

ABSTRACT
Hypertension is one of the major health problems leading to the development of cardiovascular diseases. Despite a rapid expansion in global hypertension prevalence, molecular mechanisms leading to hypertension are not fully understood largely due to the complexity of pathogenesis involving several factors. Salt intake is recognized as a leading determinant of blood pressure, since reduced dietary salt intake is related to lower morbidity and mortality, and hypertension in relation to cardiovascular events. Compared with salt-resistant populations, salt-sensitive individuals exhibit high sensitivity in blood pressure responses according to changes in salt intake. In this setting, the kidney plays a major role in the maintenance of blood pressure under the hormonal control of the renin–angiotensin–aldosterone system. In the present review, we summarize the current overview on the molecular mechanisms for modulation of blood pressure associated with renal ion channels/transporters including sodium–hydrogen exchanger isoform 3 (NHE3), Na+-K+-2Cl– cotransporter (NKCC2), sodium–chloride cotransporter (NCC), epithelial sodium channel (ENaC) and pendrin expressed in different nephron segments. In particular, recent studies on experimental animal models with deletion of renal ion channels led to the identification of several crucial physiological mechanisms and molecules involved in hypertension. These findings could further provide a potential for novel therapeutic approaches applicable on human patients with hypertension.
Keywords: blood pressure regulation, hypertension, renin–angiotensin–aldosterone system (RAAS), renal salt transport
INTRODUCTION
Hypertension is a major health problem in people over 50 years old, associated with cardiovascular events [1]. According to population-representative studies, the global prevalence of hypertension in adult people aged 30–79 years old doubled from 1990 to 2019 [2]. In particular, a remarkable increase in age-standardized prevalence was observed in low-income countries. Despite a rapid expansion in global hypertension prevalence, molecular mechanisms leading to hypertension are not fully understood largely due to the complexity of pathogenesis involving several factors such as genetic variation, dietary patterns (salt, potassium, fiber, protein, fat intake), alcohol consumption, insufficient physical activity and obesity [3, 4].
Salt (NaCl) intake is recognized as a leading determinant of blood pressure, as reduced dietary salt intake is associated with lower morbidity and mortality, and hypertension in relation to cardiovascular events [5]. It is of note that we distinguish ‘salt (NaCl)’ and ‘sodium (Na)’ intake, since sodium in conjunction with chloride is indispensable for the development of hypertension [6, 7]. The relationship between blood pressure and dietary salt intake has been also demonstrated in animal models [8, 9]. Compared with salt-resistant populations, salt-sensitive individuals exhibit high sensitivity in blood pressure responses according to changes in salt intake [10–12]. In this setting, kidney plays a pivotal role on the modulation of blood pressure via extracellular fluid volume regulation acquired through salt reabsorption activity, under the control of the renin–angiotensin–aldosterone system (RAAS).
The human adult kidney typically filters approximately 20–25 moles of sodium per day, and more than 99% of filtered sodium is reabsorbed into the circulation through sodium transporters/exchangers located along different nephron segments [13, 14]. Na+/K+-ATPase ubiquitously expressed at the basolateral membrane along the nephron provides an essential driving force for the sodium entry from the apical side through an electrochemical gradient [14]. However, sensing of sodium and the regulation of the rate-limiting step of trans-epithelial sodium transport is located on the entry side at the apical membrane [15]. Sodium–hydrogen exchanger isoform 3 (NHE3) is chiefly responsible for the apical sodium transport in the proximal tubules (PT) where almost 70% of sodium is absorbed [16]. Approximately 25% of salt reuptake is through Na+-K+-2Cl− cotransporter (NKCC2) expressed on the apical membrane of the thick ascending limb of the loop of Henle (TAL) [17]. Aldosterone-sensitive distal nephron (ASDN) finely tunes salt reabsorption through several transporters including the thiazide-sensitive sodium-chloride cotransporter (NCC) at distal convoluted tubules (DCT) [18], and amiloride-sensitive epithelial sodium channel (ENaC) in principal cells (PC) of connecting tubule and collecting duct (CD) [19]. In addition, recent studies demonstrated a participation of intercalated cells (ICs) for salt uptake through coupling of pendrin and Na+-dependent Cl−/2HCO3− exchanger (NDCBE) [20]. In the present review, we summarize the current overview on molecular mechanisms for modulation of blood pressure associated with renal ion channels/transporters described above.
THE ROLES OF NHE3 IN THE CONTROL OF BLOOD PRESSURE
NHE3 is encoded by the solute carrier family 9 member A3 (SLC9A3) gene [21], belonging to the Na+/H+ exchanger family composed of nine isoforms. The N-terminal region of NHE family exchangers is located in the extracellular part and plays a role in solute exchange, while the C-terminus is involved in the hormonal regulations of the NHE family [22, 23]. In the kidney, NHE3 is mainly expressed in the brush border of PT together with sodium-glucose cotransporters (SGLT) and sodium-phosphate cotransporter (Napi-2), and exchanger activity of NHE3 directly or indirectly contributes to the absorption of Na+, Cl−, HCO3− and water (Fig. 1). In addition to the major role in the systemic acid–base balance homeostasis, renal NHE3 is responsible for more than 50% of reabsorption of filtered sodium. NHE3 is also expressed in the gastrointestinal tract and involved in absorption of the majority of ingested sodium [24]. Indeed, mutations in the SLC9A3 gene could lead to a rare genetic disorder, congenital secretory sodium diarrhoea 8 [25].
Figure 1:

Sodium transporters, exchangers and ion channels along the nephron. NHE3, sodium-hydrogen exchanger isoform 3; Napi-2a, sodium-phosphate cotransporter 2a; SGLT2, sodium-glucose cotransporter; NKCC2, Na+-K+-2Cl− cotransporter; ROMK, renal outer medullary potassium channel; ClC-Kb, chloride channel Kb; NCC, sodium-chloride cotransporter; Kir4.1, inward-rectifying potassium channel 4.1; ENaC, epithelial sodium channel; NDCBE, sodium-driven chloride/bicarbonate exchanger; vH+-ATPase, vacuolar H+-ATPase.
Accumulating evidence obtained from animal models targeting NHE3 revealed a fundamental role of NHE3 on the maintenance of blood pressure as well as involvement in angiotensin II (Ang II)-induced hypertension [26] (Table 1). A mouse model with global NHE3 deletion exhibited slight diarrhoea, mild acidosis and reduced blood pressure with significantly high level of plasma aldosterone [24]. Renal expression of renin and Cl−/HCO3− exchanger AE1 mRNAs are elevated. Severe absorptive defect was evident in the intestine, and both ENaC activity and H+-K+-ATPase mRNA were significantly increased in the colon, which might be part of compensatory response. Targeted proteomics approach on the kidney of NHE3 knockout mice revealed significant enhancements of NaPi-2 in PT and cleaved γ-ENaC in CD in addition to the considerable reduction in glomerular filtration [27]. Woo et al. generated and characterized the tgNhe3−/− mouse, in which NHE3 expression in global NHE3 deletion was rescued transgenically only in the small intestines using the intestinal fatty acid binding protein (IFABP) promoter [28]. In tgNhe3−/− mice, mild to moderate diarrhoea and increased faecal Na+ excretion were observed. Basal systolic blood pressure and mean intra-arterial blood pressure as well as glomerular filtration, urine volume and urinary sodium/potassium/chloride excretions were significantly lower in tgNhe3−/− mice compared with its littermates. These phenotypes were associated with considerably enhanced plasma Ang II and aldosterone levels in tgNhe3−/− mice [28–30]. Another mouse model targeting NHE3 in the kidney, namely Pax8-Cre/NHE3-floxed mouse, showed significantly higher urinary pH with no evidence for metabolic acidosis, while glomerular filtration, food and fluid consumption were similar to those in control mice. Normal body salt and fluid, as well as acid and base balances in these mice, suggested the compensatory mechanism for the NHE3 deletion in the kidney. However, intra-arterial blood pressure was considerably lower in Pax8-Cre/NHE3-floxed mice, and increased sensitivity to dietary salt (20%) was marked [31, 32]. Recently, a mouse model with PT-specific NHE3 deletion was generated through a SGLT2-Cre/NHE3-floxed approach (PT-Nhe3−/−) [33–35]). These mice demonstrated normal functions and structures in gastrointestinal tract, which was abnormally altered in global Nhe3−/− and to a lesser extent in tgNhe3−/− mouse models [24, 28, 30]. PT-Nhe3−/− mice showed similar 24 h faecal Na+ excretion to control mice, with no sign of diarrhoea. Notably, an inducible intestinal epithelial cell-specific NHE3 knockout mouse model exhibited marked abnormality in intestinal absorptive ability and diarrhoea [36, 37]. Therefore, in agreement with similar phenotypes observed in global and in intestine-specific Nhe3−/− mice as well as human patients with absent or mutated NHE3 protein [38], it is likely that diarrhoea is due to the defect in intestinal NHE3. PT-Nhe3−/− mice showed significantly increased 24-h urine volume, urinary sodium and potassium excretions with a drop of 12–15 mmHg in blood pressure compared with wild-type (WT) mice. Therefore, deletion of NHE3 in the PT is sufficient to alter the blood pressure. Molecular mechanisms and hormonal regulations in both renal and intestinal NHE3 require further investigations as they might be distinctively regulated [26]. In addition, spontaneous hypertensive rats (SHR) demonstrate the altered NHE3 levels suggesting the involvement in the pathology of hypertension. Expression and activity of NHE3 in PT were increased both in the pre-hypertensive and adults stages of SHR [39].
