

ABSTRACT
Autosomal dominant polycystic kidney disease (ADKPD) is the most frequent type of polycystic kidney disease. It is inherited through family members, with an incidence of approximately 1:400 to1:1000.Typically, individuals with ADKPD are identified between their fourth and fifth decade of life. ADKPD occurs as a results of mutation in one of the two genes, PDK1 and PDK2.Patients with PKD1 experience renal failure at an earlier onset than those with PKD2. We report on a 2 year-old-boy with hepatosplenomegaly and signs of portal hypertension. Both kidneys appeared normal until the age of 8, when multiple cysts developed, this being typical of ADKPD. Suspecting ADKPD, we performed whole exome sequencing, thereby confirming a mutation of c.6730 673del p.(Ser 2244Hisfs*17). The investigations of all family members found other individuals affected by ADKPD.
Key words: Autosomal polycystic kidney disease ADKPD, congenital hepatic fibrosis CHF
INTRODUCTION
The most common type of inherited polycystic kidney disease, known as ADKPD, typically presents in adults, but it is now well-recognized in children of all ages. A variety of polycystic kidney diseases are accompanied by congenital hepatic fibrosis, but rarely ADKPD[1,2,3,4]. A rare autosomal recessive condition known as Congenital hepatic fibrosis (CHF) is characterized bybile duct distortion and periportal fibrosis in varying degrees. It is usually assumed to be a characteristic for autosomal recessive polycystic kidney disease.
Manifestations of portal hypertension such as hypersplenism and variceal bleeding in early infancy or childhood are commonly present in the diagnosis of CHF [18]. However, some of these patients may not develop any symptoms for many years, leading to a late adult CHF diagnosis. This case report describes a large family with ADKPD, whose 8-year-old son is the only member who manifests congenital hepatic fibrosis with portal hypertension, based on the results of imaging techniques (ultrasonography, hepatic elastography), liver biopsy and a genetic test.
CASE REPORT
An 8-year-old boy was referred to our service of pediatric gastro hepatology with hepatosplenomegaly having a 6 year duration without a clear diagnosis. At the age of two, he presented easy bruising over the lower limbs. At the same time was found that he had thrombocytopenia and mild hepatosplenomegaly, demonstrated also by an abdominal ultrasound.
He was the third boy of an Albanian couple without consanguinity. According to his mother the pregnancy was normal and delivery was by a cesarian section. The neonatal period was uneventful. He was vaccinated according to our national schedule. No history of other diseases appeared until he reached two years old. This is when the first presentations were some bruising on legs and mild hepatosplenomegaly. During frequent hospitalizations, autoimmune hepatitis was excluded along with, viral hepatitis (Cytomegalovirus, Epstein-Barr virus, TORCH), metabolic disease like Gaucher (dry blood spot test), and alpha-1 antitrypsin deficiency. Furthermore, a bone marrow examination showed normal marrow with no evidence for any pathology.
At the age of six, an ultrasound confirmed a hepatosplenomegaly meanwhile a liver elastography showed fibrosis at stage F2. An enlarged liver and spleen were found during an abdominal exam, both of which were located 4 to 5 cm under the costal margin. Following admission, laboratory results indicated PT 90%, RBC = 3.85 X 106/µL, Hb 9.2 g/L and platelet count of 77 k/uL. His liver function tests were normal and his serum ferritin level was also in the normal range.
The last abdominal ultrasound demonstrated an enlarged liver and spleen (liver right lobe 130 mm; left lobe 110mm; caudate lobe 40mm, and spleen with dimension 125mm). Doppler image revealed the presence of esophageal varices. An upper endoscopy confirmed the presence of grade 1 esophageal varices (see Figure 1). Serial abdominal ultrasonography were done, but only the last one revealed the presence of numerous millimetric cysts on both kidneys. There were some cysts at the right kidney reaching 9.2 mm in diameter (see Figure 2). It was at this point that the family’s history of polycystic kidney disease was noticed. The paternal grandmother died of renal disease of unknown cause. His paternal aunt continues hemodialysis and is known to have polycystic kidney disease. Thus, in this context we decided to screen all family members by abdominal ultrasound. The patient’s father, and the patient’s brothers even though asymptomatic, demonstrated several bilateral renal cysts, while the liver had a normal morphology(see Figure 3). ADKPD was suspected, based on the pedigree of the family(see Figure 4).
Figure 1.

