

ABSTRACT
Viruses in the family Rhabdoviridae display remarkable genomic variation and ecological diversity. This plasticity occurs despite the fact that, as negative sense RNA viruses, rhabdoviruses rarely if ever recombine. Here, we describe nonrecombinatorial evolutionary processes leading to genomic diversification in the Rhabdoviridae inferred from two novel rhabdoviruses of freshwater mussels (Mollusca: Bivalvia: Unionida). Killamcar virus 1 (KILLV-1) from a plain pocketbook (Lampsilis cardium) is closely related phylogenetically and transcriptionally to finfish-infecting viruses in the subfamily Alpharhabdovirinae. KILLV-1 offers a novel example of glycoprotein gene duplication, differing from previous examples in that the paralogs overlap. Evolutionary analyses reveal a clear pattern of relaxed selection due to subfunctionalization in rhabdoviral glycoprotein paralogs, which has not previously been described in RNA viruses. Chemarfal virus 1 (CHMFV-1) from a western pearlshell (Margaritifera falcata) is closely related phylogenetically and transcriptionally to viruses in the genus Novirhabdovirus, the sole recognized genus in the subfamily Gammarhabdovirinae, representing the first known gammarhabdovirus of a host other than finfish. The CHMFV-1 G-L noncoding region contains a nontranscribed remnant gene of precisely the same length as the NV gene of most novirhabdoviruses, offering a compelling example of pseudogenization. The unique reproductive strategy of freshwater mussels involves an obligate parasitic stage in which larvae encyst in the tissues of finfish, offering a plausible ecological mechanism for viral host-switching.
IMPORTANCE Viruses in the family Rhabdoviridae infect a variety of hosts, including vertebrates, invertebrates, plants and fungi, with important consequences for health and agriculture. This study describes two newly discovered viruses of freshwater mussels from the United States. One virus from a plain pocketbook (Lampsilis cardium) is closely related to fish-infecting viruses in the subfamily Alpharhabdovirinae. The other virus from a western pearlshell (Margaritifera falcata) is closely related to viruses in the subfamily Gammarhabdovirinae, which until now were only known to infect finfish. Genome features of both viruses provide new evidence of how rhabdoviruses evolved their extraordinary variability. Freshwater mussel larvae attach to fish and feed on tissues and blood, which may explain how rhabdoviruses originally jumped between mussels and fish. The significance of this research is that it improves our understanding of rhabdovirus ecology and evolution, shedding new light on these important viruses and the diseases they cause.
KEYWORDS: mussel, fish, ecology, evolution, Rhabdoviridae, Alpharhabdovirinae, Gammarhabdovirinae, Unionidae, Margaritiferidae, Novirhabdovirus
INTRODUCTION
Rhabdoviruses (Rhabdoviridae) are distinctive for their broad host range, diverse ecologies, and varied pathogeneses. Rhabdoviruses infect hosts ranging from mammals to plants, are transmitted directly or by invertebrate vectors, and range from benign commensals to etiologic agents of lethal hemorrhagic diseases in humans and other animals (1, 2). A commensurate degree of genomic plasticity underlies this phenotypic variation (3). Nearly all rhabdovirus genomes contain the canonical N (nucleocapsid), P (phosphoprotein), M (matrix), G (glycoprotein), and L (RNA-directed RNA polymerase; RdRp) gene arrangement, in that order, and many also contain additional open reading frames encoding accessory proteins and noncoding regions that vary markedly in length, even among closely related rhabdoviruses (2, 3). This variation occurs despite the fact that, like all negative sense RNA viruses, rhabdoviruses rarely if ever recombine (4, 5).
Rhabdovirus systematics considers genome architecture, accessory genes, phylogenies based on L protein sequences, cellular tropism, and ecology in delineating taxonomic boundaries (2). Three rhabdoviral subfamilies are thereby currently recognized: Alpharhabdovirinae, Betarhabdovirinae, and Gammarhabdovirinae (2). In addition, seven recognized genera infecting arthropods and nematodes form a clade but are not currently classified to subfamily (2). Rhabdovirus diversity almost certainly exceeds that circumscribed by current taxonomy; for example, approximately 100 relatives of known rhabdoviruses are described but not yet classified (2). Within classified taxa, the gammarhabdoviruses are highly divergent from other subfamilies, so much so that phylogenies of L protein amino acid sequences tend to ally them more closely with viruses in the families Paramyxoviridae, Pneumoviridae, and Filoviridae than with other rhabdoviruses, reflecting deep evolutionary relationships within the order Mononegavirales (1, 6). The gammarhabdoviruses do, however, retain the canonical N-P-M-G-L genomic architecture and bullet-shaped virion morphology, as well as other rhabdoviral genomic and transcriptional regulatory features, by virtue of which they remain rhabdoviruses taxonomically (6).
Rhabdoviruses infecting finfish have been classified in two subfamilies, the Alpharhabdovirinae and Gammarhabdovirinae (6). Viruses in the alpharhabdoviral genera Sprivivirus, Perhabdovirus, Siniperhavirus, and Scophravirus infect diverse families of freshwater and marine fishes and cause morbidity, mass mortality and hemorrhagic disease (6). Viruses in these genera form a monophyletic group within the Alpharhabdovirinae together with viruses in the genera Vesiculovirus and Ledantevirus, which infect terrestrial mammals and arthropods, and Cetarhavirus, which infect marine mammals (6). In contrast, the subfamily Gammarhabdovirinae contains only one genus, Novirhabdovirus, all members of which infect finfish. Novirhabdoviruses cause hemorrhagic, ulcerative, and other diseases that pose serious threats to wild fisheries and aquaculture worldwide (6). The defining genomic characteristic of the novirhabdoviruses is the NV (non-virion) gene between G and L, which is not essential for viability but contributes to efficient replication, immune evasion and virulence (7–12). The evolutionary history of the novirhabdoviruses has remained understudied due to the absence of other described gammarhabdoviruses with which to compare members of this genus.
Here, we provide new evidence of evolutionary processes that have led to genomic and ecological variation within the Rhabdoviridae. In so doing, we describe two novel rhabdoviruses of freshwater mussels (Mollusca: Bivalvia: Unionida) that offer unique illustrations of these processes. One virus is a close relative of the finfish-infecting alpharhabdoviruses and offers a novel example of gene duplication (paralogy). The other virus is a close relative of the novirhabdoviruses and is, to our knowledge, the first member of the Gammarhabdovirinae described from a host other than finfish. The novel gammarhabdovirus also exhibits a striking example of pseudogenization, which may be a common mechanism for generating genomic variation in the rhabdoviruses, especially with respect to length variation in noncoding regions.
RESULTS
Sequencing and virus identification.
Sequencing of hemolymph from two mussels from Indiana and Oregon, USA, resulted in 1,311,555 and 3,076,010 reads of average lengths 161 and 123 nucleotides, respectively, after quality and length trimming. De novo assembly of these reads and additional sequencing to fill gaps and confirm low-coverage regions yielded two contiguous sequences (contigs) with homology to sequences of known rhabdoviruses: a 12,008-nucleotide (nt) contig with average coverage of 1,196 from the Indiana sample, and a 16,076-nucleotide contig with average coverage of 6,433 from the Oregon sample, each containing a coding-complete rhabdoviral genome and partial termini. We name the first virus killamcar virus 1 (KILLV-1; GenBank OQ368743) based on its identification in Kilmore Creek in a plain pocketbook (Lampsilis cardium, family Unionidae). We name the second virus chemarfal virus 1 (CHMFV-1; GenBank OQ368744) based on its identification in the Chehalis River in a western pearlshell (Margaritifera falcata, family Margaritiferidae).
Evolutionary relationships and genome plasticity.
