

Abstract
Background
Evidence suggests that LPA risk genotypes are a possible contributor to the clinical diagnosis of familial hypercholesterolemia (FH). This study aimed at determining the prevalence of LPA risk variants in adult individuals with FH enrolled in the Italian LIPIGEN (Lipid Transport Disorders Italian Genetic Network) study, with (FH/M+) or without (FH/M−) a causative genetic variant.
Methods and Results
An lp(a) [lipoprotein(a)] genetic score was calculated by summing the number risk‐increasing alleles inherited at rs3798220 and rs10455872 variants. Overall, in the 4.6% of 1695 patients with clinically diagnosed FH, the phenotype was not explained by a monogenic or polygenic cause but by genotype associated with high lp(a) levels. Among 765 subjects with FH/M− and 930 subjects with FH/M+, 133 (17.4%) and 95 (10.2%) were characterized by 1 copy of either rs10455872 or rs3798220 or 2 copies of either rs10455872 or rs3798220 (lp(a) score ≥1). Subjects with FH/M− also had lower mean levels of pretreatment low‐density lipoprotein cholesterol than individuals with FH/M+ (t test for difference in means between FH/M− and FH/M+ groups <0.0001); however, subjects with FH/M− and lp(a) score ≥1 had higher mean (SD) pretreatment low‐density lipoprotein cholesterol levels (223.47 [50.40] mg/dL) compared with subjects with FH/M− and lp(a) score=0 (219.38 [54.54] mg/dL for), although not statistically significant. The adjustment of low‐density lipoprotein cholesterol levels based on lp(a) concentration reduced from 68% to 42% the proportion of subjects with low‐density lipoprotein cholesterol level ≥190 mg/dL (or from 68% to 50%, considering a more conservative formula).
Conclusions
Our study supports the importance of measuring lp(a) to perform the diagnosis of FH appropriately and to exclude that the observed phenotype is driven by elevated levels of lp(a) before performing the genetic test for FH.
Keywords: cardiovascular risk, clinical diagnosis, familial hypercholesterolemia, lipoprotein(a)
Subject Categories: Genetics, Clinical Studies
Nonstandard abbreviations and acronyms
- FH
familial hypercholesterolemia
- FH/M−
negative at FH genetic test
- FH/M+
positive at FH genetic test
- LDLR
LDL receptor
- LIPIGEN
Lipid Transport Disorders Italian Genetic Network
- lp(a)
lipoprotein(a)
- LPA
lipoprotein(a) coding gene
Clinical Perspective.
What Is New?
In the 4.6% of patients with clinically diagnosed familial hypercholesterolemia, the phenotype was not explained by a monogenic or polygenic cause but by the genotype associated with high lp(a) [lipoprotein(a)] levels.
The adjustment of low‐density lipoprotein cholesterol levels based on lp(a) concentration reduced from 68% to 42% the proportion of subjects with low‐density lipoprotein cholesterol levels ≥190 mg/dL (or from 68% to 50% considering a more conservative formula).
What Are the Clinical Implications?
In patients with a clinical suspicion of familial hypercholesterolemia, lp(a) should be measured to perform an appropriate diagnosis and to exclude the possibility that the observed phenotype is driven by elevated levels of lp(a) before performing the genetic test.
The differentiation of familial hypercholesterolemia, elevated lp(a), or the combination of both is crucial for an accurate risk stratification.
In the future, patients with both elevated lp(a) and familial hypercholesterolemia condition may be shown to benefit from interventions to reduce both low‐density lipoprotein cholesterol and lp(a) plasma levels.
Familial hypercholesterolemia (FH) is a genetic inherited disorder mainly caused by variants in genes encoding for the LDL receptor (LDLR), apoB (apolipoprotein B), or PCSK9 (proprotein convertase subtilisin/kexin type 9). FH is characterized by lifelong exposure to elevated low‐density lipoprotein cholesterol (LDL‐C) levels and subsequent premature cardiovascular disease development. 1 Depending upon the diagnostic criteria used, a causative variant in candidate genes can be detected in 40% to 80% of clinically defined patients with FH. In all other cases, the cause for the clinical phenotype of hypercholesterolemia remains undefined. 2 In some cases, these patients likely carry a cluster of common polymorphisms affecting several loci associated with raised LDL‐C levels, and a polygenic cause can be suspected. 3 In all other cases, the presence of unknown genetic or environmental factors could be assumed, which results in a FH phenotype consistent with that observed in monogenic FH. This clinical and genetic heterogeneity can make the FH diagnosis very complex.
It has been already described that patients clinically diagnosed with FH have elevated levels of lp(a) [lipoprotein(a)], 4 , 5 which is a causal genetic risk factor for atherosclerotic cardiovascular disease, but the reasons for this remain unclear. It has been suggested, however, that high lp(a) cholesterol could be responsible for clinical FH. 4 , 6
Lp(a) levels are 75% to 95% heritable and predominantly determined by single‐nucleotide variants at the LPA gene and copy number variants specifically in the kringle IV type 2 domain. 7 To date, 2 specific genetic variants in the LPA gene have been shown to significantly influence lp(a) plasma values: rs3798220 (Ile4399→Met) and rs10455872 (intronic A/G polymorphism). 8 A single copy of the rs10455872‐G allele, which is common in European populations, is known to associate with extremely elevated lp(a) that can result in a phenotype similar to FH. 9 This evidence points out the possibility that LPA genotypes are a possible cause of clinical FH, especially in those individuals presenting a low LDL‐C polygenic predisposition.
The objective of this study was to determine the prevalence of LPA risk variants in a large cohort of Italian subjects clinically diagnosed with FH, by comparing this distribution in subjects with or without causative variants.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
This analysis was performed in patients enrolled in the LIPIGEN (Lipid Transport Disorders Italian Genetic Network) study, an observational, multicenter, retrospective, and prospective ongoing study started in 2012 and aimed at identifying and registering patients with FH in Italy. 10 Detailed information about the procedures of LIPIGEN study has been previously published. 10 The LIPIGEN project and its amendments were approved by the Ethics Committee of the Coordinating Unit (Ethical Committee IRCCS MultiMedica, Via Milanese 300—20099, Sesto San Giovanni, Milan, September 13, 2011 and June 23, 2015) and by ethics committees of each participating institution. Eligible patients who were able to understand the study procedures and who voluntarily agreed to participate provided written informed consent.
