

ABSTRACT
Doxorubicin (DOX) resistance in breast cancer (BC) poses a huge challenge for the therapeutic effect on BC. Lnc KCNQ1OT1 play crucial roles in chemotherapy resistance. However, the role and mechanism of lnc KCNQ1OT1 in DOX resistance BC have not been investigated, which merits further exploration. Based on MCF‐7 and MDA-MB-231 cells, MCF‐7/DOX and MDA-MB-231/DOX cells were established using gradient concentrations of DOX. IC50 values and cell viability were determined using MTT. Cell proliferation was investigated by colony formation. Flow cytometry was performed to examine cell apoptosis and cell cycle. Gene expression was examined using qRT-PCR and western blot. The interactions among METTL3, lnc KCNQ1OT1, miR-103a-3p, and MDR1 were validated with MeRIP-qPCR, RIP, and dual-luciferase reporter gene assays. The results showed that Lnc KCNQ1OT1 was highly expressed in DOX-resistant BC cells, and lnc KCNQ1OT1 depletion could enhance DOX sensitivity in BC cells and DOX-resistant BC cells. Besides, lnc KCNQ1OT1 was modulated by MELLT3 in the manner of m6A modification. MiR-103a-3p could interact with lnc KCNQ1OT1 and MDR1. Overexpression of MDR1 abolished the impacts of lnc KCNQ1OT1 depletion on DOX resistance in BC. In conclusion, our results unveiled that in BC cells and DOX-resistant BC cells, lnc KCNQ1OT1 could be mediated by METTL3 through m6A modification to elevate and stabilize its expression, further inhibiting miR-103a-3p/MDR1 axis to promote DOX resistance, which might provide novel thought to overcome DOX resistance in BC.
KEYWORDS: METTL3, M6a, Lnc KCNQ1OT1, breast cancer, doxorubicin resistance
Introduction
According to an authoritative published study, the number of breast cancers (BCs) diagnosed in 2020 has reached approximately 2.3 million, and the mortality of BC was estimated roughly 0.69 million worldwide [1]. At present, therapeutic methods for BC have been developed in several respects, such as mammectomy and chemoradiotherapy drugs. Doxorubicin (DOX) is a broad-spectrum chemotherapy agent, which can effectively suppress the progression of BC [2]. However, the therapeutic effects of DOX on a part of patients of BC were not effective because of drug resistance. Therefore, how to attenuate DOX resistance to BC deserves further investigations.
LncRNAs are important components of non-coding RNA and play crucial roles in biological functions, such as tumour growth and drug resistance [3,4]. For example, ANXA2P2 was elevated in cisplatin (DDP)-resistant cervical cancer cells, and its knockdown could suppress the growth of DDP-resistant cervical cancer cells and enhance the sensitivity of cisplatin to DDP-resistant cervical cancer cells [5]. Lnc KCNQ1OT1 affecting drug resistance to multiple cancers has been explored by several studies. In nasopharyngeal carcinoma, lnc KCNQ1OT1 promoted cisplatin resistance and tumour progression through miR‑454/USP47 axis [6]. In additional, lnc KCNQ1OT1 was reported to be highly expressed and accelerated BC progression [6–8]. However, lnc KCNQ1OT1 to drug resistance in BC has not been investigated. Currently, N6-methyladenosine (m6A) broadly exists in RNA modification to mediate the expression and stability of RNA [9]. The ‘writers’ m6 A methyltransferase-like 3 (METTL3) was found higher expression and adriamycin resistance in BC [10], indicating METTL3 might be involved in drug resistance to affect BC progression. As previously described, the METTL3-mediated MALAT1 expression by m6A modification promoted the adriamycin resistance to BC [11]. In this study, we would explore whether METTL3 mediate m6A modification of lnc KCNQ1OT1 to influence DOX resistance in BC.
Massive investigations have evidenced that the regulatory mechanism of competing endogenous RNA (ceRNA) such as lncRNA-miRNA-mRNA axis plays important roles in various diseases [12,13]. Previous studies revealed that miR-103a-3p was low expression in BC and was recommended as a diagnostic and prognostic biomarker [14,15]. MiR-103a-3p was involved with cisplatin resistance to non-small cell lung carcinoma [16]. Additionally, plentiful studies have demonstrated that multidrug resistance protein 1(MDR1) is identified as a key and independent risk factor of DOX resistance in cancers including BC [17,18]. In combination with bioinformatics analysis, the binding sites among lnc KCNQ1OT1, miR-103a-3p, and MDR1 were predicted by the starbase website. Hence, we conjectured that lnc KCNQ1OT1/miR-103a-3p/MDR1 axis might regulate DOX resistance in BC.
To sum up, we propose a notion that METTL3 upregulates and stabilizes lnc KCNQ1OT1 by m6A modification to enhance DOX resistance in BC via modulating miR-103a-3p/MDR1 axis. In this study, we employed BC cells and DOX-resistant BC cells as experimental subjects to conduct a series of experiments, which will lay the foundations for our hypothesis.
Methods
Cell culture and treatments
MCF10A (Human breast epithelial cells, ATCC, Manassas, VA, USA), MCF-7, and MDA-MB-231 (BC cells, Cell Bank of Chinese Academy of Science, Shanghai, China) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 100 units/mL penicillin and 100 mg/mL streptomycin (Invitrogen, Carlsbad, CA, USA) under condition of 5% CO2 flow at 37°C. DOX‐resistant MCF‐7 (MCF‐7/DOX) and DOX‐resistant MDA-MB-231 (MDA-MB-231/DOX) cells were generated by continuous exposure of MCF‐7 or MDA-MB-231 cells to gradient concentrations of DOX (Sigma‐Aldrich, St Louis, MO, USA) [19]. In addition, in this study, 1.5 μg/mL DOX was applied for the treatment of BC cells, and 30 μg/mL DOX was used to treat DOX-resistant BC cells.
The determination of cell viability and IC50 value
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay was employed to detect cell viability and IC50 (half maximal inhibitory concentration) values of DOX. The cells were implanted onto 96-well plates and cultured for 24 h. The transfected cells were treated with various concentrations of DOX (10, 20, 40, 80 ng/mL). About 20 μL MTT reagent solution (Beyotime) was supplied to each well and incubated for 4 h. Afterwards, the formazan crystals were dissolved using dimethyl sulphoxide. The absorbance at 490 nm was determined by a microplate reader (Bio-Tek, USA). The data were used to calculate the cell viability and IC50 value.
Colony formation assay
Cell proliferation was determined by colony formation. Shortly, MCF‐7/DOX and MDA-MB-231/DOX were implanted onto 6‐well plates (100 cells/well). Two weeks later, the cells were fixated with methanol for 15 min and were then stained with 0.1% crystal violet. Finally, the colony number of cells was calculated and documented under a microscope.
Flow cytometry
Flow cytometry was employed to detect cell apoptosis rate and cell cycle. Referring to manufacturer’s instruction, Annexin V-FITC/propidium iodide (PI) Apoptosis Detection Kit (YEASEN) was selected for evaluation of cell apoptosis rate. About 5 μL of Annexin V-FITC (labelled as early apoptotic cells) and 10 μL of PI (labelled late apoptotic and necrotic cells) Staining Solution were added in the cell suspension for 15 min.
For the detection of cell cycle, cells were fixed with ethanol for 48 h at 4°C and subsequently stained with PI. Finally, cell apoptosis rate and cell cycle were evaluated by flow cytometry (BD, NJ, USA).
