

Summary
Metastasis is the major cause of cancer death and the development of therapy resistance is common. The tumor microenvironment can confer chemotherapy resistance (chemoresistance), but little is known about how specific host cells influence therapy outcome. We show that chemotherapy induces neutrophil recruitment and Neutrophil Extracellular Trap (NET) formation, which reduces therapy response in mouse models of breast cancer lung metastasis. We reveal that chemotherapy-treated cancer cells secrete IL-1β, which in turn triggers NET formation. Two NET-associated proteins are required to induce chemoresistance: integrin-αvβ1, which traps latent TGFβ, and matrix metalloproteinase 9, which cleaves and activates the trapped latent TGFβ. TGFβ activation causes cancer cells to undergo epithelial-to-mesenchymal transition and correlates with chemoresistance. Our work demonstrates that NETs regulate activities of neighboring cells by trapping and activating cytokines and suggests that chemoresistance in the metastatic setting can be reduced or prevented by targeting the IL-1β-NET-TGFβ axis.
Graphical Abstract
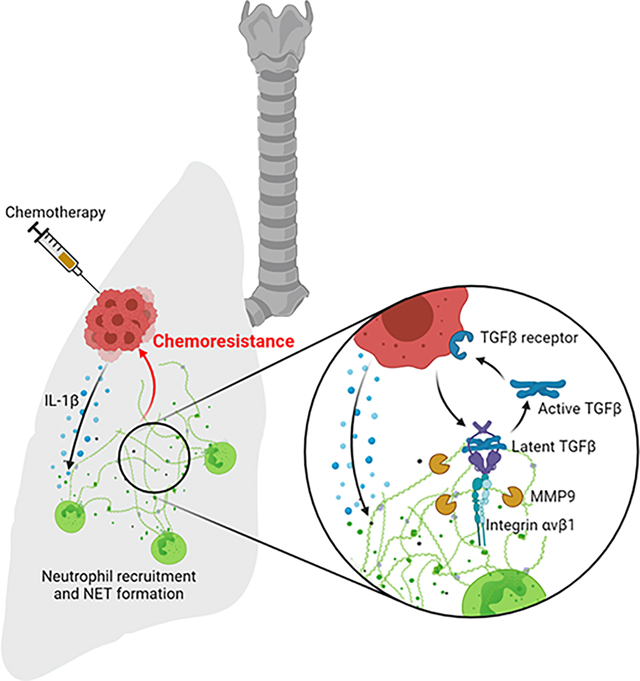
Mousset et al. show that chemotherapy promotes neutrophil extracellular trap (NET) formation which reduces therapy efficacy against breast cancer lung metastasis. NETs induce a TGFβ-dependent epithelial-mesenchymal transition (EMT) in cancer cells and pharmacological targeting of the NET/TGFβ-axis ameliorates chemotherapy efficacy.
Introduction
Breast cancer accounts for the highest incidence and death rates in women worldwide, and metastasis is responsible for most breast cancer-related deaths 1. Most patients with metastases are treated with chemotherapy, but in the majority of cases, resistance to chemotherapy develops and it is a major challenge in treating metastatic cancers 2,3. While cancer cell intrinsic factors, such as its genetic and epigenetic traits, play a role in therapy resistance, it is now well established that the tumor microenvironment (TME) modulates therapy response 4. Cancer extrinsic resistance is mediated by growth factors, cytokines and extracellular cellular matrix (ECM) proteins secreted by host cells. Among host cells, inflammatory cells, such as monocytes and macrophages, provide cues for the behavior of cancer cells during cancer progression 5 and have emerged as potent regulators of the therapeutic response in primary cancer 6–11. Interestingly, chemotherapy has itself been linked to inflammation, which is considered a common, persistent, and potentially debilitating complication of chemotherapy 12–14. Altogether, while inflammation has been linked to cancer progression, it is still unclear whether and how chemotherapy-induced inflammation is responsible for chemoresistance, and if so, by which mechanisms.
Despite the fact that neutrophils constitute the largest percentage of leukocytes in human blood and that they have well recognized roles in cancer progression, little is known about their roles in chemoresistance 15,16. Outside of cancer, neutrophils can kill harmful microorganisms via (i) phagocytosis; (ii) degranulation of cytotoxic enzymes and proteases into the extracellular space; or (iii) formation of neutrophil extracellular traps (NETs). NETs are scaffolds of DNA associated with cytotoxic enzymes and proteases — such as Neutrophil Elastase (NE) and Matrix MetalloProteinase 9 (MMP9) — that are released into the extracellular space. NETs are generated through a signaling process that involves citrullination of histones by the Protein Arginine Deiminase 4 (PAD4) enzyme, chromatin decondensation, and disintegration of the nuclear membrane 17. Contents from the neutrophil’s secretory granules — including NE and MMP9 — associate with the decondensed chromatin. The plasma membrane ruptures, and the protease-associated chromatin fibers are finally released into the extracellular space. Recently, we and others determined that NETs have pro-metastatic activities 18–22. In addition, NETs play a role in diseases involving tissue damage, including infectious and noninfectious inflammatory diseases, such as COVID-19, rheumatoid arthritis, and atherosclerosis 23,24. Neutrophils are rapidly recruited to damaged tissue and, through the release of NETs, neutrophils crosstalk with both innate and adaptive immune cells during acute and chronic inflammation 25. Chemotherapy, when effective, causes cell damage and death, and accordingly, high neutrophil infiltration is associated with poor response to chemotherapy in several cancer types 25–28. In this context, chemokine (C-X-C motif) ligand 1 (CXCL1), a neutrophil chemoattractant, has specifically been linked to chemoresistance in the metastatic setting 29. Identifying and understanding the potential roles of neutrophils and NETs, and more generally inflammation, in chemoresistance, could lead to the identification of novel ways to improve both chemotherapy efficacy and survival for patients with cancer.
Results
Neutrophil recruitment triggered by chemotherapy reduces treatment response of lung metastases
To determine how chemotherapy modifies the metastatic lung environment, syngeneic Balb/c mice were injected intravenously with 410.4 murine breast cancer cells and metastases were allowed to form for 7 days. Then, mice were treated with cisplatin or Adriamycin/Cyclophosphamide (AC) chemotherapies (Figure 1A). While chemotherapy caused no significant changes in the lungs of non-tumor bearing mice, we observed increased neutrophil recruitment in metastatic lungs as determined by flow cytometry (Figure 1B) and confirmed by immunofluorescence (Figures 1C and S1A). Interestingly, these changes in the lungs were not accompanied by differences in the number and the percentage of neutrophils, detected by flow cytometry, in the blood and in the bone marrow (Table S1). The marked increase in neutrophil infiltration in the lungs, led us to hypothesize that neutrophil recruitment might modulate therapy response. To test this hypothesis, we depleted neutrophils using Ly6G antibodies (Figures 1D, S1B and S1C) 30 which dramatically improved both cisplatin and AC treatment responses (Figures 1E, 1F and 1G). Neutrophil chemotaxis is mediated by the expression of C-X-C motif Chemokine Receptor 2 (CXCR2) 15. We found that in vitro cisplatin treatment upregulated two CXCR2 ligands, CXCL1 and CXCL5, in cancer cells (Figure S1D). Furthermore, conditioned media (CM) from cisplatin-treated 410.4 cancer cells (410.4 Cis CM) promoted neutrophil chemotaxis in vitro and this was abrogated by a CXCR2 inhibitor, and by CXCL1 and CXCL5 blocking antibodies (Figures S1E–G). In mice, the pharmacological inhibition of CXCR2 reduced chemotherapy-induced neutrophil recruitment in the lungs and improved chemotherapy treatment response (Figures 1E, 1H, 1I, S1H and S1I). Of note, neither neutrophil depletion nor CXCR2 inhibition given alone were able to significantly reduce metastases (Figures 1D–I).
Figure 1. Neutrophils recruited during chemotherapy counteract treatment efficacy.
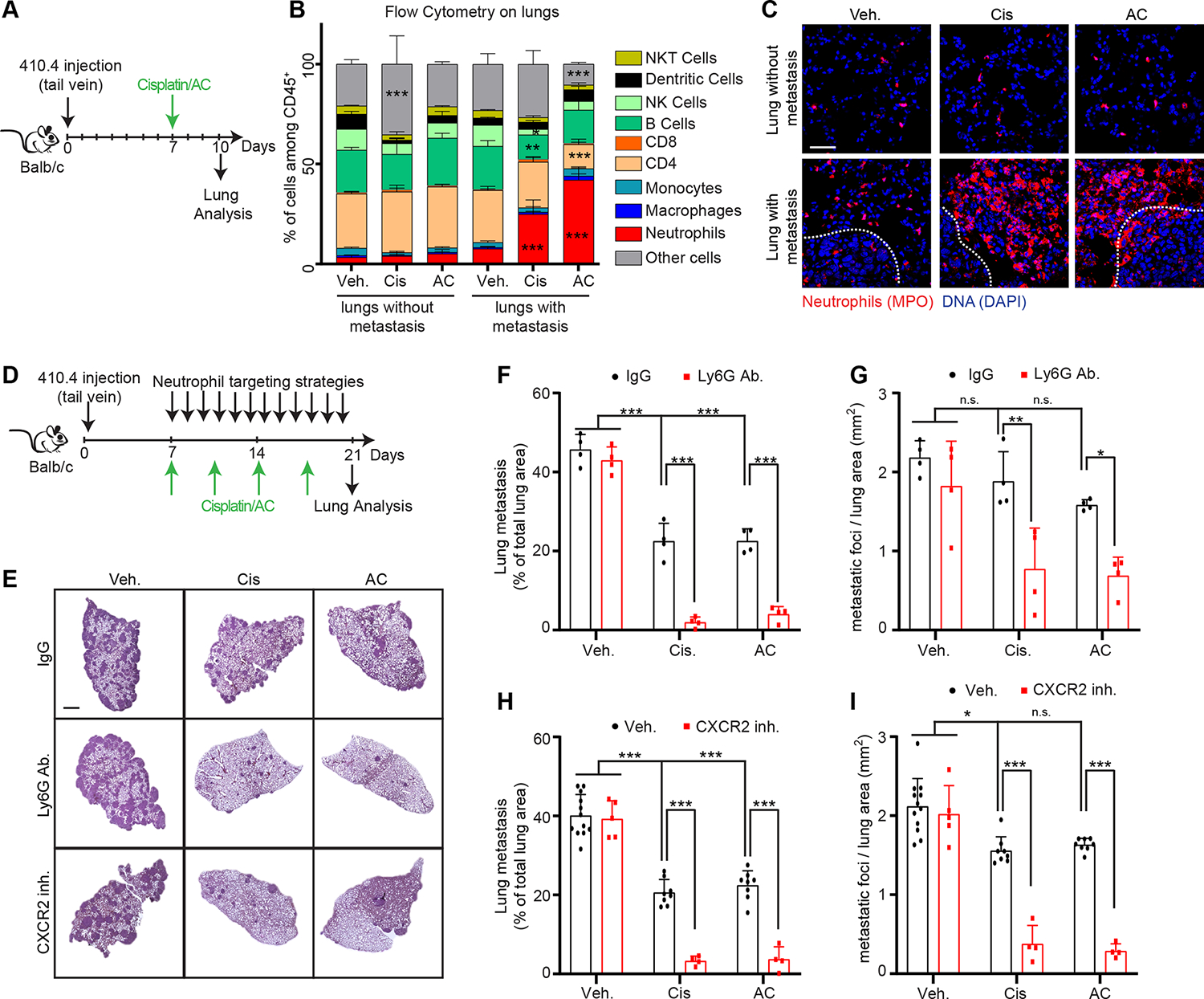
(A) Schematic showing experimental design. (B) Flow cytometry analysis showing the immune composition of lungs from mice injected or not with 410.4 cancer cells and treated as indicated in (A) (n=3 mice per group; mean±SD). (C) Representative immunostaining of neutrophils (red, myeloperoxidase [MPO]) and DNA (blue, DAPI) in the lungs of mice treated as indicated in (A). The tumor-stroma interface is outlined with a white dashed line. Scale bar: 50 μm. (D) Schematic showing experimental design. (E) Representative images of Hematoxylin and Eosin (H&E) staining of lungs from animals injected intravenously with 410.4 cells and treated as indicated in 1D. Scale bar: 1 mm. (F-I) Mice injected intravenously with 410.4 cells were treated as indicated, and the size (F, H) and the number of metastatic foci (G, I) in the lungs were quantified from H&E staining (in (F, G) n=4 mice per group; in (H, I) n=12 mice for the Veh. group, n=8 mice for the Cis and AC groups, n=5 mice for the CXCR2 inh. group and n=4 mice for the other groups; mean±SD).
Two-way ANOVA followed by Dunnett’s procedure where the Veh. treated non-tumor bearing mice group is the reference was used for (B), and two-way ANOVA followed by Tukey’s procedure was used for (F, G, H, I). See also Figure S1 and Table S1.
NETs promote chemoresistance of breast cancer lung metastases
To determine how neutrophils modulated chemotherapy response, we designed a co-culture system where 410.4 cancer cells were cultured with freshly isolated bone-marrow derived neutrophils (Figure 2A). In vitro co-culturing the breast cancer cells with primary neutrophils reduced the response to chemotherapy (Figures 2A and 2B). Interestingly, we noticed that neutrophils formed NETs when co-cultured with the 410.4 cells, but only in the co-cultures treated with chemotherapy (Figure 2A). However, neutrophils cultured alone did not form NETs when treated with chemotherapy (Figures S1J and S1K). Due to prior research showing that NETs promote cancer progression 31, we hypothesized that NETs modulated the therapy response. Indeed, treatment with a PAD4 inhibitor to block NET formation or DNase I to digest the NET-DNA scaffold, overcame neutrophil-mediated chemoresistance but had no effect on cancer cell numbers in the untreated conditions (Figure 2B). We have previously reported that some cancer cells (e.g., D2.A1 and 4T1) induce neutrophils to form NETs while others do not (e.g., D2.0R and 4T07) 18,32, and decided to test whether these differences could modulate the effect of NETs on chemotherapy efficacy. Interestingly, PAD4 inhibition ameliorated chemotherapy efficacy of all these cell lines in vitro (Table S2). Of note, chemotherapy had no effect on D2.0R cells when they were cultured on Matrigel to mimic cellular dormancy 18 (Table S2). Further supporting a direct role of the NETs on chemoresistance, the addition of NET-containing conditioned media (CM, induced with Phorbol Myristate Acetate [PMA], NET CM) also caused breast cancer cell resistance to chemotherapy (Figure 2C). In contrast, CM from unstimulated neutrophils (PMN CM) and CM from degranulated neutrophils (obtained using complement 5a; PMN C5a CM) had no effect on response to chemotherapy (Figure 2C). As observed for the co-culture experiments, targeting NETs during PMA-induced NET formation also overcame the chemoresistance induced by the NET CM in vitro (Figure 2C).
Figure 2. NETs formed during chemotherapy counteract treatment efficacy.
