

Abstract
Rat sarcoma (RAS), as a frequently mutated oncogene, has been studied as an attractive target for treating RAS‐driven cancers for over four decades. However, it is until the recent success of kirsten‐RAS (KRAS)G12C inhibitor that RAS gets rid of the title “undruggable”. It is worth noting that the therapeutic effect of KRASG12C inhibitors on different RAS allelic mutations or even different cancers with KRASG12C varies significantly. Thus, deep understanding of the characteristics of each allelic RAS mutation will be a prerequisite for developing new RAS inhibitors. In this review, the structural and biochemical features of different RAS mutations are summarized and compared. Besides, the pathological characteristics and treatment responses of different cancers carrying RAS mutations are listed based on clinical reports. In addition, the development of RAS inhibitors, either direct or indirect, that target the downstream components in RAS pathway is summarized as well. Hopefully, this review will broaden our knowledge on RAS‐targeting strategies and trigger more intensive studies on exploiting new RAS allele‐specific inhibitors.
Keywords: allelic RAS mutation, cancer, personalized therapy, RAS inhibitor
Personalized therapeutic strategy targeting specific mut‐RAS‐induced cancers is essential. Cancer type, RAS mutation type, activation of downstream pathways, and clinicopathological background should be comprehensively considered when designing strategies for drug combinations.
1. INTRODUCTION
Rat sarcoma (RAS) proteins belong to the small‐molecule G protein family. As binary molecular switch, RAS GTPases cycle between guanosine triphosphate (GTP)‐bound active form and guanosine diphosphate (GDP)‐bound inactive form, facilitating “on” or “off” state in signal transduction. There are three canonical RAS genes, kirsten‐RAS (KRAS), neuroblastoma‐RAS (NRAS), and harvey‐RAS (HRAS), encoding four RAS proteins (KRAS4A, KRAS4B, NRAS, and HRAS). Among RAS genes, KRAS could encode two proteins (KRAS4A and KRAS4B) due to alternative RNA splicing. RAS and its downstream pathways control a variety of activities, such as cell proliferation, survival, migration, etc. 1
Since the discovery of mutated oncogenic RAS in human cancer in 1982, it became a common sense that RAS mainly drives the formation and development of tumor through point mutations. 2 , 3 , 4 Oncogenic RAS family mutations have been found in about 27% of all human cancers 5 , 6 , 7 , 8 , 9 ; thus, intensive research effort has been made to study RAS protein structure and biochemistry and develop anti‐mutated RAS drugs for cancer therapy. 10 However, the study is full of failures and obstacles for the following reasons: (1) RAS protein structure is smooth globular with shallow depressions, which lack well‐defined hydrophobic pockets for targeting study. (2) As small GTPases, there are also studies that attempt to competitively inhibit RAS binding to GTP, based on the fact that small G protein RAS needs to bind to GTP to form an activated form. However, there is picomolar binding strength between RAS and GTP, and the concentration of GTP in the cytoplasm reaches ∼0.5 mmol/L. 11 , 12 , 13 , 14 Therefore, the development of competitive GTP‐binding inhibitors is almost impossible. Thus, the development of oncogenic RAS inhibitors had been stuck in bottleneck until recent success in specific KRASG12C inhibitors. 15 , 16 , 17 , 18
Subsequent clinical data manifest that KRASG12C inhibitor treatment efficacy differs in cancers with different oncogenic RAS mutations or even in different cancer types with KRASG12C mutation. Therefore, knowledge of the molecular structural property, biochemistry, and biology will lay a foundation for designing an allelic‐specific mutated RAS inhibitor for personalized cancer treatment.
Based on the mutation sites and important domains affecting RAS function, this review summarizes the structural biochemical differences of different mutations, so as to provide reference for the development of inhibitors targeting specific mutations. Furthermore, in order to achieve better clinical treatment with inhibitors, the review also lists the clinicopathological features among cancers commonly carrying RAS mutations and responses to treatment strategies, providing reference for the appropriate selection of personalized treatment for cancers with RAS mutations.
2. PHYSIOLOGICAL ACTIVITIES OF FUNCTIONAL RAS DOMAINS
RAS is a member of the small‐molecule G protein family that is activated by GTP binding. Different RAS paralogs share similar structural compositions: a highly conserved G domain (aa 1–165) and a hypervariable region (aa 166–178/179). 19 , 20 , 21
The G domain is composed of an effector lobe (aa 1–86) and an allosteric lobe (87–165). 22 The former contains two switch regions: switch I and II, along with a P‐loop. 23 , 24 , 25 The latter contains regulatory sites that result in conformational change in switch II once it binds calcium or acetate. 23
Before playing a role, the C‐terminal CAAX box of RAS (comprising cysteine (C) and aliphatic (a) and variable (X) amino acids) (CAAX) tail of RAS needs to undergo a series of modifications. 26 , 27 , 28 As shown in Figure 1, after being modified by several enzymes, such as RAS and a‐factor converting enzyme 1 (RCE1), prenylcysteine carboxyl methyltransferase (pcCMT), and palmitoyltransferases (PAT), CAAX is able to associate with the cytoplasmic leaflet of cellular membrane, and then RAS is recruited to the plasma membrane and activated by receptor tyrosine kinase (RTK) signals and subsequently recruits downstream effector molecules to achieve signal transduction. 29 , 30 , 31 , 32 , 33 In the process, the cysteine in the CAAX tail is first linked to a farnesyl group (15‐carbon) via the thioether bond by FTase. Alternatively, the cysteine of CAAX would also be modified with a geranyl group (20‐carbon) by geranylgeranyltransferase I (GGTase I) if FTase was blocked by inhibitors. RCE1 then excises the AAX amino acids by recognizing farnesyl cysteine and conjugates cysteine with a carboxyl group. The carboxyl group is subsequently modified by pcCMT to produce carbomethoxy, which increases the lipophilicity of RAS and helps RAS bind to the membrane. After this, the modification processes of different paralogs begin to distinguish each other. In addition to farnesyl cysteine, the remaining cysteine residues of NRAS and HRAS would be palmitoylated by PAT, which is located in the Golgi to facilitate their transport to membrane. Unlike the former, KRAS4B will not be modified by PAT because there is no remaining cysteine residue available. However, the polybase region of KRAS4B will bind membrane through electrostatic interaction. In addition, KRAS4A has both a discontinuous polybase region and excess cysteine available for palmitoylation, which help KRAS4A choose whether or not to be modified with PAT. 30
FIGURE 1.
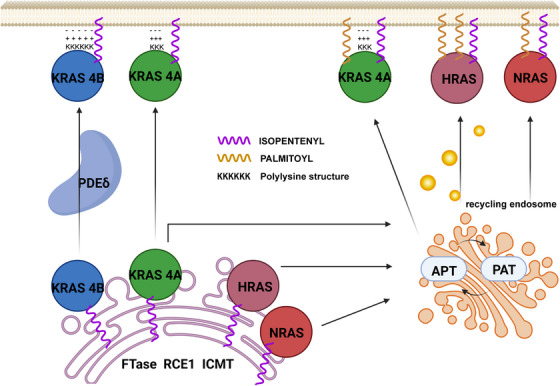
The process of Rat sarcoma (RAS) binding to plasma membrane after post‐translational modification. CAAX motifs at C‐terminal of RAS, consisting of cysteine, aliphatic amino acids, and a variable amino acid, help RAS localize to specific plasma membrane microdomains and subsequently pass signals to the downstream. 30 The post‐translational modification of CAAX is achieved by farnesylation, hydrolysis by RAS and a‐factor converting enzyme 1 (RCE1) and carboxymethylation by prenylcysteine carboxyl methyltransferase (pcCMT), followed by palmitoylation by palmitoyltransferases (PAT) in Golgi (harvey‐RAS (HRAS), neuroblastoma‐RAS (NRAS), KRAS4A). The difference between KRAS4B and other paralogs is that the CAAX of KRAS4B is able to bind to membrane depending on lysine residues. 31 , 32 The figure was made using Biorender.
After binding to the plasma membrane, the RAS protein would be activated by the upstream RTK signal and then converted into an active form for downstream signaling. RAS activity switches between RAS–GTP (activated) and RAS–GDP (inactive) binding forms, which is regulated by guanine nucleotide exchange factors (GEFs) and GTPase‐activating proteins (GAPs). Recently, however, RAS has been found to be activated by cytoplasmic RTK granules rather than RTKs located in the plasma membrane. Tulpule et al. 34 found that oncogenic RTKs, losing their lipid membrane‐targeting sequences, could form membrane‐less cytoplasmic protein granules, in which the RTK granules activate RAS in a lipid membrane‐independent manner. Besides, RAS activity can be regulated by other modifications, such as phosphorylation, 35 , 36 , 37 ubiquitination, 38 , 39 , 40 lysine acetylation, 41 , 42 , 43 and lysine methylation. 44
In cells, once an upstream signal is received, GEF (such as SOS) forms a complex with RAS binding to GDP while promoting the dissociation of GDP from RAS. Then, GTP would replace GEF in the complex to form the active GTP‐bound RAS to pass the signal downstream. 45 , 46 Normally, the activation of RAS signaling pathway is transient under physiological conditions. After the signal transmission is complete, GAP could boost RAS intrinsic GTPase activity to dissociate from GTP and restore it to GDP‐bound form, thereby closing related signaling pathways. 47 However, when a specific point mutation of RAS occurs, the intrinsic GTPase activity or GAP‐binding ability of RAS would be attenuated. Thus, the existence duration of RAS–GTP, along with the activation duration of the downstream signaling pathway, is prolonged, resulting in abnormal cell proliferation as well as tumor occurrence and development. 48 , 49 , 50 , 51
3. GENERAL DAMAGE INDUCED BY RAS MUTATIONS AT HOT‐SPOT SITES
There are mainly three RAS paralogs with different mutation frequencies in cancer, including KRAS (85%), NRAS (11%), and HRAS (3%). 52 , 53 When oncogenic amino acid substitution mutations occur, changes in RAS protein structure and function lead to persistently activated downstream signaling pathways. Clinically, particular isoform, mutation frequency, and amino acid substitution preference at each site complicate RAS mutation occurrence in various cancers. 54 The types of RAS mutations and their frequency in related cancers are shown in Figure 2.
FIGURE 2.
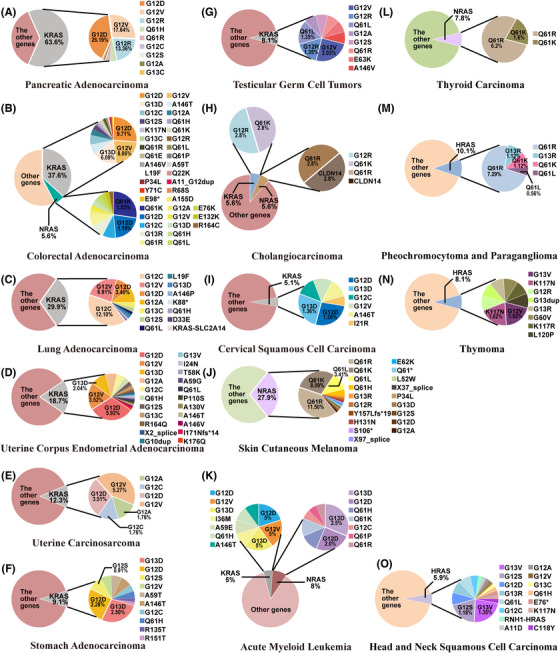
The frequency of RAS mutations in human cancers. The data come from TCGA PanCancer Atlas Studies. The activating mutations of RAS occur predominantly at codons 12, 13, and 61 and kirsten‐RAS (KRAS) has the most tendency to be mutated in the three paralogs. Clinically, RAS mutations are most prevalent in pancreatic adenocarcinoma (PC) (70%), colorectal cancer (CRC) (40%), and non‐small cell lung cancer (NSCLC) (30%). (A) PC; (B) colorectal adenocarcinoma; (C) lung adenocarcinoma; (D) uterine corpus endometrial adenocarcinoma; (E) uterine carcinosarcoma; (F) stomach adenocarcinoma; (G) testicular germ cell tumors; (H) cholangiocarcinoma; (I) cervical squamous cell carcinoma; (J) skin cutaneous melanoma; (K) acute myeloid leukemia; (L) thyroid carcinoma; (M) pheochromocytoma and paraganglioma; (N) thymoma.
RAS mutations occur with high frequency at G12, G13, and Q61, 55 , 56 , 57 causing unique changes in RAS domain conformation 58 , 59 and facilitating persistence of RAS–GTP‐binding form. 54 Extensive studies had been conducted on the structural and functional influences of these special sites on RAS, such as GDP/GTP binding, intrinsic GTPase activity, and effector interaction. The biochemical function of each hot‐spot mutation site and the damage to physiological activities of RAS caused by site‐related mutations are summarized below, including G12, G13, and Q61.
3.1. GTP hydrolysis rate
RAS forms a transition state during either GAP‐mediated or RAS‐intrinsic hydrolysis for GTP. 60 , 61 , 62 , 63 Although the transition state form during RAS–GTP hydrolysis is still controversial, 64 current studies support that G12, G13 (located in the P‐loop), and Q61 (located in the switch II region) once mutated, the transition state form would be disturbed to some extent, impairing the ability of RAS to hydrolyze GTP. This is likely to be one of the reasons for the oncogenic properties of mutated RAS. The related mechanisms of GTP hydrolysis rate changes caused by G12 and G13 mutations are complicated.
In the normal GAP‐mediated hydrolysis, Arg789 (also known as arginine finger) of GAP interacts with the carbon atom in the main chain at G12 of RAS. 65 , 66 , 67 , 68 However, when G12 is mutated to other amino acids, the extra side chains of alternative conflict with the interaction, which stops the formation of transition state complex of G12 mutant and GAP, resulting in a lower rate of the GAP‐dependent hydrolysis of GTP compared to wild type. 69 , 70 Similarly, G13 mutants with large side chains are unable to form transition state complexes with GAP, while the mutants with small side chains are able to form transition states. 69 In addition, there are data showing that mutations in G12 and G13 (G13D) affect the interaction between hydrolysis catalytic residues Q61 of RAS, R789 of GAP and GTP, thereby weakening GAP‐mediated GTP hydrolysis. 59 , 71 Another report mentions that the G13V mutant could hinder fast hydrolytic steps by increasing the flexibility of the region of RAS binding to GTP γ‐phosphate group, catalytic water and Arg789. 72 Surprisingly, Rabara et al. 73 found that KRAS mutation at G13 is sensitive to GTP hydrolysis stimulated by NF1 (a GAP), which is different from G12 and Q61.
The intrinsic RAS GTPase activity, relative to GAP‐dependent GTP hydrolysis activity, 74 may determine the interaction duration between RAS and downstream objective response rate (RAF)‐related pathways. 19 The affinity of RAF for KRAS is higher than that of P120GAP and NF1, and more importantly, their binding protein domains of RAS overlap. 75 At the same time, these differences in affinities may also be one of the reasons for biological behavior differences among mutant forms. 76 When RAS binds to RAF–ras binding domain (RBD), the combination of Y32 residue located in switch I of RAS and Q61 residue interacts with the γ‐phosphate group in GTP through a bridged water molecule to form a transition state complex. 77 , 78 Although the model of specific transition state formation has not yet been determined, it is found that once other amino acids, such as alanine and valine, replace glycine at 12‐position, the steric hindrance caused by oversized side chain would hinder the formation of transition states or reduce the stability. 79 Moreover, according to previous reports, G13 is also involved in the hydrogen bond interaction in GTP hydrolysis, and the large side chain due to mutation would cause instability of the transition state. 59
Compared with G12 and G13, Q61 plays a direct and important role in both intrinsic and GAP‐mediated GTP hydrolysis. Q61 regulates nucleophilic water molecules close to the γ‐phosphate of GTP, and the intrinsic hydrolysis rate of RAS significantly reduces once Q61 is mutated. 19 , 80 Thus, the hydrolysis rates in Q61 mutants are the lowest. 76 However, unlike the G12 mutations, the hydrolysis rate of the Q61 mutations could be significantly increased by GAP, 81 although the mechanism is still unclear.
