

ABSTRACT
Marine herbivorous fish that feed primarily on macroalgae, such as those from the genus Kyphosus, are essential for maintaining coral health and abundance on tropical reefs. Here, deep metagenomic sequencing and assembly of gut compartment-specific samples from three sympatric, macroalgivorous Hawaiian kyphosid species have been used to connect host gut microbial taxa with predicted protein functional capacities likely to contribute to efficient macroalgal digestion. Bacterial community compositions, algal dietary sources, and predicted enzyme functionalities were analyzed in parallel for 16 metagenomes spanning the mid- and hindgut digestive regions of wild-caught fishes. Gene colocalization patterns of expanded carbohydrate (CAZy) and sulfatase (SulfAtlas) digestive enzyme families on assembled contigs were used to identify likely polysaccharide utilization locus associations and to visualize potential cooperative networks of extracellularly exported proteins targeting complex sulfated polysaccharides. These insights into the gut microbiota of herbivorous marine fish and their functional capabilities improve our understanding of the enzymes and microorganisms involved in digesting complex macroalgal sulfated polysaccharides.
IMPORTANCE This work connects specific uncultured bacterial taxa with distinct polysaccharide digestion capabilities lacking in their marine vertebrate hosts, providing fresh insights into poorly understood processes for deconstructing complex sulfated polysaccharides and potential evolutionary mechanisms for microbial acquisition of expanded macroalgal utilization gene functions. Several thousand new marine-specific candidate enzyme sequences for polysaccharide utilization have been identified. These data provide foundational resources for future investigations into suppression of coral reef macroalgal overgrowth, fish host physiology, the use of macroalgal feedstocks in terrestrial and aquaculture animal feeds, and the bioconversion of macroalgae biomass into value-added commercial fuel and chemical products.
KEYWORDS: fish gut microbiome, polysaccharide utilization, sulfatase, macroalgal digestion, kyphosid
INTRODUCTION
The feeding activity of herbivorous fish on tropical coral reefs profoundly affects coral abundance and stress recovery by suppressing competing macroalgae (1–3). Reef fish that consume macroalgae are often modeled by species from the genus Kyphosus, which have elongated digestive tracts divided into multiple sections harboring distinct microbial taxa (4–6). Like other marine herbivores, distal hindgut regions of these fish contain abundant fermentative bacteria capable of digesting complex carbohydrates (7–12). Although dietary preferences are known to vary among kyphosid species and ontogenic stages of individual animals, macroalgae are believed to be the primary adult food source, based on visual observation of stomach contents (13, 14). However, the full range of potential dietary diversity among individual animals over time has not yet been established using molecular methods.
The red and brown macroalgae consumed by kyphosid fish contain large quantities of highly sulfated polysaccharides, including structural carrageenans, porphyrans, and fucoidans (reviewed in references 15 and 16). As a result, free sulfate concentrations in fish hindgut environments can reach or exceed levels of 100 mM (17). Bacterial isolates capable of deconstructing sulfated macroalgal polysaccharides have been cultured from epiphytic adherents to algal surfaces (18–24), detritovores in seawater and marine sediments (25–28), and host-associated gut bacteria from sea urchins (29), sea cucumbers (30), marine snails (31), and surgeonfishes (32), as well as macroalga-consuming humans (33). However, only one cultured isolate, from the Bacillota family Erysipelotrichaceae, is currently available from the digestive systems of macroalgivorous fish (34).
16S rRNA-based molecular methods have identified a number of bacterial species in hindgut compartments of kyphosid fish (35, 36) that are taxonomically related to those present in plant-digesting terrestrial herbivores, including Alistipes-related Bacteroidota, Clostridia-related Bacillota, and Desulfovibrio-related Deltaproteobacteria. Although numerous cultured isolates, genomes, and metagenome-assembled genomes (MAGs) have been obtained for these taxa in terrestrial animals (37, 38), few laboratory cultures, complete genomes, or high-quality MAGs are currently available for their relatives within herbivorous fish gut microbiota (34, 36). As a result, strain-specific functional adaptations that might account for their success in marine hosts with macroalgal diets have not yet been established.
The diverse galactose, mannose, xylose, fucose, rhamnose, arabinose, and uronic acid subunits of macroalgal polysaccharides occur in a wide variety of different ratios, sulfation states, and branching patterns (39–41). Processing these complex carbohydrates into simpler sugars for incorporation into central metabolic pathways requires sulfate removal either before, concurrently with, or after glycosidic bond cleavage (31, 42, 43), but determining precise hydrolysis mechanisms can be difficult for substrates requiring coordinated action by multiple different enzymes. Enzyme characterization ambiguities can also arise in cases of natural substrate promiscuity, potentially confounding experimental determinations based on simplified oligosaccharide model compounds instead of full-length polymers (44). Additional challenges arise in trying to predict functional specificities from genomic, metagenomic, and/or transcriptomic sequence data for protein families with few experimentally characterized examples.
Bacterial genes encoding cooperative functional activities are often located in close genomic proximity to each other, providing opportunities for coordinated regulation. Well-studied glycohydrolase gene clusters known as polysaccharide utilization loci (PUL) and carbohydrate-active enzyme gene clusters (CGCs), may also include sulfatases, sugar binding proteins, transporters, and regulatory elements (45, 46). PUL and CGCs can be characterized bioinformatically by sequence comparison to the CAZy database of carbohydrate-active enzymes (47), and the associated PUL database (48), using either bacterial isolate genomes or culture-independent metagenomes (23). A large database of sulfatase sequences is also available (49), but its protein family classifications are based on broad evolutionary relatedness, rather than experimentally determined substrate specificities.
The breakdown of naturally occurring sulfated polysaccharides via exported extracellular enzymes is known to occur in cultured bacteria isolated from macroalgal surfaces (50), but the extent to which these observations can be applied to the microbiomes of macroalga-eating vertebrates is unknown. A better understanding of enzyme candidate diversity, species of origin, and subcellular localization can lead to practical applications in aquaculture and animal husbandry: for example, by enhancing digestibility of macroalgal feed sources through probiotic supplements (51–56) and/or reducing ruminant methane contributions to global warming by adding small amounts of seaweed to their diets (57). This information would also be useful in identifying key organisms and enzyme variants for biotechnological processing of marine algal feedstocks for the production of value-added products such as biofuels (58), as well as pharmaceutical production of biologically active glycan compounds such as fucoidans, with medicinally valuable anti-inflammatory, anticancer, antiviral, antioxidation, anticoagulant, antithrombotic, and antiangiogenic effects (43, 59).
The current study uses metagenomic analysis to survey microbially encoded proteins related to macroalgal digestion in the gut microbiota of three sympatric Hawaiian reef fishes, Kyphosus vaigiensis, Kyphosus cinerascens, and Kyphosus hawaiiensis (60, 61). Networks of expanded CAZyme and sulfatase protein families occurring in close genomic proximity have been used to obtain insights into the diversity, taxonomic distribution, and operon context of microbial enzymes enabling these fish to digest a broad variety of diverse macroalgal polysaccharides.
RESULTS
Taxonomic composition of metagenomic samples.
The 16 fish gut metagenomic samples assembled using metaSPAdes yielded a total of 1.478 Tbp of assembled nucleotides in contigs of >2 kb, encoding 1,432,202 predicted proteins (see Table S1B in the supplemental material). Microbial community compositions of these samples were assessed using unassembled reads with Kraken2, enabling relative abundance estimates for eukaryotic, archaeal, and viral taxa as well as bacteria.
High-level taxonomic classifications for individual samples (Fig. 1) were generally consistent with 16S rRNA gene abundances reported from the same kyphosid fish samples (35), although some additional variability was observed between individual fishes that had previously been masked by averaging. Bacteroidota and Bacillota were highly abundant in all hindgut samples, with Alpha-, Beta-, and Deltaproteobacteria, Spirochaetota, Verrucomicrobiota, and Archaea distributed at lower abundances across all gut regions. Interfish differences were observed for Gammaproteobacteria, dominating midgut samples from K. vaigiensis and K. hawaiiensis but not K. cinerascens, and Eukaryota, present at much higher abundances in fish 8 (juvenile K. vaigiensis).
FIG 1.

Relative abundance of fish gut-associated microbial taxa. Relative abundances are based on Kraken2 analysis of unassembled Illumina NovaSeq reads. Sample ID numbers indicate fish numbers (Table S1), followed by gut region code, in progressively distal order. Abbreviations: GI, midgut; HG, hindgut.
Taxonomic associations of fish gut-associated bacterial clades were further explored using predicted protein sequences of the single-copy DNA-directed RNA polymerase subunit beta (rpoB) gene on assembled contigs. An amino acid sequence tree (Fig. 2) shows the largest numbers of rpoB genes recovered were taxonomically classified as Bacillota (50), followed by Bacteroidota (39), Gammaproteobacteria (19), Spirochaetota (7), Deltaproteobacteria (6), Verrucomicrobiota (5), Alphaproteobacteria (4), and Melainabacteria (1). No rpoB sequences were detected for Archaea, consistent with the low coverage of this taxonomic group observed in unassembled reads.
FIG 2.
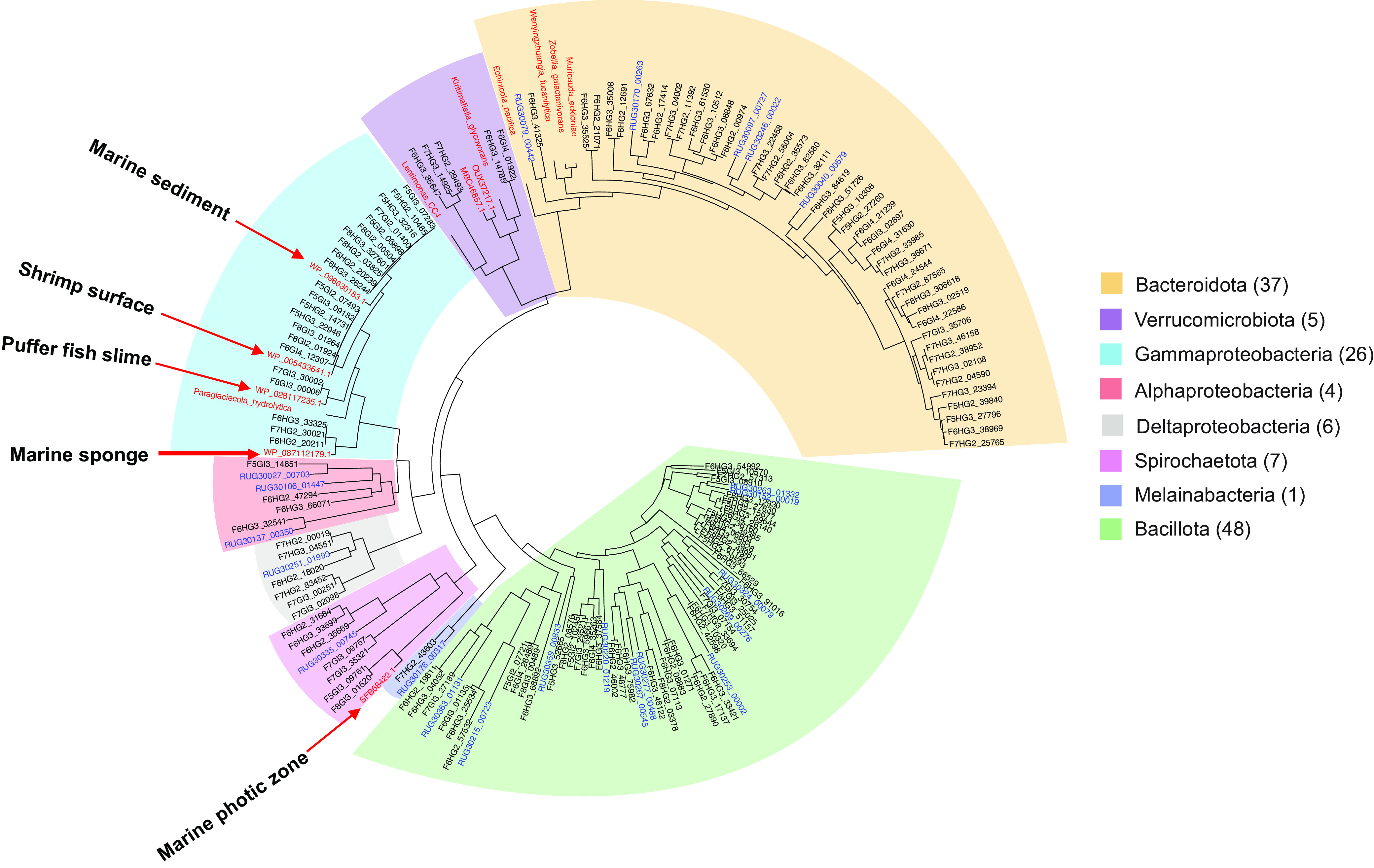
Single-copy rpoB gene bacterial taxonomy. Predicted protein sequences from this study are shown in black, identified by fish number, gut compartment, and Prokka annotation number. Sequences in red are from GenBank nr, with selected species known to digest sulfated macroalgal polysaccharides highlighted in boldface as outgroups. Blue text indicates predicted rpoB sequences from terrestrial ruminant MAGs (not currently included in GenBank as protein sequences) prefixed with GenBank genome identifier codes from reference 38, followed by Prokka annotation numbers from this study (e.g., RUGXXXXX). Additional information about these sequences is provided in Table S2.
