was added dropwise to a suspension of NaBH4 (182 mg, 4.77 mmol) in MeOH (10 mL) under ice cooling conditions. After stirring at rt for 1 h, the reaction mixture was quenched with H2O. The resulting precipitate was filtrated in vacuo to give alcohol 22 (848 mg, 93%) as a yellow solid. mp 177–179 °C (CHCl3). IR (ATR) ν = 3745 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 4.53 (d, J = 8.2 Hz, 2H), 5.74 (t, J = 8.2 Hz, 1H), 7.64 (t, J = 8.2 Hz, 1H), 7.74 (t, J = 8.2 Hz, 1H), 7.95 (d, J = 8.2 Hz, 1H), 8.04 (d, J = 8.2 Hz, 1H), 8.22 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 65.5, 123.1, 127.1, 127.3, 127.6, 128.0, 129.9, 133.5, 137.7, 147.9. MS m/z: 285 (M+). HRMS (EI): calcd for C10H8NOI 284.9651; found 284.9663.
3.5. 2-Iodo-3-methoxymethylquinoline (23)
A solution of alcohol 22 (600 mg, 2.11 mmol) in THF (10 mL) was added dropwise to a suspension of 60% NaH (336 mg, 8.42 mmol) in THF (5 mL) under ice cooling. After stirring at the same temperature for 15 min, MeI (710 mg, 5.00 mmol) was added to the reaction mixture and this was then stirred at rt for 1 h. After quenching with H2O, the reaction mixture was extracted with EtOAc. The organic layer was washed with brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:9 v/v) to produce 3-methoxymethylquinoline 23 (543 mg, 86%) as a yellow solid. mp 45–46 °C (EtOAc-hexane). 1H-NMR (400 MHz, CDCl3) δ 3.58 (s, 3H), 4.55 (s, 2H), 7.57 (t, J = 8.1 Hz, 1H), 7.70 (t, J = 8.1 Hz, 1H), 7.82 (d, J = 8.1 Hz, 1H), 8.04–8.06 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ 58.9, 76.4, 122.2, 127.3, 127.6, 128.4, 129.9, 134.2, 134.3 (2C), 148.7. MS m/z: 299 (M+). HRMS (EI): calcd for C11H10NOI 298.9807; found 298.9807.
3.6. Methyl 2-(2-Formylphenyl)ethynylquinoline-3-carboxylate (24a)
We added a solution of 2-ethynylbenzaldehyde 19 (62 mg, 0.48 mmol) in THF (1 mL) to a solution of 2-iodoquinoline 21 (100 mg, 0.32 mmol), CuI (6 mg, 0.032 mmol), PdCl2(PPh3)2 (7 mg, 0.0096 mmol) and Et3N (2 mL, 14.47 mmol) in THF (5 mL). The reaction mixture was stirred at 60 °C for 1 h. After cooling to ambient temperature, the reaction mixture was filtrated through Celite pad, washed with EtOAc, and the filtrate was evaporated in vacuo. The residue was purified using column chromatography (EtOAc/hexane 1:4 v/v) to give 2-alkynylbenzaldehyde 24a (43 mg, 44%) as a yellow solid. mp 131–132 °C (EtOAc-hexane). IR (ATR) ν = 1689, 1709 cm−1. 1H-NMR (400 MHz, CDCl3) δ 4.03 (s, 3H), 7.53 (t, J = 7.8 Hz, 1H), 7.62–7.68 (m, 2H), 7.85–7.89 (m, 2H), 7.93 (d, J = 7.8 Hz, 1H), 8.01 (d, i = 7.8 Hz, 1H), 8.19 (d, J = 7.8 Hz, 1H), 8.87 (s, 1H), 10.86 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 52.8, 88.5, 94.6, 125.1, 125.9, 126.0, 127.0, 128.4, 128.6, 129.3, 129.6, 132.5, 133.7, 134.0, 136.9, 140.0, 141.0, 149.1, 165.2, 192.4. MS m/z: 315 (M+). HRMS (EI): calcd for C20H13NO 315.0895; found 315.0887.
3.7. 2-(3-Methylquinolin-2-yl)ethynylbenzaldehyde (24b)
The same procedure as above was carried out with 2-iodoquinoline 20a (700 mg, 2.60 mmol) to give 2-alkynylbenzaldehyde 24b (500 mg, 71%) as a white solid. mp 129–131 °C (EtOAc-hexane). IR (ATR) ν = 1689 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.71 (s, 3H), 7.52–7.57 (m, 2H), 7.64 (t, J = 7.8 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.99–8.02 (m, 2H), 8.11 (d, J = 7.8 Hz, 1H), 10.75 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 19.8, 88.2, 94.3, 125.5, 126.8, 127.4, 127.7 (2C), 128.9, 129.2, 129.4, 132.5, 133.7, 134.0, 135.5, 136.2, 143.6, 146.7, 191.1. MS m/z: 271 (M+). HRMS (EI): calcd for C19H13NO 271.0997; found 271.0988.
