

ABSTRACT
Arthritogenic alphaviruses, including chikungunya virus (CHIKV), preferentially target joint tissues and cause chronic rheumatic disease that adversely impacts the quality of life of patients. Viruses enter target cells via interaction with cell surface receptor(s), which determine the viral tissue tropism and pathogenesis. Although MXRA8 is a recently identified receptor for several clinically relevant arthritogenic alphaviruses, its detailed role in the cell entry process has not been fully explored. We found that in addition to its localization on the plasma membrane, MXRA8 is present in acidic organelles, endosomes, and lysosomes. Moreover, MXRA8 is internalized into cells without a requirement for its transmembrane and cytoplasmic domains. Confocal microscopy and live cell imaging revealed that MXRA8 interacts with CHIKV at the cell surface and then enters cells along with CHIKV particles. At the moment of membrane fusion in the endosomes, many viral particles are still colocalized with MXRA8. These findings provide insight as to how MXRA8 functions in alphavirus internalization and suggest possible targets for antiviral development.
IMPORTANCE The globally distributed arthritogenic alphaviruses have infected millions of humans and induce rheumatic disease, such as severe polyarthralgia/polyarthritis, for weeks to years. Alphaviruses infect target cells through receptor(s) followed by clathrin-mediated endocytosis. MXRA8 was recently identified as an entry receptor that shapes the tropism and pathogenesis for multiple arthritogenic alphaviruses, including chikungunya virus (CHIKV). Nonetheless, the exact functions of MXRA8 during the process of viral cell entry remain undetermined. Here, we have provided compelling evidence for MXRA8 as a bona fide entry receptor that mediates the uptake of alphavirus virions. Small molecules that disrupt MXRA8-dependent binding of alphaviruses or internalization steps could serve as a platform for unique classes of antiviral drugs.
KEYWORDS: MXRA8, chikungunya virus, entry, receptor
INTRODUCTION
Arthropod-borne alphaviruses are enveloped, single-stranded, positive-sense RNA viruses that belong to the Togaviridae family. Alphaviruses are comprised of two groups: the “New World” alphaviruses that primarily cause encephalitis and the “Old World” arthritogenic alphaviruses that predominantly are associated with rheumatic disease in humans (1, 2). The arthritogenic alphaviruses include chikungunya virus (CHIKV), Mayaro virus (MAYV), Ross River virus (RRV), and o’nyong-nyong virus (ONNV) and cause infection with symptoms ranging from fever, rash, and myalgia to severe arthralgia and arthritis (3–6). Although infection with arthritogenic alphaviruses generally has a low fatality rate, chronic muscle and joint pain can endure for months to years (7), which has a substantial impact on the quality of life.
After a mosquito bite, CHIKV replicates in the skin and then disseminates to various organs and joints (8, 9). In animal models and humans, viral antigens and RNA are detected in the muscles, bones, articular tissues, and joint space. CHIKV infection of cells is facilitated by the two transmembrane viral glycoproteins (10) E1 and E2, which are arranged as trimeric heterodimers on mature virions. The viral E1 and E2 proteins bind to cellular attachment factors and receptors, after which the particle is internalized via clathrin-mediated endocytosis. Membrane fusion within early endosomes is mediated by the viral E1 protein. While several candidate attachment and entry factors for CHIKV have been reported, including prohibitins (PHB) (11, 12), glycosaminoglycans (GAGs) (13), T cell immunoglobulin and mucin domain-1 (TIM-1) (14), and ATP synthase β-subunit (ATPSβ) (15), their physiological relevance remains uncertain.
Recently, using a CRISPR-Cas9 genome screen, we identified the plasma membrane protein MXRA8 (also known as limitrin, ASP3, or DICAM) as an entry receptor for multiple arthritogenic alphaviruses, including CHIKV (16). The evidence for this included the following: (i) MXRA8 promotes virion binding and internalization, (ii) purified MXRA8 protein and anti-MXRA8 antibodies block alphavirus infection, (iii) ectopic expression of MXRA8 enhances viral infection, (iv) MXRA8 expression promotes infection of arthritogenic alphaviruses but not encephalitic alphaviruses or other unrelated RNA viruses, (v) MXRA8 directly binds the E1/E2 dimer with high-affinity (17, 18) and can engage all heterodimers on the surface of the virion, and (vi) MXRA8 expression is required for viral pathogenesis and infection in multiple animal species (19, 20). Together, these findings support the conclusion that MXRA8 is a key entry receptor that shapes the tropism and pathogenesis of several arthritogenic alphaviruses. Nonetheless, the exact functions of MXRA8 during the process of viral cell entry remain undetermined.
Here, we evaluated in detail the internalization pathway of MXRA8 from the cell surface. We observed colocalization of MXRA8 with CHIKV from virion binding and trafficking to membrane fusion using multiple imaging approaches. CHIKV virions directly interact with MXRA8 at the cell surface and remain colocalized up to the moment of fusion with the endosomal membrane.
RESULTS
MXRA8 localizes at acidic organelles, endosomes, and lysosomes.
To begin to determine the intracellular localization of MXRA8, we first used a Lysotracker dye to test whether it is localized to acidic organelles. Murine embryonic fibroblasts (MEFs) that were gene edited for Mxra8 (ΔMxra8) (16) were complemented with Mxra8-turbo green fluorescent protein (turboGFP), where the cytoplasmic tail of MXRA8 is fused with the fluorescent protein turboGFP to allow visualization in living cells. The complementation of Mxra8-turboGFP in ΔMxra8 cells restored CHIKV infectivity to levels comparable to wild-type (WT) cells (Fig. 1A). Lysotracker Red DND-99 was used to label the acidic compartments in cells. Notably, the GFP+ signal largely colocalized with the Lysotracker-labeled organelles (Fig. 1B and C), suggesting that within cells, complemented MXRA8 is preferentially located in acidic compartments.
FIG 1.

MXRA8 localizes to acidic organelles and endolysosomes. (A) ΔMxra8 MEFs were complemented with C-terminally GFP-tagged Mxra8 (Mxra8-GFP) and infected with CHIKV 181/25 to analyze the expression of intracellular E2 protein; KO, knockout. (B) Cells were stained with Lysotracker Red DND-99 that labels acidic organelles. (C) Spots-based colocalization was analyzed using Imaris software. The percentage of colocalized Lysotracker spots with MXRA8-GFP (left column) of the total number of Lysotracker spots or the percentage of colocalized MXRA8-GFP spots with Lysotracker (right column) of the total number of MXRA8-GFP spots was calculated. (D) Endogenous MXRA8 was upregulated by a CRISPR-based promoter activation strategy in MEFs and was detected by Western blotting. (E and F) Cell surface expression of upregulated MXRA8 as detected by flow cytometry (E) and confocal imaging (F). (G to J) The endogenously upregulated MXRA8 partially colocalizes with the early endosomal marker EEA1 (G and H) or the lysosomal marker LAMP1 (I and J). Cells were fixed, permeabilized for intracellular staining of EEA1 or LAMP1, and stained with fluorophore-labeled secondary antibodies. Orthogonal views and spot-based colocalization were processed using Imaris software. The percentage of colocalized EEA1 (H, left column) with MXRA8 of the total number of EEA1 and the percentage of colocalized MXRA8 (H, right column) with EEA1 of the total number of MXRA8 were calculated. The colocalization of LAMP1 with MXRA8 was calculated similarly (J). (K) Uptake of Alexa Fluor 488-labeled transferrin and FITC-labeled dextran in WT and ΔMxra8 MEFs. The Alexa Fluor 555-labeled wheat germ agglutinin was used to mark the plasma membrane. (L) Flow cytometry analysis of FITC-labeled transferrin or dextran in WT and ΔMxra8 MEFs. The percentage or geometric mean fluorescence intensity (GMFI) of FITC+ cells was measured. Data shown are an average of two independent experiments and are normalized to the control of individual experiments (mean ± SD). Images in B, F, G, I, and K are representative of at least three independent experiments; scale bar, 5 μm.
