

Abstract
Many drugs are poorly water-soluble and suffer from low bioavailability. Metal-phenolic network (MPN), a hydrophilic thin layer such as tannic acid (TA)-FeIII network, has been recently used to encapsulate hydrophobic drugs to improve their bioavailability. However, it remains challenging to synthesize nanocapsules of a wide variety of hydrophobic drugs and to scale up the production in a continuous manner. Here, we present a microfluidic synthesis method to continuously produce TA-FeIII network nanocapsules of hydrophobic drugs. We hypothesize that nanocapsules can continuously be formed only when the microfluidic mixing timescale is shorter than the drug’s nucleation timescale. The hypothesis was tested on three hydrophobic drugs - paclitaxel, curcumin, and vitamin D with varying solubility and nucleation timescale. The proposed mechanism was validated by successfully predicting the synthesis outcomes. The microfluidically-synthesized nanocapsules had well-controlled sizes of 100–200 nm, high drug loadings of 40–70%, and a throughput of up to 70 mg hr−1 per channel. The release kinetics, cellular uptake, and cytotoxicity were further evaluated. The effect of coating constituents on nanocapsule properties were characterized. Fe content of nanocapsules was reported. The stability of nanocapsules at different temperatures and pHs were also tested. The results suggest that the present method can provide a quantitative guideline to predictively design a continuous synthesis scheme for hydrophobic drug encapsulation via MPN nanocapsules with scaled-up capability.
Keywords: continuous synthesis, hydrodynamic flow focusing, mixing timescale, nucleation timescale, drug nanocrystal core, metal-phenolic network coating
Graphical Abstract
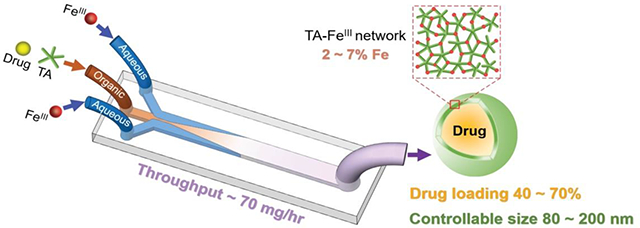
Tannic acid-FeIII network nanocapsules of hydrophobic drugs can be continuously synthesized by controlling the flow condition of mixing on microfluidic platforms.
1. Introduction
Metal-phenolic networks (MPNs), supramolecular structures formed by the rapid coordination of natural polyphenols with metal ions, have gained popularity in various biological and healthcare applications. The flexibility and compatibility of MPNs with a wide range of biomaterials make them attractive coating materials for the delivery of imaging and therapeutic agents [1–6]. Moreover, MPNs can serve as nontoxic and degradable coatings to encapsulate biological entities such as proteinosomes, microbes, and mammalian cells [7–9]. Recently, MPN nanocapsule has been employed as a promising nanoparticle (NP) formulation of hydrophobic drugs with enhanced bioavailability, colloidal stability, high drug loading, and easily functionalized surface with other ligands [10–12].
MPN nanocapsules have previously been produced by batch nanoprecipitation methods to encapsulate hydrophobic drugs, such as paclitaxel [10], carfilzomib [11], curcumin [13], rapamycin [14], chlorin e6 [15], SN-38 [16], simvastatin [17], andrographolide [17], and cabazitaxel [18]. The batch methods enable one-pot synthesis by inducing drug nanoprecipitation under manual mixing, combined with the postprocessing step of sonication. However, batch synthesis parameters, such as mixing orders and volumes of reagents, need to be tuned to form MPN nanocapsules of highly hydrophobic drugs such as vitamin D [12]. Moreover, manual operation results in time-consuming and labor-intensive work to scale from benchtop production to larger volume batches, which hinders the broader application of this nanocapsule formulation. To increase the throughput of MPN nanocapsule synthesis, microfluidics can provide a feasible solution for a continuous synthesis with scale-up potential.
Several microfluidic mixing methods, including hydrodynamic flow focusing (HFF), T-junction, and staggered herringbone micromixer (SHM), have already been applied to produce polymeric NPs, lipid NPs, and inorganic NPs [19–25]. In HFF, a thin fluid stream forms surrounded by a different fluid to achieve fast mixing between the two fluids. In T-junction, two fluids flow directly toward each other with a perpendicular turbulent output flow, which requires high flow rates. The SHM utilizes repeated patterns of grooves to generate chaotic mixing at low flow rates. Although these methods have been reported to synthesize various NPs, the translation from batch to microfluidic synthesis of MPN nanocapsules, which have drug core with MPN shell structure, still requires mechanistic understanding of synthesis processes to enable continuous production of nanocapsules at an increased throughput.
In this study, we develop a microfluidic continuous synthesis scheme of TA-FeIII network (TFN) nanocapsules of a wide range of hydrophobic drugs. This method is based on the rationale that the nanocapsule formation is governed by two competing processes - mixing and nucleation. The mixing process is controlled by flow conditions of hydrodynamic flow focusing [26–28] on the microfluidic platform. The nucleation process is determined by drug type. We hypothesize that nanocapsules will continuously form only when the microfluidic mixing timescale is shorter than the drug’s nucleation timescale. This hypothesis is tested by examining the TFN nanocapsule of three different hydrophobic drugs while varying the flow conditions of hydrodynamic flow focusing. We further characterize the structure, stability, drug loading, Fe content, drug release kinetics, cellular uptake, and cytotoxicity of nanocapsules synthesized by this method. The results are discussed to establish a mechanistic guideline to adjust the process parameters for reliable MPN nanocapsule formation for a wide range of hydrophobic drugs and potential ways to optimize the continuous synthesis for increased throughput.
2. Methods
2.1. Chemicals and reagents
Tannic acid (ACS reagent grade, ≥ 95%), iron (III) chloride (reagent grade, ≥ 97%), vitamin D (meets USP testing specifications, ≥ 97%), curcumin (curcuma longa, powder, purity (HPLC) ≥ 65%), Rhodamine B (≥95% (HPLC)), MTT Cell Growth Assay Kit (CT01), isopropanol (meets USP testing specifications), HCI (1 N), paraformaldehyde (reagent grade, crystalline), and dimethyl sulfoxide (molecular biology) were purchased from Sigma-Aldrich (St. Louis, MO). Paclitaxel (purity (HPLC) ≥ 99.5%) was a generous gift from Samyang Biopharm (Seongnam, Republic of Korea). Hoechst 33342 Fluorescent Stain (H3570), CellMask™ Green Plasma Membrane Stain (C37608), Dil dye (1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindocarbocyanine Perchlorate (‘DiI’; DiIC18(3))), and electron microscopy sciences formvar/carbon film 10 nm/1nm thick on a square 300 mesh copper grid was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Ethanol (200 Proof, anhydrous, meets USP specs) was purchased from Decon Labs, Inc (King of Prussia, PA). 50% dextrose (injection, USP) was purchased from VetOne (Boise, ID). 18.2 MΩ Deionized (DI) water was obtained from a Milli-Q ultrafiltration system (Millipore, Billerica, MA). 20 kDa MWCO, 0.5 mL (Catalog number: 88402) was purchased from Thermo Fisher. PDMS (SYLGARD™ 184 Silicone Elastomer Kit) was purchased from Dow (Midland, MI).
2.2. Synthesis of the TFN nanocapsules by batch method
Drug and tannic acid were dissolved in organic solvent (ethanol for vitamin D or paclitaxel, DMSO for curcumin) to 20 mg mL−1 and 80 mg mL−1, respectively. FeCl3 was dissolved in DI water to 10 mg mL−1. The drug solution (10 μL) was mixed with a tannic acid solution (12 μL). Aqueous FeCl3 solution (6 μL) was added to 974 μL of DI water (4°C). The diluted FeCl3 solution was then added to the mixture of drug and tannic acid to start interfacial supramolecular assembly of tannic acid and FeIII under probe sonication (Sonics Vibracell probe sonicator, Newtown, CT, USA) for 2 min, with 4 sec on, 2 sec off at 40% amplitude and ice bath, creating nanocapsules. The batch nanocapsules were collected by centrifugation at 16,100 rcf (relative centrifugal force) for 20 min. Then the pellets were washed with DI water and collected again by centrifugation at 16,100 rcf for 20 min. For transmission electron microscopy (TEM) characterization, the nanocapsules were freeze-dried in 5% dextrose (D5W). Dextrose served as a cryoprotectant to prevent damage to samples during the freezing process [12]. For drug loading determination, the nanocapsules were freeze-dried in DI water, as described previously [12].
2.3. Fabrication of the microfluidic chip
The mold of the microfluidic chip was fabricated by soft lithography with an SU8-coated silicon substrate. As described previously [29, 30], PDMS was prepared by mixing the PDMS base with the curing agent at the ratio of 10:1. The degassed PDMS was poured over the mold and baked at 80 °C for 3 h on a hot plate. After polymerization, the PDMS slab was removed from the silicon mold using a razor blade. The thickness of the PDMS chips was around 5 mm. Holes of 2 mm in diameter were punched at three inlets and the outlet of the channel using the 2mm standard biopsy punches (Disposable Biopsy Punches, Integra™ Miltex®, Integra Lifesciences Corp., NJ). The punched PDMS slab was rinsed by isopropyl alcohol and DI water, dried under an N2 stream, and bonded to a glass slide using a plasma treatment device (BD-20AC Laboratory Corona Treater, Electro-Technic Products, IL). The chip was then heated at 120 °C on a hot plate for 2 h to strengthen the bounding between the PDMS and the glass. The tubing of 2.29 mm in outer diameter and 0.76 mm in inner diameter (Wall Excelon Mini-Bore Tubing, US Plastic Corp., OH) was inserted into the holes at the inlets and the outlet of the chip. To secure the tubing insertion port and avoid leakage, uncured PDMS was poured on top of the chip, which covers the tubing ports. After being baked at 80 °C for 3 h on a hot plate, a new layer of PDMS was formed, which secures the tubing insertion port. The resistance of PDMS chips to organic solvents depends on the absorption of solvent by PDMS which can swell the PDMS [31]. Ethanol and DMSO used in this study are not absorbed by PDMS and do not deseal the PDMS-glass bonding of the microfluidic chips.
2.4. Synthesis of the TFN nanocapsules by the microfluidic method
Stock solutions were prepared as follows: vitamin D was dissolved in ethanol to 20 mg mL−1, paclitaxel was dissolved in ethanol to 20 mg mL−1, curcumin was dissolved in DMSO to 20 mg mL−1, tannic acid was dissolved in ethanol or DMSO to 80 mg mL−1, and FeCl3 was dissolved in DI water to 10 mg mL−1. In this study, the mixing ratio of aqueous solution to organic solution ranged from 10:1 to 40:1, while yielding a drug concentration of 0.1 – 0.3 mg mL−1, as well as a TA concentration of 0.4 – 1.2 mg mL−1 after mixing. To enable these studies, the stock solutions of drugs and TA were prepared at concentrations of 20 and 80 mg mL−1, respectively. The organic solution introduced to the central channel of the microfluidic chip was prepared by mixing the drug stock solution, the tannic acid solution, and the organic solvent (ethanol or DMSO). The mixing ratio depends on what drug and TA concentrations need to be tested. The aqueous solution introduced to the side channels of the microfluidic chip was prepared by mixing the FeCl3 stock solution with DI water. The mixing ratio depends on what the FeIII concentration needs to be tested. The organic solution was delivered by a single-channel syringe pump (NE-1000 One Channel Syringe Pump, New Era Pump Systems Inc., NY), and the aqueous solution was delivered by a dual-channel syringe pump (NE-4000 Programmable 2 Channel Syringe Pump, New Era Pump Systems Inc., NY). The mixing ratio of the aqueous antisolvent to the drug-containing organic solvent can be described by the flow ratio of the aqueous solution (introduced to the side inlets) to the organic solution (introduced to the central inlet). For all three drugs, the tested flow ratio ranged from 10 to 40, and the total flow rate ranged from 4 to 220 mL hr−1. The microfluidically synthesized nanocapsules were collected by centrifugation at 16,100 rcf for 20 min. For drug loading determination, the nanocapsules were freeze-dried in DI water.
2.5. Characterization of the flow field on the microfluidic platform
To visualize the flow field in the microfluidic chip, Rhodamine B was dissolved in ethanol to the concentration of 200 μM, which was introduced to the central inlet of the microfluidic chip. DI water was introduced to both side inlets. The flow rates of ethanol and water were controlled by the syringe pumps. The imaging system consisted of a fluorescence microscope (IX71, Olympus, PA) equipped with a TRITC filter, a CCD camera (Aqua, QImaging, CA), and a fluorescent illuminator (X-Cite 120Q, Excelitas Technologies, MA), which recorded the fluorescence of Rhodamine B to show the profiles of hydrodynamically focused central streams. The flow field in the microfluidic channel was also computationally analyzed by Ansys Fluent (Ansys Inc., PA). Hexahedral grids were generated by Ansys ICEM. All walls of the microfluidic channels were set as non-slip boundary conditions. Velocity inlet boundary condition was applied to three inlets, and pressure outlet boundary condition was applied to the outlet of the microfluidic channel. Standard turbulence model was used to simulate the solvent and antisolvent flow patterns in the mixing channel. A species transport model was used to simulate the mixing between the organic and the aqueous phases.
