

Abstract
Background
Colorectal cancer (CRC) is the third leading cause of cancer‐related deaths worldwide. Studies have shown that the DNA damage response (DDR) mutation is strongly associated with microsatellite instability (MSI) status and is an indication for patients with CRCs receiving immune checkpoint inhibitor (ICI) treatment. However, DDR mutation in microsatellite stable (MSS) CRC remains unclear.
Methods
In this study, Fisher's exact test, Student'st‐test, Wilcoxon rank‐sum test and Cox proportional hazards regression model were performed, and a p value of < 0.05 was considered statistically significant.
Results
The most common gene alterations were APC (77%), TP53 (73%), KRAS (48%), and PIK3CA (25%). The mutationfrequency of APC and TP53 in left‐sided CRC was significantly higher than that for right‐sided CRC, while the mutation frequency of PIK3CA, ACVR2A, FAT4, and RNF43 in right‐sided CRC was significantly higher than that for left‐sided CRC. DDR mutations occurred in100% of MSI CRCs and in 83.77% of MSS CRCs, with the most frequently mutated DDR genes being ARID1A (7.5%), ATM (5.7%,) and BRCA2 (2.6%). When right‐ and left‐sided CRCs were compared, no significant difference was observed for DDR genes and pathways. A survival analysis indicated that the DDR mutation was not associated with overall survival (OS) in MSS CRCs, while left‐sided patients with homologous recombination repair (HRR) pathway mutations had a significantly prolonged OS compared with right‐sided CRCs.
Conclusions
Here, we found that stage and grade were statistically significant independent prognostic factors in the left‐sided CRC and the right‐sided CRC, recommending treatment for these patients stratified by stage. For the future, utilizing DDR gene defects for expanding treatment options and improving prognosis is an issue worth exploring.
Keywords: DDR mutations, genomic landscape, MSS CRC, prognosis
In this study, we utilized next generation sequencing (NGS) to investigate genomic alterations in colorectal cancer (CRC), with a particular emphasis on comparing DNA damage response (DDR) mutations in left‐ and right‐sided CRC. Furthermore, we assessed the correlation between DDR mutations and prognosis for microsatellite stable (MSS) CRC.
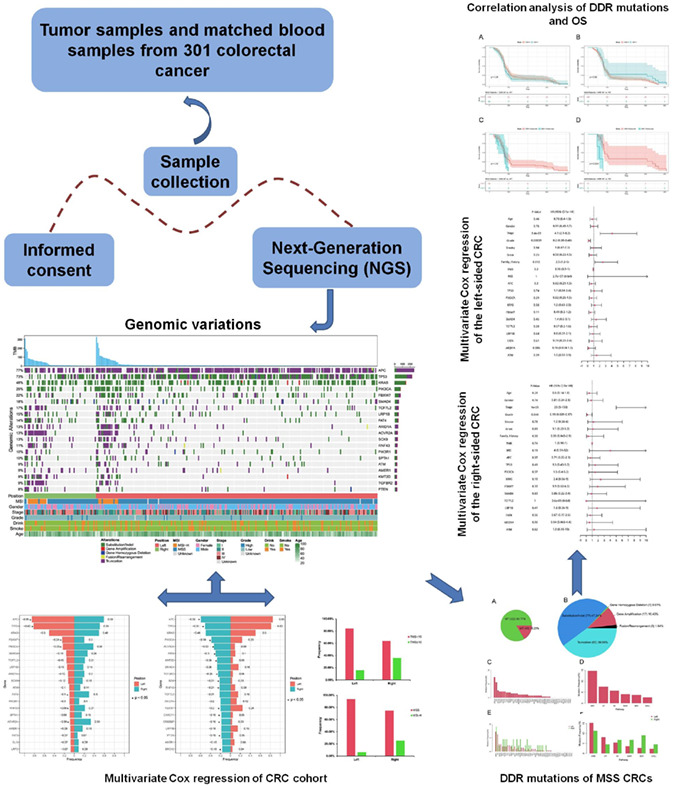
1. INTRODUCTION
Colorectal cancer (CRC) is a malignancy characterized by the abnormal growth of large intestine tissue. 1 CRC is one of the most common cancers, with an incidence rate that ranks third in the world, and is more prevalent in men than in women. 2 Despite effective cancer screening technology and modern medicine, the incidence and mortality of CRC have both increased in China. 3 Therefore, identifying novel diagnostic and prognostic biomarkers and exploring potentially relevant targets for the treatment of CRC are important goals.
