

Abstract
The present review aims to summarize available knowledge on the role of the ubiquitin-proteasome system (UPS) in the pathogenesis of scleroderma and scleroderma-related disease mechanisms. This will provide the reader with a more mechanistic understanding of disease pathogenesis and help to identify putative novel targets within the UPS for potential therapeutic intervention. Due to the heterogenous manifestations of scleroderma, we will primarily focus on conserved mechanisms that are involved in the development of lung scleroderma phenotypes.
2. Pathogenesis of scleroderma
Scleroderma is a heterogenous autoimmune inflammatory disorder characterized by progressive fibrosis of the skin and internal organs. The American College of Rheumatology describes the three pathogenic hallmarks of scleroderma as fibroblast dysfunction, small vessel vasculopathy, and production of autoantibodies, leading to excess extracellular matrix deposition and fibrosis 1. In the United States, scleroderma affects nearly 100,000 people, with an incidence estimated to be ~20/million people with a prevalence of ~240/million 2–4. Clinically, the consequences of irreversible skin and organ fibrosis can be devastating leading to long term disability and increased mortality, with the greatest effect from interstitial lung disease and pulmonary hypertension 5–7. Alterations in several molecular processes have been identified in the initiation and progression of scleroderma-related organ fibrosis, including activation of the innate immune system, autoimmune responses of the adaptive immune system, and excessive transforming growth factor β (TGFβ) signaling driving fibrotic tissue remodeling 8–10. The pathophysiology of SSc-ILD is complex and incompletely understood, but shares the common features of organ fibrosis. The process of fibrosis is thought to follow four general phases: injury, activation of effector cells, synthesis of extracellular matrix (ECM) components, and ECM deposition with inadequate resorption 11. In scleroderma, both endothelial and epithelial injury are histopathological characteristics of the disease 12,13. While the inciting injury or antigens are incompletely understood, these insults are associated with the presence of inflammation, activation of the innate immune system, and activation of the adaptive immune system 9,14–16. These events activate tissue fibroblasts, which are the primary effector cells that synthesize and deposit excessive ECM components 17,18, which eventually culminates in organ fibrosis. This paradigm is an oversimplification of the complex pathological changes in SSc-ILD, as infiltrating immune cells, epithelial cell differentiation, and impaired injury resolution are all additional mechanisms that contribute to the phenotype 18. These pathogenic changes are dependent on a complex network of genetic, transcriptomic, and post-translational factors leading to the clinical phenotype of organ fibrosis. One important post-translational mechanism regulating protein levels is the ubiquitin-proteasome system (UPS), which selectively targets proteins for degradation 19,20. This review will focus on the role of the UPS and protein degradation in scleroderma to evaluate this disposal system as a new therapeutic target for systemic sclerosis with a particular focus on the activation of the innate and adaptive immune systems, as well as the profibrotic response that is driven by fibroblasts.
3. The ubiquitin-proteasome system
The UPS is the main non-lysosomal protein degradation pathway in the cell. It degrades proteins that are covalently tagged with a polyubiquitin chain into small peptides which are then used for amino acid recycling but also for MHC class I antigen presentation 21,22. Ubiquitination is carried out in a multi-step process involving E1-activating enzymes, E2-conjugating enzymes, and E3 Ligases. In humans there exist two E1 enzymes, about 30 E2s, and over 700 E3 ligases (Figure 1). The substrate ubiquitin acceptor site is typically a lysine, whereby the epsilon-amino group (ε-NH+3) forms an isopeptide bond with the carboxyl group (COO−) of the C-terminal glycine of ubiquitin. After a substrate is modified by a single ubiquitin moiety, it may be altered further by the transfer of additional ubiquitin moieties linked to one of seven lysine residues of ubiquitin, creating poly-ubiquitinated or multi-ubiquitinated linear or branched chains. The degree and type of ubiquitination serve different purposes 23. The present understanding is that substrates that contain K48-linked polyubiquitin chains are primarily degraded by the 26S proteasome, while substrates linked with K63-polyubiquitin chains function to regulate such diverse cellular activities as kinase activation and protein trafficking 24. K48-mediated protein ubiquitination and subsequent degradation by the proteasome is a critical regulatory mechanism controlling stability of proteins involved in inflammatory, metabolic, neurologic, hematologic, oncologic, and age-related diseases 20,25. Ubiquitinated substrates are also subject to removal of the 8.5 kDa ubiquitin moiety catalyzed by one of several families of deubiquitination enzymes.
Figure 1:

Protein degradation is mediated by the ubiquitin proteasome system. Ubiquitin is transferred to E1 activating enzymes in an ATP-dependent fashion, followed by ubiquitin transfer to E2 conjugating enzymes. E3 ligases recognize “degrons” on substrate proteins created by modifications such as phosphorylation. These ligases link substrate proteins to the ubiquitin-transferring machinery, and with further ubiquitination events (K48-linked poly-ubiquitin), substrates are shuttled to the proteasome for degradation.
Proteins that are modified by K48-linked polyubiquitin chains are proteolytically cleaved by the 26S proteasome consisting of one or two 19S regulator complexes bound to the symmetric and barrel-shaped 20S catalytic core 26. While the multimeric 19S regulators confers binding of ubiquitinated substrates, their deubiquitination and ATP-dependent unfolding of proteins, the 20S core contains three distinct active sites that proteolytically cleave the unfolded amino acid chain after tunneling into the proteolytic chambers 27,28. These active sites come in two flavors, the standard subunits beta 1, beta 2 and beta 5, and the inducible immunoproteasome subunits beta1i (LMP2), beta2i (MECL-1), and beta5i (LMP7) forming the standard 20S (s20S) and the immunoproteasome (i20S), respectively 29. Immunoproteasomes are constitutively present in immune cells but can be induced in every parenchymal cell type upon stimulation with Interferon (IFN) γ or tumor necrosis factor (TNF) α 30. Although, s20S and i20S do not largely differ in their overall structure 31, they diverge in their cleavage site preference thus generating distinct sets of peptides upon substrate degradation. Immunoproteasomes thereby enable the more efficient generation of MHC class I ligands and have been shown to improve MHC I mediated immune responses 32. In addition, accumulating evidence suggests a role for immunoproteasomes in the production of inflammatory cytokines, Th1 and Th17 differentiation, B cell maturation, autoimmune responses, and alveolar macrophage polarization 30,33. The mechanisms whereby the immunoproteasome is mechanistically involved in these diverse processes are, however, still enigmatic.