3.2. Nucleotide exchange rate
Compared with wild type, G12 and Q61 mutated RAS, the nucleotide exchange rate in the RASG13D is significantly higher, and it is more important that this rapid nucleotide exchange is spontaneous through an SOS‐independent way. 81 Chen et al. 82 G13D mutation causes the destabilization of the hydrogen bond formed between D30 in SWI switch and GDP, facilitating RAS–GDP to GTP transformation. Additionally, an increase in the structural flexibility of the switch domains SW1 and SW2 in mutant may result in a decrease in the stability of the nucleotide‐binding pocket. For the reasons described above, nucleotide exchange with a rapid rate starts up. It is found that the G13D mutation is rare in HRAS and NRAS compared with other mutations, and there is a speculation that KRAS may have a specific residue background different from the other paralogs to support the instability of the G13D active site. 83 Unfortunately, there are few data for other types of mutations at G13.
As for G12 and Q61 mutations, Smith et al. 81 found that without SOS1 mediation, the nucleotide exchange rate of G12V is 1.8 times slower than that of wild type, while the exchange rate of Q61L is 2.4 times faster, and the former requires more SOS1 assistance to reach the wild‐type exchange rate.
3.3. Affinity differences in downstream effectors
When mutated, the binding affinity of RAS to different downstream effectors changes, which means that the biases for downstream signaling may change.
There are data showing that the affinities of ARAF and BRAF to wild‐type RAS are stronger than the affinity of G12V mutant, while the binding preference of RGL1 and RALGDS (two GEFs of RAL GTPases) for RASG12V increases. 84 In addition, compared to KRASWT‐overexpressing cells, the Akt signaling of HBECsiP53 cells with KRASG12C overexpression is lower and Ral signaling and anchorage‐independent growth are stronger, whereas the phosphorylated Akt levels are higher and Ral activation is lower when KRASG12D is overexpressed. 85
In conclusion, signal transduction bias and variation in downstream signal stringency arising from different RAS mutations suggest that it is necessary to selectively use inhibitors targeting mutation‐preferred pathways in facing different mutations in clinical treatment.
4. CHARACTERISTICS OF SPECIFIC RAS MUTATIONS
Although the previous section shows the general impact of RAS mutations associated with respective hot‐spot sites, each mutation has specific biochemical characteristics. For example, the development of G12C inhibitor is based on the feature that G12C is in the GDP‐binding form for a longer time than KRASWT or other mutations. 86 Therefore, we introduce the biochemical characteristics of common oncogenic RAS mutations in this section. In addition, in order to emphasize the significance of specific mutation, pathological differences brought by different mutations, such as the tendency of co‐mutations, transformation capacity, and clinicopathological features, are also listed.
4.1. Differences in structure and biochemical signature
The function of protein depends on its structure. Table 1 summarizes the structural differences of the following RAS mutations, expecting to be helpful for the design of allele‐specific inhibitors.
TABLE 1.
Structural differences of RAS mutations.
| Allele | Difference | Potential biochemical impact | References |
|---|---|---|---|
| KRASG12C | Cysteine insertion may improve the electrostatic environment around γ‐phosphate of GTP | Intrinsic hydrolysis rate close to wild type | 76 |
| More exposed nucleotide binding site | An increased propensity for the conformational transition of KRAS from inactive to active | 59 | |
| R789 of GAP away from GTP in KRASG12C–GTP–GAP complex | A decrease in the rate of GAP‐mediated hydrolysis | 59 , 76 | |
| Broken hydrogen bond between Q61 and the γ‐phosphate of GTP | |||
| KRASG12D | The atom OE1 of Q61 side chain away from GTP in KRASG12D–GTP–GAP complex | A decrease in the rate of GAP‐mediated hydrolysis | 59 |
| More exposed nucleotide‐binding site | An increased propensity for the conformational transition of KRAS from inactive to active | 59 | |
| Deviation of SII residues toward the α3 helix and disruption of the hydrogen bonding network in SII | Slow GTP hydrolysis rate | 87 | |
| The electrostatic repulsion between the carboxylate group on the side chain of aspartic acid and the γ‐phosphate of GTP | The binding force of KRASG12D to GppNp is weaker than that of KRASG12V or KRASWT | 88 | |
| KRASG12V | The atom OE1 of Q61 side chain away from GTP in KRASG12V–GTP–GAP complex | A decrease in the rate of GAP‐mediated hydrolysis | 59 |
| KRASG12R | The flexibility of Q61 affected and the cooperation of Q61 with nucleophilic water broken | A decrease in GAP‐dependent hydrolysis rate | 76 |
| The side chain of arginine creates a steric conflict with the Y32 residue, and displaces adjacent coordinated water | A decrease in intrinsic hydrolysis rate | 76 | |
| KRASG12A | Alanine insertion may change the electrostatic environment around the γ‐phosphate of GTP and reorder the solvent | A decrease in intrinsic hydrolysis rate | 76 |
| KRASG13D | The arginine finger of GAP is blocked from accessing phosphate of GTP | A decrease in GAP‐dependent hydrolysis rate | 73 |
| KRASG13R | The arginine finger of GAP is blocked from accessing phosphate of GTP | A decrease in GAP‐dependent hydrolysis rate | 73 |
| KRASQ61H | The atom ND1 of H61 side chain away from GTP in KRASG12V–GTP–GAP complex | A decrease in the rate of GAP‐mediated hydrolysis | 59 |
| HRASQ61L | An increase in the flexibility of the SII domain, while in the opposite with RAF–RBD binding | Reduced intrinsic hydrolysis rate and stronger RAF/MEK/ERK signal | 89 , 90 |
Abbreviations: GTP, guanosine triphosphate; HRAS, harvey‐RAS; KRAS, kirsten‐RAS; NRAS, neuroblastoma‐RAS; RAF, Serine/threonine‐protein kinase RAF; RAS, rat sarcoma; RBD, RAS binding domain.
By comparing various biochemical indicators of KRAS mutants, Hunter et al. 76 found that small structural differences due to mutation led to specific biochemical signatures. For example, alteration in nucleotide exchange rate is most notable in KRASG13D. According to Table 1, KRASG13D enables faster nucleotide exchange independent of GEFs. As for the intrinsic hydrolysis rate, KRASG12A/G12R/Q61H/Q61L mutation brings about a drop to the lowest level. 76 On the contrary, unlike other mutants with severely impaired hydrolysis capacity, KRASG12C has no significant effect on intrinsic hydrolysis rate compared to wild type. 76 Alternatively, it is more likely to develop the GDP‐binding form KRASG12C than other mutations, which gives opportunities for the generation of direct inhibitors.
Overall, biochemical differences among RAS mutations offer an opportunity to design specific inhibitors of RAS mutants.
4.2. Differences in co‐mutation
Rabara et al. 73 reported that the KRASG13X is more sensitive to GTP hydrolysis stimulated by NF1 (a GAP) than G12 mutants and Q61 mutants and partially dependent on upstream signaling from epidermal growth factor receptor (EGFR) or other RTK, which may enable EGFR inhibitors to prevent the development of colorectal cancer (CRC) with G13D mutation. 91 In addition, Rabara et al. 73 also pointed out that cells with low‐frequency and weakly oncogenic KRAS mutations are more prone to additional mutations. In other words, their genomes have higher genetic instability. For example, KRASG13X and NRASA146T are occasionally associated with NF1 mutations, while NF1 mutations are rarely observed in cells with KRASG12X and KRASQ61X mutations. According to the above, EGFR inhibitors may be an option that is worth considering in the treatment of cancers with KRASG13x and normal NF1 activity.
4.3. Differences in transformation capacity
The transformation capacities of different mutants vary. By comparing a large number of c‐Ha‐ras1 mutants, Seeburg et al. 92 showed that the transformation abilities of HRASG12R and HRASG12V mutations are stronger than those of other mutations at codon 12. Additionally, by transfecting NIH3T3 cells with plasmids expressing wild‐type or mutated KRAS, Smith et al. 93 observed that the transformation ability of mutations at codon 12 was slightly stronger than that of mutations at codons 13, 61, 146, and 117.
4.4. Clinicopathological varieties in different RAS mutations
In addition to the above, the clinicopathologic features of cancers carrying different RAS mutations are also different. Table 2 summarizes the pathological differences in various RAS mutation‐induced cancers.
TABLE 2.
Clinicopathological differences among cancers.
| Cancer | Mutation | Difference | References |
|---|---|---|---|
| PC | KRASG12D | Lower survival rate and poorer prognosis after first‐line gemcitabine therapy and shorter OS in patients with locally advanced and/or metastatic PDAC | 94 |
| Little response to the combination of cobimetinib and gemcitabine in PDAC | 95 | ||
| A strong association with early distant metastasis after radical tumor resection and shorter postoperative OS and PFS in PDAC | 96 | ||
| KRASG12V | Little response to the combination of cobimetinib and gemcitabine in PDAC | 95 | |
| CRC | KRASG12D | Poor response to FOLFOX treatment and high risk of disease recurrence | 97 |
| Shorter OS compared with patients with KRASWT | 98 | ||
| KRASG12X | For patients with CRLM undergoing radical liver resection, KRAS G12 mutations is associated with shorter OS than G13 mutations, especially G12V or G12S. Besides, G12D and G12V are related to shorter OS and PFS | 99 , 100 | |
| KRASG12V | A higher risk of relapse and death | 88 , 101 | |
| Poorer survival in BRAFWT CRC | 102 | ||
| KRASG13D | Multiple metastatic sites as the disease progressed | 103 | |
| A tendency to lead lymph node metastasis, more common in advanced cancer and associated with higher PFS | 104 | ||
| KRASG12X | Associated with mucus histotype and favoring signaling pathways involved in regulating mucin production in colonic mucosal cells | 104 | |
| KRASG12C | Shorter OS, higher basal EGFR activation, and reduced immune profile | 105 | |
| NSCLC | KRASG12C | Shorter PFS treated with PD‐L1 inhibitors among patients with high PD‐L1 expression | 106 |
| KRASG12V | Worse OS, PFS, and recurrence rates | 107 | |
| Shorter survival, shorter duration of response to initial chemotherapy, and shorter OS after immunotherapy in patients with advanced NSCLC with KEAP1/NFE2L2 co‐mutation | 108 | ||
| More pleural–pericardial metastases after thoracic surgery compared with other mutations | 107 |
Abbreviations: CRC, colorectal cancer; CRLM, colorectal liver metastases; EGFR, epidermal growth factor receptor; NSCLC, non‐small cell lung cancer; OS, overall survival; PC, pancreatic adenocarcinoma; PDAC, pancreatic ductal carcinoma; PD‐L1, programmed death‐1‐ligand 1; PFS, progression‐free survival.
5. DEVELOPMENT OF DIRECT MUT‐RAS INHIBITORS
5.1. Non‐mutation‐specific inhibition ways targeting oncogenic RAS
The structure of RAS has been studied in depth. However, it was not until recent years that inhibitors that bind directly and block the abnormal activities of oncogenic RAS were developed. The first hurdle in targeting RAS mutations is the lack of deep binding pockets. RAS is similar to globular protein with smooth surface and shallow depressions, lacking a clear hydrophobic pocket and obscuring allosteric site. 109 The other strategy is reducing the active state of RAS–GTP by competing for the GTP‐binding region. However, the binding strength of RAS with GTP reaches picomolar level and the concentration of GTP in the cytoplasm reaches as high as ∼0.5 mmol/L; thus, it is too difficult to develop drugs that compete with RAS–GTP binding. 11 , 59
After decades of efforts, some “hidden weaknesses” of RAS have gradually been uncovered. Lu et al. 110 found that the formation of activated RAS experiences multiple sub‐states, and there is a new allosteric druggable site P4 hidden in the protein, which could regulate the GTPase activity of RAS. More importantly, this site is located at the interface of the RAS domain, which interacts with downstream effectors, so targeting the interaction of RAS and downstream effectors becomes a new strategy to design drugs. In addition, some research interests have focused on two RAS regions, switch II pocket and the junction area of the switch I/II pocket, both of which are potential druggable pockets. 18 The former was found to be an important region for covalent binding of drugs during the development of KRASG12C inhibitors, which could lock KRASG12C in an inactive form after binding to drugs, 111 also named KRAS‐off inhibitors. 15 , 86 , 112 The latter, as a conserved structure shared by all three RAS paralogs, has more potential as a pan‐RAS inhibitor targeting region, but how to improve the affinity of the drug for mutant RAS and make it much higher than for the wild‐type RAS is a thorny challenge. 113 More recently, “KRAS‐on” inhibitors that target active KRAS–GTP have also been developed to block interactions of “on‐state” RAS with downstream effectors. In this strategy, cyclophilin‐A, as an intracellular chaperone protein of drug, binds to S‐IIP of RAS–GTP to form a tri‐complex of KRAS, cyclophilin‐A, and drugs, such as RMC‐6291. 114 The tri‐complex leads to the covalent RAS cross‐linkage, subsequently blocking downstream effector binding to KRAS. Similarly, in the presence of the compound RM‐018, chaperone proteins block downstream signal transduction by forming tri‐complexes to block binding of RAS with RAF. 115 This strategy provides more flexibility for investigating ways to specifically target RAS mutations.
In addition, recent studies have revealed that there are missense mutations at the same site or certain differences among mutations at different RAS sites, which may have implications for the development of drugs targeting specific RAS mutations. Unfortunately, there are few studies comparing the differences between paralogs at the same point. For example, a report shows significant phenotypic differences as well as divergence in signaling pathways of KRASG12D and NRASG12D in CRC. 116
5.2. Specific inhibitors targeting mutated RAS
In addition to the non‐specific mutation inhibitors, the recent success in developing KRASG12C inhibitors has revived interest in developing KRAS inhibitors that either directly target KRAS mutations or target key steps required for KRAS activation. The research progress for specific targeted inhibitors of KRASG12x mutations (such as G12C) is still ahead of other mutant KRAS. Regrettably, efficacious targeted inhibitors of G13D and Q61R have not been found, nor have specific inhibitors of oncogenic NRAS and HRAS mutants.
5.2.1. Drugs targeting KRASG12C
5.2.1.1. Introduction of KRASG12C inhibitors
KRASG12C with a high incidence in cancers such as non‐small cell carcinoma is a common type of KRAS mutation. 117 Recently, researchers have found that there is a new pocket adjacent to the mutated cysteine residue (cys12) on the switch II region of the GDP‐binding KRASG12C protein. The developed small‐molecule targeted drugs could bind to the allosteric pocket by forming irreversible covalent compounds with cys12. Moreover, other inhibitors preferentially bind to the KRASG12C–GDP and block SOS‐mediated nucleotide exchange and thereby inhibit the hyperactivation signals of RAS. 86 The successes in clinical trials of these inhibitors make a breakthrough in the development of KRASG12C‐specific compounds with anticancer activity in vivo for targeting the SII pocket. The information on the structure, covalent bond, and medication guidance of KRASG12C inhibitors is summarized in detail in several reviews. Dunnett‐Kane et al. 118 provide a detailed description of the therapeutic activity of several registered KRASG12C‐specific inhibitors and summarizes their clinical trial stages and trial designs. In addition, interestingly, in cancer cells that are insensitive or resistant to a covalent KRASG12C inhibitor ARS1620, Zhang et al. 119 reported that ARS1620‐modified peptides in major histocompatibility complex I (MHC‐I) complex could serve as neoantigens. Subsequently, these neoantigens are recognized by recombinant antibody P1A4. The P1A4 induces cytolytic T‐cell response, so ARS1620‐resistant KRAS G12C mutant cells are killed in vitro. The strategy provides inspiration for reducing clinical resistance of single G12C inhibitors.
Therapeutic effect of RAS G12C inhibitors
Since the successful development of G12C mutation inhibitors was reported, various G12C inhibitors have been rapidly put into clinical trials. Among G12C inhibitors, sotorasib (AMG510) and adagrasib (MRTX849) are representative agents that have been shown to have direct therapeutic effects in various cancers with G12C mutations. Although targeted inhibitors of KRASG12D have also been developed, there are no clinical trial data. Therefore, this section will focus on the clinical progress of G12C inhibitors.
