

Abstract
With the rapid development of nanotechnology, the potential adverse health effects of nanoparticles have caught more attention and become global concerns. However, the underlying mechanisms in metal nanoparticle-induced toxic effects are still largely obscure. In this study, we investigated whether exposure to nickel nanoparticles (Nano-Ni) and titanium dioxide nanoparticles (Nano-TiO2) would alter autophagy and apoptosis levels in normal human bronchial epithelial BEAS-2B cells and the underlying mechanisms involved in this process. Our results showed that the expressions of autophagy- and apoptosis-associated proteins were dysregulated in cells exposed to Nano-Ni. However, exposure to the same dose of Nano-TiO2 had no significant effects on these proteins. In addition, exposure to Nano-Ni, but not Nano-TiO2, led to nuclear accumulation of HIF-1α and decreased phosphorylation of mTOR in BEAS-2B cells. Inhibition of HIF-1α by CAY10585 abolished Nano-Ni-induced decreased phosphorylation of mTOR, while activation of mTOR by MHY1485 did not affect Nano-Ni-induced nuclear accumulation of HIF-1α. Furthermore, both HIF-1α inhibition and mTOR activation abolished Nano-Ni-induced autophagy but enhanced Nano-Ni-induced apoptosis. Blockage of autophagic flux by Bafilomycin A1 exacerbated Nano-Ni-induced apoptosis, while activation of autophagy by Rapamycin effectively rescued Nano-Ni-induced apoptosis. In conclusion, our results demonstrated that Nano-Ni exposure caused increased levels of autophagy and apoptosis via the HIF-1α/mTOR signaling axis. Nano-Ni-induced autophagy has a protective role against Nano-Ni-induced apoptosis. These findings provide us with further insight into Nano-Ni-induced toxicity.
Keywords: Nickel nanoparticles, Autophagy, Apoptosis, HIF-1α, mTOR
1. Introduction
Metal nanoparticles possess unique physicochemical properties, such as high surface area, high magnetic properties, high surface energy, low melting point, low ignition point, etc. (Mo et al., 2019; Zhang et al., 1998; Zhang et al., 2003). They are wildly used in industry as chemical catalysts, magnetics, solar cells, and so on (Bajpai et al., 2012; Bhattacharjee et al., 2018; Du et al., 2021; Wu et al., 2018b). The wide range of industrial applications of metal nanoparticles has undoubtedly increased the risk of environmental and occupational exposure, raising public concerns about the potential health effects brought by them. Thus, it is necessary to evaluate the potentially toxic effects of metal nanoparticles and their underlying mechanisms to avoid their adverse effects on biological systems and human health. Although it has been demonstrated that the toxicity of metal nanoparticles depends mainly on composition, size, shape, and concentration (Griffitt et al., 2008; Wang et al., 2008), the detailed mechanisms are still unclear. The toxic effects of some metal nanoparticles may be related to their physicochemical properties. For example, our previous studies have shown that nickel (Nano-Ni), cobalt (Nano-Co), and titanium dioxide (Nano-TiO2) nanoparticles were dramatically different in their ability to cause lung injury and inflammation despite having the same size (Zhang et al., 1998). Differences in free-radical-generation activity might underlie the differences in the inflammatory response to these three metal nanoparticles (Zhang et al., 1998). Previous reports also have shown that exposure to nickel nanoparticles causes adverse health effects on workers, even including death. For example, a 26-year-old female chemist exhibited allergic symptoms after unprotected exposure to Nano-Ni powder (Journeay and Goldman 2014), and a 38-year-old healthy male died of adult respiratory distress syndrome (ARDS) 13 days after exposure to Nano-Ni (Phillips et al., 2010). Nano-Ni with diameters less than < 25 nm were identified in his alveolar macrophages on pathological autopsy (Phillips et al., 2010). However, the mechanisms underlying the toxic effects of Nano-Ni require further investigation.
Autophagy is an intracellular and conserved catabolic process by which impaired organelles and proteins in the cytoplasm are sequestered into double-membraned autophagosomes and subsequently degraded upon fusion with a lysosome (Cao et al., 2020; Kuma et al., 2004; Yuan et al., 2017). Although previous studies have shown that autophagy is involved in various nanoparticle-induced toxic effects (Li et al., 2011; Mohammadinejad et al., 2019), different nanoparticles have different effects on the autophagic flux. For example, silica, zinc oxide, and copper oxide nanoparticles could hinder autophagy by disrupting the lysosomal system or damaging the mitochondrial integrity, thus promoting the occurrence of autophagic cell death (Liu et al., 2020; Stern et al., 2012; Xiao et al., 2021; Yu et al., 2014). In contrast, gold and silver nanoparticles exert cytoprotective effects by inducing the expression of autophagy-associated genes, thus presenting great potential in tumor therapy (Li et al., 2020; Lin et al., 2018). Furthermore, exposure to 40, 50, and 60 μg/mL of graphene oxide, a two-dimensional carbon nanosheet, could stimulate autophagic response and block autophagic flux by disrupting lysosomal function in a rat pheochromocytoma-derived PC12 cell line (Feng et al., 2018). Besides the chemical nature of the metal nanoparticles, autophagy response might also be vastly diverse due to the distinguished properties of nanoparticles such as size, surface modification, functionalization, chemical structure, and so on (Minocha and Mumper 2012; Xie et al., 2019; Zhou et al., 2022). Current evidence suggests that autophagy is involved in various pulmonary diseases including acute lung injury, lung inflammatory diseases, asthma, pulmonary fibrosis, etc. (Ryter et al., 2012; Zhang et al., 2022). Our previous studies have shown that Nano-Ni instilled intratracheally into mice or rats caused severe and persistent lung injury, pulmonary inflammation, lung fibrosis, emphysema, and alveolar proteinosis (Mo et al., 2019; Mo et al., 2020; Mo et al., 2021; Zhang et al., 1998; Zhang et al., 2003). However, the role of autophagy in Nano-Ni-induced cell outcome is still unclear; currently published studies showed contradictory results. For example, one previous study showed that exposure to Nano-Ni caused increased methionine oxidation and decreased cell viability in human alveolar epithelial A549 cells, which might be due to suppression of methionine repair enzymes MSRA and MSRB3 and decreased production of LC3, an autophagy marker (Feng et al., 2015b), while another study showed that Nano-Ni exposure remarkably upregulated mitochondrial autophagy-associated proteins PINK1 and Parkin in mouse spermatogonia (GC-1 cells), which then promoted reproductive toxicity by interacting with Drp1-mediated mitochondrial division (Liu et al., 2022).
Hypoxia-inducible factor 1α (HIF-1α) is an unstable alpha subunit, which can form a heterodimer, HIF-1, with another stable beta subunit (HIF-1β) (Kaelin and Ratcliffe 2008). HIF-1α is rapidly ubiquitinated and degraded through the von Hippel–Lindau protein (pVHL), a substrate recognition subunit of E3 ubiquitin ligase, under normoxic conditions. However, HIF-1α translocates from the cytoplasm to the nucleus when the oxygen supply is limited and then participates in the transcriptional regulation of multiple cellular events (Ziello et al., 2007). HIF-1α has been confirmed to be dramatically upregulated in response to exposure to Ni, Nano-Ni, and Nano-NiO in vitro and in vivo (Glista-Baker et al., 2012; Mo et al., 2021; Salnikow et al., 1999; Saquib et al., 2020). Our previous studies showed that Nano-Ni-caused HIF-1α nuclear accumulation was involved in Nano-Ni-induced tight junction loss, chemokine expression, MMP-2 and MMP-9 upregulation, and cell transformation (Glista-Baker et al., 2012; Mo et al., 2021; Wan et al., 2011; Yuan et al., 2021). HIF-1α has also been identified as one of the key inducers of autophagy through inhibiting the mTOR complex or enhancing BNIP3/BNIP3L, which then restores the autophagy-initiating role of Beclin 1 (Ashkenazi et al., 2017; Gozuacik and Kimchi 2004). In particular, mTOR acts as a major regulator of autophagic activity, and targeted regulation by mTOR complex 1 (mTORC1) of multiple kinases, transcription factors, or signaling molecules associated with autophagy has been implicated as a central mechanism of autophagy (Dossou and Basu 2019). However, whether Nano-Ni-caused HIF-1α nuclear accumulation is also involved in Nano-Ni-induced autophagy and cell death through the interaction with mTOR signal remains to be further elucidated.
In the present study, we explored whether exposure to Nano-Ni caused altered levels of autophagy and apoptosis in normal human bronchial epithelial cells (BEAS-2B) and the potential mechanisms underlying these effects. We hypothesized that Nano-Ni-induced cellular autophagy has a potentially protective role in Nano-Ni-induced apoptosis through the HIF-1α/mTOR signaling axis. At first, we examined whether exposure to Nano-Ni induced apoptosis and autophagy by determining the protein expression levels of Bax, Bcl-2, cleaved caspase-3, LC3B-II/LC3B-I, Beclin 1, and p62. We then evaluated the levels of nuclear HIF-1α and mTOR phosphorylation in BEAS-2B after exposure to Nano-Ni. The role of HIF-1α/mTOR in Nano-Ni-induced apoptosis and autophagy was determined by using a HIF-1α inhibitor, CAY10585 or an mTOR agonist, MHY1485. Finally, we explored the role of Nano-Ni-induced autophagy in Nano-Ni-induced apoptosis. Nano-TiO2 was used as a negative control since our previous studies have shown that exposure to Nano-TiO2 did not cause HIF-1α nuclear accumulation (Wan et al., 2011; Yuan et al., 2021; Yuan et al., 2022).
