

Abstract
In this Letter we describe structure-activity relationship (SAR) studies conducted in five distinct regions of a new 2-amino-N-phenylacetamides series of Slack potassium channel inhibitors exemplified by recently disclosed high-throughput screening (HTS) hit VU0606170 (4). New analogs were screened in a thallium (Tl+) flux assay in HEK-293 cells stably expressing wild-type human (WT) Slack. Selected analogs were screened in Tl+ flux versus A934T Slack and other Slo family members Slick and Maxi-K and evaluated in whole-cell electrophysiology (EP) assays using an automated patch clamp system. Results revealed the series to have flat SAR with significant structural modifications resulting in a loss of Slack activity. More minor changes led to compounds with Slack activity and Slo family selectivity similar to the HTS hit.
Keywords: Slack, KNa1.1, Slo2.2, KCNT1, MMPSI, EIMFS, 2-amino-N-phenylacetamide
Malignant migrating partial seizure of infancy (MMPSI), also known as epilepsy of infancy with migrating focal seizures (EIMFS), is a rare, serious form of infantile epilepsy.1 Patients with MMPSI are typically pharmacoresistant with seizure onset developing within the first six months of life and a 25% mortality rate within the first year. Approximately half of MMPSI cases have been linked to multiple de novo gain-of-function (GOF) heterozygous missense mutations in the KCNT1 gene.2, 3 KCNT1 encodes for the sodium-activated potassium channel known as KNa1.1 or Slack (Sequence Like A Calcium-Activated K+ Channel). Slack is a member of the Slo family (Slo2.2) of K+ channels along with Slo1 (Maxi-K), Slo2.1 (Slick), and Slo3. Slack channels are essential regulators of electrical activity and distributed throughout the central nervous system, where they play important roles in affecting neuronal excitability.3–7 Slack channels are characterized by 4 subunits with six hydrophobic transmembrane domains (S1-S6) and a pore-forming domain between S5 and S6. Also present are an extended C-terminal cytoplasmic domain, containing a regulator of potassium conductance (RCK) domain and a nicotinamide adenine dinucleotide-binding (NAD+) domain.2, 6, 8–11
Several recent reports have documented more than 30 mutations in Slack. The GOF mutations correlated with MMPSI are associated with an increase in Slack current.3, 12–15 In addition to MMPSI, KCNT1 GOF mutations are associated with other epilepsy disorders, including autosomal dominant sleep-related hypermotor epilepsy (ADSHE),16 early-onset epileptic encephalopathy (EOEE),17 myoclonic-atonic epilepsy,18 temporal lobe epilepsy with intellectual disability,19 and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE).20, 21 Multiple potential mechanisms by which these GOF mutations in Slack channels lead to increased neuronal excitability and rate of firing have been put forth. For example, activation of Slack channels leads to increased amplitude of AHP, resulting in shorter action potentials and increased neuronal firing frequency.12, 22 Likewise, GOF Slack mutations may reduce the excitability of inhibitory interneurons, ultimately leading to decreased inhibitory tone.8, 13 Finally, Slack also has non-conducting functions, and interacts with downstream signaling pathways that may be disrupted by mutations.22, 23
No broadly effective and safe drug therapy option is presently available for the treatment of MMPSI. Drugs such as quinidine, bepridil, and cofilium modulate Slack currents in vitro, and attempts have been made to repurpose them for KCNT1-associated epilepsies; however, these compounds are hampered by a lack of selectivity, toxicity, and suboptimal drug metabolism and pharmacokinetic (DMPK) properties.4, 8, 14, 24–30 Notably, recent reports have described new compounds from diverse chemotypes that inhibit Slack function (Figure 1).31 For example, virtual screening of a commercial library employing a cryo-electron microscopy-derived structure of chicken KNa1.1 identified six Slack inhibitors with micromolar potency, each one predicted to function via blockade of the channel pore.32 Compounds 1-2 are representative analogs with reported IC50 values of 3.2 and 3.0 µM, respectively, in a whole-cell patch clamp assay using HEK-293 cells expressing human wild-type (WT) Slack channels. In addition, a high-throughput screen (HTS) and ensuing optimization effort identified orally available 1,2,4-oxadiazole 3, which normalizes the EEG phenotype in a mouse model of Kcnt1 GOF.33 Compound 3 was a potent Slack inhibitor with a reported IC50 of 40 nM in an automated patch clamp assay in HEK-TREX cells stably expressing human WT Slack channels. Finally, we recently reported the results of our own HTS utilizing a thallium (Tl+) flux assay in HEK-293 cells stably expressing human WT and selected mutant Slack channels. The 2-amino-N-phenylacetamide VU0606170 (4) was identified through this campaign and possessed low micromolar potency for both WT and A934T Slack in Tl+ flux and whole-cell automated patch-clamp assays. VU0606170 (4) has an attractive selectivity profile and decreased the firing rate in an overexcited, spontaneously firing cortical neuronal culture consistent with an antiepileptic effect.34 In this Letter, we report the results of structure-activity relationship (SAR) studies within the 2-amino-N-phenylacetamide chemotype exemplified by 4.
Figure 1.
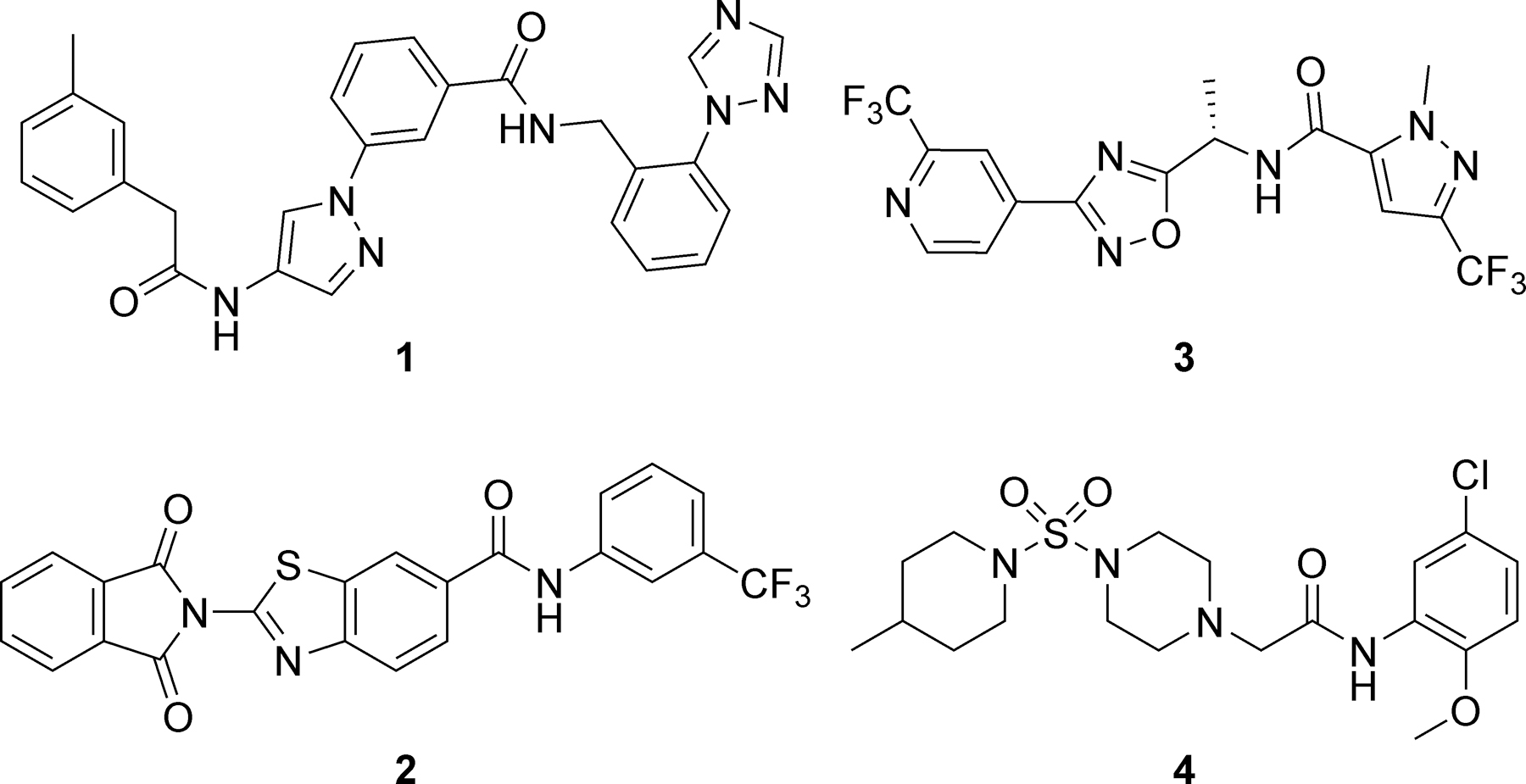
Recently reported small molecule inhibitors of Slack channels: predicted pore blockers 1-2, in vivo tool 3, and HTS hit 4 (VU0606170).
