

Abstract
We have analyzed a set of new and existing strong mutations in the myospheroid gene, which encodes the βPS integrin subunit of Drosophila. In addition to missense and other null mutations, three mutants behave as antimorphic alleles, indicative of dominant negative properties. Unlike null alleles, the three antimorphic mutants are synthetically lethal in double heterozygotes with an inflated (αPS2) null allele, and they fail to complement very weak, otherwise viable alleles of myospheroid. Two of the antimorphs result from identical splice site lesions, which create a frameshift in the C-terminal half of the cytoplasmic domain of βPS. The third antimorphic mutation is caused by a stop codon just before the cytoplasmic splice site. These mutant βPS proteins can support cell spreading in culture, especially under conditions that appear to promote integrin activation. Analyses of developing animals indicate that the dominant negative properties are not a result of inefficient surface expression, or simple competition between functional and nonfunctional proteins. These data indicate that mutations disrupting the C-terminal cytoplasmic domain of integrin β subunits can have dominant negative effects in situ, at normal levels of expression, and that this property does not necessarily depend on a specific new protein sequence or structure. The results are discussed with respect to similar vertebrate β subunit cytoplasmic mutations.
INTRODUCTION
The integrin family of cell surface receptors is important for adhesion between the extracellular matrix and the cytoskeleton (Yamada and Miyamoto, 1995; Dedhar and Hannigan, 1996). Binding of extracellular ligands and/or the formation of integrin complexes can trigger integrin association with a number of cellular components, including both cytoplasmic proteins and other membrane proteins. This association makes strong connections between the matrix and cytoskeleton, and also transmits signals that can regulate cellular functions such as proliferation, differentiation, and migration, often in cooperation with information from other cell surface receptors (Dedhar and Hannigan, 1996; Howe et al., 1998). Signals also can be transmitted in the opposite direction, as it is clear that events inside the cell can regulate the extracellular binding activities of the integrin αβ heterodimers (Fernandez et al., 1998; Hughes and Pfaff, 1998).
The PS integrins of Drosophila are important for a variety of embryonic and postembryonic morphogenetic events (Stark et al., 1997; Brown et al., 2000). Like most vertebrate integrins, the PS1 and PS2 integrins are receptors for extracellular matrix components (Gotwals et al., 1994). To date, five different αPS subunit genes have been identified: mew, encoding αPS1; inflated, encoding αPS2; scab, encoding αPS3; and two other as yet poorly characterized αPS3-like genes (Hynes and Zhao, 2000). At least the first three of these encode polypeptides that combine with βPS subunits, encoded by the myospheroid (mys) gene, to generate PS1 (αPS1βPS), PS2 (αPS2βPS), or PS3 (αPS3βPS) integrins.
The primary sequences of integrins indicate a high degree of structural conservation. The PS integrins are no exception, and their sequences are as similar to vertebrate integrins as these are to one another (Gotwals et al., 1994). One structural constraint probably is associated with the interactions of α and β subunits that must be important in propagating conformational changes between the short cytoplasmic tails of the protein and the extracellular ligand binding domains (Humphries, 1996). In any case, integrin structure and function appear to be strongly conserved in evolution, and therefore basic information gleaned from studies of one integrin is likely to be applicable to others.
Much of what we know about integrin structure-function derives from studies involving site-directed mutagenesis followed by transfection into cultured cells for functional assays. Although very successful, this approach has limitations. For example, expression levels in transfected cells are often artificially high or unbalanced, and only phenotypes manifested by cultured cells can easily be examined. Also, it is impractical to sample more than a relatively small number of mutagenic changes. These limitations are ameliorated by the standard “forward” genetic approach of random mutagenesis followed by selection of mutants in situ, in developing animals. Mutations affecting required functions that are specific to certain cell types can be identified, and the animal tells the experimenter which mutations alter function, without regard to preconceived notions as to the functions of specific residues. Of course, “blind” genetic screens also have drawbacks; the important point is that this complementary approach has the potential to provide insights that would not readily be forthcoming from directed mutagenesis studies.
Genetic screens of this sort are not easily feasible with vertebrates, although β subunit mutations are revealed in human clinical syndromes, such as leukocyte adhesion deficiency (β2) and Glanzmann thrombasthenia (β3). Random mutagenesis screens can more easily be accomplished in cell culture (Baker et al., 1997), or with invertebrates such as Drosophila melanogaster and Caenorhabditis elegans. Drosophila provides a particularly good system for pursuing screens for integrin mutations, because the PS integrin genes are well characterized genetically and molecularly, and many different integrin-dependent functions have been defined during fly development (Brown et al., 2000).
Previously, a number of mutant alleles of myospheroid (βPS) were generated and partially characterized (Wright, 1960, 1968; Costello and Thomas, 1981; Newman and Wright, 1981; Wieschaus et al., 1984; Leptin et al., 1989; Bunch et al., 1992; Zusman et al., 1993; Roote and Zusman, 1995). Some are null for protein function, others retain at least some protein function (hypomorphs), and one antimorphic allele (mysXR04) has been described that, in complementation tests, is worse than a null allele. We have generated new strong alleles of myospheroid and have characterized these and existing alleles with respect to their molecular lesions and genetic properties. Most importantly, we have identified and analyzed additional alleles that behave genetically as antimorphs, suggesting that these proteins have dominant negative properties. The properties of these antimorphic mutants are compared with those of the human splicing variant β1B, which has molecular similarities and has been shown to have dominant negative properties when assayed in transfected tissue culture cells (Altruda et al., 1990; Balzac et al., 1994; Retta et al., 1998).
MATERIALS AND METHODS
General
All flies were grown on the food described in Condie and Brower (1989). Marker mutations not specifically referenced are described in Lindsley and Zimm (1992).
Screen for New myospheroid Mutants
Some of the new strong alleles described herein were by-products of a screen for mutations in mew, the gene encoding αPS1, as described in Brower et al. (1995); both mew and myospheroid are on the X chromosome. Other alleles were from a similar screen set up specifically to identify myospheroid alleles (Figure 1). Briefly, males with the proximal FRT18A recombination site on the X chromosome (along with the cuticle markers yellow and forked36a) were mutagenized with ethylmethanesulfonate (EMS) (Lewis and Bacher, 1968), and crossed to females with the same X chromosome FRT, as well as a heat shock-inducible FLPase on the second chromosome. Somatic recombination was induced by a heat shock of the F1 larvae to generate clones of cells homozygous for the mutagenized X chromosome. Animals with wing blisters from mutant clones (a known PS integrin phenotype) were selected, and those harboring myospheroid mutations were identified by complementation tests.
Figure 1.

(A) Mosaic screen to recover new strong mutations in the myospheroid gene. See text for details. (B) Wing blister resulting from clone of homozygous mysG1 mutant cells.
Phenotype and Genetic Assays
To score viability of various combinations of alleles, eggs were laid at the appropriate temperature, and vials were thinned to prevent overcrowding of larvae. Progeny were scored at least once a day, and if any animals from any single vial were scored, all subsequent progeny from that vial were counted, to guard against genotypic differences in developmental rates.
To quantitate the severity of dorsal herniation, eggs were collected from balanced stocks of the various mutants, and aged at 25oC to allow wild-type animals to hatch. The mutant embryos were then dechorionated in bleach (Ashburner, 1989a) and scored blind under a dissecting microscope. For each genotype, 100–200 embryos were scored. For photography, embryos were mounted in Hoyers medium and photographed with phase contrast optics.
Mutant Sequencing and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Heterozygous adults or hemizygous mutant embryos were homogenized and genomic DNA template was isolated using QIAGEN′s QiaAmp tissue kit (Valencia, CA). PCR primers were designed to yield three overlapping PCR products, covering the myospheroid coding region (exons 2–7; Yee, 1993; Zusman et al., 1993). The first fragment began 139 base pairs before the initiating AUG and the third fragment continued 62 base pairs after the UAG stop codon. The introns were amplified but the largest (between exons 3 and 5) were not sequenced in their entirety. The resulting PCR fragments were purified using QIAGEN′s QiaQuick PCR purification kit and sequenced directly by the University of Arizona Genomic Analysis and Technology Core (Tucson, AZ).
For RT-PCR of mysXR04, 30 hemizygous mutant embryos were used to generate mRNA with QIAGEN′s RNeasy mini kit. The complementary strand was generated using a myospheroid-specific primer and SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA). PCR primers were designed to then generate a 273-base pair fragment that included the intron between the sixth and seventh exons, as well as a potential downstream splice acceptor site. To examine the possible use of a potential downstream splice site, cDNAs from five developmental stages, embryo through adult, of wild-type flies were used for RT-PCR as described above. To maximize the likelihood of generating sufficient amounts of this novel potential fragment, amplified DNA was digested with HinfI at various times during the amplification procedure, taking advantage of a restriction site specific to the common, larger PCR fragment.
To search for the presence of a potential novel splice site in mysP9, mRNA was isolated from 15 mysP9/FM7i and y sn3 v FRT18A adults by using QIAGEN′s RNeasy mini kit. The complementary strand was generated using QIAGEN′s Omniscript reverse transcriptase and a specific primer located 62 base pairs after the UAG stop codon. Two sets of internal primers were used to generate a PCR fragment that spanned the splice site. The fragments from both mysP9/FM7i and control DNA were examined on a 1.5% agarose 0.5× Tris borate-EDTA gel stained with ethidium bromide. The remainder of the PCR products was purified using QIAGEN′s QIAquick PCR purification kit and sequenced.
