

Abstract
Chromosome conformation capture (3C) is used to detect three-dimensional chromatin interactions. Typically, chemical crosslinking with formaldehyde (FA) is used to fix chromatin interactions. Then, chromatin digestion with a restriction enzyme and subsequent religation of fragment ends converts three-dimensional (3D) proximity into unique ligation products. Finally, after reversal of crosslinks, protein removal, and DNA isolation, DNA is sheared and prepared for high-throughput sequencing. The frequency of proximity ligation of pairs of loci is a measure of the frequency of their colocalization in three-dimensional space in a cell population.
A sequenced Hi-C library provides genome-wide information on interaction frequencies between all pairs of loci. The resolution and precision of Hi-C relies on efficient crosslinking that maintains chromatin contacts and frequent and uniform fragmentation of the chromatin. This paper describes an improved in situ Hi-C protocol, Hi-C 3.0, that increases the efficiency of crosslinking by combining two crosslinkers (formaldehyde [FA] and disuccinimidyl glutarate [DSG]), followed by finer digestion using two restriction enzymes (DpnII and DdeI). Hi-C 3.0 is a single protocol for the accurate quantification of genome folding features at smaller scales such as loops and topologically associating domains (TADs), as well as features at larger nucleus-wide scales such as compartments.
Summary
Hi-C 3.0 is an improved Hi-C protocol that combines formaldehyde and disuccinimidyl glutarate crosslinkers with a cocktail of DpnII and DdeI restriction enzymes to increase the signal-to-noise ratio and the resolution of chromatin interaction detection.
Introduction
Chromosome conformation capture has been used since 20021. Fundamentally, every conformation capture variant relies on the fixation of DNA-protein and protein-protein interactions to preserve 3D chromatin organization. This is followed by DNA fragmentation, usually by restriction digestion, and, finally, religation of nearby DNA ends to convert spatially proximal loci into unique covalent DNA sequences. Initial 3C protocols used PCR to sample specific, “one-to-one” interactions. Subsequent 4C assays allowed the detection of “one-to-all” interactions2, while 5C detected “many-to-many” interactions3. Chromosome conformation capture came to full fruition after implementing next-generation, high-throughput sequencing (NGS), which allowed detection of “all-to-all” genomic interactions using, genome-wide Hi-C4 and comparable techniques such as 3C-seq5, TCC6 and Micro-C7,8 (see also review by Denker and De Laat9).
In Hi-C, biotinylated nucleotides are used to mark 5′ overhangs after digestion and before ligation (Figure 1). This allows for the selection of properly digested and religated fragments using streptavidin-coated beads, setting it apart from GCC10. An important update to the Hi-C protocol was implemented by Rao et al.11, who performed the digestion and religation in intact nuclei (i.e., in situ) to reduce spurious ligation products. Moreover, substituting HindIII digestion with MboI (or DpnII) digestion reduced the fragment size and increased the resolution potential of Hi-C. This increase allowed the detection of relatively small-scale structures and a more precise genomic localization of points of contact, such as DNA loops between small cis-elements, e.g., loops between CTCF-bound sites generated by loop extrusion11,12. However, this potential comes at a cost. First, a two-fold increase in resolution requires a four-fold (22) increase in sequencing reads13. Second, the small fragment sizes increase the possibility of mistaking undigested neighboring fragments for digested and religated fragments14. As mentioned, in Hi-C, digested and religated fragments differ from undigested fragments by the presence of biotin at the ligation junction. However, proper biotin removal from unligated ends is required to assure that only ligation junctions are pulled down14,15.
Figure 1: Hi-C procedure in six steps.
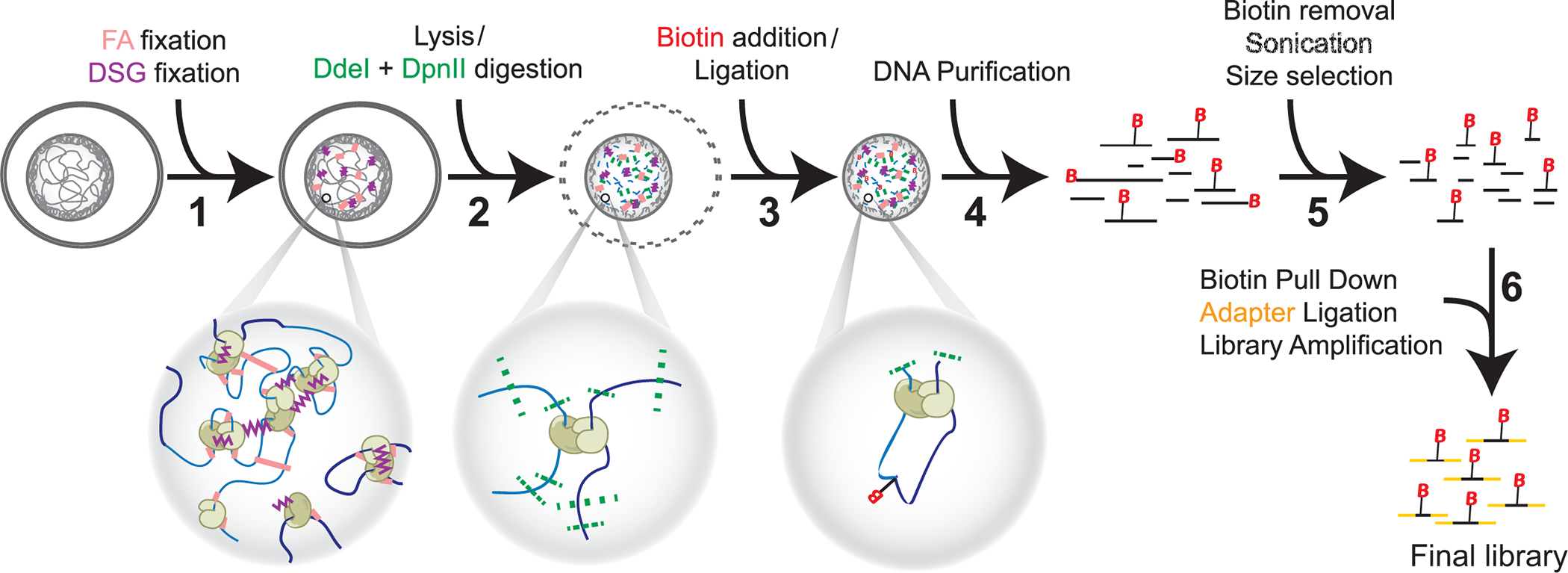
Cells are fixed first with FA, and then DSG (1). Then, lysis precedes a double digestion with DdeI and DpnII (2). Biotin is added by overhang fill-in and proximal blunted ends are ligated (3) before DNA purification (4). Biotin is removed from unligated ends before sonication and size selection (5). Finally, pull-down of biotin allows for adapter ligation and library amplification by PCR (6). Abbreviations: FA = formaldehyde; DSG = disuccinimidyl glutarate; B = Biotin.
With the decreasing cost of NGS, it becomes feasible to study chromosome folding in greater detail. To decrease the size of DNA fragments, and thereby increase resolution, the Hi-C protocol can be adapted to use more frequently cutting restriction enzymes16 or to use combinations of restriction enzymes17,18,19. Alternatively, MNase7,8 in Micro-C and DNase in DNase Hi-C20 can be titrated to achieve optimal digestion.
A recent systematic evaluation of the fundamentals of 3C methods showed that the detection of chromosome folding features at every length scale greatly improved with sequential crosslinking with 1% FA followed by 3 mM DSG17. Furthermore, Hi-C with HindIII digestion was the best option for detecting large-scale folding features, such as compartments, and that Micro-C was superior at detecting small-scale folding features such as DNA loops. These results led to the development of a single, high-resolution “Hi-C 3.0” strategy, that uses the combination of FA and DSG crosslinkers followed by double digestion with DpnII and DdeI endonucleases21. Hi-C 3.0 provides an effective strategy for general use because it accurately detects folding features across all length scales17. The experimental portion of the Hi-C 3.0 protocol is detailed here and typical results that can be expected after sequencing are shown.
Protocol
1. Fixation by crosslinking
1. Formaldehyde fixation: starting from cells in monolayer
-
Have cells seeded in appropriate medium to harvest 5 × 106 cells per 150 mm plate.
NOTE: Users can choose any preferred container that ensures optimal growth of any mammalian cell line. Additionally, cells can be isolated from tissue.
Aspirate the medium with a Pasteur pipette coupled to a vacuum trap from 150 mm plate, wash 2x with ~10 mL HBSS.
Immediately before crosslinking, prepare a 1% FA crosslinking solution in a 50 mL tube by combining 22.5 mL of HBSS and 625 μL of 37% FA to a final 1% concentration. Mix gently by rocking. CAUTION: Use fume hood; formaldehyde is toxic.