Table 1:
Main findings in mouse models targeting renal sodium transporters and modulators.
| Target protein | Animal model | Phenotypes | Reference |
|---|---|---|---|
| NHE3 (Slc9a3) | Nhe3 −/− | Slight diarrhoea, mild acidosis, low blood pressure, high plasma aldosterone, intestinal absorptive defect, high renal expression of renin and AE1 mRNA, high activity of ENaC and H+-K+-ATPase mRNA in the colon, lower response of blood pressure to Ang II in fusion | [24, 27] |
| tgNhe3−/− (transgenic rescue of small intestinal NHE3) | Mild to moderate diarrhoea, increased faecal Na+ excretion, low systolic blood pressure and mean intra-arterial blood pressure, low glomerular filtration rate, low urine volume, low urinary sodium/potassium/chloride excretions, high plasma Ang II and aldosterone levels, lower response of blood pressure to Ang II in fusion | [28–30] | |
| Pax8-Cre/NHE3-floxed (renal tubulus-specific NHE3 deletion) | Higher urinary pH, low intra-arterial blood pressure, increased sensitivity to dietary salt | [31, 32] | |
| PT-Nhe3−/− (proximal tubule-specific deletion of NHE3) | High 24-h urine volume, high urinary sodium and potassium excretions, low blood pressure (12–15 mmHg lower than WT), lower response of blood pressure to Ang II in fusion | [33–35] | |
| NKCC2 (Slc12a1) | NKCC2−/− | Signs of extracellular volume depletion 1 day after birth, failure to thrive, small body, marked dehydration, renal insufficiency, high plasma potassium, metabolic acidosis, hydronephrosis, high plasma renin concentration, no survival before weaning | [51] |
| NCC (Slc12a3) | NCC−/− | Mild perturbations of sodium and fluid volume homeostasis, normal blood pressure, normal acid–base and plasma electrolyte concentrations, normal serum aldosterone levels, development of hyportension hypotension in response to 2 weeks of sodium depletion, structural remodelling in DCT, increased abundance of cleaved γ-ENaC | [27, 72, 73] |
| Kir4.1 (Kcnj10) | KS-Kir4.1 KO (kidney-specific knockout of Kir4.1) | Increased basal urinary Na+ excretion, no significant natriuretic effect of HCTZ, hypokalaemia and metabolic alkalosis under normal and low potassium condition | [75] |
| WNK4 (Wnk4) | WNK4−/− | Higher plasma pH, lower plasma Na+ and Cl–, lower systolic blood pressure under low-salt diet, considerable decrease of total and phosphorylated NCC | [85] |
| WNK1 (Wnk1) | Wnk1+/− (Wnk1 heterozygote) | Low blood pressure, while homozygous Wnk1–/– died during embryonic development | [83] |
| SPAK (Stk39) | SPAK−/− | Hypotension, hypokalemia, hypomagnesemia, hypocalciuria | [91] |
| α-ENaC (scnn1a) | αENaC−/− | Lethal respiratory distress syndrome, metabolic acidosis | [99, 100] |
| β-ENaC (scnn1b) | βENaC−/− | Delayed liquid clearance at birth, salt wasting, lethal hyperkalaemia, metabolic acidosis | [99] |
| γ-ENaC (scnn1g) | γENaC−/− | Low urinary potassium, high urinary sodium, metabolic acidosis, died between 24–36 h after birth, slow lung fluid clearance at birth | [98, 99] |
| Pendrin (Slc26a4) | Slc26a4−/− | Enhanced urinary volume and chloride excretion in response to moderate salt restriction, hypotension under salt depletion, impaired bicarbonate secretion in CD, acidic urine pH and elevated serum HCO3 concentration | [119–121] |
| Tg[E];Tg[R];Slc26a4Δ/Δ conditional transgenic | Lower blood pressure in response to acute pendrin ablation | [121] | |
| TgB1-hPDS (overexpression of pendrin in intercalated cells) | Hypertension, delayed increase in urinary NaCl under high-salt diet | [130] | |
| Pendrin/NCC (slc26a4/slc12a3) | Pendrin/NCC double knockout (dKO) | Significantly lower blood pressure, renal failure and metabolic alkalosis under basal condition, severe volume depletion and renal failure | [122] |
| NDCBE (Slc4a8) | Ndcbe−/− | Mild perturbations of Na+ homeostasis, no changes in blood pressure | [129] |
| NDCBE/NCC (Slc4a8/Slc12a3) | Ndcbe/Ncc double knockout (dKO) | Hypokalemia, upregulation of ENaC and Ca2+-activated K+ channel BKCa under basal conditions, remarkable intravascular volume depletion induced by salt restriction | [129] |
NHE3 is regulated by multiple factors including Ang II, parathyroid hormone (PTH), insulin, dopamine, glucocorticoids, protein kinase A (PKA), cAMP and cGMP. Among them, Ang II is one of the main regulators of NHE3 through modulation of NHE3 trafficking between apical membrane and cytoplasm [40–42]. The fundamental role of NHE3 in the development of Ang II–induced hypertension was demonstrated by Ang II infusion on WT, global Nhe3−/−, tgNhe3−/− and PT-Nhe3−/− mice. Responses to Ang II infusion in all knockout mouse models were significantly impaired, with almost 50% lower blood pressure values with respect to WT [26, 43]. These observations further support the hypothesis that NHE3 could serve as a potential therapeutic target to treat human hypertension. So far, a few NHE3 inhibitors were tested in vivo (Table 2). SAR218034 was demonstrated to lower the blood pressure associated with enhanced faecal sodium excretion when administrated in SHR [44]. Treatment with non-systemic drug tenapanor targeting intestinal NHE3 to block the absorption of ingested sodium showed minimal systemic effects [44, 45]. Absorbable drug AVE-0657 induced natriuresis and significantly impaired hypertensive response in Ang II–infused, 2% high salt-fed C57BL/6 J mice without changes in fecal Na+ excretion [26, 34, 35]. Finally, NHE3-mediated sodium transport has been considered fundamental also for the natriuretic effect promoted by SGLT2 inhibitors. Indeed, empagliflozin reduces NHE3 transport activity in rats, showing a tight relation between SGLT2 and NHE3 transporters [46]. This has been recently highlighted in a mice model mimicking the Fanconi syndrome secondary to glycogen storage disease 1b [47].
Table 2:
Putative: therapeutic targets for hypertension and their potential drugs discussed in the present manuscript.
| Target | Potential drugs | Observed effect | Reference |
|---|---|---|---|
| NHE3 | SAR218034 | Reduction in blood pressure in Spontaneously Hypertensive rats | [44] |
| Tenapanor | Blockage of absorption of ingested sodium with minimal systemic effects | [44, 45] | |
| AVE-0657 | Induction of natriuresis, impaired hypertensive response in Ang II–infused, 2% high salt-fed C57BL/6 J mice without changes in faecal Na+ excretion | [26, 34, 35] | |
| Empagliflozin (SGLT2 inhibitor) | Reduction of NHE3 transport activity in rats and mice | [46, 47] | |
| SPAK (CUL3/KLHL3–WNK1/4–SPAK/OSR1 regulatory cascade) | STOCK1S-50699 STOCK2S-26 016 | Inhibition of SPAK interaction to WNK, and inhibition on phosphorylation of SPAK and NCC | [92] |
| ZT-1a | Inhibitory effect on NCC phosphorylation in SPAK-dependent manner in mouse kidney | [93] |
THE ROLES OF NKCC2 ON THE CONTROL OF BLOOD PRESSURE
NKCC2 encoded by the SLC12A1 gene belongs to the cation-chloride cotransporters (CCCs). The members of CCCs mediate the coupling of Cl− with Na+ and/or K+ across the plasma membrane. Transmembrane domain of CCCs is responsible for ion translocation, while intracellular N- and C-terminal domains play a role in transport and trafficking activities [48]. NKCC2 is a kidney-specific, Na+-dependent Na+-K+-Cl− cotransporter mainly expressed in the TAL. At this site, apically expressed NKCC2 generates a hyperosmotic renal medulla through a countercurrent multiplier mechanism. In tandem to NKCC2 activity, renal outer medullary potassium channel (ROMK) transports potassium out of the cells for the maintenance of the luminal potassium concentration (Fig. 1). Loss-of-function mutations in either NKCC2 or ROMK have been associated with the blood pressure disorder Bartter syndrome (BS) type I and II, respectively. Mutations in kidney-specific chloride channel ClC-Kb and its essential β-subunit Barttin could also lead to distinguishable BS corresponding to type III and IV (Fig. 2) [49].