To further investigate the intracellular localization of MXRA8, we stained cells with antibodies to MXRA8, the endosomal marker EEA1, or the lysosomal marker LAMP1 in WT MEFs and analyzed the cells by using confocal microscopy. However, we were unable to detect clear signal of endogenous MXRA8, likely because of its low expression level. To overcome this limitation, we used CRISPR-based activation of Mxra8 gene expression (21). A single guide RNA (sgRNA) was designed to bind to the promoter sequence of the Mxra8 gene in the presence of deactivated Cas9 (dCas9) to enhance expression of endogenous MXRA8, as confirmed by Western blotting (Fig. 1D), flow cytometry (Fig. 1E), and confocal microscopy (Fig. 1F). We then used these CRISPR-activated cells expressing higher levels of endogenous MXRA8 to characterize its localization with EEA1 and LAMP1, respectively. We observed that within cells, MXRA8 partially colocalizes with EEA1 and LAMP1 (Fig. 1G to J). These results establish that MXRA8 is located in acidic organelles, including endosomes and lysosomes. We also confirmed that loss of MXRA8 expression did not impact normal clathrin-mediated endocytosis and micropinocytosis as assessed by the uptake of transferrin or dextran in ΔMxra8 cells, respectively; the uptake of transferrin or dextran in ΔMxra8 cells was comparable to that observed in WT cells (Fig. 1K and L).
MXRA8 is internalized into endosomes and lysosomes from the cell surface.
To determine whether MXRA8 can be internalized from the plasma membrane into endosomes and lysosomes, the endogenously Mxra8-activated MEFs were incubated with anti-MXRA8 antibody at 37°C to allow receptor-antibody cotrafficking into cells. Any residual anti-MXRA8 antibody on the surface was blocked using an unconjugated secondary antibody. The cells were then processed for intracellular staining of EEA1+ or LAMP1+ organelles. Surface antibody-bound MXRA8 was internalized and partially colocalized with the endosomal marker EEA1 and the lysosomal marker LAMP1 (Fig. 2A to D).
FIG 2.
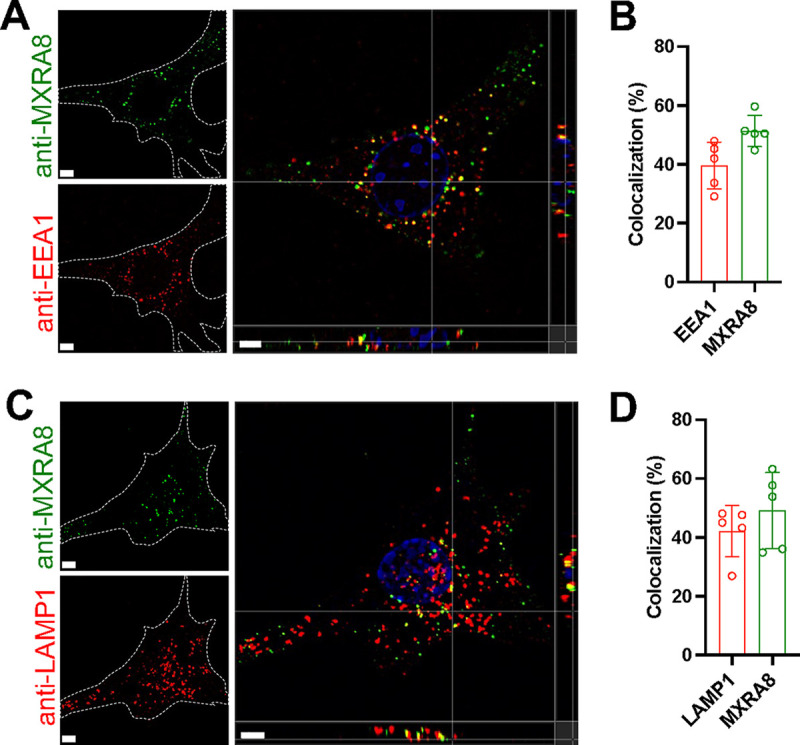
Antibody-labeled MXRA8 is internalized into early endosomes and lysosomes. Mxra8-activated MEFs were incubated with anti-MXRA8 antibody at 37°C for 30 min. Cells were washed and fixed with paraformaldehyde. Uninternalized anti-MXRA8 antibody at the cell surface was blocked with unlabeled goat anti-hamster IgG (H+L). Cells were rinsed, fixed again with paraformaldehyde, permeabilized, and stained for the early endosomal marker EEA1 (A and B) or the lysosomal marker LAMP1 (C and D), followed by staining with fluorophore-labeled secondary antibodies to detect EEA1, LAMP1, and internalized antibody-bound MXRA8. Orthogonal views (A and C) and spot-based colocalization (B and D) were processed using Imaris software. The percentage of colocalized EEA1 (B, left column) with MXRA8 of the total number of EEA1 and the percentage of colocalized MXRA8 (B, right column) with EEA1 of the total number of MXRA8 were calculated. The colocalization of LAMP1 with MXRA8 was calculated similarly (D). Representative images are from at least three independent experiments; scale bar, 5 μm.
To investigate whether the transmembrane domain and/or cytoplasmic domain is required for MXRA8 internalization, we first generated stable cell lines using ΔMxra8 MEFs that were complemented with WT or cytoplasmic domain-deleted (ΔC-tail) forms of MXRA8. Antibody-based visualization of MXRA8 was conducted similar to as described in Fig. 2. Similar to the results from endogenously Mxra8-activated MEFs, in cells complemented with WT or ΔC-tail alleles, MXRA8 was internalized and colocalized with the endosomal marker EEA1 (Fig. 3A and B). Similarly, when both the transmembrane and cytoplasmic domains were deleted and fused with glycosylphosphatidylinositol (GPI) lipid anchors, surface MXRA8 was still internalized and transported to EEA1+ endosomes (Fig. 3A and B). WT and truncated MXRA8 proteins also partially colocalized with LAMP1 (Fig. 3C). We note that all complemented MEFs showed robust surface expression of MXRA8 by flow cytometry (Fig. 3D) and restored CHIKV infection to levels comparable to those observed in WT cells (Fig. 3E).
FIG 3.