Due to the need for further research, studies are currently being conducted on genomic alterations in the DNA damage response (DDR) pathway. In this context, DDR genes mutations are emerging as novel targets for cancer therapy. The DDR pathway's function is to accurately correct and repair DNA damage in a timely manner in order to preserve cell genome integrity, so as to inhibit cell aging, apoptosis, and carcinogenesis, and to ensure normal life activities. 4 , 5 Based on DNA lesions, DDR comprises eight pathways: mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER), homologous recombination repair (HRR), nonhomologous end‐joining (NHEJ), checkpoint factors (CPF), Fanconi anemia (FA), and translesion DNA synthesis (TLS). 6 DDR deficiencies in many cancers offer new opportunities for targeted, precision therapy. Poly (ADP‐ribose) polymerase‐inhibitors (PARPi) are currently applied for the treatment of HRR (BRCA1/2, BRD4, PTEN, or other HRR related genes) defective cancers such as ovarian cancer, 7 pancreatic cancer, 8 and prostate cancer. 9 Additionally, once a failure to maintain genomic integrity and stability is established, DDR alterations may induce a hyper‐mutated phenotype with a higher tumor mutation burden (TMB) or a microsatellite instability‐high (MSI‐H) status, established as a predictive biomarker for clinical benefit from immune checkpoint inhibitor (ICI) treatment. 10 , 11 For instance, Wang et al. 12 revealed that mutations within the DDR pathways of HRR‐MMR or HRR‐BER were associated with increased TMB, neoantigen load, and increased levels of immune gene expression signatures and served as potential predictors of superior survival outcomes in response to immune checkpoint blockades. 12
In CRC, the role of DDR alterations is still widely unknown and data regarding their clinical impacts are scarce. In recent years, a subset of studies has revealed germline and/or DDR defects in CRC, with a prevalence between 13.8% and 36%. 13 , 14 , 15 Regardless of MSI status, the median (mTMB) of CRC with DDR alterations was found to be higher, as well as the positive rate of PD‐L1. 15 Additionally, DDR mutations have been correlated with improved overall survival (OS) in CRCs treated with ICIs. 13 A recent study indicated that DDR‐related ATM or BRCA2 somatic mutations are promising biomarkers for assessing the response of stage III CRC patients to oxaliplatin‐based chemotherapy. 16 However, at present, there is a lack of studies that systematically compare DDR mutations between left‐ and right‐sided CRC, and little is known about the prognostic impact of DDR mutations in microsatellite stable (MSS) CRC patients.
Therefore, the present study systematically compared DDR mutations between left‐ and right‐sided CRC and investigated the correlation between DDR mutations and prognosis for MSS CRC.
2. MATERIALS AND METHODS
2.1. Patients and tumor samples
Tumor samples from 301 CRC patients were collected at The First Affiliated Hospital of Guangxi Medical University and The First Affiliated Hospital of Kunming Medical University Hospital from 2014 to 2019. Pathological sections were cut from formalin‐fixed paraffin‐embedded (FFPE) tumor blocks for subsequent use. To confirm that samples contained the highest tumor cell purity (>50%), prior to sequencing, FFPE tumor samples were evaluated by pathologists. All patients signed written informed consent for the collection and use of tumor samples. This study was approved by the Ethics Committee of The First Affiliated Hospital of Guangxi Medical University.
2.2. Identification of genomic alterations and tumor mutational burden (TMB)
Formalin‐fixed, paraffin‐embedded (FFPE) tumor tissues and matched blood samples were obtained from the First Affiliated Hospital of Guangxi Medical University. At least 50 ng of cancer tissue DNA was extracted from the 40 mm FFPE and from blood samples using a DNA Extraction Kit (QIAamp DNA FFPE Tissue Kit, Qiagen) for subsequent targeted NGS‐based genomic testing (OrigiMed). Genomic mutations were detected using the NGS‐based YuanSu™ (OrigiMed) gene panel, which covers all coding exons for 450 cancer‐related genes frequently altered in solid tumors (including the 45 DDR genes). Genes were captured and sequenced, with a mean depth of 800× and with a minimum depth of coverage of ≥200×, using an Illumina NextSeq 500 (Illumina) by following the steps described in Frampton et al. 17 The quality scores of ≥40 were used for this study. Mutational variant allele frequency (VAF) was defined as the number of variant reads divided by the number of total reads and reported as a percentage. Mutations with VAF ≥1% were included for analysis.
Genomic alterations (GAs) were identified based on the described procedure of Cao et al. 18 Single‐nucleotide variants (SNVs) were identified using MuTect (v1.7). Insertion–deletions (Indels) were identified using PINDEL (v0.2.5). The functional impact of GAs was annotated using SnpEff 3.0. Copy number variation (CNV) regions were identified with Control‐FREEC (v9.7), using the following parameters: window = 50,000 and step = 10,000. Gene fusions were detected using an in‐house developed pipeline. Gene rearrangements were assessed by employing the Integrative Genomics Viewer (IGV). TMB was measured by counting coding somatic mutations, including SNVs and Indels, per megabase of the sequence examined for each patient. Since cutoffs for categorizing the TMB status of CRC have not been defined, we used criteria established in a previous study for different tumor types. 19 In this study, TMB‐L was defined as <10 mut(mutations)/Mb, and TMB‐H was defined as ≥10 mut/Mb of sequenced DNA.
2.3. Definition of DNA damage repair
To identify DDR inactivation mutation status, the DNA data of nonsynonymous copy number variants, single‐nucleotide variants, and multi‐nucleotide variants for 45 DDR genes (Table S1) were retrieved and combined. DDR pathway alternations were defined as any nonsynonymous somatic alteration (including missense, nonsense, insertion, deletion, and splice) in the protein‐coding region or the presence of homozygous deletions of at least one gene involved in the corresponding DDR pathways.
2.4. Statistical analyses
For statistical analyses, SPSS version 22.0 (SPSS Inc.) was applied. Fisher's exact test was used for the association analysis of categorical variables. Student's t test and Wilcoxon rank‐sum test were used for the association analysis of normally distributed data and nonnormally distributed data, respectively. A Kruskal–Wallis test was used for analyses of the association between multiple groups of nonparametric data. A Cox proportional hazards regression model was used for quantifying overall survival (OS). A p value of <0.05 was considered statistically significant.
