

Abstract
Alveolar type II (ATII) pneumocytes as defenders of the alveolus are critical to repairing lung injury. We investigated the ATII reparative response in coronavirus disease 2019 (COVID-19) pneumonia, because the initial proliferation of ATII cells in this reparative process should provide large numbers of target cells to amplify severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus production and cytopathological effects to compromise lung repair. We show that both infected and uninfected ATII cells succumb to tumor necrosis factor-α (TNF)-induced necroptosis, Bruton tyrosine kinase (BTK)-induced pyroptosis, and a new PANoptotic hybrid form of inflammatory cell death mediated by a PANoptosomal latticework that generates distinctive COVID-19 pathologies in contiguous ATII cells. Identifying TNF and BTK as the initiators of programmed cell death and SARS-CoV-2 cytopathic effects provides a rationale for early antiviral treatment combined with inhibitors of TNF and BTK to preserve ATII cell populations, reduce programmed cell death and associated hyperinflammation, and restore functioning alveoli in COVID-19 pneumonia.
Keywords: COVID-19 pneumonia, BTK, lung repair, necroptosis, PANoptosis, pyroptosis, SARS-CoV-2, TNF, type II pneumocytes
Death in fatal COVID-19 pneumonia of SARS-CoV-2 infected and uninfected ATII pneumocytes is mediated by autochthonous expression of TNF-induced necroptic, BTK-induced pyroptotic, and apoptotic pathways in ATII cell PANoptosomes.
By the third year of the coronavirus disease 2019 (COVID-19) pandemic, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus has cumulatively infected more than 750 million people worldwide with nearly 7 million reported deaths from COVID-19 pneumonia and associated comorbidities [1]. While the severe and fatal outcomes from SARS-CoV-2 infection and COVID-19 pneumonia have been greatly reduced by vaccination, they have not been altogether prevented, particularly in the elderly and other high-risk groups, and remain an ongoing threat with each new variant. Thus, identifying the mechanisms underlying disease progression in COVID-19 pneumonia is of great importance to guide development of effective treatments.
In broad outline, there is already a rationale for treating COVID-19 pneumonia as a 2-stage process initiated by SARS-CoV-2 infection and destruction of alveolar type I and II (ATI and ATII) pneumocytes, to be targeted with antiviral treatment; and a second stage in which pathological progression is mediated by hyperinflammatory mechanisms, to be targeted by inhibitors of inflammation such as steroids [2, 3]. However, further advances in treating this second stage of COVID-19 pneumonia will require a deeper understanding of specific mechanisms of hyperinflammatory lung injury, particularly the role of tumor necrosis factor-α (TNF-α) [4–8], because blocking TNF decreased hospitalization for COVID-19 pneumonia in people with rheumatoid arthritis undergoing treatment with TNF inhibitors [9], and blocking Bruton tyrosine kinase (BTK), because inhibitors of BTK to block nuclear factor-κB (NF-κB) production of proinflammatory cytokines and activation of the NLRP3 inflammasome improved oxygenation in severe COVID-19 pneumonia [10].
We investigated the role of TNF-α and BTK in fatal COVID-19 pneumonia because of these reported therapeutic benefits, focusing on ATII cells, the so-called defenders of the alveolus [11] because, in addition to supplying surfactant to keep alveoli patent on expiration, ATII cells proliferate in response to lung injury, differentiate to replace ATI cells lost to infection and other forms of lung injury, and then are largely cleared by apoptosis to restore a patent functional alveolus for gas exchange. This ATII cell reparative process, initially driven by proliferation, suggests that SARS-CoV-2 would be able to target large numbers of susceptible cell to amplify virus production and cytopathic effects, thereby reducing the population of ATII cells available for repair in the first stage of severe pneumonia. Because ATII cells can themselves produce TNF-α [11], we also thought that investigating ATII cells might yield insights into the mechanisms of hyperinflammation in the second stage of COVID-19 pneumonia.
METHODS
Human Participants
Specimens were collected from autopsies at the beginning of the COVID-19 pandemic in Milan, Italy. All the study patients had COVID-19 confirmed by a positive real-time reverse-transcription polymerase chain reaction on a nasopharyngeal swab. Health care providers obtained witnessed consent from families for limited autopsies after the death. Research using autopsy tissue for this project was approved by the local institutional review board, Comitato Etico Interaziendale Area 1. Patient demographics, clinical and laboratory data, treatment, and course of infection are summarized in Supplementary Table 1.
Immunohistochemistry
Five-micron selections were deparaffinized and rehydrated followed by heat-induced epitope retrieval. Endogenous peroxidases and tissues were blocked before adding the primary antibodies and subsequently horseradish peroxidase or alkaline phosphatase substrates. Key resources are listed in Supplementary Table 2.
RNAscope in Situ Hybridization
SARS-CoV-2 RNA was visualized by RNAscope in situ hybridization (ISH) with antisense probes and reagents from Advanced Cell Diagnostics as previously published [12]. SARS-CoV-2 antisense probes VnCoV2019S 21631-23303 of NC 045512.2, which hybridizes specifically to the 5′ end of SARS-CoV-2 spike RNA, and VnCoV-N 28275-29204 of MN908947.3 were added before continuing with the RNAscope 2.5 Red detection kit. Warp Red chromogen was added to visualize the RNA.
