

Abstract
Transthyretin (TTR) dissociation is the rate limiting step for both aggregation and subunit exchange. Kinetic stabilizers, small molecules that bind to the native tetrameric structure of TTR, slow TTR dissociation and inhibit aggregation. One such stabilizer is the non-steroidal anti-inflammatory drug (NSAID), diflunisal, which has been repurposed to treat TTR polyneuropathy. Previously, we compared the efficacy of diflunisal, tafamidis, tolcapone, and AG10 as kinetic stabilizers for transthyretin. However, we could not meaningfully compare diflunisal because we were unsure of its plasma concentration after long-term oral dosing. Herein, we report the diflunisal plasma concentrations measured by extraction, reversed phase HPLC separation, and fluorescence detection after long-term 250 mg BID oral dosing in two groups: a placebo-controlled diflunisal clinical trial group and an open-label Japanese polyneuropathy treatment cohort. The measured mean diflunisal plasma concentration from both groups was 281 μM ±144 μM (mean ± standard deviation). Thus, quantification of TTR kinetic stabilization using subunit exchange was carried out at 100, 200, 300, and 400 μM diflunisal concentrations, all observed in patients after 250 mg BID oral dosing. A 250 μM diflunisal plasma concentration reduced the wild-type TTR dissociation rate in plasma by 95%, which is sufficient to stop transthyretin aggregation, consistent with the clinical efficacy of diflunisal for ameliorating transthyretin polyneuropathy.
Background
Transthyretin (TTR) tetramer dissociation is the rate-limiting step for both subunit exchange and aggregation, as the generation of monomers are required for both processes (1–7). TTR kinetic stabilizers slow TTR tetramer dissociation in a dose-dependent fashion, inhibiting TTR aggregation (8–13). While we recently reported a blinded potency comparison of the TTR kinetic stabilizers tafamidis, AG10, and tolcapone at pharmacologically relevant concentrations by subunit exchange in human plasma (13), a caveat of this study is that we could not meaningfully compare the repurposed non-steroidal anti-inflammatory drug (NSAID), diflunisal, because we were unsure of its plasma concentration after long-term oral dosing (14–20).
Diflunisal (250 mg BID) is established to reduce the rate of progression of neurologic impairment in patients with transthyretin familial amyloid polyneuropathy (FAP) based on results from a placebo-controlled randomized clinical trial (21). Here we report the quantification of the diflunisal concentrations in the plasma samples from participants in the placebo-controlled diflunisal clinical trial using methodology previously utilized for tafamidis (12, 21, 22). We also quantified diflunisal levels in the plasma of Japanese polyneuropathy patients treated with oral diflunisal (250 mg BID) in an open-label study (16). Knowing the diflunisal plasma concentration ranges in the clinical trial samples (250 mg BID) and in the Japanese open-label study (250 mg BID) allowed us to add diflunisal back to healthy plasma to determine the extent of wild type (WT) TTR kinetic stabilization over this range of diflunisal plasma concentrations.
Materials and Methods
Detailed experimental procedures can be found in the Supplementary Material for diflunisal extraction from human plasma, for the high-performance liquid chromatography quantification of plasma diflunisal concentration, and for performing the subunit exchange assay to assess kinetic stabilization of transthyretin.
Results
Diflunisal plasma concentrations achieved by 250 mg BID oral dosing
Diflunisal plasma levels were measured in 50 plasma samples from the placebo-controlled clinical trial (Figure 1A and 1B) (21). These polyneuropathy patients were either on 250 mg diflunisal BID or placebo. Twenty-three samples exhibited a peak at ~14.3 min in the HPLC chromatogram (Figure 1C, red filled circles). We confirmed that this peak was diflunisal using liquid chromatography/negative ion mass spectrometry (m/z 248.9). The remainder of the patient samples showed no signal, consistent with these patients being in the placebo group. The majority of samples (21/23) exhibited diflunisal plasma concentrations ranging from ≈ 100 to 400 μM (Figure 1C, red filled circles), approximating the trough and average plasma concentrations upon chronic 250 mg BID dosing of hereditary ATTR amyloidosis patients (19, 20, 23). The FDA package insert cites a mean plasma concentration of 56 μg/mL, or 224 μM, for the 250 BID diflunisal dose. Among the placebo-controlled clinical trial samples, the mean diflunisal plasma concentration was 274.9 μΜ. Higher diflunisal concentrations were exhibited by two samples (≈ 600 μM and ≈ 760 μM), likely approximating the peak concentrations realized by oral dosing, which is achieved 2–3 h after oral dosing (19, 20, 23).
