

Summary
Inflammasomes are critical sentinels of the innate immune system that respond to threats to the host through recognition of distinct molecules, known as pathogen- or damage-associated molecular patterns (PAMPs/DAMPs), or disruptions of cellular homeostasis, referred to as homeostasis-altering molecular processes (HAMPs) or effector-triggered immunity (ETI). Several distinct proteins nucleate inflammasomes, including NLRP1, CARD8, NLRP3, NLRP6, NLRC4/NAIP, AIM2, pyrin, and caspases-4/5/11. This diverse array of sensors strengthens the inflammasome response through redundancy and plasticity. Here, we present an overview of these pathways, outlining mechanisms of inflammasome formation, subcellular regulation, and pyroptosis, and discuss the wide-reaching effects of inflammasomes in human disease.
Keywords: Inflammasome, IL-1β, IL-18, pyroptosis, NLRP1, CARD8, NLRP3, NLRP6, NLRC4, AIM2, Pyrin, Caspase-1, Caspase-4, Caspase-5, Caspase-11, GSDMD
Introduction
In 2001, a member of the nucleotide-binding domain (NBD), leucine-rich repeat (LRR)-containing (NLR) protein family was found to stimulate caspase-1 activation and cause cell death1. Later, another group described how another member of the NLR family exhibited a similar function when coexpressed with an adaptor protein referred to as ASC (short for Apoptosis-associated Speck-like protein containing a Caspase activation and recruitment domain) and caspase-1, causing both cell death and IL-1β release2. Simultaneously, a biochemical study provided foundational information by demonstrating the formation of an NLR containing a pyrin domain (PYD) 1 (NLRP1)-dependent supramolecular organizing center (SMOC)3 governing IL-1β processing, which the authors called the inflammasome4. Since these seminal studies, our understanding of the inflammasome and the pattern recognition receptors (PRRs) that nucleate its formation expanded substantially, though many aspects governing this critical innate immune signaling hub remain active areas of research.
Canonical inflammasomes are comprised of an activated PRR, ASC, and caspase-1. Several proteins nucleate inflammasome formation, including many in the NLR protein family, which contain a unique NBD referred to as NACHT, a name derived from proteins containing the domain: NLR family apoptosis inhibitory protein (NAIP), MHC class II transcription activator (CIITA), incompatibility locus protein from Podospora anserina (HET-E), and telomerase associated protein (TP-1)5. NLRs containing N-terminal PYDs are referred to as NLRPs, while NLRs with N-terminal caspase activation and recruitment domains (CARDs) are NLRCs. However, proteins outside of this family also nucleate inflammasomes. Inflammasome formation initiates autoproteolytic processing of caspase-1, instigating cleavage of its cellular substrates. These substrates include pro-IL-1β, pro-IL-18, and gasdermin D (GSDMD)6–11. After cleavage by caspase-1, the GSDMD N-terminus (GSDMD-NT) then oligomerizes to form pores in the plasma membrane, which mediate membrane depolarization and cell rupture. GSDMD pores also serve as conduits for IL-1β and IL-18 secretion, intrinsically linking release of these cytokines to disruption of cellular physiology12,13. Thus, inflammasome formation is a fundamental upheaval of cellular state that generates a robust pro-inflammatory response, serving as a key sentinel for pathogenic threats and damage to the host.
PRRs that nucleate inflammasomes respond to both pathogen-derived and host-derived molecules and activities. PRRs may bind directly to molecules from pathogens, or pathogen-associated molecular patterns (PAMPs), to initiate inflammasome formation. Examples of PAMPs that nucleate inflammasomes include bacterial products such as flagellin or lipopolysaccharide (LPS) or viral RNA. Similarly, PRRs may also detect the activities of pathogen effector proteins, a mechanism known as effector-triggered immunity (ETI)14. ETI was originally hypothesized as a mechanism of host defense in plants, in which a cellular R protein ‘guarded’ cellular targets of bacterial effectors to mediate disease resistance15. However, these principles are also observed in mammalian innate immunity, including the inflammasome response. Inflammasomes also form in response to host-derived signals. Disruption within the cellular environment is sufficient to activate inflammasome PRRs through recognition of homeostasis-altering molecular processes (HAMPs), such as disturbance of cellular ion gradients or the cytoskeleton16. Likewise, damage to the host causes production of damage-associated molecular patterns (DAMPs), such as extracellular DNA or ATP, which also initiate inflammasome responses3. While some PRRs are highly specific for certain PAMPs/DAMPs/HAMPs or effector activities, other PRRs respond to a broad range of stimuli, and specific molecular signals that consistently mediate receptor activation remain elusive. Thus, inflammasome nucleation is a process with features of both plasticity and specificity that enables the host cells to respond to a wide range of signals. Furthermore, several cellular processes regulate inflammasome formation and pyroptosis, which converge to generate a range of responses arising from inflammasome formation. Though space constraints prevent a description of the less studied inflammasomes, the mechanisms of the major inflammasomes and their consequences in disease are described in detail below.
Threat Recognition and Inflammasome Nucleation
Diverse PAMPs/DAMPs and threats to cellular homeostasis nucleate canonical and non-canonical inflammasomes (Table 1). Inflammasome formation typically depends on two distinct signals: (I) primes the cell for inflammasome activation and (II) acts directly on a receptor/sensor to prompt inflammasome formation and pyroptosis. Together, these signals I and II define the nature of inflammasome and pyroptotic responses, as detailed below.
Table 1:
Diversity and redundancy of inflammasome responses in different diseases and tissues
| Inflammasome | Cell Types | Disease and Pathogen Associations | Activation Mechanism(s) | |
|---|---|---|---|---|
| Molecules Detected (PAMPs/DAMPs) | Processes Detected (HAMPs/ETI) | |||
| NLRP1 | Murine myeloid cells30,55, human airway epithelial cells39,41, human keratinocytes25,48,237 | B. anthracis (murine)30, S. flexneri (murine)37, viral infections (e.g., HRV, SARS-CoV-2, SFV)39–41,49, vitiligo236, psoriasis237, atopic dermatitis238, MSPC48, FKLC48, JRRP239, arthritis240, dyskeratosis240 | Viral dsRNA49 | DPP8/9 inhibition25,43, ubiquitination by S. flexneri IpaH7.837, cleavage by B. anthracis LT34,35, cleavage by viral proteases39–41, ribotoxic and cellular stress50,51,53 |
| CARD8 | Human T cells26,46, human myeloid cells29 | HIV-142 | DPP8/9 inhibition25,29,46, cleavage by HIV-1 protease42 | |
| NLRP3 | Myeloid cells (monocytes, neutrophils, macrophages, DCs)63,138,299, oligodendroc ytes291, astrocytes67, endothelial cells216, retinal pigmented epithelial cells69 | Viral infections (e.g., IAV, SARS-CoV-2)62,63,248–250,256–258, bacterial infections (e.g., L. monocytogenes, L. pneumophila)62,63,144,179, fungal infections (e.g., C. albicans, A. fumigatus)245, atherosclerosis277, T2D281,285,286, obesity285,286,288, NAFLD288, chronic kidney disease289, AD291,292,297, AMD293,294, PD295,296, CAPS232, gout266, cancer70,270,273,276, RA269 | Nucleic acids (e.g., oxidized DNA, viral RNA)59,60,62, monosodium urate crystals266, cGAMP138, LPC67, alum, silica17, β-glucans, hyphae, mannan, zymosan, Aβ fibers297, α-synuclein295, ceramides286, saturated fatty acids285, oxLDL277, cholesterol crystals277 | K+ efflux (e.g., bacterial toxins, viral pore-forming proteins, P2X7R activation by ATP, GSDMD pore formation)56, Ca2+ flux56, Cl− efflux56, TGN disruption57,198, mitochondrial or lysosomal dysfunction57, loss of ER-endosome membrane contact sites205,206, metabolic shifts (e.g., fatty acid synthesis)58, hyperosmotic stress66 |
| NLRP6 | Myeloid cells85, colonic epithelial cells76–78, colonic goblet cells79 | Bacterial infections (e.g., L. monocytogenes, C. rodentium)85, viral infections (e.g., ECMV, MHV)84,87, colitis78, cancer76,77 | LTA85, viral RNA84,87 | |
| NAIP/NLRC4 | Myeloid cells88,89, intestinal epithelial cells110–114, astrocytes67, retinal pigmented epithelial cells69 | Bacterial infections (e.g., L. pneumophila, S. typhimurium, P. aeruginosa)88–90,93,94, AMD69, MAS241,242, colitis243, periodic fever syndromes242 | hNAIP: bacterial flagellin and T3SS needle and rod proteins95,97–100; mNAIP1: T3SS needle protein98; mNAIP2: T3SS rod protein95,96; mNAIP5/6: bacterial flagellin95,96; DDX17: SINE RNA69 LPC67 | Hyperosmotic stress66 |
| AIM2 | Myeloid cells116–118, keratinocytes126, T regulatory cells152 | Bacterial infections (e.g., L. monocytogenes, F. tularensis)125,127–129, viral infections (e.g., CMV, VACV, HPV, SARS-CoV-2)125,126,256, Plasmodium infection64, A. fumigatus infection245, radiation-induced tissue damage132, polyarthritis134, atherosclerosis135, cancer130,131,274, EAE149,152, colitis150,151, SLE153 | dsDNA116–118 | |
| Pyrin | Myeloid cells154,155 | FMF154,155, bacterial infection (e.g., Y. pestis, C. difficile)161,167,168,247 | Disruption of the actin cytoskeleton (e.g., bacterial toxins, loss of function mutation in Wdr1)161,166 | |
| Caspase-4/5/11 | Myeloid cells175, epithelial cells175, keratinocytes175, endothelial cells300 | Bacterial infections (e.g., L. pneumophila, S. typhimurium)176–179 | LPS175,180,181, oxPAPC193 | |
Abbreviations used: AD, Amyloid-associated Alzheimer’s disease; AIM2, absent in melanoma 2; AMD, Age-related macular degeneration; ATP, adenosine triphosphate; CAPS, Cryopyrin-associated periodic syndrome; CARD8, caspase-activation and recruitment domain-containing protein 8; CINCA, Chronic infantile neurological cutaneous and articular; cGAMP, cyclic GMP-AMP; CMV, cytomegalovirus; DAMP, damage-associated molecular pattern; DDX17, DEAD box helicase 17; dsDNA, double-stranded DNA; dsRNA, double-stranded RNA; DPP8/9, dipeptidyl peptidase 8/9; EAE, Experimental autoimmune encephalomyelitis; ECMV, encephalomyocarditis virus; ER, endoplasmic reticulum; ETI, effector-triggered immunity; FCAS, familial cold autoinflammatory syndrome; FKLC, Familial keratosis lichenoides chronica; FMF, Familial Mediterranean fever syndrome; GSDMD, gasdermin D; HAMP, homeostasis-altering molecular process; HPV, human papillomavirus; HRV, human rhinovirus; IAV, Influenza A virus; JRRP, Juvenile-onset recurrent respiratory papillomatosis; LPS, lipopolysaccharide; LPC, lysophosphatidylcholine; LT, lethal toxin; LTA, lipoteichoic acid; MAS, Macrophage activation syndrome; MHV, mouse hepatitis virus; MSPC, Multiple self-healing palmoplantar carcinoma; MWS, Muckle–Wells syndrome; NAFLD, Nonalcoholic fatty liver disease; NAIP, NLR family apoptosis inhibitory protein; NLRC4, NLR family CARD-containing protein 4; NLRP1, NLR family Pyrin-containing protein 1; NLRP3, NLR family Pyrin-containing protein 3; NLRP6, NLR family Pyrin-containing protein 6; NOMID, Neonatal-onset multisystem inflammatory disease; oxLDL, oxidized low density lipoprotein; oxPAPC, oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine; P2X7R, P2X7 receptor; PAMP, pathogen-associated molecular pattern; PD, Parkinson’s disease; RA, rheumatoid arthritis; SARS-CoV-2, severe acute respiratory coronavirus 2; SFV, Semliki Forest virus; SINE RNAs: Short interspersed nuclear element RNAs; SLE, systemic lupus erythematosus; T2D, Type 2 diabetes; T3SS, type 3 secretion system; TGN, trans Gogli Network; UV, Ultraviolet; VACV, vaccinia virus; Wdr1, WD repeat containing protein 1
Priming
Typically, prior to inflammasome formation, a stimulus rewires the cellular state to license pathway activation, a stimulus known as a signal I or priming signal. Early work demonstrated that transcriptional upregulation of inflammasome components and cytokines, through stimulation nuclear factor-κB (NF-κB) gene transcription by toll-like receptor (TLR) agonists and tumor necrosis factor (TNF), promoted NLRP3 activation17,18. Because of this, transcriptional upregulation is utilized as an indicator of potential inflammasome activity, yet transcriptional upregulation alone is insufficient for inflammasome activation. Furthermore, other work has demonstrated that transcriptional priming is not strictly required for inflammasome activation19,20 and has functions outside of transcriptional upregulation, including alteration of post-translational modifications (PTMs) on NLRP3 and other inflammasome components to promote activation21,22. As such, inflammasome activation is more dependent on priming in some contexts versus others. For instance, the inflammasome-secreted cytokines pro-IL-1β and pro-IL-18 have different expression patterns that influence sensitivity to priming signals. Pro-IL-18 is constitutively expressed in many cell types, while pro-IL-1β expression requires induction through TLR agonists, pro-inflammatory cytokines, and other inflammatory mediators23. Accordingly, IL-18 release is less dependent on priming than IL-1β24. Similarly, cell type and sensor identity play an important role in priming. While priming is necessary for NLRP3 inflammasome activation in murine bone marrow-derived macrophages (BMDMs) and other myeloid cell types, LPS stimulation of human monocytes leads to an alternative mechanism of inflammasome activation through direct interactions with TLR4 SMOCs, negating the requirement for additional priming signal20. Likewise, activation of the NLRP1 or CARD8 inflammasome by Val-boroPro (VbP) does not require a priming signal for IL-1β release25,26. Therefore, the requirements for priming of inflammasome pathways prior to activation are context-dependent and vary between different cell types, stimuli, and sensors.
NLRP1 and CARD8
NLRP1 was the first identified inflammasome-nucleating protein4 but only recent work expanded our understanding of this enigmatic sensor. Similar to other NLRPs, human NLRP1 (hNLRP1) has an N-terminal PYD, NACHT domain, and LRRs. Yet unlike other NLRs, these domains are followed by a function-to-find domain (FIIND) and a C-terminal CARD (Figure 1). The FIIND undergoes autoproteolytic cleavage that generates two fragments (N and C terminal), a process necessary for NLRP1 inflammasome responses27,28. In humans, CARD8 also contains this FIIND-CARD motif and nucleates inflammasomes25,29. Divergent from humans, mice have three paralogs of NLRP1, Nlrp1a, Nlrp1b, and Nlrp1c, that lack N-terminal PYDs and no paralog of CARD8. Expression and alleles of these paralogs vary between murine strains, as does their responsiveness to different stimuli.
Figure 1: Distinct and shared mechanisms of murine NLRP1b (mNLRP1b) and human NLRP1 (hNLRP1) and human CARD8.
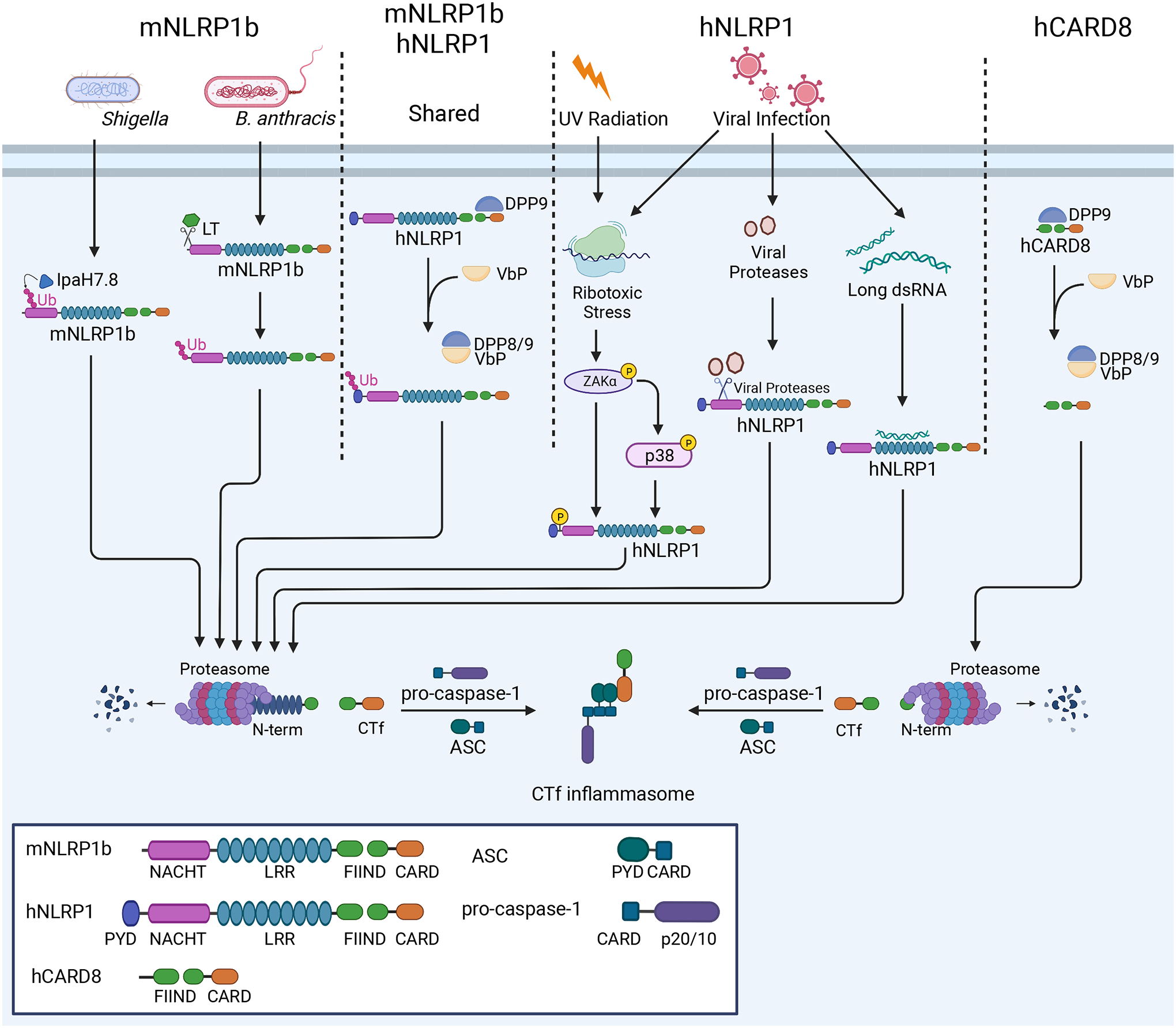
NLRP1 activation mechanisms vary between specific alleles in the murine population and between species. Some mNLRP1b alleles are sensitive to B. anthracis LT and S. flexneri effector IpaH7.8, which cleave or ubiquitinate mNLRP1b, respectively, instigating proteasomal degradation of the mNLRP1b N-terminus and formation of the mNLRP1b CTf inflammasome. Both mNLRP1b and hNLRP1 respond to DPP8/9 inhibition through Val-boroPro (VbP), leading to Ctf inflammasome formation. hNLRP1, but not mNLRP1b, responds to viral proteases, ribotoxic stress, and viral RNA to promote CTf inflammasome formation. hCARD8 responds to Val-boroPro similarly to mNLRP1b and hNLRP1 and forms CTf inflammasome.