Figure 2:

Rare genetic blood pressure disorders associated with mutations in genes encoding sodium transporters, exchangers, ion channels and their regulators expressed along the nephron.
Patients with BS show hypokalaemic alkalosis, hypercalciuria, polyuria and low blood pressure with early presentation of severe volume depletion [50]. Six homozygous mutations in NKCC2 (G193R, A267S, G319R, A508T, del526N and Y998X) identified in patients with BS type I showed low expression levels and impaired sodium transport activities compared with WT channel when expressed in Xenopus oocytes [49]. A mouse model with deletion of NKCC2 (NKCC2−/− mice) presented with extracellular volume depletion 1 day after birth and failed to thrive, not surviving up to weaning [51] (Table 1). In contrast to BS, elevated NKCC2 activity is associated with hypertension, which may also involve the increased ability to conserve water, increased glomerular capillary hydraulic pressure and predilection to glomerular injury [52]. Administration of calcineurin inhibitor cyclosporine A enhanced NKCC2 phosphorylation, salt retention and hypertension along with stimulation of renin and suppression of renal cyclooxygenase 2 (COX2) in Wistar rats [53].
The expression and activity of NKCC2 in TAL are regulated by multiple hormones including vasopressin, PTH, calcitonin, glucagon, as well as β-adrenergic agonists including isoproterenol and norepinephrine. These hormones increase intracellular cAMP levels which may ultimately modulate NKCC2 activity in terms of surface expression and phosphorylation [54]. In addition to stimulation of surface expression of NKCC2, vasopressin was shown to enhance the phosphorylation of N-terminal threonines in mice [55].
NKCC2 is inhibited by loop diuretics such as bumetanide, furosemide and torsemide as they probably bind to the extracellular ion translocation pathway [56]. The cyclic guanosine monophosphate (cGMP) generated upon stimulation by atrial natriuretic peptides or nitric oxide also inhibits NaCl reabsorption in TAL through reduced apical NKCC2 abundance mediated by phosphodiesterase 2 (PDE2) [54, 57].
Like other members of the CCCs, phosphorylation and dephosphorylation at key serine/threonine residues is a major regulatory mechanism to modulate NKCC2. Kinases such as Protein Kinase A (PKA), SPS1-related proline/alanine-rich kinase (SPAK) and oxidative stress-responsive kinase 1 (OSR1) have been reported to phosphorylate NKCC2 [58–61]. Elevated NKCC2 activity and chloride reabsorption were associated with abnormally elevated salt reabsorption in TAL of Dahl salt-sensitive rats (DSS) rats under basal condition, suggesting the contribution of NKCC2 to hypertension [62]. Phosphorylation at Thr96 and Thr101 were enhanced in DSS compared with Dahl salt-resistant rats on normal salt diet. Hyperphosphorylation was associated with enhanced SPAK phosphorylation, suggesting increased activity [63]. Free radical superoxide enhances NaCl absorption in TAL by increasing the surface expression of NKCC2 via Protein kinase C (PKC) without modulating NKCC2 phosphorylation or its upstream kinases SPAK in Sprague–Dawley rats [64].
Previously we showed that expression of NKCC2 at TAL was significantly enhanced at pre-hypertensive phase (23–25 days old) in the Milan hypertensive strain (MHS) of rat which harbours mutations in three genes encoding adducin proteins (α-F316Y, β-Q529R and γ-Q572K). However, those rats still retained similar blood pressure levels compared with age-matched Milan normotensive strain (MNS), as reduced protein levels of NCC and α-ENaC in the downstream nephron segments probably compensated the effect of NKCC2 upregulation [65, 66] (Fig. 3). In contrast, MHS rats in established hypertension stage (3 months old) showed upregulation of NCC coupled with increased chloride channel (ClC-K) protein level in DCT, with only a slight reduction of α- and β-ENaC in outer medulla [67]. Later on, Carmosino and colleagues showed that regulatory phospho-threonines (96, 101 and 111) in NKCC2 were significantly increased in the kidney of MHS rats and associated with increased NKCC2 activation implicated in the pathogenesis of hypertension in this strain of rats (Fig. 4). Elevated NKCC2 activity in MHS rats could be mediated by SPAK phosphorylation at serine 325, which was significantly increased in MHS rats [68].
Figure 3:
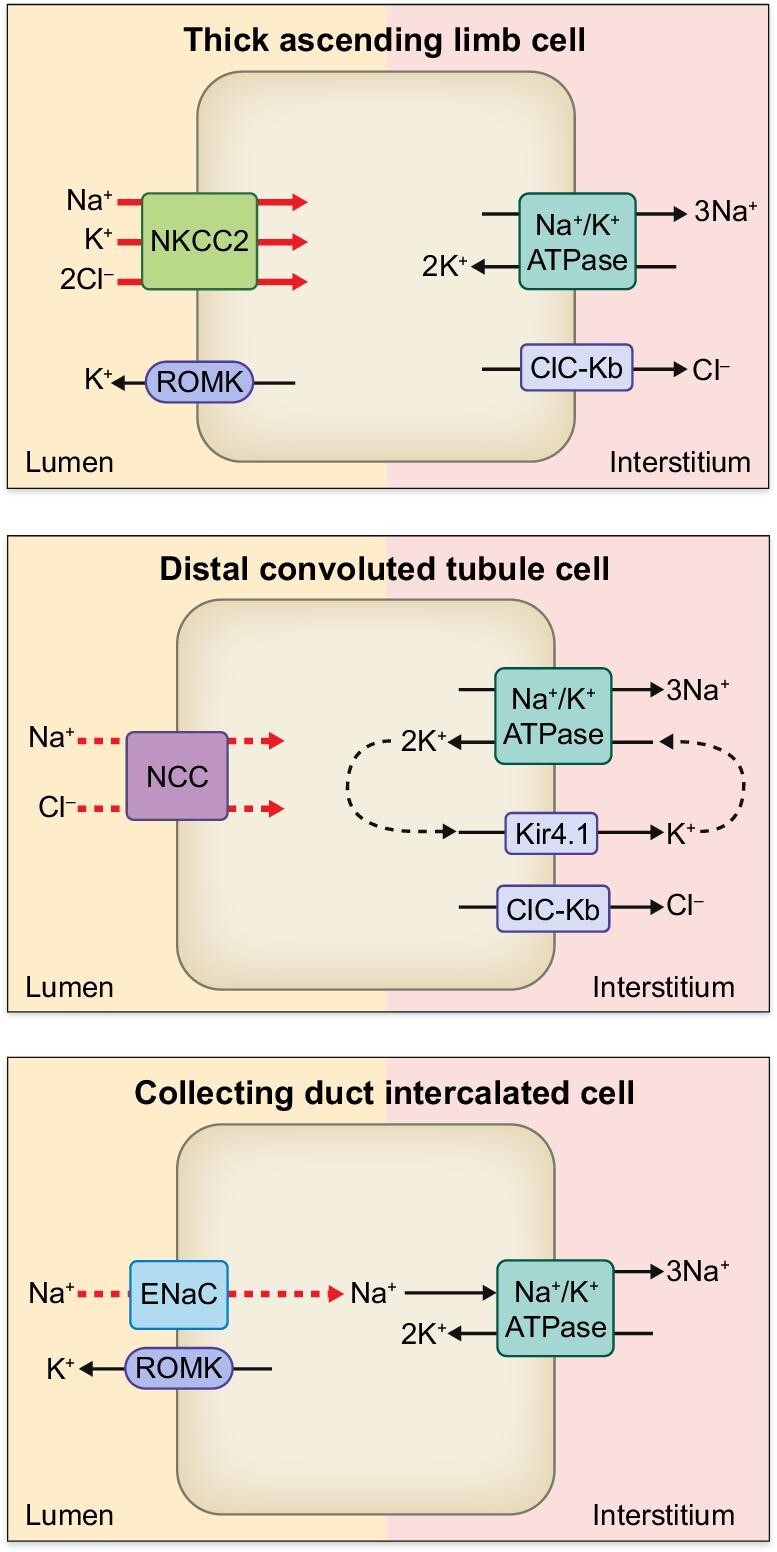
Sodium transporters, exchangers and ion channels along the nephron of MHS at pre-hypertensive phase (23–25 days old) [65]. Although NKCC2 at TAL is significantly enhanced (red thick arrow), MHS at this age retains comparable blood pressure with respect to MNS presumably due to the compensatory mechanism involving downregulation of NCC and ENaC (red dashed arrow) at the downstream nephron segments.