Figure 1.

(A) Method to quantify diflunisal concentration and its metabolites in human plasma. A standard diflunisal concentration curve was produced by adding known diflunisal amounts to a pooled healthy plasma sample ex-vivo (see Supplementary Information for more details). Diflunisal was extracted from the macromolecules comprising a 40 μL plasma sample using 200 μL of acetonitrile containing 1% (w/v) trichloroacetic acid. Ten μL of the microcentrifuged supernatant (one solution phase) was injected onto a Waters Symmetry reversed-phase C18 column (3.5 μm, 4.6 × 75 mm) eluted with a gradient of 0–100% solution B over 20 min (solution A: 99.9% water, 0.1% trifluoroacetic acid; solution B: 99.9% acetonitrile, 0.1% trifluoracetic acid) using a flow rate of 1 ml/min. The excitation and emission wavelengths employed for fluorescence detection were λex=254 nm and λem=424 nm. (B) Diflunisal was extracted from patient plasma samples and analysed exactly the same as for the standard curve generation. The HPLC chromatogram from a patient sample shows the diflunisal peak at ~14.3 min and the two indicated diflunisal-glucuronide metabolites at ~10.1 and ~11.1 min (C) Measured diflunisal concentration in polyneuropathy patient plasma samples after long-term treatment with diflunisal (250 mg BID). Depicted in red filled circles are the plasma diflunisal concentrations from the placebo-controlled trial and in blue filled circles are the plasma diflunisal concentrations from the Japanese open-label study. The mean diflunisal plasma concentrations for the two groups are 275.0 μM (red) and 289.4 μM (blue). (D) Gender and age comparisons of mean diflunisal-plasma concentration amongst polyneuropathy patient samples from the combined cohorts. There is no significant difference in mean diflunisal-plasma concentration between genders (male: 272.0 μM, female: 298.0 μM). There is a significant difference when comparing age (under 65: 217.7 μM, over 65: 332.1 μM; p = .006). p Values were calculated using an unpaired t-test. Horizontal lines represent the mean and error bars represent the standard deviation.
Diflunisal concentrations were also measured in 23 plasma samples from Japanese individuals participating in a diflunisal (250 mg BID) open-label clinical study at the Shinshu University School of Medicine (16). The mean diflunisal plasma concentration in this cohort was 287.1 μM (Figure 1C, blue filled circles). The majority of samples exhibited diflunisal plasma concentrations comparable with the clinical trial, ranging from ≈ 100 to 500 μM; however, one sample possessed a higher concentration outside this range (≈ 670 μM; Figure 1C, blue filled circles).
We observed two additional peaks in the HPLC chromatograms at ~10.1 and ~11.1 min that were identified as the diflunisal metabolites, diflunisal acyl glucuronide and diflunisal phenolic glucuronide, both exhibiting a molecular weight of 426.32 (Figure 1B). Both metabolites gave [M-H]− peaks of 425.0, confirming their identity (24). While the plasma diflunisal peak at ~14.3 min reflected the majority of the total HPLC chromatographic area (69.8 – 85.8%) in treated patients, the metabolite peak at ~10.1 min ranged from 6.5 – 18.9% of the total chromatographic peak area and the metabolite peak at ~11.1 min constituted 5.8 – 15.4% of the total chromatographic peak area.