RNAscope Enzyme-Labeled Fluorescence
Intracellular viral RNA and virions were visualized following the same protocol for the RNAscope 2.5 Red detection kit, but replacing Warp Red staining with the ELF 97 endogenous phosphatase substrate.
Scanscope, Image Acquisition, and Analysis
Immunohistochemistry (IHC)-stained slides were imaged using whole-tissue scanning by an Aperio Versa 8 (Leica Biosystems) with 20×/0.80NA, 40×/0.85NA, and oil 63×/1.30NA objectives. Digitized images were viewed and annotated using Aperio Webscope. IHC- and ISH-stained slides were imaged using brightfield whole-tissue scanning and digitized images were viewed and annotated using Aperio eSlide Manager. Monochrome images for fluorescence in situ hybridization (FISH) and immunofluorescence-stained slides were captured for each fluorophore using an Olympus DP80. A pseudocolor was applied using the constrained iterative deconvolution algorithm (Olympus) for each marker and fused into a composite image for viewing. Image processing and analysis for IHC- and ISH-stained slides were performed using the area quantification and deconvolution modules of Halo image analysis platform (Indica Labs, Inc).
RESULTS
SARS-CoV-2 Infection of Bronchiolar and ATI and ATII Cells in Early-Stage COVID-19 Pneumonia
We undertook a collaborative study of SARS-CoV-2 replication and pathology in autopsy lung tissues collected during the first wave of the pandemic in Italy from 4 unvaccinated, immunologically naive, and elderly patients who succumbed with a short hospitalized course of 0–16 days to primary infection caused by the ancestral variant of SARS-CoV-2 (Supplementary Table 1). The lung tissue pathology ranged from early focal infection as the initial stage of COVID-19 pneumonia [13] to late-stage COVID-19 pneumonia with diffuse alveolar damage and hemorrhage. We found evidence of focal pneumonia in the tissues from an asymptomatic 88-year-old woman who died of a cardiac arrhythmia. Abundant SARS-CoV-2 genomic RNA was detectable by ISH in (1) fused terminal bronchiolar and ATI and ATII cells, identified by their morphology and anatomic location (Figure 1A and 1B); (2) large clusters of fused ATII cells lining and lying within alveolar spaces (Figure 1C); and (3) large desquamated syncytial mats of lysed cells (Figure 1D). At each location, viral RNA (vRNA) was concentrated in round bodies (RBs) that we subsequently discovered to be distinctive components of a latticework that mediates necroptosis and viral cytopathic effects.
Figure 1.

SARS-CoV-2 replication and cytopathic effects in bronchiolar epithelium and ATI and ATII pneumocytes. RNAscope in situ hybridization on autopsy lung tissue: red, SARS-CoV-2 viral RNA, and hematoxylin counterstain. A, SARS-CoV-2 RNA in fused cells in a bifurcation of terminal bronchiolar epithelium. Viral RNA is concentrated in small RBs. B, Alveoli lined by thin fused SARS-CoV-2 RNA+ ATI cells. Arrow points to a SARS-CoV-2 RNA+ cell, identified morphologically as an ATII cell. C, Syncytial clusters of fused SARS-CoV-2 RNA+ ATII cells with viral RNA in dark-staining RBs. D, Large desquamated syncytial mat with viral RNA in RBs. Abbreviations: AT, alveolar type; RB, round body; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
SARS-CoV-2 Production, Spread, and Cytopathic Effects
We characterized SARS-CoV-2 virus production with ISH and ELF-97 staining that renders virions visible by light and immunofluorescence microscopy, as previously described [14, 15]. Most visualized SARS-CoV-2 virions were detected in RBs, similar to the finding for vRNA, in lysed fused cells lining the terminal bronchioles and alveoli (Figure 2). The hub and spoke distribution of these virus aggregates (Figure 2A) visually traced replication and spread from branching terminal bronchioles into lung parenchyma, consistent with spread of virus from the upper airways into the lung. The images of SARS-CoV-2 in a continuous alveolar lining traced infection and spread, fusion, and lysis of contiguous ATI cells (Figure 2B); similarly, virus spread and fusion of spatially contiguous cells generated focal clusters of virus-producing cells and large syncytia of virus-producing cells with virus aggregated in RBs (Figure 2C); and lysis of these syncytia generated mats of cells with virus concentrated in RBs (Figure 2D).
Figure 2.

SARS-CoV-2 spread to and replication in the lung. Individual SARS-CoV-2 virions detected by in situ hybridization with ELF-97 substrate appear green and are approximately 0.25 μm. Nuclei stained blue with DAPI. Large numbers of virions are amassed in RBs of varying size. A, Virus production and spread in bronchiolar epithelium leave a visible trace of the hub and spoke mode of spread from branching terminal bronchioles into the lung parenchyma (encircled). B, Virus is concentrated in RBs in fused lysed ATI cells lining alveolar walls. C, Virus spread and fusion of spatially contiguous ATII cells generate focal clusters of virus-producing cells. D, Syncytial mat of virus RBs in lysed epithelium. Abbreviations: AT, alveolar type; RB, round body; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
SARS-CoV-2 Replicates and Spreads in Type I Interferon-Negative ATII Cells
SARS-CoV-2 replicates and spreads in this early-stage pneumonia in spatially contiguous ATII cells identified by napsin A staining of vRNA+ cells (Figure 3A). Virus production and spread followed a similar pattern of infection of cells in close spatial proximity and fusion of contiguous cells to generate clusters of virus-producing cells (Figure 3B). While type I interferon (IFN) was expressed in many cells in the lung sections, the vRNA+ cells were uniformly negative for type I IFN (Figure 3C).