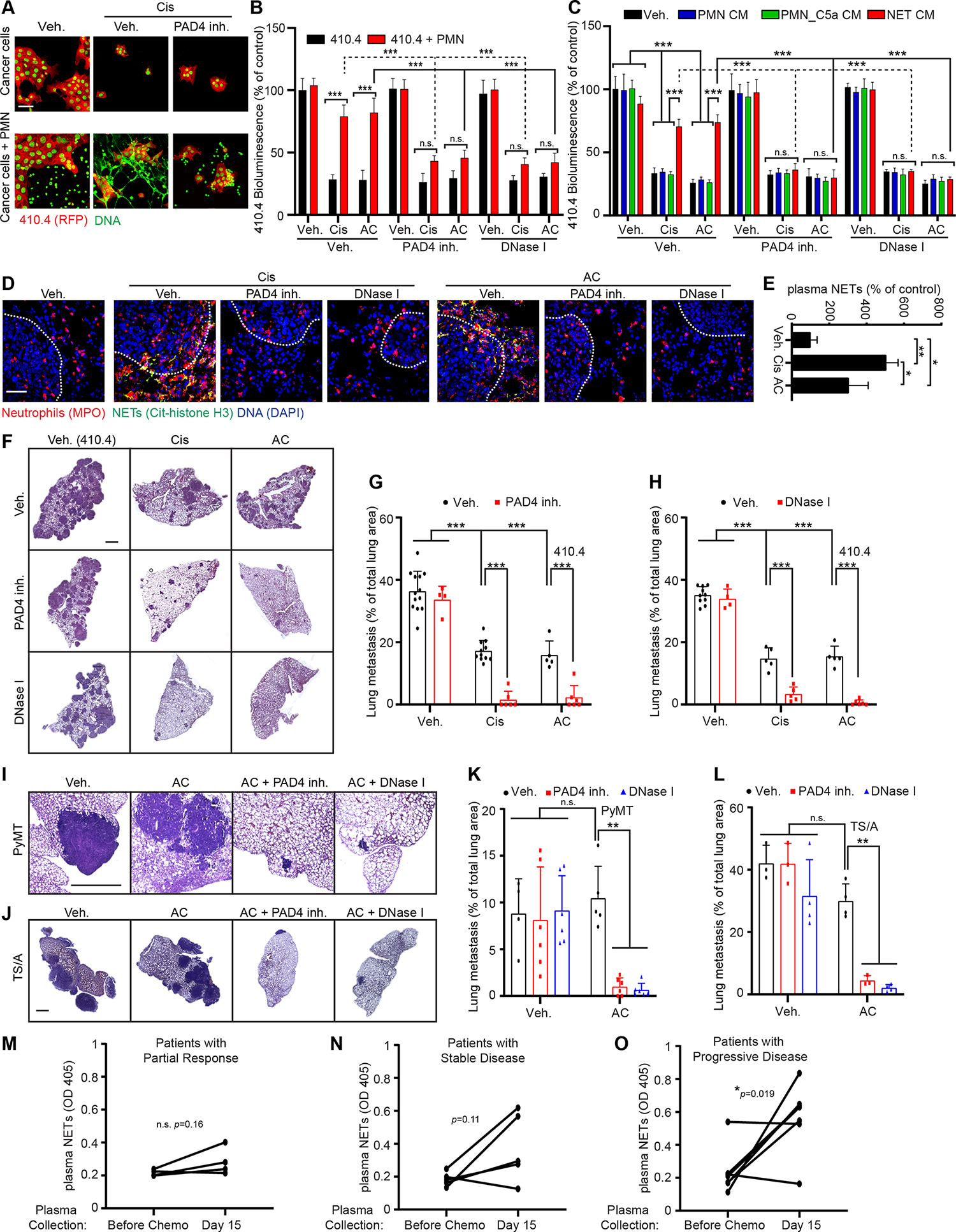
(A) mCherry-expressing 410.4 cells were cultured either alone or with neutrophils and treated as indicated for 48 hours. Images show immunostaining of mCherry (red) and DAPI (green). Cancer cells are displayed in red with a green nuclei and neutrophils/NETs in green. Scale bar: 50 μm. (B) Bioluminescence imaging (BLI) signal from luciferase-expressing 410.4 cells cultured as indicated at day 2 (n=3; mean±SD). (C) BLI signal from luciferase-expressing 410.4 cells at day 2 after indicated treatments. PAD4 inh. and DNase I were used during the neutrophil culture to block or digest NET formation, respectively (n=3; mean±SD). (D) Representative immunostaining of neutrophils (red, MPO), NETs (green, citrullinated histone H3 [cit-H3]), and DNA (blue, DAPI) in the lungs of mice treated as indicated. Scale bar: 50 μm. (E) ELISA quantification of NETs in the plasma of mice treated as indicated (n=3 mice per group; mean±SD). (F, I, J) Representative images of H&E staining of lungs from animals injected intravenously with 410.4 (F), PyMT (I) or TS/A (J) cells and treated as indicated. Scale bar: 1 mm. (G, H, K, L) Mice injected intravenously with 410.4 (G, H), PyMT (K) or TS/A (L) cells were treated for 21 days as indicated, and the lung metastatic burden was quantified from H&E staining (in (G) n=13 mice for the Veh. group, n=11 mice for the Cis group, n=5 for the AC group, n=4 mice for the PAD4 inh. group and n=6 mice for the other groups; in (H) n= 10 mice for the Veh. group, n=5 mice for the Cis, AC and DNase I + Cis groups, n=4 mice for the DNase I group and n=6 mice for the other groups; mean±SD; in (K) n=4 mice for the Veh. group, n=5 mice for the AC group and n=6 mice for the other groups; in (L) n=3 mice for the Veh., PAD4 inh. and AC + PAD4 inh. groups and n=4 mice for the other groups). (M-O) ELISA quantification of NETs in the plasma of patients treated as indicated. See also Table S5.
Two-way ANOVA followed by Tukey’s procedure was used for (B, C, G, H, K, L), one-way ANOVA followed by Tukey’s procedure was used for (E) and a two-sided t-test was used for (M-O). See also Figures S1, S2 and S3, Tables S2, S3, S4, S5 and S6.
In vivo, chemotherapy treatment also led to NET formation in the lungs (as detected by co-localized staining for citrullinated histone H3 and myeloperoxidase [MPO]) and the presence of NETs in the blood (as detected by ELISA from plasma samples) at day 21, four days after the last treatment (Figure 2D and 2E). We next collected plasma and lungs at different time points after chemotherapy treatment: in the lungs, increased neutrophil infiltration was first evident two days after chemotherapy, while NETs were detected in both lungs and plasma after three days, and present until five days after treatment (Figures S1L–Q). These results support that chemotherapy did not directly act on neutrophils to cause NETs. To determine whether NETs contributed to chemoresistance in vivo, we targeted NETs in our mouse model of lung metastasis (Figures 1D). Targeting NETs with either a PAD4 inhibitor or DNase I enhanced chemotherapy efficacy (Figures 2F–H, S1R and S1S). Long term PAD4 inhibition and DNase I treatment not only eliminated NETs in the metastatic lungs and in the plasma, but also reduced neutrophil recruitment to the lungs (Figures 2D, S1T–X). However, while short term PAD4 inhibition only led to a non-significant decrease of neutrophil recruitment, long term PAD4 inhibition decreased CXCL1 and CXCL5 levels in the metastatic lungs (Figures S2A–F). These results are consistent with previous reports suggesting that the presence of NETs promotes further neutrophil recruitment 18,21,33. Inhibiting NETs in the absence of chemotherapy had no effect on the metastatic burden of mice bearing 410.4 cancer cells (Figures 2F–H, S1R and S1S). This is consistent with the inability of 410.4 cells to directly induce pro-metastatic NETs, while inhibiting NETs reduces metastases of cancer cells that induce NETs (e.g., the murine breast cancer cell line 4T1) 32,34. We confirmed the specificity of NETs in chemoresistance by using Nexinhib20, a degranulation inhibitor. Nexinhib20 did not block chemotherapy-induced NET formation and accordingly did not ameliorate chemotherapy efficacy in vivo (Figures S2G–K). We next confirmed our results using two additional metastatic breast cancer cell lines, the TS/A and PyMT cell lines. In vitro, PAD4 inhibition counteracted neutrophil-mediated chemoresistance of TS/A and PyMT cancer cells (Figures S2L and S2M). In vivo, chemotherapy treatment of mice injected intravenously with TS/A and PyMT cells also led to NET formation in the lungs and targeting of NETs with either a PAD4 inhibitor or DNase I enhanced chemotherapy efficacy (Figures 2I–L and S2N–U).
The inhibitory effects of neutrophils and NETs on chemotherapy response at the lung metastatic site, prompted us to assess the effects of NETs on chemotherapy response of primary tumors (Figure S3A). Only a few neutrophils were present in the primary tumors formed by 410.4 cells (Figure S3B). We detected no NETs regardless of whether the mice were treated with chemotherapy (Figures S3B and S3C) and targeting NETs with a PAD4 inhibitor did not affect chemotherapy response at the primary site (Figures S3D). However, PAD4 inhibition ameliorated chemotherapy efficacy against spontaneous lung metastasis of mice bearing a primary tumor (Figures S3E–J). Interestingly, chemotherapy was more effective against primary tumors when compared to its efficacy against lung metastasis (Figures S3K). To confirm the effects of NETs on lung metastasis specifically, we next injected 410.4 cells both in the mammary fat pad and in the tail vein of the same mice. Using this model, we confirm our results that chemotherapy treatment induced neutrophil recruitment and NET formation specifically in the lungs and that targeting NETs improved greatly chemotherapy response of lung metastasis (Table S3). In this model, chemotherapy was also more efficient at the primary site than in metastatic lungs (Table S3). Thus, the effects of NETs on chemotherapy response are specific to lung metastatic disease in mice. Interestingly, CXCL1 and 5 were increased in the metastatic lungs but not in the primary tumor after chemotherapy treatment, suggesting that the absence of these factors might be responsible for the absence of neutrophil recruitment within the primary tumor following chemotherapy, which could explain chemotherapy selectivity between metastasis and primary tumors. (Table S4).
To evaluate the translational relevance of our findings, we analyzed plasma NETs levels from 16 patients with metastatic breast cancer before and two weeks after the first cycle of their second line chemotherapy. Our results showed a significative increase of plasma NETs level after chemotherapy in patients with chemoresistant disease (i.e. having progressive disease as best response), while the level of plasma NETs remained low in patients experiencing a partial tumor response (Figure 2M–O, Table S5). However, we observed two subgroups within the group of patients with stable disease (Figure 2M–O, Table S5). Additionally, we analyzed 18 primary tumors samples from patients with breast cancer before and after chemotherapy and NETs were not detected and only a few neutrophils were detected before and after chemotherapy (Figures S3L, S3M and Table S6). Altogether these results support the data obtained in mice indicating that, in patients, the presence of NETs correlates with chemoresistance of metastasis.
A known, severe complication of cisplatin administration is Acute Kidney Injury (AKI) 35, which limits the use of this drug in the clinics and can require patients to stop treatment. Evidence of AKI was observed in our mouse model with an increased concentration of plasma-creatinine and Blood Urea Nitrogen (BUN) following cisplatin treatment but not AC treatment (Figures S3N and S3O). NETs were detected in the kidney after cisplatin treatment and inhibiting NETs reduced kidney damage (Figures S3P–V and Table 1). Suggesting that cisplatin toxicity via NET formation is independent of tumor burden, we noticed that whereas chemotherapy did not trigger neutrophil recruitment and NET formation in the lungs of non-tumor bearing mice (Figures S3W and S3X), cisplatin treatment increased plasma NETs even in non-tumor bearing mice (Figures S3Y and Table 1). Accordingly, cisplatin also induced NET formation and kidney injury in non-tumor bearing mice and mice bearing a primary tumor and PAD4 inhibition reduced cisplatin-induced kidney damage in non-tumor bearing mice (Table 1). We did not detect NETs in the liver and in the spleen of mice following chemotherapy treatment, suggesting a specificity toward kidney injury (Table 1). Altogether, we found that inhibition of NETs dramatically increased the response of metastases to chemotherapy, and overcame kidney injury complications of the therapy, complications that can require cessation of treatment.
Table 1:
NETs participate in cisplatin-induced kidney injury
| Kidney neutrophils | Kidney damage | Plasma NETs | Liver neutrophils | Spleen neutrophils | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Conditions | Neutrophils | NETs | BUN | Creatinine | Neutrophils | NETs | Neutrophils | NETs | ||
| Non-tumor bearing mice | Veh. | - | - | ++ | ++ | - | - | - | - | - |
| Cis | ++ | ++ | ++ | ++ | + | - | - | - | - | |
| AC | N/A | N/A | N/A | N/A | N/A | - | - | - | - | |
| Cis + PAD4 inh. | + | - | - | - | - | N/A | N/A | N/A | N/A | |
| Cis + NLRP3 inh. | + | - | - | - | - | N/A | N/A | N/A | N/A | |
| Cis + IL1β Ab | + | - | - | - | - | N/A | N/A | N/A | N/A | |
| Cis + TGFβR1 inh. | ++ | ++ | ++ | ++ | + | N/A | N/A | N/A | N/A | |
| Cis + MMP9 inh. | ++ | ++ | ++ | ++ | + | N/A | N/A | N/A | N/A | |
| Mice with a primary tumor | Veh. | - | - | - | - | - | N/A | N/A | N/A | N/A |
| Cis | ++ | ++ | ++ | ++ | + | N/A | N/A | N/A | N/A | |
| AC | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| Mice with lung metastases | Veh. | - | - | - | - | - | - | - | - | - |
| AC | - | - | - | - | + | - | - | + | - | |
| Cis | ++ | ++ | ++ | ++ | ++ | - | - | + | - | |
| Cis + PAD4 inh. | + | - | - | - | - | N/A | N/A | N/A | N/A | |
| Cis + NLRP3 inh. | ++ | - | - | - | - | N/A | N/A | N/A | N/A | |
| Cis + IL1β ab. | + | - | - | - | - | N/A | N/A | N/A | N/A | |
| Cis + TGFβR1 inh. | ++ | ++ | ++ | ++ | + | N/A | N/A | N/A | N/A | |
| Cis + TGFβ ab. | N/A | N/A | ++ | ++ | + | N/A | N/A | N/A | N/A | |
| Cis + MMP9 inh. | ++ | ++ | ++ | ++ | + | N/A | N/A | N/A | N/A | |
| Cis + ITGαvβ1 | N/A | N/A | ++ | ++ | + | N/A | N/A | N/A | N/A | |
Non-tumor bearing mice, mice injected intravenously with 410.4 cancer cells and mice injected with 410.4 cancer cells in the mammary fat pad were treated for two weeks as indicated. Kidney, Liver and Spleen Neutrophils and NETs were quantified based on immunostaining for myeloperoxidase and Citrullinated-H3 respectively. For neutrophils: “-” indicates the basal level of neutrophils; “+” indicates a 2 to 3-fold increase; “++” indicates more than a 3-fold increase. For NETs: “-” indicates the absence of NETs, and “++” indicates the presence of NETs. For plasma NETs: “-” indicates the basal level of neutrophils; “+” indicates a 2 to 4-fold increase; “++” indicates more than a 4-fold increase. For BUN: “-” indicates a concentration lower than 25mg/dL and “++” indicates a concentration above 65 mg/dL. For creatinine: “-” indicates a concentration lower than 0.25 mg/dL and “++” indicates a concentration above 0.75 mg/dL. (n=3 mice per group; mean±SD). See also Figure S3, S4, S5, S7 and S8.
Chemotherapy triggers NLRP3-mediated IL-1β secretion in cancer cells, leading to NET formation and chemoresistance
We next sought to determine the mechanism driving NET formation after chemotherapy. Neither cisplatin nor AC chemotherapy directly induced primary neutrophils to form NETs (Figures S1J and S1K). However, CM from chemotherapy-treated cancer cells (410.4 Cis CM and 410.4 AC CM) promoted NET formation (Figures 3A and S4A), suggesting the presence of a NET-inducing, secreted factor released by cancer cells after chemotherapy. To identify candidate factors, we used a proteome cytokine array on the CM from chemotherapy-treated cancer cells (Figure 3B). As before (Figure S1D), our analysis showed upregulation of the neutrophil chemoattractants CXCL1 and CXCL5 but also revealed upregulation of interleukin-1β (IL-1β), specifically in the CM of chemotherapy-treated cancer cells (Figure 3B). Importantly, neutralization of CXCL1 and CXCL5 and inhibition of CXCR2 did not block NET formation induced by 410.4 Cis CM and 410.4 AC CM (Table S7). Consistent with prior reports 36–38, recombinant IL-1β triggered NET formation in vitro (Figures S4B and S4C). Furthermore, secretion of IL-1β by chemotherapy-treated cancer cells (410.4, TS/A, PyMT, D2.0R and 4T07 cancer cells) was confirmed by enzyme-linked immunoabsorbent assay (ELISA) and neutralization of IL-1β with an IL-1β blocking antibody inhibited NET formation induced by chemotherapy-treated cancer cells CM (Figures 3C, 3D, S4D–J and Table S2). Additionally, the IL-1β blocking antibody improved chemotherapy response of all these cell lines in the co-culture assay in vitro (Figure 3E and Table S2). As reported before, the CM from untreated D2.A1 and 4T1 cells was sufficient to induce NET formation, but the ability of these cells to induce NET formation was not dependent on IL-1β (Table S2). While both 4T1 and D2.A1 cells secreted IL-1β in response to chemotherapy, neutralization of IL-1β did not counteract NETs formation and therefore did not counteract chemoresistance in vitro (Table S2). This is in accordance with the fact that 4T1 cells secrete granulocyte colony stimulating factor (G-CSF) which induces NETs at the steady state 32. Interestingly, the IL-1β blocking antibody did not increase the chemotherapy response of 410.4 cancer cells when using NET CM, where the NETs had already been formed (Figure 3F). These data support a role for IL-1β in NET formation but not in the downstream effects causing chemotherapy resistance. Also, in the context of lung metastasis, the IL-1β blocking antibody inhibited NET formation, reduced neutrophil recruitment (Figures 3G–J) and improved chemotherapy response (Figures 3K–M). As observed with the PAD4 inhibitor (Figures S2A–D), short term inhibition of IL-1β only led to a non-significant decrease of neutrophil recruitment in the lungs (Figures S4K–M). However, the effect of IL-1β blockade on neutrophil recruitment was not direct as IL-1β blockade did not block neutrophil chemotaxis toward chemotherapy-treated cancer cells CM in vitro (Figure S4N). Interestingly, long term inhibition of PAD4 decreased the levels of IL-1β which were induced by chemotherapy within the metastatic lungs (Figure S4O). In addition, IL-1β blockade counteracted cisplatin-induced NET formation in the kidney of non-tumor bearing mice and mice with lung metastasis and improved kidney function following cisplatin treatment (Table 1, Figures S4P and S4Q).
