

Abstract
Background
Myocardial infarction (MI) is a cardiovascular disease with high morbidity and mortality. PI16 (peptidase inhibitor 16), as a secreted protein, is highly expressed in heart diseases such as heart failure. However, the functional role of PI16 in MI is unknown. This study aimed to investigate the role of PI16 after MI and its underlying mechanisms.
Methods and Results
PI16 levels after MI were measured by enzyme‐linked immunosorbent assay and immunofluorescence staining, which showed that PI16 was upregulated in the plasma of patients with acute MI and in the infarct zone of murine hearts. PI16 gain‐ and loss‐of‐function experiments were used to investigate the potential role of PI16 after MI. In vitro, PI16 overexpression inhibited oxygen–glucose deprivation–induced apoptosis in neonatal rat cardiomyocytes, whereas knockdown of PI16 exacerbated neonatal rat cardiomyocyte apoptosis. In vivo, left anterior descending coronary artery ligation was performed on PI16 transgenic mice, PI16 knockout mice, and their littermates. PI16 transgenic mice showed decreased cardiomyocyte apoptosis at 24 hours after MI and improved left ventricular remodeling at 28 days after MI. Conversely, PI16 knockout mice showed aggravated infract size and remodeling. Mechanistically, PI16 downregulated Wnt3a (wingless‐type MMTV integration site family, member 3a)/β‐catenin pathways, and the antiapoptotic role of PI16 was reversed by recombinant Wnt3a in oxygen–glucose deprivation–induced neonatal rat cardiomyocytes. PI16 also inhibited HDAC1 (class I histone deacetylase) expression, and overexpression HDAC1 abolished the inhibition of apoptosis and Wnt signaling of PI16.
Conclusions
In summary, PI16 protects against cardiomyocyte apoptosis and left ventricular remodeling after MI through the HDAC1‐Wnt3a‐β‐catenin axis.
Keywords: cardiomyocyte apoptosis, HDAC1, myocardial infarction, PI16, remodeling
Subject Categories: Fibrosis, Myocardial Biology, Cell Signalling/Signal Transduction
Nonstandard Abbreviations and Acronyms
- HDAC1
class I histone deacetylase
- NRCM
neonatal rat cardiomyocyte
- OGD
glucose–oxygen deprivation
- PI16
peptidase inhibitor 16
Clinical Perspective.
What Is New?
PI16 (peptidase inhibitor 16) expression is increased in the plasma of patients with acute myocardial infarction (MI) and in the infarct zone of murine hearts, consistent with the result of cardiomyocytes after glucose–oxygen deprivation.
PI16 protects against cardiomyocyte apoptosis and cardiac remodeling by inhibiting HDAC1 (class I histone deacetylase) and downstream Wnt3a/β‐catenin signaling pathway.
What Are the Clinical Implications?
PI16 expression is elevated in the plasma of patients with MI, which indicates that PI16 could be used as a potential biomarker for MI.
We demonstrate the protective role of PI16 in left ventricular injury and remodeling after MI by suppressing the HDAC1‐Wnt3a (wingless‐type MMTV integration site family, member 3a)‐β‐catenin axis, which means PI16, as an endogenous HDAC1 inhibitor, may provide a promising strategy for MI.
Myocardial infarction (MI) is an adverse cardiovascular event with a high mortality rate and poor prognosis. Acute coronary occlusion leads to loss of cardiomyocytes and subsequently to ventricular remodeling including myofibroblast activation, residual myocardium hypertrophy, and scar formation. 1 , 2 , 3 Although existing surgical and pharmaceutical treatments have greatly improved the prognosis, the overall disease burden of MI is still high. Therefore, it is particularly important to find new therapeutic targets and mechanisms.
PI16 (peptidase inhibitor 16) was first identified as a binding protein of prostate secretory protein of 94 amino acids and was associated with prostate cancer. 4 , 5 Recent studies have found that PI16 is functionally expressed in the heart, 6 the meningeal sheath surrounding the dorsal root ganglion, and the specific subset of memory Tregs. 7 , 8 In the heart, PI16 expression is weak under physiological states but is highly upregulated in heart diseases such as heart failure. PI16 can attenuate TAC (transverse aortic constriction)‐induced cardiomyocyte hypertrophy 6 and inhibit protease and chemokine activities, 9 which indicates that PI16 has broad effects on heart tissue.
The Wnt (wingless‐type MMTV integration site family) signaling pathway is a highly conserved pathway that plays significant roles in cell proliferation, differentiation, and death. The level of Wnt signaling is typically low in adult hearts, but this pathway is activated in many pathologies such as MI, valvular disease, cardiac arrhythmias, and heart failure. 10 Three major Wnt signaling pathways have been demonstrated: the canonical Wnt/β‐catenin pathway, the noncanonical planar cell polarity pathway, and the Wnt/Ca2+ pathway. 11 The canonical Wnt/β‐catenin pathway is reported to aggravate myocardial injury and cardiac remodeling after MI, 12 , 13 which provides a potential therapeutic target for MI.
HDAC (class I histone deacetylase) is a key protein that regulates epigenetic modification and plays an important role in heart disease. HDAC1 inhibitors have been proven to significantly reduce the infarct size and effectively inhibit chronic remodeling after MI. 14 , 15 It has been reported that PI16 can reduce the expression of HDAC1 to inhibit cardiac fibroblast fibrosis, 16 but the effect of PI16 on cardiomyocytes and MI remains unclear. This study elucidates that PI16 protects against myocardium apoptosis and left ventricular (LV) remodeling after MI by inhibiting the HDAC1‐Wnt3a (wingless‐type MMTV integration site family, member 3a)‐β‐catenin signaling axis.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Statement
The study was approved by the ethics committee of the First Affiliated Hospital of Nanjing Medical University. We obtained written informed consent from all patients.
All experiments were authorized by the Institutional Animal Care and Use Committee of Nanjing Medical University (IACUC‐1710006).
Animals
PI16 transgenic (PI16‐Tg) mice and PI16 knockout (PI16−/−) mice were generated by Cyagen Biosciences (Guangzhou, China). PI16‐Tg mice on the FVB background were generated through microinjection with a recombinant PI16 vector containing an EF1‐α (elongation factor‐1 α) promoter. PI16−/− mice on the C57BL/6J background were generated by microinjection with a recombinant PI16 vector containing a Neo insertion under the IRES (internal ribosome entry site)/lacZ promoter. Six‐ to 8‐week‐old PI16‐Tg mice and PI16−/− mice and their littermate controls were used in our study. All experiments were approved by the Animal Ethical and Welfare Committee of Nanjing Medical University. All mice were raised at the animal core facility of Nanjing Medical University.
MI Model
Mice were anesthetized with pentobarbital (50 mg/kg, i.p.) and placed on a warm blanket. Respiration was maintained with a rodent respirator via tracheal intubation. A small incision was performed between the fourth and fifth intercostal spaces, and the pericardium was carefully incised to expose the left anterior descending coronary artery. MI was induced by permanent ligation of the left anterior descending coronary artery with 7‐0 suture. Then, the chest was closed, followed by suturing the muscle and skin. Mice were euthanized at 24 hours and 28 days after MI to harvest the hearts.
To assess the infarct size, mice were anesthetized with pentobarbital (50 mg/kg, i.p.) and were perfused with 1% Evans blue dye (Sigma‐Aldrich). The area at risk was shown as nonblue areas with no Evans blue perfusion. Then, the excised heart was frozen at −20 °C for 20 minutes and cut into 1‐ to 2‐mm slices, which were subsequently incubated in 1% 2,3,5‐triphenyl‐2H‐tetrazolium chloride (Sigma‐Aldrich) at 37 °C for 20 minutes to visualize the infarct zone (white) and the viable myocardium (red). Finally, all slices were fixed in 4% paraformaldehyde and were photographed. Image‐Pro Plus was used to quantify the total left ventricle areas, area at risk, and infarct areas.
Cell Isolation, Culture, and Treatment
Hearts were harvested from 1‐ to 3‐day‐old Sprague‐Dawley rats. Then, the hearts were washed with ADS (NaCl, HEPES, NaHPO4, glucose, KCl and MgSO4·7H2O diluted in ddH2O; all reagents from Sigma‐Aldrich) and cut into small pieces. The tissue was digested with type II collagenase (15 mg/mL) (Gibco) at 120 rpm and 37 °C for 30 minutes. Then, the cell suspension was seeded into 10‐cm dishes and cultured for 1 hour at 37 °C in 5% CO2 to isolate neonatal rat cardiomyocytes (NRCMs) with neonatal rat cardiac fibroblasts. Isolated NRCMs were cultured in DMEM containing 10% horse serum, 5% fetal bovine serum, and 1% penicillin–streptomycin (Gibco) at 37 °C in 5% CO2.
The adenovirus was designed by Genechem (Shanghai, China), and adenovirus transfection was performed according to the manufacturer's instructions. Briefly, NRCMs were transfected with Ad‐GFP/Ad‐PI16 (1×107 pfu/mL), Ad‐shCtrl/Ad‐shPI16 (2×107 pfu/mL), or Ad‐HDAC1 (1×107 pfu/mL) for 4 hours and then cultured with complete medium for 48 hours. NRCMs were incubated in glucose‐free DMEM without serum and placed in a box with Anaeropack‐anaero (Mitsubishi Gas Chemical) for 8 hours to establish the oxygen–glucose deprivation (OGD) model. The Wnt signaling agonist recombinant Wnt3a (100 ng/mL) (MedChemExpress) and Wnt signaling inhibitor IWR‐1‐endo (10 μM) were added to the medium for 2 hours before OGD.
Enzyme‐Linked Immunosorbent Assay
Human blood samples were obtained with the approval of the Ethics Committee of First Affiliated Hospital of Nanjing Medical University (2021‐SR‐289). A total of 24 blood samples were obtained from patients without acute MI or related diseases, and 24 blood samples were obtained from patients with acute MI within 7 days. Venous blood was collected in vacuum tubes, and plasma was stored at −80 °C. The PI16 concentration in the plasma was measured by an ELISA kit (Cloud‐Clone; China) for human PI16 according to the manufacturer's instructions.
Histology and Immunohistochemistry Staining
The heart samples were fixed in 4% paraformaldehyde overnight and then embedded in paraffin. Approximately 4‐μm tissue sections were used for immunohistochemistry staining. Masson's trichrome staining, Sirius red staining, and hematoxylin and eosin staining were performed following standard methods. Images were captured by using a Nikon Eclipse 50i (Nikon, Japan). Image‐Pro‐Plus software was used for quantification of fibrosis, and the size of the myocardium was analyzed by ImageJ software.