Desiring to rapidly evaluate the potential tractability of VU0606170 (4), we decided to pursue SAR development in five regions of the scaffold (Figure 2). Where amenable, we employed late-stage penultimate intermediates that facilitated the synthesis of small compound libraries. Scheme 1 was used to evaluate SAR in the eastern ring of the scaffold. Commercial sulfamoyl chloride 5 was reacted with 1-Boc-piperazine 6 to afford sulfamide 7. Cleavage of the protecting group under acidic conditions gave secondary amine 8 as the trifluoroacetic acid salt. Alkylation of 8 with ethyl bromoacetate was accomplished with mild heating to provide ester 9. Hydrolysis of the ester with aqueous lithium hydroxide afforded acid and penultimate intermediate 10. Coupling with a variety of aryl amines provided final amide analogs 11-29 and 31-36. Synthesis of analog 30 via amide coupling proved ineffective. Thus, 2,5-dichloroaniline 37 was alkylated with chloroacetyl chloride to afford 38, which was subsequently reacted with intermediate 8 to provide 30.
Figure 2.
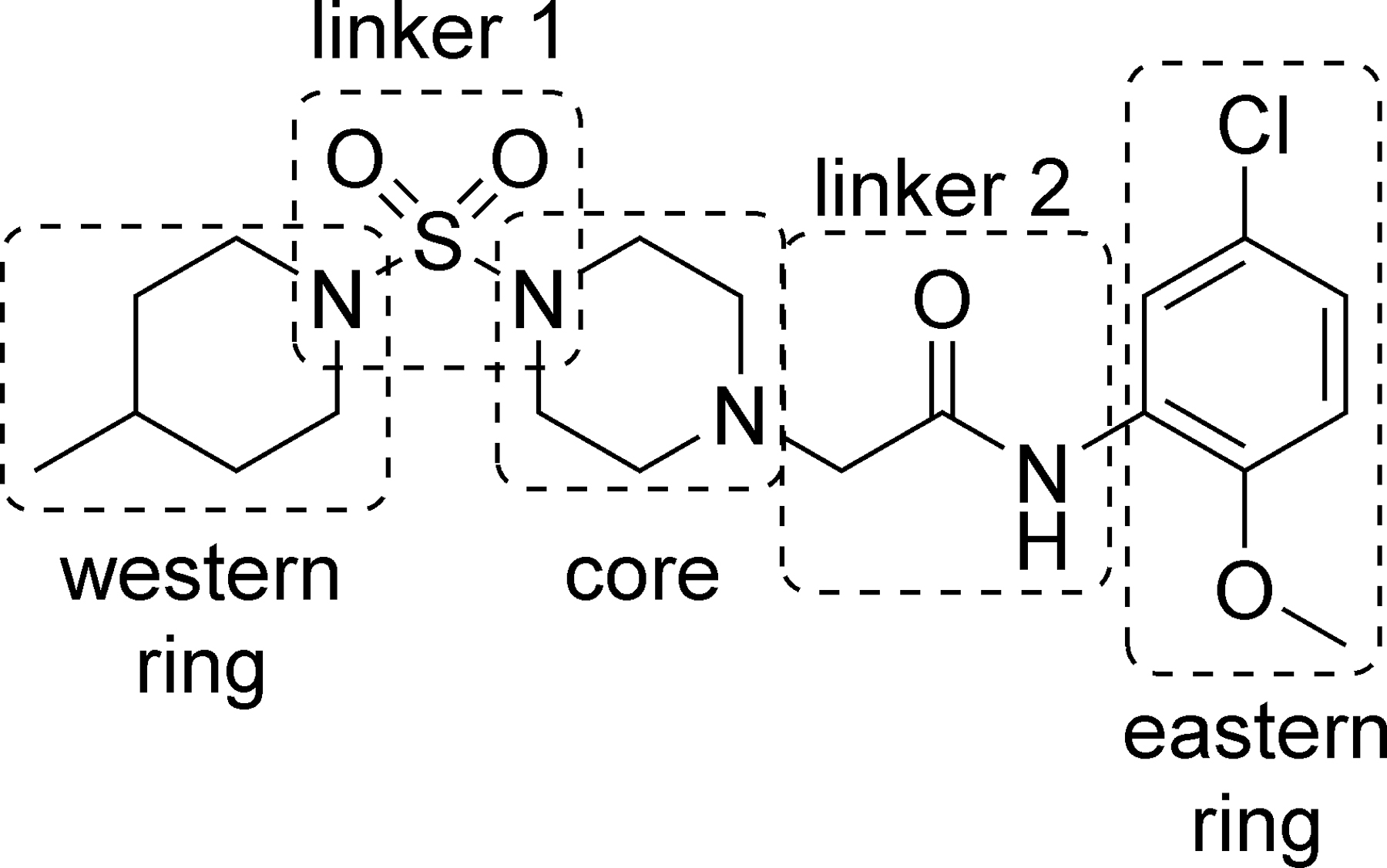
Regions for SAR development in the VU0606170 scaffold.
Scheme 1.