Figure 3. Chemotherapy triggers the secretion of IL-1β by cancer cells, which drives NET formation.
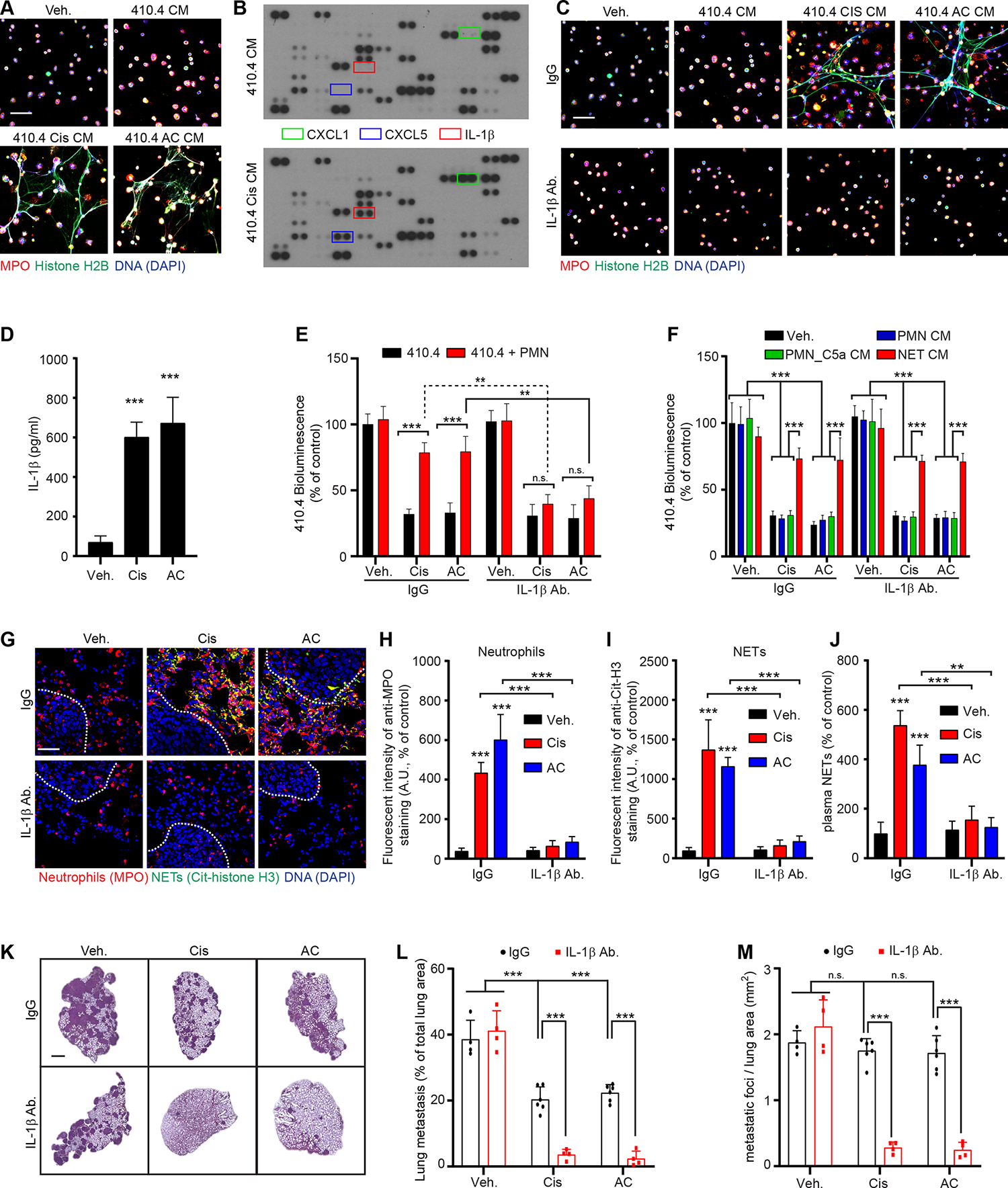
(A) Immunostaining of mouse neutrophils cultured as indicated. DAPI (blue), anti-MPO (red), and anti-histone H2B (green) staining were used to assess NET formation. Scale bar: 50 μm. (B) Protein content was investigated in the CM from vehicle- and chemotherapy-treated 410.4 cancer cells using protein arrays. (C) Immunostaining of mouse neutrophils cultured as indicated. DAPI (blue), anti-MPO (red), and anti-histone H2B (green) staining were used to assess NET formation. Scale bar: 50 μm. (D) ELISA quantification of IL-1β in the CM from 410.4 cells treated as indicated (n=3; mean±SD). (E) BLI from luciferase-expressing 410.4 cells cultured as indicated at day 2 (n=3; mean±SD). (F) BLI signal from luciferase-expressing 410.4 cells at day 2 after indicated treatments (n=3; mean±SD). (G) Representative immunostaining of neutrophils (red, MPO), NETs (green, cit-H3), and DNA (blue, DAPI) in the lungs of mice treated as indicated. Scale bar: 50 μm. (H, I) Quantification of images represented in (G) from the lungs of mice treated as indicated and stained for MPO (neutrophils, H) and cit-H3 (NETs, I) (n=3 mice per group; mean±SD). (J) ELISA quantification of NETs in the plasma of mice treated as indicated (n=3 mice per group; mean±SD). (K) Representative images of H&E staining of lungs from animals injected intravenously with 410.4 cells and treated as indicated. Scale bar: 1 mm. (L, M) Mice injected intravenously with 410.4 cells were treated for 21 days as indicated, and the size (L) and the number of metastatic foci (M) in the lungs were quantified from H&E staining (n=6 mice for the Cis and AC groups and n=4 mice for the other groups; mean±SD).
One-way ANOVA followed by Dunnett’s procedure where the Veh. group is the reference was used for (D), two-way ANOVA followed by Tukey’s procedure was used for (E, F, H, I, J, L, M). See also Figure S4 and Table S7.
It was unclear why chemotherapy-treated cancer cells would secrete IL-1β. IL-1β is generally secreted after activation of inflammasomes, large protein complexes assembled when cells sense danger, e.g., components such as those from dead cells 39. The assembly of the inflammasome complex activates caspase-1, which then cleaves pro-IL-1β to generate mature IL-1β 40. We determined that NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome and caspase-1 inhibition inhibited IL-1β secretion by chemotherapy-treated cancer cells (Figure 4A). NLRP3 inflammasome activation requires two signals: (i) a priming signal, which leads to Nuclear Factor-κ-light-chain-enhancer of activated B cells (NF-κB)-mediated inflammasome and pro-IL-1β transcription; and (ii) a danger signal, which leads to inflammasome assembly and caspase-1 activation. Constitutive activation of NF-κB signaling pathway in cancer cells has been well described in the past 41 and was identified in 410.4 cancer cells with the constitutive phosphorylation of IκB in the absence of any external signal (Figure S4R). Adenosine triphosphate (ATP) binding to its purigenic receptor P2X 7 (P2RX7) is a well-known NLRP3 danger signal and cancer cell death induced by chemotherapy causes the release of ATP into the extracellular space 42. We confirmed that chemotherapy treated 410.4 cancer cells released ATP (Figure 4B). In addition, we determined that both NF-κB and P2RX7 inhibition blocked chemotherapy-induced IL-1β secretion but had no effect on ATP release (Figures 4C and S4S). We therefore hypothesized that ATP released by dying chemotherapy-treated tumor cells triggers NLRP3 inflammasome activation in neighboring non-dying cancer cells. To test this hypothesis, we first generated the CM from cisplatin-treated cancer cells in which we identified the presence of ATP (Figure 4B). We next used this CM to treat naive cancer cells and used immunofluorescence for Apoptosis-associated Speck-like protein containing a CARD (ASC) assembly as a readout of inflammasome activation (Figures 4D and S4T) 43. CM from cisplatin-treated cancer cells triggered ASC assembly, and it was dependent on NF-κB, P2RX7, and NLRP3 activity. As expected, caspase 1 inhibition did not block ASC assembly as caspase-1 only mediates the cleavage of pro-IL-1β into IL-1β following ASC assembly (Figures 4D and S4T). Similarly to the blocking of IL-1β secretion, pharmacological inhibition of NF-κB, NRLP3, P2RX7 and caspase-1 counteracted the ability of cisplatin-treated cancer cells to promote NET formation (Figures 4E and S4U). In vitro, NLRP3 inhibition improved chemotherapy efficacy in the co-culture system (Figure 4F) but had no effect when NETs were already formed (Figure S4V). Additionally, NLRP3 inhibition blocked chemotherapy-induced NET formation in vivo and significantly improved both treatment efficacy and kidney function (Figures 4G–L, S4W–Y, and Table 1). Altogether, our results suggest that as chemotherapy induces cancer cell death, the dying cancer cells respond by releasing CXCL1 and CXCL5, which promote neutrophil recruitment, and ATP, which triggers NLRP3 inflammasome activation and IL-1β secretion from neighboring cancer cells. IL-1β then promotes NET formation and these NETs inhibit chemotherapy efficacy.
Figure 4. Chemotherapy triggers NLRP3 activation in cancer cells which drives IL-1β secretion.
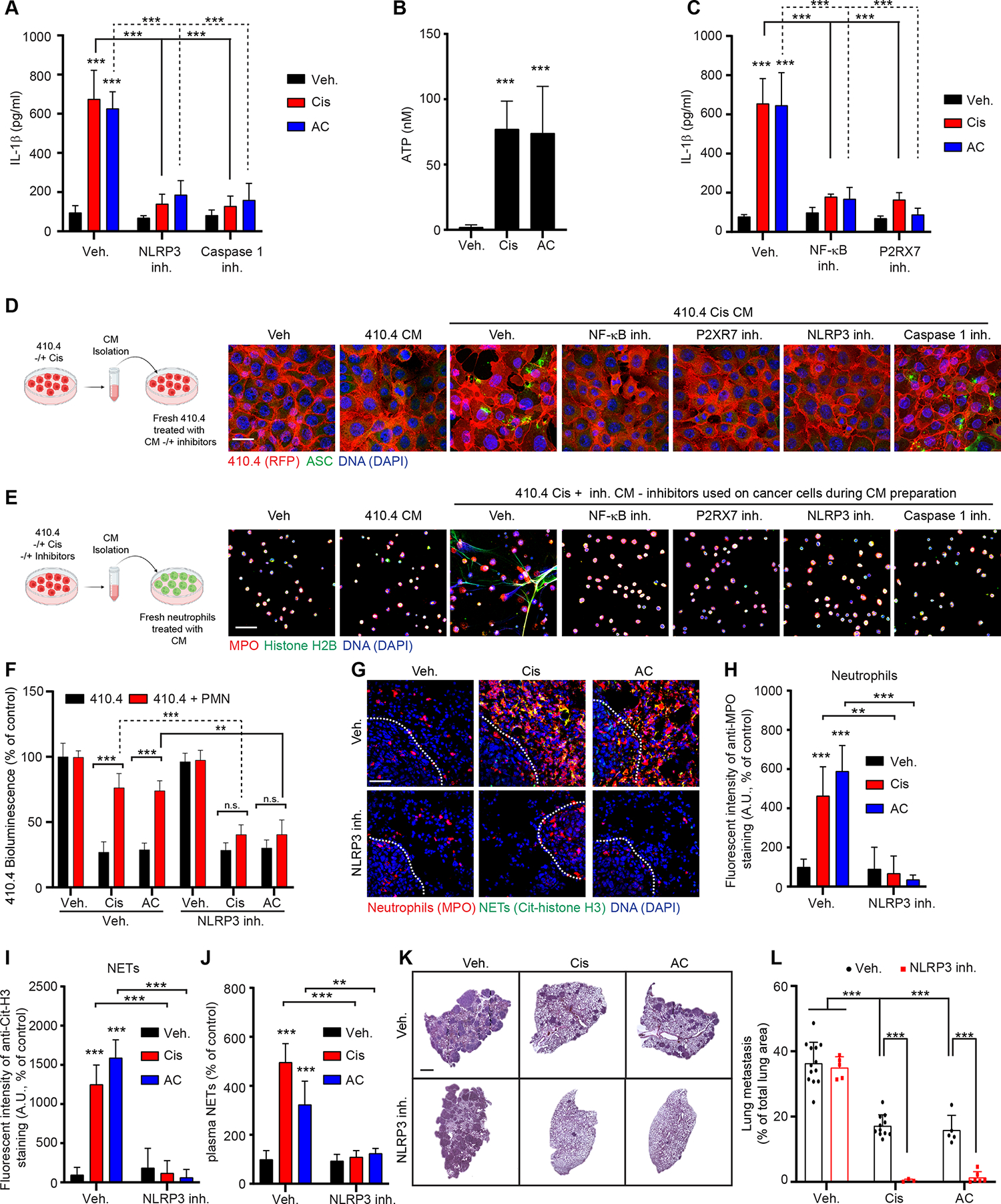
(A) ELISA quantification of IL-1β in the CM from 410.4 cells treated as indicated (n=3; mean±SD). (B) ATP quantification in the CM from 410.4 cells treated as indicated (n=3; mean±SD). (C) ELISA quantification of IL-1β in the CM from 410.4 cells treated as indicated (n=3; mean±SD). (D) Immunostaining of 410.4 cancer cells cultured as indicated on the left scheme and the different panels. DAPI (blue) and anti-ASC (green) staining were used to assess inflammasome oligomerization. Scale bar: 50 μm. Schematic created with BioRender.com. (E) Immunostaining of mouse neutrophils cultured as indicated on the left scheme and different panels. DAPI (blue), anti-MPO (red), and anti-histone H2B (green) staining were used to assess NET formation. Scale bar: 50 μm. Schematic created with BioRender.com. (F) BLI from luciferase-expressing 410.4 cells as indicated at day 2 (n=3; mean±SD). (G) Representative immunostaining of neutrophils (red, MPO), NETs (green, cit-H3), and DNA (blue, DAPI) in the lungs of mice treated as indicated. Scale bar: 50 μm. (H, I) Quantification of images represented in (G) from the lungs of mice treated as indicated and stained for MPO (neutrophils, H) and cit-H3 (NETs, I) (n=3 mice per group; mean±SD). (J) ELISA quantification of NETs in the plasma of mice treated as indicated. (n=3 mice per group; mean±SD). (K) Representative images of H&E staining of lungs from animals injected intravenously with 410.4 cells treated as indicated. Scale bar: 1 mm. (L) Mice injected intravenously with 410.4 cells were treated as indicated, and the lung metastatic burden was quantified from H&E staining (n=13 mice for the veh. group, n=11 mice for the Cis group, n=5 mice for the AC and NLRP3 inh. group, n=3 mice for the NLRP3 inh. + Cis group and n=6 mice for the NRLP3 inh. + AC group; mean±SD). The same Veh., Cis and AC groups were used for mice with 410.4 cells and treated with the NLPR3 inh. and the PAD4 inh.
Two-way ANOVA followed by Tukey’s procedure was used for (A, C, F, H, I, J, L), and one-way ANOVA followed by Dunnett’s procedure where the Veh. group is the reference was used for (B). See also Figure S4.