Figure 3.
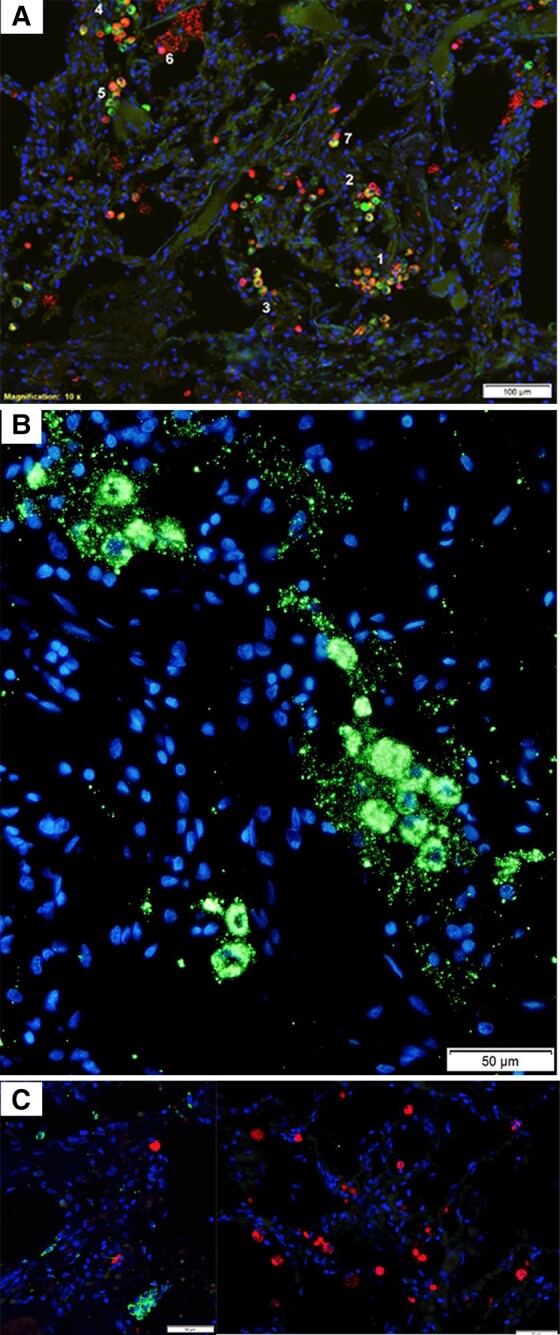
SARS-CoV-2 replication and spread in type I interferon-negative ATII pneumocytes. A, Red cells are viral RNA+; green cells are napsin A+ ATII cells; red-green cells are infected ATII cells; nuclei stained blue with DAPI. The viral RNA+ ATII cells in numbered regions 1–5 are spatially contiguous, consistent with spread of infection to susceptible type II cells in close spatial proximity. Region 6 shows a napsin A− viral RNA+ cell overlying lysed viral RNA+ epithelial syncytium. Region 7 shows a viral RNA+ napsin A+ and napsin A− cell conjugate, consistent with acquisition of viral RNA in macrophages by phagocytosis. B, SARS-CoV-2 virions and intracellular RNA, green. Virus spread and fusion of spatially contiguous ATII cells generates focal clusters of virus-producing cells. C, SARS-CoV-2 RNA+ cells, red; type I interferon, green; nuclei stained blue with DAPI. Viral RNA+ cells are type I interferon−, and interferon+ cells are viral RNA−. Abbreviations: AT, alveolar type; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
SARS-CoV-2 Cytopathic Effects and TNF-Induced Necroptosis
To assess the role of TNF and hyperinflammation in COVID-19 pneumonia, we stained lung tissues from each patient to detect TNF+ cells, and, in the focal pneumonia lung tissue, combined detection of TNF with ISH to assess the spatial relationship to SARS-CoV-2 infection. We discovered in this way that there were large numbers of TNF+ SARS-CoV-2 RNA+ ATII cells in which vRNA and TNF colocalized in latticework bodies (LBs) corresponding to the previously described RBs. The LBs also housed necroptotic pathway components that mediated necroptosis as the mechanism underlying SARS-CoV-2 cytopathic effects in ATII cells. In this process, LBs became porous and this porous latticework was associated with emptying of cell components, cell enlargement, and eventually rupture, leaving disrupted cells and pores in the alveoli and residual cell membranes lining alveolar walls (Figure 4A).
Figure 4.

