

ABSTRACT
The decomposition of wood and detritus is challenging to most macroscopic organisms due to the recalcitrant nature of lignocellulose. Moreover, woody plants often protect themselves by synthesizing toxic or nocent compounds which infuse their tissues. Termites are essential wood decomposers in warmer terrestrial ecosystems and, as such, they have to cope with high concentrations of plant toxins in wood. In this paper, we evaluated the influence of wood age on the gut microbial (bacterial and fungal) communities associated with the termites Reticulitermes flavipes (Rhinotermitidae) (Kollar, 1837) and Microcerotermes biroi (Termitidae) (Desneux, 1905). We confirmed that the secondary metabolite concentration decreased with wood age. We identified a core microbial consortium maintained in the gut of R. flavipes and M. biroi and found that its diversity and composition were not altered by the wood age. Therefore, the concentration of secondary metabolites had no effect on the termite gut microbiome. We also found that both termite feeding activities and wood age affect the wood microbiome. Whether the increasing relative abundance of microbes with termite activities is beneficial to the termites is unknown and remains to be investigated.
IMPORTANCE Termites can feed on wood thanks to their association with their gut microbes. However, the current understanding of termites as holobiont is limited. To our knowledge, no studies comprehensively reveal the influence of wood age on the termite-associated microbial assemblage. The wood of many tree species contains high concentrations of plant toxins that can vary with their age and may influence microbes. Here, we studied the impact of Norway spruce wood of varying ages and terpene concentrations on the microbial communities associated with the termites Reticulitermes flavipes (Rhinotermitidae) and Microcerotermes biroi (Termitidae). We performed a bacterial 16S rRNA and fungal ITS2 metabarcoding study to reveal the microbial communities associated with R. flavipes and M. biroi and their impact on shaping the wood microbiome. We noted that a stable core microbiome in the termites was unaltered by the feeding substrate, while termite activities influenced the wood microbiome, suggesting that plant secondary metabolites have negligible effects on the termite gut microbiome. Hence, our study shed new insights into the termite-associated microbial assemblage under the influence of varying amounts of terpene content in wood and provides a groundwork for future investigations for developing symbiont-mediated termite control measures.
KEYWORDS: wood-feeding termites, Reticulitermes flavipes, Microcerotermes biroi, plant defenses, terpenoids, core-microbiome, ectosymbionts, endosymbionts, bacteria, fungi
INTRODUCTION
Termites (Isoptera) are one of the few animal lineages capable of feeding on lignocellulose, the most common polymer on Earth. They play an important role in nutrient recycling in tropical ecosystems, where they are among the most abundant animals (1–3). Termites are traditionally divided into two informal groups based on their gut symbionts which aid plant material digestion. The “lower” termites include 9 of the 10 termite families and primarily rely on hypermastigian protists (Parabasalia), bacteria, and archaea to digest wood. In contrast, the “higher” termites, consisting only of the family Termitidae, have lost their gut protists and are primarily associated with bacteria and archaea (4) and, in the case of Macrotermitinae, with the symbiotic fungus Termitomyces (5). While “lower” termites feed solely on wood or grasses, many lineages of termitids feed on other substrates, especially rotten wood and the organic matter present in soil (6–9).
Termites thrive on wood, a nutritionally limiting substrate often rich in secondary metabolites (10). Many host trees are chemically defended with various phenolic compounds and terpenoids produced constitutively or upon attack by insects or pathogens (11). High concentrations of these defensive compounds, such as monoterpenes, olefins, and diterpenes, are entomotoxic and contribute to the plant’s resistance to insect infestation (12, 13). In response, insects have evolved strategies to overcome toxic plant compounds (14, 15). In addition to aiding in the degradation of complex dietary polymers (16, 17) and providing vitamins such as essential amino acids (18, 19), insect-associated microbes often manipulate and degrade toxic plant secondary metabolites (20–24).
Over the past decade, the contribution of symbiotic microorganisms to insect ecology has come to the forefront (25–27). Insect feeding habits play a vital role in shaping gut microbiotas. For example, cockroaches feeding on a low-protein, high-fiber diet exhibit reduced acetate and lactate production in their gut, resulting in low abundances of streptococci and lactobacilli (28). Similarly, a high-cellulose diet increased the protozoan population in the gut of Periplaneta americana (29). As noted above, “lower” termites harbor symbiotic protists which provide cellulolytic enzymes that help in wood digestion. In contrast, “higher” termites lack symbiotic protists and solely depend on their gut bacteria, archaea, and associated fungi to decompose cellulose (30, 31). Wood-feeding Termitidae host bacterial communities distinct from those of soil-feeding Termitidae (32, 33), producing larger amounts of lignocellulases (33, 34). The plant secondary metabolites content in wood is another factor which potentially influences gut microbial communities, a factor that has been poorly investigated so far (32). Termite gut bacterial and fungal symbionts may play an active role in detoxifying plant secondary metabolites, as is the case in bark beetles (35–39).
The present study aims to determine the influence of tree secondary metabolites (terpenes) on the gut microbial communities of wood-feeding termites. We studied two termite species, the “lower” termite Reticulitermes flavipes (Rhinotermitidae) and the “higher” termite Microcerotermes biroi (Termitidae), which we fed with Norway spruce wood of various ages. As pieces of Norway spruce age, their secondary metabolite content dwindles (40), allowing the investigation of secondary metabolite impact on the microbial assemblages associated with “lower” and “higher” termites. In this study, we characterized microbial communities using high-throughput amplicon sequencing targeting bacterial 16S rRNA and fungal ITS2. We compared the gut microbial communities of termite groups fed on woods of various ages with different concentrations of secondary metabolites. We used the microbial assemblages of the wood used to feed termites as wood controls and the unfed termites as termite controls. With this experimental design, we determined the impact of tree secondary metabolites on termite gut microbial communities.
RESULTS
Plant secondary metabolites.
The wood terpene content decreased as the wood aged (Fig. 1). Fresh wood (FW_C) contained a larger amount of α-pinene (106.9 ± 26.7 μg/g), β-pinene + β-myrcene (219.9 ± 55 μg/g), 3-carene (33 ± 8.2 μg/g), limonene (19.6 ± 4.9 μg/g), and camphene (2.8 ± 0.7 μg/g) followed by standard timber (SW_C) that contained α-pinene (69.5 ± 17.4 μg/g), β-pinene + β-myrcene (38 ± 9.5 μg/g), 3-carene (1.6 ± 0.4 μg/g), limonene (3.3 ± 0.8 μg/g), and camphene (2.8 ± 0.7 μg/g). Terpene concentration was the lowest in old wood (OW_C), with only α-pinene (0.46 ± 0.12 μg/g), β-pinene + β-myrcene (0.44 ± 0.11 μg/g), and camphene (0.32 ± 0.08 μg/g) being detectable (Fig. 1). These values correspond to the average of two independent determinations in dry weight form (DW) for each type of wood, expressed together with the 95% confidence interval, based on the uncertainty of gas chromatography coupled with mass spectrometry (GC-MS) determination, calculated for each analyte during validation. Unfortunately, statistical significance could not be estimated due to the lack of sufficient replications for each type of wood during the GC-MS determination of terpene content. However, even with two replicates, the wood samples showed distinct differences in terpene content.
FIG 1.

Terpenic compounds content (μg/g DW) (DW, dry weight) present in Norway spruce wood upon storage. FW_C represents freshly cut spruce wood, SW_C denotes commercially available spruce timber felled 3 to 4 years ago, and OW_C represents old wood that was cut circa 120 years ago. Values are the average of two independent analytical determinations of compound concentrations in pooled homogenized (n = 6) samples of wood. Error bars represent the uncertainty (U) of the whole analytical procedure, including extraction and GC-MS (gas chromatography coupled with mass spectrometry) instrumental analysis. U is based on relative standard deviation (RSD) obtained from 9 independent determinations of compounds concentration in one homogenized wood sample. U is calculated using the equation U = 2 × RSD.
Illumina sequencing.
The Illumina pair-end sequencing performed on the MiSeq platform yielded a total of 2,347,945 bacterial 16S sequences and 779,354 fungal internal transcribed spacer (ITS) sequences after the initial quality check and removal of chimeric sequences. The reads which passed quality assessment obtained from termites, infested wood, and uninfested control wood samples were used for downstream bioinformatic analyses. The completeness of the sequencing is illustrated by the rarefaction curves (Fig. S1in the Supplemental file 1).
Bacterial diversity.
The operational taxonomic unit (OTU) clustering performed at a 97% similarity cutoff documented the presence of 3,633 bacterial OTUs that were assigned to 722 bacterial genera belonging to 30 phyla (Supplemental File 2, part 1). The bacterial communities of wood pieces infested by R. flavipes revealed 12 dominant phyla with relative abundance of ≥1% (Fig. 2a), while those infested by M. biroi revealed 9 dominant phyla (Fig. 2b). Within the control wood, the relative abundance of Proteobacteria (44.46%) was higher in old wood (OW_C) than in other types of wood controls (SW_C, 27.58%; FW_C, 29.23%). FW_C had the highest relative abundance of Planctomycetes (6.42%) and the lowest relative abundance of Actinobacteria (18.23%) and Firmicutes (8.10%) among the wood controls. The termite-infested wood microbiome was characterized by a higher relative abundance of Proteobacteria than the microbiomes of uninfested wood controls (Fig. 2). Proteobacteria and Actinobacteria were the dominant phyla in both infested and uninfested wood, followed by Firmicutes and Bacteroidetes. Elusimicrobia and Mycoplasmatota were only observed in infested wood samples and might have been introduced by termites during feeding activities (Fig. 2; Supplemental File 2, part 1).