2.6. . Characterization of batch and microfluidic nanocapsules
The nanocapsules prepared by either batch or microfluidic method were suspended in phosphate buffer (10 mM, pH 7.4), and their sizes and PDI were measured by a Malvern Zetasizer Nano ZS90 (Worcestershire, UK) based on dynamic light scattering (DLS). Nanocapsule morphology was observed by Tecnai transmission electron microscopy (FEI, Hillsboro, OR, USA) after negative staining with 1% uranyl acetate. The TFN nanocapsules was incubated in 0.8 mL of ethanol and 0.2 mL of water for 1 h to selectively dissolve paclitaxel or vitamin D, collected by centrifugation at 16,100 rcf for 20 min, and imaged by TEM to visualize the TA-FeIII coating. For curcumin, 0.6 mL of DMSO with 0.2 mL of ethanol and 0.2 mL of water was used. Scanning electron microscopy (SEM) was used to observe the formation of vitamin D crystals over nanometer size when nanocapsules of vitamin D were not successfully produced at lower aqueous-organic mixing ratios of 10:1 and 20:1 using a microfluidic channel of 300 μm in width. The samples were placed on a carbon tape attached to an aluminum stud and coated with platinum for SEM analysis using an FEI NOVA NanoSEM scanning electron microscope. To confirm the formation of TA-FeIII complex on the surface of the drug nanocore, TA, TA-FeIII complex, paclitaxel, and nanocapsule of paclitaxel were analyzed by Fourier transform infrared (FTIR) spectroscopy in the range 800 − 4500 cm−1 at a resolution of 4 cm−1. All samples were lyophilized and analyzed by Thermo Nicolet Nexus 470 FTIR (Madison, WI, USA) installed with Smart-iTR (diamond crystal).
2.7. HPLC analysis of paclitaxel, vitamin D, and curcumin content
The nanocapsules prepared from either batch or microfluidic method were precisely weighed and redispersed in 0.8 mL ethanol with 0.2 mL water for 1 h to extract paclitaxel and vitamin D. For curcumin, 0.6 mL of DMSO with 0.2 mL ethanol and 0.2 mL water was used. The drugs in the supernatant were collected by centrifugation (16,100 rcf, 20 minutes) and quantified by the Agilent 1100/1260 HPLC system equipped with Ascentis C18 column (25 cm × 4.6 mm, particle size: 5 μm). For paclitaxel, the sample injection volume was 20 μL, and the mobile phase was a 50:50 volume mixture of water and ACN and ran at 1 mL min−1. The column temperature was not controlled. Paclitaxel was detected by a UV detector at a wavelength of 227 nm. For both vitamin D and curcumin, the following procedure was used. The sample injection volume was 50 μL. The mobile phase was a 75:25 volume mixture of methanol and ACN and ran at 1.5 mL mL−1. The column temperature was maintained at 40 °C. Vitamin D and curcumin were detected by a UV detector at a wavelength of 265 nm and 420 nm, respectively. Paclitaxel, vitamin D, and curcumin were dissolved in the same manner as the nanoparticles above 50 – 800 μg mL−1 and analyzed in the same condition to establish a calibration curve. The drug contents in particles were calculated as the drug amount divided by the particle mass.
2.8. In vitro release kinetics
The TFN nanocapsules of paclitaxel produced by microfluidic or batch methods, equivalent to 195 μg paclitaxel, were dispersed in 0.5 mL of 5% dextrose solution (D5W), phosphate buffered saline (10 mM phosphate, pH 7.4) containing 0.2% Tween 80, or acetate buffered saline (10 mM acetate, pH 5.5) containing 0.2% Tween 80. The suspension was loaded in a Slide-A-Lyzer™ MINI Dialysis Device with a molecular weight cut-off of 20 kDa. The dialysis well was inserted in a polypropylene tube containing 15 mL of D5W, phosphate buffered saline (pH 7.4), or acetate buffered saline (pH 5.5), all supplemented with 0.2% Tween 80. The tube was incubated at 37 °C under constant rotation. At each time point, 100 μL of the release medium in the polypropylene tube was sampled and replaced with fresh medium. The collected samples were filtered and analyzed by HPLC. After 216 hours, the PTX remaining in the dialysis device was extracted by ethanol to check the mass balance. 5% dextrose was used for release kinetics tests as a dispersant. It is a clinically used isotonic solution for parenteral injection. TFN particles were found to be dispersed best in 5% dextrose in our previous study [12].
2.9. Preparation of dye-doped nanocapsules
Stock solutions were prepared as follows: paclitaxel was dissolved in ethanol to 20 mg mL−1, Dil dye was dissolved in ethanol to 5 mg mL−1, tannic acid was dissolved in ethanol to 80 mg mL−1, and FeCl3 was dissolved in DI water to 10 mg mL−1. The organic solution was prepared by mixing 400 μL paclitaxel stock solution, 500 μL tannic acid solution, 16 μL Dil dye stock solution, and 84 μL ethanol. The aqueous solution was prepared by mixing the FeCl3 stock solution with DI water to the final concentration of 0.06 mg mL−1. The batch dye-doped nanocapsules was prepared by pipet mixing of 24 μL organic solution with 976 μL aqueous solution followed by sonication (details of batch procedures described in section 2.2). The microfluidic dye-doped nanocapsules was prepared at the aqueous-organic flow ratio of 40:1 (details of microfluidic procedures described in section 2.4).
2.10. Cell culture and uptake assay
Human pancreatic cancer cells of Panc10.05 and Panc1 were maintained in DMEM/F12 medium (Invitrogen, NY) supplemented by 10% v/v fetal bovine serum, 2 mM L-glutamine, and 100 μg mL−1 penicillin/streptomycin. Panc10.05 or Panc1 cells were cultured in 25 cm2 T-flasks at 37°C and 5% CO2, and routinely harvested at 80–90% confluency by using 0.05% trypsin and 0.53 mM EDTA, used for experiments or subculture. The cells were maintained up to 15th passage during the experiments. For the cellular uptake assay, 5 × 105 Panc10.05 cells were placed in a 35 mm glass-bottom Petri dish and allowed to adhere overnight. The medium was removed, and 2 mL of fresh medium was added. Dil dye-doped TFN nanocapsules prepared by batch or microfluidic method were suspended in D5W (10 μL, containing 1 mg mL−1 paclitaxel) and added to the dish. The cells were then incubated for 4, 12, and 24 hours. Before observation with a Nikon A1Rsi confocal microscope, the cells were washed three times with PBS, stained with CellMask Green and Hoechst 33342, and then fixed with 4% paraformaldehyde. For confocal imaging, the nucleus, the cell membrane, and the Dil-doped nanocapsules were excited by a 405 nm laser, a 488 nm laser, and a 559 nm laser, respectively.
2.11. Cytotoxicity of TFN nanocapsules
Panc10.05 or Panc1 cells were seeded in 96-well plates at a density of 5000 cells per well and allowed to adhere overnight. Panc10.05 cells were incubated with drug-free nanocapsules (pTAs), free paclitaxel, or TFN nanocapsules of paclitaxel at the concentrations ranging from 0.001 to 10 μg mL−1 calculated by paclitaxel content at 37 °C and 5% CO2 for 24 hr. Panc1 cells were incubated with pTAs, free curcumin, or TFN nanocapsules of curcumin at the concentration of 5 – 160 μM calculated by curcumin content at 37 °C and 5% CO2 for 48 hr. After the incubation, the cells were washed with PBS 3 times, and 100 μL fresh medium was added to each well. Then cells were cultured for another 24 hours. After incubation, 100 μL fresh medium was changed to each well, and 10 μL MTT was added to each well, followed by a 4 h incubation for cleavage at 37 °C. Then 100 μL isopropanol with 0.04 N HCl was added to each well and mixed thoroughly with the medium by repeated pipetting. The result was read with a test wavelength of 570 nm and a reference wavelength of 630 nm using a Microplate Spectrophotometer. Experiments were performed in triplicate and presented as means ± SD.
2.12. Analysis of Fe content of TFN nanocapsules
The Fe content of TFN nanocapsules was analyzed using Thermo Scientific Element II ICP-MS with a nebulizer of Teledyne Cetac Aridus II. Ar was used as the plasma gas. The samples of TFN nanocapsules were prepared as suspensions in 5% dextrose. 2 mL 70% HNO3 and 0.5 mL H2O2 were added to 100 μL of each sample. The samples were digested in a microwave digestor with the program “organic”, followed by dilution to 50 mL with H2O. A control blank of 2 mL 70% HNO3 + 0.5 mL H2O2 was included to go through the same digestion and dilution processes. 5 ppb Indium was used as an internal standard by adding 250 μL 1 ppm Indium to 50 mL 2% HNO3. 0 – 1000 ppb standard iron was analyzed in the same condition to establish a calibration curve.
2.13. Colloidal stability tests of TFN nanocapsules
To evaluate the size stability in the presence of serum, TFN nanocapsules equivalent to 50, 100, and 200 μg of PTX were suspended in 1 mL of 50% FBS and incubated for 24 hr at 37°C. The particle size distribution was measured before and after the incubation. To evaluate the colloidal stability in long-term storage, TFN nanocapsules equivalent to 200 μg of PTX were suspended in 1 mL D5W and stored for 3 weeks at 4°C. The particle size was measured by a Malvern Zetasizer Nano ZS90 every 7 days.
2.14. Statistical analysis
Statistical analysis of all data was performed using GraphPad Prism 9 (La Jolla, CA). Statistical significance was assessed by unpaired t-test or one-/two-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant. (*: p < 0.05; **: p < 0.01; ***: p < 0.001; and ****: p < 0.0001)
3. Results
3.1. Design of timescale-based microfluidic synthesis of TA-FeIII network nanocapsules
A microfluidic synthesis scheme adapting nanoprecipitation [32–35] was designed to achieve a continuous synthesis of TA-FeIII network (TFN) nanocapsules of hydrophobic drugs, as illustrated in Figure 1. This scheme consists of three steps – flow focusing, mixing for nucleation, and particle growth with coating. Drug and TA in the organic solvent were introduced to the central inlet. The antisolvent, FeCl3 in water, was introduced to both side inlets. The central organic stream is squeezed by the outer aqueous stream, which flows at a much higher flow rate. The mixing, i.e., the solvent exchange between the central organic phase and the surrounding aqueous phase, induces supersaturation of drug molecules and the formation of prenucleation clusters [36, 37] prior to the nucleation. The nucleation can be initiated before or after the mixing is completed and generates drug nuclei. Immediately following the nucleation, the particle growth and the TFN coating proceed simultaneously. TA and FeIII instantaneously generate a supramolecular complex and starts to assemble on the particle surface while the particle is still consuming the rest of the drug molecules to grow in size. Besides nucleation, spinodal decomposition may occur during a nanoprecipitation process [36]. However, for the nanoprecipitation of small molecule drugs, a large metastable zone must be crossed to induce spinodal decomposition in the antisolvent mixing process, during which nucleation is very likely to occur and dominate the nanoprecipitation [38]. Thus, we adapt the classical nucleation theory.
Figure 1.
