

Abstract
Biomaterial systems such as hydrogels enable localized delivery and post-injection modulation of cellular therapies in a wide array of contexts. Biomaterials as adjuvants has been an active area of investigation, but the study of functionalized biomaterials supporting immunosuppressive cell therapies for tolerogenic applications is still nascent. Here, we developed a 4-arm poly(ethylene-glycol)-maleimide (PEG-4MAL) hydrogel functionalized with interleukin-10 (IL-10) to improve the local delivery and efficacy of a cell therapy against autoimmune disease. The biophysical and biochemical properties of PEG-4MAL hydrogels were optimized to support dendritic cell (DC) viability and an immature phenotype. IL-10-functionalized PEG-4MAL (PEG-IL10) hydrogels exhibited controlled IL-10 release, extended the duration of DC support, and protected DCs from inflammatory assault. After incorporation in PEG-IL10 hydrogels, these DCs induced CD25+FoxP3+ regulatory T cells (Tregs) during in vitro co-culture. These studies serve as a proof-of-concept for improving the efficacy of immunosuppressive cell therapies through biomaterial delivery. The flexible nature of this system enables its widespread application across a breadth of other tolerogenic applications for future investigation.
Keywords: Dendritic cells, immunosuppression, multiple sclerosis, hydrogel, tolerance
Graphical Abstract
1. Introduction
Multiple Sclerosis (MS) is an autoimmune inflammatory disease that affects over 400,000 people in the United States and 1.2 million globally1, with annuals costs between $8,528 and $54,244 per patient2. MS is characterized by axonal demyelination in the central nervous system (CNS) and resultant neurological deficits3,4. Neurodegeneration due to MS can lead to loss of motor control, cognitive deficits, and eventual paralysis4. Currently, there is no cure for MS, and present treatment strategies aim only to slow the progression of neurodegeneration in patients through inhibition of neuroinflammation. The severity and progression of MS have been shown to be dependent on the balance between the number and functional capacity of inflammatory auto-reactive T cells (Teff) and regulatory T cells (Tregs)5.
Dendritic cells (DCs) are professional antigen-presenting cells that are potent mediators of immunity6. Dendritic cells bridge the innate and adaptive immune systems and direct antigen-specific immune responses by educating T cells, B cells, and other lymphocytes7. Many current pharmaceutical treatments target DCs due to their ability to control effector T cell responses8. Interleukin-10 (IL-10) treated DCs (DC10s) are regulatory IL-10-secreting DCs that have been shown to present self-antigens in a regulatory context and function to induce Tregs9,10. More recently, DCs have been targeted11 or employed as a cell therapy to re-educate the immune system in an antigen-specific manner12,13. However, a major challenge cited by these clinical trials and other academic studies is the delivery of cells to the target site14,15. Some groups have shown that using very large numbers or multiple doses of DCs can induce tolerance in mice with various autoimmune conditions12,16,17, but the high cost of processing an inflated cell number limits the clinical translation of DCs as a cell therapy.
Biomaterials have been utilized to improve the delivery of cells and biomolecules in diverse applications18–22. Delivery vehicles loaded with bioactive agents are also able to control cell fate by providing sustained treatments to incorporated cells or by modulating endogenous cells that may interact with the therapy23–25. Whereas much effort has been dedicated to the development of adjuvant materials for immune activation, there has been much less focus on the development of biomaterials supporting immunosuppressive or tolerogenic responses. The therapeutic capacity of DCs in tolerogenic applications is dependent on the ability of DCs to remain immunosuppressive26,27. It has been shown that DCs can be activated by mechanical stress, adjuvanting epitopes, and inflammation28. Thus, the selected biomaterial carrier must be mechanically tunable, non-adjuvanting, and biochemical-cue enabled to support tolerogenic DC function and protect DCs from the heightened inflammation characteristic to autoimmune patients.
Here, we developed an IL-10-functionalized PEG-4MAL hydrogel for the delivery of DCs in immunosuppressive or tolerogenic applications. We utilize PEG-4MAL hydrogels, which are non-immunostimulatory, modular, protease-degradable, and injectable via in-situ crosslinking. The maleimide end groups also allow for functionalization with bioactive agents (e.g., IL-10) to support tolerance or immunosuppression post-injection. The biophysical and biochemical properties of PEG-4MAL hydrogels were optimized to support DC viability and maintain an immature (and immunosuppressive) phenotype. Hydrogels were covalently functionalized with IL-10, and gels displayed controlled release of bioactive IL-10 over 4 days with near complete release expected ~8 days. Hydrogels consisting of IL-10-functionalized PEG-4MAL (PEG-IL10) extended the duration of DC viability and immaturity and protected DCs from inflammatory stimuli. Further analysis in vitro suggested extensive infiltration of endogenous lymphocytes into hydrogels and CD25+FoxP3+ Treg induction by PEG-IL10 treated DCs. This study serves as a proof-of-concept delivery strategy for DCs and the flexible design of these hydrogels enables adaptation of this platform for other tolerogenic applications.
2. Experimental / Materials and methods
2.1. Animals.
All animal procedures were conducted under the approval of the Institutional Animal Care and Use Committee (IACUC) of the Georgia Institute of Technology or Emory University. Male and female C57BL/6J mice (8–12-week old, Jackson Laboratories) were housed and maintained in the Physiological Research Laboratory (PRL) at the Georgia Institute of Technology.
2.2. Differentiation of Dendritic Cells.
Murine bone marrow derived DCs were processed using a method previously described with minor modifications29,30. Bone marrow (BM) was harvested from 8-week-old male C57BL/6J mice, red blood cells (RBCs) were lysed, and the remaining cells were washed twice in phosphate-buffered saline (PBS) with 10% fetal bovine serum (FBS). Cells were then cultured (1.5×106 cells/mL) in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS supplemented with 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4), refreshed every three days, for six days. On Day 6, non-adherent cells were removed, and differentiated adherent DCs were released from plates using CellStripper (Corning) for 15 minutes at 37°C/5% CO2. These DCs were then treated with 50 ng/mL IL-10 (DC10), 10 nM vitamin D3 (VD3-DCs), 50 ng/mL TNF-α and 10 ng/mL IFN-γ (stimulated DCs, STIM-DC), 1 μg/mL lipopolysaccharide, LPS (mature DCs, mDC) or no treatment (immature DC, iDC) and cultured (106 cells/mL) in 24-well plates for 24 hours at 37°C/5% CO2.