2. Materials and methods
2.1. Characterization and preparation of metal nanoparticles
Nickel nanoparticles (Nano-Ni) and titanium dioxide nanoparticles (Nano-TiO2) were provided by INABTA and Co., Ltd., Vacuum Metallurgical Co., Ltd. (Japan). The detailed characterization of Nano-Ni and Nano-TiO2 was described in our previous studies (Mo et al., 2019; Wan et al., 2011; Yuan et al., 2021). Briefly, the mean diameters of Nano-Ni and Nano-TiO2 in the powder are 20 nm and 28 nm determined by transmission electron microscopy (TEM), and their mean hydrodynamic sizes are 250 nm and 280 nm determined by dynamic light scattering (DLS). The specific surface area is 43.8 m2/g for Nano-Ni and 45 m2/g for Nano-TiO2. The nanoparticles were suspended in normal saline at a concentration of 1 mg/mL. To reduce agglomeration, nanoparticle suspension was ultrasonicated for at least 10 min prior to each experiment.
2.2. Cell culture and treatment
Human bronchial epithelial BEAS-2B cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA, cat#. CRL-9609) and cultured in RPMI1640 (Mediatech, Manassas, VA, USA) supplemented with 10% FBS (Mediatech) and 1% penicillin/streptomycin (Mediatech) in a humidified incubator at 37 °C with 5% CO2.
For mechanistic studies, cells were pretreated with inhibitors or agonists [(1) CAY10585, 30 μM, cat#. B2277, BioVision Inc., Milpitas, CA, USA; (2) MHY1485, 10 μM, cat#. 9419, BioVision Inc., Milpitas, CA, USA; (3) Bafilomycin A1, 5 nM, cat#. 88899–55-2, MilliporeSigma, Burlington, MA, USA; or (4) Rapamycin, 100 nM, cat#. 53123–88-9, Enzo Clinical Labs, Farmingdale, NY, USA] for 4 h prior to 20 μg/mL of Nano-Ni exposure for another 24 h.
2.3. Protein extraction and Western blot
Total proteins were extracted from the cells by using RIPA buffer (Santa Cruz Biotechnology, USA) for the detection of autophagy-associated proteins, p-mTOR, mTOR, and apoptosis-associated markers. Nuclear proteins were isolated by using NE-PER® Nuclear and Cytoplasmic Extraction Reagent (Thermo Fisher Scientific, Rockford, IL, USA) for the detection of HIF-1α protein. The protein concentration was determined by the Bradford method by using the spectrophotometer (Beckman Coulter, Brea, CA, USA). 30 μg protein of each sample was loaded and separated on SDS-PAGE and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, CA, USA). The PVDF membrane loaded with the proteins was blocked with TBST-dissolved 5% skim milk for 2 h at room temperature (RT) and then incubated with primary antibody overnight at 4 °C with gentle shaking. The membrane was then washed with TBST and incubated with the corresponding secondary antibody conjugated with horseradish peroxidase (HRP) at RT for 1 h. SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA) was used for chemiluminescence, and the bands were captured by using CL-XPosure™ film (Thermo Scientific). Coomassie Brilliant Blue staining was used to validate the equal loading of nuclear protein samples. β-actin was selected as the internal reference for cytosolic proteins. For Coomassie Brilliant Blue staining, the gel was rinsed briefly with distilled water twice after SDS-PAGE, stained with 0.1% Coomassie Brilliant Blue R-250 (Bio-Rad, Hercules, CA, USA) in 45% methanol and 10% acetic acid solution for one hour at room temperature, and destained with 10% acetic acid until the clear bands were observed. NIH ImageJ software (http://imagej.nih.gov/ij/) was applied to quantify the results. The antibodies used in this study were listed in Table 1.
Table 1.
The list of antibodies used in this study.
| Antibody | Supplier | Cat. No. | Dilution |
|---|---|---|---|
| LC3B | Abcam | ab51520 | 1:2000 |
| Beclin 1 | Abcam | ab62557 | 1:2000 |
| SQSTM1/p62 | Abcam | ab56416 | 1:2000 |
| HIF-1α | BD Biosciences | 610959 | 1:1000 |
| p-mTOR | Cell Signaling Technology | 2971 | 1:1000 |
| mTOR | Cell Signaling Technology | 2972 | 1:1000 |
| Bax | Cell Signaling Technology | 5023 | 1:1000 |
| Bcl-2 | Cell Signaling Technology | 15071 | 1:1000 |
| Caspase-3 | Cell Signaling Technology | 9662 | 1:1000 |
| Cleaved caspase-3 | Cell Signaling Technology | 9661 | 1:1000 |
| β-actin | Cell Signaling Technology | 58169 | 1:1000 |
| Anti-mouse IgG (HRP) | Cell Signaling Technology | 7076 | 1:1000 |
| Anti-rabbit IgG (HRP) | Cell Signaling Technology | 7074 | 1:1000 |
Note: Abcam, Burlingame, CA, USA; BD Biosciences, San Jose, CA, USA; and Cell Signaling Technology, Beverly, MA, USA.
2.4. Hoechst 33342 staining
BEAS-2B cells were seeded into each chamber of a 4-well LAB-TEK® II chamber slide (Nalge Nunc International, IL, USA) at a density of 5 × 104 cells/chamber. When the cells covered 70%-80% of the slide surface, the cells were treated with 20 μg/mL of Nano-Ni or Nano-TiO2 for 24 h. After treatment, the cells were rinsed with 1x PBS, fixed with 4% paraformaldehyde for 30 min at RT, and washed again with 1x PBS. The cells were then dyed with 1 μg/mL of Hoechst 33342 (Thermo Scientific) for 15 min at dark. The slides were examined under a fluorescent microscope (Nikon, Japan) and the images were captured. Cells without any treatments were used as controls.
2.5. Statistical analysis
SigmaPlot 13.0 software purchased from Systat Software, Inc. (San Jose, CA, USA) was used for data management and statistical analysis. All data were presented as mean ± SE. One-way or two-way ANOVA was applied for the analysis of differences among groups. p < 0.05 was considered statistically significant.
3. Results
3.1. Nano-Ni exposure induced apoptosis in BEAS-2B cells
The cytotoxicity of Nano-Ni and Nano-TiO2 on BEAS-2B cells was determined by MTS assay and alamarBlue™ assay, respectively, and the results were reported in our recent publication (Mo et al., 2021). 30 and 40 μg/mL of Nano-Ni exposure led to a significant reduction in BEAS-2B cell viability, while exposure to 0 – 40 μg/mL of Nano-TiO2 had no remarkable cytotoxic effects on BEAS-2B cells. To identify the potential biological effects of Nano-Ni other than cytotoxicity, two non-cytotoxic doses (10 and 20 μg/mL) were adopted in this study to investigate the effects of Nano-Ni on human bronchial epithelial cells.
To validate whether exposure to metal nanoparticles could result in apoptosis of BEAS-2B cells, Western blot was performed to determine the expression levels of apoptosis-associated proteins in cells exposed to 10 and 20 μg/mL of Nano-TiO2 or Nano-Ni for 24 h. Our results showed that exposure of BEAS-2B cells to Nano-Ni caused significant upregulation of pro-apoptotic proteins Bax and cleaved Caspase-3, but a remarkable downregulation of anti-apoptotic protein Bcl-2. Exposure to Nano-TiO2 did not cause any statistically significant expression changes in these genes (Fig. 1A and C). When the cells were treated with 20 μg/mL of Nano-Ni for 0, 6, 12, 24, and 48 h, a time-dependent increase in the expressions of Bax and cleaved Caspase-3 and a decrease in the expression of Bcl-2 were observed (Fig. 1B and D).
Fig. 1. Nano-Ni exposure induced apoptosis in BEAS-2B cells.

BEAS-2B cells were exposed to 10 and 20 μg/mL of Nano-Ni or Nano-TiO2 for 24 h (A & C) or exposed to 20 μg/mL of Nano-Ni for 0, 6, 12, 24, and 48 h (B & D). Cells without treatment were used as a control. Western blot (A & B) was performed to determine the expression of apoptosis-associated proteins (Bax, Bcl-2, caspase-3, and cleaved caspase-3) and the normalized grayscale values of the immuno-bands were from at least three independent experiments (C & D). (E) Hoechst 33342 staining was performed to identify the apoptotic cells induced by 20 μg/mL of Nano-Ni or Nano-TiO2 exposure for 24 h. All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the same dose of Nano-TiO2-treated group, P < 0.05.
In addition, Hoechst 33342 staining was used to visualize the nuclei of the cells. The fluorescent dye Hoechst 33342 penetrates the cell membrane and releases blue fluorescence when embedded in double-stranded DNA. Apoptotic cells have a higher fluorescence intensity than normal cells due to the compact chromatin of apoptotic nuclei and increased permeability of the cell membrane that allows more Hoechst 33342 dye to enter apoptotic cells (Sandhu et al., 1985). Our results showed that more nuclei with significantly higher blue fluorescence were observed in BEAS-2B cells treated with 20 μg/mL of Nano-Ni for 24 h as compared to that in the control cells or the cells treated with Nano-TiO2 (Fig. 1E). These results suggest that exposure to Nano-Ni caused BEAS-2B cell apoptosis.