We found no significant difference when we compared the mean diflunisal plasma concentration in the combined treatment groups by gender (male: 293.1 ± 131.6 μM; female: 298.0 ± 169.5 μM) (Figure 1D). However, we did see a significant difference in the mean diflunisal plasma concentrations between treated patient samples when we compared by age (under 65: 208.5 ± 112.0 μM; over 65: 329.6 ± 143.3 μM; p = 0.004) (Figure 1D). The mean diflunisal plasma concentration of 281 ± 144 μM (mean ± SD) was derived from quantifying the concentration of diflunisal from the plasma samples of the combined treatment groups (Figure 1C, red and blue filled circles). Patient demographics were similar amongst the two groups. In the Clinical Trial cohort, 9/23 patients were under 65 and 13/23 were male. Among the Japanese group, 9/23 patients were under 65 and 15/23 were male. The most prevalent mutation in both groups was V30M (13/23 in the Clinical trial group and 14/23 in the Japanese cohort). Other mutations harbored by the patients were: E42G, T60A, Y114C, H90N, T58H, T60A, D38A, S50R, I107V, S50I, and E54K.
Subunit exchange assay to assess TTR kinetic stability
Subunit exchange between endogenous plasma WT TTR (~3.7 μM) and recombinant dual-FLAG-tagged Cys10Ala TTR (FT2-C10A TTR) added at a final concentration of 1 μM to pooled healthy human plasma samples was monitored over seven days, at final diflunisal concentrations of 100, 200, 300, and 400 μM versus vehicle control (DMSO) (Figure 2). In order to determine the concentration of endogenous tetrameric TTR in our pooled healthy plasma sample (3 donors, see Supplementary Material), we compared the integrated area of Peak 1 (endogenous native TTR tetramer) with that of Peak 5 (FT2-C10A-TTR) before subunit exchange occurred. The ratio afforded a value of 3.7 μM for the endogenous TTR tetramer concentration. We had planned to quantify diflunisal-mediated TTR stabilization in the plasma of the diflunisal familial polyneuropathy clinical trial participants by subunit exchange, however the high viscosity of the plasma samples, likely due to plasma thawing and the plasma heating during shipping, precluded this. The apparent rate constant for TTR subunit exchange (kex) determined in this study in the absence of any kinetic stabilizer is 0.0151 h−1, consistent with the rate constant from prior subunit exchange data (kex = 0.0149 h−1) (13). The plasma TTR subunit exchange rate constant (kex) at a final diflunisal plasma concentration of 100 μM decreased to 0.0027 h−1, at 200 μM diflunisal it decreased to kex = 0.0010 h−1, at 300 μM diflunisal it decreased to kex = 0.0006 h−1, and at a 400 μM diflunisal concentration the subunit exchange rate decreased to 0.0003 h-1. In our previous paper (13), we assessed subunit exchange between plasma WT TTR and dual-FLAG-tagged WT TTR (FT2-WT TTR; employing a final concentration = 1 μM) at 1, 5, 10, 20, and 30 μM final diflunisal plasma concentrations, all concentrations below those that are pharmacologically relevant at the standard 250 mg BID dose, but were germane to the prior publication in that they enabled direct comparisons with the plasma concentrations of tolcapone, tafamidis, and AG10 achieved by oral dosing (13). From the data reported by Nelson et al. (13) we were able to conclude that the primary determinant of a kinetic stabilizer’s efficacy at a given concentration for inhibiting TTR tetramer dissociation in plasma is the spread between its dissociation constant from the first TTR thyroxine binding site engaged (KD1) (10) and its dissociation constant from albumin (KD,Alb = 1.2 ± 0.1 μM for diflunisal) (13). The KD,Alb / KD1 ratio decreases in the order of AG10 (KD,Alb / KD1=1980), tafamidis (KD,Alb / KD1=580) and diflunisal (KD,Alb / KD1=16) (13).
Figure 2.