Ultrasound of liver(left). The axial plane of the colour Doppler image shows discrete esophageal varices. Upper endoscopy (right) showing grade 1 esophageal varices.
Figure 2.
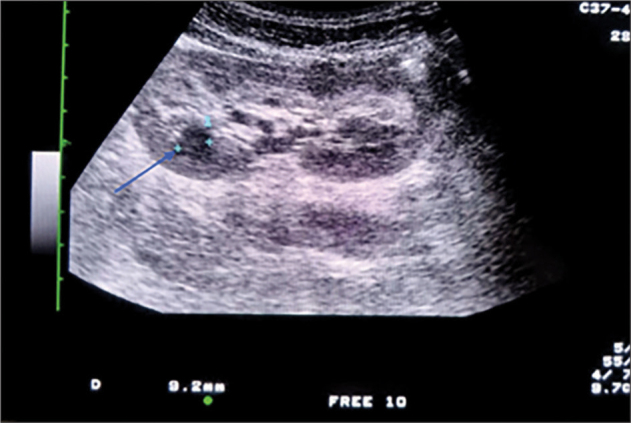
Ultrasound imaging of kidney. The arrow shows the 9.2 mm renal cyst.
Figure 3.

Ultrasound imaging of kidney cysts A) Father Enlarged hyperechogenic kidney with loss of corticomedullary differentiation and multiple renal cysts and the larger cyst 15 mm B) The older brother - Enlarged kidneys with multiple small cysts and one large cyst 11 mm (arrow) C) & D) The other brothers - Enlarged polycystic kidneys with loss of corticomedullary differentiation and multiple kidney cysts in the kidneys (arrows).
Figure 4.
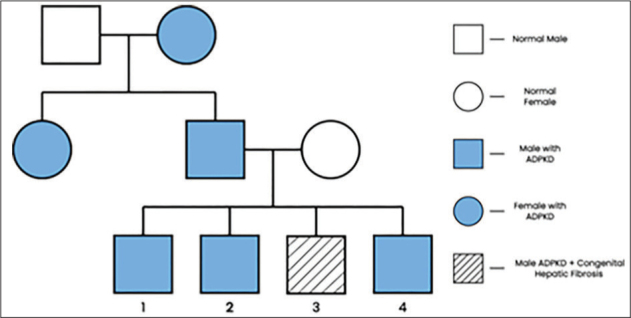
Pedigree of family with ADPKD.
A liver biopsy was indicated for the patient. In the histopathologic evaluation, the parenchyma of the tissue was divided into lobules by severe fibrosis of the portal tracts and several bile ducts with irregular shapes. The results supported the diagnosis of congenital hepatic fibrosis.
A genetic test (whole exome sequencing) was performed and the results confirmed the diagnosis of Autosomal Dominant Polycystic kidney disease Type 1. A heterozygous pathogenic variant was identified in the PKD1 gene. The PDK1 variant c.6730_673del p.(Ser2244Hisfs* 17) creates a shift in the reading frame starting at codon 2244. The new reading frame ends in a stop codon 16 position downstream. This variant was confirmed by Sanger sequencing.
Further studies with cranial magnetic resonance angiography and echocardiography did not reveal any other abnormality. Currently, the patient is 8 years old and is generally healthy. His liver function results as INR, albumin and transaminase are normal but platelets count remain low. He needs routine endoscopic evaluation of the progression of portal hypertension and varices bleeding.
DISCUSSION
Autosomal dominant polycystic kidney disease (AD-KPD) is the most frequent inherited renal cystic diseases that is identified by the growth of renal cysts and a variety of extra renal presentations. The report on the family shows good evidence of multiple renal cysts, transmitted by the family in a clear autosomal dominant pattern. In addition, one of the members of the third generation presents Congenital hepatic fibrosis. Despite being generally related to ARPKD, a few cases also associate CHF to ADKPD [1-9]. The significance of their occurrence is uncertain; these conditions have different modes of inheritance.
A similar case is reported by Tazeeler [9] in a 19 year-old woman, whose family’s involvement with ADKPD was known. She firstly presented with portal hypertension and was eventually shown to have renal cysts and a family history of ADKPD. Manifestations of portal hypertension such as splenomegaly and variceal bleeding in early infancy or during childhood led to a diagnosis of CHF. Interestingly, some of these patients may be asymptomatic for a significant period of time, resulting to an unexpected CHF in adulthood [20].