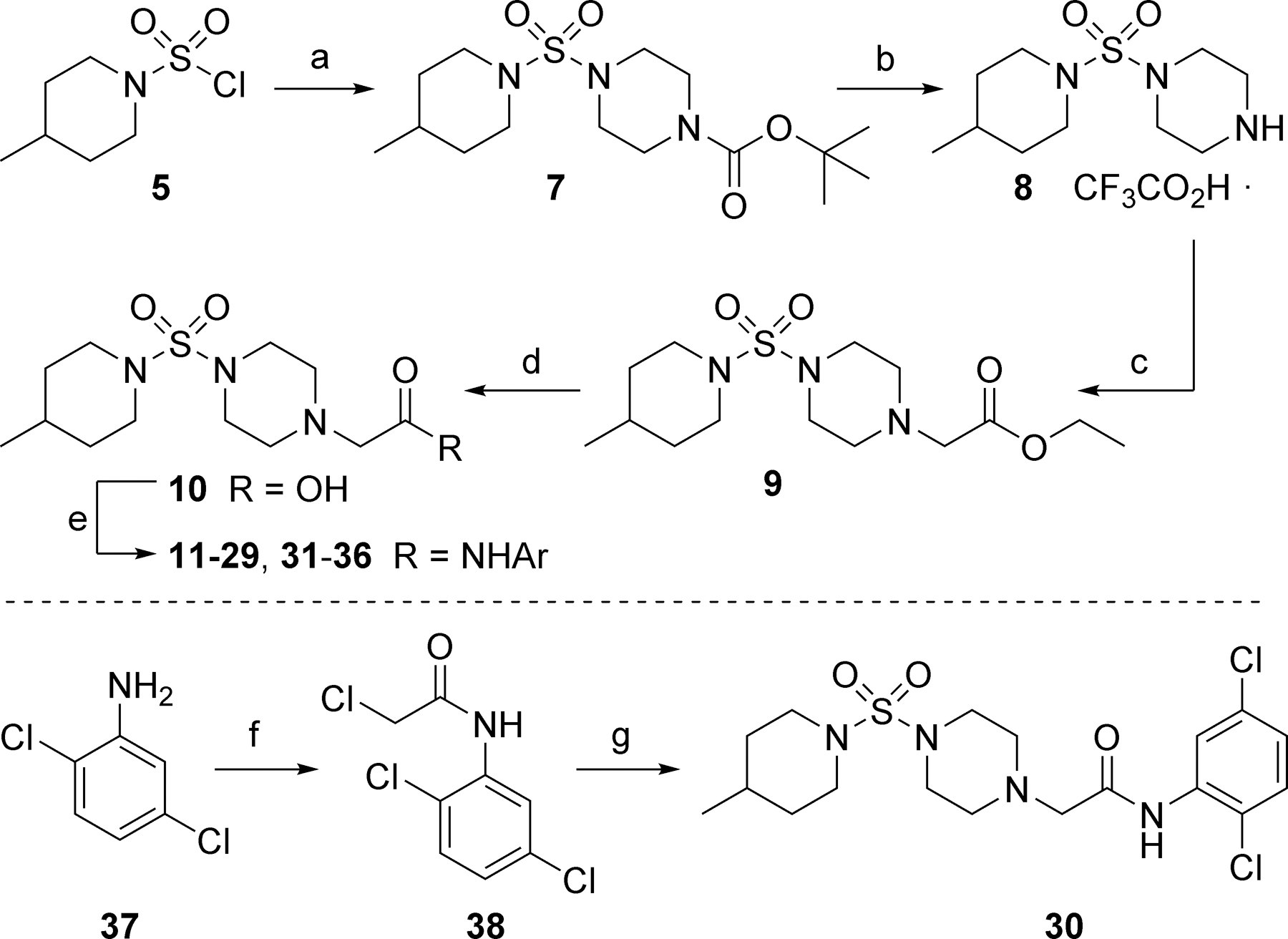
Reagents and conditions: (a) 1-Boc-piperazine (6), DIEA, CH2Cl2, 81%; (b) TFA, CH2Cl2, 97%; (c) Ethyl bromoacetate, K2CO3, DMF, 40 ºC, 92%; (d) LiOH, H2O, THF, 99%; (e) ArNH2, HATU, DIEA, DMF, 4–64%; (f) chloroacetyl chloride, NEt3, DMAP, CH2Cl2; (g) 8, K2CO3, KI, DMF, 50 ºC, 7% (2 steps).
Screening of all new compounds versus WT Slack in our Tl+ flux assay utilized optimized buffer conditions that were slightly modified from those previously published to afford a larger signal amplitude (see Supporting Information).34 An initial scan at all positions employing common functional groups quickly alerted us to the potential for mode switching in the scaffold as most of the analogs were weak activators of the channel (Table 1). Such an observation is consistent with compounds in this chemotype modulating Slack function by a mechanism other than simple pore blockade. A few analogs (15, 18, 19, and 22) displayed a mixed profile, appearing to behave as either weak inhibitors or inactive at higher concentrations and weak activators at low concentrations. Although the reason for this profile is not known, a possible explanation is that there are two binding sites in the channel and occupancy of the first site is weakly activating while occupancy of both is inhibiting. Alternatively, there may be two distinct binding sites, one activating and the other inhibiting. Given that 2-methoxy analog 21 was the only compound that maintained the inhibitory profile observed with VU0606170 (4), we redirected our efforts toward analogs that maintained the 2,5-disubstituted pattern of 4 with either the 2-methoxy group (24-28) or the 5-chloro group (29-35) held constant (Table 2).
Table 1.
Eastern amide analogs from initial scan
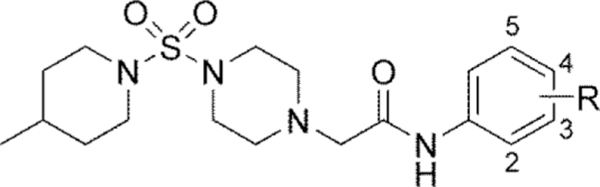
| ||||
|---|---|---|---|---|
| No. | R | Modea | IC50 / EC50 (µM)b | Efficacy (%)b,c |
| 4 | 2-OMe, 5-Cl | Inh | 2.4 | 100 |
| 11 | H | Act | >10d | 21 |
| 12 | 2-F | Act | >10d | 30 |
| 13 | 3-F | Act | 4.6 | 13 |
| 14 | 4-F | Act | >10d | 30 |
| 15 | 2-Cl | not determinede | ||
| 16 | 3-Cl | Act | 7.6 | 38 |
| 17 | 4-Cl | Act | >10d | 58 |
| 18 | 2- Me | not determinede | ||
| 19 | 3- Me | not determinede | ||
| 20 | 4- Me | Act | 4.1 | 13 |
| 21 | 2-OMe | Inh | >10d | 85 |
| 22 | 3-OMe | not determinede | ||
| 23 | 4-OMe | Act | >10d | 15 |
Inh = inhibitor; Act = activator; NA = not active
Concentration-response curve (CRC) from Tl+ flux assay in HEK-293 cells expressing WT Slack
Amplitude of response in the presence of 30 µM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
CRC does not plateau within the concentration range tested
Compounds appear to be weak inhibitors/inactive at high concentrations and weak activators at low concentrations
Table 2.
Eastern amide analogs from focused scan
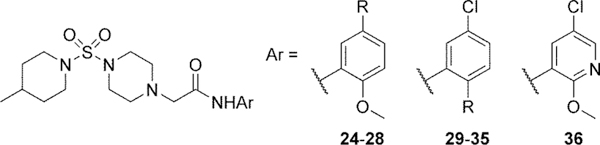
| ||||
|---|---|---|---|---|
| No. | R | Modea | IC50 / EC50 (µM)b | Efficacy (%)b,c |
| 24 | F | Inh | 4.9 | 102 |
| 25 | Br | Inh | 1.4 | 52 |
| 26 | Me | Inh | >10d | 82 |
| 27 | CF3 | Inh | 2.4 | 89 |
| 28 | t-Bu | not determinede | ||
| 29 | F | Act | 3.8 | <10 |
| 30 | Cl | inactive | ||
| 31 | Me | Inh | >10d | 44 |
| 32 | OEt | Inh | 7.1 | 98 |
| 33 | O(n-Pr) | Inh | 4.3 | 95 |
| 34 | O(i-Pr) | Inh | 5.1 | 92 |
| 35 | OCH2(c-Pr) | Inh | 7.5 | 59 |
| 36 | — | inactive | ||
Inh = inhibitor; Act = activator; NA = not active
CRC from Tl+ flux assay in HEK-293 cells expressing WT Slack
Amplitude of response in the presence of 30 µM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
CRC does not plateau within the concentration range tested
Compound appears to be a weak inhibitor/inactive at high concentrations and weak activator at low concentrations
Replacement of the 5-chloro group with other electron-withdrawing substituents maintained the inhibitory profile with bromo (25) and trifluoromethyl (27) analogs showing similar potency to VU0606170 (4). Notably, while the potency was similar, 25 demonstrated approximately half of the efficacy observed with 4 and 27 as evidenced by clear plateauing of the CRC well above the maximum observed with the control compound. Replacement of the 2-methoxy group with halogens (29 and 30) or methyl (31) was not effective. Analogs 32-35 explored lengthening and branching of the alkyl ether substituent. In each case, the compounds were inhibitors; however, potency was 2–3 fold lower than 4. Finally, analog 36 demonstrated the lack of tolerance for an aromatic nitrogen atom.