Qrt-PCR
The RNAs were obtained from MCF10A cells, BC cells, and DOX-resistant BC cells using TRIzol reagent (Invitrogen, Carlsbad, USA). The Prime Script Reverse Transcription Reagent Kit (Takara, Dalian, China) was used to synthesize cDNA sequences. Using the SYBR Premix Ex Taq II Kit (Takara), qRT-PCR was performed on the ABI7500 system (Applied Biosystems, CA, USA). The primers were generalized as follows:
lnc KCNQ1OT1 (F), 5‘‑TTGGTAGGATTTTGTTGAGG-3’;
lnc KCNQ1OT1 (R), 5‘‑CAACCTTCCCCTACTACC-3’;
miR-103a-3p (F), 5‘-GCAGCAGCATTGTACAGGG-3’;
miR-103a-3p (TR), 5‘-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGAC
TCATAG-3’;
MDR1 (F), 5‘-TCGTTTCCTTTAGGTCTTTCCAC-3’;
MDR1 (R), 5‘-CTTCTTCTTTGCTCCTCCATTGC-3’;
U6 (F), 5’-CTCGCTTCGGCAGCACA-3’;
U6 (R), 5’-AACGCTTCACGAATTTGCGT-3’;
GAPDH (F), 5’-TGGAAGGACTCATGACCACA-3’;
GAPDH (R), 5’-TTCAGCTCAGGGATGACCTT-3’.
All THE data were calculated by using 2−ΔΔCt formula. The GAPDH and U6 were regarded as reference genes.
Western blot
Proteins were isolated from BC cells and DOX-resistant BC cells using RIPA buffer (Beyotime). Next, an equal amount of protein (20 μg) was added into the sample loading hole and were segregated by SDS-PAGE. Then, the isolated proteins were transferred onto PVDF membranes. After the membranes were blocked by non-fat milk for 1 h, the primary antibodies MDR1 (1:2000, #ab170904, Abcam, Cambridge, UK), cyclin D1 (1:200, #ab16663), Bax (1:5000, #ab32503), Bcl-2 (1:1000, #ab32124), and β-actin (1:5000, #ab6276) were incubated on the membranes overnight at 4°C. The HRP-conjugated secondary antibody was adopted to immerse PVDF membrane for 1 h. ECL chemiluminescent reagent (Beyotime) were adopted to evaluate protein bands and the densitometry analysis was tested by Image J.
Cell transfection
Small hairpin RNA targeting lnc KCNQ1OT1 (sh-lnc KCNQ1OT1), pcDNA3.1, pcDNA3.1-METTL3, pcDNA3.1-MDR1, miR-103a-3p inhibitor/mimics, small interfering RNA against METTL3 (si-METTL3), and negative control (si-NC, sh-NC, NC inhibitor, NC mimics, empty pcDNA3.1 vector) were purchased from by GenePharma (Shanghai, China). Using Lipofectamine 2000 (Invitrogen), the above sequences, and plasmids were transfected into BC cells and DOX‐resistant BC cells according to the manufacturer’s protocol.
Dual luciferase reporter gene assay
The sequences of lnc KCNQ1OT1 or MDR1 binding with miR-103a-3p were cloned into pGL3 vectors (Beyotime) for constructing reporter vectors lnc KCNQ1OT1 (WT/MUT) or MDR1 (WT/MUT). MiR-103a-3p mimics were transfected into DOX-resistant BC cells coupled with lnc KCNQ1OT1 (WT/MUT) or MDR1 (WT/MUT) by lipofectamine 3000. The luciferase activity was tested by a dual-luciferase reporter assay system (Promega).
RNA stability
As for the detection of RNA stability of lnc KCNQ1OT1. Actinomycin D (2 μg/mL) was added to prevent the process of gene transcription in cells. Next, RT-qPCR was performed to evaluate lnc KCNQ1OT1 mRNA level after 0, 1, 2, and 3 h.
M6A immunoprecipitation (MeRIP) and RNA immunoprecipitation (RIP) assay
MeRIP and RIP were used to validate the interaction between METTL3 and lnc KCNQ1OT1. A BersinBio RIP assay kit (BersinBio, Guangzhou, China) and m6A RNA Enrichment Kit (EPIGENTEK, Farmingdale, NY, USA) were conducted following the manufacturer’s protocol. Cell lysates were incubated with magnetic beads and anti-m6A (Abcam) or anti-METTL3 (Abcam) or anti-AGO2 (Abcam) or anti-IgG (Abcam). Lnc KCNQ1OT1 with m6A modification, lnc KCNQ1OT1, and miR-103a-3p enrichment were evaluated by RT-qPCR.
Statistical analysis
The data were presented as mean ± SD. Statistical analysis was processed by GraphPad Prism 6 (San Diego, CA, United States) using Student’s paired t-test between two groups and one-way ANOVA among three or more than three groups. All data were derived from three independent experiments. The p-value less than 0.05 was identified to be statistically significant.
Results
Lnc KCNQ1OT1 and MDR1 were highly expressed in DOX-resistant BC cells, while miR-103a-3p was lowly expressed
Here, BC cells (MCF-7 and MDA-MB-231) and DOX-resistant BC cells (MCF-7/DOX and MDA-MB-231/DOX) served as experimental subjects. A series of concentrations of DOX (0.5, 1, 1.5, 2, 20, 40, 80 μg/mL) were added to the cells to determine IC50 values of DOX (Table 1). The results revealed that IC50 of DOX resistant BC cells was notably higher than that of BC cells. In addition, cell viability of DOX resistant BC cells was stronger than that of BC cells under DOX treatment (Figure 1a). Subsequently, 1.5 μg/mL DOX was selected to intervene following experiments. Clone formation exhibited that with DOX addition, the number of DOX-resistant BC cells was apparently more than that of corresponding BC cells (Figure 1b). Inversely, the apoptosis rate of DOX-resistant BC cells was lower relative to corresponding BC cells (Figure 1c). These results revealed that DOX intervention has limited influence on DOX-resistant BC cells. Subsequently, lnc KCNQ1OT1, miR-103a-3p and MDR1 levels were determined. In DOX-resistant BC cells, lnc KCNQ1OT1, and MDR1 levels were dramatically elevated and miR-103a-3p level was distinctly declined, which were compared to BC cells (Figure 1d-e), indicating lnc KCNQ1OT1, miR-103a-3p, and MDR1 might be implicated in DOX resistant BC.
Table 1.
IC50 values of different cell lines under DOX treatment.
| Cell line | IC50 |
|---|---|
| MCF-7 | 1.25 μg/mL |
| MDA-MB-231 | 1.53 μg/mL |
| MCF-7/DOX | 33.71 μg/mL |
| MDA-MB-231/DOX | 27.45 μg/mL |
Figure 1.
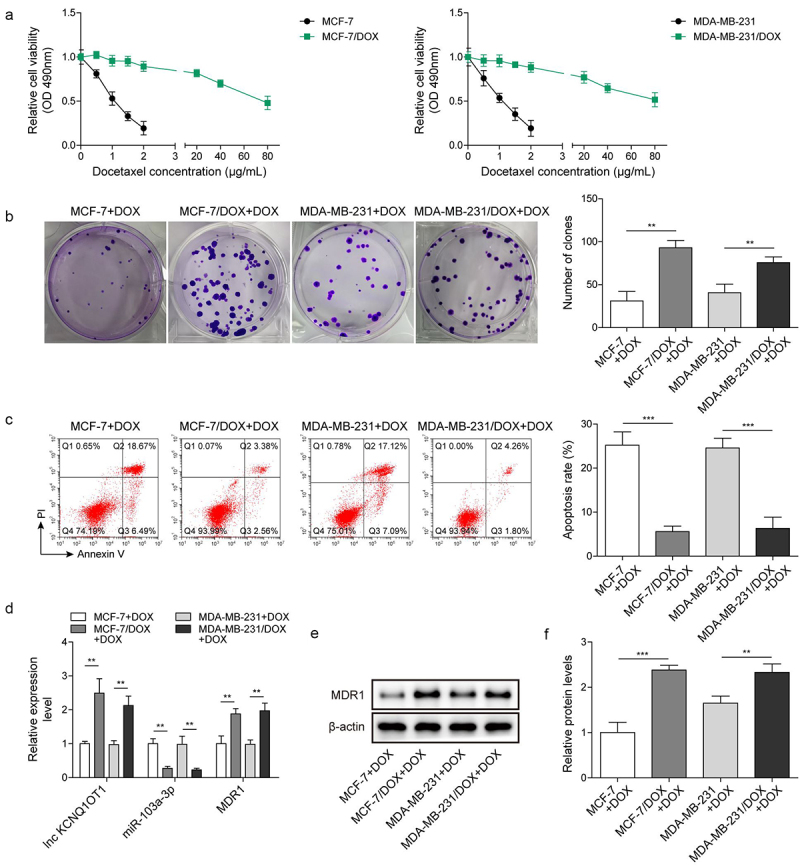
Lnc KCNQ1OT1 and MDR1 level were highly expressed in DOX-resistant BC cells, while miR-103a-3p level was lowly expressed. Note: a, IC50 values of BC cells and DOX-resistant BC cells were determined by MTT. b, cell proliferation was detected using clone formation. c, the apoptosis rate was measured with flow cytometry. d, lnc KCNQ1OT1, miR-103a-3p, and MDR1 expression was tested using qRT-PCR. e, western blot was performed to measure MDR1 expression.