Early work identified Bacillus anthracis Lethal Toxin (LT) as a mediator of pyroptosis in murine macrophages30, a process abrogated by proteasome inhibitors31 and inhibitors of N-end rule-mediated degradation32. Later, specific alleles33 of Nlrp1b were identified to mediate this response through cleavage of its N-terminus34,35 (Figure 1). This N-terminal cleavage event led to ubiquitination and N-end rule-mediated functional degradation of the NLRP1 N-terminus, freeing the C-terminal fragment (CTf) to mediate inflammasome formation36,37. Similar to cleavage by LT, the Shigella flexneri protein IpaH7.8 E3 ubiquitin ligase ubiquitinates the murine NLRP1b N terminus to promote its functional degradation and mediate CTf inflammasome nucleation37. Though it should be noted that neither of these bacterial products appear to affect hNLRP1 directly. However, we know that functional degradation of NLRP1 is a common mechanism of activation of this PRR across species (Figure 1). For example, the addition of artificial cleavage sites in the N-termini of murine NLRP1a (mNLRP1a), mNLRP1b, and hNLRP1 promotes inflammasome activation and IL-1β release38. More recently, several viral proteases were shown to cleave the N-terminus of hNLRP1, leading to its functional degradation and formation of a CTf inflammasome39–41. Similarly, the human immunodeficiency virus (HIV)-1 protease was shown to cleave CARD8, nucleating inflammasomes in T cells and myeloid cells42.
Though pathogen-derived NLRP1 stimuli differ across species and alleles, all functional NLRP1 alleles respond to the inhibition of dipeptidyl peptidases 8 and 9 (DPP8/9)25,43 (Figure 1). Cryo-EM studies revealed DPP9 forms an inhibitory, ternary (2:1) complex with both full-length human NLRP1 and the human NLRP1 CTf44,45. The CTf occupies the DPP9 active site, and upon DPP9 inhibition by Val-boroPro (VbP), the CTf is freed leading to inflammasome formation. The related PRR, CARD8, is similarly sequestered by DPP9 and forms an inflammasome upon DPP8/9 inhibitor treatment25,29,46 (Figure 1). Regulation of NLRP1 may be a critical function of DPP9, as spontaneous inflammation resulting from mutation or loss of DPP9 is rescued by loss of NLRP1 and other inflammasome components47.
Though CARD8 and hNLRP1 have seemingly redundant activation mechanisms, the dependence of the inflammasome response to VbP varies by cell type in humans. CARD8 mediates this response in T cells and myeloid cells26,29,46, while hNLRP1 mediates this response in epithelial cell types25. While mNLRP1b functions in myeloid cells, hNLRP1 has emerged as an epithelial innate immune sensor, forming inflammasomes in human keratinocytes25,48 and airway epithelial cells39,41. In epithelial cells, hNLRP1 detects diverse stimuli, including viral protease activity39–41, RNA virus replication49–51, ultraviolet (UV) radiation51, ribotoxic stress50,51, and shifts in cellular redox potential52. Detection of UV radiation and ribotoxic stress occur through activation of the p38/MAPK and ZAKα kinases, which activate hNLRP1 through hyperphosphorylation of the linker region between the hNLRP1 pyrin and NACHT domains (Figure 1)50,51. Activation of these kinases through cellular stress also mediates NLRP1 detection of the DNA mimetic poly(dA:dT)53. Though one report indicates the hNLRP1 N terminus directly binds viral RNA to initiate inflammasome formation49, many infectious stimuli of NLRP1 and CARD8 mediate responses by instigating functional degradation of the N-terminus, which can occur through its cleavage39,40 or ubiquitination36,37. This detection of pathogen activities, rather than binding specific PAMPs or DAMPs, is a form of ETI that has led some to speculate that NLRP1 may act as a guard protein15,54. In this model, the N-terminus of NLRP1 or CARD8 acts as a “decoy” or “tripwire” that when modified by pathogen effectors is degraded to release the inflammasome-nucleating CTf15,37,42,55. Similarly, NLRP1 and CARD8 detect changes in cellular homeostasis through recognition of HAMPs, including phosphorylation50,51 and DPP9 inhibition25,43,46. Thus, these sensors detect diverse signals to mediate inflammasome formation. Future work will shed further light on the mechanisms of NLRP1/CARD8 activation, their sensory capacity in tissue injury and infection, and the role that DPP9 plays in these processes. For more information on NLRP1 and CARD8, readers can refer to an excellent recent review55.
NLRP3
Highly expressed in myeloid cell types, NLRP3 is the best-studied inflammasome-nucleating sensor. While many different stimuli mediate NLRP3 activation, these mechanisms converge on changes in ion flux56, organelle dysfunction57, and shifts in metabolic flux58. While some work has linked NLRP3 activation to direct binding of nucleic acids59,60, a recent study indicated that NLRP3 is the primary mediator of the inflammasome response to double-stranded DNA (dsDNA) in human monocytes, activated through ion gradient depolarization initiated by cyclic GMP-AMP synthase (cGAS)- stimulator of interferon genes (STING)-driven lytic cell death61. Indeed, NLRP3 is reported to respond to the gamut of innate immune stimuli, recognizing HAMPs from cellular dysfunction, binding PAMPs/DAMPs in the form of nucleic acids, and mediating ETI by responding to pore-forming toxins that disrupt cellular ion gradients62,63. How such diverse signals converge on a singular mechanism to activate NLRP3 remains an active area of research. NLRP3 activation also has been observed as functioning in concert with other inflammasome PRRs, including AIM264,65 and NLRC466,67, in specific contexts. While some work indicates that NLRP3 directly interacts with other PRRs to create a dual inflammasomes68,69, these observations using microscopy and gene-deletion experiments may reflect pathway redundancy rather than direct molecular interaction. Regardless, these properties highlight NLRP3 as a critical mediator of inflammasome formation. For further discussion of NLRP3 biology, we refer interested readers to several superb reviews62,63,70,71.
Recently, several cryo-EM studies revealed the inactive and active structures of NLRP372–74. In its inactive form, NLRP3 forms a large multimeric cage73,74. Murine NLRP3 forms a dodecameric cage that shields the NLRP3 PYD to inhibit premature activation and promotes NLRP3 association with the trans-Golgi network (TGN)74, while inactive human NLRP3 forms a similar decameric structure73. In its active conformation, human NLRP3 forms a decameric disc structure, freeing the PYD to interact with ASC and nucleate the inflammasome72. Future work will determine how different stimuli initiate conformational changes between these active and inactive structures, which may offer new therapeutic targets.
NLRP6
First observed to stimulate caspase-1 and NF-κB activation in overexpression systems75, NLRP6 is reported to act as an inflammasome nucleating PRR in addition to other functions. NLRP6 is highly expressed in the colonic epithelium78–80 and the gastrointestinal immune environment76–78. Loss of NLRP6 was found to abrogate mucus production in colonic goblet cells79, promote the development of a colitogenic microbiota in dextran sodium sulfate (DSS)-induced colitis78, and maintain the colonic microbiota through a relationship with microbiota-derived metabolites80. However, this role in maintaining the colonic microbiota is debated, as others were unable to recapitulate these findings81–83. In response to pathogens, loss of NLRP6 increased murine susceptibility to viral infection84 and colonic tumorigenesis76,77. Findings in response to colonic bacterial infection show that NLRP6 protects the host against some bacterial infections79, while it reduces host defense against others85,86. These studies in animal models highlight the nuanced and context-dependent role of NLRP6 in various diseases.
Furthering this notion, mechanisms of NLRP6 activity vary between studies and ligands described (Figure 2). Some report a suppressive role for NLRP6 in dampening NF-κB and MAPK responses to Listeria monocytogenes infection86. However, other phenotypes are dependent also on ASC, caspase-1, and IL-18, implicating an NLRP6 inflammasome79,80,85. In Gram-positive bacterial infection, this response is mediated by lipoteichoic acid (LTA) binding to NLRP6 to nucleate inflammasomes85,87. However, in RNA virus infection, NLRP6 detects viral RNA through an interaction with the RNA helicase DHX15 to promote IFN responses84,87. A recent study suggested that NLRP6 may accomplish both of these activities through liquid-liquid phase separation (LLPS)87. In this work, NLRP6 bound dsRNA or LTA directly to promote LLPS and form liquid condensates with ASC or DHX15 with the former driving inflammasome formation and the latter promoting interferon responses87. Therefore, more research is necessary to clarify the molecular cues driving the wide range of reported actions of the NLRP6 inflammasome and other SMOCs it nucleates.
Figure 2: Mechanisms of NLRP6 and NLRC4 inflammasome activation.
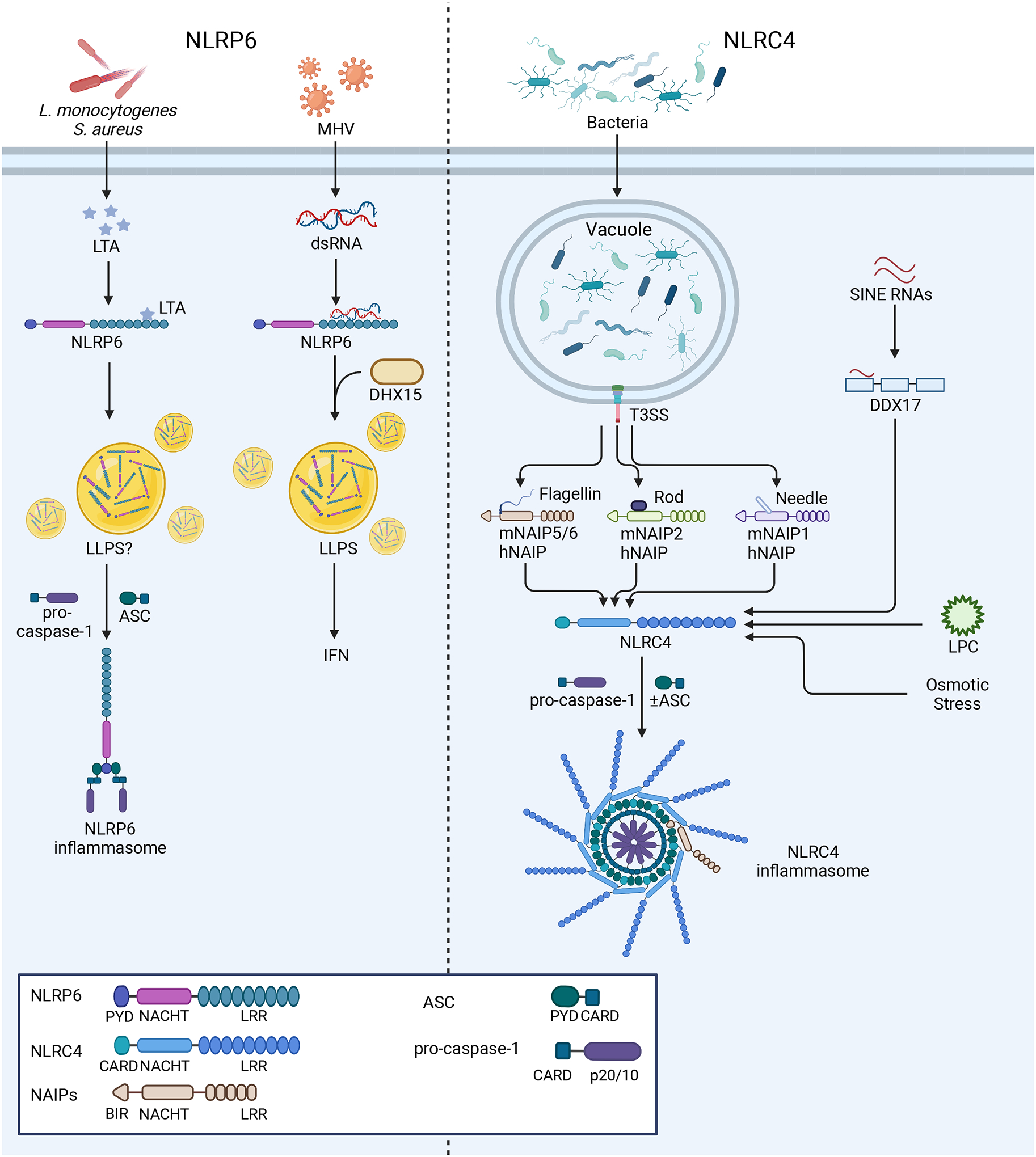
Left: NLRP6 responds to both LTA from bacterial infections and viral RNA from mouse hepatitis virus (MHV). In response to LTA, NLRP6 nucleates an inflammasome, while the response to RNA is mediated by DHX15 and promotes IFN production. LLPS is reported to govern both of these responses. Right: NLRC4 and hNAIP or mNAIP1–6 form inflammasomes upon recognition of flagellin or T3SS components from bacterial infections, as described in the text. More recent reports suggest that NLRC4 may also recognize sterile stimuli, including SINE RNAs through DDX17 activity, osmotic stress, and LPC. NLRC4 inflammasomes may form with or without ASC.
NLRC4/NAIP
NLRC4 was the first inflammasome sensor demonstrated to cause both caspase-1 activation and cell death1 and plays a key role in the innate immune response to bacterial pathogens88–90. Early work demonstrated that loss of NLRC4 blocked cell death following Salmonella typhimurium infection88 and identified that cytosolic delivery of bacterial flagellin was sufficient to initiate this response91,92. However, NLRC4 does not bind flagellin directly. Rather, it relies on the sensory activity of NAIPs, specifically Naip1-6 in mice and a single NAIP in humans. Murine Naip2 and Naip5 were initially identified as candidate genes for Legionella pneumophila pathogenesis or resistance93, a function later found to also depend on NLRC494. Murine NAIP5 (mNAIP5), mNAIP6, and human NAIP (hNAIP) bind bacterial flagellin in the cytosol95–97, which then interacts with NLRC4 to form an inflammasome. In addition to flagellin, NAIPs recognize components of bacterial type III secretion systems (T3SS). Individual murine NAIPs recognize distinct T3SS protein ligands, namely the rod protein, detected by mNAIP295,96, and needle protein, detected by mNAIP198. However, hNAIP recognizes both the T3SS rod and needle proteins in addition to flagellin95,98–100(Figure 2). Cryo-EM structures revealed how NAIPs nucleate the assembly of the NLRC4 inflammasome. Multiple groups showed that a single copy of mNAIP2 bound to the S. typhimurium rod protein nucleates a multi-subunit disk structure containing a single mNAIP2 and 10 monomers of NLRC4101,102, corroborating earlier EM work that indicated a similar arrangement for the mNAIP5/NLRC4 inflammasome103. These higher-order assemblages leave the CARD of NLRC4 free to recruit caspase-1 and promote its oligomerization, demonstrating how these inflammasomes can form in the absence of ASC. While ASC is not strictly required for NLRC4 inflammasome formation, loss of ASC leads to inefficient caspase-1 cleavage and diminished IL-1β release, indicating that ASC enhances these responses104,105. Some work has implicated phosphorylation of NLRC4 at serine 533 as a requirement for its activation106, while the structure of the autoinhibited, inactive NLRC4 also contains this PTM107. Yet, others have not observed this requirement108,109, leaving the role of this PTM unclear106,107.
While expressed in macrophages and other myeloid cell types, NLRC4 is also expressed in intestinal epithelial cells (IECs). In murine models of infection, loss of NLRC4 expression in the epithelium but not the bone marrow led to a loss of control of S. typhimurium and Citrobacter rodentium infection110,111. In the intestinal epithelium, NLRC4 promotes pyroptosis and IL-18 release in response to S. typhimurium and mediates cell death and expulsion of infected IECs111,112. Caspase-1 was dispensable in this process, which could also utilize caspase-8112. Similarly, NLRC4 prevented Shigella colonization and mediated infected cell expulsion in IEC monolayer cultures, and loss of NLRC4 in the intestinal epithelium led to shigellosis in mice113,114. Taken together, these studies point toward a pivotal role for NLRC4 in the innate immune and cell death responses in the intestinal epithelium.