To explore SAR in the western ring and associated linker 1, we employed chemistry outlined in Scheme 2. Reaction of 1-Boc-piperazine 6 with ethyl bromoacetate under basic conditions afforded ester 39. Hydrolysis of the ester with lithium hydroxide provided lithium salt intermediate 40. Coupling of 40 with 5-chloro-2-methoxyaniline proceeded with HATU. Removal of the Boc protecting group gave secondary amine 42 as its trifluoroacetic acid salt. Reaction of 42 with sulfamoyl chlorides provided sulfamides 43 and 44, while reaction with sulfonyl chlorides yielded sulfonamides 45-48. To access urea analogs 50-55, we first converted 42 to 4-nitrophenylcarbamate 49. Treatment of 49 with various cyclic secondary amines under microwave heating afforded the desired ureas 50-55.35
Scheme 2.
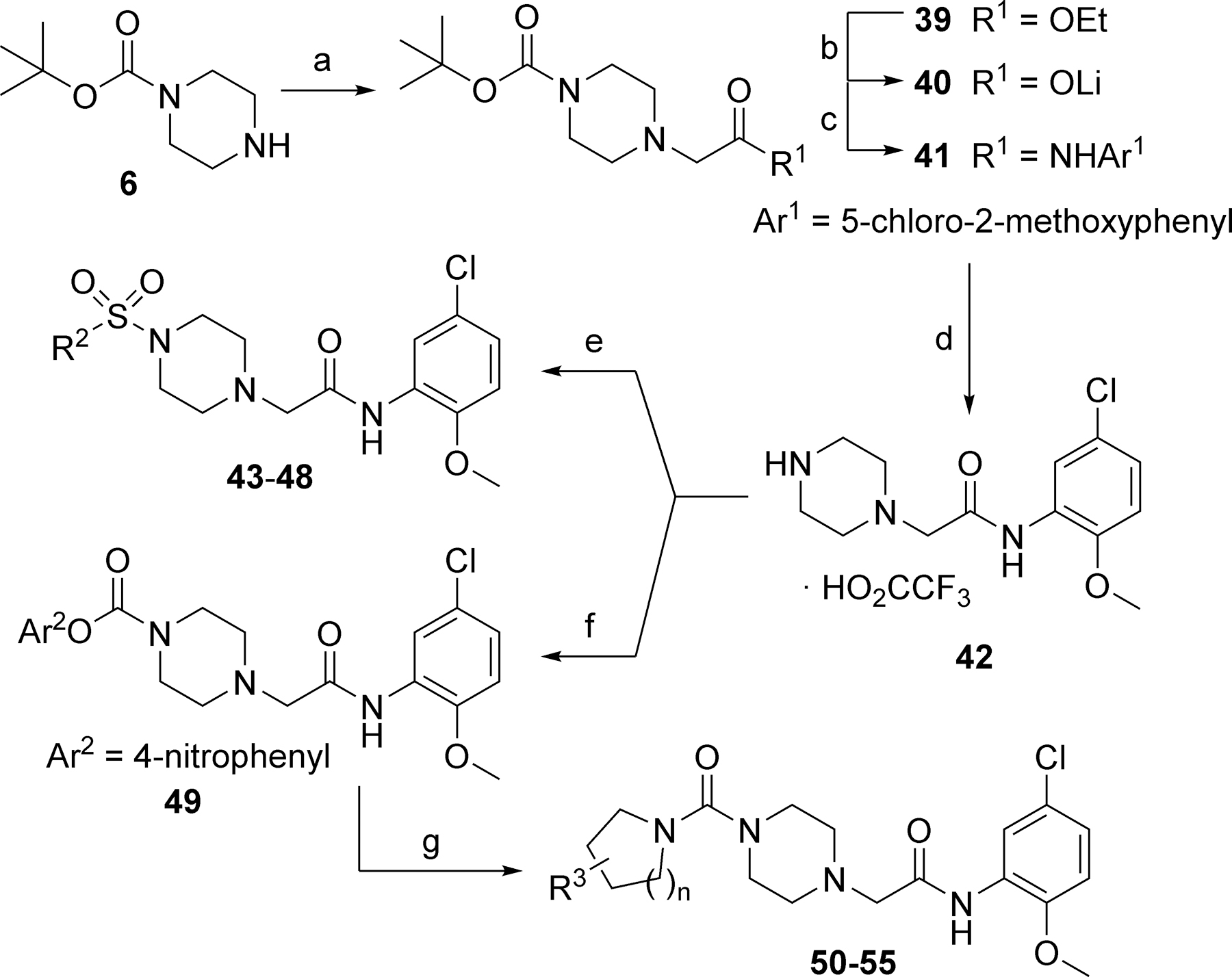
Reagents and conditions: (a) Ethyl bromoacetate, K2CO3, DMF, 68%; (b) LiOH, H2O, THF, 98%; (c) 5-chloro-2-methoxyaniline, HATU, DIEA, DMF, 61%; (d) TFA, CH2Cl2, 100%; (e) R2SO2Cl, DIEA, CH2Cl2; 41–58%; (f) 4-nitrophenyl chloroformate, CH2Cl2, 0 ºC; (g) cyclic secondary amine, NMP, µwave, 200 ºC, 25 min, 32–51% (2 steps).
The most modest changes in the western ring (43-45) were not tolerated (Table 3). Benzene and 4-chlorobenzene sulfonamides 46 and 48 exhibited potency on par with 4; however, efficacy was diminished considerably. Surprisingly, 4-methylbenzene sulfonamide 47 was inactive. For the most part, the urea analogs were weak activators of the channel (50, 51, 54, and 55) or displayed a mixed profile (53). Only analog 52 functioned as a weak inhibitor.
Table 3.
Western ring and linker 1 analogs
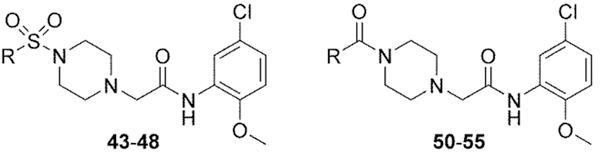
| ||||
|---|---|---|---|---|
| No. | R | Modea | IC50 / EC50 (µM)b | Efficacy (%)b,c |
| 43 | piperidin-1-yl | inactive | ||
| 44 | 4-morpholino | inactive | ||
| 45 | cyclohexyl | inactive | ||
| 46 | phenyl | Inh | 1.1 | 32 |
| 47 | 4-methylphenyl | inactive | ||
| 48 | 4-chlorophenyl | Inh | 2.3 | 16 |
| 50 | 3,3-difluoropyrrolidin-1-yl | Act | >10d | <10 |
| 51 | piperidin-1-yl | Act | >10d | 10 |
| 52 | 4-propylpiperidin-1-yl | Inh | >10d | 58 |
| 53 | 4-isopropylpiperidin-1-yl | not determinede | ||
| 54 | 4,4-dimethylpiperidin-1-yl | Act | 9.0 | 23 |
| 55 | azepan-1-yl | Act | >10d | 17 |
Inh = inhibitor; Act = activator; NA = not active
CRC from Tl+ flux assay in HEK-293 cells expressing WT Slack
Amplitude of response in the presence of 30 µM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
CRC does not plateau within the concentration range tested
Compound appears to be a weak inhibitor/inactive at high concentrations and weak activator at low concentrations
We previously reported that relocation of the carbonyl group on linker 2 from the carbon adjacent to the aniline nitrogen to the carbon adjacent to the piperazine nitrogen was not tolerated.34 In fact, we employed this regioisomer of VU0606170 known as VU0849686 as a negative control. Still, we were interested in exploring substitution of the linker and investigated stereospecific methylated analogs via Scheme 3. Following a method described in the patent literature,36 we converted commercially available chiral alcohol 56 to its corresponding triflate 57, which was reacted with secondary amine 8 to provide 58 with inversion of stereochemistry. Hydrolysis of the methyl ester was accomplished with 6N hydrochloric acid and heating to provide acid 59 as its hydrochloride salt. Coupling with 5-chloro-2-methoxyaniline and 2-methoxy-5-(trifluoromethyl)aniline afforded analogs 60 and 61, respectively. The same sequence was repeated utilizing methyl (S)-2-hydroxypropanoate instead of 56 to afford the R-enantiomer analogs 62 and 63 (Table 4). No evidence of significant preference for one enantiomer versus the other was noted. In both cases the trifluoromethyl analogs (61 and 63) were slightly more potent than the chloro analogs (60 and 62); however, all four compounds were 1.5 to 2-fold less potent than the hit VU0606170 (4).