To crosslink the cells, pour 23.125 mL of the 1% FA solution to each 15 cm plate.
Incubate at room temperature for 10 min and gently rock the plates by hand every 2 min.
Add 1.25 mL of 2.5 M glycine (128 mM final) and gently swirl the plate to quench the crosslinking reaction.
Incubate at room temperature for 5 min and continue incubation on ice for at least 15 min to stop crosslinking.
Scrape the cells from the plates with a cell scraper or rubber policeman.
Transfer the cell suspension to a 50 mL conical tube with a pipet. Centrifuge at 1,000 × g for 10 min at room temperature and discard the supernatant by aspiration.
Wash the cell pellet once with 10 mL of Dulbecco’s phosphate-buffered saline (DPBS), using a pipet to resuspend. Then, centrifuge at 1,000 × g for 10 min at room temperature. Proceed immediately to DSG crosslinking.
NOTE: Be careful when washing the cell pellet as cell pellets can be loose, and cells may be lost.
2. Formaldehyde fixation: starting from cells in suspension
-
Have cells seeded in appropriate medium to harvest 5 × 106 cells per vessel.
NOTE: Users can choose any preferred container that ensures optimal cell growth of any mammalian cell line.
Immediately before harvest, count the cells and transfer 5 × 106 cells to a 50 mL conical tube.
Gently pellet the cells by centrifuging at 300 × g for 10 min at room temperature.
-
Prepare 1% FA crosslinking solution by adding 1.25 mL of 37% FA to 45 mL of HBSS and mix by inverting the tube several times.
NOTE: Add the entire 1.25 mL of FA without splitting the amount. CAUTION: Formaldehyde is highly toxic.
Resuspend the cell pellet in the 46.25 mL of 1% FA crosslinking solution prepared in the previous step by pipetting up and down.
Incubate at room temperature for exactly 10 min on rotator, rocker, or by gentle manual inversion of the tube every 1–2 min.
Quench the crosslinking reaction by adding 2.5 mL of 2.5 M glycine (128 mM final) and mix well by inverting the tube.
Incubate for 5 min at room temperature, and then on ice for at least 15 min to stop crosslinking completely.
Centrifuge at room temperature to pellet the crosslinked cells at 1,000 × g for 10 min and discard the supernatant by aspiration.
-
Wash the cells once with 10 mL of DPBS, and then centrifuge at 1,000 × g for 10 min at room temperature. Discard the supernatant completely using a pipet and immediately proceed to DSG crosslinking.
NOTE: Be careful when washing the cell pellet as cell pellets can be loose, and cells may be lost.
3. Crosslinking with disuccinimidyl glutarate
-
Resuspend the pelleted cells in 9.9 mL of DPBS before adding 100 μL of 300 mM DSG (3 mM final). Mix by inversion.
NOTE: DSG is moisture-sensitive. It is important to prepare a fresh stock of 300 mM DSG in DMSO on the day of crosslinking. CAUTION: DSG in DMSO is highly toxic.
Crosslink the cells at room temperature for 40 min on a rotator.
Add 1.925 mL of 2.5 M glycine (400 mM final), invert to mix, and incubate at room temperature for 5 min.
-
Centrifuge the cells at 2,000 × g for 15 min at room temperature.
NOTE: Be careful when removing the supernatant from loose cell pellets.
-
Resuspend the pellet in 1 mL of 0.05% bovine serum albumin (BSA)-DPBS and transfer to a 1.7 mL tube.
NOTE: The addition of BSA can help to reduce cell clumping.
-
Centrifuge the cells at 2,000 × g for 15 min at 4 °C and remove the supernatant by pipet.
NOTE: To avoid losing the pellet, quickly and completely remove the supernatant.
Snap-freeze the pellet in liquid nitrogen and store at −80 °C or immediately proceed to the next step.
2. Chromosome conformation capture
1. Cell lysis and chromatin digestion
-
Resuspend (pipet) the crosslinked cell aliquots (~5 × 106 cells) in 1 mL of ice-cold lysis buffer (recipe in Supplemental Table S1) containing 10 μL of protease inhibitor cocktail and transfer to a dounce homogenizer for a 15 min incubation on ice.
NOTE: Add protease inhibitors to the lysis buffer immediately before use.
Slowly move pestle A up and down 30 times to homogenize the cells on ice and incubate on ice for 1 min to allow the cells to cool down, before another 30 strokes.
-
Transfer the lysate to a 1.7 mL microcentrifuge tube.
NOTE: Keep the suspension moving because sometimes cells stick in the pipette tip.
Centrifuge the lysed suspension at 2,500 × g for 5 min at room temperature.
Discard the supernatant and flick or vortex the wet pellet to resuspend. Remove as much of the supernatant as possible to obtain a yoghurt-like substance with minimal clumps.
-
Resuspend the pellet in 500 μL of ice-cold 1x Restriction Buffer (from 10x; see recipe in Supplemental Table S1) and centrifuge for 5 min at 2,500 × g. Repeat this step for a second wash.
NOTE: The pellet in 1x Restriction Buffer is more granular than the previous pellet in lysis buffer.
Resuspend the cells in a final volume of 360 μL of 1x Restriction Buffer by pipetting after adding ~340 μL to the carryover volume of the pellet, which depends on the cell size.
Set aside 18 μL of each lysate for testing the Chromatin Integrity (CI). Store the CI samples at 4 °C.
-
Add 38 μL of 1% sodium dodecyl sulfate (SDS) to each Hi-C tube (total volume of 380 μL) and mix carefully by pipetting without introducing bubbles.
CAUTION: SDS is toxic.
Incubate the samples at 65 °C without shaking for exactly 10 min to open up the chromatin.
Immediately place the tubes on ice and prepare the Digestion Mixture with Triton X-100 to quench the SDS as described in Table 1.
-
Add 107 μL of the Digestion Mixture to the Hi-C-tube (487 μL total) to digest the chromatin overnight (~16 h) at 37 °C in a thermomixer with interval shaking (e.g., 900 rpm, 30 s on, 4 min off).
NOTE: The addition of Triton to a final concentration of 1% serves to quench the SDS.
Table 1:
Digestion reagents
| Reagent | Amount |
|---|---|
| Restriction Buffer (10×) | 10 μL |
| Triton X-100 (10%) | 49 μL |
| DdeI (10 U/μL) | 40 μL |
| DpnII (50 U/μL) | 8 μL |
| TOTAL | 107 μL |
2. Biotinylation of DNA ends
After overnight digestion, transfer the samples to 65 °C for 20 min to deactivate the remaining endonuclease activity.
During the incubation, prepare a fill-in master mix as shown in Table 2.
After incubation, place the samples immediately on ice.
Set aside a 10 μL Digestion Control (DC) for each sample and store at 4 °C.
Remove the condensation from the lid with a pipet or by spinning. To each sample, add 58 μL of biotin fill-in mix (total sample volume 535 μL) and pipet gently without forming bubbles.
Incubate the samples at 23 °C for 4 h in a thermomixer (e.g., 900 rpm, 30 s on, 4 min off).
Table 2:
Biotin Fill-In reagents
| Reagent | Amount |
|---|---|
| Restriction Buffer * (10×) | 5.8 μL |
| 10 mM dCTP | 1.5 μL |
| 10 mM dGTP | 1.5 μL |
| 10 mM dTTP | 1.5 μL |
| 0.4 mM biotin-14-dATP* | 37.5 μL |
| Ultrapure water | 0.2 μL |
| Klenow DNA polymerase (5 U/μL) | 10 μL |
| TOTAL | 58 μL |
Note that changing enzymes may require different buffers and biotinylated dNTPs.
3. Ligation of proximal DNA fragments
Prepare the ligation mix as shown in Table 3 while the biotin fill-in is incubating.
Add 665 μL of the ligation mix to each sample (total sample volume 1,200 μL). Mix gently by pipetting.
Incubate the samples at 16 °C for 4 h in a thermomixer with interval shaking (e.g., 900 rpm, 30 s on, 4 min off). Store these samples containing covalently linked chromatin at 4 °C for a few days.
Table 3:
Ligation reagents
| Reagent | Amount |
|---|---|
| Ultrapure water | 243 μL |
| T4 ligation buffer (5×) | 240 μL |
| Triton X-100 (10%) | 120 μL |
| BSA (10 mg/ml) | 12 μL |
| T4 DNA ligase (1U/μL) | 50 μL |
| TOTAL | 665 μL |
4. Reversal of crosslinking
Bring the volumes of the CI and DC samples to 50 μL with 1x Tris Low EDTA (TLE; see the recipe in Supplemental Table S1).