Lnc KCNQ1OT1 silencing promoted the suppressing effects of DOX on BC cells through elevating miR-103a-3p
To explore the function of lnc KCNQ1OT1on BC cells (MCF-7 and MDA-MB-231) upon DOX treatment, MCF-7 and MDA-MB-231 cells were transfected with sh-lnc KCNQ1OT1. Of note, the concentration of DOX to treat BC cells was 1.5 μg/mL. qRT-PCR displayed that lnc KCNQ1OT1 was successful knockdown in MCF-7 and MDA-MB-231 cells and miR-103a-3p expression was obviously upregulated by sh-lnc KCNQ1OT1 transfection (Figure S1a). Cell viability of BC cells was prominently decreased by sh-lnc KCNQ1OT1 depletion compared to that of sh-NC transfection (Figure S1b). Lnc KCNQ1OT1 knockdown inhibited cell clone formation and elevated cell apoptosis rate of BC cells (Figure S1c-d). Concomitantly, apoptosis-related proteins were detected. We found that Bax was notably elevated, while Bcl-2 was evidently reduced in BC cells with sh-lnc KCNQ1OT1 transfection (Figure S1e). Altogether, sh-lnc KCNQ1OT1 enhanced DOX-induced suppression of BC cells via regulating miR-103a-3p.
Lnc KCNQ1OT1 depletion restrained DOX resistance in DOX-resistant BC cells via elevating miR-103a-3p
To investigate the function of lnc KCNQ1OT1 on DOX-resistant BC cells, MCF-7/DOX and MDA-MB-231/DOX were transfected with sh-lnc KCNQ1OT1 to silence lnc KCNQ1OT1 and then followed DOX treatment. Of note, 30 μg/mL DOX was applied to treat DOX-resistant BC cells. Lnc KCNQ1OT1 expression was evidently downregulated, and miR-103a-3p expression was notably upregulated by sh-lnc KCNQ1OT1 transfection (Figure 2a). Cell viability of DOX resistant BC cells was apparently suppressed by sh-lnc KCNQ1OT1 silencing (Figure 2b). Besides, lnc KCNQ1OT1 knockdown attenuated the ability of cell clone formation and promoted cell apoptosis rate of DOX resistant BC cells (Figure 2c-d). Meanwhile, Bax was observably increased, while Bcl-2 was evidently reduced in DOX-resistant BC cells by sh-lnc KCNQ1OT1 transfection (Figure 2e). These results suggested that lnc KCNQ1OT1 depletion resulted in enhanced DOX-sensitivity to DOX-resistant BC cells. Additionally, starbase predicted a putative binding site between lnc KCNQ1OT1 and miR-103a-3p. Luciferase activity in WT-lnc KCNQ1OT1 group was suppressed by miR-103a-3p mimics but failed to change luciferase activity in MUT-lnc KCNQ1OT1 group, RIP experiment further verified the targeting–binding relationship between lnc KCNQ1OT1 and miR-103a-3p (Figure 2f). These data suggested the interaction between lnc KCNQ1OT1 and miR-103a-3p. Taken together, Knockdown of lnc KCNQ1OT1 elevated miR-103a-3p to repress DOX resistance in DOX resistant BC cells.
Figure 2.
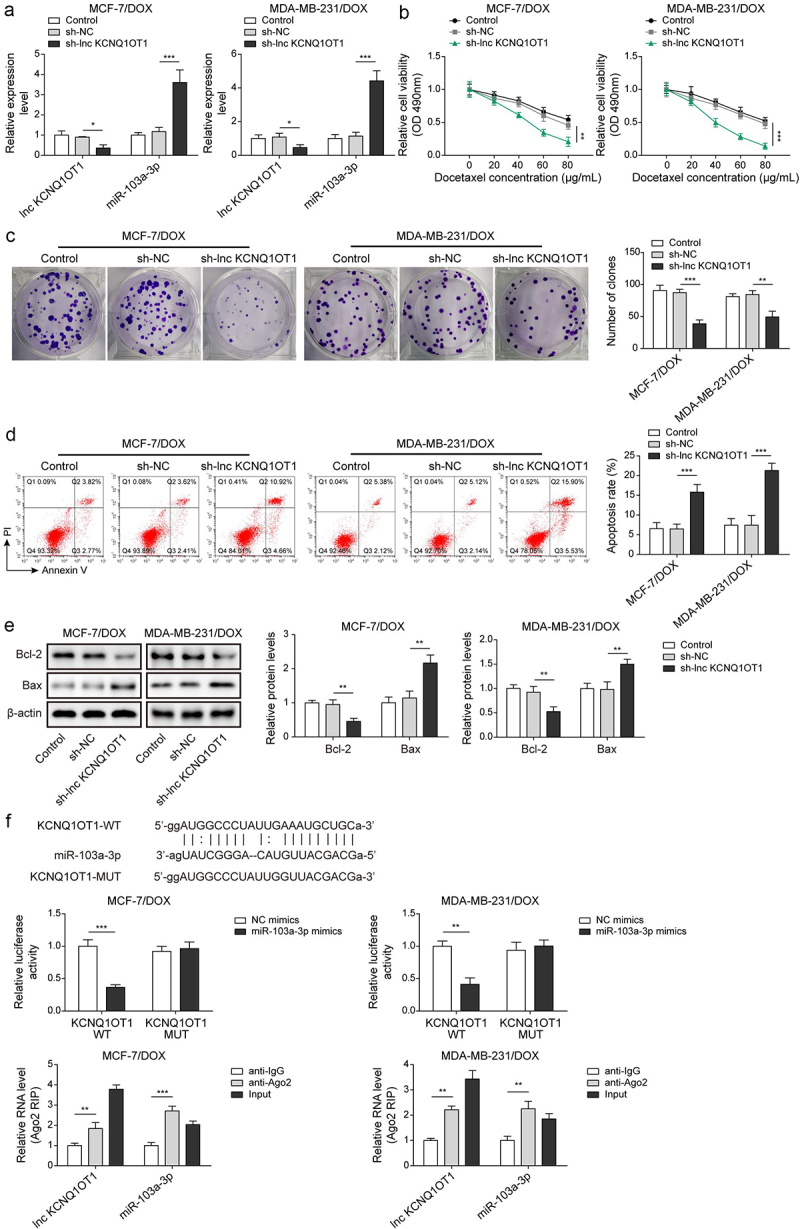
Lnc KCNQ1OT1 depletion restrained DOX resistance in BC cells and DOX-resistant BC cells via elevating miR-103a-3p.Note: sh-lnc KCNQ1OT1 was transfected into DOX-resistant BC cells. a, qRT-PCR was employed to detect transfection efficiency and miR-103a-3p expression. b, cell viability was determined by MTT. c, the capability of cell proliferation was detected using clone formation. d, the apoptosis rate was measured with flow cytometry. e, western blot was performed to measure Bax, Bcl-2 expression. f, the mutual binding site between lnc KCNQ1OT1 and miR-103a-3p was predicted by starbase, and the interplay between lnc KCNQ1OT1 and miR-103a-3p was validated by dual-luciferase reporter gene assay and RIP assay.