3.8. 2-(3-Methoxymethylquinolin-2-yl)ethynylbenzaldehyde (24c)
The same procedure as above was carried out with 2-iodoquinoline 23 (612 mg, 2.05 mmol) to give 2-alkynylbenzaldehyde 24c (519 mg, 84%) as a white solid. mp 97–98 °C (EtOAc-hexane). IR (ATR) ν = 1693 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.57 (s, 3H), 4.87 (s, 2H), 7.52–7.61 (m, 2H), 7.65 (t, J = 7.8 Hz, 1H), 7.74 (t, J = 7.8 Hz, 1H), 7.76–7.82 (m, 2H), 8.01 (t, J = 7.8 Hz, 1H), 8.14 (t, J = 8.6 Hz, 1H), 8.27 (s, 1H), 10.72 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 59.0, 71.7, 88.6, 93.2, 125.3, 127.5, 127.6, 127.7, 127.8, 129.1, 129.6, 130.0, 132.9, 133.8, 134.1, 134.6, 136.5, 141.7, 147.6, 191.3. MS m/z: 301 (M+). HRMS (EI): calcd for C20H15NO2 301.1103; found 301.1121.
3.9. Methyl 2-(2-Hydroxyiminomethylphenyl)ethynylquinoline-3-carboxylate (25a)
A mixture of benzaldehyde 24a (70 mg, 0.22 mmol), NH2OH·HCl (30 mg, 0.44 mmol), and AcONa (36 mg, 0.44 mmol) in EtOH (5 mL) was stirred at rt for 1 h. After the removal of the solvent, the residue was diluted with H2O and then filtrated off to give crude oxime 25a (50 mg, 68%) as a yellow solid. The product was recrystallized from EtOAc-hexane. mp 171–173 °C (EtOAc-hexane). IR (ATR) ν = 3047, 1709 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 3.99 (s, 3H), 7.43–7.52 (m, 2H), 7.71–7.77 (m, 2H), 7.91–7.97 (m, 2H), 8.11 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 8.73 (s, 1H), 9.03 (s, 1H), 11.67 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 53.3, 89.6, 93.8, 121.1, 125.1, 125.7, 126.2, 128.9, 129.0, 129.7, 130.1, 130.7, 133.4, 133.6, 135.3, 140.2, 140.6, 146.6, 148.9, 165.7. MS m/z: 330 (M+). HRMS (EI): calcd for C20H14N2O3 330.1004; found 330.1011.
3.10. 2-(3-Methylquinolin-2-yl)ethynylbenzaldehyde Oxime (25b)
The same procedure as above was carried out with benzaldehyde 24b (500 mg, 1.84 mmol) to give oxime 25b (280 mg, 53%) as a red solid. mp 179–181 °C (EtOAc-hexane). IR (ATR) ν = 3051 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 2.65 (s, 3H), 7.50–7.52 (m, 2H), 7.61 (t, J = 7.8 Hz, 1H), 7.72–7.76 (t, J = 7.8 Hz, 2H), 7.89–7.93 (m, 2H), 8.01 (d, J = 7.8 Hz, 1H), 8.29 (s, 1H), 8.64 (s, 1H), 11.71 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 19.8, 89.9, 93.6, 121.0, 125.4, 127.80, 127.83, 128.1, 128.9, 130.0, 130.2, 130.6, 132.9, 133.6, 134.9, 136.1, 143.9, 146.4, 146.8. MS m/z: 286 (M+). HRMS (EI): calcd for C19H14N2O 286.1106; found 286.1115.
3.11. 2-(3-Methoxymethylquinolin-2-yl)ethynylbenzaldehyde Oxime (25c)
The same procedure as above was carried out with benzaldehyde 24c (150 mg, 0.50 mmol) to give oxime 25c (137 mg, 86%) as a red solid. mp 151–153 °C (EtOAc-hexane). IR (ATR) ν = 3055 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 3.47 (s, 3H), 4.79 (s, 2H), 7.48–7.55 (m, 2H), 7.65 (t, J = 7.5 Hz, 1H), 7.74–7.76 (m, 1H), 7.80 (t, J = 7.5 Hz, 1H), 7.90 (d, J = 7.5 Hz, 1H), 8.02–8.05 (m, 2H), 8.41 (s, 1H), 8.66 (s, 1H), 11.72 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 58.2, 71.0, 89.4, 92.1, 120.3, 124.9, 126.9, 127.7, 128.0, 128.4, 129.6, 130.1, 130.2, 132.7, 133.1, 134.5, 134.7, 141.6, 145.9, 146.9. MS m/z: 316 (M+). HRMS (EI): calcd for C20H16N2O2 316.1212; found 316.1216.
3.12. 3-(3-Methylquinolin-2-yl)isoquinolin-1-one (27b) and 4-Acetoxy-3-(3-methylquinolin-2-yl)isoquinoline (28b)
A solution of oxime 25b (40 mg, 0.14 mmol) in 1,2-dichlorobenzene (4 mL) was stirred at 80 °C for 1 h. After the removal of the solvent, Ac2O (4 mL) was added to the residue, and the mixture was stirred at 110 °C for 3 h. After the removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give isoquinolone 27b (2 mg, 6%) and the 4-acetoxyisoquinoline 28b (15 mg, 33%).