MXRA8 internalization occurs independently of its transmembrane and cytoplasmic domains as shown by antibody labeling. (A and B) ΔMxra8 MEFs were complemented with WT MXRA8, MXRA8 lacking its cytoplasmic domain (ΔC-tail), or MXRA8 lacking both transmembrane and cytoplasmic domains but fused with rodent herpesvirus Peru (RHVP)-encoded or placental alkaline phosphatase (PLAP)-encoded GPI anchors. Cells were incubated with anti-MXRA8 antibody at 37°C for 30 min. After blocking of uninternalized anti-MXRA8 on the cell surface, the internalized MXRA8 and early endosome marker EEA1 were stained for confocal imaging. Orthogonal views (A) and spot-based colocalization (B) were processed using Imaris software. The percentage of colocalized EEA1 (B, left column) with MXRA8 of the total number of EEA1 and the percentage of colocalized MXRA8 (B, right column) with EEA1 of the total number of MXRA8 were calculated. One representative image from at least three independent experiments is shown; scale bar, 5 μm. (C) Internalized MXRA8 and the lysosomal marker LAMP1 were stained for confocal imaging. Spot-based colocalization was analyzed using Imaris software. The percentage of colocalized LAMP1 (C, left column) with MXRA8 of the total number of LAMP1 and the percentage of colocalized MXRA8 (C, right column) with LAMP1 of the total number of MXRA8 were calculated. (D) Cell surface expression of MXRA8 in complemented cells that express different forms by flow cytometry. A representative image of at least two independent experiments is shown. (E) CHIKV infection as measured by E2 antigen expression in complemented MEFs by flow cytometry. Data are an average of two independent experiments performed in triplicate (mean ± SD).
Next, we labeled MXRA8 on the cell surface with biotin and tracked the kinetics of internalization and degradation at 37°C. The residual biotin-labeled MXRA8 on the cell surface was stripped with glutathione, followed by immunoprecipitation with streptavidin beads and Western blotting. In ΔMxra8 MEFs complemented with WT Mxra8, biotin-labeled MXRA8 was detected within cells as early as 5 min, and by 10 min, the largest amount of internalized biotin-labeled MXRA8 was observed (Fig. 4A). Similarly, in ΔMxra8 cells complemented with ΔC-tail (Fig. 4B) or glycosylphosphatidylinositol (GPI)-anchored (Fig. 4C) MXRA8, the internalized MXRA8 was detected at 10 min after incubation, albeit at lower levels than WT MXRA8. These tracking data suggest that surface MXRA8 is internalized, and this occurs independent of its transmembrane and cytoplasmic domains. The endocytosed MXRA8 in the lysosome may undergo degradation. To assess the kinetics of MXRA8 degradation after internalization, the lysosomal protease inhibitor leupeptin was added throughout the experiment. Notably, leupeptin partially prevented the degradation of internalized MXRA8 at 5 min of incubation, and this effect was observed at least for 15 min (Fig. 4D). These experiments confirm that biotin-labeled MXRA8 at the cell surface is internalized to the lysosome.
FIG 4.
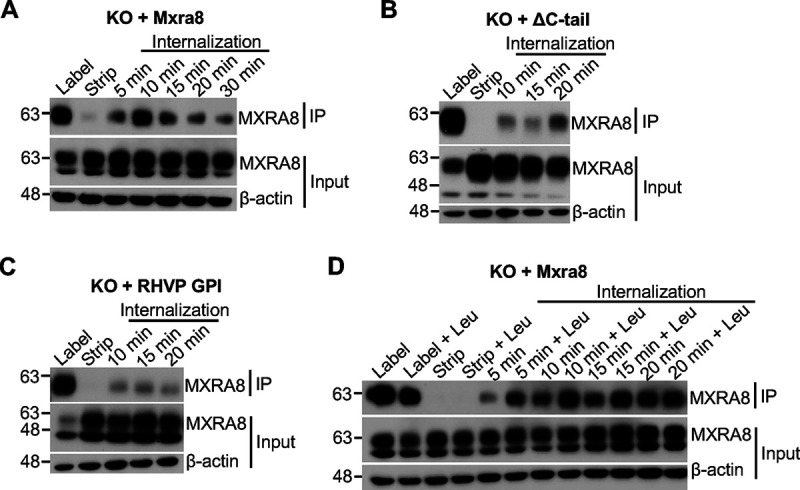
MXRA8 internalization occurs independently of transmembrane and cytoplasmic domains as shown by biotin labeling. (A) Internalization of biotin-labeled surface MXRA8 in ΔMxra8 MEFs complemented with WT MXRA8. After biotin labeling of plasma membrane proteins, cells were incubated at 37°C to allow internalization, followed by stripping of surface biotinylated proteins. The internalized biotin-labeled proteins were immunoprecipitated and Western blotted. (B) Internalization of biotin-labeled surface MXRA8 in ΔMxra8 MEFs complemented with ΔC-tail MXRA8. (C) Internalization of biotin-labeled surface MXRA8 in ΔMxra8 MEFs complemented with GPI-anchored MXRA8. (D) Degradation of internalized MXRA8 could be inhibited by the lysosomal protease inhibitor leupeptin (Leu) in ΔMxra8 MEFs complemented with WT MXRA8. Leupeptin was present throughout the experiment. Representative blots of two or three independent experiments are shown; IP, immunoprecipitation.
Surface MXRA8 and virions are cointernalized into cells.
To investigate the role of MXRA8 during CHIKV virion entry, we analyzed internalization of MXRA8 and virus particles using our antibody-based labeling approach. To avoid possible inhibitory effects on virus binding of the anti-MXRA8 antibody, a Myc tag plus a linker were inserted after the N-terminal signal peptide of MXRA8 and used for complementation of ΔMxra8 MEFs (Fig. 5A). The insertion of the Myc-tag enabled the detection of MXRA8 on the cell surface with an anti-Myc antibody and had only subtle effects on CHIKV infection (Fig. 5B and C). After virion binding at 4°C, intact cells were incubated with anti-Myc and anti-E2 antibodies to label MXRA8 and virions on the cell surface, respectively. Using confocal microscopy, we detected colocalization of MXRA8 and virions on the plasma membrane from orthogonal and three-dimensional (3D) views (Fig. 5D). After internalization at 37°C and stripping of the cell surface-bound virions using proteinase K, cells were fixed and permeabilized for intracellular staining of Myc-tagged MXRA8 and virions. Colocalization of MXRA8 and CHIKV E2 protein was observed (Fig. 5E), and this was intracellular, as stripping of surface virions and tagged MXRA8 protein with proteinase K was highly efficient (Fig. 5F). Thus, CHIKV particles bind to MXRA8 on the cell surface, internalize, and then colocalize within cells.
FIG 5.

Myc-tagged MXRA8 colocalizes with CHIKV virions. ΔMxra8 MEFs were complemented with Myc-tagged MXRA8. (A) Insertion of a Myc tag plus a linker in MXRA8. The amino acid sequence is indicated. (B) The expression of Myc-tagged MXRA8 in ΔMxra8 MEFs. The complemented cells were stained with anti-Myc tag or isotype control antibody and were subjected to flow cytometry analysis. (C) Functional assessment of Myc-tagged MXRA8 in ΔMxra8 MEFs. The complemented cells were infected with CHIKV for 8 h, followed by E2 antigen detection using flow cytometry. Data shown are an average of two independent experiments performed in triplicate (mean ± SD). (D) Myc-MXRA8 colocalizes with CHIKV virions at the cell surface. Cells were incubated with virus particles at 4°C for 30 min and incubated with anti-E2 and anti-Myc primary antibodies at 4°C for 30 min. After washing, cells were fixed and stained with fluorophore-labeled secondary antibodies. Orthogonal and 3D views of colocalization are presented. (E) Myc-MXRA8 colocalizes with CHIKV particles in the cytoplasm. Cells were incubated with viral particles and antibodies at 37°C for 15 min and subsequently placed on ice. Surface virions and antibodies were removed by treatment with protease K. Cells were fixed, permeabilized, and stained with fluorophore-labeled secondary antibodies. Orthogonal and 3D views of colocalization are presented. (F) As a control, cells before fixation were treated with proteinase K to remove bound virus particles and antibodies, followed by fixation and surface staining with secondary antibodies to monitor stripping efficiency. In D to F, one representative image from at least three independent experiments is shown; scale bar, 5 μm. (G) Myc-tagged MXRA8 proteins were labeled with anti-Myc antibody to assess the internalization efficiency in the presence or absence of virions. Cells in suspension were processed similar to as shown in E and analyzed by flow cytometry. The geometric mean fluorescence intensity (GMFI) of anti-Myc antibody-positive cells was measured. Data shown are an average of two independent experiments performed in triplicate. Data were analyzed by unpaired t test and are presented as mean ± SD; *, P < 0.05.
