

Abstract
Chimeric antigen receptor (CAR) T cell therapy relies on T cells that are guided by synthetic receptors to target and lyse cancer cells. CARs bind to cell surface antigens through a scFv (binder), the affinity of which is central to determining CAR T cell function and therapeutic success. CAR T cells targeting CD19 were the first to achieve dramatic clinical responses in patients with relapsed/refractory B cell malignancies and to be approved by the U.S. Food and Drug Administration (FDA). We report cryo-EM structures of CD19 antigen with the binder FMC63 which is used in four FDA-approved CAR T cell therapies (Kymriah, Yescarta, Tecartus, Breyanzi), and the binder SJ25C1 which has also been used extensively in multiple clinical trials. We use these structures for molecular dynamics simulations which guide creation of lower or higher affinity binders, and ultimately produce CAR T cells endowed with distinct tumor recognition sensitivities. The CAR T cells exhibit different antigen density requirements to trigger cytolysis and differ in their propensity to prompt trogocytosis upon contacting tumor cells. Our work shows how structural information can be applied to tune CAR T cell performance to specific target antigen densities.
One-Sentence Summary:
Structures of CAR binders with CD19 antigen are used to engineer new binders which tune CAR T cells to distinguish between different antigen densities.
Introduction:
Chimeric antigen receptors (CARs) are synthetic receptors for antigen that reprogram the specificity and function of T cells and other immune effector cells (1). These transmembrane receptors typically comprise an extracellular single-chain variable fragment (scFv) with specificity for a cell-surface antigen that is expressed by a particular cancer cell type, and a cytoplasmic region assembled from immune receptor protein domains and designed to initiate T cell activation and sustain T cell effector functions and persistence (Fig. 1A) (1–3). CAR T cell therapy is deployed by isolating T cells from the patient and integrating therein the CAR cDNA (4). CARs targeting the B cell lineage marker CD19 (5) have provided remarkable clinical outcomes in patients with relapsed and refractory B cell malignancies (6). CD19 CAR therapy has become a paradigm for cancer immunotherapy based on genetic engineering of therapeutic immune responses (6–9). Of the more than 700 CAR therapy trials listed on the clinicaltrials.gov website about 40% target CD19 (9). There are at present four CD19-specific CAR T cell products approved by the US FDA (10, 11), all of which utilize the CD19-specific scFv FMC63 (12) but differ in their signaling and hinge domains (13, 14). The original CD19 scFv used to demonstrate the feasibility and efficacy of CD19 CAR therapy (5) is SJ25C1 (15), which has also been successfully used in patients with refractory acute lymphoblastic leukemia (16).
Fig. 1. Structures of CAR binders in complex with CD19.
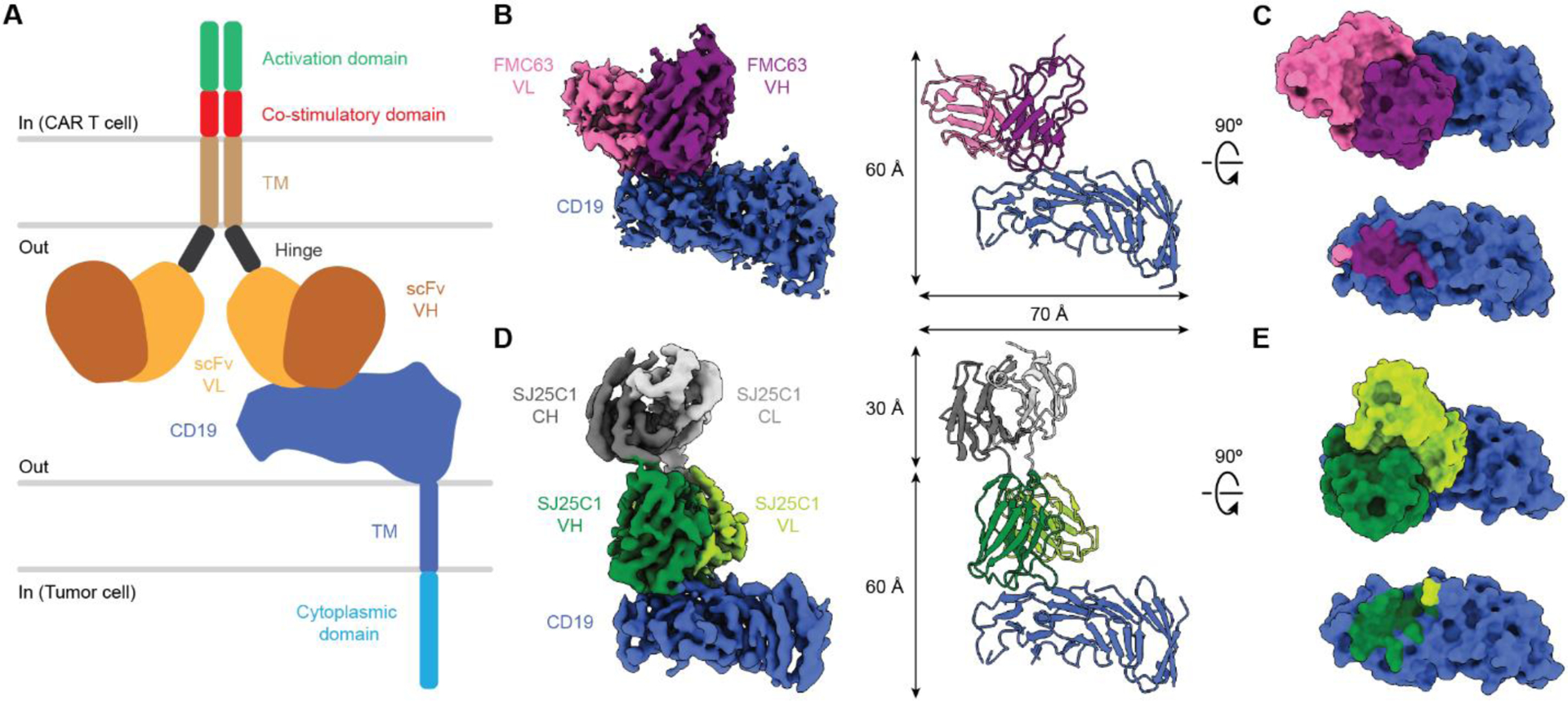
(A) Illustration of a CAR on a CAR-T cell in complex with CD19 antigen displayed on a tumor cell. (B) Cryo-EM density map (left) and molecular model (right) of FMC63 scFv in complex with CD19. (C) Surface view of the FMC63-CD19 complex as shown perpendicular to the membrane (top). CD19 is also shown in surface view with the footprint of FMC63 colored by contact with the VL or VH region (bottom). (D) Cryo-EM density map (left) and molecular model (right) of SJ25C1 Fab in complex with CD19. (E) Surface view of the SJ25C1-CD19 complex as shown perpendicular to the membrane (top) but with the Fab constant regions (CH and CL) omitted for clarity. CD19 is also shown in surface view with the footprint of SJ25C1 colored by contact with the VL or VH region (bottom). In (C) and (E), the footprints of the binders on CD19 correspond to CD19 residues within 4 Å of each respective binder. TM, transmembrane; CL, light chain constant domain; CH, heavy chain constant domain.
A persistent challenge in CAR T cell development is calibration of the affinity and/or specificity of the scFv (binder) portion of the receptor (3, 17). A high-affinity binder can lead to on-target/off-tumor toxicities in which CARs successfully engage cancer cells where target antigen is over-expressed, but also bind to healthy cells with lower antigen levels (18–20). In contrast, an exceedingly low-affinity binder may not engage sufficient target antigen to activate the CAR T cells (20, 21). For an antigen like CD19 which is abundant on cancerous cells it was conventionally thought that higher affinity CARs would equate to better CAR performance. However, this straightforward model appears oversimplified. A recently developed low-affinity anti-CD19 CAR (22) showed enhanced antileukemic effect in vivo in pediatric patients, which was proposed to be a function of increased cytotoxicity and increased proliferation of the CAR T cells. In addition, we previously demonstrated that CAR T cell trogocytosis (one cell gnawing another) has a major role in abating the performance of anti-CD19 CAR T cells (23), which has been recently extended to CAR NK cells (24). Both studies showed that acquisition of the target antigen by the T cell or NK cell exposes the latter to be lysed by other CAR T cells (fratricide killing). In the CAR NK study it was concluded that the degree of trogocytosis was influenced by tumor antigen density and CAR binder affinity (24). Altogether these studies further underscore the relevance of lowering the affinity for the targeted antigen without impairing tumor recognition, including for CD19. Challenges with binder engineering have motivated a variety of strategies to identify new binders through large-scale screens (25, 26) or by integrating multiple binders into a single CAR (27, 28). Notably, structural biology approaches have not been emphasized in CAR T cell development, but the increasing power of experimental and computational structural methods makes it possible to understand binder-antigen engagement mechanisms for existing CARs, and to rationally improve their binders.
In this study, we investigated the antigen engagement mechanisms for two different CAR binders, both of which target CD19. We studied FMC63 which is used in four FDA-approved CARs (Tisagenlecleucel (Kymriah), Axicabtagene ciloleucel (Yescarta), Brexucabtagene autoleucel (Tecartus), Lisocabtagene maraleucel (Breyanzi)) (29), and SJ25C1 which has also been used extensively in clinical trials (16). Their variable region sequences are unrelated yet the binders show similar performance in the clinic (30). SJ25C1 and FMC63 have long been thought to bind overlapping epitopes in CD19 (12, 31), but their binding sites have not been hitherto identified. We hypothesized that visualizing the structures would unlock the ability to rationally design mutant binders with tuned affinities. First, we solved cryo-electron microscopy (cryo-EM) structures of the two binders in complex with the soluble ectodomain of CD19 to identify the residues mediating the binder-antigen interactions. We then used these structures for molecular dynamics (MD) simulations to calculate the contribution of each binder residue, and each CD19 residue, to the interaction interfaces. Next, we integrated these data to design a series of mutant binders and measured their affinities using surface plasmon resonance (SPR). Finally, we selected a subset of the binder mutants and incorporated them into new CARs, generated CAR T cells, and compared their cancer cell killing performance. Altogether this study provides antigen engagement mechanisms for two binders in clinical use and translates these results to generate new CAR T cells with distinct physiological attributes. Results from this study are of immediate utility and present a broadly applicable template to aid the design of optimized CAR T cell therapies.