On the one hand, sotorasib was approved by the Food and Drug Administration as a second‐line treatment for locally advanced or metastatic non‐small cell lung cancer (NSCLC) containing the KRASG12C mutation. 120 In the CodeBreaK100 phase I trial, 960 mg was identified as the recommended phase II dose by estimating the response of approximately 30 patients in the dose‐escalation cohort. 121 After 11.7 months as the median follow‐up time, 32.2% of the patients in the subgroup with NSCLC had a confirmed response with a disease control rate of 88.1%. The median progression‐free survival (mPFS) for patients with NSCLC was 6.3 months. In the phase II portion of CodeBreaK100, among the 126 enrolled patients, 37.1% of the patients had a confirmed response with a disease control rate of 80.6% with a median follow‐up of 15.3 months. The mPFS was 6.8 months and the median overall survival (mOS) was 12.5 months. The rate of treatment‐related adverse events was 69.8%, including 19.8% probability of occurrence of grade 3 event and 0.8% probability of occurrence of grade 4 event. 112
Also, sotorasib has shown monotherapy clinical activity in KRASG12C‐mutated CRC in the CodeBreaK100 phase I trial, although the treatment is less effective. With a median follow‐up of 12.8 months, the response rate among patients was 7.1% with a disease control rate of 66.7%. 121
On the other hand, the phase I/II KRYSTAL‐1 trial explored the therapeutic effect of adagrasib monotherapy. 122 A dose of 600 mg twice daily was identified as the recommended phase II dose by estimating the response of 25 patients in the dose‐escalation cohort. Among 15 evaluable patients with KRASG12C NSCLC treated with adagrasib 600 mg bid, 53.3% of the patients had a confirmed response, with a median duration of response of 16.4 months. The mPFS was 11.1 months and the 12‐month survival rate was 66.7%.
Adagrasib is also being studied in CRC with KRASG12C. 123 With a median follow‐up of 12.8 months, the response rate among patients was 22%, with a disease control rate of 87%, and the mPFS was 5.6 months.
Resistance mechanism of G12C inhibitor monotherapy
In the clinical application of G12C inhibitors, it has been observed that there are differences in drug‐resistance involved pathways in different cancer types. In NSCLC, upregulation of MEK and ERK is found to develop for resistance of sotorasib. Differently, in CRC, the phosphorylation of EGFR leads to the resistance to G12C inhibitors. 94 , 124 , 125 This suggests that for CRC carrying G12C, the combination of G12C inhibitors and EGFR inhibitors may be clinically more effective than G12C inhibitor monotherapy.
5.2.2. Drugs targeting KRASG12D
There are several studies on specific inhibitors targeting KRASG12D. KRASG12D commonly occurs in digestive system cancers, especially pancreatic cancer and CRC. The lack of a highly reactive residue, such as the cysteine residue at position 12, renders targeted KRASG12D drug design in a distinct approach. Vatansever et al. 58 performed computational simulations on the kinetic data of KRASG12D to explore the changes in the domain of mutated RAS, aiming to lay a foundation for subsequent targeted drug research. Similarly, through computational analysis of protein structures, Feng et al. 126 found P110, the connection site adjacent to proline 110, and a small molecule named KAL‐21404358 that binds to this site (K D = 100 μM). In addition, the compound 3144 developed by Stockwell et al. 127 could target KRASG12D with micromolar affinity. Interestingly, when screening small‐molecule compounds targeting KRAS switch I/II pocket, Kessler et al. 113 reported that a compound named BI‐2852, in contrast to covalent inhibitors, could bind to both the active and inactive forms of KRAS with nanomolar affinity. Recently, a specific inhibitor targeting KRASG12D, named MRTX1133, has been shown to bind KRASG12D in a non‐covalent binding form (K D = ∼0.2 pM). And the selectivity of MRTX1133 binding to KRASG12D is 700‐fold higher than that of binding to KRASWT. 128 In addition to small‐molecule compounds, there has been some progress in the development of peptides as another form of targeted drugs. For example, KRpep‐2d and its derived peptide KS‐58 are developed to selectively bind to the KRASG12D–GDP with sub‐nanomolar K D values and inhibit nucleotide exchange of KRASG12D. 129 Table 3 summarizes the characteristics of mutated KRAS inhibitors (except G12C).
TABLE 3.
Features of non‐G12C KRAS inhibitors.
| Targeted mutation | Name | Structure | Reactive functional group | Targeted position in RAS | K D | Reference |
|---|---|---|---|---|---|---|
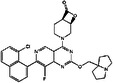
| G12D | KAL‐21404358 |
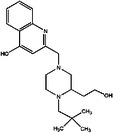
|
Quinolinol and piperazinyl group | Proline 110 | 100 μM | 126 |
| 3144 |
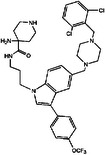
|
N/A | S39, D38, E37, and I36 | ∼20 μM | 130 | |
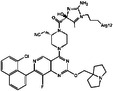
| BI‐2852 |
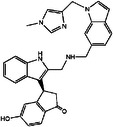
|
Isoindolinone | SI/II pocket | 750 nM | 113 | |
| MRTX1133 |
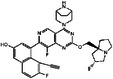
|
N/A | S‐II pocket | ∼0.2 pM | 128 | |
| KS‐58 |
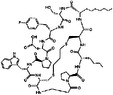
|
Amide bond cyclization and main chain cyclization | S‐II pocket | 100 nM | 129 | |
| G12V | H‐REV107 | N/A | L65, Y66, D67, G70, D72, K73, and Y74 | SI/II pocket and P‐loop | 30 μM | 131 |
| TKR15 | N/A | Thiourea and benzotrifluoride group | N/A | N/A | 132 | |
| G12S | G12Si‐5 |
|
β‐Lactone | S‐II pocket (Ser12) | 26 μM | 133 |
| G12R | N/A |
|
α,β‐Diketoamide | S‐II pocket (ε‐N and η‐N of R12) | N/A | 134 |
The structure of the chemical formula in Table 3 is drawn with ChemDraw.
5.2.3. Drugs targeting KRASG12V
Currently, the clinical trials for specific inhibitors of KRASG12V are rare. Han et al. 131 developed a derived peptide based on H‐REV107 in vivo that could interact with KRASG12V–GDP to form a stable complex, thereby blocking the activating function of KRAS and inhibiting phosphorylation level of MEK/ERK.
In addition, there is a report finding that tyrosine kinase receptor 15 (TKR15), a thiourea derivative, is able to inhibit cell proliferation and induce apoptosis in A549 cells by targeting KRASG12V. 132
5.2.4. Drugs targeting other KRAS mutations
In the development of KRASG12R inhibitors, Zhang et al. 134 screened α,β‐diketoamide from common electrophilic ligands based on the nucleophilicity of arginine residue in KRASG12R. The research reveals that the ligand is able to selectively bind to KRASG12R in a covalent form, providing reference for the development of inhibitors targeting KRASG12R.
In the development of KRASG12S inhibitors, Zhang et al. 133 also chose the nucleophilicity of serine residue as the weakness of KRASG12S. The potential of β‐lactone derivatives as ligands for KRASG12S is revealed, and Ser12 in SIIP is acylated by the carbonyl group of β‐lactone during ligand formation of a covalent complex with KRASG12S.
6. INDIRECT MUT‐RAS INHIBITION: INTERVENTION IN DOWNSTREAM PATHWAYS
RAS, as an important component in regulating cell growth and proliferation, is involved in a large network of related cascaded signaling pathways. In the development of anticancer drugs, it is essential to understand the signaling pathways associated with RAS. 115 , 135 Overactivation of the RAF/MEK/ERK and phosphatidylinositol‐3‐kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) pathways enhances growth, survival, and metabolism of cancer cells. Thus, the signaling pathways have been identified as promising therapeutic targets for cancer therapy. 136 This section focuses on the RAS downstream pathways shown in Figure 3 that are closely associated with the development of cancers with RAS mutation. The impact of pathways for specific cancer types is summarized as well.
FIGURE 3.
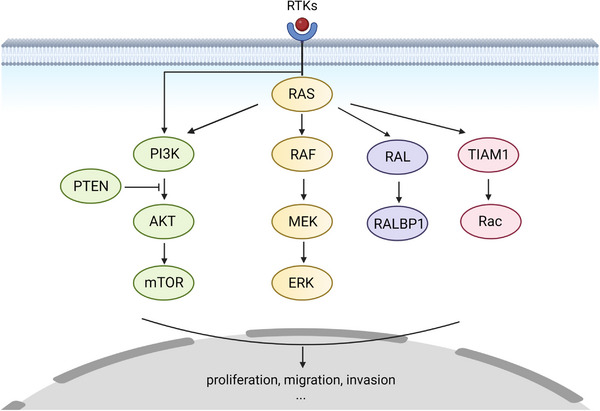
Downstream signaling pathway of KRAS. After receiving the signal of epidermal growth factor, receptor tyrosine kinases (RTKs) such as EGFR will recruit RAS, targeting the membrane and activating it. Therefore, phosphorylation activation signals are passed in the downstream cascades, which contain the RAF–MEK–ERK, PI3K–protein kinase B (AKT), RAL, and TIAM1 pathways. These cascades regulate cell proliferation, migration, and invasion. The figure was made using Biorender.
6.1. Intervention in RAF–MEK–ERK (MAPK) pathway
6.1.1. Components of MAPK pathway
The RAS/mitogen‐activated protein kinase (MAPK) pathway, as a classical downstream signaling pathway of RAS, is closely related to variety of cancers. MAPK is a phosphorylated activation effector cascade composed of RAF, MEK, and ERK protein kinases, regulating cell growth, survival, proliferation, and differentiation. 137 , 138 , 139 , 140 RAF is the direct effector of activated RAS as the beginning of cascade signals. And there are three proteins in RAF family, ARAF, BRAF, and CRAF. In cascade signaling, RAF is recruited through its RBD domain selectively binding to the active GTP–RAS on the plasma membrane. 141 , 142 Subsequently, the cysteine‐rich domain (CRD) domain of RAF selectively binds to the farnesyl groups of modified RAS, which further promotes the recruitment of RAF to cell membrane. 143 , 144 On the basis of RBD binding to RAS, CRD also interacts with phosphatidyl serine in the plasma membrane. 145 During the recruitment, affected by the increase in local concentration and conformational change of RAF, 146 , 147 its homologous dimerization and heterodimerization with any other RAF family member will be induced, which also promotes the activating phosphorylation of RAF. 148 After activating the downstream MEK/ERK, RAF kinase reverts to an inactive conformation due to complex regulation, such as dephosphorylation by kinases, 149 autoinhibition by amino‐terminal domain, and negative feedback regulation of ERK. 150 , 151 , 152 However, once RAF is mutated, the above complex regulation of RAF activation will be disrupted. Oncogenic BRAF mutations are common in cancer. 153 One class of BRAF mutants, BRAF‐V600 mutants, will lose its autoinhibitory activity and also be activated continuously without relying on RAS or dimerizing. Another class of mutants will similarly be RAS independently activated, which still relies on dimerization for complete activation. The third mutants, stimulating BRAFWT to form heterodimers, are kinase‐impaired proteins and need RAS‐binding activation and dimerization. 154 , 155 , 156 , 157
Similar to RAF, a region of the N‐terminal lobe (negative regulatory region) of MEK kinase also exists for autoinhibition. 158 In the process of MEK activation by RAF phosphorylation, kinases such as kinase suppressor of RAS (KSR) act as scaffold proteins to assist the assembly of RAF–MEK–ERK complex and promote RAF to activate MEK. 159 , 160 After the complex forms, the serine residues of MEK (S218 and S222 of MEK1, S222 and S226 of MEK2) are diphosphorylated rapidly for MEK activation. 161 , 162 Subsequently, tyrosine and threonine residues of downstream ERK (T202/Y204 of ERK1 and T183/Y185 of ERK2) are dephosphorylated. 163 Unlike RAF kinase, MEK/ERK mutations are rare in human tumors. 164 , 165
6.1.2. The role of MAPK in cancer
In driving the development of RAS‐mutated cancers, the activity of MAPK kinases varies among different tumors. Furthermore, RAF subtypes have different influences on RAS‐driven tumors. For example, BRAF is dispensable for NSCLC with KRASG12V or KRASG12D, caused by the other RAF kinases presenting a compensatory effect without increased expression. On the contrary, CRAF is essential to mediate oncogenic signaling in the same cancer. 166 , 167 However, CRAF deficiency does not affect tumor development of pancreatic ductal carcinoma (PDAC) with KRASG12D or KRASG12V, while concomitant ablation of EGFR and CRAF completely prevent PDAC development driven by KRASG12V/Trp53 mutation. 168 , 169 However, the selection of CRAF as a therapeutic target tends to rely on ablation rather than inhibition of kinase activity. On the one hand, clinical inhibition of CRAF kinase activity requires prevention of inhibition of BRAF kinase activity, thereby avoiding increased toxicity. 170 On the other hand, CRAF ablation regulates apoptotic pathway‐related proteins independent of kinase activity or MAPK pathways, which may support CRAF ablation to inhibit tumor development. 171 In addition to apoptosis‐related proteins, CRAF regulates many proteins in a kinase‐independent manner. For example, CRAF inhibits the kinase activity of ROKα, and the ablation of RAF1 induces the regression of squamous tumors via ROKα‐mediated cellular differentiation. 172
Unlike RAF, systemic ablation of MEK or ERK will induce the unacceptable toxicities in adult mice, although preventing tumor development. 170 Interestingly, in the CRC model with KRASG12V, heterogeneity in intracellular ERK phosphorylation has been observed. ERK levels are generally higher in cancer cells adjacent to stromal cells at the invasive front and lower in more central areas of cancer specimens. 173
6.1.3. Clinical effect of MAPK inhibitors
Although the MAPK is a linear cascade, the regulation of each element involves multiple kinases. Therefore, in the treatment with inhibitors, the regulation of feedback pathways in vivo after drug administration should be considered to avoid drug resistance.
Significant efforts have been made to develop inhibitors targeting the MAPK pathway, especially BRAF and MEK inhibitors. However, only the BRAF inhibitors vemurafenib and dabrafenib and the MEK inhibitors trametinib and cobimetinib have been approved only for BRAF‐V600E/K metastatic melanomas. 174 In KRAS‐mutated tumors, BRAF inhibitors promote heterodimerization of BRAF and CRAF, thereby activating the MAPK pathway and helping secondary tumor development. 175 , 176 , 177 Recently, some novel pan‐RAF inhibitors have been investigated to solve the problem of promoting dimerization. LY3009120 inhibits MEK1/2 phosphorylation by inhibiting kinase activity in BRAF–CRAF heterodimers and retards the development of tumors carrying KRAS mutations while inducing a more significant dimerization. 178 However, when used as a single agent, the required dose of RAF inhibitor being effective in KRAS‐mutated models is significantly higher than that in the BRAF‐V600E model. Therefore, it is necessary to continue upgrading effective drugs or treat with a combination of drugs.
Similar to RAF inhibitors, cobimetinib also fails to inhibit KRAS mutation‐carrying tumors due to RAF feedback activation of MEK phosphorylation. 179 In contrast, trametinib appears to inhibit the proliferation of KRAS mutant A549 cells effectively by impairing the RAF–MEK interaction. 180 However, its resistance still exists in KRAS mutant NSCLC, which is caused by compensatory activation of FGFR1. 181 As a result, MEK inhibitors are increasingly used in combination with other inhibitors to avoid drug resistance due to the feedback regulatory mechanism induced by their use alone.