In brief, patients with hypercholesterolemia attending lipid clinics were enrolled in the registry if they had a clinical diagnosis of FH. The clinical diagnosis may be guided by clinical scores, such as the Dutch Lipid Clinic Network Score, 11 and ultimately based on the decision of the lipid specialist, supported by an evocative lipid profile or by a familial history of premature cardiovascular disease. After the visit, patients fulfilling the aforementioned criteria are referred for genetic testing of the appropriate candidate genes.
Statistical Analysis
In the present analysis, we included adult (aged ≥18 years) patients with FH, with genetic testing performed in a centralized laboratory searching for possible causative variants in candidate genes and evaluating the 2 common lp(a)–raising single nucleotide polymorphisms (rs3798220 and rs10455872). We defined patients as genetic‐positive FH (those with at least 1 causative variant on LDLR [FH/M+]), and genetic‐negative FH (ie, without causative variants on LDLR [FH/M−]). Subjects presenting only variants of uncertain significance were excluded from the analysis (393 individuals, 18.8% of the sample), as well as individuals with homozygous FH. For each subject, a polygenic score (LDL‐C‐score) including 12 common LDL‐C–raising single nucleotide polymorphisms (rs2479409 [PCSK9 gene], rs629301 [CELSR2 gene], rs1367117 [APOB gene], rs4299376 [ABCG8 gene], rs1564348 [SLC22A1 gene], rs1800562 [HFE gene], rs3757354 [MYLIP gene], rs11220462 [ST3GAL4 gene], rs8017377 [NYNRIN gene], rs6511720 [LDLR gene], rs429358 [APOE gene], rs7412 [APOE gene]), selected by Talmud et al 12 from the Global Lipid Genetic Consortium for being significantly associated with LDL‐C with a P value cutoff of <5×10−8, was also evaluated. Briefly, for each individual, we calculated the specific gene scores using the weighted sum of the risk allele (ie, the LDL‐C‐raising allele). The weights used were the corresponding per‐risk‐allele beta coefficients reported by the Global Lipid Genetic Consortium, as proposed by Talmud et al 12 Because the beta‐coefficients reported by the Global Lipid Genetic Consortium are for the effect of each minor allele, where the effect is LDL lowering, the other allele is considered the risk allele and the absolute effect size is used as the weight. An LDL‐C score value ≥1.08 (the cutoff that identifies the 80th percentile of the distribution of the score in their general population 12 ) was considered as indicative of a high probability of having polygenic hypercholesterolemia. An lp(a) genetic score was calculated for each participant by summing the number risk‐increasing alleles inherited at rs3798220 and rs10455872 variants. Four different groups of subjects were then created based on genetic test response and LPA genotype (lp(a)/FH groups).
Baseline LDL‐C levels were defined as those before initiation of cholesterol‐lowering medication. Among the subgroup of patients with measured lp(a) levels available, lp(a)‐adjusted LDL‐C was calculated as recommended by Dahlen 13 , 14 as follows (in mg/dL): total cholesterol–(triglycerides/5)–high‐density lipoprotein cholesterol–[0.3×lp(a)], and also as recommended by Yeang et al 14 as follows (in mg/dL): total cholesterol–(triglycerides/5)–high‐density lipoprotein cholesterol–[0.173×lp(a)], which is a more conservative correction.
Continuous variables are presented as mean±SD, or as median and interquartile range, whereas categorical variables are presented as cases and percentage rate. Chi‐square tests were used for contingency analyses. Normally distributed data were analyzed with unpaired Student's t tests or 1‐way ANOVA, whereas nonnormal data were analyzed with Wilcoxon‐Mann–Whitney test as appropriate. Analyses were performed in SAS software version 9.4 (SAS Institute, Cary, NC).
Statistical significance was claimed when 2‐sided P values were ≤0.05.
RESULTS
We reported data on 1695 subjects with clinically diagnosed FH. Considering the presence of a causative variant, the polygenic score, and the genetic score for lp(a) as the cause of the FH phenotype (Figure), a total of 54.87% of the sample had a causative variant (FH/M+), 11.80% had only a high polygenic score, and 4.60% had only an lp(a) score ≥1 (representing 10.2% among individuals with FH/M−), whereas in the 3.24% of the total sample both these conditions were present. The remaining 25.49% were subjects with FH/M− for whom neither polygenic cause nor high lp(a) genetic score were found.
Figure . Distribution of patients with clinically diagnosed FH by genotype.
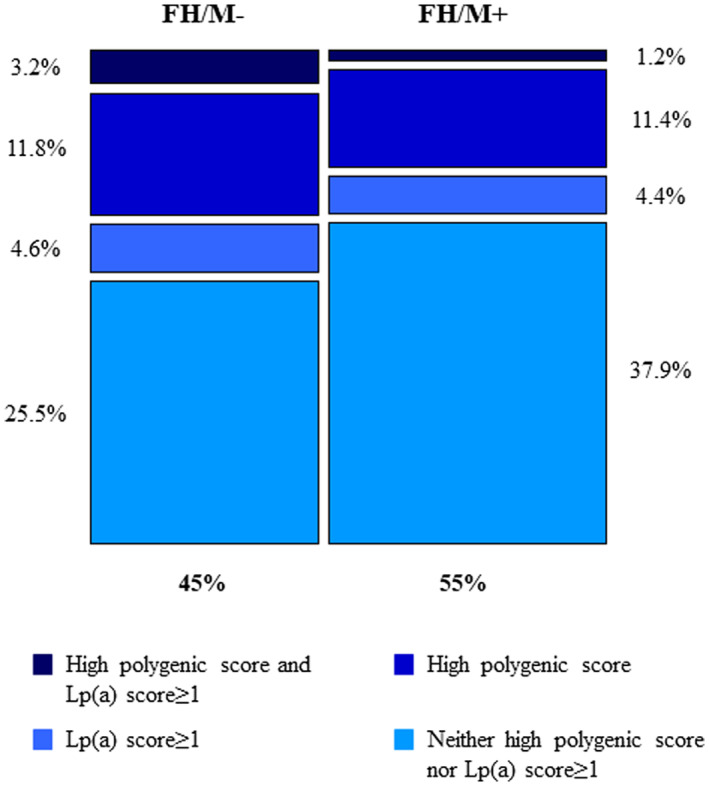
The figure describes the distribution of patients with clinically diagnosed FH according to the results of the genetic test (FH/M+: 54.87% of the sample; FH/M−: 45.13%), and based on the presence of a causative variant, polygenic score, or lp(a) genotype. FH/M− indicates negative familial hypercholesterolemia genetic test; FH/ M+, positive familial hypercholesterolemia genetic test; and lp(a), lipoprotein A.
