

Abstract
The extracellular matrix (ECM) is an intricate network composed of various multi-domain macromolecules like collagen, proteoglycans, and fibronectin, etc., that form a structurally stable composite, contributing to the mechanical properties of tissue. However, matricellular proteins are non-structural, secretory extracellular matrix proteins, which modulate various cellular functions via interacting with cell surface receptors, proteases, hormones, and cell-matrix. They play essential roles in maintaining tissue homeostasis by regulating cell differentiation, proliferation, adhesion, migration, and several signal transduction pathways. Matricellular proteins display a broad functionality regulated by their multiple structural domains and their ability to interact with different extracellular substrates and/or cell surface receptors. The expression of these proteins is low in adults, however, gets upregulated following injuries, inflammation, and during tumor growth. The marked elevation in the expression of these proteins during atherosclerosis suggests a positive association between their expression and atherosclerotic lesion formation. The role of matricellular proteins in atherosclerosis development has remained an area of research interest in the last two decades and studies revealed these proteins as important players in governing vascular function, remodelling, and plaque formation. Despite extensive research, many aspects of the matrix protein biology in atherosclerosis are still unknown and future studies are required to investigate whether targeting pathways stimulated by these proteins represent viable therapeutic approaches for patients with atherosclerotic vascular diseases. This review summarizes the characteristics of distinct matricellular proteins, discusses the available literature on the involvement of matrix proteins in the pathogenesis of atherosclerosis and suggests new avenues for future research.
Keywords: Atherosclerosis, matricellular proteins, inflammation, vascular smooth muscle cells, macrophages
1. Introduction
Although great progress has been made in the field of medicine, cardiovascular disease (CVD) remains the leading cause of morbidity and mortality globally [1]. Approximately, 19 million deaths worldwide were associated with CVD in the year 2020, which indicates an almost 18.7% rise in CVD-related mortalities in comparison to the year 2010 [2]. In the year 2020, between the age of 30–79 years, approximately 28% people (over a billion) had abnormal carotid intima-media thickness of 1 mm or above, and 21% people (approximately 816 million) and 1.5% individuals (approximately 58 million) were detected with carotid plaques and carotid stenosis, respectively. The incidence of these pathologies increases with age, and men are more susceptible to developing these diseases than women [3]. The World Health Organization in 2019 predicted that by 2030, almost 23.6 million people will die annually due to CVD.
The underlying cause of the majority of CVD is atherosclerosis, in which lipid accumulation takes place in the middle- and large-sized arteries. This lipid accumulation causes inflammation that ultimately may lead to clinical complications, myocardial infarction (MI), and stroke. Structurally, blood vessels are made up of three layers namely tunica intima, tunica media, and tunica adventitia. Tunica intima, the innermost layer, is constituted by a single layer of endothelial cells that facilitate the frictionless flow of blood, while tunica media is made up of elastic and vascular smooth muscle cells (VSMCs) that regulate the diameter of blood vessels [4]. Adventitial fibroblasts, immune cells, blood capillaries, and lymphatic vessels intricated together with ECM form tunica adventitia [5]. Atherosclerosis is characterized by intimal thickening and the formation of plaques, particularly at sites with endothelial cell injury and disturbed laminar flow [6]. It is mainly affected by the nature of the blood flow, as the regions that are subjected to laminar shear stress are protected from atherosclerosis and endothelial cells present at those sites have higher expression of atheroprotective genes [7]. Various inflammatory factors interact with vascular cells like endothelial cells, smooth muscle cells (SMCs), adventitial fibroblasts, resident macrophages, etc., and drive the progression of this devastating disease [8]. The abnormalities in the ECM structure and cellular behaviour lead to different vascular modalities. Matricellular proteins, a family of non-structural proteins that regulate the function of various ECM proteins, have emerged as important players in regulating vascular structure and function [9]. Over the past years, the involvement of various matricellular proteins in atherosclerosis development has been investigated. In this review, we will discuss the types of various matricellular proteins and summarize their role in the pathogenesis of atherosclerosis.
2. Development of atherosclerosis
The formation of fatty streaks is the first step in the development of atherosclerosis. It begins in the early ages usually in childhood and adolescence. In the subendothelial space and underlying smooth muscles, smaller cholesterol crystals start to deposit which leads to the initiation of plaque formation. Damage to the endothelial layer of arteries due to high blood pressure, smoking, elevated levels of circulating lipoproteins and diabetes makes the intimal layer leaky, which promotes the transport of plasma low-density lipoprotein (LDL) and TG-rich lipoproteins to the subendothelial space either by trans-endothelial transport or diffusion at cell-cell junctions [10, 11] and these lipids undergo oxidation. This leads to the secretion of various proinflammatory molecules and increased expression of adhesion molecules on endothelial cells, which attract immune cells including T cells, neutrophils, monocytes, and mast cells, etc. [12, 13]. Monocytes bind to activated endothelial cells, transmigrate across the intima, and get differentiated into macrophages. These macrophages phagocytose lipids and become cholesterol-lengorged macrophages or foam cells. Further, in response to proinflammatory molecules and chemokines, VSMCs in the media transform from a contractile to a proliferative state and migrate into the intima, and subendothelial space, which leads to the formation of bulge resulting in the reduction of blood flow [14]. The continuous accumulation and coalescence of lipids lead to cell apoptosis and necrosis, which distorts and later disrupts the normal structure of intima. Failure to remove apoptotic cells results in the formation of lipid-rich necrotic cores, which is covered by fibrous cap [15]. In advanced atheroma, unchecked activity of proteolytic enzymes causes the weakening of the fibrous cap at certain places. This increases the chances of plaque rupture, thrombus formation, and blockage of blood supply to the heart or brain [15, 16]. Laboratory studies and clinical observations have greatly advanced our knowledge regarding the mechanisms involved in the pathogenesis of atherosclerosis. The pathogenesis of atherosclerosis has been reviewed earlier [17–20].
3. Matricellular proteins
The historic concept of “matricellular” was first given by Bornstein et al. while working on two novel proteins namely thrombospondin 1 (THBS1) and SPARC, which later became prototype members of this family known as matricellular proteins [21]. The term “matricellular” was coined in the year 1995 and consists of proteins that are a part of the ECM. ECM acts as a critical niche that regulates various cellular processes including survival, proliferation and migration, and thus contributes to the fundamental physiologic processes such as development, tissue homeostasis, and tissue remodeling [22]. This dynamic nature of ECM is regulated by secretory non-structural matricellular proteins in contrast to the classical ECM proteins such as collagens, fibronectin and laminin, etc., that play structural roles [23, 24]. Matricellular proteins modulate various cellular functions via interacting with cell surface receptors, proteases, hormones, and cell-matrix [25]. These proteins contain domains, which can enzymatically alter matrix components, modulate or sequester the activities of various growth factors. They are known to serve by bridging the functional gap between growth factors, proteases, cytokines, and macromolecules, and eventually function as modulators and adaptors of cell-matrix interactions [26–28]. The members of this family include thrombospondins, CCN proteins, SPARC, periostin, osteopontin, tenascins, etc. [29–32].
4. Matricellular proteins in atherosclerosis
Most of the matricellular proteins have higher expression during embryogenesis, which diminishes abruptly after birth and becomes low to absent during normal adult life. Their expression again upregulates during tumor growth, vascular pathologies and after tissue injury [31], suggesting their essential role in the migration and proliferation of malignant cells, wound healing, and ECM remodeling. In this section, we will describe the scientific evidence for the presence and function of various matricellular proteins involved in the pathogenesis of atherosclerosis (Table 1 and Fig. 1).
Table 1:
Studies investigating the role of matricellular proteins in atherosclerosis.
| Protein | Role | Study type | Cell/animal model type | Diet// concentration | References |
|---|---|---|---|---|---|
| THBS1 |
|
In vitro | Human aortic SMCs | 5 μg/mL | [228] |
| In vitro | Bovine pulmonary artery SMCs | 100 nM | [229] | ||
|
In vitro | Bovine aortic endothelial cells | 2.2 nM | [64, 67, 230, 231] | |
| In vitro | Human umbilical vein endothelial cells | 100 pM | [231] | ||
|
In vitro | Human aortic SMCs | 2.2 nM | [64] | |
|
In vitro | Human aortic SMCs | 2.2 nM | [70, 232, 233] | |
| Rat aortic SMCs | 2.2 nM | ||||
|
In vivo | Apoe−/− and Apoe−/−/Thbs1−/− mice | Normocholesterolemic chow diet Murine recombinant leptin (5 μg/g body weight, 3 weeks before sacrifice) |
[58] | |
|
In vivo | Apoe−/− and Apoe−/−/Thbs1−/− mice | Normocholesterolemic chow diet (6 months and 9 months) | [57] | |
|
In vivo | Thbs1−/− mice | Normal chow diet (3 weeks) | [74] | |
| COMP |
|
In vitro | Human arterial SMCs | 20 μg/mL | [86] |
|
In vitro | Bovine aortic SMCs | COMP overexpressed by using adeno associated virus and silenced by using SiRNA | [94] | |
|
|||||
|
In vitro | Rat aortic SMCs | Comp overexpressed by using adeno associated virus and silenced by using SiRNA | [49] | |
|
In vivo | Comp−/− mice | Regular chow diet and CaCl2 | [234] | |
|
In vivo | Apoe−/− and Comp−/−/Apoe−/− mice | High fat diet and periadventitial collar injury | [87] | |
| CCN1 |
|
In vitro | Human umbilical vein endothelial cells | 1, 10 & 100 ng/ mL | [235] |
|
In vitro | Human coronary arterial endothelial cells | CCN1 overexpression was done by treating cells with 0.5 μg of mouse CCN1 cDNA in the presence of Lipofectamine® (Invitrogen). CCN1 silencing was done by using SiRNA | [125] | |
|
In vitro | Rat aortic VSMCs | 0.5–20 μg/mL | [120] | |
|
In vitro | Murine macrophages | 1.25, 2.5, 5, and 10 ng/mL | [123] | |
|
In vitro | Splenic macrophage cell line I-13.35 | 10 μg/mL | [236] | |
|
In vivo | Apoe−/− mice | Regular chow diet and intraperitoneal injections of CCN1 (10 μg/day/kg body weight, 4 weeks) | [123] | |
|
In vivo | C57BL/6 wild type mice | Regular chow diet | [235] | |
|
In vivo | Apoe−/− mice | High fat diet (8 weeks and 16 weeks) | [125] | |
|
In vivo | Sprague–Dawley rats | Standard lab diet | [120] | |
| RSPO2 |
|
In vitro | Human dermal lymphatic endothelial cells | 100 ng/mL | [159] |
| In vivo | Apoe−/− mice | Western diet and periadventitial Lgr4-ECD (Rspo2’s decoy receptor) | |||
| Osteopontin |
|
Clinical | Human aorta samples | - | [237] |
|
Clinical | Human blood samples | - | [238] | |
|
In vivo | Apoe−/−/Ldlr−/− /Spp1−/− triple knockout mice | High fat diet | [239] | |
|
In vivo | Apoe−/−/Spp1−/− mice (36-week-old) | Normal chow diet | [145] | |
| Vitronectin |
|
Clinical | Human blood samples | - | [177] |
|
In vitro | Human aortic SMCs | [179] | ||
| Tenascin C |
|
Clinical | human coronary and inteinal mammary arteries | - | [200] |
|
Clinical | Hman aorta samples and CATHGEN cardiovascular study | - | [240] | |
|
In vivo | Apoe−/−/Tnc−/− mice | High fat diet | [205] | |
| Galectin 1 |
|
Clinical | Human blood samples | - | [220] |
|
In vivo | pAAV/D377Y-mPCSK9 injected Lgals1−/− mice | High fat diet | [225] | |
|
In vitro | Human peripheral blood mononuclear cells | 10 μg/mL | [241] |
Figure 1: Role of matricellular proteins in the development of atherosclerosis.
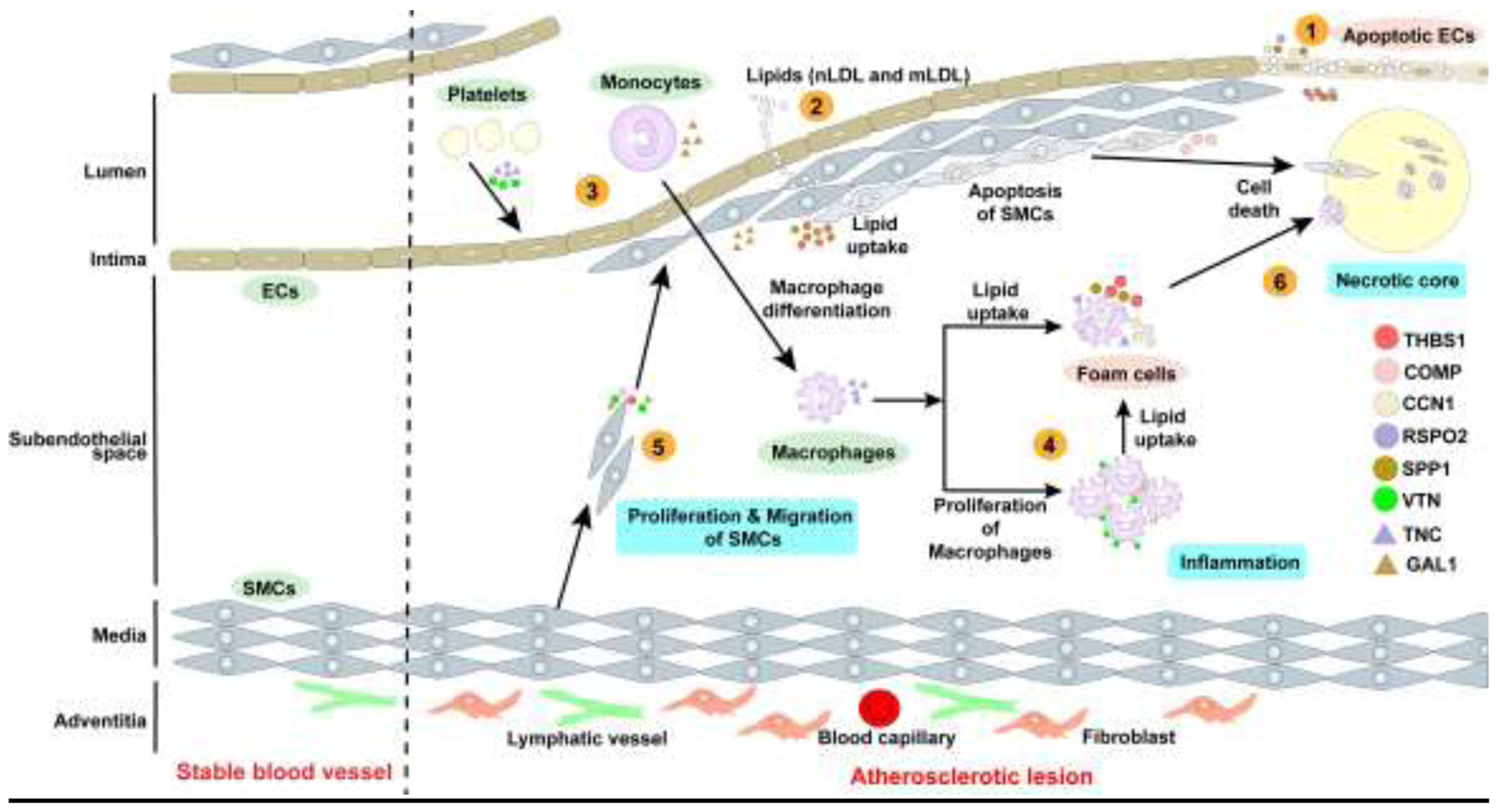
Matricellular proteins play roles in various stages of atherosclerosis development, which include: 1. Endothelial damage - Damage to endothelial cells induces endothelial cell inflammation and promotes adhesion and transmigration of monocytes across intima. 2. Lipoprotein entry into subendothelial space - Leaky intima promotes the transport of plasma LDL to the subendothelial space, where LDL particles undergo oxidative and acetylated modifications. 3. Leukocyte recruitment- Endothelial cells with increased expression of adhesion molecules release chemo-attractants which attract monocytes and T lymphocytes. 4. Foam cell formation- Monocytes differentiate into macrophages and engulf native and modified LDL and become foam cells. 5. Plaque progression-Contractile VSMCs in the media dedifferentiate into a synthetic state and migrate into the intima and subendothelial space to form a fibrous cap. 6. Necrotic core formation-Accumulation and coalescence of lipids lead to cell apoptosis and necrosis. Failure to remove apoptotic cells results in the formation of lipid-rich necrotic core.
