

ABSTRACT
Ewing sarcoma (ES), described as a diffuse endothelioma of the bone, is divided into two categories: osseous and extraosseous, which mainly affects adolescents. Extraosseous Ewing Sarcomas (EES) are rare tumors originating from soft tissues. Their clinical presentation depends mainly on the primary location of the tumor and are highly chemosensitive and radiosensitive. The purpose of this study was to describe the clinical characteristics and outcomes of 3 children with EES and uncommon presentation treated in our Unit. The diagnosis of EES was confirmed by biopsy and cytogenetic analysis with fluorescence in situ hybridization (FISH). Surgical excision was planned as primary treatment, followed by adjuvant chemotherapy according to EURO-E.W.I.N.G protocol. To date, all patients are alive, 1, 3 and 4 years after completion of treatment, with no signs of recurrence or metastasis.
Key-words: cytogenetics, extraosseous Ewing sarcoma, surgical resection
INTRODUCTION
Extraosseous Ewing Sarcoma (EES) is a highly aggressive, poorly differentiated small round cell tumor accounting for 20-30% of Ewing Sarcoma (ES) cases. EES belongs to the histological group of Ewing Sarcoma Family Tumors (ESFT) originating from neuroectodermal and mesenchymal cells along with classical osseous ES, small-cell tumor of the thoracopulmonary region (Askin tumor) and soft-tissue-based primitive neuroectodermal tumors (PNETs) (1–4). In accordance with findings from adult studies (5), the characteristics of patients, as well as prognosis, differ between ES and EES. In the EES group a higher mean age and lower percentage of males and Caucasians were observed. Extraskeletal origin was a favorable prognostic factor. Importantly, no significant differences in genomic features were noted (6).
The clinical manifestation varies largely depending on the site of origin, with a rapidly growing mass causing pain being the most common symptom. Commonly affected extraskeletal sites include the paravertebral spaces, lower extremities, head, neck and pelvis, while distant metastases are detected in up to 25% of cases, most commonly in the lungs (1,7).
The recommended treatment is local therapy (surgery and/or radiotherapy) plus chemotherapy (1–3). Surgery appears to play a more vital role in EES compared to osseous ES, since complete resection is a prognostic factor for survival (8, 9). Chemotherapy is a key element in the treatment of EES, with both neoadjuvant and adjuvant therapy showing comparable results in patients with localized disease (10–12). EES is radiosensitive, but the use of radiotherapy has been reduced over the years mainly due to the advances in surgical technique and the radiation-associated risks (secondary malignancies) (13).
We describe 3 patients with an uncommon presentation of EES, who were successfully treated in our Unit.
CASE REPORT 1
A 13-year-old boy presented to the emergency department with persistent pain located in the right hemithorax. There was no history of trauma and no signs or symptoms of infection. A week prior to admission, the patient had noticed a stabbing pain in his right hemithorax and was referred by his pediatrician for a chest X-ray, which revealed a homogeneous opacification of the right lung. On physical examination, a healthy young male with a normal posture was observed. On percussion, a dull sound was found in the right upper zone of the chest, while auscultation revealed normal cardiac sounds without murmurs and normal breathing sounds on the left and lower right side of the chest.
Abdominal examination did not reveal palpable masses or other abnormalities. Also, no palpable lymph nodes were found. Subsequent computed tomography (CT) revealed an expansive lobular mass (86x47x42mm) in the right hemithorax in continuity with the parietal pleura containing fluid-filled cysts (Figures 1 a and b). No bone involvement or distant metastatic disease were detected. Molecular analysis revealed chimeric transcripts of (t[11;22][q24;q12]), EWS-ERG (t[21;22][q22;q12]) and EWS-WT1(t[11;22][p13;q12]). Using immunohistochemistry, tumor cells were positive for Vimentin and CD99+, and lacked CD45, CK8/18, TdT and desmin staining. The morphology, in combination with immunophenotype, was compatible with EES. After total resection of the mass, chemotherapy according to EURO-E.W.I.N.G. 99 protocol was initiated and completed without adverse events. After 3 years of follow-up, there are no signs of recurrence.