27b: a white solid. mp 214–215 °C (EtOAc-hexane). IR (ATR) ν = 1628 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.88 (s, 3H), 7.55–7.60 (m, 2H), 7.68–7.76 (m, 3H), 7.80 (d, J = 8.2 Hz, 1H), 8.10 (s, 1H), 8.13 (d, J = 8.2 Hz, 1H), 8.50 (d, J = 8.2 Hz, 1H), 10.64 (br s, 1H). 13C-NMR (100 MHz, CDCl3) δ 22.5, 108.8, 126.6, 126.7, 127.4, 127.6, 127.7, 127.8, 128.9, 129.1, 129.7, 132.6, 136.2, 137.6, 139.3, 145.7, 148.7, 162.6. MS m/z: 286 (M+). HRMS (EI): calcd for C19H14N2O 286.1106; found 286.1109.
28b: an orange oil. IR (ATR) ν = 1770 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.09 (s, 3H), 2.48 (s, 3H), 7.55 (t, J = 7.5 Hz, 1H), 7.65–7.72 (m, 2H), 7.77–7.82 (m, 2H), 7.97 (d, J = 7.5 Hz, 1H), 8.08–8.12 (m, 3H), 9.28 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 19.4, 20.5, 121.2, 126.8, 126.9, 127.5, 128.0, 128.7, 129.2, 129.5, 131.0, 131.1, 137.0, 140.5, 143.9, 146.1, 149.4, 156.2, 168.7. MS m/z: 328 (M+). HRMS (EI): calcd for C21H16N2O2 328.1212; found 328.1222.
3.13. 3-(3-Methoxymethylquinolin-2-yl)isoquinoline N-Oxide (26c)
A solution of oxime 25c (150 mg, 0.47 mmol) in 1,2-dichlorobenzene (10 mL) was stirred at 80 °C for 1 h. After the removal of the solvent, the residue was crystallized from Et2O to give N-oxide 26c (109 mg, 73%) as a white solid. mp 222–223 °C (CHCl3). 1H-NMR (400 MHz, CDCl3) δ 3.32 (s, 3H), 4.56 (d, J = 12.8 Hz, 1H), 4.84 (d, J = 12.8 Hz, 1H), 7.60–7.69 (m, 3H), 7.74 (t, J = 8.2 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.92 (d, J = 8.2 Hz, 1H), 8.01 (s, 1H), 8.15 (d, J = 8.2 Hz, 1H), 8.37 (s, 1H), 8.90 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 58.6, 71.5, 124.6, 125.7, 127.2, 127.5, 127.6, 128.2, 129.0, 129.4, 129.6 (3C), 129.7, 132.4, 134.9, 136.3, 146.4, 147.2, 151.2. MS m/z: 316 (M+). HRMS (EI): calcd for C20H16N2O2 316.1212; found 316.1222.
3.14. 3-(3-Methoxymethylquinolin-2-yl)isoquinolin-1-one (27c) and 4-Acetoxy-3-(3-methoxymethylquinolin-2-yl)isoquinoline (28c)
A solution of N-oxide 26c (30 mg, 0.095 mmol) in Ac2O (2 mL) was heated at 50 °C under microwave irradiation for 3.5 h. After the removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give isoquinolone 27c (22 mg, 73%) and 4-acetoxyisoquinoline 28c (7 mg, 21%).
27c: a white solid. mp 156–157 °C (EtOAc-hexane). IR (ATR) ν = 1624 m−1. 1H NMR (400 MHz, CDCl3) δ 3.62 (s, 3H), 4.79 (s, 2H), 7.34 (s, 1H), 7.54–7.63 (m, 2H), 7.68–7.73 (m, 2H), 7.79 (t, J = 8.2 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 8.16 (d, J = 8.2 Hz, 1H), 8.35 (s, 1H), 8.49 (d, J =8.2 Hz, 1H), 10.50 (br s, 1H). 13C-NMR (100 MHz, CDCl3) δ 58.2, 72.6, 108.9, 126.7, 127.3, 127.4, 127.5 (2C), 127.7, 127.8, 128.2, 129.2, 130.7, 132.6, 135.9, 137.7, 139.5, 146.7, 150.0, 162.6. MS m/z: 316 (M+). HRMS (EI): calcd for C20H16N2O2 316.1212; found 316.1208.
28c: a yellow solid. mp 106–108 °C (EtOAc-hexane). IR (ATR) ν = 1774 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.13 (s, 3H), 3.41 (s, 3H), 4.73 (s, 2H), 7.58 (t, J = 8.2 Hz, 1H), 7.69–7.73 (m, 2H), 7.80 (t, J = 8.2 Hz, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 8.2 Hz, 1H), 8.09 (d, J = 8.2 Hz, 1H), 8.13 (d, J = 8.2 Hz, 1H), 8.46 (s, 1H), 9.25 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 20.6, 58.6, 71.7, 121.4, 126.9, 127.5, 127.6, 127.7, 128.2, 129.1, 129.3, 129.5, 131.1, 131.2, 131.9, 134.6, 140.7, 143.0, 146.5, 149.1, 154.2, 168.7. MS m/z: 358 (M+). HRMS (EI): calcd for C22H18N2O3 358.1317; found 358.1323.