Add 10 μL of 10 mg/mL Proteinase K to CI and DC samples.
Incubate at 65 °C overnight with interval shaking (e.g., 900 rpm, 30 s on, 4 min off). Alternatively, perform a 30 min reversal of crosslinking for these controls during DNA purification of the Hi-C samples.
To each Hi-C sample, add 50 μL of 10 mg/mL Proteinase K and incubate at 65 °C for at least 2 h with interval shaking (e.g., 900 rpm, 30 s on, 4 min off).
-
Add another 50 μL of 10 mg/mL Proteinase K to each Hi-C tube (total sample volume 1,300 μL) and continue incubating at 65 °C overnight. Store at 4 °C until DNA purification.
NOTE: Splitting up Proteinase K incubations ensures total protein digestion.
5. DNA purification
Allow the tubes to cool from 65 °C down to room temperature.
Transfer each sample to a 15 mL conical tube and add a 2.6 mL (2x volume) of phenol:chloroform:isoamyl alcohol to each tube. CAUTION: Phenol:chloroform:isoamyl alcohol is a very toxic irritant and is potentially carcinogenic.
Vortex each tube for 1 min and then transfer its contents into a 15 mL phase-lock tube.
Centrifuge the samples for 5 min at maximum speed (1,500–3,500 × g) in a benchtop centrifuge.
-
Carefully pour the aqueous phase into a 35 mL ultracentrifuge tube and add ultrapure water to a final volume of 1,250 μL.
NOTE: Use tubes to fit the available ultracentrifuge or split into multiple microcentrifuge tubes.
Add a 1/10th volume (~125 μL) of 3 M sodium acetate and mix well by inversion.
Add a 2.5x volume (~3.4 mL) of ice-cold 100% ethanol to each sample, balance the tubes for ultracentrifugation by adding ice-cold 100% ethanol, and mix well by inversion.
Incubate the tubes on dry ice for ~15 min (avoid solidification).
-
Centrifuge the tubes at 18,000 × g for 30 min at 4 °C.
NOTE: For angled rotors: mark the tubes for where the pellet will be.
-
Using a pipette, remove and discard the supernatant completely from the non-pellet side.
NOTE: At this point, the pellet should become visible and can be marked on the tube, because it may not be clearly visible after drying in the next step.
Air-dry the samples for approximately 10 min or until they are visibly dry.
Solubilize each pellet in 450 μL of 1x TLE by pipetting or swirling and transfer to 0.5 mL Centrifugal Filter Unit (CFU) with a 3 kDa molecular weight cut-off.
-
Centrifuge the CFU at maximum speed for 10 min and discard the flowthrough. Wash each ultracentrifuge tube with an additional 450 μL of 1x TLE and transfer to its CFU for another wash.
NOTE: Washing the CFU this way limits DNA loss while reducing salt concentration.
Centrifuge the CFU at maximum speed for 10 min and discard the flowthrough.
Add 80 μL of 1x TLE to the column and turn the column around into a new collection tube before centrifuging for 2 min at maximum speed to obtain a final volume of ~100 μL.
Add 1 μL of RnaseA (1 mg/mL; 10-fold dilution of 10 mg/mL stock) to each sample and incubate at 37 °C on a heat block, in a water bath, or a thermomixer for at least 30 min.
After the Rnase treatment, remove the samples from 37 °C and store at 4 °C until the quality control step.
6. Checking chromatin quality, enzyme digestion, and sample ligation
-
Cool the CI and DC samples to room temperature after reversing the crosslinks in step 2.4.5. Then, transfer to a prespun 2 mL phase-lock tube.
NOTE: Make sure the phase lock content is centrifuged to a pellet (maximum speed for 2 min).
Add 200 μL of phenol:chloroform:isoamyl alcohol and mix the samples by vortexing for 1 min.
Centrifuge the tubes for 5 min at maximum speed.
Transfer the aqueous phase from each sample (~50 μL) to a new 1.7 mL microfuge tube.
Add 1 μL of Rnase A (from 1 mg/mL) and incubate at 37 °C for at least 30 min.
-
Load the samples onto a 0.8% agarose gel as recommended in Table 4.
NOTE: The results expected from a quality control are shown in Figure 2.
-
Quantify the DNA by densitometry from the gel or using Qubit or Nanodrop.
NOTE: Accurate quantification ensures the correct input volume in the next part of the protocol. Use several standards with a known quantity to construct a standard curve.
Table 4:
Gel loading parameters for quality and size selection assessment
| Sample | Contents |
|---|---|
| Quality assessment gel | |
| 1 kb Ladder (50 ng/ul) | 50 ng (1 μL) |
| 100 ng (2 μL) | |
| 150 ng (3 μL) | |
| 200 ng (4 μL) | |
| 250 ng (5 μL) | |
| Chromatin Integrity (CI) | 20 μL + 4 μL 6× dye |
| Digestion Control (DC) | 20 μL + 4 μL 6× dye |
| Hi-C Library | 1 μL Hi-C Library + 9 μL water + 2 μL 6×dye |
| Size selection assessment gel | |
| 1 kb Ladder (50 ng/ul) | 50 ng (1 μL) |
| 100 ng (2 μL) | |
| 150 ng (3 μL) | |
| 200 ng (4 μL) | |
| Upper Fraction | 7 μL sample + 3 μL water + 2 μL 6× dye |
| Lower Fraction | 3 μL sample + 7 μL water + 2 μL 6× dye |
| Lower Fraction | 1 μL Hi-C Library + 9 μL water + 2 μL 6× dye |
Figure 2: Agarose gel showing typical postDNA purification quality control results.
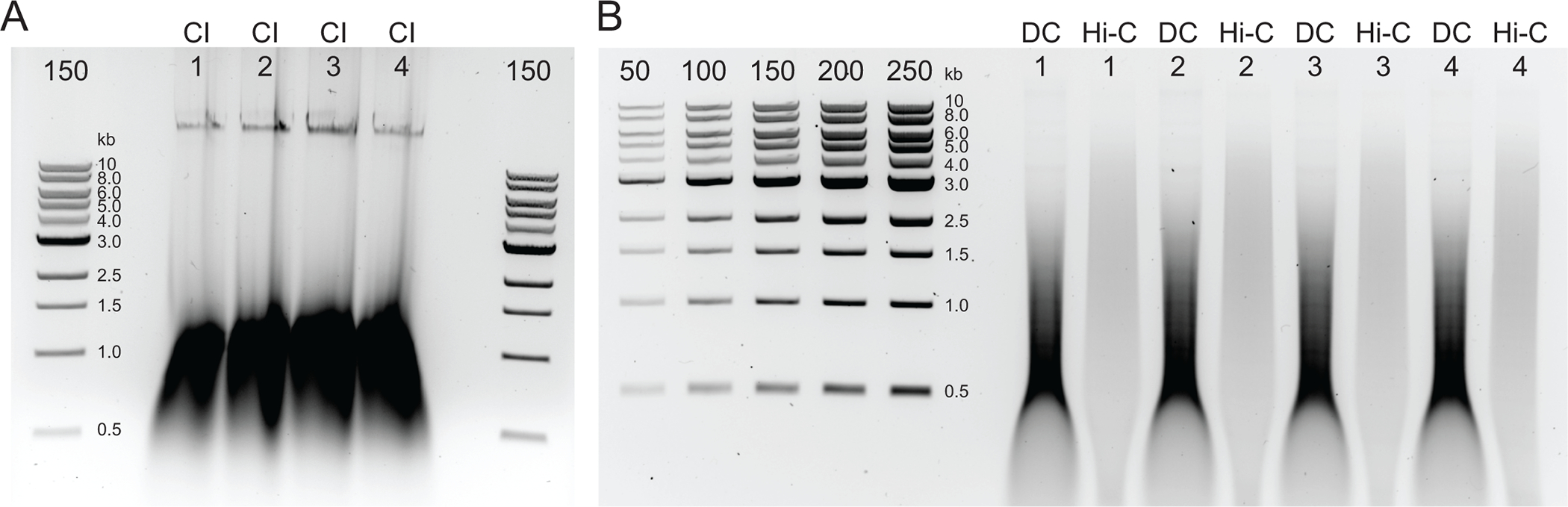
(A) The CI control should indicate a band of high molecular weight DNA. (B) The DC and Hi-C samples show a range of DNA sizes. The Hi-C sample, having been combined into larger fragments, should be of higher molecular weight than the DC. The concentration range of markers allows for generating a standard curve. Note, in this example, the CI was loaded on a separate gel, but it is advised to load and run all samples and controls together. Abbreviations: CI = chromatin integrity; DC = digestion control; Hi-C = proximity-ligated.