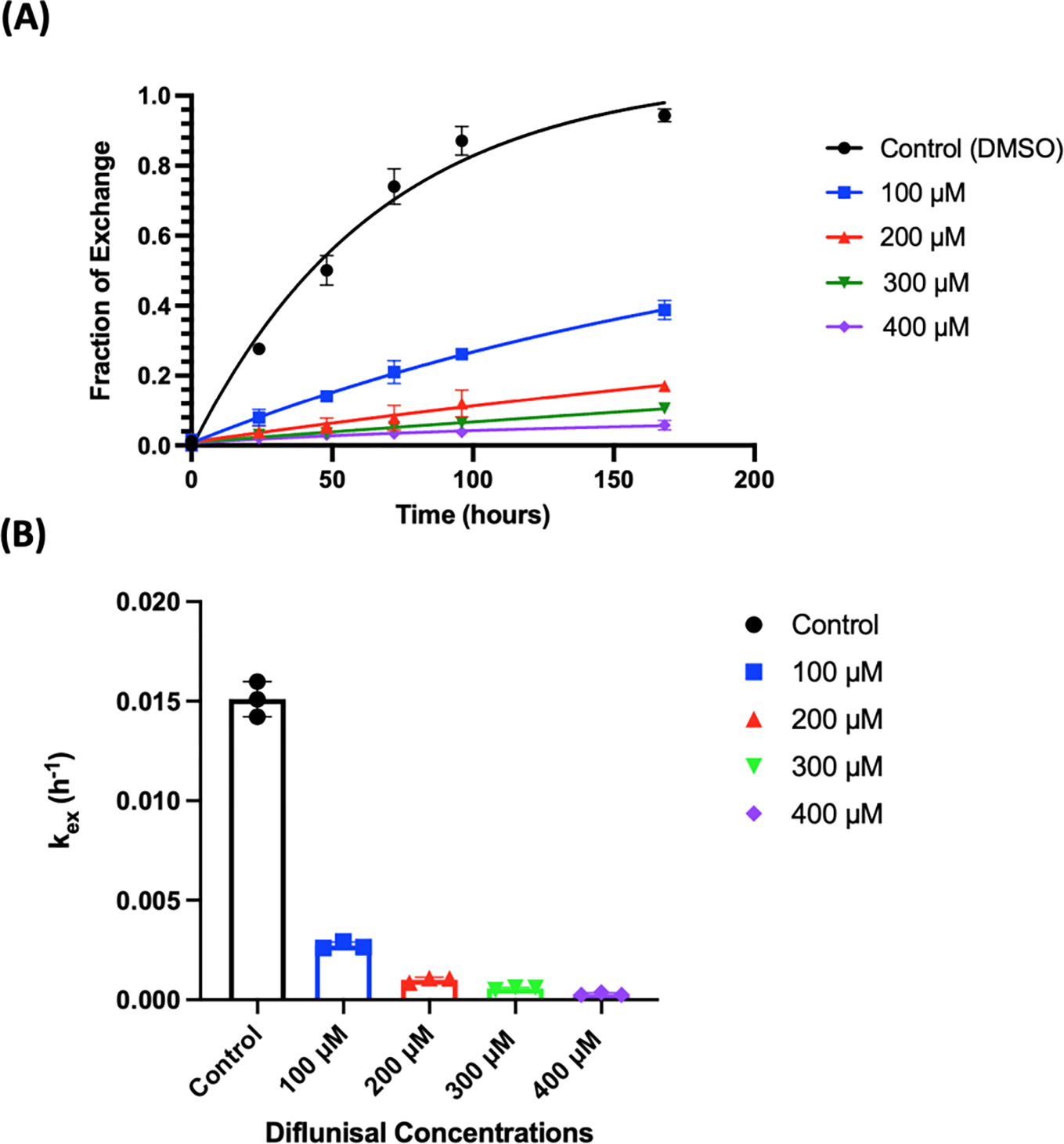
Time courses of subunit exchange between endogenous plasma WT TTR (~ 3.7 μM tetramer) and added dual-FLAG-tagged Cys10Ala TTR (FT2-C10A TTR; 1 μM tetramer) at the indicated diflunisal concentrations. (A) The x-axis shows time in hours and the y-axis depicts the fraction of exchange calculated from the area under peak 3 in the ion-exchange chromatograms. All timepoints were measured in biological triplicate. (B) The decrease in the plasma TTR subunit exchange rate constant (increase in pharmacological kinetic stabilization) correlates with the increase in plasma diflunisal concentrations.