3.2. The effects of Nano-Ni exposure on autophagy in BEAS-2B cells
Autophagy is an important cell survival mechanism. Nevertheless, there are still few studies focused on the potential function of autophagy in Nano-Ni-induced cytotoxicity. To investigate whether exposure of BEAS-2B cells to Nano-Ni would induce autophagy, the expression levels of autophagy-associated proteins, microtubule-associated protein light chain 3 B (LC3B) and Beclin 1, were examined by Western blot. When autophagy is activated, the hydrophilic LC3B-I would be lipidated to form LC3B-II, thus the ratio of LC3B-II/LC3B-I is an ideal marker for the initiation of autophagy (Lystad et al., 2019). Beclin 1 is a mammalian homolog of yeast Atg6 and plays a pivotal role in autophagy through the mediation of the localization of other autophagy-associated proteins to the autophagosomal membrane (Kang et al., 2011). Our results demonstrated that exposure of BEAS-2B cells to both 10 and 20 μg/mL of Nano-Ni caused an increase in the LC3B-II/LC3B-I ratio and the Beclin 1 protein level; however, exposure of BEAS-2B cells to Nano-TiO2 did not cause such effects (Fig. 2A and C). For the time-response study, BEAS-2B cells were treated with 20 μg/mL of Nano-Ni for 0, 6, 12, 24, and 48 h. The results showed that exposure to Nano-Ni caused a time-dependent increase in the LC3B-II/LC3B-I ratio and Beclin 1 protein level (Fig. 2B and D). These results suggest that Nano-Ni exposure may initiate autophagy in human bronchial epithelial cells.
Fig. 2. Nano-Ni exposure induced autophagy in BEAS-2B cells.

BEAS-2B cells were exposed to 10 and 20 μg/mL of Nano-Ni or Nano-TiO2 for 24 h (A & C) or exposed to 20 μg/mL of Nano-Ni for 0, 6, 12, 24, and 48 h (B & D). Cells without treatment were used as a control. Western blot (A & B) was performed to measure the expression of autophagy-associated proteins (LC3B-I, LC3B-II, Beclin 1, and p62), and normalized grayscale values of the immuno-bands were from at least three independent experiments (C & D). All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the same dose of Nano-TiO2-treated group, P < 0.05.
The expression level of another autophagy-associated protein, sequestosome 1 (SQSTM1/p62) (abbreviated as p62), was also determined. P62 is a selective autophagic cargo receptor that recognizes LC3, whose accumulation is often considered a hallmark of fusion failure of autophagosomes and lysosomes (Yao et al., 2021). Our results demonstrated that exposure of BEAS-2B cells to 20 μg/mL of Nano-Ni for 24 or 48 h caused a significant increase in p62 protein level (Fig. 2), indicating that Nano-Ni exposure might also disrupt autophagosome-lysosome fusion or cargo degradation in the cells.
3.3. Nano-Ni exposure induced decreased phosphorylation of mTOR and its role in Nano-Ni-induced autophagy and apoptosis
The role of mTOR as an inhibitory regulator in autophagy has been widely recognized (González and Hall 2017). Here we examined the phosphorylation level of mTOR in BEAS-2B cells after Nano-Ni exposure. Our results showed that there was a dose- or time-dependent decrease in the phosphorylation level of mTOR in BEAS-2B cells after Nano-Ni exposure; however, exposure of the cells to Nano-TiO2 did not cause this change (Fig. 3).
Fig. 3. Nano-Ni exposure caused decreased phosphorylation of mTOR in BEAS-2B cells.

BEAS-2B cells were exposed to 10 and 20 μg/mL of Nano-Ni or Nano-TiO2 for 24 h (A & B) or exposed to 20 μg/mL of Nano-Ni for 0, 6, 12, 24, and 48 h (C & D). Cells without treatment were used as a control. Western blot (A & C) was performed to measure the levels of mTOR and phosphorated mTOR (p-mTOR) and normalized grayscale values of the immuno-bands were from at least three independent experiments (B & D). All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the same dose of Nano-TiO2-treated group, P < 0.05.
To assess the role of mTOR phosphorylation in Nano-Ni-induced autophagy and apoptosis, MHY1485, a small molecular agonist of mTOR, was employed to globally upregulate mTOR phosphorylation in BEAS-2B cells. The cells were pretreated with MHY1485 for 4 h prior to 20 μg/mL of Nano-Ni exposure for 24 h. The results demonstrated that MHY1485 pretreatment abolished Nano-Ni-induced decreased phosphorylation of mTOR and increased lipidation transition of LC3B (Fig. 4). In addition, pretreatment of the cells with MHY1485 enhanced Nano-Ni-induced cell apoptosis which was reflected by the increased expression level of cleaved caspase-3 (Fig. 4). Our results suggest that phosphorylation of mTOR is involved in Nano-Ni-induced autophagy and apoptosis.
Fig. 4. Activation of mTOR attenuated Nano-Ni-induced autophagy but enhanced Nano-Ni-induced apoptosis in BEAS-2B cells.

BEAS-2B cells were pretreated with 10 μM of mTOR agonist, MHY1485, for 4 h prior to exposure to 20 μg/mL of Nano-Ni for another 24 h. Cells without any treatments were used as a control. Western blot (A) was performed to measure the protein expression levels of mTOR, phosphorated mTOR (p-mTOR), LC3B-I, LC3B-II, caspase-3, and cleaved caspase-3, and normalized grayscale values of the immuno-bands were from at least three independent experiments (B). (C) Hoechst 33342 staining was performed to identify the apoptotic cells. All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the Nano-Ni only group, P < 0.05.
3.4. Nano-Ni exposure induced HIF-1α nuclear accumulation and its role in Nano-Ni-induced autophagy and apoptosis
Our previous in vitro and in vivo studies have demonstrated that Nano-Ni exposure causes HIF-1α nuclear accumulation (Mo et al., 2021; Wan et al., 2011; Yuan et al., 2021). Accumulation of HIF-1α in nuclei has been tightly linked to the occurrence of autophagy (Mazure and Pouyssegur 2010), thus, we investigated the role of HIF-1α in Nano-Ni-induced alteration of mTOR phosphorylation, autophagy, and apoptosis. When the cells were exposed to 10 and 20 μg/mL of Nano-Ni, but not Nano-TiO2, for 24 h or 20 μg/mL of Nano-Ni for 6, 12, 24, and 48 h, nuclear accumulation of HIF-1α was observed (Fig. 5), which was consistent with our previous results.
Fig. 5. Nano-Ni exposure induced nuclear accumulation of HIF-1α in BEAS-2B cells.

BEAS-2B cells were exposed to 10 and 20 μg/mL of Nano-Ni or Nano-TiO2 for 24 h (A & B) or exposed to 20 μg/mL of Nano-Ni for 0, 6, 12, 24, and 48 h (C & D). Cells without any treatment were used as a control. Western blot (A & C) was performed to measure the level of HIF-1α, and normalized grayscale values of the immuno-bands were from at least three independent experiments (B & D). Equal nuclear protein loading was verified by Coomassie Brilliant Blue staining. All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the same dose of Nano-TiO2-treated group, P < 0.05.
To study whether HIF-1α was involved in Nano-Ni-induced autophagy and apoptosis, CAY10585, a small molecular inhibitor of HIF-1α, was applied to pretreat cells prior to Nano-Ni exposure. CAY10585 facilitates the degradation of HIF-1α by upregulating pVHL whereas it does not affect HIF-1β. Our results showed that CAY10585 pretreatment effectively attenuated the nuclear accumulation of HIF-1α in the BEAS-2B cells exposed to Nano-Ni and reduced the ratio of LC3B-II/LC3B-I which was elevated by Nano-Ni (Fig. 6), suggesting that Nano-Ni-induced HIF-1α nuclear accumulation is involved in Nano-Ni-induced increased autophagy. On the other hand, pretreatment of the cells with CAY10585 enhanced Nano-Ni-induced increased expression of cleaved caspase-3 (Fig. 6), indicating nuclear accumulation of HIF-1α by Nano-Ni exposure is also involved in Nano-Ni-induced apoptosis.
Fig. 6. HIF-1α inhibition suppressed Nano-Ni-induced autophagy but enhanced Nano-Ni-induced apoptosis in BEAS-2B cells.