3.15. 4-Hydroxy-3-(3-methoxymethylquinolin-2-yl)isoquinoline (29)
A solution of 4-acetoxyisoquinoline 28c (28 mg, 0.078 mmol) in THF (3 mL) was added dropwise to a suspension of LiAlH4 (8 mg, 0.20 mmol) in THF (3 mL) under ice cooling, and then stirred at 70 °C for 75 min. After quenching with H2O, the reaction mixture was filtrated through a Celite pad and washed with H2O and EtOAc. Next, the filtrate was extracted with EtOAc. The organic layer was washed with brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 3:15 v/v) to give 4-hydroxyquinoline 29 (22 mg, 87%) as an orange solid. mp 139–140 °C (EtOH). IR (ATR) ν = 3055 cm−1. 1H-NMR (400 MHz, CDCl3) δ 1.25 (br s, 1H), 3.66 (s, 3H), 5.47 (s, 2H), 7.56 (t, J = 7.8 Hz, 1H), 7.67–7.75 (m, 3H), 7.88 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 7.8 Hz, 1H), 8.06 (d, J = 7.8 Hz, 1H), 8.51 (d, J = 7.8 Hz, 1H), 8.66 (s, 1H), 8.81 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 58.8, 73.3, 123.0, 125.6, 126.6, 126.7, 126.7, 127.6, 128.8, 128.9, 129.1, 129.6, 130.1, 132.6, 135.6, 139.9, 141.7, 156.2, 156.3. MS m/z: 316 (M+). HRMS (EI): calcd for C20H16N2O2 316.1212; found 316.1216.
3.16. Rosettacin (5)
We added dropwise conc. H2SO4 (0.5 mL) under ice cooling to a suspension of isoquinolone 27c (13 mg, 0.041 mmol) in EtOH (1 mL) and the solution was stirred at 110 °C for 12 h. After cooling to ambient temperature, the solvent was evaporated in vacuo. The residue was diluted with H2O (1 mL), and then was alkalified with 1 M aqueous NaOH. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with Na2SO4, and evaporated in vacuo. The residue was washed with Et2O and filtrated in vacuo to give rosettacin (5) (10 mg, 88%) as a yellow solid. mp 292–294 °C (EtOAc-hexane, lit. [26] mp 288 °C). IR (ATR) ν = 1651 cm−1. 1H-NMR (400 MHz, CDCl3) δ 5.39 (s, 2H), 7.57–7.65 (m, 2H), 7.68 (s, 1H), 7.74 (t, J = 8.2 Hz, 1H), 7.79–7.83 (m, 2H), 7.92 (d, J = 8.2 Hz, 1H), 8.23 (d, J = 8.2 Hz, 1H), 8.35 (s, 1H), 8.56 (d, J = 8.2 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 49.5, 101.1, 126.1, 127.3 (2C), 127.5 (2C), 128.0 (2C),128.8, 129.5, 130.2, 130.7, 132.5, 137.5, 140.0, 148.9, 153.7, 161.1 MS m/z: 284 (M+). HRMS (EI): calcd for C19H12N2O 284.0950; found 284.0952.
3.17. 3-Methoxymethyloxymethyl-2-[(3-methoxymethylquinolin-2-yl)ethynyl]benzaldehyde (31)
We added a solution of 2-ethynylbenzaldehyde 30 (82 mg, 0.40 mmol) in THF (1 mL) to a solution of 2-iodoquinoline 23 (100 mg, 0.33 mmol), CuI (3.0 mg, 0.016 mmol), PdCl2(PPh3)2 (23 mg, 0.033 mmol) and Et3N (1 mL, 21.63 mmol) in THF (2 mL). The reaction mixture was stirred at 60 °C for 30 min. After cooling to ambient temperature, the reaction mixture was filtrated through Celite pad, washed with EtOAc, and the filtrate was evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 1:9 v/v) to give 2-alkynylbenzaldehyde 31 (109 mg, 87%) as a yellow solid. mp 87–88 °C (EtOAc-hexane). IR (ATR) ν = 1693 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.43 (s, 3H), 3.56 (s, 3H), 4.84 (s, 2H), 4.87 (s, 2H), 5.01 (s, 2H), 7.54–7.61 (m, 2H), 7.74 (t, J = 7.8 Hz, 1H), 7.85 (d, J = 7.8 Hz, 2H), 7.95 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 7.8 Hz, 1H), 8.28 (s, 1H), 10.77 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 55.6, 58.8, 67.1, 71.6, 85.5, 96.4, 98.2, 123.7, 126.6, 127.5, 127.6, 127.8, 129.2, 129.4, 130.0, 132.8, 132.9, 134.4, 136.7, 141.4, 142.1, 147.6, 191.5. MS m/z: 375 (M+). HRMS (EI): calcd for C23H21NO4 375.1471; found 375.1477.