Immunofluorescence and Protein Expression
For embryo staining, chromosomes with myospheroid alleles were balanced over an FM7c chromosome containing a P[w+, actin-lacZ] insert (Davis et al., 1996); simultaneous staining for β-galactosidase and βPS therefore allowed the unequivocal identification of hemizygous mutant embryos before overt myospheroid phenotypes became evident. Embryos were collected for 1.5 h at 25°C, and aged 23 h at 18°C, so that the majority of embryos were at stage 15–16, before muscles of the myospheroid embryos begin to detach. Embryos were then fixed and permeablized by using a protocol slightly modified from that of Tim Karr (Ashburner, 1989a). Embryos were incubated in three different antibody solutions, separated by washes. Blocking, washes, and antibody dilutions were in phosphate-buffered saline, pH 7, 1–10% fetal calf serum, 1% Triton-X 100. Antibody incubations were for 4 h at room temperature or overnight at 4oC, with constant agitation. In order, the incubations were in rabbit anti-β-galactosidase (Harlan Sera-Lab, Crawley Down, Sussex, United Kingdom), the mouse monoclonal anti-myospheroid CF.6G11 (Brower et al., 1984), and finally a combination of goat anti-mouse fluorescein isothiocyanate (Jackson ImmunoResearch Labs, West Grove, PA) and goat anti-rabbit Texas Red (ICN Biomedicals, Cleveland, OH). In some experiments, an incubation in a mouse anti-Scr (Glicksman and Brower, 1988) was included as a control for antibody permeability. After the final wash, embryos (and the discs below) were mounted (in 30% 1 mM Tris pH 9.0, 70% glycerol, to which was added 2% n-propyl gallate to reduce bleaching) and examined in a standard Zeiss immunofluorescence or Leitz confocal microscope.
For staining wing imaginal discs, clones of homozygous myospheroid mutant cells were generated by somatic recombination in heterozygous animals. The procedure for clone induction was similar to that for the screen, except that known alleles were crossed to the FRT, FLPase-containing line, and heat shocks were applied daily to induce multiple clones per disk. (Clone induction with this protocol is typically so extensive that all of the animals of the correct genotype die as pupae or nonpupating larvae.) Wing discs were stained with the mouse monoclonal anti-myospheroid CF.6G11 and goat anti-mouse fluorescein isothiocyanate (Jackson ImmunoResearch Laboratories) as previously described (Brower et al., 1984).
RNAi Treatment of Cells
Production and use of double-stranded, interfering RNA (RNAi) was similar to that described in Clemens et al. (2000). To produce a DNA fragment containing 681 base pairs of 3′-untranslated sequence specific for the endogenous myospheroid gene (the 3′-untranslated sequence in the transgenes has been replaced with sequences from the Drosophila tubulin α-1 gene; Bunch and Brower, 1992), genomic DNA from S2 cells was amplified by PCR with the primers mys3′d1 (CGGAAATCAGAAGGAACCC) and mys3′u2 (GTTAAGTATCCCAATTCTGAC). This fragment was then amplified using similar primers that also contained a 5′ T7 RNA polymerase binding site (GAATTAATACGACTCACTATAGGGAGA). The PCR products were purified using a QIAquick PCR purification kit (QIAGEN) and used as templates to produce double-stranded RNA via the MEGASCRIPT T7 transcription kit (Ambion, Austin, TX). The double-stranded RNA was ethanol precipitated and resuspended in water, incubated at 65°C for 30 min followed by slow cooling to room temperature, and then stored at −20°C until use. Preliminary experiments showed similar effects when the final concentration of RNAi in the growth medium was 3–30 μg/ml. We used a final concentration of 15 μg of RNAi/ml medium in the experiments reported herein.
For treatment of cells with RNAi, 1 × 106 cells were washed and suspended in 0.66 ml of HyQ-CCM3 serum-free medium (Hyclone Laboratories, Logan, UT) per well of a six-well culture dish. RNAi (30 μg) was added and the cells were incubated for 30 min at room temperature followed by the addition of 1.34 ml of M3 + 12.5% fetal calf serum + 0.2 μM methotrexate. Experiments were done 5 d or more after initial exposure to RNAi. Every 5 d cells were diluted in fresh medium containing 15 μg of RNAi/ml.
The effectiveness of RNAi treatment was tested on cells transfected with a gene expressing the αPS2 subunit from a heat shock promoter (Bunch and Brower, 1992), without a transgene expressing the βPS subunit. Therefore, expression of PS2 integrin is dependent solely on the endogenous βPS gene. If these cells are heat shocked to induce high expression of αPS2, RNAi treatment results in an 80–90% reduction in integrin expression levels, as measured by flow cytometry; thus, RNAi treatment does not eliminate steady-state βPS integrin expression. However, under the conditions of our cell spreading and expression experiments, where cells are cleared of preexisting integrins during the heat shock by the addition of dispase/collagenase, and then analyzed after 3–4 h of recovery, RNAi treatment results in a virtually complete inhibition of integrin expression (our unpublished data). It should also be noted that under the experimental conditions used in this article, there are also high levels of competing βPS subunits expressed from the heat shock-driven transgenes.
Cell Spreading Assays
Cell culture techniques and methods for transfection of cells have been previously described, as have Schneider's line 2 cells transfected with integrin transgenes under the regulation of the heat shock protein 70 promoter (Bunch and Brower, 1992; Zavortink et al., 1993). Some of the current transformed cell lines were transfected using the CellFECTIN technique (Invitrogen). PCR was used to generate mutations in the pHSβPS plasmid that correspond to the protein encoded by the mysG1 mutation and a cytoplasmic truncation that is missing exon 7 (mysΔ7). In all experiments, the transgenes corresponded to the “c” isoform of αPS2 and the “4A” isoform of βPS (Graner et al., 1998).
The ligand used in cell spreading assays is RBB-Tig. RBB-Tig is a bacterial fusion protein that contains 53 amino acids of the Drosophila extracellular matrix protein tiggrin (residues 1964–2016, including the RGD sequence and 25 amino acids upstream and downstream), fused to a histidine tag, from the pTrcHisB vector (Xpress SystemTM; Invitrogen). This fusion protein is as active in promoting cell spreading as the previously described tiggrin fusion protein and tiggrin itself (Fogerty et al., 1994). Fusion protein was purified by affinity chromatography on Ni-NTA agarose (QIAexpress; QIAGEN). Ninety-six well tissue culture plates or slides were incubated with 500 ng/ml RBB-Tig diluted in phosphate-buffered saline (PBS) for either 1 h at room temperature or overnight at 4°C, blocked with 10% dried milk in PBS for 1 h at room temperature, and washed three times with PBS.
Standard cell spreading assays involved pretreatment with dispase/collagenase before integrin expression and assays for cell spreading in serum-free medium. These were done as previously described (Bunch and Brower, 1992; Zavortink et al., 1993) with one change. The dispase/collagenase treatment to remove existing integrins and extracellular matrix was done at 37°C at the same time as the heat shock, which induces expression of the integrin transgenes. Staining cells for surface integrins demonstrated that this treatment quantitatively removes the existing heterodimers from the surface of the cells. For RNAi-treated cells, RNAi was included in the cell-spreading medium (M3 + 2 mg/ml bovine serum albumin). The number of spread cells was determined by microscopy 3–4 h after heat shock and plating. Results of cell spreading assays are expressed as the averages of three experiments with SEs. For the “normal growth conditions” assay, cells were suspended and heat shocked without protease treatment, and replated into regular growth medium with serum. Spreading was assayed overnight on plates without RBB-Tig, or 3–4 h postinduction on plates coated with RBB-Tig.
To examine integrin expression levels, cells were incubated with biotinylated anti-αPS2 integrin monoclonal antibodies (CF.2C7) and R-phycoerythrin-streptavidin (Molecular Probes, Eugene, OR) in M3 medium + 10% fetal calf serum on ice, followed by dilution with 1 ml of 2% formaldehyde in PBS. Cells were analyzed by flow cytometry at the Arizon Research Labs-Biotechnology Cell Sorting Facility (University of Arizona).
RESULTS
mysXR04 Is a Weak Dominant Negative In Situ
A number of strong mutant alleles of myospheroid have been isolated previously, and some of these have been shown to make little or no βPS protein. By using these alleles, the myospheroid null embryonic lethal phenotype has been described in detail, along with some genetic interactions with other integrin mutants (Wright, 1960, Wieschaus et al., 1984; Newman and Wright, 1981; Wieschaus and Noell, 1986; Leptin et al., 1989; Bunch et al., 1992; Zusman et al., 1993; Roote and Zusman, 1995). In the course of these studies, it was noticed that one EMS-generated allele, mysXR04, displays antimorphic (stronger than the null phenotype) genetic properties (Wilcox, 1990; Bunch et al., 1992; Brabant and Brower, 1993). Specifically, a double heterozygote of mysXR04 and a null allele for the αPS2-encoding gene (inflated) can be synthetically lethal. It is also true that the embryonic lethal phenotype of mysXR04 hemizygous embryos (from heterozygous mothers) can be different from zygotic null embryos; most obviously, they often have a “tail up” phenotype, and the dorsal herniation characteristic of βPS loss is often more severe. This phenotype has also been noted in embryos that lack both maternal and zygotic βPS function (Wieschaus and Noell, 1986; Leptin et al., 1989; Roote and Zusman, 1995), consistent with the hypothesis that mysXR04 could exert negative effects on the maternally contributed wild-type βPS.