Scheme 3.
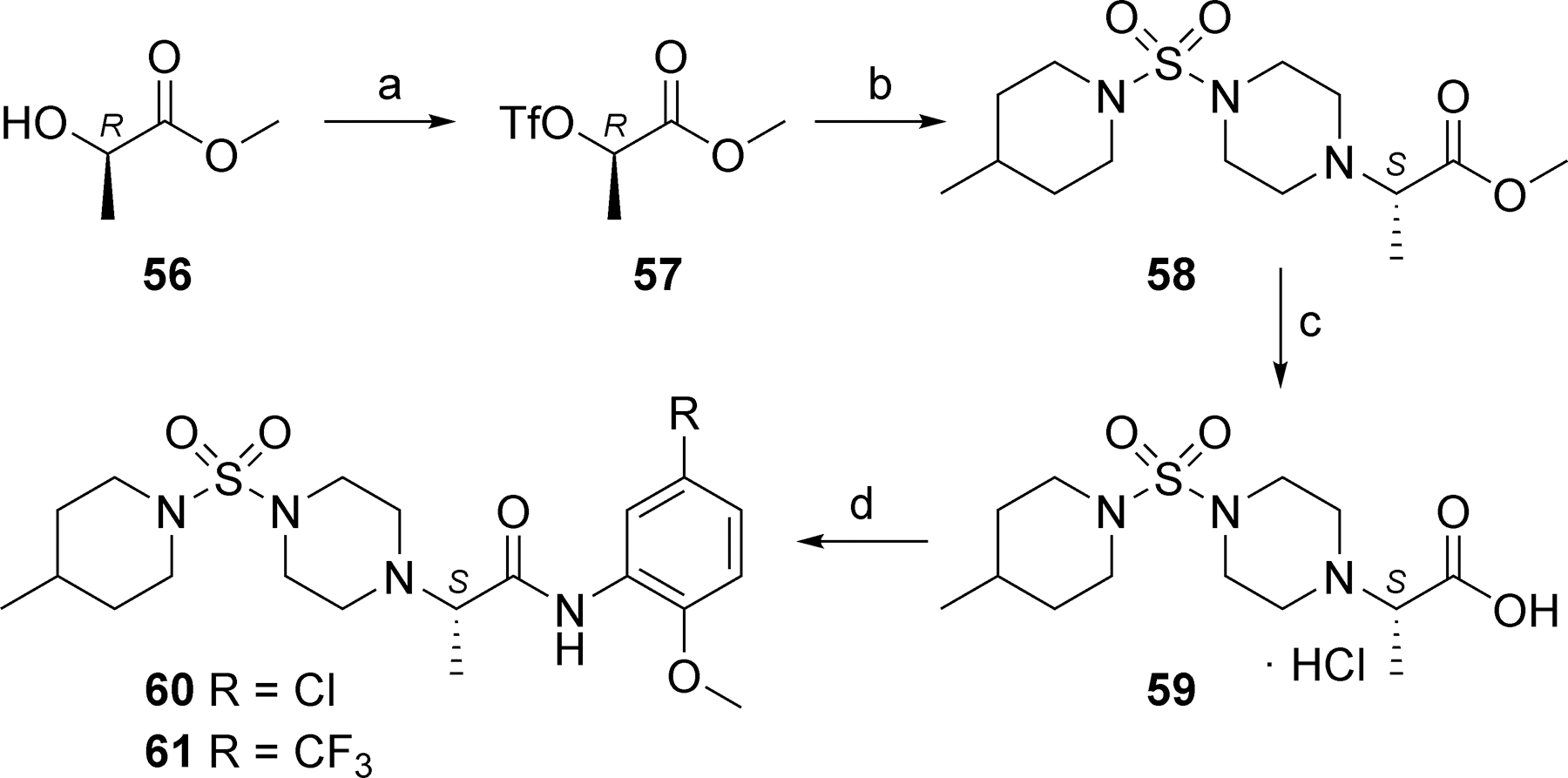
Reagents and conditions: (a) Tf2O, 2,6-lutidine, CH2Cl2, −70 ºC to r.t., 53%; (b) 8, K2CO3, CH2Cl2, H2O, 0 ºC to r.t., 71%; (c) 6N HCl, 100 ºC, 56%; (d) 5-chloro-2-methoxyaniline or 2-methoxy-5-(trifluoromethyl)aniline, HATU, DIEA, DMF, 35% (60), 63% (61).
Table 4.
Methylated linker 2 analogs
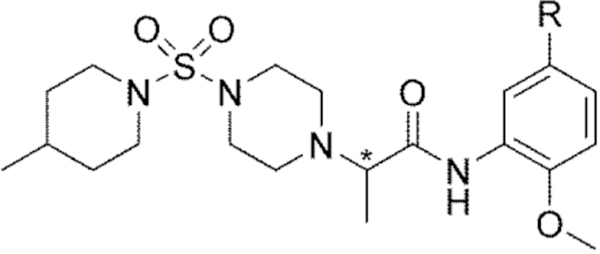
| |||||
|---|---|---|---|---|---|
| No. | * | R | Modea | IC50 / EC50 (µM)b | Efficacy (%)b,c |
| 60 | S | Cl | Inh | 4.4 | 93 |
| 61 | S | CF3 | Inh | 3.4 | 95 |
| 62 | R | Cl | Inh | 4.9 | 98 |
| 63 | R | CF3 | Inh | 3.5 | 100 |
Inh = inhibitor; Act = activator; NA = not active
CRC from Tl+ flux assay in HEK-293 cells expressing WT Slack
Amplitude of response in the presence of 30 µM test compound as a percentage of the maximum response for VU0606170
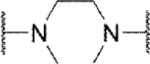
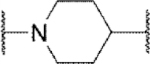
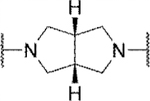
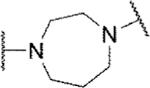
In the core of the scaffold, we sought to evaluate the impact of substitution as well as the tolerability for various piperazine isosteres.35, 37, 38 In some cases the core monomers were not symmetric (e.g., 64 in Scheme 4), and we thus employed complementary routes to arrive at two distinct analogs. Specifically, monomer 64 was converted to sulfamide 65 as described previously. Removal of the protecting group to afford 66 and subsequent alkylation with 67 gave analog 68. The same three-step sequence was also carried out in reverse order to arrive at analog 71. The same complementary approach was likewise followed for monomers 72 and 73, while symmetric monomers 74-76 were simply reacted in the manner used to prepare analog 71. Two additional core analogs (80 and 85) followed slightly modified procedures (Scheme 5). In the case of acyclic analog 80, the use of a protecting group was avoided by employing an excess of diamine 77. The remaining steps were as previously described. Preparation of piperidine analog 85 utilized previously outlined chemistry but was initiated with starting material 82, which already contained an ester group.
Scheme 4.