Results:
Structures of FMC63 and SJ25C1 in complex with soluble CD19
To obtain structures of the FMC63-CD19 and SJ25C1-CD19 complexes it was necessary to express and purify both binders and CD19. We first produced the FMC63 scFv using the Sf9 insect cell expression system (32) and produced soluble CD19 in HEK 293S cells (33). Soluble CD19 (31 kDa) contains the entire ectodomain of the protein but lacks the transmembrane helix and the unstructured cytosolic region. We chose soluble CD19 instead of full-length CD19 because it allowed us to investigate the protein without using detergents to solubilize the transmembrane helix, which was desirable because detergents reduce contrast in cryo-EM images (34). For brevity, we refer to soluble CD19 as CD19. Purified FMC63-CD19 complex (fig. S1A) was imaged by cryo-EM and the structure solved by single particle analysis. The cryo-EM density map was refined to a global resolution of 3.05 Å (fig. S1, S3). The map contained two copies of the FMC63-CD19 complex and one has slightly higher local resolution so we selected it for structural modeling (fig. S1C).
We next pursued the SJ25C1-CD19 complex. Although SJ25C1 is an scFv we decided to transfer it into an antigen binding fragment (Fab) format. The Fab format preserves the structure and binding capacity of scFvs but doubles the mass of the binder from ~25 kDa (scFv) to ~50 kDa (Fab). The motivation was to aid cryo-EM single particle alignment, which generally becomes easier as the target mass increases (35). The SJ25C1 Fab was produced in HEK 293F cells and used to form the SJ25C1-CD19 complex (fig. S2A). The complex was imaged using cryo-EM and the structure refined to a resolution of 3.40 Å (fig. S2, S3).
To begin our structural analysis we compared how each binder is oriented on CD19. FMC63 makes contact on the extracellular-facing side of CD19, and at the region of CD19 distal to the membrane (Fig. 1B, C). The heavy variable (VH) domain forms most of the binder’s “footprint” on CD19, with an additional contribution from the light variable (VL) domain (Fig. 2A–D). The complementarity-determining regions (CDRs) of the VH and VL both make contact with CD19. Specifically, CDRL1 and CDRL2 from VL and CDRH3 from VH converge on a region of CD19 we designated loop 1 (residues 154–166) (Fig. 2B, C). In turn, CDRH2 and CDRH3 of FMC63 form multiple contacts with a CD19 region we called loop 2 (residues 214–224) (Fig. 2D). Inspection of the binder side chains suggested that contacts with CD19 were formed by residues Y70 and H88 on the VL CDR and S214, T216, Y218, Y260 and Y261 on the VH CDR (Fig. 2B–D).
Fig. 2. Binding interface between CAR binders and CD19.
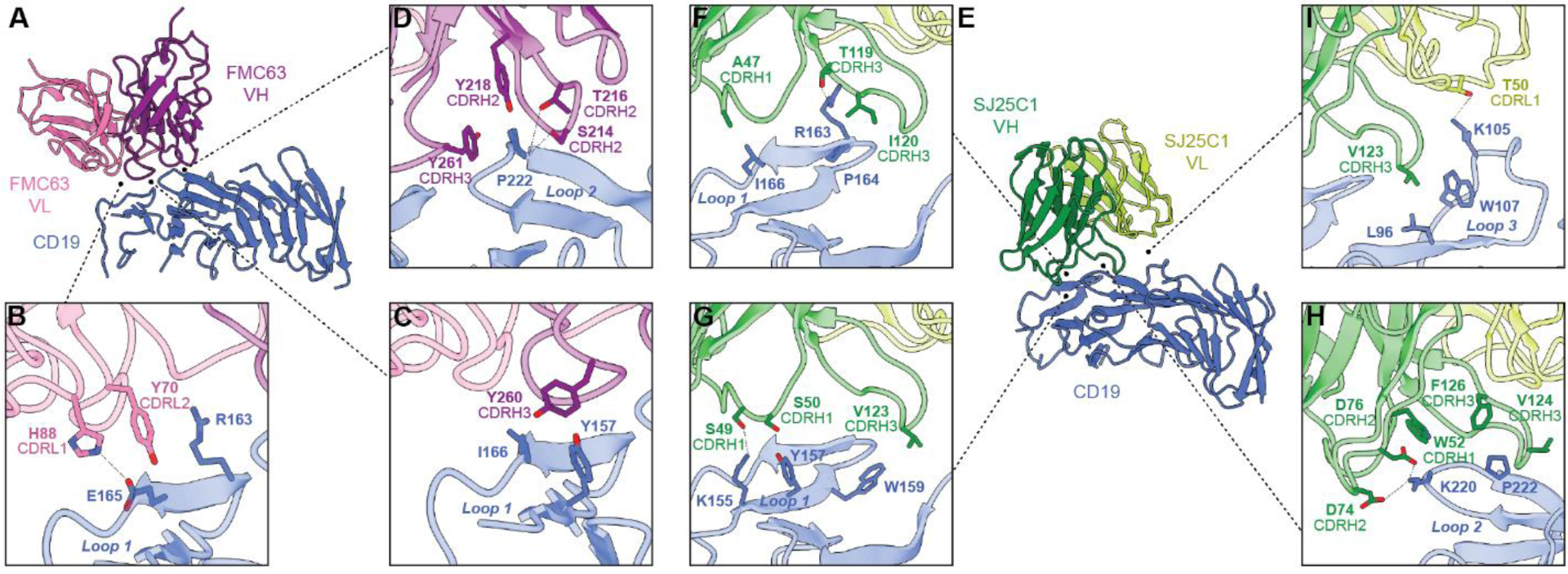
(A to D) Molecular model of FMC63 in complex with CD19 (A). The VL domain of FMC63 contacts CD19 with residues of CDRL1 and CDRL2 (B). The VH domain forms most of the contacts for FMC63, and these are mediated by residues in CDRH2 and CDRH3 (C and D). (E to I) Molecular model of SJ25C1 in complex with CD19 (E). The VH domain forms extensive contacts with CD19 through CDRH1, CDRH2, and CDRH3 (F to I), with more limited contact from CDRL1 of the VL domain (I).
Evaluation of the SJ25C1 binding orientation on the surface of CD19 showed that relative to FMC63 it is rotated in-plane by ~90° clockwise, as viewed from the extracellular space (Fig. 1D, E). Like FMC63, the SJ25C1 binder principally contacts CD19 using its VH CDRs (Fig. 2E). All three VH CDRs form contacts with CD19, but CDR3H3 forms the majority of contacts which are across CD19 loop 1, loop 2, and loop 3 (the latter loop corresponding to residues 95–109) (Fig. 2E–I). The VL of SJ25C1 also makes a contact using CDRL1, which binds to CD19 loop 3 (Fig. 2I). We identified residues A47, S49, S50, W52, D74, D76, T119, I120, V123, V124 and F126 on the VH CDR and T50 on the VL CDR as likely mediators of the binder’s interaction with CD19 (Fig. F–I).
This analysis showed that although the two binders adopt different orientations on CD19, they bind the same overall patch on CD19 (Fig. 1C, E). This observation motivated us to compare FMC63 and SJ25C1 complexes to available structures of soluble CD19 bound to Fabs from antibodies B43 (36) and Coltuximab (37), which have CDR sequences unrelated to those of FMC63 and SJ25C1. Strikingly, all four binders recognize a similar region on CD19 and their VH domains contribute most of the interactions (fig. S6A–H). We noted that SJ25C1 has a similar binding orientation as B43, even though these two antibodies are not related (fig. S6B, C) (15). Altogether, the structural analysis suggests that the region of CD19, comprised of loops 1–3, may be particularly antigenic.
Affinity tuning of FMC63 and SJ25C1
The structures of FMC63 and SJ25C1 with CD19 provided a framework in which to design binder mutants with modified affinity. However, the sheer number of contacts the binders form with CD19 (Fig. 2) presented a challenge in deciding precisely which sites to mutate. We reasoned that by using MD simulations we could refine the list of binder residues which are most important to CD19 interaction, and thereby narrow the number of residues to later subject to mutagenesis. In this way, by including the MD step in our workflow we aimed to increase the efficiency of the search for viable mutant binders. We used all-atom MD simulations followed by Molecular Mechanics-Generalized Born Surface Area (MM/GBSA) analysis (38) to calculate binding energy for the FMC63-CD19 and SJ25C1-CD19 complexes as well as the binding energy contributions for each residue forming contact between the binders and CD19. For both binder-CD19 structures we ran multiple MD trajectory replicates on the complexes (fig. S4 and see also Methods). In all the runs, the profiles of the root-mean-square-deviation (RMSD) of the protein systems showed only low amplitude fluctuations after the initial equilibration phase (fig. S4A, B), suggesting that the cryo-EM structures, and specifically the interfacial regions, remained stable during MD. We then used MM/GBSA to approximate per-residue binding energy decompositions (39). This yielded a list of binder residues ranked by their binding energy contributions to the CD19 interactions (Fig. 3A, B). For completeness, we also extended our structural analysis of the B43-CD19 (36) and Coltuximab-CD19 (37) complexes (fig. S6C, D, G, H) to include MD simulations (fig. S5). The MD trajectories exhibit profiles similar to those for the FMC63-CD19 and SJ25C1-CD19 complexes (fig. S4), suggesting that all four complexes are comparatively stable.
Fig. 3. Affinity measurements of CAR binder mutants.