While most work focused on the response to bacterial pathogens, recent studies implicate NLRC4 in mechanisms of sterile inflammation. Many different cell types form inflammasomes in response to DAMPs via NLRC4, including lysophosphatidylcholine (LPC) in microglia and astrocytes67, hyperosmotic stress in BMDMs66, and short interspersed nuclear element (SINE) RNAs in retinal pigmented epithelial cells69. Unlike other NLRC4 inflammasome assemblies, the RNA helicase DDX17 was reported to mediate NLRC4 response to SINE RNAs, rather than NAIPs, suggesting the NLRC4 inflammasome may interface with multiple PRRs to mediate inflammasome responses. Notably, all of these DAMPs activated both NLRC4 and NLRP366,67,69. Understanding the molecular requirements of NLRC4 activation, the extent of signals that activate NLRC4, and the role of NLRC4 in different cell types will be important areas for further research.
AIM2
AIM2 (absent in melanoma 2) was first described as a tumor suppressor115 but was later found to bind cytosolic dsDNA to nucleate an inflammasome in human cell lines and murine BMDMs116–118. AIM2 is a member of the IFN-inducible HIN-200 family of proteins with an N-terminal PYD and a C-terminal HIN-200 domain. Prior to DNA binding, AIM2 exists in an autoinhibited conformation with interactions between the PYD and HIN-200 domain119. The HIN-200 domain binds dsDNA in a sequence-independent manner but requires a minimal length reported between 70–80bp for recognition120,121. Structural data reveals that the AIM2 HIN-200 domain oligomerizes along dsDNA121,122, which then frees the AIM2 PYD to form helical assemblies that bind ASC to form an inflammasome123,124. Since its identification as a DNA sensor, AIM2 was found to mediate inflammasome responses to bacterial and viral pathogens, including cytomegalovirus (CMV)125, vaccinia virus (VACV)125, human papillomavirus (HPV)126, Francisella tularensis125,127, and L. monocytogenes125,128,129 (Figure 3). AIM2 also detects self-derived DNA with noted roles in the immune response to tumorigenesis130,131, radiation-induced tissue damage132, and the DNA-damage response in mouse models of neurodevelopment133, polyarthritis134, and atherosclerosis135.
Figure 3: Nucleation of the AIM2, pyrin, and non-canonical inflammasomes.
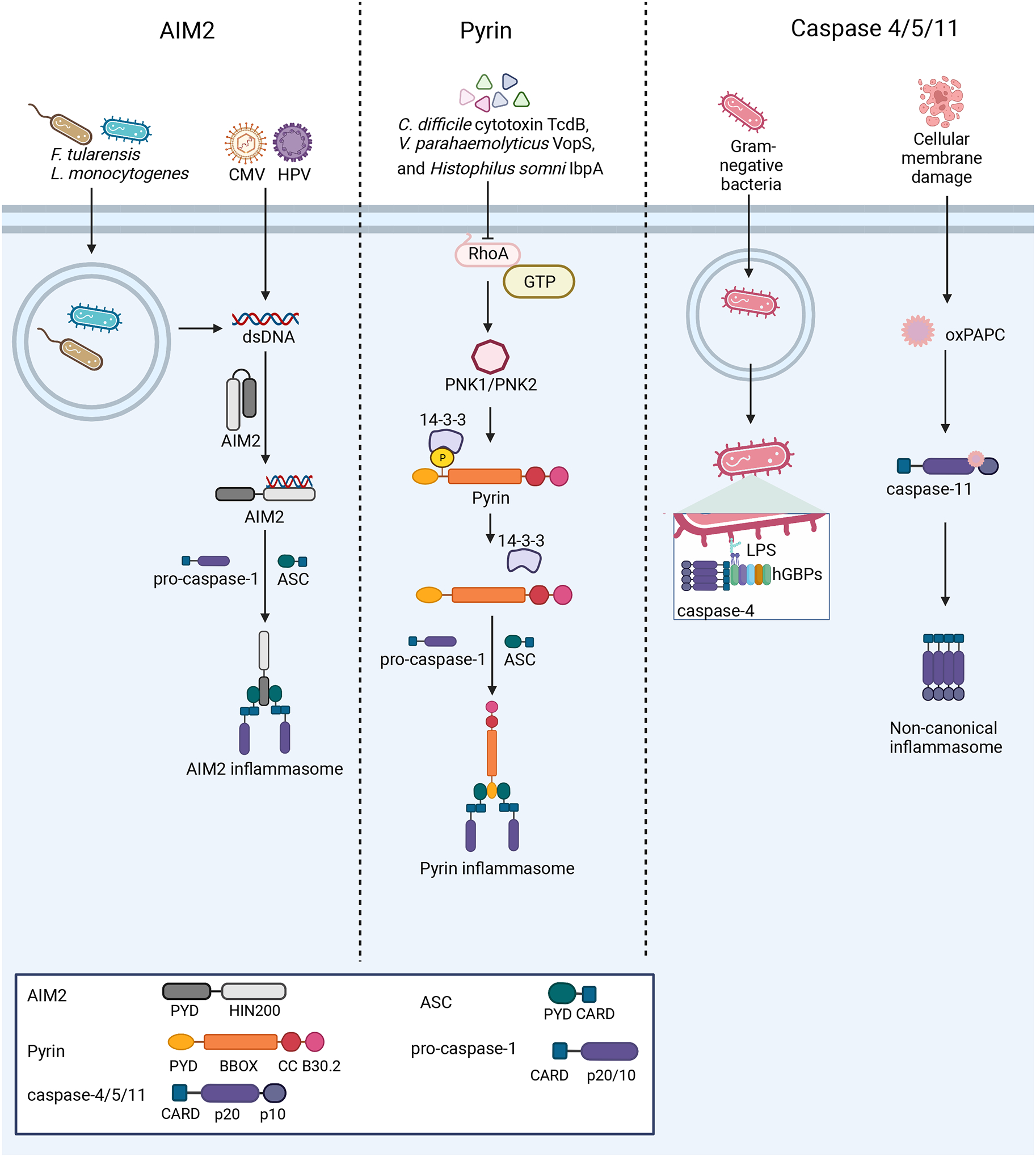
Left: AIM2 detects dsDNA from a variety of sources, including bacterial and viral infections. Upon dsDNA binding, AIM2 oligomerizes along the dsDNA and interacts with ASC to form the inflammasome. Center: At steady-state, pyrin is kept in an inactive conformation through phosphorylation by PKN1/2 and interaction of 14-3-3 proteins with this PTM. Several bacterial toxins activate the pyrin inflammasome through inactivation of the RhoA GTPase, which in turn causes pyrin to be dephosphorylated and nucleate inflammasome formation. Right: Murine caspase-11 and human caspases-4/5 act as PRRs. Guanylate-binding proteins (GBPs) assemble on the surface of Gram-negative bacteria after escape from the vacuole, exposing Lipid A moieties from LPS. This GBP coat then serves as a platform for caspase-4 recruitment and activation186–188. The binding of LPS or oxPAPC by these caspases promotes their activation and downstream inflammasome formation.
Crosstalk with other innate immune sensors and cellular homeostasis regulate AIM2 activity. Notably, AIM2 interfaces with another DNA sensing pathway, the cGAS-STING pathway, which initiates a type I IFN response to cytosolic dsDNA136. cGAS-STING signaling can promote AIM2 inflammasome priming through IFN-stimulated upregulation of AIM2137,138. Conversely, AIM2 inflammasomes inhibit cGAS-STING signaling139–142. Mechanistically, cGAS-STING signaling is abrogated by AIM2 inflammasomes through caspase-1-mediated cleavage of cGAS140 and inhibition of cGAS activity through disruption of intracellular K+ levels caused by GSDMD pore formation139. AIM2 also coordinates with NLRP3, and multiple reports indicate both proteins are required for inflammasome responses in many contexts, including bacterial infection143,144, Plasmodium infection64, and STING agonist stimulation138. AIM2 is also implicated in a hybrid cell death pathway consisting of simultaneous activation of pyroptosis, apoptosis, and necroptosis, called PANoptosis145, in which AIM2 is reported to form a complex with pyrin and ZBP1 to execute this form of inflammatory cell death146. Furthermore, changes in cellular metabolism govern AIM2 responses. For instance, excessive synthesis of cholesterol can trigger mitochondrial DNA (mtDNA) release and AIM2 activation147. Similarly, another study reported that upregulation of the M2 isoform of pyruvate kinase to promote glycolysis in macrophages initiates both AIM2 and NLRP3 inflammasome responses148.
AIM2 has roles in immunity outside of the inflammasome response. In the murine azoxymethane/DSS (AOM/DSS) model of colorectal cancer, AIM2 loss led to a greater tumor burden than the loss of ASC, and AIM2 was found to suppress AKT activation through interaction with DNA-dependent protein kinase (DNA-PK) to control tumor development130 and suppress intestinal stem cell proliferation131. Another group found AIM2 suppression of DNA-PK-AKT signaling in microglia decreased inflammation in experimental autoimmune encephalomyelitis (EAE)149. However, two other groups described conventional DNA and inflammasome-mediated roles of AIM2 in murine DSS colitis150,151. AIM2 also enhances the stability of T regulatory cells in EAE through suppression of AKT-mTOR signaling and alteration of immune metabolism152. Furthermore, AIM2 was shown to bind neutrophil extracellular traps, leading to DNase-resistant nucleoprotein fibers that can serve as autoantigens in systemic lupus erythematosus153. These studies suggest that AIM2 has inflammasome-independent functions and may function as a rheostat in various inflammatory processes. Future work will determine how both inflammasome-dependent and -independent functions of AIM2 inform the immune response and its relevance in human biology.
Pyrin
Pyrin is encoded by MEFV and was first identified as the genetic determinant of Familial Mediterranean fever (FMF)154,155. Pyrin was found to interact with ASC in a yeast-two hybrid screen156 and is considered a member of the tripartite motif-containing (TRIM) family of proteins. However, unlike canonical TRIM proteins, pyrin has an N-terminal PYD rather than a RING E3 ubiquitin ligase domain. This N-terminal PYD mediates its interaction with ASC, while the following zinc finger (bBox), coiled-coil (CC), and C-terminal B30.2/SPRY domains regulate Pyrin activation. Though its role in inflammasome formation was debated for many years157–159, missense mutations linked to FMF in pyrin led to gain-of-function inflammasome activation in mice independent of NLRP3160. Later, pyrin was shown to sense inactivation of the RhoA GTPase mediated by bacterial toxins161, solidifying its role as an inflammasome-nucleating innate immune sensor.
Pyrin associates with actin filaments162 and senses perturbations in the actin cytoskeleton through its relationship with RhoA. RhoA and other Rho GTPases regulate actin and microtubule dynamics to control cell shape, motility, and polarity163. Intracellular pathogens may disrupt these cytoskeletal dynamics to promote replication, as observed with bacterial toxins161, activating pyrin inflammasome formation. A loss-of-function mutation in Wdr1, an actin depolymerization cofactor, led to spontaneous pyrin inflammasome activation164. Mechanistically, pyrin is maintained in an inactive state through phosphorylation of Ser208/242 in humans and Ser205/241 in mice by the RhoA-dependent kinases PKN1/2165,166. 14-3-3 proteins bind these PTMs, placing pyrin in an inactive state164,166. Disruption of RhoA function leads to pyrin dephosphorylation and loss of association with 14-3-3 proteins. However, pathogens triggering this response may also evade it, as different Yersinia species inhibit pyrin detection through YopM-mediated promotion of PKN1/2-mediated phosphorylation167,168. Pyrin also associates with microtubules166. Recently, a study demonstrated that microtubule-dependent trafficking of pyrin through HDAC6 activity was necessary for inflammasome formation169, highlighting the complex relationship between pyrin and the cytoskeleton. Through the detection of changes in RhoA function, pyrin initiates inflammasome formation upon disruption of cellular homeostasis, sensing HAMPs and mediating ETI (Figure 3). Future work will determine the range of pathogens and cytoskeletal disruptions sensed by this critical innate immune sensor.
Non-canonical caspases
Originally identified as caspase-1 homologs170–173, human caspases-4 and −5 and murine caspase-11 are unique among the inflammasome sensors, acting as both PRRs and effectors. Similar to other caspases, caspases-4/5 and caspase-11 are zymogens activated through autoproteolysis and trigger cell death in overexpression systems170,171,173,174. However, rather than binding ASC, the CARDs of these caspases directly bind LPS to mediate their oligomerization and autoproteolytic cleavage175, forming a non-canonical inflammasome. These PRRs play an important role in the immune response to bacteria176–179 and intracellular LPS175,180,181 (Figure 3).
Non-canonical caspases have distinct requirements for activation. Unlike caspase-1, caspase-11 expression is induced by type I IFNs182,183, and loss of type I IFN signaling diminishes caspase-11 activation during bacterial infection184. This response is also critical for caspase-11 access to its ligand, because IFN-inducible GTPases that lyse pathogen-containing vacuoles mediate caspase-11 sensing of LPS in the cytosol185. Indeed, access to ligands is critical for non-canonical inflammasome formation. Human guanylate binding protein-1 (hGBP1) facilitates caspase-4 LPS detection through a “detergent-like” mechanism that exposes lipid A from the outer membrane of gram-negative bacteria186–188. Another mechanism of cytosolic LPS entry is through host proteins, as uptake of high-mobility group box 1 (HMGB1)-bound LPS mediates caspase-11 activation189. Furthermore, caspases-4/5/11 do not process proIL-1β into its mature form177,190 but cause cell death through cleavage of GSDMD191,192. Because caspases-4/5/11 do not directly cleave IL-1β, the release of this cytokine depends on subsequent caspase-1 activation. Mechanistically, caspase-11 induces K+ efflux, through cleavage of GSDMD and subsequent pore formation, to in turn activate NLRP3 and IL-1β release6,192. Understanding the complex relationship between NLRP3 and non-canonical inflammasome activation is an important topic for further research.
Non-canonical caspases have PRR activity beyond LPS detection. Caspase-11 was shown to bind oxidized phospholipids derived from 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (PAPC) (oxPAPC), a class of DAMPs released from dying cells, to elicit IL-1β release and pyroptosis193. However, another group reported that oxPAPC binds caspase-4 and caspase-11 to inhibit noncanonical inflammasome activation194. These discrepancies may be due to differences between specific oxPAPCs moieties195, and future work is needed to resolve these conflicting reports. Furthermore, caspase-11 activation is linked to actin cytoskeleton dynamics. Loss of caspase-11 led to defects in cellular migration, through interaction with actin-interacting protein 1 (Aip1)196. Another study observed that caspase-11 promoted fusion of the Legionella pneumophila-containing phagosome with the lysosome through Aip1-mediated remodeling of filamentous actin197. As a whole, these studies suggest that the non-canonical inflammasomes may have roles outside of LPS detection.
Cellular Orchestration of Inflammasomes and Pyroptosis
Following inflammasome formation, caspase-1 activation mediates the processing and release of pro-IL-1β and pro-IL-18 and pyroptotic cell death. Several regulatory events govern each step of inflammasome formation and execution of cell death following activation of an inflammasome sensor, as described below.
Cellular coordination of inflammasomes with organelles
Inflammasomes form a singular, macromolecular structure within the cell following PRR activation, and several cellular events coordinate the formation of these SMOCs to potentiate cytokine release and pyroptotic cell death. While these regulatory events are studied mostly in the context of the NLRP3 inflammasome (Figure 4), cell biology may govern many aspects of other receptors as they condense into singular inflammasomes.
Figure 4: Cellular orchestration of the NLRP3 inflammasome.
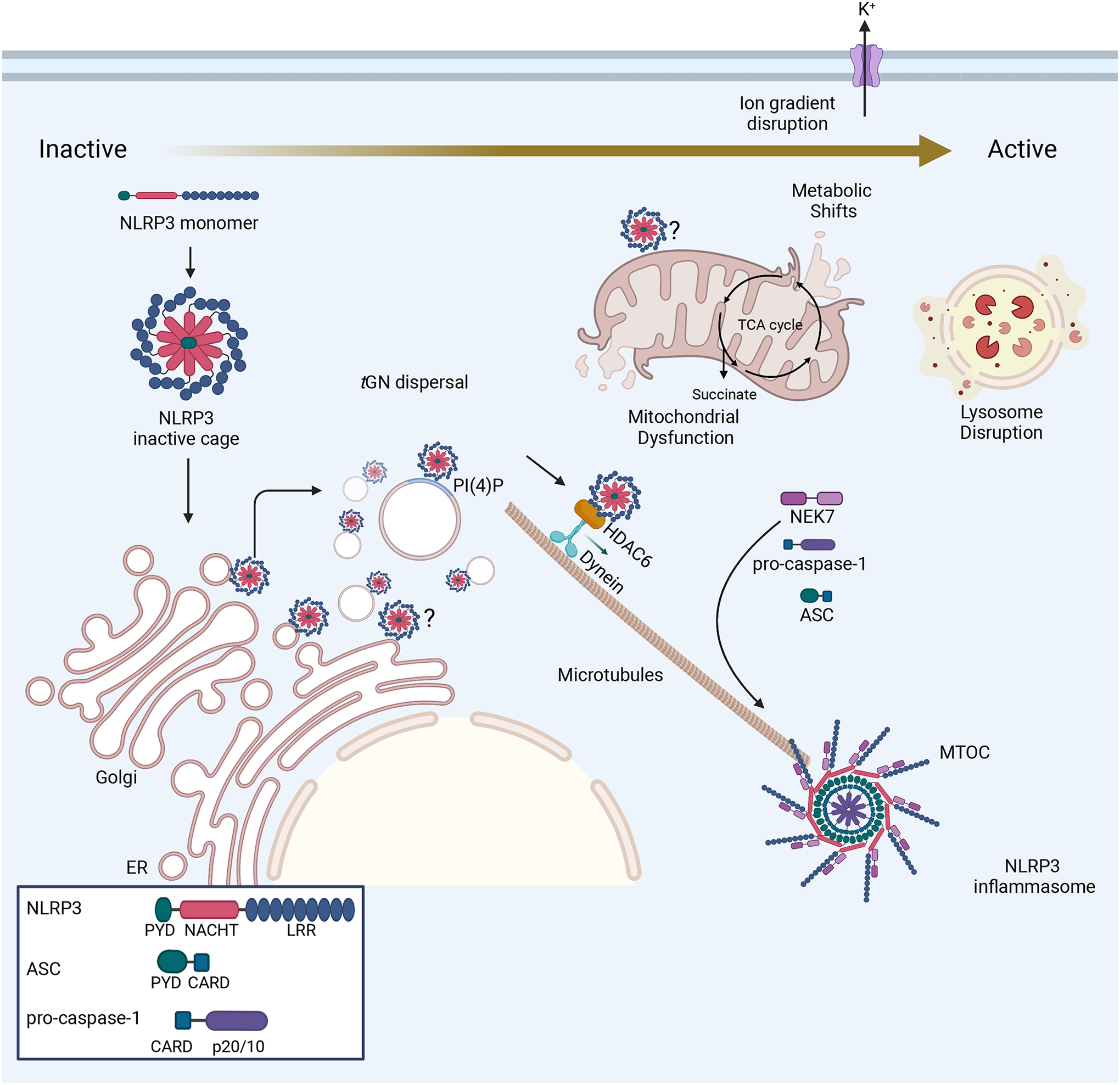
Prior to activation, NLRP3 exists in an inactivate cage conformation and is reported to associate with many different organelles, including the mitochondria and the ER. Recent work revealed that NLRP3 associates with the TGN through electrostatic interactions with the membrane component lipid PI(4)P74,198, and a new study describes how these PI(4)P+ vesicles are endosomal in origin, displaying markers of early endosomes and the TGN205,206. Upon its activation through ion gradient disruption, organelle dysfunction, or metabolic shifts, NLRP3 mediates the dispersal of the TGN and traffics along microtubules in a manner dependent on HDAC6 and dynein. At the MTOC, NLRP3 associates with NEK7, and NLRP3 monomers assemble into a decameric inflammasome72.