A maximum likelihood phylogenetic tree based on alignments of complete L protein sequences of the Rhabdoviridae shows that KILLV-1 clusters in the subfamily Alpharhabdovirinae within a well-supported subclade that includes finfish-infecting viruses in the genera Perhabdovirus, Siniperhavirus, and Scophravirus (to which it is most closely related) and cetacean-infecting viruses in the genus Cetarhavirus (Fig. 1; Table S1). Pairwise N, G, and L gene nucleotide and amino acid similarities between KILLV-1 and viruses in the genus Scophravirus are approximately equal to those within the scophraviruses, consistent with KILLV-1 being a close phylogenetic outgroup to this genus (Table S2). This subclade is, in turn, most closely related to viruses in the genera Vesiculovirus, Ledantevirus, and the remaining finfish-infecting alpharhabdoviral genus Sprivivirus, although with lower bootstrap support (Fig. 1).
FIG 1.

Maximum likelihood phylogenetic tree of rhabdoviruses based on inferred amino acid sequence of complete L gene ORFs (range of lengths 1874 to 2233 amino acids). Italicized taxa are recognized genera, with triangles proportional in depth to the maximum divergence within each genus. Colored shading indicates the three recognized rhabdovirus subfamilies, and gray shading indicates genera currently unassigned to subfamily. Viruses identified in freshwater mussels (KILLV-1 and CHMFV-1) are in bold type. Colored circles indicate host type(s) associated with each taxon. Ellipses are shaded in proportion to bootstrap values based on 1,000 replicates; only values ≥50% are shown. Scale bar indicates amino acid substitutions per site. Details of viruses and sequences are given in Table S1.
The KILLV-1 genome is similar to that of viruses within these other genera (Fig. 2), with the notable exception of an additional long (1,389 nt) and apparently transcribed ORF (GNS) between G and L (Fig. 2). The canonical N, P, M, G, and L genes and noncoding regions of KILLV-1 are comparable in length and GC content to other viruses within its subclade (Table S3). Individual phylogenies based on N, G, and L support the sister taxon relationship between KILLV-1 and the scophraviruses, with the L phylogeny reflecting established relationships among taxa (2, 6) (Fig. 2). Comparison of the KILLV-1 genome to rhabdoviruses currently unassigned to genus (2) using tblastx (13) revealed a maximum of 52.0% amino acid identity to the L protein of American dog tick rhabdovirus 2 (MF962659) with a query cover of 35% (E-value 0), indicating that KILLV-1 is not closely related to any as-yet unclassified rhabdoviruses.
FIG 2.
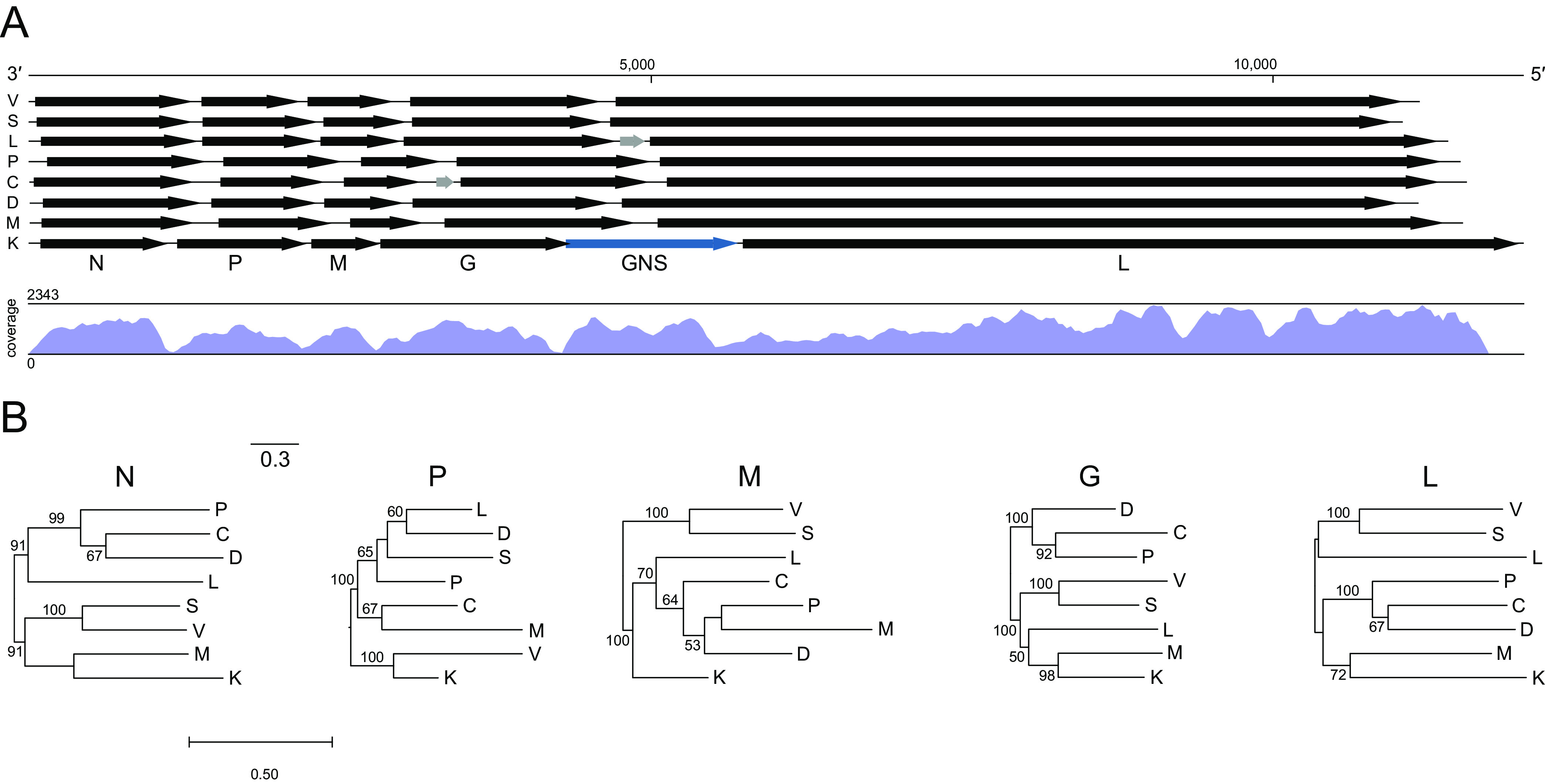
Genome organization, sequence coverage, and gene-specific phylogenies of killamcar virus 1 (KILLV-1) and exemplar viruses of seven genera within the subfamily Alpharhabdovirinae. Viruses are Siniperca chuatsi rhabdovirus (C; Siniperhavirus), dolphin rhabdovirus (D; Cetarhavirus), Le Dantec virus (L; Ledantevirus), Scophthalmus maximus rhabdovirus (M; Scophrhavirus), perch rhabdovirus (P; Perhabdovirus), spring viremia of carp virus (S; Sprivivirus), vesicular stomatitis Indiana virus (V; Vesiculovirus), and KILLV-1 (K; currently unassigned) (Table S1). Arrows (A) indicate open reading frames (scale = nucleotides). Black arrows indicate canonical rhabdovirus gene ORFs in the order nucleoprotein (N), phosphoprotein (P), matrix (M), glycoprotein (G), and RNA-directed RNA polymerase (L); gray arrows indicate accessory genes; and blue arrow indicates the KILLV-1 duplicated glycoprotein gene (GNS). Letters beneath ORFs refer to KILLV-1. Map shows coverage of the KILLV-1 genome. Maximum likelihood phylogenies (B) are shown for each of the canonical rhabdoviral gene ORF nucleotide sequences. Bootstrap values above branches are based on 1,000 replicates (only values ≥50% are shown). Scale bar indicates nucleotide substitutions per site.
Based on the same maximum likelihood phylogenetic tree, CHMFV-1 forms a close outgroup to viruses in the genus Novirhabdovirus, the only currently recognized genus in the Gammarhabdovirinae, all of which infect finfish (Fig. 1). The evolutionary distance between CHMFV-1 and the novirhabdoviruses based on this phylogeny is less than the maximum divergence within many recognized rhabdoviral genera in other subfamilies. However, pairwise N, G, and L nucleotide and amino acid similarities between CHMFV-1 and viruses in the genus Novirhabdovirus are nearly uniformly lower than those within the novirhabdoviruses, consistent with CHMFV-1 being outgroup to this genus (Table S2).