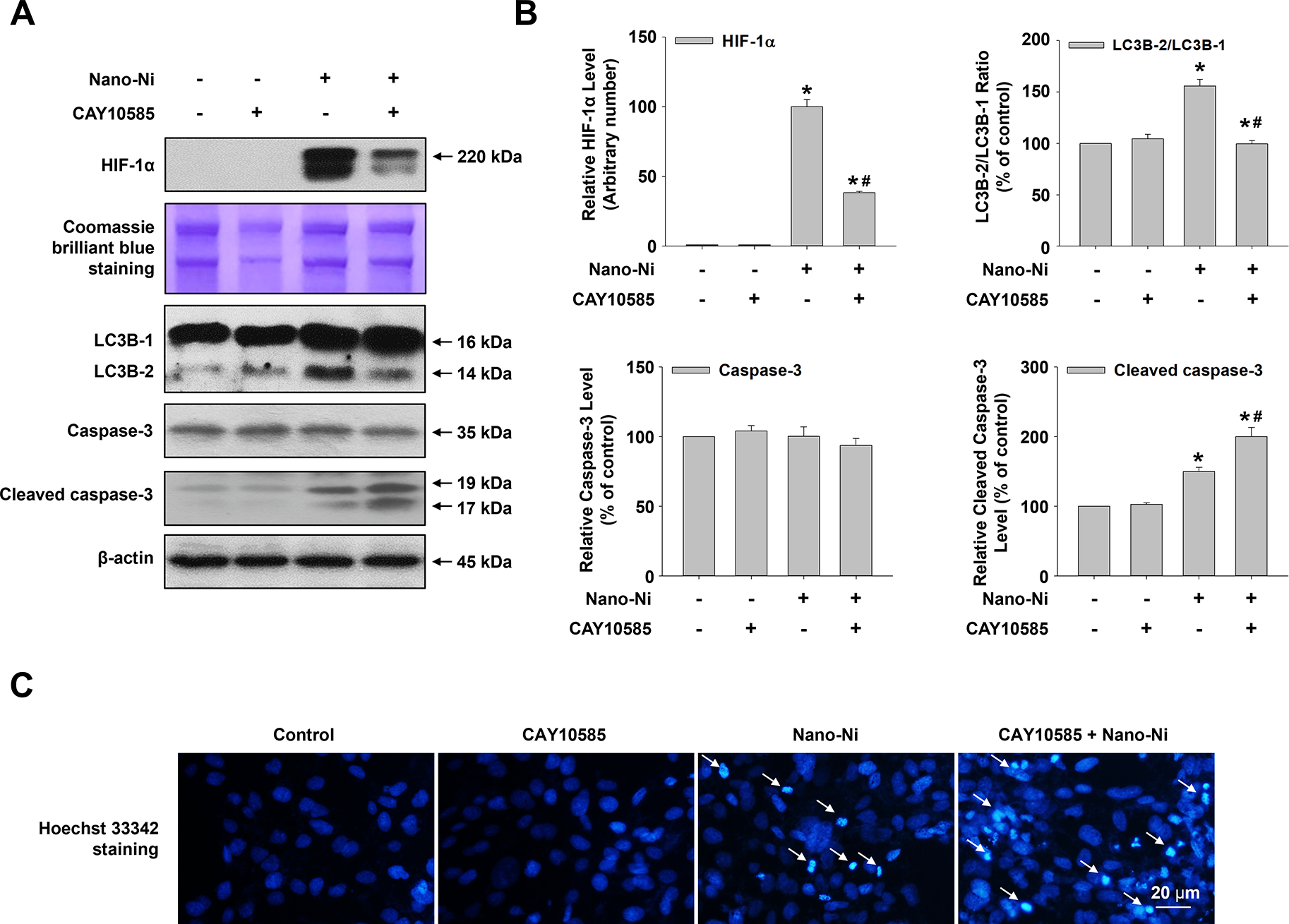
BEAS-2B cells were pretreated with 30 μM of CAY10585, a HIF-1α inhibitor, for 4 h prior to exposure to 20 μg/mL of Nano-Ni for another 24 h. Cells without treatment were used as a control. Western blot (A) was performed to measure the protein levels of HIF-1α, LC3B-I, LC3B-II, caspase-3, and cleaved caspase-3, and normalized grayscale values of the immuno-bands were from at least three independent experiments (B). Equal protein loading was verified by Coomassie Brilliant Blue staining. (C) Hoechst 33342 staining was performed to identify the apoptotic cells. All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the Nano-Ni only group, P < 0.05.
3.5. Nano-Ni-induced autophagy and apoptosis were through HIF-1α-mediated inactivation of mTOR
The above results showed that exposure of BEAS-2B cells to Nano-Ni caused decreased phosphorylation of mTOR but increased nuclear accumulation of HIF-1α and both were involved in Nano-Ni-induced alteration of autophagy and apoptosis. Previous studies showed that hypoxia could inhibit the phosphorylation of mTOR (Cosin-Roger et al., 2017; Huang et al., 2022). To understand the interaction between mTOR and HIF-1α in Nano-Ni-induced autophagy and apoptosis in BEAS-2B cells, we used HIF-1α inhibitor, CAY10585, and mTOR agonist, MHY1485, to pretreat cells prior to Nano-Ni exposure, then determined the protein levels of p-mTOR and HIF-1α. Our results showed that HIF-1α inhibitor, CAY10585, pretreatment significantly inhibited Nano-Ni-induced decreased phosphorylation of mTOR (Fig. 7A and B), whereas mTOR agonist, MHY1485, pretreatment did not alter Nano-Ni-induced nuclear accumulation of HIF-1α (Fig. 7C and D), suggesting HIF-1α is at the upstream of HIF-1α/mTOR axis, which is involved in Nano-Ni-induced autophagy and apoptosis.
Fig. 7. HIF-1α was at the upstream of mTOR.

BEAS-2B cells were pretreated with 30 μM of CAY10585 (a HIF-1α inhibitor) (A & B) or 10 μM of MHY1485 (an mTOR agonist) (C & D) for 4 h prior to exposure to 20 μg/mL of Nano-Ni for another 24 h. Cells without any treatments were used as a control. Western blot (A & C) was performed to measure the levels of mTOR, phosphorated mTOR (p-mTOR), and HIF-1α, and normalized grayscale values of the immuno-bands were from at least three independent experiments (B & D). Equal cytosolic protein loading was verified by β-actin, while equal nuclear protein loading was verified by Coomassie Brilliant Blue staining. All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the Nano-Ni only group, P < 0.05.
3.6. Nano-Ni-induced autophagy played a protective role in Nano-Ni-induced apoptosis in BEAS-2B cells
The role of autophagy in cell death has long been likened to a double-edged sword, as autophagy, originally considered a cell survival mechanism, has been shown to drive cell death when it occurs excessively (Shintani and Klionsky 2004). Our above results have shown that pharmacological inhibition of HIF-1α nuclear accumulation by using CAY10585, or activation of mTOR signaling by using MHY1485 could block the formation of autophagosomes but enhance Nano-Ni-induced apoptosis (Figs. 4 & 6), suggesting autophagy might be a potential protector in Nano-Ni-induced apoptosis of BEAS-2B cells. To further confirm this, autophagic flux inhibitor Bafilomycin A1 (Baf A1) and autophagy activator Rapamycin (Rapa) were used to pretreat cells prior to Nano-Ni exposure. Bafilomycin A1 disrupts autophagic flux by blocking autophagosome-lysosome fusion, while Rapamycin is an autophagy activator targeting mTOR (Wang et al., 2022). As shown in Figure 8, both Bafilomycin A1 and Rapamycin pretreatments significantly increased LC3B-II/LC3B-I ratio and also significantly enhanced Nano-Ni-induced LC3 lipidation (Fig. 8).
Fig. 8. The efficacy of Bafilomycin A1 and Rapamycin.

BEAS-2B cells were pre-treated with 5 nM of Bafilomycin A1 (Baf A1) (an autophagic flux blocker) or 100 nM of Rapamycin (Rapa) (an autophagy activator) for 4 h prior to exposure to 20 μg/mL of Nano-Ni for another 24 h. Cells without treatment were used as a control. LC3B-I and LC3B-II protein expressions were measured by Western blot (A & C), and normalized grayscale values of the immuno-bands were from at least three independent experiments (B & D). All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the Nano-Ni only group, P < 0.05.
The expression levels of apoptosis-associated proteins were also determined by Western blot after BEAS-2B cells were pretreated with Bafilomycin A1 and Rapamycin prior to Nano-Ni exposure. Our results indicated that pretreatment of the cells with autophagic flux inhibitor, Bafilomycin A1, significantly enhanced Nano-Ni-induced apoptosis, which was reflected by increased expression of Bax and cleaved caspase-3 and decreased expression of Bcl-2 (Fig. 9A and C). However, pretreatment of the cells with autophagy activator, Rapamycin, effectively abolished Nano-Ni-induced apoptosis; decreased expression of Bax and cleaved caspase-3 and increased expression of Bcl-2 were observed (Fig. 9B and D). The results were confirmed by Hoechst 33342 staining (Fig. 9E). Taken together, our results suggest that an increased level of autophagy by Nano-Ni exposure has a protective effect on Nano-Ni-induced apoptosis.
Fig. 9. Nano-Ni-induced autophagy has a protective role against Nano-Ni-induced apoptosis in BEAS-2B cells.

(A-D) BEAS-2B cells were pre-treated with 5 nM of Bafilomycin A1(Baf A1) (an autophagic flux blocker) or 100 nM of Rapamycin (Rapa) (an autophagy activator) for 4 h prior to exposure to 20 μg/mL of Nano-Ni for another 24 h, and apoptosis-associated proteins (Bax, Bcl-2, caspase-3, and cleaved caspase-3) were measured by Western blot (A & B), and normalized grayscale values of the immuno-bands were from at least three independent experiments (C & D). (E) Hoechst 33342 staining was performed to identify the apoptotic cells in BEAS-2B cells with Baf A1 or Rapa pretreatment for 4 h prior to 20 μg/mL of Nano-Ni exposure for another 24 h. Cells without any treatments were used as a control. All quantitative results were presented as mean ± SE. * Significant difference as compared with the control group, P < 0.05; # Significant difference as compared with the Nano-Ni only group, P < 0.05.
4. Discussion
Nickel nanoparticles (Nano-Ni) have been increasingly used in industrial and biomedical fields, raising the urgency to understand and study their potential health effects. The airway epithelium functions as the first barrier against environmental insults, including inhaled bacteria, pathogens, and particles (Aghapour et al., 2018). Damage to epithelial cells not only exposes the subepithelial layers to exogenous influencers but also alters the normal function of epithelial cells, which eventually contributes to the development of various lung diseases such as allergy, inflammation, fibrosis, cancer, etc. (DeLong et al., 2019; Mo et al., 2020; Roach et al., 2019). In this study, we explored the effects of Nano-Ni on normal human bronchial epithelial BEAS-2B cells and the mechanisms involved in these effects. Our results demonstrated that Nano-Ni exposure caused increased levels of autophagy and apoptosis through the HIF-1α/mTOR pathway, and Nano-Ni-induced autophagy has a protective role in Nano-Ni-induced apoptosis.