3. RESULTS
3.1. Patient characteristics
For this study, a total of 301 CRC patients were recruited, of which 240 had a left‐sided CRC diagnosis and 61 had a right‐sided CRC diagnosis. One hundred and twenty‐one patients were younger than 55 years old, and 180 patients were older than 55. One hundred and twenty‐one (40.2%) of patients were females and 180 (59.8%) were males. Based on tumor stage, there were 33 (11.0%) patients at Stage I, 99 (33.0%) patients at Stage II, 120 (39.9%) patients at Stage III, and 47 (15.5%) patients at Stage IV. The tumor stage for two (0.6%) patients was unknown. The tumor for 280 (93.0%) patients was at low grade, 16 (5.3%) patients had high‐grade tumors, and tumor grade for the remaining 5 (1.7%) patients was unknown. Seventy‐one (23.6%) patients had a history of smoking, 64 (21.3%) had a history of alcohol consumption, and 59 (19.6%) had a family history. A follow‐up for the 301 patients indicated that 133 (41.2%) patients did not progress, 4 patients (1.3%) had a recurrence, 158 (52.5%) patients had metastasis, and 6 (2.0%) patients had no progression. At the last follow‐up, 188 (62.5%) patients survived, 51 (16.9%) died, and 62 (20.6%) patients had an unknown survival status. Sixty (20%) patients were defined as TMB‐H (TMB ≥10 mut/Mb), while 241 (80%) patients were defined as TMB‐L (TMB < 10 mut/Mb). The mTMB of right‐sided CRC was 7.7 muts/Mb, whereas the mTMB of left‐sided CRC was 5.4 muts/Mb. The frequency of TMB‐H in right‐sided CRC was higher than that in left‐sided CRC (36.1% vs. 15.8%, respectively, p < 0.001, Figure S1A). Additionally, 30 (10.2%) patients were defined as MSI‐H, 265 (88%) patients were defined as MSS, and 6 (2.0%) patients had an unknown MSI status. The frequency of MSI‐H in left‐sided CRC was 6.4%, while was 25.4% in right‐sided CRC (Figure S1B). The frequency of MSI‐H in right‐sided CRC was higher than that in left‐sided CRC (25.4% vs. 6.36%, respectively, p < 0.001). Detailed clinical characteristics for each patient are provided in Table 1.
TABLE 1.
Clinical characteristics of the 301CRCs.
| Total | Left | Right | OR (95% CI) | p value | |
|---|---|---|---|---|---|
| Age | |||||
| ≤55 | 121 | 96 | 25 | Reference | |
| >55 | 180 | 144 | 36 | 0.9601 (0.5229–1.783) | 0.8849 |
| Gender | |||||
| Female | 121 | 91 | 30 | Reference | |
| Male | 180 | 149 | 31 | 0.6321 (0.3448–1.1587) | 0.1432 |
| Stage | |||||
| I | 33 | 30 | 3 | Reference | |
| II | 99 | 77 | 22 | 2.8383 (0.7659–15.8919) | 0.1253 |
| III | 120 | 93 | 27 | 2.8868 (0.7991–15.9107) | 0.1351 |
| IV | 47 | 38 | 9 | 2.3448 (0.524–14.639) | 0.3413 |
| Unknown | 2 | 2 | NA | ||
| Grade | |||||
| Low | 280 | 224 | 56 | Reference | |
| High | 16 | 11 | 5 | 1.8139 (0.4744–5.9497) | 0.3367 |
| Unknown | 5 | 5 | NA | ||
| Smoke | |||||
| No | 230 | 182 | 48 | Reference | |
| Yes | 71 | 58 | 13 | 0.8503 (0.3942–1.7336) | 0.7366 |
| Drink | |||||
| No | 237 | 187 | 50 | Reference | |
| Yes | 64 | 53 | 11 | 0.7768 (0.3403–1.6476) | 0.5999 |
| Family History | |||||
| No | 233 | 194 | 39 | Reference | |
| Yes | 59 | 42 | 17 | 2.008 (0.9692–4.0531) | 0.0422 |
| Unknown | 9 | 4 | 5 | ||
| Progress | |||||
| No | 133 | 109 | 24 | Reference | |
| Recurrent | 4 | 2 | 2 | 4.472 (0.31–64.6123) | 0.1629 |
| Metastatic | 158 | 124 | 34 | 1.2444 (0.6697–2.3401) | 0.556 |
| Unknown | 6 | 5 | 1 | ||
| Survival | |||||
| Alive | 188 | 147 | 41 | Reference | |
| Dead | 51 | 40 | 11 | 0.986 (0.4183–2.1784) | 1 |
| Unknown | 62 | 53 | 9 | ||
| TMB | |||||
| <10 | 241 | 202 | 39 | Reference | |
| ≥10 | 60 | 38 | 22 | 2.9855 (1.5109–5.8432) | 0.001 |
| MSI | |||||
| MSS | 265 | 221 | 44 | Reference | |
| MSI‐H | 30 | 15 | 15 | 4.985 (2.1047–11.8488) | 1.00 E‐04 |
| Unknown | 6 | 4 | 2 | ||
3.2. Genetic profiling of CRC
Tumor samples from the 301 CRC patients were sequenced using NGS technology. Genetic profiling is provided in Figure 1A. A total of 2881 variations from 466 genes, including 1801 (62.51%) substitutions/indels, 252 (8.75%) gene amplifications, 779 (27.04%) truncations, 30 (1.04%) fusions/rearrangements, and 19 (0.66%) gene homozygous deletions were detected in the 301 CRC patients. The landscape of genetic alterations was mapped. The most common gene alterations for the 301 CRC patients were APC (77%), TP53 (73%), KRAS (48%), PIK3CA (25%), FBXW7 (22%), SMAD4 (18%), TCF7L2 (17%), LRP1B (15%), FAT4 (14%), ARID1A (13%), ACVR2A (13%), SOX9 (13%), RNF43 (11%), PIK3R1 (10%), and SPTA1 (10%) (Figure 1A). The results of comutation analysis have shown in Figure S2. The most common gene alterations for the 80 right‐sided CRC patients and the 121 left‐sided CRC patients were also mapped (Tables S2 and S3 and Figure 1B,C, respectively). The APC, TP53, and KRAS genes were highly mutated in both left‐ and right‐sided CRCs. By comparing the mutation frequency of highly mutated genes, we found that the mutation frequency of APC and TP53 in left‐sided CRC was significantly higher than that in right‐sided CRC, while the mutation frequency of PIK3CA, ACVR2A, FAT4, and RNF43 in right‐sided CRC was significantly higher than that in left‐sided CRC. The multivariate Cox regression of the left‐sided and right‐sided CRC cohort was performed, respectively. In the right‐sided and left‐sided CRC cohort, stage, grade, age, gender, smoking history, drinking history, TMB, MSI status, and top high‐frequented mutated genes were included. The multivariate Cox regression showed that stage, grade, and family history were statistically significant independent prognostic factors in the left‐sided CRC (Figure 2A), and stage and grade were statically significant in the right‐sided CRC (Figure 2B).