The CHMFV-1 genome differs from that of the novirhabdoviruses in several important ways. First, all CHMFV-1 ORFs except for L and all noncoding regions are all substantially longer than their homologs in the novirhabdoviruses (Fig. 3; Table S4). Second, the GC content of all CHMFV-1 genes and noncoding regions is notably lower than for the novirhabdoviruses (Table S4). Third, the CHMFV-1 genome contains two accessory genes (U1 and U2) between M and G with no detectable homology to other proteins in NCBI databases, and this is a feature not shared with any classified novirhabdoviruses (Fig. 3). Individual phylogenies based on all five canonical gene ORFs support an outgroup relationship between CHMFV-1 and the novirhabdoviruses, with the L phylogeny reflecting established relationships among viruses within the genus (2, 14) (Fig. 3). Comparison of the CHMFV-1 genome to rhabdoviruses currently unassigned to genus (2) using tblastx (13) revealed a maximum of 32.7% amino acid identity to Diachasmimorpha longicaudata rhabdovirus (KP735609) with a query cover of 8% (E-value 2e-35), indicating that CHMFV-1 is not closely related to any as-yet unclassified rhabdoviruses.
FIG 3.
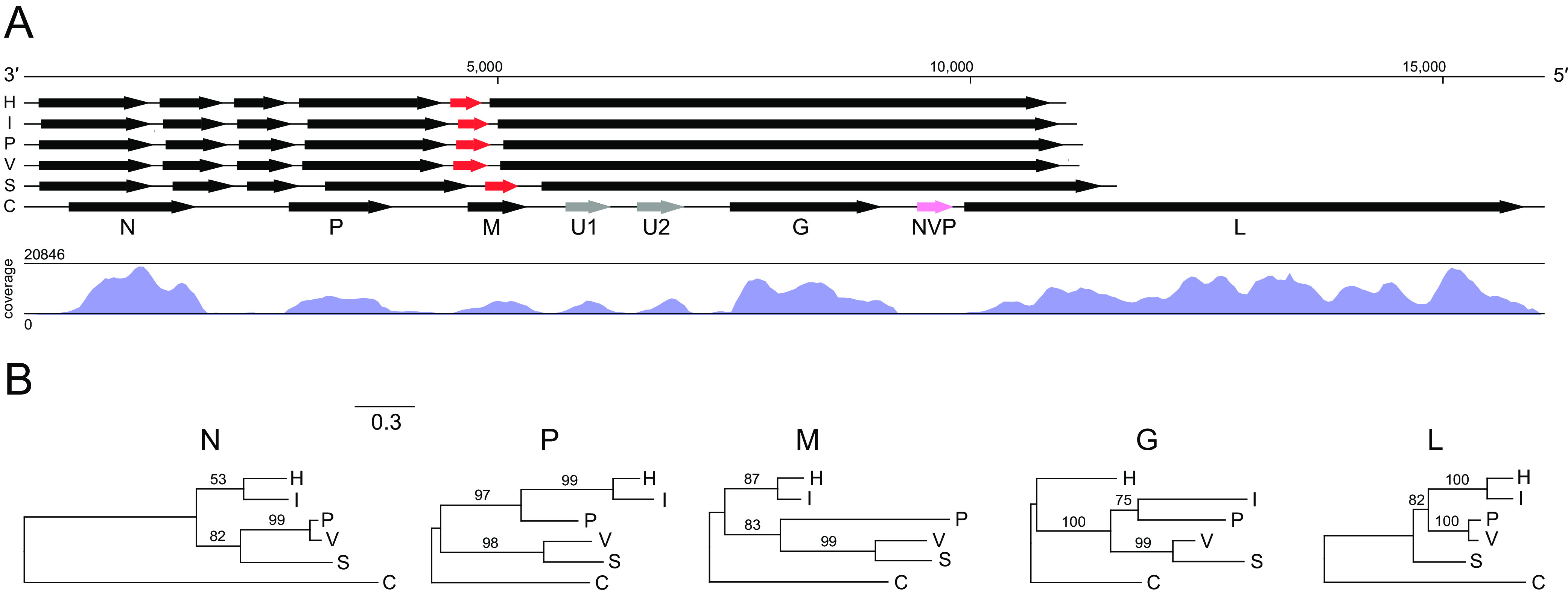
Genome organization, sequence coverage, and gene-specific phylogenies of chemarfal virus 1 (CHMFV-1) and viruses within the subfamily Gammarhabdovirinae, genus Novirhabdovirus. Viruses are hirame rhabdovirus (H), infectious hematopoietic necrosis virus (I), Paralichthys olivaceus rhabdovirus (P), viral hemorrhagic septicemia virus (V), snakehead rhabdovirus (S), and CHMFV-1 (C) (Table S2). Arrows (A) indicate open reading frames (scale = nucleotides). Black arrows indicate canonical rhabdovirus gene ORFs in the order nucleoprotein (N), phosphoprotein (P), matrix (M), glycoprotein (G), and RNA-directed RNA polymerase (L); gray arrows indicate accessory genes, red arrows indicate novirhabdovirus NV genes; and pink arrow indicates the CHMFV-1 NVP pseudogene. Letters beneath ORFs refer to CHMFV-1. Map shows coverage of the CHMFV-1 genome. Maximum likelihood phylogenies (B) are shown for each of the canonical rhabdoviral gene ORF nucleotide sequences. Bootstrap values above branches are based on 1,000 replicates (only values ≥50% are shown). Scale bar indicates nucleotide substitutions per site. Details of viruses and sequences are given in Tables S1 and S3.
Transcriptional regulation.
Sequences of KILLV-1 from hemolymph of an individual L. cardium show a clear pattern of high coverage (maximum 2,343-fold) in the coding regions and low coverage (minimum 7-fold) in the noncoding regions, indicating active transcription of the ORFs (Fig. 2A). Similarly, sequences of CHMFV-1 from hemolymph of an individual M. falcata show a clear pattern of very high coverage (maximum 20,846-fold) in the coding regions and low coverage (minimum 5-fold) in the noncoding regions, also indicating active transcription of the ORFs (Fig. 3A).
Transcriptional regulatory sequences are highly conserved among KILLV-1 and its relatives in the Alpharhabdovirinae (Fig. 4). Specifically, transcription termination/polyadenylation (TTP) and transcription initiation (TI) consensus sequences are highly conserved among all KILLV-1 genes, being identical to those of viruses in the genera Vesiculovirus, Sprivivirus, Perhabdovirus, Siniperhavirus, and Cetarhavirus, and differing by only single nucleotides from those of viruses in the genera Ledantevirus and Scophravirus (Fig. 4). Although less highly conserved, KILLV-1 intergenic sequences (IS) are similar in nucleotide composition and length to those of viruses in the aforementioned genera (Fig. 4).
FIG 4.

Transcriptional regulatory minimum sequences of killamcar virus 1 (KILLV-1) and exemplar viruses of seven genera within the subfamily Alpharhabdovirinae: Siniperca chuatsi rhabdovirus (SCRV), dolphin rhabdovirus (DRV), Le Dantec virus (LDV), Scophthalmus maximus rhabdovirus (SMRV), perch rhabdovirus (PRV), spring viremia of carp virus (SVCV), and vesicular stomatitis Indiana virus (VSIV) (Table S1). For each virus, putative transcription termination/polyadenylation (TTP), intergenic sequence (IS), and transcription initiation (TI) sequences are shown. Colored shading indicates nucleotides within TTP and TI that differ from the KILLV-1 consensus.
CHMFV-1 regulatory sequences are highly similar to those of novirhabdoviruses (Fig. 5), with the intriguing exception of regulatory sequences between NVP and L (see below). Specifically, the CHMFV-1 TI consensus sequence is identical to that of all novirhabdoviruses, and the CHMFV-1 TTP consensus sequence is identical to three of five novirhabdoviruses (VHSV, PORV, and HIRRV) and differs from TTP consensus sequences of the remaining two novirhabdoviruses (IHNV and SHRV) by only a single nucleotide (Fig. 5). Furthermore, the CHMFV-1 IS consensus sequence is one nucleotide in length (A) and is identical in this respect to IS consensus sequences of all novirhabdoviruses except SHRV (Fig. 5). However, CHMFV-1 IS sequences flanking NVP differ from the consensus, with a putative 26-base insertion in the IS between G and NVP and a point substitution (A → U) in the IS between NVP and L.