Among 765 subjects with FH/M− and 930 subjects with FH/M+, 632 and 835 individuals presented 0 copies of the variant (lp(a) score=0), respectively, and 133 (17.4%) and 95 (10.2%) were characterized by 1 copy of either rs10455872 or rs3798220 or 2 copies of rs10455872 or rs3798220 (lp(a) score ≥1). Demographic and clinical data of lp(a)/FH groups are shown in the Table 1. We found that subjects with FH/M− had higher levels of lp(a) than patients in the FH/M+ group (median values 41 mg/dL [9–103] versus 19 mg/dL [8–41], Wilcoxon‐Mann–Whitney test <0.0001), with increasing lp(a) concentrations among subjects with the same FH genetic background based on increasing number of lp(a) alleles (P<0.0001 for patients with FH/M− and P=0.001 for patients with FH/M+).
Table 1.
Clinical, Demographic, and Biochemical Profile of Included Subjects
| FH/M− and lp(a) score=0 | FH/M− and lp(a) score≥1 | P value | FH/M+ and lp(a) score=0 | FH/M+ and lp(a) score≥1 | P value | |
|---|---|---|---|---|---|---|
| N | 632 | 133 | 835 | 95 | ||
| Age at baseline, y; mean (SD) | 49.08 (13.59) | 49.47 (13.77) | 0.76 | 41.25 (14.92) | 43.83 (15.16) | 0.11 |
| Female sex, % | 53.81 | 60.15 | 0.18 | 54.32 | 51.58 | 0.61 |
| lp(a), mg/dL; median (IQR)† | 16.95 (7.00–72.75) | 89.00 (62.00–136.00) | <0.0001 | 19.00 (8.00–34.00) | 64.00 (27.00–113.50) | 0.001 |
| Pretreatment low‐density lipoprotein cholesterol, mg/dL; mean (SD) | 219.38 (54.54) | 223.47 (50.40) | 0.43 | 265.13 (58.26) | 264.43 (60.09) | 0.91 |
| Triglycerides, mg/dL; median (IQR) | 129.00 (90.00–180.00) | 118.50 (89.00–158.00) | 0.20 | 97.00 (69.00–136.00) | 100.00 (78.00–138.00) | 0.45 |
| Lipid‐lowering therapy, % | 25.47 | 37.59 | 0.005 | 27.07 | 34.74 | 0.11 |
| First‐degree relative with premature CHD | 36.17 | 43.65 | 0.11 | 42.75 | 43.33 | 0.92 |
| Clinical history of premature CHD | 7.53 | 10.69 | 0.23 | 8.00 | 12.63 | 0.12 |
| Clinical history of premature cerebral or peripheral vascular disease | 4.39 | 6.11 | 0.40 | 3.03 | 4.21 | 0.53 |
CHD indicates coronary heart disease; FH, familial hypercholesterolemia; FH/M+, patients with genetic‐positive FH; FH/M−, patients with genetic‐negative FH; and lp(a), lipoprotein(a). To convert lp(a) values to nanomoles per liter, multiply by 2.15.
N=100 (FH/M− & lp(a) score=0), N=38 (FH/M− & lp(a) score≥1), N=166 (FH/M+ & lp(a) score=0), and N=13 (FH/M+ & lp(a) score≥1).
FH/M− subjects had also lower mean levels of pretreatment LDL‐C than FH/M+ individuals (t‐test for difference in means between FH/M− and FH/M+ groups <0.0001); however, FH/M− subjects with Lp(a) score≥1 had higher mean (SD) pretreatment LDL‐C levels (223.47 [50.40] mg/dL) compared to FH/M− subjects with Lp(a) score=0 (219.38 [54.54] mg/dL), although not statistically significant. Moreover, patients with FH/M− characterized by 1 copy of either rs10455872 or rs3798220, or 2 copies of either rs10455872 or rs3798220, presented higher prevalence of clinical history of premature coronary heart disease (10.69% versus 7.53%, P=0.23) compared with negative FH individuals presented 0 copies of the variants; a similar trend was observed for cerebral/peripheral vascular disease (6.11% versus 4.39%, P=0.40). Even considering only FH/M+ subjects, having the Lp(a) score≥1 resulted in a higher prevalence of clinical history of premature coronary (12.63% versus 8.00%, P=0.12) or cerebral/peripheral vascular disease (4.21% versus 3.03%, P=0.53) compared with positive FH individuals presented 0 copies of the variants.
Among individuals with baseline lp(a) measurement available (138 patients with FH/M− and 179 with FH/M+), applying the adjustment based on lp(a) levels, LDL‐C levels appeared drastically lower in subjects with lp(a) score ≥1 (−14% in FH/M− and −9% in FH/M+, on average), whereas they were only slightly different in subjects with lp(a) score=0 (−6% in FH/M− and −3% in FH/M+ on average; Table 2). Using the correction recommended by Yeang et al, LDL‐C levels percentage reduction became −8%, −5%, −4%, and −2%, respectively. Considering an LDL‐C ≥190 mg/dL as a threshold for the clinical suspicion of FH, 1 this implied that the condition would be recognized in 68% of individuals with FH/M− who had at least 1 copy of either rs10455872 or rs3798220 variants presented; however, this percentage decreased to 42% (or 50% considering a more conservative formula) when the assessment was made considering lp(a)‐adjusted LDL‐C values.
Table 2.