Our previous study showed that exposure of BEAS-2B cells to Nano-Ni at concentrations up to 20 μg/mL for 24 h did not cause any cytotoxicity (Mo et al., 2021). Therefore, non-cytotoxic doses of Nano-Ni (10 and 20 μg/mL) were chosen to investigate the potential effects of Nano-Ni on BEAS-2B cells and the possible mechanisms involved in these effects. Nano-TiO2 was used as a control, which did not cause any significant cytotoxicity at any of the experimental doses up to 40 μg/mL (Mo et al., 2021). Although it is not easy to estimate the degree of human health effects at these concentrations or the quantity of inhaled metal nanoparticles that might be comparable to these dose-response studies, metal nanoparticles may accumulate in the lungs over time. Exposure to a dose that is lower than the cytotoxic dose can help identify potential health effects of metal nanoparticles other than those due to cytotoxicity. In addition, previous studies indicated that Nano-Ni showed a low mobilization of soluble nickel ions and could be taken up by BEAS-2B cells stably and rapidly (Di Bucchianico et al., 2018; Pietruska et al., 2011), which ensured the biological effects observed in this study were most likely due to the interaction between the cells and Nano-Ni rather than the ions released from Nano-Ni.
Previous studies have shown that autophagic response may be influenced by the size, surface charge, and concentrations of nanoparticles (Chou et al., 2017; Zhou et al., 2022). And the role of autophagy in nanoparticle-induced cellular outcomes is very complex because autophagy may have close crosstalk with other cell death and signaling pathways (Guo et al., 2020; Jia et al., 2020; Mohammadinejad et al., 2019). Nanoparticles may activate autophagy that may serve a cytoprotective role with the goal of survival, on the contrary, they may also induce autophagic flux blockage, resulting in an accumulation of damaged DNA, proteins, and organelles that in turn may increase cell death or proliferation and gene mutation (Peynshaert et al., 2014). Although several studies demonstrated that exposure to nickel or nickel oxidate nanoparticles caused autophagy alteration (Cho et al., 2020; Feng et al., 2015b; Gao et al., 2022; Saquib et al., 2020), it is unclear how Nano-Ni induced autophagy and its role in Nano-Ni-induced cell outcomes. In this study, we found that exposure to Nano-Ni initiated autophagy, which was reflected by increased LC3B-II/LC3B-I ratio and Beclin 1 protein expression in BEAS-2B cells exposed to Nano-Ni. LC3B-II/LC3B-I ratio is a typical marker for autophagy activation since cytosolic LC3B-I is conjugated to phosphatidylethanolamine to form LC3B-II when autophagy is activated (Lystad et al., 2019), while Beclin 1 plays a pivotal role in autophagy through the mediation of the localization of other autophagy-associated proteins to the autophagosomal membrane (Kang et al., 2011).
In this study, we also found that exposure of BEAS-2B cells to 20 μg/mL of Nano-Ni for 24 and 48 h caused a significant increase in p62 protein, which might suggest that exposure to Nano-Ni might also interrupt the autophagic flux since the accumulation of p62 is often considered as a hallmark of fusion failure of autophagosomes and lysosomes (Yao et al., 2021). However, increased p62 protein level is not always related to the blockage of autophagic flux. For example, upregulation of SQSTM1 concomitant with increased autophagic flux was observed during retinoic acid-induced differentiation of AML cells (Trocoli et al., 2014). And other autophagy-independent mechanisms underlying p62 protein accumulation after Nano-Ni exposure cannot be completely excluded. The accumulation of p62 protein might be attributed to the impairment of lysosomal or ubiquitinated degradation systems (Kravic et al., 2020). In addition, besides its critical role in autophagy involved in cellular detoxification and stress relief, p62 also acts as a multifunctional domain protein in the signaling hub in the control of cell growth and cancer (Moscat and Diaz-Meco 2012). Nano-Ni exposure has been shown to activate transcription factors such as NF-κB and MAPKs (Gu et al., 2016), which might cause the upregulation of p62 expression independently on the autophagic system (Kim et al., 2014; Trocoli et al., 2014). High levels of p62 in epithelial cells are also considered necessary and sufficient for tumor initiation and are independent of its autophagy-related functions (Moscat et al., 2016), which may be involved in Nano-Ni-induced malignant transformation of BEAS-2B cells (Mo et al., 2021). However, the detailed function of p62 protein accumulation in BEAS-2B cells exposed to Nano-Ni needs to be further investigated.
Previous studies demonstrated that Nano-Ni induced apoptosis in a variety of lung epithelial-derived cell lines such as human alveolar epithelial adenocarcinoma cell line (A549), human large-cell lung carcinoma cell line (NCI-H460), and normal human bronchial epithelial cell line (BEAS-2B) (Di Bucchianico et al., 2018; Magaye et al., 2016; Pietruska et al., 2011). Exposure of BEAS-2B cells to 10 μg/mL of Ni2+ or Nano-Ni or 5 μg/mL of Nano-NiO caused significantly increased number of apoptotic cells (Di Bucchianico et al., 2018). In the present study, we observed that exposure of BEAS-2B cells to Nano-Ni also caused increased apoptosis, which was reflected by increased expression of pro-apoptotic proteins Bax and cleaved caspase-3 and decreased expression of anti-apoptotic protein Bcl-2.
Both autophagy and apoptosis are important processes for the maintenance of cellular and organismal homeostasis, yet the relationship between the two needs to be viewed in a dialectical manner (Fernández et al., 2015; Maiuri et al., 2007). It is well known that autophagic activity is required for the removal of toxic proteins that damage organelles in cells, thus providing part of the bioenergetic substrates necessary for cell survival (Mizushima and Komatsu 2011). However, growing evidence also demonstrated that cell death could also be stimulated by high levels of autophagy activity, which is known as autophagic cell death (Kroemer and Levine 2008). To explore the potential role of autophagy in Nano-Ni-induced apoptosis, pharmacological inhibitor (Bafilomycin A1) and activator (Rapamycin) of autophagic flux were used to pretreat cells prior to Nano-Ni exposure. We found that blockage of autophagic flux in BEAS-2B cells prior to Nano-Ni exposure caused a significant increase in apoptosis, but activation of autophagy prior to Nano-Ni exposure ameliorated Nano-Ni-induced apoptosis, suggesting that Nano-Ni-induced autophagy might have a protective role in Nano-Ni-induced apoptosis in BEAS-2B cells.
It has been extensively demonstrated that activation of mTOR, a serine/threonine kinase, can inhibit autophagy. mTOR phosphorylates Ser757 on the autophagy-initiating kinase Ulk1 to prevent Ulk1 activation, thus preventing autophagy induction (Kim et al., 2011). Decreased phosphorylation of mTOR has been suggested to be an essential molecular signal for autophagy augmentation (Liu and Sabatini 2020; Munson and Ganley 2015). Previous studies have shown that NiCl2 administration could be invoked in the induction of kidney injury by inhibiting the Akt/AMPK/mTOR pathway to activate autophagy (Guo et al., 2021; Yin et al., 2021), and Nano-Ni-induced apoptosis in testis GC-1 spg cells was through the suppression of PI3K/AKT/mTOR signaling pathway (Kong et al., 2021). Although these studies have shown that exposure to nickel compounds could inhibit mTOR signaling, the detailed mechanism is still unclear. In the present study, we demonstrated that Nano-Ni exposure caused a significant decrease in the phosphorylated mTOR protein level in BEAS-2B cells. And pretreatment of the cells with mTOR agonist MHY1485 effectively abolished Nano-Ni-induced increased level of autophagy, but significantly enhanced Nano-Ni-induced increased cleaved caspase-3, an apoptosis-associated protein. MHY1485 binds to mTOR and stimulates its action, thus potently inhibiting autophagy by suppressing fusion between autophagosomes and lysosomes (Yang et al., 2020). In addition, mTOR activation by MHY1485 has been shown to inhibit autophagic flux and augment Benzoylbenzoyl-ATP (BzATP)-induced endoplasmic reticulum (ER) stress-associated apoptosis in rat primary chondrocyte (Li et al., 2021), and also enhanced radiation-induced apoptotic and senescent phenotype in tumor cells (Li et al., 2021). Therefore, mTOR signaling-regulated autophagy may be involved in apoptosis and other cell fates induced by chemicals, radiation, or various environmental risk factors. A previous study did show that failure to initiate autophagy program leads to cell death (Jiang et al., 2016).
HIF-1α-mediated autophagy is regarded as one of the major catabolic programs for cellular adaptation to hypoxic conditions (Kroemer et al., 2010). Under hypoxic conditions, HIF-1α-mediated autophagy facilitates the clearance of damaged mitochondria and prevents the production of ROS and the consequent oxidative stress, which was proposed as a critical strategy to maintain cellular activity during tumor progression (Abdrakhmanov et al., 2021; Dan Dunn et al., 2015). Mechanistically, nuclear accumulation of HIF-1α due to degradation failure or endogenous upregulation would transcriptionally induce the expression of BNIP3/BNIP3L, which then interferes with the Beclin 1-Bcl-XL/Bcl-2 interaction and releases Beclin 1 to trigger autophagy (Qureshi-Baig et al., 2020; Wu et al., 2018a). Previous studies showed that inhibition of HIF-1α by CAY10585 stimulated apoptosis in photoreceptors, Müller cells, by inhibiting HIF-1α-mediated mitochondrial autophagy (Sun et al., 2021). And HIF-1α-deficient Hela cells tend to be more sensitive to N-4-hydroxyphenyl-retinamide (fenretinide or 4-HRP) to induce apoptosis due to autophagy inhibition. 4-HRP is a synthetic retinoid derivative with potential use in preventing and treating solid tumors, lymphoma, and myeloid leukemia (Liu et al., 2010). Our current and previous studies have shown that Nano-Ni exposure caused HIF-1α nuclear accumulation in BEAS-2B and other cells (Feng et al., 2015a; Mo et al., 2021; Wan et al., 2011; Yuan et al., 2021). Pre-treatment of the cells with a HIF-1α inhibitor, CAY10585, significantly reduced Nano-Ni-induced autophagic activity, but caused a significant enhancement in the expression of pro-apoptotic protein, suggesting that Nano-Ni-induced HIF-1α nuclear accumulation may be involved in Nano-Ni-induced autophagy and apoptosis.