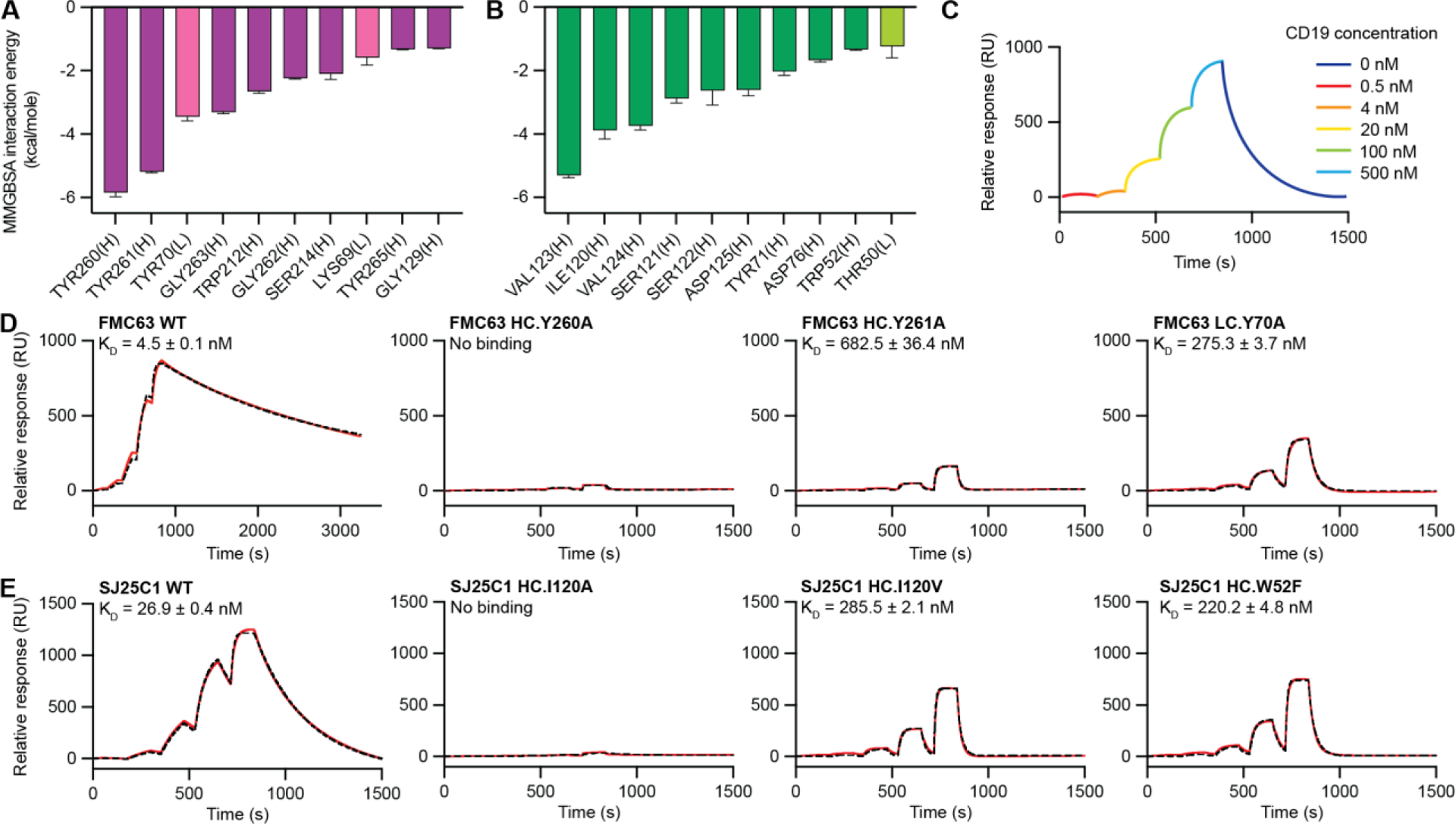
(A and B) Binding energies for residues from FMC63-CD19 (A) and SJ25C1-CD19 (B) complexes obtained from MM/GBSA analysis of the MD trajectories. Residues were ordered by decreasing magnitude of per-residue binding energy at the interface with CD19 (left to right). The binding energies are quantified by averaging six independent MM/GBSA simulation decompositions for each residue. Error bars represent means ± SEM. Residues from the VL and VH domain are labeled with “(L)” and “(H),” respectively. Data for the VL domain residues are shown using lighter colored bars. (C) Illustrated SPR sensorgram depicting the experimental protocol used to measure CD19 binding to Fabs. Single-cycle kinetics was performed by injecting CD19 protein at five concentrations (0.5, 4, 20, 100, and 500 nM) followed by one long dissociation step and reported with response units (RUs). (D and E) SPR sensorgrams of binding kinetics between CD19 and FMC63 (D) and SJ25C1 (E) binders. Experimental data are shown in red. Fitted curves calculated using 1:1 kinetics Langmuir binding model are shown with black dashed lines. Dissociation constant (KD) for each binder is displayed as an average of triplicates with SEM.
The highest-ranking residues for FMC63 are Y70, Y260 and Y261 (Fig. 3A). This result is consistent with our structural analysis showing all three residues contact loops 1 and 2 of CD19 through hydrophobic interactions, while Y70 also forms a cation-π interaction with R163 of CD19 (Fig. 2B–D). It is also consistent with the observation that tyrosine plays a dominant role in specificity and affinity in antibodies (40) and binders created de novo by in silico methods (41). Of the FMC63 residues identified in structural analysis (Fig. 2A–D), all but three appeared in the top ten ranked by MD (Fig. 3A). Based on these results we designed three mutants in which either Y70, Y260 or Y261 were converted to alanine. We reasoned that the change to an alanine side chain would largely ablate each tyrosine’s interactions with CD19, without having a deleterious effect on the overall protein backbone geometry, and hypothesized this approach would yield binders with reduced affinity. The mutants are FMC63Y70A, FMC63Y260A and FMC63Y261A (table S1).
In SJ25C1 the MM/GBSA analysis identified I120, V123 and V124 as forming the top three most significant binder contacts on CD19 (Fig. 2F–I, 3B). We reasoned that to decrease the interactions between these residues and CD19 we should mutate the sites to have smaller side chains. Accordingly, we substituted glycine at all three positions (SJ25C1I120G, SJ25C1V123G, SJ25C1V124G), and also substituted alanine and valine at position I120 (SJ25C1I120A, SJ25C1I120V) (table S1). Residue W52 was another prominent candidate (Fig. 3B) for substitution because tryptophan frequently appears at binding energy “hotspots” in protein-protein interactions (42). We substituted phenylalinine (SJ25C1W52F) to decrease the size of the side chain but preserve its hydrophobic character (table S1). Finally, we selected binder residue S121 for mutagenesis because it ranked fourth on the list of most significant contacts with CD19 and has relatively high binding energy. Unlike other sites targeted in this study, S121 appears to make contact with CD19 via the protein backbone, not its side chain (fig S11). We substituted alanine (SJ25C1S121A) with the hypothesis that this would increase mobility of the backbone and thereby decrease affinity (table S1).
Having designed mutants for FMC63 and SJ25C1 we next sought to measure their affinities (KD) for CD19 relative to the unmodified binders (wildtype, WT) using SPR. The affinity for wildtype FMC63 is 4.5 nM and for SJ25C1 is 26.9 nM (Fig. 3D, E). The value for FMC63 is in an agreement with the literature (12, 22) and to our knowledge no value for SJ25C1 has been reported. The low nanomolar affinities of the two binders and their similar CD19 binding sites is consistent with their overall similar performance in CAR T cells (23). Notably, FMC63 has an especially slow dissociation rate such that even after ~0.5 hour a significant fraction of the proteins were still in complex (Fig. 3D). We confirmed that CD19 alone does not bind the SPR chip (fig. S9A), which rules out non-specific binding of CD19 as an explanation for the slow dissociation time observed for CD19 with FMC63. Thus, the slow dissociation of CD19 from FMC63 is an inherent property of the binder.
SPR measurements of the FMC63 mutant FMCY260A showed virtually no signal and a binding affinity could not be confidently calculated (Fig. 3D). In contrast, both FMC63Y70A and FMC63Y261A gave defined SPR signals and affinities were measured as 275.3 and 682.5 nM, respectively (Fig. 3D). Next, we measured binding for the two SJ25C1 I120 mutants (SJ25C1I120A, SJ25C1I120V) and found that although alanine is too small to preserve CD19 interaction (Fig. 3E), valine is large enough to preserve binding and the substitution successfully weakens the affinity to 285.5 nM (Fig. 3E). The SJ25C1W52F mutant also has a decreased affinity (220.2 nM) (Fig. 3E). The remaining SJ25C1 mutants did not give the hypothesized reduction in affinity. The glycine mutants (SJ25C1V120G, SJ25C1I123G, SJ25C1V124G) essentially lost the ability to bind CD19 (fig. S9B) while SJ25C1I121A showed an increase in affinity (fig. S11A). We speculate that glycine is too flexible to support the surrounding peptide backbone geometry, and that an allosteric effect was obtained at position 121 since SJ25C1 contacts CD19 via the protein backbone at this site, rather than via the side chain (fig. S11B). In considering these results we note that the target binder sites were selected for mutagenesis and SPR because of their positions in the structures, and because MD simulations ranked them as the most significant contributors to CD19 binding. In summary, the substitutions weakened or abrogated binding, and one of them increased binding affinity, consistent with the selected sites making a large contribution to antigen interaction (Fig. 3A, B).
Characterization of CAR T cells incorporating FMC63 and SJ25C1 binder mutants
Through our mutational screen we successfully identified FMC63 mutant binders (FMC63Y70A, FMC63Y261A) and SJ25C1 mutant binders (SJ25C1I120V, SJ25C1W52F) with weaker affinity for CD19 relative to their respective wildtype binders (Fig. 3D, E). We hypothesized that CAR T cells incorporating these binders would retain the ability to kill cancer cells, but that their weaker CD19 affinity would limit their ability to kill those CD19+ cells displaying lower levels of CD19 surface antigen. We also hypothesized that the FMC63 and SJ25C1 mutant binders for which CD19 binding was not detected by SPR (Fig. 3, fig. S9), would not be able to kill cells displaying CD19. For these experiments we established a panel of CAR T cells bearing CARs composed of a wildtype or mutant binder, a CD28 co-stimulatory signalling domain and a CD3-ζ chain activating domain (fig. S7A) (13). To assess the CAR T cell cytolytic ability, we used a panel of cells derived from NALM6, a well-established CD19+ leukemia cell line (5) displaying either high (27,000 molecules, NALM6/WT), medium (2,000 molecules, NALM6/12–39), or low (200 molecules, NALM6/12–4) surface density of CD19 and are a model for the range of CD19 surface expression across healthy and cancerous blood cells (21).
We first analyzed CAR T cells incorporating FMC63WT and FMC63 mutants. Fluorescence-activated cell sorting (FACS) was used to verify that the CAR T cells all display CAR surface protein at similar levels (Fig. 4A). We also used FACS to evaluate the capacity of each CAR T cell type to bind surface CD19 (Fig. 4B). We found that the FMC63Y70A CAR binds CD19 at ~50% of the wildtype level (Fig. 4B), while the FMC63Y261A CAR binds an even lower percentage of cells (Fig. 4B), and the FMC63Y260A CAR shows no evidence of target cell binding. Altogether, the cell binding results for FMC63 CARs are in agreement with SPR binding data (Fig. 3D). We next evaluated the cytolytic performance of WT and mutant CAR T cells and all showed robust cytolytic activity against high density CD19 cells, although FMC63Y260A CAR T cells exhibited lower activity compared to the other FMC CAR T cells (Fig. 4C). We were surprised that FMC63Y260A CAR has cytolytic activity at all because SPR and CD19 FACS binding data gave no evidence of soluble CD19 binding for this mutant (Fig. 3C–D, 4B). However, we reasoned that the KD of FMC63Y260A is outside the detection limit of our SPR experimental setup, and based on the SPR biochemical conditions is likely weaker than 5 μM (Fig. 3C, D and see also Methods). We concluded that interaction between FMC63Y260A CAR T cells and NALM6/WT (Fig. 4C) was enhanced by an avidity effect (43, 44) compensating for the inherently low affinity of the FMC63Y260A binder. At medium CD19 density FMC63Y261A CAR T cell activity declined and FMC63Y260A CAR T cell activity was lost (Fig. 4D). At the lowest antigen density FMC63Y70A CAR T cell activity dropped to slightly below FMC63WT, and FMC63Y261A CAR T cell activity was close to baseline (Fig. 4E).