FIGURE 1.
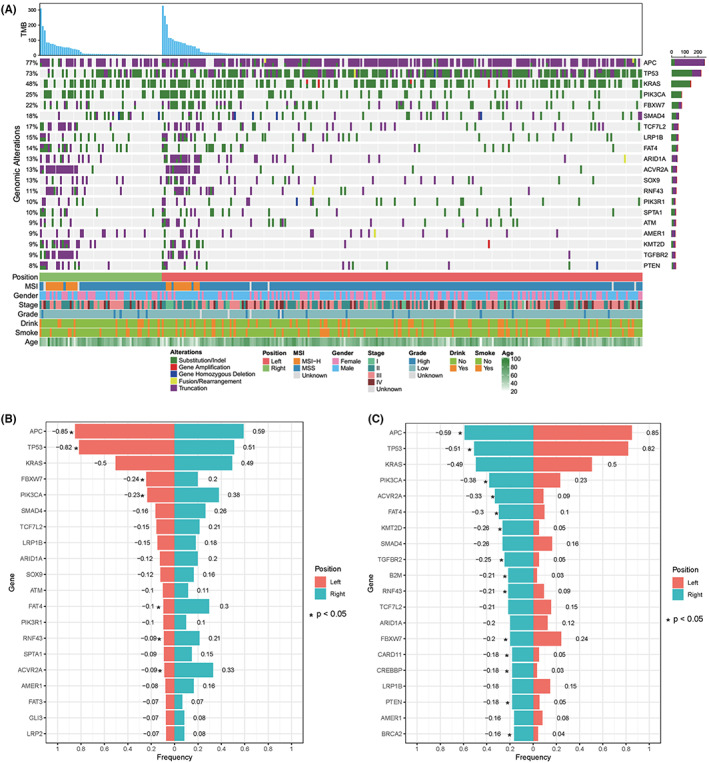
(A) The genomic variations of 301 CRCs. The panel shows the matrix of mutations colored by mutation type. The first row provides TMB values. Each column denotes an individual tumor and each row represents a gene. The right panel provides the gene name of the mutations and the left panel provides the proportion of mutations. Green: Substitution/Indel; Red: Gene amplification; Blue: Gene homozygous deletion; Yellow: Fusion/Rearrangement; Purple: Truncation. (B‐C) Genes with a significant difference in mutation frequency between left‐ and right‐sided CRC.
FIGURE 2.
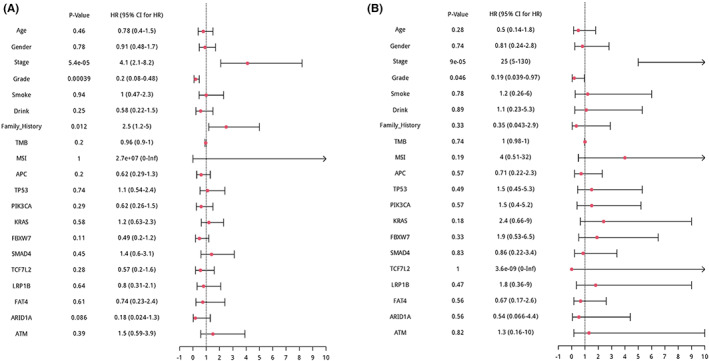
The multivariate Cox regression of CRC cohort. (A) Multivariate Cox regression of the left‐sided CRC. (B) Multivariate Cox regression of the right‐sided CRC.