Figure 4:
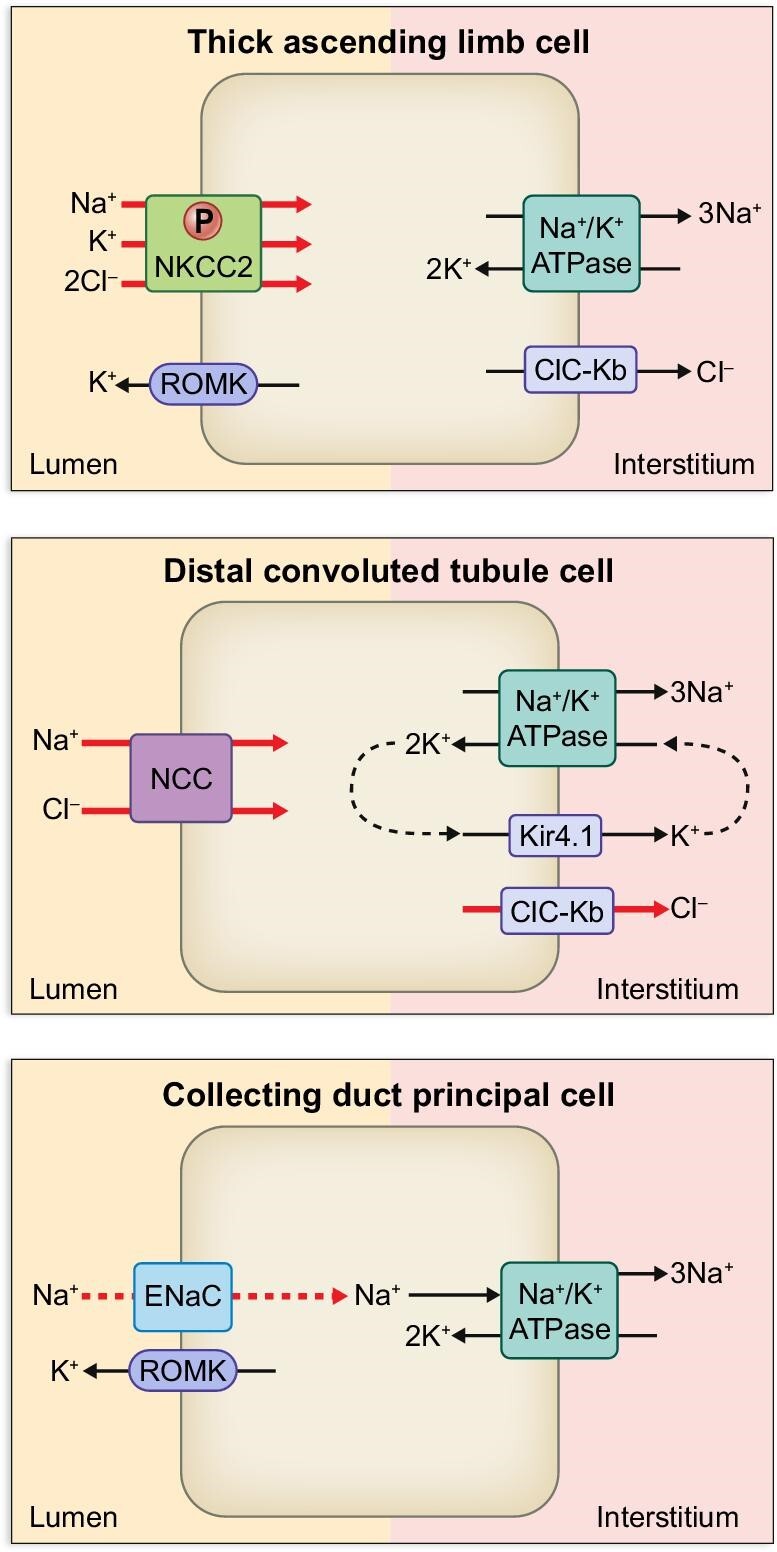
Sodium transporters, exchangers and ion channels along the nephron of MHS at 3 months old [67]. At this stage, MHS develops hypertension due to the marked upregulation of NCC in conjunction with ClC-Kb (red thick arrows). Increased phosphorylation of NKCC2 at TAL is also implicated in the pathogenesis of hypertension in this strain [68].
THE ROLES OF NCC IN THE CONTROL OF BLOOD PRESSURE
Another member of CCCs, thiazide-sensitive NCC, plays a role in fine-tuning of salt reabsorption in DCT (Fig. 1) [69]. Loss-of function mutations in the gene encoding NCC are associated with Gitelman syndrome, an autosomal recessive form of salt wasting, low blood pressure, hypokalemic metabolic alkalosis, hypomagnesemia and hypocalciuria (Fig. 2) [70, 71]. Mouse model with deletion of NCC (NCC−/−) showed similar phenotypes as Gitelman's syndrome, with mild perturbations of sodium and fluid volume homeostasis (Table 1). NCC knockout mice retained comparable blood pressure, acid–base balance, plasma electrolyte concentrations and serum aldosterone levels with respect to control mice under basal conditions, however they developed hypotension in response to 2 weeks of sodium depletion [72]. Structural remodelling in DCT were evident in NCC-deficient mice, with absence of early DCT but intact preservation of late DCT which expresses ENaC, TRPV5 and Na+-Ca2+ exchanger [73]. Renal transporter-profiling on the kidney of NCC knockout mice revealed increased abundance of cleaved γ-ENaC as a compensatory response, while other sodium transporters were unchanged [27]. Recently, low potassium diet was implicated in the activation of NCC at DCT leading to sodium retention and thereby increased blood pressure [74]. Studies on mice with kidney-specific knockout of inward-rectifying potassium channel 4.1 (Kir4.1) revealed that Kir4.1 plays an essential role on the activation of NCC in response to hypokalemia [75] (Table 1). At the basolateral side, Kir4.1 works as a sensor for circulating potassium levels, and potassium efflux by Kir4.1/Kir5.1 contributes to the maintenance of Na+/K+-ATPase activity through recycling of potassium (Fig. 1). Kir4.1/Kir5.1 is a primary determinant of membrane potential and intracellular chloride concentration, which mediates the WNK-dependent regulation of NCC [75, 76]. Indeed, loss-of-function mutations in KCNJ10 gene encoding Kir4.1 lead to EAST/SeSAME syndrome, resembling to Gitelman syndrome presenting with hypokalemic metabolic alkalosis [77–79].
In contrast to Gitelman syndrome, mutations in regulators of NCC could result in Gordon syndrome, also known as Familial hyperkalaemic hypertension syndrome or pseudohypoaldosteronism type II, which is an autosomal dominant inherited form of low-renin hypertension associated with hyperkalaemia and hyperchloremic metabolic acidosis (Fig. 2) [80]. Initially, mutations in genes encoding With No lysine (K) serine/threonine kinases WNK1 and WNK4 in DCT have been implicated in the causing mechanism of Gordon syndrome. Later on, two genes CUL3 and KLHL3 emerged as being responsible for 80% of families with Gordon syndrome [81]. Since phosphorylation modulates the activity of NCC, WNK1/4 were expected as kinases to activate NCC by phosphorylation. However, recent studies revealed more complex mechanisms for NCC. In fact, WNK1/4 do not phosphorylate NCC, but downstream serine/threonine kinases SPAK and OSR1, which in turn phosphorylate NCC for channel activation [82]. Mice with targeted disruption of WNK4 or WNK1 heterozygous (Wnk1+/−) exhibited hypotension [83, 84] (Table 1). WNK4−/− mice showed almost complete absence of phospho- and total NCC levels which were not compensated by a significantly enhanced WNK1 level, suggesting that WNK4 is the principal WNK involved in NCC regulation [85]. WNK1 interacts with WNK4 through its kinase domain and inhibits WNK4 [86]. CUL3 and KLHL3 code for a hydrophobic scaffold protein in an ubiquitin-E3 ligase Cullin3 and an adaptor protein Kelch3, respectively. These proteins form a CUL3–KLHL3 E3 ligase complex involved in the endosomal degradation of WNK4 [87, 88]. Subsequently, mutations in WNK1, WNK4, KLHL3 and CUL3 are associated with abnormal accumulation of WNK4 leading to Gordon syndrome [88, 89].
Although thiazide diuretics are effective anti-hypertensive drugs targeting NCC, the use of the thiazides have been implied in the increased risk of Type 2 diabetes due to metabolic disturbances [90]. Since NCC is regulated by the CUL3/KLHL3–WNK1/4–SPAK/OSR1 regulatory pathway, targeting the molecules involved in this cascade could represent as a therapeutic strategy for hypertension. Indeed, deficiency of SPAK in mice showed reduction in blood pressure with reduced NCC protein abundance and activity [91] (Table 1). Two novel compounds STOCK1S-50699 and STOCK2S-26016 were shown to inhibit the interaction of SPAK to WNK. Two compounds exhibited dose-dependent inhibitory effects on phosphorylation of endogenously expressed SPAK and NCC in mpkDCT cells [92] (Table 2). A selective SPAK inhibitor ZT-1a developed through scaffold-hybrid strategy showed an inhibitory effect on NCC phosphorylation in SPAK-dependent manner in mouse kidney [93]. Since the strategy to target CUL3/KLHL3–WNK1/4–SPAK/OSR1 cascade holds potential for anti-hypertensive therapy, improvements in terms of selectivity on kinase isoforms to avoid the undesirable side effects are the major challenging to be addressed.