3.18. 3-Methoxymethyloxymethyl-2-[(3-methoxymethylquinolin-2-yl)ethynyl]benzaldehyde Oxime (32)
A mixture of benzaldehyde 31 (247 mg, 0.66 mmol), NH2OH·HCl (92 mg, 1.32 mmol), and AcONa (108 mg, 1.32 mmol) in EtOH (7 mL) was stirred at rt for 2 h. After removal of solvent, the residue was diluted with H2O, and then filtrated off to give crude oxime 32 (208 mg, 81%) as a red solid. The product was recrystallized from EtOAc-hexane. mp 130–131 °C (EtOAc-hexane). IR (ATR) ν = 3734 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 3.35 (s, 3H), 3.47 (s, 3H), 4.77 (s, 2H), 4.83 (s, 2H), 4.87 (s, 2H), 7.51–7.55 (m, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.66 (t, J = 7.8 Hz, 1H), 7.79–7.86 (m, 2H), 8.04–8.06 (m, 2H), 8.44 (s, 1H), 8.70 (s, 1H), 11.71 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 54.9, 58.1, 67.0, 70.9, 86.6, 95.8, 96.8, 118.9, 123.8, 127.0, 127.8, 128.0, 128.4, 129.8, 130.2, 132.7, 134.6, 135.0, 141.3, 141.5, 146.0, 146.9. MS m/z: 390 (M+). HRMS (EI): calcd for C23H22N2O4 390.1580; found 390.1590.
3.19. 5-Methoxymethyloxymethyl-3-(3-methoxymethylquinolin-2-yl)isoquinoline N-Oxide (33)
A solution of oxime 32 (60 mg, 0.15 mmol) in 1,2-dichlorobenzene (4 mL) was stirred at 180 °C for 12 h. After removal of solvent, the residue was purified by column chromatography (MeOH) to give N-oxide 33 (21 mg, 36%). The obtained 33 contained a small amount of impurity and could not be further purified.
3.20. 5-Methoxymethyloxymethyl-3-(3-methoxymethylquinolin-2-yl)isoquinolin-1-one (34) and 4-Acetoxy-5-methoxymethyloxymethyl-3-(3-methoxymethylquinolin-2-yl)isoquinoline (35)
A solution of N-oxide 33 (15 mg, 0.038 mmol) in Ac2O (1 mL) was heated at 50 °C under microwave irradiation for 24 h. After removal of the solvent, the residue was purified by column chromatography (EtOAc/hexane 1:1 v/v) to give isoquinolone 34 (8 mg, 54%) and the 4-acetoxyisoquinoline 35 (3 mg, 20%).
34: a yellow solid. mp 113–114 °C (EtOAc-hexane). IR (ATR) ν = 1628 cm−1. 1H-NMR (400 MHz, CDCl3) δ 3.42 (s, 3H), 3.62 (s, 3H), 4.77 (s, 2H), 4.82 (s, 2H), 4.97 (s, 2H), 7.54 (t, J = 7.8 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.67 (s, 1H), 7.75–7.82 (m, 2H), 7.89 (d, J = 7.8 Hz, 1H), 8.17 (d, J = 7.8 Hz, 1H), 8.36 (s, 1H), 8.49 (d, J = 7.8 Hz, 1H), 10.61 (s, 1H). 13C-NMR (100 MHz, CDCl3) δ 29.7, 55.5, 58.4, 66.9, 72.7, 95.7, 105.4, 127.2, 127.3, 127.4, 127.7, 127.8, 128.3, 129.3, 130.7, 133.2, 133.8, 136.1, 136.5, 139.6, 146.8, 150.1, 162.8. MS m/z: 390 (M+). HRMS (EI): calcd for C23H22N2O4 390.1580; found 390.1574.
35: an orange oil. IR (ATR) ν = 1770 cm−1. 1H-NMR (400 MHz, CDCl3) δ 1.95 (s, 3H), 3.35 (s, 3H), 3.44 (s, 3H), 4.63 (s, 2H), 4.71 (s, 2H), 5.19 (s, 2H), 7.58 (t, J = 7.8 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.71 (t, J = 7.8 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 8.04 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.48 (s, 1H), 9.23 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 21.0, 55.4, 58.7, 67.8, 70.9, 94.8, 126.9, 127.66, 127.71, 127.8, 128.1, 129.0, 129.2, 129.9, 130.9, 132.3, 132.4, 132.6, 134.4, 140.7, 145.0, 146.2, 149.8, 154.4, 168.7. MS m/z: 432 (M+). HRMS (EI): calcd for C25H24N2O5 432.1685; found 432.1694.
3.21. Acuminatine (7)
We added dropwise conc. H2SO4 (2 mL) under ice cooling to a suspension of isoquinolone 34a (30 mg, 0.077 mmol) in EtOH (4 mL) and then stirred the mixture at 110 °C for 16 h. After cooling to an ambient temperature, the solvent was evaporated in vacuo. The residue was diluted with H2O (2 mL) and then was alkalified with 1 M aqueous NaOH. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with water and brine, dried with Na2SO4, and evaporated in vacuo. The residue was purified by column chromatography (EtOAc/hexane 4:6 v/v) to give acuminatine (7) (18 mg, 79%). mp 290–292 °C (EtOAc-hexane). IR (ATR) ν = 1770 cm−1. 1H-NMR (400 MHz, CDCl3) δ 2.74 (s, 3H), 5.38 (s, 2H), 7.47 (t, J = 7.8 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.79–7.83 (m, 2H), 7.91 (d, J = 7.8 Hz, 1H), 8.24 (d, J = 8.7 Hz, 1H), 8.35 (s, 1H), 8.43 (d, J = 7.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 19.6, 49.4, 98.1, 125.5, 126.4, 127.1, 127.3, 128.0, 128.1, 128.9, 129.4, 130.3, 130.9, 133.3, 135.0, 136.6, 139.6, 148.9, 153.9, 161.3. MS m/z: 298 (M+). HRMS (EI): calcd for C20H14N2O 298.1106; found 298.1111.