3.3. DDR mutation landscape in MSS CRC patients
MSI‐H is significantly positively correlated with TMB‐H and is a known prognostic biomarker and immunotherapy biomarker in CRC. In our cohort, 30 CRC patients with MSI‐H all carried DDR mutations. Therefore, we excluded MSI‐H cases and analyzed the DDR mutation landscape in MSS CRC patients. Detailed characteristics of MSS CRC with DDR mutations are provided in Table 2. A total of 163 DDR gene somatic mutations, including 77 (47.24%) substitutions/indels, 65 (39.88%) truncations, 17 (10.43%) gene amplifications, 3 (1.84%) fusions/rearrangements, and 1 (0.61%) gene homozygous deletion were detected in 83.77% (222/265) of CRC patients (Figure 3A,B). Frequencies for every DDR gene mutation are summarized in Figure 2C. The most frequently mutated DDR genes were ARID1A (7.5%, 20/245), ATM (5.7%, 15/265), BRCA2 (2.6%, 7/265), PRD52 (2.3%, 6/259), POLE (2.3, 6/259), FANCM (2.3%, 6/259), and POLB (2.3%, 6/259) (Figure 3C). Frequencies of mutations in HRR, CP, FA, MMR, BER, NHEJ, and pathways were 14.72% (39/265), 7.5% (20/265), 5.7% (15/265), 4.1% (11/265), 3.8% (10/265), and 2.6% (7/265), respectively (Figure 3D). The frequency of mutated DDR genes and pathways was additionally compared between left‐ and right‐sided CRCs. As shown in Figure 3E,F, no significant difference was observed in DDR genes and pathways.
TABLE 2.
Detailed characteristics of MSS CRC with DDR mutations.
| Total | DDR_MT | DDR_WT | OR (95% CI) | p value | |
|---|---|---|---|---|---|
| Age | |||||
| ≤55 | 97 | 81 | 16 | Reference | |
| >55 | 168 | 141 | 27 | 0.9695 (0.4712–2.0486) | 1 |
| Gender | |||||
| Female | 107 | 85 | 22 | Reference | |
| Male | 158 | 137 | 21 | 0.5935 (0.2909–1.2067) | 0.1283 |
| Stage | |||||
| I | 29 | 22 | 7 | Reference | |
| II | 82 | 67 | 15 | 0.706 (0.2325–2.3209) | 0.5888 |
| III | 107 | 90 | 17 | 0.5962 (0.2025–1.9156) | 0.4091 |
| IV | 45 | 41 | 4 | 0.3119 (0.0601–1.3848) | 0.0974 |
| Unknown | 2 | 2 | 0 | ||
| Grade | |||||
| Low | 245 | 203 | 42 | Reference | |
| High | 16 | 15 | 1 | 0.3232 (0.0075–2.2144) | 0.4836 |
| Unknown | 4 | 4 | 0 | ||
| Smoke | |||||
| No | 202 | 164 | 38 | Reference | |
| Yes | 63 | 58 | 5 | 0.3732 (0.1094–1.0144) | 0.0494 |
| Drink | |||||
| No | 208 | 174 | 34 | Reference | |
| Yes | 57 | 48 | 9 | 0.9597 (0.3781–2.2246) | 1 |
| Family history | |||||
| No | 211 | 179 | 32 | Reference | |
| Yes | 49 | 39 | 10 | 1.4322 (0.5784–3.3042) | 0.3905 |
| Unknown | 5 | 4 | 1 | ||
| Progress | |||||
| No | 112 | 94 | 18 | Reference | |
| Recurrent | 4 | 3 | 1 | 1.731 (0.0314–22.9851) | 0.5161 |
| Metastatic | 143 | 122 | 21 | 0.8993 (0.4288–1.9018) | 0.8611 |
| Unknown | 6 | 3 | 3 | ||
| Survival | |||||
| Alive | 161 | 133 | 28 | Reference | |
| Dead | 48 | 42 | 6 | 0.6797 (0.2153–1.8275) | 0.5087 |
| Unknown | 56 | 47 | 9 | ||
| TMB | |||||
| <10 | 237 | 197 | 40 | Reference | |
| ≥10 | 28 | 25 | 3 | 0.592 (0.1092–2.0858) | 0.5885 |
FIGURE 3.
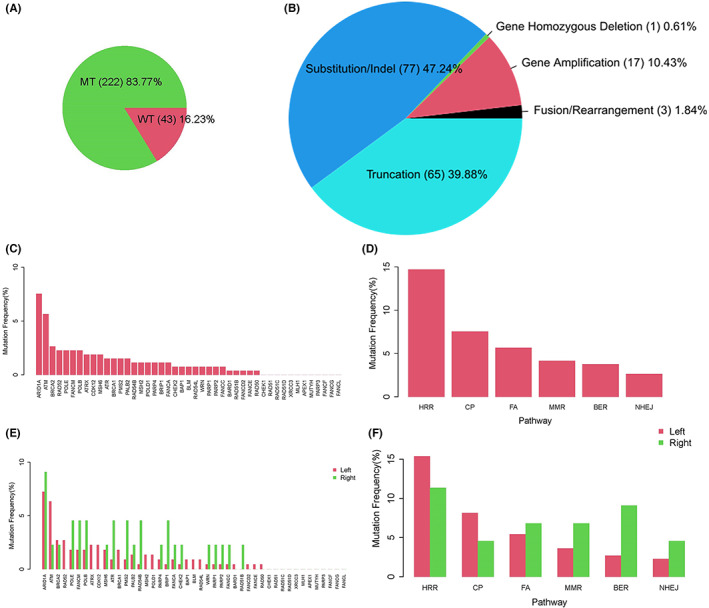
DDR mutations of MSS CRCs. (A) The mutation rate of DDR genes in MSS CRCs. (B) Gene mutation type and proportion. (C) DDR‐mutated genes and the mutation frequency of each DDR gene. (D) The mutations of DDR pathways and the mutation frequency of each DDR pathway. (E) A comparison of the mutational frequency of DDR genes between left‐ and right‐sided CRC. (F) A comparison of the mutational frequency of DDR pathways between left‐ and right‐sided CRC.