THE ROLES OF ENaC ON THE CONTROL OF BLOOD PRESSURE
ENaC is expressed on the apical membrane of PC of ASDN and participates in the fine-tuning of sodium reabsorption [18]. At this site, lumen electronegativity provides a driving force for sodium absorption through ENaC in parallel with potassium secretion via ROMK (Fig. 4). Other sodium transporters including NCC and NDCBE require simultaneous chloride absorption independent of potassium excretion [94, 95]. ENaC belongs to cation-selective, ligand-gated degenerin/ENaC (DEG/ENaC) superfamily implicated in sensory functions [96, 97]. Other members in this superfamily include acid-sensing ion channel (ASIC) in mammals and FMRFamide-gated Na+ channel (FaNaC) in invertebrate [97]. Functional ENaC channels form heteromeric trimer composed of three subunits (α, β and γ), encoded by SCNN1A, SCNN1B or SCNN1G genes. Another subunit, δ-ENaC, has been also identified with different tissue distribution pattern [97]. Although ASIC1 is functional as a homotrimer, ENaC requires a heterotrimer composed of α or δ, β and γ to form functional channel. In addition, ENaC knockout mice models demonstrated that α, β and γ subunits are crucial for the survivals [98–100] (Table 1).
Expression levels and channel activity of ENaC are modulated by several factors including SGK1, Nedd4-2, proteases (furin, prostasin and kallikrein), hormones including aldosterone, angiotensin II, vasopressin and endothelin, as well as shear stress, ATP, Na+ and nitric oxide [96]. In addition, ENaC is inhibited by either amiloride via binding to ENaC or potassium-sparing diuretics including spironolactone, canrenone and eplerenone through blocking the binding of aldosterone to mineralocorticoid receptor (MR) [101].
Mutations in genes encoding ENaC subunits could lead to blood pressure disorders as described as Liddle syndrome or pseudohypoaldosteronism type 1B (PHA1B) (Fig. 2).
Liddle syndrome is an autosomal dominant inherited form of hypertension caused by elevated renal sodium reabsorption due to gain-of-function mutations in mostly SCNN1B and SCNN1G genes [102]. Patients with Liddle syndrome are characterized by hypokalemic hypertension, low plasma renin and aldosterone levels with metabolic alkalosis. ENaC is negatively regulated by E3 ubiquitin-protein ligases NEDD4 family proteins [103]. Each ENaC subunit contains PY (Proline Tyrosine) motif at the intracellular C-terminus serving as a binding site for NEDD4 proteins. Nedd4-2 catalyzes the ubiquitination of ENaC at cell membrane, prompting the internalization of the channels and eventual proteasomal degradation. The mutations in SCNN1B and SCNN1G genes identified in Liddle syndrome are mostly missense mutations within the PY motif or nonsense/frameshift mutations leading to elimination of the PY motif [102]. Alteration or elimination of PY motif impairs interaction between Nedd4-2 and ENaC thus disrupting regulated internalization, resulting in excessive apical ENaC expression and enhanced Na+ reabsorption at the distal nephron [104]. In addition to the abnormal accumulation of ENaC at cell surface, enhanced single-channel open probability [105, 106] or aberrant proteolytic channel activation [107] could result in hypertension in Liddle syndrome. Functional expression of truncated variant β-R564X in Xenopus oocyte showed a significantly enhanced single-channel open probability under high- and low-Na+ conditions [105]. Missense γ-N530S mutation located in the extracellular loop showed a similar cell surface expression compared with WT channel but two-fold increased channel activity in Xenopus oocytes [106]. β-R566X and γ-K576X increased the cleaved form of α-ENaC at the cell surface when three ENaC subunits were co-expressed in HEK 293T cells [107]. Despite the presence of PY motif in all ENaC subunits, only one mutation in α-ENaC has been identified for Liddle syndrome so far [108]. α-C479R located at the extracellular domain participates in disulfide bond together with C394 residue. Both C479R and C394S variants exhibited increased amiloride-sensitive ENaC current in Xenopus oocyte.
In contrast to Liddle syndrome, PHA1B is described as autosomal-recessive inherited form of salt-wasting due to resistance to aldosterone [97]. Patients with PHA1B are characterized by low blood pressure, hyponatremia, hyperkalaemia, metabolic acidosis, anorexia and dehydration, with high plasma aldosterone and renin activity starting from their infancy [109]. While autosomal dominant PHA1A is due to mutations in the gene encoding MR, PHA1B is caused by the loss-of-function mutations in genes encoding ENaC subunits. Studies on three PHA1B patients demonstrated that mild PHA symptom is associated with a missense mutation in α-ENaC (G327C), while two mutations, a frameshift mutation in α-ENaC and a splice site mutation in intron 12 of the β-ENaC, were related to more severe phenotypes [110].
Recently we have identified key molecules in salt sensitivity, including glutamyl aminopeptidase (ENPEP), plasminogen activator, urokinase (PLAU), epidermal growth factor (EGF) and Xaa-Pro aminopeptidase 2 precursor (XPNPEP2) through urine proteomic analyses on salt-sensitive and salt-resistant hypertensive patients. Since these molecules are involved in the regulation of ENaC, the development of hypertension in salt-sensitive patients could be associated with ENaC-dependent sodium reabsorption along the distal tubule [111].
Finally, Pitzer and colleagues have suggested that ENaC-dependent activation of inflammasome in antigen-presenting cells could contribute the development of salt-sensitive hypertension [112, 113]. In antigen-presenting dendritic cells (DCs), upon increase in extracellular concentration of sodium due to high salt diet, sodium enters DCs via ENaC, and in turn, intracellular Ca2+ level is increased through the activity of Na+/Ca2+ exchanger. Elevated Ca2+ activates PKC, which phosphorylates p47phox to trigger the activation of NADPH-oxidase, leading to superoxide and reactive oxygen species productions. Formations of IsoLGs and IsoLG-protein adducts stimulated by superoxide and reactive oxygen species induce the activation of NLRP3 inflammasome, which enhances proinflammatory cytokine interleukin (IL)-1β maturation via activation of Caspase-1. Activated DCs promote the production of IL-17A and interferon gamma (IFN-γ) in T cells, accelerating sodium retention following infiltration to the kidney. These findings reveal a novel mechanism for ENaC in immune cells to contribute to salt-induced inflammation and ultimately salt-sensitive hypertension.
THE ROLES OF PENDRIN ON THE CONTROL OF BLOOD PRESSURE
It has long been believed that intercalated cells (ICs) of distal nephron have the sole role of acid–base homeostasis through H+ and HCO3− handling by the activities of vacuolar H+-ATPase (vH+-ATPase), Cl−/HCO3− exchangers kAE1 and pendrin. However, recent studies have shown that ICs also participate in salt reabsorption [18]. Pendrin is encoded by the SLC26A4 gene and is mainly expressed in kidney, thyroid and inner ear. Pendrin generally mediates entry of chloride anion into the cells in exchange for release of bicarbonate ion or iodide [114, 115]. In the kidney, pendrin localizes at apical membrane of β-ICs, non-α and non-β ICs of the connecting tubules and cortical CD. In the mice cortical CD, two cycles of pendrin molecules were coupled with one cycle of NDCBE to generate electroneutral thiazide-sensitive NaCl absorption (Fig. 1) [116]. In contrast to other types of cells, basolaterally expressed vH+-ATPase energizes salt reabsorption in ICs [20].
Inactivating mutations of pendrin could lead to Pendred syndrome (Fig. 2), which is associated with sensorineural deafness, hearing loss and goiter [117]. Studies on genetically modified mice targeting pendrin highlighted the role of pendrin in blood pressure modulation (Table 1). In addition to inner-ear defects as observed in Pendred syndrome [118], Slc26a4−/− mice demonstrated enhanced urinary volume and chloride excretion compared with WT mice in response to moderate NaCl restriction. Furthermore, strict salt depletion induced a hypotensive effect in Slc26a4−/− mice [119]. Another Slc26a4−/− mouse model showed impaired bicarbonate secretion in CD associated with acidic urine pH and elevated serum HCO3− concentration [120]. Trepiccione et al. generated a conditional transgenic mouse model in which expression of pendrin can be switched on in vivo by doxycycline. Acute deletion of pendrin resulted in a marked drop in blood pressure without affecting the acid–base balance or blood K+ concentration [121] (Table 1). Single deletion of pendrin or NCC exhibited volume contraction or hypotension during salt depletion, while showing only a mild degree of salt wasting at basal condition [122]. This has prompted a hypothesis that these two transporters are under the control of high aldosterone levels. Indeed, expression of MR was also confirmed in ICs [123]. Phosphorylation on MR at S843 was almost exclusively detected in ICs in vivo in the kidney [124]. The inactive phosphorylated form of MR is converted to active dephosphorylated form by Ang II via mTOR signalling, leading to aldosterone-dependent upregulation of pendrin [125]. The mTOR pathway is involved in pendrin regulation and congenital hypothyroidism in thyroid follicular cells [126]. Dietary salt restriction or Ang II infusion upregulated NCC and pendrin expressions accompanied by increased plasma aldosterone levels in control mice. Salt depletion did not change blood pressure in control mice, but considerably reduced blood pressure in pendrin-knockout mice [127] (Table 1). Furthermore, pendrin was upregulated by an analogue of aldosterone deoxycorticosterone in control mouse kidney [128]. Pendrin/NCC double knockout mice showed significantly lower blood pressure compared with WT and single NCC or pendrin knockout mice, associated with renal failure and metabolic alkalosis under basal condition [122] (Table 1). Deletion of NDCBE caused only mild perturbations of Na+ homeostasis with no significant alterations in blood pressure with respect to control mice [129]. NDCBE/NCC double-knockout (dKO) mice developed hypokalemia together with upregulations of ENaC and the Ca2+-activated K+ channel BKCa under basal conditions. Salt restriction induced remarkable intravascular volume depletion in NDCBE/NCC dKO mice. In contrast to pendrin/NCC dKO mice with severe volume depletion and renal failure, NDCBE/NCC dKO mice exhibited milder renal phenotypes. While deletion of NDCBE retains salt absorption ability through ENaC/pendrin mechanism, pendrin ablation could have impacts on salt reabsorption through distinct transport pathways involving ENaC/pendrin and NDCBE/pendrin [129].