The closest database relatives to kyphosid metagenome rpoB genes suggest that transient ingested environmental bacteria may be more abundant in midgut samples, with hindgut regions containing higher numbers of sequences more closely related to persistent taxa from gastrointestinal compartments in other animal hosts (Table S2). Predicted fish hindgut RpoB proteins from Bacteroidota, Bacillota, Deltaproteobacteria, and Alphaproteobacteria clades most closely matched sequences from terrestrial ruminant MAGs, rather than marine environmental or bacterial isolate genomes. In contrast, midgut-associated Gammaproteobacteria RpoB protein sequences most closely resembled marine environmental Vibrionaceae, including one shallow-branching clade matching Vibrio campbellii at 95.8% amino acid identity (WP_005433641.1). Surprisingly, no matches were found for previously described macroalga-degrading Gammaproteobacteria from the Alteromonadaceae and Flavobacteriaceae families, such as Paraglaciecola agarilytica (23) or Formosa agariphila (22).
Fish gut RpoB proteins from relatively novel taxa included Verrucomicrobiota sequences most closely related to marine bacteria of the Kiritimatiellaceae family (Kiritimatiella glycovorans [76% identity]) and alga-associated bacteria from genus Lentimonas (WP_162027363.1 [63% identity]), as well as a Spirochaetota clade distantly related to mammalian pathogen Brevinema andersonii (SFB68422.1 [84% identity]). Brevinema-related taxa have also been observed in 16S rRNA gene amplification studies of trout (62), tilapia (63), salmon (64), seahorses (65), and kyphosids (35), as well as environmental metagenomes from sulfidic artesian boreholes (66).
Macroalgal components of fish diets.
Eukaryotic components of fish gut samples were initially assessed based on the presence of 18S rRNA markers in assembled metagenomic contigs, but this method proved relatively insensitive, detecting only highly abundant taxa. 18S rRNA sequences assembled from metagenomic data were limited to those of kyphosid fish hosts, along with single-celled protozoa (Trichomonadea, Entamoeba, Giardia, and Plasmodium) and multicellular worm taxa (Enenterum, Acanthocephalus, Enoplus, and Opisthadena) often associated with fish pathology (Table S3).
Eukaryotic components of gut samples were subsequently analyzed using Kraken2 classifications of unassembled metagenomic reads. Despite relatively low levels of red, brown, and green algal sequences in total metagenomic DNA, the use of very-large-input data sets (millions of reads for most samples) enabled sensitive quantification of their relative abundances. This methodology was helpful in avoiding potential issues associated with variability in 18S rRNA gene copy numbers, incomplete metagenomic assembly, and potential degradative fragmentation of nucleotide sequences. Even though the reference sequence library used by Kraken2 does not include exact matches for every possible environmental species of interest, solid taxonomic assignments can be made at broader granularities based on closest available relatives (67).
A summary of phylum-level algal matches (Fig. 3) shows that metagenomic reads from red (Rhodophyta) and green (Chlorophyta) algal lineages were more abundant than those from brown algae (Phaeophyceae) in all cases, except the most proximal gut sample from fish 7 (F7GI2). Brown algal abundance declined sharply in more distal gut sections of this fish, consistent with consumption of a meal composed primarily of brown algae during a single, relatively brief time period. Finer-grained examinations revealed that algal sequences from the phylum Phaeophyceae in all fish samples were dominated by matches to the order Ectocarpales (>70% overall), with relatively few examples from Sphacelariales, Dictyotales, Fucales, and Laminariales (Table S4A).
FIG 3.

Relative abundance of dietary algal taxa. Relative abundances of algal taxa were calculated based Kraken2 analysis of the same sets of unassembled Illumina NovaSeq reads shown in Fig. 1.
Red algal matches encompassed a much broader range of taxonomic diversity, including not only macroalgae from the orders Ceramiales, Gracilariales, Gigartinales, and Bangiales, but also sequences most closely related to unicellular forms classified as Cyanidales and Porphyridiales (Table S4B). The most prominent green algal sequences matched unicellular, filamentous, and colonial classes Chlorophyceae, Trebouxiophyceae, and Mamiellophyceae, with much lower representation of parenchymatous lineages from class Ulvophyceae (Table S4C). These results are consistent with prior observations of both kyphosids and other marine reef fish consuming not only macroalgae, but also uncharacterized microalgal assemblages described as “turf algae” and “epilithic algal matrix” (13, 68, 69).
Macroalgal polysaccharide hydrolysis.
Functional annotations of predicted kyphosid fish gut microbiome proteins (see Data Set S1 in the supplemental material) were consistent with general metabolic characteristics of microbial taxa related to those observed in Fig. 1, as well as those recently described in Kyphosus sydneyanus (36, 70). Although relative abundances of predicted protein functions in metagenomic assemblies do not necessarily correspond with biological activity, expanded numbers of metagenomically predicted genes from specific protein families can indicate potential adaptive capacity.
High levels of structural microheterogeneity in naturally occurring marine macroalgal polysaccharides (15) might be expected to promote expansion and diversification of microbial enzyme families involved in their hydrolysis. To distinguish between marine macroalga-specific enzymes and more general carbohydrate digestion activities common to saccharolytic bacteria from nonalgivorous hosts with high-carbohydrate diets, predicted protein annotations for kyphosid fish gut metagenomes were compared to a set of 391 MAGs from terrestrial ruminant digestive systems (38). The most frequently encountered nonhypothetical gene function descriptions in adult kyphosid fish metagenomes were arylsulfatases (1.9% of all predicted proteins). In contrast, arylsulfatases comprised only 0.13% of annotated proteins in MAGs from terrestrial ruminants with no dietary exposure to macroalgal polysaccharides. (Data Set S1).
Although the annotations produced by Prokka are not comprehensive (for example, laminarinases, cellulases, and fucosidases were labeled with only broader, more generic keywords), these results did confirm enrichment of predicted proteins described as agarases, carrageenases, porphyranases, and arylsulfatases in the fish metagenomes (P < 0.05). Carrageenases, porphyranases, and agarases were observed at consistently higher frequencies in hindgut versus midgut samples, compared to more variable compartmental distributions for alginate lyases and arylsulfatases (see Fig. S1 in the supplemental material).
Candidate enzymes for macroalgal digestion were subsequently characterized in more detail by classification according to the CAZy reference database (Fig. 4; Data Set S2). Quantitative family abundances, normalized for total numbers of predicted proteins from each gut compartment and species sample, were used to identify significant protein family expansions and diversifications in the context of total available protein repertoire.
FIG 4.
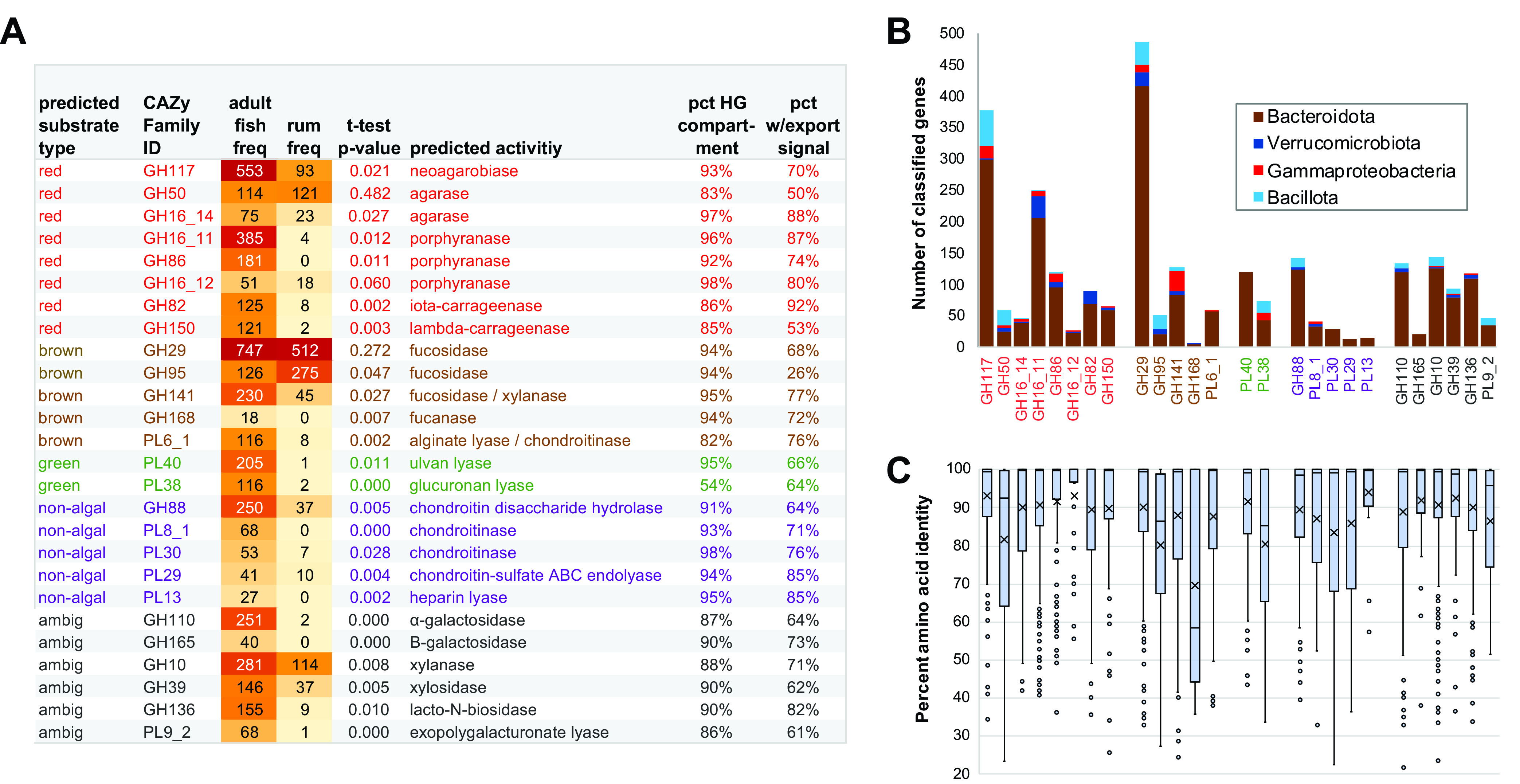
Polysaccharide hydrolase family enrichment. (A) Text colors denote predicted substrates based on CAZy annotations: red, red algae; brown, brown algae; green, green algae; purple, nonalgae; gray, ambiguous. Abbreviations: norm, normalized per 1 million predicted metagenomic proteins; HG, hindgut; rum, ruminant. Full data and significance calculations are provided in Data Set S2. (B) Taxonomic distribution of families enriched in fish relative to terrestrial ruminants, based on the top BLAST matches to database relatives. (Note that not all candidates had database matches.) (C) Intra-CAZy family diversity within fish gut metagenomes. The top of the box indicates the 75% percentile boundary, the bottom of the box indicates the 25% percentile boundary, the horizontal line indicates the median, X indicates the mean value, the whisker indicates the standard deviation, and dots indicate the outlier points.