Figure 1.

Sagittal (a) and transverse (b) CT depiction of lobular mass (86x47x42mm) in the right hemithorax in continuity with the parietal pleura in a 13-year-old male.
CASE REPORT 2
A 14-year–old girl was referred to our Unit following complete resection of a palpable abdominal mass. One month prior to surgery, the patient observed a swelling in the right hypochondrium. There was no history of pain, fever, weight loss, fatigue, excessive sweating or gastrointestinal complaints. An abdominal ultrasound revealed a hypoechoic, round lesion, approximately 30mm in diameter, without internal vasculature. There was no attachment to the diaphragm or chest wall. Initial MRI imaging (Figure 2) and staging revealed no skeletal involvement or metastatic disease. Complete resection of the mass was performed and histopathological examination demonstrated small, round tumor cells with oval nucleus, surrounded by eosinophilic cytoplasm. Immunohistochemistry revealed positivity for vimentin, CD99 and bcl2. Molecular biological examination was also performed but no transcripts t(11;22), t(21;22) or t(X;18) were detected. After radical surgical excision the patient underwent adjuvant chemotherapy according to EURO-E.W.I.N.G.99 protocol. To date, no recurrence has been observed, 4 years after the end of chemotherapy.
Figure 2.
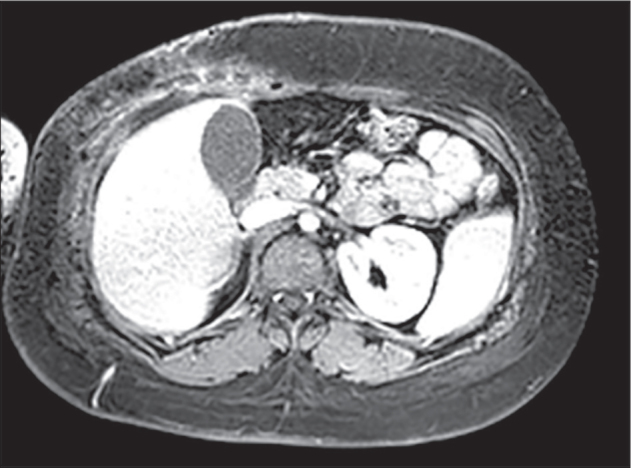
MRI imaging showing a mass (Ewing Sarcoma) in the right hypochondrium in a 14-year-old girl.
CASE REPORT 3
A 12 year-old girl was referred to our Unit due to a painless growing mass located on her right forearm, which was initially detected 3 months ago. Physical examination revealed a palpable mass of 12x5mm on the aspect of the right proximal forearm, with no signs of inflammation, and no other clinical findings. She underwent hematology/ biochemistry tests and chest X-Ray, which were normal. An ultrasound was performed, revealing a soft-tissue mass in the subcutaneous tissue of the flexor surface of the right forearm near the elbow joint with increased vascularization. A small nodule, 6mm in diameter, in the right lower lobe was detected on the chest CT. The patient underwent resection of the mass which was completely removed. Histopathological examination showed a tumor with a nodular growth pattern.
The monomorphic infiltrate consisted of small tumor cells with scanty cytoplasm and round nuclei. Immunohistochemistry revealed tumor cells with positivity for vimentin, CD99 and bcl2. Molecular biological examination was also performed but no transcripts t(11;22), t(21;22), t(X;18) were detected. She underwent chemotherapy according to EURO-E.W.I.N.G.99 chemotherapy protocol. So far, 18 months after the end of chemotherapy, there is no evidence of recurrence or metastases and she has an excellent quality of life.
DISCUSSION
EES was first described in 1969. It is a rare, malignant mesenchymal tumor similar to its intraosseous counterpart (14). Genetic studies have demonstrated reciprocal translocation of t(11;22)(q24;q12) in about 95% of patients, with a majority of the remainder demonstrating t(21;22) (q22;q12) (15, 16).