A mouse model overexpressing pendrin in ICs developed hypertension accompanied by delayed increase in urinary NaCl under high-salt diet (Table 1). Since replacement of NaCl with NaHCO3 did not have significant changes in blood pressure, hypertension in these mice was chloride-dependent. Pendrin-driven chloride reabsorption stimulates the sodium uptake from ENaC and NDCBE although these sodium transporters are downregulated due to vascular volume expansion in these mice [130].
Clinical data from patients harbouring mutations in pendrin further conferred the involvement of pendrin in blood pressure regulation. Demographic and biochemical data analyses on patients with bi-allelic SLC26A4 mutations showed that subjects with impaired pendrin function are likely to be resistant to high blood pressure. In addition, recent identification of patients with mutations in pendrin presented the Gitelman-like syndrome demonstrating low blood pressure, metabolic alkalosis and renal salt-losing with hypokalemia [125, 131, 132].
CONCLUSION
Hypertension and associated diseases have become extremely common especially in western countries. While the pathogenesis underlying the development of hypertension is still to be addressed, several crucial physiological mechanisms and molecules involved in hypertension have been identified thanks to the studies on animal models targeting renal transporters. These findings could further provide a potential for novel therapeutic approaches applicable for human patients with hypertension.
ACKNOWLEDGEMENTS
We thank Fondazione Terzo Pilastro Internazionale and Biogem, Biology and Molecular Genetics Institute for promoting the study.
Contributor Information
Yoko Suzumoto, Biogem, Biology and Molecular Genetics Institute, Ariano Irpino (AV), Italy.
Laura Zucaro, Biogem, Biology and Molecular Genetics Institute, Ariano Irpino (AV), Italy; Department of Mental, Physical Health and Preventive Medicine, University of Campania ‘Luigi Vanvitelli’, Naples, Italy.
Anna Iervolino, Biogem, Biology and Molecular Genetics Institute, Ariano Irpino (AV), Italy; Department of Translational Medical Sciences, University of Campania ‘Luigi Vanvitelli’, Naples, Italy.
Giovambattista Capasso, Biogem, Biology and Molecular Genetics Institute, Ariano Irpino (AV), Italy.
FUNDING
Not applicable.
AUTHORS’ CONTRIBUTIONS
Y.S. and G.C. designed the study. Y.S. and L.Z. wrote the manuscript. Y.S., A.I. and G.C revised the manuscript, and G.C. supervised the work. All the authors read and agreed with the final version of manuscript.
DATA AVAILABILITY STATEMENT
No new data were generated or analysed in support of this research.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
REFERENCES
- 1. Fuchs FD, Whelton PK. High blood pressure and cardiovascular disease. Hypertension 2020;75:285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 2021;398:957–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol 2020;16:223–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whelton PK, He J, Appel LJet al. Primary prevention of hypertension: clinical and public health advisory from the National High Blood Pressure Education Program. J Am Med Assoc 2002;288:1882–8. [DOI] [PubMed] [Google Scholar]
- 5. Whelton PK, He J. Health effects of sodium and potassium in humans. Curr Opin Lipidol 2014;25:75–9. [DOI] [PubMed] [Google Scholar]
- 6. Kurtz TW, Morris RCJ. Dietary chloride as a determinant of “sodium-dependent” hypertension. Science 1983;222:1139–41. [DOI] [PubMed] [Google Scholar]
- 7. Whitescarver SA, Ott CE, Jackson BAet al. Salt-sensitive hypertension: contribution of chloride. Science 1984;223:1430–2. [DOI] [PubMed] [Google Scholar]
- 8. Joe B, Shapiro JI. Molecular mechanisms of experimental salt-sensitive hypertension. J Am Heart Assoc 2012;1:e002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harris PJ, Young JA. Dose-dependent stimulation and inhibition of proximal tubular sodium reabsorption by angiotensin II in the rat kidney. Pflugers Arch 1977;367:295–7. [DOI] [PubMed] [Google Scholar]
- 10. Morimoto A, Uzu T, Fujii Tet al. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet 1997;350:1734–7. [DOI] [PubMed] [Google Scholar]
- 11. Weinberger MH, Fineberg NS, Fineberg SEet al. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension 2001;37:429–32. [DOI] [PubMed] [Google Scholar]
- 12. de Wardener HE, He FJ, MacGregor GA. Plasma sodium and hypertension. Kidney Int 2004;66:2454–66. [DOI] [PubMed] [Google Scholar]
- 13. Downie ML, Lopez Garcia SC, Kleta Ret al. Inherited tubulopathies of the kidney. Clin J Am Soc Nephrol 2021;16:620–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kleta R, Bockenhauer D. Salt-losing tubulopathies in children: what's new, what's controversial? J Am Soc Nephrol 2018;29:727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Capasso G, Cantone A, Evangelista Cet al. Channels, carriers, and pumps in the pathogenesis of sodium-sensitive hypertension. Semin Nephrol 2005;25:419–24. [DOI] [PubMed] [Google Scholar]
- 16. Wang X, Armando I, Upadhyay Ket al. The regulation of proximal tubular salt transport in hypertension: an update. Curr Opin Nephrol Hypertens 2009;18:412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Castrop H, Schießl IM. Physiology and pathophysiology of the renal Na-K-2Cl cotransporter (NKCC2). Am J Physiol Physiol 2014;307:F991–1002. [DOI] [PubMed] [Google Scholar]
- 18. Chambrey R, Trepiccione F. Relative roles of principal and intercalated cells in the regulation of sodium balance and blood pressure. Curr Hypertens Rep 2015;17:538. [DOI] [PubMed] [Google Scholar]
- 19. Capolongo G, Suzumoto Y, D'Acierno Met al. ERK1,2 signalling pathway along the nephron and its role in acid-base and electrolytes balance. Int J Mol Sci 2019;20:4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chambrey R, Kurth I, Peti-Peterdi Jet al. Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci USA 2013;110:7928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brant SR, Bernstein M, Wasmuth JJet al. Physical and genetic mapping of a human apical epithelial Na+/H+ exchanger (NHE3) isoform to chromosome 5p15.3. Genomics 1993;15:668–72. [DOI] [PubMed] [Google Scholar]
- 22. Burckhardt G, Di Sole F, Helmle-Kolb C. The Na+/H+ exchanger gene family. J Nephrol 2002;15:S3–21. [PubMed] [Google Scholar]
- 23. Pedersen SF, Counillon L. The SLC9A-C mammalian Na(+)/H(+) exchanger family: molecules, mechanisms, and physiology. Physiol Rev 2019;99:2015–13. [DOI] [PubMed] [Google Scholar]
- 24. Schultheis PJ, Clarke LL, Meneton Pet al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet 1998;19:282–5. [DOI] [PubMed] [Google Scholar]
- 25. Bogdanic E, Müller T, Heinz-Erian Pet al. Further delineation of SLC9A3-related congenital sodium diarrhea. Mol Genet Genomic Med 2022;10:e2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nwia SM, Li XC, Leite APOet al. The Na+/H+ exchanger 3 in the intestines and the proximal tubule of the kidney: localization, physiological function, and key roles in angiotensin II-induced hypertension. Front Physiol 2022;13:861659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brooks HL, Sorensen AM, Terris Jet al. Profiling of renal tubule Na+ transporter abundances in NHE3 and NCC null mice using targeted proteomics. J Physiol 2001;530:359–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Woo AL, Noonan WT, Schultheis PJet al. Renal function in NHE3-deficient mice with transgenic rescue of small intestinal absorptive defect. Am J Physiol Renal Physiol 2003;284:F1190–8. [DOI] [PubMed] [Google Scholar]
- 29. Noonan WT, Woo AL, Nieman MLet al. Blood pressure maintenance in NHE3-deficient mice with transgenic expression of NHE3 in small intestine. Am J Physiol Regul Integr Comp Physiol 2005;288:R685–91. [DOI] [PubMed] [Google Scholar]
- 30. Li XC, Shull GE, Miguel-Qin Eet al. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension in NHE3-deficient mice with transgenic rescue of NHE3 in small intestines. Physiol Rep 2015;3. doi: 10.14814/phy2.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fenton RA, Poulsen SB, de la Mora Chavez Set al. Caffeine-induced diuresis and natriuresis is independent of renal tubular NHE3. Am J Physiol Renal Physiol 2015;308:F1409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fenton RA, Poulsen SB, de la Met al. Renal tubular NHE3 is required in the maintenance of water and sodium chloride homeostasis. Kidney Int 2017;92:397–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li XC, Soleimani M, Zhu Det al. Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) promotes the pressure-natriuresis response and lowers blood pressure in mice. Hypertension 2018;72:1328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li XC, Zhu D, Chen Xet al. proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) in the kidney attenuates Ang II (angiotensin II)-induced hypertension in mice. Hypertension 2019;74:526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhuo JL, Soleimani M, Li XC. New insights into the critical importance of intratubular Na(+)/H(+) exchanger 3 and its potential therapeutic implications in hypertension. Curr Hypertens Rep 2021;23:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xue J, Thomas L, Tahmasbi Met al. An inducible intestinal epithelial cell-specific NHE3 knockout mouse model mimicking congenital sodium diarrhea. Clin Sci (Lond) 2020;134:941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xue J, Thomas L, Murali SKet al. Enhanced phosphate absorption in intestinal epithelial cell-specific NHE3 knockout mice. Acta Physiol (Oxf) 2022;234:e13756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Janecke AR, Heinz-Erian P, Yin Jet al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum Mol Genet 2015;24:6614–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crajoinas RO, Lessa LMA, Carraro-Lacroix LRet al. Posttranslational mechanisms associated with reduced NHE3 activity in adult vs. young prehypertensive SHR. Am J Physiol Renal Physiol 2010;299:F872–81. [DOI] [PubMed] [Google Scholar]