FIG 2.

Relative abundance of bacterial communities. (a and b) Relative abundance of bacterial phyla (abundance of ≥1% in at least one sample) within termites (Reticulitermes flavipes and Microcerotermes biroi) feeding on different substrates, termite-infested wood, and their controls. “Others” denotes the total relative abundance of other phyla. (c and d) Heatmap illustrating the top 45 bacterial genera with relative abundance of ≥1% in at least one sample documented in Reticulitermes and Microcerotermes before and after feeding on different substrates, termite-infested wood, and their controls. Color gradient from red to green through black represents the relative abundance of bacterial operational taxonomic units (OTUs) present in each sample type. Red color, low abundance; green color, high abundance. FW_C, fresh wood control; SW_C, standard wood control; OW_C, old wood control; FW_R_W, Reticulitermes-infested fresh wood; SW_R_W, Reticulitermes-infested standard wood; OW_R_W, Reticulitermes-infested old wood; R_C_T, Reticulitermes control termite before feeding; FW_R_T, Reticulitermes feeding on fresh wood; SW_R_T, Microcerotermes feeding on standard wood; OW_R_T, Reticulitermes feeding on old wood; Cellu_R_T, Reticulitermes feeding on cellulose; FW_M_W, Microcerotermes-infested fresh wood; SW_M_W, Microcerotermes-infested standard wood; OW_M_W, Microcerotermes-infested old wood; M_C_T, Microcerotermes control termite before feeding; FW_M_T, Microcerotermes feeding on fresh wood; SW_M_T, Microcerotermes feeding on standard wood, OW_M_T, Microcerotermes feeding on old wood; Cellu_M_T, Microcerotermes feeding on cellulose.
R. flavipes feeding on all substrates showed higher relative abundances of Proteobacteria, Bacteroidetes, Actinobacteria, and Euryarchaeota than the control workers sampled before the feeding experiment started (R_C_T). In contrast, Spirochaeta (34.14%), Elusimicrobia (27.17%), and Mycoplasmatota (4.31%) were abundant in R_C_T (Fig. 2a; Supplemental File 2, part 1). Similarly, feeding on wood substrates and cellulose increased the relative abundance of Fibrobacteres, Bacteroidetes, and Acidobacteria in M. biroi compared to their termite controls (M_C_T) (Fig. 2b). However, the relative abundance of Proteobacteria and Firmicutes was the highest in M_C_T (Fig. 2b). Comparing the two termite species the relative abundance of Elusimicrobia (27.17%), Bacteroidetes (12.2%), Euryarchaeota (4.51%), and Mycoplasmatota (4.31%) were higher in R. flavipes controls (R_C_T), while the relative abundance of Spirochaeta (57.43%), Proteobacteria (27.58%), Firmicutes (9.27%), and Fibrobacteres (2.21%) were higher in M. biroi (M_C_T) (Fig. 2, Supplemental File 2). The relative abundances of the top 45 bacterial genera observed in the two termite species fed with different substrates, their infested wood, along with the controls, are represented as heatmaps (Fig. 2c and d). These differentially abundant bacterial genera include Spirochaeta, Endomicrobium, “Candidatus Armantifilum,” “Candidatus Symbiothrix,” Dysgonomonas, Fibrobacter, Sphingomonas, Bacteroides, Ralstonia, Methylobacterium, Roseburia, Methanobrevibacter, Ruminiclostridium, Propionibacterium, Burkholderia, etc. (Supplemental File 2, parts 2 and 3).
The Shannon index revealed that the gut bacterial diversity of M. biroi controls (M_C_T) was significantly lower than those of the R. flavipes controls (R_C_T) (Tukey’s post hoc analysis; P < 0.05); however, no significant differences were observed among termites feeding on different substrates (Fig. S2a in the Supplemental file 1). M. biroi had higher bacterial richness (Chao1 index) than R. flavipes (Fig. S2b in the Supplemental file 1). Bacterial diversity was higher in the control wood samples than in wood samples infested by termites. The highest bacterial diversity was found in the SW_C group. Interestingly, termite feeding activities reduced the bacterial diversity (Shannon) and increased the bacterial richness (Chao1) in all types of wood samples (Fig. S3 in the Supplemental file 1).
The overall β-diversity represented by the nonmetric multidimensional scaling (NMDS) plot showed distinct separation of the bacterial communities of both termite species (M. biroi and R. flavipes) (Fig. 3) (permutational multivariate analysis of variance [PERMANOVA], termite_species; dfnum = 1, dfden = 38, F = 133.18, P = 0.001). Moreover, the effect of the substrate type depended on the termite species (PERMANOVA, termite_species: substrate_type; dfnum = 4, dfden = 38, F = 2.47, P = 0.021). The bacterial community composition associated with termites fed on spruce wood of different ages, cellulose, and control termites differed between termite species (Tables S1, S5 and S6 in the Supplemental file 3). No significant variation was observed among the gut bacterial communities of M. biroi feeding on different types of wood. However, the bacterial communities of M. biroi feeding on cellulose (Cellu_M_T) and control M. biroi samples (M_C_T) significantly differed from that of M. biroi feeding on different types of spruce wood substrates. A similar trend was observed in R. flavipes (Fig. 3a, Table S1 in the Supplemental file 3).
FIG 3.

Nonmetric multidimensional scaling (NMDS) plot. The variation in the bacterial communities’ present in (a) the two different termites (Reticulitermes flavipes and Microcerotermes biroi) feeding on different substrates and their control. (b) Different wood substrates infested by the termites and control uninfested wood. Samples are denoted by different shapes (square, control; circle, M. biroi; triangle, R. flavipes); colors denote substrate types.
Termite-feeding activities altered the wood microbiome. The control wood samples clustered separately in the NMDS plot (Fig. 3b). Furthermore, the wood samples infested by M. biroi and R. flavipes grouped in distinct clusters (PERMANOVA, termite_species; dfnum = 2, dfden = 39, F = 15.04, P = 0.001). The effect of wood type on bacterial community structure depended on the termite species (PERMANOVA, termite_species: wood_type; dfnum = 4, dfden = 39, F = 2.44, P = 0.001). Therefore, the bacterial communities of wood samples infested with termites differed significantly from those of wood samples free of termites (Fig. 3b; Table S2, S5 and S6 in the Supplemental file 3).
Fungal diversity.
The fungal OTU clustering documented 1,499 OTUs assigned to six phyla (Supplemental File 2, part 4). Among these, Ascomycota was prevalent in all samples, followed by Basidiomycota, Mucoromycota, and Chytridiomycota. A lower relative abundance of Basidiomycota was observed in wood samples after R. flavipes infestation. In contrast, standard (SW_M_W) and old wood (OW_M_W) samples infested by M. biroi showed a high relative abundance of Basidiomycota compared to control wood samples. Basidiomycota was proportionally more abundant in the guts of both M. biroi and R. flavipes fed with all types of wood substrates than in their unfed controls. In contrast, the relative abundance of Basidiomycota decreased in termites fed with cellulose (Supplemental File 2, part 4).
The gut microbiome of R. flavipes contained a high proportion of Eurotiales (55.44%), Onygenales (25.36%), and Agaricales (5.31%) before the feeding experiments started (R_C_T) (Fig. 4a). The fungal order GS23 was absent in control wood samples but was observed in termite-infested wood samples. Additionally, Hypocreales, Trichospornales, and Ophiostomatales were proportionally more abundant in both the R. flavipes-infested wood samples and in the guts of termites fed with all types of substrates than in the controls (Fig. 4a). Similarly, the relative abundance of Sordariales was higher in M. biroi-infested fresh wood (FW_M_W, 37.21%) and old wood (OW_M_W, 26.83%) samples than in control wood samples (FW_C, 0.93%; OW_C, 0.96%) (Fig. 4b). However, the standard wood control samples (SW_C) showed a higher relative abundance of Pleosporales (17.54%), Capnodiales (12.14%), Diaporthales (16.46%), and Agaricales (11.43%) than the infested wood samples (SW_M_W). Saccharomycetales (30.59%) were dominant in uninfested fresh wood controls (FW_C), whereas Helotiales (19.11%) and Coniochaetales (22.48%) were prevalent in the old wood controls. The relative abundance of Trichosporonales increased in M. biroi-infested wood samples (Fig. 4b). Furthermore, Eurotiales (45.26%) and Malasseziales (2.44%) were predominant in the M. biroi controls (M_C_T) and decreased in relative abundance in all types of wood substrates infested by M. biroi. In contrast, the relative abundance of several fungal orders, such as Hypocreales, Sordariales, Saccharomycetales, Botryosphaeriales, and Ustilaginales, was higher in wood samples infested by M. biroi (Fig. 4b). Nevertheless, among termite mycobiomes, the relative abundance of Eurotiomycetes was the highest in both control termites (M_C_T and R_C_T) (Supplemental File 2, part 4). Interestingly, the relative abundance of Eurotiomycetes in the gut of R. flavipes (80.82%) was nearly double that found in M. biroi (45.3%). The heatmap represented the top 39 fungal genera with a relative abundance of ≥1% in at least one sample (Fig. 4c and d). The differentially abundant fungal genera included Trichoderma, Meyerozyma, Apiotrichum, Trichosporon, Fusarium, Alternaria, Sporothrix, Cadophora, Malassezia, and Talaromyces (Supplemental File 2, parts 5 and 6).