2.3. PEG-4MAL Hydrogel Synthesis.
To form hydrogels, PEG-4MAL macromer (20 kDa; Laysan Bio) was dissolved in PBS containing 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4)31,32. The cell adhesive peptide GRGDSPC (RGD; >95% purity; GenScript) was dissolved (1.0 mM) in PBS containing 10 mM HEPES and added to the PEG-4MAL to produce a solution of RGD-functionalized PEG-4MAL. After 10 minutes of incubation, the protease-degradable crosslinking peptide GCRDVPMSMRGGDRCG (VPM; GenScript) was added (4.0 mM) and hydrogels were polymerized at 37°C/5% CO2 for 5 min. For non-degradable hydrogels, a non-degradable reducing agent dithiothreitol (DTT; ThermoFisher) was used as a crosslinker in place of VPM. For hydrogels incorporating DCs, cells were added to the RGD-functionalized PEG-4MAL solution prior to the addition of crosslinker. DC-laden hydrogels were cultured in DMEM with 10% FBS supplemented with 20 ng/mL GM-CSF and IL-4. Samples were maintained in 24-well plates at 37°C/5% CO2, refreshing media every 2 days until needed for analysis.
2.4. Rheology.
Hydrogels (10 μL) were cast in cylinder-shaped, Sigmacote-treated (Sigma-Aldrich) silicone molds. Once fully crosslinked, hydrogels were removed and swelled overnight in PBS (pH 7.4) at 4°C. Hydrogels were then removed and examined using a rheometer (MCR-302, Anton Paar; CP10–2). Samples were maintained at 37°C while a frequency sweep (100 to 0.1 rad/s) was performed at a constant strain of 1%. Storage modulus (G’) and loss modulus (G”) were determined by averaging values acquired within the linear range. These PEG-4MAL hydrogels are viscoelastic materials that can be treated as linearly elastic solids based on constant G’ values and lower G” values. Relative mesh sizes of hydrogels were estimated using Rubber Elasticity Theory:
where G’ is the storage modulus in Pa,A is the Avogadro’s constant (mol−1) R is the gas constant (cm3 Pa K−1 mol−1) and T is the temperature (K)
Mesh sizes are reported in nanometers, which theoretically represents the distance between two junctions of polymers.
2.5. Hydrogel Digestion.
To recover cells from hydrogels, culture media was removed and 1 mL collagenase II (2 mg/mL; Gibco)31,33,34 was added to each well of PEG-4MAL hydrogels incorporating DCs. Hydrogels were incubated at 37°C/5% CO2 for 15 minutes. Contents of wells were then pipetted directly into fresh media with 25% FBS to quench collagenase II activity. Cells were collected by centrifugation at 300g and prepared for flow cytometry.
2.6. Flow Cytometry.
Dendritic cells were processed for flow cytometry using a method previously described with minor modifications29,30. Cells were washed with PBS and then resuspended in 100 μL of fluorescence-activated cell sorting (FACS) Buffer (Hank’s Buffered Salt Solution, HBSS; 1% bovine serum albumin, BSA; 1 mM ethylenediaminetetraacetic acid, EDTA). For DC samples, TruStain FcX (anti-mouse CD16/32; BioLegend) was added at the manufacturer’s recommended dose, and cells were incubated on ice for 10 minutes. Cells were then stained with fluorescently-stained monoclonal antibodies against FITC anti-IAb (AF6–120.1; BD), APC anti-CD86 (GL-1; BioLegend), BV785 anti-CD11c (N418; BioLegend), or PE anti-PD-L1 (MIH7; BioLegend) at the manufacturer’s recommended dose for 30 minutes on ice, protected from light. Cells were washed twice with FACS buffer and analyzed on a BD FACSAria III cytometer. Propidium iodide (PI) was added 5 minutes before analysis to assess viability. DC samples were analyzed based on the gating strategy in Figure S1. For co-culture samples of DCs with T cells, cells were stained with 1 μL/sample of Zombie Violet Fixable Viability Kit (BioLegend) for 20 minutes. Cells were then washed with FACS Buffer. TruStain FcX™ (anti-mouse CD16/32; BioLegend) was added, and cells were incubated on ice for 10 minutes. Cells were then stained with fluorescently-labeled monoclonal antibodies, APC/Fire750 anti-CD3 (17A2; BioLegend), APC anti-CD4 (GK1.5; BioLegend), PerCP-Cy5.5 anti-CD8a (53–6.7; BioLegend), PE-Cy7 anti-CD25 (PC61; BioLegend), or PE-CF594 anti-CD279 (J43; BD), at the manufacturer’s recommended dose for 30 minutes on ice, protected from light. Cells were then fixed and permeabilized using True-Nuclear Transcription Factor Buffer Set (BioLegend) according to the manufacturer’s protocol. Cells were stained with the fluorescent dye-labeled monoclonal antibody PE anti-FoxP3 (MF23; BD) for 30 minutes and were then washed twice with Perm Buffer (BioLegend) and once with FACS buffer before being analyzed on a BD LSRFortessa flow cytometer. Co-culture samples were analyzed based on the gating strategy in Figure S2.
2.7. Confocal Microscopy.
Cell viability was visualized by staining with Calcein-AM and ethidium homodimer-1 (EthD-1) and imaging on a confocal microscope (Nikon Eclipse Ti-E C2+). Media was removed and PEG-4MAL hydrogels incorporating DCs were washed twice with 1X PBS by gentle pipetting. Samples were then stained with 1 μM Calcein-AM and 1 μM EthD-1 in media without FBS for 20 minutes, protected from light. Hydrogels were then washed two more times with 1X PBS and media without FBS was added for imaging.
2.8. Cytokine Multiplexing.
Supernatants were collected from samples at the end of culture, cleared of cells by centrifugation and stored at −80oC until use. Samples were thawed to room temperature and treated according to the manufacturer’s protocol for Bio-Plex Pro Mouse Cytokine 23-plex Assay (Bio-Rad). Samples were then analysed on a Luminex MAGPIX. A linear range of cytokine concentrations was determined to optimal sample dilution according to cytokines of interest. Cytokines tested are categorized in Table S1 based on their effects being inflammatory, pleiotropic, tolerogenic, or unrelated to DCs.
2.9. Thiolation of IL-10.
Lysine residues, the expected sites for thiolation on IL-10, were targeted using Traut’s reagent (Sigma Aldrich). Recombinant murine IL-10 (Peprotech) was thiolated with 30 molar excess of Traut’s reagent in PBS with 5 mM EDTA (Gibco) at pH 8.0 for 1 hour, according to manufacturer’s protocol. Protein samples were then washed twice by diluting with PBS (pH 8.0) and passing through a 10 kDa cellulose filter (Amicon Ultra; Millipore) at 14,000g. To quantify the extent of thiolation, samples were stained for free thiols using the Measure-iT Thiol Quantification Kit (ThermoFisher). The fluorescence intensity (Ex/Em: 494/517nm) of thiolated IL-10 (HS-IL-10), native IL-10, blank wells, and reduced glutathione standards were quantified on a spectrophotometer, according to manufacturer’s protocol (Synergy H4; BioTek).