3.4. DDR mutation was not associated with clinical prognosis in MSS CRC
We investigated whether or not DDR somatic mutations were associated with improved survival in MSS CRC patients. The presence of DDR somatic mutations was not significantly associated with better OS (p = 0.26) for MSS patients in our cohort (Figure 4A). Specifically, MSS patients with mutations in the HRR pathway did not display better OS (p = 0.08) (Figure 4B). Further analysis regarding left‐ and right‐sided CRC revealed no significant difference (p = 0.09) in OS between left‐ and right‐sided CRCs with DDR mutations (Figure 4C), whereas left‐sided CRC patients with HRR pathway mutations that were relatively independent of the KRAS mutation (p = 0.211), had a significantly prolonged OS compared with right‐sided CRC (p = 0.0091) (Figure 4D).
FIGURE 4.
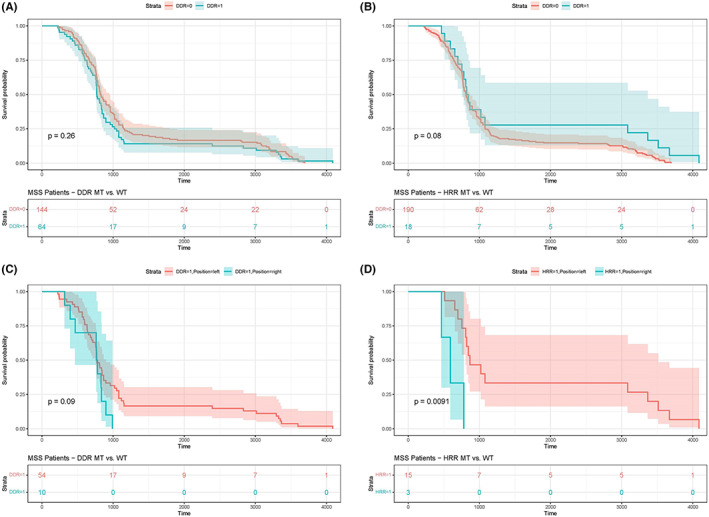
A correlation analysis of DDR mutations and prognosis. (A) DDR mutations in MSS CRC were not significantly related to OS. (B) HRR mutations in MSS CRC were not significantly related to OS. (C) No significant difference in OS among patients with DDR mutations in left‐ and right‐sided MSS CRC was determined. (D) Left‐sided MSS CRC patients with HRR mutations had a better OS compared with right‐sided MSS CRC.
4. DISCUSSION
CRC is a highly heterogeneous malignancy with diverse clinical features, therapeutic responses, and prognosis. Therefore, identifying clinical or molecular biomarkers with predictive and prognostic values is necessary. In this study, we investigated the mutation landscape of 301 Chinese CRC patients and compared mutation profiles between left‐ and right‐sided CRC.
The genomic landscape of CRC has been well studied, and, in general, the genomic landscape of CRC remains relatively stable, with the most frequently mutated genes being APC, TP53, KRAS, PIK3CA, and SMAD4. 20 , 21 Our study further confirmed that the most common gene alterations for CRC patients are APC (77%), TP53 (73%), KRAS (48%), PIK3CA (25%), FBXW7 (22%), and SMAD4 (18%). We additionally compared genetic mutation profiles between right and left‐sided CRC; and observed a higher mutation frequency for APC and TP53 and a lower mutation frequency for PIK3CA, ACVR2A, FAT4, and RNF43 in left‐sided CRC as compared to right‐sided CRC. Our results are highly consistent with a recent study which indicated that the mutation frequencies of TP53 and APC in left‐sided CRC are significantly higher than that in right‐sided CRC, whereas the mutation frequency of PIK3CA is lower than that in right‐sided CRC. 22 , 23 APC encodes a tumor suppressor protein that combines with β‐catenin within the cytoplasm in the form of protein complexes and negatively regulates the β‐catenin and Wnt signaling pathways, thus preventing excessive cell proliferation. 24 Different APC mutations lead to different levels of WNT/b‐catenin signaling pathway activation and are associated with the characteristics of different tumor sites in CRC. 25 TP53 is one of the most common tumor suppressor genes, both in CRC and in other tumor types. 26 In CRC, mutations in TP53 are associated with inferior survival. 27 PIK3CA is involved in the PI3K/Akt signaling pathway and is associated with high mutation rates in CRC 28 ; its somatic activating mutation also plays an important role during tumorigenesis. 29 Enriched mutations of TP53 and APC in left‐sided CRC and enriched mutations of PIK3CA in right‐sided CRC indicate the heterogeneity of CRC tumorigenesis and development.