MiR-103a-3p overexpression/inhibition impacted DOX-mediated influences on BC cells through modulating MDR1
Here, we investigated whether miR-103a-3p affects DOX-mediated impacts on BC cells. BC cells (MCF-7 and MDA-MB-231) were transfected with miR-103a-3p mimics or inhibitor and then followed by DOX treatment. MiR-103a-3p expression was elevated or reduced by miR-103a-3p mimics or inhibitor in BC cells (Figure S2a, 2e). Simultaneously, MDR1 expression was suppressed by miR-103a-3p mimics while it was elevated by miR-103a-3p inhibitor in BC cells (Figure S2a, 2e). Moreover, miR-103a-3p mimics could attenuate cell viability, migration, and cell cycle, while promoting cell apoptosis of BC cells, whereas miR-103a-3p silencing could acquire opposite results (Figure S2b-2d). Additionally, the apoptosis-related protein Bax was increased, and Bcl-2 was inhibited as well as cycle-related protein cyclin D1 was reduced by miR-103a-3p mimics; however, miR-103a-3p inhibitor had the opposite effects to miR-103a-3p mimics in BC cells (Figure S2e). Overall, miR-103a-3p could regulate cell growth in DOX-treated BC cells by mediating MDR1.
MiR-103a-3p overexpression/inhibition affected DOX resistance of DOX-resistant BC cells through modulating MDR1
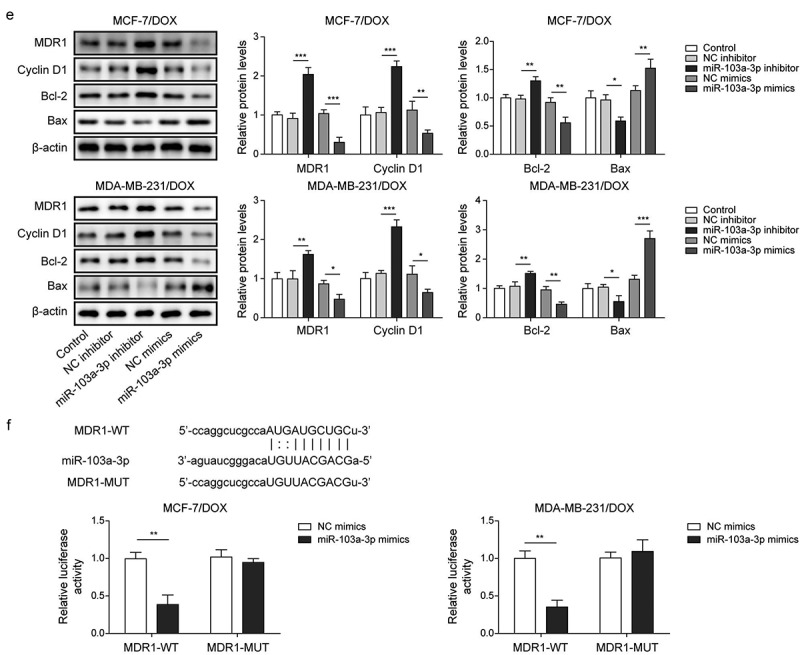
Next, the roles of miR-103a-3p in DOX-resistant BC cells as well as its downstream target were investigated. Firstly, DOX-resistant BC cells (MCF-7/DOX and MDA-MB-231/DOX) were transfected with miR-103a-3p mimics or inhibitors and then followed by DOX treatment. We observed that miR-103a-3p mimics or inhibitors resulted in elevated or reduced miR-103a-3p expression in DOX-resistant BC cells (Figure 3a, 3e). However, MDR1 expressions acquired opposite alterations (Figures 3a, 3e). Besides, miR-103a-3p mimics suppressed cell viability, migration, cell cycle, and promoted cell apoptosis of DOX-resistant BC cells compared to that of NC-inhibitor group, whereas miR-103a-3p silencing could acquire opposite results (Figure 3b-d). Meanwhile, the apoptosis-related proteins Bax was enhanced and Bcl-2 was decreased as well as cycle-related protein cyclin D1 was down-regulated by miR-103a-3p mimics in DOX-resistant BC cells, whereas miR-103a-3p inhibitor had the opposite effects (Figure 3e). Additionally, the Starbase website predicted a binding site between miR-103a-3p and MDR1 and the interaction between the two was supported by the results of dual-luciferase reporter gene assay (Figure 3f). In total, miR-103a-3p negatively mediated MDR1 level to enhance DOX’s sensibility of DOX-resistant BC cells.
Figure 3.
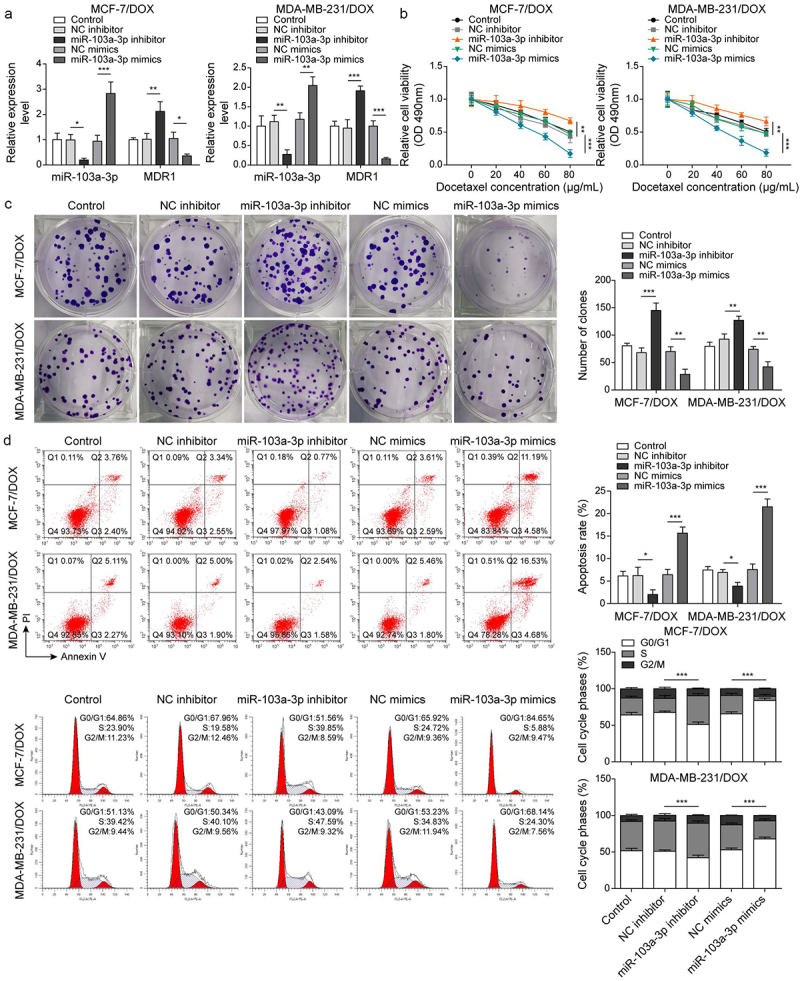
MiR-103a-3p overexpression/knockdown affected DOX resistance of BC cells and DOX-resistant BC cells through modulating MDR1.Note: DOX-resistant BC cells suffered miR-103a-3p mimics/inhibitor. a, qRT-PCR was employed to detect transfection efficiency and MDR1 expression. b, cell viability was determined by MTT. c, cell proliferation was detected using clone formation. d, the apoptosis rate and cell cycle were measured using flow cytometry. e, western blot was performed to measure MDR1, cyclin D1, Bax, and Bcl-2 expressions. f, the mutual binding site between miR-103a-3p and MDR1 was predicted by starbase, and the interplay between miR-103a-3p and MDR1 was consolidated by dual-luciferase reporter gene assay.
Figure 3.
(Continued).