2.10. Conjugation to PEG-4MAL.
Conjugation of HS-IL-10 to PEG-4MAL was achieved by reacting both components at a molar ratio of 1000:1 (PEG-4MAL:HS-IL-10) in PBS (pH 7.0) for 30 minutes35. To visualize the PEGylated IL-10, reacted protein was immediately loaded onto polyacrylamide gels (4–12% Bis-Tris; Invitrogen) and placed into a Novex Mini-Cell (Invitrogen) which was run under non-denaturing conditions35. Separated proteins were stained with SyproRuby (Invitrogen) and imaged on a Molecular Imager Gel Doc XR System (BioRad).
2.11. MC/9 Cell Bioactivity Assay.
A murine liver mast cell line responsive to soluble IL-1036,37, MC/9 cells (ATCC), were expanded in DMEM with 10% FBS, refreshing media every 2–3 days. To assess the bioactivity of PEG-IL10 compared to native IL-10, MC/9 cells were collected and plated in DMEM with 3% FBS at 25,000 cells/well in a 96-well plate. Cells were treated with 5 ng/well co-stimulatory IL-4 (Peprotech) and either 2.5 ng/well IL-10 (Peprotech) or PEG-IL10. After 48 hours of incubation, either cell counting kit-8 (CCK-8) (absorbance 240 nm; Sigma Aldrich) or AlamarBlue (Ex/Em: 530/590 nm; ThermoFisher) was added to each well. The proliferation of MC/9 cells in response to treatment was measured on a spectrophotometer (Synergy H4; BioTek) based on cellular metabolic byproducts. The experiment was repeated with titrated treatments to construct half-maximal effective concentration (EC50) curves for native and PEGylated IL-10.
2.12. IL-10 Release Study.
Recombinant murine IL-10 (Peprotech) was first tagged with amine-reactive AlexaFluor-488 (AF488) according to the manufacturer’s protocol. Briefly, AF488 was dissolved in 10 mg/mL dimethyl sulfoxide and slowly added to IL-10 in 0.1 M sodium bicarbonate buffer. After incubation at room temperature for 1 hour, 1.5 M hydroxylamine was added to stop the reaction and tagged IL-10 was separated using a 10 kDa filter (Amicon Ultra; Millipore). Fluorophore-tagged IL-10 was then thiolated and PEGylated, as described above. Hydrogels consisting of PEG-IL10 were synthesized and swelled in PBS (pH 7.4). Hydrogels with IL-10 tethered or IL-10 entrapped were plated in PBS or PBS with collagenase II (2 mg/mL). Supernatants were collected intermittently and stored at −80oC until use. Once all hydrogels had fully degraded, supernatants were collected and AF488 fluorescence (Ex495/Em519) was measured on a spectrophotometer (Synergy H4; BioTek).
2.13. DC Resistance to Re-Stimulation.
Immature DCs were incorporated in control PEG or PEG-IL10 hydrogels as described above. After incubation for 24 hours, groups were treated with a moderate stimulus (150 ng/mL TNF-α and 30 ng/mL IFN-γ, “3X STIM”) or a severe stimulus (2 μg/mL LPS, “2x LPS”). Both iDCs and DC10s with and without stimulus treatment served as controls. After an additional 48 hours, hydrogels were digested and analysed via flow cytometry, as described above.
2.14. Cell Infiltration into PEG-4MAL Hydrogels in vitro.
Cells were harvested from BM and differentiated into DCs, as described previously. On Day 6, iDCs were stained with CellTracker Red CMTPX (Life Technologies) according to the manufacturer’s protocol. Hydrogels with or without iDCs were cast into an 8-chamber imaging slide (Ibidi). Silicone moulds were used to maintain a standard shape and height for each hydrogel. Following lysis of red blood cells (RBC), splenocytes from the same mouse were incubated for 6 days in T25 flasks with IL-2 (30 U/mL). On day 6, splenocytes were stained with CellTracker Green CMFDA (Life Technologies) according to the manufacturer’s protocol. Stained splenocytes were then seeded on top of hydrogels for 6 hours. The infiltration of splenocytes into hydrogels was evaluated by confocal microscopy.
2.15. Co-Incubation of Dendritic Cells with Autologous Splenocytes in vitro.
Cells were harvested from BM and differentiated into DCs, as described previously. After lysing RBCs, splenocytes from the same mouse were plated at 5×106 cells/mL in 24-well plates (2 mL/well) with IL-2 (30 U/mL), ovalbumin (OVA)257–264 (5 μg/mL), and OVA323–339 (5 μg/mL) for 7 days. On Day 7, treated DCs were pulsed for 2–4 hours with OVA257–264 (10 μg/mL), and OVA323–339 (10 μg/mL). Treated DCs were then collected and plated in U-bottom 96-well plates with 100 μL of OVA-primed autologous splenocytes (1×106 cells/mL) for 3 days. Splenocytes were stained with carboxyfluorescein succinimidyl ester (CFSE) prior to being incubated with DCs. On Day 10, DCs and splenocytes were collected and processed for flow cytometry for detection of CFSE fluorescence.
2.16. Statistical Analysis.
Experimental values are reported as mean and standard deviation for all samples. Statistical analyses for normally distributed data sets were done using one or two-way analysis of variance (ANOVA) coupled with Sidak’s post-hoc pairwise tests using GraphPad (Prism). If datasets failed the Brown-Forsythe test (due to significant differences in variances among treatment groups), Welch’s correction and Dunnett’s post-hoc pairwise test were used. Unless otherwise specified, DC10 groups served as the control for comparison tests. p-values <0.05 were considered statistically significant. Error bars represent the standard deviation.
3. Results
3.1. PEG-4MAL Hydrogels Support DC Viability and an Immature Phenotype.
Towards designing a biomaterial delivery system tuned specifically for DC delivery, it was first necessary to identify the relationships between PEG-4MAL (Fig. 1A) polymer characteristics and the biophysical properties of the hydrogels. Cell-laden hydrogels were synthesized by functionalizing PEG-4MAL with RGD, mixing with cells, and then adding VPM crosslinker (Fig. 1B). An expected increase in hydrogel stiffness is represented by significantly higher values of G’ and G” with increasing polymer weight percentage (Fig. 1C and D). The mesh size for different hydrogel formulations was calculated using storage modulus based on Rubber Elasticity Theory38,39. Hydrogel mesh size decreases with increasing polymer weight percentage, as the hydrogel polymer network becomes denser (Fig. 1E).
Figure 1.

Schematic of A) PEG-4MAL chemical structure and B) process of hydrogel synthesis. Hydrogel characterization of PEG-4MAL hydrogels using a cone-and-plate rheometer. C) Storage modulus and D) loss modulus of PEG-4MAL hydrogels with varying polymer weight percentages. E) Mesh sizes of hydrogels with varying polymer weight percentages, calculated by Rubber Elastic Theory.