As for ERK inhibitors, the drug resistance tends to develop after monotherapy with ERK1/2 inhibitor over a period of time. 182 , 183 One of the mechanisms is the compensatory effect of ERK5 in place of the inactivated ERK1/2. 184 In response, inhibitors against ERK5 have been developed. On the other hand, ERK inhibition blocks negative feedback of ERK and induces feedforward activation of upstream RTK, thereby inducing activation of alternative pathways such as the PI3K/AKT pathway to maintain tumor cell survival. Therefore, inhibitors targeting both ERK1/2 and other kinases are also developed for treatment. 185 For example, Gao et al. 186 found that an indole‐substituted pyrimidine derivative inhibits the activities of AKT and ERK1/2, thereby inhibiting tumor growth and extending the survival time of tumor‐bearing mice.
As a significant downstream signaling pathway of RAS, MAPK has become a popular therapeutic target. However, the efficacy of MAPK inhibitors in monotherapy of tumors with RAS mutation is not ideal. According to the above, one of the reasons is that the MAPK pathway is abnormally activated after the suppression of components. The mechanisms of reduced effectiveness or resistance of inhibitors vary according to the target and how the inhibitor works. On the other hand, the toxicity caused by inhibitors is also worthy of attention. 187 Therefore, rather than monotherapy, combinations of drugs targeting the MAPK pathway have been studied more frequently in the clinical treatment of cancers carrying RAS mutations.
6.2. Intervention in PI3K/PTEN/AKT/mTOR pathway
6.2.1. Components of PI3K pathway
Similar to MAPK pathway, PI3K pathway is an effector cascade dependent on phosphorylated activation. PI3K is divided into three classes: I, II, and III. Class I PI3Ks are expressed in various cell types and are related to development of cancers. 188 , 189 , 190 , 191 , 192 Therefore, class I PI3Ks are selected to be mainly discussed in this section. Class I PI3Ks are heterodimeric proteins that are grouped into two subtypes: IA and IB. PI3K IA proteins are composed of a regulatory subunit (p85α, p85β, p50α, p55α, p55γ) and a 110‐kDa catalytic subunit (p110α, p110β, p110δ). PI3K IAs act as downstream kinases of TKRs and G protein‐coupled receptors (GPCRs). PI3K IBs have a p110γ catalytic subunit binding p101 or p87 as regulatory subunit. Different from PI3K IAs, PI3K IBs are activated by GPCRs. 193 , 194 , 195 Class I PI3Ks are activated through different upstream mechanisms, which mainly contain: (1) the regulatory subunit p85 binding to phospho‐YXXM motifs (X indicates any amino acid) of the RTK, thereby triggering activation of the catalytic subunit p110 196 ; (2) growth factor receptor‐bound protein 2 (GRB2) binding to phospho‐YXN motifs of the RTK in advance and to the scaffolding protein GAB, which in turn can bind to p85 197 ; and (3) GRB2 binding to RTK and subsequently activating SOS, RAS in turn, and finally activating p110 independently of p85. 198
After being activated, catalytic subunit of PI3Ks transfers the phosphate group to PIP2 to produce PIP3. In the course, phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a negative regulator, acts as a phosphatase to dephosphorylate PIP3 and convert it back to PIP2. Back to the downstream signaling pathway, PIP3 recognizes downstream proteins with a pleckstrin homology domain, such as PDK1 and AKT, and recruits them to the cell membrane. 199 , 200 In the cascade, AKT is partly activated by phosphorylation of PDK1 at T308. Subsequently, mTORC2 will fully activate AKT through phosphorylation of AKT at S473. 201 , 202 Then, many downstream effectors, which are involved in protein synthesis, cellular proliferation, apoptosis, and cell survival, such as mTOR, are phosphorylated by AKT. 203 , 204 , 205 , 206 , 207 , 208
6.2.2. The role of PI3K pathway in cancer
PI3K pathway activated by RAS is essential for lung carcinogenesis driven by KRASG12D. 209 However, the established tumors are less dependent on PI3K signaling and PI3K/mTOR inhibition only leads to partial tumor regression. 209 , 210 Similarly, p110α of PI3K inactivation dose dependently can prevent mouse lethality and the occurrence of cancers induced by KRASG12D. p110α activity is also required for in vivo superactivation of KRASG12D and other signaling pathways. 168 , 211
6.2.3. Clinical effect of PI3K pathway inhibitors
PI3K pathway has also been selected as a target for cancer therapy due to the discovery of overactivation of PI3K in a variety of cancers and its significance for proliferation and survival of cancer cells. However, in the course of treatment, problems such as abnormal activation of feedback, compensation activation, drug resistance, and toxicity of PI3K pathway inhibitors are found. 212 , 213 , 214
6.3. Intervention in other pathways
As a downstream effector of RAS, RAL GTPase mediates various cellular activities to regulate tumor invasion, proliferation, and resistance to cell death, which are executed by the RAL effectors, including RALBP1, Sec5, Exo8426, Filamin, PLD1, and ZONAB. 215 , 216 , 217 , 218 , 219 , 220 , 221 RAL is grouped into two subtypes: RALA and RALB. The former plays a major role in cancer development and metastasis. In NSCLC, growth of cancer cells carrying KRASG12C is more sensitive to RAL depletion. 222 As for in pancreatic cancers, RALA is essential for tumor growth, and RALA and RALB are both required for tumor invasion. 223 , 224
In addition of RAL, TIAM1 can bind to RAS through its RAS‐binding domain, causing synergistic formation of Rac‐GTP in a PI3K‐independent manner, thereby activating Rac to induce activation of the NF‐kappa B transcription factor and promotion of cancer cell survival. 225 However, in epithelial MDCK cells, Tiam1–Rac and RAS signaling seemingly oppose each other, since RASG12V‐induced epithelial–mesenchymal transition is negatively affected by Tiam1–Rac signaling. 226 , 227
7. COMBINATION STRATEGIES ADAPTED TO SPECIFIC MUTATIONS IN DIFFERENT CANCERS
After a long period of struggle in developing RAS‐targeted drugs, researchers have turned their attention to the RAS downstream signaling pathway inhibitors, in order to indirectly inhibit RAS activity. RAS would be considered as an important component of the signaling pathway that regulates cell growth and proliferation. The upstream activation network of RAS is quite complex, and the downstream signaling pathway network is highly complicated as well, both of which lead to intricate cross‐talk between RAS‐related pathways. Therefore, protein targeting in a pathway in the RAS‐related signaling network often fails to achieve the anticipated therapeutic purpose. Indeed, it is found in clinical treatment that specific targeted therapy or specific drug combination has an unexpected effect on the cancers caused by different RAS mutations. In this section, we mainly list the treatment of several cancers commonly carrying KRAS mutations, such as pancreatic cancer, NSCLC, and CRC. Table 4 summarizes the progress of clinical trials on direct KRAS inhibitors for cancers carrying known KRAS mutations with published results in the last 5 years. In addition, as a supplement, Table 5 shows ongoing clinical trials for cancers with KRAS mutations (except for KRASG12C).
TABLE 4.
Efficacy of clinical trials adapted to cancers with mutated KRAS in last 5 years.
| Cancer | Mutation | Drug | Study phase | Response rate (N) | DR (months) | PFS (months) | OS (months) | Reference |
|---|---|---|---|---|---|---|---|---|
| PC | G12C | Sotorasib | I/II | 21% (38) | N/A | 4.0 | 6.9 | 228 |
| G12R | Selumetinib sulfate | II | 0% (8) | N/A | 3.0 | N/A | NCT03040986 | |
| CRC | G12C | Sotorasib | I | 7.1% (42) | N/A | 4.0 | N/A | 121 |
| Adagrasib | I/II | 50% (2) | 4.2 | N/A | N/A | 122 | ||
| Adagrasib | I/II | 19% (43) | 4.3 | 5.6 | N/A | 229 | ||
| Adagrasib with cetuximab | 46% (28) | 7.6 | 6.9 | |||||
|
JNJ‐74699157 (ARS‐3248) |
I | 25% (4) | N/A (safety deficiency) | 230 | ||||
| NSCLC | G12C | AMG510 (sotorasib) | I | 90% (10) | 1.9–5.9 | N/A | N/A | 231 |
| Sotorasib | I | 32.2% (59) | 10.9 | 6.3 | N/A | 121 | ||
| II | 37.1% (126) | 11.1 | 6.8 | 12.5 | 112 | |||
| Docetaxel | III | 13.2% (129) | 6.8 | 4.5 | 11.3 | 232 | ||
| Sotorasib | 28.1% (158) | 8.6 | 5.6 | 10.6 | ||||
| Adagrasib | I/IIb | 53.3% (15) | 16.4 | 11.1 | N/A | 122 | ||
| Adagrasib | II | 42.9% (116) | 8.5 | 6.5 | 12.6 | 233 | ||
| JNJ‐74699157 (ARS‐3248) | I | 60% (5) | N/A (safety deficiency) | 230 | ||||
| G12D | Bortezomib with acyclovir | II | 41.2% (17) | N/A | 1 | 13 | NCT01833143 | |
Abbreviations: CRC, colorectal cancer; DR, duration of response; NSCLC, non‐small cell lung cancer; OS, overall survival; PC, pancreatic adenocarcinoma; PFS, progression‐free survival.
TABLE 5.
Ongoing clinical trials for cancers with KRAS mutations (except for G12C).
| Trial ID | Tested interventions | Treatment setting | Phase | Status |
|---|---|---|---|---|
| NCT05533463 | HRS‐4642 | Advanced KRASG12D mutant solid tumors | I | Recruiting |
| NCT05737706 | MRTX1133 | KRASG12D mutant solid tumors | I/II | Recruiting |
| NCT04853017 | ELI‐002 2P | PDAC with KRASG12D and KRASG12R | I | Recruiting |
| NCT05631899 | KRAS‐EphA‐2‐CAR‐DC abraxane cyclophosphamide PD‐1 antibody | Solid tumors with KRASG12V | I | Recruiting |
| NCT04146298 |
Cyclophosphamide fludarabine PD‐1 antibody Biological: G12V‐specific TCR transduced autologous T cells |
Advanced PDAC with KRASG12V | I/II | Recruiting |
| NCT04620330 | Avutometinib (VS‐6766) or/and defactinib | NSCLC with KRASG12V | II | Recruiting |
Note: The data originated from https://clinicaltrials.gov.
Abbreviations: NSCLC, non‐small cell lung cancer; PD‐1, programmed death‐1; PDAC, pancreatic ductal carcinoma; TCR, T cell receptor.
7.1. Therapy of KRAS‐mutated pancreatic cancer
7.1.1. Therapy of pancreatic adenocarcinoma with KRASG12D
First of all, considering the successful development of inhibitors targeting KRASG12D mutation, the therapeutic effect of KRASG12D inhibitors on pancreatic adenocarcinoma (PC) is preferentially discussed. As a non‐covalent KRASG12D inhibitor, MRTX1133 inhibits phosphorylation levels of ERK1/2 and cell viability in KRASG12D mutant cell lines (IC50 = ∼5 nM). Based on this, MRTX1133 exhibited obvious tumor regression (≥30%) in KRASG12D mutant PDAC models. 128 In addition to the efficacy, it is also found that combination of MRTX1133 and inhibition of EGFR or PI3Kα, which are members of potential feedback or bypass pathways, improves the antitumor activity in PDAC. As another means of targeting G12D mutations, combination of siG12D‐LODER with gemcitabine and FOLFIRINOX exhibits good tolerance and certain inhibition of disease progression in patients with locally advanced pancreatic cancer with KRASG12D. 234 Similarly, silence of KRASG12D by CRISPR‐CasRx appears to suppress the tumor growth, enhance the sensitivity of gemcitabine in PDAC, increase the survival of mice, and show obvious toxicity. 235
From the downstream pathway inhibitor perspective, trametinib (MEKi) and ruxolitinib (STAT3i) help nivolumab (programmed death‐1 [PD‐1] inhibitor) improve antitumor activity and survival in mice carrying Ptf1aCre/+ , LSL‐KrasG12D/+ , and Tgfbr2flox/flox tumors. More importantly, the combination strategy results in a clinical benefit for a patient undergoing chemotherapy‐refractory metastatic PDAC. 236
Despite the above‐mentioned therapies, immunotherapy is also receiving much attention. A study revealed that inhibition of interleukin‐6 enhances the antitumor effect of anti‐programmed death‐1‐ligand 1 (PD‐L1) checkpoint inhibitor therapy. The treatment combination delayed tumor progression (p < 0.001) and increased OS in engineered PDAC model with KRASG12D (p = 0.0012). 237
Recently, antidiabetic drug metformin was excavated to have antitumor activity in PDAC induced by KRASG12D. In LSL‐KrasG12D/+ , Trp53fl/+ , and Pdx1‐Cre (KPC) mouse model, metformin has ability of blocking tumor growth, inhibiting the incidence of abdominal invasion, and increasing the OS. 238 In addition, Si‐HSF1 was found to increase chemosensitivity to gemcitabine in vivo. 239
7.1.2. Therapy of PC with KRASG12V
In terms of targeting G12V drugs, Ghufran et al. 240 have designed peptides inhibiting KRASG12V, which are speculated to have the ability to inhibit G12V activity and reduce the progression of cancer. In spite of targeting RAS itself, a dual FT and geranylgeranyltransferase‐1 inhibitor named FGTI‐2734 can inhibit the growth of xenografts derived from four patients with pancreatic cancer with mutant KRAS (two G12D and two G12V) tumors. 241
In the aspect of immunotherapy, it has been reported that the combination of anlotinib and PD‐1 inhibitor and chemotherapy exhibits a long‐term partial response and good tolerance in a young patient suffering from PDAC with liver metastasis. 242
In other ways, sequential administration of cell‐cycle kinases CDK4 and CDK6 inhibitors after taxane treatment reduced cell proliferation in PDAC mouse model with both KRASG12V and Cdkn2a‐null mutations. 243 Diego‐González et al. 244 found that lipoplexes of si‐FOSL‐1 and si‐YAP reduce the tumor growth in mice carrying tumors induced by pancreatic tumoral cell lines (KRASG12V and p53 knockout) through peri‐tumoral injection.
7.1.3. Therapy of PC with KRASG12R
Pancreatic cancers with KRASG12R are studied less than the former. Recently, it was found that KRASG12R is defective for interaction with p110α of PI3K, while PI3Kγ will support macropinocytosis in KRASG12R mutant PDAC in a compensatory way. 245 , 246 As for the therapy taken in PC with KRASG12R, the administration of selumetinib as MEK1/2 inhibitor appears to have little effect. 247 This suggests that single‐agent MEK inhibition is unable to meet the therapeutic needs of patients suffering from pancreatic cancer with G12R. And the combination of cobimetinib (MEK1/2 inhibitor) and gemcitabine improves PFS and OS after treatment in patients with KRASG12R compared with pancreatic cancer patients with KRASG12D/G12V. 95
7.2. Therapy of KRAS‐mutated CRC
7.2.1. Therapy of CRC with KRASG12D
Different from the therapeutic effect in pancreatic cancer, MRTX1133 has a lower inhibitory effect in CRC carrying KRASG12D. 128 In contrast to PDAC, KRAS mutations are usually not considered an initial driver in CRC, which may be one of the reasons for the limited effect of KRASG12D inhibitors in CRC patients. 248 Similarly, the G12D‐targeting pathway is peptide KRpep‐2d. Similarly, peptide KRpep‐2d, a G12D‐targeting inhibitor, had no significant antitumor effect on the PDX model, while oxaliplatin showed a significant inhibitory effect. 249 The failure of KRpep‐2d is suspected to be related to bioavailability and stability. 250 Additionally, the combination of binimetinib, hydroxychloroquine, and bevacizumab makes a 17% reduction in lung metastasis size in FOLFOX‐resistant patients after 6 weeks treatment with this combination with FOLFOX. 251
7.2.2. Therapy of CRC with KRASG12V
The combination of low‑dose trametinib (10 nM) and ABT263 (Bcl‑xL inhibitor) was found to inhibit tumor growth against a KRASG12V xenograft. 252 In spite of targeting MEK and Bcl‑xL, miR‐4689 is also found to exhibit a potent growth inhibitory and proapoptotic effect by directly targeting KRASG12V and AKT1. 253
7.2.3. Therapy of CRC with KRASG13D
Phase III clinical trial evidence suggests that CRCs with the KRASG13D may benefit from EGFR inhibitors, such as cetuximab, in contrast to the other most common KRAS mutations. 254 Therefore, the therapy of CRC with KRASG13D mainly revolves around cetuximab. 255 , 256 , 257 Chu et al. 258 revealed that 4‐AAQB could resensitize KRAS mutant cells to cetuximab before cells were treated with cetuximab. In another study, the combination of first‐line chemotherapy drugs such as FOLFOX and cetuximab improved OS and PFS after treatment in chemotherapy‐refractory CRC patients with KRASG13D, while no response was observed in CRC cell lines with KRASG12X/Q61X mutations or KRASWT CRC cell lines with BRAF mutations or no expression of PTEN or EGFR proteins. 259 , 260
Resistance of EGFR inhibitors in CRC with KRASG13D tends to depend on tumor suppressor NF1. NF1 can convert GTP–KRASG13D to GDP–KRASG13D, and the resistance may be caused by impaired interaction between KRASG13D and NF1. 254 , 261
7.2.4. Therapy of CRC with other KRAS alleles
The combination of irinotecan and cetuximab is administered in patients with KRASWT mCRC who responded to first‐line chemotherapy with cetuximab and developed a certain therapeutic effect after cetuximab in second‐ and third‐line treatment. 262
The combination of KRASG12C inhibitor and EGFR inhibitor targets the rare CRC that carries the G12C mutation. There is a lower response rate to KRASG12C inhibitors alone in CRC patients than in NSCLC patients because of RTK–SHP2‐mediated adaptive RAS reactivation. The combination of cetuximab can enhance the efficacy of AMG510 in KRASG12C‐mutated CRC PDX model. 125 , 263
7.3. Therapy of KRAS‐mutated NSCLC
7.3.1. Therapy of NSCLC with KRASG12C
Benefiting from the successful development of KRASG12C inhibitors, the current therapeutic strategy for NSCLC with G12C mutation is to combine KRASG12C inhibitors with other related pathway inhibitors, targeting immune checkpoints, EGFR, SHP2, SOS1, MEK, PI3K, mTOR, CDK4/6 or others, to improve the therapeutic effect. 264 , 265 , 266 , 267 , 268 , 269 Nevertheless, the problem of resistance to G12C inhibitors needs to be focused.