4. Targeting the UPS in scleroderma
Targeting of the UPS in fibrotic diseases by small molecules is feasible on several levels: i) specifically interfering with defined E3 ligases, ii) inhibition of deubiquitinating enzymes, iii) catalytic inhibition of the proteasome or specific immunoproteasome active sites. The targeting of E3 ligases offers conceptual advantages when compared to, for example, the extensive array of compounds targeting protein kinases, some of which are in clinical use. Unlike protein kinases where several competitive inhibitors target structurally similar active sites that bind ATP, E3 ligases share distinct binding pockets or pharmacophores that do not depend on metabolite binding but are involved in substrate-ligase complex interactions 34. As an example for an E3 ligase inhibitor, we would like to refer to the newly developed compound BC-1215, that exerts cytokine blocking activity by inhibiting the actions of the pro-inflammatory E3 ligase subunit, Fbxo3 35. Whether Fbxo3 inhibitors could reduce cytokine-driven inflammation in scleroderma will require further studies using relevant preclinical models 36. Another compound, pevonedistat, that targets neddylation, a process required for activity of some ubiquitin E3 ligases, was shown to reduce scleroderma graft-versus host disease in a murine model 37. Collectively, these observations provide a rationale for selectively targeting checkpoints in pro-fibrotic and pro-inflammatory pathways driven by E3 ligases in scleroderma.
While designing therapies to interfere with E3 ligase activity have distinct advantages, there are also known limitations. There are several hundred known E3 ligases that target thousands of cellular proteins. As each specific E3 ligase generally targets more than one substrate for degradation, there is potential for off target effects 38. For example, the E3 ligase SCFFBXL2 has been shown to target several different substrates 39, so interfering with SCFFBXL2 activity might affect protein levels of all of its substrates. Additionally, the entirety of the E3-substrate interactome is only partially characterized, and the majority of E3-substrate interactions have not been characterized 40,41. These factors present significant challenges in designing therapies aimed to affect a specific substrate protein by modulating activity of the E3 ligase that targets it.
In addition to E3 ligase inhibition, a variety of small molecule inhibitors have been developed serving as catalytic inhibitors of the proteasome. These compounds bind covalently or non-covalently to the catalytic sites of the 20S proteasome with different specificities thereby reversibly or irreversibly inhibiting its protease activities 42. One prominent example for a reversible proteasome inhibitor is the first FDA-approved inhibitor Bortezomib (Velcade©) which has been successfully applied for the treatment of multiple myeloma patients since 2003 43. However, reports on adverse systemic effects and reported nonspecific off-target effects limit the use of this compound. In addition, tumor cells develop resistance against catalytic proteasome inhibitors either by regulating proteasome levels in the cell or by acquired mutation of the active site 44–46. Irreversible inhibitor binding induces sustained proteasome inhibition, as recovery of proteasome activity requires de novo synthesis of 20S core particles. However, these molecules exhibit a negative pharmacodynamic profile since they also inhibit proteasomes of healthy and non-malignant cells when administered intravenously 47. All proteolytic subunits bind to the inhibitors via a common mechanism involving the nucleophilic addition of their Thr1 hydroxyl group to the inhibitor analogously to the nucleophilic attack of peptides for degradation 48. Of note, the composition of side chains of the peptide scaffold of the inhibitor - but not the reactive group or the peptide backbone - defines the substrate specificity of the inhibitor. Besides covalent inhibitors, different classes of molecules interacting with the proteasome catalytic subunits in a non-covalent fashion have been generated, such as cyclic or noncyclic peptides 48. Several inhibitors of specific catalytic subunits were developed, such as the β5-specific inhibitor oprozomib (ONX 0912). In addition, several immunoproteasome specific inhibitors have recently been developed which represents another milestone in proteasome inhibitor discovery (Figure 2): ONX 0914 or PR-924 which targets LMP7 49, LMP2-specific inhibitors such as UK-101, LU-001i and KZR-504 50–52, and the immunoproteasome-specific inhibitor LU-005i 53. These inhibitors specifically inhibit either defined active sites of the immunoproteasome or act as pan-immunoproteasome inhibitors. They thus confer cell-specific activity as the immunoproteasome is constitutively expressed only in immune cells. The immunoproteasome constitutes generally about 50% of the overall proteasome content even in immune cells. Therefore, immunoproteasome inhibitors are generally well tolerated and show a comparatively large therapeutic window for treatment of diseases that involve unwanted activation of immunoproteasomes such as in autoimmunity 33. A novel concept of proteasome inhibition involves the competitive or non-competitive inhibition of the binding of proteasome activators to the outer alpha rings of the 20S catalytic core as proposed by Gaczynska & Osmulski recently 54. However, there are no drugs available yet to test this concept in systemic sclerosis.
Figure 2:

Targeting of the immunoproteasome by site-specific immunoproteasome inhibitors reflecting the current state of drug development.
5. The UPS in scleroderma related innate immune receptor pathways
The innate immune system sits at the interface between the host and the environment, and it is the first line of defense against both invading pathogens or host-derived “danger” signals 55. Pattern recognition receptors (PRR) on innate immune cells sense invading pathogen associated molecular pattern (PAMP) or host-derived damage associated molecular pattern (DAMP) molecules, causing activation of inflammatory signaling pathways and secretion of pro-inflammatory cytokines. In scleroderma, a number of ligands of both endogenous and microbial origin have been shown to promote the release of inflammatory mediators through binding to and activation of PRRs. Consequently, activation of the innate immune system and release of pro-inflammatory cytokines serves as a major stimulus for subsequent wound healing responses that underlie the fibrosis seen in the disease 9,16,56. There is much interest in therapeutic approaches to disrupt these pathways, including targeting defined E3 ligases that regulate PRR’s. Below we review PRR’s implicated in scleroderma and the corresponding E3 ligases known to regulate them. Therapies designed to modulate these E3 ligases would aim to reduce aberrant innate immune activation and the subsequent inflammatory signaling that follows (Figure 3).