CAZy families targeting red algal agars (GH117 and GH50), porphyrans (GH86), and carrageenans (GH82 and GH150), green algal ulvans (PL40 and PL38), and nonsulfated alginates from brown algae (PL6_1) were particularly abundant among fish metagenomes. Fish-specific families annotated as chondroitinases (GH88, PL8_1, PL30, and PL29) and heparin lyases (PL13) are most likely associated with the breakdown of host-associated extracellular matrix components. Fish metagenomes also contained large numbers of sequences from families annotated as galactosidases (GH110 and GH165) and xylanases (GH10), as well as several families annotated as hydrolyzing polysaccharides containing multiple different monomers (GH39, GH136, and PL9_2), but potential involvement of these families in macroalgal decomposition could not be unambiguously determined. In agreement with annotation keyword results, the great majority of sequences from CAZy families enriched in kyphosid fish relative to terrestrial ruminants and described as hydrolyzing sulfated macroalgal polysaccharides were obtained from hindgut compartments.
CAZy glycoside hydrolase families are based on shared amino acid sequence similarity, protein structural features, and catalytic mechanisms, but often do not distinguish between enzymes acting on chemically similar substrates, especially those with different branching patterns (47, 71, 72). Polyspecific CAZy families are particularly common among glycohydrolases annotated as having 1,3 β-glucosidase and/or 1,4 β-glucosidase activities, including laminarinases and cellulases (Table S5). Families GH16_3 and GH16_21, historically described as laminarinases despite having a substantially broader range of actual substrates (73), were slightly more abundant in fish gut than terrestrial ruminant metagenomes, but these differences were not statistically significant. Other CAZy families annotated as hydrolyzing nonsulfated, glucose-containing polysaccharides (e.g., GH3, GH5, GH8, GH9, and GH64) were more abundant in metagenomes of terrestrial ruminants than fish. However, model enzymes in these families hydrolyze not only algal polysaccharides (74), but also nonalgal glycans from bacterial capsule polysaccharides (75) and the cell walls of plants (76) and fungi (77). These ambiguities preclude confident substrate predictions for these particular CAZy families based on sequence data alone.
Two recently described CAZy families (GH107 and GH168) include enzymes experimentally demonstrated to hydrolyze sulfated brown algal fucans (23, 43). Although neither of these two families was detected in terrestrial ruminant metagenomes, kyphosid fish samples included several predicted sequences from family GH168 (Fig. 4). Additionally, fucosidase family GH141, present in multiple copies in PUL of known fucan-degrading bacteria (26), was highly expanded in fish gut metagenomes. In contrast, fucosidases from family GH29 were abundant in both kyphosid fish and terrestrial ruminant metagenomes, and family GH95 fucosidases were more abundant in the terrestrial samples, suggesting activity profiles that are not specific for marine macroalgae.
Bacterial export signal sequences were detected in the majority of sequences from fish-expanded CAZy families (Fig. 4A). In Gram-positive bacteria such as Bacillota, proteins containing signal sequences are translocated across the cell membrane and exported to the extracellular environment (78). In Gram-negative bacteria such as Bacteroidota, signal sequences mediate initial translocation across the inner cytoplasmic membrane to the periplasmic space, which may or may not be followed by secretion across the outer membrane (79). Although N-terminal signal sequences could be missing in some predicted proteins due to incomplete metagenomic assembly, potential assembly truncation should not adversely affect reliability of the amino acid similarity comparisons presented in Fig. 4C, which show that median percentages of identity within families GH95 (86%), PL38 (85%) and GH168 (58%) were much lower than those for other families (95 to 99%). These more heterogeneous CAZy families might need to be divided into narrower subfamilies as more experimental data become available in the future.
Taxonomic distributions of fish gut-expanded CAZy enzyme families were estimated by protein sequence comparisons to classified relatives in GenBank nr (Fig. 4B). Predicted proteins from families GH168, GH165, and PL40, targeting brown and green algal polysaccharides, along with families PL30, PL29 and PL13, most likely digesting host tissues, exclusively matched database sequences from Bacteroidota-related clades. Predicted carrageenan-degrading family GH82 enzymes were confined to taxa identified as either Bacteroidota or Verrucomicrobiota, while other macroalgal-degrading families also contained some matches to Bacillota and Gammaproteobacteria. Although not all enzyme candidates could be assigned to a particular bacterial taxon, these results confirm the dominant role of Bacteroidota as elite complex carbohydrate digesters as previously described in other environments (80).
Polysaccharide desulfation.
Negatively charged sulfate residues in polysaccharides can shield glycosidic bonds from enzymatic cleavage (26), stabilizing carbohydrate backbones against degradation and impeding transport of partially degraded external intermediates into bacterial cells. To identify enzyme families that might help overcome these barriers for marine-specific macroalgal substrates, all predicted metagenomic arylsulfatases were classified according to SulfAtlas database categories (81), and relative abundances of these sulfatase families were compared between kyphosid fish and terrestrial ruminant metagenome samples.
Relative abundances of the most highly expanded SulfAtlas families in fish gut metagenomes are shown in Fig. 5 and Data Set S2. Low or undetectable levels of these families in terrestrial herbivores suggest a high degree of marine specificity (Fig. 5A). As in CAZy families, the majority of taxonomically classifiable sulfatase sequences were associated with Bacteroidota, followed distantly by Verrucomicrobiota and Gammaproteobacteria (Fig. 5B). Only one sulfatase family (S1_27) included members classified as Bacillota. All expanded SulfAtlas families except S1_13, the only group exclusively associated with Gammaproteobacteria, were highly concentrated in hindgut regions. With the exception of family S1_46, most fish gut proteins contained export signal sequences. S1_46 sequences were also unusual in their level of intraclass heterogeneity, with a median amino acid identity of 72% versus 93 to 100% for other expanded SulfAtlas families (Fig. 5C).
FIG 5.
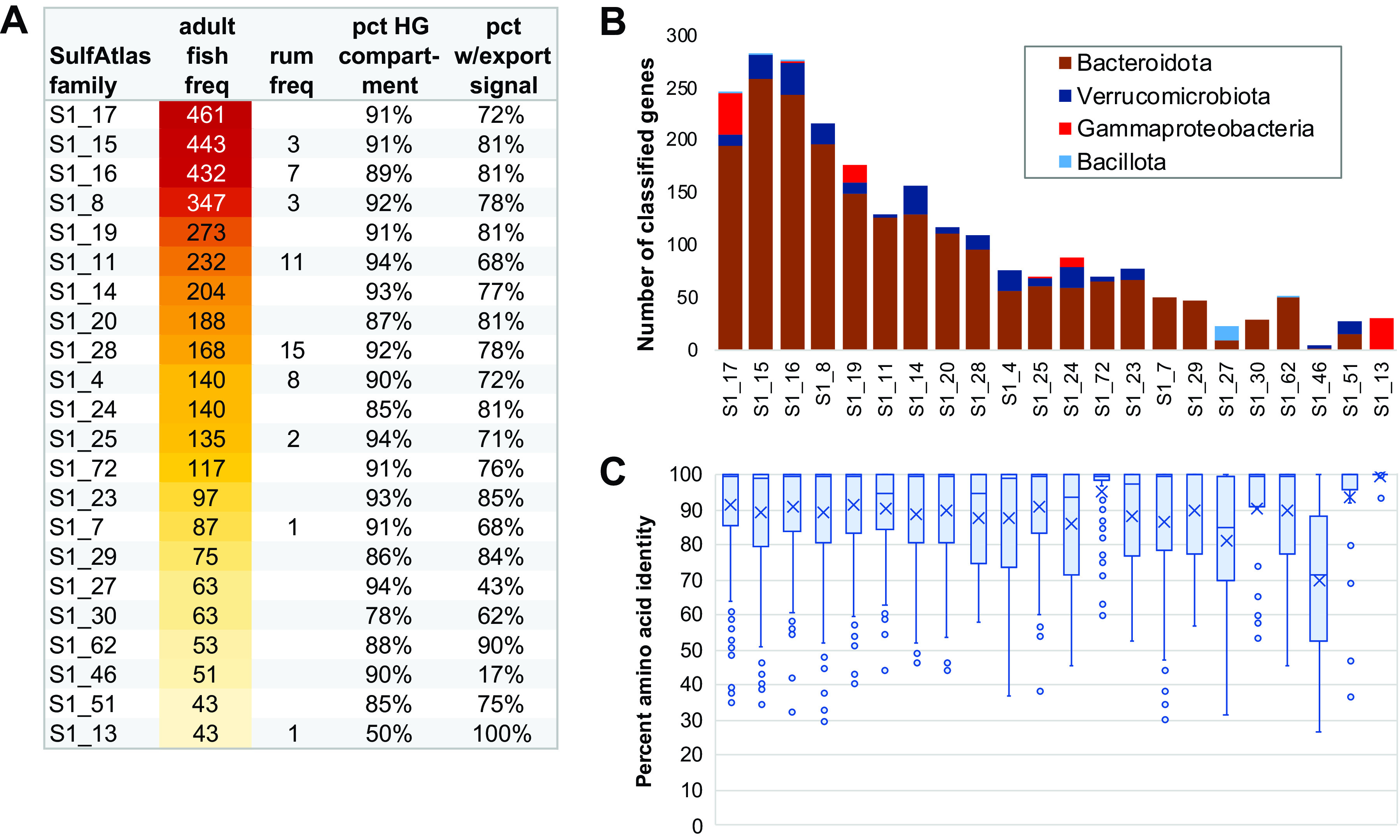
(A) SulfAtlas enzyme family enrichment. Abbreviations: avg norm, group average normalized per 1 million predicted metagenomic proteins; HG, hindgut; rum, ruminant. Full data are provided in Data Set S2. (B) Taxonomic assignments of fish sulfatase class genes matching GenBank nr sequences with minimum blastp E value scores of <1e−5 and alignments covering >30% of database protein length; (C) Intra-SulfAtlas family diversity within fish gut metagenomes. Box-whisker plot elements are defined in the legend to Fig. 4C.
SulfAtlas families S1_7 and S1_81 have previously been demonstrated to include endo-4S-κ-carrageenan sulfatases (21, 42, 49, 82), while S1_19 family members have shown activity against both endo-4S-κ- and endo-4S-ι-carrageenans (21, 83). In this study, metagenomic frequencies for family S1_19 (n = 273) were considerably higher than those for S1_7 (n = 87) or S1_81 (n = 32). These results are consistent with expansion of CAZy families targeting more highly sulfated GH82 (ι-) and GH150 (λ-) carrageenases, rather than simpler κ-carrageenases. Further research will be required to determine the role of other, as yet functionally uncharacterized, SulfAtlas families in accommodating sulfated polysaccharide diversity potentially associated with fish microbiomes encountering a wide variety of red macroalgal taxa.
Colocalization of sulfatase and polysaccharide degradation enzyme classes.
CAZy and SulfAtlas classified genes occurring in close proximity to each other on the same contig are potential candidates for colocalization within a common PUL. Although not all metagenomic contigs are long enough to encompass full-sized PULs, which can include as many as 25 adjacent genes (48), the median separation distance for the 1,453 unique CAZy/SulfAtlas gene pairs detected in fish gut metagenomes was only 4 genes apart, with 98% separated by 25 or fewer genes (Fig. S2). In contrast, the median intervening distance for CAZy and SulfAtlas family pairs colocalized on terrestrial ruminant metagenomic contigs was 15 genes, with only 62% falling within a 25-gene distance limit.
CAZy-SulfAtlas family pairs identified by proximity screening can be visualized as a network of nodes, with edges depicting colocalization frequencies (Fig. 6). Similarities and differences in potential PUL associations are highlighted by subnetworks, such as those including only abundant CAZy (Fig. 6B) or SulfAtlas (Fig. 6C) families, as well as those limited to connections associated with individual enzyme families (Fig. 7 and Fig. 8).
FIG 6.