Fig. 4. CD19-interacting residues differentially shape CAR T cell function.
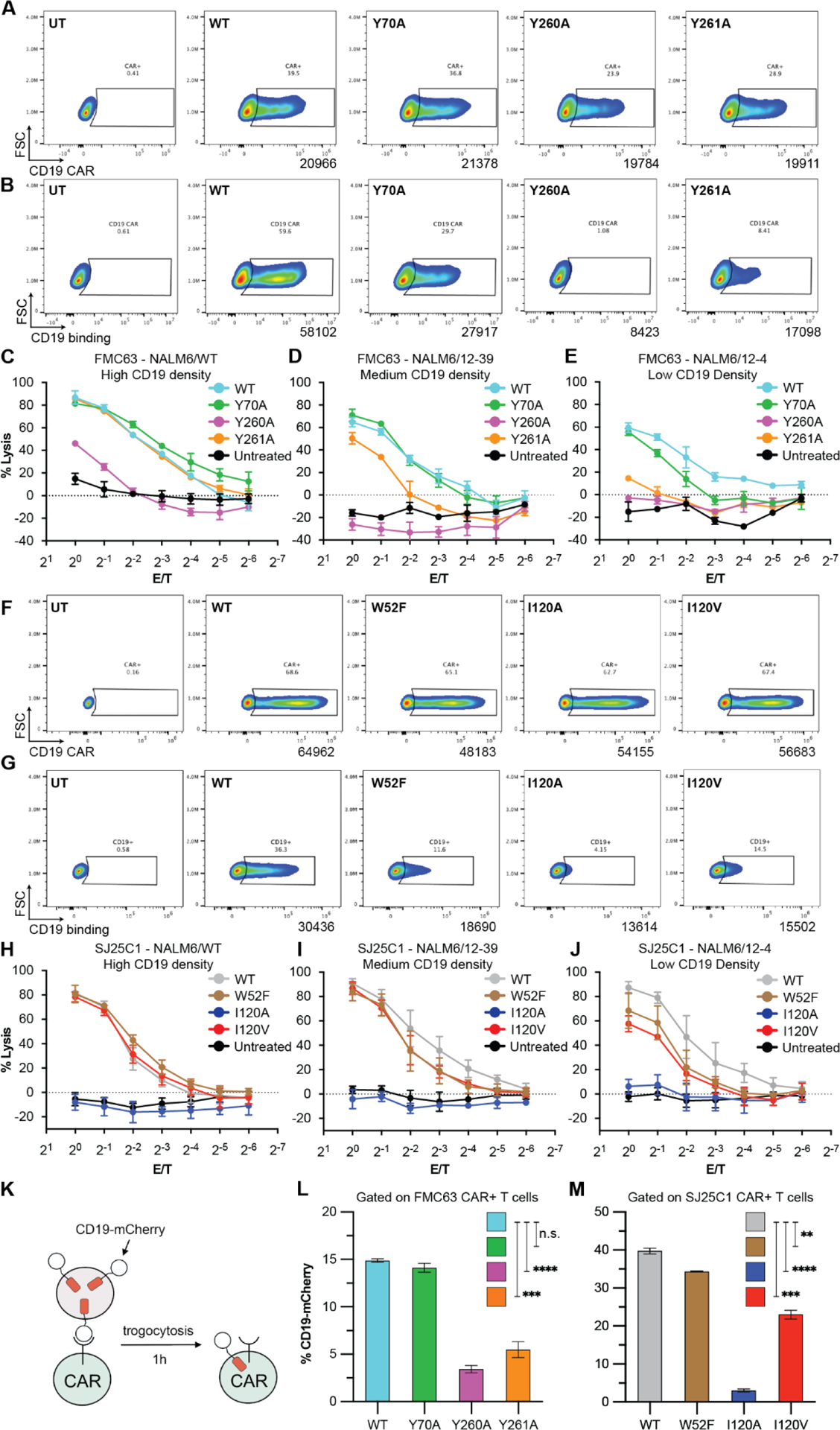
(A and F) CAR expression analyses for FMC63 (A) and SJ25C1 (F) CAR T cells. Data are from two independent experiments using different T cell donors. (B and G) Binding activity for FMC63 (B) and SJ25C1 (G) CAR T cells. Data for FMC63 CAR T cells are from two independent experiments using different T cell donors (one replicate in each experiment). Data for SJ25C1 CAR T cells are from two independent experiments using different T cell donors (two replicates in each experiment). (C to E and H to J) CAR T cell cytotoxic activity against NALM6/WT (~27,000 CD19 molecules; C), NALM6/12–39 (~2000 CD19 molecules; D), or NALM6/12–4 (~200 CD19 molecules; E) target cells. In (C) to (E), data are means ± SEM (n = 3). In (H) to (J), data are means ± SEM (n = 5, with three replicates from T cell donor 1 and two replicates from T cell donor 2). (K) Illustration of the trogocytosis assay. (L and M) Percentage of FMC63 (L) and SJ25C1 (M) CAR T cells (both CD4 and CD8 cells) with CD19-mCherry signal after coculture with CD19-mCherry–expressing NALM6 cells. Unpaired t test P value for FMC63 CAR T cells (L) or SJ25C1 CAR T cells (M). Data are representative of two independent experiments using two different T cell donors, with each experiment performed with three replicates (n = 3). Error bars are SEM.
Next, we investigated CAR T cells incorporating SJ25C1WT and SJ25C1 mutants. FACS analyses confirmed that all CAR T cells display similar amounts of CAR surface protein (Fig. 4F), and that the ability of the SJ25C1 CAR T cells to bind CD19 (Fig. 4G) matched the performance of the SJ25C1 binders in SPR experiments (Fig. 3E). Specifically, SJ25C1WT, SJ25C1W52F and SJ25C1I120V all bind CD19 with progressively decreasing ability. Cytolysis experiments showed CAR T cells with SJ25C1WT, SJ25C1W52F and SJ25C1I120V have robust and nearly equivalent cytolytic ability against target cells with high and medium CD19 density (Fig. 4H–I). However, their cytolytic performance diverged against target cells with low CD19 density (Fig. 4J). CAR T cells with SJ25C1WT were most cytolytic, followed by SJ25C1W52F and SJ25C1I120V (Fig. 4J). This ranking of cytolytic activity supports the hypothesis that weaker binder affinity offers greater sensitivity to antigen density. Meanwhile, CAR T cells with SJ25C1I120A were unable to kill target cells even at the highest CD19 density. As expected from SPR data (fig. S9), CAR T cells derived from the three SJ25C1 glycine mutants (I120G, V123G, V124G) were unable to bind CD19 and also showed no cytolytic activity (fig. S9).
We hypothesised that CAR T cells with diminished CD19 binding activity would also have reduced CD19 trogocytosis (the transfer of the target antigen from the tumor cell to the CAR T cell) (23) and cytokine release. To assay trogocytosis we used NALM6 cells expressing a CD19-mCherry fusion which allowed tracking of target cell material before and after transfer to the CAR T cell (23) (Fig. 4K, fig. S10). We found that while FMC63Y70A CAR T cells uptake tumor antigen similarly to FMC63WT, both FMC63Y260A and FMC63Y261A CAR T cells showed greatly attenuated CD19 uptake, mirroring the lower affinity of their binders (Fig 3C–D, Fig. 4L). Similarly, CAR T cells with SJ25C1WT and SJ25C1W52F transferred more CD19 antigen than CAR T cells with SJ25C1I120A and SJ25C1I120V (Fig. 4M). Cytokine release was evaluated by measuring release of two effector cytokines, IFN-γ and TNF-α (fig. S8). We found that only the FMC63Y260A and SJ25C1I120A CAR T cells showed significant impairment of cytokine release upon exposure to NALM6/WT cells (fig. S8A, C). This response is in line with the cytolytic activity described above and confirmed the ability of FMC63Y70A, FMC63Y261A, SJ25C1W52F and SJ25C1I120V to direct not only cytolysis but also effective cytokine secretion. For completeness we also assayed CAR T cells with SJ25C1S121A, the binder with stronger affinity than its parent wildtype (fig. S11), and found that the cells exhibit similar trogocytosis and cytolysis compared to wildtype (fig. S11). Altogether, these functional experiments demonstrate that tuning CAR binder affinity allows for tailoring CAR T cell sensitivity to target antigen, trogocytosis, and cytokine release, all of which are key performance metrics of CAR therapies.
Discussion:
In this study we aimed to characterize the CD19 engagement mechanisms for FMC63, which is used in FDA-approved CAR T cell therapies (29), and SJ25C1, which has been used in multiple clinical trials as well (16). In addition, we aimed to use this structural information to guide the design of new CARs. We find that the binders target the same site on CD19 albeit with different binding orientations. Moreover, close inspection of MD simulation data of CD19 with FMC63, SJ25C1 and two non-CAR binders, B43 and Coltuximab (fig. S4, S5), revealed that the same three CD19 residues (Arg163, Lys220, Pro222) are the top three most significant contributors to interaction with FMC63, SJ25C1 and B43 (fig. S6). This finding is in agreement with CD19 deep sequencing results where Arg163 and Pro222 were identified as determinants for FMC63 binding (45). In addition, our results may explain why the CD19 mutation Arg163Leu is associated with patient relapse after treatment with FMC63-based CAR T cell therapy treatment (46). We note that the mutation Leu174Val is also associated with therapeutic resistance but it is ~23 Å from the FMC63 binding site (fig. S12), suggesting the mutation may impact FMC63 CAR performance through allostery or some unknown physiological mechanism. We conclude that the site formed by Arg163, Lys220 and Pro222 is highly antigenic, which may be related to the site’s location on “top” of CD19 where it is exposed and relatively distant from the membrane (Fig. 1). The CD19 binding epitope for FMC63 which we defined in this study is in close agreement with the site mapped out previously using deep mutational scanning (45). The key differences are that in the previous work Lys220 on CD19 was not identified as part of the FMC63 binding epitope, and Trp159 was proposed to be in the epitope but we find it is not. We speculate that mutation of Trp159 may impact FMC63 binding through an allosteric effect. Our general conclusion about the antigenicity of site on “top” of CD19 is also consistent with an earlier epitope mapping study (45) which concluded that FMC63 and the CD19 antibodies 4G7 and 3B19 all bind partially overlapping regions of CD19. The overlapping binding site for these four independent CD19 binders underscores the general challenge of creating panels of immunoglobulin binders targeting diverse sites on a given antigen (47), and highlights the value of fine-tuning binders using structural and computational methods.