Patients with EES often observe a rapidly growing soft tissue mass, with about 1/3 being painful due to compression of adjacent structures (1, 2, 17). They often occur in the second decade of life with some studies reporting a mean age of 15 years and with a mild predominance for males and Caucasians. There is no evidence that familiar or environmental factors influence the incidence of the disease (10, 11, 18). According to these findings, the main symptoms of our patients included pain and swelling, while the presence of other symptoms depended on the location of the tumor.
Our findings demonstrate the variability in primary tumor sites. There are increasing reports of EES from diverse sites whose origin has been attributed to ectopic neural and neuroectodermal proliferations. Although the paravertebral spaces, lower extremities, head and neck and pelvis represent the most commonly affected sites, rare locations of EES include the omentum (19, 20), kidney (21), heart (22), uterus (23), lung (24, 25), chest wall (26) and gastrointestinal track (27–29).
It is also noteworthy that EESs present metastases at the time of diagnosis at a rate ranging from 10% to 25% (11), depending on the study. This is a fact that complicates the prognosis of the disease. In our study, one of the patients was diagnosed with possible lung metastasis. Localized tumors appear to have a survival rate of 75%, while metastatic disease has an unfavorable prognosis (30).
The National Comprehensive Cancer Network (NCCN) has published guidelines for the treatment of bone cancers including ES, and the authors suggest that any member of the Ewing tumor family can be treated according to the same algorithm. The recommended treatment is local therapy (surgery and/or radiotherapy) plus chemotherapy (31, 32). The gold standard of treatment for localized disease remains surgery, although the disease-free survival rate in patients with sarcoma without margin-negative surgery is worse (8, 9, 11). All 3 patients in our report had first-line surgery with negative margins. Post-surgery, the most appropriate course of chemotherapy was planned. Regarding the use of chemotherapy, it plays a critical role in the treatment of EES. Chemotherapy may be provided after surgery to improve overall survival rates and reduce the chance of the tumor recurrence (33). The third approach to combating Ewing Tumors is the addition of radiotherapy to the treatment plan. These malignancies have been proven to be radiosensitive, however radiation hazards in children and young adolescents are always present and require careful consideration. Surgery also provides better results in local control and survival. Indications for post-operative radiotherapy are non-radical or marginal resections and poor histological response (13,31). The patients described were treated according to the EURO-E.W.I.N.G 99 chemotherapy protocol, without the need for radiotherapy. A prevalent prognostic factor that is of great interest in multiple studies is the surgical resection of the tumor and the surgical margins. A complete resection with surgical margins clear of any malignancy constitutes a positive prognostic factor (8,9,11). All of our patients had complete resection and negative surgical margins in accordance with the above data and all are alive.
The improved understanding of EES molecular biology, made possible by the recent advances in the field of systems biology, has opened new perspectives for the treatment of this aggressive malignancy through the implementation of precision medicine. Insulin-like growth factor-1-receptor (IGF-1R), CD99 and the EWS-FLI-1 fusion product play key roles in malignant transformation by inducing cell growth and inhibiting apoptosis and have emerged as promising therapeutic targets (2, 34).