Prior to activation, NLRP3 preferentially associates with cell membranes through electrostatic interactions with membrane phospholipids74,198,199. NLRP3 associates with multiple organelles, including the endoplasmic reticulum (ER)200, the mitochondria199,201–203 and the TGN74,198,204. Likewise, NLRP3 stimuli, including bacterial toxins and viral infection, are reported to cause the dispersal of the TGN in an NLRP3-dependent manner74,198,204. Interactions with phosphatidylinositol-4-phosphate (PI(4)P) direct NLRP3 localization through a polybasic motif in an N-terminal linker region between the PYD and NACHT domains, a process necessary for inflammasome formation and pyroptosis74,198. Two recent reports demonstrated that the PI(4)P+ vesicles that NLRP3 is recruited to following activation are endosomal in origin, containing canonical markers of the early endosome in addition to markers found on endosomes and TGN205,206. The authors demonstrated that NLRP3 stimuli disrupt ER-endosome membrane contact sites, leading to PI(4)P accumulation and disruption of endosome-TGN trafficking. Following these events, the NLRP3 inflammasome forms at the microtubule organizing center (MTOC), a trafficking event dependent on the α-tubulin deacetylase HDAC6169. At the MTOC, NLRP3 is reported to interact with the mitotic kinase NEK7207–209. While studies in murine cells indicate NEK7 is required for NLRP3 activation207–209, investigation in human macrophages revealed that IKK-β priming promoted NLRP3 recruitment to PI(4)P and that NEK7 was dispensable in the presence of IKK-β post-translational priming210. Collectively, these studies demonstrate that NLRP3 activation is intrinsically tied to organelle biology and highlight the essential role that membrane component lipids and trafficking play in this process. These observations with NLRP3 mandate a better understanding of how organelle biology and spatial regulation govern the formation of all other inflammasomes.
Membrane rupture during pyroptosis
Pyroptosis is a lytic form of cell death that disrupts plasma membrane integrity and releases pro-inflammatory cytokines. The pore-forming protein GSDMD mediates this lytic cell death following inflammasome activation. Prior to cleavage, GSDMD exists in an autoinhibited conformation with its C-terminus preventing the activity of the cytotoxic N-terminus. Caspase-1 and caspases-4/5/11 cleave GSDMD at its interdomain linker to free the N-terminus6–8, causing conformation change that mediates interaction with membrane component lipids, including PIPs, phosphatidylserine, and cardiolipin8,9. This allows the GSDMD-NT to form a 31-fold to 34-fold oligomeric pore in the plasma membrane211. Reported to measure roughly 12–20nm in diameter10,11, the GSDMD pore facilitates membrane depolarization through disruption of ion gradients and release of selected cargoes, including mature IL-1β and IL-1812,13. The GSDMD pore excludes molecules larger than its diameter and acidic molecules, as its interior surface is negatively charged211. This leads to the preferential release of mature IL-1β and IL-18 over their pro-forms, as mature IL-1β and IL-18 are more basic than their precursors211,212. Thus, GSDMD pore formation is a critical step in the execution of pyroptosis (Figure 5).
Figure 5: Regulation of cell membrane rupture following inflammasome activation.
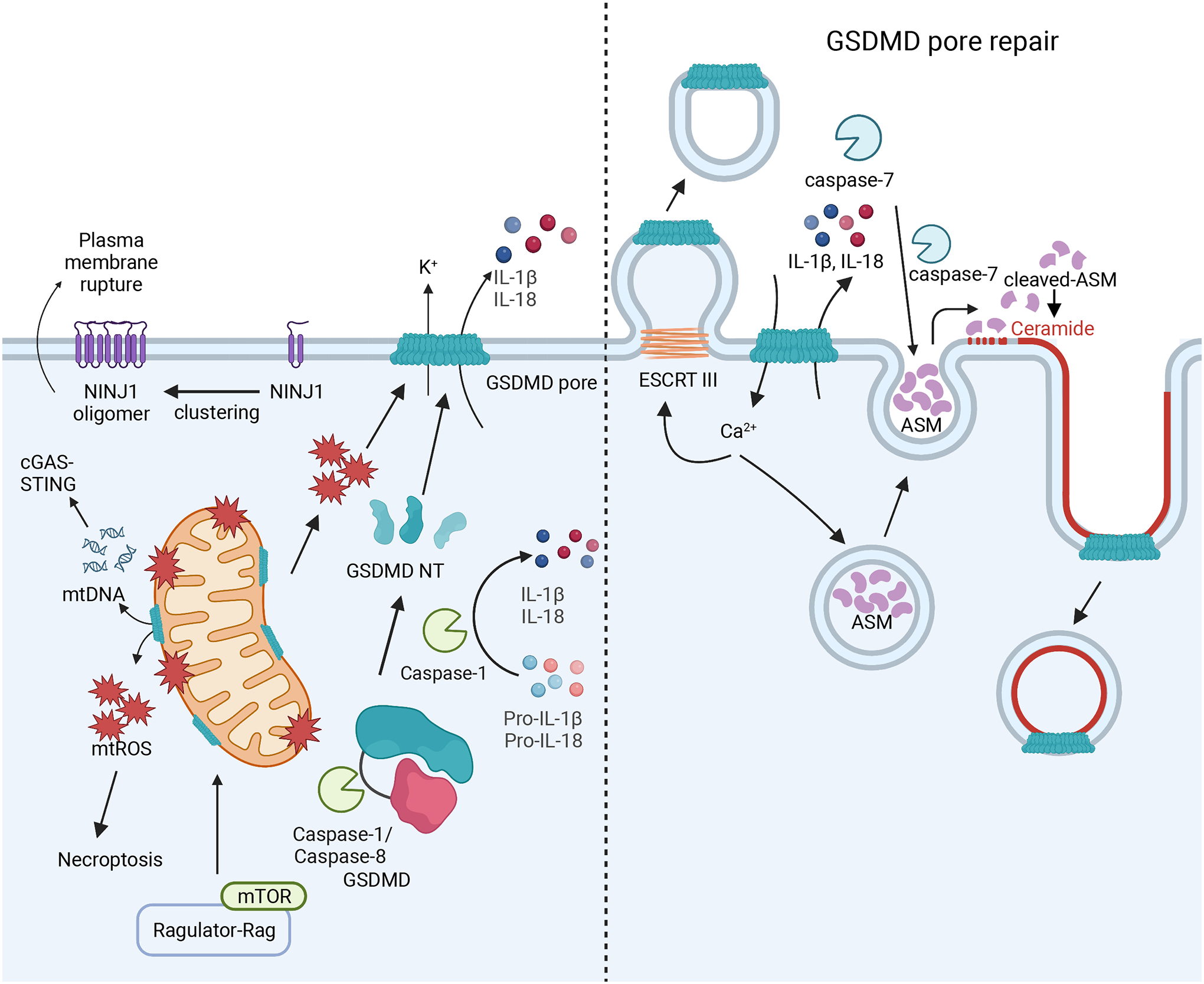
Left: Caspase-1 cleaves IL-1β/IL-18 and GSDMD to free its N-terminal (GSDMD-NT) fragment. The GSDMD-NT forms a pore on the plasma membrane, releasing IL-1β/IL-18 and disrupting cellular ion gradients to execute pyroptosis. Mitochondrial ROS downstream of Ragulator-Rag-mTORC1 activity regulates GSDMD-NT pore formation and loss of these signals prevents GSDMD NT oligomerization. GSDMD-NT pores may also form on the mitochondria through interaction with cardiolipin, causing the release of mtDNA that can activate cGAS/STING signaling and ROS that can trigger necroptosis. Inflammasomes also promote NINJ1 oligomerization in the plasma to perpetuate membrane rupture and cell death. Right: GSDMD-NT pores can be repaired through ESCRT-III-mediated cell membrane budding, triggered by Ca2+ influx. GSDMD-NT pores may also be repaired through caspase-7 activity. Caspase-7 and ASM are released into the extracellular space, where caspase-7 cleaves ASM to activate it. This causes the enrichment of ceramide on the plasma membrane through ASM activity, which promotes internalization of the microdomains on the plasma membrane that contain GSDMD pores. Both of these mechanisms lead to decreased prevalence of GSDMD pores on the plasma membrane and may explain how some cells do not die following inflammasome activation.
GSDMD pore formation is regulated by other cellular pathways. For instance, fumarate accumulation leads to the succination of GSDMD at Cys191 in humans (Cys192 in mice), limiting GSDMD processing213. Similarly, while protease processing governs GSDMD activation, the mTOR Ragulator-Rag complex promotes its oligomerization through reactive oxygen species (ROS) production214. Additionally, the GSDMD pore can permeabilize the mitochondrial outer membrane, which can alter cell death processes by activating necroptosis through the release of mitochondrial ROS (mtROS)215 or the cGAS-STING pathway through mtDNA release216. Furthermore, during Yersinia infection in macrophages, activated caspase-8 mediates GSDMD cleavage to initiate pyroptotic cell death217,218.
GSDMD pores do not permit the release of larger molecules associated with membrane rupture, such as lactate dehydrogenase (LDH) and other large DAMPs. While membrane rupture was considered a passive event, a recent study identified that ninjurin-1 (NINJ1) was essential for pyroptotic LDH release219. NINJ1 is a widely expressed cell-surface protein that mediates plasma membrane rupture in lytic cell death, including necroptosis and pyroptosis. Upon inflammasome activation, NINJ1 oligomerizes through interactions between conserved α-helices to facilitate membrane rupture, though the specific mechanism that triggers NINJ1 oligomerization remains unclear. NINJ1 oligomerization facilitates the release of large DAMPs, like HMGB1 and LDH. Of note, Ninj1−/− BMDMs had intact GSDMD pores and secreted similar amounts of IL-1β and IL-18 to WT BMDMs, indicating NINJ1 is dispensable for pro-inflammatory cytokine release219. Thus, GSDMD and NINJ1 coordinate membrane permeabilization and rupture following inflammasome activation. Future work will determine if other proteins mediate membrane rupture and the regulatory processes governing this process.
Pyroptosis and Cell Death
Recent work suggests that GSDMD pore formation, IL-1β release, and the initiation of pyroptosis do not necessitate cell death. One group reported that stimulation with oxPAPC led to caspase-11 activation in dendritic cells (DCs), causing IL-1β secretion but not cell death193. This phenotype was specific to bone marrow-derived DCs, as IL-1β release from BMDMs was not observed in this study with cytosolic delivery of oxPAPC. Similar observations were made with OatA-deficient Staphylococcus aureus infection of BMDM, where cells retained mitochondrial potential and the ability to phagocytose12. Others found that N-acetylglucosamine (NAG), a subunit of bacterial peptidoglycan, activated NLRP3 inflammasomes but did not cause cell death in BMDMs220. Similarly, Salmonella infection in neutrophils promoted NLRC4 inflammasome activation, leading to IL-1β release without promoting pyroptotic cell death221. Thus, both cell type and the nature of stimuli influence cell death outcomes downstream of inflammasome formation.
One mechanism of muting cell death is through membrane repair (Figure 5). In BMDMs, Ca2+ influx stimulated by GSDMD-NT pore formation was found to recruit endosomal sorting complexes required for transport (ESCRT)-III machinery to patch the perforated plasma membrane and inhibition of this process increased cell death and decreased membrane integrity222. Another recent study showed that caspase-7 cleaved and activated acid sphingomyelinase (ASM) to generate ceramide to mediate membrane repair by promoting endocytic uptake of GSDMD pores223. Taken together, these studies suggest that GSDMD pores in the plasma membrane can be repaired to decrease the rate of cell death, though more work is necessary to understand the full extent of regulation of GSDMD pores and their execution of cell death.
Cross-talk between pyroptosis and other cell death pathways
Recent studies have highlighted the plasticity between cell death pathways and suggest pyroptosis is not exclusively inflammasome-induced. Other members of the gasdermin (GSDM) family form pores on the plasma membrane in other cell death contexts. GSDME expression can switch TNF- or chemotherapy-induced apoptosis to lytic pyroptosis via cleavage by caspase-3 and plasma membrane pore formation224. Similarly, granzyme B released by CD8+ T and natural killer (NK) cells mediated GSDME cleavage in tumor target cells225, and granzyme A released from cytotoxic lymphocytes was shown to cleave GSDMB to promote pyroptosis in target cells226. Finally, several inflammasome-nucleating proteins are instigated in PANoptosis, a hybrid form of cell death pathways, apoptosis, pyroptosis, and necroptosis145, including NLRP3227 and AIM2146.
Function of inflammasomes in disease
Inflammasomes have wide-reaching roles in human health, including genetic diseases, infections, and sterile inflammation (Table 1). Inflammasome responses support the development of adaptive immunity by providing inflammatory cytokines that promote T cell responses, a topic recently reviewed in detail228. This section is not exhaustive but rather serves to speak to the breadth of inflammasome activity in disease. For further information, we encourage the readers to refer to more specialized reviews on this topic228–231.
Genetic Diseases
Inflammasomes require extensive regulation to ensure that pyroptotic cytokine release only occurs in appropriate contexts. Because of this, mutations in inflammasome components may cause heritable autoinflammatory disorders, including periodic fever syndromes, collectively referred to as “inflammasomopathies.” Discoveries that gain-of-function mutations in specific inflammasome receptors are linked to heritable diseases were foundational to the characterization of these genes and to the development of treatments to curtail their activities.
NLRP3 mutations are linked to a disease continuum, collectively referred to as cryopyrin-associated periodic fever syndromes (CAPS) or cryopyrinopathies232, which cause spontaneous NLRP3 activation. This spectrum of diseases ranges from familial cold autoinflammatory syndrome (FCAS), characterized by fever after cold exposure232, to Muckle-Wells syndrome (MWS), typified by rash, limb pain, and periodic fevers232, to the severe chronic infantile neurologic cutaneous articular (CINCA) syndrome or neonatal onset multi-system inflammatory disease (NOMID), classified by neonatal onset of chronic fevers, rashes, and headaches that can cause sterile meningitis and other life-threatening conditions232. Over 250 mutations in NLRP3 are associated with these conditions, a majority in the NACHT domain (https://infevers.umai-montpellier.fr/web/search.php?n=4). Left untreated, these diseases drastically affect quality of life and promote the development of secondary conditions, including hearing loss and amyloidosis.
Development of anti-IL-1 therapies were major advances in the treatment of CAPS232. These therapies include the IL-1 receptor antagonist (IL-1Ra) anakinra, a recombinant IL-1Ra that blocks IL-1R signaling; the anti-IL-1 ‘Trap’ rilonacept, an IL-1R and Fc fusion protein that acts as a soluble decoy receptor to abrogate IL-1 signaling; and the anti-IL-1β monoclonal antibody canakinumab, a monoclonal antibody that binds to circulating IL-1β to block signaling233. With anakinra treatment as the current standard of care232, these therapies drastically improve patient quality of life and decrease disease-associated damage.
Mutations in MEFV, the gene encoding pyrin, are linked to FMF syndrome, a genetic periodic fever syndrome marked by self-limited fever and serositis episodes154,155. Over 300 mutations in MEFV are linked to FMF (https://infevers.umai-montpellier.fr/web/search.php?n=1) with a majority in the C-terminal B.30.2/SPRY domain, which are thought to cause gain-of-function. A key study demonstrated that the addition of the human B30.2 domain harboring FMF mutations to murine Mefv led to spontaneous inflammation in mice160. Since 1972, colchicine, an inhibitor of microtubule polymerization, has been the cornerstone of FMF treatment, though the development of anti-IL-1 therapeutics has offered relief to those resistant to colchicine therapy234.
Other inflammasome sensors are linked to genetic diseases. Single nucleotide polymorphisms (SNPs) in NLRP1 are linked to various skin conditions, including vitiligo, psoriasis, and atopic dermatitis235–238. Mutations in NLRP1 are linked also to multiple self-healing palmoplantar carcinoma and familial keratosis lichenoides chronica (FKLC) and were shown to promote spontaneous NLRP1 oligomerization, leading to a gain-of-function48. NLRP1 gain-of-function mutations are also linked to juvenile-onset recurrent respiratory papillomatosis (JRRP)239 and NLRP1-associated autoinflammation with arthritis and dyskeratosis240. Similarly, gain of function mutations in NLRC4 are linked to macrophage activation syndrome (MAS), enterocolitis, and periodic fevers241–243, and a disease caused by one such NLRC4 mutation has been successfully treated using a combination of anakinra and IL-18 binding protein (BP)244. Thus, gain-of-function mutations in inflammasome sensors can lead to genetic diseases of varying severity (Table 1), and many of these diseases are effectively treated by anti-IL-1 therapies, though their efficacy is not universal.