In addition to the above-mentioned phenotypes, we have characterized another indicator of the antimorphic properties of mysXR04. Following on the findings of Bunch et al. (1992), we have analyzed the complementation behavior of dozens of weak (hypomorphic) alleles of myospheroid, and find that most show significant viability alone or in combination with null alleles, especially at low temperatures, but that these typically are completely lethal when trans-heterozygous with mysXR04 (unpublished data). Table 1 gives complementation data for mysb7, one of the weakest hypomorphic alleles.
Table 1.
Antimorphic complementation behavior of mysXR04, mysG1, and mysP9
| myslethal allele | myslethal/mysb7 % viabilitya | myslethal if+/mys+ ifnull % viabilitya |
|---|---|---|
| CC6 | 57 (110) | n.d. |
| F1 | 80 (96) | 115 (48) |
| F2 | 104 (74) | 112 (68) |
| G4 | 106 (49) | 100 (159) |
| G12 | 90 (79) | 96 (112) |
| M2 | 92 (95) | 92 (159) |
| N13 | 115 (55) | 68 (57) |
| U3 | 68 (133) | n.d. |
| W11 | 63 (102) | n.d. |
| XR04 | 0 (187) | 0 (214) |
| G1 | 0 (161) | 0 (159) |
| P9 | 0 (104) | 0 (111) |
n.d., not determined. (N), number of animals in control class. The crosses with mysb7 were done at 28°. Tests with the inflated null allele were at 25°.
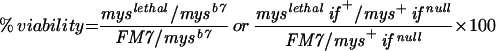 from a cross in which the numerator and denominator classes should segregate in equal numbers (see MATERIALS AND METHODS).
from a cross in which the numerator and denominator classes should segregate in equal numbers (see MATERIALS AND METHODS).
Additional Myospheroid Alleles
We performed genetic screens that isolated additional mutant alleles of myospheroid. Male flies were fed the mutagen EMS and crossed to females to generate F1 animals. During larval development, somatic recombination was induced on the myospheroid-containing X chromosomes, to generate small clones of cells homozygous for the mutagenized X chromosome (Figure 1). The adult F1 flies were then screened for wing blisters, known to be caused by strong mutations in myospheroid. Potential mutations in βPS were confirmed by complementation testing with other alleles of myospheroid, and, as described below, by sequencing. (See MATERIALS AND METHODS and Brower et al., 1995, for screen details.)
Although we have not characterized the full range of embryonic lethal phenotypes for all of the new alleles generated from this screen, they all show the dorsal herniation typical of null alleles of myospheroid. We have examined some alleles in more detail, including mysG1, mysG4, mysG12, mysM2, and mysP9. No obvious deviations from the previously described null phenotype (Wright, 1960) have been noted, except for mysG1 and mysP9. These alleles can display the tail up and extreme herniation phenotype (Figure 2), similar to that described for mysXR04. Because the phenotypes are variable, we sought to quantify the penetrance of the severe herniation. As shown in Figure 3, embryos of mysG1 and mysP9 were both significantly more severe than the null alleles.
Figure 2.

Embryo cuticle preparations from myospheroid mutants. The embryos show small, medium, and large dorsal holes (arrows) from the null mutant mysXG43 (A and B) and the antimorphic mysP9 (C) mutant. Frequencies of these phenotypes are quantitated in Figure 3. Note the anterior dorsal movement of the posterior spiracles (arrowheads) in the mysP9 embryo (C) relative to the wild type (D). Bar, ∼0.1 mm.
Figure 3.

Dorsal herniation phenotype of myospheroid mutants. Hemizygous mutant embryos from the myospheroid alleles indicated, and the double mutant mysXB87 ifB4, were dechorionated and scored for the size of the dorsal hole. Larger holes are more frequent in the antimorphic and double mutants.
Embryos doubly mutant for null alleles of myospheroid and inflated (αPS2) also were much more likely to exhibit a large dorsal hole (Figure 3). Because there is no maternally contributed inflated gene product (Bogaert et al., 1987; Roote and Zusman, 1995), the double mutant will be similar to a complete zygotic and partial maternal myospheroid mutant. The similarity of the double mutants and the myospheroid antimorphs supports the notion that the antimorphs lead to a reduction in the maternal wild-type myospheroid function.
All of the strong myospheroid alleles were tested for suggestions of dominant negative behavior in crosses with weak alleles (Table 1). Again, mysG1 and mysP9 consistently behaved similarly to the antimorphic mysXR04 in these assays, whereas other alleles behaved similarly to known null mutations. Like mysXR04, mysG1 and mysP9 also were synthetically lethal when in a double heterozygote with an inflated (αPS2) null allele.
Dominant Negative Alleles Have Similar Molecular Lesions
We sequenced the coding regions of the genomic DNA for all of the new myospheroid alleles, as well as older alleles that displayed strong, apparently null phenotypes. As shown in Table 2, many of the alleles contain premature stop codons, deletions, frameshifts, or splicing mutations that would be expected to lead to a complete lack of functional βPS protein, and for at least some of the alleles, this expectation has been confirmed by immunofluorescence (see below) or Western analysis (Leptin et al., 1989; Bunch et al., 1992). Only two alleles, mysG4 and mysG12, are characterized by missense mutations. One of these, mysG4, alters the second serine of the essential DXSXS MIDAS motif, whereas mysG12 changes the aspartate in a DYPS(hydrophobic) motif that is highly conserved in β subunits. One other myospheroid allele that is 100% lethal, mysXN101, also results from a missense mutation (C627>S) in the extracellular stalk domain; however, the lethal phenotype of this allele indicates that it retains some wild-type function (our unpublished data).
Table 2.
Null and antimorphic alleles of myospheroid
| mys allele | Nucleotide change | Predicted amino acid change | Notes |
|---|---|---|---|
| Nulls | |||
| CC6 | C224>A | S75>stop | |
| F1 | G955>A Δ961–975 |
D319>N, Δ322–326 | |
| F2 | Rearrangement or deletion? | Cannot amplify 3′ fragment for sequencing | |
| G4 | C587>T | S196>F | Protein localizes properly to muscle attachments |
| G12 | G14>A G1066>A |
R5>K D356>N |
Protein localizes properly to muscle attachments |
| M2 | C196>T | Q66>stop | No protein detected |
| N13 | Large internal deletion | Frameshift after residue 127 | |
| U3 | C2296>T | Q766>stop | |
| W11 | C1180>T | Q394>stop | |
| XB87 | G>A at exon 3 splice acceptor | No protein detected | |
| XG43 | Δ2110–2222 | Frameshift after residue 704 | No protein detected |
| Antimorphs | |||
| G1 | G>A at exon 7 splice acceptor | Frameshift after residue 825 | All phenotypes similar in this class; protein |
| P9 | G2469>A | W823>stop | expressed on surface |
| XR04 | G>A at exon 7 splice acceptor | Frameshift after residue 825 |
ND, not determined.
The three alleles showing weak dominant negative effects all have molecular lesions midway through the cytoplasmic domain of the βPS subunit. Both the mysXR04 and mysG1 chromosomes contain identical G>A mutations in the splice acceptor site before the seventh exon (Figure 4). Because these two mutations were generated independently in different genetic backgrounds (as verified by molecular polymorphisms in the two mutant strains), we can be confident that the unusual antimorphic phenotype seen in both lines results from this specific molecular lesion. The mysP9 chromosome contains a nonsense mutation in exon 6, in the third codon upstream of the above-mentioned splice site.
Figure 4.

(A) Nucleotide sequence around the boundaries of the cytoplasmic intron of myospheroid. Exon sequence in caps, intron in lowercase. The G>A mutation in mysXR04 (and mysG1) is indicated; this results in the seventh exon beginning with the underlined G. The potential downstream splice acceptor site is indicated by double underlining. To search for mRNAs that might use this site, this entire region was amplified by RT-PCR. Amplification of products using the conventional splice site was inhibited by cutting at the HinfI site indicated in bold (see text). (B) Predicted amino acid sequence of the cytoplasmic domains of wild-type βPS and that from mysXR04 and mysP9. The frameshift induced by the aberrant splicing in mysXR04 replaces the membrane-distal 21 residues with 25 essentially random amino acids (underlined). The first cytosine of exon 7 is a thymidine in mysG1; this polymorphism is silent in the parental strain, but changes to first residue encoded by the mutant exon 7 from alanine to valine.