FIG 5.

Transcriptional regulatory minimum sequences of chemarfal virus 1 (CHMFV-1) and viruses in the subfamily Gammarhabdovirinae, genus Novirhabdovirus: viral hemorrhagic septicemia virus (VHSV), Paralichthys olivaceus rhabdovirus (PORV; not be confused with Porton virus, another rhabdovirus), infectious hematopoietic necrosis virus (IHNV), hirame rhabdovirus (HIRRV), snakehead rhabdovirus (SHRV), and CHMFV-1 (Table S2). For each virus, putative transcription termination/polyadenylation (TTP), intergenic sequence (IS), and transcription initiation (TI) sequences are shown. Colored shading indicates nucleotides within TTP and TI that differ from the CHMFV-1 consensus.
Paralogy.
A notable feature of the KILLV-1 genome is a large ORF between G and L (Fig. 2; Table S3). This gene shows clear similarity to the KILLV-1 G gene, suggesting that it resulted from a gene duplication event, making KILLV-1 similar in this regard to all recognized members of the genera Ephemerovirus and Alphapaprhavirus and to one member of the genus Hapavirus (Ngaingan virus) (15–17). Unlike these other viruses, however, the KILLV-1 G and GNS genes overlap, with the AUG start codon of the GNS ORF 40 nt upstream of the TAA termination codon of the G ORF, causing GNS to be encoded in frame +2 relative to G. A maximum likelihood phylogenetic tree of G and GNS genes from members of the Alpharhabdovirinae shows that these putative paralogs form sister taxa in only one of four cases (Hubei lepidoptera virus 2 in the genus Alphapaphravirus) (Fig. 6). A maximum likelihood phylogenetic tree of all recognized ephemeroviruses shows clear evidence of duplication and parallel evolution, indicated by two well defined clades representing G and GNS and nearly identical topologies within each clade (Fig. 7). In both trees (Fig. 6 and 7), branches leading to GNS are longer than corresponding branches leading to G, indicating a greater amount of evolutionary change along GNS lineages than along corresponding G lineages.
FIG 6.

Maximum likelihood phylogenetic tree of viruses in the subfamily Alpharhabdovirinae based on aligned glycoprotein gene sequences. Viruses with putative G gene duplicates (GNS; arrows) are colored; like coloring indicates sequences from the same virus. Genus names are in parentheses. Because all currently recognized ephemeroviruses contain both G and GNS genes, only one exemplar member of this genus (BEFV) was included (see Fig. 7). Uncolored viruses are exemplars of genera with only a single (i.e., nonduplicated) G gene. Numbers above branches are bootstrap values based on 1,000 replicates; only values ≥50% are shown. Scale bar indicates nucleotide substitutions per site. Details of viruses and sequences are given in Table S1.
FIG 7.

Maximum likelihood phylogenetic tree of glycoprotein genes (G) and their paralogs (GNS) of viruses in the genus Ephemerovirus. All currently recognized ephemeroviruses contain putative G gene duplicates (GNS) and were therefore included. Like coloring indicates sequences from the same virus. Numbers above branches are bootstrap values based on 1,000 replicates; only values ≥50% are shown. Scale bar indicates nucleotide substitutions per site. Viruses and GenBank accession numbers are: Hayes Yard virus (HYV, MH507506); Puchong virus (PUCV, MH507505); Berrimah virus (BRMV, NC_025358); bovine ephemeral fever virus (BEFV, NC_002526); Kimberley virus (KIMV, NC_025396); New Kent County virus (NKCV, MF615270); Kotonkan virus (KOTV, NC_017714); Koolpinyah virus (KOOLV, NC_028239); Yata virus (YATV, NC_028241), Obodhiang virus (OBOV, NC_017685), and Adelaide River virus (ARV, NC_028246).
Selection analyses using BUSTED (18) with synonymous rate variation uncovered no evidence of gene-wide episodic diversifying selection along the GNS branches of the alpharhabdovirus G/GNS phylogeny (Fig. 6; P = 0.500). The same analysis also found no evidence of gene-wide episodic diversifying selection along the GNS branches of the Ephemerovirus G/GNS phylogeny (Fig. 7.; P = 0.500). However, analyses using RELAX (19) found strong evidence of relaxed selection along the GNS branches of the alpharhabdovirus G/GNS phylogeny (Fig. 6; K = 0.64; likelihood ratio = 21.61; P < 0.001) and along the GNS branches of the Ephemerovirus G/GNS phylogeny (Fig. 7; K = 0.77; likelihood ratio = 11.29; P = 0.001).
Pseudogenization.
A key feature of the CHMFV-1 genome is the presence of an NV-like sequence between the G and L genes. This sequence lacks a start codon but is uninterrupted by stop codons. Sequence coverage within this region is low (minimum 9-fold), similar to that of CHMFV-1 noncoding regions (Fig. 3), indicating that it is probably not actively transcribed or is transcribed at a very low level. The minimum change necessary to reconstitute a start codon for a maximum-length ORF within this region is a single nucleotide substitution at position 385 of the region (ATT to ATG). A start codon at this position would resurrect an intact putative ORF of 369 nucleotides, which is precisely the same length as the NV genes of VHSV, PORV, and SHRV (the IHNV and HIRRV NV genes are each 336 nt). We therefore name the CHMFV-1 NV-like sequence NVP, to indicate its likely origin through pseudogenization of an ancestral gene encoding a functional NV-like protein. Attempts to align NVP with novirhabdoviral NV genes were unsuccessful, likely due to very low conservation among NV genes (20) and accumulated random mutations in NVP since its pseudogenization. As described above, transcriptional regulatory sequences of CHMFV-1 differ from those of the novirhabdoviruses only in regions flanking NVP (Fig. 5), where loss of functional transcriptional signals might be expected due to relaxed purifying selection following pseudogenization.
Virus isolation.
Cytopathic effect (CPE) was observed and supernatants were passaged seven times on CHMFV-1-inoculated fathead minnow skin (FHMSkin) cells and eight times on epithelioma papulosum cyprini (EPC) cells, both of which are highly permissive for finfish-infecting rhabdoviruses (21). However, cell death was not observed earlier with each passage, as would be expected if viral replication were occurring, and lag times increased with each reset. Real-time quantitative PCR (RT-qPCR) assays revealed decreasing virus genome concentration with each passage, and the rate of decrease corresponded to the dilution factor, indicating dilution of inoculum rather than virus growth. Thus, virus isolation attempts were unsuccessful and CPE had likely resulted from cytotoxicity. Material suitable for isolation of KILLV-1 was unfortunately not available.
DISCUSSION
Evolutionary processes other than intermolecular recombination have led to variation in genome size and complexity in the Rhabdoviridae, underlying the broad phenotypic diversity among viruses in this family and their important pathogenic effects on hosts ranging from humans to plants (3). Here, we describe novel examples of two such processes, gene duplication and pseudogenization, in newly described rhabdoviruses of freshwater mussels, a host taxon in which rhabdoviruses have not previously been described, to our knowledge. Phylogenetic inferences and analyses of transcriptional regulation yield further insights into the evolutionary history of finfish-infecting rhabdoviruses in the subfamilies Alpharhabdovirinae and Gammarhabdovirinae.
Gene duplication is well documented in rhabdoviruses and is thought to play a major role in the evolution of genome size and complexity (3, 16, 22, 23). The KILLV-1 G and GNS genes provide an example of this phenomenon that is phylogenetically independent from other previously documented examples. Duplication of an ancestral glycoprotein gene must have occurred subsequent to the divergence of KILLV-1 from viruses in the genus Scophravirus, which lack this feature. The KILLV-1 G and GNS genes also overlap, which is the first documented example of overlapping rhabdoviral glycoprotein paralogs. Gene overlap in RNA viruses is thought to evolve due to selection for genetic novelty that is balanced by constraints on genome size (24–27). In the case of KILLV-1, the transcription termination signal for G is located entirely within the coding region of GNS, offering an elegant example of how these opposing selective forces operate.