4.1. Thrombospondins (THBSs)
In the year 1978, Lawler et al. characterized, decoded the microscopic appearance and the subcellular locations of THBS proteins [33]. The THBS family consists of the 5 subtypes viz THBS1 to 5 based on their molecular architecture and oligomerization status, which are encoded by five distinct genes namely THBS1 to 5 [34]. THBS1 is the most studied among all THBS proteins and plays significant roles in inflammatory responses, platelet aggregation, regulation of angiogenesis during tumor growth and wound healing. Most of these properties are also shared by THBS2 [35, 36]. The last member, THBS5 also called as cartilage oligomeric matrix protein (COMP) is involved in chondrocyte differentiation, attachment and in assembling cartilage ECM complex [37]. THBS1 and THBS2 function by binding to various receptors including syndecans [38, 39], LRP-1 [39], CD36 [40], CD47 [41], calreticulin [42] and integrins (α9β1, α6β1, αvβ3 and αIIββ3) [43–47]. On the other hand, THBS4 binds to CD44 [48] and THBS5 modulates cellular processes via binding to integrins (α7β1, αvβ3, α5β1, α5β3) [49, 50] and CD47 [51].
4.1.1. Thrombospondin 1 (THBS1):
THBS1 levels are low in healthy blood vessels; however, elevated THBS1 expression has been associated with CVD including injury-induced neointima formation and atherosclerosis [52–54]. After vascular injury, the expression of Thbs1 was found to be elevated in diabetic rats [55]. Consistently, the blockade of Thbs1-mediated signaling increased re-endothelialization and decreased neointima formation in the carotid arteries of rats following balloon-induced injury [56]. These studies have pointed out that inhibition of Thbs1-mediated signaling may be therapeutically beneficial in vascular injury-induced neointima formation. On the other hand, Apoe−/−/Thbs1−/− double knockout mice were observed to have increased atherosclerotic plaque maturation compared with control mice; however, this increase was observed only in the advanced stage of atherosclerosis [57]. In the initial stage, loss of Thbs1 reduced atherosclerotic lesion formation, while in the advanced stage, Thbs1 deletion was associated with elevated inflammation, defective phagocytosis and enhanced ECM remodeling that caused accelerated plaque maturation and necrosis [57].
Ganguly et al. reported reduced atherosclerotic plaque area and decreased collagen accumulation in aortic roots of Apoe−/−/Thbs1−/− double knockout mice compared with Apoe−/− mice after leptin stimulation [58]. Further, the deletion of Thbs1 in mice significantly decreased disturbed flow-stimulated carotid artery stiffness in comparison to wild-type controls, indicating that disturbed flow promotes arterial stiffening through Thbs1-mediated signal transduction [59]. Moreover, hypoxia as found in atherosclerotic arteries has been shown to upregulate THBS1 expression in coronary artery SMCs, which in turn promotes their migration. This migration-stimulating effect of hypoxia was inhibited with THBS1-neutralizing antibody treatment, suggesting the involvement of THBS1 in the pathogenesis of atherosclerosis [60, 61]. Roth et al. demonstrated that THBS1-neutralizing antibody blocks PDGF- and cholesterol-stimulated aortic SMC proliferation, hinting the role of THBS1 in PDGF- and cholesterol-stimulated aortic SMC proliferation [62]. THBS has also been shown to bind with very-low-density lipoprotein (VLDL), which may facilitate THBS and VLDL incorporation into nascent atherosclerotic plaques [63].
THBS1 induces its downstream effects via binding to its cognate receptor CD47 (also known as the integrin-associated receptor) [41] and governs leukocyte function, vascular resistance, and intracellular signal transduction in endothelial cells and VSMCs [64]. It inhibits endothelial nitric oxide (NO) production, regulates vascular tone, and maintains systemic blood and cardiac dynamics under stress [65–68]. In addition, THBS1-CD47 signaling regulates thrombosis/hemostasis, immune responses, and mitochondrial function [69]. In VSMCs, THBS1-mediated CD47 activation stimulates Nox1-derived reactive oxygen species production, which inhibits VSMC-dependent vasorelaxation and induces vascular dysfunction [70]. It also enhances fluid-phase phagocytosis of LDL by macrophages and causes foam cell formation, a key characteristic of atherogenesis (Fig. 2a). In macrophages, THBS1-induced Nox1 activation stimulates dephosphorylation of actin-binding protein cofilin leading to cytoskeletal rearrangements and increased LDL uptake [71]. LIM-kinases and testicular protein kinases are known to cause cofilin inactivation by phosphorylating it at Ser-3 residue. On the other hand, slingshot protein phosphatases (SSH) dephosphorylate cofilin to induce its activation and regulate actin polymerization [72]. Contrary to the effects of THBS1 on cofilin activation in macrophages, VSMC-specific deletion of Thbs1 in mice reduced cofilin phosphorylation via upregulating SSH1 protein expression, improved mechanotransduction, restored elastic lamina smooth muscle cell connections and decreased thoracic aortic aneurysm formation, suggesting the detrimental role of Thbs1 in the pathogenesis of thoracic aortic aneurysm [73]. In another study, Yamashiro et al. reported upregulated Thbs1 expression in VSMCs following mechanical stress and demonstrated that Thbs1 via binding to cell surface integrin αvβ1 aids in the maturation of the focal adhesion-actin complex, reduces small GTPase Rap2 activity, promotes nuclear shutting of Yes-associated protein and stimulates downstream signaling [74]. Deletion of Thbs1 in mice inhibited this signaling cascade, led to impaired aortic remodeling in response to transverse aortic constriction-induced pressure overload and inhibited neointima formation upon complete left carotid artery (LCA) ligation [74]. These studies indicate the important role of Thbs1 in the pathogenesis of vascular pathologies via dysregulating actin cytoskeleton remodeling. It has been shown that exposure of disturbed flow to endothelial cells causes endothelial-to-mesenchymal transition (EndoMT), which promotes atherosclerosis development [75, 76]. Elevated Thbs1 levels are also linked with EndoMT following complete LCA ligation suggesting the potential role of Thbs1 in EndoMT-mediated atherogenesis [77]. A recent study by Singla et al. reported that THBS1 inhibits lymphangiogenesis by CD47-mediated mechanisms and lymphatic endothelial cell-specific Cd47 deletion promotes arterial lymphatic vessel formation and attenuates atherosclerosis [78]. These data suggest the role of Thbs1-Cd47 axis in reducing removal of cholesterol from the arterial wall via adventitial lymphatics and promoting atherogenesis.
Figure 2:
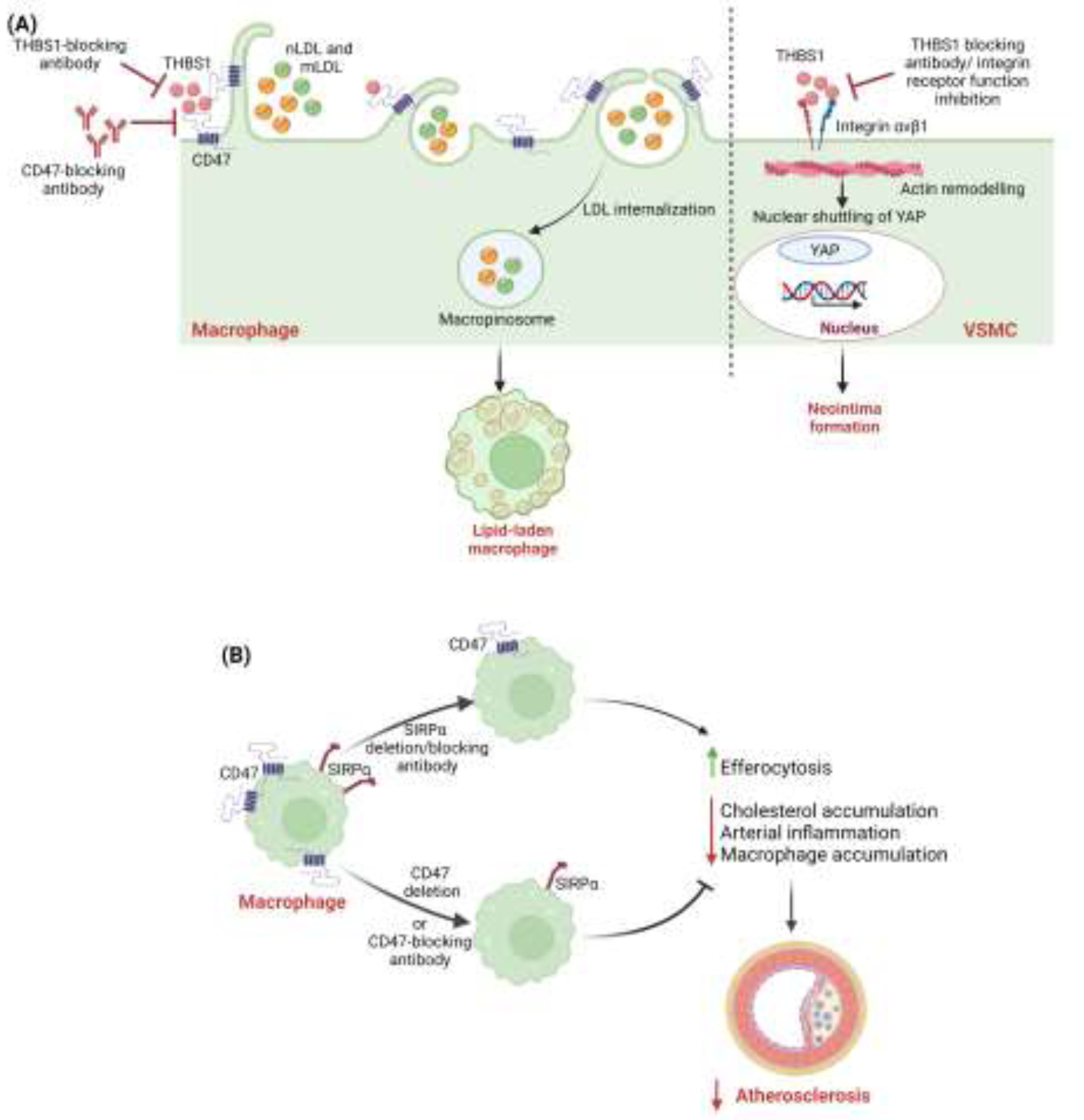
(A) THBS1 via CD47 activation induces foam cell formation in macrophages. THBS1 binds with CD47 receptors in macrophages, stimulates receptor-independent macropinocytic internalization of native LDL (nLDL) and modified LDL (mLDL), and contributes to foam cell formation. Modulation of THBS1/CD47 signaling may work as therapeutic target for treating atherosclerosis. THBS1 secreted in response to mechanical stress, binds to VSMC integrin αvβ1 which helps in the maturation of the focal adhesion–actin complex, mediating activation of nuclear shuttling of YAP and ultimately leading to neointima formation. THBS1 targeted therapies using THBS1-blocking antibodies or integrin receptor function inhibitors may also serve as a potential therapeutic target for treating atherosclerosis. (B) Differential effects of myeloid cell-Sirpa and -Cd47 deletion on macrophage efferocytosis and atherosclerosis. Loss of Sirpa signaling in macrophages stimulates efferocytosis, reduces cholesterol accumulation, promotes lipid efflux, and suppresses atherosclerosis. Conversely, myeloid cell-Cd47 deletion inhibits efferocytosis, impairs cholesterol efflux, augments cellular inflammation, and promotes plaque formation.
In addition to serving as a receptor for THBS1, CD47 binds with signal regulatory protein α (SIRPα) present on phagocytic cells including macrophages and dendritic cells [79]. CD47 is ubiquitously expressed on viable cells and interacts with phagocyte SIRPα to prevent the efferocytic removal of live cells [80]. However, the surface expression of CD47 downregulates on apoptotic cells allowing efferocytosis of those cells by phagocytes and inhibits necrosis-induced inflammation. This CD47-SIRPα signaling is an important immune checkpoint that helps to maintain tissue integrity and homeostasis [81]. Recently, Singla et al. demonstrated the differential effects of myeloid cell-specific Sirpa and Cd47 deficiencies on atherosclerosis development [82]. Loss of myeloid cell Sirpa signaling stimulated efferocytosis, enhanced lipid efflux, decreased cholesterol accumulation, and attenuated oxidized LDL-induced inflammation in macrophages. Further, it inhibited atherosclerotic lesion formation and necrotic core formation in hypercholesterolemic mice. Conversely, deletion of Cd47 in myeloid cells impaired efferocytosis and cholesterol efflux, augmented cellular inflammation, and promoted plaque formation [82] (Fig. 2b). Thus, interaction triangle of Thbs1-Cd47-Sirpα has several pathogenic consequences that promote atherogenesis and severity of disease. Thbs1 also mediates its effects by interaction with cell surface receptor Cd36 [83]. Thbs1 through Cd36-mediated signaling enhances proliferation of VSMCs via upregulating the expression of cell cycle promoter cyclin A. Ablation of Cd36 in VSMCs significantly decreases cell proliferation and neointimal hyperplasia in injured carotid arteries of Apoe−/−/Cd36−/− mice compared with Apoe−/− mice [84].
4.1.2. Thrombospondin 5 /cartilage oligomeric matrix protein (COMP):
Hedbom et al. reported the presence of COMP in bovine and rodent cartilaginous tissue [85]. Later, its expression was detected in cultured human VSMCs and also found localized in SMCs present in the medial layer of non-atherosclerotic arteries as well as in intimal SMCs of human atherosclerotic and restenotic arteries [86]. It aids in the maintenance of VSMC contractile phenotype and mediates protective effects against injury-induced VSMC dedifferentiation and neointima formation in Sprague-Dawley rats through interaction with α7β1 integrin [49]. Comp expression was also found in both inflammatory and/or fibrous atherosclerotic plaques in mice [87]. Additionally, Riessen et al. reported that VSMCs adhere strongly to COMP-coated surfaces, and it aids in the migration of VSMCs [86]. These observations suggest the possible role of COMP in vasculogenesis and vascular diseases like atherosclerosis via regulating SMC migration and adhesion. Hultman et al. in their recent study showed significantly increased COMP expression in atherosclerotic plaques from symptomatic patients compared with lesions from asymptomatic patients [88]. Further, COMP levels were positively associated with plaque lipid- and CD68-positive areas, but negatively with collagen, elastin, and SMC contents. A study has demonstrated elevated circulating levels of COMP in patients with coronary heart disease compared with control patients and observed a positive correlation between COMP levels and coronary artery calcium score, suggesting serum COMP levels can serve as a screening biomarker for coronary calcification [89]. Sandstedt et al. reported the presence of COMP neoepitope, a highly conserved sequence across several species, in carotid arteries of atherosclerotic patients. COMP neoepitope levels in plasma and endarterectomy samples were found significantly increased in comparison to control subjects. They also detected its presence in SMCs, endothelial cells, and foam cells in carotid stenosis and suggested that degradation of COMP and generation of a specific COMP fragment COMP neoepitope may be associated with atherosclerosis progression [90]. These findings indicate an association of arterial COMP expression with plaque vulnerability in humans.