FIG 4.

Relative abundance of fungal communities. (a and b) Relative abundance of fungal order with relative abundance of ≥1% in at least one sample within termites (R. flavipes and M. biroi) upon feeding on different substrates, termite-infested wood and their controls. “Others” denotes the total relative abundance of rest of the fungal orders present. (c and d) Heatmap illustrating the top 39 fungal genera documented in Reticulitermes and Microcerotermes before and after feeding on different substrates, termite-infested spruce wood, and control wood. Color gradient from red to green through black represents the relative abundance of fungal OTUs present in each sample type. Red color denotes low abundance, green color signifies high abundance. Group abbreviations are the same as in Fig. 2.
The Shannon diversity index showed that the gut fungal communities of R. flavipes and M. biroi did not significantly differ. In addition, no significant differences were observed among termites feeding on different substrates (Tukey’s post hoc analysis; P > 0.05) (Fig. S4a in the Supplemental file 1). Similarly, none of the experimental factors significantly affected the fungal richness (Chao1 index) in the guts of R. flavipes and M. biroi (Fig. S4b in the Supplemental file 1). Fungal diversity was significantly higher in the uninfested control wood samples than in the samples infested by R. flavipes and M. biroi (FW_C, SW_C, and OW_C) and decreased with wood age (Fig. S5a in the Supplemental file 1). Fungal richness decreased with wood age across termite species and controls (Fig. S5b in the Supplemental file 1).
Fungal community composition differed significantly between termite species (M. biroi and R. flavipes); however, the difference was not as marked as for bacterial communities (Fig. 5a) (PERMANOVA, termite_species; dfnum = 1, dfden = 38, F = 15.04, P = 0.001). Substrate type influenced fungal community composition in a species-specific manner (PERMANOVA, termite_species: substrate_type; dfnum = 4, dfden = 38, F = 2.44, P = 0.001). The fungal community composition of M. biroi controls (M_C_T) significantly differed from that of M. biroi fed on standard wood (SW_M_T), old wood (OW_M_T), and cellulose (Cellu_M_T) (Table S3 in the Supplemental file 3). Similarly, the fungal community composition of R. flavipes controls was significantly different from those of other treatments (Table S3 in the Supplemental file 3). The difference in fungal community composition between controls and treatments was more prominent in M. biroi than in R. flavipes (Fig. 5a, Table S3 in the Supplemental file 3).
FIG 5.

NMDS plot. The variation in the fungal communities present in (a) the two termites feeding on different substrates and their control, and (b) different wood substrates either infested by the termites or control uninfested wood. Sample types are denoted by different symbols as in Fig. 3.
The fungal community composition of wood was strongly affected by termite activity. Control wood samples clustered separately from infested wood samples, while wood samples infested with R. flavipes and M. biroi largely overlapped in the NMDS plot (PERMANOVA, termite_species; dfnum = 2, dfden = 38, F = 14.08, P = 0.001) (Fig. 5b). Termite feeding activity strongly reduced the effect of wood age on fungal community composition (PERMANOVA, termite_species: wood_type; dfnum = 4, dfden = 38, F = 2.62, P = 0.001). Control wood samples showed a clear successional pattern with fungal communities gradually changing from FW_C, through SW_C, to OW_C (Fig. 5b, Table S4 in the Supplemental file 3). In contrast, the fungal community composition of wood samples on which termites fed did not markedly change with wood age (Fig. 5b, Table S4 in the Supplemental file 3). Additionally, the fungal community composition significantly varied between termite bodies and wood as estimated using Hellinger-transformed OTU table based on Bray-Curtis dissimilarity distances (Table S6 in the Supplemental file 3).
Core gut microbiome in R. flavipes and M. biroi.
We identified the core gut microbial community consistently associated with termites feeding on all types of substrates. The core gut bacterial community of R. flavipes included 452 OTUs belonging to 133 bacterial genera (Fig. 6a; Supplemental File 2, part 7), while the core gut fungal community included 79 OTUs belonging to 31 fungal genera (Fig. 6b; Supplemental File 2, part 8). Similarly, the core gut microbial community of M. biroi included 448 OTUs assigned to 120 bacterial genera (Fig. 6c; Supplemental File 2, part 9) and 92 OTUs belonging to 32 fungal genera (Fig. 6d; Supplemental File 2, part 10). In particular, the core gut bacterial microbiome of both termite species included “Candidatus Armantifilum,” “Candidatus Symbiothrix,” Clostridium, Desulfovibrio, Desulfosarcina, Dysgonomonas, Endomicrobium, Fibrobacter, Massilia, Methanobrevibacter, Methylobacterium, Methanobacterium, Paludibacter, Paraburkholderia, Parabacteroides, Pseudomonas, Ralstonia, Rickettsia, Roseospira, Ruminococcus, Siphonobacter, Spirochaeta, Taibaiella, Tangfeifania, Tannerella, Treponema, Tyzzerella, and Wolbachia (Supplemental File 2, parts 7 and 9). The core gut fungal microbiome of both termite species included Malassezia, Meyerozyma, Trichoderma, Fusarium, Aspergillus, Penicillium, Debaryomyces, Hawksworthiomyces, Scytalidium, Trichosporon, Lasiodiplodia, and Alternaria (Supplemental File 2, parts 8 and 10). In addition to the core OTUs shared by both termite species, we also identified unique OTUs that may have been acquired during feeding (Fig. 6; Supplemental File 2, parts 7 to 10).
FIG 6.
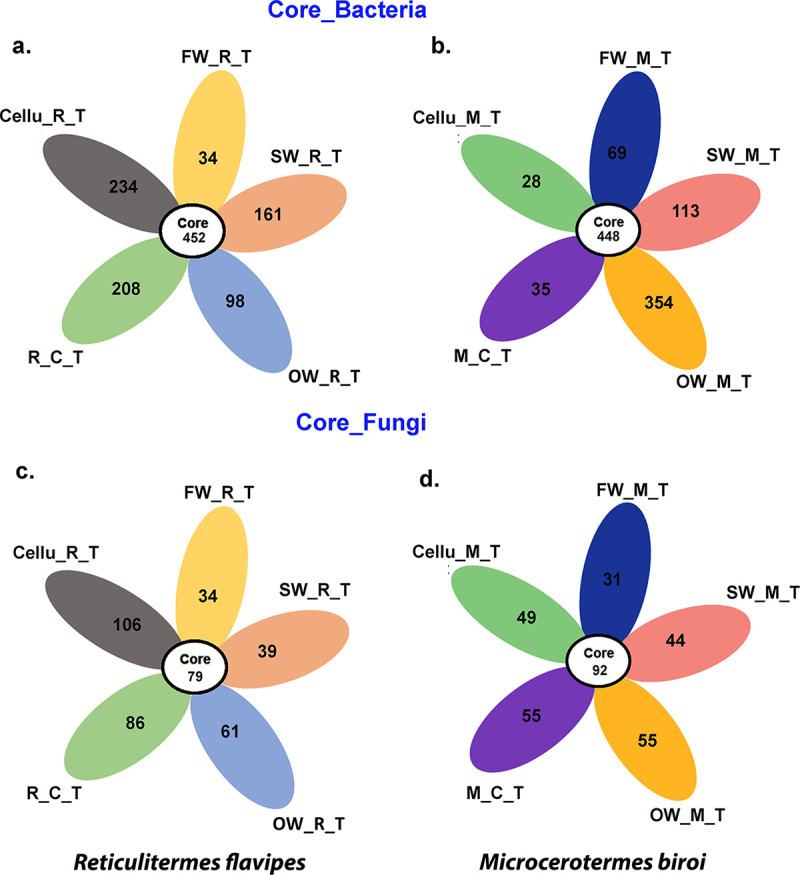
Flower diagram representing the core and unique OTUs. (a and b) Core and unique bacterial OTUs shared among R. flavipes and M. biroi upon feeding on different substrates. (c and d) Common and unique fungal OTUs shared among Reticulitermes and Microcerotermes upon feeding on different substrates. Group abbreviations are the same as in Fig. 2.
DISCUSSION
The termite gut microbiome is not affected by wood age.
The terpene content in Norway spruce wood has decreased by more than 300 times over 120 years of timber use. Our data showed that the terpene content influenced the gut microbial assemblage of both R. flavipes and M. biroi. The relative abundance of bacterial genera such as Pseudomonas, Massilia, and Rhizobium was high in wood samples infested by termites (Fig. 1), suggesting they contribute to important functions, such as detoxification of toxic plant secondary metabolites (35, 41–44). The high relative abundance of Fibrobacteres in the gut of M. biroi is likely linked to their involvement in lignocellulose degradation (45). Furthermore, our data documented the prevalence of spirochetes in both R. flavipes and M. biroi, although the bacterial genera Spirochaeta and Treponema showed significant differences in their relative abundance between these two species. Overall, the relative abundance of Spirochaeta was higher in M. biroi than in R. flavipes. Several studies have documented the importance of spirochetes in the termite gut for their involvement in various metabolic processes such as nitrogen fixation, acetogenesis, and degradation of lignocellulose (46–51). The relatively lower abundance of spirochetes in R. flavipes compared to M. biroi could reflect the presence of lignocellulolytic protists in the guts of “lower” termites (52).