Our analyses of Ephemerovirus G and GNS genes offer new insights into the evolution of paralogs. The ephemerovirus phylogeny in Fig. 7 shows nearly identical topologies in the G and GNS clades, supporting a single duplication event near the origin of the Ephemerovirus lineage followed by parallel evolution of paralogs. In the G/GNS phylogenies of both the alpharhabdoviruses (Fig. 6) and the ephemeroviruses (Fig. 7), GNS branches are nearly always longer than corresponding G branches. Analyses of selection clearly show this trend to have resulted from relaxed selection in GNS. Under this mechanism, duplicated genes retain some functions of their paralogs but not others, leading to “specialization” or “subfunctionalization,” reduced stabilizing selection in the duplicated genes, and increased rates of neutral substitution (28, 29). Our analyses strongly support a scenario of glycoprotein gene duplication followed by relaxed selection due to subfunctionalization of paralogs, which has previously been described in eukaryotes and DNA viruses but not in RNA viruses (30–32).
The CHMFV-1 genome is noteworthy for several reasons. First, the CHMFV-1 coding complete genome is approximately 41% longer than the average genome size of the novirhabdoviruses. This difference results from nearly universally longer ORFs (with the exception of the highly conserved L ORF) and noncoding regions in CHMFV-1 than in corresponding regions of the novirhabdoviruses, and from the presence in CHMFV-1 of two accessory protein genes. Second, the CHMFV-1 coding complete genome has approximately 69% lower GC content than the average GC content of the novirhabdoviruses. This difference results from far lower GC content in all ORFs and noncoding regions of CHMFV-1 than in corresponding regions of the novirhabdoviruses.
Third, and perhaps most interestingly, CHMFV-1 contains NVP, a pseudogenized version of the novirhabdovirus NV gene. Low sequence coverage of NVP and mutated transcriptional regulatory sequences associated with NVP indicate that it is likely not transcribed. A similar phenomenon has been documented for rabies virus (RABV), in which a remnant protein gene was inferred within the 423-base noncoding region between G and L (33). However, all three reading frames within the RABV remnant gene contain stop codons. In the case of CHMFV-1, NVP contains no internal stop codons and requires only a single nucleotide change to reconstitute an ORF of 369 nucleotides, which is identical in length to the NV gene ORFs of VHSV, PORV, and SHRV. Phylogenetic relationships within the Gammarhabdovirinae imply that the last common ancestor of CHMFV-1 and the novirhabdoviruses had a functional NV gene and that loss of a start codon and nonfunctionalization of transcriptional regulatory sequences occurred along the CHMFV-1 lineage, making the gene vestigial. This observation is important because it indicates that NV originated prior to the last common ancestor of the novirhabdoviruses.
KILLV-1 clusters within a subclade containing all alpharhabdoviral genera infecting finfish (Fig. 1 and 2). It is most closely related to viruses in the newly recognized genus Scophravirus, which infect turbot (Scophthalmus maximus) and redfin culter (Culter erythropterus) (6). Phylogenetic distance between KILLV-1 and the scophraviruses is greater than the maximum divergence within recognized genera in this subclade, with the exception of Ledantevirus (Fig. 1), suggesting that KILLV-1 likely merits classification within a new genus. Transcriptional regulatory sequences of viruses within this subclade are highly conserved, with KILLV-1 TTP and TI consensus sequences identical to those of viruses in six of eight genera (Fig. 4). A host switch between mollusk and finfish may therefore be inferred, although the direction of that event remains unclear.
CHMFV-1 is a close phylogenetic outgroup of the novirhabdoviruses (Fig. 1 and 3). Transcriptional regulatory sequences are highly conserved between CHMFV-1 and the novirhabdoviruses: TI consensus sequences are identical in CHMFV-1 and all novirhabdoviruses, and CHMFV-1 TTP consensus sequences are identical to those of VHSV, PORV, and HIRRV, differing from those of IHNV and SHRV by only a single nucleotide (Fig. 5). Based on L gene phylogeny alone, CHMFV-1 might be considered a member of the genus Novirhabdovirus, given that it is less divergent from recognized novirhabdoviruses than are congeneric viruses in other subfamilies (Fig. 1). However, CHMFV-1 has a markedly different genome size and architecture, host, and lack of a functional NV gene, all of which suggest that CHMFV-1 likely merits classification within a new genus. These findings are significant because, to date, Novirhabdovirus has been the only recognized genus within the Gammarhabdovirinae, and novirhabdoviruses universally infect finfish (6). As with KILLV-1, a host switch between mollusk and finfish may be inferred for CHMFV-1, although again the direction of that event remains unclear.
Rhabdoviruses are ubiquitous among invertebrates, but to our knowledge KILLV-1 and CHMFV-1 represent the first rhabdoviruses definitively identified in mollusks. Two rhabdovirus-like sequences (Caledonia dog whelk rhabdo-like virus 1 and Caledonia dog whelk rhabdo-like virus 2) have been reported in pooled samples of Nucella lapillus, a predatory marine snail (34). In the case of Caledonia dog whelk rhabdo-like virus 1, blastp of a partial (8223 nucleotide) sequence of the L gene (MF190042.1) yields hits to viruses in multiple non-rhabdoviral families, including a top hit to hymenopteran arli-related virus (QPL15345; E-value 8e-11) and a more distant hit to Behai rhabdo-like virus 2 (YP_009333449; E-value 8e-6), which, despite its original name, has since been reclassified into the family Artoviridae, genus Peropuvirus (35). In the case of Caledonia dog whelk rhabdo-like virus 2, amplification using RT-negative PCR suggests that this partial (3780 nucleotide) sequence of the N gene may be an endogenized viral element (34).
The unique reproductive and dispersal strategy of freshwater mussels may provide a mechanism for transmission of unionid viruses to and from finfish, adding to our knowledge of how rhabdoviruses switch hosts in general. Mussels in the family Unionidae, tribes Quadrulini and Lampsilini (to which L. cardium belongs) have evolved elaborate “mantle lures” (36, 37). These specialized structures mimic the morphology and behavior of small fish, sometimes in exquisite detail (36). When a predatory fish strikes the mantle lure of a gravid female, the mussel expels larvae (glochidia) from specialized structures (marsupial gills) (37). These larvae attach to the gills or fins of the fish, encyst and derive nutrients from fish blood and tissues, complete metamorphosis and excyst, then drop off to begin their sessile, filter-feeding life stage (37). The process is similar for mussels in the family Margaritiferidae, to which M. falcata belongs, except that these mussels lack mantle lures and instead release masses of glochidia (conglutinates) into the water column (36). During broadcast spawning events, M. falcata releases white, dendritic masses of glochidia that attach to the gills of host fish as they filter water or consume the masses (36, 38). In both the Unionidae and the Margaritiferidae, this obligate parasitic stage lasts for up to several weeks (39). Direct contact with fish blood and tissues during this period might be expected to facilitate transmission of a vertically transmitted mussel virus to a finfish or of a blood-borne finfish virus to a mussel.
L. cardium primarily uses fishes in the family Centrarchidae (black basses and sunfish) as larval hosts, with infection of other host fishes and even salamanders possible (40–42). In contrast M. falcata uses fishes in the family Salmonidae such as Pacific salmon and trout (genus Oncorhynchus) as larval hosts, as do most other mussels in the genus Margaritifera (43). Finfish-infecting viruses in the Alpharhabdovirinae (to which KILLV-1 is closely related) infect marine and freshwater fishes in diverse families, including centrarchids, but only one of 10 described viruses (LTRV; Perhabdovirus) infects salmonids (6). In contrast, three of four recognized novirhabdoviruses (to which CHMFV-1 is closely related) infect salmonids: IHNV infects only salmonids, VHSV infects salmonids and other fishes, and HIRRV typically infects non-salmonids but has been found in rainbow trout (O. mykiss) (6). Thus, the host ranges of the finfish-infecting alpharhabdoviruses and the novirhabdoviruses roughly correspond to the families of finfish used as larval hosts by mussels in the tribe Lampsilini (Unionidae) and the family Margaritiferidae, respectively. This observation concords with the idea that, over their evolutionary history, rhabdoviruses have occasionally infected distantly related host species then subsequently diversified across related hosts inhabiting similar environments (44).