Infectious Diseases
Both adaptive immune responses and deleterious inflammatory responses to microorganisms, including bacteria, viruses, and fungi, rely on and are affected by inflammasome activation and subsequent pyroptosis. Though we only discuss it briefly here, this expansive topic was covered by multiple recent reviews228,229,245.
Several PRRs mediate inflammasome responses during infection (Table 1). Often, a single pathogen nucleates multiple inflammasomes, whether through the production of multiple PAMPs/DAMPs/HAMPs or ETI. One example is the intracellular bacterial pathogen L. pneumophila, which activates caspase-11/4/5 by binding LPS178,179,197, NLRC4/NAIP through detection of flagellin95,96, and NLRP3 indirectly through activation of caspase-11179. Similarly, NLRP6 recognition of LTA initiates inflammasomes upon L. monocytogenes85 infection, which also activates AIM2 and NLRP3128,144. Furthermore, anthrax LT mediates N-terminal cleavage of murine NLRP1b34,35, yet in mice lacking sensitive Nlrp1b alleles, B. anthracis nucleates NLRP3 inflammasome activation through RIPK-1 and caspase-8246. Detection of infection by multiple PRRs ensures inflammasome formation occurs, as pathogen evasion mechanisms may circumvent any singular receptor. However, in instances where pathogen evasion mechanisms bypass several inflammasomes, single receptors may play a key role in pathogen clearance. In this regard, host genetics may sway disease outcomes. For instance, recent evidence suggests that outbreaks of Y. pestis promoted the inheritance of hyperactive pyrin alleles which overcome YopM-mediated inhibition of pyrin inflammasome formation247. Thus, overlapping sensors create a layered security system for pathogen detection, promoting favorable host outcomes in the face of immune evasion.
However, inflammasome activation in microbial infection can be a double-edged sword. For instance, during influenza A virus (IAV) infection, NLRP3 initiates a beneficial inflammatory response, promoting innate and adaptive immune responses to fight off infection248–250. By contrast, excessive inflammasome activity results in an overzealous inflammation that can cause the host’s demise. Notably, administration of the NLRP3 inhibitor, MCC950251, early after IAV infection leads to hyper-susceptibility to the virus, while treatment at a later time reduces lung inflammation252. In addition to activating the NLRP3 inflammasome, the IAV proteins NS1253 and PB1-F2254 inhibit NLRP3 activation. Similarly, multiple mechanisms of activating NLRP3 during IAV infection have been defined, including ion gradient depolarization through the activity of the IAV protein M2255, and crosstalk with other cell death pathways through interaction with ZBP1227. Such contrasting functions compounded by pathogen evasion and crosstalk with other cell death pathways illustrate the complexity of inflammasome’s role in infection, as it coordinates a powerful defense against infection but uncontrolled responses may cause unresolved inflammatory disease.
One timely example of these principles is the inflammasome response to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in Coronavirus disease 2019 (COVID-19). In airway epithelial cells, the SARS-CoV-2 Nsp5 protease was suggested to both activate and inhibit NLRP1 inflammasome activation, leading to no IL-1β release from these ACE2+ viral host cells41. In human monocytes, Fcγ receptor-mediated entry of SARS-CoV-2 instigates modest AIM2 and NLRP3 inflammasome responses256, while other studies suggested other mechanisms of SARS-CoV-2 replication-mediated inflammasome activation in monocytes and macrophages257,258. More recently, DNA DAMPs produced by the SARS-CoV-2-infected airway epithelium were shown to stimulate robust IL-1β release from leukocytes in co-culture259, demonstrating that inflammasome activation during viral infection extends beyond recognition of viral replicative activity within host cells. Thus, different mechanisms of pathogen encounter may shape the potential for inflammasome formation in a tissue-specific manner, and some of these responses may be more critical for protection than others. Treatment with anakinra shows efficacy in moderate to severe COVID-19 when given to patients with elevated serum levels of soluble urokinase plasminogen receptor (suPAR)260,261. Identifying which inflammasomes mediate protective versus pathologic responses in COVID-19 would allow for targeting of only detrimental inflammation, which may show broader treatment efficacy in different patient groups. Overall, understanding which inflammasomes guide disease outcomes in humans is essential for the development of effective inflammasome-targeting therapeutics.
Sterile Inflammation
Inflammasomes also play a key role in sterile inflammation. DAMPs produced by cellular stress or injury can mediate both inflammasome priming and activation. DAMPs that prime the inflammasome include HMGB1, DNA, and IL-1α262,263, while DAMPs that activate inflammasomes include ATP264,265, uric acid crystals266, and DNA132–135. Many DAMPs, such as HMGB1 and ATP, are released following lytic cell death, a major contributor to sterile inflammation. Similarly, mitochondrial dysfunction is linked to sterile inflammasome activation through ROS production, mtDNA release, and metabolic shifts57,58,267, and ER stress and other organelle dysfunction may also activate inflammasomes57. Thus, many molecular mechanisms mediate inflammasome responses in sterile disease.
Sustained or overzealous inflammasome activation is implicated in chronic inflammatory diseases, such as inflammatory bowel disease and dermatitis, and autoimmune diseases, including multiple sclerosis, systemic lupus erythematosus, and rheumatoid arthritis230,268,269 (Table 1). The many inflammatory and autoimmune diseases associated with inflammasome activation are discussed in several outstanding recent reviews71,230,268,269. The contributions of inflammasomes in many inflammatory and autoimmune diseases are shown clearly in murine models, where inflammasome components are targeted by gene deletion, and elevated inflammasome components and/or IL-1β are found in many corresponding human diseases. However, direct demonstration of the significance of these responses in human diseases is challenging to verify. Alleviation of symptoms by pharmacologic inhibition of inflammasomes would constitute important confirmation of the importance of this pathway in human diseases, but the causal effect of genetic mutations in inflammasome genes on human diseases remains the most powerful validation of their importance.
Inflammasomes also mediate important aspects of the immune response to cancer. ATP release from dying tumor cells promotes NLRP3 activation to generate CD8+ T cell anti-tumor immune responses270. Likewise, non-canonical inflammasome agonists reported to induce IL-1β release but not cell death in DCs also promoted anti-tumor CD8+ T cell responses in murine cancer models271. Conversely, inflammasome responses are also observed to have detrimental effects in cancer models. For instance, overexpression of human IL-1β in mice leads to the spontaneous development of tumors272, and NLRP3 expression in specific murine cancer models hinders NK cell anti-metastatic function273. Similarly, silencing AIM2 in DCs enhances antigen-specific T cell anti-tumor responses through STING activation142, while AIM2 inflammasome activation by DNA from phagocytosed tumor cells converts macrophages into an immunosuppressive phenotype274. Similarly, NLRP1275, NLRP3276, and NLRP676,77 are all implicated in the control of colitis-associated tumorigenesis in mice. Thus, the inflammasome response in cancer is as nuanced as the disease itself. For further discussion, readers can refer to a recent review70.
Inflammasomes also contribute to metabolic diseases. NLRP3 deletion alleviates the development of atherosclerotic plaques in different animal models277, and oxidized low-density lipoprotein (oxLDL), which is associated with atherosclerosis278, activates NLRP3 by functioning as both signals I and II277. The importance of this activation extends to obesity279 and insulin resistance280 in type 2 diabetes (T2D)281. In human macrophages, endothelial cells, or vascular smooth muscle cells, TNF, which is elevated in obesity, primes inflammasomes282, increasing NLRP3 expression in the liver and adipose tissue of old mice or obese mice283. IL-1β disrupts insulin signaling and decreases the expression of insulin receptor substrate 1 (IRS1), resulting in decreased insulin-mediated glucose uptake and insulin resistance284. In models of diet-induced obesity, high-fat diet activates NLRP3 in adipose tissue macrophages (ATMs) to secrete IL-1β, leading to insulin resistance in adipocytes285. Ceramide produced by ATMs in obese mice can also trigger the inflammasome286. Similarly, saturated fatty acids can activate the NLRP3 inflammasome285, and this activation pathway has been shown in T2D287, nonalcoholic fatty liver disease (NAFLD)288, and chronic kidney disease289. However, despite all of these associations, blockade of IL-1 signaling has not shown success in treating diabetes, and in a murine model, treatment with the NLRP3 inhibitor, MCC950, did not prevent the onset of T2D pathology290. Thus, the specific role of the inflammasome in metabolic diseases in humans remains unclear and an active area of research.
The NLRP3 inflammasome is also implicated in neurologic diseases291, including amyloid-associated Alzheimer’s disease292, age-related macular degeneration293,294, and Parkinson’s disease295,296. Inflammasome activation in neurodegenerative diseases is mediated by DAMPs, including mitochondrial dysfunction, lysosomal damage, and extracellular ATP291. However, specific protein moieties accumulated in some of these diseases, including amyloid-β (Aβ) fibers and oligomers in Alzheimer’s disease297 and α-synuclein in Parkinson’s disease295, activate NLRP3, linking the inflammasome directly to disease pathology. Similarly, the accumulation of Alu RNA elements in the retinal pigmented epithelium activates the NLRP3/NLRC4 inflammasomes in mouse models of age-related macular degeneration69,293. Though apparent in mouse models, causal links to human neurogenerative diseases warrant further investigation. Overall, inflammasomes have far-reaching effects in sterile inflammatory diseases, and in many instances, contribute to disease pathology. The therapeutic blockade of inflammasomes using either biologics or small molecule inhibitors is a robust area of research covered by outstanding reviews62,71,298.
Conclusion
Inflammasomes are critical initiators of inflammation and adaptive immunity with far-reaching effects on human health and disease. Inflammasome activation is an innate immune response that fundamentally alters the cellular state, through the initiation of pyroptosis, cell death, and inflammatory cytokine release. A host of PRRs govern inflammasome activation in the cytosol and respond to threats to cellular integrity and infection in the form discrete molecules, namely PAMPs/DAMPs, and changes in cellular processes, specifically HAMPs and ETI. Some inflammasome sensors are well-characterized, such as NLRP3, while our understanding of other sensors is more nascent, such as NLRP6. Yet, all of these PRRs have clear roles in different diseases and tissues, mandating more extensive study. Similarly, some stimuli, such as dsDNA and viral RNA, are reported to activate multiple inflammasomes in different contexts, thus understanding which PRRs respond to specific moieties in different disease states is an important topic for further investigation. Regulation of inflammasomes is not limited to their nucleation, as both cell biology and metabolic state govern inflammasome formation and the execution of cell death. Observations of inflammasomes in living cells and regulation of pyroptosis through modulation of GSDMD pores show that cell death outcomes can be altered and are context-dependent. Similarly, cross-talk between cell death pathways highlights the nuance and plasticity in cell fate decisions. Thus, while our understanding of inflammasomes has expanded significantly since their discovery, many questions remain regarding the activation and execution of inflammasome formation and pyroptosis and the role of inflammasomes in human diseases. Because inflammasomes have both detrimental and beneficial responses in human diseases, developing a toolkit of therapeutics that target individual inflammasome PRRs will allow for refined disease interventions. Achieving such a goal will require a holistic understanding of different inflammasome pathways and subsequent cell death responses in different disease contexts.
Inflammasomes are critical initiators of inflammation and adaptive immunity with far reaching effects on human health and disease. This review provides a 360-degree view on the mechanisms of inflammasome formation, subcellular regulation, and their effects on cell death and disease.
Acknowledgements:
This work is supported by NIH grants R56 AI158314, R01 AI158314, AI029564, AI141333, DK094779, U19 AI067798 and R35 CA232109 to J.P.-Y.T. Figures were created using Biorender.com. We would like to thank Ting lab members for helpful discussion, especially Brian P. Fay and Dr. Michael W. Linhoff for recommendations regarding the figures. We would also like to acknowledge those whose work was not included due to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
J.P.-Y.T. is a cofounder of IMMvention Therapeutix which works on inflammasome inhibitors.
References
- 1.Poyet J-L, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, and Alnemri ES (2001). Identification of Ipaf, a Human Caspase-1-activating Protein Related to Apaf-1*. J Biol Chem 276, 28309–28313. 10.1074/jbc.c100250200. [DOI] [PubMed] [Google Scholar]
- 2.Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, Geddes BJ, Briskin M, DiStefano PS, and Bertin J (2002). PYPAF7, a Novel PYRIN-containing Apaf1-like Protein That Regulates Activation of NF-κB and Caspase-1-dependent Cytokine Processing*. J Biol Chem 277, 29874–29880. 10.1074/jbc.m203915200. [DOI] [PubMed] [Google Scholar]
- 3.Kagan JC, Magupalli VG, and Wu H (2014). SMOCs: supramolecular organizing centres that control innate immunity. Nature Reviews Immunology 14, 821–826. 10.1038/nri3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinon F, Burns K, and Tschopp J (2002). The Inflammasome A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol Cell 10, 417–426. 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 5.Koonin EV, and Aravind L (2000). The NACHT family - a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem. Sci 25, 223–224. [DOI] [PubMed] [Google Scholar]
- 6.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 7.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang D-C, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 10.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 113, 7858–7863. 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, and Hiller S (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. The EMBO Journal 35, 1766–1778. 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2018). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44.e6. 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, and Broz P (2018). The Gasdermin‐D pore acts as a conduit for IL‐1β secretion in mice. Eur J Immunol 48, 584–592. 10.1002/eji.201747404. [DOI] [PubMed] [Google Scholar]
- 14.Fischer NL, Naseer N, Shin S, and Brodsky IE (2020). Effector-triggered immunity and pathogen sensing in metazoans. Nat Microbiol 5, 14–26. 10.1038/s41564-019-0623-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones JDG, Vance RE, and Dangl JL (2016). Intracellular innate immune surveillance devices in plants and animals. Science 354. 10.1126/science.aaf6395. [DOI] [PubMed] [Google Scholar]
- 16.Liston A, and Masters SL (2017). Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol 17, 208–214. 10.1038/nri.2016.151. [DOI] [PubMed] [Google Scholar]
- 17.Franchi L, Eigenbrod T, and Núñez G (2009). Cutting Edge: TNF-α Mediates Sensitization to ATP and Silica via the NLRP3 Inflammasome in the Absence of Microbial Stimulation. J Immunol 183, 792–796. 10.4049/jimmunol.0900173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. (2009). Cutting Edge: NF-κB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J Immunol 183, 787–791. 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schroder K, Sagulenko V, Zamoshnikova A, Richards AA, Cridland JA, Irvine KM, Stacey KJ, and Sweet MJ (2012). Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 217, 1325–1329. 10.1016/j.imbio.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 20.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AAB, Cooper MA, Graf T, and Hornung V (2016). Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 44, 833–846. 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 21.Py BF, Kim M-S, Vakifahmetoglu-Norberg H, and Yuan J (2013). Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity. Mol Cell 49, 331–338. 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, and Alnemri ES (2012). Non-transcriptional Priming and Deubiquitination Regulate NLRP3 Inflammasome Activation*. J Biol Chem 287, 36617–36622. 10.1074/jbc.m112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garlanda C, Dinarello CA, and Mantovani A (2013). The Interleukin-1 Family: Back to the Future. Immunity 39, 1003–1018. 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gritsenko A, Yu S, Martin-Sanchez F, Diaz-del-Olmo I, Nichols E-M, Davis DM, Brough D, and Lopez-Castejon G (2020). Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front Immunol 11, 565924. 10.3389/fimmu.2020.565924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong FL, Robinson K, Teo DET, Tan K-Y, Lim C, Harapas CR, Yu C-H, Xie WH, Sobota RM, Au VB, et al. (2018). Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J Biol Chem 293, 18864–18878. 10.1074/jbc.ra118.004350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linder A, Bauernfried S, Cheng Y, Albanese M, Jung C, Keppler OT, and Hornung V (2020). CARD8 inflammasome activation triggers pyroptosis in human T cells. Embo J 39, e105071. 10.15252/embj.2020105071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, Bertin J, Bertin JJ, and Gough PJ (2012). Autolytic Proteolysis within the Function to Find Domain (FIIND) Is Required for NLRP1 Inflammasome Activity*. J Biol Chem 287, 25030–25037. 10.1074/jbc.m112.378323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, and Reed JC (2011). CARD8 and NLRP1 Undergo Autoproteolytic Processing through a ZU5-Like Domain. Plos One 6, e27396. 10.1371/journal.pone.0027396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A, et al. (2018). DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med 24, 1151–1156. 10.1038/s41591-018-0082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyden ED, and Dietrich WF (2006). Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38, 240–244. 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 31.Tang G, and Leppla SH (1999). Proteasome Activity Is Required for Anthrax Lethal Toxin To Kill Macrophages. Infect Immun 67, 3055–3060. 10.1128/iai.67.6.3055-3060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wickliffe KE, Leppla SH, and Moayeri M (2008). Killing of macrophages by anthrax lethal toxin: involvement of the N‐end rule pathway. Cell Microbiol 10, 1352–1362. 10.1111/j.1462-5822.2008.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terra JK, Cote CK, France B, Jenkins AL, Bozue JA, Welkos SL, LeVine SM, and Bradley KA (2010). Cutting Edge: Resistance to Bacillus anthracis Infection Mediated by a Lethal Toxin Sensitive Allele of Nalp1b/Nlrp1b. J Immunol 184, 17–20. 10.4049/jimmunol.0903114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chavarría-Smith J, and Vance RE (2013). Direct Proteolytic Cleavage of NLRP1B Is Necessary and Sufficient for Inflammasome Activation by Anthrax Lethal Factor. Plos Pathog 9, e1003452. 10.1371/journal.ppat.1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, and Moayeri M (2012). Anthrax Lethal Factor Cleavage of Nlrp1 Is Required for Activation of the Inflammasome. Plos Pathog 8, e1002638. 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA, et al. (2019). N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85. 10.1126/science.aau1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, and Vance RE (2019). Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364. 10.1126/science.aau1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chavarría-Smith J, Mitchell PS, Ho AM, Daugherty MD, and Vance RE (2016). Functional and Evolutionary Analyses Identify Proteolysis as a General Mechanism for NLRP1 Inflammasome Activation. Plos Pathog 12, e1006052. 10.1371/journal.ppat.1006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson KS, Teo DET, Tan KS, Toh GA, Ong HH, Lim CK, Lay K, Au BV, Lew TS, Chu JJH, et al. (2021). Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 370, 1–14. 10.1126/science.aay2002. [DOI] [PubMed] [Google Scholar]