LDL Cholesterol Profile (Before Initiation of Cholesterol‐Lowering Medication and Lp(a)‐Adjusted Values) Among Lp(a)/FH Groups
| FH/M− and lp(a) score=0 | FH/M− and lp(a) score≥1 | P value | FH/M+ and lp(a) score=0 | FH/M+ and lp(a) score≥1 | P value | |
|---|---|---|---|---|---|---|
| No. | 100 | 38 | 166 | 13 | ||
| Pretreatment LDL‐C, mg/dL; mean (SD) | 217.58 (62.72) | 213.80 (51.15) | 0.74 | 275.01 (560.23) | 260.49 (48.32) | 0.40 |
| Patients with pre‐treatment LDL‐C≥190 mg/dL; % | 67.00 | 68.42 | 0.87 | 95.18 | 92.31 | 0.65 |
| Lp(a)‐adjusted LDL‐C*; mean (SD) | 203.75 (62.68) | 183.52 (50.47) | 0.08 | 266.64 (59.22) | 236.42 (54.92) | 0.08 |
| Patients with Lp(a)‐adjusted LDL‐C* ≥190 mg/dL; % | 55.00 | 42.11 | 0.18 | 93.37 | 92.31 | 0.88 |
| Lp(a)‐adjusted LDL‐C†; mean (SD) | 209.60 (62.10) | 196.34 (49.92) | 0.24 | 270.18 (59.46) | 246.61 (51.18) | 0.17 |
| Patients with Lp(a)‐adjusted LDL‐C† ≥190 mg/dL; % | 59.00 | 50.00 | 0.34 | 94.58 | 92.31 | 0.73 |
FH indicates familial hypercholesterolemia; FH/M+, patients with genetic‐positive FH; FH/M−, patients with genetic‐negative FH; LDL‐C, low‐density lipoprotein cholesterol; and lp(a), lipoprotein(a).
Lp(a) corrected levels of LDL‐C calculated based on Dahlen formula. 13
Lp(a) corrected levels of LDL‐C calculated based on the study by Yeang et al. 14
DISCUSSION
Despite considerable recent progress in the knowledge of the genetic basis of hypercholesterolemia, a causative variant is not found in many individuals with a phenotype and a clinical history consistent with that of FH. It has been suggested that, in some of these subjects, the FH phenotype can be explained by the presence of several genetic variants, which are individually associated with a slight impact on LDL‐C, but if present concomitantly can considerably raise LDL‐C values. 12 , 15 , 16 , 17 Considering the 80th percentile score in the general population as indicative of a high probability of having hypercholesterolemia of polygenic cause, in our cohort this characteristic was present in one third of the subjects with FH/M−. Of note, a high polygenic score was also found in 23% of subjects with FH/M+, which is in agreement with recent literature that has shown that the 2 criteria are independent and are both able to modulate LDL‐C levels in an additive manner. 2 Moreover, the distribution illustrated in the Figure shows how the pathogenetic picture in a population clinically diagnosed with FH is particularly complex and heterogeneous, emphasizing the need for a comprehensive clinical and genetic assessment.
We also found that in 1 out of 5 subjects with FH/M−, 1 copy of either rs10455872 or rs3798220 or 2 copies of either rs10455872 or rs3798220 were detected, a frequency that was double compared with subjects with FH/M+. Some studies 4 , 18 , 19 , 20 reported higher lp(a) levels in subjects with FH/M− compared with those with FH/M+; with a similar approach, our analysis described difference in LPA genotype in the population with FH. This genetic feature, associated with higher lp(a) levels, determined an increase in the reported LDL‐C levels, although comparisons were not statistically significant. Our subanalysis showed that patients with lp(a) score ≥1 had even lower LDL‐C levels compared with patients with lp(a) score=0 when the adjustment by lp(a) levels was applied. Although this approach could be limited by the inter‐ and intraindividual variation in the lp(a) mass, 14 the results strongly support the need to account for the contribution of lp(a) concentration to the estimated level of LDL‐C not to misidentify individuals with clinical diagnosis of FH.
Patients with lp(a) score ≥1, despite comparable (or lower, when adjusted) levels of LDL‐C, presented with a worse cardiovascular clinical profile, although statistical tests were not significant, possibly due to the low sample size. This aspect may also contribute to suggest a clinical diagnosis of FH, remarking the role of elevated lp(a) in a phenotype similar to that of subjects with confirmed FH. This evidence warrants further studies on larger samples. Interestingly, a similar trend to higher prevalence of premature coronary, cerebrovascular, or peripheral disease associated with lp(a) score ≥1 was observed within the FH/M+ cohort, suggesting a role for lp(a) in increasing cardiovascular risk that is independent not only from the effect of monogenic variants but also from LDL‐C levels. Accordingly, Ellis et al evaluated 316 patients admitted to a coronary care unit, identifying elevated lp(a) and the FH condition in 27.0% and 11.6% of patients, respectively, and both disorders in 4.4% of individuals. They also found that elevated lp(a) alone conferred a 1.9‐fold, FH alone a 3.2‐fold, and the combination of both disorders a 5.3‐fold increased risk of premature coronary artery disease. 21
Overall, in our sample of subjects with a clinical diagnosis of FH, after excluding the presence of a causative variant and the polygenic cause, a genotype associated with high lp(a) levels could explain the observed phenotype in 4.6% of cases. Possibly, these are subjects in whom hypercholesterolemia and cardiovascular risk are essentially driven by high lp(a) values, and for this reason, in the future, they may potentially benefit from therapies targeting this lipoprotein.
The recently published European Atherosclerosis Society Consensus Statement about the role of lp(a) in atherosclerotic cardiovascular disease clearly described a continuous relationship between lp(a) concentration and absolute atherosclerotic cardiovascular disease risk, 22 with an lp(a) level of 100 mg/dL being associated with a 2‐fold risk of atherosclerotic cardiovascular disease in the general population, irrespective of baseline absolute risk. From this point of view, as also suggested by our results, the assessment of lp(a) levels is of 2‐fold importance in the setting of FH. First, the measurement of lp(a) allows the FH diagnosis to be refined in cases where it is essentially based on LDL‐C levels. Second, regardless of the genetic background on LDLR, lp(a) levels should be used to refine cardiovascular risk assessment, better stratify patients, 23 and identify those requiring more intensive and timely intervention.