4. Conclusions
We developed a synthetic route to create the pentacyclic scaffold of the aromathecin family by constructing the indolizidine moiety after isoquinolone synthesis. The key step of our isoquinolone synthesis employed a thermal cyclization of 2-alkynylbenzaldehyde oxime to the isoquinoline N-oxide, followed by the performance of a Reissert–Henze-type reaction. Under the optimum reaction conditions for the Reissert–Henze-type reaction step, microwave irradiation-assisted heating of the purified N-oxide in Ac2O at 50 °C reduced the formation of the 4-acetoxyisoquinoline byproduct to deliver the desired isoquinolone at a moderate yield. The employed eight-step sequence afforded rosettacin at a 23.8% overall yield. Furthermore, the synthesis of acuminatine was achieved by applying the developed strategy. Future research will focus on the use of this strategy to efficiently synthesize a variety of polycyclic compounds, including alkaloids with promising therapeutic profiles.
Acknowledgments
This work was partly supported by Fukuyama University Grant for Academic Research Projects (Grant Number: GARP2022-223 (S.M.)).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28104059/s1, Copies of the 1H NMR and 13C NMR spectra for compounds 20–35, rosettascin (5), and acuminatine (7).
Author Contributions
Conceptualization, T.C. and T.N.; formal analysis, S.M. and T.N.; investigation, S.M., M.E., K.S., T.Y., H.B. and T.N.; resources, T.M. and N.H.; writing—original draft preparation, T.C. and T.N.; writing—review and editing, T.C., T.N., T.M. and N.H. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in Supplementary Material.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
Funding Statement
This research received no external funding.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Wall M.E., Wani M.C., Cook C.E., Palmer K.H., McPhail A.T., Sim G.A. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata. J. Am. Chem. Soc. 1966;88:3888–8890. doi: 10.1021/ja00968a057. [DOI] [Google Scholar]
- 2.Walraven H.G.M., Pandit U.K. A facile two synthon approach to the camptothecin skeleton. Tetrahedron. 1980;36:321–327. doi: 10.1016/0040-4020(80)80022-6. [DOI] [Google Scholar]
- 3.Pinho P.M.M., Pinto M.M.M., Kijjoa A., Pharadai K., Díaz J.G., Herz W. Protoberberine alkaloids from Coscinium fenestratum. Phytochemistry. 1992;31:1403–1407. doi: 10.1016/0031-9422(92)80301-T. [DOI] [Google Scholar]
- 4.Pailee P., Prachyawarakorn V., Mahidol C., Ruchirawat S., Kittakoop P. Protoberberine Alkaloids and Cancer Chemopreventive Properties of Compounds from Alangium salviifolium. Eur. J. Org. Chem. 2011;2011:3809–3814. doi: 10.1002/ejoc.201100423. [DOI] [Google Scholar]
- 5.Kawato Y., Aonuma M., Hirota Y., Kuga H., Sato K. Intracellular Roles of SN-38, a Metabolite of the Camptothecin Derivative CPT-11, in the Antitumor Effect of CPT-11. Cancer Res. 1991;51:4187–4197. [PubMed] [Google Scholar]
- 6.Rowinsky E.K., Grochow L.B., Hendricks C.B., Ettinger D.S., Forastiere A.A., Hurowitz L.A., McGuire W.P., Sartorius S.E., Lubejko B.G., Kaufmann S.H. Phase I and pharmacologic study of topotecan: A novel topoisomerase I inhibitor. J. Clin. Oncol. 1992;10:647–656. doi: 10.1200/JCO.1992.10.4.647. [DOI] [PubMed] [Google Scholar]
- 7.Ban H.-J., Oh I.-J., Kim K.-S., Ju J.-Y., Kwon Y.-S., Kim Y.-I., Lim S.-C., Kim Y.-C. Clinical Efficacy of Belotecan (CKD-602), Newly Developed Camptothecin Analog, in the 2nd Line Treatment of Relapsed Small Cell Lung Cancer. Tuberc. Respir. Dis. 2009;66:93–97. doi: 10.4046/trd.2009.66.2.93. [DOI] [Google Scholar]
- 8.Adams D.J., Dewhirst M.W., Flowers J.L., Gamcsik M.P., Colvin O.M., Manikumar G., Wani M.C., Wall M.E. Camptothecin analogues with enhanced antitumor activity at acidic pH. Cancer Chemother. Pharmacol. 2000;46:263–271. doi: 10.1007/s002800000157. [DOI] [PubMed] [Google Scholar]