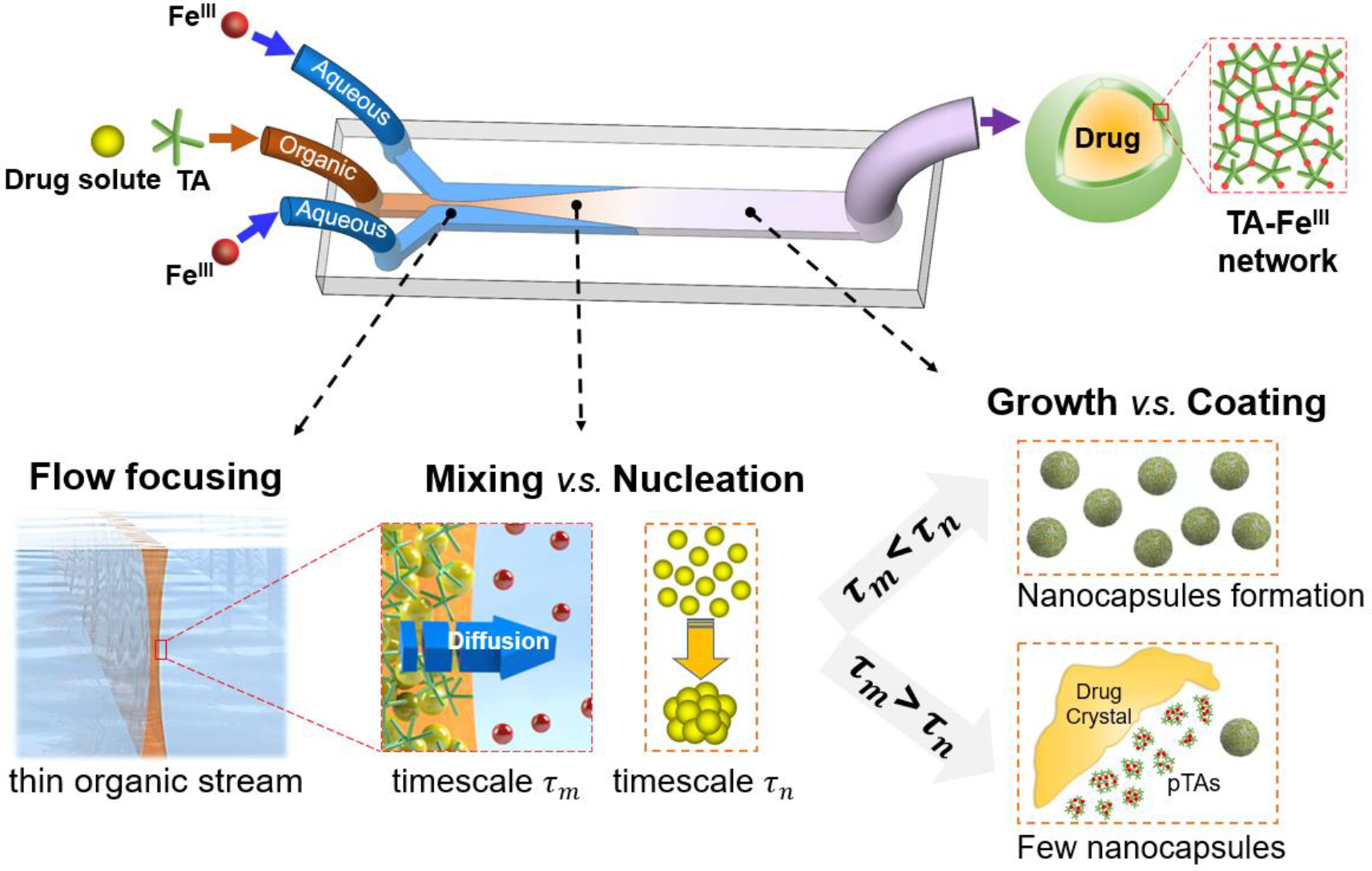
Schematic illustration of the microfluidic synthesis of TA-FeIII network nanocapsule of hydrophobic drugs. Three major steps of the antisolvent-based synthesis of nanocapsules in microfluidics: flow focusing, mixing - nucleation, and particle growth - coating. Hypothesized synthesis mechanism is based on the prediction of the mixing timescale of microfluidics and the nucleation timescale of a drug is characterized by the diffusion length of the organic center stream. A shorter than leads to the formation of nanocapsules. A longer than results in the dominant formation of drug crystals above the nanometer range. pTA indicates the TA and FeIII coordination complexes with no encapsulated drug.
To produce nanocapsules of various hydrophobic drugs, we hypothesize that this microfluidic synthesis scheme can be expanded by considering two competing timescales. The hydrodynamic flow focusing controls the width of the central organic stream, and hence the mixing timescale, , which is a measure of the solvent exchange efficiency based on the diffusion length between the organic and aqueous phases. The nucleation timescale, , is correlated with the time elapsed from the creation of supersaturation to the initiation of the nucleation [39], which may vary with drugs depending on their molecular structures and solid-state physical properties [40, 41]. When the central organic stream is thin so that the condition of is satisfied, a large number of nuclei are rapidly formed, followed by limited particle growth [42–44]. This leads to the formation of uniform nanocapsules. On the contrary, as the central organic stream becomes thicker, increases so that the nucleation rate is slow. This causes the nuclei formation is not sufficient for the depletion of drug solutes. Abundant drug solutes are still present after nucleation, resulting in the dominant formation of drug crystals above the nanometer size range. Instead of nanocapsules, TA and FeIII coordination complexes are formed with no encapsulated drug, indicated as pTAs. A more specific explanation of the different nucleation dynamics is schematically provided in Figure S1. Based on the estimation of during the mixing by hydrodynamic flow focusing and of the drugs, we postulate a hypothesis that nanocapsules can reliably be formed only when is shorter than . This will lay a mechanistic guideline for the continuous manufacturing of TFN nanocapsules of hydrophobic drugs.
3.2. Timescale-guided synthesis of TFN nanocapsules of hydrophobic drugs
In order to demonstrate that microfluidic flow focusing enables controllable , which matches drugs’ for the synthesis of nanocapsules, we investigated two major parameters of channel size and flow ratio determining the efficiency of microfluidic mixing, i.e., the solvent exchange between the central stream (ethanol or DMSO) and the surrounding stream (water). The channel size refers to the width of the mixing channel. The flow ratio, , refers to the ratio of the aqueous antisolvent flow rate to the organic solvent flow rate. Figure 2A shows examples of the hydrodynamic flow focusing along a 300 μm channel at the flow ratios of and 40. Both the experiment and simulation results show that the width of the central organic stream significantly increased when decreased from 40 to 10, which indicated the increasing with the decreasing . The central stream was imaged via the red fluorescence of rhodamine B, lacking detailed mass fraction information. The flow field simulation provided the contours of the mass fraction, which were utilized to estimate the diffusion length (see Figure S2, Supporting Information). The flow rate in the mixing channel was nondimensionalized as the Reynolds number, , a dimensionless quantity characterizing flow patterns in microfluidics [45, 46], which is expressed as , where and are the density and viscosity of the fluid, respectively, is the flow speed, and is the characteristic length of the channel. In Figure 2A, both the experiment and simulation were conducted at . The central streams were curved because vortices were generated across the microchannel, which is shown by the simulated streamlines.
Figure 2.
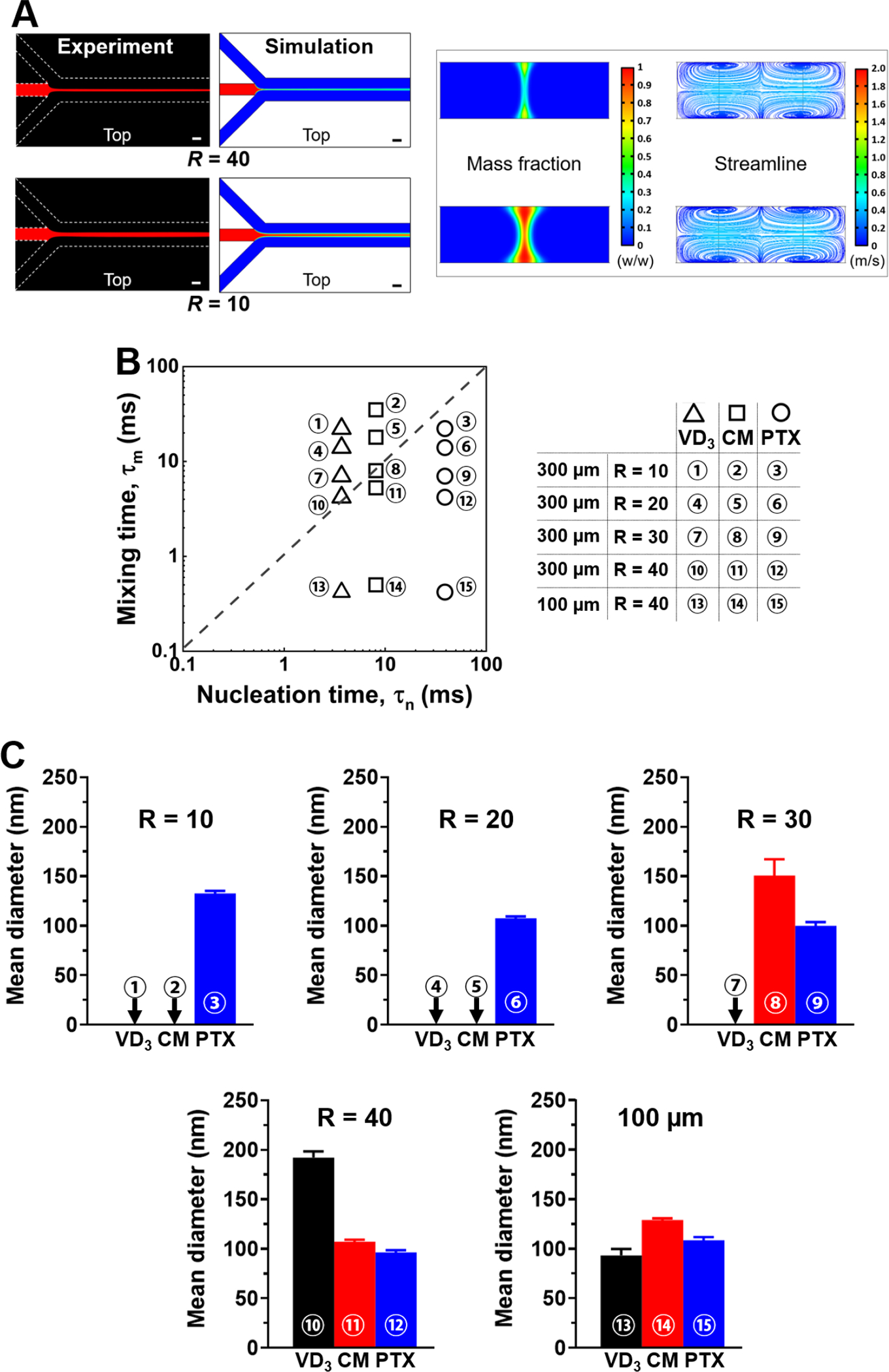
Timescale-based prediction of synthetic feasibility of nanocapsules of different hydrophobic drugs at different microfluidic conditions. (A) Experiment visualization and computational analysis showing the flow focusing configuration with different flow ratios. Scale bar: 100 μm. (B) Timescale-based prediction chart. of three drugs of VD3, CM and PTX are plotted together with of microfluidics. To vary , the tested conditions include different flow ratios of , and 40 using a 300 μm channel, as well as using a 100 μm channel. (C) Experiment results show the synthesis outcomes at the 15 conditions plotted in Figure 2B. The particle size was characterized by the mean diameter obtained from DLS measurement.
Three hydrophobic drugs with varying solubility were studied, including vitamin D (VD3) (water solubility < 1 ng mL−1 at room temperature), curcumin (CM), and paclitaxel (PTX) (> 1 μg mL−1). To test the timescale-based prediction of synthetic feasibility of different nanocapsules, the microfluidic channel size and the flow ratio were selected to cover the conditions of both and for these drugs. of VD3, CM, and PTX were approximately estimated to be 4, 8, and 39 ms, respectively, at drug concentrations > 0.1 mg mL−1. was estimated considering supersaturation ratios and solubility of drugs as described in the Supporting Information (see ‘Section of the nucleation timescale estimation’). was defined as , where is the diffusion length of flow focusing conditions and is the diffusion coefficient of drugs [47].
All experimental conditions studied are plotted in Figure 2B. To validate our hypothesis, we used microfluidic channels of different widths of 100 and 300 μm to test the synthesis of nanocapsules. For the 300 μm channel, as decreased from 40 to 10, approximately increased from 4 to 20 ms for ethanol (solvent for VD3 and PTX); and it approximately increased from 5 to 35 ms for DMSO (solvent for CM). The solvent of DMSO induced longer as it has higher density and viscosity and lower diffusivity than ethanol. PTX was located in the region of irrespective of . CM was located in the region at , approached at , and moved to the region at or 10. VD3 was located in the region of at and 30, but closely approached at . The comparison of of the drugs with of 100 – 600 μm microfluidic channels is provided in the Supporting Information (see Figure S2).
The experiment result using a 300 μm channel in Figure 2C shows the effect of flow ratio on the diameter of nanocapsules of VD3, CM, and PTX. We confirmed PTX nanocapsules with a controlled size of around 100 nm in diameter were successfully synthesized at all flow ratios . We were also able to synthesize CM nanocapsules of around 100 nm at , but the size increased to around 150 nm at and nanocapsules were not produced at or 10 . Compared to PTX or CM, VD3 nanocapsules were synthesized with a larger size of around 200 nm even at , which can be attributed to the slightly longer than its . Due to the short of VD3, nanocapsules were not successfully produced at , or 10 ; instead, irregular drug crystals over nanometer size, and clusters of excess TA and FeIII coordination complexes (pTAs) were produced. Without a drug, TA and FeIII formed clusters of small pTA particles around 10 nm in diameter (Figure S3A, Supporting Information), which were distinct from the drug-loaded nanocapsules. TEM and SEM images of the assemblies produced at the conditions of are provided in Figure S3B and S3C, respectively.