NETs activate TGFβ signaling pathway in cancer cells, causing EMT and chemoresistance
To identify the signaling pathways mediating chemoresistance following NET formation, we performed RNA-sequencing of 410.4 cells stimulated with NET CM. This analysis identified Transforming Growth Factor β (TGFβ) signaling pathway and EMT related signaling pathways as two of the most significant pathways to be increased (Figure 5A). EMT causes chemoresistance in several cancer types, including breast cancer, and Transforming Growth Factor β (TGFβ) is a major inducer of EMT 44. qRT-PCR and Western blot analysis revealed that NETs and TGFβ induced a similar upregulation of most mesenchymal markers analyzed in the cancer cells, but NETs and TGFβ induced the downregulation of only Claudin 1 among the epithelial markers tested (Figures 5B, S5A and S5B). Accordingly, NETs induced SMAD family member 2 (SMAD2) phosphorylation in cancer cells, indicating TGFβ signaling activation, and TGFβ Receptor 1 (TGFβR1) inhibition inhibited NET-mediated SMAD2 phosphorylation and EMT genes expression (Figure 5B). In addition, targeting TGFβ signaling improved chemotherapy response of cancer cells in vitro (Figures 5C and 5D) and in vivo (Figures 5E–K, S5C–F). Within the lung tissue, chemotherapy increased the expression of the mesenchymal marker N-cadherin and decreased the expression of the epithelial marker Claudin 1 in cancer cells, only in the presence of NETs (Figures 5L–O). Indeed, this switch was abrogated when targeting NETs with a PAD4 inhibitor and DNase I (Figures 5L–O). The same effect was observed after TGFβR1 inhibition, consistent with the role of TGFβ in NET-induced EMT (Figures 5L–O). Interestingly, while recombinant TGFβ had no effect on NET formation in vitro, targeting TGFβ led to decreased neutrophil recruitment and NET formation in vivo (Figures S5G–P). Interestingly, TGFβR1 inhibition did not block chemotaxis of neutrophils toward 410.4 AC CM in vitro (Figure S5Q) and short-term inhibition of TGFβR1 in vivo did not block the effect of chemotherapy on neutrophil recruitment and NET formation (Figures S5R–T). This suggests that targeting TGFβ signaling indirectly limits lung inflammation which further improves chemotherapy treatment. This is in accordance with the pleiotropic role of TGFβ signaling in immunity and cancer 45. Accordingly, TGFβR1 inhibition decreased the levels of CXCL1, CXCL5 and IL-1β in the metastatic lungs following two weeks of chemotherapy (Figures S5U–W). Interestingly, TGFβ-targeting strategies did not block cisplatin-induced NET formation in the kidney and did not reduce cisplatin-induced kidney damage, suggesting that the effect of NETs on kidney injury are not TGFβ dependent (Figures S5X–AA and Table 1). Altogether, these results indicate that NETs induce a TGFβ-dependent EMT in cancer cells which correlates with chemoresistance.
Figure 5. NETs promote chemoresistance through a TGFβ-dependent EMT in cancer cells.
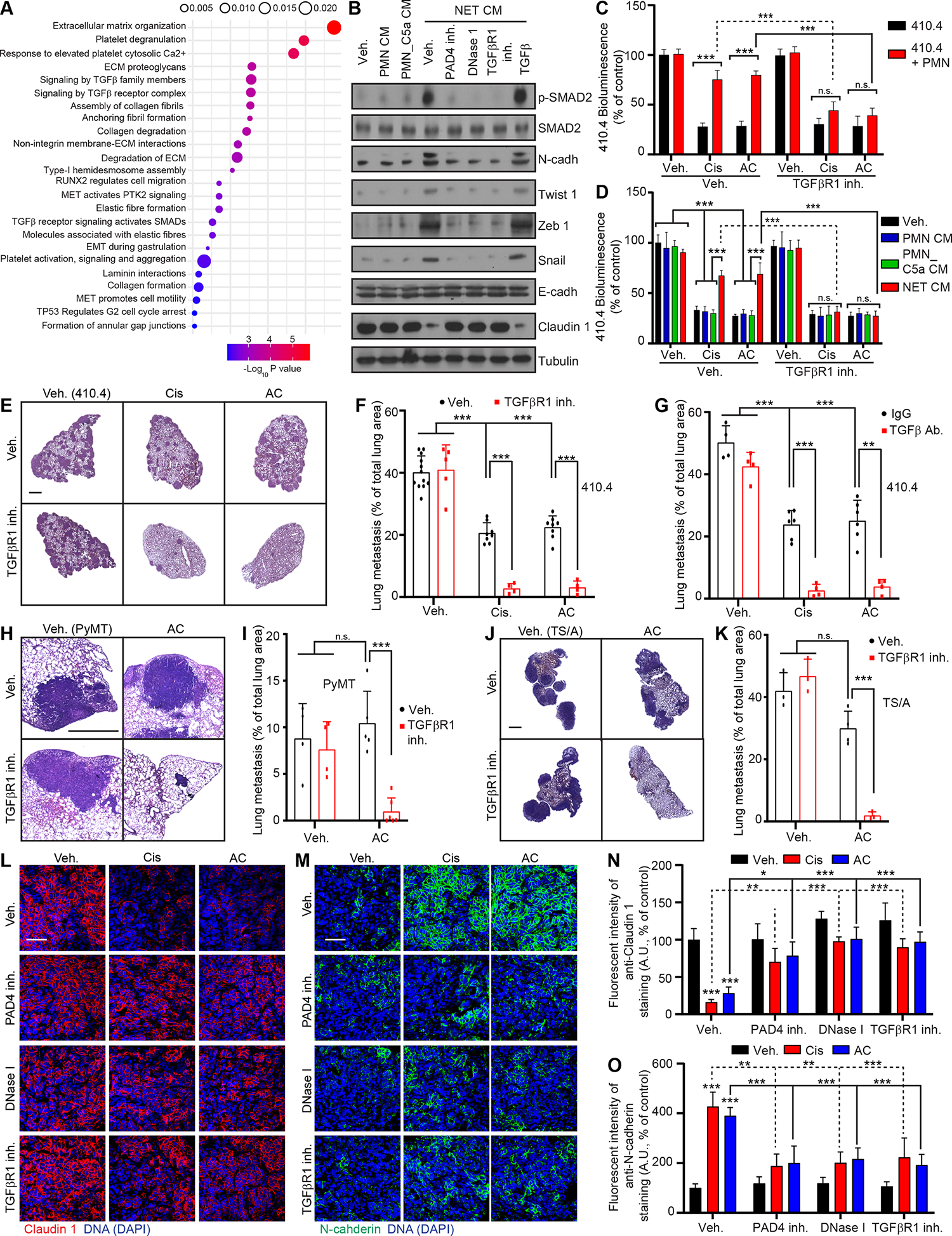
(A) Gene ontology analysis of the genes that were uniquely up-regulated in 410.4 cells treated with NET CM for 48 hours. (B) Western blot for protein listed in 410.4 cells after 48 hours of indicated treatments. Results are representative of 2 independent experiments. (C) BLI signal from luciferase-expressing 410.4 cells cultured as indicated at day 2 (n=3; mean±SD). (D) BLI signal from luciferase-expressing 410.4 cells at day 2 after indicated treatments (n=3; mean±SD). (E, H, J) Representative images of H&E staining of lungs from animals injected intravenously with 410.4 (E), PyMT (H) or TS/A (J) cells treated as indicated. Scale bars: 1 mm. (F, G, I, K) Mice injected intravenously with 410.4 (F, G), PyMT (I) or TS/A (K) cells were treated for 21 days as indicated, and the lung metastatic burden was quantified from H&E staining (in (F) n=12 mice for the Veh. group, n=8 mice for the Cis and AC groups, n=5 mice for the TGFβR1 inh. group, n=4 mice for the other groups (the same Veh., Cis and AC groups were used for mice with 410.4 cells and treated with the CXCR2 inh. and the TGFβR1 inh.); in (G) n=6 mice for the Cis and AC groups and n=4 mice for the other groups; in (I) n= 4 mice for the Veh. and TGFβR1 inh. groups, n=5 mice for the AC group and n=6 mice for the AC + TGFβR1 inh. group (the same Veh. and AC groups were used for mice with PyMT cells and treated with the PAD4 inh., DNase I and the TGFβR1 inh.); in (K) n=4 mice for the AC group and n=3 mice for the other groups (the same Veh. and AC groups were used for mice with TS/A cells and treated with the PAD4 inh., DNase I and the TGFβR1 inh.); mean±SD). (L, M) Representative immunostaining of Claudin 1 (red, L), N-cadherin (green, M), and DNA (blue, DAPI) in the lung metastasis of mice injected intravenously with 410.4 cells and treated as indicated. Scale bar: 50 μm. (N, O) Quantification of images represented in (L) and (M) (n=3 mice per group; mean±SD).
Two-way ANOVA followed by Tukey’s procedure was used for (C, D, F, G, I, K, N, O). See also Figure S5.
NET-associated MMP9 activates latent TGFβ which counteracts chemotherapy efficacy
Our results suggested that TGFβ plays a role in NET-mediated chemoresistance. TGFβ is synthesized and secreted as a latent complex which is then processed to its active form 46. Using an ELISA test to detect both total- and active- TGFβ, we found a large amount of latent-TGFβ in the CM from 410.4 cancer cells, but not in the CM from neutrophils including after NET formation (Figure 6A). Active-TGFβ was barely detectable in all CM (Figure 6A). Chemotherapy did not change the levels of active and total TGFβ present in the CM from cancer cells (Figure 6A). However, when we incubated the CM from 410.4 cells with the CM from neutrophils, NETs activated latent TGFβ released by 410.4 cells (Figure 6B). NETs did not modulate transcription of TGFβ family members or receptors in 410.4 cells (Figure S6A) but targeting of NETs abrogated the activation of latent TGFβ (Figures 6B).
Figure 6. NET-associated MMP9 activates latent TGFβ which counteracts chemotherapy efficacy.
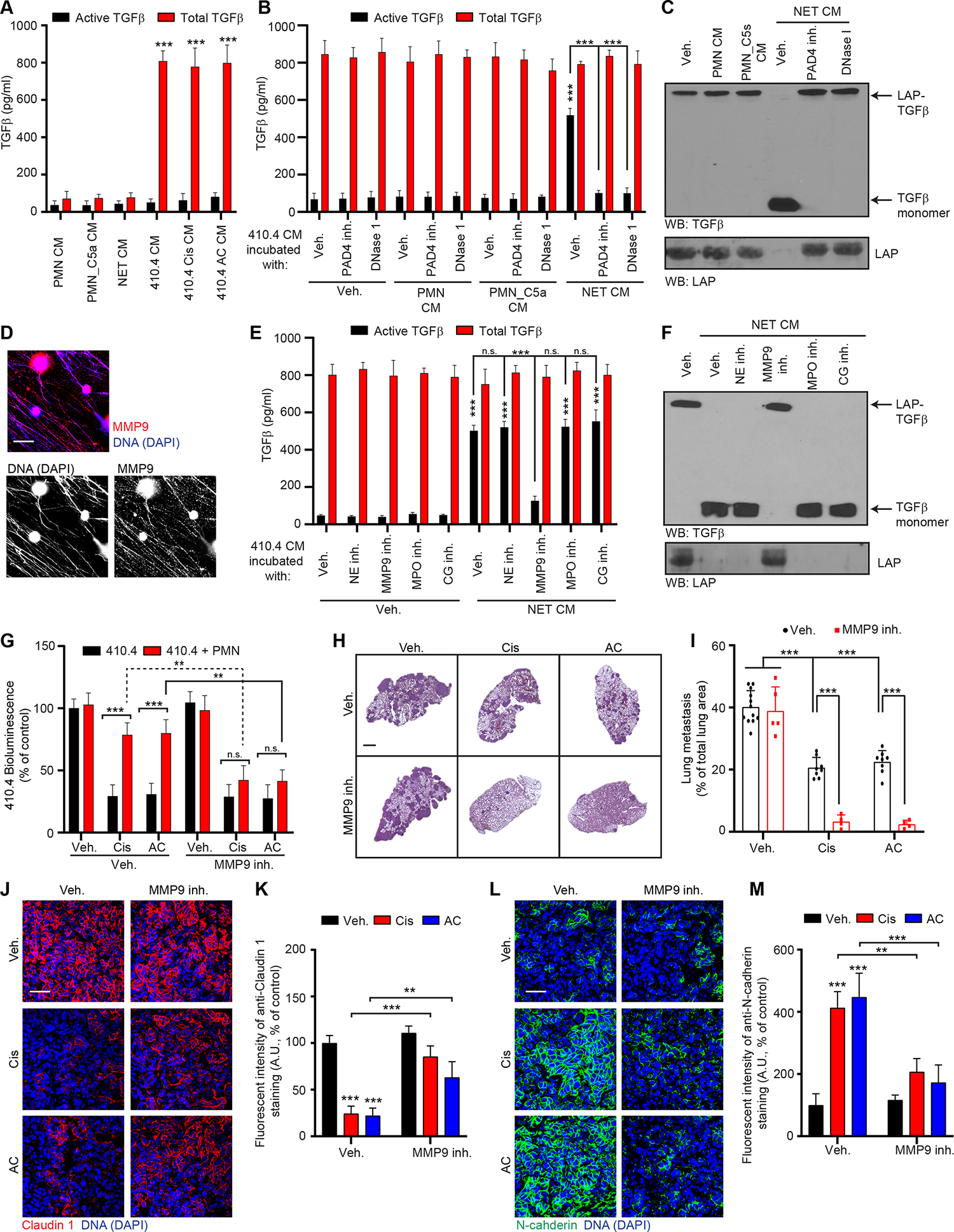
(A) ELISA quantification of total and active TGFβ secreted in indicated CM (n=3; mean±SD). (B) ELISA quantification of total and active TGFβ in the CM from 410.4 cells incubated as indicated for 24 hours. PAD4 inh. and DNase I were used during the neutrophil culture to block or digest NET formation, respectively (n=3; mean±SD). (C) Western blot for TGFβ and LAP in the CM of 410.4 cells after 24 hours of indicated treatments. Results are representative of 2 independent experiments. (D) Immunostaining of mouse neutrophils cultured with IL-1β. DAPI (blue) and MMP9 (red) localized on the DNA scaffold of NETs. Scale bar: 50 μm. (E) ELISA quantification of total and active TGFβ in the CM from 410.4 cells incubated as indicated for 24 hours (n=3; mean±SD). (F) Western blot for TGFβ and LAP in the CM of 410.4 cells after 24 hours of indicated treatments. Results are representative of 2 independent experiments. (G) BLI signal from luciferase-expressing 410.4 cells cultured as indicated at day 2 (n=3; mean±SD). (H) Representative images of H&E staining of lungs from animals treated as indicated. Scale bar: 1 mm. (I) Mice injected intravenously with 410.4 cells were treated for 21 days as indicated, and the lung metastatic burden was quantified from H&E staining (n=12 mice for the Veh. group, n=8 mice for the Cis and AC groups, n=5 mice for the MMP9 inh. group and n=4 mice for the other groups; mean±SD). The same Veh., Cis and AC groups were used for mice with 410.4 cells and treated with the CXCR2 inh., the TGFβR1 inh. and the MMP9 inh. (J, L) Representative immunostaining of Claudin 1 (red, J), N-cadherin (green, L), and DNA (blue, DAPI) in the lungs of mice injected intravenously with 410.4 cells and treated as indicated. Scale bar: 50 μm. (K, M) Quantification of images represented in (J) and (L) from the lungs of mice injected intravenously with 410.4 cells, treated as indicated and stained for Claudin 1 (K) and N-cadherin (M) (n=3; mean±SD).
Two-way ANOVA followed by Tukey’s procedure was used for (A, B, E, G, I, K, M). See also Figures S6 and S7.
Latent TGFβ is secreted either as a Small Latent Complex (SLC), associated with Latency Associated Protein (LAP) or as a Large Latent Complex (LLC), associated with LAP and Latent TGFβ Binding Protein (LBTP) 47. LBTP was not detected in cell lysate, ECM, or CM from 410.4 cancer cells, but the LAP-TGFβ complex was detectable in all three isolates of the cancer cells (Figures S6B and S6C). In agreement with the ELISA results (Figure 6B), Western Blot revealed that incubation of cancer cell CM with NETs led to the degradation of LAP and the release of active TGFβ and targeting NETs abrogated LAP degradation (Figure 6C). In contrast, CM from unstimulated and degranulated neutrophils had no effect (Figures 6B and 6C). Activation of latent TGFβ into active TGFβ was also observed when culturing cancer cells with NET CM or after chemotherapy treatment when using a co-culture system and targeting NETs abrogated the activation of TGFβ also under these conditions (Figures S6D and S6E).
NETs are characterized by the association of neutrophil proteases with the DNA scaffold 48,49. MMP9 can proteolytically activate TGFβ and is present within the NET-DNA scaffold (Figure 6D) 18,50. Using inhibitors against some of the major NET-associated proteases - Neutrophil Elastase (NE), MMP9 and Cathepsin G - and MPO (which indirectly can activate TGFβ activation 51), we found that only MMP9 inhibition inhibited NET-mediated TGFβ activation (Figures 6E, 6F, S6F and S6G) and chemoresistance in vitro (Figures 6G and S6H). MMP9 inhibition also sensitized cancer cells to cisplatin and AC chemotherapy in vivo (Figures 6H, 6I and S6I). Accordingly, MMP9 inhibition counteracted NET-mediated EMT in vitro (Figure S7A) and in vivo (Figures 6J–M). MMP9 inhibition additionally decreased the number of neutrophils and NETs in the lungs (Figures S7B–E) but did not block the ability of chemotherapy-treated cancer cells to promote NET formation in vitro (Figures S7F and S7G). In addition, as observed with TGFβR1 inhibition, MMP9 inhibition did not block the chemotaxis of neutrophils toward 410.4 AC CM in vitro (Figure S7H), and short-term inhibition of TGFβR1 in vivo did not block the effect of chemotherapy on neutrophil recruitment and NET formation (Figures S7I–K). These results suggest that TGFβ signaling indirectly limits inflammation within the metastatic lungs. As observed with TGFβ-inhibiting strategies, MMP9 inhibition did not reduce cisplatin-induced NET formation within the kidney and did not reduce kidney damage (Figures S7L and S7M and Table 1). Altogether, our results show that NET-associated MMP9 can activate latent TGFβ, secreted by cancer cells, and that this activation induces EMT in cancer cells and correlates with chemoresistance.