Fitting the data obtained herein from the subunit exchange between WT TTR in plasma and FT2-C10A TTR added to a final concentration of 1 μM in the presence of 100, 200, 300 and 400 μM final diflunisal plasma concentrations (data summarized in Figure 2) (13), allows us to more accurately model TTR subunit exchange at pharmacologically relevant diflunisal concentrations. Given the KD1 = 75 nM and KD2 = 1.1 μM TTR•diflunisal dissociation equilibrium constants, we find that the best fit value of KD, Alb is 1.4 ± 0.1 μM. This value was determined using a previously published method which can be found in the Supplemental Materials section of Nelson et al (13). Thus, by using a pharmacologically relevant range of diflunisal concentrations, our KD,Alb / KD1 becomes 18.7, similar to the value 16 we calculated with the 1 – 30 μM diflunisal concentration range (13). Based on this value of KD,Alb and the known KD1 and KD2 for diflunisal dissociation from TTR, we calculate that dissociation of TTR can be limited to 10% of its normal rate at a diflunisal concentration of 172 μM (188 μM was the value extracted from the 1 – 30 μM diflunisal subunit exchange data (13)). For comparison, 10% of the normal subunit exchange rate is achieved with 12.0 μM tafamidis and 5.7 μM AG10 plasma concentrations. Dissociation of WT TTR can be limited to 5% of its normal rate at diflunisal, tafamidis and AG10 concentrations of 250 μM, 20.7 μM, and 8.5 μM, respectively.
Discussion and Conclusion
These data and prior data (13) demonstrate that any desired level of TTR stabilization can be attained in human plasma by adjusting the kinetic stabilizer oral dose, which determines the kinetic stabilizer plasma concentration, safety attributes permitting. Collectively, our results herein and previously reported (13) clearly demonstrate that oral dosing of 500 mg diflunisal / day, 61 mg tafamidis / day, and 1600 mg of AG10 / day achieves comparable WT TTR kinetic stabilization, i.e., these daily doses limit the dissociation of WT TTR to 5% of its normal rate at diflunisal, tafamidis and AG10 plasma concentrations of 250 μM, 20.7 μM, and 8.5 μM, respectively (13). Owing to this, differences in safety, pharmacokinetics, adsorption, metabolism, tissue distribution, and tissue pharmacodynamics become important characteristics to consider when deciding which kinetic stabilizer to use. As the diflunisal dose used for TTR kinetic stabilization is below doses typically required to treat arthritis, diflunisal has proven to be remarkably well tolerated in patients with ATTR amyloid polyneuropathy (16, 21, 25). Nonetheless, potential NSAID-related decreases in renal blood flow and increased gastrointestinal bleeding risk warrant monitoring. While it remains unclear what minimal reduction in tetramer dissociation rate correlates with a maximum clinical response, published literature is starting to provide some guidance (16, 17, 21, 26–28). We previously found that the average tafamidis concentration in patients with TTR polyneuropathy dosed with the Vyndaqel 20 mg once daily formulation of tafamidis was 8.2 μM, and that this concentration was sufficient to achieve a clinical response in about two-thirds of these patients (12). This concentration of tafamidis would suppress the WT TTR dissociation rate to about 16% of normal (less dissociation rate suppression would be observed in heterozygous polyneuropathy patients having predominantly heterotetramers in solution that bind the kinetic stabilizers less well, e.g., V30M heterozygotes; this would almost certainly be the case for diflunisal as well). Suppressing the WT TTR dissociation rate to about 16% of normal can be achieved at a plasma diflunisal concentration of 124 μM, which is achieved in the vast majority of the patients evaluated in this study.