We also investigated the relationship between HIF-1α and mTOR by using a HIF-1α inhibitor and an mTOR agonist. HIF-1α has been shown to negatively regulate mTOR by an increase in AMP and the activation of AMPK (Pouysségur et al., 2006). In addition, hypoxia-mediated HIF-1α accumulation could downregulate mTOR phosphorylation and upregulate autophagy activation via inducing the expression of p62 and the reduction of NLRP3 protein levels, which then ameliorates intestinal inflammation in Crohn’s patients and murine colitis models (Cosin-Roger et al., 2017). A more direct link between HIF-1α and mTOR was established in Drosophila, in which HIF-1α directly targeted Scylla (REDD1/RTP801 in mammals) to activate the TSC complex, resulting in mTOR inhibition (Reiling and Hafen 2004). In the present study, inhibition of HIF-1α significantly restored the level of phosphorylated mTOR in cells treated with Nano-Ni, whereas mTOR agonist pretreatment showed no observable effect on the nuclear accumulation of HIF-1α, suggesting that HIF-1α may negatively regulate mTOR. However, the exact mechanism that how HIF-1α regulates mTOR needs to be further investigated.
5. Conclusions
In summary, our results showed that exposure to Nano-Ni rather than Nano-TiO2 caused increases in autophagy and apoptosis levels, nuclear accumulation of HIF-1α, but decreased phosphorylation level of mTOR in BEAS-2B cells. Nuclear accumulation of HIF-1α led to the reduction of phosphorylated mTOR expression in BEAS-2B cells with Nano-Ni exposure. Both inhibition of HIF-1α and activation of mTOR downregulated autophagy levels while promoting Nano-Ni-induced apoptosis in the cells. In addition, pretreatment of the cells with an autophagic flux blocker or agonist prior to Nano-Ni exposure exacerbated or ameliorated Nano-Ni-induced apoptosis, respectively. Our findings suggest that Nano-Ni-induced autophagy has a protective role against Nano-Ni-induced apoptosis through the HIF-1α/mTOR signaling axis (Fig. 10). Our results may provide a further understanding of cytotoxic effects caused by metal nanoparticles.
Fig. 10. Schematic graph and potential mechanisms of Nano-Ni-induced autophagy and apoptosis in BEAS-2B cells.

Exposure of BEAS-2B cells to Nano-Ni induced HIF-1α nuclear accumulation, and inhibition of HIF-1α by CAY10585 ameliorated Nano-Ni-induced decreased phosphorylation of mTOR. Inhibition of HIF-1α by CAY10585 or activation of mTOR by MHY1485 abolished Nano-Ni-induced increased autophagy and enhanced Nano-Ni-induced increased apoptosis. Blockade of autophagic flux by Bafilomycin A1 (Baf A1) enhanced Nano-Ni-induced apoptosis, while activation of autophagy by Rapamycin (Rapa) abolished Nano-Ni-induced apoptosis. All these results suggest that Nano-Ni-induced autophagy has a protective role against Nano-Ni-induced apoptosis through the HIF-1α/mTOR pathway.
Highlights.
Nano-Ni, but not Nano-TiO2, caused increased autophagy and apoptosis in BEAS-2B cells.
Nano-Ni led to HIF-1α nuclear accumulation and decreased mTOR phosphorylation.
HIF-1α may negatively regulate mTOR.
Nano-Ni-induced autophagy has a protective role against apoptosis via HIF-1α/mTOR signaling.
Acknowledgements
This work was partly supported by NIH (ES023693, ES028911, and HL147856), KSEF-148-RED-502–16-381, and Kentucky Lung Cancer Research Program to Dr. Qunwei Zhang. This work was also partly supported by P30ES030283 from the National Institute for Environmental Health Sciences (NIEHS).
Abbreviations
- Baf A1
Bafilomycin A1
- Bax
Bcl-2-associated X
- Bcl-2
B-cell lymphoma 2
- HIF-1α
hypoxia inducible factor-1α
- HRE
hypoxia response element
- LC3B
microtubule-associated proteins 1A/1B light chain 3B
- mTOR
mammalian target of rapamycin
- Nano-Ni
nickel nanoparticles
- Nano-TiO2
titanium dioxide nanoparticles
- p62
sequestosome 1 (SQSTM1/p62)
- PVDF
polyvinylidene difluoride
- Rapa
rapamycin
- VHL
von Hippel-Lindau
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethics approval and consent to participate
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data and materials
All data and materials are included in the manuscript.
References
- Abdrakhmanov A, Yapryntseva MA, Kaminskyy VO, Zhivotovsky B, Gogvadze V, 2021. Receptor-Mediated Mitophagy Rescues Cancer Cells under Hypoxic Conditions. Cancers (Basel) 13. doi: 10.3390/cancers13164027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH, 2018. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am J Respir Cell Mol Biol 58, 157–169. doi: 10.1165/rcmb.2017-0200TR. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, Squitieri F, Hardenberg MC, Imarisio S, Menzies FM, Rubinsztein DC, 2017. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545, 108–111. doi: 10.1038/nature22078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai R, Roy S, kulshrestha N, Rafiee J, Koratkar N, Misra DS, 2012. Graphene supported nickel nanoparticle as a viable replacement for platinum in dye sensitized solar cells. Nanoscale 4, 926–930. doi: 10.1039/c2nr11127f. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee D, Sheet SK, Khatua S, Biswas K, Joshi S, Myrboh B, 2018. A reusable magnetic nickel nanoparticle based catalyst for the aqueous synthesis of diverse heterocycles and their evaluation as potential anti-bacterial agent. Bioorg Med Chem 26, 5018–5028. doi: 10.1016/j.bmc.2018.08.033. [DOI] [PubMed] [Google Scholar]
- Cao Z, Song M, Liu Y, Pang J, Li Z, Qi X, Shu T, Li B, Wei D, Chen J, Li B, Wang J, Wang C, 2020. A novel pathophysiological classification of silicosis models provides some new insights into the progression of the disease. Ecotoxicol Environ Saf 202, 110834. doi: 10.1016/j.ecoenv.2020.110834. [DOI] [PubMed] [Google Scholar]
- Cho YL, Tan HWS, Saquib Q, Ren Y, Ahmad J, Wahab R, He W, Bay BH, Shen HM, 2020. Dual role of oxidative stress-JNK activation in autophagy and apoptosis induced by nickel oxide nanoparticles in human cancer cells. Free Radic Biol Med 153, 173–186. doi: 10.1016/j.freeradbiomed.2020.03.027. [DOI] [PubMed] [Google Scholar]
- Chou CC, Chen W, Hung Y, Mou CY, 2017. Molecular Elucidation of Biological Response to Mesoporous Silica Nanoparticles in Vitro and in Vivo. ACS Appl Mater Interfaces 9, 22235–22251. doi: 10.1021/acsami.7b05359. [DOI] [PubMed] [Google Scholar]
- Cosin-Roger J, Simmen S, Melhem H, Atrott K, Frey-Wagner I, Hausmann M, de Vallière C, Spalinger MR, Spielmann P, Wenger RH, Zeitz J, Vavricka SR, Rogler G, Ruiz PA, 2017. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun 8, 98. doi: 10.1038/s41467-017-00213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Dunn J, Alvarez LA, Zhang X, Soldati T, 2015. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol 6, 472–485. doi: 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong RK, Comer J, Mathew EN, Jaberi-Douraki M, 2019. Comparative Molecular Immunological Activity of Physiological Metal Oxide Nanoparticle and its Anticancer Peptide and RNA Complexes. Nanomaterials (Basel) 9. doi: 10.3390/nano9121670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bucchianico S, Gliga AR, Åkerlund E, Skoglund S, Wallinder IO, Fadeel B, Karlsson HL, 2018. Calcium-dependent cyto- and genotoxicity of nickel metal and nickel oxide nanoparticles in human lung cells. Part Fibre Toxicol 15, 32. doi: 10.1186/s12989-018-0268-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossou AS, Basu A, 2019. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers (Basel) 11. doi: 10.3390/cancers11101422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Yu J, Jin D, Chiu PWY, Zhang L, 2021. Independent Pattern Formation of Nanorod and Nanoparticle Swarms under an Oscillating Field. ACS Nano, 10.1021/acsnano.0c08284. doi: 10.1021/acsnano.0c08284. [DOI] [PubMed] [Google Scholar]
- Feng L, Zhang Y, Jiang M, Mo Y, Wan R, Jia Z, Tollerud DJ, Zhang X, Zhang Q, 2015a. Upregulation of Gadd45α after exposure to metal nanoparticles: the role of hypoxia inducible factor 1α. Environ Toxicol 30, 490–499. doi: 10.1002/tox.21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng PH, Huang YL, Chuang KJ, Chen KY, Lee KY, Ho SC, Bien MY, Yang YL, Chuang HC, 2015b. Dysfunction of methionine sulfoxide reductases to repair damaged proteins by nickel nanoparticles. Chem Biol Interact 236, 82–89. doi: 10.1016/j.cbi.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Feng X, Chen L, Guo W, Zhang Y, Lai X, Shao L, Li Y, 2018. Graphene oxide induces p62/SQSTM-dependent apoptosis through the impairment of autophagic flux and lysosomal dysfunction in PC12 cells. Acta Biomater 81, 278–292. doi: 10.1016/j.actbio.2018.09.057. [DOI] [PubMed] [Google Scholar]
- Fernández A, Ordóñez R, Reiter RJ, González-Gallego J, Mauriz JL, 2015. Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J Pineal Res 59, 292–307. doi: 10.1111/jpi.12264. [DOI] [PubMed] [Google Scholar]
- Gao Q, Chang X, Yang M, Zheng J, Gong X, Liu H, Li K, Wang X, Zhan H, Li S, Feng S, Sun X, Sun Y, 2022. LncRNA MEG3 restrained pulmonary fibrosis induced by NiO NPs via regulating hedgehog signaling pathway-mediated autophagy. Environ Toxicol 37, 79–91. doi: 10.1002/tox.23379. [DOI] [PubMed] [Google Scholar]