Recent advances in computational structure prediction raised the question of whether the FMC63-CD19 and SJ25C1-CD19 structures could be accurately predicted using such methods. We used AlphaFold2 (48) to generate model predictions and compared them to our cryo-EM structures (fig. S13). While AlphaFold2 gave good results for the overall scFv proteins, it generated partially disordered CD19 ectodomains. Critically, the FMC63-CD19 model from AlphaFold2 gave an incorrect interface between the scFv and CD19. In turn, the SJ25C1-CD19 result showed an implausible interaction between CD19 and the scFv in which the binder forms an arbitrary interface with CD19, with its CDRs directed into the surrounding solution rather than contacting the antigen. Overall this is consistent with the assessment that computational tools cannot yet routinely predict heteromeric protein complexes, and especially protein-antibody complexes (49), and that experimental approaches such as cryo-EM, X-ray crystallography, and epitope mapping will continue to be essential for understanding such systems.
Our study showed that decreasing FMC63 and SJ25C1 affinity yielded CARs with distinctive features that could be rationalized from the change in binding affinity. This suggests that alterations in CAR binder affinity will be a predictive of key aspects of CAR performance. FMC63Y70A (275.3 nM KD) elicited an effector cytokine response comparable to FMC63WT (4.5 nM KD) and effectively lysed NALM6/WT cells presenting a high density of CD19 molecules, but the mutant binder showed diminished activity against NALM6/12–4 which displays only low numbers of CD19 molecules (Fig. 4C, E). In turn, the weaker binder FMC63Y261A could still engage target cells with 2,000 but not 200 CD19 molecules (Fig. 4D–E). Although its binding was not detectable by SPR, FMC63Y260A still showed modest cytolytic activity if target antigen density reached 27,000 copies (Fig. 4C). Interestingly, FMC63Y261A showed a decrease in trogocytosis while retaining the ability to efficiently engage and lyse NALM6/WT cells (Fig. 4C, L). This finding is relevant to ongoing CAR development since high trogocytosis has been shown to impair CAR T cell function (23). While FMC63WT, FMC63Y70A (Fig. 4L) and SJ25C1WT (23) showed strong trogocytosis, a mutant of FMC63 with an affinity of 14 nM was recently shown to decrease trogocytosis and improve CAR T cell expansion (50). The general findings from experiments with SJ25C1WT and its lower-affinity mutants are consonant with those of FMC63 and, in aggregate, suggest it is possible to optimize binder affinity to diminish trogocytosis without abolishing antigen recognition.
In summary, we show the ability to rationally design affinity-tuned CARs to achieve a desired level of antigen sensitivity. Affinity tuning is desirable to target high antigen density tumor cells while averting recognition of low antigen density in normal cells, or conversely to maximize antigen detection and CAR T cell activation when tumor cells express low antigen density. Our approach combines cryo-EM, MD, and SPR which together provide a platform to visualize binder-antigen complexes, identify binder sites for mutagenesis, and experimentally measure binder affinity. All three methods are rapid, robust, and widely available. Consequently, we anticipate the strategy will find broad utility in optimizing new and existing CARs, and may substitute for screening large numbers of scFvs that may unknowingly be limited and redundant in their epitope diversity and biophysical features including affinity range. Furthermore, it is highly complementary to emerging methods in protein structure prediction (48) and generation (51, 52). A structure-guided strategy for lowering binder affinity may be particularly valuable for attenuating on-target/off-tumor cytotoxicity for CARs targeting solid tumor antigens such as HER2 and mesothelin.
Materials and Methods
Study design
The sample size selection for cryo-EM data collection and analysis followed standard practice and was guided by the structural resolutions needed to answer the target questions in the study. Specifically, in this study we targeted resolutions of 3–4 Å and acquired a quantity of data sufficient to achieve these resolutions. At this resolution range we were able to model side chains to characterize the binder-antigen interactions and perform computational analysis. The overall objective of our study was to determine the CD19 antigen recognition mechanism for select CAR T cell binders and translate that knowledge to the development of lower-affinity binders. The design of the study used cryo-EM for structure determination, MD simulation and MM/GBSA analysis to quantify the energetics of binder-antigen interaction, SPR to characterize the affinity of wildtype and mutant binders, and cellular assays to evaluate binder performance in CAR T cells. The study did not require use of subjects or experimental units in experimental groups. The study did not employ blinding. Replicates for each experiment are provided in figure legends.
Expression, purification and digestion of soluble CD19
The pTT5 vector containing a fusion of IgGκ signal peptide, 8 × histidine tag, AviTag, the small ubiquitin-like modifier- (SUMO) star fusion protein, a human rhinovirus (HRV) 3C cleavage site and CD19-SF05 mutant (M75V, R76S, F85S) was a gift from Dr. Michael W. Traxlmayr (33). The plasmid was used for transient protein expression in HEK293S cells as follows. Flasks of HEK293S GnTI− suspension cells (ATCC catalog number CRL-3022) were cultured in FreeStyle suspension media (Gibco) supplemented with 2% fetal bovine serum (Gibco) and Anti-Anti (Gibco) at 37°C and 8% CO2, and a total of 1 μg plasmid DNA and 2 μg of PEI (Polysciences) per ml culture volume was added when the suspension cells reached a density of 1.7 × 106 cells/ml. After 48 hours post-transduction, valproic acid (Sigma) was added into the suspension cells to a final concentration of 5 mM. The supernatants were clarified and harvested 120 hours post-transfection by centrifugation at 6,200 × g for 20 min at 4°C, and filtered through a 0.45 μm filter.
The supernatants were loaded on pre-equilibrated HisTrap HP column (Cytiva) with purification buffer containing 20 mM Tris (pH 8.0) and 300 mM NaCl. The column was washed with 2 column volumes of purification buffer and 2 column volume of purification buffer containing 5 mM imidazole. Protein was eluted with successive additions of 2 column volumes of purification buffer containing 40 mM imidazole then 250 mM imidazole. The main elution fractions were concentrated to 700 μl using a centrifugal filter unit (10 kDa MWCO, Amicon) and injected on a Superose 6 Increase 10/300 GL column (Cytiva) equilibrated with buffer containing 20 mM Tris (pH 8.0) and 300 mM NaCl. The peak fractions were verified by SDS-PAGE gel, then pooled and concentrated. The concentrated proteins were digested with Recombinant PreScission Protease (rPSP, GenScript) at a ratio of 60 μg protein per 0.6 IU rPSP overnight at 4°C. The mixture was loaded on pre-equilibrated TALON resin (Takara) with purification buffer and was washed with 3 column volumes of purification buffer. Both the flow-through and washed fractions were collected, combined and loaded onto a Superdex 200 Increase 10/300 GL (Cytiva) with running buffer containing either purification buffer for cryo-EM experiments or PBST (Cytiva) for SPR experiments. The digested peak fraction was collected, analyzed by SDS-PAGE gel, concentrated and flash frozen in liquid nitrogen for further use.
Expression and purification of FMC63-scFv
To generate the FMC63-scFv a DNA sequence encoding a fusion of the GP67 secretion peptide (32), FMC63-scFv, a human rhinovirus (HRV) 3C cleavage site and an 8 × histidine tag was cloned into the pFastBac1 expression vector. The construct was transformed into DH10Bac cells (Gibco) to generate bacmid, which was used to produce P1 baculovirus by transfecting Sf9 insect cells in ESF 921 media (Expression Systems) and further amplified to P2 baculovirus. After 96 hours post-transduction the supernatants were clarified and harvested by centrifugation at 680 × g for 30 min and at room temperature, and pH balanced by addition of Tris pH 8.0. Chelating agents in the Sf9 media were quenched by addition of 10 mM CaCl2 (32) and the mixture was incubated with stirring for 1 hour at room temperature. The supernatants were clarified by centrifugation at 8,000 × g for 30 min and at 4°C, then filtered through a 0.45 μm filter.
The supernatants were loaded on a HisTrap HP column (Cytiva) pre-equilibrated with high-salt purification buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl and 10 mM imidazole. The column was washed with 5 column volumes of high-salt purification buffer and 5 column volumes of low-salt purification buffer containing 20 mM Tris (pH 8.0), 100 mM NaCl and 10 mM imidazole. The protein was eluted with 4 column volumes of elution buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl and 250 mM imidazole. The main elution fractions were collected, combined and loaded onto a Superdex 200 Increase 10/300 GL (Cytiva) column with running buffer containing 20 mM Tris (pH 8.0) and 500 mM NaCl. The peak fractions were evaluated by SDS-PAGE, then pooled and concentrated.
Expression and purification of SJ25C1-Fab
The SJ25C1-Fab was engineered using a mouse IgG as a template. The sequences for the template light and heavy chains are [UniProt ID: P01834] and [UniProt ID: P01857], respectively. The SJ25C1-Fab light chain was designed by replacing the template VL domain with the VL domain from SJ25C1-scFv. The SJ25C1-Fab heavy chain was designed by removing the Fc portion from the template heavy chain and replacing the template VH domain with the VH domain from SJ25C1-scFv. An N-terminal signal peptide [UniProt ID: P01750] was added to both chains of the SJ25C1-Fab. A C-terminal 8 × histidine tag was added to the Fab heavy chain. The two chains were cloned into the pEZT-BM expression vector (53).
The Fab was generated by transiently transfecting the light and heavy chain plasmids in Expi293F cells (Gibco) using the ExpiFectamine 293 Reagent (Gibco) according to the manufacturer’s protocol. The supernatants were clarified and harvested 5 – 7 days post-transfection by centrifugation at 5,000 × g for 20 min at 4°C, then filtered through a 0.45 μm filter. The whole purification was performed in the cold room at 4°C. The supernatants were loaded on TALON resin (Takara) pre-equilibrated with purification buffer containing 20 mM Tris (pH 8.0) and 300 mM NaCl. The column was washed with 2 column volumes of purification buffer and 2 column volumes of purification buffer containing 5 mM imidazole. Protein was eluted with successive additions of 2 column volumes of purification buffer containing 40 mM imidazole then 250 mM imidazole. The main elution fractions were concentrated to 700 μl using a centrifugal filter unit (10 kDa MWCO, Amicon) and injected on a Superose 6 Increase 10/300 GL column (Cytiva) equilibrated with purification buffer. The peak fractions were evaluated by SDS-PAGE, then pooled and concentrated.
Protein complex formation and cryo-EM sample preparation
To form the FMC63-CD19 complex, CD19 was mixed with FMC63-scFv in a ratio of 1:1.7 (w/w). To form the SJ25C1-CD19 complex, CD19 was mixed with SJ25C1-Fab in a ratio of 1:2 (w/w). For each complex, the mixture was injected on a Superdex 200 Increase 10/300 GL column (Cytiva) with running buffer containing 20 mM Tris (pH 8.0) and 30 mM NaCl. The peak fractions were verified by SDS-PAGE gel, then pooled and concentrated. Cryo-EM samples were prepared by adding a 3 μl droplet of FMC63-CD19 complex (2.2 mg/ml) with 3 mM fluorinated Fos-Choline-8 (Anatrace) or SJ25C1-CD19 complex (2.1 mg/ml) with 3 mM fluorinated Fos-Choline-8 (Anatrace) to plasma-treated UltrAuFoil 1.2/1.3 300 mesh grids (Quantifoil) (54), followed by blotting and plunge freezing using a Vitrobot Mk IV (Thermo Fisher) set for 1 sec blot time and −1 blot force.