Figure 3:
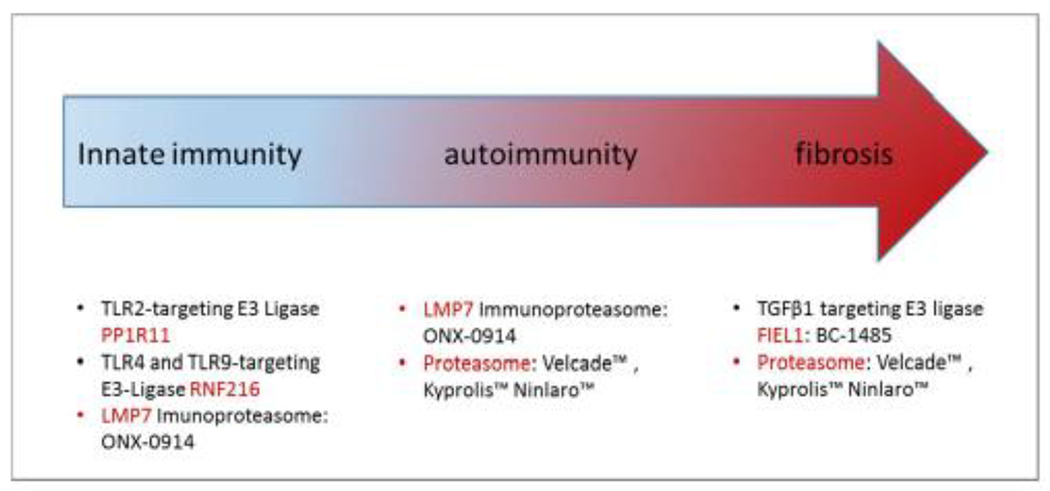
Targeting the UPS in systemic sclerosis. Diagram illustrating small molecule compounds against various targets that mediate innate immune function, autoimmunity, or the fibrotic response in scleroderma. These compounds could be potentially useful in this disorder but require further evaluation.
TLR2:
Toll-like receptor 2 (TLR2) is a cell-surface pattern recognition receptor that classically recognizes peptidoglycan of gram-positive bacteria 57. In addition, TLR2 is also activated by the endogenous acute phase reactant protein serum amyloid A (SAA), triggering TLR2-dependent inflammatory signaling and cytokine release 58,59. Importantly, SAA is elevated in the sera of scleroderma patients 60 and may serve as an endogenous stimulus driving TLR2 activation. SAA increased IL-6 secretion in a TLR2-dependent manner in dermal fibroblasts 61, suggesting that the SAA/TLR2 signaling axis may be a contributor to the inflammatory component characteristic of scleroderma. TLR2 was further implicated as a contributor to inflammation in scleroderma in a large population study, where a rare genetic polymorphism in TLR2 (Pro631His) was associated with high levels of anti-topoisomerase antibodies in serum and development of pulmonary arterial hypertension in a large cohort of scleroderma patients 62. In the same study, monocyte-derived dendritic cells from patients with the TLR2 Pro631His genotype secreted higher levels of proinflammatory cytokines TNFα and IL-6 62, further suggesting a role for TLR2 signaling as contributor to scleroderma-related chronic inflammation. Thus, TLR2-driven processes may be a novel approach to limit inflammation in scleroderma. Our laboratory has recently identified the novel E3 Ligase PP1R11 as a negative regulator of TLR2 signaling 63. PP1R11 is a member of the RING (Really Interesting New Gene) finger E3 ligases. PP1R11 ubiquitinates TLR2 and targets it for proteasomal degradation. PP1R11 over-expression reduces TLR2-dependent cytokine production, while PP1R11 inhibition augmented TLR2 signaling. Hence, PP1R11 has not been examined in the context of TLR2-related signaling in scleroderma but may represent a novel target for future studies (Figure 3).
TLR4:
Toll-Like Receptor 4 (TLR4) has also been implicated in the pathogenesis of scleroderma. TLR4 senses lipopolysaccharide (LPS) from gram-negative bacteria, but is also activated by several endogenous ligands, including extracellular matrix components up-regulated in scleroderma such as hyaluronic acid (HA), fibronectin extra Domain A, and Tenascin C 64,65. Several studies have examined the role of TLR4 signaling in scleroderma. Bhattacharyya et. al showed that chronic activation of TLR4 signaling drives a pro-fibrotic phenotype by promoting collagen synthesis and inhibiting profibrotic responses 66, while Takahashi et al discovered that TLR4−/− mice are protected using a bleomycin-induced scleroderma mouse model 67. Dendritic cells from scleroderma patients also secrete increased amounts of proinflammatory cytokines compared to healthy controls 68. Taken together, these studies suggest that chronic TLR4 over-activation in scleroderma directly contributes to a pro-fibrotic phenotype by up-regulating ECM components and also by augmenting pro-fibrotic signaling through TGFβ. Hence, attenuation of TLR4 signaling may be a novel anti-inflammatory strategy in scleroderma. TLR4 is regulated by the UPS, the E3 ligase(s) targeting TLR4 may be novel future drug targets for investigation. TLR4 is ubiquitinated and targeted for degraded by the E3 ligase RNF216 69. RNF216 over-expression reduces TLR4 levels and TLR4-dependent signaling, while RNF216 knockdown abrogates TLR4-dependent effects, including cytokine secretion. However, the role of RNF216 in scleroderma requires additional investigation (Figure 3).
TLR9:
Toll-like Receptor 9 (TLR9) is an intracellular TLR, located in endosomes. TLR9 is classically activated by bacterial CpG DNA, but also binds to and is activated by several endogenous ligands, including host mitochondrial DNA 70. TLR9 expression is up-regulated in myofibroblasts from scleroderma skin biopsies; additionally, scleroderma dermal fibroblasts have increased TLR9 levels and increase pro-fibrotic genes expression in response to CpG DNA 71. Additionally, Farina et. al also showed that TLR9 activation through Epstein-Barr Virus infection induces a pro-fibrotic phenotype in scleroderma fibroblasts 72. Thus, over-activation of the TLR9 signaling axis may augment pro-fibrotic signaling pathways, contributing to the pathogenesis of scleroderma. Similar to TLR2 and TLR4, attenuation of TLR9 signaling would also represent a reasonable strategy to reduce inflammation and fibrosis in scleroderma. Interestingly, TLR9 is also targeted by the E3 ligase RNF216 for ubiquitination and proteasomal degradation 69. Both TLR4 and TLR9 share a cytoplasmic TIR domain, and given that RNF216 ubiquitinates both proteins, TLR4 and TLR9 may share a common “degron” sequence, that could mediate recruitment of RNF216. Such molecular signatures within Toll receptors that mediate substrate-E3 ligase interaction could be a basis for design of small molecule activators that modulate the fibrotic process in scleroderma. Taken together, study of the E3 ligases regulating TLR signaling are active areas of investigation. Agents designed to modulate E3 ligase activity in TLR signaling thus might represent unique targets for therapeutic intervention.