In conclusion, EES belong to a very rare and aggressive tumor family. Despite the limitations of having a small number of patients and the rarity of such tumors, the point is that early recognition and treatment reduce morbidity and mortality. Continuous research is needed to further improve our understanding of the disease and to adjust the treatment protocols in the hope of ameliorating prognosis.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
REFERENCES
- 1.Abboud A, Masrouha K, Saliba M, Haidar R, Saab R, Khoury N, et al. Extraskeletal Ewing sarcoma: Diagnosis, management and prognosis (Review) Oncol Lett. 2021;21(5):1. doi: 10.3892/ol.2021.12615. . ; ( ): –. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ludwig JA Ewing sarcoma: historical perspectives, current state-of-the-art, and opportunities for targeted therapy in the future Curr Opin Oncol. 2008;20(4):412. doi: 10.1097/CCO.0b013e328303ba1d. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 3.Galyfos G, Karantzikos GA, Kavouras N, Sianou A, Palogos K, Filis K Extraosseous Ewing Sarcoma: Diagnosis, Prognosis and Optimal Management Indian J Surg. 2016;78(1):49. doi: 10.1007/s12262-015-1399-0. . . ; ( ): –. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grier HE The Ewing family of tumors. Ewing’s sarcoma and primitive neuroectodermal tumors Pediatr Clin North Am. 1997;44(4):991. doi: 10.1016/s0031-3955(05)70541-1. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 5.Applebaum MA, Worch J, Matthay KK, Goldsby R, Neuhaus J, West DC, et al. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma Cancer. 2011;117(13):3027. doi: 10.1002/cncr.25840. . ; ( ): –. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cash T, McIlvaine E, Krailo MD, Lessnick SL, Lawlor ER, Laack N, et al. Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children’s Oncology Group Pediatr Blood Cancer. 2016;63(10):1771. doi: 10.1002/pbc.26096. . ; ( ): –. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Galindo C, Navid F, Khoury J, Matthew Krasin . Ewing sarcoma family of tumors. In: Pappo A, editor. Pediatric Bone and Soft Tissue Sarcomas. Berlin, Heidelberg: Springer-Verlag; 2006. pp. 181–217. . , , : - [Google Scholar]
- 8.Covelli HD, Beekman JF, Kingry RL Extraskeletal Ewing’s sarcoma: prolonged survival with recurrence after operation South Med J. 1980;73(9):1294. . . ; ( ): –. . [PubMed] [Google Scholar]
- 9.Rud NP, Reiman HM, Pritchard DJ, Frassica FJ, Smithson WA Extraosseous Ewing’s sarcoma. A study of 42 cases Cancer. 1989;64(7):1548. doi: 10.1002/1097-0142(19891001)64:7<1548::aid-cncr2820640733>3.0.co;2-w. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 10.Paulussen M, Ahrens S, Dunst J, Winkelmann W, Exner GU, Kotz R, et al. Localized Ewing tumor of bone: final results of the cooperative Ewing’s Sarcoma Study CESS 86 J Clin Oncol. 2001;19(6):1818. doi: 10.1200/JCO.2001.19.6.1818. . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 11.Balamuth NJ, Womer RB Ewing’s sarcoma Lancet Oncol. 2010;11(2):184. doi: 10.1016/S1470-2045(09)70286-4. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 12.Bacci G, Balladelli A, Forni C, Ferrari S, Longhi A, Bacchini P, et al. Adjuvant and neoadjuvant chemotherapy for Ewing sarcoma family tumors in patients aged between 40 and 60: report of 35 cases and comparison of results with 586 younger patients treated with the same protocols in the same years Cancer. 2007;109(4):780. doi: 10.1002/cncr.22456. . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 13.Dunst J, Schuck A Role of radiotherapy in Ewing tumors Pediatr Blood Cancer. 2004;42(5):465. doi: 10.1002/pbc.10446. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 14.Tefft M, Vawter GF, Mitus A Paravertebral “round cell” tumors in children Radiology. 1969;92(7):1501. doi: 10.1148/92.7.1501. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 15.Downing JR, Head DR, Parham DM, Douglass EC, Hulshof MG, Link MP, et al. Detection of the (11;22)(q24;q12) translocation of Ewing’s sarcoma and peripheral neuroectodermal tumor by reverse transcription polymerase chain reaction Am J Pathol. 1993;143(5):1294. . ; ( ): –. . [PMC free article] [PubMed] [Google Scholar]
- 16.Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG Nat Genet. 1994;6(2):146. doi: 10.1038/ng0294-146. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 17.Ahmad R, Mayol BR, Davis M, Rougraff BT Extraskeletal Ewing’s sarcoma Cancer. 1999;85(3):725. . . ; ( ): –. . [PubMed] [Google Scholar]