NET-associated ITGαvβ1 traps latent TGFβ
NET-mediated proteolytic activation of TGFβ was required for cancer cell resistance to treatment and digesting NETs with DNase I improved treatment efficacy in vivo and in vitro (Figures 2B, 2C, 2H, 2K, 2L and S1S). Yet, we previously showed that DNase I digestion of NETs does not reduce NET-MMP9 activity 18. Therefore, we tested whether the NET-DNA scaffold would contribute to TGFβ activation through different means. While 410.4 cancer cells expressed the NET-DNA receptor ccdc25, siRNA against ccdc25 20 did not counteract neutrophil-mediated chemoresistance in vitro (Figures S8A and S8B). We therefore decided to focus on integrins (ITG) which are large transmembrane proteins, but parts of them are also found in NETs 52. Moreover, integrins, including ITGαvβ1, have been shown to bind latent TGFβ through the LAP-RGD domain 46,47. We hypothesized that latent TGFβ was trapped and then processed within the NET-DNA scaffold. We found that ITGαvβ1 and MMP9 were both present within the NET-DNA scaffold by immunofluorescence (Figures 7A and 7B). A potent and highly specific inhibitor of ITGαvβ1 has been developed 53, and this inhibitor, as well as blocking antibodies against ITGαvβ1 and a RGD peptide inhibited TGFβ activation and LAP degradation (Figures 7C, 7D and S8C), indicating that NET-associated ITGαvβ1 is necessary for TGFβ activation by NET-associated MMP9. Next, using NETs incubated with CM from chemotherapy treated cancer cells, we showed that TGFβ, secreted by cancer cells, was trapped within the NET-DNA scaffold and this trapping was prevented when targeting ITGαvβ1 (Figures 7E and 7F). In addition, and as observed with TGFβR1 and MMP9 inhibition, ITGαvβ1 inhibition counteracted NET-induced EMT in vitro (Figure S8D). Accordingly, targeting ITGαvβ1 improved chemotherapy response both in vitro (Figures 7G and S8E) and in vivo (Figures 7H–J) and reduced neutrophil recruitment and NET formation (Figures S8F–I). However, ITGαvβ1 targeting did not block NET formation induced by chemotherapy-treated cancer cell CM in vitro (Figures S8J and S8K). Targeting ITGαvβ1 in vivo did not limit cisplatin-induced kidney injury (Figures S8L, S8M and Table 1) suggesting that the activation of TGFβ by NETs is not responsible for NET-mediated kidney injury. Altogether, our results suggest that NETs act as a scaffold that traps cancer-cell derived latent TGFβ, which can then be cleaved efficiently by NET-associated MMP9 and released as active TGFβ to promote chemoresistance.
Figure 7. NET-associated ITGαvβ1 traps latent TGFβ.
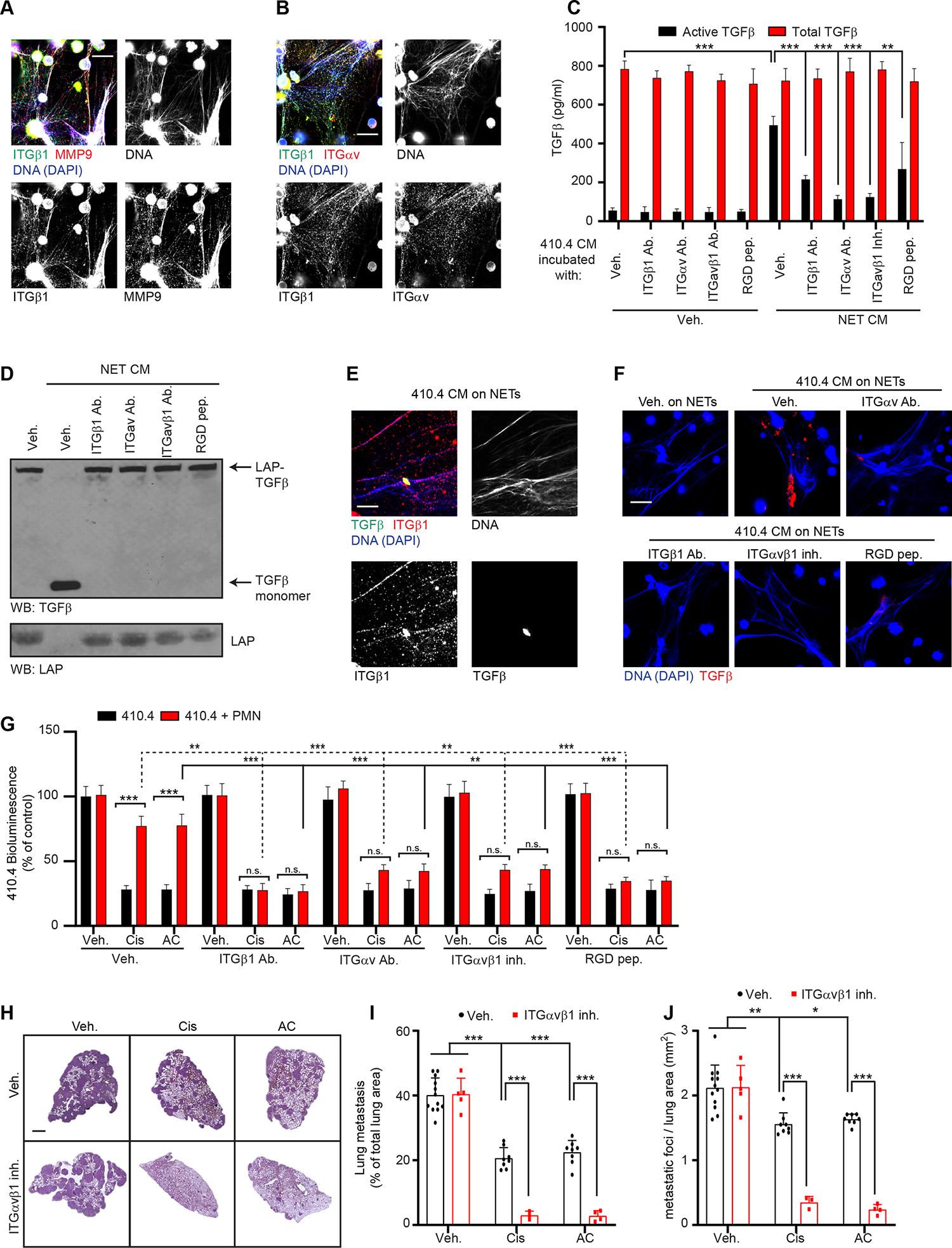
(A) Immunostaining of mouse neutrophils cultured with IL-1β. DAPI (blue), ITGβ1(green) and MMP9 (red). Scale bar: 50 μm. (B) Immunostaining of mouse neutrophils cultured with IL-1β. DAPI (blue), ITGβ1 (green) and ITGαv (red). Scale bar: 50 μm. (C) ELISA quantification of total and active TGFβ in the CM from 410.4 cells incubated as indicated for 24 hours (n=3; mean±SD). (D) Western blot for TGFβ and LAP in the CM of 410.4 cells after 24 hours of indicated treatments. Results are representative of 2 independent experiments. (E, F) NETs were formed from IL-1β-treated murine neutrophils and 410.4 CM were incubated on top of the NETs for 30 min with the indicated treatment before fixation and immunostaining. (E) DAPI (blue), TGFβ (green) and ITGβ1 (red). (F) DAPI (blue) and TGFβ (red). Scale bar: 50 μm. (G) BLI signal from luciferase-expressing 410.4 cells cultured as indicated at day 2 (n=3; mean±SD). (H) Representative images of H&E staining of lungs from animals treated as indicated. Scale bar: 1 mm. (I, J) Mice injected intravenously with 410.4 cells were treated for 21 days as indicated, and the size (I) and the number of metastatic foci (J) in the lungs were quantified from H&E staining. (n=12 mice for the Veh. group, n=8 mice for the Cis and AC groups, n=5 mice for the ITGαvβ1 inh. group, n=3 for the ITGαvβ1 + Cis group and n=4 mice for the ITGαvβ1 + AC group; mean±SD). The same Veh., Cis and AC groups were used for mice with 410.4 cells and treated with the CXCR2 inh., the TGFβR1 inh., the MMP9 inh. and the ITGαvβ1 inh.
Two-way ANOVA followed by Tukey’s procedure was used for (C, G, I, J). See also Figure S8.
Discussion
The major obstacles to the cure of metastatic breast cancer are metastasis and the emergence of resistance to chemotherapy, two phenomena that are closely linked clinically 54. In this context, we found that chemotherapy induces NET formation, which promotes chemoresistance of breast cancer lung metastasis in mice. Interestingly, it was recently shown that NETs promote radiation resistance to bladder cancer through unknown mechanisms 55 and that radiation injury to the healthy lung tissue enhances neutrophil-dependent metastatic colonization in mice 56. While recent studies identified that NETs can promote EMT 57–61, we here unveil the molecular mechanism of this promotion, via the binding and the activation of latent TGFβ by two NET-associated proteins: 1) ITGαvβ1, which traps latent TGFβ; and 2) MMP9, which cleaves and activates latent TGFβ. While we focused on cancer cells as the main source of latent TGFβ in our study, we cannot exclude that stromal cells within the metastatic TME also release TGFβ that can be trapped and activated by NETs. TGFβ has pleiotropic effects on many cell types in vivo, with both physiological and pathological consequences 62; therefore this function of NETs could have broad implications in development and disease progression.
Recently, NETs have emerged as targets in the treatment of non-infectious diseases such as cancer 17 highlighting the importance of identifying the mechanisms responsible for NET formation. The chemotherapy did not act directly on the neutrophils, rather our study reveals that the release of ATP from chemotherapy-treated, dying cancer cells drives NLRP3-mediated IL-1β secretion by other cancer cells, and that IL-1β in turn triggers NET formation. This is consistent with prior reports highlighting the importance of IL-1β in NET formation 36–38. In addition, our study re-affirms the role of NETs as a proteolysis scaffold, as we and others described previously 18,63. Our data suggest that the NET scaffold, through associated integrins, acts as a specialized local site that allows NET-proteases to cleave their substrate efficiently.
Interestingly, NET inhibition had no effect on the efficacy of chemotherapy against murine primary breast tumor, where no NETs were detected, either before or after chemotherapy. Accordingly, we did not detect any NETs but only a few neutrophils in the primary tumors from breast cancer patients before and after chemotherapy. These results are consistent with previous studies that have shown that neutrophils and NETs are scarce within the primary breast tumors both in mice and humans, and that neutrophils do not influence primary tumor growth in mice 20,32. It is possible that neutrophils do not mediate pro-tumoral activities at the breast primary tumor site because their recruitment is limited by the primary tumor microenvironment, in which CXCL1/CXCL5 were not upregulated after chemotherapy in mice. While the TME plays a crucial role in cancer progression, its role changes in primary tumors when compared to metastatic sites, especially regarding immune cell population 64. Altogether, our results suggest that resistance mechanisms differ between the primary tumor and metastatic sites, indicating that different treatment protocols could be needed to treat the patients. It is increasingly clear that neutrophils are key players in breast cancer metastasis, especially in the lungs 18,20,21,32,34 but further studies will be needed to investigate the importance of the NET-ITGαvβ1/MMP9-TGFβ axis in other metastatic organs such as the bones, the liver and the brain, but also in other cancer that metastasize to the lungs such as colorectal, bladder and prostate cancer.
Acute kidney injury (AKI) is an important limitation for cisplatin use, as there is approach treatment to reduce the onset of AKI. Interestingly, we identified NET-forming neutrophils in the kidneys of mice treated with cisplatin and showed that targeting NETs improved kidney function. This is consistent with the recently identified role of NETs in ischemic AKI 65. Additionally, our study demonstrated that NLRP3 and IL-1β targeting-strategies improved kidney function, and interestingly, NRLP3 and IL-1β cytokines were reported to play a role in AKI 66,67. Targeting TGFβ did not improve kidney function, indicating that the mechanisms by which NETs promote chemoresistance and cause kidney damage are different.
Our work identified a significative increase of plasma NETs level 15 days after chemotherapy treatment specifically in patients with progressive disease. This suggest that the detection of NETs in the blood of patients could be used as a biomarker to identify individuals where therapy responses against metastasis are impaired and could be improved by targeting the NETs/αvβ1/MMP9/TGFβ axis identified here.
In conclusion, our study identifies upstream and downstream effectors of the chemoprotective role of NETs in lung metastasis as therapeutic targets to improve chemotherapy efficacy, which could have immediate relevance to thousands of cancer patients.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Jean Albrengues (jalbrengues@unice.fr).
Materials availability
This study did not generate new unique reagents.
Data and Code availability.
RNA-seq data generated for this study are available through the Gene Expression Omnibus (GEO: GSE218119).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Animals
Six to eight-week-old immunocompetent female BALB/c and C57BL/6 mice were purchased from Charles River Laboratories. All mice had a Virus Antibody Free (AVF) immune status (pathogen free). Animals were housed 4 mice per cages in a room with controlled temperature and humidity under a 12h light/12h dark cycle. Mice were fed with food and autoclaved water provided by the animal facility of the Institute for Research on Cancer and the Aging, Nice. All procedures were conducted in accordance with French laws and European recommendations following a protocol approved by the French Ministry of Education and Research and by the local ethical committee for animal experimentation (CIEPAL Azur; Projects APAFIS#23972-2020011415206448 v2, APAFIS#29349-2021012013049209 v4).
Patient samples
Human breast cancer tissue
Formalin fixed and paraffin embedded tumor tissue from cancer patients before and after treatment with neoadjuvant chemotherapy were selected retrospectively by database extraction from the archives of the Department of Pathology at Odense University Hospital. Clinical and pathological characteristics of the patient tumors are presented in Table S6. All tissue samples were collected in compliance with informed consent policy. All clinical samples were coded to maintain patient confidentiality and studies were approved by the Ethics Committee of the Region of Southern Denmark (approval #S-20210115). Paraffin-embedded tissues were used for detection of neutrophils and NETs.
Patients and blood sampling
Patients in this study were included in the prospective single arm phase II clinical trial “MONDRIAN” (clinicaltrials.gov: NCT04720729), treated and followed at Institut Curie Hospitals. The trial was approved by an ethical committee (“CPP Ouest I”) on December 4th, 2020. In addition, the consenting to study protocol itself, patients consented to the re-use of plasma samples for other researches. Main eligibility criteria were as follows: patients aged ≥ 18 years old with HER2-negative metastatic breast cancer (MBC) (triple negative breast cancer (TNBC) or hormone receptors (HR) positive, HER2-negative breast cancer), performance status (PS) of 0–2, eligible to a second line of chemotherapy for MBC and written informed consent. All patients were enrolled at the beginning of the second line of chemotherapy. The second line of treatment was managed by clinical and radiological evaluations (RECIST v1.1). For each patient, blood samples were collected at day 1 (D1) prior to treatment start and day 15 (D15) after the first cycle of chemotherapy. All information regarding the patients is presented in Table S5.
Cell lines and cell culture
410.4 murine cancer cells were obtained from Tobias Pukrop (University Hospital Regensburg, Regensburg, Germany)68. D2.OR and D2.A1 murine cancer cells were obtained from Jeffrey Green (National Institutes of Health)69. PyMT murine cancer cells were obtained from Bodu Liu (Cold Spring Harbor Laboratory, Cold Spring Harbor, USA). 4T07 murine cells were obtained from Erik Sahai (The Francis Crick Institute, London, UK)70. TS/A murine cancer cells were purchased from Merckmillipore (#SCC177). 4T1 murine cancer cells were purchased from ATT (#CRL-2539). 293T cells were purchased from ATCC (#CRL-3216). TS/A, PyMT, 4T07, D2.OR, D2.A1 4T1 and 293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum (FCS) (FCII, Hyclone), 2 mM L-Glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin. The PyMT cell line was isolated from the primary tumor cells obtained from one MMTV-PyMT mouse on the C57BL/6J background. The isolated cells were cultured in DMEM supplemented with 10% FCS until spontaneous immortalization. Immortalized cells were then sorted based on CD326 expression using Flow cytometry, and the sorted cells were then maintained in complete medium. To generate mCherry- and luciferase-expressing cells, a pGIPz vector containing cDNA for mCherry and luciferase was used. Lentiviral supernatants were collected from a 293T packaging cell line transfected with the vector. Cancer cells were infected with the viral supernatants overnight in the presence of 10 μg/mL polybrene and selected with 1 μg/mL puromycin. To generate 4T07 GFP-expressing cells, a pLJM1 Puro vector containing cDNA for GFP was used. Lentiviral supernatants were collected from a 293T packaging cell line transfected with the vector. Cancer cells were infected with the viral supernatants overnight in the presence of 8 μg/mL polybrene and selected with 5 μg/ml puromycin. Cells were tested repeatedly for mycoplasma over the course of the study and were never positive. Neutrophils and lung cells were isolated freshly as described below.