Typical kidney and liver manifestation of ADKPD developed in the fourth and fifth decade of life in patients. Although, there are cases describing the presence of renal symptoms in ADKPD even in infancy and early childhood [5]. Similarly, as in our case, portal hypertension with CHF might be the first presenting signs of ADKPD [10-13]. While to the contrary in ARPKD, renal disease frequently precedes the symptoms related to CHF and complication of portal hypertension [14-17]. ADKPD may be present in childhood due to variability attributable to mutation at more than one locus. The first locus PK1 is present on chromosome 16 responsible for 85% of the cases. The penetrance of the autosomal dominant gene of ADKPD is almost 100%, so we needed to investigate the previous generation in the family.
It is important to highlight that patients with ADKPD may grow large kidney cysts similar to those found in ARKPD, thus a family history is extremely important for a proper diagnosis of ADKPD with CHF [18, 19]. In addition to the renal cystic abnormalities in ADKPD, several extra renal changes may be noted such as cysts in the liver, ovary, and seminal vesicles, abdominal hernias, cerebral or aortic aneurysms [21, 22, and 23]. It is recommended to monitor via ultrasound for ADKPD in these patients and their family members. This case report highlights the value of an accurate family history of polycystic kidney disease in a child with these manifestations. Finding a normal sized kidney at infancy is not unusual, since the expecting incidence of kidney alteration is in the next decade of life.
Presentation with hepatosplenomegaly can mislead to a late diagnosis. It is suggested that CHF should not be considered as a distinctive condition, as it can still be encountered in patients with ADKPD [24]. Due to such variability, we should emphasize the significance of a detailed family history of polycystic kidney disease in order to determine the type (dominant or recessive disease), as well as understanding other interrelated conditions.
CONCLUSION
ADKPD presented by congenital hepatic fibrosis in early childhood remains a challenge for every pediatrician. A prompt familial history along with image studies, endoscopy and liver histology can lead to an early and accurate diagnosis. Genetic tests can finally avoid any diagnostic suspicion.
Footnotes
Author Contribution: L.S. drafted the manuscript. VV, SS designed the manuscript. B.S, E.D, D.SH and PC read and approved the final version of the manuscript.
Parental consent: The patient’s parents signed an informed consent for publication of thiscase.
REFERENCES
- 1.Kerr DN, Harrison CV, Sherlock S, Walker RM. Congenital hepatic fibrosis Q J Med. 1961;30:91. ( ) . : –. . [PubMed] [Google Scholar]
- 2.Zhu B, Du Z, Wang Z, Li Y, Zhang J, Zhu H. Congenital hepatic fibrosis in children and adults: clinical manifestations, management, and outcome— case series and literature review Gastroenterol Res Pract. 2020;2020:1. doi: 10.1155/2020/8284274. ( ) . : –. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sommerschild HC, Langmark F, Maurseth K Congenital hepatic fibrosis: report of two new cases and review of the literature Surgery. 1973;73:53. . . ; : - . [PubMed] [Google Scholar]
- 4.Bradford WD, Bradford JW, Porter FS, Sidbury JB Cystic disease of liver and kidney with portal hypertension: a cause of sudden unexpected hematemesis ClinPediatr. 1968;7:299. doi: 10.1177/000992286800700515. . . ; : - . [DOI] [PubMed] [Google Scholar]
- 5.Gaisford W, Bloor K Congenital polycystic disease of kidneys and liver, portal hypertension-portal anastomosis Proc R Soc Med. 1968;61:304. . . ; : . [PMC free article] [PubMed] [Google Scholar]
- 6.Clermont RJ, Maillard J-N, Benhamou J-p, Fauvert R Fibrosehepatiquecongenitale Can Med Assoc J. 1967;97:1272. . . ; : - . [PMC free article] [PubMed] [Google Scholar]