3. Hi-C sequencing library preparation
1. Removal of biotin from unligated ends
-
Prepare biotin removal reactions as shown in Table 5.
NOTE: Typically, 10 μg of DNA is sufficient, but up to 30 μg can be used.
Distribute 2 × 65 μL aliquots from each 130 μL reaction into two PCR tubes.
-
Transfer to a thermocycler or PCR machine and incubate as described in Table 5.
NOTE: Samples can be stored here at 4 °C (days to weeks), −20 °C (long term), or immediately carried over to sonication.
Table 5:
Reagents and cycling parameters for removal of biotin from unligated ends
| Reagent | Amount |
| Hi-C DNA sample | 5–15 μg |
| Pre-PCR Buffer (10×) | 13 μL |
| 1 mM dATP | 3.25 μL |
| 1 mM dGTP | 3.25 μL |
| T4 DNA polymerase (3,000 U/mL) | 13 μL |
| Ultrapure water | to 130 μL |
| TOTAL | 130 μL |
| Cycling parameters | |
| Temperature | Time |
| 20 ºC | 4 hours |
| 75 ºC | 20 minutes |
| 4 ºC | hold |
2. Sonication
Pool the sample duplicates from the biotin removal step (130 μL total sample volume) into a 130 μL sonicator tube for sonication.
-
Sonicate the samples using the parameters given in Table 6 to achieve a tight narrow distribution below 500 bp.
NOTE: Different sonicator types can be used, but for a narrow fragment distribution (100–500 bp), sonicator settings may require optimization.
Table 6:
Parameters for sonication
| Parameter (Covaris soncator) | M220 | E220, E220 evolution |
|---|---|---|
| Peak Incident Power (Watt) | 50 W | 175 W |
| Duty Cycle/Duty factor | 20% | 10% |
| Intensity | - | 5 |
| Cycles per Burst | 200 | 200 |
| Treatment time (sec) | 180 | 180 |
3. Size selection with magnetic beads
Pipet the sonicated DNA from the sonicator tube(s) into a 1.7 mL low binding tube.
Bring each sample to a total volume of 500 μL with 1x TLE. Try to get the volume as close as possible to 500 μL, since the ratio of sample to magnetic beads mixture is essential for size selection.
-
Add 400 μL of magnetic beads mixture to each tube to attain a ratio of magnetic beads mixture to the sample volume of 0.8.
NOTE: Under these conditions, beads capture DNA fragments >300 bp, which will be the Upper Fraction. The supernatant will contain fragments <300 bp, which will be the Lower Fraction.
Mix the tubes by vortexing and incubate for 10 min at room temperature on a rotator. For this and other size selection ratios, make sure that the full volume of the sample mixes well. Look for the “erratic mode” that some rotors have, which works well for smaller volumes.
Incubate for 5 min at room temperature on a magnetic particle separator (MPS).
-
While incubating, add 500 μL of the magnetic beads mixture to a fresh 1.7 μL low binding tube for each sample.
NOTE: These tubes will be used for the next size selection of the lower fraction by generating a magnetic beads mixture to sample ratio of 1.1:1.
Leave the tubes on the MPS for 5 min.
-
Remove the supernatant from the beads and resuspend with 150 μL of the magnetic beads mixture.
NOTE: This step avoids saturation of the beads with DNA by increasing the number of beads without increasing the volume.
-
Transfer the supernatant from step 3.3.5 to the labeled tube prepared to select the Lower Fraction (step 3.3.8).
NOTE: 150 μL of magnetic beads mixture + 400 μL of 0.8x magnetic beads mixture (550 μL total) divided by 500 μL of initial sample = 1.1x magnetic beads mixture to sample ratio.
-
Mix the Lower Fraction tubes by vortexing and incubate for 10 min at room temperature on a rotator.
NOTE: The beads will bind DNA fragments >100 bp, resulting in a final bead-bound fraction of 100–300 bp.
Put the Lower Fraction tubes on the MPS for 5 min (room temperature).
Remove the supernatant and centrifuge the tubes briefly to further remove the supernatant as much as possible.
Wash the beads from both Fractions twice using 200 μL of 70% ethanol and reclaim the beads for 5 min on the MPS each time.
-
After a quick spin in a centrifuge, remove the ethanol completely and further dry the beads on the MPS.
NOTE: Dry until the alcohol is completely evaporated. The pellet should look like dark chocolate without cracking (can take ~10 min).
Resuspend both fractions in 50 μL of 1x TLE buffer. Incubate at room temperature for 10 min and tap or flick the tubes every other minute to stimulate mixing and elution.
Separate the beads from the supernatant on the MPS for 5 min for both fractions.
-
Keep the supernatant from each sample. Pipet the supernatant into a 1.7 mL low binding tube.
NOTE: The samples can be kept at 4 °C for a few days or at −20 °C for long term.
-
Run a 2% agarose gel as in Table 4 to determine the sample quality and quantity. See Figure 3 for an example of such a gel.
NOTE: If from experience, sonication is highly reproducible, one can skip this gel and proceed to end repair immediately. It is recommended that users hold on to the Upper Fractions until after titration PCR. Suboptimal DNA quantities that would require a lot of PCR amplification can be rescued from material in the Upper Fraction.
Quantify the amount of DNA from the gel directly after generating a standard curve from the known DNA ladder input or by using a Qubit or Nanodrop.
Figure 3: Agarose gel showing typical post-size selection results.
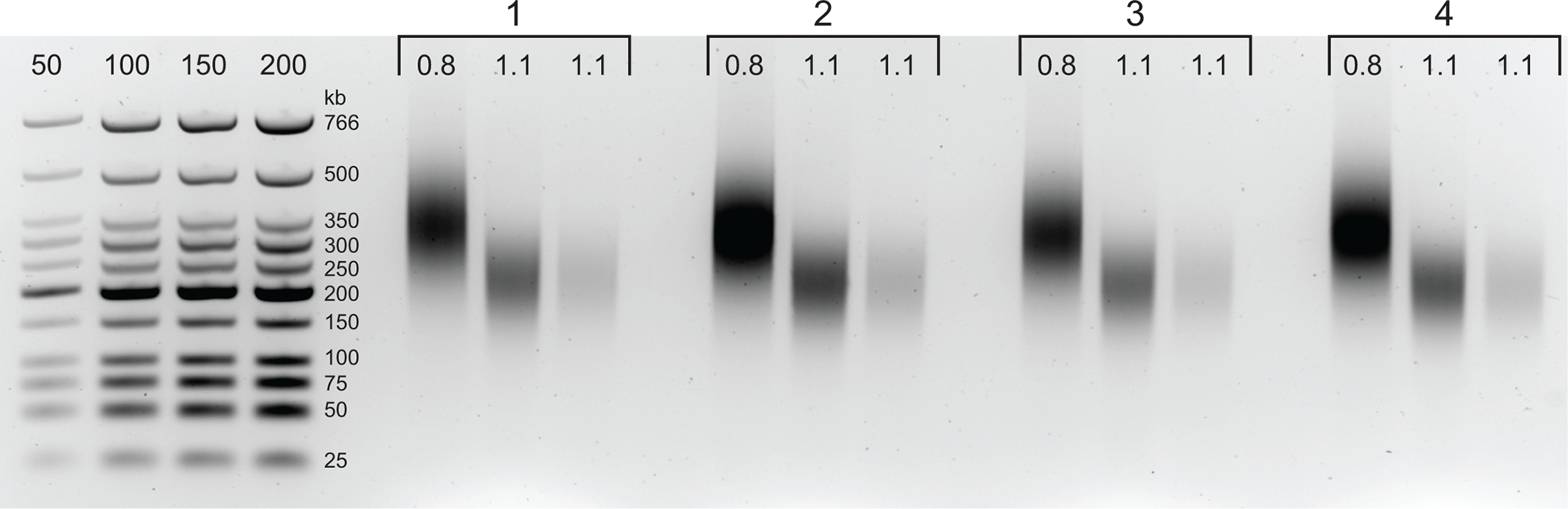
The Upper and Lower Fractions for four samples (numbered 1–4) of DpnII-DdeI Hi-C are shown. The first lane for each sample contains the Upper fraction, derived from a 0.8x magnetic beads mixture, and the second and third lanes contain a dilution of the Lower Fraction derived from a 1.1x magnetic beads mixture.
4. End repair
Prepare the end repair mix as in Table 7 (amount per reaction given).
Transfer the remaining 46 μL of eluted DNA from the Lower Fraction to PCR tubes and add 24 μL of the prepared end repair mix. Incubate in a PCR machine, as proposed in Table 7.
Once the program is completed, keep the samples at 4 °C until pull-down.