7.3.2. Therapy of NSCLC with KRASG12V
In the inhibition of RAS‐related pathway, it is showed that cotreatment with trametinib and osimertinib resensitizes the EGFR mutant NSCLC cell line with KRASG12V to osimertinib. 270 A clinical study revealed that patients in the treatment of taxane cooperating with bevacizumab had higher objective response rate (ORR) than those treated with taxane alone. 271 In another study, the combination of selumetinib and docetaxel improved PFS and ORR in patients with locally advanced or metastatic KRASG12V NSCLC (stage IIIB/IV), while no significant trend differences were observed due to the small statistical sample base. 272
7.3.3. Therapy of NSCLC with KRASG12D
One patient with high tumor mutational burden and positive PD‐L1 expression with EGFRL858R and KRASG12D mutations received therapy with a combination of bevacizumab, camrelizumab, and pemetrexed and achieved remission over 17 months. 273 , 274 In another clinical study, a series of therapy of panitumumab concomitant with radiation therapy and concurrent chemotherapy with paclitaxel and carboplatin was settled. During therapy, KRASG12D lung cancer clone of the patient with stage III NSCLC appeared to disappear. 275
8. CONCLUSIONS AND PERSPECTIVE
Cancer therapies performed by targeting allelic RAS mutations in specific clinical contexts could be regarded as the milestone in personalized cancer treatment. After decades of exploration, there are advanced understandings of the structural differences, biochemical characteristics, and downstream signaling preferences of RAS mutations. These studies provide a solid foundation for designing effective targeted therapy for cancers harboring specific RAS mutations. Especially, successful development of KRASG12C inhibitors demonstrates the feasibility of developing specific therapeutic strategies for each RAS mutation. However, there are few studies on the structure and biochemical differences of other RAS mutants, such as KRASQ61L, as well as HRAS and NRAS mutants. This may also be one of the reasons for the lack of effective inhibitors targeting other mutants other than KRASG12C. In addition, there are currently few publicly recognized and effective treatment strategies for different cancer types with different RAS mutations at different stages. Hopefully, this review would provide some insights into individualized RAS inhibitor exploration and the refinement of drug combination for personalized cancer treatment strategy.
In the treatment of cancers induced by mutated RAS, it is necessary to identify the types of RAS mutations and the overactivated signaling pathways in specific cancer tissues. The experimental examples mentioned above have shown the differences in pathology of mut‐RAS‐induced cancer and related signaling pathways, which will lead to the effectiveness of the same treatment strategies. Table 6 summarizes the association of the development of various tumors with RAS mutations and downstream pathway proteins.
TABLE 6.
Role of RAS pathways in related cancers.
| Cancer | Mutation | Role of mutation in cancer | Essential pathway component | Role of component in cancer |
|---|---|---|---|---|
| PC | KRAS mutation | Inducing early PC 276 | – | – |
| Promoting tumor metastasis and aggressiveness 277 , 278 | RAL | Essential for tumor growth and invasion 223 , 224 | ||
| Maintaining tumor 279 | – | – | ||
| KRASG12D/G12V | – | CRAF | Dispensable for tumor development 168 , 169 | |
| KRASG12C/G12D/Q61X | Increased autophagic flux after suppression of KRAS 280 , 281 | ERK | Increased autophagic flux after suppression of ERK 280 , 281 | |
| CRC | KRASG12X/Q61X | KRAS mutations are usually not considered an initial driver 248 , 254 | MAPK and PI3K/Akt | Low activation states in the primary tumors 282 |
| KRASG13D | CRC with G13D will response to EGFR inhibitors 254 | – | – | |
| NSCLC | KRASG12C/G12V | – | RAL | Increased activation 85 |
| – | AKT | Decreased growth factor‐dependent AKT activation 282 | ||
| KRASG12D/G12V | CRAF | Essential for tumor development 166 , 167 | ||
| KRASG12C | – | RAL | Tumor inhibition after RAL depletion 222 | |
| KRASG12D | – | PI3K | Increased activation 282 | |
| – | MEK | No obvious activation 282 | ||
| KRASQ61H | Increased dependence of tumor development on MAPK 283 | RAF/MEK | Increased activation and essential for tumor development 283 | |
| Endometrial cancer | KRAS mutation | Inducing early EC 284 and development | PI3K | Increased activation 285 |
| Promoting type I EC aggressiveness 286 |
MAPK PI3K |
Increased FGFR‐dependent activation 287 | ||
| Neuroblastoma | NRASQ61V | Promoting tumor development 288 | MAPK | Increased activation 288 |
| Breast cancer | HRASG12V | Inducing proliferation signal 289 | PI3K | Increased activation 289 |
| KRASG12V | Maintaining mesenchymal characteristics and metastatic behavior 290 | mTORC1 | Increased activation 291 |
Abbreviations: AKT, protein kinase B; CRC, EC, endometrial cancer; colorectal cancer; MAPK, mitogen‐activated protein kinase; NSCLC, non‐small cell lung cancer; PC, pancreatic adenocarcinoma; PI3K, phosphatidylinositol‐3‐kinase.
In addition to the treatment options of targeting RAS and downstream pathway proteins, new treatments have also been developed in recent years. First, there are studies exploring new therapeutic strategies from the perspective of energy metabolism. In cancers caused by RAS overactivation, energy metabolism‐related features such as increased glycolysis rate 292 and increased mitochondrial autophagy 293 adapted to cancer cell growth would be detected. Therefore, there are studies attempting to inhibit the development of RAS‐related cancers by inhibiting the above pathways. Bryant et al. 280 found the phenomenon of increased autophagy flux after inhibition of KRAS and MAPK pathways and demonstrated that autophagy inhibitor chloroquine and ERK inhibitor could synergically enhance the antitumor activity of KRAS‐driven PDAC. Therefore, the combination of MAPK inhibitor and autophagy inhibitor may be an effective treatment for PDAC. Secondly, the research on inhibiting RAS oligomerization is carried out. After RAS is localized in the membrane, oligomerization or dimerization of RAS occurs, which is necessary for effective RAS‐driven signaling. 294 Based on the phenomenon, nano‐antibody NS1 was developed to disrupt HRAS–KRAS autocorrelation by directly binding to the α4–α5 interface, reducing downstream pathway activation and inhibiting cell proliferation while maintaining RAS localization and GTPase activity unaffected. 295 In addition, there are other novel therapeutic methods, such as proteolysis‐targeting chimeras (PROTACs) 296 and small‐molecule RNA interference technology, 234 which also provide more selectivity for the treatment of RAS‐related cancers.
With the continuous development of mutant RAS inhibitors and other novel therapies, the drug combinations available for clinical use are more diverse. It is hoped that personalized cancer treatment research will make more targeted use of these drug combinations to continuously optimize the clinical therapeutic effect.
AUTHOR CONTRIBUTIONS
C.L. wrote the paper. D.Y., X.L., Z.S., M.D., and Y.L. revised the manuscript. H.Y. and X.C. made the figures. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare they have no conflicts of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
This work was supported by grants from Beijing Municipal Natural Science Foundation (7232283), National Natural Science Foundation of China (NSFC 82130113), and Central Universities (2020‐JYB‐ZDGG‐040).
Liu C, Ye D, Yang H, et al. RAS‐targeted cancer therapy: Advances in drugging specific mutations. MedComm. 2023;4:e285. 10.1002/mco2.285
Contributor Information
Xia Li, Email: xia.li_2010@genetics.ac.cn.
Mei Ding, Email: mding@genetics.ac.cn.
Yonggang Liu, Email: liuyg0228@163.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Chavan TS, Muratcioglu S, Marszalek R, et al. Plasma membrane regulates Ras signaling networks. Cell Logist. 2016;5(4):e1136374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225‐249. [DOI] [PubMed] [Google Scholar]
- 3. Cox AD, Der CJ. Ras history. Small GTPases. 2014;1(1):2‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu P, Wang Y, Li X. Targeting the untargetable KRAS in cancer therapy. Acta Pharmaceut Sin B. 2019;9(5):871‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129(7):1287‐1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barbacid M. ras GENES. Annu Rev Biochem. 1987;56(1):779‐827. [DOI] [PubMed] [Google Scholar]
- 7. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK‐activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gimple RC, Wang X. RAS: striking at the core of the oncogenic circuitry. Front Oncol. 2019;9:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Westcott PMK, To MD. The genetics and biology of KRAS in lung cancer. Chin J Cancer. 2013;32(2):63‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pagba CV, Gupta AK, Naji AK, et al. KRAS inhibitor that simultaneously inhibits nucleotide exchange activity and effector engagement. ACS Bio Med Chem Au. 2022;2(6):617‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang SK, Picard L‐P, Rahmatullah RSM, et al. Mapping the conformational landscape of the stimulatory heterotrimeric G protein. Nat Struct Mol Biol. 2023;30(4):502‐511. [DOI] [PubMed] [Google Scholar]
- 14. Smyth LA, Collins I. Measuring and interpreting the selectivity of protein kinase inhibitors. J Chem Biol. 2009;2(3):131‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lito P, Solomon M, Li L‐S, Hansen R, Rosen N. Allele‐specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim SM, Westover KD, Ficarro SB, et al. Therapeutic targeting of oncogenic K‐Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed. 2014;53(1):199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patricelli MP, Janes MR, Li L‐S, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6(3):316‐329. [DOI] [PubMed] [Google Scholar]
- 18. Ostrem JML, Shokat KM. Direct small‐molecule inhibitors of KRAS: from structural insights to mechanism‐based design. Nat Rev Drug Discov. 2016;15(11):771‐785. [DOI] [PubMed] [Google Scholar]
- 19. Buhrman G, Holzapfel G, Fetics S, Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc Natl Acad Sci U S A. 2010;107(11):4931‐4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clarke S, Tamanoi F. Fighting cancer by disrupting C‐terminal methylation of signaling proteins. J Clin Investig. 2004;113(4):513‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Silvius JR. Mechanisms of Ras protein targeting in mammalian cells. J Membr Biol. 2002;190(2):83‐92. [DOI] [PubMed] [Google Scholar]
- 22. Wittinghofer A, Vetter IR. Structure–function relationships of the G domain, a canonical switch motif. Annu Rev Biochem. 2011;80(1):943‐971. [DOI] [PubMed] [Google Scholar]
- 23. Marcus K, Mattos C. Direct attack on RAS: intramolecular communication and mutation‐specific effects. Clin Cancer Res. 2015;21(8):1810‐1818. [DOI] [PubMed] [Google Scholar]
- 24. Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha‐ and beta‐subunits of ATP synthase, myosin, kinases and other ATP‐requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1(8):945‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saraste M, Sibbald PR, Wittinghofer A. The P‐loop—a common motif in ATP‐ and GTP‐binding proteins. Trends Biochem Sci. 1990;15(11):430‐434. [DOI] [PubMed] [Google Scholar]
- 26. Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63(1):133‐139. [DOI] [PubMed] [Google Scholar]
- 27. Abraham SJ, Muhamed I, Nolet R, Yeung F, Gaponenko V. Expression, purification, and characterization of soluble K‐Ras4B for structural analysis. Protein Expr Purif. 2010;73(2):125‐131. [DOI] [PubMed] [Google Scholar]
- 28. Welman A, Burger MM, Hagmann J. Structure and function of the C‐terminal hypervariable region of K‐Ras4B in plasma membrane targeting and transformation. Oncogene. 2000;19(40):4582‐4591. [DOI] [PubMed] [Google Scholar]
- 29. O'Bryan JP. Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacol Res. 2019;139:503‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti‐RAS drug discovery? Clin Cancer Res. 2015;21(8):1819‐1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manolaridis I, Kulkarni K, Dodd RB, et al. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature. 2013;504(7479):301‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hancock John F, Parton Robert G. Ras plasma membrane signalling platforms. Biochem J. 2005;389(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ahearn IM, Haigis K, Bar‐Sagi D, Philips MR. Regulating the regulator: post‐translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13(1):39‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tulpule A, Guan J, Neel DS, et al. Kinase‐mediated RAS signaling via membraneless cytoplasmic protein granules. Cell. 2021;184(10):2649‐2664.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang M‐T, Holderfield M, Galeas J, et al. K‐Ras promotes tumorigenicity through suppression of non‐canonical Wnt signaling. Cell. 2015;163(5):1237‐1251. [DOI] [PubMed] [Google Scholar]
- 36. Bunda S, Heir P, Srikumar T, et al. Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc Natl Acad Sci U S A. 2014;111(36):E3785‐E3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alvarez‐Moya B, López‐Alcalá C, Drosten M, Bachs O, Agell N. K‐Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K‐Ras activity and function. Oncogene. 2010;29(44):5911‐5922. [DOI] [PubMed] [Google Scholar]
- 38. Dohlman HG, Campbell SL. Regulation of large and small G proteins by ubiquitination. J Biol Chem. 2019;294(49):18613‐18623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baker R, Lewis SM, Sasaki AT, et al. Site‐specific monoubiquitination activates Ras by impeding GTPase‐activating protein function. Nat Struct Mol Biol. 2013;20(1):46‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baker R, Wilkerson EM, Sumita K, et al. Differences in the regulation of K‐Ras and H‐Ras isoforms by monoubiquitination. J Biol Chem. 2013;288(52):36856‐36862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Knyphausen P, Lang F, Baldus L, Extra A, Lammers M. Insights into K‐Ras 4B regulation by post‐translational lysine acetylation. Biol Chem. 2016;397(10):1071‐1085. [DOI] [PubMed] [Google Scholar]
- 42. Yang MH, Nickerson S, Kim ET, et al. Regulation of RAS oncogenicity by acetylation. Proc Natl Acad Sci U S A. 2012;109(27):10843‐10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yin G, Kistler S, George SD, et al. A KRAS GTPase K104Q mutant retains downstream signaling by offsetting defects in regulation. J Biol Chem. 2017;292(11):4446‐4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cornett EM, Ferry L, Defossez P‐A, Rothbart SB. Lysine methylation regulators moonlighting outside the epigenome. Mol Cell. 2019;75(6):1092‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jančík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010;2010:150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pylayeva‐Gupta Y, Grabocka E, Bar‐Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brownbridge GG, Lowe PN, Moore KJ, Skinner RH, Webb MR. Interaction of GTPase activating proteins (GAPs) with p21ras measured by a novel fluorescence anisotropy method. Essential role of Arg‐903 of GAP in activation of GTP hydrolysis on p21ras. J Biol Chem. 1993;268(15):10914‐10919. [PubMed] [Google Scholar]
- 48. Mader S, Pantel K. Liquid biopsy: current status and future perspectives. Oncol Res Treat. 2017;40(7‐8):404. [DOI] [PubMed] [Google Scholar]
- 49. Sulzmaier FJ, Ramos JW. RSK isoforms in cancer cell invasion and metastasis. Cancer Res. 2013;73(20):6099‐6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278(1):16‐27. [DOI] [PubMed] [Google Scholar]
- 51. Yamamoto T, Kozawa O, Tanabe K, et al. Involvement of p38 MAP kinase in TGF‐β‐stimulated VEGF synthesis in aortic smooth muscle cells. J Cell Biochem. 2001;82(4):591‐598. [DOI] [PubMed] [Google Scholar]