Freshwater mussels are among the world’s most imperiled taxa and are declining precipitously, but the causes of their decreasing numbers remain enigmatic (45, 46). Previous studies show that some mass mortality events in wild freshwater mussels are associated with viruses and bacteria (47–50), but the mechanism and direction of association are unclear. At present, we do not know whether KILLV-1 and CHMFV-1 cause disease in mussels, finfish, or other species. Mass mortality caused by Lea plague virus (Arenaviridae) has been documented in triangleshell (Hyriopsis cumingii) farmed for freshwater pearl production, but the high density of these mussels in captivity could contribute to epidemic spread and virulence (51, 52). Further research into the pathogenicity of KILLV-1 and CHMFV-1 must likely await their isolation, which we unfortunately did not accomplish as part of this study. Indeed, a major barrier to progress in the field of freshwater mussel biology and virology is a lack of any freshwater mussel cell lines (53), which members of our team have attempted for years to create but without success. Until adequate tools and reagents become available, ascertaining the role of viruses in the global decline of freshwater mussels will likely remain problematic.
MATERIALS AND METHODS
Locations and samples.
Freshwater mussels were sampled as part of an effort to identify pathogens associated with mass mortality events in these highly imperiled mollusks (54). The L. cardium individual from which KILLV-1 was identified was collected from Kilmore Creek near Mulberry, Indiana, on July 2, 2019 (mussel ID: PP32). The M. falcata individual from which CHMFV-1 was identified was collected from the Chehalis River near Oakville, Washington, on September 26, 2018 (mussel ID: CWS06), wrapped in wet paper towels, and shipped overnight to the La Crosse Fish Health Center for processing. Hemolymph was collected nonlethally as previously described (49). Briefly, the epithelium of the anterior adductor muscle was disinfected using 70% isopropanol, and a 1 mL tuberculin syringe was used to remove approximately 0.5 mL hemolymph from the anterior adductor muscle sinus. Samples were placed in sterile cryovials, frozen immediately after collection, and stored at −80°C in the laboratory until further analysis.
Virus identification.
Viruses were identified using methods previously described for freshwater mussel hemolymph (49, 55). Briefly, 250 μL of hemolymph was centrifuged for 10 min at 10,000 × g to pellet cellular debris, then total nucleic acids were extracted from 200 μL of supernatant using the QIAamp MinElute Virus Spin Kit (Qiagen, Hilden, Germany), omitting carrier RNA. RNA was then reverse transcribed to cDNA using the Superscript IV system (Thermo Fisher Scientific, Waltham, MA, USA) with random hexamers, and libraries were prepared using the Nextera XT DNA sample preparation kit (Illumina, San Diego, CA, USA). Libraries were sequenced using the MiSeq reagent kit v3 (Illumina, San Diego, CA, USA), and sequencing adapters were trimmed using on-board Illumina software.
Resulting sequence reads were trimmed for quality (Phred score ≤ 30) and length (≤50 bases), and sequences corresponding to reagent contaminants were removed by in silico subtraction using CLC Genomics Workbench v20.0.1 (Qiagen, Hilden, Germany). Remaining reads were assembled de novo using SPAdes-meta and SPAdes-metaviral v3.15.5 (56), and resulting assemblies were assessed for quality using metaQUAST v5.2.0 (57). Contigs were then queried against viruses in the NCBI databases using BLAST+ 2.13.0 (58) and DIAMOND v2.0.15 (59), and ORFs were identified using ORFfinder (60). Due to the unusual genomic architecture of CHMFV-1, sequence analyses originally conducted at the University of Wisconsin-Madison were repeated and verified at the Naval Medical Research Center–Frederick. Contigs other than those corresponding to KILLV-1 and CHMFV-1 were not analyzed as part of this study.
Phylogenetic inference and analyses of selection.
Sequences of KILLV-1 and CHMFV-1 were aligned with those of other rhabdoviruses (Table S1) using T-Coffee (61), with minor manual adjustments, and poorly aligned regions were filtered using Trim AI (62). Maximum likelihood phylogenetic trees were inferred using PhyML 3.0 (63, 64) with smart model selection (65) and 1,000 bootstrap replicates to assess statistical support. Trees were displayed using Dendroscope v3.7.6 (66). To test for gene-wide episodic diversifying selection and selection relaxation, BUSTED (18) and RELAX (19), respectively, were implemented within Datamonkey 2.0 (67).
Virus isolation.
Hemolymph samples positive for CHMFV-1 via metagenomics were inoculated onto EPC and FHMSkin cells, which are permissive for finfish-infecting rhabdoviruses (21, 68). Briefly,3 to 10 μL of 0.8/0.2 μm filtered hemolymph was placed onto confluent cell monolayers in 96-well plates. Cells were then incubated at 15°C and monitored weekly, and wells showing CPE were serially passaged (21). Unfortunately, hemolymph samples containing KILLV-1 were depleted during molecular analyses, so isolation could not be attempted for this virus.
To assess viral replication in culture, a RT-qPCR was designed targeting a 108 bp portion of the CHMFV-1 L gene. Reactions (20 μL) were run on a Bio-Rad CFX96 Touch real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA) using the GoTaq Probe-1 Step RT-qPCR System (Promega, Madison, WI) with 700 nM (each) forward (5′-GTGTGACATCACTAGGCCTTAC-3′) and reverse (5′-GGGAGTTCCGACATTCTTACTG-3′) primers and 350 nM probe (5′-FAM/TCAATCCTG/ZEN/CCCAAACATACTCGCT/IABkFQ-3′). Cycling parameters were: 45°C for 15 min, 95°C for 2 min, followed by 45 cycles of (95°C for 15 sec, 60°C for 1 min). RT-qPCR sensitivity (limit of detection of 10 copies per reaction) and quantitation performance (limit of quantification of 100 copies per reaction) (69) were determined using a synthetic gBlock oligonucleotide (IDT, Coralville, IA) matching the target sequence plus 60 nucleotides on each end (300 nucleotides total) (70). Viral RNA was then extracted from 200 μL of supernatant using the QIAamp UltraSens Virus Kit (Qiagen, Germantown, MD) and used as template (2.5 μL) in reactions run in triplicate.
Data availability.
Virus genome sequence data generated as part of this study are available in GenBank under accession numbers OQ368743.1 (KILLV-1) and OQ368744.1 (CHMFV-1).
ACKNOWLEDGMENTS
We thank Liz Bockstiegel, Nancy Bodecker, Megan Bradley, Serena Ciparis, and Miranda Plumb for assisting with field work; Christopher Dunn for assistance with laboratory analyses; and Chase Nelson for advice on statistical methods. We also thank Gael Kurath and one anonymous reviewer for constructive comments on the manuscript. We are grateful to the Xerces Society for the Conservation of Invertebrates, the Indiana Department of Natural Resources, the Oregon Department of Fish & Wildlife, and the Washington State Department of Natural Resources for administrative and logistical support.
The project was supported by the U.S. Fish and Wildlife Service under awards 140F0319P0126 and F21AC00707 (T.L.G., principal investigator [PI]), by the Armed Forces Health Surveillance Division (AFHSD), Global Emerging Infections Surveillance (GEIS) Branch, under award ProMIS ID P0167_22_NM and Navy WUN A1417 (K.B.L., PI). The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of Defense, Department of the Navy, nor the U.S. Government. Several of the authors are U.S. Government employees. This work was prepared as part of their official duties. Title 17 U.S.C. § 105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties. Usage of trade names does not imply endorsement by the U.S. Government.
Footnotes
Supplemental material is available online only.
Contributor Information
Tony L. Goldberg, Email: tony.goldberg@wisc.edu.