Protein Extraction and Western Blotting
The total protein of heart samples and NRCMs was harvested in radioimmunoprecipitation assay lysis buffer containing protease inhibitor and phosphatase inhibitor cocktail (Roche). The lysate was centrifuged at 12 000 rpm and 4 °C for 20 minutes. The protein concentration was then measured using Pierce BCA protein assay kit (Beyotime Biotechnology). The supernatants were added with 5× loading buffer and then were boiled for 8 minutes to denature the protein. Protein samples were separated by 10% to 12% SDS‐PAGE and transferred to polyvinylidene difluoride membranes (Millipore), which were then blocked with 5% bovine serum albumin solution. The membranes were incubated with primary antibodies at 4 °C overnight and horseradish peroxidase–linked secondary antibodies for 2 hours at room temperature. After being washed in TBST, the blots were scanned with a ChemiDoc MP imager (Bio‐Rad). Images were quantified by Image Lab software. The primary antibodies used were as follows: anti‐GAPDH (catalog number 5174), anti‐tubulin (catalog number 2146), anti‐Bax (Bcl2‐Associated X protein) (catalog number 2772), anti‐cleaved caspase 3 (catalog number 9654), anti‐β‐catenin (catalog number 8480), and anti‐HDAC1 (catalog number 5356), which were all purchased from Cell Signaling Technology. Anti‐Wnt3a (catalog number 26744‐1‐AP) and anti‐MMP2 (matrix metalloproteinase 2) (catalog number 10373‐2‐AP) antibodies were purchased from Proteintech. An anti‐B‐cell lymphoma‐2 (Bcl2) antibody (catalog number BF9103) was purchased from Affinity Biosciences. An anti‐PI16 antibody (catalog number 121554) was purchased from GeneTex. An anti‐p‐β‐catenin antibody (catalog number AF5749) was purchased from Beyotime Biotechnology, and an anti‐collagen I antibody (catalog number WL0088) was purchased from Wanleibio. The secondary antibodies used were horseradish peroxidase–conjugated anti‐rabbit and anti‐mouse secondary antibodies (catalog numbers 7074 and 7076, respectively), which were purchased from Cell Signaling Technology.
Real‐Time Polymerase Chain Reaction
Total mRNA extraction was performed according to standard protocols. A PrimeScript RT Reagent Kit (Takara) was used to perform reverse transcription for cDNA production. Real‐time polymerase chain reactions were performed on an ABI system using Hieff UNICON qPCR SYBR Green Master Mix (Yeasen). Target genes were normalized to the reference housekeeping gene GAPDH, and the relative mRNA expression levels were calculated by the 2−ΔΔCq method. Fold differences were then calculated for each treatment group using cycle threshold (CT) values normalized to the control. Primer sequences are detailed in Table S1.
TdT‐mediated dUTP Nick‐End Labeling Assay
A TdT‐mediated dUTP nick‐end labeling (TUNEL) Apoptosis Detection Kit (Alexa Fluor 488) (Yeasen, China) was used to detect apoptotic cells in the heart tissue and NRCMs. All procedures followed the manufacturer's instructions.
Immunofluorescence Staining
Paraffin sections were placed at 65 °C for 20 minutes and subsequently soaked in xylene and gradient ethanol for dewaxing. Then, the sections were permeabilized with 0.5% Triton for 20 minutes at room temperature and repaired in sodium citrate solution in 95 °C water for 10 hours. After naturally cooling to room temperature, sections were incubated with blocking buffer (5% BSA plus 10% goat serum and 0.05% Triton X‐100 in PBS) for 1 hour at room temperature. Then, the sections were incubated with primary antibodies at 4 °C overnight and secondary antibodies for 1 hour at room temperature. Finally, sections were dried and sealed with antifade mounting medium containing DAPI. A Carl Zeiss Axioskop microscope (Carl Zeiss) was used to capture fluorescent images. The primary antibodies used were anti‐PI16 (1:200, catalog number 121554; GeneTex), anti‐α‐actinin (1:200; catalog number A7811; Sigma‐Aldrich), and anti‐HDAC1 (1:200, catalog number 10197‐1‐AP; Proteintech). The secondary antibodies used were Cy3 goat anti‐rabbit, Cy3 goat anti‐mouse, Alexa Fluor 488 goat anti‐rabbit, and Alexa Fluor 488 goat anti‐mouse (1: 400, Jackson ImmunoResearch).
Nuclear Protein Extraction
Nuclear protein was obtained by using a Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology) per manufacturer's protocols.
Statistical Analysis
All measurement data are from 3 independent experiments and are shown as the mean±SEM. Statistical analysis was performed with GraphPad Prism 9.0. First, the data were analyzed for normality test by using the Shapiro‐Wilk test. For the data that passed the normality test, the Student t test was used for 2‐group comparison, and ANOVA with Bonferroni post hoc analysis was used for multiple groups. P<0.05 was considered statistically significant.
RESULTS
PI16 Was Upregulated in Acute MI
To investigate the levels of PI16 in acute MI, we first examined the PI16 concentration in the plasma of patients with or without acute MI by ELISA, and found that PI16 was upregulated in the plasma of patients with acute infarction (Figure 1A). To explore the dynamic changes of PI16 in the progression after MI, we next tested the protein levels of PI16 in the murine heart at different times after MI. Western blotting analysis showed that PI16 was beginning to be upregulated at 1 day after MI and peaked at 7 days after MI (Figure 1B). Immunofluorescence staining further confirmed the upregulation of PI16 both in the infarct zone of murine hearts at 24 hours after MI and in OGD‐induced NRCMs (Figure 1C through 1E). In summary, PI16 is increased in acutely infarcted hearts and plasma, suggesting that PI16 may be involved in the pathological process after MI.
Figure 1. PI16 was upregulated after AMI.
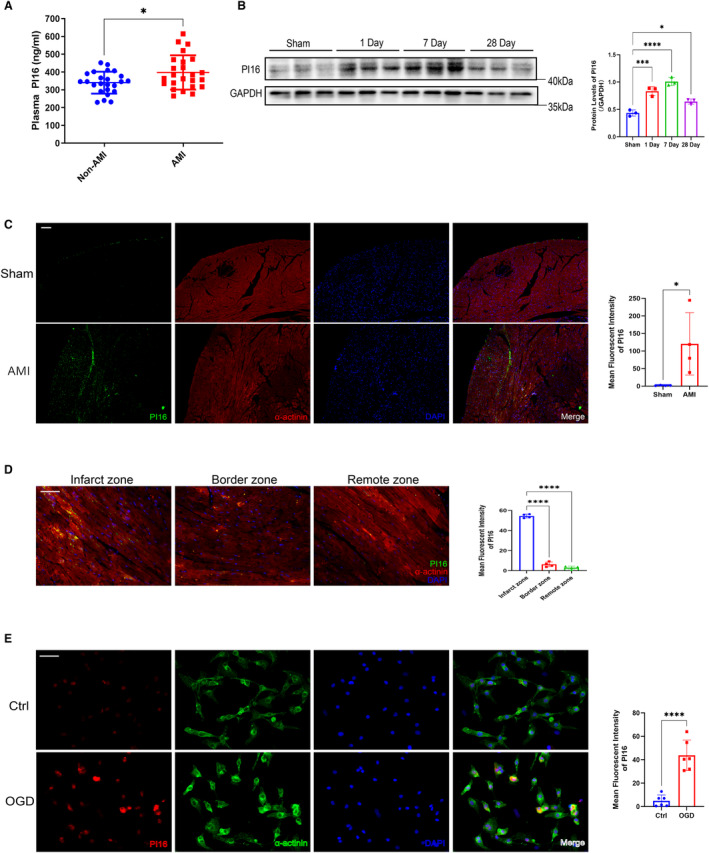
A, PI16 levels in the plasma of patients with or without AMI were assessed by ELISA (n=24). B, Protein levels of PI16 in the murine hearts at 1, 7, and 28 days after MI (n=3). C, Representative images of PI16 and α‐actinin in the murine hearts at 24 hours after MI (n=4). Scale bar, 100 μm. D, Representative images of PI16 in the infarct zone, border zone, and remote zone of the murine hearts (n=4). Scale bar, 50 μm. E, Representative images of PI16 and α‐actinin in NRCMs with or without OGD induction (n=6). Scale bar, 50 μm. Data are presented as the mean±SEM and are representative of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. AMI indicates acute myocardial infarction; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; MI, myocardial infarction; NRCM, neonatal rat cardiomyocyte; OGD, oxygen–glucose deprivation; and PI16, peptidase inhibitor 16.
PI16 Alleviated OGD‐Induced NRCM Apoptosis In Vitro
To study the potential role of PI16, we first examined the effect of PI16 on OGD‐induced NRCMs using Ad‐PI16 and Ad‐shPI16 to overexpress or knockdown PI16 in NRCMs. Overexpression of PI16 ameliorated OGD‐induced NRCM apoptosis, as shown by the downregulation of apoptosis‐associated proteins (Bax, Bax/Bcl2, and cleaved caspase 3) and TUNEL‐positive cardiomyocytes (Figure 2A and 2B). Reversely, knockdown of PI16 significantly aggravated OGD‐induced NRCM apoptosis compared with controls (Figure 2C and 2D). These in vitro results indicate the antiapoptotic role of PI16 in OGD‐induced NRCMs.
Figure 2. PI16 alleviated OGD‐induced NRCM apoptosis in vitro.