- 52. Lin DTS, Davis NG, Conibear E. Targeting the Ras palmitoylation/depalmitoylation cycle in cancer. Biochem Soc Trans. 2017;45(4):913‐921. [DOI] [PubMed] [Google Scholar]
- 53. Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Cancer Res. 2020;80(14):2969‐2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Prior IA, Lewis PD, Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Res. 2012;72(10):2457‐2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou B, Der CJ, Cox AD. The role of wild type RAS isoforms in cancer. Semin Cell Dev Biol. 2016;58:60‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. The APGC, The APGC, André F, et al. AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7(8):818‐831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Khan I, Rhett JM, O'Bryan JP. Therapeutic targeting of RAS: new hope for drugging the “undruggable”. Biochim Biophys Acta. 2020;1867(2):118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vatansever S, Erman B, Gümüş ZH. Comparative effects of oncogenic mutations G12C, G12V, G13D, and Q61H on local conformations and dynamics of K‐Ras. Comput Struct Biotechnol J. 2020;18:1000‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu S, Jang H, Nussinov R, Zhang J. The structural basis of oncogenic mutations G12, G13 and Q61 in small GTPase K‐Ras4B. Sci Rep. 2016;6(1):21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Johnson CW, Reid D, Parker JA, et al. The small GTPases K‐Ras, N‐Ras, and H‐Ras have distinct biochemical properties determined by allosteric effects. J Biol Chem. 2017;292(31):12981‐12993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mazhab‐Jafari MT, Marshall CB, Smith MJ, et al. Oncogenic and RASopathy‐associated K‐RAS mutations relieve membrane‐dependent occlusion of the effector‐binding site. Proc Natl Acad Sci U S A. 2015;112(21):6625‐6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bandaru P, Shah NH, Bhattacharyya M, et al. Deconstruction of the Ras switching cycle through saturation mutagenesis. eLife. 2017;6:e27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gremer L, Merbitz‐Zahradnik T, Dvorsky R, et al. Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum Mutat. 2011;32(1):33‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Du X, Frei H, Kim S‐H. The mechanism of GTP hydrolysis by Ras probed by Fourier transform infrared spectroscopy. J Biol Chem. 2000;275(12):8492‐8500. [DOI] [PubMed] [Google Scholar]
- 65. Khrenova MG, Grigorenko BL, Kolomeisky AB, Nemukhin AV. Hydrolysis of guanosine triphosphate (GTP) by the Ras·GAP protein complex: reaction mechanism and kinetic scheme. J Phys Chem B. 2015;119(40):12838‐12845. [DOI] [PubMed] [Google Scholar]
- 66. Grigorenko BL, Nemukhin AV, Shadrina MS, Topol IA, Burt SK. Mechanisms of guanosine triphosphate hydrolysis by Ras and Ras‐GAP proteins as rationalized by ab initio QM/MM simulations. Proteins: Struct Funct Bioinform. 2007;66(2):456‐466. [DOI] [PubMed] [Google Scholar]
- 67. Khrenova MG, Grigorenko BL, Nemukhin AV. Theoretical vibrational spectroscopy of intermediates and the reaction mechanism of the guanosine triphosphate hydrolysis by the protein complex Ras‐GAP. Spectrochim Acta Part A Mol Biomol Spectrosc. 2016;166:68‐72. [DOI] [PubMed] [Google Scholar]
- 68. McCormick F, Holmes KC, Michell RH, et al. Coupling of ras p21 signalling and GTP hydrolysis by GTPase activating proteins. Philos Trans R Soc Lond B Biol Sci. 1997;336(1276):43‐48. [DOI] [PubMed] [Google Scholar]
- 69. Gremer L, Gilsbach B, Reza Ahmadian M, Wittinghofer A. Fluoride complexes of oncogenic Ras mutants to study the Ras–RasGAP interaction. Biol Chem. 2008;389(9):1163‐1171. [DOI] [PubMed] [Google Scholar]
- 70. Kratz CP, Niemeyer CM, Zenker M. An unexpected new role of mutant Ras: perturbation of human embryonic development. J Mol Med. 2007;85(3):227‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tichauer RH, Favre G, Cabantous S, Landa G, Hemeryck A, Brut M. Water distribution within wild‐type NRas protein and Q61 mutants during unrestrained QM/MM dynamics. Biophys J. 2018;115(8):1417‐1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Khrenova MG, Mironov VA, Grigorenko BL, Nemukhin AV. Modeling the role of G12V and G13V Ras mutations in the Ras‐GAP‐catalyzed hydrolysis reaction of guanosine triphosphate. Biochemistry. 2014;53(45):7093‐7099. [DOI] [PubMed] [Google Scholar]
- 73. Rabara D, Tran TH, Dharmaiah S, et al. KRAS G13D sensitivity to neurofibromin‐mediated GTP hydrolysis. Proc Natl Acad Sci U S A. 2019;116(44):22122‐22131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gibbs JB, Sigal IS, Poe M, Scolnick EM. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci U S A. 1984;81(18):5704‐5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Marshall M. Interactions between Ras and Raf: key regulatory proteins in cellular transformation. Mol Reprod Dev. 1995;42(4):493‐499. [DOI] [PubMed] [Google Scholar]
- 76. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer‐associated KRAS mutations. Mol Cancer Res. 2015;13(9):1325‐1335. [DOI] [PubMed] [Google Scholar]
- 77. Filchtinski D, Sharabi O, Rüppel A, Vetter IR, Herrmann C, Shifman JM. What makes Ras an efficient molecular switch: a computational, biophysical, and structural study of Ras‐GDP interactions with mutants of Raf. J Mol Biol. 2010;399(3):422‐435. [DOI] [PubMed] [Google Scholar]
- 78. Gebregiworgis T, Kano Y, St‐Germain J, et al. The Q61H mutation decouples KRAS from upstream regulation and renders cancer cells resistant to SHP2 inhibitors. Nat Commun. 2021;12(1):6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Privé GG, Milburn MV, Tong L, et al. X‐ray crystal structures of transforming p21 ras mutants suggest a transition‐state stabilization mechanism for GTP hydrolysis. Proc Natl Acad Sci U S A. 1992;89(8):3649‐3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Buhrman G, Wink G, Mattos C. Transformation efficiency of RasQ61 mutants linked to structural features of the switch regions in the presence of Raf. Structure. 2007;15(12):1618‐1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Smith MJ, Neel BG, Ikura M. NMR‐based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A. 2013;110(12):4574‐4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen J, Wang L, Wang W, Sun H, Pang L, Bao H. Conformational transformation of switch domains in GDP/K‐Ras induced by G13 mutants: an investigation through Gaussian accelerated molecular dynamics simulations and principal component analysis. Comput Biol Med. 2021;135:104639. [DOI] [PubMed] [Google Scholar]
- 83. Johnson CW, Lin Y‐J, Reid D, et al. Isoform‐specific destabilization of the active site reveals a molecular mechanism of intrinsic activation of KRas G13D. Cell Rep. 2019;28(6):1538‐1550.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Smith MJ, Ikura M. Integrated RAS signaling defined by parallel NMR detection of effectors and regulators. Nat Chem Biol. 2014;10(3):223‐230. [DOI] [PubMed] [Google Scholar]
- 85. Ihle NT, Byers LA, Kim ES, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104(3):228‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Janes MR, Zhang J, Li L‐S, et al. Targeting KRAS mutant cancers with a covalent G12C‐specific inhibitor. Cell. 2018;172(3):578‐589.e17. [DOI] [PubMed] [Google Scholar]
- 87. Vatansever S, Erman B, Gümüş ZH. Oncogenic G12D mutation alters local conformations and dynamics of K‐Ras. Sci Rep. 2019;9(1):11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Al‐Mulla F, Milner‐White EJ, Going JJ, Birnie GD. Structural differences between valine‐12 and aspartate‐12 Ras proteins may modify carcinoma aggression. J Pathol. 1999;187(4):433‐438. [DOI] [PubMed] [Google Scholar]
- 89. Fetics Susan K, Guterres H, Kearney Bradley M, et al. Allosteric effects of the oncogenic RasQ61L mutant on Raf‐RBD. Structure. 2015;23(3):505‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Buhrman G, Kumar VSS, Cirit M, Haugh JM, Mattos C. Allosteric modulation of Ras‐GTP is linked to signal transduction through RAF kinase. J Biol Chem. 2011;286(5):3323‐3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. McFall T, Diedrich JK, Mengistu M, et al. A systems mechanism for KRAS mutant allele‐specific responses to targeted therapy. Sci Signal. 2019;12(600):eaaw8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c‐Ha‐ras1 genes mutated at codon 12. Nature. 1984;312(5989):71‐75. [DOI] [PubMed] [Google Scholar]
- 93. Smith G, Bounds R, Wolf H, Steele RJC, Carey FA, Wolf CR. Activating K‐Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours—implications for personalised cancer medicine. Br J Cancer. 2010;102(4):693‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bournet B, Muscari F, Buscail C, et al. KRASG12D mutation subtype is a prognostic factor for advanced pancreatic adenocarcinoma. Clin Transl Gastroenterol. 2016;7(3):e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ardalan B, Azqueta J, Sleeman D. Cobimetinib plus gemcitabine: an active combination in KRAS G12R‐mutated pancreatic ductal adenocarcinoma patients in previously treated and failed multiple chemotherapies. J Pancreatic Cancer. 2021;7(1):65‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Guo S, Shi X, Shen J, et al. Preoperative detection of KRAS G12D mutation in ctDNA is a powerful predictor for early recurrence of resectable PDAC patients. Br J Cancer. 2020;122(6):857‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zocche DM, Ramirez C, Fontao FM, Costa LD, Redal MA. Global impact of KRAS mutation patterns in FOLFOX treated metastatic colorectal cancer. Original research. Front Genet. 2015;6:116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yuan Y, Liu Y, Wu Y, et al. Clinical characteristics and prognostic value of the KRAS mutation in Chinese colorectal cancer patients. Int J Biol Markers. 2021;36(2):33‐39. [DOI] [PubMed] [Google Scholar]
- 99. Margonis GA, Kim Y, Spolverato G, et al. Association between specific mutations in KRAS codon 12 and colorectal liver metastasis. JAMA Surg. 2015;150(8):722‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li W, Liu Y, Cai S, et al. Not all mutations of KRAS predict poor prognosis in patients with colorectal cancer. Int J Clin Exp Pathol. 2019;12(3):957‐967. [PMC free article] [PubMed] [Google Scholar]
- 101. Andreyev HJN, Norman AR, Clarke PA, Cunningham D, Oates JR. Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90(9):675‐684. [DOI] [PubMed] [Google Scholar]
- 102. Imamura Y, Morikawa T, Liao X, et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild‐type colorectal cancers. Clin Cancer Res. 2012;18(17):4753‐4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zanatto RM, Santos G, Oliveira JC, et al. Impact of KRAS mutations in clinical features in colorectal cancer. Arq Bras Cir Dig. 2020;33(3):e1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bazan V, Migliavacca M, Zanna I, et al. Specific codon 13 K‐ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K‐ras mutations are associated with mucinous histotype. Ann Oncol. 2002;13(9):1438‐1446. [DOI] [PubMed] [Google Scholar]
- 105. Henry JT, Coker O, Chowdhury S, et al. Comprehensive clinical and molecular characterization of KRASG12C‐mutant colorectal cancer. JCO Precis Oncol. 2021;5:PO.20.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Arbour KC, Rizvi H, Plodkowski AJ, et al. Treatment outcomes and clinical characteristics of patients with KRAS‐G12C‐mutant non‐small cell lung cancer. Clin Cancer Res. 2021;27(8):2209‐2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Renaud S, Seitlinger J, Falcoz P‐E, et al. Specific KRAS amino acid substitutions and EGFR mutations predict site‐specific recurrence and metastasis following non‐small‐cell lung cancer surgery. Br J Cancer. 2016;115(3):346‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Arbour KC, Jordan E, Kim HR, et al. Effects of co‐occurring genomic alterations on outcomes in patients with KRAS‐mutant non‐small cell lung cancer. Clin Cancer Res. 2018;24(2):334‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. McCormick F. KRAS as a therapeutic target. Clin Cancer Res. 2015;21(8):1797‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lu S, Jang H, Muratcioglu S, et al. Ras conformational ensembles, allostery, and signaling. Chem Rev. 2016;116(11):6607‐6665. [DOI] [PubMed] [Google Scholar]
- 111. Kettle JG, Bagal SK, Bickerton S, et al. Structure‐based design and pharmacokinetic optimization of covalent allosteric inhibitors of the mutant GTPase KRASG12C. J Med Chem. 2020;63(9):4468‐4483. [DOI] [PubMed] [Google Scholar]
- 112. Skoulidis F, Li BT, Dy GK, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384(25):2371‐2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kessler D, Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A. 2019;116(32):15823‐15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Nichols R, Yang YC, Cregg J, et al. Abstract 3595: RMC‐6291, a next‐generation tri‐complex KRASG12C(ON) inhibitor, outperforms KRASG12C(OFF) inhibitors in preclinical models of KRASG12C cancers. Cancer Res. 2022;82:3595. [Google Scholar]
- 115. Tanaka N, Lin JJ, Li C, et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch‐II pocket mutation and polyclonal alterations converging on RAS–MAPK reactivation. Cancer Discov. 2021;11(8):1913‐1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Haigis KM, Kendall KR, Wang Y, et al. Differential effects of oncogenic K‐Ras and N‐Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40(5):600‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Aran V, Omerovic J. Current approaches in NSCLC targeting K‐RAS and EGFR. Int J Mol Sci. 2019;20(22):5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Dunnett‐Kane V, Nicola P, Blackhall F, Lindsay C. Mechanisms of resistance to KRAS(G12C) inhibitors. Cancers. 2021;13(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zhang Z, Rohweder PJ, Ongpipattanakul C, et al. A covalent inhibitor of K‐Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell. 2022;40(9):1060‐1069.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lee A. Sotorasib: a review in KRAS G12C mutation‐positive non‐small cell lung cancer. Target Oncol. 2022;17(6):727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hong DS, Fakih MG, Strickler JH, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383(13):1207‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ou S‐HI, Jänne PA, Leal TA, et al. First‐in‐human phase I/IB dose‐finding study of adagrasib (MRTX849) in patients with advanced KRASG12C solid tumors (KRYSTAL‐1). J Clin Oncol. 2022;40(23):2530‐2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Parikh K, Banna G, Liu SV, et al. Drugging KRAS: current perspectives and state‐of‐art review. J Hematol Oncol. 2022;15(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Suzuki S, Yonesaka K, Teramura T, et al. KRAS inhibitor resistance in MET‐amplified KRASG12C non‐small cell lung cancer induced by RAS‐ and non‐RAS‐mediated cell signaling mechanisms. Clin Cancer Res. 2021;27(20):5697‐5707. [DOI] [PubMed] [Google Scholar]
- 125. Amodio V, Yaeger R, Arcella P, et al. EGFR blockade reverts resistance to KRASG12C inhibition in colorectal cancer. Cancer Discov. 2020;10(8):1129‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Feng H, Zhang Y, Bos PH, Chambers JM, Dupont MM, Stockwell BR. K‐RasG12D has a potential allosteric small molecule binding site. Biochemistry. 2019;58(21):2542‐2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Shah S, Brock EJ, Ji K, Mattingly RR. Ras and Rap1: a tale of two GTPases. Semin Cancer Biol. 2019;54:29‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hallin J, Bowcut V, Calinisan A, et al. Anti‐tumor efficacy of a potent and selective non‐covalent KRASG12D inhibitor. Nat Med. 2022;28(10):2171‐2182. [DOI] [PubMed] [Google Scholar]