Method details
Lung metastasis model and treatments
To generate lung metastasis, we intravenously injected 410.4 (500,000), TS/A (80,000) or PyMT (500,000) mouse cancer cells in 100 μL of PBS into eight-week-old female BALB/c mice. After 7 days (for 410.4 and TS/A cells), or 28 days (for PyMT cells) chemotherapy treatments were started twice a week with cisplatin (2.5 mg/kg) or Adriamycin/Cyclophosphamide (AC, 1mg/kg and 30 mg/kg, respectively). Inhibitors were injected intraperitoneally daily using compounds targeting PAD4 (20mg/kg), CXCR2 (1mg/kg), NLRP3 (40 mg/kg), MMP9 (50 mg/kg), TGFβR1 (10mg/kg), and ITGαVβ1 (70 mg/kg). DNase (300 U/mouse) treatment was used to digest the DNA strands of NETs. Antibodies were injected intraperitoneally 3 times a week using anti-Ly6G (200 μg/mouse) and its IgG control (200 μg/mouse); anti-IL-1β (200μg/mouse) and its IgG control (200μg/mouse); anti-TGFβ (200μg/mouse) and its IgG control (200μg/mouse). Two weeks after treatments were started, mice were sacrificed for analysis otherwise stated in figure legends.
Primary tumor model and treatments
To generate mammary tumors, we injected 410.4 mouse cancer cells (1.2 million cells per mouse) in 100 μL of Matrigel/PBS (1:1 ratio) into the mammary fat pad of eight-week-old female BALB/c mice. After 5 weeks, chemotherapy treatment was started twice a week with Adriamycin/cyclophosphamide (AC, 1mg/kg and 30 mg/kg, respectively). PAD4 inhibitor was injected intraperitoneally daily (20mg/kg) and two weeks after chemotherapy treatment, mice were sacrificed, the tumor and lung removed for analysis. Tumors were pictured on millimeter paper to quantify tumor size using ImageJ.
Simultaneous lung metastasis and primary tumor model and treatment (For Table S3).
410.4 mouse cancer cells were injected the same day in the tail vein (300 000 cells per mouse in 100μL PBS) and in the mammary fad pad (2.5 million cells per mouse in 100μL Matrigel/PBS 1:1) of eight-week-old female BALB/c mice. After 2 weeks, chemotherapy treatment was started twice a week with Adriamycin/cyclophosphamide injected intraperitoneally (AC, 1mg/kg and 30 mg/kg, respectively) and inhibitors were injected intraperitoneally daily using PAD4 inhibitor (20mg/kg). Two weeks after chemotherapy treatment, mice were sacrificed, the tumor and lung removed for analysis.
Isolation of mouse neutrophils
Neutrophils were harvested from six to eight-week-old female BALB/c mice. The bone marrow of the femurs and tibias was isolated in sterile Hank’s buffered salt solution (HBSS 1X) without Ca2+/Mg2+. Bone marrow cells were flushed in HBSS and after centrifugation (400g, 5 min, 4°C), the cells were resuspended for 3 min on ice in an ammonium-chloride-potassium (ACK) buffer, and centrifuged (400g, 5 min, 4°C) before being washed twice with HBSS 1X (400g centrifugation was used for the washing steps, 5 min, 4°C). Neutrophils were then separated from mononuclear cells by plating 2 mL of the cell suspension onto a Percoll gradient consisting of 3 mL of 81% Percoll under 3 mL of 62% Percoll, followed by centrifugation at 700g for 20 min at 4°C. The middle layer containing the neutrophils was washed twice in HBSS 1X and cells were resuspended in serum-free DMEM.
Activation of neutrophils and preparation of neutrophil-conditioned media
Primary isolated neutrophils were cultured in 24-well plates (250,000 per well) containing 500 μL of serum-free DMEM and were activated overnight with PMA to induce NETs or with recombinant complement 5a (C5a) to induce degranulation. The study used conditioned media (CM) from unstimulated neutrophils (PMN CM), NET-containing conditioned media (CM, induced with PMA and referred as NET CM) and CM from degranulated neutrophils (obtained using complement 5a, PMN_C5a CM). GSK484 was added to inhibit PAD4 and therefore NET production and DNase I to digest NET-DNA. The next day, the neutrophil CM were collected, centrifuged at 450g for 10 min to remove neutrophils, and used to treat the cancer cell culture. To assess NET formation, an immunofluorescence was performed using histone H2B and myeloperoxidase on neutrophils cultured overnight on poly-L-lysine-coated coverslips in serum-free DMEM or CM (with vehicle or cytokines and/or inhibitors as indicated).
Effect of chemotherapy on cancer cells in vitro
Luciferase-expressing cancer cells (2,000) were plated in 96-well culture plates and incubated in complete media at 37°C overnight. For experiments with neutrophil CM, media were replaced the next day with 100 μL of neutrophil CM with the indicated treatment. For co-culture experiment, the next day, media were replaced, and neutrophils (10 000 per well) were plated with cancer cells in a serum-free medium with the indicated treatment. After 2 days, 100 μL of medium containing 5 μg/mL of luciferin were added and bioluminescence was measured (BLI) using a plate reader. To induce dormancy, D2.0R cells were cultured on Matrigel as described before 18. To quantify the number of GFP-4T07 cancer cells, GFP intensity was measured at excitation/emission wavelengths of 488/520 nm using a plate reader.
NETs quantification using sytox green
Neutrophils (100,000) were seeded onto 96-well plates and incubated in serum free DMEM or CM (with vehicle or cytokines and/or inhibitors as indicated) for 4 hours. Sytox green (impermeable DNA dye, 50 nM) was then added to the plate and after 5 minutes, fluorescence intensity was measured at excitation/emission wavelengths of 488/520 nm using a plate reader.
Conditioned media preparation
Cancer cells were grown in complete medium, washed twice with PBS, and subsequently incubated at 37°C in a serum-free medium with cisplatin, AC or as indicated in Figure 3E. After 48 hours, CM were collected, centrifuged at 500g for 5 min to remove cell debris and the supernatant were stored at −80°C. The CM generated from 410.4 cancer cells treated with cisplatin was referred as 410.4 Cis CM while the CM generated from cells treated with AC was referred as 410.4 AC CM. The same nomenclature was used for TS/A and PyMT cells.
Cytokine array
A Proteome Profiler Mouse XL Cytokine Array test was performed following the manufacturer’s instructions.
Chemotaxis assay
Chemotaxis was assayed using a boyden chamber with a 0.3 μm pore size cell culture insert. The upper and the lower wells were separated by a light-tight polyethylene terephthalate (PET) membrane. Serum-free DMEM or CM containing the indicated neutralizing antibodies and inhibitors were applied to the lower wells, and the cells (50,000) were seeded in each of the upper wells and incubated overnight at 37°C with 5% CO2. The next day, membranes were fixed in 4% PFA for 15 min, washed twice with PBS and stained for 10 min with Sytox green (5μM). Membranes were then mounted between glass slides and cover slips using mounting media. The number of migrating cells was then manually counted using confocal microscopy and a 20x objective.
Flow cytometry analysis
After treatment, the mice were sacrificed, and the lungs were removed. Lungs were then mechanically dissociated and digested at 37°C for 30 min with 2% FCS RPMI containing Dispase (2.5 U/mL), Collagenase D (0.1 mg/mL), DNase I (25 U/mL) and Liberase DL (0.2 mg/mL). Suspensions were then passed through a 70μm nylon cell strainer with RPMI and centrifuged at 600g for 1 min at 4°C. The supernatant was removed, and the cells were incubated at 4°C for 15 min in ice-cold Fluorescence Activated Cell Sorter (FACS) buffer (2% FCS and 0.5 mM EDTA) with diluted Fc Block (1:25). After one wash in FACS buffer, the cells were incubated with conjugated antibodies for 30 min at 4°C in the dark which allowed the detection of different immune cell populations. Cells were then washed twice in FACS buffer and resuspended in 450 μL of FACS buffer before being analysed using a CytoFLEX operated by Cytexpert software. The different immune cell populations were detected among CD45 positive cells. Among TCRβ cells, CD8 Lymphocytes were detected as CD8+ cells, and CD4 Lymphocytes were detected as CD4+ cells. B cells were detected as CD19+ cells. NK cells were detected as NKp46+ cells. Dendritic cells were detected as CD11c+ and CD11b− cells. Among CD11b+ cells, monocytes were detected as Ly6C+ cells, neutrophils as Ly6G+ cells, and macrophages as F4/80+ cells. For blood analysis, blood was obtained from cardiac blood collection using a syringe with a 25G needle. Blood was then directly transferred in a 15ml EDTA-coated tube (0.5 M EDTA, pH=8), incubated with 10 ml of ACK lysis buffer for 5 min at room temperature and centrifuged for 5 min at 400g. Cells were then resuspended in 3ml of ACK lysis buffer for 2 minutes at room temperature, centrifuged (400g, 5 min) and resuspended in FACs buffer (using the volume of blood collected originally). For bone marrow cells analysis, bone marrow cells were flushed in HBSS and after centrifugation (400g, 5 min, 4°C), the cells were resuspended for 3 min on ice in an ammonium-chloride-potassium (ACK) buffer, and centrifuged (400g, 5 min, 4°C) before being washed twice with HBSS 1X (400g centrifugation was used for the washing steps, 5 min, 4°C) and resuspended in FACs buffer (10 million/ml). Blood and bone marrow cells suspensions were then incubated with CD45, Ly6G, Ly6C and CD11b antibodies for FACS analyses, following the same protocol described as above for lung cells.
Antibodies
For Western Blot analysis, antibodies against Latent Associated Protein, TGFβ, LBTP, pIκB, IκB, pSMAD2, SMAD2, Twist1, N-cadherin, E-cadherin, Twist1, Zeb1, Snail, Claudin 1 and tubulin were used.
For immunofluorescence staining of in vitro cultures, antibodies against myeloperoxidase and Histone H2B were used to assess NET formation, and an antibody against ASC was used to assess inflammasome oligomerization. Antibodies against ITGβ1, ITGαv, MMP9 and TGFβ were used to assess the presence of these proteins on NETs.
For immunofluorescence on tissue samples, antibodies against myeloperoxidase and citrullinated histone H3 were used to detect NETs and antibodies against N-cadherin and Claudin1 were used to detect the epithelial/mesenchymal status of cancer cells.
The neutralizing/blocking antibodies used in vitro were: anti-integrin β1 antibody clone Ha2/5, anti-integrin αV, anti-mouse CXCL5, CXCL1 and IL-1β and were used at 10 μg/mL.
For NET-ELISA, an anti-neutrophil elastase antibody and an anti-DNA peroxidase conjugated antibody were used.
For flow cytometry analysis, antibodies against CD4 (1:50), CD8a (1:100), TCR β (1:50), CD19 (1:50), CD45 (1:50), NKp46 (1:20), F4/80 (1:20), CD11b (1:50), CD11c (1:50), Ly6C (1:50), Ly6G (1:50), and CD107a (1:20) were used.
Immunostaining of tissue samples
For paraffin-embedded tissue, the lungs were fixed overnight at 4°C in 4% PFA, rinsed with PBS and transferred into 70% ethanol, processed using conventional methods, embedded in paraffin, and sectioned at 8 μm. Paraffin-embedded tissue sections were deparaffinized and rehydrated, and antigen retrieval was performed in EDTA buffer (10 mM Tris Base, 1 mM EDTA solution, 0.05% Tween 20, pH=9.0). Sections were blocked with Fc Receptor blocker and incubated with 1X blocking buffer (5% donkey serum, 2.5% BSA, 0.1% Triton X-100 in PBS). Then, sections were incubated overnight at 4°C with anti-myeloperoxidase (1:100) and anti-citrullinated histone H3 antibodies (1:250) (to detect neutrophils and NETs), and with anti-claudin1 (1:150) or anti-N-cadherin (1:150) (to detect EMT phenotype), in 0.5X blocking buffer. After three washes with PBS, the sections were incubated with the suitable fluorochrome-conjugated secondary antibodies (1:150) in 0.5X blocking buffer for 45 min in the dark at room temperature. After two washes with PBS and one with water, sections were counterstained with DAPI and rinsed in water, and the slides were mounted onto coverslips using mounting media.
Hematoxylin and Eosin staining
Paraffin-embedded tissue sections were first deparaffinized and rehydrated. The slides were then incubated with hematoxylin (15 min), an ammonia solution (0.08 % in water for 10 sec), and eosin (30 sec) and washed with tap water between each step. After dehydration, the slides were mounted onto cover slips using mounting media. Metastasis areas and foci were then quantified with ImageJ software.
Plasma sample collection from mice
Plasma samples were collected from cardiac blood using a syringe with a 25 G needle and placed into EDTA tubes. Whole blood was centrifuged at 4°C at 1300g for 10 min, and the top plasma layer was collected.
Plasma sample collection from human and storage
Blood samples were collected in Cell-Free DNA BCT tubes. Plasma was isolated by centrifugation at 1500g for 10 min, RT and then stored at −80°C before use.
Enzyme-linked immunosorbent assay (ELISA)
For NETs, 96-well Enzyme ImmunoAssay/Radio ImmunoAssay (EIA/RIA) plates were coated overnight at 4°C with an anti-elastase antibody (1:250) in 15 mM of Na2CO3, 35 mM of NaHCO3, at pH 9.6. The next day, the wells were washed three times with PBS, blocked in 5% BSA for two hours at room temperature, and washed three times with PBS. Then, 50 uL of plasma samples were added to the wells, incubated for two hours at room temperature on a shaker, and plates were washed three times with wash buffer (1% BSA, 0.05% Tween 20 in PBS). Next, an anti-DNA-peroxidase conjugated antibody (1:50) in 1% BSA in PBS was added to the wells for 2 hours at room temperature, and the wells were washed five times with wash buffer before the addition of 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulphonic acid). Optical density was read 40 min later at 405 nm using a plate reader.
An IL-1β kit and a TGF-β1 kit were used on CM as indicated and following the manufacturer’s instructions.
For creatinine and urea analysis, ELISA kits were used to evaluate the plasma concentration of creatinine and urea, following the manufacturer’s instructions.
An IL-1β kit, a CXCL1 and a CXCL5 kits were used on normal lung, metastatic lungs, mammary fat pad and breast primary tumor cell lysate as indicated and following the manufacturer’s instructions. Tissue lysate was isolated by washing the tissues once with cold PBS, cutting them into smaller pieces, and homogenizing thoroughly in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris HCl [Ph=7.4], 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM NaF, and 2 mM EDTA) with protease inhibitors. For 10 mg tissue, 500 μL of RIPA buffer was used. After 30 min on ice, the samples were centrifuged at 10,000 rpm for 20 min at 4 °C, and protein concentration was determined on the supernatant using a bicinchoninic acid (BCA) protein assay. 200 μg of proteins were used for each ELISA assay.
ATP quantification
ATP was quantified in CM using an ATP determination kit following the manufacturer’s instructions.
Cell culture reagents
The following reagents were used as stated, unless otherwise mentioned in figure legends. Phorbol 12-myristate 13-acetate (PMA) was used at 20 nM; recombinant mouse complement 5a was used at 100 ng/mL. Recombinant IL-1β and TGFβ-1 were used at 1 ng/mL. Cisplatin was used at 10 μM and Adriamycin/Cyclophosphamide (referred as AC) were used at 0.1 μM and 1 μM respectively. The PAD4 inhibitor GSK484 was used at 10 μM to inhibit NET formation, and 1.5 units/mL of DNase I was used to digest NET scaffolds. The NRPL3 inhibitor MCC950 was used at 5 μM; AC-YVAD-CMK was used at 100 μM to inhibit caspase 1 and Bay11-7082 was used at 10 μM to inhibit NF-κB activity; Sivelestat, MMP9 inhibitor 1, Cathepsin G inhibitor 1, 4-Aminoebnzoic Acid hydrazide were used at 10 μM to inhibit NE, MMP9, Cathepsin G and MPO activity respectively, RGD peptide was used at 1 mM to counteract integrin β1-dependant adhesion; αVβ1 integrin-IN-1 TFA (HY-100445A, Medchem Express) was used at 100 μM to inhibit ITGαvβ1 activity; anti-Integrin β1 antibodies clone Ha2/5 and anti-integrin αv were used at 10 μg/mL to inhibit integrin β1 and αv activity respectively; anti-mouse CXCL5, CXCL1 and IL-1β were used at 10 μg/mL.