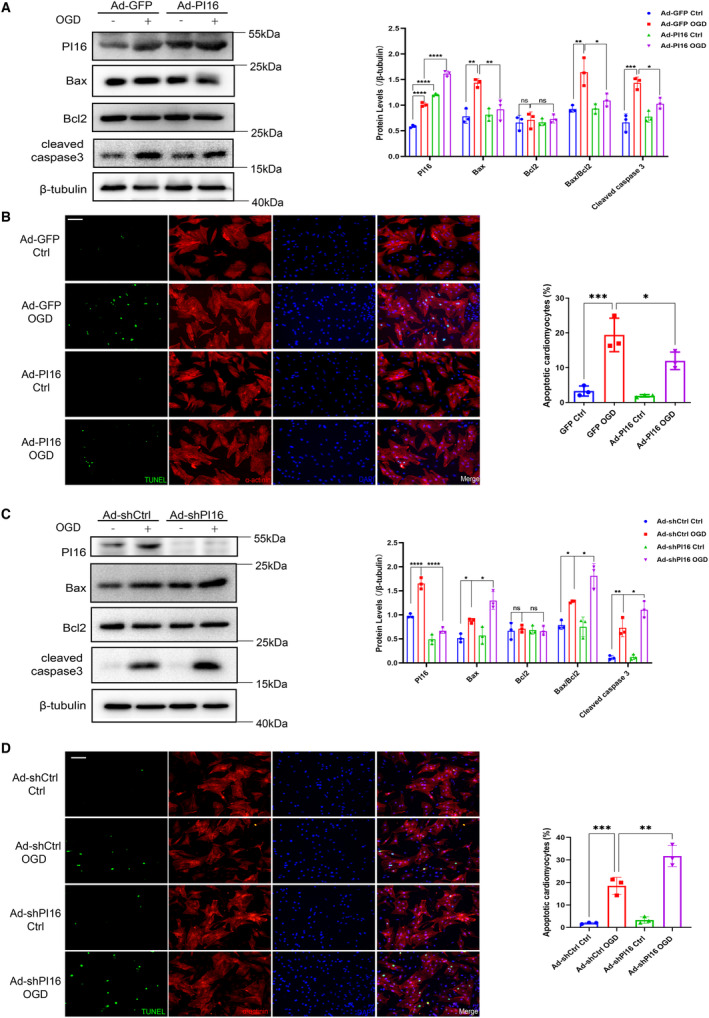
A, Protein levels and quantification of apoptosis‐associated markers, including Bax, Bcl2, Bax/Bcl2, and cleaved caspase 3, in OGD‐induced NRCMs with or without PI16 overexpression. B, Representative images of TUNEL‐positive cardiomyocytes induced by OGD with or without PI16 overexpression. Scale bar, 50 μm. C, Protein levels and quantification of apoptosis‐associated markers in OGD‐induced NRCMs with or without PI16 interference. D, Representative images of TUNEL staining in OGD‐induced NRCMs with or without PI16 knockdown. Scale bar, 50 μm. Data are presented as the mean±SEM and are representative of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Bax indicates Bcl2‐Associated X protein; Bcl2, B‐cell lymphoma‐2; Ctrl indicates control; GFP, green fluorescent protein; NRCM, neonatal rat cardiomyocyte; ns, no significance; OGD, oxygen–glucose deprivation; PI16, peptidase inhibitor 16; shCtrl, short hairpin of control; shPI16, short hairpin of peptidase inhibitor 16; and TUNEL, TdT‐mediated dUTP nick‐end labeling.
PI16 Inhibited the Loss of Cardiomyocytes and LV Dysfunction at 24 Hours After MI
To further verify the protective role of PI16 in acute MI, we performed left anterior descending coronary artery ligation on PI16‐Tg mice and their wild‐type (WT) littermates. At 24 hours after MI, echocardiography showed that PI16‐Tg mice had a higher ejection fraction and fractional shortening than WT mice (Figure 3A). We further found that PI16‐Tg mice exhibited a smaller infarct size (white) than WT mice, although the 2 groups had comparable area at risk (red) based on Evans blue dye and 2,3,5‐triphenyl‐2H‐tetrazolium chloride staining (Figure 3B), suggesting decreased loss of cardiomyocytes in infarcted PI16‐Tg mice. We then evaluated apoptosis‐associated protein levels and found that the expression levels of Bax, Bax/Bcl2, and cleaved caspase 3 were significantly higher in WT mice than in PI16‐Tg mice at 24 hours after MI (Figure 3C). TUNEL staining also showed that PI16‐Tg mice had a reduced proportion of apoptotic myocardium in the infarct area compared with WT mice (Figure 3D). These results demonstrate that overexpression of PI16 attenuates myocardial loss and cardiac dysfunction in acute MI by suppressing myocardial apoptosis.
Figure 3. PI16 inhibited the loss of cardiomyocytes and LV dysfunction at 24 hours after MI.
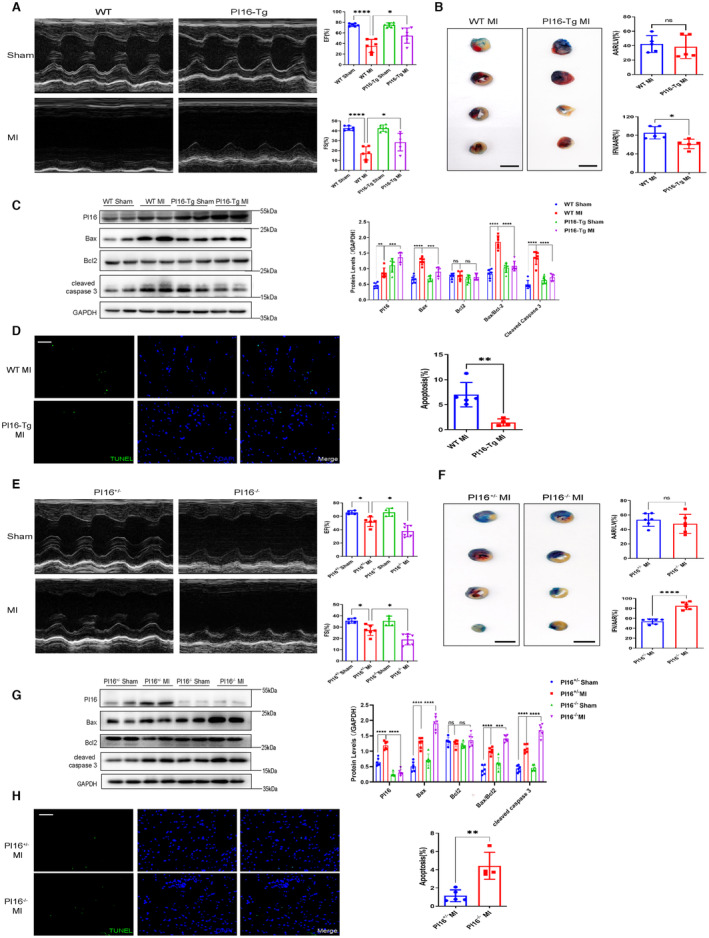
A and E, Representative M‐mode images and echocardiographic data, including ejection fraction and fraction shortening from PI16‐Tg and WT mice subjected to MI or sham surgery after 24 hours (n=6). B, Representative images and quantification of heart sections with Evans blue (blue) and TTC staining (red) in WT and PI16‐Tg mice 24 hours after being subjected to MI (n=5). Scale bar, 5 mm. C, Representative Western blotting and quantitative analysis of PI16, Bax, Bcl‐2, Bax/Bcl2, and cleaved caspase 3 in heart tissue from PI16‐Tg and WT mice 24 hours after being subjected to MI or sham surgery (n=6). D, Representative images of TUNEL staining and quantitative data in the infarcted zone from WT and PI16‐Tg mice 24 hours after MI (n=5). Scale bar, 50 μm. E, Representative M‐mode images and echocardiographic data from PI16+/− and PI16−/− mice 24 hours after being subjected to MI or sham surgery (n=6). F, Representative heart images and quantitation of Evans blue and TTC staining from PI16+/− and PI16−/− mice 24 hours after being subjected to MI (n=5). Scale bar, 5 mm. G, Representative Western blotting and quantitation of apoptosis‐associated proteins in the hearts from PI16+/− and PI16−/− mice (n=6). H, Representative images of TUNEL staining and quantitative data from infarcted PI16+/− and PI16−/− hearts (n=4–5). Scale bar, 50 μm. Data are presented as the mean±SEM and are representative of 3 independent experiments. B, D, F, and H, A Student t test was used to assess the difference. A, C, E, and G, One‐way ANOVA was used to assess the difference. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. AAR indicates area at risk; Bax, Bcl2‐Associated X protein; Bcl2, B‐cell lymphoma‐2; EF, ejection fraction; FS, fractional shortening; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; IFN, infarcted zone; LV, left ventricular; MI, myocardial infarction; ns, no significance; PI16−/−, peptidase inhibitor 16 knockout mice; PI16‐Tg, peptidase inhibitor 16 transgenic mice; TTC, 2,3,5‐triphenyl‐2H‐tetrazolium chloride; TUNEL, TdT‐mediated dUTP nick‐end labeling; and WT, wild‐type.
Next, we determined the role of PI16 deletion in acute MI by performing left anterior descending coronary artery ligation on global PI16−/− mice and their control littermates (PI16+/− mice). At 24 hours after MI, PI16−/− mice showed deteriorated cardiac function with decreased ejection fraction and fractional shortening compared with PI16+/− mice (Figure 3E). PI16 ablation exacerbated MI‐induced cell death by increasing the infarct area in PI16−/− mice (Figure 3F). We also found enhanced proapoptotic protein expression and more TUNEL‐positive nuclei in PI16−/− mice than in PI16+/− mice (Figure 3G and 3H). Together, these data demonstrate that PI16 deficiency aggravates MI‐induced cardiac injury and dysfunction.
PI16 Overexpression Improved LV Remodeling at 28 Days After MI
Because PI16 has a beneficial role in early‐phase MI, we next investigated whether PI16 attenuated LV remodeling at the late stage of MI. At 28 days after MI, compared with WT mice, PI16‐Tg mice showed higher ejection fraction and fractional shortening, together with decreased LV end‐systolic diameter and LV end‐diastolic diameter (Figure 4A). Because cardiac fibrosis and viable cardiomyocyte hypertrophy are primary pathological processes of heart remodeling after MI, we further quantified the percentage of fibrosis by Masson's trichrome staining and Sirius red staining, both of which showed markedly reduced fibrosis (from approximately 40% to 20%) in PI16‐Tg mice compared with WT mice. The size of the cardiomyocytes, as assessed by hematoxylin and eosin staining, was also decreased in PI16‐Tg mice (Figure 4B). PI16 blocked the protein levels of collagen I and MMP2 (Figure 4C) and downregulated the mRNA levels of fibrosis‐associated genes, including collagen I, MMP2, TGF‐β (transforming growth factor‐β), and CTGF (connective tissue growth factor) (Figure 4D), compared with those in WT mice. In addition, we found that the ratio of heart weight to body weight was lower in P16‐Tg mice at 28 days after MI (Figure 4E). The mRNA levels of ANP (atrial natriuretic peptide) and BNP (brain natriuretic peptide), which are markers of cardiac hypertrophy, were decreased in PI16‐Tg mice compared with WT mice (Figure 4F). These data provide evidence that PI16 overexpression alleviates adverse LV remodeling and heart failure at the sequelae stage of MI.