- 129. Sakamoto K, Masutani T, Hirokawa T. Generation of KS‐58 as the first K‐Ras(G12D)‐inhibitory peptide presenting anti‐cancer activity in vivo. Sci Rep. 2020;10(1):21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Welsch ME, Kaplan A, Chambers JM, et al. Multivalent small‐molecule pan‐RAS inhibitors. Cell. 2017;168(5):878‐889.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Han CW, Jeong MS, Ha SC, Jang SB. A H‐REV107 peptide inhibits tumor growth and interacts directly with oncogenic KRAS mutants. Cancers. 2020;12(6):1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zhang Y, Meng X, Tang H, Cheng M, Yang F, Xu W. Design, synthesis, and biological evaluation of novel substituted thiourea derivatives as potential anticancer agents for NSCLC by blocking K‐Ras protein‐effectors interactions. J Enzyme Inhib Med Chem. 2020;35(1):344‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhang Z, Guiley KZ, Shokat KM. Chemical acylation of an acquired serine suppresses oncogenic signaling of K‐Ras(G12S). Nat Chem Biol. 2022;18(11):1177‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhang Z, Morstein J, Ecker AK, Guiley KZ, Shokat KM. Chemoselective covalent modification of K‐Ras(G12R) with a small molecule electrophile. JACS. 2022;144(35):15916‐15921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314. [DOI] [PubMed] [Google Scholar]
- 137. Roux Philippe P, Blenis J. ERK and p38 MAPK‐activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Warne PH, Vician PR, Downward J. Direct interaction of Ras and the amino‐terminal region of Raf‐1 in vitro. Nature. 1993;364(6435):352‐355. [DOI] [PubMed] [Google Scholar]
- 139. Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras⋅GTP with Raf‐1 and mitogen‐activated protein kinase kinase. Science. 1993;260(5114):1658‐1661. [DOI] [PubMed] [Google Scholar]
- 140. Xu S, Robbins D, Frost J, Dang A, Lange‐Carter C, Cobb MH. MEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen‐activated protein kinase. Proc Natl Acad Sci U S A. 1995;92(15):6808‐6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M. Complex formation between RAS and RAF and other protein kinases. Proc Natl Acad Sci U S A. 1993;90(13):6213‐6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhang X‐F, Settleman J, Kyriakis J, et al. Normal and oncogenic p21ras proteins bind to the amino‐terminal regulatory domain of c‐Raf‐1. Nature. 1993;364(6435):308‐313. [DOI] [PubMed] [Google Scholar]
- 143. Luo Z, Diaz B, Marshall MS, Avruch J. An intact Raf zinc finger is required for optimal binding to processed Ras and for Ras‐dependent Raf activation in situ. Mol Cell Biol. 1997;17(1):46‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Williams JG, Drugan JK, Yi G‐S, Clark GJ, Der CJ, Campbell SL. Elucidation of binding determinants and functional consequences of Ras/Raf‐cysteine‐rich domain interactions. J Biol Chem. 2000;275(29):22172‐22179. [DOI] [PubMed] [Google Scholar]
- 145. Hekman M, Hamm H, Villar AV, et al. Associations of B‐ and C‐Raf with cholesterol, phosphatidylserine, and lipid second messengers: preferential binding of Raf to artificial lipid rafts. J Biol Chem. 2002;277(27):24090‐24102. [DOI] [PubMed] [Google Scholar]
- 146. Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Plasma membrane nanoswitches generate high‐fidelity Ras signal transduction. Nat Cell Biol. 2007;9(8):905‐914. [DOI] [PubMed] [Google Scholar]
- 147. Hibino K, Shibata T, Yanagida T, Sako Y. A RasGTP‐induced conformational change in C‐RAF is essential for accurate molecular recognition. Biophys J. 2009;97(5):1277‐1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Freeman AK, Ritt DA, Morrison DK. The importance of Raf dimerization in cell signaling. Small GTPases. 2013;4(3):180‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. von Kriegsheim A, Pitt A, Grindlay GJ, Kolch W, Dhillon AS. Regulation of the Raf–MEK–ERK pathway by protein phosphatase 5. Nat Cell Biol. 2006;8(9):1011‐1016. [DOI] [PubMed] [Google Scholar]
- 150. Holderfield M, Merritt H, Chan J, et al. RAF inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell. 2013;23(5):594‐602. [DOI] [PubMed] [Google Scholar]
- 151. Dougherty MK, Müller J, Ritt DA, et al. Regulation of Raf‐1 by direct feedback phosphorylation. Mol Cell. 2005;17(2):215‐224. [DOI] [PubMed] [Google Scholar]
- 152. Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B‐Raf signaling. Mol Cell Biol. 2010;30(3):806‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ullah R, Yin Q, Snell AH, Wan L. RAF–MEK–ERK pathway in cancer evolution and treatment. Semin Cancer Biol. 2022;85:123‐154. [DOI] [PubMed] [Google Scholar]
- 154. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16(5):281‐298. [DOI] [PubMed] [Google Scholar]
- 155. Wan PTC, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF–ERK signaling pathway by oncogenic mutations of B‐RAF. Cell. 2004;116(6):855‐867. [DOI] [PubMed] [Google Scholar]
- 156. Mercer KE, Pritchard CA. Raf proteins and cancer: B‐Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653(1):25‐40. [DOI] [PubMed] [Google Scholar]
- 157. Ikenoue T, Hikiba Y, Kanai F, et al. Different effects of point mutations within the B‐Raf glycine‐rich loop in colorectal tumors on mitogen‐activated protein/extracellular signal‐regulated kinase kinase/extracellular signal‐regulated kinase and nuclear factor κB pathway and cellular transformation. Cancer Res. 2004;64(10):3428‐3435. [DOI] [PubMed] [Google Scholar]
- 158. Fischmann TO, Smith CK, Mayhood TW, et al. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry. 2009;48(12):2661‐2674. [DOI] [PubMed] [Google Scholar]
- 159. Brennan DF, Dar AC, Hertz NT, et al. A Raf‐induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011;472(7343):366‐369. [DOI] [PubMed] [Google Scholar]
- 160. Wortzel I, Seger R. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer. 2011;2(3):195‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Zheng CF, Guan KL. Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO. 1994;13(5):1123‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Roskoski R. MEK1/2 dual‐specificity protein kinases: structure and regulation. Biochem Biophys Res Commun. 2012;417(1):5‐10. [DOI] [PubMed] [Google Scholar]
- 163. Roskoski R. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66(2):105‐143. [DOI] [PubMed] [Google Scholar]
- 164. Gao Y, Chang MT, McKay D, et al. Allele‐specific mechanisms of activation of MEK1 mutants determine their properties. Cancer Discov. 2018;8(5):648‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Jaiswal BS, Durinck S, Stawiski EW, et al. ERK mutations and amplification confer resistance to ERK‐inhibitor therapy. Clin Cancer Res. 2018;24(16):4044‐4055. [DOI] [PubMed] [Google Scholar]
- 166.Blasco Rafael B, Francoz S, Santamaría D, et al. c‐Raf, but Not B‐Raf, is essential for development of K‐Ras oncogene‐driven non‐small cell lung carcinoma. Cancer Cell. 2011;19(5):652‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C‐Raf is required for the initiation of lung cancer by K‐RasG12D. Cancer Discov. 2011;1(2):128‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Eser S, Reiff N, Messer M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene‐driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406‐420. [DOI] [PubMed] [Google Scholar]
- 169. Blasco MT, Navas C, Martín‐Serrano G, et al. Complete regression of advanced pancreatic ductal adenocarcinomas upon combined inhibition of EGFR and C‐RAF. Cancer Cell. 2019;35(4):573‐587.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Sanclemente M, Francoz S, Esteban‐Burgos L, et al. c‐RAF ablation induces regression of advanced Kras/Trp53 mutant lung adenocarcinomas by a mechanism independent of MAPK signaling. Cancer Cell. 2018;33(2):217‐228.e4. [DOI] [PubMed] [Google Scholar]
- 171. Matallanas D, Birtwistle M, Romano D, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2(3):232‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Ehrenreiter K, Kern F, Velamoor V, et al. Raf‐1 addiction in Ras‐induced skin carcinogenesis. Cancer Cell. 2009;16(2):149‐160. [DOI] [PubMed] [Google Scholar]
- 173. Brandt R, Sell T, Lüthen M, et al. Cell type‐dependent differential activation of ERK by oncogenic KRAS in colon cancer and intestinal epithelium. Nat Commun. 2019;10(1):2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Kim G, McKee AE, Ning Y‐M, et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res. 2014;20(19):4994‐5000. [DOI] [PubMed] [Google Scholar]
- 175. Oberholzer PA, Kee D, Dziunycz P, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with Raf inhibitors. J Clin Oncol. 2011;30(3):316‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Callahan MK, Rampal R, Harding JJ, et al. Progression of RAS‐mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367(24):2316‐2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild‐type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431‐435. [DOI] [PubMed] [Google Scholar]
- 178. Peng S‐B, Henry James R, Kaufman Michael D, et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti‐tumor activities in RAS or BRAF mutant cancers. Cancer Cell. 2015;28(3):384‐398. [DOI] [PubMed] [Google Scholar]
- 179. Hatzivassiliou G, Haling JR, Chen H, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS‐ versus BRAF‐driven cancers. Nature. 2013;501(7466):232‐236. [DOI] [PubMed] [Google Scholar]
- 180. Lito P, Saborowski A, Yue J, et al. Disruption of CRAF‐mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25(5):697‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Arbour KC, Manchado E, Bott MJ, et al. Phase 1 clinical trial of trametinib and ponatinib in patients with NSCLC harboring KRAS mutations. JTO Clin Res Rep. 2022;3(1):100256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Maik‐Rachline G, Seger R. The ERK cascade inhibitors: towards overcoming resistance. Drug Resist Updates. 2016;25:1‐12. [DOI] [PubMed] [Google Scholar]
- 183. Jha S, Morris EJ, Hruza A, et al. Dissecting therapeutic resistance to ERK inhibition. Mol Cancer Ther. 2016;15(4):548‐559. [DOI] [PubMed] [Google Scholar]
- 184. Wang G, Zhao Y, Liu Y, et al. Discovery of a novel dual‐target inhibitor of ERK1 and ERK5 that induces regulated cell death to overcome compensatory mechanism in specific tumor types. J Med Chem. 2020;63(8):3976‐3995. [DOI] [PubMed] [Google Scholar]
- 185. Mendoza MC, Er EE, Blenis J. The Ras–ERK and PI3K–mTOR pathways: cross‐talk and compensation. Trends Biochem Sci. 2011;36(6):320‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Gao X, Cen L, Li F, et al. Oral administration of indole substituted dipyrido[2,3‐d]pyrimidine derivative exhibits anti‐tumor activity via inhibiting AKT and ERK1/2 on hepatocellular carcinoma. Biochem Biophys Res Commun. 2018;505(3):761‐767. [DOI] [PubMed] [Google Scholar]
- 187. Pan X, Pei J, Wang A, et al. Development of small molecule extracellular signal‐regulated kinases (ERKs) inhibitors for cancer therapy. Acta Pharma Sin B. 2022;12(5):2171‐2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Martelli AM, Chiarini F, Evangelisti C, et al. Targeting the liver kinase B1/AMP‐activated protein kinase pathway as a therapeutic strategy for hematological malignancies. Expert Opin Ther Targets. 2012;16(7):729‐742. [DOI] [PubMed] [Google Scholar]
- 189. Blanco‐Aparicio C, Renner O, Leal JFM, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28(7):1379‐1386. [DOI] [PubMed] [Google Scholar]
- 190. Cervello M, McCubrey JA, Cusimano A, Lampiasi N, Azzolina A, Montalto G. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012;3(3):236‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Martelli AM, Chiarini F, Evangelisti C, et al. Two hits are better than one: targeting both phosphatidylinositol 3‐kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget. 2012;3(4):371‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Martelli AM, Evangelisti C, Chappell W, et al. Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia. 2011;25(7):1064‐1079. [DOI] [PubMed] [Google Scholar]
- 193. McCubrey JA, Steelman LS, Kempf CR, et al. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways.. J Cell Physiol. 2011;226(11):2762‐2781. [DOI] [PubMed] [Google Scholar]
- 194. Chappell WH, Steelman LS, Long JM, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011;2(3):135‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Steelman LS, Chappell WH, Abrams SL, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy‐implications for cancer and aging. Aging. 2011;3(3):192‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Diop A, Santorelli D, Malagrinò F, et al. SH2 domains: folding, binding and therapeutical approaches. Int J Mol Sci. 2022;23(24):15944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Pawson T. Specificity in signal transduction: from phosphotyrosine‐SH2 domain interactions to complex cellular systems. Cell. 2004;116(2):191‐203. [DOI] [PubMed] [Google Scholar]
- 198. Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3‐kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U S A. 2001;98(11):6074‐6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Reuter CWM, Morgan MA, Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism‐based treatment for hematologic malignancies? Blood. 2000;96(5):1655‐1669. [PubMed] [Google Scholar]
- 200. Castellano E, Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer. 2011;2(3):261‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the Rictor–mTOR complex. Science. 2005;307(5712):1098‐1101. [DOI] [PubMed] [Google Scholar]
- 202. Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27(41):5486‐5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Mayo LD, Donner DB. A phosphatidylinositol 3‐kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98(20):11598‐11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2–p53 pathway revisited. J Biomed Res. 2013;27(4):254‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Tsuruta F, Masuyama N, Gotoh Y. The phosphatidylinositol 3‐kinase (PI3K)–Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem. 2002;277(16):14040‐14047. [DOI] [PubMed] [Google Scholar]
- 206. Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell. 1997;91(2):231‐241. [DOI] [PubMed] [Google Scholar]
- 207. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261‐1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Castellano E, Sheridan C, Thin May Z, et al. Requirement for interaction of PI3‐kinase p110α with RAS in lung tumor maintenance. Cancer Cell. 2013;24(5):617‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR‐amplified lung cancer. J Clin Investig. 2006;116(10):2695‐2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Baer R, Cintas C, Dufresne M, et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild‐type PI 3‐kinase p110α. Genes Dev. 2014;28(23):2621‐2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143‐153. [DOI] [PubMed] [Google Scholar]
- 213. Zhang M, Jang H, Nussinov R. PI3K inhibitors: review and new strategies. Chem Sci. 2020;11(23):5855‐5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Hanker AB, Kaklamani V, Arteaga CL. Challenges for the clinical development of PI3K inhibitors: strategies to improve their impact in solid tumors. Cancer Discov. 2019;9(4):482‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Frankel P, Aronheim A, Kavanagh E, et al. RalA interacts with ZONAB in a cell density‐dependent manner and regulates its transcriptional activity. EMBO J. 2005;24(1):54‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Bodemann BO, Orvedahl A, Cheng T, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell. 2011;144(2):253‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Chien Y, Kim S, Bumeister R, et al. RalB GTPase‐mediated activation of the IκB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127(1):157‐170. [DOI] [PubMed] [Google Scholar]
- 218. Ikeda M, Ishida O, Hinoi T, Kishida S, Kikuchi A. Identification and characterization of a novel protein interacting with Ral‐binding protein 1, a putative effector protein of Ral. J Biol Chem. 1998;273(2):814‐821. [DOI] [PubMed] [Google Scholar]
- 219. Takai Y, Sasaki T, Matozaki T. Small GTP‐binding proteins. Physiol Rev. 2001;81(1):153‐208. [DOI] [PubMed] [Google Scholar]