- 40. Leong PKK, Yang LE, Holstein-Rathlou N-Het al. Angiotensin II clamp prevents the second step in renal apical NHE3 internalization during acute hypertension. Am J Physiol Renal Physiol 2002;283:F1142–50. [DOI] [PubMed] [Google Scholar]
- 41. Riquier-Brison ADM, Leong PKK, Pihakaski-Maunsbach Ket al. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol 2010;298:F177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li XC, Zhuo JL. Phosphoproteomic analysis of AT1 receptor-mediated signaling responses in proximal tubules of angiotensin II-induced hypertensive rats. Kidney Int 2011;80:620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li XC, Leite APO, Zheng Xet al. Proximal tubule-specific deletion of angiotensin II Type 1a receptors in the kidney attenuates circulating and intratubular angiotensin II-induced hypertension in PT-Agtr1a(-/-) mice. Hypertension 2021;77:1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Linz D, Wirth K, Linz Wet al. Antihypertensive and laxative effects by pharmacological inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut. Hypertension 2012;60:1560–7. [DOI] [PubMed] [Google Scholar]
- 45. Spencer AG, Labonte ED, Rosenbaum DPet al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014;6:227ra36. [DOI] [PubMed] [Google Scholar]
- 46. Borges-Júnior FA, Silva Dos Santos D, Benetti Aet al. Empagliflozin inhibits proximal tubule NHE3 activity, preserves GFR, and restores euvolemia in nondiabetic rats with induced heart failure. J Am Soc Nephrol 2021;32:1616–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. D'Acierno M, Resaz R, Iervolino Aet al. Dapagliflozin prevents kidney glycogen accumulation and improves renal proximal tubule cell functions in a mouse model of glycogen storage disease type 1b. J Am Soc Nephrol 2022;33:1864–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Markadieu N, Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch 2014;466:91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Starremans PGJF, Kersten FFJ, Knoers NVAMet al. Mutations in the human Na-K-2Cl cotransporter (NKCC2) identified in Bartter syndrome type I consistently result in nonfunctional transporters. J Am Soc Nephrol 2003;14:1419–26. [DOI] [PubMed] [Google Scholar]
- 50. Simon DB, Karet FE, Hamdan JMet al. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 1996;13:183–8. [DOI] [PubMed] [Google Scholar]
- 51. Takahashi N, Chernavvsky DR, Gomez RAet al. Uncompensated polyuria in a mouse model of Bartter's syndrome. Proc Natl Acad Sci USA 2000;97:5434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aviv A, Hollenberg NK, Weder A. Urinary potassium excretion and sodium sensitivity in blacks. Hypertension 2004;43:707–13. [DOI] [PubMed] [Google Scholar]
- 53. Blankenstein KI, Borschewski A, Labes Ret al. Calcineurin inhibitor cyclosporine A activates renal Na-K-Cl cotransporters via local and systemic mechanisms. Am J Physiol Renal Physiol 2017;312:F489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol Renal Physiol 2011;301:F1143–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Giménez I, Forbush B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem 2003;278:26946–51. [DOI] [PubMed] [Google Scholar]
- 56. Zhao Y, Roy K, Vidossich Pet al. Structural basis for inhibition of the cation-chloride cotransporter NKCC1 by the diuretic drug bumetanide. Nat Commun 2022;13:2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ares GR, Caceres P, Alvarez-Leefmans FJet al. cGMP decreases surface NKCC2 levels in the thick ascending limb: role of phosphodiesterase 2 (PDE2). Am J Physiol Renal Physiol 2008;295:F877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gunaratne R, Braucht DWW, Rinschen MMet al. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci USA 2010;107:15653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 2002;277:50812–9. [DOI] [PubMed] [Google Scholar]
- 60. Richardson C, Sakamoto K, de los Heros Pet al. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 2011;124:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in blood pressure control and hypertension. Curr Opin Nephrol Hypertens 2019;28:474–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alvarez-Guerra M, Garay RP. Renal Na-K-Cl cotransporter NKCC2 in Dahl salt-sensitive rats. J Hypertens 2002;20:721–7. [DOI] [PubMed] [Google Scholar]
- 63. Ares GR, Haque MZ, Delpire Eet al. Hyperphosphorylation of Na-K-2Cl cotransporter in thick ascending limbs of Dahl salt-sensitive rats. Hypertension 2012;60:1464–70. [DOI] [PubMed] [Google Scholar]
- 64. Haque MZ, Ortiz PA. Superoxide increases surface NKCC2 in the rat thick ascending limbs via PKC. Am J Physiol Renal Physiol 2019;317:F99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Capasso G, Rizzo M, Evangelista Cet al. Altered expression of renal apical plasma membrane Na+ transporters in the early phase of genetic hypertension. Am J Physiol Renal Physiol 2005;288:F1173–82. [DOI] [PubMed] [Google Scholar]
- 66. Bianchi G, Ferrari P, Staessen JA. Adducin polymorphism detection and impact on hypertension and related disorders. Hypertension 2005;45:331–40. [DOI] [PubMed] [Google Scholar]
- 67. Capasso G, Rizzo M, Garavaglia MLet al. Upregulation of apical sodium-chloride cotransporter and basolateral chloride channels is responsible for the maintenance of salt-sensitive hypertension. Am J Physiol Renal Physiol 2008;295:F556–67. [DOI] [PubMed] [Google Scholar]
- 68. Carmosino M, Rizzo F, Ferrari Pet al. NKCC2 is activated in Milan hypertensive rats contributing to the maintenance of salt-sensitive hypertension. Pflugers Arch 2011;462:281–91. [DOI] [PubMed] [Google Scholar]
- 69. Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 2005;85:423–93. [DOI] [PubMed] [Google Scholar]
- 70. Bettinelli A, Bianchetti MG, Girardin Eet al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr 1992;120:38–43. [DOI] [PubMed] [Google Scholar]
- 71. Simeoni M, Columbano V, Suzumoto Yet al. Looking beyond entecavir to discover Gitelman syndrome in a 50 year-old man. QJM 2020;739–40. [DOI] [PubMed] [Google Scholar]
- 72. Schultheis PJ, Lorenz JN, Meneton Pet al. Phenotype resembling Gitelman's syndrome in mice lacking the apical Na+-Cl- cotransporter of the distal convoluted tubule. J Biol Chem 1998;273:29150–5. [DOI] [PubMed] [Google Scholar]
- 73. Loffing J, Vallon V, Loffing-Cueni Det al. Altered renal distal tubule structure and renal Na(+) and Ca(2+) handling in a mouse model for Gitelman's syndrome. J Am Soc Nephrol 2004;15:2276–88. [DOI] [PubMed] [Google Scholar]
- 74. Terker AS, Zhang C, McCormick JAet al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 2015;21:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang MX, Cuevas CA, Su XTet al. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int 2018;93:893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Su X-T, Klett NJ, Sharma Aet al. Distal convoluted tubule chloride concentration is modulated via potassium channels and transporters. Am J Physiol Physiol 2020;319:F534–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bockenhauer D, Feather S, Stanescu HCet al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 2009;360:1960–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Scholl UI, Choi M, Liu Tet al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 2009;106:5842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Suzumoto Y, Columbano V, Gervasi Let al. A case series of adult patients affected by EAST/SeSAME syndrome suggests more severe disease in subjects bearing KCNJ10 truncating mutations. Intractable Rare Dis Res 2021;10:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hadchouel J, Delaloy C, Fauré Set al. Familial hyperkalemic hypertension. J Am Soc Nephrol 2006;17:208–17. [DOI] [PubMed] [Google Scholar]
- 81. Boyden LM, Choi M, Choate KAet al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012;482:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Alessi DR, Zhang J, Khanna Aet al. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 2014;7:re3. [DOI] [PubMed] [Google Scholar]