Figure 4. PI16 overexpression improved cardiac function and remodeling at 28 days after MI.

A, Representative M‐mode images and echocardiographic data, including EF, FS, LVDs, and LVDd, from PI16‐Tg and WT mice 28 days after being subjected to MI or sham surgery (n=5–8). B, Representative images and quantitation of Masson's trichrome, Sirius red, and hematoxylin and eosin staining from PI16‐Tg and WT mice 28 days after being subjected to MI or sham surgery (n=5–6). Scale bar, 500 μm for Masson and Sirius red staining, 50 μm for hematoxylin and eosin staining. C, Protein levels and quantitation of collagen I and MMP2 in the hearts of PI16‐Tg and WT mice 28 days after being subjected to sham surgery or MI (n=6). D, The mRNA levels of collagen I, MMP2, TGF‐β, and CTGF in P16‐Tg and WT hearts at 28 days (n=4–6). Ratios of heart weight to body weight (n=5–8) (E) and the mRNA levels of hypertrophic markers, including ANP and BNP, assessed by real‐time polymerase chain reaction (F) in PI16‐Tg mice and WT mice 28 days after being subjected to sham or MI (n=4–6). Data are presented as the mean±SEM and are representative of 3 independent experiments. One‐way ANOVA was used to assess the difference. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ANP indicates atrial natriuretic peptide; BNP, brain natriuretic peptide; CTGF, connective tissue growth factor; EF, ejection fraction; FS, fractional shortening; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; LVDd, left ventricular end‐diastolic dimension; LVDs, left ventricular end‐systolic dimension; MI, myocardial infarction; MMP2, matrix metalloproteinase 2; PI16‐Tg, peptidase inhibitor 16 transgenic mice; TGF‐β, transforming growth factor‐β; and WT, wild‐type.
Deficiency of P116 Deteriorated LV Remodeling at 28 Days After MI
We next investigated whether PI16 deletion exacerbated the outcome of MI. PI16−/− mice had impaired cardiac function at 28 days after MI, with deteriorating ejection fraction, fractional shortening, LV end‐systolic diameter, and LV end‐diastolic diameter compared with PI16+/− mice (Figure 5A). Masson's trichrome staining, Sirius red staining, and hematoxylin and eosin staining showed more collagen deposition and more hypertrophic cardiomyocytes in PI16−/− mice (Figure 5B). In addition, the protein and mRNA levels of fibrotic markers (collagen I, MMP2, TGF‐β, CTGF) were higher in the infarcted PI16−/− hearts (Figure 5C and 5D). PI16−/− mice also showed a higher heart weight to body weight (Figure 5E) and increased mRNA levels of atrial natriuretic peptide and brain natriuretic peptide (Figure 5F) than PI16+/− mice 28 days after MI. These data demonstrate that PI16 deficiency exacerbates MI‐induced LV remodeling.
Figure 5. Deficiency of P116 deteriorated LV remodeling 28 days after MI.
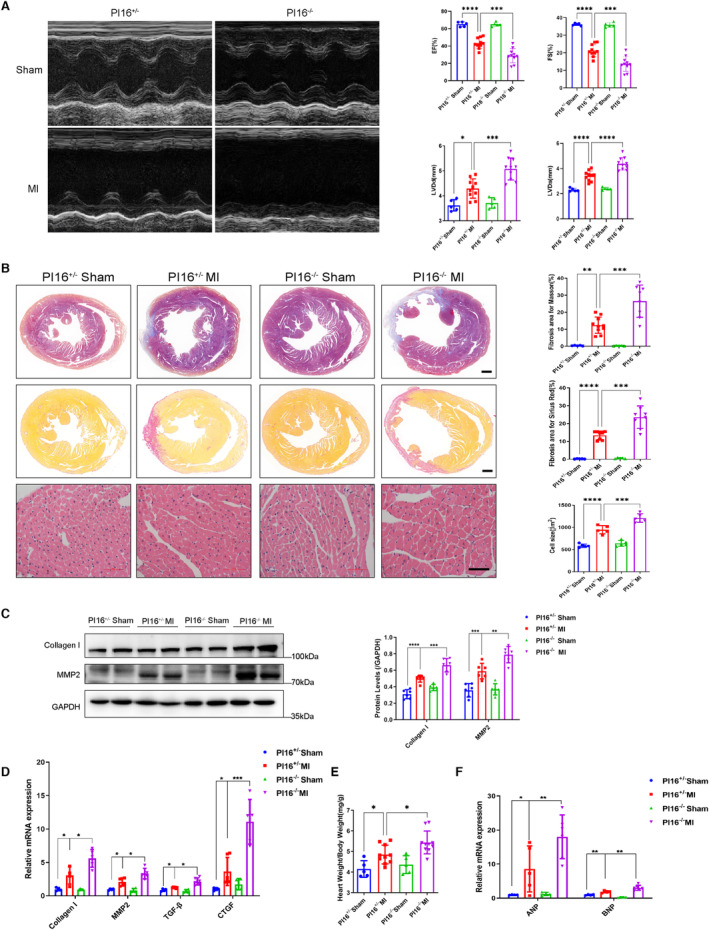
A, Representative M‐mode images and echocardiographic data, including EF, FS, LVDs, and LVDd, from PI16+/− and PI16−/− mice 28 days after being subjected to MI or sham surgery (n=5–10). B, Representative images and quantitation of Masson's trichrome, Sirius red and hematoxylin and eosin staining from PI16+/− and PI16−/− mice 28 days after being subjected to MI or sham surgery (n=5–10). Scale bar, 500 μm for Masson and Sirius red staining, 100 μm for hematoxylin and eosin staining. Protein levels and quantitation of collagen I and MMP2 (C) and the mRNA levels of collagen I, MMP2, TGF‐β, and CTGF (D) in PI16+/− nd PI16−/− hearts at 28 days (n=4–6). Ratios of heart weight to body weight (n=5–10) (E) and the mRNA levels of hypertrophic markers, including ANP and BNP, assessed by real‐time polymerase chain reaction (F) in PI16+/− and PI16−/− mice 28 days after being subjected to a sham surgery or MI (n=4–6). Data are presented as the mean±SEM and are representative of 3 independent experiments. One‐way ANOVA was used to assess the difference. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ANP indicates atrial natriuretic peptide; BNP, brain natriuretic peptide; CTGF, connective tissue growth factor; EF, ejection fraction; FS, fractional shortening; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; LV, left ventricular; LVDd, left ventricular end diastolic dimension; LVDs, left ventricular end‐systolic dimension; MI, myocardial infarction; MMP2, matrix metalloproteinase 2; PI16−/−, peptidase inhibitor 16 knockout mice; and TGF‐β, transforming growth factor‐β.
P116 Downregulated the Canonical Wnt Signaling Pathway
Growing evidence has shown that the Wnt signaling pathway plays an important role in the cell injury and heart remodeling of MI 10 , 17 ; therefore, we examined the changes of canonical Wnt/β‐catenin signaling pathway under the condition of overexpressing and knocking down PI16 in NRCMs. We measured protein levels of the key molecules of canonical Wnt signaling, including Wnt3a, FZD2 (frizzled 2), β‐catenin, and p‐β‐catenin, and found that overexpression of PI16 inhibited ligand Wnt3a, receptor FZD2, and β‐catenin levels after OGD compared with the Ad‐GFP OGD group (Figure 6A), whereas knockdown of PI16 significantly enhanced Wnt3a, FZD2, and β‐catenin levels compared with the Ad‐shctrl OGD group (Figure 6B). We also found that p‐β‐catenin, which is degraded by ubiquitination after phosphorylation, was increased in PI16‐overexpressing NRCMs and decreased in PI16‐knockdown NRCMs compared with controls after OGD (Figure 6A and 6B). To determine whether suppressing Wnt3a/β‐catenin signaling is essential for PI16 to inhibit apoptosis, we next used recombinant Wnt3a (rWnt3a) to activate the canonical Wnt signaling pathway. Western blotting showed that rWnt3a abolished the antiapoptotic effect of PI16 (Figure 6C), suggesting that PI16 protects against cardiomyocyte apoptosis by inhibiting Wnt3a/β‐catenin signaling.
Figure 6. PI16 downregulated the canonical Wnt signaling pathway.
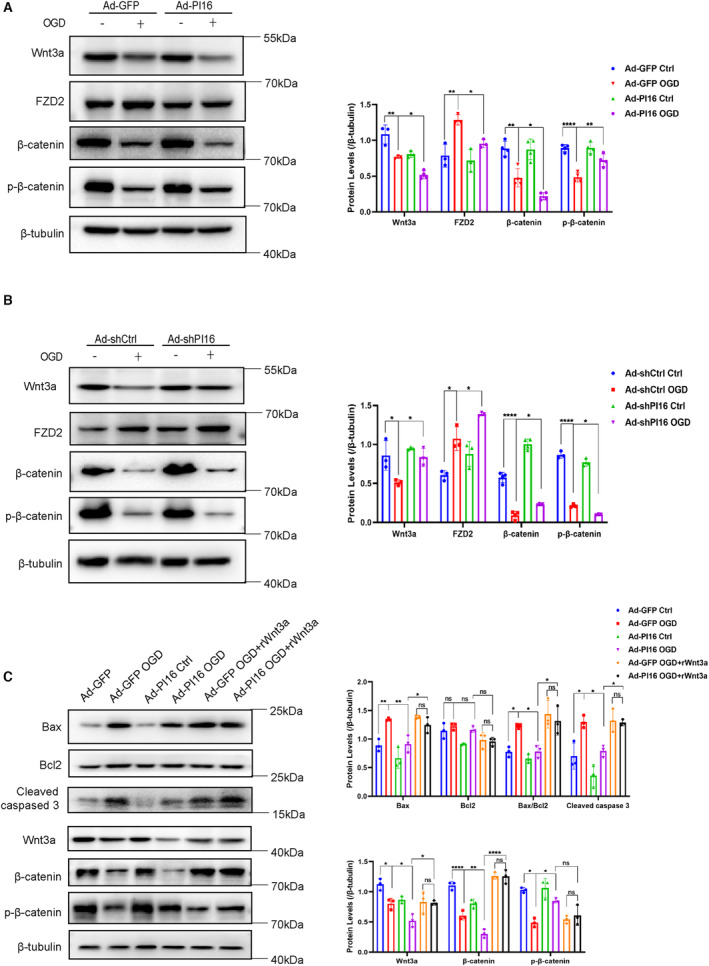
The protein levels and quantitation of Wnt3a, FZD2, β‐catenin, and p‐β‐catenin in OGD‐induced NRCMs transfected with Ad‐GFP/Ad‐PI16 (A) or Ad‐shCtrl/Ad‐shPI16 (B). C, Protein levels and quantitation of apoptosis‐associated proteins and Wnt pathway‐associated proteins in Ad‐GFP/Ad‐PI16‐transfected NRCMs treated with or without rWnt3a after OGD induction. Data are the mean±SEM. One‐way ANOVA was used to assess the difference. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. FZD2 indicates Frizzled 2; GFP, green fluorescent protein; NRCM, neonatal rat cardiomyocyte; ns, no significance; OGD, oxygen–glucose deprivation; p‐β‐catenin, phosphor‐β‐catenin; PI16, peptidase inhibitor 16; rWnt3a, recombinant Wnt3a; shCtrl, short hairpin of control; shPI16, short hairpin of peptidase inhibitor 16; and Wnt,wingless‐type MMTV integration site family.