Lnc KCNQ1OT1 indirectly elevated MDR1 expression to promote DOX resistance of BC cells and DOX-resistant BC cells
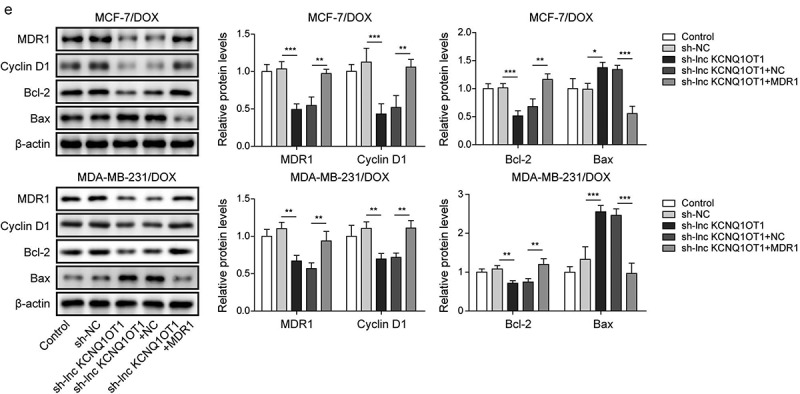
To reveal whether MDR1 affects lnc KCNQ1OT1-mediated DOX-resistance in DOX-resistant BC cells, DOX-resistant BC cells (MCF-7/DOX and MDA-MB-231/DOX) were transfected with sh-lnc KCNQ1OT1 or together with MDR1 overexpression vector following DOX treatment. qRT-PCR displayed that lnc KCNQ1OT1 knockdown reduced lnc KCNQ1OT1 and MDR1 but increased miR-103a-3p. However, MDR1 overexpression reversed lnc KCNQ1OT1 silencing-mediated reduction of MDR1 expression but failed to affect lnc KCNQ1OT1 and miR-103a-3p expression (Figures 4a and 4f). Additionally, lnc KCNQ1OT1 knockdown suppressed cell viability, cell clone, and cell cycle as well as promoted cell apoptosis in DOX-resistant BC cells, which were abolished by MDR1 overexpression (Figure 4b-d). Meanwhile, MDR1 overexpression abolished sh-lnc KCNQ1OT1-mediated elevation of Bax and reduction of Bcl-2 and cyclin D1 (Figure 4e). In conclusion, the knockdown of lnc KCNQ1OT1 could improve DOX’s sensibility of DOX-resistant BC cells through downregulating MDR1.
Figure 4.
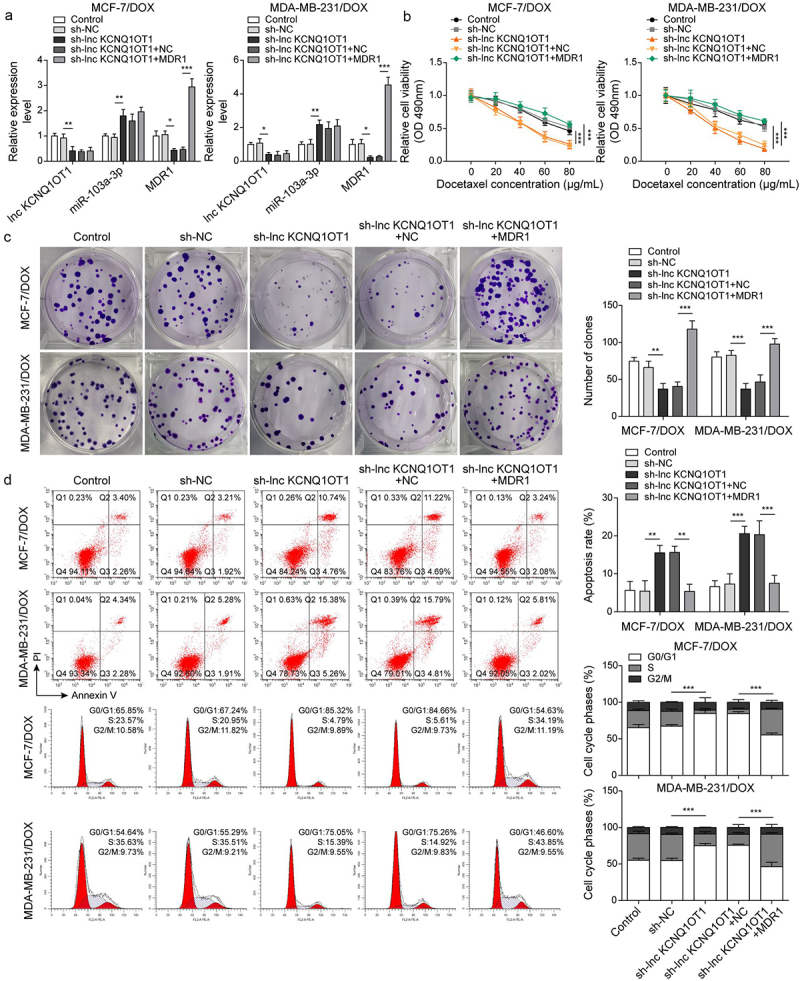
Lnc KCNQ1OT1 indirectly elevated MDR1 expression to promote DOX resistance of BC cells and DOX-resistant BC cells.Note: DOX-resistant BC cells were transfected with sh-lnc KCNQ1OT1 alone or along with oe-MDR1. a, qRT-PCR was employed to detect lnc KCNQ1OT1, miR-103a-3p, and MDR1 expressions. b, cell viability was determined by MTT. c, cell proliferation was detected using clone formation. d, the apoptosis rate and cell cycle were measured with flow cytometry. e, western blot was performed to measure MDR1, cyclin D1, Bax, and Bcl-2 expressions.
Figure 4.
(Continued).
METTL3 modulated m6A modification of lnc KCNQ1OT1 to facilitate DOX resistance of DOX-resistant BC cells
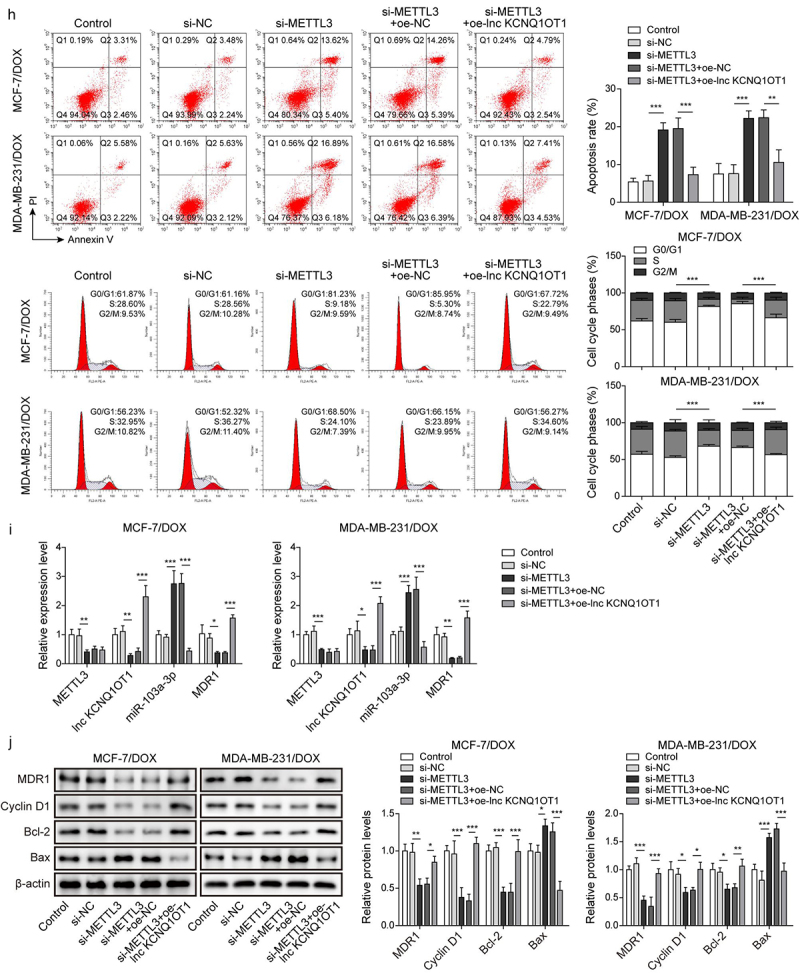
As previously reported, METTL3 mediating m6A modification of lncRNA is a prevalent phenomenon during biological regulation [20]. Therefore, we detected METTL3 expression through qRT-PCR in BC cells and DOX-resistant BC cells. The results exhibited that the expression of METTL3 in BC cells (MCF-7 and MDA-MB-231) was higher than that in normal breast cells (MCF10A). Noticeably, METTL3 expression was apparently increased in MCF-7/DOX and MDA-MB-231/DOX cells compared to that in MCF-7 and MDA-MB-231 cells (Figure 5a), indicating the importance of METTL3 on DOX resistance in DOX-resistant BC cells. Subsequently, MCF-7/DOX and MDA-MB-231/DOX cells were selected for subsequent experiments. si-METTL3 and oe-METTL3 were transfected into DOX-resistant cells to decline and increase METTL3 expression, respectively (Figure 5b). Concomitantly, lnc KCNQ1OT1 expression was also suppressed by METTL3 knockdown and was elevated by METTL3 overexpression (Figure 5b). In addition, we detected RNA stability of lnc KCNQ1OT1. DOX-resistant BC cells were treated with actinomycin D. The results revealed that the METTL3 knockdown accelerated the degradation of lnc KCNQ1OT1, but the METTL3 overexpression suppressed the degradation of lnc KCNQ1OT1 (Figure 5c). Moreover, we found that lnc KCNQ1OT1 was modified by m6A in DOX-resistant BC cells, which was determined by MeRIP-qPCR (Figure 5d). Meanwhile, the METTL3 knockdown resulted in decreased m6A modification of lnc KCNQ1OT1 (Figure 5d). As expected, RIP exhibited that lnc KCNQ1OT1 was apparently enriched by anti-METTL3 (Figure 5e). The above results indicated that METTL3 interacted with lnc KCNQ1OT1 and mediated lnc KCNQ1OT1 m6A modification. To further probe how METTL3/lnc KCNQ1OT1 