Analysis of Dominant Negative Mutant mRNAs
The three cytoplasmic mutants all have mutations that have the potential to alter mRNA splicing, and so we characterized the transcripts that result from these lesions. The mutated guanosine in mysXR04 and mysG1 is required for proper mRNA splicing, and from examination of nearby sequences, three splicing outcomes seemed possible for the mutant mRNA: 1) The splice would shift one nucleotide to the next guanosine, creating a frameshift. 2) Splicing would occur at a nearby downstream sequence (24 nucleotides away) that resembles a consensus acceptor sequence (Figure 4); this would create an in-frame deletion of eight residues, centered on the first of the two NPXY motifs present in integrin β subunits (see DISCUSSION). 3) Splicing would not occur nearby, presumably resulting in a string of missense residues. To distinguish between these possibilities, we performed RT-PCR and sequence analyses of mRNA from mysXR04 mutant embryos. PCR primers were designed to generate a 273-base pair fragment (in wild type) that includes the splice junction and the potential downstream splice acceptor site. Only a single gel fragment was visualized for mysXR04. The sequence of the fragment indicated a splicing shift deleting one nucleotide of the mysXR04 cDNA (corresponding to possibility 1 above). The frameshift introduced by the aberrant splice causes exon 7 to encode 25 novel residues (Figure 4). In mysG1, a nucleotide polymorphism present in the parental strain, and which is silent in the normal reading frame, changes the alanine at the start of the new sequence to valine. Despite this minor difference, we will refer to the frameshifted βPS subunits generically as the βPSXR04-G1 protein. This mutant protein is missing both NPXY motifs.
At first sight, the mysP9 mutation appears to be a straightforward stop codon, leading to a protein truncated three residues before the mysXR04-G1 frameshift. However, closer examination of the nucleotide sequence reveals the possibility of a cryptic splice donor sequence just upstream, and the mysP9 mutation improves the potential intron consensus sequence at this site. To see whether mysP9 might lead to significant use of this new potential splice site, we did RT-PCR analysis of mRNA from heterozygous mysP9 animals. We found no trace of the smaller (263 vs. 273 base pairs) aberrantly spliced mRNA on agarose gels or upon direct sequencing of the PCR products. There also was no indication that expression level of the mutant mRNA was reduced. Thus, the mysP9 mutation apparently leads to a straightforward truncation (Figure 4).
As mentioned above, examination of the nucleotide sequence of the wild-type gene indicated the potential for mRNA splicing that would precisely delete eight amino acids, centered on the first NPXY motif. We were curious to see whether the potential downstream splice site might be used in wild-type animals, at some restricted time or place in development. To examine this possibility, cDNA from five developmental stages, embryo through adult, of wild-type animals was used for RT-PCR as mentioned above. We anticipated that the potential alternatively spliced cDNA might be relatively rare, and PCR amplification can be a competitive process. To maximize the likelihood of generating sufficient amounts of the fragment in question, we digested the amplified DNA at various times during the amplification procedure, taking advantage of a HinfI restriction site present only in the common, larger PCR fragment (Figure 4). Several variations of this scheme were tried, but a new fragment size was never seen.
Expression of Mutant Proteins
We examined a number of the mutant alleles by immunofluorescence for expression of surface integrin. As expected, the nonsense mysM2 mutant shows no surface PS integrin in late-stage embryos (our unpublished data); loss of PS integrins has previously been reported for the deletion and splicing mutants mysXG43 and mysXB87 (Leptin et al., 1989; Bunch et al., 1992). Both missense alleles, mysG4 and mysG12, express surface integrin at levels comparable to wild-type embryos (Figure 5), and show the typical accumulation of protein at embryonic muscle attachment sites. (The embryos shown in Figure 5 are fixed before the contractions that lead to the muscle detachment characteristic of the mutant phenotype.)
Figure 5.

Confocal immunofluorescence sections of the hypoderm and underlying muscles of stage 15–16 embryos. The animals are stained with an antibody against the βPS subunit, which is seen to be concentrated in a metameric array of dots corresponding to muscle attachment sites. The wild type (wt) is a heterozygote for wild type over the nonsense allele mysM2; homozygotes for mysM2 show no staining. Bar, ∼50 μm.
The frameshifted βPSXR04-G1 protein also appears to be expressed in embryos, and localized at muscle attachments. To look more thoroughly for possible expression reductions in these mutants, we examined surface integrin expression on the epithelium of larval imaginal discs containing clones of homozygous mysG1 cells in a heterozygous background. These clones were generated similarly to those used in the mosaic screen for mutant alleles, except that the larvae were heat shocked more extensively (daily) to induce larger numbers of clones. As shown in Figure 6, control animals with the mysM2 nonsense allele typically (13 of 15 wing discs) showed multiple patches of cells without detectable integrin expression. However, no patches of reduced expression were detected in mysG1 discs grown and heat shocked at the same time (51 wing discs examined). Thus, the weakly dominant negative allele mysG1 does not lead to large-scale reduction in integrin expression in the disk epithelium.
Figure 6.

Third instar wing imaginal discs stained with antibody against βPS. These animals are heterozygous for myospheroid mutations, and have been treated to induce recombinant clones of homozygous mutant cells. (A) Heterozygote for the nonsense mutation mysM2; the clonal patches show no surface expression of PS integrins (asterisks). (B) Disk from mysG1 heterozygote cross, which shows no patches of reduced integrin expression. Bar, ∼100 μm.
Dominant Negative Mutants Support Cell Spreading in Culture
To test the ability of mutant proteins to function in a simple cell spreading assay, we transformed Drosophila S2 cells with cDNAs for αPS2 (the c splice variant) and βPS (the 4A splice variant) proteins corresponding to the mysG1 mutant and a mutant similar to mysP9, but truncated precisely at the exon 6–7 splice boundary (designated mysΔ7). This latter protein is three residues longer than that encoded by mysP9. All transformed genes were under the control of a heat shock promoter, and for each the endogenous 3′-untranslated sequences were replaced with tubulin α1 3′ sequence (Bunch and Brower, 1992; Graner et al., 1998).
S2 cells produce small but significant amounts of endogenous βPS, and cells transformed with αPS2 alone show a significant ability to spread on RBB-Tig, an RGD-containing fragment of the PS2 ligand tiggrin (Fogerty et al., 1994). This spreading is reduced by ∼90% when the cells are treated with double-stranded RNAi (Carthew, 2001) against the 3′-untranslated region of the endogenous βPS transcript (Figure 7). When cells are transformed with both αPS2 and βPS genes, and subjected to the protease clearing and heat shock induction protocol, RNAi has little effect, indicating that virtually all of the spreading is due to expression from the transformed genes. This RNAi control was used for all of the results reported herein.
Figure 7.

Cell spreading by mutant β subunits. S2 cells were transformed with αPS2 and various forms of βPS, and assayed for spreading on a fragment of the PS2 ligand tiggrin. For these experiments, cells were pretreated with protease to remove surface proteins and ECM before integrin induction. RNAi was added to inhibit expression of endogenous βPS (filled bars). Untransformed S2 cells do not spread. Cells transformed with αPS2 only can spread using the low levels of endogenous βPS, but this is reduced by RNAi treatment. Spreading of cells transformed with wild-type or mutant βPS is not significantly reduced by RNAi. The difference between wild-type and the two βPS mutants reflects differences in expression level.
Both the mysG1 and mysΔ7 proteins were expressed well on S2 cells in combination with αPS2. We tested the ability of the mutant proteins to mediate cell spreading under two different assay conditions. In one, cells are treated with protease to clear surface proteins and ECM, heat shocked to induce integrins, and allowed to spread on RBB-Tig. In this assay, both mysG1- and mysΔ7-containing integrins promote spreading that is comparable to that seen with wild-type βPS (Figure 7). Although slight cell line variations were observed, these variations could be ascribed to minor differences in the expression levels of the integrins (our unpublished data).
Although the mutant integrins can mediate spreading in the above-mentioned assay, we noticed that these cells spread very poorly during regular culture, relative to cells expressing wild-type β subunits. To quantitate this, we performed a second assay similar to regular growth conditions, in which the cells were not pretreated with protease but were suspended, heat shocked to induce high-level integrin expression, and allowed to spread on plates overnight in regular growth medium containing serum. (Although not shown, qualitatively similar results were obtained if uncleared cells were spread on RBB-Tig for 3–4 h.) Under these conditions, the mutant integrins promote cell spreading very poorly, relative to wild type (Figure 8), despite the fact that these cells are expressing more integrin than is seen after protease clearing and induction (our unpublished data). Interestingly, in this assay the activity of the mutant βPS subunits can be largely restored if combined with αPS2 subunits containing a mutation of the conserved cytoplasmic GFFXR motif (GFFNR>GFANA) that promotes integrin activation (O'Toole et al., 1994; Hughes et al., 1996).
Figure 8.

Cell spreading in different assay conditions. Integrin induction in transformed S2 cells followed protease treatment as before (open bars) or was in the absence of protease (filled bars). In the latter case, the spreading assay was in normal growth medium, with serum, on uncoated tissue culture plates. activ-G1 and activ-truncation indicate cells that also express a mutant αPS2 that promotes integrin activation. The mutant β subunits support spreading poorly except in conditions (cleared cells or activating αPS2 subunits) that appear to promote artificially high activation. This is true in spite of the fact that the normal growth G1 and truncated cells express integrin at higher levels than any of the cleared or activating αPS2 lines.
The morphology of spread cells on RBB-Tig may also be influenced by expression of the mysG1- and mysΔ7-containing integrins. Although a variety of morphologies are typical even in wild type, the mutants tend to display more regions of extreme peripheral thinning (Figure 9). These are characteristics also observed in S2 cells containing mutants that affect the ability of the cell to regulate integrin activity, such as the activating αPS2 mutants (our unpublished data).
Figure 9.

Drosophila S2 cells that were cleared, induced to express wild-type or mysG1 mutant βPS in association with αPS2, and allowed to spread on high concentrations of a recombinant fragment of tiggrin. The mutant cells have a tendency to display more thin, clear areas at the periphery (asterisks).