PI16 Impacted the Wnt Signaling Pathway by Inhibiting HDAC1
We further investigated how PI16 downregulated the Wnt3a/β‐catenin pathway. We found that overexpression of PI16 decreased Wnt3a mRNA levels, whereas interference with PI16 increased Wnt3a mRNA levels after OGD (Figure S1A and S1B), which suggested that PI16 could regulate Wnt3a expression at the transcriptional level. Because epigenetic modification, especially histone acetylation modification, plays an important role in transcriptional regulation, and previous studies have shown that PI16 inhibits HDAC1 expression in Ang II (angiotensin II)‐induced neonatal rat cardiac fibroblasts, 16 we hypothesized that PI16 may play a key role in the inhibition effects of PI16 on Wnt signaling and apoptosis. To verify this hypothesis, we first examined HDAC1 levels in the hearts from PI16‐Tg and PI16−/− mice at 24 hours after MI by immunofluorescence staining, and found that HDAC1 was decreased in the PI16‐Tg mice and increased in the PI16−/− mice compared with their littermates at 24 hours after MI (Figure 7A and 7B). We next tested the protein levels of HDAC1 in the nuclei of NRCMs after OGD, and our results showed that PI16 overexpression decreased nuclear HDAC1 levels, and interference of PI16 increased nuclear HDAC1 levels in the NRCMs after OGD. Interestingly, we also found that PI16 was expressed in the nuclei of NRCMs and was upregulated after OGD induction, suggesting that PI16 may regulate HDAC1 by entering the nucleus (Figure 7C and 7D). Then we assessed the mRNA levels of HDAC1, and found that PI16 decreased HDAC1 mRNA levels in the PI16‐Tg mice after MI and in the NRCMs transfected with Ad‐PI16 after OGD, contrary to the results in PI16−/− mice and NRCMs transfected with Ad‐shPI16 (Figure 7E). These results indicated that PI16 could downregulate HDAC1 expression at the transcriptional level by nuclear translocation.
Figure 7. PI16 impacted Wnt signaling pathways by inhibiting HDAC1.
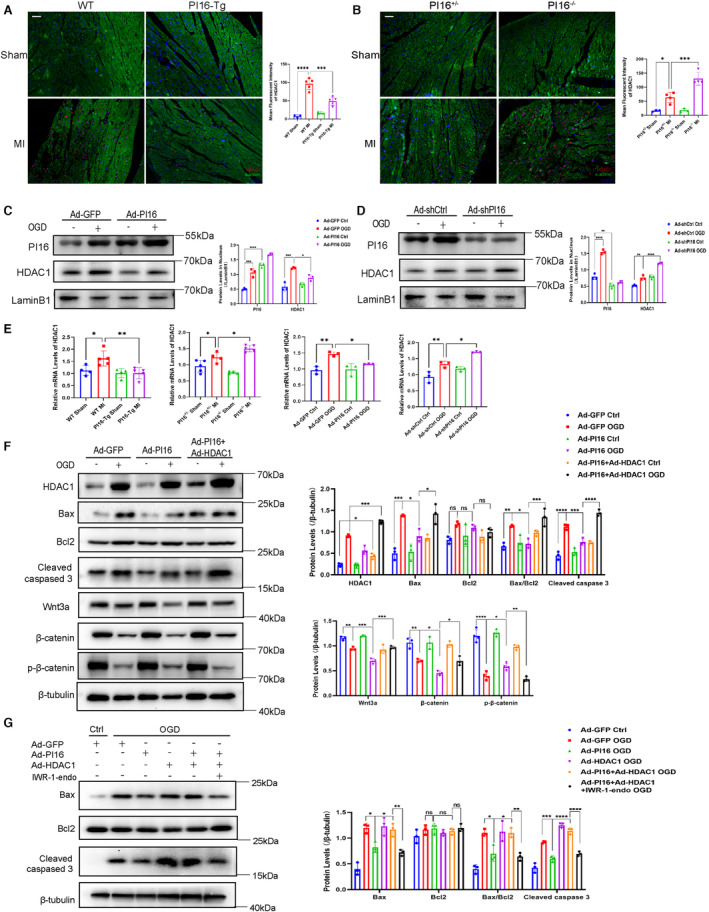
Representative immunofluorescent images of HDAC1 in the PI16‐Tg/WT (A) and PI16+/−/PI16−/− (B) hearts 24 hours after being subjected to MI or sham surgery (n=3–4). Scale bar, 50 μm. The nuclear protein levels and quantitation of HDAC1 and PI16 in the Ad‐GFP/Ad‐PI16‐transfected (C) and Ad‐shCtrl/Ad‐shPI16‐transfected (D) NRCMs after OGD. E, The mRNA levels of HDAC1 in the PI16‐Tg/WT /PI16+/−/PI16−/− hearts (n=4–5) after MI and NRCMs transfected with Ad‐PI16/Ad‐shPI16 after OGD (n=3). F, Protein levels and quantitation of apoptosis‐associated proteins and Wnt pathway‐associated proteins in NRCMs transfected with Ad‐GFP/Ad‐PI16/Ad‐HDAC1 after OGD. G, Protein levels and quantitation of apoptosis‐associated proteins in Ad‐GFP/Ad‐PI16‐transfected NRCMs treated with or without IWR‐1‐endo after OGD. Data are presented as the mean±SEM and are representative of 3 independent experiments. One‐way ANOVA was used to assess the difference. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, FZD2 indicates Frizzled 2; GFP, green fluorescent protein; HDAC1, class I histone deacetylase; IWR‐1‐endo, the inhibitor of Wnt signaling; MI, myocardial infarction; NRCM, neonatal rat cardiomyocyte; ns, no significance; OGD, oxygen–glucose deprivation; p‐β‐catenin, phosphor‐β‐catenin; PI16, peptidase inhibitor 16; PI16−/−, peptidase inhibitor 16 knockout mice; PI16‐Tg, peptidase inhibitor 16 transgenic mice; shCtrl, short hairpin of control; shPI16, short hairpin of peptidase inhibitor 16; Wnt,wingless‐type MMTV integration site family; and WT, wild‐type.
To determine whether HDAC1 was involved in the regulation of apoptosis by PI16, we cotransfected NRCMs with Ad‐PI16 and Ad‐HDAC1, and found that HDAC1 abolished the antiapoptotic effect of PI16 and reversed the weak Wnt3a/β‐catenin signaling in PI16‐overexpressing NRCMs after OGD (Figure 7F), indicating that PI16 improved apoptosis by reducing HDAC1 levels. Finally, to confirm whether the downregulation of HDAC1 levels by PI16 is dependent on the canonical Wnt signaling pathway to alleviate cardiomyocyte apoptosis, we used IWR‐1‐endo, an antagonist of the Wnt/β‐catenin pathway, to inhibit Wnt signaling transduction. Western blotting showed that IWR‐1‐endo reversed the aggravation of apoptosis induced by HDAC1 overexpression after OGD (Figure 7G). Together, these results demonstrate that PI16 plays an antiapoptotic role after myocardial infarction by inhibiting the HDAC1‐Wnt3a‐β‐catenin signaling axis (Figure 8).
Figure 8. Schematic illustration of PI16 protecting from apoptosis and left ventricular remodeling by inhibiting the HDAC1‐Wnt‐β‐catenin axis.
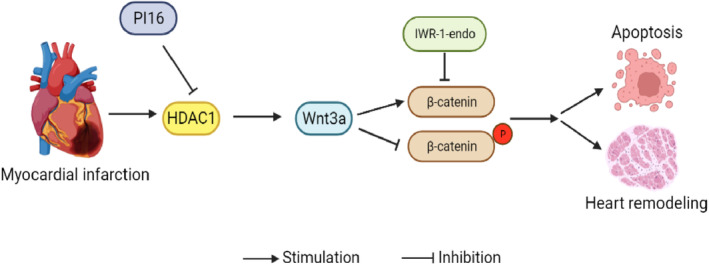
The expression of HDAC1 in cardiomyocytes is upregulated after MI, which enhances canonical Wnt/β‐catenin pathway by increasing Wnt3a expression, leading to cardiomyocyte apoptosis and cardiac remodeling. PI16 inhibits the expression of HDAC1 and thus suppresses the activation of the downstream Wnt/β‐catenin pathway, thereby protecting against injury and remodeling after MI. HDAC1 indicates class I histone deacetylase; IWR‐1‐endo, the inhibitor of Wnt signaling; MI, myocardial infarction; PI16, peptidase inhibitor 16; and Wnt,wingless‐type MMTV integration site family.
DISCUSSION
In this study, we first demonstrated that PI16 protected against the loss of cardiomyocytes and LV remodeling after MI. The beneficial role of PI16 is dependent on inhibiting HDAC1 and its downstream Wnt signaling pathway.
Only weak PI16 expression is observed in the adult murine heart and in the isolated NRCMs; however, PI16 is strongly increased under conditions of cardiac stress. PI16 is markedly upregulated in β1‐adrenergic receptor transgenic mice and the TAC‐induced heart failure model. 6 PI16 is also upregulated in Ang II‐induced hypertrophic hearts. 16 In our study, we detected PI16 expression in murine hearts in the period of MI from 1 day to 28 days, and found that PI16 was elevated throughout the course of early and late MI. PI16 began to rise at 1 day after MI and peaked at 7 days after MI, which was consistent with our observation that PI16 was upregulated in the plasma of patients with acute MI within 7 days. Although PI16 is mainly expressed by cardiac fibroblasts under physiological conditions, 9 we found that PI16 was colocalized with cardiomyocytes in the infarct zone and was dramatically increased in OGD‐induced NRCMs independent of neonatal rat cardiac fibroblasts, indicating that PI16 is highly activated and secreted by cardiomyocytes after loss of perfusion in response to MI.