In summary, TLR2, TLR4, and TLR9 are innate immune receptors implicated in scleroderma, and preclinical data indicate that increased expression and hyperactivation of these receptors contribute to both inflammation and fibrosis. The mechanisms responsible for TLR overexpression in scleroderma are unknown. One unexplored hypothesis is that TLR overexpression is the result of a reduction in their ubiquitination and degradation, causing increased protein levels and augmented signaling in response to ligands. Thus, examining the role of PP1R11 and RNF216, which target TLR2 and TLR4 and TLR9, respectively, are two new areas for future investigation in scleroderma.
6. The immunoproteasome in scleroderma
Beyond targeting single E3 ligases involved in PRR-mediated innate immune signaling, catalytic inhibition of the immunoproteasome might represent a unique therapeutic approach for systemic sclerosis. Emerging evidence suggests that immunoproteasomes are involved in shaping innate immune responses at different levels 30,73. Secretion of pentraxin-3, a specific pattern recognition protein that is secreted by neutrophils and macrophages to opsonize pathogens and dying cells 74, was found to be reduced in LMP7 deficient macrophages and upon inhibition of LMP7 75. Of note, serum levels of pentraxin-3 were recently show to be elevated systemic sclerosis (SSc) patients compared to healthy controls and correlated with disease severity and ulcer formation 76. Immunoproteasomes also directly affect macrophage function and may thereby modulate scleroderma pathogenesis: the absence of the immunoproteasome subunit LMP7 or specific inhibition of LMP7 in alveolar macrophages augmented IL-4 or IL-13-driven macrophage polarization towards an M2 phenotype 77. Polarization of bone-marrow derived macrophages, however, was not affected by LMP7 deficiency 78. The absence of immunoproteasome subunits LMP7 and MECL1 also had a pronounced impact on the transcriptome of dendritic cells and altered the maturation of DCs 79. Taken together, these data indicate that immunoproteasomes shape the innate immune response of various cell types such as macrophages and dendritic cells. In addition, immunoproteasomes are known to regulate the production of pro-inflammatory cytokines such as IL-6, IFN-γ, TNF-α, GM-CSF, and IL-23 thereby affecting innate immune signaling 33,80. Several of these cytokines are known to be involved in the pathogenesis of scleroderma and therapeutic strategies aiming at blocking of these pathways have been tested although with differing outcomes 36. Inhibition of immunoproteasomes might represent a novel approach to target inflammatory cytokine signaling in SSc (Figure 3).
7. UPS in autoimmunity
The contribution of the proteasome and namely the immunoproteasome to T and B cell mediated autoimmune responses in scleroderma has not been investigated. The immunoproteasome plays a prominent role in the generation and presentation of MHC class I epitopes 81. Mice lacking one, two or all immunoproteasome subunits show severe deficiency in MHC I surface expression and epitope generation 73,82. Accordingly, immunoproteasomes play an important role in CD8 T cell mediated adaptive immune responses against infected cells and in autoimmunity 83,84. Indeed, some evidence has suggested a role for the immunoproteasome in CD8 T cell mediated autoimmune responses such as in type 1 diabetes and multiple sclerosis 85,86. Immunoproteasome deficient mice develop early-stage multiorgan autoimmunity including symptoms of type I diabetes following irradiation and bone marrow transplantation which is mediated by autoreactive CD8 T cells 87. Accordingly, immunoproteasome subunits may protect the inflamed tissue against autoimmune CD8 T cell responses 84,88. Indeed, rare mutations in the immunoproteasome PSMB8 gene have been identified in severe autoinflammatory disorders 89–93. Moreover, recent studies have identified recessive mutations of immunoproteasome and proteasome genes resulting in altered proteolytic activity of the proteasome and sustained production of type 1 interferons in patients with the rare, genetic autoinflammatory CANDLE syndrome (Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature) 94. Taken together these data suggest that the absence or mutation of immunoproteasome subunits contributes to the onset of autoinflammatory and autoimmune diseases. Intact immunoproteasome function may thus be required to protect from the development of autoimmune activation that might contribute to the pathobiology of SSc. On the contrary, aberrant immunoproteasome expression has been observed in several other human autoimmune disorders 95–98 and experimental models of autoimmunity 99,100 and appears to drive autoimmune pathogenesis 33. It is assumed that uncontrolled immunoproteasome activation affects autoimmune reactions mainly at the level of Th1/Th2 and Treg/Th17 differentiation 33,84. Two hallmark studies showed that the inhibition of the immunosubunit LMP7 or its deficiency suppresses the differentiation of pro-inflammatory Th1 and Th17 subsets but increased generation of anti-inflammatory regulatory T Tregs in vitro 49,101. Moreover, several pro-inflammatory cytokines (IL-6, IFN-γ, TNF-α, GM-CSF, and IL-23) are suppressed by impaired immunoproteasome function. This makes the immunoproteasome a novel target for autoimmune disorders. Accordingly, specific immunoproteasome inhibitors have been successfully tested in various experimental models of autoimmunity as reviewed elsewhere 33,102. To date, several site-specific immunoproteasome inhibitors have been developed103, most notably the LMP7-specific inhibitor ONX-0914 (KZR-616) which will be tested in a Phase 1b/2, multi-center study in patients with Systemic Lupus Erythematosus or Lupus Nephritis for clinical safety and efficacy (https://clinicaltrials.gov).
Proteasome and immunoproteasome function has also been shown important for B-cell mediated humoral responses 104,105. Proteasome inhibition is particularly efficient in secretory cells such as plasma cells which has led to the approval of several proteasome inhibitors such as Velcade™, Kyprolis™, and Ninlaro™ for the treatment of multiple myeloma (see here for examples of clinical trials with proteasome inhibitors). This observation led to the application of proteasome and specific immunoproteasome inhibitors in allograft rejection in humans and several experimental models, however, with mixed results 106–110. It might be worth considering this therapeutic concept also for B-cell driven autoimmune responses including SSc as suggested previously for autoimmune diseases with renal manifestations 111.