- Glista-Baker EE, Taylor AJ, Sayers BC, Thompson EA, Bonner JC, 2012. Nickel nanoparticles enhance platelet-derived growth factor-induced chemokine expression by mesothelial cells via prolonged mitogen-activated protein kinase activation. Am J Respir Cell Mol Biol 47, 552–561. doi: 10.1165/rcmb.2012-0023OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González A, Hall MN, 2017. Nutrient sensing and TOR signaling in yeast and mammals. Embo j 36, 397–408. doi: 10.15252/embj.201696010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A, 2004. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23, 2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- Griffitt RJ, Luo J, Gao J, Bonzongo JC, Barber DS, 2008. Effects of particle composition and species on toxicity of metallic nanomaterials in aquatic organisms. Environ Toxicol Chem 27, 1972–1978. doi: 10.1897/08-002.1. [DOI] [PubMed] [Google Scholar]
- Gu Y, Wang Y, Zhou Q, Bowman L, Mao G, Zou B, Xu J, Liu Y, Liu K, Zhao J, Ding M, 2016. Inhibition of Nickel Nanoparticles-Induced Toxicity by Epigallocatechin-3-Gallate in JB6 Cells May Be through Down-Regulation of the MAPK Signaling Pathways. PLoS One 11, e0150954. doi: 10.1371/journal.pone.0150954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Yin H, Zuo Z, Yang Z, Yang Y, Wei L, Cui H, Deng H, Chen X, Chen J, Zhu Y, Ouyang P, Geng Y, Du Z, Tang H, Wang F, Fang J, 2021. Oxidative stress-mediated apoptosis and autophagy involved in Ni-induced nephrotoxicity in the mice. Ecotoxicol Environ Saf 228, 112954. doi: 10.1016/j.ecoenv.2021.112954. [DOI] [PubMed] [Google Scholar]
- Guo L, He N, Zhao Y, Liu T, Deng Y, 2020. Autophagy Modulated by Inorganic Nanomaterials. Theranostics 10, 3206–3222. doi: 10.7150/thno.40414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Phelan JD, Preite S, Gomez-Rodriguez J, Johansen KH, Shibata H, Shaffer AL 3rd, Xu Q, Jeffrey B, Kirby M, Anderson S, Yang Y, Gossa S, McGavern DB, Staudt LM, Schwartzberg PL, 2022. In vivo CRISPR screens reveal a HIF-1alpha-mTOR-network regulates T follicular helper versus Th1 cells. Nat Commun 13, 805. doi: 10.1038/s41467-022-28378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Hao SL, Yang WX, 2020. Nanoparticles induce autophagy via mTOR pathway inhibition and reactive oxygen species generation. Nanomedicine (Lond) 15, 1419–1435. doi: 10.2217/nnm-2019-0387. [DOI] [PubMed] [Google Scholar]
- Jiang LB, Lee S, Wang Y, Xu QT, Meng DH, Zhang J, 2016. Adipose-derived stem cells induce autophagic activation and inhibit catabolic response to pro-inflammatory cytokines in rat chondrocytes. Osteoarthritis Cartilage 24, 1071–1081. doi: 10.1016/j.joca.2015.12.021. [DOI] [PubMed] [Google Scholar]
- Journeay WS, Goldman RH, 2014. Occupational handling of nickel nanoparticles: a case report. Am J Ind Med 57, 1073–1076. doi: 10.1002/ajim.22344. [DOI] [PubMed] [Google Scholar]
- Kaelin WG Jr., Ratcliffe PJ, 2008. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30, 393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, Tang D, 2011. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18, 571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL, 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Hong SK, Wu PK, Richards AL, Jackson WT, Park JI, 2014. Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp Cell Res 327, 340–352. doi: 10.1016/j.yexcr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Wu Y, Hu W, Liu L, Xue Y, Liang G, 2021. Mechanisms underlying reproductive toxicity induced by nickel nanoparticles identified by comprehensive gene expression analysis in GC-1 spg cells. Environ Pollut 275, 116556. doi: 10.1016/j.envpol.2021.116556. [DOI] [PubMed] [Google Scholar]
- Kravic B, Behrends C, Meyer H, 2020. Regulation of lysosome integrity and lysophagy by the ubiquitin-conjugating enzyme UBE2QL1. Autophagy 16, 179–180. doi: 10.1080/15548627.2019.1687217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Levine B, 2008. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9, 1004–1010. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Mariño G, Levine B, 2010. Autophagy and the integrated stress response. Mol Cell 40, 280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N, 2004. The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Li H, Li Y, Jiao J, Hu HM, 2011. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat Nanotechnol 6, 645–650. doi: 10.1038/nnano.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chen J, Fan H, Cai R, Gao X, Meng D, Ji Y, Chen C, Wang L, Wu X, 2020. Initiation of protective autophagy in hepatocytes by gold nanorod core/silver shell nanostructures. Nanoscale 12, 6429–6437. doi: 10.1039/c9nr08621h. [DOI] [PubMed] [Google Scholar]
- Li Z, Huang Z, Zhang H, Lu J, Wei Y, Yang Y, Bai L, 2021. IRE1-mTOR-PERK Axis Coordinates Autophagy and ER Stress-Apoptosis Induced by P2X7-Mediated Ca(2+) Influx in Osteoarthritis. Front Cell Dev Biol 9, 695041. doi: 10.3389/fcell.2021.695041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Liu Y, Wu H, Huang Z, Ma J, Guo C, Gao F, Jin P, Wei P, Zhang Y, Liu L, Zhang R, Qiu L, Gu N, Wen L, 2018. Key Role of TFEB Nucleus Translocation for Silver Nanoparticle-Induced Cytoprotective Autophagy. Small 14, e1703711. doi: 10.1002/smll.201703711. [DOI] [PubMed] [Google Scholar]
- Liu GY, Sabatini DM, 2020. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 21, 183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Lu W, Dong J, Wu Y, Tang M, Liang G, Kong L, 2022. Study of the mechanism of mitochondrial division and mitochondrial autophagy in the male reproductive toxicity induced by nickel nanoparticles. Nanoscale 14, 1868–1884. doi: 10.1039/d1nr05407d. [DOI] [PubMed] [Google Scholar]
- Liu XW, Su Y, Zhu H, Cao J, Ding WJ, Zhao YC, He QJ, Yang B, 2010. HIF-1α-dependent autophagy protects HeLa cells from fenretinide (4-HPR)-induced apoptosis in hypoxia. Pharmacol Res 62, 416–425. doi: 10.1016/j.phrs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Liu Z, Lv X, Xu L, Liu X, Zhu X, Song E, Song Y, 2020. Zinc oxide nanoparticles effectively regulate autophagic cell death by activating autophagosome formation and interfering with their maturation. Part Fibre Toxicol 17, 46. doi: 10.1186/s12989-020-00379-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lystad AH, Carlsson SR, de la Ballina LR, Kauffman KJ, Nag S, Yoshimori T, Melia TJ, Simonsen A, 2019. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat Cell Biol 21, 372–383. doi: 10.1038/s41556-019-0274-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magaye R, Gu Y, Wang Y, Su H, Zhou Q, Mao G, Shi H, Yue X, Zou B, Xu J, Zhao J, 2016. In vitro and in vivo evaluation of the toxicities induced by metallic nickel nano and fine particles. J Mol Histol 47, 273–286. doi: 10.1007/s10735-016-9671-6. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G, 2007. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8, 741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Mazure NM, Pouyssegur J, 2010. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol 22, 177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Minocha S, Mumper RJ, 2012. Effect of carbon coating on the physico-chemical properties and toxicity of copper and nickel nanoparticles. Small 8, 3289–3299. doi: 10.1002/smll.201200478. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M, 2011. Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Mo Y, Jiang M, Zhang Y, Wan R, Li J, Zhong CJ, Li H, Tang S, Zhang Q, 2019. Comparative mouse lung injury by nickel nanoparticles with differential surface modification. J Nanobiotechnology 17, 2. doi: 10.1186/s12951-018-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Zhang Y, Wan R, Jiang M, Xu Y, Zhang Q, 2020. miR-21 mediates nickel nanoparticle-induced pulmonary injury and fibrosis. Nanotoxicology 14, 1175–1197. doi: 10.1080/17435390.2020.1808727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Zhang Y, Zhang Y, Yuan J, Mo L, Zhang Q, 2021. Nickel nanoparticle-induced cell transformation: involvement of DNA damage and DNA repair defect through HIF-1α/miR-210/Rad52 pathway. J Nanobiotechnology 19, 370. doi: 10.1186/s12951-021-01117-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadinejad R, Moosavi MA, Tavakol S, Vardar D, Hosseini A, Rahmati M, Dini L, Hussain S, Mandegary A, Klionsky DJ, 2019. Necrotic, apoptotic and autophagic cell fates triggered by nanoparticles. Autophagy 15, 4–33. doi: 10.1080/15548627.2018.1509171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, 2012. p62: a versatile multitasker takes on cancer. Trends Biochem Sci 37, 230–236. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Karin M, Diaz-Meco MT, 2016. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 167, 606–609. doi: 10.1016/j.cell.2016.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson MJ, Ganley IG, 2015. MTOR, PIK3C3, and autophagy: Signaling the beginning from the end. Autophagy 11, 2375–2376. doi: 10.1080/15548627.2015.1106668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peynshaert K, Manshian BB, Joris F, Braeckmans K, De Smedt SC, Demeester J, Soenen SJ, 2014. Exploiting intrinsic nanoparticle toxicity: the pros and cons of nanoparticle-induced autophagy in biomedical research. Chem Rev 114, 7581–7609. doi: 10.1021/cr400372p. [DOI] [PubMed] [Google Scholar]