TNF-induced necroptosis in SARS-CoV-2–infected ATII cells. A, Graphic of TNF-induced necroptosis. SARS-CoV-2–infected ATII cells express TNF and downstream components of the necroptotic pathway, including NINJ1, in a latticework of RBs that also contain SARS-CoV-2 RNA and virus. Activation of the TNF necroptotic pathway generates a porous latticework through which cell contents and virus are emptied, leaving residual cell membranes lining alveolar walls, and disrupted cells and porous latticework in alveoli. B–D, TNF+ cells and structures: brown, SARS-CoV-2 viral RNA; red, hematoxylin counterstain. B, Stages of porous latticework formation. Top, densely stained large TNF+ vRNA+ bodies in a cell in which emptying of cell contents blurs the red-brown staining at the cell margins (orange arrowheads). Middle, brown arrowheads point to the cell border lined by darkly stained TNF+ bodies. Red arrowhead points to TNF+ vRNA+ pores emerging from the disrupted cell margins. Lower, red arrow points to vRNA emptying from a TNF+ pore. C, Syncytial mass of TNF+ vRNA+ cells lining alveolar space. Orange arrowheads point to blurred staining of vRNA and TNF emptying from the porous latticework. The porous latticework within the cells is composed of the RBs in Figure 1 and Figure 2, now seen as a TNF pore surrounding vRNA. D, Remnants of fused cells lining an alveolar wall. The red double-headed arrow points to TNF+ vRNA+ pores of varying size. Abbreviations: AT, alveolar type; RB, round body; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TNF, tumor necrosis factor; vRNA, viral RNA.
This reconstruction of TNF-associated necroptosis of infected ATII cells mediated by and through the latticework was based on images of an asynchronous process at a single point in time in autopsy tissues. We designated intact deeply stained TNF+ LBs as the initial stage of the process, and the next stage as a visibly porous latticework through which cell components emptied (Figure 4B). Because of the spatial proximity of the large numbers of ATII cells that fill and line alveoli in the reparative process, the cells fuse to generate clusters of cells in which vRNA is enclosed by or empties from TNF pores (Figure 4C). In the end stages of the process, only syncytial masses of the porous latticework and cell remnants with TNF+ vRNA-containing pores remain (Figure 4D).
In addition to TNF-mediated necroptosis of infected cells, many uninfected ATII cells in this early-stage COVID-19 pneumonia were also undergoing TNF-associated necroptosis (Supplementary Figure 1) mediated by and through the latticework that housed the downstream components of the TNF-necroptotic pathway, RIPK3, MLKL-p, and NINJ1, a cell surface protein recently shown to mediate plasma membrane rupture during necroptotic and pyroptotic cell death [16–18] (Supplementary Figure 1B–1D).
Limited Viral Replication but Extensive Cytopathology in Type I Interferon-Positive Uninfected ATII Cells in Late-Stage Pneumonia
SARS-CoV-2 RNA was detectable in late-stage pneumonia but was reduced by 79- to 673-fold in late-stage pneumonia compared to the early-stage focal pneumonia (Supplementary Figure 2 SARS-CoV-2 RNA panel), and this reduction was correlated with expression of type I IFN in SARS-CoV-2− CK7+ ATII cells (Supplementary Figure 2, Type I IFN and CK7 panels). Although the ATII cells were not infected, they nonetheless showed extensive cytopathology typical of necroptosis and pyroptosis mediated by and through the latticework. However, because of the proliferation of ATII cells in the response to lung injury, CK7+ ATII cells and cell remains were only reduced approximately 1.5-fold compared to control lung. By contrast, in pyroptosis, CK7 staining of recognizable ATII cells was barely detectable and was reduced more than 50-fold compared to control lung (Supplementary Figure 2, CK7 panel).
Necroptosis of Uninfected ATII Cells in the Second Phase of COVID-19 Pneumonia
In the second phase of COVID-19 pneumonia when most of the ATII cells are not infected, the extensive cytopathology, fusion, and loss of uninfected ATII cells can be attributed to TNF-induced necroptosis, mediated through the latticework. The graphic overview (Figure 5A) again summarizes TNF-induced necroptosis in uninfected type II cells in which LBs house TNF and the necroptotic pathway components including NINJ1, cell contents are emptied via a porous latticework, the cells enlarge and are disrupted to release their contents into alveolar space, leaving residual cell membranes lining and detached from alveolar walls. Again, because the reparative process lines and fills alveoli with contiguous ATII cells, cell fusion propagates, amplifies, and creates a distinctive necroptotic cytopathology in COVID-19 pneumonia of alveoli lined by fused TNF+ multinucleated giant cells and syncytia (Figure 5B–5D). The latticework that mediates ATII necroptosis in turn creates a distinctive pathology of cells with (1) TNF+ or TNF-necroptotic pathway component-positive LBs and pores (Figure 5C and 5D and Supplementary Figure 3A, 3E–3G); (2) fused cell latticework (Supplementary Figure 3J); (3) enlarged fused cells with porous latticework (Supplementary Figure 3K); and (4) disrupted cells and latticework in alveoli and residual latticework in cell membranes lining alveoli (Supplementary Figure 3B, 3D, 3G–3I, and 3L).
Figure 5.