The bacterial lineages “Candidatus Azobacteroides” and Endomicrobium identified in the present study are known as termite gut symbionts associated with gut protists (53–55). Endomicrobium (phylum Elusimicrobia), identified as a predominant bacterial genus in the guts of “lower” termites (R_C_T) (56), is mainly involved in amino acid synthesis, glucose fermentation, nitrogen fixation, and recycling of nitrogenous wastes (57–60). In contrast, “Candidatus Azobacteroides” is closely associated with gut protists performing H2 uptake and nitrogen fixation (59, 61). Furthermore, the ectosymbionts “Candidatus Symbiothrix” and “Candidatus Armantifilum,” belonging to Bacteroidetes, are known to colonize the cell surface of gut protists and perform lignocellulose digestion and nitrogen fixation within the host termite (62, 63). Consequently, bacterial communities associated with termites contribute to diverse metabolic functions complementing the host metabolism.
The present study documented the fungal communities associated with termites feeding on various wood substrates (Fig. 5). Members of fungal orders such as Trichosporonales, Eurotiales, Saccharomycetales, and Malaseziales were observed in both R. flavipes and M. biroi (64, 65). Many fungal genera documented in this study (Fig. 5c and d) have been previously reported in different termite species (18, 66, 67), suggesting that they are associated with termites in a stable fashion. They have also reported in other insects, such as beetles (68, 69) and cockroaches (70). The high abundance of the yeasts Debaryomyces, Meyerozyma, and Malassezia suggests that they are typical termite gut inhabitants (64, 65, 71). Filamentous fungi and yeast communities inhabiting the insect gut (72, 73) are known to play important roles in the decomposition of lignocelluloses, aiding in wood digestion (65, 74), detoxifying plant allelochemicals (38, 39), and providing supplementary nutrients (75). The observed fungal genera Trichoderma, Penicillium, Scytalidium, Lasiodiplodia, and Hawksworthiomyces have been reported to exhibit lignocellulolytic activities (76–79). Furthermore, Fusarium associated with termites might participate in amino acid metabolism and the recycling of nitrogenous waste products (80).
The overall microbial diversity of M. biroi and R. flavipes showed substantial variations (Fig. 4a). However, feeding on wood pieces of different ages did not significantly alter the termite gut microbiome. These results indicate that the core gut microbial community of termites is stable and remains unaffected by varying concentrations of host tree allelochemicals.
Wood age and termite feeding activities affect the wood microbiome.
The plant terpenoid concentration decreased according to the following sequence: freshly cut wood (FW_C), standard wood (SW_C), and old wood (OW_C), in which terpenoid compounds were almost absent (Fig. S1). The bacterial communities present in wood mainly comprised Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes (81). However, their relative abundance changed with the wood age. Furthermore, the overall microbial community composition within the control and termite-infested wood samples differed, reflecting the influence of termite-feeding activities (Fig. 1 and 5). The high abundance of several bacterial genera such as Burkholderia, Erwinia, Massilia, Novosphingobium, Paraburkholderia, Pseudomonas, Rhizobium, and Sphingobium observed in termite-infested fresh wood might reflect their ability to detoxify or tolerate toxic plant secondary metabolites (35, 41–44). Furthermore, several bacterial genera such as Acetatifactor, Anaerostipes, Atopobium, “Candidatus Azobacteroides,” Dysgonomonas, Endomicrobium, Mycoplasma, Pasteuria, Parabacteroidetes, Paludibacter, Pseudohalocynthiibacter, Ralstonia, Roseospira, Siphonobacter, and Tangfeifania were absent in uninfested control wood samples but were introduced by termites during feeding. Most of these bacterial lineages were termite gut symbionts, some of which were associated with their gut protists (53–55). The amplicon DNA sequencing approach cannot differentiate between live bacteria and DNA originating from dead cells. These termite gut symbiont DNA sequences likely originated from dead cells or remnant DNA deposited by termites on their galleries. Furthermore, in termite-infested wood, fungal communities belonging to the orders Eurotiales, Sordariales, Hypocreales, Trichospornales, and Ophiostomatales were prevalent. The fungal genera Apiotrichum, Fusarium, Hawksworthiomyces, Lasiodiplodia, Sporothrix, Trichosporon, and Trichoderma were highly abundant in termite-infested wood, possibly contributing to lignocellulose degradation and detoxification of secondary metabolites (76, 77, 79, 82). Additionally, our data document the presence of the fungal orders Ustilaginales, Rhizophydiales, and GS23 in termite-infested wood, which were absent from control wood samples, suggesting that they were transferred from termites during feeding. Consequently, the control and termite-infested wood samples formed distinct groups in the NMDS plot, showing significant differences in overall microbial community composition (Fig. 4b). Although we interpreted the effect of wood age on microbial communities as being driven by terpenoid concentration, other factors such as temperature, moisture content, and nutrient availability also vary with wood age and could affect the wood microbiome.
The core gut microbiome of termites.
Our findings document that the core gut microbiome of termites is unaffected by the feeding substrate. This was true for both bacterial and fungal communities and for both termite species investigated in this study. Many of these observed core microbes, such as Clostridium, Fusarium, Malassezia, Penicillium, Ruminococcus, Tyzzerella, and Trichoderma, have been reported as core members of the gut microbiome in other wood-feeding insects, e.g., bark beetles, in which they perform vital metabolic functions for their hosts (83, 84).
Conclusion.
The present study revealed that the gut microbial community differed significantly between the two termite species. A stable core gut microbiome was identified in both R. flavipes and M. biroi. This core microbiome was not altered by the feeding substrate. The termite gut microbiome was not markedly influenced by the wood age, which is characterized by varying concentrations of secondary metabolites. Termite feeding activities considerably altered the wood microbiome, while wood age and hence the concentration in plant allelochemicals had minor effects. Our results also indicate that termite-feeding activities modified the wood microbiome, possibly increasing the proportion of microbes beneficial to the termites, such as those participating in the initial detoxification of toxic plant metabolites, decomposition of lignocellulose, and nitrogen assimilation.
MATERIALS AND METHODS
Termite sampling.
The wood-feeding “lower” termite R. flavipes (R) was obtained from David Sillam-Dussès (University Paris 13, France), while the “higher” wood-feeding termite, M. biroi (M), was brought to Prague from the village of Wanang (Madang, Papua New Guinea; lat –5.22772°, long 145.07983°). We made termite groups composed of 100 workers and 2 soldiers each (reflecting the soldier proportion in both tested species [85]), which we placed into Petri dishes (85-mm diameter) containing 20 g fine sand, 5 mL tap water, and a piece of wood. The experiment was set at 27°C (±0.5°C), and the Petri dishes were placed into larger boxes lined with wet filter paper to maintain humidity at 100% for the 10 days of the experiment. Termites were given a single piece of Norway spruce wood (20 × 3 × 3 mm). Three types of wood were provided: freshly cut wood (FW_C; harvested within 24 h of tree logging), standard timber (SW_C; 3 to 4 years of age), and old timber (OW_C; rafters 120 years old obtained during the university building reconstruction). In addition, we established groups of termites provided with 2 g microcrystalline cellulose (Merck, product no. CAS 9004-34-6) instead of wood as positive feeding controls. We also used uninfested wood fragments (six replicates) and unmanipulated termite samples (from stock colonies, four replicates) as controls. A total of five replicates were performed for each wood type. All samples were collected into RNAlater and stored at −80°C until DNA extraction.
Analyses of terpenoids.
The content of selected secondary metabolites (terpenes) was measured for the three types of wood using GC-MS. Two replicates were analyzed for each wood type. Wood samples were initially frozen in liquid nitrogen and homogenized into a fine powder using a Mixer Mill Retsch MM 400 with a sterile steel bead (5 mm) for 15 min at 30 oscillations/sec. Next, 200 mg of wood powder was placed into a 20 mL headspace vial, extracted with 2 mL N-hexane in an ultrasonic bath for 10 min, and then filtered into a 2-mL vial for GC analysis. One microliter of the extract was injected into the gas chromatograph coupled to a time-of-flight mass spectrometer (GC-TOF-MS) (Leco Pegasus 4D, Leco, USA). The temperature-programmed injector was used in a split mode with a 1:10 split ratio. A 30-m (0.25-mm internal diameter, 0.25-μm film thickness) Rxi-5MS (Restec, USA) column was used for separation. The temperature program for the GC oven was as follows: 40°C for 1 min, then raised to 210°C at a rate of 10°C/min, followed by an increase to 320°C at 20°C/min with a hold time of 2 min. The total GC run time was 26 min. The mass spectrometer was operated at a mass range of 35 to 500 m/z with an acquisition speed of 10 Hz. The similarity of the deconvoluted mass spectrum with spectra from the NIST library was used for compound identification. The compounds of interest (α- and β-pinene, β-myrcene, camphene, 3-carene, and limonene) were quantified using an external calibration curve based on commercially available standards obtained from Sigma-Aldrich (USA). Since β-pinene and β-myrcene coeluted on the used column, these compounds were reported together. Results correspond to the average values from two independent determinations for each type of wood in DW form, expressed together with a 95% confidence interval, based on the uncertainty of GC-MS determination, calculated for each analyte during validation (86).
DNA extraction, amplification, and sequencing.