Limitations
Some limitations have to be acknowledged. We have to point out that differences were often not statistically significant. This could be due to the low number of subjects in each group; thus further studies are warranted to confirm our observations. In the LIPIGEN study, the measurement of lp(a) values was not mandatory and for many centers belonging to the LIPIGEN Network it was not a routine practice. Therefore, the measured values were available for only a subgroup of subjects, which did not allow us to make an in‐depth assessment of this parameter. In addition, the methodology was not the same in all centers. However, the genetic score we applied to identify the subjects most likely to have elevated lp(a) levels was shown to explain about 40% of the variability in lp(a) levels 8 ; the use of the score allowed us to have a larger sample size, as well as to overcome the limitations linked to the heterogeneity of the measurement methods. 24
CONCLUSIONS
Our results highlight the importance of measuring lp(a) to perform the diagnosis of FH appropriately and to exclude the possibility that the observed phenotype is driven by elevated levels of lp(a) before performing the genetic test that is often requested based on the presence of elevated LDL‐C levels.
Clinical and laboratory investigations for differentiating FH, elevated lp(a), or the combination of both are also important for accurate risk stratification and treatment strategies, as in the future patients with both elevated lp(a) and FH condition may be shown to benefit from interventions to reduce both LDL‐C and lp(a) plasma levels.
APPENDIX
The LIPIGEN Study Group
Department of Translational and Precision Medicine, Sapienza University of Rome.—A.O. Policlinico Umberto I, Rome, Italy (Marcello Arca, Laura D'Erasmo); Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialties, University of Palermo, Palermo, Italy (Maurizio Averna, Angelo Baldassare Cefalù); Rare Diseases and Medical Genetic Unit, Ospedale Pediatrico Bambino Gesù, IRCCS, Rome, Italy (Andrea Bartuli, Paola Sabrina Buonuomo); SCDU Endocrinologia, Diabetologia e Malattie del Metabolismo, Dipartimento di Scienze Mediche, Università degli Studi di Torino, Turin, Italy (Andrea Benso, Guglielmo Beccuti); Università di Parma, Centro Dislipidemie in Età Evolutiva, U.O. Pediatria e Neonatologia, Ospedale Guglielmo da Saliceto, Piacenza, Italy (Giacomo Biasucci, Maria Elena Capra); S.S. Diabetologia e Malattie Metaboliche, U.C.O. Clinica Medica, ASUGI, Università di Trieste, Trieste, Italy (Gianni Biolo, Pierandrea Vinci); Ambulatorio Dislipidemie, U.O. Medicina Interna, Ospedale dell'Angelo di Mestre, Venice, Italy (Luca Bonanni); U.O. di Medicina Interna Cardiovascolare, Ambulatorio Dislipidemie, Università dI Bologna, IRCCS S.Orsola, Bologna, Italy (Claudio Borghi, Sergio D'Addato); U.O.C. Malattie Endocrine e Centro Regionale per il Diabete Mellito, ASST Bergamo Ovest, Treviglio, Bergamo, Italy (Antonio Carlo Bossi, Giancarla Meregalli); Ambulatorio Dislipidemie, Centro per lo Studio e la Prevenzione dell'Arteriosclerosi, Fondazione IRCCS Ca′ Granda, Ospedale Maggiore Policlinico e Dipartimento di Scienze Cliniche e di Comunità, Università degli Studi di Milano, Milan, Italy (Adriana Branchi); U.O.C. Cardiologia Clinica a Direzione Universitaria e U.T.I.C., A.O.R.N. “Sant'Anna e San Sebastiano”, Caserta, Italy; Dipartimento di Scienze Mediche Traslazionali Università degli studi della Campania “Luigi Vanvitelli”, Naples, Italy (Paolo Calabrò); U.O. Medicina interna metabolica, Lipidology Centre, Baggiovara Hospital, AOU of Modena and University of Modena and Reggio Emilia, Modena, Italy (Francesca Carubbi, Fabio Nascimbeni); Clinica Medica, Centro di alta specializzazione per la prevenzione dell'Aterosclerosi, centro di eccellenza ESH per l'ipertensione arteriosa, centro di riferimento regionale per le Dislipidemie, Ospedale Policlinico S.S. Annunziata, Chieti, Italy (Francesco Cipollone, Marco Bucci); Centro Dislipidemie e Aterosclerosi, Ospedale di Trento, APSS (Azienda Provinciale per i Servizi Sanitari‐Trento), Trento, Italy (Nadia Citroni); Dipartimento Scienze Cliniche, Internistiche, Anestesiologiche e Cardiovascolari—Sapienza Università, A.O. Policlinico Umberto I, Rome, Italy (Maria Del Ben, Francesco Baratta); Dipartimento di Medicina dei Sistemi, Università degli Studi di Roma Tor Vergata, Rome, Italy (Massimo Federici, Martina Montagna); Università dell'Aquila—Dipartimento MeSVA—UOC Medicina Interna e Nefrologia—Centro Ipertensione Arteriosa e Prevenzione Cardiovascolare—Ospedale San Salvatore—L'Aquila, Italy (Claudio Ferri, Serena Notargiacomo); ASSTRhodense Garbagnate Milanese, Garbagnate Milanese, Milan, Italy (Anna Maria Fiorenza, Emanuela Colombo); Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università degli studi di Napoli Federico II and CEINGE Biotecnologie Avanzate s.c.a.r.l., Naples, Italy (Giuliana Fortunato, Maria Donata Di Taranto); Center for Endocrine and Metabolic Diseases, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy (Andrea Giaccari, Simona Moffa); Department of Emergency and Organ Transplantation, Section of Internal Medicine, Endocrinology, Andrology and Metabolic Diseases, University of Bari Aldo Moro, Bari, Italy (Francesco Giorgino, Sergio Di Molfetta); Pediatric Endocrinology, Department of Public Health and Pediatric Sciences, Turin University, Turin, Italy (Ornella Guardamagna, Luisa De Sanctis); U.O. Medicina Interna 2, Centro per le malattie da arteriosclerosi, AORN Cardarelli, Naples, Italy (Arcangelo Iannuzzi, Raimondo Cavallaro); Dipartimento di Medicina Clinica e Chirurgia Università degli Studi di Napoli Federico II, Naples, Italy (Gabriella Iannuzzo, Marco Gentile); U.O.C. Pediatria, Azienda Ospedaliero Universitaria di Modena (Lorenzo Iughetti, Patrizia Bruzzi); AOU San Luigi Gonzaga, Orbassano, Turin, Italy (Salvatore Lia); ASL VCO—SOC Cardiologia, Ospedale Castelli, Verbania, Italy (Alessandro Lupi); Department of Clinical and Experimental Medicine—Lipid Center—University Hospital G. Martino, Messina, Italy (Giuseppe Mandraffino, Arianna Toscano); Dipartimento medicina sperimentale e clinica, Università di Firenze, AOU Careggi, Firenze (Rossella Marcucci, Martina Berteotti); Ambulatorio ipertensione dislipidemie, U.O. Medicina Generale, ASST Valle Olona, Ospedale di Gallarate, Gallarate, Italy (Lorenzo Maroni, Fabiana Locatelli); Dipartimento Scienze Mediche Chirurg., Università degli Studi Magna Graecia, Catanzaro, Italy (Tiziana Montalcini); Centro Dislipidemie ASST Grande Ospedale Metropolitano Niguarda, Milan, Italy (Giuliana Mombelli); Dipartimento di Scienze Biomediche, Università degli Studi di Cagliari e Centro per le Malattie Dismetaboliche e l'Arteriosclerosi, Associazione. Onlus Cagliari (Sandro Muntoni, Davide Baldera); Istituto Auxologico Italiano, IRCCS Ospedale San Luca, Milan, Italy (Gianfranco Parati); Centro per lo Studio e il Trattamento delle Malattie del Metabolismo, Aterosclerosi e Nutrizione Clinica, Azienda OspedalieraUniversitaria S. Anna di Ferrara. Dipartimento di Medicina Traslazionale e per la Romagna—Università degli Studi di Ferrara, Ferrara, Italy (Angelina Passaro); UOSD ‘Prevenzione cardiovascolare’, Dipartimento di Scienze Mediche, Azienda Sanitaria Locale Frosinone, Frosinone, Italy (Valerio Pecchioli); U.O. Clinica Pediatrica, Servizio Clinico Dislipidemie per lo Studio e la Prevenzione dell'Aterosclerosi in età Pediatrica, ASST‐Santi Paolo e Carlo, Milan, Italy (Cristina Pederiva, Giuseppe Banderali); AOU San Giovanni di Dio e Ruggi d'Aragona, Salerno, Italy (Antonio Pipolo, Debora D'Elia); Sez. Medicina Interna, Angiologia e Malattie da Arteriosclerosi, Dipartimento di Medicina e Chirurgia, Università degli Studi di Perugia, Perugia, Italy (Matteo Pirro, Vanessa Bianconi); IRCCS Ospedale policlinico San Martino UOSD Dietetica e Nutrizione Clinica, Dipartimento di Medicina Interna, Università di Genova, Genoa, Italy (Livia Pisciotta, Elena Formisano); Department of Clinical and Experimental Medicine, University of Catania, Ospedale Garibaldi, Catania, Italy (Francesco Purrello, Roberto Scicali); SOC Malattie Metaboliche e Diabetologia, ASL AT, Asti, Italy (Elena Repetti, Elena Cantino); U.O. Endocrinologia, Diabetologia e Malattie del Metabolismo, Centro regionale specializzato per la diagnosi e terapia delle dislipidemie e aferesi terapeutica, A.O. Universitaria Integrata di Verona, Verona, Italy (Elisabetta Rinaldi, Elena Sani); Clinica Medica e Geriatrica, Dipartimento di Scienze Cliniche e Molecolari, Università Politecnica delle Marche e IRCCS‐INRCA, Ancona, Italy (Riccardo Sarzani, Francesco Spannella); U.O. Lipoaferesi, Centro Regionale di Riferimento per la diagnosi e cura delle Dislipidemie Ereditarie, Fondazione Toscana “G. Monasterio”; Pisa, Italy (Francesco Sbrana, Beatrice Dal Pino); U.O. di Medicina Interna e Geriatria “C. Frugoni” e Centro di Assistenza e Ricerca Malattie Rare, A.O. Universitaria Policlinico Consorziale, Università degli Studi di Bari “Aldo Moro”, Bari, Italy (Patrizia Suppressa, Veronica Cocco); Arcispedale S. Maria Nuova—Azienda Ospedaliera di Reggio Emilia, Reggio Emilia, Italy (Chiara Trenti, Emanuele Alberto Negri); U.O. Ambulatorio Prevenzione Aterosclerosi IRCCS Centro Cardiologico Monzino, Milan, Italy (Josè Pablo Werba, Alessandra Romandini); Dipartimento di Medicina, Università di Padova, Padua, Italy (Sabina Zambon, Alberto Zambon); Servizio di Diabetologia e Malattie Metaboliche “Ospedale P. Pederzoli”—Casa di Cura Privata S.p.A., Peschiera del Garda, Italy (Maria Grazia Zenti, Giulia Fainelli); Centro per lo Studio dell'Aterosclerosi, IRCCS Multimedica, Sesto San Giovanni, Italy (Fabio Pellegatta, Liliana Grigore); SSD Endocrinologia e Diabetologia ASL TO3, Turin, Italy (Katia Bonomo); Centro per lo Studio e il Trattamento delle Malattie del Metabolismo, Aterosclerosi e Nutrizione Clinica, Azienda Ospedaliera‐Universitaria S. Anna di Ferrara, Ferrara, Italy (Eleonora Capatti); UOC Cardiologia, Ospedale dell'Angelo di Mestre, Venice, Italy (Ada Cutolo); Azienda Ospedaliera di Rilievo Nazionale AORN Dei Colli, “V. Monaldi”, Unit of Inherited and Rare Cardiovascular Diseases, CCMR Regione Campania, Naples, Italy (Fabio Fimiani); Università di Milano‐Bicocca, Istituto Auxologico Italiano, IRCCS Ospedale San Luca, Milan, Italy (Simonetta Genovesi); APSS (Azienda Provinciale per i Servizi Sanitari‐Trento), Trento, Italy (Sandro Inchiostro); Centro Grossi Paoletti, Dipartimento di Scienze Farmacologiche e Biomolecolari, Università degli Studi di Milano (Chiara Pavanello); Dipartimento Medicina sperimentale e clinica, Università degli Studi Magna Graecia, Catanzaro, Italy (Roberta Pujia); ASL VCO, SOC Cardiologia, Ospedale San Biagio, Domodossola, Italy (Alon Schaffer); Department of Internal Medicine, University of Genoa, Genoa, Italy (Stefano Bertolini); Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy (Sebastiano Calandra); Epidemiology and Preventive Pharmacology Service (SEFAP), Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy. IRCCS MultiMedica, Sesto San Giovanni (MI), Italy (Alberico Luigi Catapano, Manuela Casula, Elena Olmastroni); Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Modena, Italy (Patrizia Tarugi); SISA Foundation, Milan, Italy (Marta Gazzotti); IRCCS MultiMedica, Sesto San Giovanni (MI), Italy (Federica Galimberti).