- 220. Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8(2):133‐140. [DOI] [PubMed] [Google Scholar]
- 221. Kashatus DF. Ral GTPases in tumorigenesis: emerging from the shadows. Exp Cell Res. 2013;319(15):2337‐2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Guin S, Ru Y, Wynes MW, et al. Contributions of KRAS and RAL in non‐small‐cell lung cancer growth and progression. J Thorac Oncol. 2013;8(12):1492‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Lim K‐H, Brady DC, Kashatus DF, et al. Aurora—a phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2010;30(2):508‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Lim K‐H, O'Hayer K, Adam SJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16(24):2385‐2394. [DOI] [PubMed] [Google Scholar]
- 225. Lambert JM, Lambert QT, Reuther GW, et al. Tiam1 mediates Ras activation of Rac by a PI(3)K‐independent mechanism. Nat Cell Biol. 2002;4(8):621‐625. [DOI] [PubMed] [Google Scholar]
- 226. Zondag GCM, Evers EE, ten Klooster JP, Janssen L, van der Kammen RA, Collard JG. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial–mesenchymal transition. J Cell Biol. 2000;149(4):775‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Sander EE, van Delft S, ten Klooster JP, et al. Matrix‐dependent Tiam1/Rac signaling in epithelial cells promotes either cell–cell adhesion or cell migration and is regulated by phosphatidylinositol 3‐kinase. J Cell Biol. 1998;143(5):1385‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Strickler JH, Satake H, George TJ, et al. Sotorasib in KRAS p.G12C‐mutated advanced pancreatic cancer. N Engl J Med. 2022;388(1):33‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Yaeger R, Weiss J, Pelster MS, et al. Adagrasib with or without cetuximab in colorectal cancer with mutated KRAS G12C. N Engl J Med. 2022;388(1):44‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Wang J, Martin‐Romano P, Cassier P, et al. Phase I study of JNJ‐74699157 in patients with advanced solid tumors harboring the KRAS G12C mutation. Oncologist. 2022;27(7):536‐e553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Zou Jiayun YTWY. Precision therapy of metastatic colorectal cancer with RAS mutation. Cancer Res Prevent Treat. 2021;48(08):820‐824. [Google Scholar]
- 232. de Langen AJ, Johnson ML, Mazieres J, et al. Sotorasib versus docetaxel for previously treated non‐small‐cell lung cancer with RASG12C mutation: a randomised, open‐label, phase 3 trial. Lancet. 2023;401(10378):733‐746. [DOI] [PubMed] [Google Scholar]
- 233. Jänne PA, Riely GJ, Gadgeel SM, et al. Adagrasib in non‐small‐cell lung cancer harboring a KRASG12C mutation. N Engl J Med. 2022;387(2):120‐131. [DOI] [PubMed] [Google Scholar]
- 234. Golan T, Khvalevsky EZ, Hubert A, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015;6(27):24560‐24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Jiang W, Li H, Liu X, et al. Precise and efficient silencing of mutant KrasG12D by CRISPR‐CasRx controls pancreatic cancer progression. Theranostics. 2020;10(25):11507‐11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Datta J, Dai X, Bianchi A, et al. Combined MEK and STAT3 inhibition uncovers stromal plasticity by enriching for cancer‐associated fibroblasts with mesenchymal stem cell‐like features to overcome immunotherapy resistance in pancreatic cancer. Gastroenterology. 2022;163(6):1593‐1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Thomas AM, Reena S, Jason RP, et al. IL‐6 and PD‐L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67(2):320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Chen K, Qian W, Jiang Z, et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol Cancer. 2017;16(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Qin T, Chen K, Li J, et al. Heat shock factor 1 inhibition sensitizes pancreatic cancer to gemcitabine via the suppression of cancer stem cell‐like properties. Biomed Pharmacother. 2022;148:112713. [DOI] [PubMed] [Google Scholar]
- 240. Ghufran M, Khan HA, Ullah M, et al. In silico strategies for designing of peptide inhibitors of oncogenic K‐Ras G12V mutant: inhibiting cancer growth and proliferation. Cancers. 2022;14(19):4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Kazi A, Xiang S, Yang H, et al. Dual farnesyl and geranylgeranyl transferase inhibitor thwarts mutant KRAS‐driven patient‐derived pancreatic tumors. Clin Cancer Res. 2019;25(19):5984‐5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Wang Y, Wang B, Xiang L, et al. Case report: anlotinib combined with PD‐1 inhibitor and sequential GA regimen or FOLFIRINOX chemotherapy in treatment of KRAS G12V mutated pancreatic ductal adenocarcinoma with liver metastasis: a case and literature review. Front Immunol. 2022;13:1016647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Salvador‐Barbero B, Álvarez‐Fernández M, Zapatero‐Solana E, et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 2020;37(3):340‐353.e6. [DOI] [PubMed] [Google Scholar]
- 244. Diego‐González L, Fernández‐Carrera A, Igea A, et al. Combined inhibition of FOSL‐1 and YAP using siRNA‐lipoplexes reduces the growth of pancreatic tumor. Cancers. 2022;14(13):3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Hobbs GA, Baker NM, Miermont AM, et al. Atypical KRASG12R mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 2020;10(1):104‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Hobbs GA, Der CJ. KRASG12R‐independent macropinocytosis in pancreatic cancer. In: Commisso C, ed. Macropinocytosis: Functions and Mechanisms. Springer International Publishing; 2022:205‐221. [DOI] [PubMed] [Google Scholar]
- 247. Kenney C, Kunst T, Webb S, et al. Phase II study of selumetinib, an orally active inhibitor of MEK1 and MEK2 kinases, in KRASG12R‐mutant pancreatic ductal adenocarcinoma. Invest New Drugs. 2021;39(3):821‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Drosten M, Barbacid M. KRAS inhibitors: going noncovalent. Mol Oncol. 2022;16(22):3911‐3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Li W, Chen W, Wang J, et al. A PDX model combined with CD‐DST assay to evaluate the antitumor properties of KRpep‐2d and oxaliplatin in KRAS (G12D) mutant colorectal cancer. Heliyon. 2022;8(12):e12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Lubell WD. Peptide‐based drug development. Biomedicines. 2022;10(8):2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Orlov SV, Urtenova MA, Sviridenko MA, Nesterov DV, Sokolova TN, Imyanitov EN. Rapid improvement of the performance status and reduction of the tumor size in KRAS‐mutated colorectal cancer patient receiving binimetinib, hydroxychloroquine, and bevacizumab. Case Rep Oncol. 2020;13(2):985‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Kitazawa M, Miyagawa Y, Koyama M, et al. Drug sensitivity profile of minor KRAS mutations in colorectal cancer using mix culture assay: the effect of AMG‐510, a novel KRAS G12C selective inhibitor, on colon cancer cells is markedly enhanced by the combined inhibition of MEK and BCL‐XL. Mol Clin Oncol. 2021;15(1):148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Hiraki M, Nishimura J, Takahashi H, et al. Concurrent targeting of KRAS and AKT by MiR‐4689 is a novel treatment against mutant KRAS colorectal cancer. Mol Ther Nucleic Acids. 2015;4(3):e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. McFall T, Schomburg NK, Rossman KL, Stites EC. Discernment between candidate mechanisms for KRAS G13D colorectal cancer sensitivity to EGFR inhibitors. Cell Commun Signal. 2020;18(1):179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Bando H, Yoshino T, Yuki S, et al. Clinical outcome of Japanese metastatic colorectal cancer patients harbouring the KRAS p.G13D mutation treated with cetuximab + irinotecan. Jpn J Clin Oncol. 2012;42(12):1146‐1151. [DOI] [PubMed] [Google Scholar]
- 256. Osumi H, Shinozaki E, Osako M, et al. Cetuximab treatment for metastatic colorectal cancer with KRAS p.G13D mutations improves progression‐free survival. Mol Clin Oncol. 2015;3(5):1053‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Segelov E, Waring P, Desai J, et al. ICECREAM: randomised phase II study of cetuximab alone or in combination with irinotecan in patients with metastatic colorectal cancer with either KRAS, NRAS, BRAF and PI3KCA wild type, or G13D mutated tumours. BMC Cancer. 2016;16:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Chu YC, Tsai T‐Y, Yadav VK, et al. 4‐Acetyl‐antroquinonol B improves the sensitization of cetuximab on both Kras mutant and wild type colorectal cancer by modulating the expression of Ras/Raf/miR‐193a‐3p signaling axis. Int J Mol Sci. 2021;22(14):7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Messner I, Cadeddu G, Huckenbeck W, et al. KRAS p.G13D mutations are associated with sensitivity to anti‐EGFR antibody treatment in colorectal cancer cell lines. J Cancer Res Clin Oncol. 2013;139(2):201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy‐refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304(16):1812‐1820. [DOI] [PubMed] [Google Scholar]
- 261. McFall T, Stites EC. A mechanism for the response of KRASG13D expressing colorectal cancers to EGFR inhibitors. Mol Cell Oncol. 2020;7(2):1701914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Masuishi T, Tsuji A, Kotaka M, et al. Phase 2 study of irinotecan plus cetuximab rechallenge as third‐line treatment in KRAS wild‐type metastatic colorectal cancer: JACCRO CC‐08. Br J Cancer. 2020;123(10):1490‐1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Jiao D, Yang S. Overcoming resistance to drugs targeting KRAS(G12C) mutation. Innovation. 2020;1(2):100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Molina‐Arcas M, Moore C, Rana S, et al. Development of combination therapies to maximize the impact of KRAS‐G12C inhibitors in lung cancer. Sci Transl Med. 2019;11(510):eaaw7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Addeo A, Banna GL, Friedlaender A. KRAS G12C mutations in NSCLC: from target to resistance. Cancers. 2021;13(11):2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Akhave NS, Biter AB, Hong DS. Mechanisms of resistance to KRASG12C‐targeted therapy. Cancer Discov. 2021;11(6):1345‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Zhang SS, Nagasaka M. Spotlight on sotorasib (AMG 510) for KRAS (G12C) positive non‐small cell lung cancer. Lung Cancer. 2021;12:115‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Palma G, Khurshid F, Lu K, Woodward B, Husain H. Selective KRAS G12C inhibitors in non‐small cell lung cancer: chemistry, concurrent pathway alterations, and clinical outcomes. NPJ Precis Oncol. 2021;5(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Corral de la Fuente E, Olmedo Garcia ME, Gomez Rueda A, Lage Y, Garrido P. Targeting KRAS in non‐small cell lung cancer. Front Oncol. 2022;11:792635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Fukuda K, Otani S, Takeuchi S, et al. Trametinib overcomes KRAS‐G12V‐induced osimertinib resistance in a leptomeningeal carcinomatosis model of EGFR‐mutant lung cancer. Cancer Sci. 2021;112(9):3784‐3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Renaud S, Guerrera F, Seitlinger J, et al. KRAS‐specific amino acid substitutions are associated with different responses to chemotherapy in advanced non‐small‐cell lung cancer. Clin Lung Cancer. 2018;19(6):e919‐e931. [DOI] [PubMed] [Google Scholar]
- 272. Jänne PA, Smith I, McWalter G, et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non‐small‐cell lung cancer. Br J Cancer. 2015;113(2):199‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Ward AB, Keeton AB, Chen X, et al. Enhancing anticancer activity of checkpoint immunotherapy by targeting RAS. MedComm. 2020;1(2):121‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Yang R, Wang D, Li X, et al. An advanced non‐small cell lung cancer patient with EGFR and KRAS mutations, and PD‐L1 positive, benefited from immunotherapy: a case report. Ann Transl Med. 2022;10(6):381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Zaorsky NG, Sun Y, Wang Z, et al. Identification of a KRAS mutation in a patient with non‐small cell lung cancer treated with chemoradiotherapy and panitumumab. Cancer Biol Ther. 2013;14(10):883‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Löhr M, Müller P, Zauner I, et al. Immortalized bovine pancreatic duct cells become tumorigenic after transfection with mutant k‐ras. Virchows Arch. 2001;438(6):581‐590. [DOI] [PubMed] [Google Scholar]
- 277. Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas‐specific blockade of transforming growth factor‐beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20(22):3147‐3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17(24):3112‐3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Ying H, Kimmelman Alec C, Lyssiotis Costas A, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Huynh MV, Hobbs GA, Schaefer A, et al. Functional and biological heterogeneity of KRASQ61 mutations. Sci Signal. 2022;15(746):eabn2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Lam KK, Tang CL, Tan E, Wong SH, Cheah PY. KRAS mutation‐independent downregulation of MAPK/PI3K signaling in colorectal cancer. Mol Oncol. 2022;16(5):1171‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Zhou Z‐W, Ambrogio C, Bera AK, et al. KRASQ61H preferentially signals through MAPK in a RAF dimer‐dependent manner in non‐small cell lung cancer. Cancer Res. 2020;80(17):3719‐3731. [DOI] [PubMed] [Google Scholar]
- 284. Banno K, Yanokura M, Iida M, Masuda K, Aoki D. Carcinogenic mechanisms of endometrial cancer: involvement of genetics and epigenetics. J Obstet Gynaecol Res. 2014;40(8):1957‐1967. [DOI] [PubMed] [Google Scholar]
- 285. Chaudhry P, Asselin E. Resistance to chemotherapy and hormone therapy in endometrial cancer. Endocr Relat Cancer. 2009;16(2):363‐380. [DOI] [PubMed] [Google Scholar]
- 286. Mustachio LM, Chelariu‐Raicu A, Szekvolgyi L, Roszik J. Targeting KRAS in cancer: promising therapeutic strategies. Cancers. 2021;13(6):1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Guo F, Zhang H, Jia Z, Cui M, Tian J. Chemoresistance and targeting of growth factors/cytokines signalling pathways: towards the development of effective therapeutic strategy for endometrial cancer. Am J Cancer Res. 2018;8:1317‐1331. (Print 2156‐6976) [PMC free article] [PubMed] [Google Scholar]
- 288. Eleveld TF, Schild L, Koster J, et al. RAS–MAPK pathway‐driven tumor progression is associated with loss of CIC and other genomic aberrations in neuroblastoma. Cancer Res. 2018;78(21):6297‐6307. [DOI] [PubMed] [Google Scholar]
- 289. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase‐3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12(22):3499‐3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Kim R‐K, Suh Y, Yoo K‐C, et al. Activation of KRAS promotes the mesenchymal features of basal‐type breast cancer. Exp Mol Med. 2015;47(1):e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Ricoult SJH, Yecies JL, Ben‐Sahra I, Manning BD. Oncogenic PI3K and K‐Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. 2016;35(10):1250‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Muñoz‐Pinedo C, El Mjiyad N, Ricci JE. Cancer metabolism: current perspectives and future directions. Cell Death Dis. 2012;3(1):e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Galmiche A, Fueller J, Santel A, et al. Isoform‐specific Interaction of C‐RAF with mitochondria. J Biol Chem. 2008;283(21):14857‐14866. [DOI] [PubMed] [Google Scholar]
- 294. Inouye K, Mizutani S, Koide H, Kaziro Y. Formation of the Ras dimer is essential for Raf‐1 activation. J Biol Chem. 2000;275(6):3737‐3740. [DOI] [PubMed] [Google Scholar]
- 295. Khan I, Spencer‐Smith R, O'Bryan JP. Targeting the α4–α5 dimerization interface of K‐RAS inhibits tumor formation in vivo. Oncogene. 2019;38(16):2984‐2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Röth S, Macartney TJ, Konopacka A, et al. Targeting endogenous K‐RAS for degradation through the affinity‐directed protein missile system. Cell Chem Biol. 2020;27(9):1151‐1163.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.