Contrarily, Fu et al. reported that Comp deficiency in mice leads to increased plaque size accompanied by increased calcification. Apoe−/−/Comp−/− mice fed with a chow diet for 12 months had enhanced atherosclerotic calcification in the innominate arteries than Apoe null control mice. Furthermore, microarray profiling of wild-type and Comp knockout macrophages revealed that Comp-deficient macrophages have atherogenic and osteogenic characteristics. The authors concluded that Comp deficiency aggravates atherosclerotic calcification by switching macrophage phenotype toward the atherogenic and osteogenic type via integrin β3 activation [91]. Additional analysis demonstrated significantly decreased COMP expression in aortic samples of aortic aneurysm patients compared with controls [92]. COMP along with blood flow fine-tunes endothelial homeostasis, as it inhibits disturbed flow-induced endothelial activation by interacting with integrin α5. Under normal as well as pathological conditions including partially ligated carotid arteries mouse models, increased endothelial cell activation was observed in the aortic arch of Comp−/− mice compared with control mice. Moreover, Comp-derived peptidomimetics (CCPep24) mimicking a specific Comp-integrin α5 interaction, protected against endothelial cell activation and atherogenesis in vivo [93]. Further, Du et al. also reported Comp as a novel endogenous inhibitor of vascular calcification employing two different rat models of vascular calcification. They revealed that its inhibitory effect is exerted partially through the direct binding of its C-terminal domain to bone morphogenetic protein-2 (Bmp-2), which prevents Bmp-2 binding to its receptor and consequently inhibits Bmp-2-induced osteogenic signaling [94]. COMP also plays a catalytic function in collagen-microfibril assembly as it brings collagen molecules together, and aids in the formation of mature fibrils [95]. It has been observed that the size of inflammatory lesions significantly decreases with defective collagen assembly [96]. Having a role in the assembly of collagen fibers, COMP reduced expression accordingly affects the structure and growth of atherosclerotic lesions and its elevated levels with respect to collagen lead to the impairment of fibrillogenesis [97]. Consistently, Bond et al. showed larger plaques with higher collagen content in brachiocephalic arteries of Comp-deficient Apoe−/− mice [87]. The changes in morphology in brachiocephalic artery plaques in mice lacking Comp could be a consequence of altered collagen metabolism [87]. Consequently, the risk of rupturing the atherosclerotic plaque increases with the degradation of collagen fibers in the vessel wall. Thus, COMP stands to be a capable molecule in collagen fibrillogenesis and shows a significant anti-atherogenic effect.
4.2. CCN Family
This family of matricellular proteins got its acronym of CCN from the first three discovered members of the family known as Connective Tissue Growth factor, Cysteine-Rich Protein and Nephroblastoma Overexpressed Gene. It consists of 6 multifunctional proteins named CCN1 to CCN6 [98]. CCN proteins regulate various cellular processes including cell adhesion, migration, chemotaxis, cell survival [99], skeletal formation and further development [99, 100], and cell proliferation [101]. These proteins drive angiogenesis by manipulating cell communication network, which integrate most of growth factors and proteins toward new vessel formation [102]. CCN1 induces its cellular effects by binding to cell surface receptors including syndecan 4 [103], αMβ2 and α6β1 [104], CCN2 binds to heparan sulfate proteoglycan [105], tropomyosin-related kinase A [106], LRP-1 [107]; CCN3 acts by binding to notch [108] and integrins (α6β1 and αvβ5). One of the important functions of CCNs appears to be as an adaptor molecule, binding growth factors, such as vascular endothelial growth factor (VEGF) and transforming growth factor-β (TGF-β) and shuttling them near cell surface via second binding partners, namely integrins or heparin sulfate proteoglycans. CCNs may directly bind to cell surface and initiate intracellular signaling cascades, which are critical for cellular growth and mobility during vascular development and aid in the progression of vascular diseases such as atherosclerosis and restenosis [109].
Earlier studies have shown upregulated CCN1 expression during inflammation, tissue repair, and wound healing [110]. CCN1 is considered to be an important player in inflammatory diseases including rheumatoid arthritis, bacterial and viral infections, vascular injury, and colon inflammation [111]. A case-control study was done to evaluate serum levels of CCN1 in rheumatoid arthritis patients, and then examined correlation among serum CCN1 levels, carotid intima-media thickness, and predisposition to subclinical carotid atherosclerosis. Serum CCN1 levels were found significantly elevated in rheumatoid arthritis patients compared with healthy controls and observed positively correlated with carotid intima-media thickness [112]. Similarly, a significant positive correlation between CCN1 expression and myocardial infarct size was found among ST-elevation MI patients [113]. Additionally, CCN1 levels were determined in type 2 diabetic patients and related to the occurrence of peripheral artery disease. The results demonstrated that patients with more advanced peripheral artery disease had significantly higher CCN1 levels and these levels were positively associated with disease severity [114]. Further, it has been shown associated with fibroblast migration and monocyte adhesion [110, 115], myocardial angiogenesis and remodeling of the vascular bed following myocardial injury [116].
CCN1 expression in normal endothelial cells, VSMCs and fibroblasts is very low [117], however, in atherosclerotic plaques particularly in neovascularized regions, its expression is highly elevated [118]. Additional studies reported increased CCN1 levels in human arteriosclerotic carotid and coronary arteries as well as arteriosclerotic aortas of Apoe-deficient mice [115, 119]. In a rat carotid artery injury model, rapid elevation in the expression of this protein was observed in the media and neointima regions, and Ccn1 knockdown suppressed balloon injury-induced neointima formation [120]. These observations are further supported by the findings that CCN1 immunoreactivity is present in the heart tissues of patients who died of sudden cardiac death and revealed that CCN1 expression significantly associates with myocardial ischemia, interstitial edema and atheromatosis of coronary arteries [121]. Further, immunological studies identified elevated CCN1 expression in neointimal lesions and suggested a possible role of CCN1 in lysophosphatidic acid-induced vascular neointima formation [122]. Zhoa et al showed predominant expression of Ccn1 in Apoe−/− mice aortic foamy macrophages and reported worsened hyperlipidemia, enhanced systemic inflammation, and augmented atherosclerosis in Ccn1-treated Apoe−/− mice compared with control mice. Additionally, Ccn1 treatment impaired macrophage reverse cholesterol transport capacity as well as reduced the expression of proteins associated with cholesterol clearance including ABCG5, ABCG8, liver X receptor α, cholesterol 7α-hydrolase and LDL receptor in mouse liver and exacerbated hepatic lipid accumulation [123].
Disturbed blood flow-induced CCN1 expression in endothelial cells has been shown to regulate endothelial cell functional phenotypes. CCN1-α6β1 mediates shear stress-induced NF-κβ activation and expression of atheroprone genes in endothelial cells. Activation of NF-κβ by CCN1/α6β1 works through a positive-feedback loop and enhances the production of CCN1 and α6β1 [124]. Besides, a mutation in Ccn1 gene that prevents binding of Ccn1 with its receptor integrin α6β1 attenuates partial left carotid artery ligation-stimulated atherosclerosis in Apoe−/− mice. During atherosclerosis development, Ccn1 also regulates tumor necrosis factor-α (TNF-α)-induced vascular endothelial cell apoptosis via p53 and NF-κβ activation [125]. Thus, CCN1 is a critical pathophysiological regulator mediating the endothelial dysfunction induced by disturbed blood flow and can be targeted for therapeutic restoration of endothelial function to prevent atherosclerosis [126].
4.3. Secreted phosphoprotein 1 (SPP1)
Secreted Phosphoprotein 1 is also called as Osteopontin. It exists both as a component of the ECM and a soluble cytokine. It regulates bone formation and ECM mineralization. It is considered as a strong predictor of calcification, biomineralization, vascular diseases like atherosclerosis and is also a prognostic indicator for inflammatory heart diseases [127, 128]. SPP1 interacts with cell surface integrins [αv (β1, β3, β5, β6)] and modulates a variety of biological processes such as migration, cell adhesion and survival, ECM remodeling and regulates vascular calcification [129, 130]. It is also highly expressed in cancer models and has shown a possibility of enhancing cancer cell survival [131].
Previous studies have investigated the role of SPP1 in atherosclerosis and examined the effects of its overexpression/deficiency on atherosclerotic lesion formation [132, 133]. Elevated SPP1 levels were observed in patients with coronary artery disease (CAD) [134]. In a pilot study, plasma SPP1 concentrations were found significantly higher in patients with confirmed presence of rupture plaque in comparison to healthy individuals [135]. A significant association was also detected between plasma SPP1 levels and increased risks of adverse outcomes after ischemic stroke [136]. SPP1 expression is localized in atherosclerotic lesions, especially in macrophages- and foam cell-positive areas, suggesting its role in the development and progression of atherosclerosis and vascular remodeling [133, 137, 138]. Under normal physiological circumstances, the circulating/tissue SPP1 levels are low but enough to maintain normal arterial physiology [139, 140].
It is now well known that SPP1 acts as a physiological inhibitor of vascular calcification. Vascular cell types comprising endothelial cells, VSMCs, and macrophages are the major source of SPP1 in the arterial wall [141]. Upregulation of Spp1 was observed in response to ischemic injury in the murine model of hindlimb ischemia [142], that in turn promotes cell adhesion, proliferation, migration, and survival assisting in the healing process [143, 144]. Matsui et al. using Spp1 knockout mice on Apoe null background showed reduced atherosclerotic lesion areas in female heterozygous (Spp1+/−) and homozygous (Spp1−/−) mice compared with wild-type (Spp1+/+) mice. However, no differences in atherosclerosis were observed in male mice demonstrating the sex-specific role of Spp1 in atherosclerotic lesion formation. Additionally, significantly increased vascular calcification was observed with Spp1 deletion in male 60-week-old mice indicating an inhibitory effect of Spp1 on vascular calcification [145]. Altogether, these findings suggest that SPP1 plays a promoting effect on atherosclerosis development and an inhibitory effect in vascular calcification. Chiba et al. examined the effects of Spp1 overexpression in hematopoietic cells on atherosclerotic lesion formation and reported that Spp1 overexpression in lymphoid tissues associates with an increase in aortic lesion size in atherogenic diet-fed mice. Further, they showed significantly higher expression of Spp1 in lesional foamy macrophages of Spp1 transgenic mice compared with such macrophages in control mice [133]. Additional experiments revealed that higher Spp1 levels in atherosclerotic lesions were not due to the deposition of serum Spp1, but mainly due to production of Spp1 by infiltrating macrophages. Consistently, Isoda et al. showed that global overexpression of Spp1 in atherogenic diet-fed mice results in larger atherosclerotic lesions compared with control non-transgenic mice [138].
In diabetic vascular disease, Spp1 secretion was also observed to increase with high glucose concentrations in the rat aortic SMCs in vitro [146]. SPP1 also regulates the recruitment of inflammatory cells and adhesion or migration of foam cells by binding to its receptors such as integrins (avb3, avb1, avb5, and a4b1) or the splice variant of CD44 v3-v6, AT1, or AT2 [147, 148]. A recent study by Yu et al. has reported that Nox4-induced Spp1 expression in VSMCs is partially responsible for AngII-stimulated aortic aneurysm and atherosclerosis [149]. Collectively, SPP1 seems to exert an important role in the formation of atherosclerotic plaque, enhances vascular inflammation, and participates in vasculopathy by increasing proliferation of endothelial and VSMCs [150].
4.4. Roof plate-specific spondins (RSPOs)
RSPOs get their name due to the discovery of RSPO1 in mouse roof plate of neural tube during development. In vertebrates, four types of RSPOs namely RSPO1–4 are identified [151]. RSPOs serve as ligands for various receptors including leucine-rich repeat containing G-protein coupled receptors 4–6 [151, 152], ZNRF3/RNF43 [153] and heparan sulfate proteoglycan. RSPOs play various physiological and pathophysiological roles. RSPOs potently bind with leucine-rich repeat-containing G protein-coupled receptors (LGRs) and prevent the degradation of Wnt receptors, thereby stimulate Wnt-stimulated signaling [154]. Wnt signaling is pivotal for developmental processes, including cell proliferation, differentiation and tissue patterning. The cardiovascular system in healthy adults has little Wnt activity, however, this signaling pathway reactivates during heart or blood vessel pathological conditions [155]. Wnt signaling has gained interest as a potential therapeutic target because of its higher expression in various pathologies [155]. Various studies reported a protective role for Wnt signaling in cholesterol trafficking and its accumulation in tissues, even the arterial wall [156–158].
The role of RSPO2 in atherosclerosis development has been recently reported by Singla et al. [159]. Elevated RSPO2 expression was observed in various cell types present in the different layers of human and mouse atherosclerotic arteries including medial SMCs, intraplaque macrophages and adventitial fibroblasts. Using in vitro and in vivo experiments, the authors revealed the inhibitory role of RSPO2 on VEGFC/VEGFR3-mediated lymphangiogenesis. Further, RSPO2 limited the activation of AKT-eNOS signaling via LGR4-mediated mechanisms. NO signaling has been shown critical for lymphangiogenesis and maintenance of lymphatic function [160], therefore RSPO2 via suppressing NO signaling in lymphatic endothelial cells prevented arterial lymphangiogenesis and impaired arterial cholesterol removal that in turn led to increased atherosclerotic lesion formation [159] (Fig. 3). Interestingly, perivascular application of LGR4-extracellular domain peptide suppressed atherosclerotic lesion formation and increased arterial lymphatic vessel density in hypercholesterolemic mice following partial left carotid artery ligation. Moreover, the authors demonstrated that RSPO2 inhibited Wnt-β-catenin signaling in lymphatic endothelial cells via diminishing NO-mediated nuclear translocation of β-catenin [159] (Fig. 3). On contrary, Carmon et al. reported the induction of Wnt signaling following RSPO2 exposure in other cell types like MDCK, HEK293T and HeLa cells, suggesting the cell-specific effects of RSPO2 treatment on Wnt-β-catenin pathway [161]. Future studies are needed to better understand the roles of RSPOs in regulating atherosclerotic lesion formation, underlying mechanisms, and RSPOs’ cell-specific effects during atherogenesis.
Figure 3: RSPO2 inhibits lymphangiogenesis and contributes to atherosclerosis development.
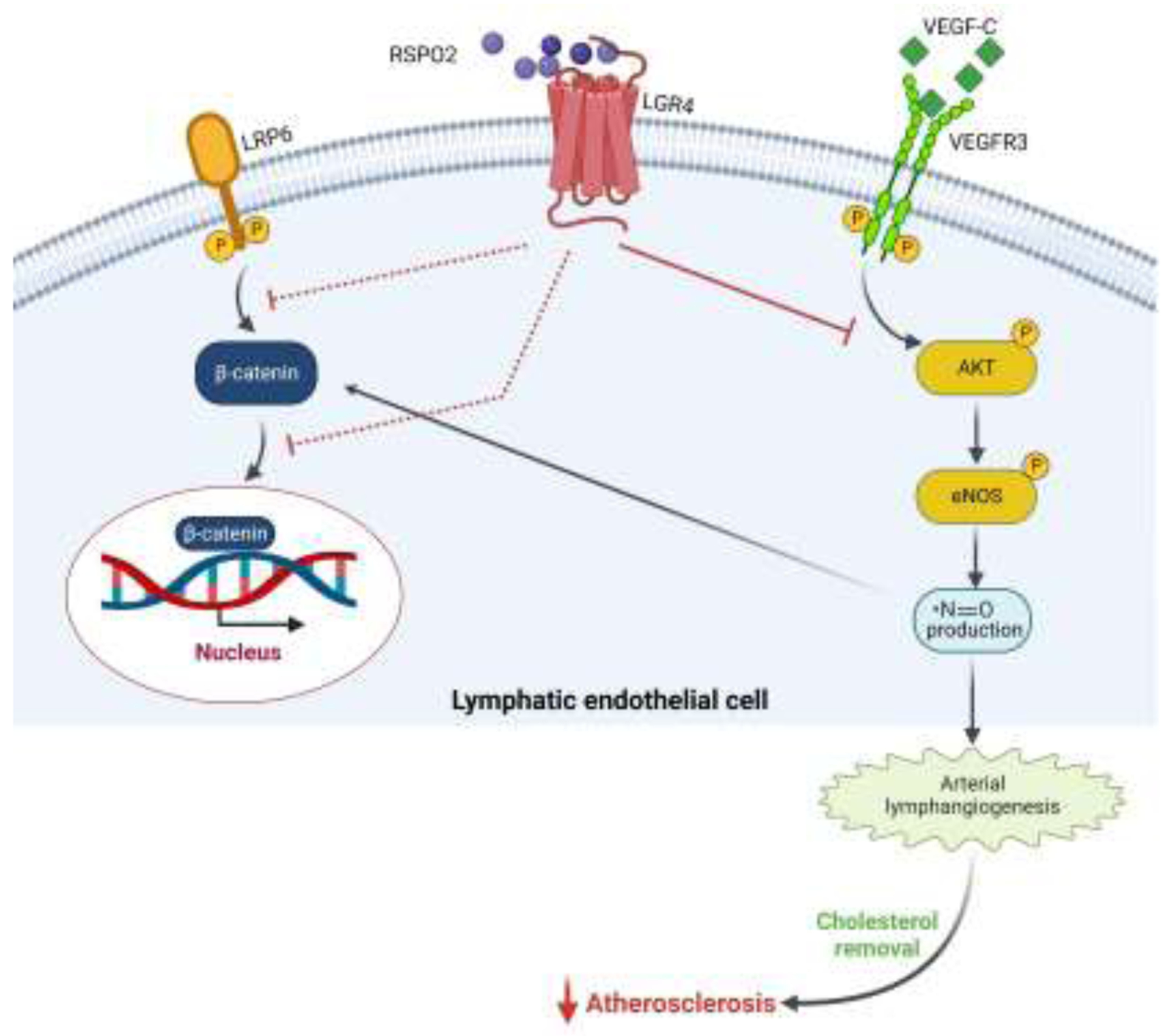
RSPO2 via binding to LGR4 receptors on lymphatic endothelial cells (LEC) hinders VEGF-C-stimulated AKT and eNOS activation, leading to impaired NO production and reduces lymphatic vessel formation. It also inhibits activation of the canonical Wnt-β-catenin pathway in LEC in a NO-dependent manner, thus decreasing lymphatic vessel-mediated LDL drainage from the arterial wall and promoting atherosclerosis.