For further validation of the proposed timescale-based mechanism, we utilized a 100 μm channel to decrease to < 0.5 ms, which was much shorter than of all three drugs. As shown in Figure 2C, PTX or CM nanocapsules produced with 100 μm channel did not decrease in size compared to 300 μm channel, as the criterion of was already satisfied by 300 μm channel. It agrees well with the proposed mechanism that as long as , further decrease of can not further decrease the nanocapsule size. In contrast, the size of VD3 nanocapsules significantly decreased from about 200 to 100 nm with a channel size decrease from 300 to 100 μm (size distribution curve provided in Figure S4), as of 300 μm channel was not shorter than . A significant zeta potential change of VD3 nanocapsules from −55 to −30 mV was also observed (see Figure S4). The flow rate was kept at for 300 μm channel, and for 100 μm channel ( exceeded the capacity of 100 μm channel). Different nucleation dynamics of three drugs are specifically explained in Supporting information (see Figure S5).
Following this timescale-guided synthetic scheme, we utilized the 300 μm channel with controlled flow conditions of and to synthesize TFN nanocapsules of VD3, CM, or PTX for TEM and HPLC analysis. The high-magnification TEM images of the microfluidic nanocapsules are shown in Figure 3A (top row). VD3 nanocapsules were around 200 nm in diameter, while CM and PTX nanocapsules were around 100 nm in diameter. TA-FeIII coatings were clearly visible after ethanol (for PTX and VD3) or DMSO (for CM) etching, which selectively dissolved the encapsulated drugs from the TA-FeIII assemblies. TA molecules and Fe ions may form nanoparticulate assemblies without encapsulating the model drugs, i.e., pTA, characterized as clusters of small particles around 10 nm in diameter (Figure S3A). TEM images of Figure 3A show the attachment of pTAs to the surface of both microfluidic and batch nanocapsules, which suggests the generation of pTAs along with the formation of TFN nanocapsules of drugs. Compared to the TEM images of the batch nanocapsules, the microfluidic nanocapsules had more regular shapes with clearer boundaries because fewer pTA particles were formed and attached to the surface of nanocapsules. The low-magnification TEM images are provided in Figure S6, showing the morphology of nanocapsules synthesized by our microfluidic method. The formation of TA-FeIII complex on the surface of the drug nanocore was confirmed by FTIR spectra (Figure S7). The coordination between TA and FeIII ions can be reflected by the shift of peak position of the phenol group in the FTIR spectrum [48, 49]. TA-FeIII complex showed a peak at 1191 cm−1 whereas non-coordinated TA’s phenol group had a peak at 1169 cm−1. The FTIR spectrum of nanocapsule resembled that of TA-FeIII complex rather than the encapsulated paclitaxel, indicating complete encapsulation of the drug in TA-FeIII shell.
Figure 3.
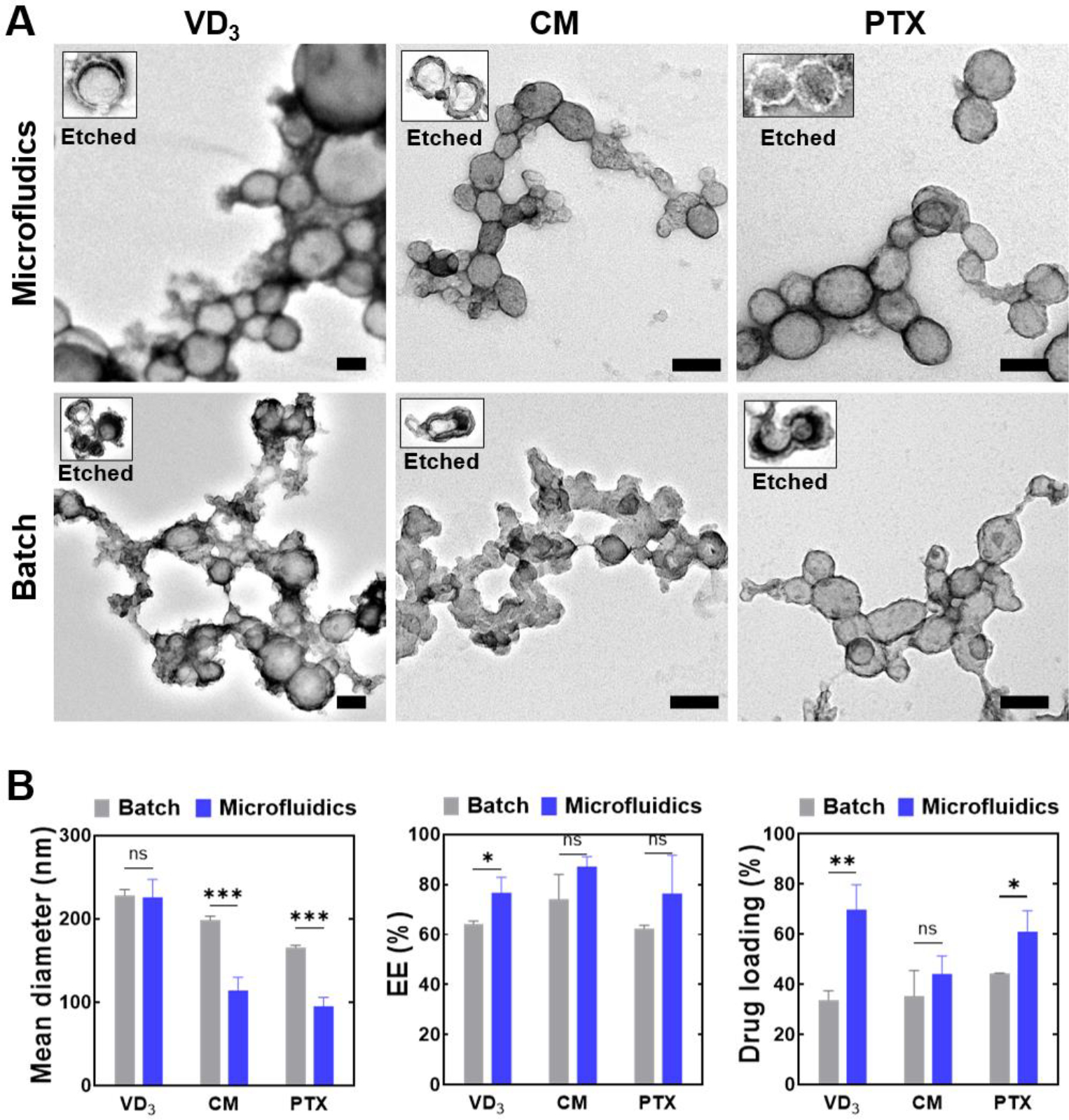
Characterization of nanocapsules synthesized by microfluidic and batch methods. (A) TEM images of microfluidically and batch-prepared TFN nanocapsules of VD3, CM and PTX. The inserts show the TFN nanocapsules after ethanol or DMSO etching. Scale bar = 100 nm applies to both the intact and etched nanocapsules. (B) Mean diameter of nanocapsules measured by DLS (left). Encapsulation efficiency (EE) of the drugs (middle). Drug loading content of drugs by HPLC analysis (right).
According to the DLS measurement shown in Figure 3B (left), microfluidic VD3 nanocapsule had a z-average of 226 ± 21 nm with the PDI of 0.14 ± 0.01, microfluidic CM nanocapsule 114 ± 16 nm (PDI of 0.20 ± 0.03), and microfluidic PTX nanocapsule 95 ± 21 nm (PDI of 0.18 ± 0.11). Batch VD3 nanocapsule had a z-average of 228 ± 7 nm (PDI of 0.08 ± 0.01), batch CM nanocapsule 199 ± 5 nm (PDI of 0.07 ± 0.02), and batch PTX nanocapsule 166 ± 2 nm (PDI of 0.10 ± 0.03). The DLS data corresponded reasonably well with the TEM images of microfluidic nanocapsules. Figure 3B (middle) shows the encapsulation efficiency (EE) calculated by the ratio of encapsulated drug amount to the total amount of added drug. The microfluidic synthesis achieved > 75% EE for all three drugs. Figure 3B (right) shows the drug loadings of batch and microfluidic synthesis. For the batch synthesis, VD3, CM, and PTX were encapsulated by TFN with a loading content of 34 ± 3.7, 35 ± 10, and 44 ± 0.2 wt%, respectively. For the microfluidic synthesis, VD3, CM, and PTX were encapsulated with a loading content of 70 ± 9.9, 44 ± 7.2, and 61 ± 8.4, wt%, respectively.
3.3. Effect of microfluidic flow rate on the synthesis quality
The flow focusing configuration, where a central thin organic stream is generated, formed at relatively low flow rates. As increased higher than 100, as shown in Figure 4A, the flow deviated from the flow-focusing configuration and formed complex mixing patterns. The strong vortices, indicated by the simulated streamlines, distorted the central organic stream and split it into four bifurcated streams, which is shown by the cross-section view of the mass fraction contours. Here we note this flow pattern as “flow bifurcations”. The flow pattern transition from flow-focusing to flow-bifurcations can be attributed to the dramatic increase of the eddy kinetic energy especially at (see Figure S8).
Figure 4.
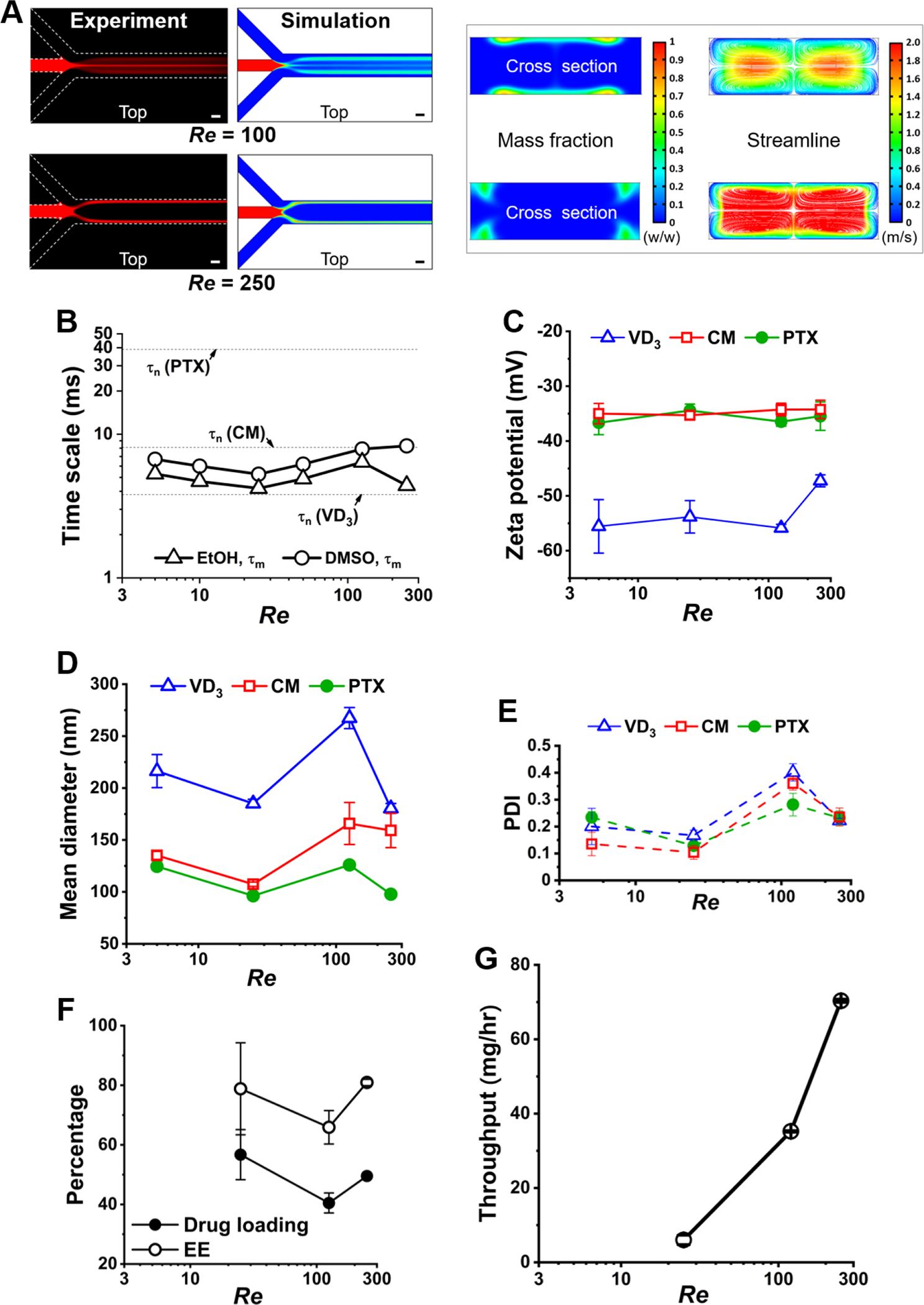
Effects of flow rate on the (A) mixing flow patterns and (B) their , (C) zeta potential, (D) nanocapsule size, (E) PDI, (F) drug loading and encapsulation efficiency (EE), as well as (G) throughput of the synthesized nanocapsules using microfluidics.