- 40.Tsu BV, Beierschmitt C, Ryan AP, Agarwal R, Mitchell PS, and Daugherty MD (2021). Diverse viral proteases activate the NLRP1 inflammasome. Elife 10, e60609. 10.7554/elife.60609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Planès R, Pinilla M, Santoni K, Hessel A, Passemar C, Lay K, Paillette P, Valadão A-LC, Robinson KS, Bastard P, et al. (2022). Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol Cell 82, 2385–2400.e9. 10.1016/j.molcel.2022.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Gao H, Clark KM, Mugisha CS, Davis K, Tang JP, Harlan GH, DeSelm CJ, Presti RM, Kutluay SB, et al. (2021). CARD8 is an inflammasome sensor for HIV-1 protease activity. Science 371. 10.1126/science.abe1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okondo MC, Rao SD, Taabazuing CY, Chui AJ, Poplawski SE, Johnson DC, and Bachovchin DA (2018). Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell Chem Biol 25, 262–267.e5. 10.1016/j.chembiol.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang M, Zhang X, Toh GA, Gong Q, Wang J, Han Z, Wu B, Zhong F, and Chai J (2021). Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature 592, 773–777. 10.1038/s41586-021-03320-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hollingsworth LR, Sharif H, Griswold AR, Fontana P, Mintseris J, Dagbay KB, Paulo JA, Gygi SP, Bachovchin DA, and Wu H (2021). DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature 592, 778–783. 10.1038/s41586-021-03350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson DC, Okondo MC, Orth EL, Rao SD, Huang H-C, Ball DP, and Bachovchin DA (2020). DPP8/9 inhibitors activate the CARD8 inflammasome in resting lymphocytes. Cell Death Dis 11, 628. 10.1038/s41419-020-02865-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harapas CR, Robinson KS, Lay K, Wong J, Traspas RM, Nabavizadeh N, Rass-Rothschild A, Boisson B, Drutman SB, Laohamonthonkul P, et al. (2022). DPP9 deficiency: An inflammasomopathy that can be rescued by lowering NLRP1/IL-1 signaling. Sci Immunol 7, eabi4611. 10.1126/sciimmunol.abi4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhong FL, Mamaï O, Sborgi L, Boussofara L, Hopkins R, Robinson K, Szeverényi I, Takeichi T, Balaji R, Lau A, et al. (2016). Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 167, 187–191.e17. 10.1016/j.cell.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 49.Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, and Hornung V (2021). Human NLRP1 is a sensor for double-stranded RNA. Science 371. 10.1126/science.abd0811. [DOI] [PubMed] [Google Scholar]
- 50.Jenster L-M, Lange K-E, Normann S, vom Hemdt A, Wuerth JD, Schiffelers LDJ, Tesfamariam YM, Gohr FN, Klein L, Kaltheuner IH, et al. (2022). P38 kinases mediate NLRP1 inflammasome activation after ribotoxic stress response and virus infection. J Exp Med 220, e20220837. 10.1084/jem.20220837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson KS, Toh GA, Rozario P, Chua R, Bauernfried S, Sun Z, Firdaus MJ, Bayat S, Nadkarni R, Poh ZS, et al. (2022). ZAKα-driven ribotoxic stress response activates the human NLRP1 inflammasome. Science 377, 328–335. 10.1126/science.abl6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ball DP, Tsamouri LP, Wang AE, Huang H-C, Warren CD, Wang Q, Edmondson IH, Griswold AR, Rao SD, Johnson DC, et al. (2022). Oxidized thioredoxin-1 restrains the NLRP1 inflammasome. Sci Immunol 7, eabm7200. 10.1126/sciimmunol.abm7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou JY, Sarkar MK, Okamura K, Harris JE, Gudjonsson JE, and Fitzgerald KA (2023). Activation of the NLRP1 inflammasome in human keratinocytes by the dsDNA mimetic poly(dA:dT). Proc National Acad Sci 120, e2213777120. 10.1073/pnas.2213777120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lacey CA, and Miao EA (2019). NLRP1 – One NLR to guard them all. Embo J 38, e102494. 10.15252/embj.2019102494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taabazuing CY, Griswold AR, and Bachovchin DA (2020). The NLRP1 and CARD8 inflammasomes. Immunol Rev 297, 13–25. 10.1111/imr.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gong T, Yang Y, Jin T, Jiang W, and Zhou R (2018). Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol 39, 393–406. 10.1016/j.it.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 57.Seoane PI, Lee B, Hoyle C, Yu S, Lopez-Castejon G, Lowe M, and Brough D (2020). The NLRP3–inflammasome as a sensor of organelle dysfunction. J Cell Biol 219, e202006194. 10.1083/jcb.202006194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hughes MM, and O’Neill LAJ (2018). Metabolic regulation of NLRP3. Immunol Rev 281, 88–98. 10.1111/imr.12608. [DOI] [PubMed] [Google Scholar]
- 59.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. (2012). Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 36, 401–414. 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X, Wong J, Ding S, Seki E, Schnabl B, et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O’Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, et al. (2017). The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 171, 1110–1124.e18. 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Swanson KV, Deng M, and Ting JPY (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature Reviews Immunology 19, 1–13. 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moretti J, and Blander JM (2021). Increasing complexity of NLRP3 inflammasome regulation. J Leukocyte Biol 109, 561–571. 10.1002/jlb.3mr0520-104rr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, Latz E, Gazzinelli RT, Golenbock DT, and Fitzgerald KA (2014). Dual Engagement of the NLRP3 and AIM2 Inflammasomes by Plasmodium-Derived Hemozoin and DNA during Malaria. Cell Reports 6, 196–210. 10.1016/j.celrep.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, Lamkanfi M, and Kanneganti T-D (2015). Concerted Activation of the AIM2 and NLRP3 Inflammasomes Orchestrates Host Protection against Aspergillus Infection. Cell Host Microbe 17, 357–368. 10.1016/j.chom.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ip WKE, and Medzhitov R (2019). Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nature Communications, 1–11. 10.1038/ncomms7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Freeman L, Guo H, David CN, Brickey WJ, Jha S, and Ting JPY (2017). NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med 214, 1351–1370. 10.1084/jem.20150237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Man SM, Hopkins LJ, Nugent E, Cox S, Glück IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, and Bryant CE (2014). Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. U.S.A 111, 7403–7408. 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang S, Narendran S, Hirahara S, Varshney A, Pereira F, Apicella I, Ambati M, Ambati VL, Yerramothu P, Ambati K, et al. (2021). DDX17 is an essential mediator of sterile NLRC4 inflammasome activation. Sci Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharma BR, and Kanneganti T-D (2021). NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol 22, 550–559. 10.1038/s41590-021-00886-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, and Latz E (2018). Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 17, 588–606. 10.1038/nrd.2018.97. [DOI] [PubMed] [Google Scholar]
- 72.Xiao L, Magupalli VG, and Wu H (2023). Cryo-EM structures of the active NLRP3 inflammasome disc. Nature 613, 595–600. 10.1038/s41586-022-05570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hochheiser IV, Pilsl M, Hagelueken G, Moecking J, Marleaux M, Brinkschulte R, Latz E, Engel C, and Geyer M (2022). Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature 604, 184–189. 10.1038/s41586-022-04467-w. [DOI] [PubMed] [Google Scholar]
- 74.Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, and Wu H (2021). NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell 184, 6299–6312.e22. 10.1016/j.cell.2021.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grenier JM, Wang L, Manji GA, Huang W-J, Al-Garawi A, Kelly R, Carlson A, Merriam S, Lora JM, Briskin M, et al. (2002). Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF‐κB and caspase‐1. Febs Lett 530, 73–78. 10.1016/s0014-5793(02)03416-6. [DOI] [PubMed] [Google Scholar]
- 76.Chen GY, Liu M, Wang F, Bertin J, and Núñez G (2011). A Functional Role for Nlrp6 in Intestinal Inflammation and Tumorigenesis. J Immunol 186, 7187–7194. 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D, and Chamaillard M (2011). Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc National Acad Sci 108, 9601–9606. 10.1073/pnas.1100981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. (2011). NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell 145, 745–757. 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang J-P, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, et al. (2014). NLRP6 Inflammasome Orchestrates the Colonic Host-Microbial Interface by Regulating Goblet Cell Mucus Secretion. Cell 156, 1045–1059. 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, et al. (2015). Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443. 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mamantopoulos M, Ronchi F, Hauwermeiren FV, Vieira-Silva S, Yilmaz B, Martens L, Saeys Y, Drexler SK, Yazdi AS, Raes J, et al. (2017). Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity 47, 339–348.e4. 10.1016/j.immuni.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 82.Lemire P, Robertson SJ, Maughan H, Tattoli I, Streutker CJ, Platnich JM, Muruve DA, Philpott DJ, and Girardin SE (2017). The NLR Protein NLRP6 Does Not Impact Gut Microbiota Composition. Cell Reports 21, 3653–3661. 10.1016/j.celrep.2017.12.026. [DOI] [PubMed] [Google Scholar]
- 83.Volk JK, Nyström EEL, van der Post S, Abad BM, Schroeder BO, Johansson Å, Svensson F, Jäverfelt S, Johansson MEV, Hansson GC, et al. (2019). The Nlrp6 inflammasome is not required for baseline colonic inner mucus layer formation or function. J Exp Med 216, 2602–2618. 10.1084/jem.20190679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, Zheng Y, Rongvaux A, Sun Q, Yang G, et al. (2015). Nlrp6 regulates intestinal antiviral innate immunity. Science 350, 826–830. 10.1126/science.aab3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, Inohara N, Chen GY, and Núñez G (2018). The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 175, 1651–1664.e14. 10.1016/j.cell.2018.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, and Kanneganti T-D (2012). NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488, 389–393. 10.1038/nature11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shen C, Li R, Negro R, Cheng J, Vora SM, Fu T-M, Wang A, He K, Andreeva L, Gao P, et al. (2021). Phase separation drives RNA virus-induced activation of the NLRP6 inflammasome. Cell 184, 5759–5774.e20. 10.1016/j.cell.2021.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, and Dixit VM (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218. 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 89.Damiano JS, Newman RM, and Reed JC (2004). Multiple Roles of CLAN (Caspase-Associated Recruitment Domain, Leucine-Rich Repeat, and NAIP CIIA HET-E, and TP1-Containing Protein) in the Mammalian Innate Immune Response. J Immunol 173, 6338–6345. 10.4049/jimmunol.173.10.6338. [DOI] [PubMed] [Google Scholar]
- 90.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, and Flavell RA (2007). Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Medicine 204, 3235–3245. 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, and Aderem A (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat Immunol 7, 569–575. 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 92.Franchi L, Amer A, Body-Malapel M, Kanneganti T-D, Özören N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, et al. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat Immunol 7, 576–582. 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 93.Growney JD, and Dietrich WF (2000). High-resolution Genetic and Physical Map of the Lgn1 Interval in C57BL/6J Implicates Naip2 or Naip5 in Legionella pneumophila Pathogenesis. Genome Res 10, 1158–1171. 10.1101/gr.10.8.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amer A, Franchi L, Kanneganti T-D, Body-Malapel M, Özören N, Brady G, Meshinchi S, Jagirdar R, Gewirtz A, Akira S, et al. (2006). Regulation of Legionella Phagosome Maturation and Infection through Flagellin and Host Ipaf*. J Biol Chem 281, 35217–35223. 10.1074/jbc.m604933200. [DOI] [PubMed] [Google Scholar]
- 95.Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, Liu L, and Shao F (2011). The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600. 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 96.Kofoed EM, and Vance RE (2011). Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595. 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kortmann J, Brubaker SW, and Monack DM (2015). Cutting Edge: Inflammasome Activation in Primary Human Macrophages Is Dependent on Flagellin. J Immunol 195, 815–819. 10.4049/jimmunol.1403100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang J, Zhao Y, Shi J, and Shao F (2013). Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc National Acad Sci 110, 14408–14413. 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grandjean T, Boucher A, Thepaut M, Monlezun L, Guery B, Faudry E, Kipnis E, and Dessein R (2017). The human NAIP-NLRC4-inflammasome senses the Pseudomonas aeruginosa T3SS inner-rod protein. Int Immunol 29, 377–384. 10.1093/intimm/dxx047. [DOI] [PubMed] [Google Scholar]
- 100.Ruiz VMR, Ramirez J, Naseer N, Palacio NM, Siddarthan IJ, Yan BM, Boyer MA, Pensinger DA, Sauer J-D, and Shin S (2017). Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc National Acad Sci 114, 13242–13247. 10.1073/pnas.1710433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, Li Y, David L, Lu A, Wang WL, et al. (2015). Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409. 10.1126/science.aac5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, Shao F, Wang H-W, Sui S-F, and Chai J (2015). Structural and biochemical basis for induced self-propagation of NLRC4. Science 350, 399–404. 10.1126/science.aac5489. [DOI] [PubMed] [Google Scholar]
- 103.Halff EF, Diebolder CA, Versteeg M, Schouten A, Brondijk THC, and Huizinga EG (2012). Formation and Structure of a NAIP5-NLRC4 Inflammasome Induced by Direct Interactions with Conserved N- and C-terminal Regions of Flagellin*. J Biol Chem 287, 38460–38472. 10.1074/jbc.m112.393512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Case CL, and Roy CR (2011). Asc Modulates the Function of NLRC4 in Response to Infection of Macrophages by Legionella pneumophila. Mbio 2, e00117–11. 10.1128/mbio.00117-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Broz P, von Moltke J, Jones JW, Vance RE, and Monack DM (2010). Differential Requirement for Caspase-1 Autoproteolysis in Pathogen-Induced Cell Death and Cytokine Processing. Cell Host Microbe 8, 471–483. 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, Louie S, Kayagaki N, Liu J, Kömüves L, et al. (2012). Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542. 10.1038/nature11429. [DOI] [PubMed] [Google Scholar]
- 107.Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, Wang R, Zhang Y, Martinon F, Miao D, et al. (2013). Crystal Structure of NLRC4 Reveals Its Autoinhibition Mechanism. Science 341, 172–175. 10.1126/science.1236381. [DOI] [PubMed] [Google Scholar]
- 108.Tenthorey JL, Chavez RA, Thompson TW, Deets KA, Vance RE, and Rauch I (2020). NLRC4 inflammasome activation is NLRP3- and phosphorylation-independent during infection and does not protect from melanoma. J Exp Med 217, e20191736. 10.1084/jem.20191736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suzuki S, Franchi L, He Y, Muñoz-Planillo R, Mimuro H, Suzuki T, Sasakawa C, and Núñez G (2014). Shigella Type III Secretion Protein MxiI Is Recognized by Naip2 to Induce Nlrc4 Inflammasome Activation Independently of Pkcδ. Plos Pathog 10, e1003926. 10.1371/journal.ppat.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nordlander S, Pott J, and Maloy KJ (2014). NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol 7, 775–785. 10.1038/mi.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, Maslowski KM, and Hardt W-D (2014). Epithelium-Intrinsic NAIP/NLRC4 Inflammasome Drives Infected Enterocyte Expulsion to Restrict Salmonella Replication in the Intestinal Mucosa. Cell Host Microbe 16, 237–248. 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 112.Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, Philip NH, Ayres JS, Brodsky IE, Gronert K, et al. (2017). NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and −8. Immunity 46, 649–659. 10.1016/j.immuni.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mitchell PS, Roncaioli JL, Turcotte EA, Goers L, Chavez RA, Lee AY, Lesser CF, Rauch I, and Vance RE (2020). NAIP–NLRC4-deficient mice are susceptible to shigellosis. Elife 9, e59022. 10.7554/elife.59022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hausmann A, Böck D, Geiser P, Berthold DL, Fattinger SA, Furter M, Bouman JA, Barthel-Scherrer M, Lang CM, Bakkeren E, et al. (2020). Intestinal epithelial NAIP/NLRC4 restricts systemic dissemination of the adapted pathogen Salmonella Typhimurium due to site-specific bacterial PAMP expression. Mucosal Immunol 13, 530–544. 10.1038/s41385-019-0247-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen I-F, Ou-Yang F, Hung J-Y, Liu J-C, Wang H, Wang S-C, Hou M-F, Hortobagyi GN, and Hung M-C (2006). AIM2 suppresses human breast cancer cell proliferation in vitro and mammary tumor growth in a mouse model. Mol Cancer Ther 5, 1–7. 10.1158/1535-7163.mct-05-0310. [DOI] [PubMed] [Google Scholar]