DISCUSSION
The most interesting of the mutant alleles described herein are the three cytoplasmic mutants that display dominant negative properties in flies, and we will refer to them collectively as mysDN alleles. Although they are viable as heterozygotes, these animals have reduced integrin function relative to animals heterozygous for null alleles. This is perhaps best illustrated by the synthetic lethality of the mysDN inflatednull double heterozygotes; reducing αPS2 expression by 50% is not sufficient to generate phenotypes in a myospheroid null heterozygote. Dominant negative qualities also are indicated by crosses with weak alleles; although mysb7/mysnull supports viability very well at high temperatures, the mysb7/mysDN combination is strongly lethal. Finally, the embryonic lethal phenotypes of the mutants are consistent with the idea that these alleles compromise the functioning of the maternally contributed wild-type βPS.
Similarity to Mammalian β Subunit Variants
The position of the cytoplasmic intron is conserved in most integrin β subunit genes (Schmitt and Brower, 2001), and just as mysXR04 and mysG1 create a frameshift at this site (caused by shifting the splice site one nucleotide), similar naturally occurring cytoplasmic splice variations have been described for human β1 and β3 (van Kuppevelt et al., 1989; Altruda et al., 1990; Languino and Ruoslahti, 1992; Kumar et al., 1997; Svineng et al., 1998; for review, see Melker and Sonnenberg, 1999). The β1C splice variant has been shown to result in loss of some cytoplasmic associations and signaling properties, and to affect cell proliferation (Languino and Ruoslahti, 1992; Fornaro et al., 1995, 1998, 2000; Meredith et al., 1995; Pfaff et al., 1998). Human β3B and β3C splice variants also generally show a decrease in cellular functions such as focal adhesion localization, focal adhesion kinase phosphorylation, and cell adhesion (Akiyama et al., 1994; LaFlamme et al. 1994; Kumar et al., 1997; Pfaff et al., 1998). Extensive analyses of one human isoform, β1B, have uncovered dominant negative properties similar to those we see, and we will compare human β1B with our Drosophila mutants in greater detail.
Like mysXR04 and mysG1, human β1B alters the protein sequence (relative to the common β1A isoform) distally from the splice site, but in this case as a result of a failure to splice and subsequent translation into the intron (Altruda et al., 1990). The β1B isoform is expressed primarily in skin and liver, where it constitutes a minority of the β1 protein (Balzac et al., 1993). To date, no function of β1B has been demonstrated in situ, but there have been a number of studies of the properties of the isoform in cultured cells (Balzac et al., 1993, 1994; Cali et al., 1998; Retta et al., 1998; Armulik et al., 2000). Overall, this work indicates that β1B is deficient in both “outside in” and “inside out” integrin signaling. For example, to mediate adhesion, the β1B-containing integrins typically require activation using nonphysiological treatments such as incubation in Mn2+ ions. And even upon adhesion or antibody cross-linking, β1B integrins are not efficient stimulators of cytoplasmic proteins such as focal adhesion kinase and paxillin.
Perhaps the most striking property of β1B is its ability to act in a dominant negative manner with respect to other cellular integrins. When transfected into cells, β1B has been reported to inhibit processes mediated by other β1 or β3 integrins, including cell spreading, motility, matrix assembly, and stimulation of specific cytoplasmic proteins (Balzac et al., 1994; Cali et al., 1998; Retta et al., 1998; see Armulik et al., 2000, for a contradictory view of the effects on αvβ3 functions). Retta et al. (1998) further report that these dominant negative properties are dependent on the “intron-encoded” residues; proteins that are simply truncated at the splice site do not inhibit the functions of endogenous integrins.
All of the experiments showing dominant negative effects of β1B rely on cell transfection, and the mysDN mutants are the first demonstration that replacement of the C-terminal portion of the β cytoplasmic domain can have dominant negative effects in situ. And because the Drosophila mutants are generated in otherwise normal chromosomes, and their expression is controlled by the wild-type regulatory machinery, it is by analogy reasonable to think that physiological levels of β1B expression could have significant repercussions for human cells in situ.
Why Dominant Negative?
How do the mysDN mutants (and by extension, human β1B) exert their unusual properties? We can rule out some possibilities from these studies. It has recently been noted that β1B contains a new double lysine motif that can reduce surface expression through trapping of the protein in the endoplasmic reticulum (Kee et al., 2000). The βPSDN proteins contain no similar double lysine, and our expression studies in situ indicate that the mutant proteins reach the cell surface without great difficulty.
Another potential explanation is that the βPSDN proteins just get in the way, by adding a pool of nonfunctional integrins that compete on the cell surface with the wild-type proteins. The simplest version of this scenario does not appear to be true, because two missense alleles (mysG4 and mysG12) that make stable but probably nonfunctional (for ligand binding) protein do not show any dominant negative genetic properties. Competition models might be workable if one proposes that the cytoplasmic mutants also lead to greatly enhanced protein stability, so that the mutant proteins would eventually be present at much higher numbers than their wild-type competitors. However, when we follow the turnover of surface integrin after a single heat pulse of our transformed S2 cells, we see no indication that the βPSDN proteins are unusually stable (our unpublished data). And, in the analogous β1B system, expression of the variant can exert dominant negative effects on β1A functions when each is expressed at equivalent levels (Balzac et al., 1994). Also, β1B-expressing cells can display phenotypes such as reduced αv expression (presumably in αvβ3 heterodimers) in focal adhesions, without β1B actually displacing the αv integrins directly (Retta et al., 1998).
Our genetic results are consistent with the previous proposal that the dominant negative mutants create a cellular signal that leads to inactivation of other surface integrins (Retta et al., 1998). This idea also is consistent with data on the activity of the dominant negative mutants themselves. In general, disruptions of β subunit NPXY motifs leads to reduced integrin activation and cytoskeletal associations, but β1B, for example, can mediate cell adhesion if activated artificially with Mn2+ (Retta et al., 1998; Armulik et al., 2000). In our S2 cell assays, we see evidence that the βPSDN proteins are only poorly able to mediate cell spreading during normal growth, but that a latent activity can be uncovered under conditions that are known (mutant α subunits) or suspected (protease pretreatment) to activate integrins. This also argues that a primary affect of the βPSDN proteins is related to the cells' ability to regulate integrin function.
The Drosophila results further allow some insights as to the structural requirements for the dominant negative properties of these cytoplasmic variants. The failure to detect inhibitory effects of subunits truncated at the splice site led Retta et al. (1998) to surmise that some contribution of the new residues of β1B is important. There is no sequence similarity between the C-terminal residues of the βPSXR04-G1 proteins and those of β1B, making it unlikely that a sequence-specific motif is involved. Moreover, we find that a truncation, mysP9, has dominant negative properties in situ similar to the frameshift mutants, in contrast to the results of Retta et al. (1998) in cell culture. Thus, it appears that in general there is no absolute requirement for a particular sequence, or any sequence after the cytoplasmic splice site, to induce dominant negative effects. It is possible that β1B and βPS are truly different in this respect, but it seems equally likely that if the human proteins were assayed in situ, where the entire range of integrin functions is required, dominant negative effects might emerge for the truncated β1 proteins.
Like β1B, the βPSDN proteins are missing both cytoplasmic NPXY motifs, which, along with neighboring residues, have been shown to be important for activation of extracellular ligand binding, intracellular signal transduction events, or processes such as adhesion or migration mediated by vertebrate integrins (Reszka et al., 1992; Filardo et al., 1995; O'Toole et al., 1995; Ylänne et al., 1995; Baker et al., 1997; Blystone et al., 1997; Chang et al., 1997; Tahiliani et al., 1997; Vignoud et al., 1997; Loo et al., 1998; Pfaff et al., 1998; Romzek et al., 1998; Sakai et al., 1998; Schaffner-Reckinger et al., 1998; Buttery et al., 1999; Kaapa et al., 1999; Mastrangelo et al., 1999; Sakai et al., 1999; Levy et al., 2000; Stroeken et al., 2000; Wennerberg et al., 2000; Boettiger et al., 2001; Ginsberg et al., 2001). Numerous studies have demonstrated requirements for specific residues in these motifs, although dominant negative effects have not generally been tested or noted. However, one anecdotal piece of evidence suggests that these motifs may be critical to the unusual genetics of our mutants and β1B. Grinblat et al. (1994) mutagenized various residues of the cytoplasmic tail of βPS, and asked whether these mutants when transformed into flies could rescue myospheroid null phenotypes. (The mutant constructs contained endogenous myospheroid regulatory elements, and were generally expressed at levels equal to or below that of the endogenous myospheroid gene.) Like some others, they found that changing the tyrosines of the NPXY motifs to phenylalanines had relatively little effect. This alteration is expected to prevent potential phosphorylation of the motifs, but not alter their ability to make a predicted β-turn structure (Haas and Plow, 1997; Ginsberg et al., 2001; Ulmer et al., 2001). However, when Grinblat et al. (1994) changed these tyrosines to alanines, they were unable to recover transformants that expressed the mutant proteins. Of course, this failure could have been due to some unknown technical glitch. However considering their success with numerous other constructs, Grinblat et al. (1994) proposed that the pair of Y>A mutations creates a toxic protein. Our results support this proposal, and combined, the studies suggest that the disruption of NPXY motifs is the critical requirement for creating these dominant negative β subunits. Finally, it appears that most of the proximal cytoplasmic tail must be intact for strong dominant negative properties. For example, Grinblat et al. (1994) were able to generate transformants with a cytoplasmic truncation that is only a few residues shorter than our dominant negative mysP9 truncation.