To investigate whether the binding of CHIKV virions facilitates internalization of MXRA8, we tracked the fate of surface MXRA8 with or without virions by using flow cytometry. These experiments showed that the addition of virions slightly enhanced the internalization of Myc-tagged WT MXRA8 beyond that seen in the presence of anti-Myc antibody alone (Fig. 5G). These data suggest that binding of virions to MXRA8 can promote receptor endocytosis.
Surface MXRA8 colocalizes with CHIKV during cell entry and through membrane fusion.
To validate the entry receptor role of MXRA8 during cell entry of CHIKV, we applied a microscopy assay that can measure the extent of viral membrane fusion in cells (22, 23). The virus membrane is labeled with the fluorescent probe 1,10-dioctadecyl-3,3,30,30-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD) under self-quenching conditions that still allow the detection of single virus particles. Membrane (hemi)fusion of CHIKV is measured based on DiD dequenching caused by dilution of the probe into the endosomal membrane (22). The DiD-labeled CHIKV particles were incubated with ΔMxra8 cells or Mxra8-GFP-complemented cells at 37°C for 20 min to allow viral entry and membrane (hemi)fusion. The total area of fluorescent spots was quantified. Notably, membrane (hemi)fusion of DiD-labeled CHIKV is enhanced in cells that express MXRA8 (Fig. 6A).
FIG 6.

MXRA8 colocalizes with DiD-labeled CHIKV virions during cell entry and through membrane fusion. (A) Microscopic cell entry assay in ΔMxra8 MEFs and cells complemented with MXRA8-GFP. Cells were incubated with DiD-labeled CHIKV particles for 20 min and subsequently washed to remove unbound particles. The extent of membrane hemifusion was measured in 15 randomly acquired microscopic snapshots per experiment. The total extent of membrane hemifusion in ΔMxra8 MEFs was normalized to that in Mxra8-GFP cells. Bars represent mean percentage of membrane fusion activity, and error bars represent standard deviations (SD) from three independent experiments, each performed in duplicate. (B) Single-particle tracking of DiD-labeled CHIKV in MXRA8-GFP cells. In total, 43 trajectories were analyzed. Only particles with a fluorescence intensity lower than 40 arbitrary units (a.u.) were selected. Time of membrane hemifusion is defined as the moment when the fluorescence intensity is increased greater than 2-fold within 1 to 2 s. (C) Total number of CHIKV virions that colocalize with MXRA8-GFP at the moment of membrane fusion. In total, 39 trajectories were analyzed. Nineteen CHIKV particles colocalized with MXRA8-GFP at the moment of membrane (hemi)fusion, of which, 10 that colocalized with MXRA8-GFP from the start of the trajectory are depicted (red). Twenty fusion-competent particles did not colocalize with MXRA8-GFP at the moment of membrane (hemi)fusion (black bar). (D) Time series of DiD-labeled CHIKV in MXRA8-GFP cells. A DiD-labeled CHIKV particle interacts with MXRA8-GFP from the start of trajectory. (E) Time series of DiD-labeled CHIKV that interacts with MXRA8-GFP after fast-directed movement. (F) Velocity and fluorescence intensity of a single virion over time. A sudden increase in fluorescence intensity indicates the moment of fusion. The arrow depicts the start of colocalization. The data are from the same particle as depicted in D. (G) Velocity and fluorescence intensity of DiD-labeled CHIKV over time. The data are from the same particle as depicted in E.
Next, we performed single virus tracking of DiD-labeled CHIKV particles in Mxra8-GFP cells to establish whether MXRA8 and virus particles cointernalize in cells. The trafficking behavior of 42 fusion-competent CHIKV particles was analyzed. The time to virus (hemi)fusion in Mxra8-GFP cells was comparable to that observed previously for CHIKV in kidney epithelial BS-C-1 cells (22); half of the virions fused within 6 min of infection, and 95% fused within 15 min of infection (Fig. 6B). From this analysis, 39 particles were used to study the colocalization with MXRA8-GFP. Three particles were excluded because they fused too close to the nucleus, an area of the cell that is too thick for imaging in one focal plane as applied in the setup here. Approximately half of the virions (N = 19, 49%) colocalized with MXRA8-GFP at the moment of membrane (hemi)fusion (Fig. 6C), which is defined as when the fluorescence intensity suddenly increases (>2-fold signal within 1 to 2 s). Of these, 10 particles (53%) colocalized with MXRA8-GFP from the start of the trajectory (Fig. 6C, red; an example is given in Fig. 6D and F), indicating cotrafficking of CHIKV virions with MXRA8. To determine whether the remaining particles (N = 9) colocalized with MXRA8-GFP before or after internalization, we investigated their trafficking behavior in greater detail. Previously, we discovered that CHIKV virions exhibit two different trafficking behaviors before membrane fusion (24), with roughly half showing fast-directed movement (FDM) along microtubules toward the perinuclear region. FDM occurs after colocalization with clathrin-coated vesicles but before colocalization with Rab5+ early endosomes. Three of 9 particles colocalized with MXRA8-GFP before FDM, suggesting that these particles are cointernalized with MXRA8 into the host cell. Of note, 3 particles colocalized with MXRA8-GFP after FDM, which suggests that these particles enter cells through an MXRA8-independent pathway and are transported toward an MXRA8-GFP+ vesicle after endocytosis (example given in Fig. 6E and G); indeed, the CHIKV-LR 2006 strain tested can infect cells through both MXRA8-dependent and MXRA8-independent pathways (16). The remaining 3 particles did not exhibit FDM, and MXRA8-GFP gradually appeared at the same location as the virion and remained present until membrane (hemi)fusion occurred; we cannot distinguish whether these particles enter cells via MXRA8-dependent or MXRA8-independent pathways, as we do not know the time point that these particles are internalized by cells in the current experimental setup.
DISCUSSION
MXRA8 functions as a specific entry receptor for arthritogenic alphaviruses, as reflected by its importance in virion binding and internalization (16). In the present study, we showed that MXRA8 is located at both the plasma membrane and in acidic organelles, including endolysosomes. Moreover, we found that some CHIKV particles cointernalize with MXRA8 at the plasma membrane, cotraffic toward endosomal organelles, and remain colocalized until acid-catalyzed membrane (hemi)fusion.
MXRA8 transits to EEA1+ endosomes independently of its transmembrane and cytoplasmic domains. Although further study is warranted, this suggests that interacting partners may facilitate MXRA8 internalization, as receptor internalization often is regulated via covalent modifications in the cytoplasmic domain of cell surface receptors (25, 26). We observed cotrafficking of CHIKV particles with MXRA8 throughout the cell entry process and in a manner that is consistent with previous studies, showing clathrin-mediated endocytosis as the principal entry route for CHIKV and other alphaviruses (22, 27). Future studies are needed to determine whether MXRA8 or a partner protein interacts with clathrin-coated vesicle formation-related proteins (e.g., adaptor-2, dynamin, clathrin, epsin, or Eps15) to facilitate viral entry. Importantly, the processes of clathrin-mediated endocytosis and micropinocytosis were not affected by MXRA8 expression.