axis affects DOX sensibility to DOX-resistant BC cells, MCF-7/DOX and MDA-MB-231/DOX cells were transfected si-METTL3 alone or along with oe-lnc KCNQ1OT1. The detailed groups were as follows: Control, si-NC, si-METTL3, si-METTL3+oe-NC, si-METTL3+ oe-lnc KCNQ1OT1. The results revealed that METTL3 silencing suppressed cell viability, cell clone formation, and cell cycle but enhanced cell apoptosis in DOX-resistant BC cells, whereas these phenomena were reversed by lnc KCNQ1OT1 overexpression (Figure 5f-h). In addition, METTL3 knockdown reduced the expression of METTL3, lnc KCNQ1OT1, and MDR1, while elevated the expression of miR-103a-3p. However, lnc KCNQ1OT1 overexpression abolished METTL3 knockdown-mediated reduction of lnc KCNQ1OT1 and MDR1 expression and elevation of miR-103a-3p expression but failed to influence METTL3 expression (Figure 5i). Western blot exhibited that the levels of MDR1, Bcl-2, and cyclin D1 were inhibited, and the level of Bax was elevated by METTL3 silencing, while lnc KCNQ1OT1 overexpression reversed these effects (Figure 5j). Altogether, METTL3 knockdown suppressed DOX resistance in DOX-resistant BC cells through declining lnc KCNQ1OT1 expression.
Figure 5.
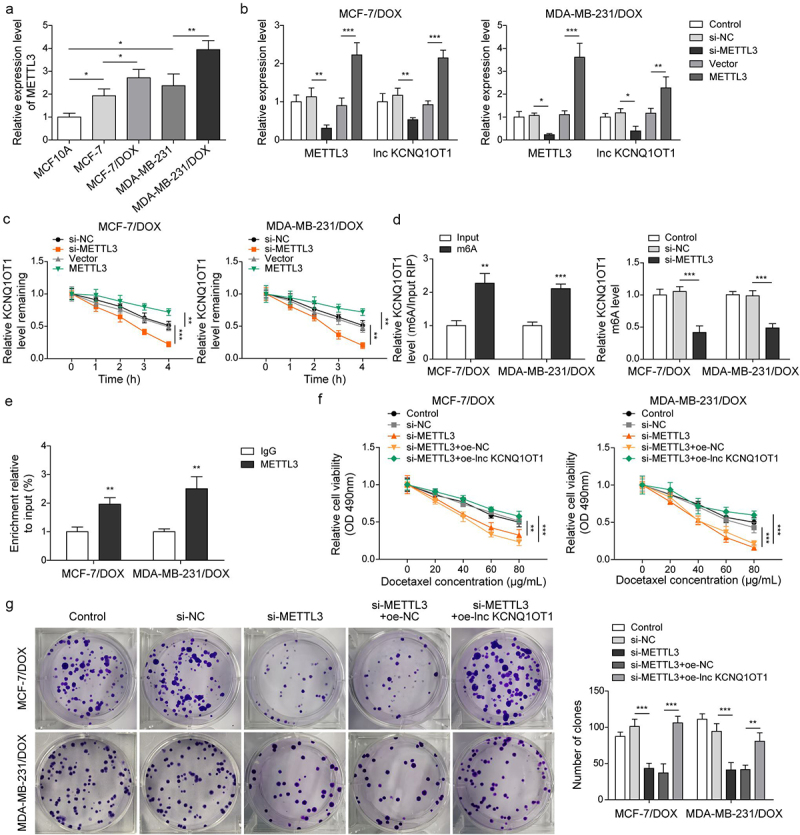
METTL3 modulated m6A modification of lnc KCNQ1OT1 to facilitate DOX resistance of DOX-resistant BC cells.Note: a, qRT-PCR was applied to examine METTL3 expression in MCF10A, MCF-7, MDA-MB-231, MCF‐7/DOX, and MDA-MB-231/DOX cells. si-METTL3 or oe-METTL3 was transfected into DOX-resistant BC cells. b, qRT-PCR was performed to measure METTL3 and lnc KCNQ1OT1 expression. c, qRT-PCR was applied to detect the stability of lnc KCNQ1OT1. d, MeRIP‐qPCR was applied to test lnc KCNQ1OT1 expression of m6A modification. e, RIP assay was adopted to evaluate the enrichment of lnc KCNQ1OT1 by anti-METTL3 in MCF‐7/DOX and MDA-MB-231/DOX cells. si-METTLE3 alone or along with oe-lnc KCNQ1OT1 were transfected into DOX-resistant BC cells. f, cell viability was determined by MTT. g, cell proliferation was detected using clone formation. h, the apoptosis rate and cell cycle were measured with flow cytometry. i, lnc KCNQ1OT1, miR-103a-3p, and MDR1 expressions were tested using qRT-PCR. j, western blot was performed to measure MDR1, cyclin D1, Bax, and Bcl-2 expressions.
Figure 5.
(Continued).
Discussion
Drug resistance is a common problem when tumours are treated with chemotherapeutics [21]. Previous studies suggested that DOX resistance significantly hinders the therapeutic effect in BC, resulting in a poor prognosis [22]. Enhancing DOX sensitivity to BC cells could suppress the progression of BC. In this study, we found lnc KCNQ1OT1, which was modified by METTL3-mediated m6A modification, promoted DOX resistance in DOX-resistant BC cells by regulating miR-103a-3p/MDR1 axis.
Massive evidence has demonstrated that lncRNAs are closely related to chemotherapy resistance in tumours [23]. For instance, lncRNA DDX11-AS1 promoted adriamycin (ADR) resistance through LIN28A-mediated ATG12 mRNA stabilization in BC [24]. Lnc KCNQ1OT1 was reported to be closely related to multiple chemotherapeutic drug resistances such as ADR and Temozolomide in tumours including acute myeloid leukaemia and gliomas [25,26]. At present, previous studies have revealed lnc KCNQ1OT1 promoted BC progression [8]. However, there was no evidence of lnc KCNQ1OT1 on DOX resistance in DOX-resistance BC. In this study, we found that lnc KCNQ1OT1 was elevated in DOX-resistant BC cells compared to BC cells. Lnc KCNQ1OT1 knockdown effectively enhanced DOX sensibility in DOX-resistant BC cells by suppressing cell viability and elevating cell apoptosis.