TNF-induced necroptosis of uninfected ATII cells. A, Graphic showing TNF, downstream necroptosis pathway components, and NINJ1 are initially expressed in latticework bodies. Activation of the pathway generates a porous latticework composed initially of darker staining latticework bodies that stain with decreasing intensity as pathway and cell contents are progressively emptied. In the process, the cells enlarge and are eventually disrupted, releasing the pores and porous latticework into alveolar space. Because ATII cells proliferate in the reparative response to lung injury, alveoli are lined and filled by contiguous ATII cells. Activation of the necroptotic pathway fuses adjacent cells to amplify and shape the cytopathology, giving rise to multinucleated giant cells, syncytia, and residual cell membranes lining and detached from alveolar walls. B, Brown-stained TNF+ cells and structures, and hematoxylin counterstain. Fused syncytial mass of TNF+ cells lining alveolar wall (traced). Arrow points to a trinucleate cell with “owl's eye” appearance created by residual clumped chromatin in a largely emptied nucleus. Asynchronous emptying results in variable clearing with the most extensive clearing in the nucleus at the top of the cell. C, Red-stained RIPK3+ cells and structures and hematoxylin counterstain. Alveolar space lined and filled by fused RIPK3+ cells. Line traces a RIPK3+ syncytium in which the latticework pores retain RIPK3 staining (red arrowhead) or are largely emptied (outlined white arrowhead) The black arrowhead points to residual cell membranes. D, Brown-stained cells and structures and hematoxylin counterstain. Trinucleate cell with latticework at various stages of emptying from early darkly staining latticework to later stages in which the intensity of staining decreases as NINJ1 and cell content empty. Abbreviations: AT, alveolar type; TNF, tumor necrosis factor; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Pyroptosis of Uninfected ATII Cells in the Second Phase of COVID-19 Pneumonia
We examined lung tissues for evidence of BTK/NLRP3 inflammasome-mediated pyroptosis of ATII cells, motivated by the reported improved oxygenation associated with BTK inhibitor suppression of blood monocyte activation. While the inferred beneficial effects were attributed to the hypothesized role of an influx of activated macrophages in COVID-19 lung injury [10], our direct examination of lung sections in fatal COVID-19 pneumonia instead revealed destruction of ATII cells by BTK/NLRP3-mediated pyroptosis.
In the graphic overview (Figure 6A) and other images shown in Figure 6 and Supplementary Figure 4, expression of autophosphorylated and activated BTK and known NLRP3 inflammasome components —caspase 1 (CASP1), interleukin 1β (IL-1β), and gasdermin D (GSD)—, as well as NINJ1, colocalize in the same latticework described for necroptosis. In the images of an asynchronous process in the autopsy tissues, expression of activated BTK first results in fusion of BTK+ cells that fill alveoli (Supplementary Figure 4A) that then undergo rapid emptying through the latticework, leaving honeycombed cavities of cell “ghosts” and walls of cell remnants (Figure 6B and Supplementary Figure 4B and 4C), which impart a moth-eaten appearance to the pyroptotic foci (Supplementary Figure 4D). Images of downstream components CASP1 and GSD and emptying of IL-β through the porous latticework similarly illustrate the formation of honeycombed cavities of cell ghosts and walls of the residues of disrupted cell membranes (Figure 6C and 6D, and Supplementary Figure 4E–4G), as well as images of filled or partially emptied latticework pores (Figure 6D and Supplementary Figure 4H). Lastly, the images of NINJ1 capture both the formation of the honeycombed cavities and cell remnants and LBs (Supplementary Figure 4I) and the distinctive differences and appearances of cells in pyroptotic destruction of ATII cells versus the enlargement and late disruption of necroptotic cells (Supplementary Figure 4J).
Figure 6.