Cryo-EM data acquisition
The FMC63-CD19 complex was imaged on a Talos Arctica (Thermo Fisher) operated at 200 kV and 36,000 × nominal magnification and equipped with a Gatan K3 camera. Movies were acquired in super-resolution mode (0.5480 Å pixel size) with a nominal defocus range of 1.1 – 2.9 μm. Each movie was 48 frames and had a total exposure time of 2.4 sec and an accumulated dose of 50.40 – 50.82 e–/Å2. A total of 2,341 movies were collected.
The SJ25C1-CD19 complex was imaged on a Talos Arctica (Thermo Fisher) operated at 200 kV and 45,000× nominal magnification and equipped with a Gatan K3 camera. Movies were acquired in super-resolution mode (0.4296 Å pixel size) with a nominal defocus range of 1.4 – 2.5 μm. Each movie was 40 frames and had a total exposure time of 1.6 sec and an accumulated dose of 53.47 – 54.04 e–/Å2. A total of 8,752 movies were collected. Data collections were managed using Leginon (55).
Image processing
Structure determination for the FMC63-CD19 complex was initiated by processing movie stacks with beam-induced motion correction and dose weighting in Relion 3.1 (56) with two-fold binning, yielding an image pixel size of 1.096 Å. These images were imported into cryoSPARC v3.3.1 (57) and used for contrast transfer function (CTF) estimation with the Patch CTF estimation module. Particles were picked with Blob picker and extracted with a box size of 320 pixels and binned to 128 pixels. Next, the particles were processed with 2D classification and ab initio reconstruction, followed by several rounds of heterogeneous refinement to obtain a homogeneous set of particles. The particles were re-extracted with a box size of 320 pixels without binning, processed with multiple rounds of ab initio reconstruction and heterogeneous refinement, and a final set of 442,863 particles were refined with non-uniform refinement to 3.05 Å.
Structure determination for the SJ25C1-CD19 complex was initiated by processing movie stacks with beam-induced motion correction and dose weighting in Relion 3.1 (56) with two-fold binning, yielding an image pixel size of 0.8592 Å. These images were used for CTF estimation with CTFFIND4.1 (58). Particles were picked with the reference-free Laplacian-of-Gaussian (LoG) tool in Relion, extracted with a box size of 300 pixels and binned to 60 pixels. Particles were then imported into cryoSPARC v3.3.1 (57) and processed with 2D classification and ab initio reconstruction, followed by several rounds of heterogeneous refinement to obtain a homogeneous particle set. The particles were re-extracted in Relion with a box size of 300 pixels without binning, and processed in cryoSPARC with ab initio reconstruction followed by multiple rounds of heterogeneous refinement, and a final set of 371,968 particles was refined with non-uniform refinement to 3.91 Å. To improve the resolution further, particle polishing was done in Relion, and ab initio reconstruction, heterogeneous refinement and non-uniform refinement were done in cryoSPARC to yield a 3.40 Å map with a final set of 351,934 particles.
Model building
For both the FMC63-CD19 and SJ25C1-CD19 complexes the soluble CD19 protein was built starting from the crystal structure of human CD19 ectodomain (PDB: 6AL5).
To model the FMC63 scFv, a homology model was generated with SWISSMODEL (59) using the crystal structure of a human scFv (PDB: 6TCS) as input. The starting models for FMC63-scFv and CD19 were docked into the cryo-EM density map using UCSF Chimera (60), refined using real-space refinement in Phenix (61) and rebuilt using Coot (62). The following regions were not resolved in the cryo-EM map so were omitted from the atomic model: residues 146–160 corresponding to the GS linker in the FMC63 scFv, and residues 21–22, 40–46, 133–152 and 175 −184 corresponding to flexible loops in CD19.
To model SJ25C1 Fab, homology models for the heavy chain and light chain were generated with SWISSMODEL (59) using a human Fab heavy chain (PDB: 5O4O) and a human Fab light chain (PDB: 6R2S) as input, respectively. The light and heavy chain models were docked into the cryo-EM map, refined and rebuilt as described above. The following regions were not resolved in the cryo-EM map so were omitted from the atomic model: residues 40–46, 133–152, 175–184 and 276–277 corresponding to the flexible regions in CD19. The resolution in the constant region of the Fab was attenuated so the heavy chain (residues 141–241) and light chain (residues 126–234) in this region were modeled as poly-alanine chains.
Molecular dynamics simulations
Structural models of the FMC63-CD19, SJ25C1-CD19, B43-CD19 (PDB: 6AL5) and Coltuximab-CD19 (PDB: 7JIC) complexes were used as inputs for all-atom MD simulations. Each system was solvated and ionized in 150 mM NaCl at pH 7.5 using CHARMM-GUI (63). The total system size was ~212,000 atoms for FMC63-CD19, ~167,000 atoms for SJ25C1-CD19, ~207,000 atoms for B43-CD19, and ~167,000 atoms for Coltuximab-CD19.
Each system was first equilibrated using the multi-step equilibration protocol provided by CHARMM-GUI. This was followed by the production simulations during which each system was run in six independent replicates for 150 ns per replicate. All the simulations were performed with OpenMM 7.4 (64) and using the latest all-atom CHARMM36m force fields for protein and ions (65). The simulations implemented a 4 fs integration timestep (with hydrogen mass repartitioning), Particle Mesh Ewald (PME) for electrostatic interactions (66), and were carried out in isothermal-isobaric (NPT) ensemble with the periodic boundary conditions, at a temperature of 310 K. The van der Waals interactions were calculated by applying a cutoff distance of 12 Å and switching the potential from 10 Å. The root mean square deviation (RMSD) analysis was calculated with VMD (67).
Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) calculations
MM-GBSA free energy calculations were carried out on 20 ns–150 ns time intervals of the all-atom trajectories, with the same settings as described previously (68). Briefly, the dielectric constant inside the protein was set to 2 and the accurate Generalized Born Molecular Volume II (GBMVII) method (69) was used to calculate the polar solvation free energies. The non-polar solvation free energy was assumed to be proportional to the solvent accessible surface area (SASA) with a coefficient of −0.0072 kcal/mol/Å2 (70). We used the Debeye-Hückel correction to account for ionic screening (71), with screening constant κ=0.316 (expressed in Å−1) (72), where [salt] is the monovalent ion concentration taken to be 0.154 mol/l. The system for each complex was composed of a binder (FMC63, SJ25C1, B43 or Coltuximab) and CD19, and per residue decomposition was performed for all binder and CD19 residues located near the binder-CD19 interface.
Production of mutant CAR binders
The binders were expressed in a Fab scaffold because, compared to scFv expression, we found Fab expression to be more efficient and robust. CAR binders were engineered in a Fab format with a C-terminal 8 × histidine tag on the Fab heavy chain and cloned into the pEZT-BM expression vector (53) as described above. The light and heavy chains of the Fab were transiently transfected into Expi293F cells (Gibco) using the ExpiFectamine 293 Reagent according to the manufacturer’s protocol. The supernatants containing Fabs were clarified and harvested 5 – 7 days post-transfection by centrifugation at 5,000 × g for 20 min at 4°C, then filtered through a 0.22 μm filter. The expression of Fabs were verified by SDS-PAGE.
Surface plasmon resonance (SPR)
Surface plasmon resonance (SPR) was performed on a Biacore 8K instrument (Cytiva). The supernatants containing Fabs with an 8 × histidine tag at the C-terminus were coupled to a sensor chip NTA (Cytiva) at a target density of 1,000 response units (RU). Single cycle kinetics was performed by injecting CD19 protein at five different concentrations (0.5 nM, 4 nM, 20 nM, 100 nM, 500 nM) followed by one dissociation step over the flow cell at a flow rate of 30 μl/min. The relative response curve was fitted with a 1:1 kinetics Langmuir binding model and the kinetic rate constants were obtained on Biacore Insight Evaluation software (Cytiva).
Cell lines
GFP-firefly-luciferase (FFL)-expressing NALM6 cells (NALM6/WT and 19-mCherry fusion-expressing NALM6/WT (23), NALM6/12–4 and NALM6/12–39 (21) were cultured in RPMI 1040 medium supplemented with 10% fetal bovine serum (HyClone), 10 mM HEPES (Invitrogen), 2 mM L-glutamine (Invitrogen), 1X NEAA (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 50 μM β-mercaptoethanol, 10U/ml Penicillin, 10 μg/ml Streptomycin (Gibco), and 2.5 μg/ml Plasmocyn (InvivoGen). Cells were split every 2–3 days and plated at 0.5×106 cells/ml.
T cell isolation, activation, and culture
T cell culture was performed as described (21). In brief, healthy donor peripheral blood mononuclear cells obtained from buffy coats (New York Blood Center) were used to isolate T cells with a Pan T cell isolation kit (Miltenyi Biotec). T cells (>90% viability) were activated with Dynabeads (1:1 beads:cell) Human T-Activator CD3/CD28 (ThermoFisher) in X-vivo 15 medium (Lonza) supplemented with 5% human serum (Gemini Bioproducts) with 5 ng/ml human recombinant IL7 (Miltenyi Biotec) and 5 ng/ml human recombinant IL15 (Miltenyi Biotec) at a density of 106 cells per ml. The medium was changed every 2 days, and cells were replated at 1–1.5 ×106 cells per ml.
Gammaretroviral vector construction, production, and transduction
Plasmids encoding the SFG γ-retroviral vector were used to clone a bicistronic construct to express a gene for a CAR containing an anti-CD19 scFv (FMC63, SJ25C1, or mutants), CD28, and CD3-ζ, simultaneously with the gene for low-affinity nerve growth factor receptor (LNGFR) (23). VSV-G pseudotyped vector supernatants derived H29 cells were used to construct stable RD114 retroviral-producing cell lines (73). T cells were transduced with RD114-derived vector supernatants by centrifugation on RetroNectin-coated plates (Takara), as described previously (73).
Flow cytometry
CAR expression was measured with Alexa-Fluor-647-conjugated goat anti-mouse F(ab’)2 antibody (AF647-GAM, Jackson ImmunoResearch). LNGFR expression was measured with either PE- or Alexa-Fluor-647-conjugated mouse anti-human LNGFR antibodies (BD Biosciences). CD3 was detected with Pacific Blue-conjugated mouse anti-human CD3 antibody (Biologend). T cells were incubated with antibodies in 100 μl MACS buffer (PBS, 0.5 mM EDTA and 0.5% BSA) at 4°C for 20–30 min, washed, and resuspended in 300 μl MACS buffer. Flow cytometric data were acquired on a 5-laser Aurora (Cytek Biosciences) and analyzed in FlowJo v10.1 (BD).