Which molecular regulatory mechanism results in increased lnc KCNQ1OT1 expression in DOX-resistant BC cells? Numerous studies have suggested that m6A modification to lncRNA is a common reason. Furthermore, m6A modification of lncRNA was extensively explored in cancers and correlated with chemotherapy resistance [27]. Here, we found that lnc KCNQ1OT1 was notably modified by m6A, which evidenced by MeRIP-qPCR. METTL3 is a crucial transmethylase that mediates lncRNAs’ m6A modification and regulates the expression of lncRNAs [28]. For instance, METTL3 stabilized and upregulated lncRNA LNCAROD through m6A modification to promote the development of head and neck squamous cell carcinoma (HNSCC) [29]. In addition, METTL3 mediated lncRNA MALAT1 m6A modification to enhance lncRNA MALAT1 expression, promoting cisplatin resistance in non-small lung cancer (NSCLC) [28]. Min Li and co-workers suggested that METTL3 compromised the influences of 5‑Fluorouracil (5‑FU) chemotherapy and contributed to 5-FU resistance in colorectal carcinoma [30]. In this work, METTL3 elevated DOX-resistant BC cells compared to BC cells. The RIP assay determined that METTL3 interacted with lnc KCNQ1OT1 and METTL3 knockdown inhibited m6A modified level of lnc KCNQ1OT1 and suppressed lnc KCNQ1OT1 expression. Moreover, METTL3 knockdown resulted in suppressed cell viability and cell cycle and enhanced cell apoptosis of DOX-resistant BC cells, which were abolished by lnc KCNQ1OT1 overexpression. These results indicated that METTL3 knockdown reduced m6A level of lnc KCNQ1OT1 and the expression of lnc KCNQ1OT1, which resulted in suppression of DOX resistance in DOX-resistant BC cells.
It is generally known that lncRNA could interact with miRNA as a sponge to affect gene expression, which is the regulatory mechanism is broadly existing biological functions [31]. For example, lncRNA NEAT1/miR-23a-3p/FOXA1 axis regulated Taxol resistance of BC [32]. In current study, lnc KCNQ1OT1/miR-103a-3p/MDR1 axis was established to mediate DOX resistance in BC. As previously described, miR-103a-3p was lower expression in BC [15,33]. In current study, miR-103a-3p expression was apparently declined in DOX-resistant BC cells. Furthermore, miR-103a-3p mimics could effectively suppressed cell viability and cell cycle and elevated cell apoptosis of BC cells and DOX-resistant BC cells. Moreover, starbase and dual luciferase reporter gene assay validated that miR-103a-3p interacted with lnc KCNQ1OT1. Lnc KCNQ1OT1 knockdown enhanced miR-103a-3p expression, indicating miR-103a-3p was regulated by lnc KCNQ1OT1. MDR1 is a well-known molecule involving chemotherapy resistance in tumours [34]. A former study found that MDR1 was highly expressed in cisplatin-resistant BC and was negatively regulated by miR-381, and targeting MDR1 suppressed cisplatin resistance in BC [35]. In our study, MDR1 expression was elevated in DOX-resistant BC cells compared with BC cells. Furthermore, MDR1 was a downstream molecular of miR-103a-3p and negatively regulated miR-103a-3p. Exogenous overexpression of MDR1 reversed lnc KCNQ1OT1 silencing-mediated suppression of DOX resistance in DOX-resistant BC cells. which was that firstly put forward the function of lnc KCNQ1OT1/miR-103a-3p/MDR1 axis in DOX-resistant BC cells (Figure 6).
Figure 6.
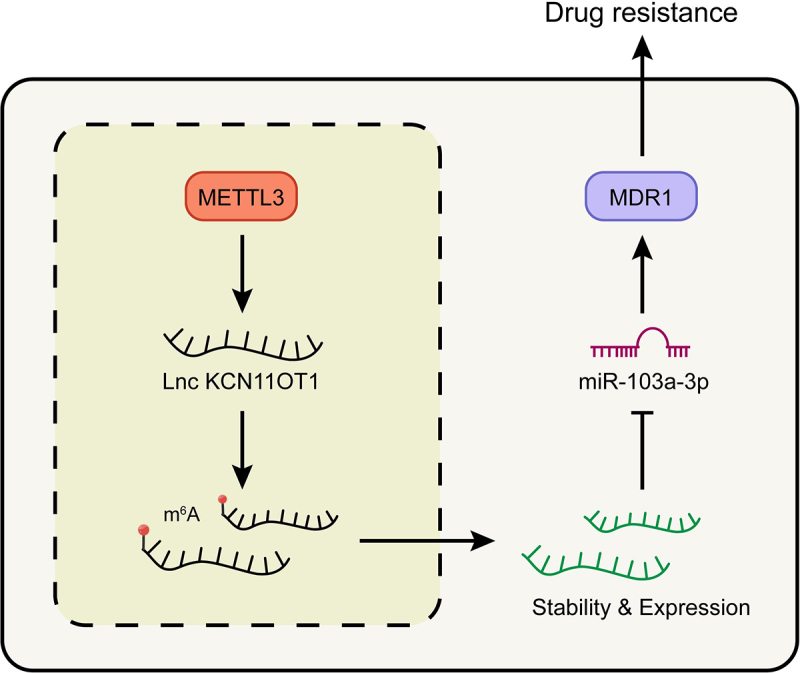
Schematic of molecular mechanism.Note: METTL3 promoted the expression of lncKCNQ1OT1 by regulating its m6A modification. While the up-regulated lncKCNQ1OT1 played the role of ceRNA in the targeted inhibition of miR-103a-3p, and MDR1, as the downstream molecule targeting miR-103a-3p, could enhance DOX resistance of breast cancer.
In conclusion, we put forward for the first time that METTL3 mediated m6A modification of lnc KCNQ1OT1 and promoted lnc KCNQ1OT1 expression, which prompted DOX resistance in DOX-resistant BC cells and accelerated the progression of BC through suppressing miR-103a-3p/MDR1 axis. Therefore, targeting METTL3/lnc KCNQ1OT1/miR-103a-3p/MDR1 axis might be effectively suppressed DOX resistance of BC. Frankly, there are still some limitations. Our data are only acquired from the cellular level, lacking validation on animal and clinical samples. Additionally, we did not probe the signalling pathways that might be involved in DOX resistance in BC. Hence, more experiments need to be carried out to further illuminate this scientific problem.
Supplementary Material
Funding Statement
This work was supported by the Natural Science Foundation of Hunan Province (2021JJ31049)
Data availability statement
All data generated or analysed during this study are included in this published article
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2217033.