- 7.Longmire WP, Mandiola SA, Gordon HE Congenital cystic disease of the liver and biliary system Ann Surg. 1971;174:711. doi: 10.1097/00000658-197110000-00014. . . ; : - . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manes JL, Kissane JM, Valdes AJ Congenital hepatic fibrosis liver cell carcinoma and adult polycystic kidneys Cancer. 1977;39:2619. doi: 10.1002/1097-0142(197706)39:6<2619::aid-cncr2820390647>3.0.co;2-4. . . ; : - . [DOI] [PubMed] [Google Scholar]
- 9.Tazelaar HENRY D., PAYNE JOHN A., PATEL NARGIS S. Congenital Hepatic Fibrosis an Asymptomatic Familial Adult-Type Polycystic Kidney Disease in a 19-yearold Woman. Rush-Presbyterian-St. Luke’s MedicalCenter; Chicago, Illinois: , [Google Scholar]
- 10.Ross DG, Travers H Infantile presentation of adult-type polycystic kidney disease in a large kindred J Pediatr. 1975;76:760. doi: 10.1016/s0022-3476(75)80303-9. . . ; : - . [DOI] [PubMed] [Google Scholar]
- 11.Kaye C, Lewy PR Congenital appearance of adult type (autosomal dominant) polycystic kidney disease J Pediat. 1974;85:807. doi: 10.1016/s0022-3476(74)80346-x. . . ; : - . [DOI] [PubMed] [Google Scholar]
- 12.Shokier MKH Expression of “adult” polycystic renal disease in the fetus and newborn Clin Genet. 1978;14:61. doi: 10.1111/j.1399-0004.1978.tb02107.x. . . ; : - . [DOI] [PubMed] [Google Scholar]
- 13.Gunay-Aygun M. Liver and kidney disease in ciliopathies Am J Med Genet C Semin Med Genet. 2009;151C:296. doi: 10.1002/ajmg.c.30225. . ; : –. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blyth H, Ockenden BG Polycystic disease of kidneys and liver presenting in childhood J Med Genet. 1971;8:257. doi: 10.1136/jmg.8.3.257. . . ; : - . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Association of congenital hepatic fibrosis with autosomal dominant polycystic kidney disease. Report of a family with review of literature Lipshitz B., Berdon W. E., Defelice A. R., Levy J. pediatrRadiol. 1993;23:131. doi: 10.1007/BF02012406. . ) : - . [DOI] [PubMed] [Google Scholar]
- 16.Zhu L., Zhao G., Jia C. F., Li Y. Congenital hepatic fibrosis with medullary sponge kidney: a case report ZhonghuaNeikeZazhi 2010;49(12):1060. , “. ,”. , vol. , no. , pp. - , . [Google Scholar]
- 17.Congenital hepatic fibrosis-is it a single clinical entity? Murray-Lyon, Ockenden G., Williams Roger. Gastroenterology. 1973;64:653. Liver Unit, King’s College Hospital London S.E. 5, England. : - , . [PubMed] [Google Scholar]
- 18.Lieberman E, Salinas-Madrigal L, Gwinn JL, Brennan LP, Fine RN, Landing BH Infantile polycystic disease of the kidneys and liver Medicine. 1971;50:277. doi: 10.1097/00005792-197107000-00003. . . ; : - . [DOI] [PubMed] [Google Scholar]
- 19.Zhang W., Gao J. R., Lang Y. H., Shao L. P. A case report of adult ADKPD with congenital hepatic fibrosis ZhonghuaShenzangbingZazhi 2011;27(7):547. , “. ,”. , vol. , no. , pp. –. , . [Google Scholar]
- 20.Parkash A, Cheema HA, Malik HS, Fayyaz Z Congenital hepatic fibrosis: clinical presentation, laboratory features and management at a tertiary care hospital of Lahore J Pak Med Assoc. 2016 NaN66(8):984. . . ; ( ): –. . [PubMed] [Google Scholar]
- 21.Wu YJ, Ding HG Hereditary polycystic kidney disease: a neglected etiology of liver cirrhosis Zhonghuaganzang Bing zazhi. 2016;24:728. doi: 10.3760/cma.j.issn.1007-3418.2016.10.003. . . ; : –. . [DOI] [PubMed] [Google Scholar]
- 22.Bergmann Carsten, et al. Polycystic kidney disease Nature reviews. Disease primers. 2018 NaN 6;4,1:50. doi: 10.1038/s41572-018-0047-y. “. .”. vol. . , doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Autosomal dominant polycystic kidney disease Torres Vicente E, Harris Peter C, Pirson Yves. Lancet. 2007;369:1287. doi: 10.1016/S0140-6736(07)60601-1. . ; : –. . [DOI] [PubMed] [Google Scholar]
- 24.Nobakht N, Hanna RM, Al-Baghdadi M, Ameen KM, Arman F, Nobahkt E, Kamgar M, Rastogi A Advances in Autosomal Dominant Polycystic Kidney Disease: A Clinical Review Kidney Med. 2020 NaN 22;2(2):196. doi: 10.1016/j.xkme.2019.11.009. . . ; ( ): - . Doi: . [DOI] [PMC free article] [PubMed] [Google Scholar]