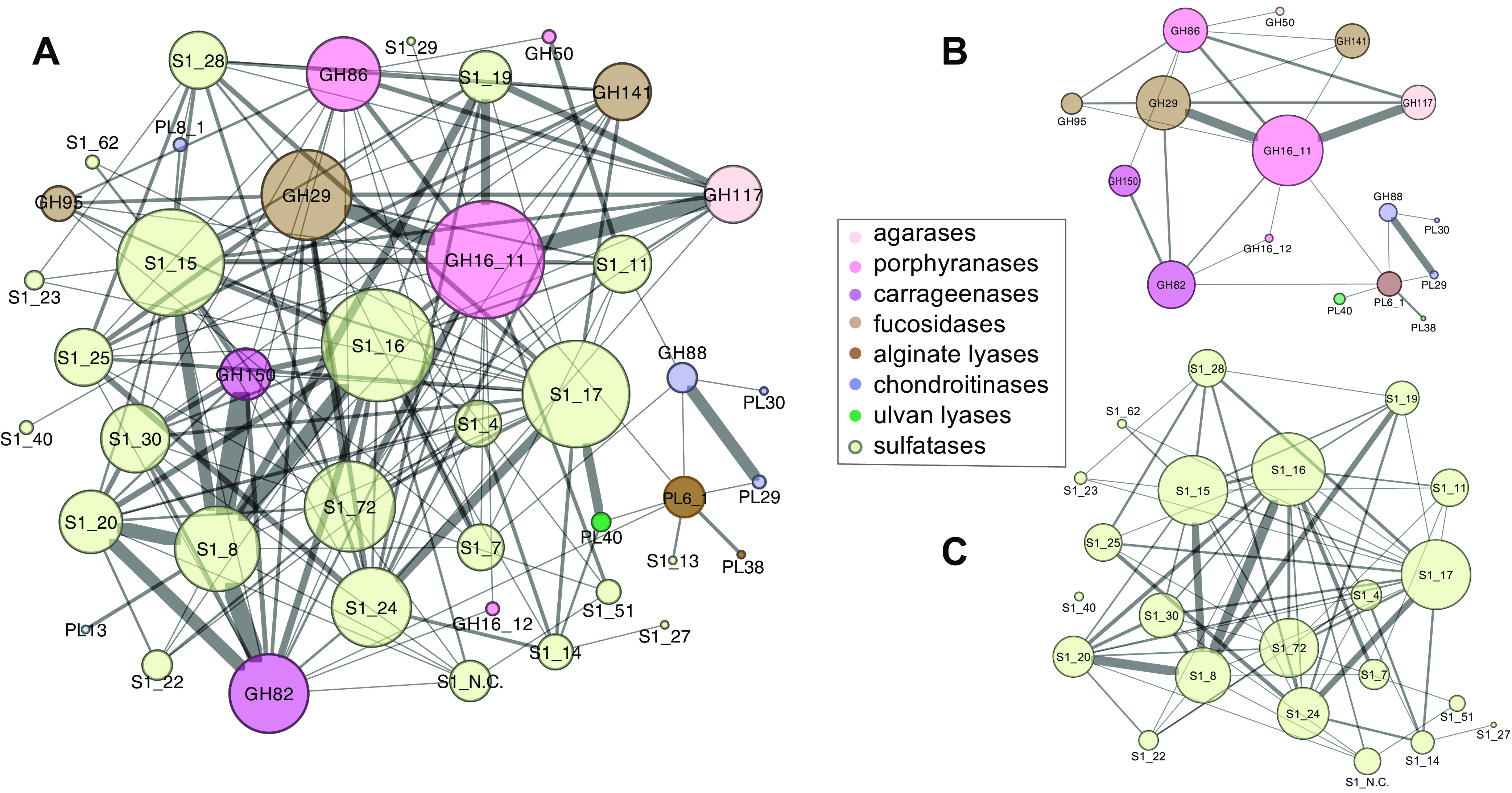
SulfAtlas and CAZy family colocalization networks. Enzyme classes enriched in fish relative to terrestrial ruminants are shown as nodes, with edges denoting colocalization on the same contig within an intervening distance of 25 or fewer genes. Node size is proportional to the total number of edges and edge thickness to connection frequency. Self-loops have been omitted to simplify visualization. (A) Full network diagram for families described in Fig. 4 and 5; (B) subnetwork including CAZy families only; (C) subnetwork including SulfAtlas families only. Raw data tables are provided in Data Set S3.
FIG 7.
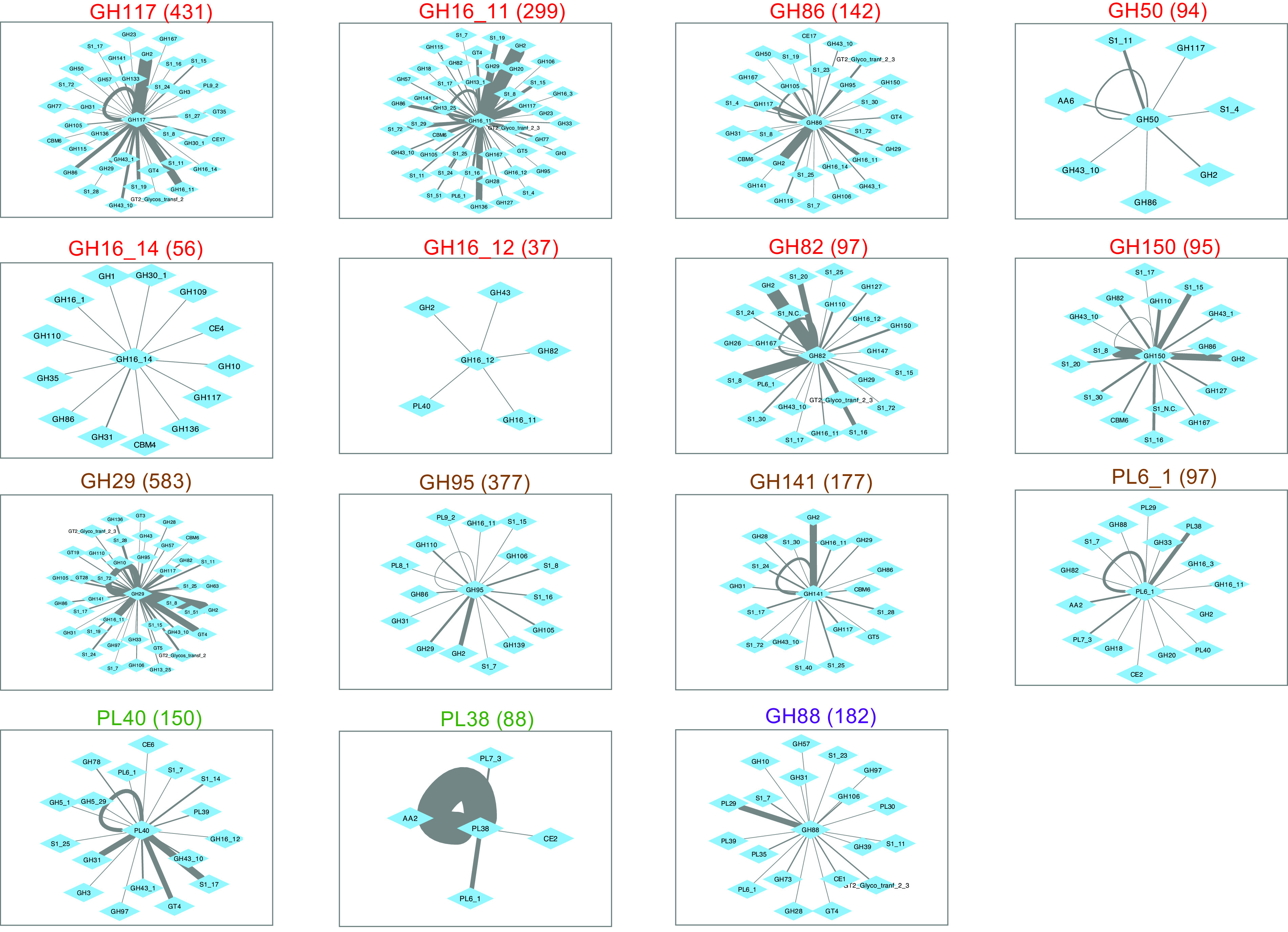
Colocalization network diagrams for individual fish-enriched CAZy families. Text colors denote predicted macroalgal substrate sources, as defined in the legend to Fig. 4. Numbers in parentheses indicate relative gene frequencies per 1 million proteins in adult fish metagenomes. The maximum intervening distance between nodes is 25 genes.
FIG 8.
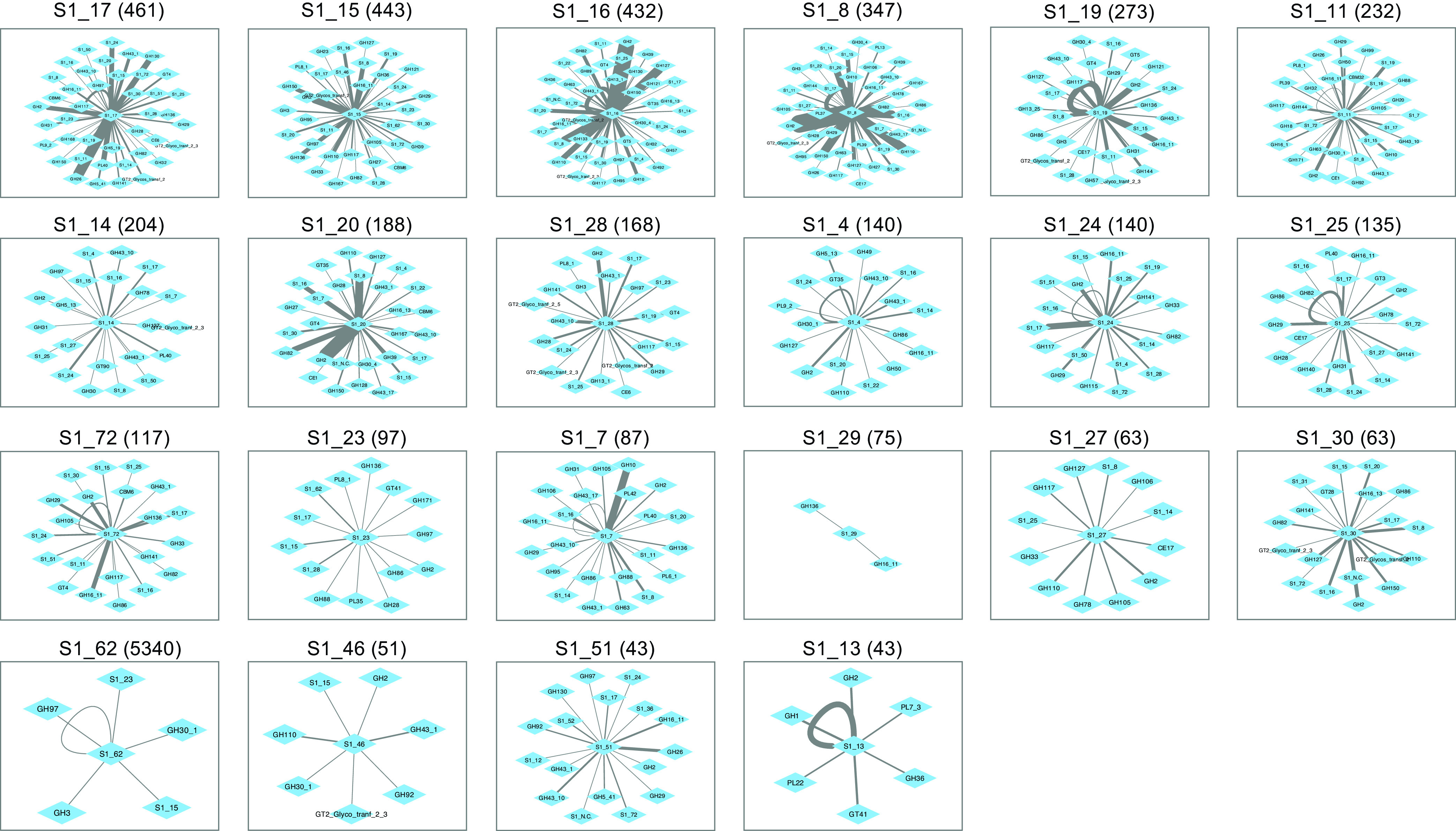
Colocalization network diagrams for individual fish-enriched SulfAtlas families. Numbers in parentheses indicate relative gene frequencies per 1 million proteins in adult fish metagenomes. The maximum intervening distance between nodes is 25 genes.