Table 7:
End Repair reagents
| Reagent | Amount |
| Ligation buffer (10×) | 7 μL |
| dNTP mix (2.5 mM) | 7 μL |
| T4 DNA polymerase (3 U/μL) | 2.5 μL |
| T4 polynucleotide kinase (10 U/μL) | 2.5 μL |
| Klenow DNA polymerase (5 U/μL) | 0.5 μL |
| Ultrapure MilliQ water | 4.5 μL |
| TOTAL | 24 μL |
| Temperature | Time |
| 20 ºC | 30 minutes |
| 75 ºC | 20 minutes |
| 4 ºC | hold |
5. Pull-down of biotinylated ligation products with streptavidin coated-beads
-
Determine the amount of streptavidin-coated beads for each library from the quantified size selection (step 3.3.19).
NOTE: These streptavidin-coated beads (10 mg/mL solution) can bind 20 μg of double-stranded DNA per mg beads (= 20 μg / 100 μL beads). Use 2 μL for each 1 μg of Hi-C DNA but no less than 10 μL.
Mix the streptavidin-coated beads and pipet the volume of beads needed for each library (calculated in the previous step) into individual 1.7 mL low binding tubes.
Resuspend the beads in 400 μL of Tween wash buffer (TWB; see recipe in Supplemental Table S1) and incubate for ~3 min at room temperature on a rotator (see instructions in step 3.3.4).
Separate the beads from the supernatant on the MPS for 1 min and remove the supernatant.
Wash the beads by pipetting another 400 μL of TWB.
Separate the beads from the supernatant on the MPS for 1 min and remove the supernatant.
Add 400 μL of 2x Binding Buffer (BB) (recipe in Supplemental Table S1) to the beads and resuspend. Additionally, add 330 μL of 1x TLE and the solution from End-repair (from step 3.4.3).
Incubate the samples for 15 min at room temperature while mixing on a rotator.
Separate the beads from the supernatant on the MPS for 1 min and remove the supernatant.
Add 400 μL of 1x BB to the beads and resuspend.
Separate the beads from the supernatant on the MPS for 1 min and remove the supernatant.
Add 100 μL of 1x TLE to wash the beads.
Separate the beads from the supernatant on the MPS for 1 min and remove the supernatant.
Finally, add 41 μL of 1x TLE to resuspend the beads.
6. A-tailing
Prepare the A-tailing mix as in Table 8.
Pipet the reactions into PCR tubes and incubate as in Table 8.
Place the PCR tubes on ice immediately after removal from thermocycler and transfer the contents to 1.7 mL low binding tubes.
Separate the beads from the supernatant on the MPS for 1 min and discard the supernatant.
Add 400 μL of 1x ligation buffer, diluted from 5x T4 DNA ligase buffer with ultrapure water.
Separate the beads from the supernatant on the MPS for 1 min, and then discard the supernatant.
Add 1x ligation buffer to a final volume of 40 μL.
Table 8:
A-tailing reagents and temperatures
| Reagent | Amount |
| Hi-C library on beads (step 3.5.14) | 41 μL |
| Pre-PCR Buffer (10×) | 5 μL |
| dATP (10 mM) | 1 μL |
| Klenow Fragment (3→5’ exo-; 5 U/μL) | 3 μL |
| TOTAL | 50 μL |
| Temperature | Time |
| 37 ºC | 30 minutes |
| 65 ºC | 20 minutes |
| 4 ºC | hold |
7. Annealing adapter oligos
-
Prepare adapter oligo stocks of 100 μM (Table 9).
NOTE: Order 250 nmoles of HPLC-purified oligos.
Anneal the adapters in PCR tubes as described in Table 9.
Use a PCR thermocycler to gradually ramp up the temperature at 0.5 °C/s to 97.5 °C. Hold at 97.5 °C for 2.5 min.
Use a PCR thermocycler to gradually ramp up temperature at 0.1 °C/s for 775 cycles (reaching 20 °C). Hold the temperature at 4 °C until further use.
Add 83 μL of 1x Annealing Buffer (recipe in Supplemental Table S1) to dilute the adapters to 15 μM. Store the adapters at −20 °C.
Table 9:
PCR Primers and Paired-End-adapter oligos with reagents annealing for annealing
| Oligo | Sequence |
| PE1.0 primer | CAAGCAGAAGACGGCATACGAGA*T |
| PE2.0 primer | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACG*A |
| Adapter-Index01 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACATCACGATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index02 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACCGATGTATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index03 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index04 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACTGACCAATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index05 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACACAGTGATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index06 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACGCCAATATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index07 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACCAGATCATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index08 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACACTTGAATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index09 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACGATCAGATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index10 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACTAGCTTATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index11 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACGGCTACATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-Index12 | /5PHOS/GATCGGAAGAGCACACGTCTGAACTCCAGTCACCTTGTAATCTCGTATGCCGTCTTCTGCTTG |
| Adapter-universal | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*T |
| Reagent | Amount |
| Adapter-Universal oligo | 20 μL |
| Adapter-Indexed oligo # | 20 μL |
| Annealing Buffer (5×) | 10 μL |
| TOTAL | 50 μL |
8. Sequencing adaptor ligation
Prepare the adapter ligation mix in a 1.7 mL low binding tube (Table 10).
Ligate for 2 h at room temperature.
Separate the beads from the supernatant on the MPS for 1 min and discard the supernatant.
Add 400 μL of TWB and pipet the beads up and down carefully before incubation on the rotator for 5 min at room temperature. Separate the beads from the supernatant on the MPS and repeat this step once more.
Separate the beads from the supernatant on the MPS (~1 min), discard the supernatant, and add 200 μL of 1x BB.
Separate the beads from the supernatant on the MPS (~1 min) and discard the supernatant.
Add 200 μL of 1x pre-PCR Buffer (from 10x; recipe in Supplemental Table S1) and transfer to a new 1.7 mL low binding tube.
Separate the beads from the supernatant on the MPS (~1 min) and discard the supernatant.
Add 20 μL of 1x pre-PCR Buffer and mix by pipetting.
Keep the tubes on ice while in use or store at 4 °C.
Table 10:
Adapter ligation reagents
| Reagent | Amount |
|---|---|
| Hi-C library on beads (step 3.6.7) | 40 μL |
| T4 ligation buffer (5×) | 2 μL |
| Sequencing adapters | 5 μL |
| T4 DNA ligase 1U/μL | 3 μL |
| TOTAL | 50 μL |
9. Optimization the PCR cycle number by titration
Set up 30 μL of master mix reactions per sample as in Table 11 (amount per reaction given).
Run the lowest number of cycles and take a 5 μL aliquot. Run additional 2–3 cycles on the remaining reaction before taking the next 5 μL aliquot. Repeat to collect four aliquots.
Use PCR parameters from Table 11 for each aliquot.
-
Add 5 μL of water and 2 μL of 6x dye to each 5 μL sample. Run on a 2% agarose TBE gel (recipe in Supplemental Table S1) with 25–150 ng of Low Molecular weight ladder. See Figure 4 for expected results.
NOTE: An optimal number of cycles for the final library PCR amplification [step 3.10] is the lowest number of cycles to get visible product on the gel minus one cycle.
Table 11:
PCR reagents and cycling parameters
| PCR reagents | ||
| Reagent | Titration (1 reaction) | Production (12 reactions) |
| PE1.0 primer (10 μM) | 0.3 μL | 3.6 μL |
| PE2.0 primer (10 μM) | 0.3 μL | 3.6 μL |
| Hi-C library on beads | 1.5 μL | 18 μL |
| dNTP (25 mM) | 0.3 μL | 3.6 μL |
| Pfu Ultra II Buffer (10×) | 3 μL | 36 μL |
| Pfu Ultra II HiFi DNA Polymerase | 0.5 μL | 6 μL |
| Ultrapure water | 24.1μL | 289.2 μL |
| TOTAL | 30 μL | 360 μL |
| PCR cycling parameters | ||
| Step | Temperature | Time |
| 1 | 98 ºC | 30 seconds |
| 2 | 98 ºC | 10 seconds |
| 3 | 65 ºC | 30 seconds |
| 4 | 72 ºC | 30 seconds |
| 5 | Steps 2–4 (N times) | |
| 6 | 72 ºC | 2 minutes |
Figure 4: Agarose gel with PCR titration results.