BTK-induced pyroptosis of uninfected ATII cells. A, Graphic showing expression of BTK and downstream pyroptotic/NLRP3 inflammasome pathway components in latticework bodies leads to cell fusion and subsequent disruption and emptying of the cells and latticework. In Supplementary Figure 4B and C, stages labeled 1–4 show an initial stage 1 of a largely intact cell and latticework RBs with cell disruption and progressive emptying of the latticework in stages 2 and 3 to create cavities and walls in stage 4, formed from cell remnants and residual cell membranes. This asynchronous process imparts a characteristic honeycombed and moth-eaten appearance to foci composed of cavities with cell remnants, lined by residual cell membranes and cells at earlier stages of pyroptosis. B, Red-stained BTK+ cells and structures and hematoxylin counterstain. Arrows point to remnants of BTK+ cells in cavities and arrowheads to residual cell membranes that constitute cavity walls. C, Brown-stained CASP1+ cells and structures, immunohistochemical staining, and hematoxylin counterstain. Arrows point to cell remnants in cavities and arrowheads point to residual cell membranes in the cavity walls. D, Red-stained gasdermin D (GSD)+ cells and structures, and hematoxylin counterstain. In the deconvoluted image, GSD is red and hematoxylin is blue. Boxes correlate the appearance of latticework bodies and pores of the images with or without deconvolution to reveal a hematoxylin-stained component in nuclear pores. Abbreviations: AT, alveolar type; BTK, Bruton tyrosine kinase; CASP1, caspase 1; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Apoptosis and Combinatorial PANoptosis
In investigating apoptosis of ATII cells, we identified intrinsic pathway apoptotic cells as caspase 9 (CASP9) and caspase 3 (CASP3) positive cells and discovered that the initiator and executioner of the pathway were expressed in the same latticework described for necroptosis and pyroptosis, but with the striking difference that many of the LBs, pores, latticework, and nuclear and cytoplasmic membranes remained visibly intact (Supplementary Figure 5A and 5B). The colocalization of necroptotic, pyroptotic, and apoptotic pathways in a common latticework structure is consistent with the idea that the latticework is a newly discovered cellular structure that corresponds to the PANoptosome that mediates multiple programmed cell death (PCD) pathways collectively designated as PANoptosis [19, 20] (Figure 7A). Locating 3 PCD pathways in the latticework also raised the possibility that individual cells might have latticework expressing more than one pathway in the same cell, and this indeed was the case, for example, CASP3+ pores and latticework in necroptotic appearing cells, designated apoptosis and necroptosis (Figure 7B and Supplementary Figure 5C). Similarly, independent expression in the latticework of apoptosis and pyroptosis created pyroptotic-appearing cells with overlying CASP3+ LBs (Figure 7C). In apoptosis plus necroptosis or pyroptosis, coexpression in the same LBs in which membrane-disrupting pathways dominate created blurred staining in cells in which the CASP3 LBs and pores have been disrupted (Figure 7B and 7C). We further documented combinatorial PANoptotic expression in the latticework by immunofluorescence staining with CASP3 and RIPK3 and show in Figure 7D examples of independent (CASP3+RIPK3−; RIPK3+CASP3−) or coexpression (CASP3+RIPK3+) of apoptosis and necroptosis in LBs and pores.
Figure 7.

Apoptosis and PANoptosis of uninfected ATII cells. A, Graphic showing intrinsic pathway apoptosis components CASP9 and CASP3 are expressed in the same latticework LBs and pores as described for necroptosis and pyroptosis pathways. In individual cells undergoing apoptosis, the latticework bodies and pores and cell membranes are largely intact. In PANoptosis, (1) the latticework may independently express apoptotic and necroptotic or pyroptotic pathways with necroptotic and pyroptotic cytopathology dominant; or (2) apoptotic and necroptotic pathways may be coexpressed in latticework bodies and pores with disruption of the pores and necroptotic cytopathology dominant. B and C, Red-stained CASP3+ cells and structures and hematoxylin counterstain. B, Apoptosis arrow points to largely intact CASP3+ latticework pores in a binucleate cell. The necroptosis arrow points to a largely CASP– necroptosis-appearing porous latticework enclosed by the line. In necroptosis + pyroptosis, coexpression of the necroptotic and apoptotic pathway in the same latticework disrupts apoptotic pores, resulting in blurred staining of released CASP3 traced by a line in the multinucleated cell at the bottom of the panel. C, Pyroptotic cell remnants and residual membranes with superimposed intact CASP3+ latticework bodies and pores (red arrowhead). Red arrow points to blurred CASP3 staining in latticework in which the pyroptotic pathway is coexpressed. D, Green-stained CASP3+ cells and structures, red-stained RIPK3+ cells and structures, and DAPI blue-stained nuclei. Green arrows point to predominantly CASP3+ apoptotic latticework; green-outlined red arrow points to separate CASP3+ and RIPK3+ latticework and pores; and green-outlined orange arrow points to porous latticework expressing both CASP3 and RIPK3. Abbreviations: AT, alveolar type; CASP, caspase; DAPI, 4′,6-diamidino-2-phenylindole; PANoptosis, pyroptosis, apoptosis, necroptosis.
The recognition of combinatorial PANoptosis in the latticework of ATII cells provided the necessary insight to interpret the originally puzzling images of apoptotic, necroptotic, and pyroptotic pathway components together in blood vessels (Supplementary Figure 6). The association of microthrombi adherent to endothelial lining damaged and disrupted by necroptosis in the blood vessels was particularly intriguing as a possible mechanism for the extensive thrombosis evident in the blood vessels of all 4 patients, including the early-stage focal pneumonia in patient 2, and possibly a general potential mechanism for pulmonary vascular coagulopathy, which might be targeted by the administration of TNF inhibitors during early-stage infection to potentially reduce COVID-19 thrombosis and microangiopathy [21, 22].
Phenotypically Diverse Macrophages Phagocytose SARS-CoV-2, Necroptotic, and Pyroptotic Cell Remnants
The principal role we assign to phenotypically diverse populations of CD68+, CD14+, and CD163+ macrophages in infection and PCD is clearing cell debris from infected and uninfected cells undergoing necroptosis and pyroptosis. We base this conclusion on the acquisition by phagocytosis of SARS-CoV-2 RNA by uninfected napsin A− (Figure 3) CD68+ macrophages (Supplementary Figure 7A–7E); phagocytosis of BTK+ pyroptotic cell remnants by CD14+ macrophages (Supplementary Figure 7F and 7G); and phagocytosis of necroptotic and pyroptotic cell bodies and pores by CD163+ macrophages (Supplementary Figure 7H).