- 18.Parkin DM, Stiller CA, Nectoux J International variations in the incidence of childhood bone tumours Int J cancer. 1993;53(3):371. doi: 10.1002/ijc.2910530305. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 19.Geens L, Van Robays J, Geert V, Van Der Speeten K An unusual location of extraosseous ewing’s sarcoma Case Rep Oncol. 2013;6(2):293. doi: 10.1159/000351836. . . ; ( ): –. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanida S, Tanioka F, Inukai M, Yoshioka N, Saida Y, Imai K, et al. Ewing’s sarcoma/peripheral primitive neuroectodermal tumor (pPNET) arising in the omentum as a multilocular cyst with intracystic hemorrhage J Gastroenterol. 2000;35(12):933. doi: 10.1007/s005350070009. . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 21.Maeda M, Tsuda A, Yamanishi S, Uchikoba Y, Fukunaga Y, Okita H, et al. Ewing sarcoma/primitive neuroectodermal tumor of the kidney in a child Pediatr Blood Cancer. 2008;50(1):180. doi: 10.1002/pbc.20831. . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 22.Buffoni I, Nuri H, Pome’ G, Sementa AR, Stagnaro N, Barra S, et al. Pediatric Extraskeletal Ewing Sarcoma Originating in the Heart: A Case Report and Review of the Literature J Pediatr Hematol Oncol. 2021;43(4):147. doi: 10.1097/MPH.0000000000001622. . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 23.Park J-Y, Lee S, Kang HJ, Kim H-S, Park S-Y Primary Ewing’s sarcoma-primitive neuroectodermal tumor of the uterus: a case report and literature review Gynecol Oncol. 2007;106(2):427. doi: 10.1016/j.ygyno.2007.04.036. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 24.Ling X, Tong J, Wang L, Yao C, Chen Z Primary pulmonary Ewing’s sarcoma: rare cause of massive hemothorax in a young girl-case report BMC Pediatr. 2021;21(1):194. doi: 10.1186/s12887-021-02672-6. . . ; ( ): . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehra S, Atwal SS, Garga UC Primary Pulmonary Ewing’s Sarcoma: Rare Cause of Superior Vena Cava Syndrome in Children J Clin Diagn Res. 2014;8(8):RD05-6. doi: 10.7860/JCDR/2014/8681.4713. . . ; ( ): . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathew D, Prince DN, Mahomed N Extra-skeletal Ewing Sarcoma of the chest wall in a child SA J Radiol. 2019;23(1):1733. doi: 10.4102/sajr.v23i1.1733. . . ; ( ): . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma A, Sethi N, Saini S, Pandia K, Jangir R Primary Ewings sarcoma in liver - A rare case report with review of literature Indian J Pathol Microbiol. 2021;64(Supplement):S136. doi: 10.4103/IJPM.IJPM_288_19. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 28.Shadhu K, Ramlagun-Mungur D, Ping X-C Ewing sarcoma of the jejunum: A case report and literature review World J Gastrointest Surg. 2021;13(5):507. doi: 10.4240/wjgs.v13.i5.507. . . ; ( ): - . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shek TW, Chan GC, Khong PL, Chung LP, Cheung AN Ewing sarcoma of the small intestine J Pediatr Hematol Oncol. 2001;23(8):530. doi: 10.1097/00043426-200111000-00013. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 30.Van den Berg H, Heinen RC, van der Pal HJ, Merks JHM Extra-osseous Ewing sarcoma Pediatr Hematol Oncol. 2009;26(4):175. doi: 10.1080/08880010902855581. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 31.Casali PG, Bielack S, Abecassis N, Aro HT, Bauer S, Biagini R, et al. Bone sarcomas: ESMO-Paed-Can-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up Ann Oncol. 2018;29(Suppl 4):iv79. doi: 10.1093/annonc/mdy310. . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 32.Biermann JS Updates in the treatment of bone cancer J Natl Compr Canc Netw. 2013;11(5 Suppl):681. doi: 10.6004/jnccn.2013.0200. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 33.El Weshi A, Allam A, Ajarim D, Al Dayel F, Pant R, Bazarbashi S, et al. Extraskeletal Ewing’s sarcoma family of tumours in adults: analysis of 57 patients from a single institution Clin Oncol. 2010;22(5):374. doi: 10.1016/j.clon.2010.02.010. . ; ( ): –. . [DOI] [PubMed] [Google Scholar]
- 34.Kontny U Regulation of apoptosis and proliferation in Ewing’s sarcoma--opportunities for targeted therapy Hematol Oncol. 2006;24(1):14. doi: 10.1002/hon.766. . . ; ( ): –. . [DOI] [PubMed] [Google Scholar]