- Phillips JI, Green FY, Davies JC, Murray J, 2010. Pulmonary and systemic toxicity following exposure to nickel nanoparticles. Am J Ind Med 53, 763–767. doi: 10.1002/ajim.20855. [DOI] [PubMed] [Google Scholar]
- Pietruska JR, Liu X, Smith A, McNeil K, Weston P, Zhitkovich A, Hurt R, Kane AB, 2011. Bioavailability, intracellular mobilization of nickel, and HIF-1α activation in human lung epithelial cells exposed to metallic nickel and nickel oxide nanoparticles. Toxicol Sci 124, 138–148. doi: 10.1093/toxsci/kfr206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouysségur J, Dayan F, Mazure NM, 2006. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- Qureshi-Baig K, Kuhn D, Viry E, Pozdeev VI, Schmitz M, Rodriguez F, Ullmann P, Koncina E, Nurmik M, Frasquilho S, Nazarov PV, Zuegel N, Boulmont M, Karapetyan Y, Antunes L, Val D, Mittelbronn M, Janji B, Haan S, Letellier E, 2020. Hypoxia-induced autophagy drives colorectal cancer initiation and progression by activating the PRKC/PKC-EZR (ezrin) pathway. Autophagy 16, 1436–1452. doi: 10.1080/15548627.2019.1687213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiling JH, Hafen E, 2004. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by downregulating S6K activity upstream of TSC in Drosophila. Genes Dev 18, 2879–2892. doi: 10.1101/gad.322704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach KA, Anderson SE, Stefaniak AB, Shane HL, Kodali V, Kashon M, Roberts JR, 2019. Surface area- and mass-based comparison of fine and ultrafine nickel oxide lung toxicity and augmentation of allergic response in an ovalbumin asthma model. Inhal Toxicol 31, 299–324. doi: 10.1080/08958378.2019.1680775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Nakahira K, Haspel JA, Choi AM, 2012. Autophagy in pulmonary diseases. Annu Rev Physiol 74, 377–401. doi: 10.1146/annurev-physiol-020911-153348. [DOI] [PubMed] [Google Scholar]
- Salnikow K, An WG, Melillo G, Blagosklonny MV, Costa M, 1999. Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis 20, 1819–1823. doi: 10.1093/carcin/20.9.1819. [DOI] [PubMed] [Google Scholar]
- Sandhu LC, Warters RL, Dethlefsen LA, 1985. Fluorescence studies of Hoechst 33342 with supercoiled and relaxed plasmid pBR322 DNA. Cytometry 6, 191–194. doi: 10.1002/cyto.990060304. [DOI] [PubMed] [Google Scholar]
- Saquib Q, Xia P, Siddiqui MA, Zhang J, Xie Y, Faisal M, Ansari SM, Alwathnani HA, Alatar AA, Al-Khedhairy AA, Zhang X, 2020. High-throughput transcriptomics: An insight on the pathways affected in HepG2 cells exposed to nickel oxide nanoparticles. Chemosphere 244, 125488. doi: 10.1016/j.chemosphere.2019.125488. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ, 2004. Autophagy in health and disease: a double-edged sword. Science 306, 990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern ST, Adiseshaiah PP, Crist RM, 2012. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol 9, 20. doi: 10.1186/1743-8977-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wen F, Yan C, Su L, Luo J, Chi W, Zhang S, 2021. Mitophagy Protects the Retina Against Anti-Vascular Endothelial Growth Factor Therapy-Driven Hypoxia via Hypoxia-Inducible Factor-1α Signaling. Front Cell Dev Biol 9, 727822. doi: 10.3389/fcell.2021.727822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trocoli A, Bensadoun P, Richard E, Labrunie G, Merhi F, Schläfli AM, Brigger D, Souquere S, Pierron G, Pasquet JM, Soubeyran P, Reiffers J, Ségal-Bendirdjian E, Tschan MP, Djavaheri-Mergny M, 2014. p62/SQSTM1 upregulation constitutes a survival mechanism that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ 21, 1852–1861. doi: 10.1038/cdd.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan R, Mo Y, Chien S, Li Y, Li Y, Tollerud DJ, Zhang Q, 2011. The role of hypoxia inducible factor-1alpha in the increased MMP-2 and MMP-9 production by human monocytes exposed to nickel nanoparticles. Nanotoxicology 5, 568–582. doi: 10.3109/17435390.2010.537791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zeng L, Su P, Ma L, Zhang M, Zhang YZ, 2022. Autophagy: a multifaceted player in the fate of sperm. Hum Reprod Update 28, 200–231. doi: 10.1093/humupd/dmab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Lu W, Tovmachenko O, Rai US, Yu H, Ray PC, 2008. Challenge in Understanding Size and Shape Dependent Toxicity of Gold Nanomaterials in Human Skin Keratinocytes. Chem Phys Lett 463, 145–149. doi: 10.1016/j.cplett.2008.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JC, Tsai HE, Liu GS, Wu CS, Tai MH, 2018a. Autophagic cell death participates in POMC-induced melanoma suppression. Cell Death Discov 4, 11. doi: 10.1038/s41420-018-0070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Xiao T, Luo Z, He R, Cao Y, Guo Z, Zhang W, Chen Y, 2018b. A micro-/nano-chip and quantum dots-based 3D cytosensor for quantitative analysis of circulating tumor cells. J Nanobiotechnology 16, 65. doi: 10.1186/s12951-018-0390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Tu B, Zhou X, Jiang X, Xu G, Zhang J, Qin X, Sumayyah G, Fan J, Wang B, Chen C, Zou Z, 2021. Autophagy deficiency exacerbates acute lung injury induced by copper oxide nanoparticles. J Nanobiotechnology 19, 162. doi: 10.1186/s12951-021-00909-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Wan B, Yang Y, Cui X, Xin Y, Guo LH, 2019. Cytotoxicity and autophagy induction by graphene quantum dots with different functional groups. J Environ Sci (China) 77, 198–209. doi: 10.1016/j.jes.2018.07.014. [DOI] [PubMed] [Google Scholar]
- Yang H, Wen Y, Zhang M, Liu Q, Zhang H, Zhang J, Lu L, Ye T, Bai X, Xiao G, Wang M, 2020. MTORC1 coordinates the autophagy and apoptosis signaling in articular chondrocytes in osteoarthritic temporomandibular joint. Autophagy 16, 271–288. doi: 10.1080/15548627.2019.1606647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao RQ, Ren C, Xia ZF, Yao YM, 2021. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy 17, 385–401. doi: 10.1080/15548627.2020.1725377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Zuo Z, Yang Z, Guo H, Fang J, Cui H, Ouyang P, Chen X, Chen J, Geng Y, Chen Z, Huang C, Zhu Y, 2021. Nickel induces autophagy via PI3K/AKT/mTOR and AMPK pathways in mouse kidney. Ecotoxicol Environ Saf 223, 112583. doi: 10.1016/j.ecoenv.2021.112583. [DOI] [PubMed] [Google Scholar]
- Yu Y, Duan J, Yu Y, Li Y, Liu X, Zhou X, Ho KF, Tian L, Sun Z, 2014. Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. J Hazard Mater 270, 176–186. doi: 10.1016/j.jhazmat.2014.01.028. [DOI] [PubMed] [Google Scholar]
- Yuan J, Han R, Esther A, Wu Q, Yang J, Yan W, Ji X, Liu Y, Li Y, Yao W, Ni C, 2017. Polymorphisms in autophagy related genes and the coal workers’ pneumoconiosis in a Chinese population. Gene 632, 36–42. doi: 10.1016/j.gene.2017.08.017. [DOI] [PubMed] [Google Scholar]
- Yuan J, Zhang Y, Zhang Y, Mo Y, Zhang Q, 2021. Effects of metal nanoparticles on tight junction-associated proteins via HIF-1α/miR-29b/MMPs pathway in human epidermal keratinocytes. Part Fibre Toxicol 18, 13. doi: 10.1186/s12989-021-00405-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Mo Y, Zhang Y, Zhang Y, Zhang Q, 2022. Nickel nanoparticles induce epithelial-mesenchymal transition in human bronchial epithelial cells via the HIF-1alpha/HDAC3 pathway. Nanotoxicology 16, 695–712. doi: 10.1080/17435390.2022.2142169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Kusaka Y, Sato K, Nakakuki K, Kohyama N, Donaldson K, 1998. Differences in the extent of inflammation caused by intratracheal exposure to three ultrafine metals: role of free radicals. J Toxicol Environ Health A 53, 423–438. doi: 10.1080/009841098159169. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Kusaka Y, Zhu X, Sato K, Mo Y, Kluz T, Donaldson K, 2003. Comparative toxicity of standard nickel and ultrafine nickel in lung after intratracheal instillation. J Occup Health 45, 23–30. doi: 10.1539/joh.45.23. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Fu Z, 2022. Role of autophagy in lung diseases and ageing. Eur Respir Rev 31. doi: 10.1183/16000617.0134-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Jin W, Sun H, Li C, Jia J, 2022. Perturbation of autophagy: An intrinsic toxicity mechanism of nanoparticles. Sci Total Environ, 10.1016/j.scitotenv.2022.153629, 153629. doi: 10.1016/j.scitotenv.2022.153629. [DOI] [PubMed] [Google Scholar]
- Ziello JE, Jovin IS, Huang Y, 2007. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med 80, 51–60. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and materials are included in the manuscript.