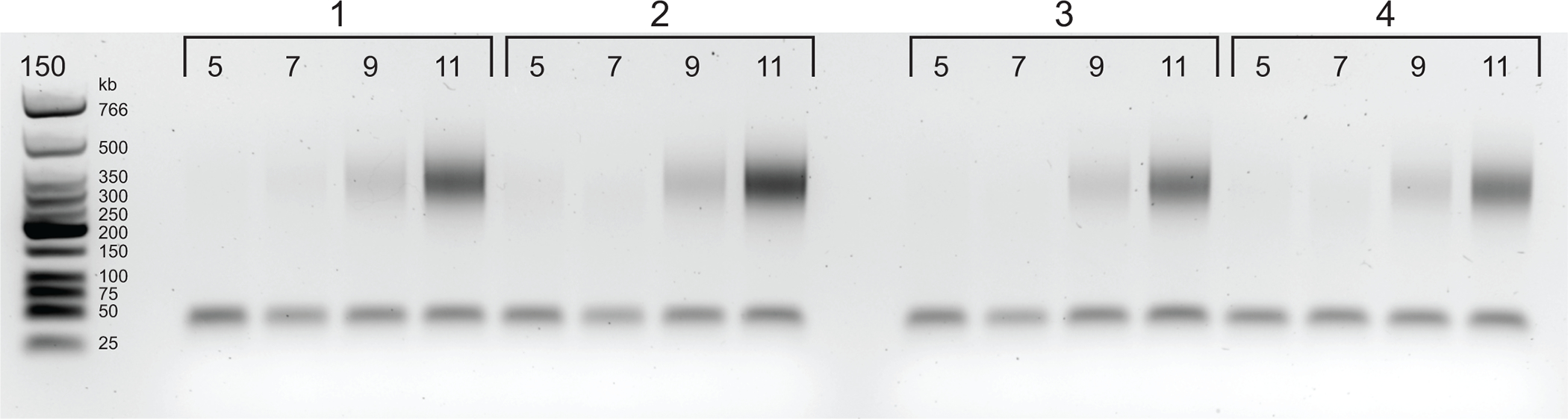
Starting from 5 cycles of PCR, samples are taken after every 2 cycles (5, 7, 9, and 11 cycles) for each of four libraries. Based on this figure, 6 cycles was chosen as the optimal cycle for each sample.
10. Final library PCR amplification
Set up 12 × 30 μL reactions to amplify each final library for sequencing as in Table 11.
Cycle the PCR reactions according to Table 11 after determining the number of cycles after PCR titration (step 3.9.3).
Once the PCR is complete, pool the replicate samples into a 1.7 mL low binding microfuge tube.
Place the tubes on the MPS and transfer the supernatant to a new 1.7 mL low binding microfuge tube.
-
Resuspend the leftover streptavidin-coated beads in 20 μL of 1x pre-PCR Buffer.
NOTE: This template can be reused when stored at 4 °C for days to weeks or long-term at −20 °C.
11. Removing primers with magnetic beads mixture
Use 1x TLE buffer (recipe in Supplemental Table S1) to adjust the volume from step 3.10.4 to exactly 360 μL.
To each sample, add 360 μL of the magnetic beads mixture and pipette up and down to mix.
On a rotator, mix the samples for 10 min at room temperature.
Separate the beads from the supernatant on the MPS at room temperature (3–5 min).
Wash the beads twice using 200 μL of 70% ethanol and reclaim the beads for 5 min on the MPS each time.
-
Quick-spin in a centrifuge and completely pipette off the ethanol. Dry the beads in air on the MPS to further evaporate the ethanol.
NOTE: The pellet should look like dark chocolate without cracking (can take ~10 min).
Add 30 μL of ultrapure water and resuspend to elute the DNA for 10 min at room temperature. Flick the tubes every 2 min to help mixing.
Separate the beads from the supernatant on the MPS for 5 min.
Collect the supernatant from each sample in a fresh 1.7 mL tube.
-
Run 1 μL of the library on a 2% agarose TBE (recipe in Supplemental Table S1) gel to obtain a fragment size distribution and to quantify the final library (Figure 5).
NOTE: ClaI digestion can only occur for DpnII-DpnII junctions and serves as a positive ligation control that should result in a lower fragment size distribution for the final library. Final libraries can be stored for a few days at 4 °C, long term at −20 °C, or immediately diluted and submitted for sequencing.
Figure 5: Final PCR products.
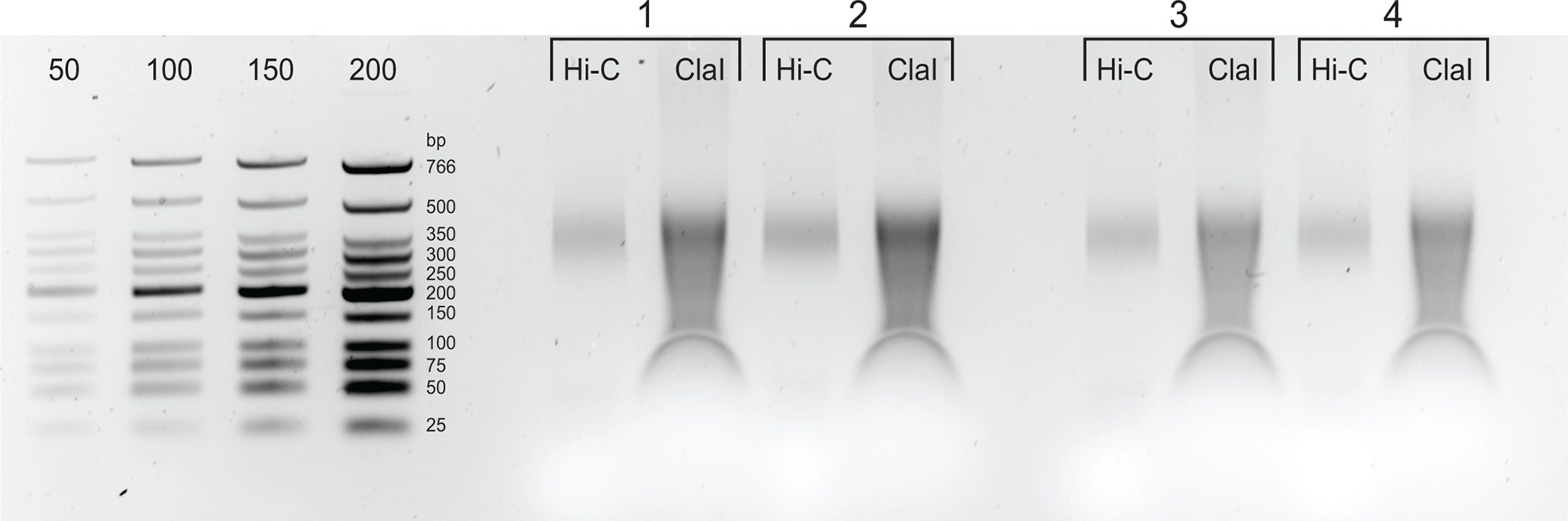
After cleaning and size-selection, PCR products (Hi-C) were loaded next to a ClaI-digested fraction of the same library (ClaI). ClaI-digested fragments indicate the presence of sought-after DpnII-DpnII ligations. Note that ClaI does not digest DpnII-DdeI junctions and, therefore, not all ligations will contribute to a size reduction from this restriction.
Representative Results
The figures in this manuscript were generated from a separate, replicate experiment of the one published previously by Lafontaine et al.21. After obtaining high-throughput sequencing data, the Open Chromatin Collective (Open2C: https://github.com/open2c) was used to process the Hi-C data. A similar pipeline can be found on the data portal of the 4D Nucleome project (https://data.4dnucleome.org/resources/data-analysis/hi_c-processing-pipeline). Briefly, the Nextflow pipeline distiller (https://github.com/open2c/distiller-nf) was implemented to (1) align the sequences of Hi-C molecules to the reference genome, (2) parse .sam alignment and form files with Hi-C pairs, (3) filter PCR duplicates, and (4) aggregate pairs into binned matrices of Hi-C interactions. These HDF5 formatted matrices, called coolers, can then be (1) viewed on a HiGlass server (https://higlass.io/) and (2) analyzed using a large set of open source computational tools present in the “cooltools” collection maintained by the Open Chromatin Collective (https://github.com/open2c/cooltools) to extract and quantify folding features such as compartments, TADs, and loops.
Some quality indicators of the Hi-C3.0 libraries can be assessed immediately after mapping read pairs to a reference genome, using a few simple metrics/indicators. First, typically ~50% of sequenced read pairs can be uniquely mapped for human cells. Due to the polymeric nature of chromosomes, most of these mapped reads (~60%–90%) represent interactions within a chromosome (cis), with interaction frequencies rapidly decaying with increasing genomic distance (distance-dependent decay). The distance-dependent decay can be visualized best in a “scaling plot”, which shows the contact probability (per chromosome arm) as a function of genomic distance. We found that the use of different crosslinkers and enzymes can alter the distance-dependent decay at long- and short-range distances17. The addition of DSG crosslinking increases the detectability of interactions at short distances when combined with enzymes such as Mnase and combinations of DpnII-DdeI that produce smaller fragments (Figure 6A).