In recent years, studies have revealed germline and/or DDR defects in CRC, with a prevalence between 13.8% and 36%. 13 , 14 , 15 In our study, we identified 100% DDR mutations in MSI CRC and 83.77% in MSS CRC. Due to our inclusion of a greater number of DDR genes (45 DDR genes) compared with previous studies, we detected a higher DDR mutation rate. We further investigated the mutation frequency of DDR genes in MSS CRC and determined that the mutation incidence of ARID1A and ATM are notably higher than for other genes, consistent with the finding of alterations in ARID1A in 8.3% of CRCs 30 and ATM in 7% of CRCs 31 from previous studies. The most frequent mutation type, ARID1A, was a truncating mutation, 30 like a frameshift mutation, that leads to DNA damage repair defects in tumor cells. 32 Preclinical studies have shown that ARID1A deficiency sensitizes CRC cells to PARP inhibitors (olaparib, rucaparib, veliparib, or BMN673) in vitro and in vivo. 33
A Phase II clinical trial (NCT02576444, OLAPCO) is currently ongoing for olaparil combination therapy in cancer patients with PTEN, PIK3CA, AKT, or ARID1A mutations or other mutations that lead to dysregulation of the PI3K/AKT pathway. ATM defects increase genomic instability by impeding the DNA double‐strand breakage (DSB) repair process but also increase tumor cell dependence on other DNA repair mechanisms, especially PARP‐mediated DNA single‐strand breakage (SSB). 34 , 35 Using the synthetic lethality mechanism, kinases (such as PARP) that inhibit the SSB repair process of ATM‐deficient tumors have potential therapeutic prospects. 34 , 35 Clinical trials are ongoing for several PARP inhibitors in patients with ATM‐deficient solid tumors (NCT01972217, NCT02693535, NCT03375307, NCT03233204, NCT03565991, and NCT03207347).
Agents targeting ATMs have drawn increasing attention from pharmaceutical companies. 36 Recent research has indicated that ALT neuroblastoma chemotherapy resistance occurs via ATM activation and is reversible with the ATM inhibitor AZD0156. Combining AZD0156 with temozolomide plus irinotecan warrants clinical testing for neuroblastoma. 37 Another ATM inhibitor, AZD1390, was verified to cross the intact blood–brain barrier, supporting the treatment of AZD1390 for glioblastoma multiforme or other brain malignancies. 38 Targeted therapy for other DDR mutations, including BRCA, ATR, ERCC2, etc., is also in progress. 39 , 40 , 41 , 42 Our results indicate that targeted therapy, especially for PARB and ATM inhibitors, has great potential for the treatment of CRC harboring DDR mutations.
In addition to the DDR mutation landscape, we also analyzed the relationship between DDR mutations and clinical prognosis in MSS CRC. Our results revealed that DDR pathway mutations, including HRR pathway mutations, were not significantly associated with better OS in MSS CRC patients. Accordingly, Sebastian et al.43 found that DDR pathway alterations were not associated with survival or progression‐free survival (PFS) in CRC patients receiving oxaliplatin‐containing chemotherapy. Song et al. 13 indicated that the DDR mutation was strongly associated with MSI status and was associated with a favorable median OS in CRC patients treated with ICI. However, in the Song et al. 13 study, no significant difference was identified in the prognosis of patients with DDR mutations with conventional treatment, indicating that DDR mutations may be a specific biomarker for predicting the efficacy of ICI immunotherapy in CRCs. Therefore, for MSS CRC, it is reasonable that DDR mutations are not significantly associated with a better prognosis.
In our study, we observed that HRR pathway mutations were significantly associated with better OS in left‐sided MSS CRC patients compared with right‐sided MSS CRC patients. However, in our cohort, the number of left‐sided CRC patients with HRR mutations was much higher than that of right‐sided CRC patients (n = 15 vs. n = 3, respectively). As such, our data can only be used as a clinical reference. A larger sample size is needed for further validation.
In conclusion, we identified the most frequently mutated DDR genes: ARID1A, ATM, and BRCA2 in CRC. Although DDR mutations do not significantly differ between left‐ and right‐sided CRC, and although no significant correlation exists between DDR mutations and prognosis in MSS CRC, we believe that DDR mutations remain a potential cancer therapeutic target for CRC treatment. MSS CRC still represents an unmet medical need. Going forward, how we can utilize DDR gene defects to expand treatment options and improve prognosis is an issue worth exploring.
AUTHOR CONTRIBUTIONS
Wei Huang: Conceptualization (equal); project administration (equal); writing – original draft (equal). Wenliang Li: Conceptualization (equal); project administration (equal); writing – original draft (equal). Ning Xu: Methodology (equal); resources (equal); software (equal). Hui Li: Methodology (equal); resources (equal); validation (equal). Zihan Zhang: Formal analysis (equal); software (equal); visualization (equal). Xiaolong Zhang: Formal analysis (equal); methodology (equal); resources (equal). Tingting He: Data curation (equal); visualization (equal). Jicheng Yao: Formal analysis (equal); methodology (equal); software (equal); visualization (equal). Mian Xu: Writing – original draft (equal). Qingqing He: Formal analysis (equal). Lijie Guo: Formal analysis (equal); writing – review and editing (equal).
FUNDING INFORMATION
This study was supported by the Yunnan Fundamental Research Projects (grant no. 2019FA039), the National Natural Science Foundation of China (grant no. 31660312), and leading medical talents in Yunnan (grant no. L‐2017001).
Supporting information
Figure S1.
Figure S2.
Table S1–S3.