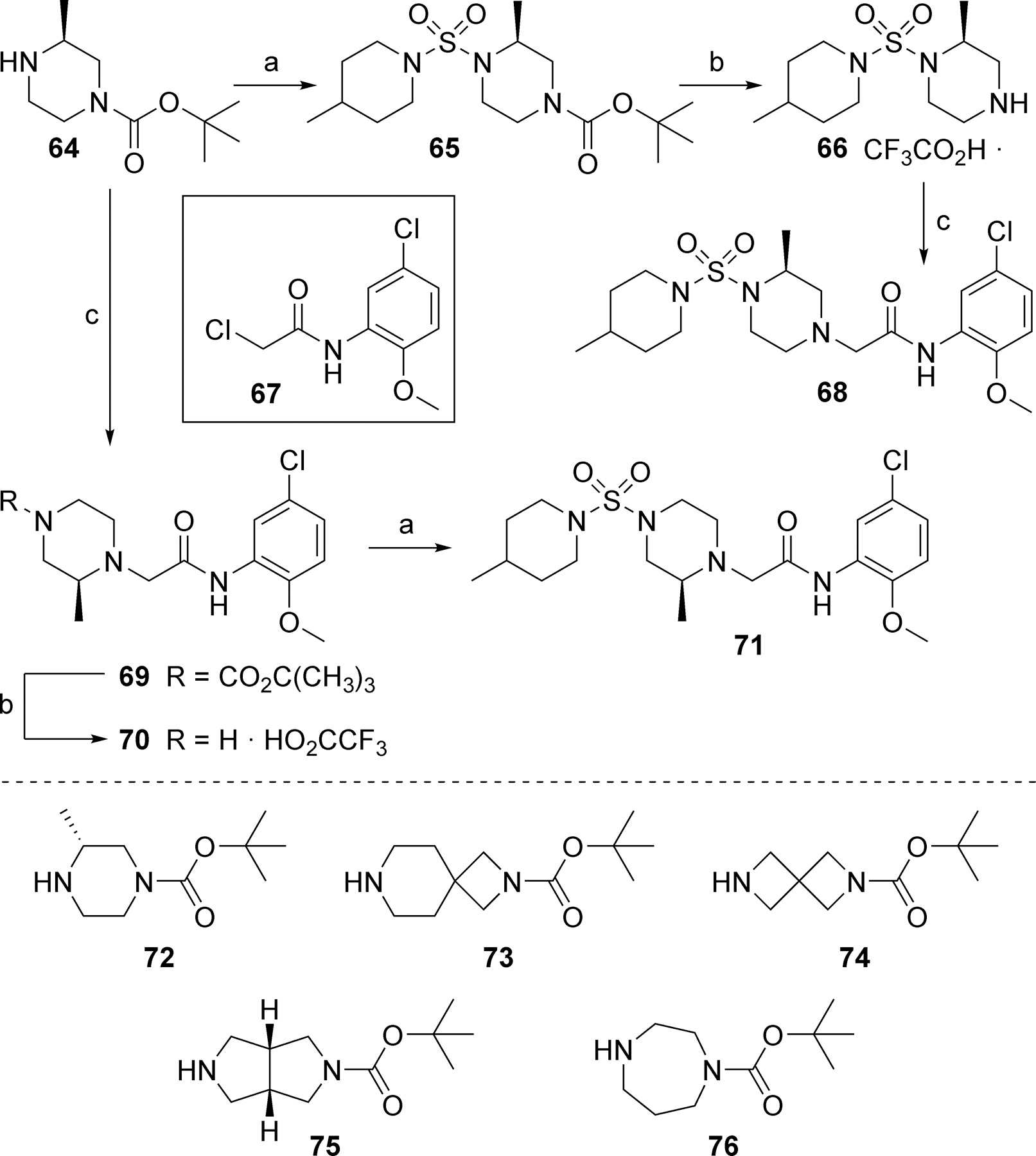
Reagents and conditions: (a) 5, DIEA, CH2Cl2, 61% (65), 98% (71); (b) TFA, CH2Cl2, 96% (66), 94% (70); (c) 67, DIEA, CH3CN, 75 ºC, 66% (68), 81% (69).
Scheme 5.
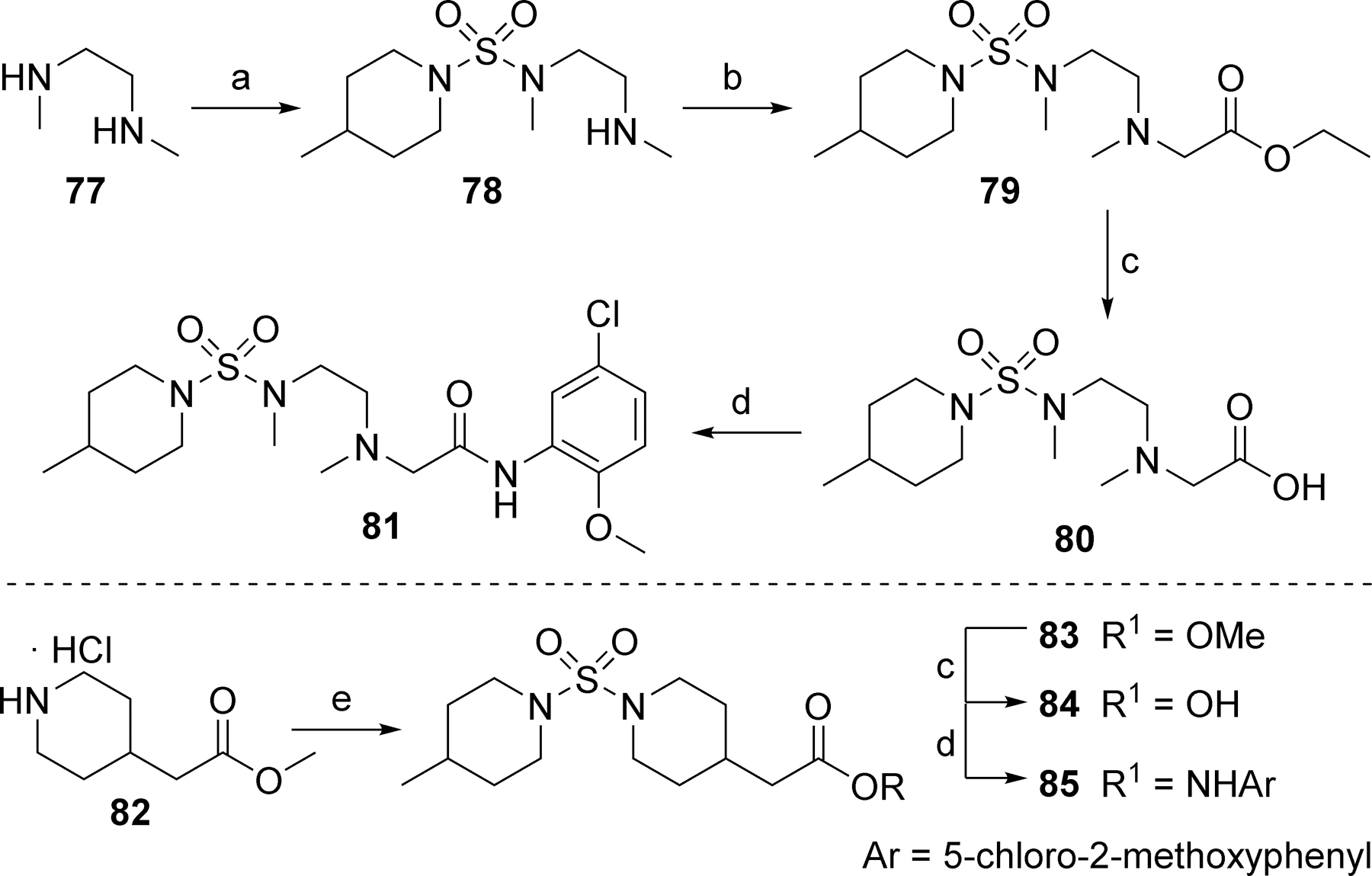
Reagents and conditions: (a) 5, CH2Cl2, 99%; (b) Ethyl bromoacetate, K2CO3, DMF, 33%; (c) LiOH, H2O, THF, 95% (80), 99% (84); (d) 5-chloro-2-methoxyaniline, HATU, DIEA, DMF, 40% (81), 24% (85); (e) 5, DIEA, CH2Cl2, 93%.