Scarcity of Essential Residues of βPS
Two missense mutations of myospheroid display the same strong phenotype seen for alleles that produce no functional βPS. Both of the “null” missense mutations change oxygenated residues in the globular head domain of βPS. mysG4 (S196>F) changes the second serine of the conserved MIDAS sequence DXSXS, which is involved in the formation of a cation-binding pocket (Xiong et al., 2001), and this serine has been shown to be required for function or stability for vertebrate β2 and β3 (Bajt and Loftus, 1994; Bajt et al., 1995; Hogg et al., 1999). The mysG12 chromosome contains two missense mutations, R5>K and D356>N. The latter change most likely is responsible for the mutant phenotype, because this residue is acidic in all sequenced β subunits, and has been shown to be essential for function in β1 (Puzon-McLaughlin and Takada, 1996). The arginine residue at position five is early in the βPS signal sequence. The change to lysine is conservative, and analysis by the PSORT algorithm (Nakai and Kanehisa, 1992) predicts little effect on the ability of the altered domain to function as a signal sequence. Moreover, we find that mysG12 (as well as mysG4) subunits are expressed well at the cell surface in situ, arguing strongly that the signal sequence alteration is not responsible for the mutant phenotype.
Not surprisingly, the most common class of genetically strong mutations is nonsense mutants, represented by five alleles. Of the 14 null or antimorphic alleles, only two result from missense mutations. One other missense allele, mysXN101, is known to be 100% lethal and have a dorsal herniation phenotype, but other studies suggest that it has some residual function (Wieschaus et al., 1984; Bunch et al., 1992; our unpublished data). Three of the strong mutations are in splice sites, and four alleles are associated with deletions or other rearrangements, mutagenic events that are expected to be relatively rare after EMS mutagenesis (Ashburner, 1989b). The most common mutation created by EMS is a G-to-A transition. If one assesses the potential results only of G-to-A transitions in the myospheroid gene, there are approximately 10 times as many sites that would create missense mutations as the sum of sites that would lead to nonsense mutations and changes that would eliminate correct splicing. The data are limited, but the results to date, in which the number of strong missense alleles is equaled by splice site mutations, suggest that relatively few of the 846 residues of βPS are absolutely essential for function. On the other hand, experiments to be described elsewhere indicate that myospheroid missense mutations that alter βPS function, but do not eliminate it, are relatively easy to generate, and just as whole animal genetics provides a sensitive assay for dominant negative properties, forward genetic screens should be useful for finer dissection of other integrin structure–function relationships.
ACKNOWLEDGMENTS
We thank Brian Coullahan and the group in the Genomic Analysis and Technology Core sequencing facility, Barb Carolus for help with fluorescence-activated cell sorting analyses, Lynn Manseau for assistance with confocal microscopy, and Mike Rhee for help in some mutant characterizations. We also are indebted to Mark Ginsberg for the activating α subunit mutant. Eric Wieschaus, Mani Ramaswami, and the Bloomington Stock Center kindly supplied fly lines. This study was supported by the National Institutes of Health (R01-GM-42474) and the Arizona Disease Control Research Commission (#10003).
Footnotes
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.01–08–0429. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.01–08–0429.
REFERENCES
- Akiyama SK, Yamada SS, Yamada KM, LaFlamme SE. Transmembrane signal transduction by integrin cytoplasmic domains expressed in single-subunit chimeras. J Biol Chem. 1994;269:15961–15964. [PubMed] [Google Scholar]
- Altruda F, Cervella P, Tarone G, Botta C, Balzac F, Stefanuto G, Silengo L. A human integrin β1 subunit with a unique cytoplasmic domain generated by alternative mRNA processing. Gene. 1990;95:261–266. doi: 10.1016/0378-1119(90)90369-3. [DOI] [PubMed] [Google Scholar]
- Armulik A, Svineng G, Wennerberg K, Fassler R, Johansson S. Expression of integrin subunit β1B in integrin β1-deficient G.D25 cells does not interfere with V3 functions. Exp Cell Res. 2000;254:55–63. doi: 10.1006/excr.1999.4722. [DOI] [PubMed] [Google Scholar]
- Ashburner M. Drosophila: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989a. [Google Scholar]
- Ashburner M. Drosophila: A Laboratory Handbook. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989b. [Google Scholar]
- Bajt ML, Goodman T, McGuire SL. β2 (CD18) mutations abolish ligand recognition by I domain integrins LFA-1 (αLβ2, CD11a/CD18) and MAC-1 (αMβ2, CD11b/CD18) J Biol Chem. 1995;270:94–98. doi: 10.1074/jbc.270.1.94. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Loftus JC. Mutation of a ligand binding domain of β3 integrin. Integral role of oxygenated residues in αIIbβ3 (GPIIb-IIIa) receptor function. J Biol Chem. 1994;269:20913–20919. [PubMed] [Google Scholar]
- Baker EK, Tozer EC, Pfaff M, Shattil SJ, Loftus JC, Ginsberg MH. A genetic analysis of integrin function: Glanzmann thrombasthemia in vitro. Proc Natl Acad Sci USA. 1997;94:1973–1978. doi: 10.1073/pnas.94.5.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzac F, Belkin AM, Koteliansky VE, Balabanov YV, Altruda F, Silengo L, Tarone G. Expression and functional analysis of a cytoplasmic domain variant of the β1 integrin subunit. J Cell Biol. 1993;121:171–178. doi: 10.1083/jcb.121.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzac F, Retta SF, Albini A, Melchiorri A, Koteliansky VE, Geuna M, Silengo L, Tarone G. Expression of β1B integrin isoform in CHO cells results in a dominant negative effect on cell adhesion and motility. J Cell Biol. 1994;127:557–565. doi: 10.1083/jcb.127.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blystone SD, Williams MP, Slater SE, Brown EJ. Requirement of integrin β3 tyrosine 747 for β3 tyrosine phosphorylation and regulation of αvβ3 avidity. J Biol Chem. 1997;272:28757–28761. doi: 10.1074/jbc.272.45.28757. [DOI] [PubMed] [Google Scholar]
- Boettiger D, Huber F, Lynch L, Blystone S. Activation of αvβ3-vitronectin binding is a multistage process in which increases in bond strength are dependent on Y747 and Y759 in the cytoplasmic domain of β3. Mol Biol Cell. 2001;12:1227–1237. doi: 10.1091/mbc.12.5.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert T, Brown N, Wilcox M. The Drosophila PS2 antigen is an invertebrate integrin that, like the fibronectin receptor, becomes localized to muscle attachments. Cell. 1987;51:929–940. doi: 10.1016/0092-8674(87)90580-0. [DOI] [PubMed] [Google Scholar]
- Brabant MC, Brower DL. PS2 integrin requirements in Drosophila embryo and wing morphogenesis. Dev Biol. 1993;157:49–59. doi: 10.1006/dbio.1993.1111. [DOI] [PubMed] [Google Scholar]
- Brower DL, Bunch TA, Mukai L, Adamson TE, Wehrli M, Lam S, Friedlander E, Roote CE, Zusman S. Nonequivalent requirements for PS1 and PS2 integrin at cell attachments in Drosophila: genetic analysis of the αPS1 integrin subunit. Development. 1995;121:1311–1320. doi: 10.1242/dev.121.5.1311. [DOI] [PubMed] [Google Scholar]
- Brower DL, Wilcox M, Piovant M, Smith RJ, Reger LA. Related cell-surface antigens expressed with positional specificity in Drosophila imaginal discs. Proc Natl Acad Sci USA. 1984;81:7485–7489. doi: 10.1073/pnas.81.23.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NH, Gregory ST, Martin-Bermudo MD. Integrins as mediators of morphogenesis in Drosophila. Dev Biol. 2000;223:1–16. doi: 10.1006/dbio.2000.9711. [DOI] [PubMed] [Google Scholar]
- Bunch TA, Brower DL. Drosophila PS2 integrin mediates RGD dependent cell-matrix interactions. Development. 1992;116:239–247. doi: 10.1242/dev.116.1.239. [DOI] [PubMed] [Google Scholar]
- Bunch TA, Salatino R, Engelsgjerd MC, Mukai L, West RF, Brower DL. Characterization of mutant alleles of myospheroid, the gene encoding the β subunit of the Drosophila PS integrins. Genetics. 1992;132:519–528. doi: 10.1093/genetics/132.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttery PC, Mallawaarachchi CM, Milner R, Doherty P, french-Constant C. Mapping regions of the β1 integrin cytoplasmic domain involved in migration and survival in primary oligodendrocyte precursors using cell-permeable homeopeptides. Biochem Biophys Res Commun. 1999;259:121–127. doi: 10.1006/bbrc.1999.0726. [DOI] [PubMed] [Google Scholar]