Figure 4B shows the effect of flow rate on in the range of . At a constant flow ratio of 40, increasing the flow rate did not significantly change at different mixing patterns, which maintained the synthesis quality of nanocapsules. Specifically, Figure 4C shows no significant effect of flow rate on zeta potential. Figure 4D shows, in the wide range of flow rates, VD3 nanocapsules maintained around 200 nm in diameter, and CM and PTX nanocapsules maintained around 100 nm in diameter. Figure 4E shows PDI maintained within 0.1 – 0.2 at both the lowest and highest flow rates. Figure 4F shows PTX loadings higher than 40% with varied flow rates. Thus, the present method can increase the synthesis throughput without losing the quality control of the nanocapsules.
To further optimize the flow conditions for both synthesis quality and throughput, we noticed an increase in nanocapsule size and PDI and a decrease in EE and drug loading at . This may be caused by organic solvent accumulation in the bifurcated streams (see Figure S8), which induced an elevated chance of particle aggregation. However, when the flow rate was further increased to , organic solvent accumulation was reduced (see Figure S8), and thus, as shown in Figure 4G, the synthesis of PTX nanocapsules fulfilled a throughput up to 70 mg hr−1 utilizing one single microfluidic channel, while maintaining a good quality of nanocapsules with ~100 nm in size, a PDI of ~0.1, ~70% EE, as well as ~50% drug loading. The summary table of TFN nanocapsules’ mean diameter, PDI, zeta potential, and drug loading produced at different microfluidic flow conditions of channel width, flow ratio, and flow rate is provided in Table S1.
3.4. Release kinetics, cellular uptake and cytotoxicity activity of TFN nanocapsule of PTX
The colloidal TFN nanocapsules prepared by the batch process have been shown to retain their integrity and demonstrate good stability in physiological media, which is favorable for drug delivery [10]. To demonstrate the drug delivery potential of the nanocapsules synthesized by our microfluidic method, we investigated the in vitro release kinetics and cellular uptake of the TFN nanocapsule of PTX. The release of the encapsulated PTX was observed over 216 hours, as shown in Figure 5A, a gradual release profile without burst release was displayed at the initial stage, and ~80% of PTX was released after 96 h. After 216 hours, the mass balance was confirmed by extracting the PTX remaining in the dialysis well. There was no significant difference in drug release profile between nanocapsules produced by the microfluidic method and those by batch method. We analyzed the drug release data using different conventional mathematical models of First Order, Higuchi, Hixson-Crowell, and Square Root of Mass [50]. The coefficient of determination (R2) and root mean square error (Sy.x) of the fitting results show that the First Order model has the best accuracy and predictability with the highest value of R2 (≥ 0.95 for both microfluidic and batch nanocapsules) and lowest value of Sy.x among the four models tested. The specific fitting results are provided in Table S2 of Supporting Information.
Figure 5.
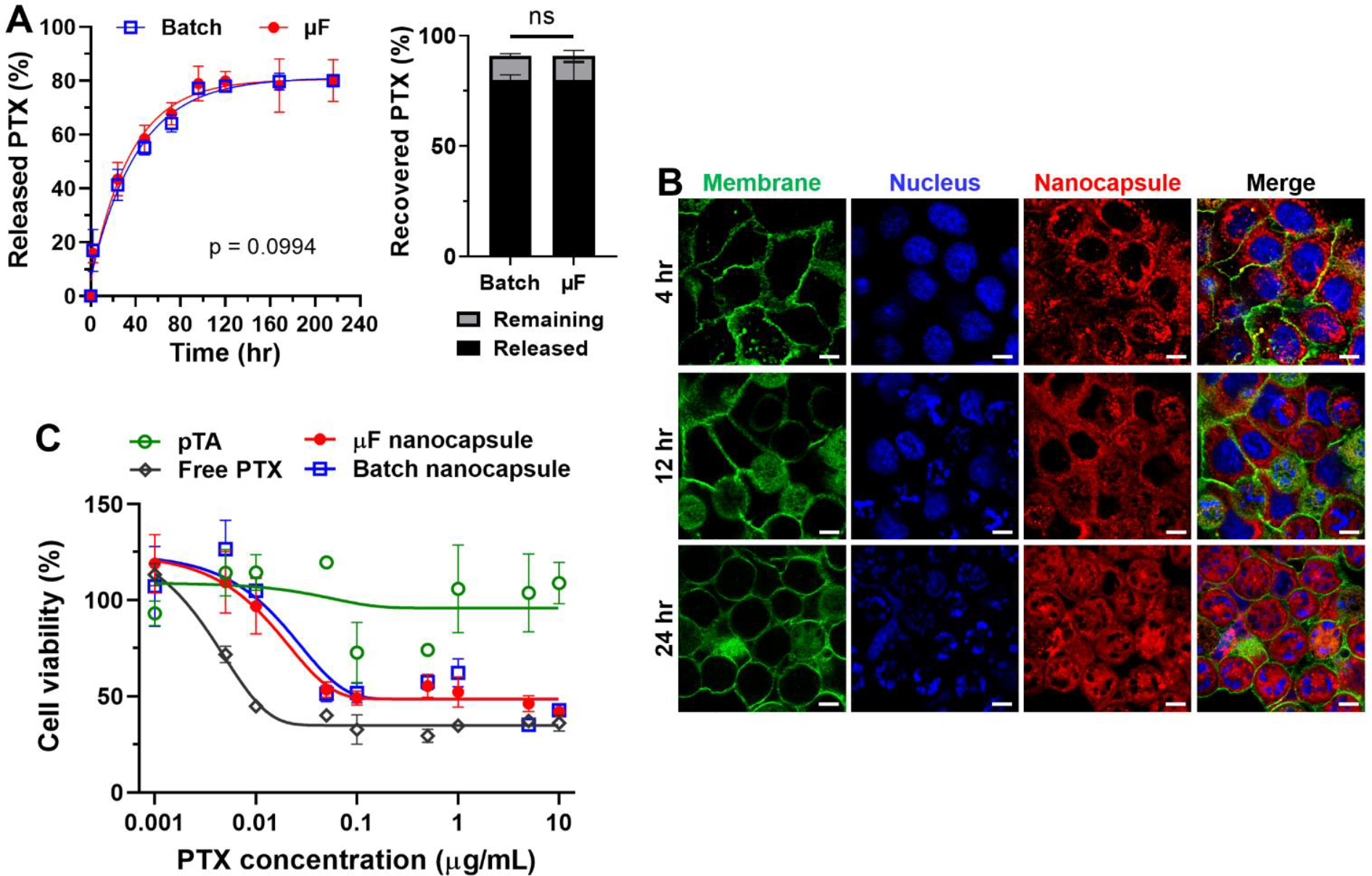
Release kinetics, cellular uptake and cytotoxicity activity of TFN nanocapsule of PTX. (A) Left: Release profile of the encapsulated PTX from nanocapsules produced by microfluidics (μF) or batch methods. Release kinetics performed with D5W as a dispersant and D5W + 0.2% Tween 80 as a release medium. No significant difference by paired t test, n=3 per group. Right: Released and remaining fraction relative to total used drug. (B) Confocal images of Panc10.05 cells (a human pancreatic cancer cell line) incubated with Dil-doped nanocapsules at various times (4, 12, and 24 hr). Blue nuclei were stained by Hoechst 33342, and green membranes were stained by CellMask. Dil-doped nanocapsules had red fluorescence. Scale bars are 10 μm. (C) Viability of Panc10.05 cells with pTA, free PTX, batch and microfluidic nanocapsules at different dosages calculated by PTX content.
To visualize cellular uptake, we prepared Dil-doped TFN nanocapsules with encapsulated PTX as described. The Dil doping did not change the size or zeta potential of the synthesized nanocapsules (see Figure S9). Figure 5B shows, after 4 hours of incubation, the localization of Dil fluorescence was observed surrounding the nuclei of Panc10.05 cells, which suggested the cellular uptake, but the cells were still observed to maintain a healthy morphology with an intact nucleus. After 12 hours of incubation, approximately 50% of the cells were observed to have the breakage of nuclei, as well as the morphology change to a circular shape. With prolonged incubation to 24 hours, all the cells changed into a circular shape with the formation of multiple nuclear envelope fragments. The confocal images of Panc10.05 cells incubated with microfluidic-produced TFN nanocapsules were comparable to those of batch-produced nanocapsules (Figure S10), suggesting comparable uptake.
The MTT cell survival assay was used to quantitatively assess the cytotoxicity of nanocapsules synthesized by batch or microfluidic methods. As shown in Figure 5C, the blank nanocapsule of pTA showed no toxicity at the dosages up to 10 μg mL−1 (calculated by PTX content) tested in this study, suggesting that the TFN used to make nanocapsules were cytocompatible. The batch and microfluidic nanocapsules exhibited identical cytotoxicity in the wide PTX concentration range of 0.001 – 10 μg mL−1. The TFN nanocapsule of PTX showed comparable cytotoxicity to free PTX at higher dosages > 5 μg mL−1, but showed lower cytotoxicity than free PTX at lower dosages < 1 μg mL−1, especially in the range of < 0.01 μg mL−1, which can be attributed to the gradual PTX release profile from nanocapsules within the 24 hours treatment period. The drug release and cytotoxicity results indicated that the microfluidic and batch nanocapsules were functionally identical. The viability of Panc1 cells with TFN nanocapsules of CM was also tested (Figure S11). The TFN nanocapsule of CM produced by the microfluidic method showed comparable cytotoxicity to free CM in the concentration range of 5 – 160 μM. Both free curcumin and curcumin nanocapsules showed significant suppression of Panc1 growth at the dosages of > 40 μM. The blank nanocapsule of pTA showed no toxicity up to the dosage of 160 μM (calculated by CM content), suggesting the cytocompatibility of TFN.
3.5. Effect of concentration of coating materials on nanocapsule properties
In the present microfluidic synthetic method, the drug core growth and the TFN coating proceed immediately after nucleation; therefore, the interaction between the two processes may affect the final size of nanocapsule. Figure 6A and 6B show the effect of concentrations of the coating materials of TA and FeCl3, respectively, on the nanocapsule size. For VD3, CM, and PTX, the mean diameter and the PDI decreased with increasing TA concentration from 0.4 to 1.0 mg mL−1 but increased with increasing FeCl3 concentration from 0.06 to 0.12 mg mL−1. The low FeCl3 concentration of 0.03 mg mL−1 decreased the size of CM nanocapsules but increased the size of VD3 and PTX nanocapsules. The high TA concentration of 1.5 mg mL−1 further decreased the size of VD3 nanocapsule, but the effect was saturated for CM and PTX. The drug concentration for Figure 6A, B was kept at 0.1 mg mL−1, and Figure 6C shows the nanocapsule size increased with increasing drug concentration from 0.1 to 0.3 mg mL−1. The summary table of TFN nanocapsules’ mean diameter, PDI, and drug loading produced with different concentrations of coating materials is provided in Table S3.
Figure 6.
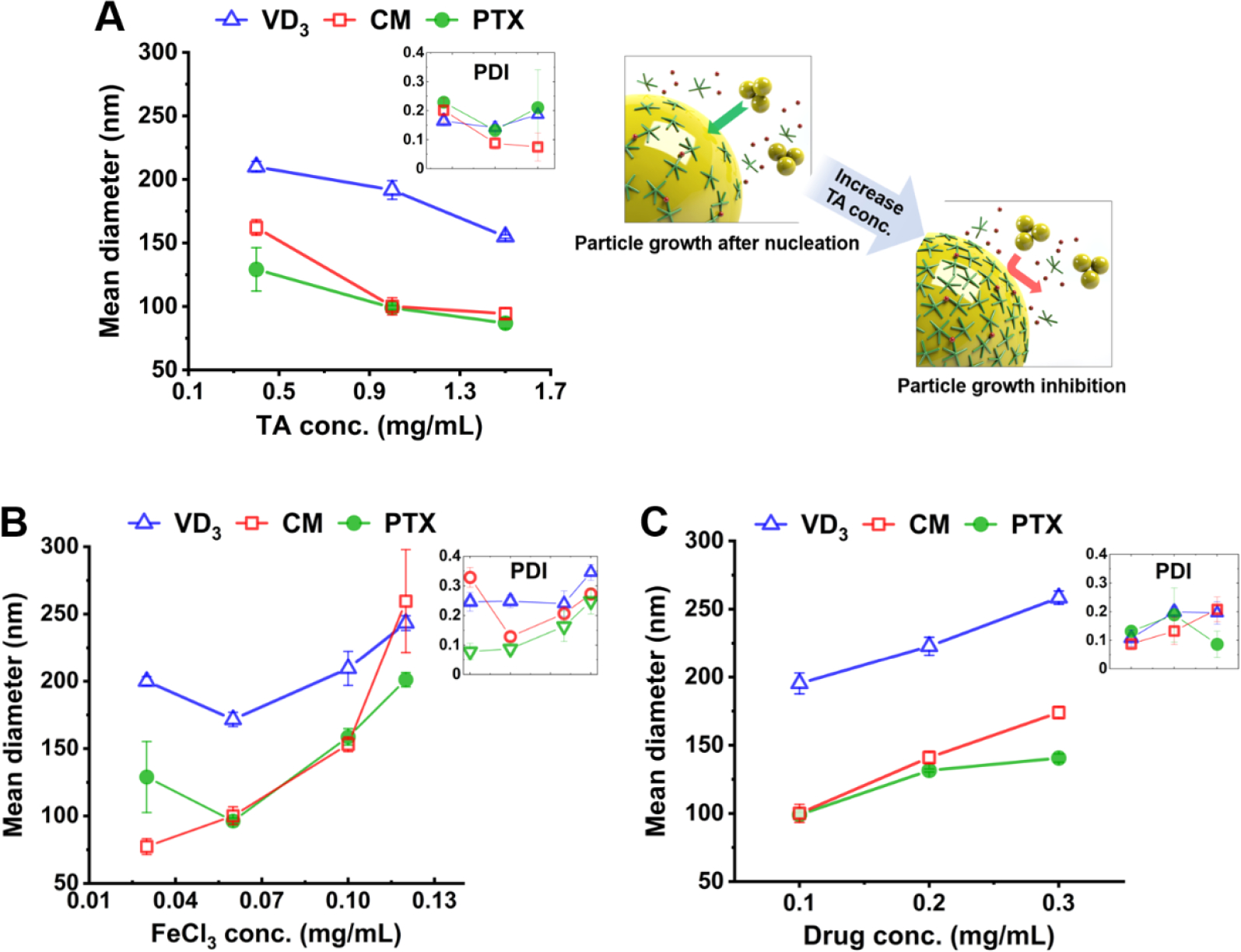
Effects of the concentrations of the coating materials of (A) TA and (B) FeCl3, and (C) drug concentration on the size of the synthesized nanocapsules.