The pathological process of MI is divided into an inflammatory phase, characterized by cardiac cell death and leukocyte infiltration, and a reparative phase consisting of myofibroblast proliferation and scar formation. 18 Knockdown of PI16 has been recently reported to promote sorafenib‐induced apoptosis of hepatocellular carcinoma cells and suppress tumor growth via the p38/MAPK (mitogen‐activated protein kinase) pathway. 19 Secreted PI16 has been shown to inhibit the activation of chemerin 9 and cathepsin K 20 and restrain subsequent proinflammatory pathways. The roles of Tregs in controlling immune homeostasis and repairing tissue after MI have been well established. 21 , 22 , 23 , 24 , 25 PI16 has been identified as a biomarker of functionally distinct human Treg subsets with high FOXP3 (forkhead box protein 3) levels, 7 which showed enhanced migration toward sites of inflammation and regulated the inflammatory response. 26 All of these studies imply the potential protective role of PI16 in early MI. In our study, we used gain‐ and loss‐of‐function experiments to prove that PI16 decreased the loss of myocardium after MI by inhibiting cardiomyocyte apoptosis. During the reparative phase of MI, PI16 significantly improved the structure and function of the heart, which may be attributed to the mild sequelae caused by the early antiapoptotic effect of PI16 and to the inhibitory effect of PI16 on myofibroblast activation. 16 We also found that the expression of MMP2 was decreased at 28 days after MI. One study reported that PI16 could suppress MMP2 activity by binding to its exposed peptide loop in human coronary artery endothelial cells. 27 Increasing evidence has shown that inhibition of MMP2 improves myocardial damage and cardiac remodeling after MI, 28 , 29 , 30 , 31 which agrees with our research.
The level of Wnt signaling is typically low in adult hearts under physiological conditions, but this pathway is activated and profoundly involved in the process of MI. 10 Multiple Wnt‐associated ligands and receptors are upregulated after heart injury, especially Wnt3a, which is recognized as an activator of canonical Wnt signaling, and FZD2, which is required for Wnt3a‐dependent accumulation of β‐catenin 32 , 33 and shows high levels in the heart. 34 , 35 , 36 In our research, PI16 overexpression markedly inhibited β‐catenin levels, and knockdown of PI16 increased β‐catenin levels in OGD‐induced NRCMs. The downregulation of β‐catenin was due to the inhibition of both Wnt3a and FZD2 levels by PI16. Hilde et al demonstrated that Wnt3a increased the transcriptional activity of FZD2, which was abolished by a Wnt3a homologous antagonist. 37 Based on the abolishment of the antiapoptotic phenotype by recombinant Wnt3a and the regulation of FZD2 via Wnt3a, we believe that PI16 suppresses the Wnt/β‐catenin pathway mainly by inhibiting Wnt3a expression.
Mounting evidence has proven that the Wnt/β‐catenin pathway exacerbates heart remodeling after MI 17 , 38 , 39 , 40 ; however, the role of canonical Wnt/β‐catenin signaling in acute MI and cardiomyocyte apoptosis is controversial. Oikonomopoulos et al found that injection of recombinant Wnt3a in murine hearts after MI led to impaired cardiac regeneration and increased cardiomyocyte death by inhibiting renewal of cardiac side population cells through IGFBP3 (insulin‐like growth factor binding protein 3). 41 Using a homologous peptide fragment of Wnt3a to block Frizzled signaling decreased infarct expansion and ameliorated heart remodeling. 37 In vitro, Wnt3a has been shown to exacerbate cardiomyocyte apoptosis induced by hypoxia 42 , 43 and increase caspase activity in NRCMs undergoing hypoxia/reoxygenation injury. 44 Furthermore, cardiomyocyte‐specific deletion of β‐catenin improved cardiac function and LV remodeling after MI by promoting cardiac progenitor cell proliferation. 12 Short‐term pharmacologic inhibition of the Wnt/β‐catenin pathway after MI reduced cardiomyocyte death and augmented cardiac repair. 45 Likewise, administration of the small molecule Wnt modulator ICG‐001, which inhibits β‐catenin‐mediated transcription, increased the ejection fraction after MI. 13 In addition, suppression of FZD2 attenuated cell apoptosis induced by hypoxia/reoxygenation, 46 , 47 and multiple secreted Frizzled‐related proteins, which competitively inhibit Wnt signaling, showed beneficial effects on infarct size and cardiac function after MI. 48 , 49 All of these studies confirm the deleterious effect of the Wnt3a/β‐catenin pathway on MI, and inhibition of canonical Wnt signaling could improve cardiac injury and remodeling after MI. However, a few studies have also demonstrated that activating the Wnt3/β‐catenin pathway inhibits cardiomyocyte apoptosis and protects against myocardial injury after MI. 50 , 51 , 52 These contradictory results may be partly due to the different models and diverse experimental times used for in vivo and in vitro studies, because Wnt signaling is a highly time‐ and context‐dependent pathway. 41 In our study, we found that PI16 inhibited OGD‐induced NRCM apoptosis by downregulating Wnt3a/β‐catenin signaling, and that recombinant Wnt3a reversed the inhibitory effect of PI16 on cardiomyocyte apoptosis, suggesting that PI16 alleviated myocardial apoptosis by suppressing the Wnt3a/β‐catenin pathway.
Because PI16 downregulated the transcriptional level of Wnt3a, we focused on the epigenetic modification of PI16. One study proved that PI16 attenuated Ang II‐induced cardiac fibrosis by targeting HDAC1. 16 In our study, HDCA1 was significantly increased in the infarct zone at 24 hours after MI and in OGD‐induced NRCMs, which was blunted by PI16. We further found that HDAC1 mRNA was also downregulated by PI16 overexpression, suggesting PI16 had an effect on HDAC1 transcription. Interestingly, although PI16 has been identified as a secreted protein, we found that PI16 can also be detected in the nucleus, especially after OGD. Secreted proteins have been extensively reported to enter the nucleus and regulate gene expression. 53 , 54 HDAC1 has also been reported to be regulated by GKN1 (gastrokine‐1), an antiamyloidogenic protein expressed and secreted by foveolar epithelial cells of the gastric mucosa, which has been reported to downregulate HDAC1 level by epigenetic modification. 55 GKN1 could induce Tip60 expression, which acetylated DNMT1 (DNA methyltransferase) and reduced the expression of HDAC1 as well as DNMT1, EZH2 (zeste homolog 2), and methyl histone H3. 56 Therefore, we hypothesized that PI16 may regulate HDAC1 level via entering the nucleus and interacting with some nuclear factors that could influence HDAC1 transcription, such as transcriptional factors, histones, or enzymes involved in epigenetic modification. It is also possible that PI16 may interact with some microRNAs and promote degradation of HDAC1 mRNA. However, more effort is needed to study the direct downstream target and mechanism of PI16 to regulate HDAC1 transcription.
Increasing studies have shown the beneficial role of HDAC1 inhibition on myocardium injury. Knockdown of HDAC1 in H9C2 cells protected against hypoxia‐induced apoptosis and swelling. 57 Reducing HDAC1 attenuated sepsis‐induced heart injury in mice. 58 HDAC1‐induced PFKM (phosphofructokinase, muscle) transcriptional repression aggravated DOX (doxorubicin)‐mediated cell apoptosis. 59 High levels of glucose induced apoptosis in cardiomyocytes by HDAC1 transcriptional inhibition of insulin‐like growth factor 1. 60 HDAC1 inhibition also improved heart remodeling and LV function after MI. 61 , 62 In our study, HDAC1 overexpression upregulated the protein levels of Bax and cleaved caspase 3 compared with those in the OGD group, indicating the proapoptotic effect of HDAC1 in OGD‐induced NRCMs. In addition, HDAC1 overexpression abolished the inhibition of apoptosis and Wnt3a/β‐catenin signaling by PI16. Thus, PI16 attenuates cardiomyocyte apoptosis by inhibiting HDAC1 and the downstream Wnt signaling pathway.
The mechanism by which HDAC1 regulates WntA/β‐catenin signaling remains unclear. One study demonstrated that a class I HDAC inhibitor reduced canonical Wnt/β‐catenin signaling and improved aortic valvular interstitial cell calcification. 63 HDAC1 significantly increased the expression of Wnt and β‐catenin in fibroblasts from patients with ankylosing spondylitis. 64 HDAC1, as a deacetylase, in addition to regulating histone deacetylation modification, can also deacetylate other proteins, such as p53. HDAC1 reduces p53 acetylation, which results in instability and degradation of p53. 65 , 66 , 67 One study proved that a PI16‐mediated decrease in HDAC1 upregulated p53 and acetylated p53 levels in Ang II‐induced fibroblasts. 16 Another recent study demonstrated that p53 induced Wnt3 transcription followed by activation of the Wnt/β‐catenin pathway, 68 which may provide new insight into HDAC1 regulation of the Wnt/β‐catenin signaling pathway. Our study showed that the proapoptotic effect of HDAC1 overexpression was reversed by Wnt inhibitor in OGD‐induced NRCMs. However, more studies are needed to determine the specific mechanism by which PI16 regulates the HDAC1 and Wnt/β‐catenin pathways.
In conclusion, our research identifies PI16 as a protective factor for myocardial apoptosis and LV remodeling by negatively regulating the HDAC1‐Wnt‐β‐catenin axis. PI16, as an endogenous HDAC1 inhibitor, may provide a promising strategy for MI.
Sources of Funding
This work was supported by grants from the National Key Research and Development Program of China (number 2019YFA0210100) and National Natural Science Foundation of China (number 82170425).
Disclosures
None.
Supporting information
Table S1
Figure S1
Acknowledgments
L.W. and A.D. performed the vitro and in vivo experiments, analyzed data, and wrote the article. Y.L. and Y.Z. participated in the in vitro study. M.Q. and H.S. participated in the animal experiments. Z.S. collected the samples of patients. W.S. and X.K. designed and supervised the study. All authors read and approved the final article.
This article was sent to Rebecca D. Levit, MD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.028866
For Sources of Funding and Disclosures, see page 16.
Contributor Information
Wei Sun, Email: weisun7919@njmu.edu.cn.