Cytotoxicity assays
The cytotoxic activity of CAR T cells was determined as previously described (21). LNGFR expression was used to determine the percentage of CAR-expressing T cells, which were prepared at 1 M/ml. FFL-expressing NALM6 cell lines WT, 12–39, and 12–4 were used as target cells, and they were resuspended at 1M/ml in T cell medium. The effector I and tumor target (T) cells were co-cultured in triplicates at the indicated effector/target (E/T) ratio using black-walled 96-well plates with 1×105 target cells in a total volume of 100 μl per well. Eighteen hours later, 100 μl luciferase substrate (Bright-Glo, Promega) was added to each well. Emitted light was detected in a luminescence plate reader, and lysis was calculated using the formula 100 × (1-(RLUsample)/(RLUtarget alone)).
CD19 binding assays
CAR T cells (0.1 million) were incubated in 100 μl MACS buffer containing Alexa-Fluor-488-conjugated recombinant hCD19-hFc (AF488-CD19, R&D Systems) at 4 °C for 1 hour. After a wash with 2 ml of MACS buffer, cells were resuspended in 300 μl MACS buffer and analyzed by FACS. Experiments with FMC63-based CAR T cells used 0.5 μg of AF488-CD19. Experiments with SJ25C1-based CAR T cells used 1.5 μg AF488-CD19.
Trogocytosis assay
CAR T cells (0.5 × 105) were co-cultured with 19-mCherry fusion-expressing NALM6/WT target cells (0.5 × 105) in 96-well plates containing 100 μl of T cell medium. After 1 hour incubation at 37 °C cells were stained with AF647-anti-LNGFR and PB-anti-CD3 antibodies. Cells were incubated for 30 min at 4 °C. Following staining, cells were washed and then analyzed by FACS. Trogocytosis was measured by the acquisition of 19-mCherry by CAR T cells (GFP- CD3+ LNGFRmed/hi+).
Cytokine analyses
To measure intracellular levels, CAR T cells were incubated with NALM6/WT at 1:1 ratio for 4 hours in the presence of Brefeldin A. T cells were first stained with mouse anti-human LNGFR antibodies (BD Biosciences) then fixed and permeabilized using Cytofix/ Cytoperm Plus kit (BD Biosciences) as per the recommendation of the manufacturer, followed by staining with BV711-conjugated mouse anti-human TNF-α Antibody (Biolegend) or BV421-conjugated mouse anti-human IFN-γ Antibody (Biolegend). Cytokine levels were determined by FACS.
Statistics
Statistical significance was determined by two-tailed t-test analysis using GraphPad Prism 9 software. Data are expressed as means +/− SEM and differences were considered significant when P<0.05, and nonsignificant (n.s.) when P>0.05. In addition, * is P<0.05, ** is P<0.01, *** is P<0.001, and **** is P<0.0001.
Supplementary Material
Fig. S1. Cryo-EM image processing workflow for the FMC63-CD19 structure.
Fig. S2. Cryo-EM image processing workflow for the SJ25C1-CD19 structure.
Fig. S3. Quality assessment of model-to-map fitting.
Fig. S4. Convergence of the interaction interface for FMC63-CD19 and SJ25C1-CD19 complexes in molecular dynamics simulations.
Fig. S5. Convergence of the interaction interface for B43-CD19 and Coltuximab-CD19 complexes in the molecular dynamics simulations.
Fig. S6. Comparison of CD19 interaction interfaces with previously published structures.
Fig. S7. Expression analysis of co-expressed LNGFR reporter in wildtype and mutant CAR T cells.
Fig. S8. Mutations in CD19-interacting binder residues affects cytokine expression upon CAR stimulation.
Fig. S9. SPR control data and characterization of SJ25C1 glycine mutants.
Fig. S10. FMC63 CAR T cell-mediated trogocytosis.
Fig. S11. Characterization of mutant binder SJ25C1S121A by SPR and in CAR T cells.
Fig. S12. Evaluation of clinical mutants associated with diminished CD19 CAR efficacy.
Fig. S13. Comparison of FMC63-CD19 and SJ25C1-CD19 models from cryo-EM and AlphaFold2.
Table S1. List of FMC63 and SJ25C1 mutants used in affinity tuning studies.
Table S2. Cryo-EM data collection and refinement statistics for FMC63-CD19 and SJ25C1-CD19.
Table S3. Raw data for MM/GBSA per-residue binding energies on CD19.
Table S4. Raw data for surface plasmon resonance experiments.
Table S5. Materials and reagents.
Acknowledgments:
We thank Michael Traxlmayr for generously providing the expression construct for soluble CD19, Daniel Mann and Carsten Sachse for suggestions on cryo-EM experiments, Henrik Molina for assistance with LC/MS experiments, and Alessio Accardi and members of his lab for discussions. Cryo-EM was performed at the NYU Langone Health Cryo-Electron Microscopy Laboratory. We acknowledge the Fisher Drug Discovery Resource Center at Rockefeller University for providing critical instrumentation and infrastructure. Molecular graphics were prepared and analysis performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. The authors acknowledge the use of the computational resources of the David A. Cofrin Center for Biomedical Information in the Institute for Computational Biomedicine at Weill Cornell Medical College. The accession codes for FMC63-CD19 are PDB-7URV and EMDB-26719, and for SJ25C1-CD19 are PDB-7URX and EMDB-26720.
Funding:
Supported by 1923 Fund (GK), Pasteur-Weizmann/Servier award (MS), Leopold Griffuel award (MS), MSK Core Grant P30 CA008748 (MS), The Leukemia and Lymphoma Society, LLS Grant ID: 7014-17 (MS), STARR Cancer Consortium grant (MS and JRM).
Footnotes
Competing interests:
The authors declare no competing interests.
Supplementary Materials
Data and materials availability:
The cryo-EM density maps and models for FMC63-CD19 and SJ25C1-CD19 have been deposited in the PDB and EMDB and will be released upon publication. Code used for simulations is available at: https://github.com/weinsteinlab/OpenMM-Slurm-Ensemble. Code used for MM/GBSA analysis is available at: https://github.com/weinsteinlab/mmgbsa.
References and Notes
- 1.Sadelain M, Rivière I, Riddell S, Therapeutic T cell engineering. Nature. 545, 423–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guedan S, Calderon H, Posey AD, Maus MV, Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev 12, 145–156 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srivastava S, Riddell SR, Engineering CAR-T cells: Design concepts. Trends Immunol. 36, 494–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Rivière I, Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol. Ther. Oncolytics 3, 16015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brentjens RJ, Latouche J-B, Santos E, Marti F, Gong MC, Lyddane C, King PD, Larson S, Weiss M, Rivière I, Sadelain M, Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med 9, 279–286 (2003). [DOI] [PubMed] [Google Scholar]
- 6.June CH, Sadelain M Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacKay M, Afshinnekoo E, Rub J, Hassan C, Khunte M, Baskaran N, Owens B, Liu L, Roboz GJ, Guzman ML, Melnick AM, Wu S, Mason CE, The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol 38, 233–244 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Chong EA, Ruella M, Schuster SJ, Lymphoma Program Investigators at the University of Pennsylvania, Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N. Engl. J. Med 384, 673–674 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Globerson Levin A, Rivière I, Eshhar Z, Sadelain M, CAR T cells: Building on the CD19 paradigm. Eur. J. Immunol 51, 2151–2163 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadelain M, CD19 CAR T cells. Cell. 171, 1471 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Saez-Ibañez AR, Upadhaya S, Partridge T, Shah M, Correa D, Campbell J, Landscape of cancer cell therapies: trends and real-world data. Nat. Rev. Drug Discov 21, 631–632 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Nicholson IC, Lenton KA, Little DJ, Decorso T, Lee FT, Scott AM, Zola H, Hohmann AW, Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol. Immunol 34, 1157–1165 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M, Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol 20, 70–75 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D, Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 18, 676–684 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Bejcek BE, Wang D, Berven E, Pennell CA, Peiper SC, Poppema S, Uckun FM, Kersey JH, Development and characterization of three recombinant single chain antibody fragments (scFvs) directed against the CD19 antigen. Cancer Res. 55, 2346–2351 (1995). [PubMed] [Google Scholar]
- 16.Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E Roshal M, Maslak P, Davila M, Brentjens RJ, Sadelain M, Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miao L, Zhang J, Huang B, Zhang Z, Wang S, Tang F, Teng M, Li Y, Special chimeric antigen receptor (CAR) modifications of T cells: A review. Front. Oncol 12, 832765 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H, Lee DA, Heimberger AB, Champlin RE, Cooper LJN, Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 75, 3505–3518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B, Zhou L, Loew A, Young RM, June CH, Zhao Y, Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 75, 3596–3607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinrichs CS, Restifo NP, Reassessing target antigens for adoptive T-cell therapy. Nat. Biotechnol 31, 999–1008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mansilla-Soto J, Eyquem J, Haubner S, Hamieh M, Feucht J, Paillon N, Zucchetti AE, Li Z, Sjöstrand M, Lindenbergh PL, Saetersmoen M, Dobrin A, Maurin M, Iyer A, Garcia Angus A, Miele MM, Zhao Z, Giavridis T, van der Stegen SJC, Tamzalit F, Rivière I, Huse M, Hendrickson RC, Hivroz C, Sadelain M, HLA-independent T cell receptors for targeting tumors with low antigen density. Nat. Med 28, 345–352 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, Albon SJ, Casanovas-Company J, Castro F, Popova B, Villanueva K, Yeung J, Vetharoy W, Guvenel A, Wawrzyniecka PA, Mekkaoui L, Cheung GW-K, Pinner D, Chu J, Lucchini G, Silva J, Ciocarlie O, Lazareva A, Inglott S, Gilmour KC, Ahsan G, Ferrari M, Manzoor S, Champion K, Brooks T, Lopes A, Hackshaw A, Farzaneh F, Chiesa R, Rao K, Bonney D, Samarasinghe S, Goulden N, Vora A, Veys P, Hough R, Wynn R, Pule MA, Amrolia PJ, Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med 25, 1408–1414 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, Eyquem J, Zhao Z, Whitlock BM, Miele MM, Li Z, Cunanan KM, Huse M, Hendrickson RC, Wang X, Rivière I, Sadelain M, CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 568, 112–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Basar R, Wang G, Liu E, Moyes JS, Li L, Kerbauy LN, Uprety N, Fathi M, Rezvan A, Banerjee PP, Muniz-Feliciano L, Laskowski TJ, Ensley E, Daher M, Shanley M, Mendt M, Acharya S, Liu B, Biederstädt A, Rafei H, Guo X, Melo Garcia L, Lin P, Ang S, Marin D, Chen K, Bover L, Champlin RE, Varadarajan N, Shpall EJ, Rezvani K, KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat. Med 28, 2133–2144 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bloemberg D, Nguyen T, MacLean S, Zafer A, Gadoury C, Gurnani K, Chattopadhyay A, Ash J, Lippens J, Harcus D, Pagé M, Fortin A, Pon RA, Gilbert R, Marcil A, Weeratna RD, McComb S, A High-Throughput Method for Characterizing Novel Chimeric Antigen Receptors in Jurkat Cells. Mol. Ther. Methods Clin. Dev 16, 238–254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochi T, Maruta M, Tanimoto K, Kondo F, Yamamoto T, Kurata M, Fujiwara H, Masumoto J, Takenaka K, Yasukawa M, A single-chain antibody generation system yielding CAR-T cells with superior antitumor function. Commun. Biol 4, 273 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA, Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY, T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res 4, 498–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao R, Hussein MS, He Y, Chimeric antigen receptor engineered T cells and their application in the immunotherapy of solid tumours. Expert Rev.Mol. Med 24, e7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majzner RG, Mackall CL, Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 8, 1219–1226 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Uckun FM, Jaszcz W, Ambrus JL, Fauci AS, Gajl-Peczalska K, Song CW, Wick MR, Myers DE, Waddick K, Ledbetter JA, Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood. 71, 13–29 (1988). [PubMed] [Google Scholar]