- 116.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey Daniel.R., Latz E, and Fitzgerald KA (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518. 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, et al. (2009). An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 10, 266–272. 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 118.Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, and Alnemri ES (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513. 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jin T, Perry A, Smith P, Jiang J, and Xiao TS (2013). Structure of the Absent in Melanoma 2 (AIM2) Pyrin Domain Provides Insights into the Mechanisms of AIM2 Autoinhibition and Inflammasome Assembly*. J Biol Chem 288, 13225–13235. 10.1074/jbc.m113.468033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam VA, Johnstone RW, et al. (2012). Structures of the HIN Domain:DNA Complexes Reveal Ligand Binding and Activation Mechanisms of the AIM2 Inflammasome and IFI16 Receptor. Immunity 36, 561–571. 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Morrone SR, Matyszewski M, Yu X, Delannoy M, Egelman EH, and Sohn J (2015). Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat Commun 6, 7827. 10.1038/ncomms8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ru H, Ni X, Zhao L, Crowley C, Ding W, Hung L-W, Shaw N, Cheng G, and Liu Z-J (2013). Structural basis for termination of AIM2-mediated signaling by p202. Cell Res 23, 855–858. 10.1038/cr.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lu A, Kabaleeswaran V, Fu T, Magupalli VG, and Wu H (2014). Crystal Structure of the F27G AIM2 PYD Mutant and Similarities of Its Self-Association to DED/DED Interactions. J Mol Biol 426, 1420–1427. 10.1016/j.jmb.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lu A, Li Y, Yin Q, Ruan J, Yu X, Egelman E, and Wu H (2015). Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov 1, 15013. 10.1038/celldisc.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11, 395–402. 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Reinholz M, Kawakami Y, Salzer S, Kreuter A, Dombrowski Y, Koglin S, Kresse S, Ruzicka T, and Schauber J (2013). HPV16 activates the AIM2 inflammasome in keratinocytes. Arch Dermatol Res 305, 723–732. 10.1007/s00403-013-1375-0. [DOI] [PubMed] [Google Scholar]
- 127.Fernandes-Alnemri T, Yu J-W, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, et al. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11, 385–393. 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sauer J-D, Witte CE, Zemansky J, Hanson B, Lauer P, and Portnoy DA (2010). Listeria monocytogenes Triggers AIM2-Mediated Pyroptosis upon Infrequent Bacteriolysis in the Macrophage Cytosol. Cell Host Microbe 7, 412–419. 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Warren SE, Armstrong A, Hamilton MK, Mao DP, Leaf IA, Miao EA, and Aderem A (2010). Cutting Edge: Cytosolic Bacterial DNA Activates the Inflammasome via Aim2. J Immunol 185, 818–821. 10.4049/jimmunol.1000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wilson JE, Petrucelli AS, Chen L, Koblansky AA, Truax AD, Oyama Y, Rogers AB, Brickey WJ, Wang Y, Schneider M, et al. (2015). Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med 21, 906–913. 10.1038/nm.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Man SM, Zhu Q, Zhu L, Liu Z, Karki R, Malik A, Sharma D, Li L, Malireddi RKS, Gurung P, et al. (2015). Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 162, 45–58. 10.1016/j.cell.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hu B, Jin C, Li H-B, Tong J, Ouyang X, Cetinbas NM, Zhu S, Strowig T, Lam FC, Zhao C, et al. (2016). The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768. 10.1126/science.aaf7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, Hurt ME, Paysour MJ, Ennerfelt HE, and Lukens JR (2020). AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature 580, 647–652. 10.1038/s41586-020-2174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jakobs C, Perner S, and Hornung V (2015). AIM2 Drives Joint Inflammation in a Self-DNA Triggered Model of Chronic Polyarthritis. Plos One 10, e0131702. 10.1371/journal.pone.0131702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, et al. (2021). The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592, 296–301. 10.1038/s41586-021-03341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 339, 786–791. 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu F, Niu Q, Fan X, Liu C, Zhang J, Wei Z, Hou W, Kanneganti T-D, Robb ML, Kim JH, et al. (2017). Priming and Activation of Inflammasome by Canarypox Virus Vector ALVAC via the cGAS/IFI16–STING–Type I IFN Pathway and AIM2 Sensor. J Immunol 199, 3293–3305. 10.4049/jimmunol.1700698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Swanson KV, Junkins RD, Kurkjian CJ, Holley-Guthrie E, Pendse AA, Morabiti RE, Petrucelli A, Barber GN, Benedict CA, and Ting JPY (2017). A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med 214, 3611–3626. 10.1084/jem.20171749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, Ghosh A, Vella AT, Vanaja SK, Sarkar SN, et al. (2018). Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 49, 413–426.e5. 10.1016/j.immuni.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, Zhou X, Gao J, Fang R, Meng G, et al. (2017). Inflammasome Activation Triggers Caspase-1- Mediated Cleavage of cGAS to Regulate Responses to DNA Virus Infection. Immunity 46, 393–404. 10.1016/j.immuni.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 141.Corrales L, Woo S-R, Williams JB, McWhirter SM, Dubensky TW, and Gajewski TF (2016). Antagonism of the STING Pathway via Activation of the AIM2 Inflammasome by Intracellular DNA. J Immunol 196, 3191–3198. 10.4049/jimmunol.1502538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fukuda K, Okamura K, Riding RL, Fan X, Afshari K, Haddadi N-S, McCauley SM, Guney MH, Luban J, Funakoshi T, et al. (2021). AIM2 regulates anti-tumor immunity and is a viable therapeutic target for melanoma. J Exp Med 218, e20200962. 10.1084/jem.20200962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wu J, Fernandes-Alnemri T, and Alnemri ES (2010). Involvement of the AIM2, NLRC4, and NLRP3 Inflammasomes in Caspase-1 Activation by Listeria monocytogenes. J Clin Immunol 30, 693–702. 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E, and Hornung V (2010). Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol 40, 1545–1551. 10.1002/eji.201040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pandian N, and Kanneganti T-D (2022). PANoptosis: A Unique Innate Immune Inflammatory Cell Death Modality. J Immunol 209, 1625–1633. 10.4049/jimmunol.2200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, and Kanneganti T-D (2021). AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 597, 415–419. 10.1038/s41586-021-03875-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dang EV, McDonald JG, Russell DW, and Cyster JG (2017). Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 171, 1057–1071.e11. 10.1016/j.cell.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, Sun X, Yang M, Billiar TR, Wang H, et al. (2016). PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun 7, 13280. 10.1038/ncomms13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ma C, Li S, Hu Y, Ma Y, Wu Y, Wu C, Liu X, Wang B, Hu G, Zhou J, et al. (2021). AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J Exp Med 218, e20201796. 10.1084/jem.20201796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ratsimandresy RA, Indramohan M, Dorfleutner A, and Stehlik C (2017). The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell Mol Immunol 14, 127–142. 10.1038/cmi.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hu S, Peng L, Kwak Y-T, Tekippe EM, Pasare C, Malter JS, Hooper LV, and Zaki Md.H. (2015). The DNA Sensor AIM2 Maintains Intestinal Homeostasis via Regulation of Epithelial Antimicrobial Host Defense. Cell Reports 13, 1922–1936. 10.1016/j.celrep.2015.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chou W-C, Guo Z, Guo H, Chen L, Zhang G, Liang K, Xie L, Tan X, Gibson SA, Rampanelli E, et al. (2021). AIM2 in regulatory T cells restrains autoimmune diseases. Nature 591, 300–305. 10.1038/s41586-021-03231-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Antiochos B, Trejo-Zambrano D, Fenaroli P, Rosenberg A, Baer A, Garg A, Sohn J, Li J, Petri M, Goldman DW, et al. (2022). The DNA sensors AIM2 and IFI16 are SLE autoantigens that bind neutrophil extracellular traps. Elife 11, e72103. 10.7554/elife.72103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Consortium TFF, Bernot A, Clepet C, Dasilva C, Devaud C, Petit J-L, Caloustian C, Cruaud C, Samson D, Pulcini F, et al. (1997). A candidate gene for familial Mediterranean fever. Nat Genet 17, 25–31. 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 155.Consortium TIF (1997). Ancient Missense Mutations in a New Member of the RoRet Gene Family Are Likely to Cause Familial Mediterranean Fever. Cell 90, 797–807. 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 156.Richards N, Schaner P, Diaz A, Stuckey J, Shelden E, Wadhwa A, and Gumucio DL (2001). Interaction between Pyrin and the Apoptotic Speck Protein (ASC) Modulates ASC-induced Apoptosis*. J Biol Chem 276, 39320–39329. 10.1074/jbc.m104730200. [DOI] [PubMed] [Google Scholar]
- 157.Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, and Kastner DL (2006). The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1β production. Proc National Acad Sci 103, 9982–9987. 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hesker PR, Nguyen M, Kovarova M, Ting JP-Y, and Koller BH (2012). Genetic Loss of Murine Pyrin, the Familial Mediterranean Fever Protein, Increases Interleukin-1β Levels. Plos One 7, e51105. 10.1371/journal.pone.0051105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Papin S, Cuenin S, Agostini L, Martinon F, Werner S, Beer H-D, Grütter C, Grütter M, and Tschopp J (2007). The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1β processing. Cell Death Differ 14, 1457–1466. 10.1038/sj.cdd.4402142. [DOI] [PubMed] [Google Scholar]
- 160.Chae JJ, Cho Y-H, Lee G-S, Cheng J, Liu PP, Feigenbaum L, Katz SI, and Kastner DL (2011). Gain-of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1β Activation and Severe Autoinflammation in Mice. Immunity 34, 755–768. 10.1016/j.immuni.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong Y-N, Peng X, Xi JJ, Chen S, et al. (2014). Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241. 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 162.Mansfield E, Chae JJ, Komarow HD, Brotz TM, Frucht DM, Aksentijevich I, and Kastner DL (2001). The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood 98, 851–859. 10.1182/blood.v98.3.851. [DOI] [PubMed] [Google Scholar]
- 163.Hodge RG, and Ridley AJ (2016). Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Bio 17, 496–510. 10.1038/nrm.2016.67. [DOI] [PubMed] [Google Scholar]
- 164.Kim ML, Chae JJ, Park YH, Nardo DD, Stirzaker RA, Ko H-J, Tye H, Cengia L, DiRago L, Metcalf D, et al. (2015). Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β. J Exp Med 212, 927–938. 10.1084/jem.20142384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Park YH, Wood G, Kastner DL, and Chae JJ (2016). Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 17, 914–921. 10.1038/ni.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gao W, Yang J, Liu W, Wang Y, and Shao F (2016). Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc National Acad Sci 113, E4857–E4866. 10.1073/pnas.1601700113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ratner D, Orning MPA, Proulx MK, Wang D, Gavrilin MA, Wewers MD, Alnemri ES, Johnson PF, Lee B, Mecsas J, et al. (2016). The Yersinia pestis Effector YopM Inhibits Pyrin Inflammasome Activation. Plos Pathog 12, e1006035. 10.1371/journal.ppat.1006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, Chae JJ, and Bliska JB (2016). The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome. Cell Host Microbe 20, 296–306. 10.1016/j.chom.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Caprio GD, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, et al. (2020). HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369. 10.1126/science.aas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Faucheu C, Blanchet A, Collard‐Dutilleul V, Lalanne J, and Diu‐Hercend A (1996). Identification of a Cysteine Protease Closely Related to Interleukin‐1β‐Converting Enzyme. Eur J Biochem 236, 207–213. 10.1111/j.1432-1033.1996.t01-1-00207.x. [DOI] [PubMed] [Google Scholar]
- 171.Munday NA, Vaillancourt JP, Ali A, Casano FJ, Miller DK, Molineaux SM, Yamin T-T, Yu VL, and Nicholson DW (1995). Molecular Cloning and Pro-apoptotic Activity of ICErelII and ICErelIII, Members of the ICE/CED-3 Family of Cysteine Proteases (*). J Biol Chem 270, 15870–15876. 10.1074/jbc.270.26.15870. [DOI] [PubMed] [Google Scholar]
- 172.Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, Greenberg AH, and Yuan J (1996). Identification and Characterization of Ich-3, a Member of the Interleukin-1β Converting Enzyme (ICE)/Ced-3 Family and an Upstream Regulator of ICE*. J Biol Chem 271, 20580–20587. 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- 173.Kamens J, Paskind M, Hugunin M, Talanian RV, Allen H, Banach D, Bump N, Hackett M, Johnston CG, Li P, et al. (1995). Identification and Characterization of ICH-2, a Novel Member of the Interleukin-1β-converting Enzyme Family of Cysteine Proteases (*). J Biol Chem 270, 15250–15256. 10.1074/jbc.270.25.15250. [DOI] [PubMed] [Google Scholar]
- 174.Kang S-J, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, Huang Z, Srinivasan A, Tomaselli KJ, Thornberry NA, et al. (2000). Dual Role of Caspase-11 in Mediating Activation of Caspase-1 and Caspase-3 under Pathological Conditions. J Cell Biology 149, 613–622. 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, and Shao F (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 176.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, and Monack DM (2012). Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291. 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 178.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, et al. (2013). Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science 339, 975–978. 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, Zamboni DS, and Roy CR (2013). Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc National Acad Sci 110, 1851–1856. 10.1073/pnas.1211521110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, et al. (2013). Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 341, 1246–1249. 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 181.Hagar JA, Powell DA, Aachoui Y, Ernst RK, and Miao EA (2013). Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 341, 1250–1253. 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schauvliege R, Vanrobaeys J, Schotte P, and Beyaert R (2002). Caspase-11 Gene Expression in Response to Lipopolysaccharide and Interferon-γ Requires Nuclear Factor-κB and Signal Transducer and Activator of Transcription (STAT) 1*. J Biol Chem 277, 41624–41630. 10.1074/jbc.m207852200. [DOI] [PubMed] [Google Scholar]
- 183.Yen J-H, and Ganea D (2009). Interferon β induces mature dendritic cell apoptosis through caspase-11/caspase-3 activation. Blood 114, 1344–1354. 10.1182/blood-2008-12-196592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, and Fitzgerald KA (2012). TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell 150, 606–619. 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Meunier E, Dick MS, Dreier RF, Schürmann N, Broz DK, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, et al. (2014). Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509, 366–370. 10.1038/nature13157. [DOI] [PubMed] [Google Scholar]
- 186.Kutsch M, Sistemich L, Lesser CF, Goldberg MB, Herrmann C, and Coers J (2020). Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. Embo J 39, e104926. 10.15252/embj.2020104926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, Heilig R, Chen KW, Lim RYH, and Broz P (2020). Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun 11, 3276. 10.1038/s41467-020-16889-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wandel MP, Kim B-H, Park E-S, Boyle KB, Nayak K, Lagrange B, Herod A, Henry T, Zilbauer M, Rohde J, et al. (2020). Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol 21, 880–891. 10.1038/s41590-020-0697-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, et al. (2018). The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49, 740–753.e7. 10.1016/j.immuni.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ramirez MLG, Poreba M, Snipas SJ, Groborz K, Drag M, and Salvesen GS (2018). Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J Biol Chem 293, 7058–7067. 10.1074/jbc.ra117.001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, Anderson K, Warming S, Zhang J, Lee WP, and Kayagaki N (2018). Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med 215, 2279–2288. 10.1084/jem.20180589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rühl S, and Broz P (2015). Caspase‐11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Eur. J. Immunol 45, 2927–2936. 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- 193.Zanoni I, Tan Y, Gioia MD, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, et al. (2016). An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236. 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, Dorfleutner A, and Stehlik C (2018). The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nature Communications, 1–16. 10.1038/s41467-018-03409-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Vladykovskaya E, Ozhegov E, Hoetker JD, Xie Z, Ahmed Y, Suttles J, Srivastava S, Bhatnagar A, and Barski OA (2011). Reductive metabolism increases the proinflammatory activity of aldehyde phospholipids[S]. J Lipid Res 52, 2209–2225. 10.1194/jlr.m013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li J, Brieher WM, Scimone ML, Kang SJ, Zhu H, Yin H, von Andrian UH, Mitchison T, and Yuan J (2007). Caspase-11 regulates cell migration by promoting Aip1–Cofilin-mediated actin depolymerization. Nat Cell Biol 9, 276–286. 10.1038/ncb1541. [DOI] [PubMed] [Google Scholar]
- 197.Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DHA, Voss OH, Doseff AI, Hassan H, Azad AK, et al. (2012). Caspase-11 Promotes the Fusion of Phagosomes Harboring Pathogenic Bacteria with Lysosomes by Modulating Actin Polymerization. Immunity 37, 35–47. 10.1016/j.immuni.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chen J, and Chen ZJ (2018). PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature, 1–26. 10.1038/s41586-018-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, et al. (2013). Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 39, 311–323. 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z, Chen S, Zhang L, Liu Q, Guo X, et al. (2018). Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity 49, 842–856.e7. 10.1016/j.immuni.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 201.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, and Akira S (2013). Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14, 454–460. 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 202.Zhou R, Yazdi AS, Menu P, and Tschopp J (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 203.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, and Germain RN (2013). The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell 153, 348–361. 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Daussy CF, Monard SC, Guy C, Muñoz-González S, Chazal M, Anthonsen MW, Jouvenet N, Henry T, Dreux M, Meurs EF, et al. (2021). The Inflammasome Components NLRP3 and ASC Act in Concert with IRGM To Rearrange the Golgi Apparatus during Hepatitis C Virus Infection. J Virol 95, e00826–20. 10.1128/jvi.00826-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhang Z, Venditti R, Ran L, Liu Z, Vivot K, Schürmann A, Bonifacino JS, Matteis MAD, and Ricci R (2023). Distinct changes in endosomal composition promote NLRP3 inflammasome activation. Nat Immunol 24, 30–41. 10.1038/s41590-022-01355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lee B, Hoyle C, Wellens R, Green JP, Martin-Sanchez F, Williams DM, Matchett BJ, Seoane PI, Bennett H, Adamson A, et al. (2023). Disruptions in endocytic traffic contribute to the activation of the NLRP3 inflammasome. Sci Signal 16, eabm7134. 10.1126/scisignal.abm7134. [DOI] [PubMed] [Google Scholar]