Cellular receptors located in endosomal compartments can be sorted to the trans-Golgi network or the endolysosomal degradation pathway or can be recycled back to the plasma membrane (25, 28, 29). We found that MXRA8 colocalizes with the endosomal marker EEA1 and the lysosome marker LAMP1. This colocalization could suggest that MXRA8 is targeted toward the endolysosomal degradation pathway. Moreover, we show that internalized MXRA8 degradation can be partially prevented using a lysosomal protease inhibitor leupeptin. Further investigation is required to understand how endosomal retrieval complexes orchestrate the sorting of internalized MXRA8 receptors.
Levels of endogenous MXRA8 expression are low, resulting in failure to clearly visualize the protein by confocal microscopy. To overcome this limitation, we used CRISPR-dCas9 and a specific sgRNA to recruit transcription activators and induce the expression of endogenous Mxra8. In parallel, we used orthogonal approaches for upregulating or ectopically expressing MXRA8 within cells. For example, to allow live cell visualization, MXRA8 was fused with the fluorescent protein GFP (MXRA8-GFP) at the C terminus or a Myc tag was inserted after the N-terminal signal peptide. The usage of a diverse set of MXRA8 expression techniques strengthens our observation of MXRA8 localization in endolysosomes and colocalization with CHIKV virions.
The use of MXRA8-GFP-expressing cells enabled us to visualize the dynamic interaction of MXRA8 with CHIKV particles during cell entry. DiD-labeled CHIKV virions were used to visualize single virus particles throughout cell entry. Our data demonstrate that MXRA8 colocalizes with virions during cell entry up to the moment of membrane fusion. This result suggests that MXRA8 functions as a cell entry receptor that promotes CHIKV binding and subsequent internalization and membrane fusion. CHIKV membrane fusion is not strictly dependent on receptor interactions, as low-pH-dependent membrane fusion can be initiated with receptor-free liposomes (22, 30, 31). Nonetheless, membrane proteins may aid in cellular infection (32, 33). High-resolution cryo-electron microscopic reconstructions of CHIKV virions bound to MXRA8 suggested that E2-E1 binding interactions might be sensitive to low pH (17). Based on our imaging data, it is tempting to speculate that MXRA8 receptor interactions might contribute to the reorganization of CHIKV envelope proteins that is required for membrane fusion in acidified endosomes. Future studies are needed to address whether MXRA8 interactions alter the pH threshold of viral fusion and the organelle/cellular location in which membrane fusion occurs.
Not all particles colocalized with MXRA8 at the moment of membrane fusion. We might underestimate the actual number of particles that colocalize with MXRA8 at the moment of membrane fusion if only a few MXRA8 molecules are present and below our imaging threshold. Alternatively, other as of yet identified receptors could contribute to virion uptake. Indeed, differences in MXRA8 dependency exist between CHIKV strains, with those in the Asian (AF15561 strain) and African (37997 strain) lineage showing more complete dependence on MXRA8 than those in the East-Central South African sublineage (16). Consistent with this observation, a genetic knockout of Mxra8 does not fully abrogate infection of CHIKV-LR 2006 in vitro or in vivo (16).
In summary, we have demonstrated that MXRA8 interacts with CHIKV virions on the plasma membrane, cointernalizes into the target cell, and, in many cases, cotraffics until fusion occurs in the late endosome. These findings provide compelling evidence for MXRA8 function as a bona fide entry receptor mediating the uptake of alphavirus virions. Small molecules that disrupt MXRA8-dependent binding of alphaviruses or internalization steps could serve as a platform for unique classes of antiviral drugs.
MATERIALS AND METHODS
Cells and viruses.
Vero cells and MEFs were cultured at 37°C in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), nonessential amino acids, and penicillin-streptomycin. BHK-21 cells (ATCC, CCL-10) were cultured at 37°C in RPMI medium supplemented with 10% FBS and penicillin-streptomycin. CHIKV 181/25 was propagated in Vero cells and titrated by focus-forming assays, as described previously (16). CHIKV LR-2006 was propagated in BHK-21 cells, purified by sucrose gradient ultracentrifugation, and titrated by viral plaque assay, as described previously (22).
Plasmid constructions.
Murine Mxra8 was amplified by PCR from the vector pCSII-EF1-IRES-Venus (16) and C-terminally fused with the fluorescent protein gene turboGFP. The fused gene was cloned into vector pLV-EF1-IRES-BLAST (Addgene, 85133) with restriction sites BamHI/EcoRI. Similarly, the ΔC-tail-truncated, placental alkaline phosphatase (PLAP) or rodent herpesvirus peru (RHVP) GPI-anchored Mxra8 was amplified by PCR from the corresponding pCSII-EF1-IRES-Venus vectors (16) and cloned into pLV-EF1-IRES-BLAST. To generate the N-terminally Myc-tagged Mxra8, Myc tag and linker sequences (gaacaaaaactcatctcagaagaggatctcggtggcggaggttct; the Myc tag is underlined) were inserted into Mxra8 after the N-terminal signal peptide (between amino acids 22 and 23) and cloned into pLV-EF1-IRES-BLAST, as described above. To induce the expression of endogenous Mxra8, sgRNA sequences (5′-ccaccaactgggcccggaca-3′) were cloned into the plasmid pXPR_502 (Addgene, 96923).
Complementation and gene activation.
The plasmids constructed above were packaged into lentivirus by cotransfection with psPAX2 (Addgene, 12260) and pMD2.G (Addgene, 12259). ΔMxra8 MEFs (16) were transduced with the lentiviruses constructed above and selected with blasticidin. To induce the expression of endogenous Mxra8, WT MEFs were transduced with lentivirus dCAS-VP64_BLAST (Addgene, 61425) and selected with blasticidin. The resistant cells were then transduced with lentivirus pXPR_502 constructed above and selected with puromycin.
Virus infection.
To analyze viral antigen expression in complemented cells, cells were infected with CHIKV 181/25 (multiplicity of infection [MOI] of 3) for 8 h, fixed with 2% paraformaldehyde, permeabilized, and processed for CHIKV E2 expression using 1 μg/mL mouse CHK-11 monoclonal antibody (34). After washing, cells were stained with goat anti-mouse IgG (H+L) conjugated to Alexa Fluor 647 (Thermo, A21235; 2 μg/mL). Cells were subjected to flow cytometry analysis and processed using FlowJo software (Tree Star).
Biotin labeling and tracking of cell surface MXRA8.