Western blot
For immunoblotting analysis, cells grown on plastic plates were lysed on ice in lysis buffer (25 mM Tris [pH=6.8], 2% sodium dodecyl sulfate (SDS), 5% glycerol, 1% β-mercaptoethanol, 0.01% bromophenol blue) and the samples were sonicated. Equal amounts of protein from each sample were loaded on SDS-polyacrylamide gel electrophoresis, separated, and transferred onto nitrocellulose. The immunoblots were incubated in blocking buffer (5% BSA, 10 mM Tris-HCl [pH=7.5], 500 mM NaCl) for 30 min at room temperature and probed with specific antibodies overnight at 4°C. Then, the immunoblots were washed three times for 10 min in Tris-buffered saline Tween 20 (TBST, 10 mM Tris-HCl [Ph=7.5], 500 mM NaCl, 0.1% Tween 20), incubated with secondary antibodies for one hour at room temperature in blocking buffer, and washed three times in TBST again. Immunodetection was performed using chemiluminescent horseradish peroxidase (HRP) substrate.
Immunofluorescence of cell cultures
Cells were fixed with 4% paraformaldehyde (PFA) for 20 min. After fixation, they were rinsed twice in PBS, incubated in 50 mM of NH4Cl for 10 min and permeabilized with 0.5% Triton X-100 for 5 min. Cells were next blocked in PBS containing 1% bovine serum albumin (BSA) for 60 min and incubated with anti-H2B (1:200), anti-myeloperoxidase (1:400) antibodies in blocking buffer overnight at 4°C. After two washes in PBS, cells and matrices were incubated in the presence of fluorochrome-conjugated secondary antibodies (1:250) for 40 min, rinsed twice in PBS, stained with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min, rinsed in water, and the coverslips were mounted onto glass slides using mounting media.
mRNA analysis
RNA was isolated from total cell lysates using a RNeasy Mini kit according to the manufacturer’s instructions. Reverse transcription was performed using 500ng cytoplasmic RNA using Superscript II reverse transcriptase. Real time PCR was performed using Fast SYBR Green Master Mix in duplicates according to the recommendations of the manufacturer on an ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA).
Primer sequences are listed in the Key resources table and Table 8.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Latent Associated Protein (LAP) antibody | Novus | Cat #NBP2-22114SS; RRID:AB_2909813 |
| TGFβ | Cell Signaling Technology | Cat #3711; RRID:AB_2063354 |
| Latent Binding TGFβ Protein (LTBP) | Abcam | Cat #ab78294; RRID:AB_1952060 |
| pIκB | Cell Signaling Technology | Cat #2859T; RRID:AB_561111 |
| IκB | Cell Signaling Technology | Cat #4814T; RRID:AB_390781 |
| tubulin | Sigma | Cat #T4026; RRID:AB_477577 |
| Twist1 | Thermo Fisher Scientific | Cat #PA5116093; RRID:AB_2900727 |
| Zeb1 | Thermo Fisher Scientific | Cat #PA595862; RRID:AB_2807664 |
| Snail | Thermo Fisher Scientific | Cat #PA5115940; RRID:AB_2900574 |
| E-cadherin | Cell Signaling Technology | Cat #3195S; ID UniProt : P12830 |
| Phospho Smad2 | Cell Signaling Technology | Cat #3108; ID UniProt : Q15796 |
| Smad2 | Cell Signaling Technology | Cat #3103; ID Uniprot : Q15796 |
| myeloperoxidase | Dako | Cat #A0398; RRID:AB_2335676 |
| Histone H2B | Abcam | Cat #ab52484; RRID:AB_1139809 |
| RFP (mcherry) | Rockland | Cat #600-401-379; RRID:AB_2209751 |
| ASC | EMD Millipore | Cat #04-147; RRID:AB_1977033 |
| ITGβ1 | BD Biosciences | Cat #555003; RRID:AB_395637 |
| ITGαv (clone RMV-7) | Invitrogen | Cat #14-0512-82; RRID:AB_467296 |
| MMP9 | Abcam | Cat #ab38898; RRID:AB_776512 |
| TGFβ | Cell Signaling Technology | Cat #3711; RRID:AB_2063354 |
| anti-myeloperoxidase antibody (used for mouse tissue) | R & D Systems | Cat #AF3667; RRID:AB_2250866 |
| anti-myeloperoxidase antibody (used for human tissue) | R & D Systems | Cat #MAB3174; RRID:AB_2250873 |
| anti-citrullinated histone H3 antibody (used for mouse and human tissue) | Abcam | Cat #ab5103; RRID:AB_304752 |
| anti-N-cadherin | Abcam | Cat #ab18203; ID Uniprot:P19022 |
| anti-Claudin1 | Thermo Fisher Scientific | Cat #71-7800; RRID:AB_2533997 |
| Donkey anti-Goat IgG secondary antibody, Alexa Fluor 647 | Invitrogen | Cat #A21445; RRID:AB_2535862 |
| Donkey anti-Rabit IgG secondary antibody, Alexa Fluor 568 | Invitrogen | Cat #A10042; RRID:AB_2534017 |
| ITGβ1 (clone Ha2/5) | BD Biosciences | Cat #555003; RRID:AB_395637 |
| ITGαv (clone RMV-7) | Invitrogen | Cat #14-0512-82; RRID:AB_467296 |
| CXCL5 | R & D Systems | Cat #Mab433; RRID:AB_2086587 |
| CXCL1 | R & D Systems | Cat #Mab453; RRID:AB_2087696 |
| IL-1β | R & D Systems | Cat #AF-401-NA; RRID:AB_416684 |
| anti-mouse Neutrophil Elastase | Santa Cruz Biotechnology | Cat #sc-9521; RRID:AB_2096537 |
| anti-human Neutrophil Elastase | Santa Cruz Biotechnology | Cat #sc-9520; RRID:AB_650173 |
| anti-DNA peroxidase conjugated | Roche | Cat#11774425001; RRID:AB_2909412 |
| CD4 (clone GK1.5) | BD Biosciences | Cat #612974; RRID:AB_2870246 |
| CD8a (clone REA601) | Miltenyi Biotec | Cat #130-120-806; RRID:AB_2752203 |
| TCR β (clone REA318) | Miltenyi Biotec | Cat #130-104-856; RRID:AB_2654019 |
| CD19 (clone T45-2342) | Miltenyi Biotec | Cat #130-112-042; RRID:AB_2655828 |
| CD45 (clone REA737) | Miltenyi Biotec | Cat #130-110-803; RRID:AB_2658224 |
| NKp46 (clone 29A1.4) | BD Biosciences | Cat #741029; RRID:AB_2740647 |
| F4/80 (clone T45-2342) | BD Biosciences | Cat #743281; RRID:AB_2741399 |
| CD11b (clone REA754) | Miltenyi Biotec | Cat #130-113-808; RRID:AB_2751173 |
| CD11c (clone REA754) | Miltenyi Biotec | Cat #130-110-837; RRID:AB_2654705 |
| Ly6C (clone REA796) | Miltenyi Biotec | Cat #130-111-920; RRID:AB_2652814 |
| Ly6G (clone 1A8) | BD Biosciences | Cat #561236; RRID:AB_10611860 |
| CD107a (clone 1D4B) | BD Biosciences | Cat #565533; RRID:AB_2739285 |
| CD326 | Biolegend | Cat #118214; RRID:AB_1134102 |
| anti-Ly6G antibody (clone 1A8) | BioXCell | Cat #BP0075-1; RRID:AB_1107721 |
| IgG for antiLy6G antibody (clone 2A3) | BioXCell | Cat #BP0089; RRID:AB_1107769 |
| anti-IL-1β antibody (clone B122) | BioXCell | Cat #BE0246; RRID:AB_2687727 |
| IgG for anti-IL-1β antibody | BioXCell | Cat #BE0091; RRID:AB_1107773 |
| anti-TGFβ antibody (clone 1D11.16.8) | BioXCell | Cat #BE0057; RRID:AB_1107757 |
| IgG for anti-TGFβ antibody (clone MOPC-21) | BioXCell | Cat #BE0083; RRID:AB_1107784 |
| Biological samples | ||
| Human plasma samples from metastatic breast cancer patients | Institut Curie, Paris, France | Table S5 |
| Paraffin-embeded breast cancer tissue | University of Southern Denmark | Table S6 |
| Chemicals, peptides, and recombinant proteins | ||
| Cisplatin | Sigma | Cat #C2210000 |
| Doxorubicin | Sigma | Cat #D1515 |
| Cyclophosphamide | Sigma | Cat #C0768 |
| PAD4 inh. | Cayman Chemical | Cat #17488 |
| CXCR2 inh. | Selleckchem | Cat #SB225002 |
| NLRP3 inh. (MCC950) | EMD Millipore | Cat #5.38120.0001 |
| MMP9 inh. (in vivo) | Medchem Express | Cat #HY-12354 |
| TGFβR1 inh. | Selleckchem | Cat #SB431542 |
| ITG αVβ1 inh. | Medchem Express | Cat #HY-100445A |
| DNAse I | Sigma | Cat #04536282001 |
| HBSS 1X without Ca2+/Mg2+ | Gibco | Cat #14175-053 |
| ACK | Gibco | Cat #A10492-01 |
| Percoll | GE Healthcare | Cat #17-0891-02 |
| serum-free DMEM (Dulbecco’s Modified Eagle Medium) | Gibco | Cat #21969-035 |
| Dharmafect 3 | Dharmacon | Cat #T-2003-02 |
| Opti-MEM | Gibco | Cat #31985-047 |
| Cytiva HyClone™ Sérum FetalClone™ II (FCII) | Cytiva | Cat #SH3006603 |
| Matrigel | Corning | Cat #356234 |
| Luciferin | Goldbio | Cat #Luck-1G |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma | Cat #P1585 |
| Recombinant mouse complement 5a (C5a) | R & D Systems | Cat #2150-C5/CF |
| Recombinant IL-1β | Peprotech | Cat #211-11B |
| Recombiant TGFβ-1 | Biolegend | Cat #763102 |
| Caspase I inh. AC-YVAD-CMK | abcam | Cat #SML0429 |
| NF-kB inh. Bay 11-7082 | Cayman Chemical | Cat#10010266 |
| NE inh. Sivelestat | Tocris | Cat #3535 |
| MMP9 inh. 1 (in vitro) | Calbiochem | Cat #44278 |
| Cathepsin G inhibitor 1 | EMD Millipore | Cat #219372 |
| Myeloperoxidase inh. (4-Aminoebnzoic Acid hydrazide) | Cayman Chemical | Cat #14845 |
| RGD peptide | abcam | Cat #A8052 |
| Sytox green | Thermo Fisher Scientific | Cat #S7020 |
| Chemiluminescent horseradish peroxidase (HRP) substrate | GE Healthcare | Cat #RPN 2109 |
| RPMI | Gibco | Cat #31870-025 |
| Dispase | Stem Cell | Cat #07913 |
| Collagenase D | Sigma | Cat#11088866001 |
| Liberase DL | Sigma | Cat #05466202001 |
| Fc block | BD Biosciences | Cat#553142 |
| PFA | Electron Microscopy Science | Cat #15714 |
| Triton X-100 | Thermo Fisher Scientific | Cat #BB151-500 |
| Fc receptor blocker | Innovex | Cat #NB309 |
| Donkey serum | Sigma | Cat #D9663 |
| BSA | Sigma | Cat #A3294 |
| DAPI | Thermo Fisher Scientific | Cat #D1306 |
| Hematoxylin | Thermo Fisher Scientific | Cat #6765008 |
| Eosin | Sigma | Cat #HT110180 |
| Polybrene | Sigma | Cat #TR-1003-G |
| Puromycine | Sigma | Cat #P8833 |
| L-Glutamine | Gibco | Cat #25030 |
| Penicillin-streptomycin | Gibco | Cat #15140-122 |
| Critical commercial assays | ||
| 96-well Enzyme ImmunoAssay/Radio ImmunoAssay (EIA/RIA) | Costar | Cat #3590 |
| ELISA kit for TGF-β1 | R & D Systems | Cat #DY1679 |
| ELISA kit for Urea | MyBiosource | Cat #MBS2611085 |
| ELISA kit for creatinine | MyBiosource | Cat #MBS763433 |
| ELISA kit for IL-1β (for tissue analysis) | R & D Systems | Cat #MLB00C |
| ELISA kit for IL-1β (for CM analysis) | Thermo Fisher Scientific | Cat #88-7013-22 |
| ELISA kit for CXCL1/KC | R & D Systems | Cat #DY453 |
| ELISA kit for CXCL5 (LIX) | R & D Systems | Cat #P313546 |
| ATP determination kit | Thermo Fisher Scientific | Cat #A22066 |
| RNeasy Mini kit | Qiagen, Turnberry Ln Valencia, CA | Cat #217004 |
| Proteome Profiler Mouse XL Cytokine Array test | R & D Systems | Cat #ARY028 |
| PierceTM BCA Protein Assay Kit | Thermofisher | Cat #23225 |
| Illumina Stranded mRNA Prep | Illumina | Cat #20040532 |
| Deposited data | ||
| RNA-seq data | This paper | GEO: GSE218119 |
| Experimental models: Cell lines | ||
| 410.4 murine cancer cells | Blazquez et al. 68 | Obtained from Dr. Tobias Pukrop (University Hospital Regensburg, Regensburg, Germany) |
| PyMT murine cancer cells | This paper | Obtained from Dr. Bodu Liu (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA) |
| TS/A murine cancer cells | Merckmillipore | Cat. # SCC177 |
| D2.OR murine cancer cells | Barkan et al. 69 | Gift from Dr. Jeffrey Green (National Institutes of Health) |
| D2.A1 murine cancer cells | Barkan et al. 69 | Gift from Dr. Jeffrey Green (National Institutes of Health) |
| 4T07 murine cancer cells | Avgustinova et al. 70 | Gift from Dr. Erik Sahai (The Francis Crick Institute, London, UK). RRID:CVCL_B383 |
| 4T1 murine cancer cells | ATCC | Cat. #CRL-2539 |
| 293T cells | ATCC | Cat. #CRL-3216 |
| Experimental models: Organisms/strains | ||
| Mouse Balb/c female | Charles River | Cat #028 |
| Mouse C57BL/6 female | Charles River | Cat #027 |
| Oligonucleotides | ||
| siRNA ON TARGET Plus Non-targeting pool target sequences: UGGUUUACAUGUCGACUAA; UGGUUUACAUGUUGUGUGA; UGGUUUACAUGUUUUCUGA; UGGUUUACAUGUUUUCCUA | Dharmacon | Cat #D-001810-10-05 |
| siRNA ON TARGET Plus mouse ccdc25 #1: -J-053396-09: GCAAGAUGAACAACGUUAA | Dharmacon | Cat #L-053396-01-0005 |
| siRNA ON TARGET Plus mouse ccdc25 #2: -J-053396-10: CCAUGUGUACCUACGAUUA | Dharmacon | Cat #L-053396-01-0005 |
| Additional primer sequences | This paper | Table S8 |
| Software and algorithms | ||
| ImageJ software | ImageJ | N/A |
| GraphPad Prism software version 7 | GraphPad | N/A |
| Cytexpert | Beckman Coulter | N/A |
| STAR | Dobin et al. 71 | N/A |
| FeatureCounts | Liao et al. 72 | N/A |
| DEseq2 | Love et al. 73 | N/A |
| ReactomePA R package | Yu et al. 74 | N/A |
| Other | ||
| 70μm nylon cell strainer | BD Falcon | Cat #352350 |
| 96-well plate | Falcon | Cat #353072 |
| Plate reader FLUOstar Omega | BMG Labtech | N/A |
| CytoFLEX | Beckman Coulter | N/A |
| Mounting media immunostaining | Electron Microscopy Sciences | Cat #17985-16 |
| Cytoseal 60 mounting media for histological staining | Thermo Fisher Scientific | Cat #8310-4 |
| Syringe 25 G | Terumo | Cat #AN*2516R1 |
| 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (antibodyTS) | Thermo Fisher Scientific | Cat #37615 |
| Superscript II reverse transcriptase | Invitrogen, Carlsbad, CA | Cat #18064-014 |
| Fast SYBR Green Master Mix | Applied Biosystems, Foster City, CA | Cat #18064-014 |
| Boyden chamber | Corning | Cat #351151 |
| 96-well Enzyme ImmunoAssay/Radio ImmunoAssay (EIA/RIA) plates | Costar | Cat #3590 |
| Cell-Free DNA BCT tubes | Streck | N/A |
Relative expression of the respective gene was determined after normalization to GAPDH and calculated using the following formula: relative expression level = 2ddCT.