- Cali G, Retta SF, Negri R, Damiano I, Gentile R, Tarone G, Nitsch L, Garbi C. β1B integrin interferes with matrix assembly but not with confluent monolayer polarity, and alters some morphogenetic properties if FRT epithelial cells. Eur J Cell Biol. 1998;75:107–117. doi: 10.1016/s0171-9335(98)80053-8. [DOI] [PubMed] [Google Scholar]
- Carthew RW. Gene silencing by double-stranded RNA. Curr Opin Cell Biol. 2001;13:244–248. doi: 10.1016/s0955-0674(00)00204-0. [DOI] [PubMed] [Google Scholar]
- Chang DD, Wong C, Smith H, Liu J. ICAP-1, a novel β1 integrin cytoplasmic domain-associated protein, binds to a conserved and functionally important NPXY sequence motif of β1 integrin. J Cell Biol. 1997;138:1149–1157. doi: 10.1083/jcb.138.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens JC, Worby CA, Simonson-Leff N, Muda M, Maehama T, Hemmings BA, Dixon JE. Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction. Proc Natl Acad Sci USA. 2000;97:6499–6503. doi: 10.1073/pnas.110149597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condie JM, Brower DL. Allelic interactions at the engrailed locus of Drosophila: engrailed protein expression in imaginal discs. Dev Biol. 1989;135:31–42. doi: 10.1016/0012-1606(89)90155-3. [DOI] [PubMed] [Google Scholar]
- Costello WJ, Thomas JB. Development of thoracic muscles in muscle-specific mutant and normal Drosophila melanogaster. Neurosci Abstr. 1981;5:543. [Google Scholar]
- Davis GW, Schuster CM, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. III. CREB is necessary for presynaptic functional plasticity. Neuron. 1996;17:669–679. doi: 10.1016/s0896-6273(00)80199-3. [DOI] [PubMed] [Google Scholar]
- Dedhar S, Hannigan GE. Integrin cytoplasmic interactions and bidirectional transmembrane signaling. Curr Opin Cell Biol. 1996;8:657–669. doi: 10.1016/s0955-0674(96)80107-4. [DOI] [PubMed] [Google Scholar]
- Fernandez C, Clark K, Burrows L, Schofield NR, Humphries MJ. Regulation of the extracellular ligand binding activity of integrins. Front Biosci. 1998;3:684–700. doi: 10.2741/a313. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Brooks PC, Deming SL, Damsky C, Cheresh DA. Requirement of the NPXY motif in the integrin β3 subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. J Cell Biol. 1995;130:441–450. doi: 10.1083/jcb.130.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogerty FJ, Fessler LI, Bunch TA, Yaron Y, Parker CG, Nelson RE, Brower DL, Gullberg D, Fessler JH. Tiggrin, a novel Drosophila extracellular matrix protein that functions as a ligand for Drosophila αPS2βPS integrins. Development. 1994;120:1747–1758. doi: 10.1242/dev.120.7.1747. [DOI] [PubMed] [Google Scholar]
- Fornaro M, Manzotti M, Tallini G, Slear AE, Bosari S, Ruoslahti E, Languino LR. β1C integrin in epithelial cells correlates with a nonproliferative phenotype: forced expression of β1C inhibits prostate epithelial cell proliferation. Am J Pathol. 1998;153:1079–1087. doi: 10.1016/s0002-9440(10)65652-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornaro M, Steger CA, Bennett AM, Wu JJ, Languino LR. Differential role of β1C and β1A integrin cytoplasmic variants in modulating focal adhesion kinase, protein kinase B/AKT, and Ras/Mitogen-activated protein kinase pathways. Mol Biol Cell. 2000;11:2235–2249. doi: 10.1091/mbc.11.7.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornaro M, Zheng DQ, Languino LR. The novel structural motif Gln795-Gln802 in the integrin β1C cytoplasmic domain regulates cell proliferation. J Biol Chem. 1995;270:24666–24669. doi: 10.1074/jbc.270.42.24666. [DOI] [PubMed] [Google Scholar]
- Ginsberg MH, Yaspan B, Forsyth J, Ulmer TS, Campbell ID, Slepak M. A membrane-distal segment of the integrin αIIb cytoplasmic domain regulates integrin activation. J Biol Chem. 2001;276:22514–22521. doi: 10.1074/jbc.M101915200. [DOI] [PubMed] [Google Scholar]
- Glicksman MA, Brower DL. Expression of the homeotic Sex combs reduced gene in Drosophila larvae. Dev Biol. 1988;127:113–118. doi: 10.1016/0012-1606(88)90193-5. [DOI] [PubMed] [Google Scholar]
- Gotwals PJ, Paine-Saunders SE, Stark KA, Hynes RO. Drosophila integrins and their ligands. Curr Opin Cell Biol. 1994;6:734–739. doi: 10.1016/0955-0674(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Graner MW, Bunch TA, Baumgartner S, Kerschen A, Brower DL. Splice variants of the Drosophila PS2 integrins differentially interact with RGD-containing fragments of the extracellular proteins Tiggrin, Ten-m, and D-laminin α2. J Biol Chem. 1998;273:18235–18241. doi: 10.1074/jbc.273.29.18235. [DOI] [PubMed] [Google Scholar]
- Grinblat Y, Zusman S, Yee G, Hynes RO, Kafatos FC. Functions of the cytoplasmic domain of the βPS integrin subunit during Drosophila development. Development. 1994;120:91–102. doi: 10.1242/dev.120.1.91. [DOI] [PubMed] [Google Scholar]
- Haas TA, Plow EF. Development of a structural model for the cytoplasmic domain of an integrin. Protein Eng. 1997;10:1395–1405. doi: 10.1093/protein/10.12.1395. [DOI] [PubMed] [Google Scholar]
- Hogg N, Stewart MP, Scarth SL, Newton R, Shaw JM, Law SKA, Klein N. A novel leukocyte adhesion deficiency caused by expressed but nonfunctional β2 integrins Mac-1 and LFA-1. J Clin Invest. 1999;103:97–106. doi: 10.1172/JCI3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe A, Aplin AE, Alahari SK, Juliano RL. Integrin signaling and cell growth control. Curr Opin Cell Biol. 1998;10:220–231. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- Hughes PE, Diaz-Gonzalez F, Leong L, Wu C, McDonald JA, Shattil SJ, Ginsberg MH. Breaking the integrin hinge; a defined structural constraint regulates integrin signaling. J Biol Chem. 1996;271:6571–6574. doi: 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- Hughes PE, Pfaff M. Integrin affinity modulation. Trends Cell Biol. 1998;8:359–364. doi: 10.1016/s0962-8924(98)01339-7. [DOI] [PubMed] [Google Scholar]
- Humphries MJ. Integrin activation: the link between ligand binding and signal transduction. Curr Opin Cell Biol. 1996;8:632–640. doi: 10.1016/s0955-0674(96)80104-9. [DOI] [PubMed] [Google Scholar]
- Hynes RO, Zhao Q. The evolution of cell adhesion. J Cell Biol. 2000;150:F89–F95. doi: 10.1083/jcb.150.2.f89. [DOI] [PubMed] [Google Scholar]
- Kaapa A, Peter K, Ylanne J. Effects of mutations in the cytoplasmic domain of integrin β1 to talin binding and cell spreading. Exp Cell Res. 1999;250:524–534. doi: 10.1006/excr.1999.4533. [DOI] [PubMed] [Google Scholar]
- Kee WJ, Li ER, Watt FM. β1B integrin subunit contains a double lysine motif that can cause accumulation within the endoplasmic reticulum. J Cell Biochem. 2000;78:97–111. doi: 10.1002/(sici)1097-4644(20000701)78:1<97::aid-jcb9>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Kumar CS, et al. Cloning and characterization of a novel integrin β3 subunit. J Biol Chem. 1997;272:16390–16397. doi: 10.1074/jbc.272.26.16390. [DOI] [PubMed] [Google Scholar]
- LaFlamme SE, Thomas LA, Yamada SS, Yamada KM. Single subunit chimeric integrins as mimics and inhibitors of endogenous integrin functions in receptor localization, cell spreading and migration, and matrix assembly. J Cell Biol. 1994;126:1287–1298. doi: 10.1083/jcb.126.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Languino LR, Ruoslahti E. An alternative form of the β1 subunit with a variant cytoplasmic domain. J Biol Chem. 1992;267:7116–7120. [PubMed] [Google Scholar]
- Leptin M, Bogaert T, Lehmann R, Wilcox M. The function of PS integrins during Drosophila embryogenesis. Cell. 1989;56:401–408. doi: 10.1016/0092-8674(89)90243-2. [DOI] [PubMed] [Google Scholar]
- Levy L, Broad S, Diekmann D, Evans RD, Watt FM. Symbol“ § 121 integrins regulate keratinocyte adhesion and differentiation by distinct mechanisms. Mol Biol Cell. 2000;11:453–466. doi: 10.1091/mbc.11.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB, Bacher F. Methods of feeding ethyl methane sulfonate (EMS) to Drosophila males. Dros Inf Serv. 1968;43:193. [Google Scholar]
- Lindsley DL, Zimm GG. The Genome of Drosophila melanogaster. San Diego, CA: Academic Press; 1992. [Google Scholar]
- Loo DT, Kanner SB, Aruffo A. Filamin binds to the cytoplasmic domain of the β1-integrin. J Biol Chem. 1998;273:23304–23312. doi: 10.1074/jbc.273.36.23304. [DOI] [PubMed] [Google Scholar]