One striking feature of gene colocalization networks was the presence of higher proximity frequencies between certain enzyme families. These enhanced links suggest potential occurrence within a common PUL that might facilitate coregulation of gene expression. Both linkage frequencies and the diversity of colocalized nodes varied widely between families. These variations were not necessarily proportional to overall metagenomic abundance, as illustrated by comparing the colocalization frequency network centered on CAZy family GH50 (94 metagenomic occurrences) with that of family GH150 (95 metagenomic occurrences) in Fig. 7 and SulfAtlas family S1_29 (75 metagenomic occurrences) with SulfAtlas family S1_30 (63 metagenomic occurrences) in Fig. 8.
Network comparisons also show enormous variation in the number of self-loops formed by neighboring genes from the same class on a single metagenomic contig, suggesting differences in the expansion of particular families by gene duplication (summarized in panels B and C of Fig. 9). The most extreme example of gene duplication was observed in family PL38 glucuronan lyases, where tandem repeats of 7 to 9 closely related sequences predicted to originate from an unknown Spirochaete lineage were found in assembled contigs from both K. cinerascens and K. hawaiiensis samples. Although no closely related proteins have been reported in other Spirochaetes, the PUL Database (48) does contain numerous examples of tandem duplications of PL38 family proteins among Bacteroidota genomes, suggesting the possibility of horizontal gene transfer into a Spirochaete lineage, followed by additional local gene duplications.
FIG 9.
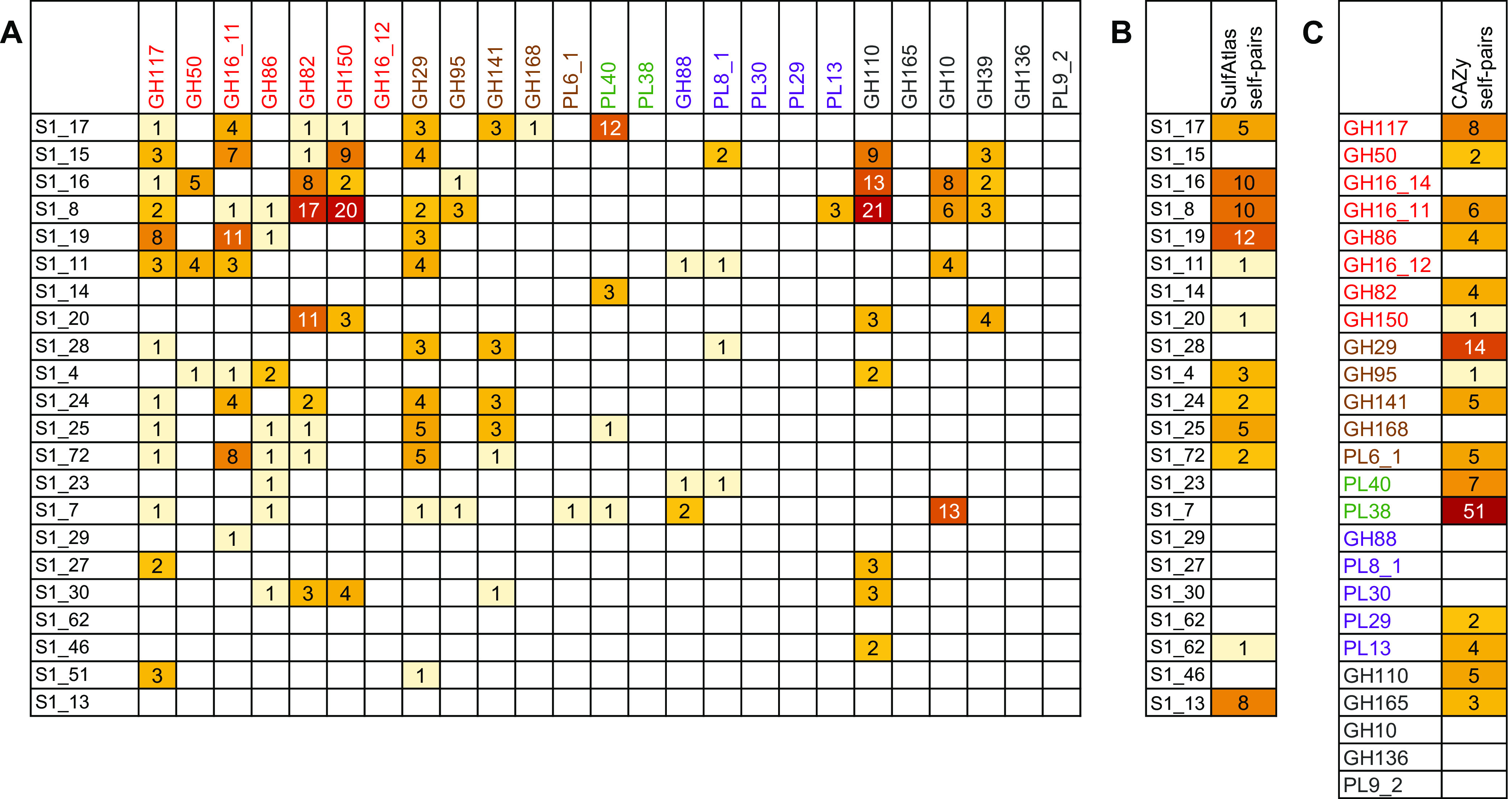
Colocalization frequencies for CAZy and SulfAtlas enzyme families. (A) Colocalization frequencies between SulfAtlas and CAZy family members; (B) self-colocalization frequencies within SulfAtlas families; (C) self-colocalization frequencies within CAZy families. Darker colors indicate higher values for the number of times each intersecting family pair or self-pair occurred on the same metagenomic contig with an intervening distance of 25 genes or less. Text colors for CAZy families denote predicted macroalgal sources, as defined in the legend to Fig. 4.
Detailed comparisons of colocalized enzyme families can reveal subtle relationships and provide hints about potential metabolic interactions that might benefit from coregulation. As an example, sulfatase families S1_16 and S1_8 were frequently colocalized on the same contig (Fig. 6C), suggesting potential complementary PUL-linked functions within a single genome. Both families were also frequently paired with polysaccharide-degrading CAZy families (Fig. 9A) that included those targeting highly sulfated ι-carrageenans (GH82) and λ-carrageenans (GH150), as well as exo-α-galactosidase family GH110, α-1,3-(3, 6)-anhydro-d-galactosidase family GH127, and generalized hexosidase family GH2. Links to CAZy families targeting agarases (GH117 and GH50) and porphyranases (GH16_11) occurred much less frequently.
Nearly all SulfAtlas families expanded in fish gut metagenomes included some examples of proximity with red algal polysaccharide-hydrolyzing CAZy families, but some were also located near CAZy families predicted to degrade brown and green algal and/or host chondroitin substrates (Fig. 9A). SulfAtlas families S1_28 and S1_25 were predominantly linked to CAZy families hydrolyzing brown algal substrates, while families S1_19, S1_20, S1_27, and S1_30, were more narrowly associated with red algal-digesting enzyme sequences. Families S1_14 and S1_17 were most often associated with green algal ulvan lyases. These distinctively different colocalization patterns suggest promising avenues for future experimental determination of sulfatase family-specific substrate ranges.
One limitation of metagenomic colocalization analysis is that not all paired enzyme features are equally informative, particularly those including diverse glycohydrolase families like GH2. At least a dozen different functional activities have been ascribed to this CAZy family, targeting not only β-galactosides, but also β-mannosides, β-glucuronosides, and β-galacturonosides. Another constraint is that predicted proteins from sparsely represented microbial taxa are often encoded on shorter, less completely assembled contigs, reducing the amount of available information about potential neighboring genes. Although this limitation can decrease detection sensitivity for rare enzyme family pairs, infrequent relationships could also be interpreted as less important to overall fish digestive processes than features showing elevated metagenomic linkage frequencies that potentially contribute more to successful metabolic strategies, resulting in higher community abundances for the organisms encoding them.
DISCUSSION
Although diet is undoubtedly a key element in determining gut microbial community composition, discovering and quantifying the full variety of dietary items a wild fish might have eaten on a particular day are challenging. Stomach contents are homogenized, fragmented, and conglomerated into amorphous mixtures, making visual identification difficult. Epibionts, biofilms, and marine sediments may be ingested unintentionally along with preferred food items, and relative quantities of the items consumed may vary for individual fish over time periods considerably briefer than those required for digestive system transit. As a result, single postmortem snapshots of stomach contents may fail to capture or recognize the full range of dietary diversity associated with individual fish.
The deep metagenomic sequencing methods used in this study have addressed these issues by quantifying molecular signatures of algal taxa as they passed through the digestive systems of individual fish, minimizing potential observational bias arising from differences in the physical sizes and deterioration states of ingested food. Dietary composition, microbial taxonomy, and predicted enzyme functions were all assessed in parallel for each sample, circumventing potential inconsistencies due to time-dependent variability in host feeding behavior. This approach has also provided evidence suggesting progressive replacement of transient, environmentally sourced microbes such as marine Vibrionaceae in more proximal gut regions with more persistent host-associated taxa during transit to distal regions.
Although the fish gut samples in this study were taken from luminal rather than epithelial or mucosal tissue sections, the most abundant Bacteroidota, Bacillota, and Deltaproteobacteria species in hindgut regions were more closely related to host-associated bacteria from terrestrial vertebrate guts than to those found in any previously described external marine source, including potential algal food items. These results are consistent with possible infiltration of incoming gut contents by bacteria from host-associated tissue microhabitats that might be physically protected from fecal stream depletion: for example, within mucus layers or crypt structures (84, 85). Multihour transit times through elongated kyphosid digestive systems (7) would provide ample opportunities for microbial community remodeling to occur within the gut lumen.
One limitation of the in-depth metagenomic techniques applied in this study is that only one adult representative was analyzed from each of the three different kyphosid species. There is no guarantee that identical dietary profiles would be obtained from other fish of the same species sampled from the same habitat or even the same fish if captured at different times. However, results spanning multiple gut regions in sympatric individuals consuming similar diets have yielded a consistent, high-level picture that greatly extends previous analyses based on observations of stomach contents alone (13, 36). These insights will inform the design of future experiments aimed at investigating the relationships between diet and microbial compositions within, as well as between, species and how these relationships might vary between individuals of different ages and sexes.
The microbiomes of wild-caught individuals from three different sympatric kyphosid species revealed consistent connections between particular microbial groups and shared functional capabilities likely to contribute to efficient processing of macroalgal polysaccharides. These connections were highlighted by both expansion of individual enzyme families and their co-occurrence in operon context. Consistent availability of especially diverse red macroalgal dietary items correlated with expanded, marine-specific enzyme families capable of hydrolyzing their characteristic galactose-rich sulfated polysaccharides. Lower metagenomic frequencies were observed for enzyme families targeting brown algal fucans and green algal ulvans, complementing molecular evidence suggesting less frequent consumption of foods containing these polysaccharides.
Previously described methods for detecting polysaccharide utilization loci (PUL) and their operon context rely heavily on sequences from well-characterized taxa (28, 86), potentially limiting discovery of novel examples from uncultured or poorly studied environmental taxa with atypical PUL architectures. Quantification of metagenomic enzyme family colocalizations avoids this limitation by identifying statistically verifiable frequency patterns replicated in multiple independent samples, revealing informative associations even in cases where only partial PUL are assembled. This technique has identified relationships among several thousand newly sequenced macroalgal degradation enzyme candidates from kyphosid fish metagenomes, providing valuable tools for exploring evolutionary mechanisms that may be responsible for acquisition and expansion of these capabilities among individual microbial taxa.