Sample and library preparation for RNA-seq
410.4 cells (200,000 per well) were plated in 6 well plate in 5% FCII DMEM media. The day after, the media was removed and replaced by NET CM or fresh control media for 48h. RNA was isolated from total cell lysates using a RNeasy Mini kit according to the manufacturer’s instructions. 500ng of total RNA was used to prepare RNA libraries. There were generated with Illumina Stranded mRNA Prep and performed following the manufacturer’s instructions. Each library was normalized to 8pM and sequences on an Illumina NextStep 2000.
Analysis of RNA-seq data
FASTQ sequencing files were processed and analyzed using DRAGEN included in the Nextseq2000 Illumina sequencer. Reads were aligned to the GRCm39/mm10 reference mouse genome using STAR 71. Gene read counts were determined using FeatureCounts 72. Differential gene expression analysis was performed using DESeq2 73, with a 0.05 false discovery rate (FDR)-adjusted p-value cutoff for significance. Signaling pathways were revealed using ReactomePA R package 74. The option “pAdjustMethod” was set to “fdr”. Raw data are available on GEO number GSE218119
siRNA Transfections
410.4 cancer cells were transfected using Dharmafect. Briefly, cells were plated at 60% confluence and subjected to transfection the following day in Opti-MEM using 100nM final concentration of siRNA. The next day, cells were detached using trypsin and used in a co-culture assay with freshly isolated neutrophils. SiRNA was purchased from Dharmacon and sequences are listed in the Key resources table.
Quantification and statistical analysis
For the quantification of the lung metastatic burden, the number of lung metastatic foci, NET-ELISA, lung neutrophils and NETs (based on immunofluorescence imaging), BUN and plasma creatinine: the vehicle, cisplatin and AC mice groups used for the following experiments were the same as the experiments were conducted at the same time to reduce the number of animals used in our study. i) mice with 410.4 cells and treated with the PAD4 inh. and the NLRP3 inh.; ii) mice with 410.4 cells and treated with the CXCR2 inh., the TGFβR1 inh., the MMP9 inh. and the ITGαvβ1 inh.; iii) Mice with PyMT cells and treated with the PAD4 inh., DNase I, and the TGFβR1 inh.; iv) Mice with TS/A cells and treated with the PAD4 inh., DNase I, and the TGFβR1 inh; v) Non tumor-bearing mice treated with the PAD4 inh., the NLRP3 inh., the TGFβR1 inh and the MMP9 inh. For the quantification of lung neutrophils using Flow Cytometry (Figures S1I and S1X), the same Veh., cisplatin and AC mice groups were used for the CXCR2 inh. and the PAD4 inh. as the experiments were conducted at the same time to reduce the number of animals used in our study.
Statistical tests are presented within figure legends. A p-value less than 0.05 was considered significant, and p-values are indicated in figures as ***P<0.001, **P<0.01, and *P<0.05. All statistical analyses were performed using GraphPad Prism software version 7, unless otherwise stated.
Supplementary Material
Chemotherapy promotes NET formation in breast cancer lung metastasis
NETs promote breast cancer lung metastasis chemotherapy resistance
NETs activate latent TGFβ, promoting EMT in cancer cells
Targeting NETs ameliorates chemotherapy efficacy against lung metastasis
Acknowledgments
We thank the animal facility, histology core, flow cytometry core and microscopy core (Molecular and Cellular Core Imaging, PICMI) from the Institute for Research on Cancer and Aging, Nice (IRCAN), and the 3D-Hub-S platform (supported by le Canceropôle PACA, la Région PACA, le Conseil Départemental 06, l’INSERM, l’ARC, and IBiSA) for technical support. The study was supported by the Canceropôle PACA (Emergence Grant to J.A. and Emergence Grant to P.L.), Association pour la Recherche sur le Cancer (#PDF20181208327 and #PJA2021060003717 to J.A and #PJA20191209728 to C.G.), Institut National du Cancer (#PLBIO21-073 to C.G., J.A., and F.M-G. and #PLBIO2018-138 to C.G.), the French National Research Agency (ANR-21-CE14-0006 to J.A., ANR-11-LABX-0028-01 to University Cote d’Azur and ANR-15-IDEX-01), Fondation pour la Recherche Médicale (#DEQ20180339183 to C.G.), the National Infrastructure France Génomique (ANR-10-INBS-09-03 and ANR-10-INBS-09-02 to G.R. and C.G.R.), the National Cancer Institute (5P01CA013106-Project 3 to D.L.S). We thank T. Pukrop (University Hospital Regensburg) for the 410.4 cells.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests
M.E. is a member of the research advisory board for brensocatib for Insmed, Inc., a member of the scientific advisory board for Vividion Therapeutics, Inc., a consultant for Protalix, Inc., and holds shares in Agios Pharmaceuticals, Inc. J.A., C.G. and A.M. have a patent related to this work (EP22306004). The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weigelt B, Peterse JL, and van ‘t Veer LJ (2005). Breast cancer metastasis: markers and models. Nat Rev Cancer 5, 591–602. 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- 2.Mansoori B, Mohammadi A, Davudian S, Shirjang S, and Baradaran B (2017). The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull 7, 339–348. 10.15171/apb.2017.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao J, Zhang M, Wang B, Zhang L, Fang M, and Zhou F (2021). Chemoresistance and Metastasis in Breast Cancer Molecular Mechanisms and Novel Clinical Strategies. Front Oncol 11, 658552. 10.3389/fonc.2021.658552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bejarano L, Jordao MJC, and Joyce JA (2021). Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov 11, 933–959. 10.1158/2159-8290.CD-20-1808. [DOI] [PubMed] [Google Scholar]
- 5.Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat Med 19, 1423–1437. 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, and Kzhyshkowska J (2019). Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 8, 1596004. 10.1080/2162402X.2019.1596004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, et al. (2012). Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 21, 488–503. 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruffell B, and Coussens LM (2015). Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472. 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quail DF, and Joyce JA (2017). Molecular Pathways: Deciphering Mechanisms of Resistance to Macrophage-Targeted Therapies. Clin Cancer Res 23, 876–884. 10.1158/1078-0432.CCR-16-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, and Joyce JA (2011). Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev 25, 2465–2479. 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. (2011). Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1, 54–67. 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vyas D, Laput G, and Vyas AK (2014). Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Onco Targets Ther 7, 1015–1023. 10.2147/OTT.S60114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klemm F, and Joyce JA (2015). Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol 25, 198–213. 10.1016/j.tcb.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greten FR, and Grivennikov SI (2019). Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 51, 27–41. 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nemeth T, Sperandio M, and Mocsai A (2020). Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov 19, 253–275. 10.1038/s41573-019-0054-z. [DOI] [PubMed] [Google Scholar]
- 16.Nolan E, and Malanchi I (2022). Connecting the dots: Neutrophils at the interface of tissue regeneration and cancer. Semin Immunol, 101598. 10.1016/j.smim.2022.101598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papayannopoulos V (2018). Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18, 134–147. 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 18.Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Kuttner V, et al. (2018). Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361. 10.1126/science.aao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mollinedo F (2019). Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol 40, 228–242. 10.1016/j.it.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F, et al. (2020). DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 583, 133–138. 10.1038/s41586-020-2394-6. [DOI] [PubMed] [Google Scholar]
- 21.Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, Wang Y, Yang S, Liang C, Liang Y, et al. (2020). Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 10.1016/j.ccell.2020.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Adrover JM, McDowell SAC, He XY, Quail DF, and Egeblad M (2023). NETworking with cancer: The bidirectional interplay between cancer and neutrophil extracellular traps. Cancer Cell. 10.1016/j.ccell.2023.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorch SK, and Kubes P (2017). An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 23, 279–287. 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- 24.Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M, et al. (2020). Neutrophil extracellular traps in COVID-19. JCI Insight 5. 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, and Mantovani A (2020). Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer 20, 485–503. 10.1038/s41568-020-0281-y. [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Chen K, Xiao X, Nie Y, Qu S, Gong C, Su F, and Song E (2016). Pretreatment neutrophil-to-lymphocyte ratio is correlated with response to neoadjuvant chemotherapy as an independent prognostic indicator in breast cancer patients: a retrospective study. BMC Cancer 16, 320. 10.1186/s12885-016-2352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J, Ni C, Ma C, Zhang L, Jing X, Li C, Liu Y, and Qu X (2017). Association of neutrophil/lymphocyte ratio and platelet/lymphocyte ratio with ER and PR in breast cancer patients and their changes after neoadjuvant chemotherapy. Clin Transl Oncol 19, 989–996. 10.1007/s12094-017-1630-5. [DOI] [PubMed] [Google Scholar]
- 28.Chae S, Kang KM, Kim HJ, Kang E, Park SY, Kim JH, Kim SH, Kim SW, and Kim EK (2018). Neutrophil-lymphocyte ratio predicts response to chemotherapy in triple-negative breast cancer. Curr Oncol 25, e113–e119. 10.3747/co.25.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al. (2012). A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178. 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daley JM, Thomay AA, Connolly MD, Reichner JS, and Albina JE (2008). Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 83, 64–70. 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 31.Masucci MT, Minopoli M, Del Vecchio S, and Carriero MV (2020). The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front Immunol 11, 1749. 10.3389/fimmu.2020.01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH, et al. (2016). Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 8, 361ra138. 10.1126/scitranslmed.aag1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adrover JM, Carrau L, Dassler-Plenker J, Bram Y, Chandar V, Houghton S, Redmond D, Merrill JR, Shevik M, tenOever BR, et al. (2022). Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight 7. 10.1172/jci.insight.157342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, and Naora H (2019). Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med 216, 176–194. 10.1084/jem.20181170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller RP, Tadagavadi RK, Ramesh G, and Reeves WB (2010). Mechanisms of Cisplatin nephrotoxicity. Toxins (Basel) 2, 2490–2518. 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, Serbulea V, Leitinger N, Ailawadi G, and Upchurch GR Jr. (2018). Novel Role of IL (Interleukin)-1beta in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler Thromb Vasc Biol 38, 843–853. 10.1161/ATVBAHA.117.309897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, Barthwal MK, and Dikshit M (2012). Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS One 7, e48111. 10.1371/journal.pone.0048111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, Drosos GI, Boumpas DT, and Ritis K (2011). Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS One 6, e29318. 10.1371/journal.pone.0029318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stutz A, Horvath GL, Monks BG, and Latz E (2013). ASC speck formation as a readout for inflammasome activation. Methods in molecular biology 1040, 91–101. 10.1007/978-1-62703-523-1_8. [DOI] [PubMed] [Google Scholar]
- 40.Kolb R, Liu GH, Janowski AM, Sutterwala FS, and Zhang W (2014). Inflammasomes in cancer: a double-edged sword. Protein & cell 5, 12–20. 10.1007/s13238-013-0001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia Y, Shen S, and Verma IM (2014). NF-kappaB, an active player in human cancers. Cancer Immunol Res 2, 823–830. 10.1158/2326-6066.CIR-14-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Seror C, Metivier D, Perfettini JL, Zitvogel L, and Kroemer G (2009). Chemotherapy induces ATP release from tumor cells. Cell Cycle 8, 3723–3728. 10.4161/cc.8.22.10026. [DOI] [PubMed] [Google Scholar]
- 43.Bryan NB, Dorfleutner A, Rojanasakul Y, and Stehlik C (2009). Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182, 3173–3182. 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brabletz T, Kalluri R, Nieto MA, and Weinberg RA (2018). EMT in cancer. Nat Rev Cancer 18, 128–134. 10.1038/nrc.2017.118. [DOI] [PubMed] [Google Scholar]
- 45.Batlle E, and Massague J (2019). Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 50, 924–940. 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Budi EH, Duan D, and Derynck R (2017). Transforming Growth Factor-beta Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol 27, 658–672. 10.1016/j.tcb.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 47.Munger JS, and Sheppard D (2011). Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol 3, a005017. 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pham CT (2006). Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6, 541–550. 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 49.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, and Zychlinsky A (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 50.Jenkins G (2008). The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol 40, 1068–1078. 10.1016/j.biocel.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 51.Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, and Chen JW (2015). Myeloperoxidase-Hepatocyte-Stellate Cell Cross Talk Promotes Hepatocyte Injury and Fibrosis in Experimental Nonalcoholic Steatohepatitis. Antioxid Redox Signal 23, 1255–1269. 10.1089/ars.2014.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, Giannias B, Berube J, Rousseau S, Ferri LE, and Spicer JD (2017). Neutrophil extracellular traps sequester circulating tumor cells via beta1-integrin mediated interactions. Int J Cancer 140, 2321–2330. 10.1002/ijc.30635. [DOI] [PubMed] [Google Scholar]
- 53.Reed NI, Jo H, Chen C, Tsujino K, Arnold TD, DeGrado WF, and Sheppard D (2015). The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci Transl Med 7, 288ra279. 10.1126/scitranslmed.aaa5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonzalez-Angulo AM, Morales-Vasquez F, and Hortobagyi GN (2007). Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol 608, 1–22. 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 55.Shinde-Jadhav S, Mansure JJ, Rayes RF, Marcq G, Ayoub M, Skowronski R, Kool R, Bourdeau F, Brimo F, Spicer J, and Kassouf W (2021). Role of neutrophil extracellular traps in radiation resistance of invasive bladder cancer. Nat Commun 12, 2776. 10.1038/s41467-021-23086-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nolan E, Bridgeman VL, Ombrato L, Karoutas A, Rabas N, Sewneth CAN, Vasquez M, Rodrigues FS, Horswell S, Faull P, et al. (2022). Radiation exposure elicits a neutrophil-driven response in healthy lung tissue that enhances metastatic colonization. Nat Cancer 3, 173–187. 10.1038/s43018-022-00336-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martins-Cardoso K, Almeida VH, Bagri KM, Rossi MID, Mermelstein CS, Konig S, and Monteiro RQ (2020). Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial-Mesenchymal Transition. Cancers (Basel) 12. 10.3390/cancers12061542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu T, Zou X, Yang C, Li L, Wang B, Li R, Li H, Xu Z, Huang D, and Wu Q (2021). Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelialmesenchymal transition. Int J Mol Med 48. 10.3892/ijmm.2021.4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pandolfi L, Bozzini S, Frangipane V, Percivalle E, De Luigi A, Violatto MB, Lopez G, Gabanti E, Carsana L, D’Amato M, et al. (2021). Neutrophil Extracellular Traps Induce the Epithelial-Mesenchymal Transition: Implications in Post-COVID-19 Fibrosis. Front Immunol 12, 663303. 10.3389/fimmu.2021.663303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jin W, Yin H, Li H, Yu XJ, Xu HX, and Liu L (2021). Neutrophil extracellular DNA traps promote pancreatic cancer cells migration and invasion by activating EGFR/ERK pathway. J Cell Mol Med 25, 5443–5456. 10.1111/jcmm.16555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia X, Zhang Z, Zhu C, Ni B, Wang S, Yang S, Yu F, Zhao E, Li Q, and Zhao G (2022). Neutrophil extracellular traps promote metastasis in gastric cancer patients with postoperative abdominal infectious complications. Nat Commun 13, 1017. 10.1038/s41467-022-28492-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akhurst RJ, and Hata A (2012). Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 11, 790–811. 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clancy DM, Henry CM, Sullivan GP, and Martin SJ (2017). Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J 284, 1712–1725. 10.1111/febs.14075. [DOI] [PubMed] [Google Scholar]
- 64.Jin MZ, and Jin WL (2020). The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther 5, 166. 10.1038/s41392-020-00280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, Kraft F, Lei Y, Fukasawa Y, Moeckel GW, et al. (2017). Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J Am Soc Nephrol 28, 1753–1768. 10.1681/ASN.2016080925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin Q, Li S, Jiang N, Jin H, Shao X, Zhu X, Wu J, Zhang M, Zhang Z, Shen J, et al. (2021). Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy 17, 2975–2990. 10.1080/15548627.2020.1848971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anders HJ (2016). Of Inflammasomes and Alarmins: IL-1beta and IL-1alpha in Kidney Disease. J Am Soc Nephrol 27, 2564–2575. 10.1681/ASN.2016020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blazquez R, Rietkotter E, Wenske B, Wlochowitz D, Sparrer D, Vollmer E, Muller G, Seegerer J, Sun X, Dettmer K, et al. (2020). LEF1 supports metastatic brain colonization by regulating glutathione metabolism and increasing ROS resistance in breast cancer. Int J Cancer 146, 3170–3183. 10.1002/ijc.32742. [DOI] [PubMed] [Google Scholar]
- 69.Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, Webster JD, Hoover S, Simpson RM, Gauldie J, and Green JE (2010). Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res 70, 5706–5716. 10.1158/0008-5472.CAN-09-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Avgustinova A, Iravani M, Robertson D, Fearns A, Gao Q, Klingbeil P, Hanby AM, Speirs V, Sahai E, Calvo F, and Isacke CM (2016). Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat Commun 7, 10305. 10.1038/ncomms10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 73.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu G, and He QY (2016). ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst 12, 477–479. 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data generated for this study are available through the Gene Expression Omnibus (GEO: GSE218119).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.