- 9.Lin L.Z., Cordell G.A. Quinoline Alkaloids from Camptotheca acuminata. Phytochemistry. 1989;28:1295–1297. doi: 10.1016/0031-9422(89)80242-0. [DOI] [Google Scholar]
- 10.Pin F., Comesse S., Daich A. Association of intramolecular furan Diels-Alder (IMFDA) reaction and N-acyliminium alkylation for the synthesis of pentacyclic precursor of aromathecins. Synlett. 2009;19:3214–3218. [Google Scholar]
- 11.Grillet F., Baumlová B., Preévost G., Constant J.-F., Chaumeron S., Bigg D.C.H., Greenea A.E., Kanazawa A. Synthesis and bioevaluation of 22-hydroxyacuminatine analogs. Bioorg. Med. Chem. Lett. 2008;18:2143–2146. doi: 10.1016/j.bmcl.2008.01.082. [DOI] [PubMed] [Google Scholar]
- 12.Xiao X., Antony S., Pommier Y., Cushman M. Total Synthesis and Biological Evaluation of 22-Hydroxyacuminatine. J. Med. Chem. 2006;49:1408–1412. doi: 10.1021/jm051116e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cinelli M.A., Morrell A., Dexheimer T.S., Scher E.S., Pommier Y., Cushman M. Design, Synthesis, and Biological Evaluation of 14-Substituted Aromathecins as Topoisomerase I Inhibitors. J. Med. Chem. 2008;51:4609–4619. doi: 10.1021/jm800259e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng K., Rahier N.J., Eisenhauer B.M., Gao R., Thomas S.J., Hecht S.M. 14-Azacamptothecin: A Potent Water-Soluble Topoisomerase I Poison. J. Am. Chem. Soc. 2005;127:838–839. doi: 10.1021/ja0442769. [DOI] [PubMed] [Google Scholar]
- 15.Fox B.M., Xiao X., Antony S., Kohlhagen G., Pommier Y., Staker B.L., Stewart L., Cushman M. Design, Synthesis, and Biological Evaluation of Cytotoxic 11-Alkenylindenoisoquinoline Topoisomerase I Inhibitors and Indenoisoquinoline-Camptothecin Hybrids. J. Med. Chem. 2003;46:3275–3282. doi: 10.1021/jm0300476. [DOI] [PubMed] [Google Scholar]
- 16.Cinelli M.A., Cordero B., Dexheimer T.S., Pommier Y., Cushman M. Synthesis and biological evaluation of 14-(aminoalkyl-aminomethyl)aromathecins as topoisomerase I inhibitors: Investigating the hypothesis of shared structure–activity relationships. Bioorg. Med. Chem. 2009;17:7145–7155. doi: 10.1016/j.bmc.2009.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song L., Tian G., He Y., Van der Eycken E.V. Rhodium(III)-catalyzed intramolecular annulation through C-H activation: Concise synthesis of rosettacin and oxypalmatime. Chem. Commun. 2017;53:12394–12397. doi: 10.1039/C7CC06860C. [DOI] [PubMed] [Google Scholar]
- 18.Song L., Tian G., Van der Eycken E.V. Rhodium(III)-catalyzed intermolecular cascade annulation through C-H activation: Concise synthesis of rosettacin. Mol. Catal. 2018;459:129–134. doi: 10.1016/j.mcat.2018.09.004. [DOI] [Google Scholar]
- 19.Song L., Zhang X., Tian G., Robeyns K., Van Meervelt L., Harvey J.N., Van der Eycken E.V. Intramolecular cascade annulation triggered by C-H activation via rhodium hydride intermediate. Mol. Catal. 2019;463:30–36. doi: 10.1016/j.mcat.2018.11.016. [DOI] [Google Scholar]
- 20.Wang G., Hu W., Hu Z., Zhang Y., Yao W., Li L., Fu Z., Huang W. Carbene-catalyzed aerobic oxidation of isoquinolinium salts: Efficient synthesis of isoquinolinones. Green Chem. 2018;20:3302–3307. doi: 10.1039/C8GC01488D. [DOI] [Google Scholar]
- 21.Raji Reddy C., Mallesh K. Rh(III)-Catalyzed Cascade Annulations To Access Isoindolo [2,1-b]isoquinolin-5(7H)-ones via C-H Activation: Synthesis of Rosettacin. Org. Lett. 2018;20:150–153. doi: 10.1021/acs.orglett.7b03509. [DOI] [PubMed] [Google Scholar]
- 22.Baguia H., Deldaele C., Romero E., Michelet B., Evano G. Copper-Catalyzed Photoinduced Radical Domino Cyclization of Ynamides and Cyanamides: A Unified Entry to Rosettacin, Luotonin A, and Deoxyvasicinone. Synthesis. 2018;50:3022–3030. [Google Scholar]