Frequently observed extracellular export signals in CAZy and SulfAtlas family proteins enriched in fish relative to terrestrial ruminants, combined with the known complexity and diversity of naturally occurring macroalgal polysaccharides, suggest potential opportunities for cooperative activity on recalcitrant substrates. Exported enzyme cooperativity could include multiple genes originating from the same strain, variants comprising the pangenomic repertoire of closely related strains, or even panmicrobiome diversity arising from widely different taxa within the gut microbial community. Future strategies for testing this hypothesis could include reconstruction of metagenome-assembled genomes (MAGs) from binned metagenomic contigs, transcriptional mapping to determine in vivo gene expression levels, in vitro substrate hydrolysis measurements using combinations of purified enzymes obtained by expression cloning, and comparisons of macroalgal degradation performance by enrichments or defined mixed cultures of live bacteria.
The work presented here extends previous kyphosid microbiome studies (35, 36, 70) by highlighting taxa likely to be most active in digestion of sulfated macroalgal polysaccharides, identifying potentially extracellularly exported, cooperative networks of macroalgal digestion enzymes, and revealing prospective functional properties of previously uncharacterized sulfatase enzyme families. The discovery of gut compartment-specific microbial adaptations to a diet rich in sulfated macroalgal polysaccharides improves our understanding of the enzymes and organisms involved in host utilization of these molecules, providing foundational resources for future investigations into suppression of coral reef macroalgal overgrowth, the role of gut microbiota in fish physiology, the use of macroalgal feedstocks and probiotics in aquaculture, the inclusion of seaweed supplements in terrestrial ruminant feedstocks, and the use of naturally occurring microbial enzymes in extraction of commercially valuable products from macroalgae.
MATERIALS AND METHODS
Metagenomic sequencing and assembly.
DNA samples were isolated from lumen contents distributed over gut compartments from three different kyphosid fish species (see Table S1A in the supplemental material), using previously described collection and processing procedures (35), in accordance with IACUC protocol S12219. Approximately 30 million 250-bp paired-end reads were generated per sample using Illumina NovaSeq 6000 technology. Reads from each individual sample were quality filtered and trimmed using Trimmomatic version 0.36 with the following parameters: adapter-read alignment settings 2:30:10, LEADING:10, TRAILING:20, HEADCROP:12, SLIDINGWINDOW:4:15, and MINLEN:200 (87). Trimmed reads from each sample were then assembled separately using metaSPAdes version 3.13 (88) with a minimum contig retention size of 2,000 nucleotides (nt).
Taxonomic classification.
Taxonomic assignments were made for unassembled reads with Kraken2 version 2.1.2 (67) using GenBank nr (accessed August 2021) as a custom reference database. Assembled metagenomic contigs were taxonomically classified using DarkHorse version 2.0_rev09 as previously described (89). DNA-directed RNA polymerase subunit beta (RpoB) protein sequences were retrieved from assembled contigs annotated with Prokka as described below. The closest sequenced database relatives were identified by top matches in blastp searches against a combined database including both GenBank nr entries and predicted proteins from terrestrial ruminant metagenome MAGs (38).
Multiple-sequence alignments of fish gut RpoB sequences of >900 amino acids and their closest database relatives were obtained using MUSCLE version 3.8.31 (90) and used to build phylogenetic trees using FastTree version 2.1.10 (91). Trees were visualized using the R package ggtree version 3.3.5 (92). Fish microbiome 18S rRNA gene sequences were identified by blastn search against the SILVA_138.1_SSURef_NR99 database (93), retaining alignments covering at least >30% of the reference sequence length with E values of 1e−5 or better. These relatively loose parameters were chosen to maximize sensitivity in assigning approximate taxonomies to both full-length and partial, incomplete metagenomic sequences.
Gene annotation.
Assembled metagenomic contigs were annotated with Prokka version 1.14 (94). Predicted proteins containing microbial extracellular export signals were identified using SignalP version 6.0f (95). Carbohydrate enzyme families were assigned using HMMSEARCH with CAZy version 10 database patterns downloaded from dbCAN2 (96), retaining matches with alignments covering at least 30% of the protein and E values of 1e−15 or better. Sulfatase enzyme categories were determined using the SulfAtlas hidden Markov model (HMM) subfamily classification tool with database version 2.3 (49). Assembled nucleotide sequences for 391 terrestrial ruminant MAGs (38) were downloaded from NCBI BioProject no. PRJEB34458 and annotated using Prokka, SignalP, CAZy, and SulfAtlas, as described above, to enable direct comparisons with assembled fish gut metagenomes.
The overlap of dominant microbial community taxa observed in the digestive systems of terrestrial vertebrate herbivores with those of kyphosid marine fish—for example Alistipes-related Bacteroidota, Clostridia-related Bacillota, and Desulfovibrio-related Deltaproteobacteria (35–38)—provides a context of evolutionarily shared microbial genomic backgrounds that can be used to facilitate discrimination between broad enzyme activities involved in digesting polysaccharides common to all Viridiplantae and those more specific to marine macroalgae. Vertebrate MAGs representing microbiota from domesticated and wild-caught herbivores were selected as potentially informative comparators based on taxonomic similarity of their microbial communities, breadth of potential polysaccharide types consumed, and previously documented similarity in gut transit times (1) and fermentative acetate turnover rates (6).
Metagenomic occurrence frequencies were tallied for CAZy and SulfAtlas database entries and selected Prokka annotation keywords and then grouped into subsets according to the species of origin (for fish, K. vaigiensis, K. cinerascens, and K. hawaiiensis; for ruminants, reindeer, red deer, sheep, goat, cattle, or mixed ruminant assembly) and normalized for total number of predicted protein sequences in each subset. Normalized values were subjected to 1-tailed, homoscedastic t tests using Microsoft Excel version 16.54 to evaluate statistical significance (P values) of observed differences between fish and ruminant data sets. Enzyme families colocalized on the same metagenomic contig were identified using a Unix command-line pipeline of custom perl scripts available on GitHub (97). Co-occurrence frequencies of enzyme pairs obtained using this pipeline were plotted as edge-weighted network diagrams using Cytoscape version 3.9.1 (98).
Data availability.
Sequence reads are available under SRA accession no. SRR19136343 through SRR19136358, assembled contigs under WGS accession no. JAMHIX000000000 through JAMHIZ000000000 and JAMHJA000000000 through JAMHJM000000000, and predicted protein sequences in Zenodo (https://zenodo.org) under DOI no. 10.5281/zenodo.6635023 and 10.5281/zenodo.6635166.
ACKNOWLEDGMENTS
We acknowledge Lucie Leonard, Keelee Martin, and the entire Ocean Era team for assistance in organizing and conducting fieldwork for sample collection. We also thank Helen Meigs and all of the local fisherfolk for assistance in field sampling.
This work was funded by the United States Government, Department of Energy, Advanced Research Projects Agency—Energy grant ARPA-E DE-FOA-0001858 to R.S.N and L.M.L.L. with contracts to C.E.N., L.W.K., and E.E.A., National Science Foundation grants OCE-1837116 and EF-2025217 to E.E.A., and National Institutes of Health NIEHS grant R01-ES030316 to E.E.A.
S.P., L.W.K., R.S.N., L.M.L.L., N.A.S., C.E.N., and E.E.A. conceived the study. W.J.S., L.W.K., A.M.P., and S.A. performed wet-lab fish gut sampling and DNA isolations. S.P. and A.O. designed and performed bioinformatic analyses and generated visualizations. Custom computer code was designed and written by S.P., who also wrote the manuscript. All authors read and approved the final version of the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Sheila Podell, Email: spodell@ucsd.edu.
Eric E. Allen, Email: eallen@ucsd.edu.
Knut Rudi, Norwegian University of Life Sciences.
REFERENCES
- 1.Horn MH. 1989. Biology of marine herbivorous fishes. Aberdeen University Press, Aberdeen, United Kingdom. [Google Scholar]
- 2.Hughes TP, Rodrigues MJ, Bellwood DR, Ceccarelli D, Hoegh-Guldberg O, McCook L, Moltschaniwskyj N, Pratchett MS, Steneck RS, Willis B. 2007. Phase shifts, herbivory, and the resilience of coral reefs to climate change. Curr Biol 17:360–365. doi: 10.1016/j.cub.2006.12.049. [DOI] [PubMed] [Google Scholar]
- 3.McCook L, Jompa J, Diaz-Pulido G. 2001. Competition between corals and algae on coral reefs: a review of evidence and mechanisms. Coral Reefs 19:400–417. doi: 10.1007/s003380000129. [DOI] [Google Scholar]
- 4.Choat JH, Clements KD. 1998. Vertebrate herbivores in marine and terrestrial environments: a nutritional ecology perspective. Annu Rev Ecol Syst 29:375–403. doi: 10.1146/annurev.ecolsys.29.1.375. [DOI] [Google Scholar]
- 5.Sturm E, Horn M. 1998. Food habits, gut morphology and pH, and assimilation efficiency of the zebraperch Hermosilla azurea, an herbivorous kyphosid fsh of temperate marine waters. Mar Biol 132:515–522. doi: 10.1007/s002270050417. [DOI] [Google Scholar]
- 6.Mountfort DO, Campbell J, Clements KD. 2002. Hindgut fermentation in three species of marine herbivorous fish. Appl Environ Microbiol 68:1374–1380. doi: 10.1128/AEM.68.3.1374-1380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fidopiastis PM, Bezdek DJ, Horn MH, Kandel JS. 2006. Characterizing the resident, fermentative microbial consortium in the hindgut of the temperate-zone herbivorous fish, Hermosilla azurea (Teleostei: Kyphosidae). Mar Biol 148:631–642. doi: 10.1007/s00227-005-0106-2. [DOI] [Google Scholar]
- 8.Nielsen S, Wilkes Walburn J, Verges A, Thomas T, Egan S. 2017. Microbiome patterns across the gastrointestinal tract of the rabbitfish Siganus fuscescens. PeerJ 5:e3317. doi: 10.7717/peerj.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones J, DiBattista JD, Stat M, Bunce M, Boyce MC, Fairclough DV, Travers MJ, Huggett MJ. 2018. The microbiome of the gastrointestinal tract of a range-shifting marine herbivorous fish. Front Microbiol 9:2000. doi: 10.3389/fmicb.2018.02000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egerton S, Culloty S, Whooley J, Stanton C, Ross RP. 2018. The gut microbiota of marine fish. Front Microbiol 9:873. doi: 10.3389/fmicb.2018.00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng W, Zhang J, Jakovlic I, Xiong F, Wu S, Zou H, Li W, Li M, Wang G. 2019. Gut segments outweigh the diet in shaping the intestinal microbiota composition in grass carp Ctenopharyngodon idellus. AMB Express 9:44. doi: 10.1186/s13568-019-0770-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ringø E, Zhou Z, Vecino JLG, Wadsworth S, Romero J, Krogdahl Å, Olsen RE, Dimitroglou A, Foey A, Davies S, Owen M, Lauzon HL, Martinsen LL, De Schryver P, Bossier P, Sperstad S, Merrifield DL. 2016. Effect of dietary components on the gut microbiota of aquatic animals. A never-ending story? Aquacult Nutr 22:219–282. doi: 10.1111/anu.12346. [DOI] [Google Scholar]