- Mastrangelo AM, Homan SM, Humphries MJ, LaFlamme SE. Amino acid motifs required for isolated beta cytoplasmic domains to regulate ‘in trans’β1 integrin conformation and function in cell attachment. J Cell Sci. 1999;112:217–229. doi: 10.1242/jcs.112.2.217. [DOI] [PubMed] [Google Scholar]
- de Melker AA, Sonnenberg A. Integrins: alternative splicing as a mechanism to regulate ligand binding and integrin signaling events. BioEssays. 1999;21:499–409. doi: 10.1002/(SICI)1521-1878(199906)21:6<499::AID-BIES6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Meredith J, Jr, Takada Y, Fornaro M, Languino LR, Schwartz MA. Inhibition of cell cycle progression by the alternatively spliced integrin β1C. Science. 1995;269:1570–1572. doi: 10.1126/science.7545312. [DOI] [PubMed] [Google Scholar]
- Nakai K, Kanehisa M. A knowledge base for predicting protein localization sites in eukaryotic cells , Genomics. 1992;14:897–911. doi: 10.1016/S0888-7543(05)80111-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman SM, Jr, Wright TRF. A histological and ultrastructural analysis of developmental defects produced by the mutation lethal(1)myospheroid, in Drosophila melanogaster. Dev Biol. 1981;86:393–402. doi: 10.1016/0012-1606(81)90197-4. [DOI] [PubMed] [Google Scholar]
- O'Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole TE, Ylänne J, Culley BM. Regulation of integrin affinity states through an NPXY motif in the β subunit cytoplasmic domain. J Biol Chem. 1995;270:8553–8558. doi: 10.1074/jbc.270.15.8553. [DOI] [PubMed] [Google Scholar]
- Pfaff M, Liu S, Erle DJ, Ginsberg MH. Integrin β cytoplasmic domains differentially bind to cytoskeletal proteins. J Biol Chem. 1998;273:6104–6109. doi: 10.1074/jbc.273.11.6104. [DOI] [PubMed] [Google Scholar]
- Puzon-McLaughlin W, Takada Y. Critical residues for ligand binding in an I domain-like structure of the integrin β1 subunit. J Biol Chem. 1996;271:20438–20443. doi: 10.1074/jbc.271.34.20438. [DOI] [PubMed] [Google Scholar]
- Reszka AA, Hayashi Y, Horwitz AF. Identification of amino acid sequences in the integrin β1 cytoplasmic domain implicated in cytoskeletal association. J Cell Biol. 1992;117:1321–1330. doi: 10.1083/jcb.117.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retta SF, Balzac F, Ferraris P, Belkin AM, Fässler R, Humphries MJ, De Leo G, Silengo L, Tarone G. β1-integrin cytoplasmic subdomains involved in dominant negative function. Mol Biol Cell. 1998;9:715–731. doi: 10.1091/mbc.9.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romzek NC, Harris ES, Dell CL, Skronek J, Hasse E, Reynolds PJ, Hunt SW, III, Shimizu Y. Use of a β1 integrin-deficient human T cell to identify β1 integrin cytoplasmic domain sequences critical for integrin function. Mol Biol Cell. 1998;9:2715–2727. [PMC free article] [PubMed] [Google Scholar]
- Roote CE, Zusman S. Functions for PS integrins in tissue adhesion, migration and shape changes during early embryonic development in Drosophila. Dev Biol. 1995;169:322–336. doi: 10.1006/dbio.1995.1147. [DOI] [PubMed] [Google Scholar]
- Sakai T, de la Pena JM, Mosher DF. Synergism among lysophosphatidic acid, beta1A integrins, and epidermal growth factor or platelet-derived growth factor in mediation of cell migration. J Biol Chem. 1999;274:15480–15486. doi: 10.1074/jbc.274.22.15480. [DOI] [PubMed] [Google Scholar]
- Sakai T, Zhang Q, Fässler R, Mosher DF. Modulation of β1A integrin functions by tyrosine residues in the β1 cytoplasmic domain. J Cell Biol. 1998;141:527–538. doi: 10.1083/jcb.141.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner-Reckinger E, Gouon V, Melchior C, Plançon S, Kieffer N. Distinct involvement of β3 integrin cytoplasmic domain tyrosine residues 747 and 759 in integrin-mediated cytoskeletal assembly and phosphotyrosine signaling. J Biol Chem. 1998;273:12623–12632. doi: 10.1074/jbc.273.20.12623. [DOI] [PubMed] [Google Scholar]
- Schmitt DN, Brower DL. Intron dynamics and the evolution of integrin β subunit genes: maintenance of an ancestral gene structure in the coral, Acropora millepora. J Mol Evol. 2001;53:703–710. doi: 10.1007/s002390010257. [DOI] [PubMed] [Google Scholar]
- Stark KA, Yee GH, Roote CE, Williams EL, Zusman S, Hynes RO. A novel α integrin subunit associates with βPS and functions in tissue morphogenesis and movement during Drosophila development. Development. 1997;124:4583–4594. doi: 10.1242/dev.124.22.4583. [DOI] [PubMed] [Google Scholar]
- Stroeken PJ, van Rijthoven EA, Boer E, Geerts D, Roos E. Cytoplasmic domain mutants of β1 integrin, expressed in β1-knockout lymphoma cells, have distinct effects on adhesion, invasion and metastasis. Oncogene. 2000;19:1232–1238. doi: 10.1038/sj.onc.1203423. [DOI] [PubMed] [Google Scholar]
- Svineng G, Fassler R, Johansson S. Identification of β1C-2, a novel variant of the integrin β1 subunit generated by utilization of an alternative splice acceptor site in exon C. Biochem J. 1998;330:1255–1263. doi: 10.1042/bj3301255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani PD, Singh L, Auer KL, LaFlamme SE. The role of conserved amino acid motifs within the integrin β3 cytoplasmic domain in triggering focal adhesion kinase phosphorylation. J Biol Chem. 1997;272:7892–7898. doi: 10.1074/jbc.272.12.7892. [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Yaspan B, Ginsberg MH, Campbell ID. NMR analysis of structure and dynamics of the cytosolic tails of integrin αIIbβ3 in aqueous solution. Biochemistry. 2001;40:7498–7508. doi: 10.1021/bi010338l. [DOI] [PubMed] [Google Scholar]
- van Kuppevelt TH, Languino LR, Gailit JO, Suzuki S, Ruoslahti E. An alternative cytoplasmic domain of the integrin β3 subunit. Proc Natl Acad Sci USA. 1989;86:5415–5418. doi: 10.1073/pnas.86.14.5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignoud L, Albiges-Rizo C, Frachet P, Block MR. NPXY motifs control the recruitment of the α5β1 integrin in focal adhesions independently of the association of talin with the β1 chain. J Cell Sci. 1997;110:1421–1430. doi: 10.1242/jcs.110.12.1421. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Armulik A, Sakai T, Karlsson M, Fassler R, Schaefer EM, Mosher DF, Johansson S. The cytoplasmic tyrosines of integrin subunit β1 are involved in focal adhesion kinase activation. Mol Cell Biol. 2000;20:5758–5765. doi: 10.1128/mcb.20.15.5758-5765.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieschaus E, Noell E. Specificity of embryonic lethal mutations in Drosophila analyzed in germ line clones. Roux's Arch Dev Biol. 1986;195:63–73. doi: 10.1007/BF00444042. [DOI] [PubMed] [Google Scholar]
- Wieschaus E, Nusslein-Volhard C, Jurgens G. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster III. Zygotic loci on the X chromosome. Roux's Arch Dev Biol. 1984;193:296–307. doi: 10.1007/BF00848158. [DOI] [PubMed] [Google Scholar]
- Wilcox M. Genetic analysis of the Drosophila PS integrins. Cell Diff Dev. 1990;32:391–400. doi: 10.1016/0922-3371(90)90055-2. [DOI] [PubMed] [Google Scholar]
- Wright TRF. The phenogenetics of the embryonic mutant, lethal myospheroid, in Drosophila melanogaster. J Exp Zool. 1960;143:77–99. doi: 10.1002/jez.1401430107. [DOI] [PubMed] [Google Scholar]
- Wright TRF. Phenogenetics of temperature sensitive alleles of lethal myospheroid in Drosophila. Proc. 12th Int. Congr Genet. 1968;1:41. [Google Scholar]
- Xiong J-P, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott D, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin αvβ3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Miyamoto S. Integrin transmembrane signaling and cytoskeletal control. Curr Opin Cell Biol. 1995;7:681–689. doi: 10.1016/0955-0674(95)80110-3. [DOI] [PubMed] [Google Scholar]
- Yee GH. Identification and characterization of integrin receptor subunits in Drosophila melanogaster. Ph.D. Thesis. Cambridge, MA: Massachusetts Institute of Technology; 1993. [Google Scholar]
- Ylänne J, Huuskonen J, O'Toole TE, Ginsberg MH, Virtanen I, Gahmberg CG. Mutation of the cytoplasmic domain of the integrin β3 subunit. J Biol Chem. 1995;270:9550–9557. doi: 10.1074/jbc.270.16.9550. [DOI] [PubMed] [Google Scholar]
- Zavortink M, Bunch TA, Brower DL. Functional properties of alternatively spliced forms of the Drosophila PS2 integrin α subunit. Cell Adhes Commun. 1993;1:251–264. doi: 10.3109/15419069309097258. [DOI] [PubMed] [Google Scholar]
- Zusman S, Grinblat Y, Yee G, Kafatos FC, Hynes RO. Analyses of PS integrin functions during Drosophila development. Development. 1993;118:737–750. doi: 10.1242/dev.118.3.737. [DOI] [PubMed] [Google Scholar]