- 23.Lerchen A., Knecht T., Koy M., Daniliuc C.G., Glorius F. A General Cp*CoIII-Catalyzed Intramolecular C-H Activation Approach for the Efficient Total Syntheses of Aromathecin, Protoberberine, and Tylophora Alkaloids. Chem. Eur. J. 2017;23:12149–12152. doi: 10.1002/chem.201702648. [DOI] [PubMed] [Google Scholar]
- 24.Li K., Ou J., Gao S. Total Synthesis of Camptothecin and Related Natural Products by a Flexible Strategy. Angew. Chem. Int. Ed. 2016;55:14778–14783. doi: 10.1002/anie.201607832. [DOI] [PubMed] [Google Scholar]
- 25.Xu X., Liu Y., Park C.-M. Rhodium(III)-Catalyzed Intramolecular Annulation through C-H Activation: Total Synthesis of (±)- Antofine, (±)-Septicine, (±)-Tylophorine, and Rosettacin. Angew. Chem. Int. Ed. 2012;51:9372–9376. doi: 10.1002/anie.201204970. [DOI] [PubMed] [Google Scholar]
- 26.Pin F., Comesse S., Sanselme M., Daiech A. A Domino N-Amidoacylation/Aldol-Type Condensation Approach to the Synthesis of the Topo-I Inhibitor Rosettacin and Derivatives. J. Org. Chem. 2008;73:1975–1978. doi: 10.1021/jo702387q. [DOI] [PubMed] [Google Scholar]
- 27.Nishiyama T., Takaiwa S., Kotouge R., Tani S., Yoshinaga R., Hamada E., Endo M., Sugino Y., Hatae N., Hibino S., et al. First asymmetric enantioselective total synthesis of phenanthridine alkaloid, (S)-(+)-asiaticumine and its enantiomer. Tetrahedron Lett. 2019;60:151278. doi: 10.1016/j.tetlet.2019.151278. [DOI] [Google Scholar]
- 28.Nishiyama T., Murakami M., Taninaka K., Hamada E., Endo M., Kinou D., Hatae N., Choshi T. Synthesis of Pyrrolo [2,3-c]quinoline Alkaloid Marinoquinolines. Heterocycles. 2021;103:300–310. [Google Scholar]
- 29.Nishiyama T., Hamada E., Ishii D., Kihara Y., Choshi N., Nakanishi N., Murakami M., Taninaka K., Hatae N., Choshi T. Total synthesis of pyrrolo [2,3-c]quinoline alkaloid: Trigonoine B. Beilstein J. Org. Chem. 2021;17:730–736. doi: 10.3762/bjoc.17.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishiyama T., Kihara Y., Takeuchi N., Mizuno S., Hieda Y., Hatae N., Choshi T. Total syntheses of carbazole alkaloid mukoenine A and pyrano [3,2-a]carbazole alkaloid girinimbine. Tetrahedron. 2022;120:132895. doi: 10.1016/j.tet.2022.132895. [DOI] [Google Scholar]
- 31.Hieda Y., Une Y., Satoh A., Tsuruga T., Choshi T. Total synthesis of the reported structure of bioactive dibenzofuran natural product karnatakafuran B. Tetrahedron. 2023;135:133327. doi: 10.1016/j.tet.2023.133327. [DOI] [Google Scholar]
- 32.Kanekiyo N., Kuwada T., Choshi T., Nobuhiro J., Hibino S. Total syntheses of β-carboline alkaloids, (R)-(–)-pyridindolol K1, (R)-(–)-pyridindolol K2, and (R)-(–)-pyridindolol. J. Org. Chem. 2001;66:8793–87988. doi: 10.1021/jo0105585. [DOI] [PubMed] [Google Scholar]
- 33.Kanekiyo N., Choshi T., Kuwada T., Sugino E., Hibino S. The first total synthesis of (R)-(–)-pyridindolol K2 and its enantiomer. Heterocycles. 2000;53:1877–1880. [Google Scholar]
- 34.Nishiyama T., Hironaka M., Taketomi M., Taguchi R., Kotouge R., Shigemori Y., Hatae N., Ishikura M., Choshi T. Total Synthesis of Two 8-Oxoprotoberberine Alkaloids: Alangiumkaloids A and B. Eur. J. Org. Chem. 2018;2018:673–678. doi: 10.1002/ejoc.201701557. [DOI] [Google Scholar]
- 35.Baruah B., Bhuyan P.J. Synthesis of some complex pyrano [2,3-b]- and pyrido [2,3-b]quinolines from simple acetanilides via intramolecular domino hetero Diels–Alder reactions of 1-oxa-1,3-butadienes in aqueous medium. Tetrahedron. 2009;65:7099–7104. doi: 10.1016/j.tet.2009.06.036. [DOI] [Google Scholar]
- 36.Wagh M.B., Shankar R., Kumar U.K.S., Gill C.H. A Concise and Convergent Synthesis of Luotonin B and E. Synlett. 2011;1:84–88. [Google Scholar]
- 37.Robinson M.M., Robison B.L. The Rearrangement of Isoquinoline-n-oxides. J. Org. Chem. 1957;20:1337–1341. [Google Scholar]
- 38.Ciufolini M.A., Roschangar F. Total Synthesis of (+)-Camptothecin. Angew. Chem. Int. Ed. 1996;35:1692–1694. doi: 10.1002/anie.199616921. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available in Supplementary Material.