These data suggest the TFN coating may play a role during the particle growth stage. As drug nuclei form in the solution, TA molecules started to accumulate on the nuclei’s surface. As illustrated in Figure 6A, more TA molecules on the nucleus surface had a bigger chance to inhibit particle growth by preventing additional drug molecules or clusters from moving inward. Also, TFN formed faster when FeIII ions were attracted to the particle surface faster. Therefore, nanocapsule size decreased with increasing TA or FeCl3 concentration. Nevertheless, excess FeCl3 (> 0.06 mg mL−1) resulted in the increase of particle size because extra FeIII ions induced particle aggregations. Thus, the nanocapsule size can be affected by how fast the TFN coating can inhibit the particle growth (i.e., effect of FeCl3 and TA concentrations), as well as how many remaining drug molecules in the solution can move toward the particle (i.e., the effect of drug concentration).
In the TFN nanocapsule preparation process, the concentrations of FeCl3 and TA had a significant effect on the Fe content of the nanocapsules (Figure 7A). The Fe content (wt/wt) of TFN nanocapsules was 2.02 ± 0.10% when prepared with 0.06 mg mL−1 FeCl3 and 1.0 mg mL−1 TA. In comparison, the Fe content significantly increased to 5.00 ± 0.32% when the TA concentration decreased to 0.4 mg mL−1; and the Fe content further increased to 6.93 ± 0.15% when the FeCl3 concentration increased to 0.1 mg mL−1. The significant decrease of Fe content with the increase of TA concentration from 0.4 to 1.0 mg mL−1 can be attributed to the increased amount of Fe3+ forming supramolecular complexes with the excess TA molecules in solution rather than the TA molecules attached to the surface of drug nanocores. With the increase of Fe content among the three cases, the drug loadings show a decreasing trend of 74.3 ± 1.1%, 68.0 ± 1.2%, and 54.7 ± 3.9% (Figure 7B). Figure 7C shows the cytotoxicity of TFN nanocapsules of PTX prepared with 0.1 mg mL−1 FeCl3 and 1.0 mg mL−1 TA yielding an Fe content of ~7%. Compared to the nanocapsules produced with 0.06 mg mL−1 FeCl3 and 1.0 mg mL−1 TA yielding an Fe content of ~2% (Figure 5C), the cytotoxicity showed no significant change to Panc10.05 cells in the PTX concentration range of 0.001 – 10 μg mL−1. Similarly, drug-free nanocapsules (pTA) showed no cytotoxicity.
Figure 7.
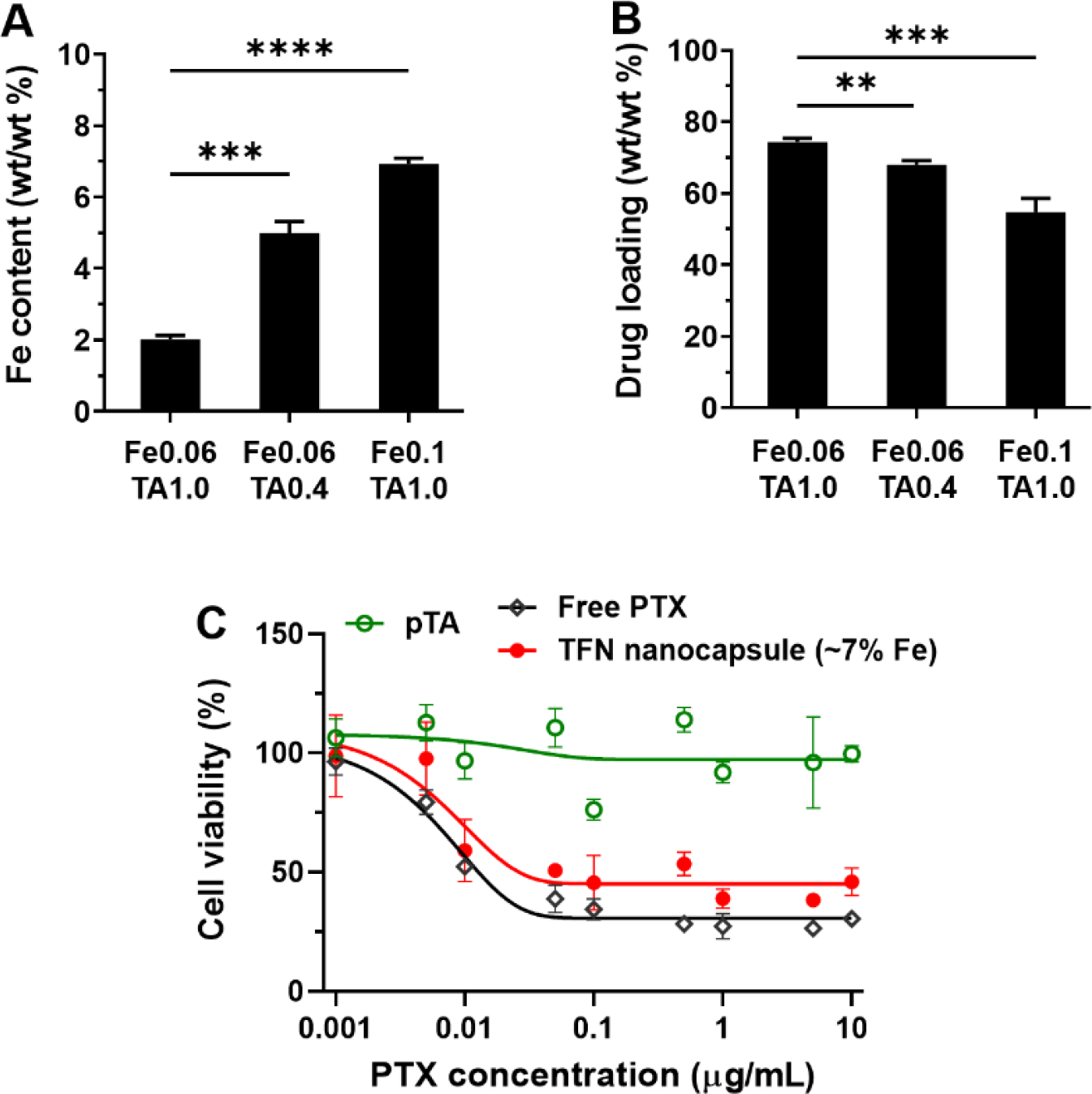
Fe content of TFN nanocapsules and its effect on drug loading and cytotoxicity. (A) Fe content and (B) drug loading of TFN nanocapsules with varied FeCl3 and TA concentrations in the preparation process. (C) Cytotoxicity of TFN nanocapsules of PTX with ~7% Fe content.
3.6. Stability of TFN nanocapsules
The stability of TFN nanocapsules of PTX produced by the microfluidic method was tested at different temperatures and pHs. Figure 8A shows that TFN nanocapsules maintained colloidal stability in 50% FBS at 37°C for 24 hours, irrespective of the concentration range of 50 to 200 μg mL−1 (calculated by PTX content). It is also found that the colloidal solution of TFN nanocapsules remained stable at 4°C during a storage period of 3 weeks (Figure 8B). In the TFN nanocapsule preparation process, the concentrations of FeCl3 and TA had a significant effect on the particle size (Figure 6), Fe content of the nanocapsules (Figure 7A), as well as drug loadings (Figure 7B). However, the long-term colloidal stability can be achieved with different compositions of coating constituents of Fe and TA. Specifically, these three FeCl3/TA concentrations generated TFN nanocapsules with the Fe content of approximately 2%, 5%, and 7% (Figure 7A), respectively, and drug loading of 74%, 68%, and 55% (Figure 7B), respectively; and these nanocapsules maintained their size over 3 weeks of storage with a mean diameter of approximately 80 nm, 100 nm, and 150 nm, respectively (Figure 8B). The TFN nanocapsules were further tested in the media with different pHs (pH 7.4 and pH 5.5). Figure 8C shows TFN nanocapsules released PTX faster at pH 5.5 than at pH 7.4. The pH dependence of the drug release may be explained by the protonation of pyrogallol groups of tannic acid, which causes the dissociation of tannic acid and Fe3+ of the TA-Fe3+ network [1, 51]. This suggests that, once taken up by cells, TFN nanocapsules will undergo accelerated disintegration in the acidity of endo/lysosomes to release the payload.
Figure 8.
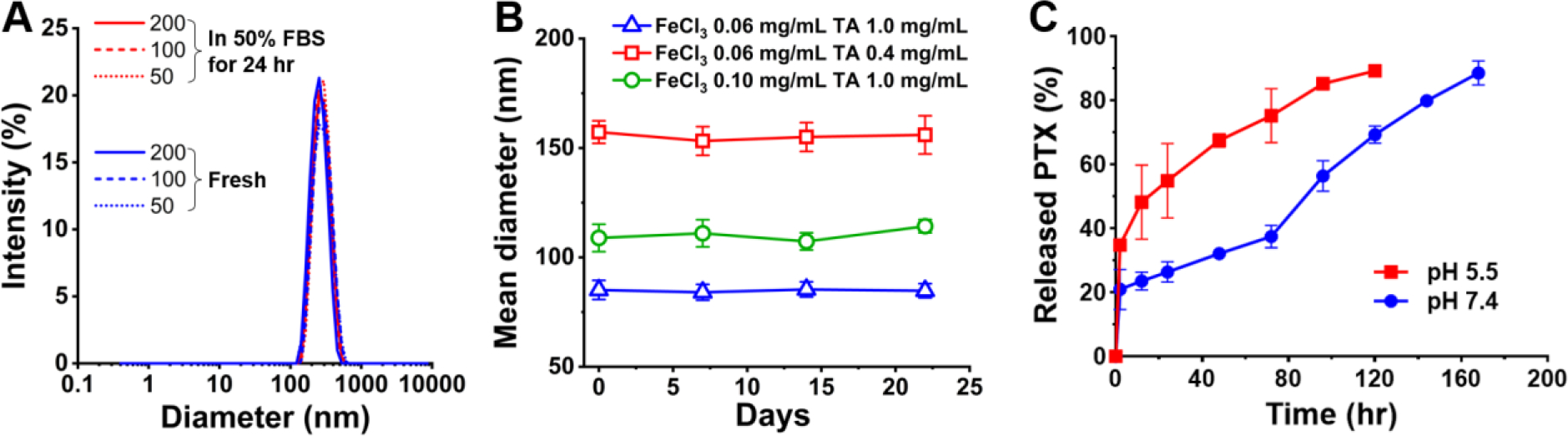
Tests of the stability of TFN nanocapsules at different temperatures and pHs. (A) Size distribution of fresh TFN nanocapsules and TFN nanocapsules incubated in 50% FBS at concentrations equivalent to 50, 100, and 200 μg mL−1 PTX for 24 hr at 37°C. (B) Long-term colloidal stability of TFN nanocapsules prepared with different concentrations of FeCl3 and TA stored at 4°C. (C) pH dependence of the release profile of the encapsulated PTX from nanocapsules. Release kinetics performed with phosphate buffered saline (pH 7.4) + 0.2% Tween 80 or acetate buffered saline (pH 5.5) + 0.2% Tween 80 as both a dispersant and a release medium.