Xiangqing Kong, Email: kongxq_njmu@outlook.com.
REFERENCES
- 1. Zhang J, Bolli R, Garry DJ, Marban E, Menasche P, Zimmermann WH, Kamp TJ, Wu JC, Dzau VJ. Basic and translational research in cardiac repair and regeneration: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2021;78:2092–2105. doi: 10.1016/j.jacc.2021.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frantz S, Hundertmark MJ, Schulz‐Menger J, Bengel FM, Bauersachs J. Left ventricular remodelling post‐myocardial infarction: pathophysiology, imaging, and novel therapies. Eur Heart J. 2022;43:2549–2561. doi: 10.1093/eurheartj/ehac223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garde SV, Basrur VS, Li L, Finkelman MA, Krishan A, Wellham L, Ben‐Josef E, Haddad M, Taylor JD, Porter AT, et al. Prostate secretory protein (PSP94) suppresses the growth of androgen‐independent prostate cancer cell line (PC3) and xenografts by inducing apoptosis. Prostate. 1999;38:118–125. doi: [DOI] [PubMed] [Google Scholar]
- 5. Reeves JR, Xuan JW, Arfanis K, Morin C, Garde SV, Ruiz MT, Wisniewski J, Panchal C, Tanner JE. Identification, purification and characterization of a novel human blood protein with binding affinity for prostate secretory protein of 94 amino acids. Biochem J. 2005;385:105–114. doi: 10.1042/BJ20040290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frost RJ, Engelhardt S. A secretion trap screen in yeast identifies protease inhibitor 16 as a novel antihypertrophic protein secreted from the heart. Circulation. 2007;116:1768–1775. doi: 10.1161/CIRCULATIONAHA.107.696468 [DOI] [PubMed] [Google Scholar]
- 7. Hope CM, Welch J, Mohandas A, Pederson S, Hill D, Gundsambuu B, Eastaff‐Leung N, Grosse R, Bresatz S, Ang G, et al. Peptidase inhibitor 16 identifies a human regulatory T‐cell subset with reduced FOXP3 expression over the first year of recent onset type 1 diabetes. Eur J Immunol. 2019;49:1235–1250. doi: 10.1002/eji.201948094 [DOI] [PubMed] [Google Scholar]
- 8. Becker E, Dedden M, Gall C, Wiendl M, Ekici AB, Schulz‐Kuhnt A, Schweda A, Voskens C, Hegazy A, Vitali F, et al. Residual homing of α4β7‐expressing β1+PI16+ regulatory T cells with potent suppressive activity correlates with exposure‐efficacy of vedolizumab. Gut. 2022;71:1551–1566. doi: 10.1136/gutjnl-2021-324868 [DOI] [PubMed] [Google Scholar]
- 9. Regn M, Laggerbauer B, Jentzsch C, Ramanujam D, Ahles A, Sichler S, Calzada‐Wack J, Koenen RR, Braun A, Nieswandt B, et al. Peptidase inhibitor 16 is a membrane‐tethered regulator of chemerin processing in the myocardium. J Mol Cell Cardiol. 2016;99:57–64. doi: 10.1016/j.yjmcc.2016.08.010 [DOI] [PubMed] [Google Scholar]
- 10. Foulquier S, Daskalopoulos EP, Lluri G, Hermans KCM, Deb A, Blankesteijn WM. WNT signaling in cardiac and vascular disease. Pharmacol Rev. 2018;70:68–141. doi: 10.1124/pr.117.013896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meyer IS, Leuschner F. The role of Wnt signaling in the healing myocardium: a focus on cell specificity. Basic Res Cardiol. 2018;113:44. doi: 10.1007/s00395-018-0705-y [DOI] [PubMed] [Google Scholar]
- 12. Zelarayan LC, Noack C, Sekkali B, Kmecova J, Gehrke C, Renger A, Zafiriou MP, Van Der Nagel R, Dietz R, De Windt LJ, et al. Beta‐catenin downregulation attenuates ischemic cardiac remodeling through enhanced resident precursor cell differentiation. Proc Natl Acad Sci USA. 2008;105:19762–19767. doi: 10.1073/pnas.0808393105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sasaki T, Hwang H, Nguyen C, Kloner RA, Kahn M. The small molecule Wnt signaling modulator ICG‐001 improves contractile function in chronically infarcted rat myocardium. PLoS ONE. 2013;8:e75010. doi: 10.1371/journal.pone.0075010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee TM, Lin MS, Chang NC. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am J Physiol Heart Circ Physiol. 2007;293:H968–H977. doi: 10.1152/ajpheart.00891.2006 [DOI] [PubMed] [Google Scholar]
- 15. Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deng M, Yang S, Ji Y, Lu Y, Qiu M, Sheng Y, Sun W, Kong X. Overexpression of peptidase inhibitor 16 attenuates angiotensin II‐induced cardiac fibrosis via regulating HDAC1 of cardiac fibroblasts. J Cell Mol Med. 2020;24:5249–5259. doi: 10.1111/jcmm.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ozhan G, Weidinger G. Wnt/beta‐catenin signaling in heart regeneration. Cell Regen. 2015;4:3. doi: 10.1186/s13619-015-0017-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang P, Jiang Z, Liu X, Yu K, Wang C, Li H, Zhong L. PI16 attenuates response to sorafenib and represents a predictive biomarker in hepatocellular carcinoma. Cancer Med. 2020;9:6972–6983. doi: 10.1002/cam4.3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lupsa N, Ersek B, Horvath A, Bencsik A, Lajko E, Sillo P, Oszvald A, Wiener Z, Remenyi P, Mikala G, et al. Skin‐homing CD8(+) T cells preferentially express GPI‐anchored peptidase inhibitor 16, an inhibitor of cathepsin K. Eur J Immunol. 2018;48:1944–1957. doi: 10.1002/eji.201847552 [DOI] [PubMed] [Google Scholar]
- 21. Weiss E, Ramos GC, Delgobo M. Myocardial‐Treg crosstalk: how to tame a wolf. Front Immunol. 2022;13:914033. doi: 10.3389/fimmu.2022.914033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blanco‐Dominguez R, De La Fuente H, Rodriguez C, Martin‐Aguado L, Sanchez‐Diaz R, Jimenez‐Alejandre R, Rodriguez‐Arabaolaza I, Curtabbi A, Garcia‐Guimaraes MM, Vera A, et al. CD69 expression on regulatory T cells protects from immune damage after myocardial infarction. J Clin Invest. 2022;132:e152418. doi: 10.1172/JCI152418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng G, Bajpai G, Ma P, Koenig A, Bredemeyer A, Lokshina I, Lai L, Forster I, Leuschner F, Kreisel D, et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T cells. Circulation. 2022;145:765–782. doi: 10.1161/CIRCULATIONAHA.121.055888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia N, Lu Y, Gu M, Li N, Liu M, Jiao J, Zhu Z, Li J, Li D, Tang T, et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation. 2020;142:1956–1973. doi: 10.1161/CIRCULATIONAHA.120.046789 [DOI] [PubMed] [Google Scholar]
- 25. Sun K, Li YY, Jin J. A double‐edged sword of immuno‐microenvironment in cardiac homeostasis and injury repair. Signal Transduct Target Ther. 2021;6:79. doi: 10.1038/s41392-020-00455-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nicholson IC, Mavrangelos C, Bird DR, Bresatz‐Atkins S, Eastaff‐Leung NG, Grose RH, Gundsambuu B, Hill D, Millard DJ, Sadlon TJ, et al. PI16 is expressed by a subset of human memory Treg with enhanced migration to CCL17 and CCL20. Cell Immunol. 2012;275:12–18. doi: 10.1016/j.cellimm.2012.04.002 [DOI] [PubMed] [Google Scholar]
- 27. Hazell GG, Peachey AM, Teasdale JE, Sala‐Newby GB, Angelini GD, Newby AC, White SJ. PI16 is a shear stress and inflammation‐regulated inhibitor of MMP2. Sci Rep. 2016;6:39553. doi: 10.1038/srep39553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hughes BG, Schulz R. Targeting MMP‐2 to treat ischemic heart injury. Basic Res Cardiol. 2014;109:424. doi: 10.1007/s00395-014-0424-y [DOI] [PubMed] [Google Scholar]
- 29. Roczkowsky A, Chan BYH, Lee TYT, Mahmud Z, Hartley B, Julien O, Armanious G, Young HS, Schulz R. Myocardial MMP‐2 contributes to SERCA2a proteolysis during cardiac ischaemia‐reperfusion injury. Cardiovasc Res. 2020;116:1021–1031. doi: 10.1093/cvr/cvz207 [DOI] [PubMed] [Google Scholar]
- 30. Lindsey ML. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat Rev Cardiol. 2018;15:471–479. doi: 10.1038/s41569-018-0022-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li T, Wei X, Evans CF, Sanchez PG, Li S, Wu ZJ, Griffith BP. Left ventricular unloading after acute myocardial infarction reduces MMP/JNK associated apoptosis and promotes FAK cell‐survival signaling. Ann Thorac Surg. 2016;102:1919–1924. doi: 10.1016/j.athoracsur.2016.05.007 [DOI] [PubMed] [Google Scholar]
- 32. Sato A, Yamamoto H, Sakane H, Koyama H, Kikuchi A. Wnt5a regulates distinct signalling pathways by binding to Frizzled2. EMBO J. 2010;29:41–54. doi: 10.1038/emboj.2009.322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dijksterhuis JP, Baljinnyam B, Stanger K, Sercan HO, Ji Y, Andres O, Rubin JS, Hannoush RN, Schulte G. Systematic mapping of WNT‐FZD protein interactions reveals functional selectivity by distinct WNT‐FZD pairs. J Biol Chem. 2015;290:6789–6798. doi: 10.1074/jbc.M114.612648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barandon L, Couffinhal T, Ezan J, Dufourcq P, Costet P, Alzieu P, Leroux L, Moreau C, Dare D, Duplaa C. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation. 2003;108:2282–2289. doi: 10.1161/01.CIR.0000093186.22847.4C [DOI] [PubMed] [Google Scholar]