Figure 6: Representative results of mapped and filtered Hi-C libraries.
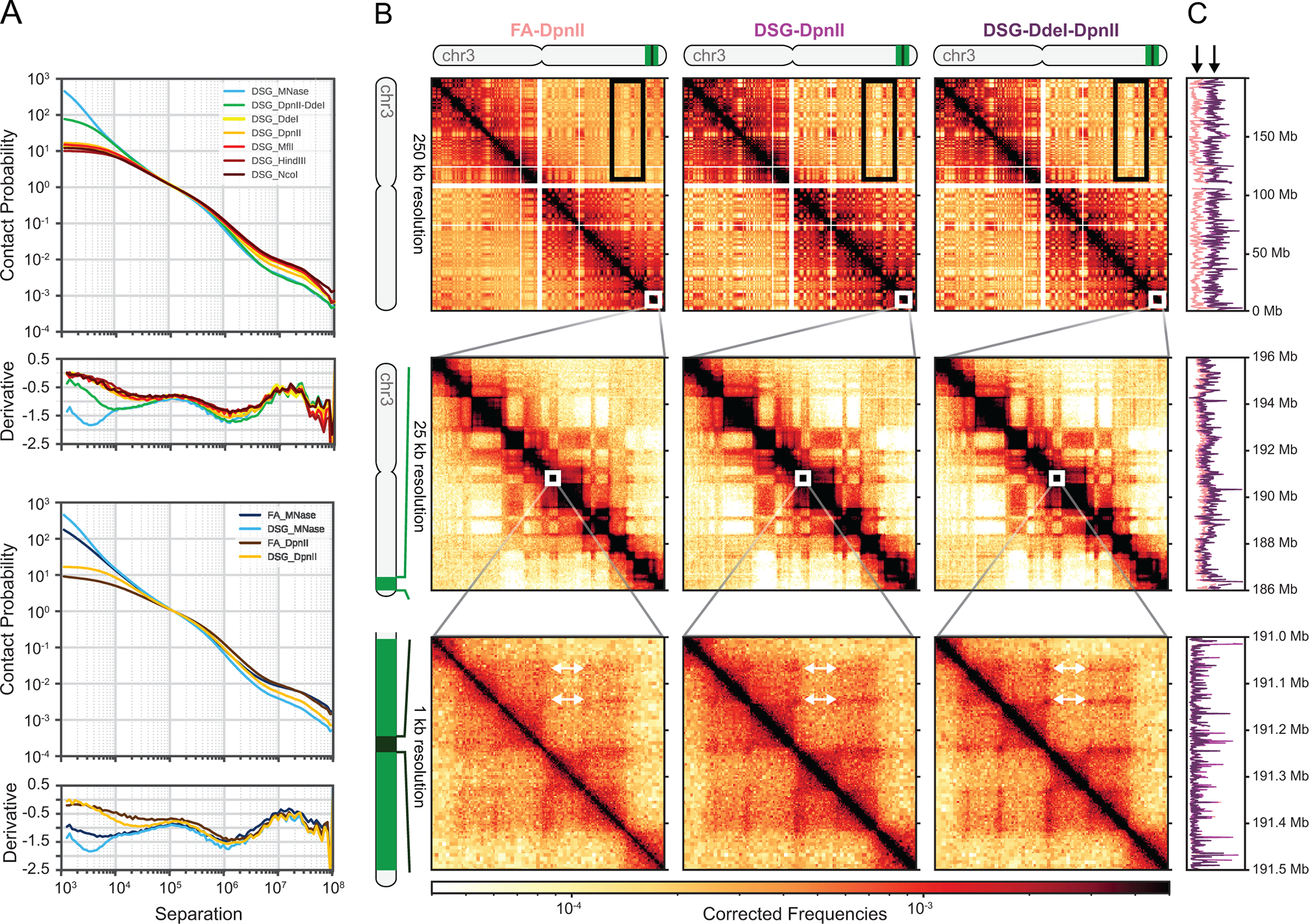
(A) Scaling plots with contact probability and its derivative for various enzymes, ordered by fragment length (top) and crosslinking with either FA or FA + DSG (bottom). Digestion with MNAse (microC) or DpnII-DdeI (Hi-C 3.0) significantly increases short range contacts (top) as does adding DSG to FA (bottom). (B) Columns show Hi-C heatmaps of DpnII digestion after just FA crosslinking and DpnII or DdeI digestion after FA+DSG crosslinking. White arrows show increasing strength of “dots” after DSG crosslinking and DdeI digestion, implying better detection of DNA loops. Rows show different parts of chromosome 3 at increasing resolution, aligning with panel C: top row: entire chromosome 3 (0–198,295,559 Mb); middle row: 186–196 Mb; bottom row: 191.0–191.5 Mb. (C) Coverage graphs for the regions depicted in A. Black arrows show the lower coverage (%cis reads) for FA-only crosslinking. Abbreviations: FA = formaldehyde; DSG = disuccinimidyl glutarate; chr = chromosome.
Distance-dependent decay can also be observed directly from 2D interaction matrices: interactions become more infrequent when located further away from the central diagonal (Figure 6B). Additionally, genomic folding features, such as compartments, TADs, and loops can be identified from Hi-C matrices and scaling plots as deviations from the general genome-wide average distance-dependent decay. Importantly, crosslinking with DSG in addition to FA decreases random ligations, which are unrestricted due to the polymer nature of chromosomes and, therefore, more likely to occur between chromosomes (in trans) (Figure 6C). Reducing random ligation leads to increased signal-to noise ratios, especially for inter-chromosomal and very long range (>10–50 Mb) intrachromosomal interactions.
Not all mapped reads are useful. A second quality indicator is the number of PCR duplicates. Exact duplicate reads are highly unlikely to occur by chance after ligation and sonication. Thus, such reads likely resulted from PCR amplification and need to be filtered out. Duplicates often arise when too many PCR cycles are required to amplify low complexity libraries. Generally, for Hi-C, most libraries only need 5–8 cycles of final PCR amplification, as determined by titration PCR (see step 3.9; Figure 4). However, libraries with sufficient complexity can be obtained even after 14 cycles of PCR amplification.
Another category of duplicate reads, so-called optical duplicates, can arise from the amplification process on Illumina sequencing platforms that use patterned flow cells (such as HiSeq4000). Optical duplicates are found from either overloading the flow cell, causing (large) clusters to be called two separate clusters, or from local reclustering of the original paired-end molecule after a first round of PCR. Because both types of optical duplicates are local, they can be identified and distinguished from PCR duplicates by their location on the flow cell. Whereas libraries with >15% PCR duplicates would need regeneration, libraries with optical duplicates can be reloaded after optimizing the loading process.
Discussion
Critical steps for cell handling
Although it is possible to use a lower number of input cells, this protocol has been optimized for ~5 × 106 cells per sequencing lane (~400 M reads) to ensure proper complexity after deep sequencing. Cells are best counted prior to fixation. For the generation of ultradeep libraries, we generally multiply the number of lanes (and cells) until the desired read-depth is reached. For optimal fixation, serum-containing medium should be replaced with PBS prior to FA fixation, and fixative solutions should be added immediately and without concentration gradients15,22. For cell harvesting, scraping is preferred over trypsinization, because the transition from a flatter to a spherical shape after trypsinization could affect the nuclear conformation. After the addition of DSG, loose and clumpy cell pellets are easily lost. Be careful when handling cells at this stage and add up to 0.05% BSA to decrease clumping.
Modifications to the method
This protocol was developed using human cells17. Yet, based on experience with chromosome conformation capture, this protocol should work for most eukaryotic cells. For a significantly lower input (~1 × 106 cells), we advise using half the volumes for the lysis and conformation capture procedures [steps 2.1–2.4]. This would also allow DNA isolation [step 2.5] to be performed in a tabletop centrifuge with 1.7 mL tubes, which could improve pelleting for low DNA concentrations. The quantification of DNA (step 2.6.6) will indicate how to proceed. For low amounts of isolated DNA (1–5 μg), we suggest skipping the size selection (step 3.3) and proceed with biotin removal after reducing the volume from 130 μL to ~45 μL with a CFU.