8. UPS in fibrotic remodeling
The rationale for using small molecules and related chemical entities in fibrotic diseases is based on the inflammatory component and key signaling elements that promote the pro-fibrotic phenotype. A mechanistic centerpiece for the pathobiology of fibrosis as seen in scleroderma is activation of the TGFβ signaling network that is modulated by ubiquitin E3 ligases 8. TGFβ activates intracellular signaling by binding to cell surface receptors TGFβR1 and TGFβR2, causing recruitment and activation of downstream intermediaries including the SMAD family of proteins 8. Ultimately, the consequences of TGFβ signaling alter gene transcription to drive collagen synthesis, cross-linking, and the secretion of other extracellular matrix components 8. Several E3 ligases have been shown to be critical regulators of TGFβ signaling, and indeed, expression of several E3 ligases, including Smurf1, Arkadia, Synoviolin, NEDD4, and Pellino1 are upregulated in various models of fibrosis, in fibroblasts, or tissues 112. These E3 ligases may play a role by mediating disposal of key proteins that antagonize TGFβ signaling, that increase TGFβ production, or enhance matrix deposition. Specifically, there is evidence that the E3 ligase, Smurf2, is reduced in scleroderma fibroblasts, but increased after TGFβ stimulation, resulting in increased actions of Smad2/Smad3 that mediate TGFβ signaling leading to fibrosis 113. Inhibitors of Smurf2 have been generated and provide an opportunity for testing in preclinical fibrosis models 114. Another E3 ligase termed Fibrosing-inducing E3 Ligase 1 (FIEL1) has recently been shown to promote fibrosis downstream of TGFβ 115. FIEL1 targets a key negative regulator of TGFβ signaling - protein inhibitor of activated STAT 4 (PIAS4) - for ubiquitination and degradation. PIAS4 reduces activity of SMAD3 through several mechanisms, attenuating pro-fibrotic pathways downstream of TGFβ 116,117. FIEL1 ubiquitination and degradation of PIAS4 augmented TGFβ pro-fibrotic signaling in vitro and FIEL1 overexpression augmented bleomycin-induced lung fibrosis, while FIEL1 silencing in ameliorated fibrosis in the same model. Thus, inhibition of FIEL1 might serve as a novel strategy to reduce TGFβ-driven fibrosis, which is a central feature of scleroderma. In this study a first-in-class small molecule inhibitor of FIEL1 was generated, termed BC-1485. BC-1485 disrupted the FIEL1–PIAS4 interaction, reduced PIAS4 ubiquitination, and increased PIAS4 protein levels. BC-1485 reduced TGFβ-dependent gene transcription in vitro, and reduced lung fibrosis in a bleomycin mouse model of injury in vivo. Thus, inhibiting the ubiquitination and degradation of PIAS4 by BC-1485 may serve as a strategy to reduce TGFβ-mediated fibrosis and may be relevant in scleroderma 115. Additional proof-of-concept and preclinical studies, however, are needed to assess whether PIAS4 is a valid target in fibrotic disease and whether chemical inhibition of this target is effective in other complementary models of fibrosis (Figure 3).
Catalytic inhibition of the proteasome has also been shown to mediate anti-fibrotic effects in several experimental models of tissue fibrosis such as of the heart, liver, kidney, skin and lung 118–120. Antifibrotic effects involve attenuation of profibrotic TGFβ signaling 118,120 (Figure 3). However, results are controversial and hampered by the adverse side-effects of ubiquitous proteasome inhibition 121–123. We have recently shown that TGFβ induced myodifferentiation of lung fibroblasts depends on an increased assembly and activation of the 26S proteasome 124. Moreover, levels of the 19S regulatory subunit Rpn6 were elevated in an experimental model of lung fibrosis and in idiopathic pulmonary fibrosis lungs. Silencing of Rpn6 impaired assembly of 26S proteasome complexes and counteracted TGFβ-mediated myodifferentiation suggesting that specific targeting of 26S proteasome assembly may thus represent a unique therapeutic approach to counteract the profibrotic effects of TGFβ. Interfering with the interaction of proteasome activators and the 20S catalytic counterpart thus emerges as a promising therapeutic strategy that might also be applied in the setting of SSc 54. The involvement of the immunoproteasome in fibrotic tissue remodeling and in scleroderma-related lung fibrosis has not been investigated so far and remains to be unraveled.
9. Conclusion and outlook
Taken together, targeting the UPS in SSc represents a novel therapeutic approach which is, however, not well investigated. There are several lines of evidence that the use of specific inhibitors of E3 ligases may be useful to interfere with defined pathobiologic mechanisms in the course of SSc such as TLR signaling and profibrotic TGFβ signaling (Figure 3). In addition, the application of newly developed immunoproteasome inhibitors may be beneficial to counteract innate and autoimmune signaling thereby providing a more specific approach of targeting defined proteasome complexes in distinct cell types compared to the use of wide-spectrum proteasome inhibitors that are hampered by their adverse side effects (Figure 3).
Acknowledgments
All authors have read the journal’s authorship agreement. The manuscript has been reviewed by and approved by all named authors. All authors have read the journal’s policy on disclosure of potential conflicts of interest. They have the following conflicts of interests to declare: S.M.: no conflict; J.E.: no conflict; R.K.M. is a consultant for Koutif Therapeutics. There has been no particular editorial support for preparation of the manuscript.