Cells seeded in 6-well plates 24 h before the experiment were chilled on ice for 10 min and labeled with 2.5 mg/mL biotin (Thermo, 21331) in phosphate-buffered saline (PBS) for 30 min on ice. Cells were quenched with 100 mM glycine in PBS for 10 min, and this was repeated three times. After washing with PBS, cells were switched to 37°C for 5 to 30 min to allow for protein internalization. Cells were placed back on ice, and the surface-bound residual biotin was stripped with 50 mM glutathione for 15 min (repeated three times), followed by treatment with 25 mM iodoacetamide for 5 min (repeated two times). To assess protein degradation, 100 μg/mL of the lysosomal protease inhibitor leupeptin was present throughout the quenching, internalization, and stripping steps. To monitor the stripping efficiency of cell surface-bound biotin, biotin-labeled cells were kept on ice and used as a stripping control. Cells were then lysed in RIPA buffer (Cell Signaling, 9806S) with a cocktail of protease inhibitors (Sigma-Aldrich, S8830) and immunoprecipitated with streptavidin agarose beads overnight at 4°C. Beads were washed three times with RIPA buffer, eluted into 5× loading buffer (Beyotime, P0015L), and incubated at 95°C for 10 min. After centrifuging at maximum speed (14,000 rpm) for 10 min, supernatants were harvested for Western blotting using hamster anti-MXRA8 monoclonal antibody (3G2.F5) (16) or rabbit anti-β-actin (Proteintech, 20536-1-AP), as described below. Nonimmunoprecipitated lysates were used as input control.
Antibody labeling and tracking of cell surface MXRA8 and virions.
Cells grown on coverslips in 24-well plates were incubated with 100 μg/mL hamster anti-mouse MXRA8 antibody (4E7.D10) (16) for 30 min at 37°C. Cells were washed four times with PBS and fixed with 4% paraformaldehyde in PBS at room temperature for 8 min. After washing, cells were blocked with 200 μg/mL unlabeled goat anti-hamster IgG (H+L; Abcam, ab5738) overnight at room temperature. Cells were rinsed three times and fixed again with 4% paraformaldehyde in PBS at room temperature for 8 min. After rinsing, cells were permeabilized with 0.05% saponin in PBS for 30 min, followed by blocking with 5% bovine serum albumin (BSA) for 1 h at room temperature. Next, cells were stained with rabbit anti-EEA1 (Cell Signaling, 3288; 1:200) or rabbit anti-LAMP1 (Abcam, ab24170; 2 μg/mL) for 1 h, followed by staining with goat anti-rabbit IgG (H+L) conjugated with Alexa Fluor 555 (Thermo, A32732; 2 μg/mL) or goat anti-hamster IgG (H+L) conjugated with Alexa Fluor 488 (Abcam, ab173003; 2 μg/mL) for 1 h. After cells were stained with 4′,6-diamidino-2-phenylindole (DAPI), z-stack images were collected using a Zeiss LSM880 confocal microscope and processed with Imaris software 9.0.1.
To determine the colocalization of virion and MXRA8 at the cell surface, ΔMxra8 MEFs complemented with Myc-tagged MXRA8 proteins were incubated with CHIKV 181/25 virions (MOI of 150) at 4°C for 30 min, followed by incubation with mouse anti-Myc antibody (Cell Signaling, 2276S; 1 μg/mL) and anti-CHIKV E2 antibody (humanized CHK-166 [34]; 1 μg/mL) at 4°C for 30 min. After washing, cells were fixed with 4% paraformaldehyde in PBS at room temperature for 10 min. Cells were then stained with goat anti-mouse IgG (H+L) conjugated with Alexa Fluor 555 (Thermo, A21424; 2 μg/mL) and goat anti-human IgG (H+L) conjugated with Alexa Fluor 488 (Thermo, A11013; 2 μg/mL). For the intracellular staining of virions and MXRA8, complemented cells were incubated with virion and anti-Myc antibody at 4°C for 30 min, followed by incubating at 37°C for 15 min. After washing, cells were treated with 400 μg/mL proteinase K at 4°C for 45 min to remove the surface virions and antibody. Cells were rinsed, fixed, and permeabilized for intracellular staining with anti-E2 antibody, followed by staining with fluorophore-conjugated secondary antibodies. After cells were stained with DAPI, the z-stack images were collected using a Zeiss LSM880 confocal microscope and processed with Imaris software 9.0.1. To examine the internalization of Myc-tagged MXRA8 proteins in the presence or absence of virions by flow cytometry, cells in suspension were processed similar to as described above, analyzed with an Attune NxT cytometer (Thermo), and processed using FlowJo software.
DiD labeling and microscopic fusion assay.
Purified CHIKV LR-2006 was labeled with the lipophilic fluorescent probe 1,10-dioctadecyl-3,3,30,30-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD; Invitrogen, D7757) under experimental conditions that do not interfere with virus infectivity, as described previously (22). DiD-labeled virus was stored at 4°C in the dark and used within 2 days. MEFs were grown on Nunc 8-well Lab-Tek II chambered coverglass slides (Thermo, 155409) and washed three times with serum-free and phenol red-free MEM before phenol red-free MEM containing 1% glucose was added. Next, cells were incubated with DiD-labeled CHIKV for 20 min at 37°C. After cells were rinsed three times to remove unbound virus with serum-free, phenol red-free MEM, fresh phenol red-free MEM containing 1% glucose was added. Fifteen snapshots of randomly selected fields were collected using a Leica Biosystems 6000B instrument. Images were analyzed using the “particle analyzer” plugin of ImageJ. We only used those particles that had a fluorescence intensity lower than 40 arbitrary units (a.u.). The total area of fluorescent spots was quantified in a.u. for each snapshot and averaged per experiment. The average fluorescence intensity of MXRA8-GFP was set to 100%. No distinction was made between fused and nonfused particles.
Single-particle tracking of DiD-labeled CHIKV.
MEFs were grown in Nunc 8-well Lab-Tek II chambered coverglass slides to obtain a confluence of ~50 to 70% on the day of tracking. Cells were rinsed three times with phenol red-free MEM, and phenol red-free MEM supplemented with 1% glucose was added to the cells. Glucose oxidase was added just before image acquisition to prevent phototoxicity (23). Cells were placed on a Leica Biosystems 6000B microscope and kept at 37°C. Next, DiD-labeled CHIKV was added in situ, and image series were recorded at 1 frame per s for 25 to 30 min. Differential inference contrast (DIC) snapshots were taken before and after imaging. Image processing and analysis were performed in ImageJ and Imaris x64, release 7.6.1. Trajectories were generated using the “particle tracking” function of Imaris by pairing peaks per frame to previously established trajectories according to proximity and similarity of intensity. For the generated trajectories, the “particle tracking” function of Imaris calculated the particle velocity. The fluorescence intensity during a trajectory was quantified using an in-house macro based on the “particle analyzer” plugin of ImageJ. A sudden (within 1 to 2 s) greater than 2-fold increase in fluorescence intensity was defined as the moment of membrane fusion. Colocalization with MXRA8-GFP was assessed by eye.
Surface staining of MXRA8.
To detect the surface expression of MXRA8, complemented cells were collected with TrypLE (Thermo, 12605010) and washed two times with ice-cold PBS. Live cells were incubated with hamster anti-mouse MXRA8 (4E7.D10; 1 μg/mL), followed by staining with 2 μg/mL goat anti-hamster IgG (H+L) conjugated with Alexa Fluor 647 (Abcam, ab173004). Cells were subjected to flow cytometry analysis and were processed using FlowJo software. For confocal microscopy imaging, cells were seeded on a coverslip in 24-well plates 24 h before the experiment and fixed with 2% paraformaldehyde in PBS for 10 min. Cells were then incubated with hamster anti-mouse MXRA8 (4E7.D10; 1 μg/mL) at 4°C overnight. After three washes, cells were incubated with goat anti-hamster IgG (H+L) conjugated with Alexa Fluor 568 (Abcam, ab175716; 2 μg/mL) for 2 h at room temperature, followed by image collection using a Zeiss LSM880 confocal microscope and processing with Imaris software 9.0.1.
Acidic organelle labeling.