Total DNA was extracted from termites and wood samples (either infested or uninfested). Termite samples were rinsed twice in 70% ethanol and washed with sterile water for surface sterilization. Each termite sample consisted of a pool of 10 workers homogenized with a Mixer Mill Retsch MM 400 and two sterile steel beads (3 mm) for 2 min at 30 oscillations/sec before DNA extraction. Similarly, the wood samples were freeze-dried in liquid nitrogen and homogenized with a Mixer Mill Retsch MM 400 using a 5-mm sterile steel bead for 15 min at 30 oscillations/sec before DNA extraction. Total DNA was isolated using the Macherey-Nagel NucleoSpin Soil kit following the manufacturer’s protocol and electrophoresed on 1% agarose gel with GelRed Nucleic Acid Stain (Biotium, USA) to check DNA integrity. The DNA concentration was quantified on Nanodrop (Thermo Fisher Scientific), and 10 ng of DNA was used as a template for downstream amplification reactions.
PCR amplification of the bacterial 16S rRNA gene targeting the V4 hypervariable region was performed using the universal, uniquely barcoded primers 515F and 806R (87), while the fungal ITS2 domain was amplified using gITS7F and ITS4R uniquely barcoded primers (88, 89). PCR amplifications were performed in triplicate using an Eppendorf Mastercycler (Eppendorf AG, Hamburg, Germany) nexus cycler. The reaction mixture included 1× PCR buffer with 1.5 mM MgCl2, 10 μM primers, 200 μM each dNTP, 10 μg/mL of bovine serum albumin, 2 U Q5 High Fidelity DNA Polymerase (New England Biolabs), and 10 ng of template DNA. PCRs were performed as follows: initial denaturation at 94°C for 5 min; followed by 30 cycles of amplification at 94°C for 45 s, 50°C for 60 s, 72°C for 45 sec (bacterial amplification), or 94°C for 45 s, 56°C for 30 s, 72°C for 30 s (fungal amplification); and a final extension step at 72°C for 10 min. A template control was also run to verify the absence of any contaminations. The amplicons were purified using the MinElute PCR Purification kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s protocol and quantified with a Qubit 2.0 Fluorometer using the dsDNA HS Quantification kit (Invitrogen). Sequencing libraries of purified amplicons adjusted at equimolar concentration were prepared using the NEBNext Ultra DNA Library Pre-kit. Libraries were sequenced on the Illumina MiSeq platform (Illumina Inc., USA) to yield 250-bp paired-end reads.
Data processing.
(i) Quality control and data filtering. Paired-end raw reads obtained from Illumina sequencing were merged to generate single reads using the fastq-join command (90) of SEED 2 (version 2.1.05) (91). Merged sequences were demultiplexed and trimmed. Quality control tests were performed for sequences with mean quality Phred scores Q < 30. Sequences with mismatches in barcodes were discarded. Bacterial 16S sequences with read lengths of <230 bp or >280 bp and fungal ITS sequences with read lengths of <40 bp were removed from the data set. All fungal sequences were extracted using ITSx (version 1.0.11) (92) before the OTU clustering step to obtain the entire fungal ITS2 region.
(ii) OTU clustering. OTUs were clustered at 97% sequence similarity using the UPARSE algorithm implemented in USEARCH version 8.1.1861 (93). Sequences identified as chimeric were excluded from downstream analyses. OTUs with less than five reads were discarded to minimize the effect of contaminations (if any) and barcode hopping during sequencing (94). We used one representative sequence for each OTU (i.e., the most abundant sequence) to perform taxonomic identification with RDP database release 11 (95) to obtain the closest BLAST hit for each OTU. We used rrnDB version 5.4 (96) to estimate the relative OTU abundance, a measure that corrects for the variation in 16S copy number per genome among bacterial species (97). Similarly, the fungal sequences were annotated using the UNITE database version 7.2 (98).
(iii) Alpha (α-) and beta (β-) diversities. Bacterial and fungal species diversity (α-diversity) was estimated using community richness (Chao1) and diversity (Shannon) indices (99). We estimated these indices for each termite species, termite-infested wood, and the controls using SEED v 2.1. The results were displayed in R (100). We identified the microbial communities shared by termites fed with different substrates.
The variability in the overall microbial community structure (β-diversity) among termites, termite-infested wood, and their controls was visualized using nonmetric multidimensional scaling analysis (101) with a Bray-Curtis distance matrix. The analyses were performed in R. The bacterial and fungal communities of termite guts and wood pieces were analyzed separately. For termite guts, a PERMANOVA analysis (102) was performed using the adonis() function of the R package vegan (103) to test the null hypothesis of no effect of the termite species and substrate type on the composition of the termite gut microbiome. Bray-Curtis dissimilarity matrices were used as responses in the analyses. The relative abundance tables were fourth-root-transformed prior to calculation of the dissimilarity matrix to reduce the influence of the more abundant taxa relative to the less dominant taxa and allow community-wide assessment of changes in taxon composition (104). Termite species (factor with two levels: M. biroi and R. flavipes), substrate type (five levels: fresh wood, standard wood, old wood, cellulose, and control), and their interaction were included as explanatory variables. Post hoc pairwise comparisons of specific combinations of termite species and substrate types were performed using a pairwise PERMANOVA implemented in the pairwiseAdonis R package (105). False-discovery rate (FDR) correction was used to adjust P values to multiple comparisons (106). Significance was assessed using 999 permutations.
Similarly, the null hypothesis of no effect of termite species and wood type on the wood microbiome composition was tested using adonis(). Response matrices were again calculated using Bray-Curtis dissimilarity of the fourth-root-transformed relative abundance tables. Termite species (factor with three levels: M. biroi, R. flavipes, and wood control), wood type (three levels: fresh wood, standard wood, and old wood), and their interaction were used as explanatory variables. Pairwise comparisons were performed using the same procedure as for termite guts.
Additionally, to ensure the robustness of our results to the choice of dissimilarity index, these analyses were repeated using a Hellinger-transformed OTU abundance table based on Bray-Curtis dissimilarity, as well as phylogeny-based UniFrac (weighted and unweighted) dissimilarity (107). UniFrac-based analysis was conducted only for bacteria since the ITS region shows higher variation than 16S and hence does not allow the building of reliable phylogenetic trees for fungi, possibly leading to erroneous results for this analysis (108).
Relative abundances of different taxa in specific experimental conditions were visualized using barplots and heatmaps. We used linear models to test the null hypothesis of no effect of termite species and substrate type on termite gut and wood fungal and bacterial α-diversity (as measured by Chao1 community richness estimator and Shannon-Wiener diversity index). Alpha diversity indices calculated for termite and wood samples were used as response variables in the models, and termite species and substrate type were used as explanatory variables, as explained previously for the multivariate PERMANOVA analyses. For pairwise comparisons of specific combinations of termite species and substrate types, Tukey’s post hoc tests were implemented using the R function TukeyHSD().
We performed a differential abundance analysis of bacterial and fungal genera in termite guts and wood pieces between experimental groups using a quasi-likelihood negative binomial generalized log-linear model to count data. This analysis was performed using the glmQLFit() function of the R package edgeR (109). As for the previously described PERMANOVA analyses, termite species, substrate type, and their interaction were included as explanatory variables for termite guts, whereas termite species, wood type, and their interaction were included as explanatory variables for wood pieces. FDR correction was used to adjust P values to multiple comparisons (50). Because one of our main objectives was to determine the influence of wood age on the termite-associated microbiome, the following contrasts (independently for Microcerotermes and Reticulitermes) were tested to identify differentially abundant taxa: control termites versus termites fed with fresh wood, standard wood, old wood, and cellulose. Similarly, we also studied differentially abundant microbial communities in termites fed with different wood substrates. Furthermore, in the case of wood pieces, our objective was to determine how termite substrate influences the termite microbiota. As such, the uninfested wood controls were compared with the infested wood for the two termite species.
Data availability.
The sequence data of the bacterial V4 hypervariable region of the 16S rRNA gene and the fungal ITS2 gene amplified from two different wood-feeding termites (R. flavipes and M. biroi), infested spruce wood, and their controls have been deposited in NCBI (SRA database) under BioProject accession no. PRJNA792414.
ACKNOWLEDGMENTS
We thank David Sillam-Dussès for providing R. flavipes material. We are grateful to the New Guinea Binatang Research Centre (Madang, Papua New Guinea) for help with the M. biroi material acquisition.
Financial support was provided by Czech Science Foundation projects no. 16-05318S and 19-28126X; by the grant “EVA 4.0,” project no. CZ.02.1.01/0.0/0.0/16_019/0000803, financed by the OP RDE; and by Faculty of Tropical AgriSciences project IGA 20223112.
Footnotes
Supplemental material is available online only.
Contributor Information
Jan Šobotník, Email: sobotnik@ftz.czu.cz.
Isaac Cann, University of Illinois Urbana-Champaign.