To develop a biomaterial delivery platform for DCs against autoimmune disease, it is vital that incorporated DCs remain viable and immature so that they can be tailored in their phenotype. Prior studies have shown that the stiffness of biological and synthetic matrices can influence the viability, differentiation, phenotype, and function of incorporated cells40–42. After 6 days of differentiation on tissue culture supports, immature DCs were incorporated in PEG-4MAL hydrogels with varying stiffnesses. Hydrogels with low polymer densities (3.5–4.5%) supported high DC viability, while higher polymer density hydrogels (6.0–10.0%) resulted in widespread cell death of incorporated cells (Fig. 2A and B). Cells that survived incorporation in higher polymer density hydrogels had elongated morphology, whereas DCs in lower polymer density gels exhibited typical round DC morphology of immature cells (Fig. 2A). The number of live cells recovered after gel digestion was higher in lower polymer density hydrogels (Fig. 2B). The cell death observed in higher polymer density, stiff hydrogels (6.0–10.0%) is likely due to the smaller mesh size of these dense hydrogels, resulting in inhibited diffusion of survival compounds or mechanical constriction of DCs. Expression of a common co-stimulatory molecule, CD86, was similar to immature DC controls after 24 hours of cell incorporation in lower polymer density hydrogels (Fig. 2C), indicating no maturation effect of incorporation. Interestingly, the maturity of recovered DCs, based on CD86 expression, was not varied among different PEG-4MAL hydrogel formulations. Thus, we selected hydrogels with 4.5% polymer density for subsequent experiments because of higher cell recovery and viability.
Figure 2.

Viability and phenotype of DCs after 24 hours of incorporation in PEG-4MAL hydrogels with various polymer weight percentages. Control groups either received no treatment (iDCs), 50 ng/mL IL-10 (DC10s), 50 ng/mL TNF-α and 10 ng/mL IFN-γ (TNF-α/IFN-γ), or 1 μg/mL LPS (mDCs). A) Calcein AM (green) and Ethidium homodimer-1 (red) assessed by confocal microscopy (scale bar = 100 μm). B) Propidium iodide (PI) negative cells recovered after collagenase II digestion and C) CD86 expression in DCs assessed by flow cytometry. White bars denote unincorporated DCs in 24-well plates and gray bars denote hydrogel-incorporated DCs. *p<0.05 via one-way ANOVA and Tukey’s comparison test for B). ****p<0.0001 and *p<0.05 via one-way ANOVA with Welch’s correction and Dunnett’s comparison test for C).
Integrin-mediated signaling resulting from cell-ECM interactions has been implicated in cell morphology, differentiation, survival, and other cell processes40,41,43–45. The effect of RGD, a ubiquitous cell adhesive peptide sequence, on DC viability and phenotype was explored. Hydrogel functionalization with the adhesive ligand RGD or the scrambled peptide RDG at concentrations 0.08 – 1.0 mM did not significantly influence the morphology, viability, nor phenotype of DCs after 24 hours in vitro (Fig. S3). However, the presence of RGD in synthetic matrices has been shown to promote ECM remodelling and vascularization through interactions with endogenous immune cells and vasculogenic cells in vivo19,46. Remodelling the ECM would favour our proposed design, such that endogenous cells could migrate into the hydrogel to be conditioned by DCs and DCs could effectively migrate to relevant tissues. Thus, RGD was incorporated at 1.0 mM, a concentration that has been shown to support these activities in vivo without significantly impacting DC viability or phenotype (as shown here in vitro).
Previous studies have demonstrated the benefit of degradable matrices on the cellular processes of incorporated and infiltrating cells41,47,48, and DCs have been shown to secrete matrix metalloproteinase (MMP)-1, MMP-2, MMP-3, and MMP-949. Hydrogels synthesized with various ratios of degradable (VPM) and non-degradable (DTT) crosslinkers had no effect on DCs viability or phenotype after 24 hours incorporation (Fig. S4). The VPM crosslinking peptide is effectively cleaved by MMP-2 and MMP-933 at physiological pH (7.4) at a relatively fast rate of 24,000 ± 1000 and 51,000 ± 3000 kcat/KM (M−1s−1), respectively34. Since DCs can secrete both MMP-2 and MMP-9, VPM-crosslinked hydrogels should be easily degradable by incorporated DCs. Fast-degrading hydrogels will enable the migration of cells necessary for DCs to modulate endogenous immune cells. Thus, VPM was selected as the crosslinker moving forward.
3.2. Thiolated IL-10 is Covalently Tethered to PEG-4MAL.
Maleimide functional groups on PEG-4MAL polymers allow a wide array of proteins and peptides to be covalently linked to the polymer backbone through free thiols found in free cysteine residues40,43,46. However, native IL-10 does not have any free cysteines and must be thiolated before tethering to PEG-4MAL35,50. Investigation of the crystal structures for IL-10 and its receptor revealed minimal overlap between IL-10/IL-10R binding sites and lysine residues on IL-1051,52. Thus, Traut’s reagent (2-iminothiolane) was used to thiolate lysine residues on IL-10. Thiolation of IL-10 with Traut’s reagent resulted in 3.68 μM of free thiols on HS-IL-10 samples whereas negligible values were detected for untreated IL-10 (Fig. 3A-C). This level of thiolation suggests that, on average, ~13 lysine residues were thiolated on each IL-10 molecule, representing numerous free thiols for reaction with the maleimide group on PEG-4MAL.
Figure 3.

A) Schematic of functionalizing IL-10 onto the backbone of PEG-4MAL macromers. Quantification of thiolation of IL-10 using Traut’s reagent and SDS-PAGE results of IL-10 PEGylation. B) Standard curve produced using glutathione. C) Concentration of free thiols on modified IL-10 proteins. D) Validation of tethering thiolated IL-10 to PEG-4MAL as shown by unmodified IL-10 at 20 kDa (black arrow) and PEGylated IL-10 at 40 kDa (orange arrow). E) Bioactivity of PEG-4MAL-IL-10 based on MC/9 cell proliferation measured by CCK-8 metabolic assay. F) Half-maximal effective concentration (EC50) curves produced by titration of treatments. G) Controlled release curve of IL-10 from PEG-IL10 hydrogels. ****p<0.0001 versus IL-10 controls via one-way ANOVA and Dunnett’s comparison test.