Anice C. Lowen, Emory University School of Medicine
REFERENCES
- 1.Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, Qin XC, Xu J, Holmes EC, Zhang YZ. 2015. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 4. doi: 10.7554/eLife.05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker PJ, Freitas-Astua J, Bejerman N, Blasdell KR, Breyta R, Dietzgen RG, Fooks AR, Kondo H, Kurath G, Kuzmin IV, Ramos-Gonzalez PL, Shi M, Stone DM, Tesh RB, Tordo N, Vasilakis N, Whitfield AE, ICTV Report Consortium . 2022. ICTV virus taxonomy profile: Rhabdoviridae 2022. J Gen Virol 103. doi: 10.1099/jgv.0.001689. [DOI] [PubMed] [Google Scholar]
- 3.Walker PJ, Firth C, Widen SG, Blasdell KR, Guzman H, Wood TG, Paradkar PN, Holmes EC, Tesh RB, Vasilakis N. 2015. Evolution of genome size and complexity in the Rhabdoviridae. PLoS Pathog 11:e1004664. doi: 10.1371/journal.ppat.1004664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han GZ, Worobey M. 2011. Homologous recombination in negative sense RNA viruses. Viruses 3:1358–1373. doi: 10.3390/v3081358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deviatkin AA, Lukashev AN. 2018. Recombination in the rabies virus and other lyssaviruses. Infect Genet Evol 60:97–102. doi: 10.1016/j.meegid.2018.02.026. [DOI] [PubMed] [Google Scholar]
- 6.Walker PJ, Bigarre L, Kurath G, Dacheux L, Pallandre L. 2022. Revised taxonomy of rhabdoviruses infecting fish and marine mammals. Animals (Basel) 12:1363. doi: 10.3390/ani12111363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurath G, Leong JC. 1985. Characterization of infectious hematopoietic necrosis virus mRNA species reveals a nonvirion rhabdovirus protein. J Virol 53:462–468. doi: 10.1128/JVI.53.2.462-468.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjooklund HV, Kurath G, Higman KH. 1997. Distribution and variation of NV genes in fish rhabdoviruses. J Gen Virol 78:113–117. doi: 10.1099/0022-1317-78-1-113. [DOI] [PubMed] [Google Scholar]
- 9.Thoulouze M-I, Bouguyon E, Carpentier C, Brémont M. 2004. Essential role of the NV protein of Novirhabdovirus for pathogenicity in rainbow trout. J Virol 78:4098–4107. doi: 10.1128/jvi.78.8.4098-4107.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ammayappan A, Vakharia VN. 2011. Nonvirion protein of novirhabdovirus suppresses apoptosis at the early stage of virus infection. J Virol 85:8393–8402. doi: 10.1128/JVI.00597-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi MK, Moon CH, Ko MS, Lee UH, Cho WJ, Cha SJ, Do JW, Heo GJ, Jeong SG, Hahm YS, Harmache A, Bremont M, Kurath G, Park JW. 2011. A nuclear localization of the infectious haematopoietic necrosis virus NV protein is necessary for optimal viral growth. PLoS One 6:e22362. doi: 10.1371/journal.pone.0022362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim MS, Kim KH. 2013. The role of viral hemorrhagic septicemia virus (VHSV) NV gene in TNF-alpha- and VHSV infection-mediated NF-kappaB activation. Fish Shellfish Immunol 34:1315–1319. doi: 10.1016/j.fsi.2013.02.026. [DOI] [PubMed] [Google Scholar]
- 13.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim D-H, Oh H-K, Eou J-I, Seo H-J, Kim S-K, Oh M-J, Nam S-W, Choi T-J. 2005. Complete nucleotide sequence of the hirame rhabdovirus, a pathogen of marine fish. Virus Res 107:1–9. doi: 10.1016/j.virusres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Walker PJ, Byrne KA, Riding GA, Cowley JA, Wang Y, McWilliam S. 1992. The genome of bovine ephemeral fever rhabdovirus contains two related glycoprotein genes. Virology 191:49–61. doi: 10.1016/0042-6822(92)90165-l. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Walker PJ. 1993. Adelaide River rhabdovirus expresses consecutive glycoprotein genes as polycistronic mRNAs: new evidence of gene duplication as an evolutionary process. Virology 195:719–731. doi: 10.1006/viro.1993.1423. [DOI] [PubMed] [Google Scholar]
- 17.Gubala A, Davis S, Weir R, Melville L, Cowled C, Walker P, Boyle D. 2010. Ngaingan virus, a macropod-associated rhabdovirus, contains a second glycoprotein gene and seven novel open reading frames. Virology 399:98–108. doi: 10.1016/j.virol.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 18.Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, Scheffler K, Kosakovsky Pond SL. 2015. Gene-wide identification of episodic selection. Mol Biol Evol 32:1365–1371. doi: 10.1093/molbev/msv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. 2015. RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol 32:820–832. doi: 10.1093/molbev/msu400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurath G. 2012. Fish novirhabdoviruses, p 89–116. In Dietzgen RG, Kuzmin IV (ed), Rhabdoviruses: molecular taxonomy, evolution, genomics, ecology, host-vector interactions, cytopathology and control. Caister Academic Press, Norfolk, UK. [Google Scholar]
- 21.Poynter SJ, Leis EM, DeWitte-Orr SJ. 2019. In vitro transcribed dsRNA limits viral hemorrhagic septicemia virus (VHSV)-IVb infection in a novel fathead minnow (Pimephales promelas) skin cell line. Fish Shellfish Immunol 86:403–409. doi: 10.1016/j.fsi.2018.11.053. [DOI] [PubMed] [Google Scholar]
- 22.Johnson N, Freuling C, Marston DA, Tordo N, Fooks AR, Müller T. 2007. Identification of European bat lyssavirus isolates with short genomic insertions. Virus Res 128:140–143. doi: 10.1016/j.virusres.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 23.Allison AB, Mead DG, Palacios GF, Tesh RB, Holmes EC. 2014. Gene duplication and phylogeography of North American members of the Hart Park serogroup of avian rhabdoviruses. Virology 448:284–292. doi: 10.1016/j.virol.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chirico N, Vianelli A, Belshaw R. 2010. Why genes overlap in viruses. Proc Biol Sci 277:3809–3817. doi: 10.1098/rspb.2010.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon-Loriere E, Holmes EC, Pagan I. 2013. The effect of gene overlapping on the rate of RNA virus evolution. Mol Biol Evol 30:1916–1928. doi: 10.1093/molbev/mst094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandes N, Linial M. 2016. Gene overlapping and size constraints in the viral world. Biol Direct 11:26. doi: 10.1186/s13062-016-0128-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson CW, Ardern Z, Goldberg TL, Meng C, Kuo CH, Ludwig C, Kolokotronis SO, Wei X. 2020. Dynamically evolving novel overlapping gene as a factor in the SARS-CoV-2 pandemic. Elife 9:e59633. doi: 10.7554/eLife.59633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes AL. 1994. The evolution of functionally novel proteins after gene duplication. Proc Biol Sci 256:119–124. doi: 10.1098/rspb.1994.0058. [DOI] [PubMed] [Google Scholar]
- 29.Lynch M, Conery JS. 2000. The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 30.Lynch M, Force A. 2000. The probability of duplicate gene preservation by subfunctionalization. Genetics 154:459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeling M, Scanlon MJ, Fowler JE. 2015. Fractionation and subfunctionalization following genome duplications: mechanisms that drive gene content and their consequences. Curr Opin Genet Dev 35:110–118. doi: 10.1016/j.gde.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Bayer A, Brennan G, Geballe AP. 2018. Adaptation by copy number variation in monopartite viruses. Curr Opin Virol 33:7–12. doi: 10.1016/j.coviro.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tordo N, Poch O, Ermine A, Keith G, Rougeon F. 1986. Walking along the rabies genome: is the large G-L intergenic region a remnant gene? Proc Natl Acad Sci USA 83:3914–3918. doi: 10.1073/pnas.83.11.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waldron FM, Stone GN, Obbard DJ. 2018. Metagenomic sequencing suggests a diversity of RNA interference-like responses to viruses across multicellular eukaryotes. PLoS Genet 14:e1007533. doi: 10.1371/journal.pgen.1007533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dietzgen RG, Jiāng D, Kuhn JH, Vasilakis N, ICTV Report Consortium . 2019. ICTV virus taxonomy profile: Artoviridae. J Gen Virol 100:1202–1203. doi: 10.1099/jgv.0.001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnhart MC, Haag WR, Roston WN. 2008. Adaptations to host infection and larval parasitism in Unionoida. J North Am Benthol Soc 27:370–394. doi: 10.1899/07-093.1. [DOI] [Google Scholar]