This work was supported in part by NIH grants UH3HL123502, 2P50AR060780, and P01HL114453 to R.K.M. This work was also supported in part by the United States Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, a Merit Review Award from the United States Department of Veterans Affairs to R.K.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.van den Hoogen F et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 65, 2737–47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayes MD et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 48, 2246–2255 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Barnes J & Mayes MD Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 24, 165–170 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Chifflot H, Fautrel B, Sordet C, Chatelus E & Sibilia J Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 37, 223–235 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Al-Dhaher FF, Pope JE & Ouimet JM Determinants of morbidity and mortality of systemic sclerosis in Canada. Semin. Arthritis Rheum 39, 269–77 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Tyndall AJ et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis 69, 1809–15 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Steen VD & Medsger TA Changes in causes of death in systemic sclerosis, 1972-2002. Ann. Rheum. Dis 66, 940–4 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lafyatis R Transforming growth factor β--at the centre of systemic sclerosis. Nat. Rev. Rheumatol 10, 706–19 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Dowson C, Simpson N, Duffy L & O’Reilly S Innate Immunity in Systemic Sclerosis. Curr. Rheumatol. Rep 19, (2017). [DOI] [PubMed] [Google Scholar]
- 10.Fuschiotti P Current perspectives on the immunopathogenesis of systemic sclerosis. ImmunoTargets Ther. 21 (2016). doi: 10.2147/ITT.S82037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rockey DC, Bell PD & Hill JA Fibrosis--A Common Pathway to Organ Injury and Failure. N. Engl. J. Med 373, 96 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Asano Y & Sato S Vasculopathy in scleroderma. Semin. Immunopathol 37, 489–500 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Abraham DJ & Varga J Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol. 26, 587–95 (2005). [DOI] [PubMed] [Google Scholar]
- 14.York MR Novel insights on the role of the innate immune system in systemic sclerosis. Expert Rev. Clin. Immunol 7, 481–9 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Lafyatis R & York M Innate immunity and inflammation in systemic sclerosis. Curr Opin Rheumatol 21, 617–622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fullard N & O’Reilly S Role of innate immune system in systemic sclerosis. Semin Immunopathol 37, 511–517 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Hoyles RK et al. Fibroblast-specific perturbation of transforming growth factor beta signaling provides insight into potential pathogenic mechanisms of scleroderma-associated lung fibrosis: exaggerated response to alveolar epithelial injury in a novel mouse model. Arthritis Rheum. 58, 1175–88 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Wells AU & Denton CP Interstitial lung disease in connective tissue disease--mechanisms and management. Nat. Rev. Rheumatol 10, 728–39 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Weathington NM, Sznajder JI & Mallampalli RK The emerging role of the ubiquitin proteasome in pulmonary biology and disease. Am. J. Respir. Crit. Care Med 188, 530–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciechanover A Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ. 12, 1178–90 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Ciechanover A & Stanhill A The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim. Biophys. Acta (2013). doi: 10.1016/j.bbamcr.2013.07.007 [DOI] [PubMed] [Google Scholar]
- 22.Finley D Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem 78, 477–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komander D & Rape M The ubiquitin code. Annu Rev Biochem 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Ravid T & Hochstrasser M Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol 9, 679–90 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reinstein E & Ciechanover A Narrative review: protein degradation and human diseases: the ubiquitin connection. Ann. Intern. Med 145, 676–84 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Collins GA & Goldberg AL The Logic of the 26S Proteasome. Cell 169, 792–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lander GC, Martin A & Nogales E The proteasome under the microscope: the regulatory particle in focus. Curr. Opin. Struct. Biol 23, 243–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groll M et al. A gated channel into the proteasome core particle. Nat. Struct. Biol 7, 1062–7 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Basler M, Kirk CJ & Groettrup M The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol 25, 1–7 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Kammerl IEIE & Meiners S Proteasome function shapes innate and adaptive immune responses. Am. J. Physiol. Lung Cell. Mol. Physiol 311, L328–36 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Huber EM et al. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148, 727–38 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Kloetzel P-MM The proteasome and MHC class I antigen processing. Biochim. Biophys. Acta 1695, 225–33 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Basler M, Mundt S, Bitzer A, Schmidt C & Groettrup M The immunoproteasome: A novel drug target for autoimmune diseases. Clin. Exp. Rheumatol 33, 74–79 (2015). [PubMed] [Google Scholar]
- 34.Liu Y & Mallampalli RK Small molecule therapeutics targeting F-box proteins in cancer. Semin. Cancer Biol 36, 105–19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen BB et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat. Immunol 14, 470–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimitroulas T, Daoussis D, Garyfallos A, Sfikakis PP & Kitas GD Molecular and cellular pathways as treatment targets for biologic therapies in systemic sclerosis. Curr. Med. Chem 22, 1943–55 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Pai C-CS, Khuat LT, Chen M, Murphy WJ & Abedi M Therapeutic Effects of a NEDD8-Activating Enzyme Inhibitor, Pevonedistat, on Sclerodermatous Graft-versus-Host Disease in Mice. Biol. Blood Marrow Transplant 23, 30–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cromm PM & Crews CM Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem. Biol 24, 1181–1190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen BB & Mallampalli RK F-box protein substrate recognition: a new insight. Cell Cycle 12, 1009–10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iconomou M & Saunders DN Systematic approaches to identify E3 ligase substrates. Biochem. J 473, 4083–4101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nalepa G, Rolfe M & Harper JW Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov 5, 596–613 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Dick LR & Fleming PE Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 15, 243–9 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Herndon TM et al. U.s. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res 19, 4559–63 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Drews O & Taegtmeyer H Targeting the ubiquitin-proteasome system in heart disease: the basis for new therapeutic strategies. Antioxid. Redox Signal. 21, 2322–2343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsvetkov P et al. Suppression of 19S proteasome subunits marks emergence of an altered cell state in diverse cancers. Proc. Natl. Acad. Sci. U. S. A 201619067 (2016). doi: 10.1073/pnas.1619067114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsvetkov P et al. Compromising the 19S proteasome complex protects cells from reduced flux through the proteasome. Elife 4, 1–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beck P, Dubiella C & Groll M Covalent and non-covalent reversible proteasome inhibition. Biol. Chem 393, 1101–20 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Kisselev AF, van der Linden W. a & Overkleeft HS Proteasome inhibitors: an expanding army attacking a unique target. Chem. Biol 19, 99–115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muchamuel T et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med 15, 781–7 (2009). [DOI] [PubMed] [Google Scholar]