DISCUSSION
In this exploration of the impact of SARS-CoV-2 infection on the proliferative reparative ATII response in fatal COVID-19 pneumonia, we discovered that ATII cells succumb to SARS-CoV-2 infection, necroptosis, pyroptosis, and PANoptosis on a scale magnified by the distinctive anatomy and nature of the ATII reparative response to acute lung injury. In that proliferative reparative response, contiguous ATII cells lining the alveoli provide the virus with abundant targets for replication and cell-to-cell spread to amplify virus production. This organization of the reparative response also amplifies the destruction of infected and uninfected ATII cells and the fusion of contiguous ATII cells to generate distinctive histopathological features of COVID-19 pneumonia, such as multinucleated giant cells and syncytia [23]. While fusion of lung cells has been attributed to SARS-CoV-2 spike protein altered activity of a scramblase that externalizes phosphatidylserine [24], we provide evidence of a more general mechanism in infected and uninfected cells of cell fusion mediated by necroptosis and pyroptosis in contiguous cells.
The concept of PANoptosis and the PANoptosome was derived from genetic, biochemical, and pharmacological lines of evidence for a molecular complex of interacting components that mediate apoptosis, necroptosis, and pyroptosis in cell populations [19, 20]. Locating the intrinsic pathway of apoptosis in the same latticework as the necroptotic and pyroptotic pathways now identifies the latticework as a cellular structure corresponding to the PANoptosome that separately initiates and executes TNF-induced necroptosis of SARS-CoV-2 infected and uninfected ATII cells, BTK-induced pyroptosis, and apoptosis in ATII populations and in individual ATII cells to generate distinctive PANoptosome cytopathologies by which PANoptosis can now be recognized in diverse cells and tissues. We further refer to PANoptosis as combinatorial to capture the capabilities of the PANoptosome to create diverse forms of PCD, and to underscore the dominance of disruptive and universal inflammatory outcomes for ATII cells that compromises the reparative response and provides a massive stimulus underlying the hyperinflammatory state in the lung in COVID-19 pneumonia.
The discovery of proinflammatory cytokines and autochthonous PCD pathways in ATII cells themselves was initially surprising but consistent with the many roles ATII cells must play as defenders of the alveolus [11]. TNF-induced necroptosis of SARS-CoV-2 ATII cells traps the virus in dead cells, and the death of ATII cells succumbing to necroptosis and pyroptosis greatly reduces the availability of susceptible cells for further virus production and spread, and emptying of damage-associated molecular pattern cell contents from necroptotic, pyroptotic, and PANoptotic cells provides a strong signal to induce innate and adaptive host defenses. Thus, PANoptosis of ATII cells in the response to SARS-CoV-2 infection is a fail-safe mechanism that prevents a pathogen escaping detection by the immune system or sparing infected cells to perpetuate infection when the pathogen has an evasion strategy for one PCD pathway, analogous to the reinforcing role in host defenses against herpes viruses of activating both apoptotic and necroptotic PCD pathways [25, 26], which falls on the benefit side of Balachandran's characterization of the benefits and perils of necroptosis in influenza A infection [27]. However, massive losses of ATII cells to PANoptosis and massive inflammatory stimulus fall on the Balachandran's and Rall's peril side of necroptosis [27] by irreversibly incapacitating the ATII reparative response.
Our study was limited to autopsy lung tissues collected during the first wave of the pandemic in Italy from 4 immunologically naive elderly patients who did not receive steroids, immunomodulating treatments, or direct acting antivirals, and thus future studies will be needed to assess the effects of vaccination, effective treatment with antiviral and anti-inflammatory agents, and steroids on the mechanisms we describe. Our study was also limited to autopsy lung tissues from a small number of individuals. Nonetheless, the in-depth analysis of samples spanning early to later stages of fatal COVID-19 pneumonia revealed new mechanisms and cell structures that mediate destruction of the defenders of the alveolus. Knowing that ATII cells succumb in COVID-19 pneumonia from its early stages onward to a combination of virus infection, TNF-induced necroptosis. and BTK-induced pyroptosis provides a rational framework for treatment to preserve the ATII reparative response with emphasis on combined therapies at an early stage to inhibit the initiators of inflammatory PCD pathways and antivirals to limit access to salvaged targets and progression of SARS-CoV-2 infection (Supplementary Figure 8).
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Supplementary Material
Contributor Information
Luca Schifanella, Department of Surgery, University of Minnesota, Minneapolis, Minnesota, USA.
Jodi Anderson, Department of Medicine, University of Minnesota, Minneapolis, Minnesota, USA.
Garritt Wieking, Department of Medicine, University of Minnesota, Minneapolis, Minnesota, USA.
Peter J Southern, Department of Microbiology and Immunology, University of Minnesota, Minneapolis, Minnesota, USA.
Spinello Antinori, III Division of Infectious Diseases, ASST Fatebenefratelli Sacco, Milan, Italy; Department of Biomedical and Clinical Sciences, Università Degli Studi di Milano, Milan, Italy.
Massimo Galli, III Division of Infectious Diseases, ASST Fatebenefratelli Sacco, Milan, Italy; Department of Biomedical and Clinical Sciences, Università Degli Studi di Milano, Milan, Italy.