- 13.Choat JH, Clements KD, Robbins WD. 2002. The trophic status of herbivorous fishes on coral reefs. I. Dietary analyses. Mar Biol 140:613–623. [Google Scholar]
- 14.Wu P, Wang T, Liu Y, Li C, Xiao Y, Xu S, Han T, Lin L, Quan Q. 2022. Differences of macroalgal consumption by eight herbivorous coral reef fishes from the Xisha Islands, China. Front Mar Sci doi: 10.3389/fmars.2022.882196. [DOI] [Google Scholar]
- 15.Arnosti C, Wietz M, Brinkhoff T, Hehemann JH, Probandt D, Zeugner L, Amann R. 2021. The biogeochemistry of marine polysaccharides: sources, inventories, and bacterial drivers of the carbohydrate cycle. Annu Rev Mar Sci 13:81–108. doi: 10.1146/annurev-marine-032020-012810. [DOI] [PubMed] [Google Scholar]
- 16.Zhang K, Zhang F, Wu YR. 2021. Emerging technologies for conversion of sustainable algal biomass into value-added products: a state-of-the-art review. Sci Total Environ 784:147024. doi: 10.1016/j.scitotenv.2021.147024. [DOI] [PubMed] [Google Scholar]
- 17.Evans DH, Claiborne JB, Currie S. 2013. The physiology of fishes, 4th ed, p 175–204. Taylor & Francis, Abingdon-on-Thames, United Kingdom. [Google Scholar]
- 18.Hehemann JH, Correc G, Thomas F, Bernard T, Barbeyron T, Jam M, Helbert W, Michel G, Czjzek M. 2012. Biochemical and structural characterization of the complex agarolytic enzyme system from the marine bacterium Zobellia galactanivorans. J Biol Chem 287:30571–30584. doi: 10.1074/jbc.M112.377184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann AJ, Hahnke RL, Huang S, Werner J, Xing P, Barbeyron T, Huettel B, Stuber K, Reinhardt R, Harder J, Glockner FO, Amann RI, Teeling H. 2013. The genome of the alga-associated marine flavobacterium Formosa agariphila KMM 3901T reveals a broad potential for degradation of algal polysaccharides. Appl Environ Microbiol 79:6813–6822. doi: 10.1128/AEM.01937-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakunina IY, Nedashkovskaya OI, Balabanova LA, Kukhlevsky AD, Belous OS. 2020. Activities of O-glycoside hydrolases and other polysaccharide-degrading enzymes of cultivable bacterial isolates of the Pacific Red Alga Ahnfeltia tobuchiensis (Kanno et Matsubara, 1932) Makienko, 1970. Russ J Mar Biol 46:387–389. doi: 10.1134/S1063074020050028. [DOI] [Google Scholar]
- 21.Ficko-Blean E, Prechoux A, Thomas F, Rochat T, Larocque R, Zhu Y, Stam M, Genicot S, Jam M, Calteau A, Viart B, Ropartz D, Perez-Pascual D, Correc G, Matard-Mann M, Stubbs KA, Rogniaux H, Jeudy A, Barbeyron T, Medigue C, Czjzek M, Vallenet D, McBride MJ, Duchaud E, Michel G. 2017. Carrageenan catabolism is encoded by a complex regulon in marine heterotrophic bacteria. Nat Commun 8:1685. doi: 10.1038/s41467-017-01832-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reisky L, Stanetty C, Mihovilovic MD, Schweder T, Hehemann JH, Bornscheuer UT. 2018. Biochemical characterization of an ulvan lyase from the marine flavobacterium Formosa agariphila KMM 3901(T). Appl Microbiol Biotechnol 102:6987–6996. doi: 10.1007/s00253-018-9142-y. [DOI] [PubMed] [Google Scholar]
- 23.Schultz-Johansen M, Bech PK, Hennessy RC, Glaring MA, Barbeyron T, Czjzek M, Stougaard P. 2018. A novel enzyme portfolio for red algal polysaccharide degradation in the marine bacterium Paraglaciecola hydrolytica S66(T) encoded in a sizeable polysaccharide utilization locus. Front Microbiol 9:839. doi: 10.3389/fmicb.2018.00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gobet A, Barbeyron T, Matard-Mann M, Magdelenat G, Vallenet D, Duchaud E, Michel G. 2018. Evolutionary evidence of algal polysaccharide degradation acquisition by Pseudoalteromonas carrageenovora 9(T) to adapt to macroalgal niches. Front Microbiol 9:2740. doi: 10.3389/fmicb.2018.02740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao B, Jin M, Li L, Qu W, Zeng R. 2017. Genome sequencing reveals the complex polysaccharide-degrading ability of novel deep-sea bacterium Flammeovirga pacifica WPAGA1. Front Microbiol 8:600. doi: 10.3389/fmicb.2017.00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sichert A, Corzett CH, Schechter MS, Unfried F, Markert S, Becher D, Fernandez-Guerra A, Liebeke M, Schweder T, Polz MF, Hehemann JH. 2020. Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat Microbiol 5:1026–1039. doi: 10.1038/s41564-020-0720-2. [DOI] [PubMed] [Google Scholar]
- 27.Wolter LA, Mitulla M, Kalem J, Daniel R, Simon M, Wietz M. 2021. CAZymes in Maribacter dokdonensis 62-1 from the Patagonian Shelf: genomics and physiology compared to related flavobacteria and a co-occurring Alteromonas strain. Front Microbiol 12:628055. doi: 10.3389/fmicb.2021.628055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alejandre-Colomo C, Francis B, Viver T, Harder J, Fuchs BM, Rossello-Mora R, Amann R. 2021. Cultivable Winogradskyella species are genomically distinct from the sympatric abundant candidate species. ISME Commun 1:51. doi: 10.1038/s43705-021-00052-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silchenko AS, Rasin AB, Zueva AO, Kusaykin MI, Zvyagintseva TN, Kalinovsky AI, Kurilenko VV, Ermakova SP. 2018. Fucoidan sulfatases from marine bacterium Wenyingzhuangia fucanilytica CZ1127(T). Biomolecules 8:98. doi: 10.3390/biom8040098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikkelsen MD, Cao HTT, Roret T, Rhein-Knudsen N, Holck J, Tran VTT, Nguyen TT, Tran VHN, Lezyk MJ, Muschiol J, Pham TD, Czjzek M, Meyer AS. 2021. A novel thermostable prokaryotic fucoidan active sulfatase PsFucS1 with an unusual quaternary hexameric structure. Sci Rep 11:19523. doi: 10.1038/s41598-021-98588-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vuillemin M, Silchenko AS, Cao HTT, Kokoulin MS, Trang VTD, Holck J, Ermakova SP, Meyer AS, Mikkelsen MD. 2020. Functional characterization of a new GH107 endo-alpha-(1,4)-fucoidanase from the marine bacterium Formosa haliotis. Mar Drugs 18:562. doi: 10.3390/md18110562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ngugi DK, Miyake S, Cahill M, Vinu M, Hackmann TJ, Blom J, Tietbohl MD, Berumen ML, Stingl U. 2017. Genomic diversification of giant enteric symbionts reflects host dietary lifestyles. Proc Natl Acad Sci USA 114:E7592–E7601. doi: 10.1073/pnas.1703070114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, Shang Q, Li G, Wang X, Yu G. 2017. Degradation of marine algae-derived carbohydrates by Bacteroidetes isolated from human gut microbiota. Mar Drugs 15:92. doi: 10.3390/md15040092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pardesi B, Roberton AM, Wollmuth EM, Angert ER, Rosendale DI, White WL, Clements KD. 2022. Tannockella kyphosi gen. nov., sp. nov., a member of the family Erysipelotrichaceae, isolated from the hindgut of the marine herbivorous fish Kyphosus sydneyanus. Int J Syst Evol Microbiol doi: 10.1099/ijsem.0.005374. [DOI] [PubMed] [Google Scholar]
- 35.Sparagon WJ, Gentry EC, Minich JJ, Vollbrecht L, Laurens LML, Allen EE, Sims NA, Dorrestein PC, Kelly LW, Nelson CE. 2022. Fine scale transitions of the microbiota and metabolome along the gastrointestinal tract of herbivorous fishes. Anim Microbiome 4:33. doi: 10.1186/s42523-022-00182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevenson SJR, Lee KC, Handley KM, Angert ER, White WL, Clements KD. 2022. Substrate degradation pathways, conserved functions and community composition of the hindgut microbiota in the herbivorous marine fish Kyphosus sydneyanus. Comp Biochem Physiol A Mol Integr Physiol 272:111283. doi: 10.1016/j.cbpa.2022.111283. [DOI] [PubMed] [Google Scholar]
- 37.Xie F, Jin W, Si H, Yuan Y, Tao Y, Liu J, Wang X, Yang C, Li Q, Yan X, Lin L, Jiang Q, Zhang L, Guo C, Greening C, Heller R, Guan LL, Pope PB, Tan Z, Zhu W, Wang M, Qiu Q, Li Z, Mao S. 2021. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 9:137. doi: 10.1186/s40168-021-01078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glendinning L, Genc B, Wallace RJ, Watson M. 2021. Metagenomic analysis of the cow, sheep, reindeer and red deer rumen. Sci Rep 11:1990. doi: 10.1038/s41598-021-81668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hehemann JH, Boraston AB, Czjzek M. 2014. A sweet new wave: structures and mechanisms of enzymes that digest polysaccharides from marine algae. Curr Opin Struct Biol 28:77–86. doi: 10.1016/j.sbi.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 40.Michel G, Czjzek M. 2013. Polysaccharide-degrading enzymes from marine bacteria, p 429–464. In Trincone A (ed), Marine enzymes for biocatalysis: sources, biocatalytic characteristics and bioprocesses of marine enzymes. Woodhead Publishing, Sawston, Cambridge, United Kingdom. [Google Scholar]
- 41.Lee WK, Ho CL. 2022. Ecological and evolutionary diversification of sulphated polysaccharides in diverse photosynthetic lineages: a review. Carbohydr Polym 277:118764. doi: 10.1016/j.carbpol.2021.118764. [DOI] [PubMed] [Google Scholar]
- 42.Prechoux A, Genicot S, Rogniaux H, Helbert W. 2016. Enzyme-assisted preparation of furcellaran-like κ-/β-carrageenan. Mar Biotechnol (NY) 18:133–143. doi: 10.1007/s10126-015-9675-3. [DOI] [PubMed] [Google Scholar]
- 43.Silchenko AS, Rasin AB, Zueva AO, Kusaykin MI, Zvyagintseva TN, Rubtsov NK, Ermakova SP. 2021. Discovery of a fucoidan endo-4O-sulfatase: regioselective 4O-desulfation of fucoidans and its effect on anticancer activity in vitro. Carbohydr Polym 271:118449. doi: 10.1016/j.carbpol.2021.118449. [DOI] [PubMed] [Google Scholar]
- 44.Liu G, Wu S, Jin W, Sun C. 2016. Amy63, a novel type of marine bacterial multifunctional enzyme possessing amylase, agarase and carrageenase activities. Sci Rep 6:18726. doi: 10.1038/srep18726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lapebie P, Lombard V, Drula E, Terrapon N, Henrissat B. 2019. Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat Commun 10:2043. doi: 10.1038/s41467-019-10068-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ausland C, Zheng J, Yi H, Yang B, Li T, Feng X, Zheng B, Yin Y. 2021. dbCAN-PUL: a database of experimentally characterized CAZyme gene clusters and their substrates. Nucleic Acids Res 49:D523–D528. doi: 10.1093/nar/gkaa742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drula E, Garron ML, Dogan S, Lombard V, Henrissat B, Terrapon N. 2022. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res 50:D571–D577. doi: 10.1093/nar/gkab1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terrapon N, Lombard V, Drula E, Lapebie P, Al-Masaudi S, Gilbert HJ, Henrissat B. 2018. PULDB: the expanded database of polysaccharide utilization loci. Nucleic Acids Res 46:D677–D683. doi: 10.1093/nar/gkx1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stam M, Lelievre P, Hoebeke M, Corre E, Barbeyron T, Michel G. 2023. SulfAtlas, the sulfatase database: state of the art and new developments. Nucleic Acids Res 51:D647–D653. doi: 10.1093/nar/gkac977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brunet M, Le Duff N, Barbeyron T, Thomas F. 2022. Consuming fresh macroalgae induces specific catabolic pathways, stress reactions and type IX secretion in marine flavobacterial pioneer degraders. ISME J 16:2027–2039. doi: 10.1038/s41396-022-01251-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yukgehnaish K, Kumar P, Sivachandran P, Marimuthu K, Arshad A, Paray BA, Arockiaraj J. 2020. Gut microbiota metagenomics in aquaculture: factors influencing gut microbiome and its physiological role in fish. Rev Aquacult 12:1903–1927. doi: 10.1111/raq.12416. [DOI] [Google Scholar]