Sources of Funding
This substudy is part of the LIPIGEN study, an initiative of the SISA Foundation supported by an unconditional research grant from Sanofi.
Disclosures
Alberico L. Catapano received research funding or honoraria for advisory boards, consultancy or speaker bureau from Aegerion, Amgen, AstraZeneca, Eli Lilly, Genzyme, Mediolanum, Merck or MSD, Pfizer, Recordati, Rottapharm, Sanofi‐Regeneron, and SigmaTau. The remaining authors have no disclosures to report.
Acknowledgments
The genetic assessment was performed in collaboration with GenInCode.The work of MC has been supported by Italian Ministry of Health‐IRCCS MultiMedica GR‐2016‐02361198. The work of ALC has been supported by Fondazione Cariplo 2015‐0524 and 2015‐0564; H2020 REPROGRAM PHC‐03‐2015/667837‐2; PRIN 2017H5F943; ERANET ER‐2017‐2364981; RP‐2017‐ERACVD‐OPERATION ERP‐2017‐23671155; Italian Ministry of Health‐IRCCS MultiMedica GR‐201102346974; SISA Lombardia and Fondazione SISA. The work of ALC and MC has been also supported by Italian Ministry of Health—Ricerca Corrente— IRCCS MultiMedica.
Elena Olmastroni, Marta Gazzotti, Manuela Casula, and Alberico L. Catapano were responsible for the study concept and design. Marta Gazzotti and Manuela Casula were responsible for study management and data collection. Elena Olmastroni, provided methodological and statistical knowledge and performed the analysis. Alberico L. Catapano contributed to the interpretation of the results. Elena Olmastroni and Manuela Casula wrote the article. Maurizio Averna, Marcello Arca, Patrizia Tarugi, Sebastiano Calandra, and Stefano Bertolini critically revised for important intellectual content and approved the final article.
This article was sent to Daniel Edmundowicz, MD, Guest Editor, for review by expert referees, editorial decision, and final disposition.
Contributor Information
Elena Olmastroni, Email: elena.olmastroni@unimi.it.
the LIPIGEN Study Group:
Marcello Arca, Laura D’Erasmo, Maurizio Averna, Angelo Baldassare Cefalù, Andrea Bartuli, Paola Sabrina Buonuomo, Andrea Benso, Guglielmo Beccuti, Giacomo Biasucci, Maria Elena Capra, Gianni Biolo, Pierandrea Vinci, Luca Bonanni, Claudio Borghi, Sergio D’Addato, Antonio Carlo Bossi, Giancarla Meregalli, Adriana Branchi, Paolo Calabrò, Francesca Carubbi, Fabio Nascimbeni, Francesco Cipollone, Marco Bucci, Nadia Citroni, Maria Del Ben, Francesco Baratta, Massimo Federici, Martina Montagna, Claudio Ferri, Serena Notargiacomo, Anna Maria Fiorenza, Emanuela Colombo, Giuliana Fortunato, Maria Donata Di Taranto, Andrea Giaccari, Simona Moffa, Francesco Giorgino, Sergio Di Molfetta, Ornella Guardamagna, Luisa De Sanctis, Arcangelo Iannuzzi, Raimondo Cavallaro, Gabriella Iannuzzo, Marco Gentile, Lorenzo Iughetti, Patrizia Bruzzi, Salvatore Lia, Alessandro Lupi, Giuseppe Mandraffino, Arianna Toscano, Rossella Marcucci, Martina Berteotti, Lorenzo Maroni, Fabiana Locatelli, Tiziana Montalcini, Giuliana Mombelli, Sandro Muntoni, Davide Baldera, Gianfranco Parati, Angelina Passaro, Valerio Pecchioli, Cristina Pederiva, Giuseppe Banderali, Antonio Pipolo, Debora D’Elia, Matteo Pirro, Vanessa Bianconi, Livia Pisciotta, Elena Formisano, Francesco Purrello, Roberto Scicali, Elena Repetti, Elena Cantino, Elisabetta Rinaldi, Elena Sani, Riccardo Sarzani, Francesco Spannella, Francesco Sbrana, Beatrice Dal Pino, Patrizia Suppressa, Veronica Cocco, Chiara Trenti, Emanuele Alberto Negri, Josè Pablo Werba, Alessandra Romandini, Sabina Zambon, Alberto Zambon, Maria Grazia Zenti, Giulia Fainelli, Fabio Pellegatta, Liliana Grigore, Katia Bonomo, Eleonora Capatti, Ada Cutolo, Fabio Fimiani, Simonetta Genovesi, Sandro Inchiostro, Chiara Pavanello, Roberta Pujia, Alon Schaffer, Stefano Bertolini, Sebastiano Calandra, Alberico Luigi Catapano, Manuela Casula, Elena Olmastroni, Patrizia Tarugi, Marta Gazzotti, and Federica Galimberti
References
- 1. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490. doi: 10.1093/eurheartj/eht273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Olmastroni E, Gazzotti M, Arca M, Averna M, Pirillo A, Catapano AL, Casula M, Groupdagger LS. Twelve variants polygenic score for low‐density lipoprotein cholesterol distribution in a large cohort of patients with clinically diagnosed familial hypercholesterolemia with or without causative mutations. J Am Heart Assoc. 2022;11:e023668. doi: 10.1161/JAHA.121.023668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Talmud PJ, Drenos F, Shah S, Shah T, Palmen J, Verzilli C, Gaunt TR, Pallas J, Lovering R, Li K, et al. Gene‐centric association signals for lipids and apolipoproteins identified via the HumanCVD BeadChip. Am J Hum Genet. 2009;85:628–642. doi: 10.1016/j.ajhg.2009.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Langsted A, Kamstrup PR, Benn M, Tybjaerg‐Hansen A, Nordestgaard BG. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol. 2016;4:577–587. doi: 10.1016/S2213-8587(16)30042-0 [DOI] [PubMed] [Google Scholar]