To identify the acidic organelles in living cells, ΔMxra8 MEFs were complemented with C-terminally turboGFP-tagged MXRA8. Cells were seeded on Lab-Tek chambered coverglass (Thermo, 155411) and cultured in phenol red-free medium. Cells were treated with LysoTracker Red DND-99 (Thermo, L7528; 50 nM) for 30 min and washed with fresh medium, followed by image collection using a Zeiss LSM880 confocal microscope and processing with Imaris software 9.0.1.
Uptake of transferrin or dextran.
WT or ΔMxra8 cells were starved with serum-free culture medium for 1 h and chilled on ice for 10 min. Cells were incubated with 50 μg/mL Alexa Fluor 488-labeled transferrin (Thermo, T13342) or 500 μg/mL fluorescein isothiocyanate (FITC)-labeled dextran (Sigma-Aldrich, 46945) for 30 min at 4°C. Cells were shifted to 37°C for 30 min, followed by stripping with acid buffer (0.1 M glycine and 150 mM NaCl, pH 3.0). For confocal microscopy imaging, cells were fixed with 2% paraformaldehyde, and images were collected using a Zeiss LSM880 confocal microscope and processed with ImageJ2 software. Alexa Fluor 555-labeled wheat germ agglutinin (Thermo, W32464) was used to mark the plasma membrane. For flow cytometry analysis, cells were harvested with trypsin, washed once with PBS, subjected to analysis with an Attune NxT cytometer (Thermo), and processed using FlowJo software.
Western blotting.
MEFs in 6-well plates were rinsed once with ice-cold PBS and lysed with RIPA buffer containing protease inhibitors. Samples were prepared in reducing buffer (50 mM Tris [pH 6.8], 10% glycerol, 2% SDS, 0.02% [wt/vol] bromophenol blue, and 100 mM dithiothreitol [DTT]). After heating (95°C for 10 min), samples were electrophoresed in 10% SDS polyacrylamide gels, and proteins were transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 5% nonfat dry powdered milk in 100 mM NaCl, 10 mM Tris (pH 7.6), and 0.1% Tween 20 (TBST) for 1 h at room temperature and probed with hamster anti-MXRA8 monoclonal antibodies (3G2.F5; 0.5 μg/mL) or rabbit anti-β-actin (Proteintech, 20536-1-AP; 1:2,000) at 4°C overnight. After washing with TBST, blots were incubated with peroxidase AffiniPure goat anti-hamster IgG (H+L; Jackson ImmunoResearch, 127-035-160) or horseradish peroxidase-conjugated goat anti-rabbit (Thermo, 31460; 1:5,000) for 1 h at room temperature, washed again with TBST, and developed using SuperSignal West Pico (Thermo, 34077) or Femto (Thermo, 34095) chemiluminescent substrate according to the manufacturer’s instructions.
Immunofluorescence microscopy.
Cells grown on coverslips in 24-well plates were washed twice with PBS, fixed with 2% paraformaldehyde in PBS for 10 min, and permeabilized with 0.1% saponin for 30 min. Next, cells were incubated with primary antibodies at 4°C overnight. After three washes, cells were incubated with the secondary antibodies for 2 h at room temperature, followed by staining with DAPI. Images were collected using a Zeiss LSM880 confocal microscope and processed with ImageJ or Imaris software 9.0.1. The primary antibodies used are as follows: rabbit anti-EEA1 (Cell Signaling, 3288S; 1:200), rabbit anti-LAMP1 (Abcam, ab24170; 2 μg/mL), hamster anti-mouse MXRA8 (4E7.D10; 1 μg/mL), mouse anti-Myc tag (Cell Signaling, 2276S; 1 μg/mL), and anti-CHIKV E2 antibody (humanized CHK-166 [34]; 1 μg/mL). The secondary antibodies include the following: goat anti-rabbit IgG (H+L) conjugated with Alexa Fluor 555 (Thermo, A32732; 2 μg/mL), goat anti-mouse IgG (H+L) conjugated with Alexa Fluor 555 (Thermo, A21424; 2 μg/mL), goat anti-hamster IgG (H+L) conjugated with Alexa Fluor 488 (Abcam, ab173003; 2 μg/mL), and goat anti-human IgG (H+L) conjugated with Alexa Fluor 488 (Thermo, A11013; 2 μg/mL).
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81974305), Shanghai Municipal Science and Technology Major Project (ZD2021CY001), Program of Shanghai Academic/Technology Research Leader (22XD1420600), and grants from the National Institutes of Health (R01-AI143673). E.M.B. was supported by the Graduate School of Medical Sciences of the University of Groningen.
R.Z., F.F., E.M.B., G.H., Y.Z., and Y.Y. performed the experiments. R.Z., F.F., E.M.B., J.M.S., and M.S.D. designed the experiments. R.Z., F.F., and E.M.B. performed data analysis. R.Z. and E.M.B. wrote the initial draft of the manuscript, with the other authors contributing to editing into the final form.
M.S.D. is a consultant for Inbios, Vir Biotechnology, Senda Biosciences, Moderna, Ocugen, and Immunome. The Diamond laboratory has received unrelated funding support in sponsored research agreements from Moderna, Vir Biotechnology, and Emergent BioSolutions.
Contributor Information
Jolanda M. Smit, Email: jolanda.smit@umcg.nl.
Michael S. Diamond, Email: diamond@borcim.wustl.edu.
Rong Zhang, Email: rong_zhang@fudan.edu.cn.