A series of experiments were conducted to determine the best conditions for covalently tethering IL-10 to PEG-4MAL, including the ratio of PEG-4MAL to IL-10, incubation time, thiolation chemistry, and IL-10 species (Fig. S5). Successful conjugation was achieved by reacting both components at a molar ratio of 1000:1 PEG-4MAL:HS-IL-10. An increase in molecular weight from ~19 kDa (IL-10; Lane 2; black arrow) to ~39 kDa (PEG-IL10; Lane 3; orange arrow) demonstrates successful PEGylation of some portion of the HS-IL-10 (Fig. 3D). The ~20 kDa shift in molecular weight for PEGylated IL-10 suggests one PEG-4MAL macromer was tethered to IL-10. For the reaction conditions tested, a faint band can be observed at ~20 kDa for PEG-IL10 samples, suggesting that not all IL-10 was PEGylated. Owing to the quantification difficulty of SDS-PAGE images, a release study was conducted to evaluate the release kinetics and PEGylation efficiency, or the fraction of IL-10 that was successfully PEGylated. Based on release studies discussed later, the PEGylation efficiency was estimated to be 40–50%.
Using PEG-IL10, hydrogels were successfully synthesized and appeared macroscopically identical to PEG-4MAL hydrogels. Incorporation of IL-10 into PEG-4MAL hydrogels did not alter the storage or loss moduli as shown by rheological studies (Fig. S6). It should be noted that 50 ng of IL-10 was incorporated for direct comparison to controls of soluble delivery, but up to ~2 mg (40,000X) of IL-10 can be incorporated with this system.
3.3. PEG-IL10 Retains its Bioactivity and can be Released from PEG-IL10 Hydrogels.
The immunosuppressive cytokine IL-10 does not need to be internalized to promote tolerogenic signalling35,50–52, but a potential concern is that the bioactivity of IL-10 is diminished due to thiolation and PEGylation modifications53. The bioactivity of PEG-IL10 was measured by the proliferation of MC/9 cells, a murine liver mast cell line commonly used in IL-10 bioactivity assays36,37. Read-outs for proliferation were comparable for native and PEGylated IL-10 at both 4 and 24 hours after stimulation (Fig. 3E), indicating that PEGylation does not affect the bioactivity of IL-10. To further validate the bioactivity of PEGylated IL10, PEG-IL10 or IL-10 was added in titrated amounts to construct EC50 curves for native and PEGylated IL-10. The observed EC50 values for PEG-IL10 and native IL-10 were nearly identical at ~0.003 ng/well (Fig. 3F). These results indicate that PEG-IL10 bioactivity is equivalent to IL-10 in its native state. Furthermore, our results suggest that thiolation is a superior method of PEGylation than acylation or reductive amination, both of which dramatically reduce its bioactivity53.
A key design parameter of these hydrogels is for IL-10 presentation to be sustained for a significant duration to offset injected-related inflammation and the elevated inflammation characteristic to subjects with autoimmune disease54. To demonstrate the controlled release of incorporated IL-10, PEG-4MAL hydrogels were synthesized with 50 ng of conjugated or unbound but physically entrapped IL-10 tagged with a fluorophore and the release of IL-10 was characterized over 5 days. Hydrogels synthesized with IL-10 (unbound) or HS-IL-10 (tethered) were incubated in collagenase II to cleave the VPM crosslinker and IL-10 release was examined. These gel formulations exhibited equivalent release profiles over 96 hours, with nearly 45 ng of protein released after just 5 hours (Fig. 3G, dashed lines). Hydrogels containing IL-10 incubated in PBS also exhibited rapid release of protein within the first 5 hours (~40 ng). In contrast, hydrogels incorporating HS-IL-10 released only ~30 ng after 5 hours (Fig. 3G, violet solid lines). For hydrogels in PBS with HS-IL-10, another ~10 ng of IL-10 was gradually released between 5–96 hours. Collagenase II was added to PBS hydrogels at 96 hours to release IL-10 still bound to hydrogels, after which another 6–8 ng of IL-10 was released from groups with bound IL-10 (Fig. 3G). At the rate of release for tethered IL-10 in PBS, extrapolation of the release curve predicts that near complete release will occur at ~8 days. Based on the mass of IL-10 released immediately, it is estimated that the extent of incorporation into PEG-IL10 hydrogels is between 40–50%. The remaining 50–60% of IL-10 is likely untethered and did not react to the maleimides within the hydrogel. These hydrogels gel in under a minute at the pH utilized herein, and unused maleimide groups are quickly quenched, likely limiting incorporation of the full dose of IL-10. Competition from fluorescent tagging of IL-10 may also hinder IL-10 incorporation, suggesting that untagged IL-10 may have a higher extent of incorporation.
3.4. PEG-IL10 Hydrogels Prolong DC Support and Protect DCs from Inflammatory Stimuli.
Because activation of tolerogenic DCs could render them therapeutically ineffective and potentially exacerbate autoimmune disease, maintaining the capacity for immunosuppression in DCs is crucial to their efficacy26,27. In multiple sclerosis, promoting viability and phenotypic stability of tolerogenic DC therapies are significant challenges in retaining their effectiveness14,15. A major advantage of biomaterial delivery is the ability to modulate cell responses post-injection, which is employed here through sustained treatment with IL-10.
Whereas the optimized PEG-4MAL hydrogel without IL-10 was capable of maintaining DC viability and an immature phenotype after 24 hours of incorporation (Fig. 2A-C), DCs will likely persist in the biomaterial longer than 24 hours in vivo. Thus, we sought to assess the potential benefit of functionalization with IL-10 at 48 hours post-incorporation. Cells recovered from PEG-IL10 hydrogels exhibited significantly higher levels of viability compared to DCs from control PEG-4MAL hydrogels after 48 hours (Fig. 4A). Dendritic cells in PEG-4MAL hydrogels were also activated to similar levels as mDCs, whereas DCs from PEG-IL10 hydrogels remained immature based on the expression of CD86 (Fig. 4B). To determine if IL-10 conjugation impacts its ability to promote DC viability, iDCs were incorporated in hydrogels with 50 ng of bound or unbound IL-10. To determine if the effect on DC viability was dependent on IL-10 presentation, PEG-4MAL hydrogels were synthesized with either unbound or tethered IL-10. The increased viability of DCs was independent of whether IL-10 was entrapped in hydrogels or covalently tethered to the polymer backbone, as shown by the similar viability observed through confocal imaging (Fig. 4C). Thus, the benefit of IL-10 on DC viability is not affected by tethering IL-10 to the polymer network, suggesting that IL-10 is bioactive when presented to DCs as PEG-IL10. These results show that PEG-IL10 hydrogels support DC viability and an immature phenotype longer than PEG-4MAL hydrogels without IL-10.
Figure 4.