- 37.Haag WR. 2012. North American freshwater mussels: natural history, ecology, and conservation, 1st ed. Cambridge University Press, New York, NY, USA. [Google Scholar]
- 38.O'Brien C, Nez D, Wolf D, Box JB. 2013. Reproductive biology of Anodonta californiensis, Gonidea angulata, and Margaritifera falcata (Bivalvia: Unionoida) in the Middle Fork John Day River, Oregon. Northwest Sci 87:59–72. doi: 10.3955/046.087.0105. [DOI] [Google Scholar]
- 39.Modesto V, Ilarri M, Souza AT, Lopes-Lima M, Douda K, Clavero M, Sousa R. 2018. Fish and mussels: importance of fish for freshwater mussel conservation. Fish Fish 19:244–259. doi: 10.1111/faf.12252. [DOI] [Google Scholar]
- 40.Coker R, Shira A, Clark H, Howard A. 1921. Natural history and propagation of fresh-water mussels. Bull Bureau Fish 37:77–181. [Google Scholar]
- 41.Watters GT. 1996. New hosts for Lampsilis cardium. Triannual Unionid Report 9. [Google Scholar]
- 42.Watters GT. 1997. Glochidial metamorphosis of the freshwater mussel Lampsilis cardium (Bivalvia: Unionidae) on larval tiger salamanders, Ambystoma tigrinum ssp. (Amphibia: Ambystomidae). Can J Zool 75:505–508. doi: 10.1139/z97-062. [DOI] [Google Scholar]
- 43.Lopes-Lima M, Bolotov IN, Do VT, Aldridge DC, Fonseca MM, Gan HM, Gofarov MY, Kondakov AV, Prie V, Sousa R, Varandas S, Vikhrev IV, Teixeira A, Wu RW, Wu X, Zieritz A, Froufe E, Bogan AE. 2018. Expansion and systematics redefinition of the most threatened freshwater mussel family, the Margaritiferidae. Mol Phylogenet Evol 127:98–118. doi: 10.1016/j.ympev.2018.04.041. [DOI] [PubMed] [Google Scholar]
- 44.Longdon B, Murray GG, Palmer WJ, Day JP, Parker DJ, Welch JJ, Obbard DJ, Jiggins FM. 2015. The evolution, diversity, and host associations of rhabdoviruses. Virus Evol 1:vev014. doi: 10.1093/ve/vev014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haag WR, Williams JD. 2014. Biodiversity on the brink: an assessment of conservation strategies for North American freshwater mussels. Hydrobiologia 735:45–60. doi: 10.1007/s10750-013-1524-7. [DOI] [Google Scholar]
- 46.Lopes-Lima M, Burlakova LE, Karatayev AY, Mehler K, Seddon M, Sousa R. 2018. Conservation of freshwater bivalves at the global scale: diversity, threats and research needs. Hydrobiologia 810:1–14. doi: 10.1007/s10750-017-3486-7. [DOI] [Google Scholar]
- 47.Richard JC, Leis EM, Dunn CD, Harris C, Agbalog RE, Campbell LJ, Knowles S, Waller DL, Putnam JG, Goldberg TL. 2022. Freshwater mussels show elevated viral richness and intensity during a mortality event. Viruses 14:2603. doi: 10.3390/v14122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richard JC, Campbell LJ, Leis EM, Agbalog RE, Dunn CD, Waller DL, Knowles S, Putnam JG, Goldberg TL. 2021. Mussel mass mortality and the microbiome: evidence for shifts in the bacterial microbiome of a declining freshwater bivalve. Microorganisms 9:1976. doi: 10.3390/microorganisms9091976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richard JC, Leis E, Dunn CD, Agbalog R, Waller D, Knowles S, Putnam J, Goldberg TL. 2020. Mass mortality in freshwater mussels (Actinonaias pectorosa) in the Clinch River, USA, linked to a novel densovirus. Sci Rep 10:14498. doi: 10.1038/s41598-020-71459-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leis E, Erickson S, Waller D, Richard J, Goldberg T. 2019. A comparison of bacteria cultured from unionid mussel hemolymph between stable populations in the Upper Mississippi River basin and populations affected by a mortality event in the Clinch River. Freshwater Mollusk Biol and Cons 22:70–80. [Google Scholar]
- 51.Zhang Z, Sufang D, Yimin X, Jie W. 1986. Studies on the mussel Hyriopsis cumingii plague. I. a new viral infectious disease. Acta Hydrobiologica Sinica 26:308–312. [Google Scholar]
- 52.Carella F, Villari G, Maio N, De Vico G. 2016. Disease and disorders of freshwater unionid mussels: a brief overview of recent studies. Front Physiol 7:489. doi: 10.3389/fphys.2016.00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshino TP, Bickham U, Bayne CJ. 2013. Molluscan cells in culture: primary cell cultures and cell lines. Can J Zool 91:391–404. doi: 10.1139/cjz-2012-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leis E, Waller D, Knowles S, Goldberg T, Putnam J, Richard J, Erickson S, Blevins E, Weinzinger J. 2018. Building a response network to investigate potential pathogens associated with unionid mortality events. Ellipsaria 20:44–45. [Google Scholar]
- 55.Goldberg TL, Dunn CD, Leis E, Waller DL. 2019. A novel picornalike virus in a Wabash Pigtoe (Fusconaia flava) from the Upper Mississippi River, USA. Freshwater Mollusk Biol Cons 22:81–84. [Google Scholar]
- 56.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mikheenko A, Saveliev V, Gurevich A. 2016. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32:1088–1090. doi: 10.1093/bioinformatics/btv697. [DOI] [PubMed] [Google Scholar]
- 58.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 60.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J. 2011. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 39:D38–51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 62.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 64.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 65.Lefort V, Longueville JE, Gascuel O. 2017. SMS: smart model selection in PhyML. Mol Biol Evol 34:2422–2424. doi: 10.1093/molbev/msx149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huson DH, Scornavacca C. 2012. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol 61:1061–1067. doi: 10.1093/sysbio/sys062. [DOI] [PubMed] [Google Scholar]
- 67.Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL. 2018. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol 35:773–777. doi: 10.1093/molbev/msx335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.USFWS and AFS-FHS. 2014. Standard procedures for aquatic animal health inspections In AFS–FHS (U.S. Fish and Wildlife Service and American Fisheries Society-Fish Health Section). In AFS–FHS (ed), FHS blue book: suggested procedures for the detection and identification of certain finfish and shellfish pathogens, 2020 edition. American Fisheries Society, Bethesda, Maryland. [Google Scholar]
- 69.Kralik P, Ricchi M. 2017. A basic guide to real time PCR in microbial diagnostics: definitions, parameters, and everything. Front Microbiol 8:108. doi: 10.3389/fmicb.2017.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Standish I, Leis E, Schmitz N, Credico J, Erickson S, Bailey J, Kerby J, Phillips K, Lewis T. 2018. Optimizing, validating, and field testing a multiplex qPCR for the detection of amphibian pathogens. Dis Aquat Organ 129:1–13. doi: 10.3354/dao03230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download jvi.00196-23-s0001.xlsx, XLSX file, 0.02 MB (16.6KB, xlsx)
Table S2. Download jvi.00196-23-s0002.xlsx, XLSX file, 0.01 MB (11.9KB, xlsx)
Table S3. Download jvi.00196-23-s0003.xlsx, XLSX file, 0.01 MB (12KB, xlsx)
Table S4. Download jvi.00196-23-s0004.xlsx, XLSX file, 0.01 MB (11.8KB, xlsx)
Data Availability Statement
Virus genome sequence data generated as part of this study are available in GenBank under accession numbers OQ368743.1 (KILLV-1) and OQ368744.1 (CHMFV-1).