- 50.de Bruin G et al. Structure-based design of beta1i or beta5i specific inhibitors of human immunoproteasomes. J Med Chem 57, 6197–6209 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Wehenkel M et al. A selective inhibitor of the immunoproteasome subunit LMP2 induces apoptosis in PC-3 cells and suppresses tumour growth in nude mice. Br. J. Cancer 107, 53–62 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson HWB et al. Discovery of Highly Selective Inhibitors of the Immunoproteasome Low Molecular Mass Polypeptide 2 (LMP2) Subunit. ACS Med. Chem. Lett 2, acsmedchemlett.6b00496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Basler M et al. Amelioration of autoimmunity with an inhibitor selectively targeting all active centers of the immunoproteasome. Br. J. Pharmacol (2017). doi: 10.1111/bph.14069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaczynska M & Osmulski PA Harnessing proteasome dynamics and allostery in drug design. Antioxid. Redox Signal 21, 2286–301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brubaker SW, Bonham KS, Zanoni I & Kagan JC Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33, 257–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lafyatis R & York M Innate immunity and inflammation in systemic sclerosis. Curr. Opin. Rheumatol 21, 617–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawai T & Akira S Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Connolly M et al. Acute serum amyloid A is an endogenous TLR2 ligand that mediates inflammatory and angiogenic mechanisms. Ann Rheum Dis 75, 1392–1398 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Cheng N, He R, Tian J, Ye PP & Ye RD Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol 181, 22–26 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lakota K et al. Serum amyloid A is a marker for pulmonary involvement in systemic sclerosis. PLoS One 10, e0110820 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Reilly S et al. Serum amyloid A induces interleukin-6 in dermal fibroblasts via Toll-like receptor 2, interleukin-1 receptor-associated kinase 4 and nuclear factor-κB. Immunology 143, 331–40 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Broen JCA et al. A Rare Polymorphism in the Gene for Toll-like Receptor 2 Is Associated With Systemic Sclerosis Phenotype and Increases the Production of Inflammatory Mediators. 64, 264–271 (2012). [DOI] [PubMed] [Google Scholar]
- 63.McKelvey AC et al. RING finger E3 ligase PPP1R11 regulates TLR2 signaling and innate immunity. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhattacharyya S et al. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci. Transl. Med 6, 232ra50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ciechomska M, Cant R, Finnigan J, van Laar JM & O’Reilly S Role of toll-like receptors in systemic sclerosis. Expert Rev. Mol. Med 15, e9 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Bhattacharyya S et al. Toll-like receptor 4 signaling augments transforming growth factor-β responses: a novel mechanism for maintaining and amplifying fibrosis in scleroderma. Am. J. Pathol 182, 192–205 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takahashi T et al. Amelioration of tissue fibrosis by toll-like receptor 4 knockout in murine models of systemic sclerosis. Arthritis Rheumatol. (Hoboken, N.J.) 67, 254–65 (2015). [DOI] [PubMed] [Google Scholar]
- 68.van Bon L et al. Distinct evolution of TLR-mediated dendritic cell cytokine secretion in patients with limited and diffuse cutaneous systemic sclerosis. Ann. Rheum. Dis 69, 1539–1547 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Chuang T-H & Ulevitch RJ Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat. Immunol 5, 495–502 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Zhang Q et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fang F et al. Toll-like Receptor 9 Signaling Is Augmented in Systemic Sclerosis and Elicits Transforming Growth Factor β-Dependent Fibroblast Activation. Arthritis Rheumatol. (Hoboken, N.J.) 68, 1989–2002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farina A et al. Epstein-Barr virus infection induces aberrant TLR activation pathway and fibroblast-myofibroblast conversion in scleroderma. J Invest Dermatol 134, 954–964 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Groettrup M, Kirk CJ & Basler M Proteasomes in immune cells: more than peptide producers? Nat. Rev. Immunol 10, 73–8 (2010). [DOI] [PubMed] [Google Scholar]
- 74.Deban L, Jaillon S, Garlanda C, Bottazzi B & Mantovani A Pentraxins in innate immunity: lessons from PTX3. Cell Tissue Res. 343, 237–49 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Paeschke A et al. The immunoproteasome controls the availability of the cardioprotective pattern recognition molecule Pentraxin3. Eur. J. Immunol 46, 619–33 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Shirai Y et al. Elevated levels of pentraxin 3 in systemic sclerosis: associations with vascular manifestations and defective vasculogenesis. Arthritis Rheumatol. (Hoboken, N.J.) 67, 498–507 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Chen S et al. Immunoproteasome dysfunction augments alternative polarization of alveolar macrophages. Cell Death Differ. 23, 1026–1037 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hewing B et al. Immunoproteasome subunit ß5i/LMP7-deficiency in atherosclerosis. Sci. Rep 7, 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Verteuil D. a et al. Immunoproteasomes Shape the Transcriptome and Regulate the Function of Dendritic Cells. J. Immunol 193, 1121–32 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Koerner J, Brunner T & Groettrup M Inhibition and deficiency of the immunoproteasome subunit LMP7 suppress the development and progression of colorectal carcinoma in mice. Oncotarget 8, 50873–50888 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kloetzel PM Generation of major histocompatibility complex class I antigens: functional interplay between proteasomes and TPPII. Nat. Immunol 5, 661–9 (2004). [DOI] [PubMed] [Google Scholar]
- 82.Kincaid EZ et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol 13, 129–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mundt S, Basler M, Buerger S, Engler H & Groettrup M Inhibiting the immunoproteasome exacerbates the pathogenesis of systemic Candida albicans infection in mice. Sci. Rep 6, 19434 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feist E, Burmester GR & Krüger E The proteasome — victim or culprit in autoimmunity. Clin. Immunol 172, 83–89 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Friese MA et al. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat. Med 14, 1227–35 (2008). [DOI] [PubMed] [Google Scholar]