- 5. Utermann G, Hoppichler F, Dieplinger H, Seed M, Thompson G, Boerwinkle E. Defects in the low density lipoprotein receptor gene affect lipoprotein (a) levels: multiplicative interaction of two gene loci associated with premature atherosclerosis. Proc Natl Acad Sci USA. 1989;86:4171–4174. doi: 10.1073/pnas.86.11.4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trinder M, DeCastro ML, Azizi H, Cermakova L, Jackson LM, Frohlich J, Mancini GBJ, Francis GA, Brunham LR. Ascertainment bias in the association between elevated lipoprotein(a) and familial hypercholesterolemia. J Am Coll Cardiol. 2020;75:2682–2693. doi: 10.1016/j.jacc.2020.03.065 [DOI] [PubMed] [Google Scholar]
- 7. Zekavat SM, Ruotsalainen S, Handsaker RE, Alver M, Bloom J, Poterba T, Seed C, Ernst J, Chaffin M, Engreitz J, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun. 2018;9:2606. doi: 10.1038/s41467-018-04668-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604 [DOI] [PubMed] [Google Scholar]
- 9. Trinder M, Uddin MM, Finneran P, Aragam KG, Natarajan P. Clinical utility of lipoprotein(a) and LPA genetic risk score in risk prediction of incident atherosclerotic cardiovascular disease. JAMA Cardiol. 2020;6:1–9. doi: 10.1001/jamacardio.2020.5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Averna M, Cefalu AB, Casula M, Noto D, Arca M, Bertolini S, Calandra S, Catapano AL, Tarugi P; LIPIGEN Group . Familial hypercholesterolemia: the Italian Atherosclerosis Society Network (LIPIGEN). Atherosclerosis Supp. 2017;29:11–16. doi: 10.1016/j.atherosclerosissup.2017.07.001 [DOI] [PubMed] [Google Scholar]
- 11. Casula M, Olmastroni E, Pirillo A, Catapano AL, Arca M, Averna M, Bertolini S, Calandra S, Tarugi P, Pellegatta F, et al. Evaluation of the performance of Dutch Lipid Clinic Network score in an Italian FH population: the LIPIGEN study. Atherosclerosis. 2018;277:413–418. doi: 10.1016/j.atherosclerosis.2018.08.013 [DOI] [PubMed] [Google Scholar]
- 12. Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, Harrison SC, Li K, Drenos F, Karpe F, et al. Use of low‐density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case‐control study. Lancet. 2013;381:1293–1301. doi: 10.1016/S0140-6736(12)62127-8 [DOI] [PubMed] [Google Scholar]
- 13. Dahlen GJ. Potential significance of Lp (a) lipoprotein clinical methodological aspects. In: Bearn AG, ed. Genetics of Coronary Heart Disease. Oslo: Institute of Medical Genetics, University of Oslo; 1992:75–88. [Google Scholar]
- 14. Yeang C, Witztum JL, Tsimikas S. Novel method for quantification of lipoprotein(a)‐cholesterol: implications for improving accuracy of LDL‐C measurements. J Lipid Res. 2021;62:100053. doi: 10.1016/j.jlr.2021.100053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mariano C, Alves AC, Medeiros AM, Chora JR, Antunes M, Futema M, Humphries SE, Bourbon M. The familial hypercholesterolaemia phenotype: monogenic familial hypercholesterolaemia, polygenic hypercholesterolaemia and other causes. Clin Genet. 2020;97:457–466. doi: 10.1111/cge.13697 [DOI] [PubMed] [Google Scholar]
- 16. Narayanaswamy M, Sharma S. Polygenic hypercholesterolemia. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022. [PubMed] [Google Scholar]
- 17. Tada H, Nohara A, Kawashiri MA. Monogenic, polygenic, and oligogenic familial hypercholesterolemia. Curr Opin Lipidol. 2019;30:300–306. doi: 10.1097/QCO.0000000000000563 [DOI] [PubMed] [Google Scholar]
- 18. Alonso R, Argueso R, Alvarez‐Banos P, Muniz‐Grijalvo O, Diaz‐Diaz JL, Mata P. Familial hypercholesterolemia and lipoprotein(a): two partners in crime? Curr Atheroscler Rep. 2022;24:427–434. doi: 10.1007/s11883-022-01019-5 [DOI] [PubMed] [Google Scholar]
- 19. Merino‐Ibarra E, Puzo J, Jarauta E, Cenarro A, Recalde D, Garcia‐Otin AL, Ros E, Martorell E, Pinto X, Franco M, et al. Hyperlipoproteinaemia(a) is a common cause of autosomal dominant hypercholesterolaemia. J Inherit Metab Dis. 2007;30:970–977. doi: 10.1007/s10545-007-0585-z [DOI] [PubMed] [Google Scholar]
- 20. Pederiva C, Capra ME, Biasucci G, Banderali G, Fabrizi E, Gazzotti M, Casula M, Catapano AL; LIPIGEN Paediatric Group ; Arca M, Averna M, et al. Lipoprotein(a) and family history for cardiovascular disease in paediatric patients: a new frontier in cardiovascular risk stratification. Data from the LIPIGEN paediatric group. Atherosclerosis. 2022;349:233–239. doi: 10.1016/j.atherosclerosis.2022.04.021 [DOI] [PubMed] [Google Scholar]
- 21. Ellis KL, Pang J, Chieng D, Bell DA, Burnett JR, Schultz CJ, Hillis GS, Watts GF. Elevated lipoprotein(a) and familial hypercholesterolemia in the coronary care unit: between Scylla and Charybdis. Clin Cardiol. 2018;41:378–384. doi: 10.1002/clc.22880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kronenberg F, Mora S, Stroes ESG, Ference BA, Arsenault BJ, Berglund L, Dweck MR, Koschinsky M, Lambert G, Mach F, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43:3925–3946. doi: 10.1093/eurheartj/ehac361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tromp TR, Ibrahim S, Nurmohamed NS, Peter J, Zuurbier L, Defesche JC, Reeskamp LF, Hovingh GK, Stroes ESG. Use of lipoprotein(a) to improve diagnosis and management in clinical familial hypercholesterolemia. Atherosclerosis. 2023;365:27–33. doi: 10.1016/j.atherosclerosis.2022.11.020 [DOI] [PubMed] [Google Scholar]
- 24. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57:526–537. doi: 10.1194/jlr.R061648 [DOI] [PMC free article] [PubMed] [Google Scholar]