A) Propidium iodide (PI) negative cells and B) CD86 expression assessed by flow cytometry after 48 hours of hydrogel incorporation. C) Calcein AM (green) and Ethidium homodimer-1 (red) assessed by confocal microscopy (scale bar = 100 μm). DCs were stimulated 24 hours after receiving various treatments or after incorporation in PEG-4MAL or PEG-4MAL-IL10 hydrogels. D) CD86 expression of DCs 48 hours after stimulation. Control groups either received no treatment (iDCs), 50 ng/mL IL-10 (DC10s), 10 nM VD3 (VD3), 50 ng/mL TNF-α and 10 ng/mL IFN-γ (TNF-α/IFN-γ), or 1 μg/mL LPS (mDCs). White bars denote unincorporated DCs in 24-well plates and gray bars denote hydrogel-incorporated DCs. ****p<0.0001 and **p<0.01 via one-way ANOVA and Dunnett’s comparison test in comparison to DC10s for B) and PEG-4MAL(−/−) for D).
Multiple sclerosis patients have increased levels of inflammatory cytokines and hyper-activated immune systems that can lead to allergic inflammation and the development of unrelated autoimmune disease55. We next sought to assess the extent to which these IL-10-presenting hydrogels could protect incorporated DCs from inflammation. Dendritic cells either remained unincorporated and received control treatments or were incorporated in hydrogels for 24 hours before stimulation with either a moderate stimulus (150 ng/mL TNF-α and 30 ng/mL IFN-γ “STIM 3x”) or a severe stimulus (2 μg/mL LPS “LPS 2x”) to assess their resistance to maturation, where “2x” and “3x” refer to the dose compared to standard treatments. Remarkably, LPS-stimulated DCs incorporated in PEG-IL10 hydrogels had CD86 expression levels that were not different from un-stimulated controls at 48 hours post-stimulation (Fig. 4D). However, LPS-stimulated DCs incorporated in PEG-4MAL hydrogels matured to levels of CD86 similar to TNF-α/IFN-γ-treated DCs and unincorporated controls (iDCs and DC10s) exhibited CD86 expression similar to mDCs (Fig. 4D) upon treatment with LPS. This data indicates that both IL-10 treatment and PEG-4MAL incorporation contribute to the ability of DCs to resist maturation in response to inflammatory stimuli. It is likely that the protection provided by biomaterial incorporation is due to the inhibited diffusion of inflammatory cytokines or survival signals provided by the synthetic ECM, which is likely mediated by the presence of RGD. The contributions to maturation resistance from IL-10 and biomaterial incorporation combined are sufficient to protect incorporated DCs from the moderate stimulus, whereas either treatment alone (DC10 and PEG-4MAL groups) does not confer the same level of resistance as the combined approach. Taken together, these studies illustrate that PEG-IL10 delivery systems improve the duration of DC viability and immaturity in vitro, suggesting that this system can improve the regulatory capacity of these cells after delivery. While these modest effects are statistically significant, the amplification of these impacts when escalating the dose of IL-10 will be investigated in future experiments.
3.5. DCs Increase FoxP3+ Tregs and Reduce CD8+ T cells in vitro.
To gain insights into the immunomodulatory process by which DC10s ameliorate symptoms of EAE in vivo, we investigated the effects of these DCs on splenocytes in a co-culture study using the model antigen ovalbumin (OVA). Dendritic cells were either treated with soluble factors or incorporated in biomaterials for 24 hours before hydrogels were digested and DCs were collected. Dendritic cells receiving were then co-incubated with autologous, OVA-primed (OVA257–264 and OVA323–339) splenocytes for 72 hours to evaluate their effect on T cells in vitro. Co-cultures with iDCs, DC10s, and vitamin-D3 (VD3) DCs at a ratio of 1:1 (DCs:T cells) had significantly higher viability of CD3+ T cells compared to splenocytes alone (Fig. 5A). For CD4+ T cells, we observed a significant increase in CD25+FoxP3+ T regulatory cells for iDCs (1:1), DC10s (1:1, 1:10), VD3 (1:1), PEG-4MAL (1:1, 1:10), and PEG-IL10 (1:1) compared to splenocytes alone, which demonstrates the ability of these DCs to induce Treg cells (Fig. 5B). Groups of CD4+ T cells with higher frequencies of CD25+FoxP3+ T regulatory cells also exhibited lower frequencies of CD4+FoxP3- effectors cells (data not shown). Given PD-1 expression on T cells inhibits T cell inflammatory activity, cells were also analyzed for PD-1 as an indicator of immunosuppression. CD4+ T cells from co-cultures with iDCs or DC10s at higher DC concentrations (1:1 or 1:10) also had a significant increase in the percentage of PD-1+ CD4+ T cells (Fig. 5C). As a measure of proliferation, expression of CFSE on CD8+ T cells was measured and found to be closely correlated to DC concentration across all treatment groups, indicating that DCs had an impact on these T cells (Fig. 5D). Similar to CD4+ T cells, CD8+ T cells mixed with iDCs (1:1) or DC10s (1:1 or 1:10) had a significant increase in PD-1+ CD8+ T cells (Fig. 5E). This data suggests that this tolerogenic DC therapy has the capacity to induce CD25+FoxP3+ Tregs and suppress CD8+ T cells in vitro.
Figure 5.

Viability and phenotype of CD3+ T cells after 72 hours of incubation with various DCs at various concentrations. A) Zombie Violet negative CD3+ cells, B) CD4+CD25+FoxP3+ T regulatory cells, C) CD4+PD-1+ T cells, D) CD8+ T cell CFSE expression, and E) CD8+PD-1+ T cells assessed by flow cytometry. ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05 via one-way ANOVA and Dunnett’s comparison test in comparison to samples with no DCs.
To further profile the state of DC-treated splenocytes, supernatants from co-cultures were collected and analyzed for cell-secreted cytokines (Table S1). We observed significantly higher concentrations of IL-10 in supernatants from iDC (1:1), DC10 (1:1), VD3 (1:1), LPS (1:1), PEG-4MAL (1:1), and PEG-IL10 (1:1) compared to splenocytes alone, demonstrating these DCs ability to produce a key immunosuppressive cytokine involved in Treg induction (Fig. 6A). With the current methodology, it is unclear how much of the cytokine measured in PEG-IL10 samples were due to DC-secreted IL-10 versus biomaterial-released PEG-IL10, or if modified PEG-IL10 is even recognized by the antibodies used herein, but IL-10 concentrations similar to iDC and DC10 controls were observed nonetheless. Concentrations of IFN-γ were significantly increased only in TNF-α/IFN-γ (1:1) co-cultures (Fig. 6B). Concentrations of IL-17 were analyzed to identify the possibility of a shift to TH17 cells, but no significant differences were observed (Fig. 6C). Concentrations of IL-5 were significantly increased across all treatment groups at high ratios (1:1) with the exception of PEG-4MAL, highlighting another functional difference between PEG-4MAL and IL-10-functionalized hydrogels (Fig. 6D). All treatment groups produced supernatants with increased levels of CCL5, to varying degrees (Fig. 6E). Concentrations of CCL2 were notably increased for iDC, DC10, mDC, and PEG-IL10 groups, while only mDC and PEG-IL10 co-cultures exhibited increased levels of CCL3 and CXCL1 (Fig. 6E). Increases in chemokines such as CCL2 and CCL5 are not implicated in tolerogenicity, but instead illustrate the extent to which these DCs can recruit other lymphocytes.