This protocol was developed specifically to ensure high-quality data after subsequent crosslinking with FA and DSG and digestion with DpnII and DdeI. However, alternative crosslinking strategies such as FA followed by EGS (ethylene glycol bis(succinimidyl succinate)), which is also used in ChIP-seq23 and ChIA-PET24, might work equally well17. Similarly, different enzyme combinations, such as DpnII and HinfI18 or MboI, MseI, and NlaIII19 can be used for digestion. When adapting enzyme combinations, be sure to use biotinylated nucleotides that can fill in the specific 5’ overhangs and use the most optimal buffers for each cocktail. DpnII comes with its own buffer and the enzyme manufacturer recommends a specific buffer for DdeI digestion. Yet, for the double-digestion with DpnII and DdeI in this protocol, Restriction Buffer is recommended because it is rated at 100% activity for both enzymes.
Troubleshooting conformation capture
The three key steps in chromosome conformation capture: crosslinking, digestion, and religation have all been performed before the results can be visualized on gel. To determine the quality of each of these three steps and discern where problems could have arisen, aliquots before (CI) and after digestion (DC) are taken and loaded on the gel along with the ligated Hi-C sample (Figure 2). This gel is used to determine the quality of the Hi-C sample and whether it will be worth continuing the protocol. Without the CI and DC, it is difficult to pinpoint potential suboptimal step(s). It is worth noting that suboptimal ligation could be due to a problem in the ligation itself, the fill-in, or a problem with crosslinking. To troubleshoot crosslinking, be sure not to use more than 1 × 107 cells per library and start with fresh crosslinking reagents and clean cells (i.e., rinsed with PBS). For ligation, make sure cells and ligation mixture are kept on ice. Add T4 DNA ligase just before the 4 h incubation at 16 °C and mix well.
Troubleshooting library preparation
If more than 10 PCR cycles are needed or no PCR product can be seen on gel after PCR titration (Figure 4), there are a few options to save the Hi-C sample. Working back from the PCR titration, the first option is to try the PCR again. If there is still not enough product, it is possible to attempt another round of A-tailing and adapter ligation (step 3.6) after washing the beads twice with 1x TLE buffer. After this additional A-tailing and adapter ligation, one can proceed to the PCR titration as before. If there is still no product, the last option is to resonicate the 0.8x fraction from step 3.3 and proceed from there.
Limitations and advantages of Hi-C3.0
It is important to realize that Hi-C is a population-based method that captures the average frequency of interactions between pairs of loci in the cell population. Some computational analyses are designed to disentangle combinations of conformations from a population25, but in principle, Hi-C is blind to differences between cells. Although it is possible to perform single-cell Hi-C26,27 and computational inferences can be made28, single-cell Hi-C is not suitable for obtaining ultrahigh-resolution 3C information. An additional limitation of Hi-C is that it only detects pairwise interactions. To detect multicontact interactions, one can either use frequent cutters combined with short-read sequencing (Illumina)16 or perform multicontact 3C29 or 4C30, using long-read sequencing from PacBio or Oxford Nanopore platforms. Hi-C derivatives to specifically detect contacts between and along sister chromatids have also been developed31,32.
Although Hi-C19 and Micro-C33 can be used to generate contact maps at subkilobase resolutions, both require a large amount of sequencing reads and this can become a costly undertaking. To get to similar or even higher resolution without the costs, enrichment for specific genomic regions (capture-C34) or specific protein interactions (ChiA-PET35, PLAC-seq36, Hi-ChIP37) can be applied. The strength and downside of these enrichment applications is that only a limited number of interactions are sampled. With such enrichments, the global aspect of Hi-C (and the option of global normalization) is lost.
Importance and potential applications of Hi-C3.0
This protocol was designed to enable high-resolution, ultradeep 3C while simultaneously detecting large-scale folding features such as TADs and compartments17 (Figure 6). This protocol starts with 5 × 106 cells per tube for each Hi-C library, which should be more than enough material to sequence one or two lanes on a flow cell to obtain up to 1 billion paired-end reads. For ultradeep sequencing, multiple tubes of 5 × 106 cells should be prepared, depending on the number of mapped reads and PCR duplicates. At the highest resolution (<1 kb), looping interactions are mostly found between CTCF sites, but promoter-enhancer interactions can also be detected. Readers can refer to Akgol Oksuz et al.17 for a detailed description of the data analysis.
Supplementary Material
Buffers and solutions.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/64001.
References
- 1.Dekker J, Rippe K, Dekker M, Kleckner N Capturing chromosome conformation. Science. 295 (5558), 1306–1311, doi: 10.1126/science.1067799 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Simonis M et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet. 38 (11), 1348–1354, doi: 10.1038/ng1896 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Dostie J et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 16 (10), 1299–1309, doi: 10.1101/gr.5571506 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289–293, doi: 10.1126/science.1181369 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadhouders R et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. doi: 10.1038/nprot.2013.018 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Kalhor R, Tjong H, Jayathilaka N, Alber F, Chen L Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat Biotechnol. 30 (1), 90–98, doi: 10.1038/nbt.2057 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell. 162 (1), 108–119, doi: 10.1016/j.cell.2015.05.048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh T-HS, Fudenberg G, Goloborodko A, Rando OJ Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. doi: 10.1038/nmeth.4025 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Denker A, de Laat W The second decade of 3C technologies: detailed insights into nuclear organization. Genes & development. 30 (12), 1357–82, doi: 10.1101/gad.281964.116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodley CD, Bertels F, Jones B, O’Sullivan JM Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genet Biol. 46 (11), 879–886, doi: 10.1016/j.fgb.2009.07.006 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665–1680, doi: 10.1016/j.cell.2014.11.021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alipour E, Marko JF Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research. 40 (22), 11202–11212, doi: 10.1093/nar/gks925 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lajoie BR, Dekker J, Kaplan N The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 72 (C), 65–75, doi: 10.1016/j.ymeth.2014.10.031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belaghzal H, Dekker J, Gibcus JH Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golloshi R, Sanders JT, McCord RP Iteratively improving Hi-C experiments one step at a time. Methods. 142, 47–58, doi: 10.1016/j.ymeth.2018.04.033 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Darrow EM et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. doi: 10.1073/pnas.1609643113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akgol Oksuz B et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18, 1046–1055, doi: 10.1038/s41592-021-01248-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghuryeid J et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol. 15 (8), doi: 10.1371/journal.pcbi.1007273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu H et al. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. 2021.10.03.462599, doi: 10.1101/2021.10.03.462599 (2021). [DOI] [Google Scholar]
- 20.Ramani V et al. Mapping 3D genome architecture through in situ DNase Hi-C. doi: 10.1038/nprot.2016.126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lafontaine DL, Yang L, Dekker J, Gibcus JH Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. doi: 10.1002/cpz1.198 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belton J-MM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58 (3), 268–276, doi: 10.1016/j.ymeth.2012.05.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truch J, Telenius J, Higgs DR, Gibbons RJ How to Tackle Challenging ChIP-Seq, with Long-Range Cross-Linking, Using ATRX as an Example. Methods in molecular biology (Clifton, N.J.). 1832, 105–130, doi: 10.1007/978-1-4939-8663-7_6 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Wang P et al. In situ Chromatin Interaction Analysis Using Paired-End Tag Sequencing. doi: 10.1002/cpz1.174 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tjong H et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization PNAS PLUS. 7, doi: 10.1073/pnas.1512577113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagano T et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547 (7661), 61–67, doi: 10.1038/nature23001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramani V et al. Massively multiplex single-cell Hi-C. Nature Methods. 14 (3), 263–266, doi: 10.1038/nmeth.4155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meng L, Wang C, Shi Y, Luo Q Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. doi: 10.1038/s41467-021-24662-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavares-Cadete F, Norouzi D, Dekker B, Liu Y, Dekker J Multi-contact 3C reveals that the human genome during interphase is largely not entangled. doi: 10.1038/s41594-020-0506-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vermeulen C et al. Multi-contact 4C: long-molecule sequencing of complex proximity ligation products to uncover local cooperative and competitive chromatin topologies. doi: 10.1038/s41596-019-0242-7. [DOI] [PubMed] [Google Scholar]
- 31.Oomen ME, Hedger AK, Watts JK, Dekker J Detecting chromatin interactions between and along sister chromatids with SisterC. Nature methods. 17 (10), 1002–1009, doi: 10.1038/S41592-020-0930-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitter M et al. Conformation of sister chromatids in the replicated human genome. Nature. 586 (7827), 139–144, doi: 10.1038/S41586-020-2744-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krietenstein N et al. Ultrastructural Details of Mammalian Chromosome Architecture. Molecular Cell. 78 (3), 554–565.e7, doi: 10.1016/j.molcel.2020.03.003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes JR et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genet. 46 (2), 205–212, doi: 10.1038/ng.2871 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Fullwood MJ et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462 (7269), 58–64, doi: 10.1038/nature08497 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang R et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345–1348, doi: 10.1038/CR.2016.137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature methods. doi: 10.1038/nmeth.3999 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Buffers and solutions.