- 207.He Y, Zeng MY, Yang D, Motro B, and Núñez G (2016). NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357. 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, et al. (2016). NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17, 250–258. 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, and Hornung V (2016). A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation*. J Biol Chem 291, 103–109. 10.1074/jbc.c115.700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schmacke NA, O’Duill F, Gaidt MM, Szymanska I, Kamper JM, Schmid-Burgk JL, Mädler SC, Mackens-Kiani T, Kozaki T, Chauhan D, et al. (2022). IKKβ primes inflammasome formation by recruiting NLRP3 to the trans-Golgi network. Immunity. 10.1016/j.immuni.2022.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang L, Fu T-M, Jacobson MP, Greka A, et al. (2021). Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593, 607–611. 10.1038/s41586-021-03478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, Holley CL, Boucher D, Shakespear MR, Kapetanovic R, et al. (2018). Interleukin-1β Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and - Independent Secretion. CellReports 24, 1425–1433. 10.1016/j.celrep.2018.07.027. [DOI] [PubMed] [Google Scholar]
- 213.Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, Wilson R, Jiang Z, Khalighinejad F, Muneeruddin K, et al. (2020). Succination inactivates gasdermin D and blocks pyroptosis. Science 369, 1633–1637. 10.1126/science.abb9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Evavold CL, Hafner-Bratkovič I, Devant P, D’Andrea JM, Ngwa EM, Boršić E, Doench JG, LaFleur MW, Sharpe AH, Thiagarajah JR, et al. (2021). Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 184, 4495–4511.e19. 10.1016/j.cell.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, Coleman AK, VanPortfliet JJ, Zhao B, Wagner AR, et al. (2022). Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 185, 3214–3231.e23. 10.1016/j.cell.2022.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, Rehman J, and Malik AB (2020). Mitochondrial DNA Activates cGAS Signaling and Suppresses YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, et al. (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc National Acad Sci 115, E10888–E10897. 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Demarco B, Grayczyk JP, Bjanes E, Roy DL, Tonnus W, Assenmacher C-A, Radaelli E, Fettrelet T, Mack V, Linkermann A, et al. (2020). Caspase-8–dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv 6, eabc3465. 10.1126/sciadv.abc3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, Sandoval W, Yan D, Kang J, Xu M, et al. (2021). NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136. 10.1038/s41586-021-03218-7. [DOI] [PubMed] [Google Scholar]
- 220.Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu NI, Coggeshall KM, Arditi M, et al. (2016). Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 166, 624–636. 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chen KW, Groß CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, and Schroder K (2014). The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1β Maturation without Pyroptosis during Acute Salmonella Challenge. Cell Reports 8, 570–582. 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 222.Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, and Broz P (2018). ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960. 10.1126/science.aar7607. [DOI] [PubMed] [Google Scholar]
- 223.Nozaki K, Maltez VI, Rayamajhi M, Tubbs AL, Mitchell JE, Lacey CA, Harvest CK, Li L, Nash WT, Larson HN, et al. (2022). Caspase-7 activates ASM to repair gasdermin and perforin pores. Nature 606, 960–967. 10.1038/s41586-022-04825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, and Shao F (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 225.Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, et al. (2020). Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420. 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y, et al. (2020). Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368. 10.1126/science.aaz7548. [DOI] [PubMed] [Google Scholar]
- 227.Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti T-D (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1, aag2045. 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Deets KA, and Vance RE (2021). Inflammasomes and adaptive immune responses. Nat Immunol 22, 412–422. 10.1038/s41590-021-00869-6. [DOI] [PubMed] [Google Scholar]
- 229.Man SM, Karki R, and Kanneganti T (2017). Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277, 61–75. 10.1111/imr.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhang Y, Yang W, Li W, and Zhao Y (2021). NLRP3 Inflammasome: Checkpoint Connecting Innate and Adaptive Immunity in Autoimmune Diseases. Front Immunol 12, 732933. 10.3389/fimmu.2021.732933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Harapas CR, Steiner A, Davidson S, and Masters SL (2018). An Update on Autoinflammatory Diseases: Inflammasomopathies. Curr Rheumatol Rep 20, 40. 10.1007/s11926-018-0750-4. [DOI] [PubMed] [Google Scholar]
- 232.Booshehri LM, and Hoffman HM (2019). CAPS and NLRP3. J Clin Immunol 39, 277–286. 10.1007/s10875-019-00638-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Abbate A, Toldo S, Marchetti C, Kron J, Tassell BWV, and Dinarello CA (2020). Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ Res 126, 1260–1280. 10.1161/circresaha.120.315937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Alghamdi M (2017). Familial Mediterranean fever, review of the literature. Clin Rheumatol 36, 1707–1713. 10.1007/s10067-017-3715-5. [DOI] [PubMed] [Google Scholar]
- 235.Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y, McFann KK, Holland PJ, Fain PR, Dinarello CA, et al. (2013). NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1β processing via the NLRP1 inflammasome. Proc National Acad Sci 110, 2952–2956. 10.1073/pnas.1222808110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, Fain PR, and Spritz RA (2007). NALP1 in Vitiligo-Associated Multiple Autoimmune Disease. New Engl J Medicine 356, 1216–1225. 10.1056/nejmoa061592. [DOI] [PubMed] [Google Scholar]
- 237.Fenini G, Karakaya T, Hennig P, Filippo MD, and Beer H-D (2020). The NLRP1 Inflammasome in Human Skin and Beyond. Int J Mol Sci 21, 4788. 10.3390/ijms21134788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Bivik C, Verma D, Winge MC, Lieden A, Bradley M, Rosdahl I, and Söderkvist P (2013). Genetic Variation in the Inflammasome and Atopic Dermatitis Susceptibility. J Invest Dermatol 133, 2486–2489. 10.1038/jid.2013.168. [DOI] [PubMed] [Google Scholar]
- 239.Drutman SB, Haerynck F, Zhong FL, Hum D, Hernandez NJ, Belkaya S, Rapaport F, de Jong SJ, Creytens D, Tavernier SJ, et al. (2019). Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc National Acad Sci 116, 19055–19063. 10.1073/pnas.1906184116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Grandemange S, Sanchez E, Louis-Plence P, Mau-Them FT, Bessis D, Coubes C, Frouin E, Seyger M, Girard M, Puechberty J, et al. (2017). A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis 76, 1191. 10.1136/annrheumdis-2016-210021. [DOI] [PubMed] [Google Scholar]
- 241.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, DiMattia MA, Zaal KJM, Sanchez GAM, Kim H, et al. (2014). An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 46, 1140–1146. 10.1038/ng.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Romberg N, Vogel TP, and Canna SW (2017). NLRC4 inflammasomopathies. Curr Opin Allergy Cl 17, 398–404. 10.1097/aci.0000000000000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Romberg N, Moussawi KA, Nelson-Williams C, Stiegler AL, Loring E, Choi M, Overton J, Meffre E, Khokha MK, Huttner AJ, et al. (2014). Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 46, 1135–1139. 10.1038/ng.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, Surrey LF, Russo P, Sleight A, Schiffrin E, et al. (2017). Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immun 139, 1698–1701. 10.1016/j.jaci.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Briard B, Malireddi RKS, and Kanneganti T-D (2021). Role of inflammasomes/pyroptosis and PANoptosis during fungal infection. Plos Pathog 17, e1009358. 10.1371/journal.ppat.1009358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Hauwermeiren FV, Opdenbosch NV, Gorp HV, de Vasconcelos N, van Loo G, Vandenabeele P, Kanneganti T-D, and Lamkanfi M (2022). Bacillus anthracis induces NLRP3 inflammasome activation and caspase-8–mediated apoptosis of macrophages to promote lethal anthrax. Proc National Acad Sci 119, e2116415119. 10.1073/pnas.2116415119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Park YH, Remmers EF, Lee W, Ombrello AK, Chung LK, Shilei Z, Stone DL, Ivanov MI, Loeven NA, Barron KS, et al. (2020). Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis. Nat Immunol 21, 857–867. 10.1038/s41590-020-0705-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, and Ting JPY (2009). The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity 30, 556–565. 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Ichinohe T, Lee HK, Ogura Y, Flavell R, and Iwasaki A (2009). Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Medicine 206, 79–87. 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Thomas PG, Dash P, Aldridge JR, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, et al. (2009). The Intracellular Sensor NLRP3 Mediates Key Innate and Healing Responses to Influenza A Virus via the Regulation of Caspase-1. Immunity 30, 566–575. 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21, 248–255. 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Tate MD, Ong JDH, Dowling JK, McAuley JL, Robertson AB, Latz E, Drummond GR, Cooper MA, Hertzog PJ, and Mansell A (2016). Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci Rep-uk 6, 27912. 10.1038/srep27912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Cheong W-C, Kang H-R, Yoon H, Kang S-J, Ting JP-Y, and Song MJ (2015). Influenza A Virus NS1 Protein Inhibits the NLRP3 Inflammasome. Plos One 10, e0126456. 10.1371/journal.pone.0126456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Cheung PH, Ye Z, Lee TT, Chen H, Chan C, and Jin D (2020). PB1‐F2 protein of highly pathogenic influenza A (H7N9) virus selectively suppresses RNA‐induced NLRP3 inflammasome activation through inhibition of MAVS‐NLRP3 interaction. J Leukocyte Biol 108, 1655–1663. 10.1002/jlb.4ab0420-694r. [DOI] [PubMed] [Google Scholar]
- 255.Ichinohe T, Pang IK, and Iwasaki A (2010). Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11, 404–410. 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Junqueira C, Crespo Â, Ranjbar S, de Lacerda LB, Lewandrowski M, Ingber J, Parry B, Ravid S, Clark S, Schrimpf MR, et al. (2022). FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature, 1–13. 10.1038/s41586-022-04702-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, Brewer JR, Han A, Steach HR, Israelow B, et al. (2022). Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature, 1–1. 10.1038/s41586-022-04802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Rodrigues TS, de Sá KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, Gonçalves AV, Perucello DB, Andrade WA, Castro R, et al. (2020). Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Medicine 218, e20201707. 10.1084/jem.20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Barnett KC, Xie Y, Asakura T, Song D, Liang K, Taft-Benz SA, Guo H, Yang S, Okuda K, Gilmore RC, et al. (2022). An epithelial-immune circuit amplifies inflammasome and IL-6 responses to SARS-CoV-2. Cell Host Microbe. 10.1016/j.chom.2022.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kyriazopoulou E, Poulakou G, Milionis H, Metallidis S, Adamis G, Tsiakos K, Fragkou A, Rapti A, Damoulari C, Fantoni M, et al. (2021). Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nature Medicine, 1–22. 10.1038/s41591-021-01499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kyriazopoulou E, Huet T, Cavalli G, Gori A, Kyprianou M, Pickkers P, Eugen-Olsen J, Clerici M, Veas F, Chatellier G, et al. (2021). Effect of anakinra on mortality in patients with COVID-19: a systematic review and patient-level meta-analysis. Lancet Rheumatology 3, e690–e697. 10.1016/s2665-9913(21)00216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, and Yang H (2006). HMGB1 SIGNALS THROUGH TOLL-LIKE RECEPTOR (TLR) 4 AND TLR2. Shock 26, 174–179. 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 263.Yang D, Han Z, and Oppenheim JJ (2017). Alarmins and immunity. Immunol Rev 280, 41–56. 10.1111/imr.12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, and Dixit VM (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 265.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galán JE, Askenase PW, et al. (2006). Critical Role for NALP3/CIAS1/Cryopyrin in Innate and Adaptive Immunity through Its Regulation of Caspase-1. Immunity 24, 317–327. 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 266.Martinon F, Pétrilli V, Mayor A, Tardivel A, and Tschopp J (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 267.Moon J-S, Hisata S, Park M-A, DeNicola GM, Ryter SW, Nakahira K, and Choi AMK (2015). mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Reports 12, 102–115. 10.1016/j.celrep.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 268.Tartey S, and Kanneganti T (2020). Inflammasomes in the pathophysiology of autoinflammatory syndromes. J Leukocyte Biol 107, 379–391. 10.1002/jlb.3mir0919-191r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Voet S, Srinivasan S, Lamkanfi M, and Loo G (2019). Inflammasomes in neuroinflammatory and neurodegenerative diseases. Embo Mol Med 11, e10248. 10.15252/emmm.201810248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. (2009). Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat Med 15, 1170–1178. 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 271.Zhivaki D, Borriello F, Chow OA, Doran B, Fleming I, Theisen DJ, Pallis P, Shalek AK, Sokol CL, Zanoni I, et al. (2020). Inflammasomes within Hyperactive Murine Dendritic Cells Stimulate Long-Lived T Cell-Mediated Anti-tumor Immunity. Cell Reports 33, 108381. 10.1016/j.celrep.2020.108381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, et al. (2008). Overexpression of Interleukin-1β Induces Gastric Inflammation and Cancer and Mobilizes Myeloid-Derived Suppressor Cells in Mice. Cancer Cell 14, 408–419. 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Chow MT, Sceneay J, Paget C, Wong CSF, Duret H, Tschopp J, Möller A, and Smyth MJ (2012). NLRP3 Suppresses NK Cell–Mediated Responses to Carcinogen-Induced Tumors and Metastases. Cancer Res 72, 5721–5732. 10.1158/0008-5472.can-12-0509. [DOI] [PubMed] [Google Scholar]
- 274.Su S, Zhao J, Xing Y, Zhang X, Liu J, Ouyang Q, Chen J, Su F, Liu Q, and Song E (2018). Immune Checkpoint Inhibition Overcomes ADCP-Induced Immunosuppression by Macrophages. Cell 175, 442–457.e23. 10.1016/j.cell.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 275.Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, Heid B, Cecere TE, and Allen IC (2015). The NLRP1 Inflammasome Attenuates Colitis and Colitis-Associated Tumorigenesis. J Immunol 194, 3369–3380. 10.4049/jimmunol.1402098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Allen IC, TeKippe EM, Woodford R-MT, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, and Ting JP-Y (2010). The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207, 1045–1056. 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Holvoet P, Mertens A, Verhamme P, Bogaerts K, Beyens G, Verhaeghe R, Collen D, Muls E, and de Werf FV (2001). Circulating Oxidized LDL Is a Useful Marker for Identifying Patients With Coronary Artery Disease. Arteriosclerosis Thrombosis Vasc Biology 21, 844–848. 10.1161/01.atv.21.5.844. [DOI] [PubMed] [Google Scholar]
- 279.Couillard C, Ruel G, Archer WR, Pomerleau S, Bergeron J, Couture P, Lamarche B, and Bergeron N (2005). Circulating Levels of Oxidative Stress Markers and Endothelial Adhesion Molecules in Men with Abdominal Obesity. J Clin Endocrinol Metabolism 90, 6454–6459. 10.1210/jc.2004-2438. [DOI] [PubMed] [Google Scholar]
- 280.Ho RC, Davy K, Davy B, and Melby CL (2002). Whole-body insulin sensitivity, low-density lipoprotein (LDL) particle size, and oxidized LDL in overweight, nondiabetic men. Metabolis 51, 1478–1483. 10.1053/meta.2002.35577. [DOI] [PubMed] [Google Scholar]
- 281.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, et al. (2010). Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol 11, 897–904. 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Barath P, Fishbein MC, Cao J, Berenson J, Helfant RH, and Forrester JS (1990). Detection and localization of tumor necrosis factor in human atheroma. Am J Cardiol 65, 297–302. 10.1016/0002-9149(90)90291-8. [DOI] [PubMed] [Google Scholar]
- 283.Bauernfeind F, Niepmann S, Knolle PA, and Hornung V (2016). Aging-Associated TNF Production Primes Inflammasome Activation and NLRP3-Related Metabolic Disturbances. J Immunol 197, 2900–2908. 10.4049/jimmunol.1501336. [DOI] [PubMed] [Google Scholar]
- 284.Jager J, Grémeaux T, Cormont M, Marchand-Brustel YL, and Tanti J-F (2007). Interleukin-1β-Induced Insulin Resistance in Adipocytes through Down-Regulation of Insulin Receptor Substrate-1 Expression. Endocrinology 148, 241–251. 10.1210/en.2006-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT-H, Brickey WJ, and Ting JP-Y (2011). Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12, 408–415. 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, and Dixit VD (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17, 179–188. 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Reynolds CM, McGillicuddy FC, Harford KA, Finucane OM, Mills KHG, and Roche HM (2012). Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells—implications for diet‐induced insulin resistance. Mol Nutr Food Res 56, 1212–1222. 10.1002/mnfr.201200058. [DOI] [PubMed] [Google Scholar]
- 288.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. (2012). Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185. 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, et al. (2010). The NLRP3 Inflammasome Promotes Renal Inflammation and Contributes to CKD. J Am Soc Nephrol 21, 1732–1744. 10.1681/asn.2010020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Kammoun HL, Allen TL, Henstridge DC, Barre S, Coll RC, Lancaster GI, Cron L, Reibe S, Chan JY, Bensellam M, et al. (2018). Evidence against a role for NLRP3-driven islet inflammation in db/db mice. Mol Metab 10, 66–73. 10.1016/j.molmet.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Heneka MT, McManus RM, and Latz E (2018). Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19, 610–621. 10.1038/s41583-018-0055-7. [DOI] [PubMed] [Google Scholar]
- 292.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng T-C, et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, et al. (2012). DICER1 Loss and Alu RNA Induce Age- Related Macular Degeneration via the NLRP3 Inflammasome and MyD88. Cell 149, 847–859. 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang A-S, Humphries MM, Lavelle EC, et al. (2012). NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med 18, 791–798. 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, and de Bernard M (2013). Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. Plos One 8, e55375. 10.1371/journal.pone.0055375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, Robertson AAB, Butler MS, Rowe DB, O’Neill LA, et al. (2018). Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 10. 10.1126/scitranslmed.aah4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, and Golenbock DT (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol 9, 857–865. 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Coll RC, Schroder K, and Pelegrín P (2022). NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci 43, 653–668. 10.1016/j.tips.2022.04.003. [DOI] [PubMed] [Google Scholar]
- 299.Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, Tardivel A, Mattmann C, and Tschopp J (2011). Differential Expression of NLRP3 among Hematopoietic Cells. J Immunol 186, 2529–2534. 10.4049/jimmunol.1002720. [DOI] [PubMed] [Google Scholar]
- 300.Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al. (2017). Caspase-11–mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest 127, 4124–4135. 10.1172/jci94495. [DOI] [PMC free article] [PubMed] [Google Scholar]