Figure 6.

Fold changes of A) IL-10 (Tregs), B) IFN-γ (TH1), C) IL-17 (TH17), and D) IL-5 (TH2) for co-culture groups at various treatments ratios. E) Heatmap representing the fold change of selected chemokines for treated groups (1:1) over splenocyte-only controls. Data shown is from 4 replicates across n=2 experiments. ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05 via one-way ANOVA and Dunnett’s comparison test.
3.6. Extensive Infiltration of Endogenous Lymphocytes is Recapitulated in vitro.
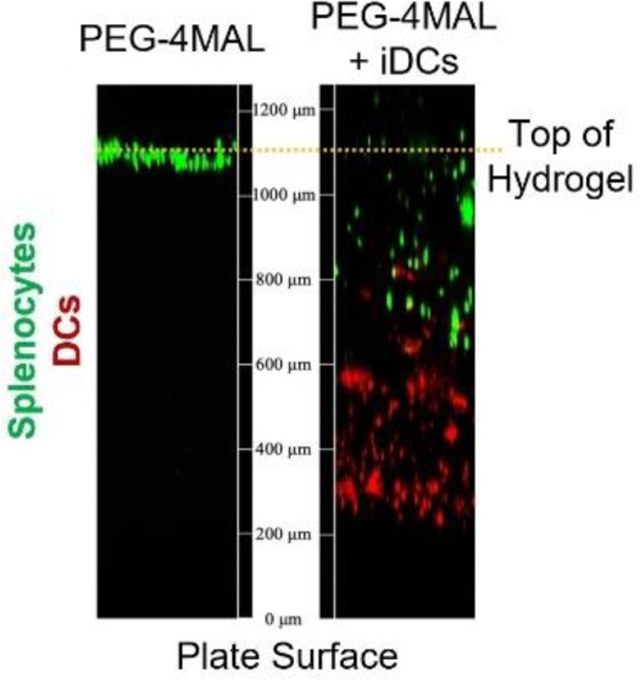
The presence of endogenous cells in the hydrogel environment post-injection suggests that the location of immunomodulation by DCs is within the delivered biomaterial niche, which has been previously reported by our group using hydrogels without IL-1056. We sought to validate the in vitro ability of endogenous immune cells to infiltrate a PEG-4MAL hydrogel by synthesizing PEG-4MAL hydrogels with or without DCs and seeding RBC-lysed splenocytes on top of the hydrogel. After 6 hours, we observed extensive infiltration of splenocytes into hydrogels with iDCs, whereas splenocyte infiltration into DC-free hydrogels was limited (Fig. 7A-C). The number of cells infiltrating hydrogels and their depth is quantified in Figure 7B and C. This data suggests that DCs incorporated in PEG-4MAL hydrogels effectively drives endogenous cell infiltration into the biomaterial. Taken together, we hypothesize that the biomaterial delivery construct functions as an immunomodulatory node, where DCs recruit and tolerize endogenous immune cells in an antigen-dependent manner. Tolerized endogenous APCs and Tregs then migrate to sites of therapeutic importance such as LNs and the CNS to mediate amelioration of autoimmune disease.
Figure 7.

A) Confocal images illustrating infiltration of lymphocytes into hydrogels after 6 hours incubation in vitro. Quantification of the number of DCs (red) and splenocytes (green) fluorescent cells in each slice for B) blank PEG-4MAL hydrogels and C) PEG-4MAL hydrogels incorporating iDCs.
4. Discussion
Whereas DCs have shown much promise as a cell therapy to treat various immune-mediated diseases, their translation to the clinic has proven problematic14,15. Poor biodistribution and post-delivery activation of cells remain significant challenges to successful translation12,16,17. Here, we developed a biomaterial towards improving the stability and efficacy of immunosuppressive DCs post-injection for tolerogenic applications. These hydrogels were engineered to support DC viability, maintain an immature DC phenotype, and functionalized with the cytokine IL-10 to support therapeutic DCs post-delivery. Our optimized vehicle resulted in an immunosuppressive biomaterial niche that maintained DC viability, protected DCs from inflammatory stimuli, and promoted infiltration of endogenous cells for conditioning. Co-culture experiments demonstrated that these DCs have the capacity to induce Tregs and suppress endogenous immune cells.
The hydrogel parameter that most impacted DCs was hydrogel polymer density, while RGD functionalization and degradability kinetics had no significant effect on DCs in vitro. Previous studies have shown that DCs seeded on 2D RGD-functionalized substrates are activated differentially with RGD concentration43, which contrasts our results. Presentation of RGD by the 3D hydrogel systems engineered herein may not activate DCs due to the functional differences between 2D and 3D substrates57, but other possible explanations include: (1) RGD concentrations studied are too low to induce DC maturation, or (2) activation-related cell signaling in DCs is not actualized due to a lack of cell adhesion with the 24 hour period of incubation. Evidently functionalization of hydrogels with adhesive peptides is important for eventual cell infiltration in vivo, but the formulation used here maintains an immature phenotype for incorporated DCs as would be desirable. With hydrogel polymer density as the only biophysical constraint, other variables that do not directly affect DC viability and immaturity enable the flexible design of our system to fit other tolerogenic applications not explored here.
The functionalization of these hydrogels with IL-10 resulted in the improvement of DC viability and phenotypic stability, protection from inflammatory stimuli, and a multi-pronged mechanism of treatment that extends to nearby endogenous immune cells. Three potential mechanistic features suggested by in vitro studies herein include: (1) incorporated PEG-IL10 is directly presented to incorporated DCs; (2) unincorporated IL-10 is immediately released and diffuses to incorporated DCs and nearby endogenous cells; and (3) as hydrogels degrade, PEG-IL10 is released from the mesh network and treats DCs and nearby endogenous cells. Utilizing thiolation to covalently tether IL-10 to PEG-4MAL resulted in bioactivity at similar levels to native IL-10, based on the commonly used MC/9 cell proliferation study. The improved viability and resistance to maturation of DCs incorporated in PEG-IL10 hydrogels were similar to native IL-10. Taken together, these direct and indirect results suggest that the modified PEG-IL10 retains its bioactivity and its ability to confer tolerance to DCs. Incorporation of IL-10 using these methods has an upper limit that is dramatically higher than the 50 ng dose tested in these studies, providing a substantial capacity for dose escalation in future studies.