References
- [1].Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: gLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–17. DOI: 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- [2].Zangouei AS, Alimardani M, Moghbeli M.. MicroRNAs as the critical regulators of Doxorubicin resistance in breast tumor cells. Cancer Cell Int. 2021;21(1):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hashemi M, Hajimazdarany S, Mohan CD, et al. Long non-coding RNA/epithelial-mesenchymal transition axis in human cancers: tumorigenesis, chemoresistance, and radioresistance. Pharmacol Res. 2022;186:106535. [DOI] [PubMed] [Google Scholar]
- [4].He J, Zhu S, Liang X, et al. LncRNA as a multifunctional regulator in cancer multi-drug resistance. Mol Biol Rep. 2021;48(8):1–15. DOI: 10.1007/s11033-021-06603-7 [DOI] [PubMed] [Google Scholar]
- [5].He S, Feng Y, Zou W, et al. The Role of the SOX9/lncRNA ANXA2P2/miR-361-3p/SOX9 Regulatory Loop in Cervical Cancer Cell Growth and Resistance to Cisplatin. Front Oncol. 2021;11:784525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yuan F, Lou Z, Zhou Z, et al. Long non‑coding RNA KCNQ1OT1 promotes nasopharyngeal carcinoma cell cisplatin resistance via the miR‑454/USP47 axis. Int J Mol Med. 2021;47(4). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [7].Ren Z, Xu Y, Wang X, et al. KCNQ1OT1 affects cell proliferation, invasion, and migration through a miR-34a/Notch3 axis in breast cancer. Environ Sci Pollut Res Int. 2022;29(19):28480–28494. DOI: 10.1007/s11356-021-18434-x [DOI] [PubMed] [Google Scholar]
- [8].Wu Y, Bi Q-J, Han R, et al. Long noncoding RNA KCNQ1OT1 is correlated with human breast cancer cell development through inverse regulation of hsa-miR-107. Biochem Cell Biol. 2020;98(3):338–344. DOI: 10.1139/bcb-2019-0271 [DOI] [PubMed] [Google Scholar]
- [9].Zhang N, Zuo Y, Peng Y, et al. Function of N6-Methyladenosine Modification in Tumors. J Oncol. 2021;2021:6461552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhao C, Ling X, Xia Y, et al. The m6A methyltransferase METTL3 controls epithelial-mesenchymal transition, migration and invasion of breast cancer through the MALAT1/miR-26b/HMGA2 axis. Cancer Cell Int. 2021; 21(1): 441. DOI: 10.1186/s12935-021-02113-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li S, Jiang F, Chen F, et al. Effect of m6A methyltransferase METTL3 -mediated MALAT1/E2F1/AGR2 axis on adriamycin resistance in breast cancer. J Biochem Mol Toxicol. 2022;36(1):e22922. DOI: 10.1002/jbt.22922 [DOI] [PubMed] [Google Scholar]
- [12].López-Urrutia E, Bustamante Montes LP, Ladrón de Guevara Cervantes D, et al. Crosstalk Between Long Non-coding RNAs, Micro-RNAs and mRnas: deciphering Molecular Mechanisms of Master Regulators in Cancer. Front Oncol. 2019;9:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Heydarzadeh S, Ranjbar M, Karimi F, et al. Overview of host miRNA properties and their association with epigenetics, long non-coding RNAs, and Xeno-infectious factors. Cell Biosci. 2021;11(1):43. DOI: 10.1186/s13578-021-00552-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu H, Bian Q-Z, Zhang W, et al. Circulating microRna‑103a‑3p could be a diagnostic and prognostic biomarker for breast cancer. Oncol Lett. 2022;23(1):38. DOI: 10.3892/ol.2021.13156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu X, Qiao K, Zhu K, et al. Long Noncoding RNA HCG18 Promotes Malignant Phenotypes of Breast Cancer Cells via the HCG18/miR-103a-3p/UBE2O/mTORC1/HIF-1α–Positive Feedback Loop. Front Cell Dev Biol. 2021;9:675082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhu H, Yang J, Yang S. MicroRNA-103a-3p potentiates chemoresistance to cisplatin in non-small cell lung carcinoma by targeting neurofibromatosis 1. Exp Ther Med. 2020;19(3):1797–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen S, Wang H, Li Z, et al. Interaction of WBP2 with ERα increases doxorubicin resistance of breast cancer cells by modulating MDR1 transcription. Br J Cancer. 2018;119(2):182–192. DOI: 10.1038/s41416-018-0119-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Elia SG, Al‐karmalawy AA, Nasr MY, et al. Loperamide potentiates doxorubicin sensitivity in triple-negative breast cancer cells by targeting MDR1 and JNK and suppressing mTOR and Bcl-2: in vitro and molecular docking study. J Biochem Mol Toxicol. 2022;36(1):e22938. DOI: 10.1002/jbt.22938 [DOI] [PubMed] [Google Scholar]
- [19].Wang Q, Liang D, Shen P, et al. Hsa_circ_0092276 promotes doxorubicin resistance in breast cancer cells by regulating autophagy via miR-348/ATG7 axis. Transl Oncol. 2021;14(8):101045. DOI: 10.1016/j.tranon.2021.101045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yi YC, Chen X-Y, Zhang J, et al. Novel insights into the interplay between m(6)A modification and noncoding RNAs in cancer. Mol Cancer. 2020;19(1):121. DOI: 10.1186/s12943-020-01233-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gatenby RA, Brown JS. The Evolution and Ecology of Resistance in Cancer Therapy. Cold Spring Harb Perspect Med. 2020;10(11):10(11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McGuirk S, Audet-Delage Y, Annis MG, et al. Resistance to different anthracycline chemotherapeutics elicits distinct and actionable primary metabolic dependencies in breast cancer. Elife. 2021;10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang X, Xie K, Zhou H, et al. Role of non-coding RNAs and RNA modifiers in cancer therapy resistance. Mol Cancer. 2020;19(1):47. DOI: 10.1186/s12943-020-01171-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Si X, Zhang G, Li M, et al. LncRNA DDX11-AS1 Promotes Chemoresistance through LIN28A-Mediated ATG12 mRNA Stabilization in Breast Cancer. Pharmacology. 2023;108(1):61–73. DOI: 10.1159/000527222 [DOI] [PubMed] [Google Scholar]
- [25].Sun H, Sun Y, Chen Q, et al. LncRNA KCNQ1OT1 contributes to the progression and chemoresistance in acute myeloid leukemia by modulating Tspan3 through suppressing miR-193a-3p. Life Sci. 2020;241:117161. [DOI] [PubMed] [Google Scholar]
- [26].Wang W, Han S, Gao W, et al. Long Noncoding RNA KCNQ1OT1 Confers Gliomas Resistance to Temozolomide and Enhances Cell Growth by Retrieving PIM1 from miR-761. Cell Mol Neurobiol. 2022;42(3):695–708. DOI: 10.1007/s10571-020-00958-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shriwas O, Mohapatra P, Mohanty S, et al. The Impact of m6A RNA Modification in Therapy Resistance of Cancer: implication in Chemotherapy, Radiotherapy, and Immunotherapy. Front Oncol. 2020;10:612337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jin D, Guo J, Wu Y, et al. M(6)a mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12(1):135. DOI: 10.1186/s13045-019-0830-6 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [29].Ban Y, Tan P, Cai J, et al. LNCAROD is stabilized by m6A methylation and promotes cancer progression via forming a ternary complex with HSPA1A and YBX1 in head and neck squamous cell carcinoma. Mol Oncol. 2020;14(6):1282–1296. DOI: 10.1002/1878-0261.12676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li M, Xia M, Zhang Z, et al. METTL3 antagonizes 5‑FU chemotherapy and confers drug resistance in colorectal carcinoma. Int J Oncol. 2022;61(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li JH, Liu S, Zhou H, et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(D1):D92–7. DOI: 10.1093/nar/gkt1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhu L, Wang F, Fan W, et al. lncRNA NEAT1 promotes the Taxol resistance of breast cancer via sponging the miR-23a-3p-FOXA1 axis. Acta Biochim Biophys Sin. 2021;53(9):1198–1206. DOI: 10.1093/abbs/gmab098 [DOI] [PubMed] [Google Scholar]
- [33].Sun Z, Zhang Q, Yuan W, et al. MiR-103a-3p promotes tumour glycolysis in colorectal cancer via hippo/YAP1/HIF1A axis. J Exp Clin Cancer Res. 2020;39(1):250. DOI: 10.1186/s13046-020-01705-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Santos SA, Paulo A. Small molecule inhibitors of multidrug resistance gene (MDR1) expression: preclinical evaluation and mechanisms of action. Curr Cancer Drug Targets. 2013;13(8):814–828. [DOI] [PubMed] [Google Scholar]
- [35].Yi D, Xu L, Wang R, et al. MiR-381 overcomes cisplatin resistance in breast cancer by targeting MDR1. Cell Biol Int. 2019;43(1):12–21. DOI: 10.1002/cbin.11071 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article