Mark T. Heise, University of North Carolina at Chapel Hill
REFERENCES
- 1.Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Powers AM, Brault AC, Shirako Y, Strauss EG, Kang W, Strauss JH, Weaver SC. 2001. Evolutionary relationships and systematics of the alphaviruses. J Virol 75:10118–10131. doi: 10.1128/JVI.75.21.10118-10131.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paquet C, Quatresous I, Solet J-L, Sissoko D, Renault P, Pierre V, Cordel H, Lassalle C, Thiria J, Zeller H, Schuffnecker I. 2006. Chikungunya outbreak in Reunion: epidemiology and surveillance, 2005 to early January 2006. Euro Surveill 11:E060202.3. doi: 10.2807/esw.11.05.02891-en. [DOI] [PubMed] [Google Scholar]
- 4.Muñoz M, Navarro JC. 2012. Mayaro: a re-emerging arbovirus in Venezuela and Latin America. Biomedica 32:286–302. doi: 10.7705/biomedica.v32i2.647. [DOI] [PubMed] [Google Scholar]
- 5.Kiwanuka N, Sanders EJ, Rwaguma EB, Kawamata J, Ssengooba FP, Najjemba R, Were WA, Lamunu M, Bagambisa G, Burkot TR, Dunster L, Lutwama JJ, Martin DA, Cropp CB, Karabatsos N, Lanciotti RS, Tsai TF, Campbell GL. 1999. O’nyong-nyong fever in south-central Uganda, 1996–1997: clinical features and validation of a clinical case definition for surveillance purposes. Clin Infect Dis 29:1243–1250. doi: 10.1086/313462. [DOI] [PubMed] [Google Scholar]
- 6.Rulli NE, Melton J, Wilmes A, Ewart G, Mahalingam S. 2007. The molecular and cellular aspects of arthritis due to alphavirus infections: lesson learned from Ross River virus. Ann N Y Acad Sci 1102:96–108. doi: 10.1196/annals.1408.007. [DOI] [PubMed] [Google Scholar]
- 7.Suhrbier A, Jaffar-Bandjee M-C, Gasque P. 2012. Arthritogenic alphaviruses—an overview. Nat Rev Rheumatol 8:420–429. doi: 10.1038/nrrheum.2012.64. [DOI] [PubMed] [Google Scholar]
- 8.Talarmin F, Staïkowsky F, Schoenlaub P, Risbourg A, Nicolas X, Zagnoli A, Boyer P. 2007. Skin and mucosal manifestations of chikungunya virus infection in adults in Reunion Island. Med Trop (Mars) 67:167–173. [PubMed] [Google Scholar]
- 9.Couderc T, Chrétien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, Touret Y, Barau G, Cayet N, Schuffenecker I, Desprès P, Arenzana-Seisdedos F, Michault A, Albert ML, Lecuit M. 2008. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog 4:e29. doi: 10.1371/journal.ppat.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jose J, Snyder JE, Kuhn R. 2009. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol 4:837–856. doi: 10.2217/fmb.09.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wintachai P, Wikan N, Kuadkitkan A, Jaimipuk T, Ubol S, Pulmanausahakul R, Auewarakul P, Kasinrerk W, Weng W-Y, Panyasrivanit M, Paemanee A, Kittisenachai S, Roytrakul S, Smith DR. 2012. Identification of prohibitin as a Chikungunya virus receptor protein. J Med Virol 84:1757–1770. doi: 10.1002/jmv.23403. [DOI] [PubMed] [Google Scholar]
- 12.Wintachai P, Thuaud F, Basmadjian C, Roytrakul S, Ubol S, Désaubry L, Smith DR. 2015. Assessment of flavaglines as potential chikungunya virus entry inhibitors. Microbiol Immunol 59:129–141. doi: 10.1111/1348-0421.12230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva LA, Khomandiak S, Ashbrook AW, Weller R, Heise MT, Morrison TE, Dermody TS. 2014. A single-amino-acid polymorphism in chikungunya virus E2 glycoprotein influences glycosaminoglycan utilization. J Virol 88:2385–2397. doi: 10.1128/JVI.03116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moller-Tank S, Kondratowicz AS, Davey RA, Rennert PD, Maury W. 2013. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J Virol 87:8327–8341. doi: 10.1128/JVI.01025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fongsaran C, Jirakanwisal K, Kuadkitkan A, Wikan N, Wintachai P, Thepparit C, Ubol S, Phaonakrop N, Roytrakul S, Smith DR. 2014. Involvement of ATP synthase β subunit in chikungunya virus entry into insect cells. Arch Virol 159:3353–3364. doi: 10.1007/s00705-014-2210-4. [DOI] [PubMed] [Google Scholar]
- 16.Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, Poddar S, Crowe JE, Doranz BJ, Fremont DH, Diamond MS. 2018. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557:570–574. doi: 10.1038/s41586-018-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basore K, Kim AS, Nelson CA, Zhang R, Smith BK, Uranga C, Vang L, Cheng M, Gross ML, Smith J, Diamond MS, Fremont DH. 2019. Cryo-EM structure of chikungunya virus in complex with the Mxra8 receptor. Cell 177:1725–1737. doi: 10.1016/j.cell.2019.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song H, Zhao Z, Chai Y, Jin X, Li C, Yuan F, Liu S, Gao Z, Wang H, Song J, Vazquez L, Zhang Y, Tan S, Morel CM, Yan J, Shi Y, Qi J, Gao F, Gao GF. 2019. Molecular basis of arthritogenic alphavirus receptor MXRA8 binding to chikungunya virus envelope protein. Cell 177:1714–1724. doi: 10.1016/j.cell.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Zhang R, Earnest JT, Kim AS, Winkler ES, Desai P, Adams LJ, Hu G, Bullock C, Gold B, Cherry S, Diamond MS. 2019. Expression of the Mxra8 receptor promotes alphavirus infection and pathogenesis in mice and Drosophila. Cell Rep 28:2647–2658. doi: 10.1016/j.celrep.2019.07.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim AS, Zimmerman O, Fox JM, Nelson CA, Basore K, Zhang R, Durnell L, Desai C, Bullock C, Deem SL, Oppenheimer J, Shapiro B, Wang T, Cherry S, Coyne CB, Handley SA, Landis MJ, Fremont DH, Diamond MS. 2020. An evolutionary insertion in the Mxra8 receptor-binding site confers resistance to alphavirus infection and pathogenesis. Cell Host Microbe 27:428–440. doi: 10.1016/j.chom.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orchard RC, Sullender ME, Dunlap BF, Balce DR, Doench JG, Virgin HW. 2019. Identification of antinorovirus genes in human cells using genome-wide CRISPR activation screening. J Virol 93:e01324-18. doi: 10.1128/JVI.01324-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoornweg TE, van Duijl-Richter MKS, Ayala Nuñez NV, Albulescu IC, van Hemert MJ, Smit JM. 2016. Dynamics of chikungunya virus cell entry unraveled by single virus tracking in living cells. J Virol 90:4745–4756. doi: 10.1128/JVI.03184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ayala-Nuñez NV, Wilschut J, Smit JM. 2011. Monitoring virus entry into living cells using DiD-labeled dengue virus particles. Methods 55:137–143. doi: 10.1016/j.ymeth.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Hoornweg TE, Bouma EM, van de Pol DPI, Rodenhuis-Zybert IA, Smit JM. 2020. Chikungunya virus requires an intact microtubule network for efficient viral genome delivery. bioRxiv. doi: 10.1101/2020.03.24.004820. [DOI] [PMC free article] [PubMed]
- 25.Kaksonen M, Roux A. 2018. Mechanisms of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 19:313–326. doi: 10.1038/nrm.2017.132. [DOI] [PubMed] [Google Scholar]
- 26.Traub LM, Bonifacino JS. 2013. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb Perspect Biol 5:a016790. doi: 10.1101/cshperspect.a016790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMahon HT, Boucrot E. 2011. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 28.Scott CC, Vacca F, Gruenberg J. 2014. Endosome maturation, transport and functions. Semin Cell Dev Biol 31:2–10. doi: 10.1016/j.semcdb.2014.03.034. [DOI] [PubMed] [Google Scholar]
- 29.Progida C, Bakke O. 2016. Bidirectional traffic between the Golgi and the endosomes—machineries and regulation. J Cell Sci 129:3971–3982. doi: 10.1242/jcs.185702. [DOI] [PubMed] [Google Scholar]
- 30.Vancini R, Wang G, Ferreira D, Hernandez R, Brown DT. 2013. Alphavirus genome delivery occurs directly at the plasma membrane in a time- and temperature-dependent process. J Virol 87:4352–4359. doi: 10.1128/JVI.03412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirchhausen T, Owen D, Harrison SC. 2014. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb Perspect Biol 6:a016725. doi: 10.1101/cshperspect.a016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hulseberg CE, Fénéant L, Szymańska KM, White JM. 2018. Lamp1 increases the efficiency of Lassa virus infection by promoting fusion in less acidic endosomal compartments. mBio 9:e01818-17. doi: 10.1128/mBio.01818-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White JM, Whittaker GR. 2016. Fusion of enveloped viruses in endosomes. Traffic 17:593–614. doi: 10.1111/tra.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MKS, Smit JM, Fremont DH, Pierson TC, Heise MT, Diamond MS. 2013. Development of a highly protective combination monoclonal antibody therapy against chikungunya virus. PLoS Pathog 9:e1003312. doi: 10.1371/journal.ppat.1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]