REFERENCES
- 1.Brune A. 2014. Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol 12:168–180. doi: 10.1038/nrmicro3182. [DOI] [PubMed] [Google Scholar]
- 2.Bar-On YM, Phillips R, Milo R. 2018. The biomass distribution on Earth. Proc Natl Acad Sci USA 115:6506–6511. doi: 10.1073/pnas.1711842115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffiths HM, Ashton LA, Evans TA, Parr CL, Eggleton P. 2019. Termites can decompose more than half of deadwood in tropical rainforest. Curr Biol 29:R118–R119. doi: 10.1016/j.cub.2019.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Inward DJ, Vogler AP, Eggleton P. 2007. A comprehensive phylogenetic analysis of termites (Isoptera) illuminates key aspects of their evolutionary biology. Mol Phylogenet Evol 44:953–967. doi: 10.1016/j.ympev.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 5.Chouvenc T, Šobotník J, Engel MS, Bourguignon T. 2021. Termite evolution: mutualistic associations, key innovations, and the rise of Termitidae. Cell Mol Life Sci 78:2749–2769. doi: 10.1007/s00018-020-03728-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourguignon T, Drouet T, Šobotník J, Hanus R, Roisin Y. 2015. Influence of soil properties on soldierless termite distribution. PLoS One 10:e0135341. doi: 10.1371/journal.pone.0135341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourguignon T, Šobotník J, Dahlsjö CA, Roisin Y. 2016. The soldierless Apicotermitinae: insights into a poorly known and ecologically dominant tropical taxon. Insect Soc 63:39–50. doi: 10.1007/s00040-015-0446-y. [DOI] [Google Scholar]
- 8.Bourguignon T, Lo N, Šobotník J, Ho SY, Iqbal N, Coissac E, Lee M, Jendryka MM, Sillam-Dusses D, Křížková B. 2017. Mitochondrial phylogenomics resolves the global spread of higher termites, ecosystem engineers of the tropics. Mol Biol Evol 34:589–597. doi: 10.1093/molbev/msw253. [DOI] [PubMed] [Google Scholar]
- 9.Bucek A, Šobotník J, He S, Shi M, McMahon DP, Holmes EC, Roisin Y, Lo N, Bourguignon T. 2019. Evolution of termite symbiosis informed by transcriptome-based phylogenies. Curr Biol 29:3728–3734.e4. doi: 10.1016/j.cub.2019.08.076. [DOI] [PubMed] [Google Scholar]
- 10.Keeling CI, Bohlmann J. 2006. Diterpene resin acids in conifers. Phytochemistry 67:2415–2423. doi: 10.1016/j.phytochem.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 11.Raffa KF, Aukema BH, Erbilgin N, Klepzig KD, Wallin KF. 2005. Interactions among conifer terpenoids and bark beetles across multiple levels of scale: an attempt to understand links between population patterns and physiological processes. Recent Adv Phytochem 39:79–118. doi: 10.1016/S0079-9920(05)80005-X. [DOI] [Google Scholar]
- 12.Werner RA. 1995. Toxicity and repellency of 4-allylanisole and monoterpenes from white spruce and tamarack to the spruce beetle and eastern larch beetle (Coleoptera: Scolytidae). Environ Entomol 24:372–379. doi: 10.1093/ee/24.2.372. [DOI] [Google Scholar]
- 13.Raffa KF, Smalley EB. 1995. Interaction of pre-attack and induced monoterpene concentrations in host conifer defense against bark beetle-fungal complexes. Oecologia 102:285–295. doi: 10.1007/BF00329795. [DOI] [PubMed] [Google Scholar]
- 14.Hammer TJ, Bowers MD. 2015. Gut microbes may facilitate insect herbivory of chemically defended plants. Oecologia 179:1–14. doi: 10.1007/s00442-015-3327-1. [DOI] [PubMed] [Google Scholar]
- 15.Ni J, Tokuda G. 2013. Lignocellulose-degrading enzymes from termites and their symbiotic microbiota. Biotechnol Adv 31:838–850. doi: 10.1016/j.biotechadv.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Douglas AE. 2009. The microbial dimension in insect nutritional ecology. Funct Ecol 23:38–47. doi: 10.1111/j.1365-2435.2008.01442.x. [DOI] [Google Scholar]
- 17.Kirk TK, Cowling EB. 1984. Biological decomposition of solid wood, p 455–487. In Rogers R (ed), Advances in chemistry vol 207. ACS Publications, Washington, DC. [Google Scholar]
- 18.Varm A, Kolli BK, Paul J, Saxena S, König H. 1994. Lignocellulose degradation by microorganisms from termite hills and termite guts: a survey on the present state of art. FEMS Microbiol Rev 15:9–28. doi: 10.1111/j.1574-6976.1994.tb00120.x. [DOI] [Google Scholar]
- 19.Hyodo F, Tayasu I, Inoue T, Azuma JI, Kudo T, Abe T. 2003. Differential role of symbiotic fungi in lignin degradation and food provision for fungus-growing termites (Macrotermitinae: Isoptera). Funct Ecol 17:186–193. doi: 10.1046/j.1365-2435.2003.00718.x. [DOI] [Google Scholar]
- 20.Chung SH, Rosa C, Scully ED, Peiffer M, Tooker JF, Hoover K, Luthe DS, Felton GW. 2013. Herbivore exploits orally secreted bacteria to suppress plant defenses. Proc Natl Acad Sci USA 110:15728–15733. doi: 10.1073/pnas.1308867110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ceja-Navarro JA, Vega FE, Karaoz U, Hao Z, Jenkins S, Lim HC, Kosina P, Infante F, Northen TR, Brodie EL. 2015. Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat Commun 6:7618. doi: 10.1038/ncomms8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammerbacher A, Schmidt A, Wadke N, Wright LP, Schneider B, Bohlmann J, Brand WA, Fenning TM, Gershenzon J, Paetz C. 2013. A common fungal associate of the spruce bark beetle metabolizes the stilbene defenses of Norway spruce. Plant Physiol 162:1324–1336. doi: 10.1104/pp.113.218610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welte CU, de Graaf RM, van den Bosch TJ, Op den Camp HJ, van Dam NM, Jetten MS. 2016. Plasmids from the gut microbiome of cabbage root fly larvae encode SaxA that catalyses the conversion of the plant toxin 2-phenylethyl isothiocyanate. Environ Microbiol 18:1379–1390. doi: 10.1111/1462-2920.12997. [DOI] [PubMed] [Google Scholar]
- 24.Gurung K, Wertheim B, Falcao Salles J. 2019. The microbiome of pest insects: it is not just bacteria. Entomol Exp Appl 167:156–170. doi: 10.1111/eea.12768. [DOI] [Google Scholar]
- 25.Six DL. 2012. Ecological and evolutionary determinants of bark beetle: fungus symbioses. Insects 3:339–366. doi: 10.3390/insects3010339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raffa KF. 2014. Terpenes tell different tales at different scales: glimpses into the chemical ecology of conifer-bark beetle-microbial interactions. J Chem Ecol 40:1–20. doi: 10.1007/s10886-013-0368-y. [DOI] [PubMed] [Google Scholar]
- 27.Douglas AE. 2015. Multiorganismal insects: diversity and function of resident microorganisms. Annu Rev Entomol 60:17–34. doi: 10.1146/annurev-ento-010814-020822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kane MD, Breznak JA. 1991. Effect of host diet on production of organic acids and methane by cockroach gut bacteria. Appl Environ Microbiol 57:2628–2634. doi: 10.1128/aem.57.9.2628-2634.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tinker KA, Ottesen EA. 2016. The core gut microbiome of the American cockroach, Periplaneta americana, is stable and resilient to dietary shifts. Appl Environ Microbiol 82:6603–6610. doi: 10.1128/AEM.01837-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brune A, Dietrich C. 2015. The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annu Rev Microbiol 69:145–166. doi: 10.1146/annurev-micro-092412-155715. [DOI] [PubMed] [Google Scholar]
- 31.Brune A, Ohkuma M. 2010. Role of the termite gut microbiota in symbiotic digestion, p 439–475. In Bignell D, Roisin Y, Lo N (ed), Biology of termites: a modern synthesis. Springer, Dordrecht, The Netherlands. [Google Scholar]
- 32.Dietrich C, Köhler T, Brune A. 2014. The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Appl Environ Microbiol 80:2261–2269. doi: 10.1128/AEM.04206-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikaelyan A, Dietrich C, Köhler T, Poulsen M, Sillam-Dussès D, Brune A. 2015. Diet is the primary determinant of bacterial community structure in the guts of higher termites. Mol Ecol 24:5284–5295. doi: 10.1111/mec.13376. [DOI] [PubMed] [Google Scholar]
- 34.Arora J, Kinjo Y, Šobotník J, Buček A, Clitheroe C, Stiblik P, Roisin Y, Žifčáková L, Park YC, Kim KY, Sillam-Dussès D, Hervé V, Lo N, Tokuda G, Brune A, Bourguignon T. 2022. The functional evolution of termite gut microbiota. Microbiome 10:78. doi: 10.1186/s40168-022-01258-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boone CK, Keefover-Ring K, Mapes AC, Adams AS, Bohlmann J, Raffa KF. 2013. Bacteria associated with a tree-killing insect reduce concentrations of plant defense compounds. J Chem Ecol 39:1003–1006. doi: 10.1007/s10886-013-0313-0. [DOI] [PubMed] [Google Scholar]
- 36.Skrodenytė-Arbačiauskienė V, Radžiutė S, Stunžėnas V, Būda V. 2012. Erwinia typographi sp. nov., isolated from bark beetle (Ips typographus) gut. Int J Syst Evol Microbiol 62:942–948. doi: 10.1099/ijs.0.030304-0. [DOI] [PubMed] [Google Scholar]