Mario Corbellino, III Division of Infectious Diseases, ASST Fatebenefratelli Sacco, Milan, Italy.
Alessia Lai, Department of Biomedical and Clinical Sciences, Università Degli Studi di Milano, Milan, Italy.
Nichole Klatt, Department of Surgery, University of Minnesota, Minneapolis, Minnesota, USA.
Timothy W Schacker, Department of Medicine, University of Minnesota, Minneapolis, Minnesota, USA.
Ashley T Haase, Department of Microbiology and Immunology, University of Minnesota, Minneapolis, Minnesota, USA.
Notes
Acknowledgments. We thank Colleen O’Neill and Tim Leonard for preparation of the manuscript and figures; Jarrett Reichel, Jacob Bjorgen, and Caitlin David for their help with image analysis; and Dr Emilian Racila for helpful discussion.
Author contributions. A. T. H., L. S., T. W. S., and N. K. conceived and designed the project. J. A., G W., P. J. S., and A. T. H. conducted the experiments, interpretation, and image analysis. A. T. H. wrote the paper with contributions from all the coauthors.
Financial support. This work was supported by the National Institutes of Health (grant numbers R01 AI134406 to A. T. H and T. W. S., R01 AI125127 to T. W. S., and R01 AI147912 to T. W. S.); and A. T. H. Regents’ Professorship, the Department of Surgery, and the Medical School, University of Minnesota. Funding to pay the Open Access publication charges for this article was provided by A.T.H. non-sponsored research account.
References
- 1. World Health Organization . World Health Organization coronavirus (COVID-19) dashboard. https://covid19.who.int/. Accessed 31 January 2023.
- 2. Horby P, Lim WS, Emberson JR, et al. . Dexamethasone in hospitalized patients with Covid-19—preliminary report. New Engl J Med 2020; 384:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van de Veerdonk FL, Giamarellos-Bourboulis E, Pickkers P, et al. . A guide to immunotherapy for COVID-19. Nat Med 2022; 8:39–50. [DOI] [PubMed] [Google Scholar]
- 4. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. . Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020; 584:463–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Del Valle DM, Kim-Schulze S, Huang H-H, et al. . An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 2020; 26:1636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hadjadj J, Yatim N, Barnabei L, et al. . Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020; 369:718–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liao M, Liu Y, Yuan J, et al. . Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 2020; 26:842–4. [DOI] [PubMed] [Google Scholar]
- 8. Karki R, Sharma BR, Tuladhar S, et al. . Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021; 184:149–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gianfrancesco M, Lyrich KL, Al-Adely S, et al. . Characteristics associated with hospitalization for COVID-19 in people with rheumatic disease: data from the COVID-19 global rheumatology alliance physician-reported registry. Ann Rheum Dis 2020; 79:859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roschewski M, Lionakis MS, Sharman JP, et al. . Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci Immunol 2020; 5:eabd0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res 2001; 2:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deleage C, Wietgrefe S, Del Prete GQ, et al. . Defining HIV and SIV reservoirs in lymphoid tissues. Pathog Immun 2016; 1:68–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hou YJ, Okuda K, Edwards CE, et al. . SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 2020; 182:429–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Z-Q, Wietgrefe SW, Li Q, et al. . Roles of substrate availability and infection of resting and activated CD4+ T cells in transmission and acute simian immunodeficiency virus infection. Proc Natl Acad Sci USA 2004; 101:5640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wietgrefe SW, Duan L, Anderson J, et al. . Detecting sources of immune activation and viral rebound in HIV infection. J Virol 2022; 96:e0088522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kayagaki N, Kornfeld OS, Lee BL, et al. . NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021; 591:131–53. [DOI] [PubMed] [Google Scholar]
- 17. Hiller S, Broz P. Active membrane rupture sours a range of cell deaths. Nature 2021; 591:36–7. [DOI] [PubMed] [Google Scholar]
- 18. Newton K, Dixit VMKayagaki N. Dying cells fan the flames of inflammation. Science 2021; 374:1076–80. [DOI] [PubMed] [Google Scholar]
- 19. Samir P, Subbarao Malireddi RK, Kanneganti T-D. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol 2020; 10:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Christgen S, Zheng M, Kesavardhana S, et al. . Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol 2020; 10:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Cobelli F, Palumbo D, Ciceri F, et al. . Pulmonary vascular thrombosis in COVID-19 pneumonia. J Cardiothorac Vasc Anes 2021; 35:3631–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ackermann M, Verleden SE, Kuehnel M, et al. . Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 2020; 383:120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Falasca L, Nardacci R, Colombo D, Lalle E, Di Caro A, Nicastri E, et al. . Postmortem findings in Italian patients with COVID-19: a descriptive full autopsy study of cases with and without comorbidities. J Infect Dis 2020; 222:1807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Braga L, Ali H, Secco I, Chiavacci E, Neves G, Goldhill D, et al. . Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 2021; 594:88–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol 2011; 12:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaiser WJ, Upton JW, Mocarski ES. Viral modulation of programmed necrosis. Curr Opin Virol 2013; 3:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balachandran S, Rall GF. Benefits and perils of necroptosis in influenza virus infection. J Virol 2020; 94:e01101–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.