- 32.Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, Schertler GFX, Granier S, Weis WI, Dror RO, Manglik A, Skiniotis G, Kobilka BK, Structure of the μ-opioid receptor-Gi protein complex. Nature. 558, 547–552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laurent E, Sieber A, Salzer B, Wachernig A, Seigner J, Lehner M, Geyeregger R, Kratzer B, Jäger U, Kunert R, Pickl WF, Traxlmayr MW, Directed Evolution of Stabilized Monomeric CD19 for Monovalent CAR Interaction Studies and Monitoring of CAR-T Cell Patients. ACS Synth. Biol 10, 1184–1198 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Autzen HE, Julius D, Cheng Y, Membrane mimetic systems in CryoEM: keeping membrane proteins in their native environment. Curr. Opin. Struct. Biol 58, 259–268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu S, Avila-Sakar A, Kim J, Booth DS, Greenberg CH, Rossi A, Liao M, Li X, Alian A, Griner SL, Juge N, Yu Y, Mergel CM, Chaparro-Riggers J, Strop P, Tampé R, Edwards RH, Stroud RM, Craik CS, Cheng Y, Fabs enable single particle cryoEM studies of small proteins. Structure. 20, 582–592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teplyakov A, Obmolova G, Luo J, Gilliland GL, Crystal structure of B-cell co-receptor CD19 in complex with antibody B43 reveals an unexpected fold. Proteins. 86, 495–500 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Susa KJ, Rawson S, Kruse AC, Blacklow SC, Cryo-EM structure of the B cell co-receptor CD19 bound to the tetraspanin CD81. Science. 371, 300–305 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiu D, Shenkin PS, Hollinger FP, Still WC, The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate born radii. J. Phys. Chem. A 101, 3005–3014 (1997). [Google Scholar]
- 39.Zoete V, Meuwly M, Karplus M, Study of the insulin dimerization: binding free energy calculations and per-residue free energy decomposition. Proteins. 61, 79–93 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, Sidhu SS, The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J. Mol. Biol 377, 1518–1528 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Cao L, Coventry B, Goreshnik I, Huang B, Sheffler W, Park JS, Jude KM, Marković I, Kadam RU, Verschueren KHG, Verstraete K, Walsh STR, Bennett N, Phal A, Yang A, Kozodoy L, DeWitt M, Picton L, Miller L, Strauch E-M, DeBouver ND, Pires A, Bera AK, Halabiya S, Hammerson B, Yang W, Bernard S, Stewart L, Wilson IA, Ruohola-Baker H, Schlessinger J, Lee S, Savvides SN, Garcia KC, Baker D, Design of protein-binding proteins from the target structure alone. Nature. 605, 551–560 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bogan AA, Thorn KS, Anatomy of hot spots in protein interfaces. J. Mol. Biol 280, 1–9 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, Scarfó I, Leick MB, Grauwet K, Berger TR, Stewart K, Anekal PV, Jan M, Joung J, Schmidts A, Ouspenskaia T, Law T, Regev A, Getz G, Maus MV, CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature. 604, 563–570 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Fernández de Larrea C, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, Purdon TJ, Ponomarev V, Wendel H-G, Brentjens RJ, Smith EL, Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape-Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 1, 146–154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klesmith JR, Wu L, Lobb RR, Rennert PD, Hackel BJ, Fine epitope mapping of the CD19 extracellular domain promotes design. Biochemistry. 58, 4869–4881 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z, Chen X, Tian Y, Li F, Zhao X, Liu J, Yao C, Zhang Y, Point mutation in CD19 facilitates immune escape of B cell lymphoma from CAR-T cell therapy. J. Immunother. Cancer 8 (2020), doi: 10.1136/jitc-2020-001150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen WC, Murawsky CM, Strategies for generating diverse antibody repertoires using transgenic animals expressing human antibodies. Front. Immunol 9, 460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature. 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yin R, Feng BY, Varshney A, Pierce BG, Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 31, e4379 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olson ML, Mause ERV, Radhakrishnan SV, Brody JD, Rapoport AP, Welm AL, Atanackovic D, Luetkens T, Low-affinity CAR T cells exhibit reduced trogocytosis, preventing rapid antigen loss, and increasing CAR T cell expansion. Leukemia. 36, 1943–1946 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ingraham J, Baranov M, Costello Z, Frappier V, Ismail A, Tie S, Wang W, Xue V, Obermeyer F, Beam A, Grigoryan G, Illuminating protein space with a programmable generative model. BioRxiv (2022), doi: 10.1101/2022.12.01.518682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, Ahern W, Borst AJ, Ragotte RJ, Milles LF, Wicky BIM, Hanikel N, Pellock SJ, Courbet A, Sheffler W, Wang J, Venkatesh P, Sappington I, Vázquez Torres S, Lauko A, De Bortoli V, Mathieu E, Barzilay R, Jaakkola TS, DiMaio F, Baek M, Baker D, Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models. BioRxiv (2022), doi: 10.1101/2022.12.09.519842. [DOI] [Google Scholar]
- 53.Morales-Perez CL, Noviello CM, Hibbs RE, Manipulation of subunit stoichiometry in heteromeric membrane proteins. Structure. 24, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson ZL, Chen J, Structural basis of substrate recognition by the multidrug resistance protein MRP1. Cell. 168, 1075–1085.e9 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B, Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH, New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 7 (2018), doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Rohou A, Grigorieff N, CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T, SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, Moriarty NW, Oeffner RD, Poon BK, Prisant MG, Read RJ, Richardson JS, Richardson DC, Sammito MD, Sobolev OV, Stockwell DH, Terwilliger TC, Urzhumtsev AG, Videau LL, Williams CJ, Adams PD, Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, MacKerell AD, Klauda JB, Im W, CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 12, 405–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang L-P, Simmonett AC, Harrigan MP, Stern CD, Wiewiora RP, Brooks BR, Pande VS, OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol 13, e1005659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, MacKerell AD, CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG, A smooth particle mesh Ewald method. J. Chem. Phys 103, 8577 (1995). [Google Scholar]
- 67.Humphrey W, Dalke A, Schulten K, VMD: visual molecular dynamics. J. Mol. Graph 14, 33–8, 27 (1996). [DOI] [PubMed] [Google Scholar]
- 68.Khelashvili G, Pillai AN, Lee J, Pandey K, Payne AM, Siegel Z, Cuendet MA, Lewis TR, Arshavsky VY, Broichhagen J, Levitz J, Menon AK, Unusual mode of dimerization of retinitis pigmentosa-associated F220C rhodopsin. Sci. Rep 11, 10536 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee MS, Feig M, Salsbury FR, Brooks CL, New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem 24, 1348–1356 (2003). [DOI] [PubMed] [Google Scholar]
- 70.Still WC, Tempczyk A, Hawley RC, Hendrickson T, Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc 112, 6127–6129 (1990). [Google Scholar]
- 71.Srinivasan J, Trevathan MW, Beroza P, Case DA, Application of a pairwise generalized Born model to proteins and nucleic acids: inclusion of salt effects. Theoretical Chemistry Accounts: Theory, Computation, and Modeling (Theoretica Chimica Acta). 101, 426–434 (1999). [Google Scholar]
- 72.Onufriev A, Bashford D, Case DA, Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 55, 383–394 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M, Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 28, 415–428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Cryo-EM image processing workflow for the FMC63-CD19 structure.
Fig. S2. Cryo-EM image processing workflow for the SJ25C1-CD19 structure.
Fig. S3. Quality assessment of model-to-map fitting.
Fig. S4. Convergence of the interaction interface for FMC63-CD19 and SJ25C1-CD19 complexes in molecular dynamics simulations.
Fig. S5. Convergence of the interaction interface for B43-CD19 and Coltuximab-CD19 complexes in the molecular dynamics simulations.
Fig. S6. Comparison of CD19 interaction interfaces with previously published structures.
Fig. S7. Expression analysis of co-expressed LNGFR reporter in wildtype and mutant CAR T cells.
Fig. S8. Mutations in CD19-interacting binder residues affects cytokine expression upon CAR stimulation.
Fig. S9. SPR control data and characterization of SJ25C1 glycine mutants.
Fig. S10. FMC63 CAR T cell-mediated trogocytosis.
Fig. S11. Characterization of mutant binder SJ25C1S121A by SPR and in CAR T cells.
Fig. S12. Evaluation of clinical mutants associated with diminished CD19 CAR efficacy.
Fig. S13. Comparison of FMC63-CD19 and SJ25C1-CD19 models from cryo-EM and AlphaFold2.
Table S1. List of FMC63 and SJ25C1 mutants used in affinity tuning studies.
Table S2. Cryo-EM data collection and refinement statistics for FMC63-CD19 and SJ25C1-CD19.
Table S3. Raw data for MM/GBSA per-residue binding energies on CD19.
Table S4. Raw data for surface plasmon resonance experiments.
Table S5. Materials and reagents.
Data Availability Statement
The cryo-EM density maps and models for FMC63-CD19 and SJ25C1-CD19 have been deposited in the PDB and EMDB and will be released upon publication. Code used for simulations is available at: https://github.com/weinsteinlab/OpenMM-Slurm-Ensemble. Code used for MM/GBSA analysis is available at: https://github.com/weinsteinlab/mmgbsa.