- 37.Dowd PF, Shen SK. 1990. The contribution of symbiotic yeast to toxin resistance of the cigarette beetle (Lasioderma serricorne). Entomol Exp Appl 56:241–248. doi: 10.1111/j.1570-7458.1990.tb01402.x. [DOI] [Google Scholar]
- 38.Tsui CK-M, Farfan L, Roe AD, Rice AV, Cooke JE, El-Kassaby YA, Hamelin RC. 2014. Population structure of mountain pine beetle symbiont Leptographium longiclavatum and the implication on the multipartite beetle-fungi relationships. PLoS One 9:e105455. doi: 10.1371/journal.pone.0105455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao T, Kandasamy D, Krokene P, Chen J, Gershenzon J, Hammerbacher A. 2019. Fungal associates of the tree-killing bark beetle, Ips typographus, vary in virulence, ability to degrade conifer phenolics and influence bark beetle tunneling behavior. Fungal Ecol 38:71–79. doi: 10.1016/j.funeco.2018.06.003. [DOI] [Google Scholar]
- 40.Bartnik C, Nawrot-Chorabik K, Woodward S. 2020. Phenolic compound concentrations in Picea abies wood as an indicator of susceptibility towards root pathogens. For Path 50:e12652. doi: 10.1111/efp.12652. [DOI] [Google Scholar]
- 41.Germida JJ. 1988. Growth of indigenous Rhizobium leguminosarum and Rhizobium meliloti in soils amended with organic nutrients. Appl Environ Microbiol 54:257–263. doi: 10.1128/aem.54.1.257-263.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balkwill D, Fredrickson J, Romine M. 2006. Sphingomonas and related genera, p 605–629. In Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (ed), The prokaryotes: a handbook on the biology of bacteria. Springer, New York, NY. [Google Scholar]
- 43.Chen W-M, Moulin L, Bontemps C, Vandamme P, Béna G, Boivin-Masson C. 2003. Legume symbiotic nitrogen fixation by β-proteobacteria is widespread in nature. J Bacteriol 185:7266–7272. doi: 10.1128/JB.185.24.7266-7272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu LT, Lu M, Sun JH. 2016. Invasive bark beetle-associated microbes degrade a host defensive monoterpene. Insect Sci 23:183–190. doi: 10.1111/1744-7917.12255. [DOI] [PubMed] [Google Scholar]
- 45.Calusinska M, Marynowska M, Bertucci M, Untereiner B, Klimek D, Goux X, Sillam-Dussès D, Gawron P, Halder R, Wilmes P, Ferrer P, Gerin P, Roisin Y, Delfosse P. 2020. Integrative omics analysis of the termite gut system adaptation to Miscanthus diet identifies lignocellulose degradation enzymes. Commun Biol 3:275. doi: 10.1038/s42003-020-1004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su L, Yang L, Huang S, Su X, Li Y, Wang F, Wang E, Kang N, Xu J, Song A. 2016. Comparative gut microbiomes of four species representing the higher and the lower termites. J Insect Sci 16:97. doi: 10.1093/jisesa/iew081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lilburn T, Kim K, Ostrom N, Byzek K, Leadbetter J, Breznak J. 2001. Nitrogen fixation by symbiotic and free-living spirochetes. Science 292:2495–2498. doi: 10.1126/science.1060281. [DOI] [PubMed] [Google Scholar]
- 48.Leadbetter JR, Schmidt TM, Graber JR, Breznak JA. 1999. Acetogenesis from H2 plus CO2 by spirochetes from termite guts. Science 283:686–689. doi: 10.1126/science.283.5402.686. [DOI] [PubMed] [Google Scholar]
- 49.Tokuda G, Mikaelyan A, Fukui C, Matsuura Y, Watanabe H, Fujishima M, Brune A. 2018. Fiber-associated spirochetes are major agents of hemicellulose degradation in the hindgut of wood-feeding higher termites. Proc Natl Acad Sci USA 115:E11996–E12004. doi: 10.1073/pnas.1810550115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breznak JA, Pankratz HS. 1977. In situ morphology of the gut microbiota of wood-eating termites [Reticulitermes flavipes (Kollar) and Coptotermes formosanus Shiraki]. Appl Environ Microbiol 33:406–426. doi: 10.1128/aem.33.2.406-426.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dröge S, Rachel R, Radek R, König H. 2008. Treponema isoptericolens sp. nov., a novel spirochaete from the hindgut of the termite Incisitermes tabogae. Int J Syst Evol Microbiol 58:1079–1083. doi: 10.1099/ijs.0.64699-0. [DOI] [PubMed] [Google Scholar]
- 52.Sethi A, Kovaleva ES, Slack JM, Brown S, Buchman GW, Scharf ME. 2013. A GHF7 cellulase from the protist symbiont community of Reticulitermes flavipes enables more efficient lignocellulose processing by host enzymes. Arch Insect Biochem Physiol 84:175–193. doi: 10.1002/arch.21135. [DOI] [PubMed] [Google Scholar]
- 53.Desai MS, Strassert JF, Meuser K, Hertel H, Ikeda-Ohtsubo W, Radek R, Brune A. 2010. Strict cospeciation of devescovinid flagellates and Bacteroidales ectosymbionts in the gut of dry-wood termites (Kalotermitidae). Environ Microbiol 12:2120–2132. doi: 10.1111/j.1462-2920.2009.02080.x. [DOI] [PubMed] [Google Scholar]
- 54.Noda S, Hongoh Y, Sato T, Ohkuma M. 2009. Complex coevolutionary history of symbiotic Bacteroidales bacteria of various protists in the gut of termites. BMC Evol Biol 9:158. doi: 10.1186/1471-2148-9-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hongoh Y, Sharma VK, Prakash T, Noda S, Taylor TD, Kudo T, Sakaki Y, Toyoda A, Hattori M, Ohkuma M. 2008. Complete genome of the uncultured Termite Group 1 bacteria in a single host protist cell. Proc Natl Acad Sci USA 105:5555–5560. doi: 10.1073/pnas.0801389105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brune A. 2012. Endomicrobia: intracellular symbionts of termite gut flagellates. Endocytobiosis Cell Res 23:11–15. https://zs.thulb.uni-jena.de/receive/jportal_jparticle_00270371. [Google Scholar]
- 57.Stingl U, Radek R, Yang H, Brune A. 2005. “Endomicrobia”: cytoplasmic symbionts of termite gut protozoa form a separate phylum of prokaryotes. Appl Environ Microbiol 71:1473–1479. doi: 10.1128/AEM.71.3.1473-1479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng H, Dietrich C, Thompson CL, Meuser K, Brune A. 2015. Population structure of Endomicrobia in single host cells of termite gut flagellates (Trichonympha spp.). Microbes Environ 30:92–98. doi: 10.1264/jsme2.ME14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hongoh Y, Sharma VK, Prakash T, Noda S, Toh H, Taylor TD, Kudo T, Sakaki Y, Toyoda A, Hattori M, Ohkuma M. 2008. Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science 322:1108–1109. doi: 10.1126/science.1165578. [DOI] [PubMed] [Google Scholar]
- 60.Izawa K, Kuwahara H, Kihara K, Yuki M, Lo N, Itoh T, Ohkuma M, Hongoh Y. 2016. Comparison of intracellular “Ca. Endomicrobium trichonymphae” genomovars illuminates the requirement and decay of defense systems against foreign DNA. Genome Biol Evol 8:3099–3107. doi: 10.1093/gbe/evw227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inoue J-I, Saita K, Kudo T, Ui S, Ohkuma M. 2007. Hydrogen production by termite gut protists: characterization of iron hydrogenases of parabasalian symbionts of the termite Coptotermes formosanus. Eukaryot Cell 6:1925–1932. doi: 10.1128/EC.00251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuki M, Kuwahara H, Shintani M, Izawa K, Sato T, Starns D, Hongoh Y, Ohkuma M. 2015. Dominant ectosymbiotic bacteria of cellulolytic protists in the termite gut also have the potential to digest lignocellulose. Environ Microbiol 17:4942–4953. doi: 10.1111/1462-2920.12945. [DOI] [PubMed] [Google Scholar]
- 63.Desai MS, Brune A. 2012. Bacteroidales ectosymbionts of gut flagellates shape the nitrogen-fixing community in dry-wood termites. ISME J 6:1302–1313. doi: 10.1038/ismej.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prillinger H, Messner R, König H, Bauer R, Lopandic K, Molnar O, Dangel P, Weigang F, Kirisits T, Nakase T, Sigler L. 1996. Yeasts associated with termites: a phenotypic and genotypic characterization and use of coevolution for dating evolutionary radiations in asco- and basidiomycetes. Syst Appl Microbiol 19:265–283. doi: 10.1016/S0723-2020(96)80053-1. [DOI] [Google Scholar]
- 65.Prillinger H, König H. 2006. The intestinal yeasts, p 319–334. In Prillinger H, König H (ed), Intestinal microorganisms of termites and other invertebrates. Springer-Verlag Berlin, Berlin, Germany. [Google Scholar]
- 66.Rajgopal S, Rajyalakshmi Rao D, Varma A. 1979. Association of fungi in termite gut. Curr Sci 48:998–999. https://www.jstor.org/stable/24082324. [Google Scholar]
- 67.Rajgopal S, Rao DR, Varma A. 1981. Fungi of the worker termite-gut, Odontotermes obesus (Rambur) from northern India. Nova Hedwigia 34:97–100. [Google Scholar]
- 68.Rojas-Jiménez K, Hernández M. 2015. Isolation of fungi and bacteria associated with the guts of tropical wood-feeding coleoptera and determination of their lignocellulolytic activities. Int J Microbiol 2015:285018. doi: 10.1155/2015/285018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ziganshina EE, Mohammed WS, Shagimardanova EI, Vankov PY, Gogoleva NE, Ziganshin AM. 2018. Fungal, bacterial, and archaeal diversity in the digestive tract of several beetle larvae (Coleoptera). Biomed Res Int 2018:6765438. doi: 10.1155/2018/6765438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salehzadeh A, Tavacol P, Mahjub H. 2007. Bacterial, fungal and parasitic contamination of cockroaches in public hospitals of Hamadan, Iran. J Vector Borne Dis 44:105–110. [PubMed] [Google Scholar]