4.5. Vitronectin (VTN)
VTN is synthesized in the liver and secreted into the plasma. It serves as a ligand for integrins αvβ1, αvβ5, αvβ3 and αIIbβ3 [162]. Like other matricellular proteins, it has a crucial role in various biological processes such as adhesion, angiogenesis and cell migration [163]. It also regulates processes including coagulation, blood fibrinolysis, complement-depended immune responses, pericellular proteolysis, etc. [164–167]. It is found to be accumulated in atherosclerotic plaque [164–167] and mediates platelet aggregation and adhesion at the vascular injury sites [168]. In contrast, reduced levels of VTN in airways of asthmatic and chronic pulmonary disease patients, contributes to the airways remodeling as seen in obstructive airway disorder [169].
Previous data suggest that the liver is a major site of VTN biosynthesis [170]. Normal plasma concentration of VTN ranges from 200 to 400 μg/mL and it constitutes 0.2–0.5% of total plasma protein [171, 172]. Expression of VTN is considered to be related to the development of atherosclerosis. Guettier et al. reported the presence and accumulation of VTN in atherosclerotic plaques [166]. It was speculated that VTN accumulation in atherosclerotic plaques resulted from its diffusion from plasma, where it is released either by activated platelets through damaged endothelium or synthesized by cells involved in atherosclerotic plaque formation [173]. VTN triggers adhesion and aggregation of platelets at the sites of injury in blood vessels, hence considered as a contributor of atherosclerosis and thrombosis [174]. It is prominent player in the development of aortic inflammation and its expression is a useful indicator in the determination of plaque burden and stability [175]. Its role was further verified by quantifying the plasma levels of VTN in patients suffering from CAD, and increased VTN levels were detected in these patients. The plasma level of VTN showed a positive correlation with the severity of the disease [176, 177]. Based on the experimental and clinical evidence, it is now established that VTN plays a very essential role in establishing the initial response to tissue injury and is thus considered as a significant biomarker of atherosclerosis [178].
VTN has been shown to be involved in adhesion and migration of SMCs, neural crest cells, and keratinocytes [179, 180]. Migration of SMCs is a contributor to the intimal thickening during atherogenesis. Interestingly, chemotactic and haptotactic activity of VTN are mediated by its receptor αVβ3 in cultured SMCs [179]. Further, Dufourcq et al. reported that treatment with neutralizing anti-Vtn antibody inhibits VSMC migration and suppresses neointima formation after vascular injury [181]. Although, in rabbits VTN was reported to be localized in atherosclerotic arteries and its role was highlighted in regard to the abnormalities in platelets like platelet activation & aggregation in CAD [182, 183]. Recent findings by Chakravarty et al. have shown a reciprocal relationship of VTN with cholesterol load and unveiled its multi-functional role beyond adhesion function. They reported that the inflammation and plaque progression is associated with systemic deficiency of Vtn in Apoe−/− mice. Imbalance in its levels facilitates the trafficking of the inflammatory cells to the plaque microenvironment, and decreased Vtn expression due to high cholesterol load and aortic inflammation leads to the formation of advanced necrotic atheroma [175].
4.6. Tenascins (TN)
This family of matricellular proteins comprises of four large glycoproteins namely tenascin W (TNW), tenascin C (TNC), tenascin R (TNR) and tenascin X (TNX) [184]. TNC expression is stimulated by various growth factors, mechanical stress and cytokines. Even having significant structural homology, these four members exhibit distinct expression patterns. TNW is mostly restricted to the skeletal system during remodeling or development phases; TNC is expressed in multiple cells and tissues depending on types of stimuli; TNR is restricted to the central nervous system (CNS) and whereas TNX is expressed in connective tissues [185]. These proteins function via interacting with receptors integrin α8β1 [186], IgCAM/contactin [187], myelin associated glycoprotein [188] and heparan sulphate proteoglycan [189].
Among all the four members, TNC is the most studied protein and reported to have a role in various CVD including the promotion of cardiac fibrosis, myocardial hypertrophy [190], cardiac dysfunction [191], and vascular diseases by showing atherogenic effects via induction of TLR4-dependent foam cell formation [192]. Significantly elevated levels of TNC were found in serum samples of CAD patients in comparison to non-CAD patients, and a positive correlation between TNC levels and severity of atherosclerosis was observed [193, 194]. Recently, Gholipour et al. reported increased TNC levels in exosomes isolated from CAD patients than non-CAD patients [195]. In a prospective observational study, higher plasma TNC levels predicted overall and cardiovascular mortality, and were associated with higher occurrence of cardiovascular events in chronic kidney disease patients [196]. Tnc expression was found increased with progression of atherosclerosis in atheroprone Apoe−/− mice [197]. Balloon catheter-induced vascular injury has also been shown to upregulate Tnc expression specifically in neointimal lesions [198]. Moreover, lipopolysaccharide (LPS) treatment induces Tnc expression in THP-1 macrophages in vitro in a dose- and time-dependent manner and it augments LPS-induced foam cell formation [199]. Besides, TNC produced by oxidized LDL-stimulated macrophages increases foam cell formation through TLR4 and scavenger receptor CD36 [192].
TNC expression was specifically detected in the macrophage-rich areas of atherosclerotic plaques [200]. TNC can act as damage-associated molecular patterns to activate macrophages and fibroblasts and induce inflammatory cytokine expression through TLR-4 receptor [201, 202]. Wang et al. demonstrated a higher expression of Tnc and annexin II in atherosclerotic plaque and their interaction facilitated macrophage migration and VEGF expression via Akt, NF-κB and ERK1/2 pathway [203]. In diabetic-acute MI patients, increased TNC expression in coronary artery atherosclerotic lesions was found to be associated with increased TGF-β, macrophage accumulation and TUNEL-positive apoptotic cells [204]. Wallner et al. reported minimal and random TNC expression in fibrotic but lipid-poor atherosclerotic plaques. In contrast, all advanced stage plaques with an organized lipid core or ruptured intimal surface has elevated expression of TNC, which was preferentially concentrated around the lipid core, shoulder regions, and ruptured area of the plaques but not in the fibrous cap. Further, TNC expression was found to be associated with the degree of inflammation present, but not with plaque size [200]. Contrary to this, Wang et al. reported the atheroprotective role of Tnc utilizing atheroprone Apoe−/− mice, in which they showed that Tnc gene deletion leads to upregulation of eotaxin, a chemokine that promotes migration and activation of eosinophils and plasma eotaxin elevation correlates with atherosclerosis development [205]. They unveiled a protective role for Tnc in atherosclerosis and suggested its potential to reduce atherosclerosis, in part by modulating Vcam-1 expression.
4.7. Galectins
Galectins is a family of β-galactoside-binding lectins that have emerged as crucial modulators of inflammatory processes [206]. They are involved in intercellular signaling, cell-cell and cell-to-matrix adhesion, apoptosis, angiogenesis, and innate and adaptive immunity [207]. Galectins are coded by the genes named Lgals. Till date, fifteen different types of galectins have been identified, out of which only two have been included in matricellular family namely galectin 1 (Gal1) and Gal3. Both Gal1 and Gal3 play important roles in regulating immune responses by serving as damage-associated molecular patterns [208, 209]. These proteins have been linked to various diseases such as cornea proliferative vitreoretinopathy [210, 211], Fuch’s endothelial corneal dystrophy [212], age-related macular degeneration [213], diabetic retinopathy [214–216].
Gal1 plays an essential role in cardiovascular pathophysiology by moderating acute and chronic inflammatory responses [217]. For instance, patients suffering from MI, heart failure, or Chagas cardiomyopathy have increased GAL1 expression in cardiomyocytes [218]. Lgals1-deficient mice after acute MI exhibited enhanced cardiac inflammation and worsened ventricular remodeling [219]. Additionally, circulating GAL1 levels associate with the severity of CAD and subsequent occurrence of major adverse cardiovascular events in patients with CAD [220]. Moreover, upregulated GAL1 expression has been reported to associate with stroke outcome in large artery atherosclerotic stroke [221]. Gal1 and Gal3 are the predominant galectins observed in atherosclerotic plaques and have specific spatial and temporal intraplaque expression patterns during atherosclerosis progression [222]. GAL1 serves as an adapter between cells and ECM and modulates adhesion, migration and proliferation of SMCs. Moiseeva et al. demonstrated increased GAL1 expression in proliferating SMCs [223]. Moreover, GAL1 fusion protein stimulated serum-induced DNA synthesis in human SMCs grown on plastic or endogenous ECM, and increased SMC adhesion to ECM. It affected SMC adhesion by interacting with β1 integrin present on cell surface and inducing outside-in signaling [223]. VSMCs-deficient in Lgals1 exhibited greater motility but adhered slower on fibronectin than wild-type control cells. Likewise, their migration was inhibited by a redox-insensitive but activity-preserved Gal1 (CSLgal1) in a glycan-dependent manner [224]. The authors concluded that Gal1 restricts VSMC migration by modulating cell-matrix interaction and focal adhesion turnover, which limits neointimal formation post vascular injury.
Interestingly, Roldán-Montero et al. reported a protective role of Gal1 in pathological vascular disorders through modulation of lipid uptake by macrophages, alterations in mitochondrial function and phenotypic switch of VSMCs. The authors observed reduced expression of Gal1 in advanced human atherosclerosis and abdominal aortic aneurysm. They showed that treatment with recombinant Gal1 prevent both atherosclerosis and abdominal aortic aneurysm in mice, which indicate it as a novel therapeutic target for attenuating the severity of chronic vascular pathologies [225].
5. Conclusions
Atherosclerotic vascular diseases are responsible for one fourth of deaths worldwide. The investigations into its pathogenesis, therapeutics and role of various endogenous factors regulating distinct aspects of atherosclerotic lesion formation are of great medical importance. Discoveries made during the last two decades have significantly advanced our understanding of the roles of various matricellular proteins in atherosclerosis. The elevated levels of these proteins significantly correlate with atherosclerosis development and are thus considered as potential biomarkers with considerable diagnostic value. However, some studies have even reported atheroprotective roles of these matricellular proteins. Experimental loss-of-function and overexpression studies have demonstrated that vascular injury via dysregulating the expression of matrix proteins contribute to atherosclerotic lesion formation. However, atherosclerosis being a complex inflammatory disease, the mechanisms stimulated/inhibited by these proteins in plaque formation needs future investigations. Further and extensive research is needed to understand the exact mechanisms by which various matricellular proteins promote endothelial cell activation, neointima formation and atherosclerotic plaque development. Performing studies using cell-specific knockout mice, gene overexpression and rescue experiments may reveal cell type-specific mechanisms involved in the disease development. This information will aid in discovering new treatment modalities for patients with atherosclerotic disease. Since THBS1 is a highly studied matricellular protein for its role in the pathogenesis of vascular pathologies, targeted therapies to block signaling stimulated by THBS1 may be tested as a potential therapeutic approach for atherosclerosis. For instance, TAX2, a CD47-derived cyclic peptide that targets THBS1 and selectively blocks THBS1:CD47 interaction can be evaluated for anti-atherogenic effects [226]. However, the shorter half-life of TAX2 in circulation remained an issue for its therapeutic use. Utilization of function-blocking anti-THBS1 monoclonal antibodies has also been considered as therapeutic strategy. C6.7 anti-Thbs1 antibody has been demonstrated to facilitate reendothelialization and decrease neointimal lesion formation in balloon-injured rat arteries [56]. Blocking THBS1:CD47 interaction using CD47 soluble receptor or CD47-blocking antibodies can also be viable therapeutic approach for the treatment of atherosclerosis. Recombinant human CD47 peptide that specifically binds and prevents THBS1’s effects, has been shown to improve vasorelaxation [227]. Further pharmacological studies are warranted to evaluate its effects in resistance vessels. Besides, integrins serve as receptors for various matricellular proteins and inhibitors blocking the integrins’ receptor function can also be evaluated for their efficacy in attenuating atherosclerotic lesion formation. Additional studies identifying the functional domains of matricellular proteins and cell surface binding partners will help in designing therapeutic peptidic inhibitors and blocking antibodies with longer half-life. Clearly, future investigations are required to better understand the role of matricellular proteins in the pathogenesis of atherosclerosis and whether inhibition of matricellular proteins-stimulated pathways in arterial wall truly represents a therapeutic target in patients with atherosclerotic vascular disease.
Highlights.
Matricellular proteins are secreted non-structural proteins that play important roles in the maintenance of tissue homeostasis.
The elevated levels of matricellular proteins correlate with atherosclerosis development.
Matricellular proteins stimulate multiple steps of atherogenesis ranging from endothelial cell activation to plaque vulnerability.
Integrins serve as receptors for various matricellular proteins. Inhibiting integrins’ receptor function may be a viable therapeutic strategy to suppress atherogenesis.
THBS1 is a highly studied matricellular protein for its role in the pathogenesis of vascular pathologies and targeted therapies to prevent THBS1-induced signaling may serve as a potential therapeutic approach for atherosclerosis.
Acknowledgments
This work was supported by the National Institutes of Health grants K99HL146954 and R00HL146954 awarded to BS. The authors acknowledge the use of BioRender for the preparation of figures.
Abbreviations
- ECM
extracellular matrix
- CVD
cardiovascular disease
- MI
myocardial infarction
- VSMCs
vascular smooth muscle cells
- SMCs
smooth muscle cells
- LDL
low-density lipoprotein
- VLDL
very-low-density lipoprotein
- THBS
thrombospondin
- SPARC
secreted protein, acidic and rich in cysteine
- COMP
cartilage oligomeric matrix protein
- LRP
low density lipoprotein-related receptor
- CD
cluster of differentiation
- PDGF
platelet-derived growth factor
- SIRPα
signal regulatory protein α
- BMP-2
bone morphogenetic protein-2
- VEGF
vascular endothelial growth factor
- TGF-β
transforming growth factor-β
- TNF-α
tumor necrosis factor α
- SPP1
secreted phosphoprotein 1
- CAD
coronary artery disease
- RSPOs
roof plate-specific spondins
- LGRs
leucine-rich repeat-containing G protein-coupled receptors
- YAP
yes-associated protein
- EndoMT
endothelial-to-mesenchymal transition
- NO
nitric oxide
- VTN
vitronectin
- TN
tenascin
- LPS
lipopolysaccharide
- VCAM-1
vascular cell adhesion molecule-1
- Gal
Galectin.