Supplementary Material
Acknowledgments
The repurposing of diflunisal and the discovery of tafamidis for ATTR amyloidosis would not have been possible without sustained NIH funding (DK 046335) for the Kelly lab. Figures were made with BioRender.com.
Footnotes
Disclosure statement
JWK and ETP discovered tafamidis, receive sales royalties and sales milestone payments.
References
- 1.Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31(36):8654–60. Epub 1992/09/15. [DOI] [PubMed] [Google Scholar]
- 2.Lai Z, Colon W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 1996;35(20):6470–82. Epub 1996/05/21. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 3.Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99 Suppl 4:16427–32. Epub 2002/09/28. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40(38):11442–52. Epub 2001/09/19. [DOI] [PubMed] [Google Scholar]
- 5.Wiseman RL, Green NS, Kelly JW. Kinetic stabilization of an oligomeric protein under physiological conditions demonstrated by a lack of subunit exchange: implications for transthyretin amyloidosis. Biochemistry. 2005;44(25):9265–74. Epub 2005/06/22. doi: 10.1021/bi050352o. [DOI] [PubMed] [Google Scholar]
- 6.Foss TR, Kelker MS, Wiseman RL, Wilson IA, Kelly JW. Kinetic stabilization of the native state by protein engineering: implications for inhibition of transthyretin amyloidogenesis. Journal of Molecular Biology. 2005;347(4):841–54. Epub 2005/03/17. doi: 10.1016/j.jmb.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 7.Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry. 2005;44(47):15525–33. Epub 2005/11/23. doi: 10.1021/bi051608t. [DOI] [PubMed] [Google Scholar]
- 8.Cho Y, Baranczak A, Helmke S, Teruya S, Horn EM, Maurer MS, Kelly JW. Personalized medicine approach for optimizing the dose of tafamidis to potentially ameliorate wild-type transthyretin amyloidosis (cardiomyopathy). Amyloid-Journal of Protein Folding Disorders. 2015;22(3):175–80. doi: 10.3109/13506129.2015.1063485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rappley I, Monteiro C, Novais M, Baranczak A, Solis G, Wiseman RL, Helmke S, Maurer MS, Coelho T, Powers ET, Kelly JW. Quantification of Transthyretin Kinetic Stability in Human Plasma Using Subunit Exchange. Biochemistry. 2014;53(12):1993–2006. doi: 10.1021/bi500171j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299(5607):713–6. Epub 2003/02/01. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 11.Bulawa CE, Connelly S, DeVit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudiniere R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(Copyright (C) 2012 American Chemical Society (ACS). All Rights Reserved.):9629–34, S/1-S/9. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monteiro C, Mesgazardeh JS, Anselmo J, Fernandes J, Novais M, Rodrigues C, Brighty GJ, Powers DL, Powers ET, Coelho T, Kelly JW. Predictive model of response to tafamidis in hereditary ATTR polyneuropathy. JCI Insight. 2019;4(12). doi: 10.1172/jci.insight.126526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson LT, Paxman RJ, Xu J, Webb B, Powers ET, Kelly JW. Blinded potency comparison of transthyretin kinetic stabilisers by subunit exchange in human plasma. Amyloid-Journal of Protein Folding Disorders. 2021;28(1):24–9. doi: 10.1080/13506129.2020.1808783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. Journal of medicinal chemistry. 2004;47(2):355–74. Epub 2004/01/09. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 15.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2006;13(4):236–49. Epub 2006/11/17. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 16.Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid-Journal of Protein Folding Disorders. 2015;22(2):79–83. doi: 10.3109/13506129.2014.997872. [DOI] [PubMed] [Google Scholar]