Huang W, Li W, Xu N, et al. Differences in DNA damage repair gene mutations between left‐ and right‐sided colorectal cancer. Cancer Med. 2023;12:10187‐10198. doi: 10.1002/cam4.5716
Wei Huang and Wenliang Li contribute to this article equally.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Kuipers EJ, Grady WM, Lieberman D, et al. Colorectal cancer. Nat Rev Dis Primers. 2015;1:15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14(2):89‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Feng RM, Zong YN, Cao SM, Xu RH. Current cancer situation in China: good or bad news from the 2018 global cancer statistics? Cancer Commun (Lond). 2019;39(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16(1):20‐33. [DOI] [PubMed] [Google Scholar]
- 5. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475‐1485. [DOI] [PubMed] [Google Scholar]
- 6. Scarbrough PM, Weber RP, Iversen ES, et al. A cross‐cancer genetic association analysis of the DNA repair and DNA damage signaling pathways for lung, ovary, prostate, breast, and colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2016;25(1):193‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ray‐Coquard I, Pautier P, Pignata S, et al. Olaparib plus bevacizumab as first‐line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416‐2428. [DOI] [PubMed] [Google Scholar]
- 8. Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for germline BRCA‐mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mateo J, Carreira S, Sandhu S, et al. DNA‐repair defects and Olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697‐1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348(6230):124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD‐1/PD‐L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36(17):1685‐1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Z, Zhao J, Wang G, et al. Comutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Cancer Res. 2018;78(22):6486‐6496. [DOI] [PubMed] [Google Scholar]
- 13. Song Y, Huang J, Liang D, et al. DNA damage repair gene mutations are indicative of a favorable prognosis in colorectal cancer treated with immune checkpoint inhibitors. Front Oncol. 2020;10:549777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arai H, Elliott A, Wang J, et al. The landscape of DNA damage response (DDR) pathway in colorectal cancer (CRC). J Clin Oncol. 2020;38(15_suppl):4064.33052759 [Google Scholar]
- 15. Li W, Zhu Z, Xu N, et al. Alterations of DNA damage repair genes in Chinese colorectal cancer patients. J Clin Oncol. 2020;38(15_suppl):e16121. [Google Scholar]
- 16. Lin PC, Yeh YM, Chan RH, et al. Sequential and co‐occurring DNA damage response genetic mutations impact survival in stage III colorectal cancer patients receiving adjuvant oxaliplatin‐based chemotherapy. BMC Cancer. 2021;21(1):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cao J, Chen L, Li H, et al. An accurate and comprehensive clinical sequencing assay for cancer targeted and immunotherapies. Oncologist. 2019;24(12):e1294‐e1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33(1):125‐136 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagahashi M, Wakai T, Shimada Y, et al. Genomic landscape of colorectal cancer in Japan: clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med. 2016;8(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang W, Li H, Shi X, et al. Characterization of genomic alterations in Chinese colorectal cancer patients. Jpn J Clin Oncol. 2021;51(1):120‐129. [DOI] [PubMed] [Google Scholar]
- 23. Takahashi Y, Sugai T, Habano W, et al. Molecular differences in the microsatellite stable phenotype between left‐sided and right‐sided colorectal cancer. Int J Cancer. 2016;139(11):2493‐2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kawahara K, Morishita T, Nakamura T, Hamada F, Toyoshima K, Akiyama T. Down‐regulation of beta‐catenin by the colorectal tumor suppressor APC requires association with Axin and beta‐catenin. J Biol Chem. 2000;275(12):8369‐8374. [DOI] [PubMed] [Google Scholar]
- 25. Christie M, Jorissen RN, Mouradov D, et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/beta‐catenin signalling thresholds for tumourigenesis. Oncogene. 2013;32(39):4675‐4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olivier M, Taniere P. Somatic mutations in cancer prognosis and prediction: lessons from TP53 and EGFR genes. Curr Opin Oncol. 2011;23(1):88‐92. [DOI] [PubMed] [Google Scholar]
- 27. Zaidi SH, Harrison TA, Phipps AI, et al. Landscape of somatic single nucleotide variants and indels in colorectal cancer and impact on survival. Nat Commun. 2020;11(1):3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cathomas G. PIK3CA in colorectal cancer. Front Oncol. 2014;4:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Samuels Y, Diaz LA Jr, Schmidt‐Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7(6):561‐573. [DOI] [PubMed] [Google Scholar]
- 30. Tokunaga R, Xiu J, Goldberg RM, et al. The impact of ARID1A mutation on molecular characteristics in colorectal cancer. Eur J Cancer. 2020;140:119‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Watanabe R, Ui A, Kanno SI, et al. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014;74(9):2465‐2475. [DOI] [PubMed] [Google Scholar]
- 33. Shen J, Peng Y, Wei L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5(7):752‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cremona CA, Behrens A. ATM signalling and cancer. Oncogene. 2014;33(26):3351‐3360. [DOI] [PubMed] [Google Scholar]
- 35. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15(8):1781‐1791. [DOI] [PubMed] [Google Scholar]
- 36. Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 2015;149:124‐138. [DOI] [PubMed] [Google Scholar]
- 37. Koneru B, Farooqi A, Nguyen TH, et al. ALT neuroblastoma chemoresistance due to telomere dysfunction‐induced ATM activation is reversible with ATM inhibitor AZD0156. Sci Transl Med. 2021;13(607):eabd5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jucaite A, Stenkrona P, Cselényi Z, et al. Brain exposure of the ATM inhibitor AZD1390 in humans‐a positron emission tomography study. Neuro Oncol. 2021;23(4):687‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 40. Gorecki L, Andrs M, Rezacova M, Korabecny J. Discovery of ATR kinase inhibitor berzosertib (VX‐970, M6620): clinical candidate for cancer therapy. Pharmacol Ther. 2020;210:107518. [DOI] [PubMed] [Google Scholar]
- 41. Yap T, O'Carrigan B, Penney MS, et al. Phase I trial of first‐in‐class ATR inhibitor M6620 (VX‐970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol. 2020;38(27):3195‐3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yap TA, Tan DSP, Terbuch A, et al. First‐in‐human trial of the Oral ataxia telangiectasia and RAD3‐related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov. 2021;11(1):80‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Table S1–S3.
Data Availability Statement
The data that support the findings of this study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.