Footnotes
Declaration of Competing Interest
All authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Dahlof B, Cardiovascular disease risk factors: epidemiology and risk assessment, Am J Cardiol 105(1 Suppl) (2010) 3A–9A. [DOI] [PubMed] [Google Scholar]
- [2].Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, Elkind MSV, Evenson KR, Eze-Nliam C, Ferguson JF, Generoso G, Ho JE, Kalani R, Khan SS, Kissela BM, Knutson KL, Levine DA, Lewis TT, Liu J, Loop MS, Ma J, Mussolino ME, Navaneethan SD, Perak AM, Poudel R, Rezk-Hanna M, Roth GA, Schroeder EB, Shah SH, Thacker EL, VanWagner LB, Virani SS, Voecks JH, Wang NY, Yaffe K, Martin SS, Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association, Circulation 145(8) (2022) e153–e639. [DOI] [PubMed] [Google Scholar]
- [3].Song P, Fang Z, Wang H, Cai Y, Rahimi K, Zhu Y, Fowkes FGR, Fowkes FJI, Rudan I, Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: a systematic review, meta-analysis, and modelling study, Lancet Glob Health 8(5) (2020) e721–e729. [DOI] [PubMed] [Google Scholar]
- [4].Tucker WD, Arora Y, Mahajan K, Anatomy, Blood Vessels, StatPearls, Treasure Island (FL), 2022. [PubMed] [Google Scholar]
- [5].Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, Riddle SR, Frid MG, The adventitia: essential regulator of vascular wall structure and function, Annu Rev Physiol 75 (2013) 23–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Moore KJ, Tabas I, Macrophages in the pathogenesis of atherosclerosis, Cell 145(3) (2011) 341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gimbrone MA Jr., Topper JN, Nagel T, Anderson KR, Garcia-Cardena G, Endothelial dysfunction, hemodynamic forces, and atherogenesis, Ann N Y Acad Sci 902 (2000) 230–9; discussion 239–40. [DOI] [PubMed] [Google Scholar]
- [8].Moore KJ, Sheedy FJ, Fisher EA, Macrophages in atherosclerosis: a dynamic balance, Nat Rev Immunol 13(10) (2013) 709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bornstein P, Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1, J Cell Biol 130(3) (1995) 503–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mundi S, Massaro M, Scoditti E, Carluccio MA, van Hinsbergh VWM, Iruela-Arispe ML, De Caterina R, Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review, Cardiovasc Res 114(1) (2018) 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang X, Sessa WC, Fernandez-Hernando C, Endothelial Transcytosis of Lipoproteins in Atherosclerosis, Front Cardiovasc Med 5 (2018) 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Quinn MT, Parthasarathy S, Fong LG, Steinberg D, Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis, Proc Natl Acad Sci U S A 84(9) (1987) 2995–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Libby P, The changing landscape of atherosclerosis, Nature 592(7855) (2021) 524–533. [DOI] [PubMed] [Google Scholar]
- [14].Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H, Atherosclerosis: process, indicators, risk factors and new hopes, Int J Prev Med 5(8) (2014) 927–46. [PMC free article] [PubMed] [Google Scholar]
- [15].Insull W Jr., The pathology of atherosclerosis: plaque development and plaque responses to medical treatment, Am J Med 122(1 Suppl) (2009) S3–S14. [DOI] [PubMed] [Google Scholar]
- [16].Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM, Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions, Arterioscler Thromb Vasc Biol 20(5) (2000) 1262–75. [DOI] [PubMed] [Google Scholar]
- [17].Engelen SE, Robinson AJB, Zurke Y-X, Monaco C, Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed?, Nature Reviews Cardiology 19(8) (2022) 522–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z, Vascular smooth muscle cells in atherosclerosis, Nature Reviews Cardiology 16(12) (2019) 727–744. [DOI] [PubMed] [Google Scholar]
- [19].Jebari-Benslaiman S, Galicia-García U, Larrea-Sebal A, Olaetxea JR, Alloza I, Vandenbroeck K, Benito-Vicente A, Martín C, Pathophysiology of Atherosclerosis, International Journal of Molecular Sciences, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Soehnlein O, Libby P, Targeting inflammation in atherosclerosis — from experimental insights to the clinic, Nature Reviews Drug Discovery 20(8) (2021) 589–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bornstein P, McPherson J, Sage H, Synthesis and Secretion of Structural Macromolecules by Endothelial Cells in Culture, in: Nossel HL, Vogel HJ (Eds.), Pathobiology of the Endothelial Cell, Academic Press; 1982, pp. 215–228. [Google Scholar]
- [22].Gerarduzzi C, Hartmann U, Leask A, Drobetsky E, The Matrix Revolution: Matricellular Proteins and Restructuring of the Cancer Microenvironment, Cancer Res 80(13) (2020) 2705–2717. [DOI] [PubMed] [Google Scholar]
- [23].Bornstein P, Matricellular proteins: an overview, Matrix Biol 19(7) (2000) 555–6. [DOI] [PubMed] [Google Scholar]
- [24].Bornstein P, Matricellular proteins: an overview, J Cell Commun Signal 3(3–4) (2009) 163–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bornstein P, Thrombospondins as matricellular modulators of cell function, Journal of Clinical Investigation 107(8) (2001) 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bornstein P, Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1., Journal of Cell Biology 130(3) (1995) 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Roberts DD, Lau LF, Matricellular Proteins, in: Mecham RP (Ed.), The Extracellular Matrix: an Overview, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 369–413. [Google Scholar]
- [28].Sage EH, Bornstein P, Extracellular proteins that modulate cell-matrix interactions. SPARC, tenascin, and thrombospondin, J Biol Chem 266(23) (1991) 14831–4. [PubMed] [Google Scholar]
- [29].Chiquet-Ehrismann R, Tenascins, a growing family of extracellular matrix proteins, Experientia 51(9–10) (1995) 853–62. [DOI] [PubMed] [Google Scholar]
- [30].Brekken RA, Sullivan MM, Workman G, Bradshaw AD, Carbon J, Siadak A, Murri C, Framson PE, Sage EH, Expression and characterization of murine hevin (SC1), a member of the SPARC family of matricellular proteins, J Histochem Cytochem 52(6) (2004) 735–48. [DOI] [PubMed] [Google Scholar]
- [31].Bornstein P, Sage EH, Matricellular proteins: extracellular modulators of cell function, Curr Opin Cell Biol 14(5) (2002) 608–16. [DOI] [PubMed] [Google Scholar]
- [32].Murphy-Ullrich JE, Sage EH, Revisiting the matricellular concept, Matrix Biol 37 (2014) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lawler JW, Slayter HS, Coligan JE, Isolation and characterization of a high molecular weight glycoprotein from human blood platelets, J Biol Chem 253(23) (1978) 8609–16. [PubMed] [Google Scholar]
- [34].Isenberg JS, Roberts DD, THBS1 (thrombospondin-1), Atlas Genet Cytogenet Oncol Haematol 24(8) (2020) 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bornstein P, Armstrong LC, Hankenson KD, Kyriakides TR, Yang Z, Thrombospondin 2, a matricellular protein with diverse functions, Matrix Biol 19(7) (2000) 557–68. [DOI] [PubMed] [Google Scholar]
- [36].Lawler J, The functions of thrombospondin-1 and-2, Curr Opin Cell Biol 12(5) (2000) 634–40. [DOI] [PubMed] [Google Scholar]
- [37].Unger S, Hecht JT, Pseudoachondroplasia and multiple epiphyseal dysplasia: New etiologic developments, Am J Med Genet 106(4) (2001) 244–50. [PubMed] [Google Scholar]
- [38].Adams JC, Kureishy N, Taylor AL, A role for syndecan-1 in coupling fascin spike formation by thrombospondin-1, J Cell Biol 152(6) (2001) 1169–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sun X, Mosher DF, Rapraeger A, Heparan sulfate-mediated binding of epithelial cell surface proteoglycan to thrombospondin, J Biol Chem 264(5) (1989) 2885–9. [PubMed] [Google Scholar]
- [40].Asch AS, Silbiger S, Heimer E, Nachman RL, Thrombospondin sequence motif (CSVTCG) is responsible for CD36 binding, Biochem Biophys Res Commun 182(3) (1992) 1208–17. [DOI] [PubMed] [Google Scholar]
- [41].Gao AG, Lindberg FP, Finn MB, Blystone SD, Brown EJ, Frazier WA, Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin, J Biol Chem 271(1) (1996) 21–4. [DOI] [PubMed] [Google Scholar]
- [42].Goicoechea S, Orr AW, Pallero MA, Eggleton P, Murphy-Ullrich JE, Thrombospondin mediates focal adhesion disassembly through interactions with cell surface calreticulin, J Biol Chem 275(46) (2000) 36358–68. [DOI] [PubMed] [Google Scholar]
- [43].Calzada MJ, Annis DS, Zeng B, Marcinkiewicz C, Banas B, Lawler J, Mosher DF, Roberts DD, Identification of novel beta1 integrin binding sites in the type 1 and type 2 repeats of thrombospondin-1, J Biol Chem 279(40) (2004) 41734–43. [DOI] [PubMed] [Google Scholar]
- [44].Calzada MJ, Sipes JM, Krutzsch HC, Yurchenco PD, Annis DS, Mosher DF, Roberts DD, Recognition of the N-terminal modules of thrombospondin-1 and thrombospondin-2 by alpha6beta1 integrin, J Biol Chem 278(42) (2003) 40679–87. [DOI] [PubMed] [Google Scholar]
- [45].Staniszewska I, Zaveri S, Del Valle L, Oliva I, Rothman VL, Croul SE, Roberts DD, Mosher DF, Tuszynski GP, Marcinkiewicz C, Interaction of alpha9beta1 integrin with thrombospondin-1 promotes angiogenesis, Circ Res 100(9) (2007) 1308–16. [DOI] [PubMed] [Google Scholar]
- [46].Lawler J, Weinstein R, Hynes RO, Cell attachment to thrombospondin: the role of ARG-GLY-ASP, calcium, and integrin receptors, J Cell Biol 107(6 Pt 1) (1988) 2351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lawler J, Hynes RO, An integrin receptor on normal and thrombasthenic platelets that binds thrombospondin, Blood 74(6) (1989) 2022–7. [PubMed] [Google Scholar]
- [48].Sadvakassova G, Dobocan MC, Congote LF, Osteopontin and the C-terminal peptide of thrombospondin-4 compete for CD44 binding and have opposite effects on CD133+ cell colony formation, BMC Res Notes 2 (2009) 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, Liu CJ, Zhu Y, Gao Y, Xu Q, Kong W, Wang X, Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin, Circ Res 106(3) (2010) 514–25. [DOI] [PubMed] [Google Scholar]
- [50].Chen FH, Thomas AO, Hecht JT, Goldring MB, Lawler J, Cartilage oligomeric matrix protein/thrombospondin 5 supports chondrocyte attachment through interaction with integrins, J Biol Chem 280(38) (2005) 32655–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rock MJ, Holden P, Horton WA, Cohn DH, Cartilage oligomeric matrix protein promotes cell attachment via two independent mechanisms involving CD47 and alphaVbeta3 integrin, Mol Cell Biochem 338(1–2) (2010) 215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Raugi GJ, Mullen JS, Bark DH, Okada T, Mayberg MR, Thrombospondin deposition in rat carotid artery injury, Am J Pathol 137(1) (1990) 179–85. [PMC free article] [PubMed] [Google Scholar]
- [53].Sajid M, Hu Z, Guo H, Li H, Stouffer GA, Vascular expression of integrin-associated protein and thrombospondin increase after mechanical injury, J Investig Med 49(5) (2001) 398–406. [DOI] [PubMed] [Google Scholar]
- [54].Riessen R, Kearney M, Lawler J, Isner JM, Immunolocalization of thrombospondin-1 in human atherosclerotic and restenotic arteries, Am Heart J 135(2 Pt 1) (1998) 357–64. [DOI] [PubMed] [Google Scholar]
- [55].Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, Topol EJ, Plow EF, Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat, Circulation 107(25) (2003) 3209–15. [DOI] [PubMed] [Google Scholar]
- [56].Chen D, Asahara T, Krasinski K, Witzenbichler B, Yang J, Magner M, Kearney M, Frazier WA, Isner JM, Andres V, Antibody blockade of thrombospondin accelerates reendothelialization and reduces neointima formation in balloon-injured rat carotid artery, Circulation 100(8) (1999) 849–54. [DOI] [PubMed] [Google Scholar]
- [57].Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, Hoylaerts MF, Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE−/− mice, Circ Res 103(10) (2008) 1181–9. [DOI] [PubMed] [Google Scholar]
- [58].Ganguly R, Khanal S, Mathias A, Gupta S, Lallo J, Sahu S, Ohanyan V, Patel A, Storm K, Datta S, Raman P, TSP-1 (Thrombospondin-1) Deficiency Protects ApoE(−/−) Mice Against Leptin-Induced Atherosclerosis, Arterioscler Thromb Vasc Biol 41(2) (2021) e112–e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kim CW, Pokutta-Paskaleva A, Kumar S, Timmins LH, Morris AD, Kang DW, Dalal S, Chadid T, Kuo KM, Raykin J, Li H, Yanagisawa H, Gleason RL Jr., Jo H, Brewster LP, Disturbed Flow Promotes Arterial Stiffening Through Thrombospondin-1, Circulation 136(13) (2017) 1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Osada-Oka M, Ikeda T, Akiba S, Sato T, Hypoxia stimulates the autocrine regulation of migration of vascular smooth muscle cells via HIF-1alpha-dependent expression of thrombospondin-1, J Cell Biochem 104(5) (2008) 1918–26. [DOI] [PubMed] [Google Scholar]
- [61].Takahashi M, Oka M, Ikeda T, Akiba S, Sato T, [Role of thrombospondin-1 in hypoxia-induced migration of human vascular smooth muscle cells], Yakugaku Zasshi 128(3) (2008) 377–83. [DOI] [PubMed] [Google Scholar]
- [62].Roth JJ, Gahtan V, Brown JL, Gerhard C, Swami VK, Rothman VL, Tulenko TN, Tuszynski GP, Thrombospondin-1 is elevated with both intimal hyperplasia and hypercholesterolemia, J Surg Res 74(1) (1998) 11–6. [DOI] [PubMed] [Google Scholar]
- [63].Muraishi A, Capuzzi DM, Tuszynski GP, Binding of thrombospondin to human plasma lipoproteins, Biochem Biophys Res Commun 193(3) (1993) 1145–51. [DOI] [PubMed] [Google Scholar]