- 71.Zhang N, Suh S-O, Blackwell M. 2003. Microorganisms in the gut of beetles: evidence from molecular cloning. J Invertebr Pathol 84:226–233. doi: 10.1016/j.jip.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 72.Blackwell M. 2017. Yeasts in insects and other invertebrates, p 397–433. In Buzzini P, Lachance M-A, Yurkov A (ed), Yeasts in natural ecosystems: diversity. Springer Cham, Cham, Switzerland. [Google Scholar]
- 73.Stefanini I. 2018. Yeast-insect associations: it takes guts. Yeast 35:315–330. doi: 10.1002/yea.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schäfer A, Konrad R, Kuhnigk T, Kämpfer P, Hertel H, König H. 1996. Hemicellulose-degrading bacteria and yeasts from the termite gut. J Appl Bacteriol 80:471–478. doi: 10.1111/j.1365-2672.1996.tb03245.x. [DOI] [PubMed] [Google Scholar]
- 75.Davis TS, Hofstetter RW, Foster JT, Foote NE, Keim P. 2011. Interactions between the yeast Ogataea pini and filamentous fungi associated with the western pine beetle. Microb Ecol 61:626–634. doi: 10.1007/s00248-010-9773-8. [DOI] [PubMed] [Google Scholar]
- 76.de França Passos D, Pereira N Jr, de Castro AM. 2018. A comparative review of recent advances in cellulases production by Aspergillus, Penicillium and Trichoderma strains and their use for lignocellulose deconstruction. Curr Opin Green Sustain Chem 14:60–66. doi: 10.1016/j.cogsc.2018.06.003. [DOI] [Google Scholar]
- 77.De Beer ZW, Marincowitz S, Duong TA, Kim J-J, Rodrigues A, Wingfield MJ. 2016. Hawksworthiomyces gen. nov.(Ophiostomatales), illustrates the urgency for a decision on how to name novel taxa known only from environmental nucleic acid sequences (ENAS). Fungal Biol 120:1323–1340. doi: 10.1016/j.funbio.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 78.Eriksson K-EL, Blanchette RA, Ander P. 2012. Microbial and enzymatic degradation of wood and wood components. Springer Science & Business Media, Berlin, Germany. [Google Scholar]
- 79.Félix C, Libório S, Nunes M, Félix R, Duarte AS, Alves A, Esteves AC. 2018. Lasiodiplodia theobromae as a producer of biotechnologically relevant enzymes. Int J Mol Sci 19:29. doi: 10.3390/ijms19020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scully ED, Geib SM, Carlson JE, Tien M, McKenna D, Hoover K. 2014. Functional genomics and microbiome profiling of the Asian longhorned beetle (Anoplophora glabripennis) reveal insights into the digestive physiology and nutritional ecology of wood feeding beetles. BMC Genomics 15:1096. doi: 10.1186/1471-2164-15-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bulgarelli D, Schlaeppi K, Spaepen S, Van Themaat EVL, Schulze-Lefert P. 2013. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64:807–838. doi: 10.1146/annurev-arplant-050312-120106. [DOI] [PubMed] [Google Scholar]
- 82.Molnar O, Schatzmayr G, Fuchs E, Prillinger H. 2004. Trichosporon mycotoxinivorans sp. nov., a new yeast species useful in biological detoxification of various mycotoxins. Syst Appl Microbiol 27:661–671. doi: 10.1078/0723202042369947. [DOI] [PubMed] [Google Scholar]
- 83.Chakraborty A, Modlinger R, Ashraf MZ, Synek J, Schlyter F, Roy A. 2020. Core mycobiome and their ecological relevance in the gut of five Ips bark beetles (Coleoptera: Curculionidae: Scolytinae). Front Microbiol 11:568853. doi: 10.3389/fmicb.2020.568853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chakraborty A, Ashraf MZ, Modlinger R, Synek J, Schlyter F, Roy A. 2020. Unravelling the gut bacteriome of Ips (Coleoptera: Curculionidae: Scolytinae): identifying core bacterial assemblage and their ecological relevance. Sci Rep 10:18572. doi: 10.1038/s41598-020-75203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haverty MI. 1977. The proportion of soldiers in termite colonies: a list and a bibliography. Sociobiology 2:199–216. [Google Scholar]
- 86.Štěpán R, Hajšlová J, Kocourek V, Tichá J. 2004. Uncertainties of gas chromatographic measurement of troublesome pesticide residues in apples employing conventional and mass spectrometric detectors. Anal Chim Acta 520:245–255. doi: 10.1016/j.aca.2004.05.045. [DOI] [Google Scholar]
- 87.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA 108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ihrmark K, Bödeker ITM, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, Strid Y, Stenlid J, Brandström-Durling M, Clemmensen KE, Lindahl BD. 2012. New primers to amplify the fungal ITS2 region: evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol 82:666–677. doi: 10.1111/j.1574-6941.2012.01437.x. [DOI] [PubMed] [Google Scholar]
- 89.Tedersoo L, Anslan S, Bahram M, Põlme S, Riit T, Liiv I, Kõljalg U, Kisand V, Nilsson H, Hildebrand F, Bork P, Abarenkov K. 2015. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 10:1–43. doi: 10.3897/mycokeys.10.4852. [DOI] [Google Scholar]
- 90.Aronesty E. 2011. ea-utils: Command-line tools for processing biological sequencing data. Available from https://github.com/ExpressionAnalysis/ea-utils. [Google Scholar]
- 91.Vetrovský T, Baldrian P, Morais D. 2018. SEED 2: a user-friendly platform for amplicon high-throughput sequencing data analyses. Bioinformatics 34:2292–2294. doi: 10.1093/bioinformatics/bty071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, De Wit P, Sánchez-García M, Ebersberger I, de Sousa F. 2013. Improved software detection and extraction of ITS1 and ITS 2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol 4:914–919. doi: 10.1111/2041-210X.12073. [DOI] [Google Scholar]
- 93.Edgar R. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 94.Thomas D, Vandegrift R, Bailes G, Roy B. 2017. Understanding and mitigating some limitations of Illumina MiSeq for environmental sequencing of fungi. bioRxiv. doi: 10.1101/184960. [DOI]
- 95.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. 2014. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stoddard SF, Smith BJ, Hein R, Roller BR, Schmidt TM. 2015. rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res 43:D593–D598. doi: 10.1093/nar/gku1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Větrovský T, Baldrian P. 2013. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Dueñas M, Grebenc T, Griffith GW, Hartmann M, Kirk PM, Kohout P, Larsson E, Lindahl BD, Lücking R, Martín MP, Matheny PB, Nguyen NH, Niskanen T, Oja J, Peay KG, Peintner U, Peterson M, Põldmaa K, Saag L, Saar I, Schüßler A, Scott JA, Senés C, Smith ME, Suija A, Taylor DL, Telleria MT, Weiss M, Larsson K-H. 2013. Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277. doi: 10.1111/mec.12481. [DOI] [PubMed] [Google Scholar]
- 99.Magurran AE. 1988. Ecological diversity and its measurement. Princeton University Press, Princeton, NJ. [Google Scholar]
- 100.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 101.Oksanen J. 2011. vegan: Community ecology package. R package version 1.17–9. Available from http://cran.r-project.org/package=vegan.
- 102.Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- 103.Oksanen J, Blanchet F, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin P, O’Hara R, Simpson G, Solymos P. 2018. vegan: Community ecology package. R package version 2.5–2. https://cran.r-project.org/web/packages/vegan/index.html.
- 104.Clarke KR, Gorley RN, Somerfield PJ, Warwick RM. 2014. Change in marine communities: an approach to statistical analysis and interpretation. https://updates.primer-e.com/primer7/manuals/Methods_manual_v7.pdf.
- 105.Martinez Arbizu P. 2017. pairwiseAdonis: Pairwise multilevel comparison using adonis. R package version 00 1. https://github.com/pmartinezarbizu/pairwiseAdonis.
- 106.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 107.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Halwachs B, Madhusudhan N, Krause R, Nilsson RH, Moissl-Eichinger C, Högenauer C, Thallinger GG, Gorkiewicz G. 2017. Critical issues in mycobiota analysis. Front Microbiol 8:180. doi: 10.3389/fmicb.2017.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aem.00361-23-s0001.pdf, PDF file, 1.1 MB (1.1MB, pdf)
Supplemental material. Download aem.00361-23-s0002.xlsx, XLSX file, 2.9 MB (2.9MB, xlsx)
Supplemental material. Download aem.00361-23-s0003.docx, DOCX file, 0.02 MB (19.5KB, docx)
Data Availability Statement
The sequence data of the bacterial V4 hypervariable region of the 16S rRNA gene and the fungal ITS2 gene amplified from two different wood-feeding termites (R. flavipes and M. biroi), infested spruce wood, and their controls have been deposited in NCBI (SRA database) under BioProject accession no. PRJNA792414.