Methyl substitution of the piperazine core on the carbon adjacent to the sulfamide nitrogen (68 and 86) significantly reduced potency (Table 5). On the other hand, methyl substitution of the other carbon was well tolerated (71 and 87), with (R)-enantiomer 87 being equipotent to hit compound 4. Acyclic analog 81 retained inhibitory activity but exhibited reduced potency, likely due to increased degrees of rotational freedom. Piperidine 85 and spirocycles 88 and 89 were weak activators while spirocycle 90 was a weak inhibitor. Octahydropyrrolo[3,4-c]pyrrole 91 was the only analog in this set devoid of activity at Slack. Finally, expansion of the ring in the form of homopiperazine 92 gave a moderately potent analog.
Table 5.
Core analogs
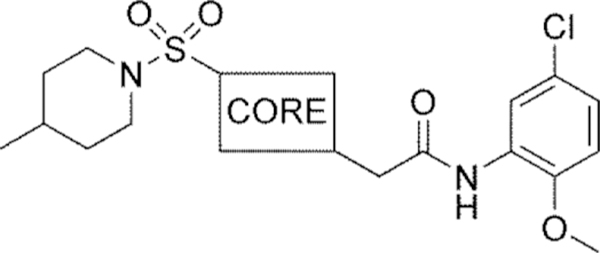
| ||||
|---|---|---|---|---|
| No. | CORE | Modea | IC50 / EC50 (µM)b | Efficacy (%)b,c |
| 68 |
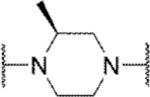
|
Inh | >10d | 20 |
| 86 |
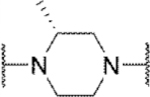
|
Inh | 13 | 58 |
| 71 |
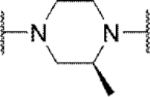
|
Inh | 3.5 | 93 |
| 87 |
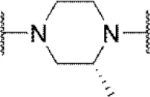
|
Inh | 2.4 | 101 |
| 81 |
|
Inh | >10d | 103 |
| 85 |
|
Act | 9.3 | 25 |
| 88 |
|
Act | >10d | 17 |
| 89 |
|
Act | 10 | 32 |
| 90 |
|
Inh | >10d | 69 |
| 91 |
|
inactive | ||
| 92 |
|
Inh | 6.2 | 106 |
Inh = inhibitor; Act = activator; NA = not active
CRC from Tl+ flux assay in HEK-293 cells expressing WT Slack
Amplitude of response in the presence of 30 µM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
CRC does not plateau within the concentration range tested
Our SAR studies in the VU0606170 (4) chemotype to date have not identified analogs that improve upon the potency of the hit compound. Still, we have identified several analogs that were equipotent or near equipotent with 4.8 Thus, we decided to take a closer look at the pharmacological profiles of select analogs. First, we screened nine select inhibitors against A934T mutant Slack, a variant commonly associated with MMPSI (Table 6).2, 3 All of the molecules functioned as inhibitors and were equipotent or more potent versus A934T as compared to WT with the majority of the molecules being slightly more potent versus A934T. Such a result may indicate that our inhibitors block an open confirmation of the channel, a phenomena observed with quinidine in other variants with Slack GOF mutations.8
Table 6.
Comparison of activity versus WT and A934T Slack for select inhibitors
| No. | WT | A934T | ||
|---|---|---|---|---|
| IC50 (µM)a | Efficacy (%)a,b | IC50 (µM)a | Efficacy (%)a,b | |
| 4 | 2.4 | 100 | 1.4 | 100 |
| 24 | 4.9 | 102 | 2.3 | 100 |
| 27 | 2.4 | 89 | 1.3 | 88 |
| 33 | 4.3 | 95 | 3.1 | 95 |
| 60 | 4.4 | 93 | 2.7 | 98 |
| 61 | 3.4 | 95 | 2.5 | 96 |
| 62 | 4.9 | 98 | 2.9 | 106 |
| 63 | 3.5 | 100 | 2.8 | 102 |
| 71 | 3.5 | 93 | 2.1 | 94 |
| 87 | 2.4 | 101 | 1.1 | 100 |
CRC from Tl+ flux assay in HEK-293 cells expressing either WT or A934T Slack
Amplitude of response in the presence of 30 µM test compound as a percentage of the maximum response for VU0606170
We next chose three of the most potent inhibitors with variations at distinct points of the scaffold for screening versus Slick (Slo2.1) and Maxi-K (Slo1 α1/β3) to assess Slo family selectivity (Table 7). Selectivity versus Maxi-K was generally very good, with 27 and 87 showing weak inhibition and 61 being inactive. Most analogs demonstrated weak inhibition of Slick. While 27 had an IC50 of 4.1 µM, efficacy was low at 29% of the positive control. Thus, these analogs displayed similar selectivity to VU0606170 (4), which is not surprising, given the small structural differences. These same three inhibitors were also examined in a whole-cell electrophysiology (EP) assays using an automated patch clamp system (SyncroPatch 384) (Table 8). Gratifyingly, potency and efficacy in this assay was on par with those observed in Tl+ flux in both WT and A934T expressing cells with each of these analogs.
Table 7.
Slo family selective for select Slack inhibitors
| No. | Slick (Slo2.1) | Maxi-K (Slo1 α1/β3) | ||
|---|---|---|---|---|
| IC50 (µM)a | Efficacy (%)a,b | IC50 (µM)a | Efficacy (%)a,b | |
| 4 | >10c | 35 | inactive | |
| 27 | 4.1 | 29 | >10c | 36 |
| 61 | >10c | 34 | inactive | |
| 87 | >10c | 50 | >10c | 32 |
CRC from Tl+ flux assay in HEK-293 cells expressing either Slick (Slo2.1) or Maxi-K (Slo1 α1/β3)
Amplitude of response in the presence of 30 µM test compound as a percentage of the maximum response for SKF96365 (Slick) or paxilline (Maxi-K)
CRC does not plateau within the concentration range tested
Table 8.
Whole-cell electrophysiology (EP) versus WT and A934T Slack for select inhibitors
| No. | WT | A934T | ||
|---|---|---|---|---|
| IC50 (µM)a | Efficacy (%)a,b | IC50 (µM)a | Efficacy (%)a,b | |
| 4 | 1.7 | 100 | 1.6 | 100 |
| 27 | 2.3 | 100 | 1.1 | 90 |
| 61 | 1.9 | 100 | 1.5 | 70 |
| 87 | 1.7 | 100 | 1.9 | 100 |
CRC from whole-cell EP in HEK-293 cells expressing either WT or A934T Slack utilizing an automated patch clamp system
Amplitude of response in the presence of 30 µM test compound as a percentage of the maximum response for VU0606170
In conclusion, we have executed SAR studies in a new series of 2-amino-N-phenylacetamide inhibitors of Slack potassium channels based on the HTS hit VU0606170. Systematic evaluation of five distinct regions of the chemotype revealed flat SAR with most substantial structural modifications proving deleterious to Slack activity. Some analogs with more modest structural deviations from the hit compound exhibit potency on par with the HTS hit. For example, chiral methyl substituents on the core (87) and linker 2 (61) and exchange of the chloro group of the hit for a trifluoromethyl group (27 and 61) gave potent analogs. Importantly, testing of selected analogs versus the A934T Slack mutant revealed potency on par with the WT, and results in whole-cell EP mirrored those obtained with Tl+ flux for the same analogs, validating our primary optimization screening approach utilizing Tl+ flux as our frontline assay. Utilization of homology models and docking studies might prove instrumental in the optimization of small molecule Slack inhibitors in the future; however, additional information about the binding site for these compounds would be required to best enable such an approach. The observed mode-switching in this scaffold argues against the compounds functioning as simple pore blockers. Hit evaluation and optimization continues in additional distinct scaffolds discovered through our HTS campaign. Reports from those efforts will be reported in the near future.