- 35. Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial‐to‐mesenchymal transition. Dis Model Mech. 2011;4:469–483. doi: 10.1242/dmm.006510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mizutani M, Wu JC, Nusse R. Fibrosis of the neonatal mouse heart after cryoinjury is accompanied by Wnt signaling activation and epicardial‐to‐mesenchymal transition. J Am Heart Assoc. 2016;5:e002457. doi: 10.1161/JAHA.115.002457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Laeremans H, Hackeng TM, Van Zandvoort MA, Thijssen VL, Janssen BJ, Ottenheijm HC, Smits JF, Blankesteijn WM. Blocking of frizzled signaling with a homologous peptide fragment of wnt3a/wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation. 2011;124:1626–1635. doi: 10.1161/CIRCULATIONAHA.110.976969 [DOI] [PubMed] [Google Scholar]
- 38. Haybar H, Khodadi E, Shahrabi S. Wnt/beta‐catenin in ischemic myocardium: interactions and signaling pathways as a therapeutic target. Heart Fail Rev. 2019;24:411–419. doi: 10.1007/s10741-018-9759-z [DOI] [PubMed] [Google Scholar]
- 39. Hahn JY, Cho HJ, Bae JW, Yuk HS, Kim KI, Park KW, Koo BK, Chae IH, Shin CS, Oh BH, et al. Beta‐catenin overexpression reduces myocardial infarct size through differential effects on cardiomyocytes and cardiac fibroblasts. J Biol Chem. 2006;281:30979–30989. doi: 10.1074/jbc.M603916200 [DOI] [PubMed] [Google Scholar]
- 40. Liu Y, Neogi A, Mani A. The role of Wnt signalling in development of coronary artery disease and its risk factors. Open Biol. 2020;10:200128. doi: 10.1098/rsob.200128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oikonomopoulos A, Sereti KI, Conyers F, Bauer M, Liao A, Guan J, Crapps D, Han JK, Dong H, Bayomy AF, et al. Wnt signaling exerts an antiproliferative effect on adult cardiac progenitor cells through IGFBP3. Circ Res. 2011;109:1363–1374. doi: 10.1161/CIRCRESAHA.111.250282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Y, Du L, Cheng S, Guo J, Zhu S, Wang Y, Gao H. Hypoxia exacerbates cardiomyocyte injury via upregulation of Wnt3a and inhibition of Sirt3. Cytokine. 2020;136:155237. doi: 10.1016/j.cyto.2020.155237 [DOI] [PubMed] [Google Scholar]
- 43. Xu L, Wang F. LINC00936 exacerbated myocardial infarction progression via miR‐4795‐3p/Wnt3a signaling pathway based on biological and imaging methods. Perfusion. 2023;38:706–716. doi: 10.1177/02676591221076788 [DOI] [PubMed] [Google Scholar]
- 44. Mirotsou M, Zhang Z, Deb A, Zhang L, Gnecchi M, Noiseux N, Mu H, Pachori A, Dzau V. Secreted frizzled related protein 2 (Sfrp2) is the key Akt‐mesenchymal stem cell‐released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sci USA. 2007;104:1643–1648. doi: 10.1073/pnas.0610024104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bastakoty D, Saraswati S, Joshi P, Atkinson J, Feoktistov I, Liu J, Harris JL, Young PP. Temporary, systemic inhibition of the WNT/beta‐catenin pathway promotes regenerative cardiac repair following myocardial infarct. Cell Stem Cells Regen Med. 2016;2:10. doi: 10.16966/2472-6990.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou SS, He F, Chen AH, Hao PY, Song XD. Suppression of rat Frizzled‐2 attenuates hypoxia/reoxygenation‐induced Ca2+ accumulation in rat H9c2 cells. Exp Cell Res. 2012;318:1480–1491. doi: 10.1016/j.yexcr.2012.03.030 [DOI] [PubMed] [Google Scholar]
- 47. Hu X, Zhou C, He G, Cheng Y, Pan M, Gao Y. Inhibition of Frizzled‐2 by small interfering RNA protects rat hepatic BRL‐3A cells against cytotoxicity and apoptosis induced by hypoxia/reoxygenation. Gastroenterol Hepatol. 2020;43:107–116. doi: 10.1016/j.gastrohep.2019.02.006 [DOI] [PubMed] [Google Scholar]
- 48. Barandon L, Casassus F, Leroux L, Moreau C, Allieres C, Lamaziere JM, Dufourcq P, Couffinhal T, Duplaa C. Secreted frizzled‐related protein‐1 improves postinfarction scar formation through a modulation of inflammatory response. Arterioscler Thromb Vasc Biol. 2011;31:e80–e87. doi: 10.1161/ATVBAHA.111.232280 [DOI] [PubMed] [Google Scholar]
- 49. He W, Zhang L, Ni A, Zhang Z, Mirotsou M, Mao L, Pratt RE, Dzau VJ. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci USA. 2010;107:21110–21115. doi: 10.1073/pnas.1004708107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shen J, Li Y, Jiao Y, Wang J, Hou X, Su Y, Liu B, Liu H, Sun Z, Xi Q, et al. Wnt 3a protects myocardial injury in elderly acute myocardial infarction by inhibiting serum cystatin C/ROS‐induced mitochondrial damage. Front Physiol. 2022;13:950960. doi: 10.3389/fphys.2022.950960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang G, Ding L, Sun G, Liu Z, Ou W, Wang B, Sun Y. LncRNA AZIN1‐AS1 ameliorates myocardial ischemia‐reperfusion injury by targeting miR‐6838‐5p/WNT3A axis to activate WNT‐beta/catenin signaling pathway. In Vitro Cell Dev Biol Anim. 2022;58:54–68. doi: 10.1007/s11626-022-00646-1 [DOI] [PubMed] [Google Scholar]
- 52. Guo X, Gu X, Hareshwaree S, Rong X, Li L, Chu M. Induced pluripotent stem cell‐conditional medium inhibits H9C2 cardiomyocytes apoptosis via autophagy flux and Wnt/beta‐catenin pathway. J Cell Mol Med. 2019;23:4358–4374. doi: 10.1111/jcmm.14327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baxter RC. Nuclear actions of insulin‐like growth factor binding protein‐3. Gene. 2015;569:7–13. doi: 10.1016/j.gene.2015.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lu J, Wu T, Zhang B, Liu S, Song W, Qiao J, Ruan H. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun Signal. 2021;19:60. doi: 10.1186/s12964-021-00741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Overstreet AC, Grayson BE, Boger A, Bakke D, Carmody EM, Bales CE, Paski SC, Murphy SF, Dethlefs CR, Shannon KJ, et al. Gastrokine‐1, an anti‐amyloidogenic protein secreted by the stomach, regulates diet‐induced obesity. Sci Rep. 2021;11:9477. doi: 10.1038/s41598-021-88928-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoon JH, Choi YJ, Choi WS, Ashktorab H, Smoot DT, Nam SW, Lee JY, Park WS. GKN1‐miR‐185‐DNMT1 axis suppresses gastric carcinogenesis through regulation of epigenetic alteration and cell cycle. Clin Cancer Res. 2013;19:4599–4610. doi: 10.1158/1078-0432.CCR-12-3675 [DOI] [PubMed] [Google Scholar]
- 57. Li Y, Zhang Z, Zhou X, Li R, Cheng Y, Shang B, Han Y, Liu B, Xie X. Histone deacetylase 1 inhibition protects against hypoxia‐induced swelling in H9c2 cardiomyocytes through regulating cell stiffness. Circ J. 2017;82:192–202. doi: 10.1253/circj.CJ-17-0022 [DOI] [PubMed] [Google Scholar]
- 58. Nong R, Qin C, Lin Q, Lu Y, Li J. Down‐regulated HDAC1 and up‐regulated microRNA‐124‐5p recover myocardial damage of septic mice. Bioengineered. 2022;13:7168–7180. doi: 10.1080/21655979.2022.2034583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou M, Sun X, Wang C, Wang F, Fang C, Hu Z. PFKM inhibits doxorubicin‐induced cardiotoxicity by enhancing oxidative phosphorylation and glycolysis. Sci Rep. 2022;12:11684. doi: 10.1038/s41598-022-15743-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu XY, Geng YJ, Liang JL, Lin QX, Lin SG, Zhang S, Li Y. High levels of glucose induce apoptosis in cardiomyocyte via epigenetic regulation of the insulin‐like growth factor receptor. Exp Cell Res. 2010;316:2903–2909. doi: 10.1016/j.yexcr.2010.07.004 [DOI] [PubMed] [Google Scholar]
- 61. Mani SK, Kern CB, Kimbrough D, Addy B, Kasiganesan H, Rivers WT, Patel RK, Chou JC, Spinale FG, Mukherjee R, et al. Inhibition of class I histone deacetylase activity represses matrix metalloproteinase‐2 and ‐9 expression and preserves LV function postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2015;308:H1391–H1401. doi: 10.1152/ajpheart.00390.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhou S, Liu P, Zhang G, Cheng Z, Wang S, Zhao J. Histone deacetylase 1 depletion alleviates coronary heart disease via the microRNA‐182‐mediated transforming growth factor β/Smad signaling pathway. J Cardiovasc Pharmacol. 2022;79:815–826. doi: 10.1097/FJC.0000000000001260 [DOI] [PubMed] [Google Scholar]
- 63. Li SJ, Kao YH, Chung CC, Cheng WL, Chen YJ. HDAC I inhibitor regulates RUNX2 transactivation through canonical and non‐canonical Wnt signaling in aortic valvular interstitial cells. Am J Transl Res. 2019;11:744–754. [PMC free article] [PubMed] [Google Scholar]
- 64. Zeng Y, He R, Liu Y, Luo T, Li Q, He Y, Fang M, Wang T. HDAC1 regulates inflammation and osteogenic differentiation of ankylosing spondylitis fibroblasts through the Wnt‐Smad signaling pathway. J Orthop Surg Res. 2022;17:343. doi: 10.1186/s13018-022-03224-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2‐HDAC1‐mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mizuguchi Y, Specht S, Lunz JG 3rd, Isse K, Corbitt N, Takizawa T, Demetris AJ. SPRR2A enhances p53 deacetylation through HDAC1 and down regulates p21 promoter activity. BMC Mol Biol. 2012;13:20. doi: 10.1186/1471-2199-13-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gu W, Luo J, Brooks CL, Nikolaev AY, Li M. Dynamics of the p53 acetylation pathway. Novartis Found Symp. 2004;259:197–205. [PubMed] [Google Scholar]
- 68. Cho YH, Ro EJ, Yoon JS, Mizutani T, Kang DW, Park JC, Il Kim T, Clevers H, Choi KY. 5‐FU promotes stemness of colorectal cancer via p53‐mediated WNT/beta‐catenin pathway activation. Nat Commun. 2020;11:5321. doi: 10.1038/s41467-020-19173-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figure S1