- 52.Wang A, Ran C, Wang Y, Zhang Z, Ding Q, Yang Y, Olsen RE, Ringo E, Bindelle J, Zhou Z. 2019. Use of probiotics in aquaculture of China—a review of the past decade. Fish Shellfish Immunol 86:734–755. doi: 10.1016/j.fsi.2018.12.026. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh K, Ray AK, Ringø E. 2019. Applications of plant ingredients for tropical and subtropical freshwater finfish: possibilities and challenges. Rev Aquacult 11:793–815. doi: 10.1111/raq.12258. [DOI] [Google Scholar]
- 54.Hoseinifar SH, Dadar M, Ringø E. 2017. Modulation of nutrient digestibility and digestive enzyme activities in aquatic animals: the functional feed additives scenario. Aquacult Res 48:3987–4000. doi: 10.1111/are.13368. [DOI] [Google Scholar]
- 55.Zhao Y, Zhao G. 2022. Decreasing ruminal methane production through enhancing the sulfate reduction pathway. Anim Nutr 9:320–326. doi: 10.1016/j.aninu.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thepot V, Campbell AH, Paul NA, Rimmer MA. 2021. Seaweed dietary supplements enhance the innate immune response of the mottled rabbitfish, Siganus fuscescens. Fish Shellfish Immunol 113:176–184. doi: 10.1016/j.fsi.2021.03.018. [DOI] [PubMed] [Google Scholar]
- 57.Sofyan A, Irawan A, Herdian H, Jasmadi Harahap MA, Sakti AA, Suryani AE, Novianty H, Kurniawan T, Darma ING, Windarsih A, Jayanegara A. 2022. Effects of various macroalgae species on methane production, rumen fermentation, and ruminant production: a meta-analysis from in vitro and in vivo experiments. Anim Feed Sci Technol 294:115503. doi: 10.1016/j.anifeedsci.2022.115503. [DOI] [Google Scholar]
- 58.Kumar M, Sun Y, Rathour R, Pandey A, Thakur IS, Tsang DCW. 2020. Algae as potential feedstock for the production of biofuels and value-added products: opportunities and challenges. Sci Total Environ 716:137116. doi: 10.1016/j.scitotenv.2020.137116. [DOI] [PubMed] [Google Scholar]
- 59.Lin Y, Qi X, Liu H, Xue K, Xu S, Tian Z. 2020. The anti-cancer effects of fucoidan: a review of both in vivo and in vitro investigations. Cancer Cell Int 20:154. doi: 10.1186/s12935-020-01233-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knudsen SW, Clements KD. 2013. Revision of the fish family Kyphosidae (Teleostei: Perciformes). Zootaxa 3751:1–101. doi: 10.11646/zootaxa.3751.1.1. [DOI] [PubMed] [Google Scholar]
- 61.Knudsen SW, Choat JH, Clements KD. 2019. The herbivorous fish family Kyphosidae (Teleostei: Perciformes) represents a recent radiation from higher latitudes. J Biogeogr 46:2067–2080. doi: 10.1111/jbi.13634. [DOI] [Google Scholar]
- 62.Brown RM, Wiens GD, Salinas I. 2019. Analysis of the gut and gill microbiome of resistant and susceptible lines of rainbow trout (Oncorhynchus mykiss). Fish Shellfish Immunol 86:497–506. doi: 10.1016/j.fsi.2018.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li M, Li L, Huang T, Liu Y, Lei A, Ma C, Chen F, Chen M. 2018. Effects of attenuated S. agalactiae strain YM001 on intestinal microbiota of tilapia are recoverable. Front Microbiol 9:3251. doi: 10.3389/fmicb.2018.03251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, Bruni L, Jaramillo-Torres A, Gajardo K, Kortner TM, Krogdahl A. 2021. Differential response of digesta- and mucosa-associated intestinal microbiota to dietary insect meal during the seawater phase of Atlantic salmon. Anim Microbiome 3:8. doi: 10.1186/s42523-020-00071-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang R, Pan X, Xu Y. 2020. Altered intestinal microbiota composition associated with enteritis in yellow seahorses Hippocampus kuda (Bleeker, 1852). Curr Microbiol 77:730–737. doi: 10.1007/s00284-019-01859-6. [DOI] [PubMed] [Google Scholar]
- 66.Karnachuk OV, Lukina AP, Kadnikov VV, Sherbakova VA, Beletsky AV, Mardanov AV, Ravin NV. 2021. Targeted isolation based on metagenome-assembled genomes reveals a phylogenetically distinct group of thermophilic spirochetes from deep biosphere. Environ Microbiol 23:3585–3598. doi: 10.1111/1462-2920.15218. [DOI] [PubMed] [Google Scholar]
- 67.Wood DE, Lu J, Langmead B. 2019. Improved metagenomic analysis with Kraken 2. Genome Biol 20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nalley EM, Donahue MJ, Heenan A, Toonen RJ. 2022. Quantifying the diet diversity of herbivorous coral reef fishes using systematic review and DNA metabarcoding. Environ DNA 4:191–205. doi: 10.1002/edn3.247. [DOI] [Google Scholar]
- 69.Clements KD, German DP, Piché J, Tribollet A, Choat JH. 2017. Integrating ecological roles and trophic diversification on coral reefs. Biol J Linnean Soc Lond 120:729–751. doi: 10.1111/bij.12914. [DOI] [Google Scholar]
- 70.Pardesi B, Roberton AM, Lee KC, Angert ER, Rosendale DI, Boycheva S, White WL, Clements KD. 2022. Distinct microbiota composition and fermentation products indicate functional compartmentalization in the hindgut of a marine herbivorous fish. Mol Ecol 31:2494–2509. doi: 10.1111/mec.16394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barras D, Moore A, Ba S. 1969. Enzyme-substrate relationships among β-glucan hydrolases, p 105–138. In Hajny GJ, Reese ET (ed), Cellulases and their applications. American Chemical Society, Washington, DC. [Google Scholar]
- 72.Nguyen STC, Freund HL, Kasanjian J, Berlemont R. 2018. Function, distribution, and annotation of characterized cellulases, xylanases, and chitinases from CAZy. Appl Microbiol Biotechnol 102:1629–1637. doi: 10.1007/s00253-018-8778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Viborg AH, Terrapon N, Lombard V, Michel G, Czjzek M, Henrissat B, Brumer H. 2019. A subfamily roadmap of the evolutionarily diverse glycoside hydrolase family 16 (GH16). J Biol Chem 294:15973–15986. doi: 10.1074/jbc.RA119.010619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Salmean AA, Duffieux D, Harholt J, Qin F, Michel G, Czjzek M, Willats WGT, Herve C. 2017. Insoluble (1→3), (1→4)-β-d-glucan is a component of cell walls in brown algae (Phaeophyceae) and is masked by alginates in tissues. Sci Rep 7:2880. doi: 10.1038/s41598-017-03081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McIntosh M, Stone BA, Stanisich VA. 2005. Curdlan and other bacterial (1→3)-β-d-glucans. Appl Microbiol Biotechnol 68:163–173. doi: 10.1007/s00253-005-1959-5. [DOI] [PubMed] [Google Scholar]
- 76.Kuge T, Nagoya H, Tryfona T, Kurokawa T, Yoshimi Y, Dohmae N, Tsubaki K, Dupree P, Tsumuraya Y, Kotake T. 2015. Action of an endo-β-1,3(4)-glucanase on cellobiosyl unit structure in barley β-1,3:1,4-glucan. Biosci Biotechnol Biochem 79:1810–1817. doi: 10.1080/09168451.2015.1046365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chaari F, Chaabouni SE. 2019. Fungal β-1,3–1,4-glucanases: production, proprieties and biotechnological applications. J Sci Food Agric 99:2657–2664. doi: 10.1002/jsfa.9491. [DOI] [PubMed] [Google Scholar]
- 78.Tsirigotaki A, De Geyter J, Sostaric N, Economou A, Karamanou S. 2017. Protein export through the bacterial Sec pathway. Nat Rev Microbiol 15:21–36. doi: 10.1038/nrmicro.2016.161. [DOI] [PubMed] [Google Scholar]
- 79.Hui X, Chen Z, Zhang J, Lu M, Cai X, Deng Y, Hu Y, Wang Y. 2021. Computational prediction of secreted proteins in Gram-negative bacteria. Comput Struct Biotechnol J 19:1806–1828. doi: 10.1016/j.csbj.2021.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Garcia-Lopez M, Meier-Kolthoff JP, Tindall BJ, Gronow S, Woyke T, Kyrpides NC, Hahnke RL, Goker M. 2019. Analysis of 1,000 type-strain genomes improves taxonomic classification of Bacteroidetes. Front Microbiol 10:2083. doi: 10.3389/fmicb.2019.02083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barbeyron T, Brillet-Gueguen L, Carre W, Carriere C, Caron C, Czjzek M, Hoebeke M, Michel G. 2016. Matching the diversity of sulfated biomolecules: creation of a classification database for sulfatases reflecting their substrate specificity. PLoS One 11:e0164846. doi: 10.1371/journal.pone.0164846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hettle AG, Hobbs JK, Pluvinage B, Vickers C, Abe KT, Salama-Alber O, McGuire BE, Hehemann JH, Hui JPM, Berrue F, Banskota A, Zhang JZ, Bottos EM, Van Hamme J, Boraston AB. 2019. Insights into the κ/ι-carrageenan metabolism pathway of some marine Pseudoalteromonas species. Commun Biol 2:474. doi: 10.1038/s42003-019-0721-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prechoux A, Genicot S, Rogniaux H, Helbert W. 2013. Controlling carrageenan structure using a novel formylglycine-dependent sulfatase, an endo-4S-iota-carrageenan sulfatase. Mar Biotechnol (NY) 15:265–274. doi: 10.1007/s10126-012-9483-y. [DOI] [PubMed] [Google Scholar]
- 84.Donaldson GP, Lee SM, Mazmanian SK. 2016. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14:20–32. doi: 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vaga S, Lee S, Ji B, Andreasson A, Talley NJ, Agreus L, Bidkhori G, Kovatcheva-Datchary P, Park J, Lee D, Proctor G, Ehrlich SD, Nielsen J, Engstrand L, Shoaie S. 2020. Compositional and functional differences of the mucosal microbiota along the intestine of healthy individuals. Sci Rep 10:14977. doi: 10.1038/s41598-020-71939-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stewart RD, Auffret MD, Warr A, Walker AW, Roehe R, Watson M. 2019. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol 37:953–961. doi: 10.1038/s41587-019-0202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Podell S, Ugalde JA, Narasingarao P, Banfield JF, Heidelberg KB, Allen EE. 2013. Assembly-driven community genomics of a hypersaline microbial ecosystem. PLoS One 8:e61692. doi: 10.1371/journal.pone.0061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu G. 2020. Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinformatics 69:e96. doi: 10.1002/cpbi.96. [DOI] [PubMed] [Google Scholar]
- 93.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(Database issue):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 95.Teufel F, Almagro Armenteros JJ, Johansen AR, Gislason MH, Pihl SI, Tsirigos KD, Winther O, Brunak S, von Heijne G, Nielsen H. 2022. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol 40:1023–1025. doi: 10.1038/s41587-021-01156-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Podell S. 2022. Metagenomic co-localization analysis pipeline. GitHub https://github.com/spodell/MCoLA. Accessed 9 June 2022.
- 98.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aem.02154-22-s0001.pdf, PDF file, 0.4 MB (390KB, pdf)
Supplemental material. Download aem.02154-22-s0002.xlsx, XLSX file, 0.01 MB (13.9KB, xlsx)
Supplemental material. Download aem.02154-22-s0003.xlsx, XLSX file, 0.02 MB (23.2KB, xlsx)
Supplemental material. Download aem.02154-22-s0004.xlsx, XLSX file, 0.01 MB (15.5KB, xlsx)
Supplemental material. Download aem.02154-22-s0005.xlsx, XLSX file, 0.02 MB (20.5KB, xlsx)
Supplemental material. Download aem.02154-22-s0006.xlsx, XLSX file, 0.01 MB (11.9KB, xlsx)
Supplemental material. Download aem.02154-22-s0007.xlsx, XLSX file, 3.0 MB (3MB, xlsx)
Supplemental material. Download aem.02154-22-s0008.xlsx, XLSX file, 2.5 MB (2.5MB, xlsx)
Supplemental material. Download aem.02154-22-s0009.xlsx, XLSX file, 0.3 MB (259.8KB, xlsx)
Data Availability Statement
Sequence reads are available under SRA accession no. SRR19136343 through SRR19136358, assembled contigs under WGS accession no. JAMHIX000000000 through JAMHIZ000000000 and JAMHJA000000000 through JAMHJM000000000, and predicted protein sequences in Zenodo (https://zenodo.org) under DOI no. 10.5281/zenodo.6635023 and 10.5281/zenodo.6635166.