- 83. Zambrowicz BP, Abuin A, Ramirez-Solis Ret al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci USA 2003;100:14109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ohta A, Rai T, Yui Net al. Targeted disruption of the Wnk4 gene decreases phosphorylation of Na-Cl cotransporter, increases Na excretion and lowers blood pressure. Hum Mol Genet 2009;18:3978–86. [DOI] [PubMed] [Google Scholar]
- 85. Takahashi D, Mori T, Nomura Net al. WNK4 is the major WNK positively regulating NCC in the mouse kidney. Biosci Rep 2014;34. doi: 10.1042/BSR20140047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yang C-L, Zhu X, Wang Zet al. Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J Clin Invest 2005;115:1379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. O'Shaughnessy KM. Gordon syndrome: a continuing story. Pediatr Nephrol 2015;30:1903–8. [DOI] [PubMed] [Google Scholar]
- 88. Ohta A, Schumacher F-R, Mehellou Yet al. The CUL3-KLHL3 E3 ligase complex mutated in Gordon's hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J 2013;451:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mabillard H, Sayer JA. The molecular genetics of Gordon syndrome. Genes (Basel) 2019;10:986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Scheen AJ. Type 2 diabetes and thiazide diuretics. Curr Diab Rep 2018;18:6. [DOI] [PubMed] [Google Scholar]
- 91. Yang S-S, Lo Y-F, Wu C-Cet al. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 2010;21:1868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mori T, Kikuchi E, Watanabe Yet al. Chemical library screening for WNK signalling inhibitors using fluorescence correlation spectroscopy. Biochem J 2013;455:339–45. [DOI] [PubMed] [Google Scholar]
- 93. Zhang J, Bhuiyan MIH, Zhang Tet al. Modulation of brain cation-Cl(-) cotransport via the SPAK kinase inhibitor ZT-1a. Nat Commun 2020;11:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pearce D, Soundararajan R, Trimpert Cet al. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 2015;10:135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Eladari D, Chambrey R, Picard Net al. Electroneutral absorption of NaCl by the aldosterone-sensitive distal nephron: implication for normal electrolytes homeostasis and blood pressure regulation. Cell Mol Life Sci 2014;71:2879–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na(+) channel regulation by extracellular and intracellular factors. Annu Rev Physiol 2018;80:263–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 2016;579:95–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Barker PM, Nguyen MS, Gatzy JTet al. Role of gammaENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. J Clin Invest 1998;102:1634–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bonny O, Hummler E. Dysfunction of epithelial sodium transport: from human to mouse. Kidney Int 2000;57:1313–8. [DOI] [PubMed] [Google Scholar]
- 100. Hummler E, Barker P, Gatzy Jet al. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet 1996;12:325–8. [DOI] [PubMed] [Google Scholar]
- 101. Pratt JH, Eckert GJ, Newman Set al. Blood pressure responses to small doses of amiloride and spironolactone in normotensive subjects. Hypertension 2001;38:1124–9. [DOI] [PubMed] [Google Scholar]
- 102. Tetti M, Monticone S, Burrello Jet al. Liddle syndrome: review of the literature and description of a new case. Int J Mol Sci 2018;19. doi: 10.3390/ijms19030812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chandran S, Li H, Dong Wet al. Neural precursor cell-expressed developmentally down-regulated protein 4-2 (Nedd4-2) regulation by 14-3-3 protein binding at canonical serum and glucocorticoid kinase 1 (SGK1) phosphorylation sites. J Biol Chem 2011;286:37830–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Furuhashi M, Kitamura K, Adachi Met al. Liddle's syndrome caused by a novel mutation in the proline-rich PY motif of the epithelial sodium channel beta-subunit. J Clin Endocrinol Metab 2005;90:340–4. [DOI] [PubMed] [Google Scholar]
- 105. Anantharam A, Tian Y, Palmer LG. Open probability of the epithelial sodium channel is regulated by intracellular sodium. J Physiol 2006;574:333–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hiltunen TP, Hannila-Handelberg T, Petäjäniemi Net al. Liddle's syndrome associated with a point mutation in the extracellular domain of the epithelial sodium channel gamma subunit. J Hypertens 2002;20:2383–90. [DOI] [PubMed] [Google Scholar]
- 107. Knight KK, Olson DR, Zhou Ret al. Liddle's syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci USA 2006;103:2805–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Salih M, Gautschi I, van Bemmelen MXet al. A missense mutation in the extracellular domain of αENaC causes Liddle syndrome. J Am Soc Nephrol 2017;28:3291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Amin N, Alvi NS, Barth JHet al. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep 2013;2013:130010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Edelheit O, Hanukoglu I, Gizewska Met al. Novel mutations in epithelial sodium channel (ENaC) subunit genes and phenotypic expression of multisystem pseudohypoaldosteronism. Clin Endocrinol (Oxf) 2005;62:547–53. [DOI] [PubMed] [Google Scholar]
- 111. Matafora V, Lanzani C, Zagato Let al. Urinary proteomics reveals key markers of salt sensitivity in hypertensive patients during saline infusion. J Nephrol 2021;34:739–51. [DOI] [PubMed] [Google Scholar]
- 112. Pitzer AL, Van Beusecum JP, Kleyman TRet al. ENaC in salt-sensitive hypertension: kidney and beyond. Curr Hypertens Rep 2020;22:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pitzer A, Elijovich F, Laffer CLet al. DC ENaC-dependent inflammasome activation contributes to salt-sensitive hypertension. Circ Res 2022;131:328–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wagner CA, Mohebbi N, Capasso Get al. The anion exchanger pendrin (SLC26A4) and renal acid-base homeostasis. Cell Physiol Biochem 2011;28:497–504. [DOI] [PubMed] [Google Scholar]
- 115. Yoshida A, Hisatome I, Taniguchi Set al. Mechanism of iodide/chloride exchange by pendrin. Endocrinology 2004;145:4301–8. [DOI] [PubMed] [Google Scholar]
- 116. Leviel F, Hübner CA, Houillier Pet al. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 2010;120:1627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wémeau J-L, Kopp P. Pendred syndrome. Best Pract Res Clin Endocrinol Metab 2017;31:213–24. [DOI] [PubMed] [Google Scholar]
- 118. Everett LA, Belyantseva IA, Noben-Trauth Ket al. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 2001;10:153–61. [DOI] [PubMed] [Google Scholar]
- 119. Wall SM, Kim YH, Stanley Let al. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension 2004;44:982–7. [DOI] [PubMed] [Google Scholar]
- 120. Amlal H, Petrovic S, Xu Jet al. Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl(-)/HCO3(-) exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am J Physiol Cell Physiol 2010;299:C33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Trepiccione F, Soukaseum C, Baudrie Vet al. Acute genetic ablation of pendrin lowers blood pressure in mice. Nephrol Dial Transplant 2017;32:1137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Soleimani M, Barone S, Xu Jet al. Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci USA 2012;109:13368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ackermann D, Gresko N, Carrel Met al. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol 2010;299:F1473–85. [DOI] [PubMed] [Google Scholar]
- 124. Shibata S, Rinehart J, Zhang Jet al. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 2013;18:660–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Shibata S. Role of pendrin in the pathophysiology of aldosterone-induced hypertension. Am J Hypertens 2019;32:607–13. [DOI] [PubMed] [Google Scholar]
- 126. Kang HS, Kumar D, Liao Get al. GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J Clin Invest 2017;127:4326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hirohama D, Ayuzawa N, Ueda Ket al. Aldosterone is essential for angiotensin II-induced upregulation of pendrin. J Am Soc Nephrol 2018;29:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Verlander JW, Hassell KA, Royaux IEet al. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 2003;42:356–62. [DOI] [PubMed] [Google Scholar]
- 129. Sinning A, Radionov N, Trepiccione Fet al. Double knockout of the Na+-driven Cl-/HCO3- exchanger and Na+/Cl- cotransporter induces hypokalemia and volume depletion. J Am Soc Nephrol 2017;28:130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Jacques T, Picard N, Miller RLet al. Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J Am Soc Nephrol 2013;24:1104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Trepiccione F, Suzumoto Y, Perna Aet al. Pure Gitelman-like syndrome secondary to SLC26A4 (pendrin) mutation. Kidney Int 2021;100:947–8. [DOI] [PubMed] [Google Scholar]
- 132. Lemoine S, Eladari D, Juillard Let al. The case | Hypokalemia and severe renal loss of sodium. Kidney Int 2020;97:1305–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analysed in support of this research.