- [64].Yao M, Roberts DD, Isenberg JS, Thrombospondin-1 inhibition of vascular smooth muscle cell responses occurs via modulation of both cAMP and cGMP, Pharmacol Res 63(1) (2011) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Isenberg JS, Frazier WA, Krishna MC, Wink DA, Roberts DD, Enhancing cardiovascular dynamics by inhibition of thrombospondin-1/CD47 signaling, Curr Drug Targets 9(10) (2008) 833–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Isenberg JS, Qin Y, Maxhimer JB, Sipes JM, Despres D, Schnermann J, Frazier WA, Roberts DD, Thrombospondin-1 and CD47 regulate blood pressure and cardiac responses to vasoactive stress, Matrix Biol 28(2) (2009) 110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bauer EM, Qin Y, Miller TW, Bandle RW, Csanyi G, Pagano PJ, Bauer PM, Schnermann J, Roberts DD, Isenberg JS, Thrombospondin-1 supports blood pressure by limiting eNOS activation and endothelial-dependent vasorelaxation, Cardiovasc Res 88(3) (2010) 471–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Soto-Pantoja DR, Kaur S, Roberts DD, CD47 signaling pathways controlling cellular differentiation and responses to stress, Crit Rev Biochem Mol Biol 50(3) (2015) 212–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Roberts DD, Miller TW, Rogers NM, Yao M, Isenberg JS, The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47, Matrix Biol 31(3) (2012) 162–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Csanyi G, Yao M, Rodriguez AI, Al Ghouleh I, Sharifi-Sanjani M, Frazziano G, Huang X, Kelley EE, Isenberg JS, Pagano PJ, Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1, Arterioscler Thromb Vasc Biol 32(12) (2012) 2966–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Csanyi G, Feck DM, Ghoshal P, Singla B, Lin H, Nagarajan S, Meijles DN, Al Ghouleh I, Cantu-Medellin N, Kelley EE, Mateuszuk L, Isenberg JS, Watkins S, Pagano PJ, CD47 and Nox1 Mediate Dynamic Fluid-Phase Macropinocytosis of Native LDL, Antioxid Redox Signal 26(16) (2017) 886–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Singla B, Lin HP, Ghoshal P, Cherian-Shaw M, Csanyi G, PKCdelta stimulates macropinocytosis via activation of SSH1-cofilin pathway, Cell Signal 53 (2019) 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yamashiro Y, Thang BQ, Shin SJ, Lino CA, Nakamura T, Kim J, Sugiyama K, Tokunaga C, Sakamoto H, Osaka M, Davis EC, Wagenseil JE, Hiramatsu Y, Yanagisawa H, Role of Thrombospondin-1 in Mechanotransduction and Development of Thoracic Aortic Aneurysm in Mouse and Humans, Circ Res 123(6) (2018) 660–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yamashiro Y, Thang BQ, Ramirez K, Shin SJ, Kohata T, Ohata S, Nguyen TAV, Ohtsuki S, Nagayama K, Yanagisawa H, Matrix mechanotransduction mediated by thrombospondin-1/integrin/YAP in the vascular remodeling, Proc Natl Acad Sci U S A 117(18) (2020) 9896–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M, Kovacic JC, Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability, Nat Commun 7 (2016) 11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liang G, Wang S, Shao J, Jin YJ, Xu L, Yan Y, Gunther S, Wang L, Offermanns S, Tenascin-X Mediates Flow-Induced Suppression of EndMT and Atherosclerosis, Circ Res 130(11) (2022) 1647–1659. [DOI] [PubMed] [Google Scholar]
- [77].Yamashiro Y, Ramirez K, Nagayama K, Hattori N, Liu YY, Matsunaga S, Tomita S, Kubota Y, Yanagisawa H, Partial endothelial-to-mesenchymal transition mediated by HIF-induced CD45 in neointima formation upon carotid artery ligation, Cardiovasc Res (2022). [DOI] [PubMed] [Google Scholar]
- [78].Singla B, Aithabathula RV, Ahn W, Csanyi G, Abstract 13606: Deletion of CD47 Specifically in Lymphatic Endothelium Augments Arterial Lymphangiogenesis and Attenuates Atherosclerosis, Circulation 146(Suppl_1) (2022) A13606–A13606. [Google Scholar]
- [79].Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, Schadt EE, Quertermous T, Betancur P, Maegdefessel L, Matic LP, Hedin U, Weissman IL, Leeper NJ, CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis, Nature 536(7614) (2016) 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chao MP, Majeti R, Weissman IL, Programmed cell removal: a new obstacle in the road to developing cancer, Nat Rev Cancer 12(1) (2011) 58–67. [DOI] [PubMed] [Google Scholar]
- [81].Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP, Role of CD47 as a marker of self on red blood cells, Science 288(5473) (2000) 2051–4. [DOI] [PubMed] [Google Scholar]
- [82].Singla B, Lin HP, Ahn W, Xu J, Ma Q, Sghayyer M, Dong K, Cherian-Shaw M, Zhou J, Huo Y, White J, Csanyi G, Loss of myeloid cell-specific SIRPalpha, but not CD47, attenuates inflammation and suppresses atherosclerosis, Cardiovasc Res 118(15) (2022) 3097–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Li Y, Qi X, Tong X, Wang S, Thrombospondin 1 activates the macrophage Toll-like receptor 4 pathway, Cell Mol Immunol 10(6) (2013) 506–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Yue H, Febbraio M, Klenotic PA, Kennedy DJ, Wu Y, Chen S, Gohara AF, Li O, Belcher A, Kuang B, McIntyre TM, Silverstein RL, Li W, CD36 Enhances Vascular Smooth Muscle Cell Proliferation and Development of Neointimal Hyperplasia, Arterioscler Thromb Vasc Biol 39(2) (2019) 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hedbom E, Antonsson P, Hjerpe A, Aeschlimann D, Paulsson M, Rosa-Pimentel E, Sommarin Y, Wendel M, Oldberg A, Heinegard D, Cartilage matrix proteins. An acidic oligomeric protein (COMP) detected only in cartilage, J Biol Chem 267(9) (1992) 6132–6. [PubMed] [Google Scholar]
- [86].Riessen R, Fenchel M, Chen H, Axel DI, Karsch KR, Lawler J, Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by human vascular smooth muscle cells, Arterioscler Thromb Vasc Biol 21(1) (2001) 47–54. [DOI] [PubMed] [Google Scholar]
- [87].Bond AR, Hultgardh-Nilsson A, Knutsson A, Jackson CL, Rauch U, Cartilage oligomeric matrix protein (COMP) in murine brachiocephalic and carotid atherosclerotic lesions, Atherosclerosis 236(2) (2014) 366–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hultman K, Edsfeldt A, Bjorkbacka H, Duner P, Sundius L, Nitulescu M, Persson A, Boyle JJ, Nilsson J, Hultgardh-Nilsson A, Bengtsson E, Goncalves I, Cartilage Oligomeric Matrix Protein Associates With a Vulnerable Plaque Phenotype in Human Atherosclerotic Plaques, Stroke 50(11) (2019) 3289–3292. [DOI] [PubMed] [Google Scholar]
- [89].Wang FF, Ha L, Yu HY, Mi L, Han JL, Gao W, Altered serum level of cartilage oligomeric matrix protein and its association with coronary calcification in patients with coronary heart disease, J Geriatr Cardiol 14(2) (2017) 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Sandstedt J, Vargmar K, Bjorkman K, Ruetschi U, Bergstrom G, Hulten LM, Skioldebrand E, COMP (Cartilage Oligomeric Matrix Protein) Neoepitope: A Novel Biomarker to Identify Symptomatic Carotid Stenosis, Arterioscler Thromb Vasc Biol 41(3) (2021) 1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Fu Y, Gao C, Liang Y, Wang M, Huang Y, Ma W, Li T, Jia Y, Yu F, Zhu W, Cui Q, Li Y, Xu Q, Wang X, Kong W, Shift of Macrophage Phenotype Due to Cartilage Oligomeric Matrix Protein Deficiency Drives Atherosclerotic Calcification, Circ Res 119(2) (2016) 261–76. [DOI] [PubMed] [Google Scholar]
- [92].Qin W, Cao Y, Li L, Chen W, Chen X, Upregulation of ADAMTS-7 and downregulation of COMP are associated with aortic aneurysm, Mol Med Rep 16(4) (2017) 5459–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lv H, Wang H, Quan M, Zhang C, Fu Y, Zhang L, Lin C, Liu X, Yi X, Chen J, Wang X, Cheng T, Ai D, Kong W, Zhu Y, Cartilage oligomeric matrix protein fine-tunes disturbed flow-induced endothelial activation and atherogenesis, Matrix Biol 95 (2021) 32–51. [DOI] [PubMed] [Google Scholar]
- [94].Du Y, Wang Y, Wang L, Liu B, Tian Q, Liu CJ, Zhang T, Xu Q, Zhu Y, Ake O, Qi Y, Tang C, Kong W, Wang X, Cartilage oligomeric matrix protein inhibits vascular smooth muscle calcification by interacting with bone morphogenetic protein-2, Circ Res 108(8) (2011) 917–28. [DOI] [PubMed] [Google Scholar]
- [95].Halasz K, Kassner A, Morgelin M, Heinegard D, COMP acts as a catalyst in collagen fibrillogenesis, J Biol Chem 282(43) (2007) 31166–73. [DOI] [PubMed] [Google Scholar]
- [96].Shami A, Gustafsson R, Kalamajski S, Krams R, Segers D, Rauch U, Roos G, Nilsson J, Oldberg A, Hultgardh-Nilsson A, Fibromodulin deficiency reduces low-density lipoprotein accumulation in atherosclerotic plaques in apolipoprotein E-null mice, Arterioscler Thromb Vasc Biol 33(2) (2013) 354–61. [DOI] [PubMed] [Google Scholar]
- [97].Heinegard D, Fell-Muir Lecture: Proteoglycans and more--from molecules to biology, Int J Exp Pathol 90(6) (2009) 575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bork P, The modular architecture of a new family of growth regulators related to connective tissue growth factor, FEBS Lett 327(2) (1993) 125–30. [DOI] [PubMed] [Google Scholar]
- [99].Perbal B, Takigawa M, CCN proteins: A new family of cell growth and differentiation regulators, 2005.
- [100].Parisi MS, Gazzerro E, Rydziel S, Canalis E, Expression and regulation of CCN genes in murine osteoblasts, Bone 38(5) (2006) 671–7. [DOI] [PubMed] [Google Scholar]
- [101].Lau LF, Lam SC, The CCN family of angiogenic regulators: the integrin connection, Exp Cell Res 248(1) (1999) 44–57. [DOI] [PubMed] [Google Scholar]
- [102].Kubota S, Takigawa M, CCN family proteins and angiogenesis: from embryo to adulthood, Angiogenesis 10(1) (2007) 1–11. [DOI] [PubMed] [Google Scholar]
- [103].Todorovic V, Chen CC, Hay N, Lau LF, The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts, J Cell Biol 171(3) (2005) 559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Lau LF, CCN1/CYR61: the very model of a modern matricellular protein, Cell Mol Life Sci 68(19) (2011) 3149–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Babic AM, Chen CC, Lau LF, Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo, Mol Cell Biol 19(4) (1999) 2958–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wahab NA, Weston BS, Mason RM, Connective tissue growth factor CCN2 interacts with and activates the tyrosine kinase receptor TrkA, J Am Soc Nephrol 16(2) (2005) 340–51. [DOI] [PubMed] [Google Scholar]
- [107].Segarini PR, Nesbitt JE, Li D, Hays LG, Yates JR 3rd, Carmichael DF, The low density lipoprotein receptor-related protein/alpha2-macroglobulin receptor is a receptor for connective tissue growth factor, J Biol Chem 276(44) (2001) 40659–67. [DOI] [PubMed] [Google Scholar]
- [108].Sakamoto K, Yamaguchi S, Ando R, Miyawaki A, Kabasawa Y, Takagi M, Li CL, Perbal B, Katsube K, The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway, J Biol Chem 277(33) (2002) 29399–405. [DOI] [PubMed] [Google Scholar]
- [109].Klenotic PA, Zhang C, Lin Z, Emerging roles of CCN proteins in vascular development and pathology, J Cell Commun Signal 10(3) (2016) 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen CC, Mo FE, Lau LF, The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts, J Biol Chem 276(50) (2001) 47329–37. [DOI] [PubMed] [Google Scholar]
- [111].Emre Y, Imhof BA, Matricellular protein CCN1/CYR61: a new player in inflammation and leukocyte trafficking, Semin Immunopathol 36(2) (2014) 253–9. [DOI] [PubMed] [Google Scholar]
- [112].Esaily HA, Serag DM, Rizk MS, Badr IT, Sonbol AA, Fotoh DS, Relationship between cellular communication network factor 1 (CCN1) and carotid atherosclerosis in patients with rheumatoid arthritis, Med J Malaysia 76(3) (2021) 311–317. [PubMed] [Google Scholar]
- [113].Mahendiran T, Klingenberg R, Nanchen D, Gencer B, Meier D, Raber L, Carballo D, Matter CM, Luscher TF, Mach F, Rodondi N, Muller O, Fournier S, CCN family member 1 (CCN1) is an early marker of infarct size and left ventricular dysfunction in STEMI patients, Atherosclerosis 335 (2021) 77–83. [DOI] [PubMed] [Google Scholar]
- [114].Feng B, Xu G, Sun K, Duan K, Shi B, Zhang N, Association of serum Cyr61 levels with peripheral arterial disease in subjects with type 2 diabetes, Cardiovasc Diabetol 19(1) (2020) 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Schober JM, Chen N, Grzeszkiewicz TM, Jovanovic I, Emeson EE, Ugarova TP, Ye RD, Lau LF, Lam SC, Identification of integrin alpha(M)beta(2) as an adhesion receptor on peripheral blood monocytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2): immediate-early gene products expressed in atherosclerotic lesions, Blood 99(12) (2002) 4457–65. [DOI] [PubMed] [Google Scholar]
- [116].Hilfiker-Kleiner D, Kaminski K, Kaminska A, Fuchs M, Klein G, Podewski E, Grote K, Kiian I, Wollert KC, Hilfiker A, Drexler H, Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation, Circulation 109(18) (2004) 2227–33. [DOI] [PubMed] [Google Scholar]
- [117].Jun JI, Lau LF, Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets, Nat Rev Drug Discov 10(12) (2011) 945–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Sigala F, Georgopoulos S, Papalambros E, Chasiotis D, Vourliotakis G, Niforou A, Kotsinas A, Kavantzas N, Patsouris E, Gorgoulis VG, Bastounis E, Heregulin, cysteine rich-61 and matrix metalloproteinase 9 expression in human carotid atherosclerotic plaques: relationship with clinical data, Eur J Vasc Endovasc Surg 32(3) (2006) 238–45. [DOI] [PubMed] [Google Scholar]
- [119].Hilfiker A, Hilfiker-Kleiner D, Fuchs M, Kaminski K, Lichtenberg A, Rothkotter HJ, Schieffer B, Drexler H, Expression of CYR61, an angiogenic immediate early gene, in arteriosclerosis and its regulation by angiotensin II, Circulation 106(2) (2002) 254–60. [DOI] [PubMed] [Google Scholar]
- [120].Matsumae H, Yoshida Y, Ono K, Togi K, Inoue K, Furukawa Y, Nakashima Y, Kojima Y, Nobuyoshi M, Kita T, Tanaka M, CCN1 knockdown suppresses neointimal hyperplasia in a rat artery balloon injury model, Arterioscler Thromb Vasc Biol 28(6) (2008) 1077–83. [DOI] [PubMed] [Google Scholar]
- [121].Papetta A, Gakiopoulou H, Agapitos E, Patsouris ES, Lazaris AC, Correlations between CCN1 immunoexpression and myocardial histologic lesions in sudden cardiac death, Am J Forensic Med Pathol 34(2) (2013) 169–76. [DOI] [PubMed] [Google Scholar]
- [122].Hao F, Zhang F, Wu DD, An D, Shi J, Li G, Xu X, Cui MZ, Lysophosphatidic acid-induced vascular neointimal formation in mouse carotid arteries is mediated by the matricellular protein CCN1/Cyr61, Am J Physiol Cell Physiol 311(6) (2016) C975–C984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zhao JF, Chen HY, Wei J, Jim Leu SJ, Lee TS, CCN family member 1 deregulates cholesterol metabolism and aggravates atherosclerosis, Acta Physiol (Oxf) 225(3) (2019) e13209. [DOI] [PubMed] [Google Scholar]
- [124].Hsu PL, Chen JS, Wang CY, Wu HL, Mo FE, Shear-Induced CCN1 Promotes Atheroprone Endothelial Phenotypes and Atherosclerosis, Circulation 139(25) (2019) 2877–2891. [DOI] [PubMed] [Google Scholar]
- [125].Zhang J, Wu G, Dai H, The matricellular protein CCN1 regulates TNF-alpha induced vascular endothelial cell apoptosis, Cell Biol Int 40(1) (2016) 1–6. [DOI] [PubMed] [Google Scholar]