- 17.Rosenblum H, Castano A, Alvarez J, Goldsmith J, Helmke S, Maurer MS. TTR (Transthyretin) Stabilizers Are Associated With Improved Survival in Patients With TTR Cardiac Amyloidosis. Circulation-Heart Failure. 2018;11(4). doi: 10.1161/circheartfailure.117.004769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tojo K, Sekijima Y, Kelly JW, Ikeda SI. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neuroscience Research. 2006;56(4):441–9. doi: 10.1016/j.neures.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Nuernberg B, Koehler G, Brune K. Pharmacokinetics of diflunisal in patients. Clin Pharmacokinet. 1991;20(1):81–9. doi: 10.2165/00003088-199120010-00006. [DOI] [PubMed] [Google Scholar]
- 20.Tempero KF, Cirillo VJ, Steelman SL. Diflunisal: a review of pharmacokinetic and pharmacodynamic properties, drug interactions, and special tolerability studies in humans. Br J Clin Pharmacol. 1977;4, Suppl. 1:31. doi: 10.1111/j.1365-2125.1977.tb04511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, Nordh E, Corato M, Lozza A, Cortese A, Robinson-Papp J, Colton T, Rybin DV, Bisbee AB, Ando Y, Ikeda S, Seldin DC, Merlini G, Skinner M, Kelly JW, Dyck PJ, Diflunisal Trial C. Repurposing Diflunisal for Familial Amyloid Polyneuropathy A Randomized Clinical Trial. Jama-Journal of the American Medical Association. 2013;310(24):2658–67. doi: 10.1001/jama.2013.283815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loewen GR, Macdonald JI, Verbeeck RK. High-Performance Liquid-Chromatographic Method For The Simultaneous Quantitation Of Diflunisal And Its Glucuronide And Sulfate Conjugates In Human-Urine. J Pharm Sci. 1989;78(3):250–5. doi: 10.1002/jps.2600780317. [DOI] [PubMed] [Google Scholar]
- 23.Patel DS, Sharma N, Patel MC, Patel BN, Shrivastav PS, Sanyal M. Sensitive and Selective Determination of Diflunisal in Human Plasma by LC-MS. Journal of Chromatographic Science. 2013;51(9):872–82. doi: 10.1093/chromsci/bms181. [DOI] [PubMed] [Google Scholar]
- 24.Verbeeck R, Tjandramaga TB, Mullie A, Verbesselt R, Verberckmoes R, Deschepper PJ. Biotransformation Of Diflunisal And Renal Excretion Of Its Glucuronides In Renal-Insufficiency. British Journal of Clinical Pharmacology. 1979;7(3):273–82. doi: 10.1111/j.1365-2125.1979.tb00932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikram A, Donnelly JP, Sperry BW, Samaras C, Valent J, Hanna M. Diflunisal tolerability in transthyretin cardiac amyloidosis: a single center’s experience. Amyloid-Journal of Protein Folding Disorders. 2018;25(3):197–202. doi: 10.1080/13506129.2018.1519507. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi R, Ono K, Shibata S, Nakamura K, Komatsu J, Ikeda Y, Ikeda T, Samuraki M, Sakai K, Iwasa K, Kayano D, Yamada M. Efficacy of diflunisal on autonomic dysfunction of late-onset familial amyloid polyneuropathy (TTR Va130Met) in a Japanese endemic area. Journal of the Neurological Sciences. 2014;345(1–2):231–5. doi: 10.1016/j.jns.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 27.Ibrahim M, Saint Croix GR, Lacy S, Fattouh M, Barillas-Lara MI, Behrooz L, Mechanic O. The use of diflunisal for transthyretin cardiac amyloidosis: a review. Heart Fail Rev.8. doi: 10.1007/s10741-021-10143-4. [DOI] [PubMed] [Google Scholar]
- 28.Lohrmann G, Pipilas A, Mussinelli R, Gopal DM, Berk JL, Connors LH, Vellanki N, Hellawell J, Siddiqi OK, Fox J, Maurer MS, Ruberg FL. Stabilization of Cardiac Function With Diflunisal in Transthyretin (ATTR) Cardiac Amyloidosis. Journal of Cardiac Failure. 2020;26(9):753–9. doi: 10.1016/j.cardfail.2019.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.