- 86.Pinkse GGM et al. Autoreactive CD8 T cells associated with beta cell destruction in type 1 diabetes. Proc. Natl. Acad. Sci. U. S. A 102, 18425–30 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zaiss DMW, Bekker CPJ, Gröne A, Lie BA & Sijts A. J. a M. Proteasome immunosubunits protect against the development of CD8 T cell-mediated autoimmune diseases. J. Immunol 187, 2302–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eleftheriadis T The existence of two types of proteasome, the constitutive proteasome and the immunoproteasome, may serve as another layer of protection against autoimmunity. Med. Hypotheses 78, 138–41 (2012). [DOI] [PubMed] [Google Scholar]
- 89.Agarwal AK et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am. J. Hum. Genet 87, 866–72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kitamura A et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Immunol 121, 4150–4160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Y et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 64, 895–907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arima K et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl. Acad. Sci 108, 14914–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McDermott A, Jacks J, Kessler M, Emanuel PD & Gao L Proteasome-associated autoinflammatory syndromes: advances in pathogeneses, clinical presentations, diagnosis, and management. Int. J. Dermatol 54, 121–129 (2015). [DOI] [PubMed] [Google Scholar]
- 94.Torrelo A CANDLe syndrome as a paradigm of proteasome-related autoinflammation. Front. Immunol 8, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Egerer T et al. Tissue-specific up-regulation of the proteasome subunit beta5i (LMP7) in Sjögren’s syndrome. Arthritis Rheum. 54, 1501–8 (2006). [DOI] [PubMed] [Google Scholar]
- 96.Krause S et al. Immunoproteasome subunit LMP2 expression is deregulated in Sjogren’s syndrome but not in other autoimmune disorders. Ann. Rheum. Dis 65, 1021–1027 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mishto M et al. Immunoproteasome LMP2 60HH variant alters MBP epitope generation and reduces the risk to develop multiple sclerosis in Italian female population. PLoS One 5, e9287 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ghannam K et al. Upregulation of Immunoproteasome Subunits in Myositis Indicates Active Inflammation with Involvement of Antigen Presenting Cells, CD8 T-Cells and IFNγ. PLoS One 9, e104048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Basler M, Dajee M, Moll C, Groettrup M & Kirk CJ Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J. Immunol. (Baltimore, Md. 1950) 185, 634–641 (2010). [DOI] [PubMed] [Google Scholar]
- 100.Belogurov A et al. Ubiquitin-independent proteosomal degradation of myelin basic protein contributes to development of neurodegenerative autoimmunity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 29, 1901–1913 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kalim KW, Basler M, Kirk CJ & Groettrup M Immunoproteasome Subunit LMP7 Deficiency and Inhibition Suppresses Th1 and Th17 but Enhances Regulatory T Cell Differentiation. J. Immunol 189, 4182–4193 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Kisselev AF & Groettrup M Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr. Opin. Chem. Biol 23, 16–22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Bruin G et al. A Set of Activity-Based Probes to Visualize Human (Immuno)proteasome Activities. Angew. Chem. Int. Ed. Engl 55, 4199–203 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Hensley SE et al. Unexpected role for the immunoproteasome subunit LMP2 in antiviral humoral and innate immune responses. J. Immunol 184, 4115–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Meister S et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 67, 1783–92 (2007). [DOI] [PubMed] [Google Scholar]
- 106.Li J et al. Immunoproteasome inhibition prevents chronic antibody-mediated allograft rejection in renal transplantation. Kidney Int. (2017). doi: 10.1016/j.kint.2017.09.023 [DOI] [PubMed] [Google Scholar]
- 107.Eskandari SK, Seelen MAJ, Lin G & Azzi JR The immunoproteasome: An old player with a novel and emerging role in alloimmunity. Am. J. Transplant 17, 3033–3039 (2017). [DOI] [PubMed] [Google Scholar]
- 108.Mundt S, Basler M, Sawitzki B & Groettrup M No prolongation of skin allograft survival by immunoproteasome inhibition in mice. Mol. Immunol 88, 32–37 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Ensor CR et al. Proteasome Inhibitor Carfilzomib-Based Therapy for Antibody-Mediated Rejection of the Pulmonary Allograft: Use and Short-Term Findings. Am. J. Transplant 17, 1380–1388 (2017). [DOI] [PubMed] [Google Scholar]
- 110.Eskandary F et al. A Randomized Trial of Bortezomib in Late Antibody-Mediated Kidney Transplant Rejection. J. Am. Soc. Nephrol (2017). doi: 10.1681/ASN.2017070818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hiepe F & Radbruch A Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat. Rev. Nephrol 12, 232–40 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Huang XL et al. E3 ubiquitin ligase: A potential regulator in fibrosis and systemic sclerosis. Cell. Immunol 306–307, 1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 113.Zuscik MJ, Rosier RN & Schwarz EM Altered negative regulation of transforming growth factor beta signaling in scleroderma: potential involvement of SMURF2 in disease. Arthritis Rheum. 48, 1779–80 (2003). [DOI] [PubMed] [Google Scholar]
- 114.Mund T, Lewis MJ, Maslen S & Pelham HR Peptide and small molecule inhibitors of HECT-type ubiquitin ligases. Proc. Natl. Acad. Sci. U. S. A 111, 16736–41 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lear T et al. Ubiquitin E3 ligase FIEL1 regulates fibrotic lung injury through SUMO-E3 ligase PIAS4. J. Exp. Med 213, 1029–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Imoto S, Sugiyama K, Yamamoto T & Matsuda T The RING domain of PIASy is involved in the suppression of bone morphogenetic protein-signaling pathway. Biochem Biophys Res Commun 319, 275–282 (2004). [DOI] [PubMed] [Google Scholar]
- 117.Imoto S et al. Regulation of transforming growth factor-beta signaling by protein inhibitor of activated STAT, PIASy through Smad3. J Biol Chem 278, 34253–34258 (2003). [DOI] [PubMed] [Google Scholar]
- 118.Fineschi S, Reith W, Guerne PA, Dayer J-M & Chizzolini C Proteasome blockade exerts an antifibrotic activity by coordinately down-regulating type I collagen and tissue inhibitor of metalloproteinase-1 and up-regulating metalloproteinase-1 production in human dermal fibroblasts. FASEB J. 20, 562–4 (2006). [DOI] [PubMed] [Google Scholar]
- 119.Meiners S et al. Downregulation of Matrix Metalloproteinases and Collagens and Suppression of Cardiac Fibrosis by Inhibition of the Proteasome. Hypertension 44, 471–477 (2004). [DOI] [PubMed] [Google Scholar]
- 120.Mutlu GM et al. Proteasomal inhibition after injury prevents fibrosis by modulating TGF-β(1) signalling. Thorax 67, 139–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meiners S, Ludwig A, Stangl V & Stangl K Proteasome inhibitors: poisons and remedies. Med. Res. Rev 28, 309–27 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Semren N et al. Validation of the 2nd generation proteasome inhibitor oprozomib for local therapy of pulmonary fibrosis. PLoS One 10, 1–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weiss CH, Budinger GRS, Mutlu GM & Jain M Proteasomal regulation of pulmonary fibrosis. Proc. Am. Thorac. Soc 7, 77–83 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Semren N et al. Regulation of 26S proteasome activity in pulmonary fibrosis. Am. J. Respir. Clin. Care Med 192, 1089–101 (2015). [DOI] [PubMed] [Google Scholar]