- [126].Mo FE, Hsu P-L, The extracellular matrix protein CCN1 mediates the endothelial dysfunction induced by disturbed flow, Atherosclerosis 263 (2017) e32–e33. [Google Scholar]
- [127].Waller AH, Sanchez-Ross M, Kaluski E, Klapholz M, Osteopontin in cardiovascular disease: a potential therapeutic target, Cardiol Rev 18(3) (2010) 125–31. [DOI] [PubMed] [Google Scholar]
- [128].Duvall CL, Taylor WR, Weiss D, Wojtowicz AM, Guldberg RE, Impaired angiogenesis, early callus formation, and late stage remodeling in fracture healing of osteopontin-deficient mice, J Bone Miner Res 22(2) (2007) 286–97. [DOI] [PubMed] [Google Scholar]
- [129].Giachelli CM, Bae N, Almeida M, Denhardt DT, Alpers CE, Schwartz SM, Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques, J Clin Invest 92(4) (1993) 1686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Chowdhury UR, Jea SY, Oh DJ, Rhee DJ, Fautsch MP, Expression profile of the matricellular protein osteopontin in primary open-angle glaucoma and the normal human eye, Invest Ophthalmol Vis Sci 52(9) (2011) 6443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Lund SA, Giachelli CM, Scatena M, The role of osteopontin in inflammatory processes, J Cell Commun Signal 3(3–4) (2009) 311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Giachelli CM, Liaw L, Murry CE, Schwartz SM, Almeida M, Osteopontin expression in cardiovascular diseases, Ann N Y Acad Sci 760 (1995) 109–26. [DOI] [PubMed] [Google Scholar]
- [133].Chiba S, Okamoto H, Kon S, Kimura C, Murakami M, Inobe M, Matsui Y, Sugawara T, Shimizu T, Uede T, Kitabatake A, Development of atherosclerosis in osteopontin transgenic mice, Heart Vessels 16(3) (2002) 111–7. [DOI] [PubMed] [Google Scholar]
- [134].Maniatis K, Siasos G, Oikonomou E, Vavuranakis M, Zaromytidou M, Mourouzis K, Paraskevopoulos T, Charalambous G, Papavassiliou AG, Tousoulis D, Osteoprotegerin and Osteopontin Serum Levels are Associated with Vascular Function and Inflammation in Coronary Artery Disease Patients, Curr Vasc Pharmacol 18(5) (2020) 523–530. [DOI] [PubMed] [Google Scholar]
- [135].Kosowski M, Basiak M, Hachula M, Okopien B, Plasma Concentrations of New Biochemical Markers of Atherosclerosis in Patients with Dyslipidemia-A Pilot Study, Medicina (Kaunas) 58(6) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Zhu Z, He Y, Shi M, Guo D, Zhang K, Ren L, Peng Y, Yang P, Chen J, Zang Y, Wang A, Xu T, Li Q, Ju Z, Geng D, Zhang Y, He J, Plasma osteopontin levels and adverse clinical outcomes after ischemic stroke, Atherosclerosis 332 (2021) 33–40. [DOI] [PubMed] [Google Scholar]
- [137].Isoda K, Nishikawa K, Kamezawa Y, Yoshida M, Kusuhara M, Moroi M, Tada N, Ohsuzu F, Osteopontin plays an important role in the development of medial thickening and neointimal formation, Circ Res 91(1) (2002) 77–82. [DOI] [PubMed] [Google Scholar]
- [138].Isoda K, Kamezawa Y, Ayaori M, Kusuhara M, Tada N, Ohsuzu F, Osteopontin transgenic mice fed a high-cholesterol diet develop early fatty-streak lesions, Circulation 107(5) (2003) 679–81. [DOI] [PubMed] [Google Scholar]
- [139].Myers DL, Harmon KJ, Lindner V, Liaw L, Alterations of arterial physiology in osteopontin-null mice, Arterioscler Thromb Vasc Biol 23(6) (2003) 1021–8. [DOI] [PubMed] [Google Scholar]
- [140].Lok ZSY, Lyle AN, Osteopontin in Vascular Disease, Arterioscler Thromb Vasc Biol 39(4) (2019) 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].O’Brien ER, Garvin MR, Stewart DK, Hinohara T, Simpson JB, Schwartz SM, Giachelli CM, Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques, Arterioscler Thromb 14(10) (1994) 1648–56. [DOI] [PubMed] [Google Scholar]
- [142].Lyle AN, Joseph G, Fan AE, Weiss D, Landazuri N, Taylor WR, Reactive oxygen species regulate osteopontin expression in a murine model of postischemic neovascularization, Arterioscler Thromb Vasc Biol 32(6) (2012) 1383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM, NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival, J Cell Biol 141(4) (1998) 1083–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Liaw L, Almeida M, Hart CE, Schwartz SM, Giachelli CM, Osteopontin promotes vascular cell adhesion and spreading and is chemotactic for smooth muscle cells in vitro, Circ Res 74(2) (1994) 214–24. [DOI] [PubMed] [Google Scholar]
- [145].Matsui Y, Rittling SR, Okamoto H, Inobe M, Jia N, Shimizu T, Akino M, Sugawara T, Morimoto J, Kimura C, Kon S, Denhardt D, Kitabatake A, Uede T, Osteopontin deficiency attenuates atherosclerosis in female apolipoprotein E-deficient mice, Arterioscler Thromb Vasc Biol 23(6) (2003) 1029–34. [DOI] [PubMed] [Google Scholar]
- [146].Takemoto M, Yokote K, Yamazaki M, Ridall AL, Butler WT, Matsumoto T, Tamura K, Saito Y, Mori S, Enhanced expression of osteopontin by high glucose. Involvement of osteopontin in diabetic macroangiopathy, Ann N Y Acad Sci 902 (2000) 357–63. [DOI] [PubMed] [Google Scholar]
- [147].Sorensen ES, Petersen TE, Identification of two phosphorylation motifs in bovine osteopontin, Biochem Biophys Res Commun 198(1) (1994) 200–5. [DOI] [PubMed] [Google Scholar]
- [148].Giachelli CM, Steitz S, Osteopontin: a versatile regulator of inflammation and biomineralization, Matrix Biol 19(7) (2000) 615–22. [DOI] [PubMed] [Google Scholar]
- [149].Yu W, Xiao L, Que Y, Li S, Chen L, Hu P, Xiong R, Seta F, Chen H, Tong X, Smooth muscle NADPH oxidase 4 promotes angiotensin II-induced aortic aneurysm and atherosclerosis by regulating osteopontin, Biochim Biophys Acta Mol Basis Dis 1866(12) (2020) 165912. [DOI] [PubMed] [Google Scholar]
- [150].Ding Y, Chen J, Cui G, Wei Y, Lu C, Wang L, Diao H, Pathophysiological role of osteopontin and angiotensin II in atherosclerosis, Biochem Biophys Res Commun 471(1) (2016) 5–9. [DOI] [PubMed] [Google Scholar]
- [151].Chen PH, Chen X, Lin Z, Fang D, He X, The structural basis of R-spondin recognition by LGR5 and RNF43, Genes Dev 27(12) (2013) 1345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Nam JS, Turcotte TJ, Smith PF, Choi S, Yoon JK, Mouse cristin/R-spondin family proteins are novel ligands for the Frizzled 8 and LRP6 receptors and activate beta-catenin-dependent gene expression, J Biol Chem 281(19) (2006) 13247–13257. [DOI] [PubMed] [Google Scholar]
- [153].Jin YR, Yoon JK, The R-spondin family of proteins: emerging regulators of WNT signaling, Int J Biochem Cell Biol 44(12) (2012) 2278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].de Lau WBM, Snel B, Clevers HC, The R-spondin protein family, Genome Biology 13(3) (2012) 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Foulquier S, Daskalopoulos EP, Lluri G, Hermans KCM, Deb A, Blankesteijn WM, WNT Signaling in Cardiac and Vascular Disease, Pharmacol Rev 70(1) (2018) 68–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Qin L, Hu R, Zhu N, Yao HL, Lei XY, Li SX, Liao DF, Zheng XL, The novel role and underlying mechanism of Wnt5a in regulating cellular cholesterol accumulation, Clin Exp Pharmacol Physiol 41(9) (2014) 671–8. [DOI] [PubMed] [Google Scholar]
- [157].Christman MA 2nd, Goetz DJ, Dickerson E, McCall KD, Lewis CJ, Benencia F, Silver MJ, Kohn LD, Malgor R, Wnt5a is expressed in murine and human atherosclerotic lesions, Am J Physiol Heart Circ Physiol 294(6) (2008) H2864–70. [DOI] [PubMed] [Google Scholar]
- [158].Boucher P, Matz RL, Terrand J, atherosclerosis: gone with the Wnt?, Atherosclerosis 301 (2020) 15–22. [DOI] [PubMed] [Google Scholar]
- [159].Singla B, Lin HP, Chen A, Ahn W, Ghoshal P, Cherian-Shaw M, White J, Stansfield BK, Csanyi G, Role of R-spondin 2 in arterial lymphangiogenesis and atherosclerosis, Cardiovasc Res 117(6) (2021) 1489–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Scallan JP, Hill MA, Davis MJ, Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling, Cardiovasc Res 107(1) (2015) 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Carmon KS, Gong X, Yi J, Thomas A, Liu Q, RSPO-LGR4 functions via IQGAP1 to potentiate Wnt signaling, Proc Natl Acad Sci U S A 111(13) (2014) E1221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Felding-Habermann B, Cheresh DA, Vitronectin and its receptors, Curr Opin Cell Biol 5(5) (1993) 864–8. [DOI] [PubMed] [Google Scholar]
- [163].Preissner KT, Seiffert D, Role of vitronectin and its receptors in haemostasis and vascular remodeling, Thromb Res 89(1) (1998) 1–21. [DOI] [PubMed] [Google Scholar]
- [164].Zhuang P, Blackburn MN, Peterson CB, Characterization of the denaturation and renaturation of human plasma vitronectin. I. Biophysical characterization of protein unfolding and multimerization, J Biol Chem 271(24) (1996) 14323–32. [DOI] [PubMed] [Google Scholar]
- [165].Mayasundari A, Whittemore NA, Serpersu EH, Peterson CB, The solution structure of the N-terminal domain of human vitronectin: proximal sites that regulate fibrinolysis and cell migration, J Biol Chem 279(28) (2004) 29359–66. [DOI] [PubMed] [Google Scholar]
- [166].Guettier C, Hinglais N, Bruneval P, Kazatchkine M, Bariety J, Camilleri JP, Immunohistochemical localization of S protein/vitronectin in human atherosclerotic versus arteriosclerotic arteries, Virchows Arch A Pathol Anat Histopathol 414(4) (1989) 309–13. [DOI] [PubMed] [Google Scholar]
- [167].Niculescu F, Rus HG, Vlaicu R, Immunohistochemical localization of C5b-9, S-protein, C3d and apolipoprotein B in human arterial tissues with atherosclerosis, Atherosclerosis 65(1–2) (1987) 1–11. [DOI] [PubMed] [Google Scholar]
- [168].Thiagarajan P, Kelly KL, Exposure of binding sites for vitronectin on platelets following stimulation, J Biol Chem 263(6) (1988) 3035–8. [PubMed] [Google Scholar]
- [169].Salazar-Pelaez LM, Abraham T, Herrera AM, Correa MA, Ortega JE, Pare PD, Seow CY, Vitronectin expression in the airways of subjects with asthma and chronic obstructive pulmonary disease, PLoS One 10(3) (2015) e0119717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Seiffert D, Keeton M, Eguchi Y, Sawdey M, Loskutoff DJ, Detection of vitronectin mRNA in tissues and cells of the mouse, Proc Natl Acad Sci U S A 88(21) (1991) 9402–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Barnes DW, Silnutzer J, Isolation of human serum spreading factor, J Biol Chem 258(20) (1983) 12548–52. [PubMed] [Google Scholar]
- [172].Conlan MG, Tomasini BR, Schultz RL, Mosher DF, Plasma vitronectin polymorphism in normal subjects and patients with disseminated intravascular coagulation, Blood 72(1) (1988) 185–90. [PubMed] [Google Scholar]
- [173].Dufourcq P, Louis H, Moreau C, Daret D, Boisseau MR, Lamaziere JM, Bonnet J, Vitronectin expression and interaction with receptors in smooth muscle cells from human atheromatous plaque, Arterioscler Thromb Vasc Biol 18(2) (1998) 168–76. [DOI] [PubMed] [Google Scholar]
- [174].Bennett JS, Berger BW, Billings PC, The structure and function of platelet integrins, J Thromb Haemost 7 Suppl 1 (2009) 200–5. [DOI] [PubMed] [Google Scholar]
- [175].Chakravarty D, Ray AG, Chander V, Mabalirajan U, Mondal PC, Siddiqui KN, Sinha BP, Konar A, Bandyopadhyay A, Systemic deficiency of vitronectin is associated with aortic inflammation and plaque progression in ApoE-Knockout mice, FASEB Bioadv 4(2) (2022) 121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Abolhasani S, Shahbazloo SV, Saadati HM, Mahmoodi N, Khanbabaei N, Evaluation of Serum Levels of Inflammation, Fibrinolysis and Oxidative Stress Markers in Coronary Artery Disease Prediction: A Cross-Sectional Study, Arq Bras Cardiol 113(4) (2019) 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Yaghoubi A, Ghojazadeh M, Abolhasani S, Alikhah H, Khaki-Khatibi F, Correlation of Serum Levels of Vitronectin, Malondialdehyde and Hs- CRP With Disease Severity in Coronary Artery Disease, J Cardiovasc Thorac Res 7(3) (2015) 113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Leavesley DI, Kashyap AS, Croll T, Sivaramakrishnan M, Shokoohmand A, Hollier BG, Upton Z, Vitronectin--master controller or micromanager?, IUBMB Life 65(10) (2013) 807–18. [DOI] [PubMed] [Google Scholar]
- [179].Brown SL, Lundgren CH, Nordt T, Fujii S, Stimulation of migration of human aortic smooth muscle cells by vitronectin: implications for atherosclerosis, Cardiovasc Res 28(12) (1994) 1815–20. [DOI] [PubMed] [Google Scholar]
- [180].Delannet M, Martin F, Bossy B, Cheresh DA, Reichardt LF, Duband JL, Specific roles of the alpha V beta 1, alpha V beta 3 and alpha V beta 5 integrins in avian neural crest cell adhesion and migration on vitronectin, Development 120(9) (1994) 2687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Dufourcq P, Couffinhal T, Alzieu P, Daret D, Moreau C, Duplaa C, Bonnet J, Vitronectin is upregulated after vascular injury and vitronectin blockade prevents neointima formation, Cardiovasc Res 53(4) (2002) 952–62. [DOI] [PubMed] [Google Scholar]
- [182].Ekmekci OB, Ekmekci H, Vitronectin in atherosclerotic disease, Clin Chim Acta 368(1–2) (2006) 77–83. [DOI] [PubMed] [Google Scholar]
- [183].Mori M, Iwasaki K, Sato R, Komine Y, Itabe H, Imanaka T, Takano T, Characterization of vitronectins in atherosclerotic lesions, J Atheroscler Thromb 3(1) (1996) 25–31. [DOI] [PubMed] [Google Scholar]
- [184].Chiquet-Ehrismann R, Chiquet M, Tenascins: regulation and putative functions during pathological stress, J Pathol 200(4) (2003) 488–99. [DOI] [PubMed] [Google Scholar]
- [185].Chiovaro F, Chiquet-Ehrismann R, Chiquet M, Transcriptional regulation of tenascin genes, Cell Adh Migr 9(1–2) (2015) 34–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Scherberich A, Tucker RP, Degen M, Brown-Luedi M, Andres AC, Chiquet-Ehrismann R, Tenascin-W is found in malignant mammary tumors, promotes alpha8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-alpha induced expression in vitro, Oncogene 24(9) (2005) 1525–32. [DOI] [PubMed] [Google Scholar]
- [187].Zisch AH, D’Alessandri L, Ranscht B, Falchetto R, Winterhalter KH, Vaughan L, Neuronal cell adhesion molecule contactin/F11 binds to tenascin via its immunoglobulin-like domains, J Cell Biol 119(1) (1992) 203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Yang H, Xiao ZC, Becker B, Hillenbrand R, Rougon G, Schachner M, Role for myelin-associated glycoprotein as a functional tenascin-R receptor, J Neurosci Res 55(6) (1999) 687–701. [DOI] [PubMed] [Google Scholar]