4. Discussion
For the continuous synthesis of MPN nanocapsules of hydrophobic drugs, as illustrated in Figure 1, we designed a microfluidic method consisting of 3 steps of hydrodynamic flow focusing, mixing for nucleation, and particle growth with coating. First, this study aimed at controlling the process of mixing - nucleation, since the nucleation dynamics of drug solutes induced by microfluidic mixing play a determinative role in the particle size. We hypothesized that the two competing timescales of mixing and nucleation dictate the nanocapsule formation. At the experimental conditions belonging to the region of plotted in Figure 2B, the formation of drug nanocapsules was achieved, whereas the nanocapsule formation was not observed at the experimental conditions belonging to the region. Hence, the chart in Figure 2B reasonably predicted the outcomes of three drug (VD3, CM, PTX) encapsulation, which establishes the guidance role of this chart. In summary, we proposed a timescale-guided mechanism to predict the feasibility of nanocapsule synthesis based on microfluidic conditions, and its validity has been verified.
TA or FeIII molecules have no significant effect on the microfluidic steps of flow focusing and mixing, but the TFN has the major function of particle stabilization after the nanocapsules are formed. Moreover, as discussed in Figure 6, TFN coating may influence the nanocapsule size by interfering with the particle growth process. The increase of TA concentration decreased the nanocapsule size, which was attributed to the inhibition of particle growth by TFN coating. 1.0 mg mL−1 was applied as the optimal TA concentration in this study since the effect of the maximum concentration of 1.5 mg mL−1 in reducing the nanocapsule size was saturated and may result in the increase of PDI. 0.06 mg mL−1 FeCl3 was optimal for the effective TFN coating while avoiding particle aggregation induced by excess FeIII. Therefore, the coating materials of 1.0 mg mL−1 TA and 0.06 mg mL−1 FeCl3 can be generally applied to this microfluidic method for nanocapsule synthesis.
One limitation of the application of microfluidics in NP production is the low synthesis throughput due to the small microfluidic dimensions [42, 52]. To address the throughput limitation, the microfluidic channels may be parallelized [53–55]. Our opinion is, for scalable manufacturing, it is crucial to maximize the throughput of a single microchannel prior to the parallelized designs. In this study, we estimated that 300 μm was the maximum channel size for the synthesis of nanocapsules of three drugs; of course, 100 μm channel was also applicable since it had a much shorter . The 300 μm channel enabled a flow rate of 210 mL hr−1, however, the 100 μm channel could only sustain a flow rate up to 2.5 mL hr−1. This huge difference of flow capacity, , was due to the dramatic increase of hydraulic resistance, , with the decrease of channel size, , expressed as [37]. As a result, compared to the 100 μm channel, the 300 μm channel was 3 times larger in size, but the flow capacity, and hence the production throughput, was more than 80 times higher. Thus, it is very meaningful to estimate the maximum microfluidic channel size for the nanocapsule synthesis, guided by the timescale-based mechanism we proposed, to scale up the production, even in the scenario of parallelized channels.
Apart from the synthesis throughput, the synthesis quality of nanocapsule size, PDI, EE and drug loading needs to be discussed when adjusting the microfluidic flow conditions. Compared to batch nanocapsules, the microfluidic nanocapsules synthesized at , shown in Figure 3 and Figure 5, generally had more regular shapes with fewer excess pTA particles, higher EE, more drug loading, and comparable drug release profile. As discussed in Figure 4, the flow rate around , in the transition flow pattern from flow-focusing to flow-bifurcations, was a region to be avoided due to the significant increase of particle size, PDI, and decrease of EE and drug loading. However, further increasing the flow rate to , good quality of nanocapsules was achieved with ~100 nm in size, a PDI of ~0.1, ~70% EE, as well as ~50% drug loading, at a throughput up to 70 mg hr−1 per channel. Hence, our timescale-based microfluidic method not only provides a quantitative guideline to predictively design a synthesis scheme of MPN nanocapsules, but also has scale-up feasibility while maintaining good synthesis quality via the optimization of the microfluidic flow conditions.
The negative charges of TFNs are attributable to the pyrogallol groups of tannic acid, which are partially deprotonated in water [1, 10, 56]. The negative charges are advantageous for avoiding opsonization and macrophage uptake of nanoparticles [57, 58]. Moreover, the TA assemblies with multiple pyrogallol groups can interact with ligands via various intermolecular forces, which allows for additional functionalization of the nanoparticle surface [11, 12]. The chelating agent in blood may remove iron ions from the TA-FeIII complex and affect the particle stability. The major iron chelating protein in human blood plasma is transferrin, present at 2 – 3 mg mL−1 [59, 60]. In this study, 50% FBS, approximately containing 2 mg mL−1 of transferrin [61, 62], was used to mimic blood. The microfluidically produced TFN nanocapsules maintained the particle size after 24 hours’ incubation in 50% FBS (Figure 8A). The stability of the batch-produced TFN nanocapsules in serum and whole blood was also tested and verified in our previous studies [11, 12].
The TFN nanocapsules were produced to be size compatible with parenteral administration; however, it may be considered for other routes in future studies. Tannic acid is considered a GRAS (generally recognized as safe) material by FDA for oral consumption [63] and used in topical products. Tannic acid was also used in an injectable product, which received FDA approval but has now been discontinued [64]. The results of Fe content and drug loadings of TFN nanocapsules of PTX indicate the weight ratio of Fe to PTX approximately ranges from 2.7% to 12.7% depending on the concentrations of coating materials in the preparation process. Thus, for a standard PTX dose of 175 mg m−2 per 3 weeks administered intravenously [65], the patients may approximately be exposed to 9 to 42 mg of Fe per 3 weeks. This is a safe dose of Fe since the safety of intravenous Fe has been verified at a median dose of 1400 mg in a single intravenous infusion per week [66]. Although the toxicity remains to be studied systematically, the doses of TA and Fe used in TFN nanocapsules are expected to be safe. The biocompatibility of TA and Fe has been reported by literature [67, 68]. TFN is reported to be stable over 10 days at physiological pH [1] but disassembled in acidic pH, where TA and Fe3+ are dissociated due to the protonation of pyrogallol moieties [1, 51]. Figure 8C shows TFN nanocapsules released PTX faster at pH 5.5 than at pH 7.4. Therefore, once TFN is placed in weakly acidic tumor microenvironment and/or taken up by cells to reach acidic lysosomes, it is expected to be disassembled into TA and Fe3+, which can be cleared from the system.
5. Conclusion
In this study, we proposed a timescale-guided microfluidic method for the continuous production of MPN nanocapsules of hydrophobic drugs. As a simple, flexible, and safe nanocarrier of hydrophobic drugs, the MPN nanocapsules have emerged as a carrier of different water-insoluble drugs in recent years. This work fulfills the translation from batch to microfluidic synthesis of MPN nanocapsules, enabling their continuous production at increased throughput. We demonstrated the efficiency of this method via the synthesis of nanocapsules of different drugs of curcumin, paclitaxel, and vitamin D. Moreover, our microfluidic method enables continuous production at a throughput up to 70 mg hr−1 per channel, without losing the quality control of the nanocapsules, to facilitate scale-up efforts. Our work on this new microfluidic method lays the foundation for scalable, well-controlled continuous production of MPN nanocapsules for the encapsulation of a wide variety of hydrophobic drugs.
Supplementary Material
Highlights.
Synthesis of metal-phenolic nanocapsules is translated from batch to microfluidics
Mechanism of encapsulating of a variety of hydrophobic drugs is established
Effect of concentrations of coating materials on nanocapsule properties are tested
Quality control at a throughput of 70 mg hr−1 exhibits scale-up potential
Acknowledgements
This work was partially supported by grants from the National Institutes of Health (U01 HL143403, R01 CA254110, U01 CA274304 to BH; R01 CA232419, R01 CA258737 to YY) and National Science Foundation (MCB-2134603 to BH). YS was partially supported by the Purdue University Center for Cancer Research (P30 CA023168) and a Program Grant from Purdue Institute of Drug Discovery.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ejima H, Richardson JJ, Liang K, Best JP, van Koeverden MP, Such GK, Cui J, Caruso F, One-step assembly of coordination complexes for versatile film and particle engineering, Science, 341 (2013) 154–157. [DOI] [PubMed] [Google Scholar]
- [2].Gao C, Wang Y, Ye Z, Lin Z, Ma X, He Q, Biomedical Micro-/Nanomotors: From Overcoming Biological Barriers to In Vivo Imaging, Advanced Materials, 33 (2021) 2000512. [DOI] [PubMed] [Google Scholar]
- [3].Li J, Zhou Y, Liu J, Yang X, Zhang K, Lei L, Hu H, Zhang H, Ouyang L, Gao H, Metal-phenolic networks with ferroptosis to deliver NIR-responsive CO for synergistic therapy, Journal of Controlled Release, 352 (2022) 313–327. [DOI] [PubMed] [Google Scholar]
- [4].Ping Y, Guo J, Ejima H, Chen X, Richardson JJ, Sun H, Caruso F, pH-responsive capsules engineered from metal–phenolic networks for anticancer drug delivery, Small, 11 (2015) 2032–2036. [DOI] [PubMed] [Google Scholar]
- [5].Abouelmagd SA, Abd Ellah NH, Amen O, Abdelmoez A, Mohamed NG, Self-assembled tannic acid complexes for pH-responsive delivery of antibiotics: role of drug-carrier interactions, International journal of pharmaceutics, 562 (2019) 76–85. [DOI] [PubMed] [Google Scholar]
- [6].He Z, Hu Y, Gui Z, Zhou Y, Nie T, Zhu J, Liu Z, Chen K, Liu L, Leong KW, Sustained release of exendin-4 from tannic acid/Fe (III) nanoparticles prolongs blood glycemic control in a mouse model of type II diabetes, Journal of Controlled Release, 301 (2019) 119–128. [DOI] [PubMed] [Google Scholar]
- [7].Li W, Bing W, Huang S, Ren J, Qu X, Mussel Byssus - Like Reversible Metal-Chelated Supramolecular Complex Used for Dynamic Cellular Surface Engineering and Imaging, Advanced Functional Materials, 25 (2015) 3775–3784. [Google Scholar]
- [8].Su D, Liu X, Wang L, Ma C, Xie H, Zhang H, Meng X, Huang Y, Huang X, Bio-inspired engineering proteinosomes with a cell-wall-like protective shell by self-assembly of a metal-chelated complex, Chemical Communications, 52 (2016) 13803–13806. [DOI] [PubMed] [Google Scholar]
- [9].Lee J, Cho H, Choi J, Kim D, Hong D, Park JH, Yang SH, Choi IS, Chemical sporulation and germination: cytoprotective nanocoating of individual mammalian cells with a degradable tannic acid–Fe III complex, Nanoscale, 7 (2015) 18918–18922. [DOI] [PubMed] [Google Scholar]
- [10].Shen G, Xing R, Zhang N, Chen C, Ma G, Yan X, Interfacial cohesion and assembly of bioadhesive molecules for design of long-term stable hydrophobic nanodrugs toward effective anticancer therapy, ACS nano, 10 (2016) 5720–5729. [DOI] [PubMed] [Google Scholar]
- [11].Taha MS, Cresswell GM, Park J, Lee W, Ratliff TL, Yeo Y, Sustained delivery of carfilzomib by tannic acid-based nanocapsules helps develop antitumor immunity, Nano letters, 19 (2019) 8333–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yuk SA, Kim H, Abutaleb NS, Dieterly AM, Taha MS, Tsifansky MD, Lyle LT, Seleem MN, Yeo Y, Nanocapsules modify membrane interaction of polymyxin B to enable safe systemic therapy of Gram-negative sepsis, Science Advances, 7 (2021) eabj1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen Y, Jia D, Wang Q, Sun Y, Rao Z, Lei X, Zhao J, Zeng K, Xu Z, Ming J, Promotion of the anticancer activity of curcumin based on a metal–polyphenol networks delivery system, International Journal of Pharmaceutics, 602 (2021) 120650. [DOI] [PubMed] [Google Scholar]