Supplementary Material
Acknowledgments
We thank the National Institute of Neurological Disorders and Stroke (R33NS109521 to C.D.W. and K.A.E.) for their support of our program in the development of small molecule inhibitors of Slack channels. Funding for the WaveFront Biosciences Panoptic kinetic imaging plate reader and the Syncropatch 768 PE platform was provided by the Office of The Director (OD) of the National Institutes of Health under the award numbers 1S10OD021734 and 1S10OD021734, respectively.
Footnotes
Supporting Information
Synthetic chemistry and pharmacological methods associated with this article can be found in the Supporting information.
References and Notes
- 1.Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia 1995;36(10): 1017–1024. [DOI] [PubMed] [Google Scholar]
- 2.McTague A, Nair U, Malhotra S, et al. Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology 2018;90(1): e55–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barcia G, Fleming MR, Deligniere A, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012;44(11): 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barcia G, Chemaly N, Kuchenbuch M, et al. Epilepsy with migrating focal seizures: KCNT1 mutation hotspots and phenotype variability. Neurology Genetics 2019;5(6): e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan A, Santi CM, Wei A, et al. The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron 2003;37(5): 765–773. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system. The Journal of comparative neurology 2002;454(3): 241–254. [DOI] [PubMed] [Google Scholar]
- 7.Joiner WJ, Tang MD, Wang LY, et al. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nature neuroscience 1998;1(6): 462–469. [DOI] [PubMed] [Google Scholar]
- 8.Rizzo F, Ambrosino P, Guacci A, et al. Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Molecular and cellular neurosciences 2016;72: 54–63. [DOI] [PubMed] [Google Scholar]
- 9.Kaczmarek LK. Slack, Slick and Sodium-Activated Potassium Channels. International scholarly research notices 2013;2013: Article ID 354262. [Google Scholar]
- 10.Zhang Z, Rosenhouse-Dantsker A, Tang QY, Noskov S, Logothetis DE. The RCK2 domain uses a coordination site present in Kir channels to confer sodium sensitivity to Slo2.2 channels. J Neurosci 2010;30(22): 7554–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci 2003;23(37): 11681–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quraishi IH, Mercier MR, McClure H, et al. Impaired motor skill learning and altered seizure susceptibility in mice with loss or gain of function of the Kcnt1 gene encoding Slack (KNa1.1) Na(+)-activated K(+) channels. Sci Rep 2020;10(1): 3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim Grace E, Kronengold J, Barcia G, et al. Human Slack Potassium Channel Mutations Increase Positive Cooperativity between Individual Channels. Cell reports 2014;9(5): 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milligan CJ, Li M, Gazina EV, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Annals of neurology 2014;75(4): 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin HC, Kim GE, Pagnamenta AT, et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Human molecular genetics 2014;23(12): 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nature Genetics 2012;44(11): 1188–1190. [DOI] [PubMed] [Google Scholar]
- 17.Ohba C, Kato M, Takahashi N, et al. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia 2015;56(9): e121–128. [DOI] [PubMed] [Google Scholar]
- 18.Routier L, Verny F, Barcia G, et al. Exome sequencing findings in 27 patients with myoclonic-atonic epilepsy: Is there a major genetic factor? Clinical Genetics 2019;96(3): 254–260. [DOI] [PubMed] [Google Scholar]
- 19.Hansen N, Widman G, Hattingen E, Elger CE, Kunz WS. Mesial temporal lobe epilepsy associated with KCNT1 mutation. Seizure 2017;45: 181–183. [DOI] [PubMed] [Google Scholar]
- 20.Borlot F, Abushama A, Morrison-Levy N, et al. KCNT1-related epilepsy: An international multicenter cohort of 27 pediatric cases. Epilepsia 2020. [DOI] [PubMed]
- 21.Moller RS, Heron SE, Larsen LH, et al. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 2015;56(9): e114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quraishi IH, Stern S, Mangan KP, et al. An Epilepsy-Associated KCNT1 Mutation Enhances Excitability of Human iPSC-Derived Neurons by Increasing Slack KNa Currents. J Neurosci 2019;39(37): 7438–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleming MR, Brown MR, Kronengold J, et al. Stimulation of Slack K(+) Channels Alters Mass at the Plasma Membrane by Triggering Dissociation of a Phosphatase-Regulatory Complex. Cell reports 2016;16(9): 2281–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshitomi S, Takahashi Y, Yamaguchi T, et al. Quinidine therapy and therapeutic drug monitoring in four patients with KCNT1 mutations. Epileptic disorders : international epilepsy journal with videotape 2019;21(1): 48–54. [DOI] [PubMed] [Google Scholar]
- 25.El Kosseifi C, Cornet M-C, Cilio MR. Neonatal Developmental and Epileptic Encephalopathies. Seminars in Pediatric Neurology 2019;32: 100770. [DOI] [PubMed] [Google Scholar]
- 26.Passey CC, Erramouspe J, Castellanos P, O’Donnell EC, Denton DM. Concurrent Quinidine and Phenobarbital in the Treatment of a Patient with 2 KCNT1 Mutations. Current therapeutic research, clinical and experimental 2019;90: 106–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patil AA, Vinayan KP, Roy AG. Two South Indian Children with KCNT1-Related Malignant Migrating Focal Seizures of Infancy - Clinical Characteristics and Outcome of Targeted Treatment with Quinidine. Annals of Indian Academy of Neurology 2019;22(3): 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia Y, Lin Y, Li J, et al. Quinidine Therapy for Lennox-Gastaut Syndrome With KCNT1 Mutation. A Case Report and Literature Review. Frontiers in neurology 2019;10: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dilena R, DiFrancesco JC, Soldovieri MV, et al. Early Treatment with Quinidine in 2 Patients with Epilepsy of Infancy with Migrating Focal Seizures (EIMFS) Due to Gain-of-Function KCNT1 Mutations: Functional Studies, Clinical Responses, and Critical Issues for Personalized Therapy. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 2018;15(4): 1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures of infancy with quinidine. Annals of neurology 2014;76(3): 457–461. [DOI] [PubMed] [Google Scholar]
- 31.Qunies AM, Emmitte KA. Small-molecule inhibitors of Slack potassium channels as potential therapeutics for childhood epilepsies. Pharmaceutical Patent Analyst 2022;11(2): 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole BA, Johnson RM, Dejakaisaya H, et al. Structure-Based Identification and Characterization of Inhibitors of the Epilepsy-Associated K(Na)1.1 (KCNT1) Potassium Channel. iScience 2020;23(5): 101100–101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griffin AM, Kahlig KM, Hatch RJ, et al. Discovery of the First Orally Available, Selective KNa1.1 Inhibitor: In Vitro and In Vivo Activity of an Oxadiazole Series. ACS Medicinal Chemistry Letters 2021;12(4): 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spitznagel BD, Mishra NM, Qunies AM, et al. VU0606170, a Selective Slack Channels Inhibitor, Decreases Calcium Oscillations in Cultured Cortical Neurons. ACS Chemical Neuroscience 2020;11(21): 3658–3671. [DOI] [PubMed] [Google Scholar]
- 35.Manka JT, Rodriguez AL, Morrison RD, et al. Octahydropyrrolo[3,4-c]pyrrole negative allosteric modulators of mGlu1. Bioorg Med Chem Lett 2013;23(18): 5091–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Åstrand ABM, Grimster NP, Kawatkar S, et al. , inventors; AstraZeneca AB, assignee. Compounds and methods for inhibiting JAK. PCT Int. Patent Appl WO 2017/050938 A1. 2017. Mar 30.
- 37.Melancon BJ, Utley TJ, Sevel C, et al. Development of novel M1 antagonist scaffolds through the continued optimization of the MLPCN probe ML012. Bioorg Med Chem Lett 2012;22(15): 5035–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lachance N, Gareau Y, Guiral S, et al. Discovery of potent and liver-targeted stearoyl-CoA desaturase (SCD) inhibitors in a bispyrrolidine series. Bioorg Med Chem Lett 2012;22(2): 980–984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.