- [189].Lethias C, Elefteriou F, Parsiegla G, Exposito JY, Garrone R, Identification and characterization of a conformational heparin-binding site involving two fibronectin type III modules of bovine tenascin-X, J Biol Chem 276(19) (2001) 16432–8. [DOI] [PubMed] [Google Scholar]
- [190].Podesser BK, Kreibich M, Dzilic E, Santer D, Forster L, Trojanek S, Abraham D, Krssak M, Klein KU, Tretter EV, Kaun C, Wojta J, Kapeller B, Goncalves IF, Trescher K, Kiss A, Tenascin-C promotes chronic pressure overload-induced cardiac dysfunction, hypertrophy and myocardial fibrosis, J Hypertens 36(4) (2018) 847–856. [DOI] [PubMed] [Google Scholar]
- [191].Abbadi D, Laroumanie F, Bizou M, Pozzo J, Daviaud D, Delage C, Calise D, Gaits-Iacovoni F, Dutaur M, Tortosa F, Renaud-Gabardos E, Douin-Echinard V, Prats AC, Roncalli J, Parini A, Pizzinat N, Local production of tenascin-C acts as a trigger for monocyte/macrophage recruitment that provokes cardiac dysfunction, Cardiovasc Res 114(1) (2018) 123–137. [DOI] [PubMed] [Google Scholar]
- [192].Liu R, He Y, Li B, Liu J, Ren Y, Han W, Wang X, Zhang L, Tenascin-C produced by oxidized LDL-stimulated macrophages increases foam cell formation through Toll-like receptor-4, Mol Cells 34(1) (2012) 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Gao W, Li J, Ni H, Shi H, Qi Z, Zhu S, Hao C, Xie Q, Luo X, Xie K, Tenascin C: A Potential Biomarker for Predicting the Severity of Coronary Atherosclerosis, J Atheroscler Thromb 26(1) (2019) 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Mehri H, Aslanabadi N, Nourazarian A, Shademan B, Khaki-Khatibi F, Evaluation of the serum levels of Mannose binding lectin-2, tenascin-C, and total antioxidant capacity in patients with coronary artery disease, J Clin Lab Anal 35(10) (2021) e23967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Gholipour A, Shakerian F, Zahedmehr A, Oveisee M, Maleki M, Mowla SJ, Malakootian M, Tenascin-C as a noninvasive biomarker of coronary artery disease, Mol Biol Rep 49(10) (2022) 9267–9273. [DOI] [PubMed] [Google Scholar]
- [196].Liabeuf S, Barreto DV, Kretschmer A, Barreto FC, Renard C, Andrejak M, Choukroun G, Massy Z, High circulating levels of large splice variants of tenascin-C is associated with mortality and cardiovascular disease in chronic kidney disease patients, Atherosclerosis 215(1) (2011) 116–24. [DOI] [PubMed] [Google Scholar]
- [197].Li Y, Liu J, Huang JW, Song JC, Ma ZL, Shi HB, In vivo MRI detection of atherosclerosis in ApoE-deficient mice by using tenascin-C-targeted USPIO, Acta Radiol 59(12) (2018) 1431–1437. [DOI] [PubMed] [Google Scholar]
- [198].Hedin U, Holm J, Hansson GK, Induction of tenascin in rat arterial injury. Relationship to altered smooth muscle cell phenotype, Am J Pathol 139(3) (1991) 649–56. [PMC free article] [PubMed] [Google Scholar]
- [199].Luo H, Wang J, Qiao C, Zhang X, Zhang W, Ma N, ATF3 Inhibits Tenascin-C-induced Foam Cell Formation in LPS-Stimulated THP-1 Macrophages by Suppressing TLR-4, J Atheroscler Thromb 22(11) (2015) 1214–23. [DOI] [PubMed] [Google Scholar]
- [200].Wallner K, Li C, Shah PK, Fishbein MC, Forrester JS, Kaul S, Sharifi BG, Tenascin-C is expressed in macrophage-rich human coronary atherosclerotic plaque, Circulation 99(10) (1999) 1284–9. [DOI] [PubMed] [Google Scholar]
- [201].Maqbool A, Spary EJ, Manfield IW, Ruhmann M, Zuliani-Alvarez L, Gamboa-Esteves FO, Porter KE, Drinkhill MJ, Midwood KS, Turner NA, Tenascin C upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4, World J Cardiol 8(5) (2016) 340–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F, Foxwell B, Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease, Nat Med 15(7) (2009) 774–80. [DOI] [PubMed] [Google Scholar]
- [203].Wang Z, Wei Q, Han L, Cao K, Lan T, Xu Z, Wang Y, Gao Y, Xue J, Shan F, Feng J, Xie X, Tenascin-c renders a proangiogenic phenotype in macrophage via annexin II, J Cell Mol Med 22(1) (2018) 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Fukumoto H, Naito Z, Asano G, Aramaki T, Immunohistochemical and morphometric evaluations of coronary atherosclerotic plaques associated with myocardial infarction and diabetes mellitus, J Atheroscler Thromb 5(1) (1998) 29–35. [DOI] [PubMed] [Google Scholar]
- [205].Wang L, Shah PK, Wang W, Song L, Yang M, Sharifi BG, Tenascin-C deficiency in apo E−/− mouse increases eotaxin levels: implications for atherosclerosis, Atherosclerosis 227(2) (2013) 267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Brinchmann MF, Patel DM, Iversen MH, The Role of Galectins as Modulators of Metabolism and Inflammation, Mediators Inflamm 2018 (2018) 9186940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Rabinovich GA, Toscano MA, Ilarregui JM, Rubinstein N, Shedding light on the immunomodulatory properties of galectins: novel regulators of innate and adaptive immune responses, Glycoconj J 19(7–9) (2002) 565–73. [DOI] [PubMed] [Google Scholar]
- [208].Elola MT, Wolfenstein-Todel C, Troncoso MF, Vasta GR, Rabinovich GA, Galectins: matricellular glycan-binding proteins linking cell adhesion, migration, and survival, Cell Mol Life Sci 64(13) (2007) 1679–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Sato S, St-Pierre C, Bhaumik P, Nieminen J, Galectins in innate immunity: dual functions of host soluble beta-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs), Immunol Rev 230(1) (2009) 172–87. [DOI] [PubMed] [Google Scholar]
- [210].Priglinger CS, Obermann J, Szober CM, Merl-Pham J, Ohmayer U, Behler J, Gruhn F, Kreutzer TC, Wertheimer C, Geerlof A, Priglinger SG, Hauck SM, Epithelial-to-Mesenchymal Transition of RPE Cells In Vitro Confers Increased beta1,6-N-Glycosylation and Increased Susceptibility to Galectin-3 Binding, PLoS One 11(1) (2016) e0146887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Alge CS, Priglinger SG, Kook D, Schmid H, Haritoglou C, Welge-Lussen U, Kampik A, Galectin-1 influences migration of retinal pigment epithelial cells, Invest Ophthalmol Vis Sci 47(1) (2006) 415–26. [DOI] [PubMed] [Google Scholar]
- [212].Wang Z, Handa JT, Green WR, Stark WJ, Weinberg RS, Jun AS, Advanced glycation end products and receptors in Fuchs’ dystrophy corneas undergoing Descemet’s stripping with endothelial keratoplasty, Ophthalmology 114(8) (2007) 1453–60. [DOI] [PubMed] [Google Scholar]
- [213].Wu D, Kanda A, Liu Y, Kase S, Noda K, Ishida S, Galectin-1 promotes choroidal neovascularization and subretinal fibrosis mediated via epithelial-mesenchymal transition, FASEB J 33(2) (2019) 2498–2513. [DOI] [PubMed] [Google Scholar]
- [214].Ridano ME, Subirada PV, Paz MC, Lorenc VE, Stupirski JC, Gramajo AL, Luna JD, Croci DO, Rabinovich GA, Sanchez MC, Galectin-1 expression imprints a neurovascular phenotype in proliferative retinopathies and delineates responses to anti-VEGF, Oncotarget 8(20) (2017) 32505–32522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Kanda A, Dong Y, Noda K, Saito W, Ishida S, Advanced glycation endproducts link inflammatory cues to upregulation of galectin-1 in diabetic retinopathy, Sci Rep 7(1) (2017) 16168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Abu El-Asrar AM, Ahmad A, Allegaert E, Siddiquei MM, Alam K, Gikandi PW, De Hertogh G, Opdenakker G, Galectin-1 studies in proliferative diabetic retinopathy, Acta Ophthalmol 98(1) (2020) e1–e12. [DOI] [PubMed] [Google Scholar]
- [217].van der Hoeven NW, Hollander MR, Yildirim C, Jansen MF, Teunissen PF, Horrevoets AJ, van der Pouw Kraan TC, van Royen N, The emerging role of galectins in cardiovascular disease, Vascul Pharmacol 81 (2016) 31–41. [DOI] [PubMed] [Google Scholar]
- [218].Seropian IM, Gonzalez GE, Maller SM, Berrocal DH, Abbate A, Rabinovich GA, Galectin-1 as an Emerging Mediator of Cardiovascular Inflammation: Mechanisms and Therapeutic Opportunities, Mediators Inflamm 2018 (2018) 8696543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Seropian IM, Cerliani JP, Toldo S, Van Tassell BW, Ilarregui JM, Gonzalez GE, Matoso M, Salloum FN, Melchior R, Gelpi RJ, Stupirski JC, Benatar A, Gomez KA, Morales C, Abbate A, Rabinovich GA, Galectin-1 controls cardiac inflammation and ventricular remodeling during acute myocardial infarction, Am J Pathol 182(1) (2013) 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Chou RH, Huang SS, Kuo CS, Wang SC, Tsai YL, Lu YW, Chang CC, Huang PH, Lin SJ, Galectin-1 is associated with the severity of coronary artery disease and adverse cardiovascular events in patients undergoing coronary angiography, Sci Rep 10(1) (2020) 20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].He XW, Li WL, Li C, Liu P, Shen YG, Zhu M, Jin XP, Serum levels of galectin-1, galectin-3, and galectin-9 are associated with large artery atherosclerotic stroke, Sci Rep 7 (2017) 40994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Lee YJ, Koh YS, Park HE, Lee HJ, Hwang BH, Kang MK, Lee SY, Kim PJ, Ihm SH, Seung KB, Chang K, Spatial and temporal expression, and statin responsiveness of galectin-1 and galectin-3 in murine atherosclerosis, Korean Circ J 43(4) (2013) 223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Moiseeva EP, Williams B, Goodall AH, Samani NJ, Galectin-1 interacts with beta-1 subunit of integrin, Biochem Biophys Res Commun 310(3) (2003) 1010–6. [DOI] [PubMed] [Google Scholar]
- [224].Tsai MS, Chiang MT, Tsai DL, Yang CW, Hou HS, Li YR, Chang PC, Lin HH, Chen HY, Hwang IS, Wei PK, Hsu CP, Lin KI, Liu FT, Chau LY, Galectin-1 Restricts Vascular Smooth Muscle Cell Motility Via Modulating Adhesion Force and Focal Adhesion Dynamics, Sci Rep 8(1) (2018) 11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Roldan-Montero R, Perez-Saez JM, Cerro-Pardo I, Oller J, Martinez-Lopez D, Nunez E, Maller SM, Gutierrez-Munoz C, Mendez-Barbero N, Escola-Gil JC, Michel JB, Mittelbrunn M, Vazquez J, Blanco-Colio LM, Rabinovich GA, Martin-Ventura JL, Galectin-1 prevents pathological vascular remodeling in atherosclerosis and abdominal aortic aneurysm, Sci Adv 8(11) (2022) eabm7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Jeanne A, Sarazin T, Charle M, Kawecki C, Kauskot A, Hedtke T, Schmelzer CEH, Martiny L, Maurice P, Dedieu S, Towards the Therapeutic Use of Thrombospondin 1/CD47 Targeting TAX2 Peptide as an Antithrombotic Agent, Arterioscler Thromb Vasc Biol 41(1) (2021) e1–e17. [DOI] [PubMed] [Google Scholar]
- [227].Yao M, Sturdivant J, Ebrahimi A, Ganguly S, Elbayoumi T, Novel Pharmaceutical Strategy for Selective Abrogation of TSP1-Induced Vascular Dysfunction by Decoy Recombinant CD47 Soluble Receptor in Prophylaxis and Treatment Models, Biomedicines 9(6) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Patel MK, Lymn JS, Clunn GF, Hughes AD, Thrombospondin-1 is a potent mitogen and chemoattractant for human vascular smooth muscle cells, Arterioscler Thromb Vasc Biol 17(10) (1997) 2107–14. [DOI] [PubMed] [Google Scholar]
- [229].Yabkowitz R, Mansfield PJ, Ryan US, Suchard SJ, Thrombospondin mediates migration and potentiates platelet-derived growth factor-dependent migration of calf pulmonary artery smooth muscle cells, J Cell Physiol 157(1) (1993) 24–32. [DOI] [PubMed] [Google Scholar]
- [230].Nevitt C, McKenzie G, Christian K, Austin J, Hencke S, Hoying J, LeBlanc A, Physiological levels of thrombospondin-1 decrease NO-dependent vasodilation in coronary microvessels from aged rats, Am J Physiol Heart Circ Physiol 310(11) (2016) H1842–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD, Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner, Proc Natl Acad Sci U S A 102(37) (2005) 13141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Tong X, Khandelwal AR, Qin Z, Wu X, Chen L, Ago T, Sadoshima J, Cohen RA, Role of smooth muscle Nox4-based NADPH oxidase in neointimal hyperplasia, J Mol Cell Cardiol 89(Pt B) (2015) 185–94. [DOI] [PubMed] [Google Scholar]
- [233].Tong X, Khandelwal AR, Wu X, Xu Z, Yu W, Chen C, Zhao W, Yang J, Qin Z, Weisbrod RM, Seta F, Ago T, Lee KS, Hammock BD, Sadoshima J, Cohen RA, Zeng C, Pro-atherogenic role of smooth muscle Nox4-based NADPH oxidase, J Mol Cell Cardiol 92 (2016) 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Wang M, Fu Y, Gao C, Jia Y, Huang Y, Liu L, Wang X, Wang W, Kong W, Cartilage oligomeric matrix protein prevents vascular aging and vascular smooth muscle cells senescence, Biochem Biophys Res Commun 478(2) (2016) 1006–13. [DOI] [PubMed] [Google Scholar]
- [235].Grote K, Salguero G, Ballmaier M, Dangers M, Drexler H, Schieffer B, The angiogenic factor CCN1 promotes adhesion and migration of circulating CD34+ progenitor cells: potential role in angiogenesis and endothelial regeneration, Blood 110(3) (2007) 877–85. [DOI] [PubMed] [Google Scholar]
- [236].Bai T, Chen CC, Lau LF, Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages, J Immunol 184(6) (2010) 3223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Hirota S, Imakita M, Kohri K, Ito A, Morii E, Adachi S, Kim HM, Kitamura Y, Yutani C, Nomura S, Expression of osteopontin messenger RNA by macrophages in atherosclerotic plaques. A possible association with calcification, Am J Pathol 143(4) (1993) 1003–8. [PMC free article] [PubMed] [Google Scholar]
- [238].Ohmori R, Momiyama Y, Taniguchi H, Takahashi R, Kusuhara M, Nakamura H, Ohsuzu F, Plasma osteopontin levels are associated with the presence and extent of coronary artery disease, Atherosclerosis 170(2) (2003) 333–7. [DOI] [PubMed] [Google Scholar]
- [239].Strom A, Franzen A, Wangnerud C, Knutsson AK, Heinegard D, Hultgardh-Nilsson A, Altered vascular remodeling in osteopontin-deficient atherosclerotic mice, J Vasc Res 41(4) (2004) 314–22. [DOI] [PubMed] [Google Scholar]
- [240].Minear MA, Crosslin DR, Sutton BS, Connelly JJ, Nelson SC, Gadson-Watson S, Wang T, Seo D, Vance JM, Sketch MH Jr., Haynes C, Goldschmidt-Clermont PJ, Shah SH, Kraus WE, Hauser ER, Gregory SG, Polymorphic variants in tenascin-C (TNC) are associated with atherosclerosis and coronary artery disease, Hum Genet 129(6) (2011) 641–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Malik RK, Ghurye RR, Lawrence-Watt DJ, Stewart HJ, Galectin-1 stimulates monocyte chemotaxis via the p44/42 MAP kinase pathway and a pertussis toxin-sensitive pathway, Glycobiology 19(12) (2009) 1402–7. [DOI] [PubMed] [Google Scholar]