- [14].Liu P, Liu X, Cheng Y, Zhong S, Shi X, Wang S, Liu M, Ding J, Zhou W, Core–Shell Nanosystems for Self-Activated Drug–Gene Combinations against Triple-Negative Breast Cancer, ACS Applied Materials & Interfaces, 12 (2020) 53654–53664. [DOI] [PubMed] [Google Scholar]
- [15].Liu Y, Ma K, Jiao T, Xing R, Shen G, Yan X, Water-insoluble photosensitizer nanocolloids stabilized by supramolecular interfacial assembly towards photodynamic therapy, Scientific reports, 7 (2017) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Taemaitree F, Koseki Y, Saito N, Suzuki R, Dao ATN, Kasai H, Self-assemble tannic acid and iron complexes on pure nanodrugs surface prevents aggregation and enhances anticancer drug delivery efficiency, Molecular Crystals and Liquid Crystals, 706 (2020) 116–121. [Google Scholar]
- [17].Huang F, Jiang X, Sallam MA, Zhang X, He W, A Nanocrystal Platform Based on Metal-Phenolic Network Wrapping for Drug Solubilization, AAPS PharmSciTech, 23 (2022) 1–11. [DOI] [PubMed] [Google Scholar]
- [18].Mu M, Liang X, Chuan D, Zhao S, Yu W, Fan R, Tong A, Zhao N, Han B, Guo G, Chitosan coated pH-responsive metal-polyphenol delivery platform for melanoma chemotherapy, Carbohydrate Polymers, 264 (2021) 118000. [DOI] [PubMed] [Google Scholar]
- [19].Okuda K, Sato Y, Iwakawa K, Sasaki K, Okabe N, Maeki M, Tokeshi M, Harashima H, On the size-regulation of RNA-loaded lipid nanoparticles synthesized by microfluidic device, Journal of Controlled Release, 348 (2022) 648–659. [DOI] [PubMed] [Google Scholar]
- [20].Ahn J, Ko J, Lee S, Yu J, Kim Y, Jeon NL, Microfluidics in nanoparticle drug delivery; From synthesis to pre-clinical screening, Advanced drug delivery reviews, 128 (2018) 29–53. [DOI] [PubMed] [Google Scholar]
- [21].Arduino I, Liu Z, Rahikkala A, Figueiredo P, Correia A, Cutrignelli A, Denora N, Santos HA, Preparation of cetyl palmitate-based PEGylated solid lipid nanoparticles by microfluidic technique, Acta Biomaterialia, 121 (2021) 566–578. [DOI] [PubMed] [Google Scholar]
- [22].Maeki M, Uno S, Niwa A, Okada Y, Tokeshi M, Microfluidic technologies and devices for lipid nanoparticle-based RNA delivery, Journal of Controlled Release, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shepherd SJ, Issadore D, Mitchell MJ, Microfluidic formulation of nanoparticles for biomedical applications, Biomaterials, 274 (2021) 120826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sato Y, Okabe N, Note Y, Hashiba K, Maeki M, Tokeshi M, Harashima H, Hydrophobic scaffolds of pH-sensitive cationic lipids contribute to miscibility with phospholipids and improve the efficiency of delivering short interfering RNA by small-sized lipid nanoparticles, Acta biomaterialia, 102 (2020) 341–350. [DOI] [PubMed] [Google Scholar]
- [25].Hickey JW, Santos JL, Williford J-M, Mao H-Q, Control of polymeric nanoparticle size to improve therapeutic delivery, Journal of Controlled Release, 219 (2015) 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Karnik R, Gu F, Basto P, Cannizzaro C, Dean L, Kyei-Manu W, Langer R, Farokhzad OC, Microfluidic platform for controlled synthesis of polymeric nanoparticles, Nano letters, 8 (2008) 2906–2912. [DOI] [PubMed] [Google Scholar]
- [27].Wang L, Liu Y, He J, Hourwitz MJ, Yang Y, Fourkas JT, Han X, Nie Z, Continuous microfluidic self-assembly of hybrid Janus-like vesicular motors: autonomous propulsion and controlled release, Small, 11 (2015) 3762–3767. [DOI] [PubMed] [Google Scholar]
- [28].Wang J, Chen W, Sun J, Liu C, Yin Q, Zhang L, Xianyu Y, Shi X, Hu G, Jiang X, A microfluidic tubing method and its application for controlled synthesis of polymeric nanoparticles, Lab on a Chip, 14 (2014) 1673–1677. [DOI] [PubMed] [Google Scholar]
- [29].Ozcelikkale A, Shin K, Noe-Kim V, Elzey BD, Dong Z, Zhang J-T, Kim K, Kwon IC, Park K, Han B, Differential response to doxorubicin in breast cancer subtypes simulated by a microfluidic tumor model, Journal of Controlled Release, 266 (2017) 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Moon H.-r., Ozcelikkale A, Yang Y, Elzey BD, Konieczny SF, Han B, An engineered pancreatic cancer model with intra-tumoral heterogeneity of driver mutations, Lab on a Chip, 20 (2020) 3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lee JN, Park C, Whitesides GM, Solvent compatibility of poly (dimethylsiloxane)-based microfluidic devices, Analytical chemistry, 75 (2003) 6544–6554. [DOI] [PubMed] [Google Scholar]
- [32].Yang H, Svard M, Zeglinski J, Rasmuson ÅC, Influence of solvent and solid-state structure on nucleation of parabens, Crystal growth & design, 14 (2014) 3890–3902. [Google Scholar]
- [33].Teychené S, Biscans B, Nucleation kinetics of polymorphs: induction period and interfacial energy measurements, Crystal Growth and Design, 8 (2008) 1133–1139. [Google Scholar]
- [34].Pal S, Madane K, Kulkarni AA, Antisolvent based precipitation: Batch, capillary flow reactor and impinging jet reactor, Chemical Engineering Journal, 369 (2019) 1161–1171. [Google Scholar]
- [35].Markwalter CE, Pagels RF, Hejazi AN, Ristroph KD, Wang J, Chen K, Li J, Prud’homme RK, Sustained release of peptides and proteins from polymeric nanocarriers produced by inverse Flash NanoPrecipitation, Journal of Controlled Release, 334 (2021) 11–20. [DOI] [PubMed] [Google Scholar]
- [36].Horn D, Rieger J, Organic nanoparticles in the aqueous phase—theory, experiment, and use, Angewandte Chemie International Edition, 40 (2001) 4330–4361. [DOI] [PubMed] [Google Scholar]
- [37].Polte J, Erler R, Thunemann AF, Sokolov S, Ahner TT, Rademann K, Emmerling F, Kraehnert R, Nucleation and growth of gold nanoparticles studied via in situ small angle X-ray scattering at millisecond time resolution, ACS nano, 4 (2010) 1076–1082. [DOI] [PubMed] [Google Scholar]
- [38].Lepeltier E, Bourgaux C, Couvreur P, Nanoprecipitation and the “Ouzo effect”: Application to drug delivery devices, Advanced drug delivery reviews, 71 (2014) 86–97. [DOI] [PubMed] [Google Scholar]
- [39].Johnson BK, Prud’homme RK, Mechanism for rapid self-assembly of block copolymer nanoparticles, Physical review letters, 91 (2003) 118302. [DOI] [PubMed] [Google Scholar]
- [40].Söhnel O, Mullin JW, Interpretation of crystallization induction periods, Journal of colloid and interface science, 123 (1988) 43–50. [Google Scholar]
- [41].Davey RJ, Schroeder SLM, Ter Horst JH, Nucleation of organic crystals—a molecular perspective, Angewandte Chemie International Edition, 52 (2013) 2166–2179. [DOI] [PubMed] [Google Scholar]
- [42].Hamdallah SI, Zoqlam R, Erfle P, Blyth M, Alkilany AM, Dietzel A, Qi S, Microfluidics for pharmaceutical nanoparticle fabrication: The truth and the myth, International journal of pharmaceutics, 584 (2020) 119408. [DOI] [PubMed] [Google Scholar]
- [43].Sun DD, Lee PI, Evolution of supersaturation of amorphous pharmaceuticals: nonlinear rate of supersaturation generation regulated by matrix diffusion, Molecular pharmaceutics, 12 (2015) 1203–1215. [DOI] [PubMed] [Google Scholar]
- [44].Thanh NTK, Maclean N, Mahiddine S, Mechanisms of nucleation and growth of nanoparticles in solution, Chemical reviews, 114 (2014) 7610–7630. [DOI] [PubMed] [Google Scholar]
- [45].Groisman A, Quake SR, A microfluidic rectifier: Anisotropic flow resistance at low Reynolds numbers, Physical review letters, 92 (2004) 094501. [DOI] [PubMed] [Google Scholar]
- [46].Shen Y, Hu L, Chen W, Xie H, Fu X, Drop encapsulated in bubble: a new encapsulation structure, Physical Review Letters, 120 (2018) 054503. [DOI] [PubMed] [Google Scholar]
- [47].Holcman D, Schuss Z, Time scale of diffusion in molecular and cellular biology, Journal of Physics A: Mathematical and Theoretical, 47 (2014) 173001. [Google Scholar]
- [48].Pucci C, Martinelli C, De Pasquale D, Battaglini M, di Leo N, Degl’Innocenti A, Belenli Gümüş M, Drago F, Ciofani G, Tannic acid–iron complex-based nanoparticles as a novel tool against oxidative stress, ACS Applied Materials & Interfaces, 14 (2022) 15927–15941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Çakar S, Özacar M, Fe–tannic acid complex dye as photo sensitizer for different morphological ZnO based DSSCs, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 163 (2016) 79–88. [DOI] [PubMed] [Google Scholar]
- [50].Bruschi ML, Strategies to modify the drug release from pharmaceutical systems, Woodhead Publishing, 2015. [Google Scholar]
- [51].Shi Q, Wu K, Huang X, Xu R, Zhang W, Bai J, Du S, Han N, Tannic acid/Fe3+ complex coated mesoporous silica nanoparticles for controlled drug release and combined chemo-photothermal therapy, Colloids and Surfaces A: Physicochemical and Engineering Aspects, 618 (2021) 126475. [Google Scholar]
- [52].Baby T, Liu Y, Middelberg APJ, Zhao C-X, Fundamental studies on throughput capacities of hydrodynamic flow-focusing microfluidics for producing monodisperse polymer nanoparticles, Chemical Engineering Science, 169 (2017) 128–139. [Google Scholar]
- [53].Forigua A, Kirsch RL, Willerth SM, Elvira KS, Recent advances in the design of microfluidic technologies for the manufacture of drug releasing particles, Journal of Controlled Release, 333 (2021) 258–268. [DOI] [PubMed] [Google Scholar]
- [54].Shepherd SJ, Issadore D, Mitchell MJ, Microfluidic formulation of nanoparticles for biomedical applications, Biomaterials, (2021) 120826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lim J-M, Bertrand N, Valencia PM, Rhee M, Langer R, Jon S, Farokhzad OC, Karnik R, Parallel microfluidic synthesis of size-tunable polymeric nanoparticles using 3D flow focusing towards in vivo study, Nanomedicine: Nanotechnology, Biology and Medicine, 10 (2014) 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shin M, Lee H-A, Lee M, Shin Y, Song J-J, Kang S-W, Nam D-H, Jeon EJ, Cho M, Do M, Targeting protein and peptide therapeutics to the heart via tannic acid modification, Nature biomedical engineering, 2 (2018) 304–317. [DOI] [PubMed] [Google Scholar]
- [57].He C, Hu Y, Yin L, Tang C, Yin C, Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles, Biomaterials, 31 (2010) 3657–3666. [DOI] [PubMed] [Google Scholar]
- [58].Blanco E, Shen H, Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nature biotechnology, 33 (2015) 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kontoghiorghes GJ, Kontoghiorghe CN, Iron and chelation in biochemistry and medicine: new approaches to controlling iron metabolism and treating related diseases, Cells, 9 (2020) 1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kawabata H, Transferrin and transferrin receptors update, Free Radical Biology and Medicine, 133 (2019) 46–54. [DOI] [PubMed] [Google Scholar]
- [61].Kakuta K, Orino K, Yamamoto S, Watanabe K, High levels of ferritin and its iron in fetal bovine serum, Comparative Biochemistry and Physiology Part A: Physiology, 118 (1997) 165–169. [DOI] [PubMed] [Google Scholar]
- [62].Ji Z, Jin X, George S, Xia T, Meng H, Wang X, Suarez E, Zhang H, Hoek EMV, Godwin H, Dispersion and stability optimization of TiO2 nanoparticles in cell culture media, Environmental science & technology, 44 (2010) 7309–7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chen J, Li J, Zhou J, Lin Z, Cavalieri F, Czuba-Wojnilowicz E, Hu Y, Glab A, Ju Y, Richardson JJ, Metal–Phenolic Coatings as a Platform to Trigger Endosomal Escape of Nanoparticles, ACS nano, 13 (2019) 11653–11664. [DOI] [PubMed] [Google Scholar]
- [64]. accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=011208 .
- [65].Shi L, Wang Y, Wang Q, Jiang Z, Ren L, Yan Y, Liu Z, Wan J, Huang L, Cen B, Transforming a toxic drug into an efficacious nanomedicine using a lipoprodrug strategy for the treatment of patient-derived melanoma xenografts, Journal of Controlled Release, 324 (2020) 289–302. [DOI] [PubMed] [Google Scholar]
- [66].Auerbach M, Gafter-Gvili A, Macdougall IC, Intravenous iron: a framework for changing the management of iron deficiency, The Lancet Haematology, 7 (2020) e342–e350. [DOI] [PubMed] [Google Scholar]
- [67].Shin M, Kim K, Shim W, Yang JW, Lee H, Tannic Acid as a Degradable Mucoadhesive Compound, ACS Biomaterials Science & Engineering, 2 (2016) 687–696. [DOI] [PubMed] [Google Scholar]
- [68].Gąsior G, Szczepański J, Radtke A, Biodegradable Iron-Based Materials—What Was Done and What More Can Be Done?, Materials, 14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.