In co-culture studies, the marked increase in the percentage of CD25+FoxP3+ Tregs for iDCs, DC10s, PEG-4MAL, and PEG-IL10 groups highlights the primary mechanism of immunomodulation postulated in vivo. These co-culture groups also secreted significantly higher levels of IL-10. DC10-based immunomodulation likely occurs through the antigen-dependent induction of Tregs and suppression of CD8+ T cells, as was illustrated here in vitro. This increase in Tregs is accompanied by an increase in PD-1 for the same groups in vitro, suggesting that PD-1 may be involved in the downstream immunomodulation observed in vivo. In the CD8+ T cell compartment, the marked increase in PD-1 combined with dramatic proliferation indicates that these cells are approaching a state of immunosuppression. Despite immunosuppression of CD8+ T cells in vitro, it is unclear whether they become benign or retain their cytotoxic profile in vivo, and further studies are needed. Given that these DCs have limited migratory potential (as shown in Fig. 7), the results herein suggest that DCs would condition endogenous cells in one of two ways in vivo: (1) directly, where tolerogenic DCs interact with endogenous T cells and induce a regulatory phenotype; or (2) indirectly, where tolerogenic DCs modulate endogenous DCs and other APCs, which then induce Tregs. DCs with elevated levels of CCL2, CCL3, CCL4, CCL5, and CXCL1 can recruit endogenous T cells, DCs, macrophages, monocytes, and other immune cells to the hydrogel site for reconditioning. Although the recruited T cell population might consist of naïve T cells, memory T cells, and effector T cells, DCs treated with IL-10 carry the capacity to induce Tregs or suppress autoreactive T cells9,10,58. Endogenous immune cells other than T cells might also be conditioned by DCs in the presence of antigen and IL-10, driving a broader profile of immunosuppression.
Conclusions
The biomaterial system developed in these studies utilizes PEG-4MAL hydrogels functionalized with an immunosuppressive cytokine to improve an immunosuppressive DC therapy through targeted delivery and regulatory phenotype support post-injection. Our immunosuppressive biomaterial niche is designed to protect DCs from inflammatory stimuli and provide a site for immunomodulation of recruited lymphocytes. For DCs, hydrogel polymer density was determined to be the primary parameter impacting DC viability, making this platform amenable to cell therapies for other tolerogenic or immunosuppressive applications. This hydrogel delivery system developed herein highlights the benefits of immunosuppressive biomaterials for the delivery of tolerogenic cell therapies. Future studies will investigate the immunomodulation provided by this PEG-IL10 hydrogel system to deliver incorporated DCs to ameliorate MS in a murine model of Experimental Autoimmune Encephalomyelitis (EAE).
Supplementary Material
Acknowledgements
Thank you to Michael D. Hunckler and Karen E. Martin for technical assistance. Thank you to core facilities personnel and technicians at the Parker H. Petit Institute for Bioengineering and Biosciences at the Georgia Institute of Technology.
Funding Sources
This research was supported by NIH R01AR062368 (AG) and NIH U01AI132817 (AG). Nicholas M. Beskid was supported by the training grant NIH 1 T32 EB021962-A1 (JEB).
Footnotes
The authors declare no competing financial interest.
Supporting Information Available
The following files are available free of charge:
“Supporting information” (pdf): includes flow cytometry gating strategies, hydrogel optimization studies referenced in this manuscript, and rheological characterization of hydrogels used herein.
References
- 1.Dilokthornsakul P; Valuck RJ; Nair KV; Corboy JR; Allen RR; Campbell JD Multiple Sclerosis Prevalence in the United States Commercially Insured Population. Neurology 2016, 86 (11), 1014–1021. DOI: 10.1212/WNL.0000000000002469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adelman G; Rane SG; Villa KF The Cost Burden of Multiple Sclerosis in the United States: A Systematic Review of the Literature. J. Med. Econ 2013, 16 (5), 639–647. DOI: 10.3111/13696998.2013.778268 [DOI] [PubMed] [Google Scholar]
- 3.Compston A; Coles A. Multiple Sclerosis. The Lancet 2008, 372 (9648), 1502–1517. DOI: 10.1016/s0140-6736(08)61620-7 [DOI] [PubMed] [Google Scholar]
- 4.Dooley MA; Hogan SL Environmental Epidemiology and Risk Factors for Autoimmune Disease. Curr. Opin. Rheumatol. 2003, 15 (2), 99–103. DOI: 10.1097/00002281-200303000-00002 [DOI] [PubMed] [Google Scholar]
- 5.Amodio G; Gregori S. Dendritic Cells a Double-Edge Sword in Autoimmune Responses. Front. Immunol. 2012, 3. DOI: 10.3389/fimmu.2012.00233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinman RM; Cohn ZA Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice. J. Exp. Med. 1973, 137 (5), 1142–1162. DOI: 10.1084/jem.137.5.1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaplin DD Overview of the Immune Response. J. Allergy Clin. Immunol. 2010, 125 (2). DOI: 10.1016/j.jaci.2009.12.980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Comabella M; Montalban X; Münz C; Lünemann JD Targeting Dendritic Cells to Treat Multiple Sclerosis. Nat. Rev. Neurol. 2010, 6 (9), 499–507. DOI: 10.1038/nrneurol.2010.112 [DOI] [PubMed] [Google Scholar]
- 9.Boks MA; Kager-Groenland JR; Haasjes MSP; Zwaginga JJ; van Ham SM; ten Brinke A. Il-10-Generated Tolerogenic Dendritic Cells Are Optimal for Functional Regulatory T Cell Induction — a Comparative Study of Human Clinical-Applicable DC. Clin. Immunol. (Amsterdam, Neth.) 2012, 142 (3), 332–342. DOI: 10.1016/j.clim.2011.11.011 [DOI] [PubMed] [Google Scholar]
- 10.Lucca LE; Axisa PP; Aloulou M; Perals C; Ramadan A; Rufas P; Kyewski B; Derbinski J; Fazilleau N; Mars LT; Liblau RS Myelin Oligodendrocyte Glycoprotein Induces Incomplete Tolerance of CD4 + T Cells Specific for Both a Myelin and a Neuronal Self‐Antigen in Mice. Eur. J. Immunol. 2016, 46 (9), 2247–2259. DOI: 10.1002/eji.201646416 [DOI] [PubMed] [Google Scholar]
- 11.Krienke C; Kolb L; Diken E; Streuber M; Kirchhoff S; Bukur T; Akilli-Öztürk Ö; Kranz LM; Berger H; Petschenka J; Diken M; Kreiter S; Yogev N; Waisman A; Karikó K; Türeci Ö; Sahin U. A Noninflammatory Mrna Vaccine for Treatment of Experimental Autoimmune Encephalomyelitis. Science 2021, 371 (6525), 145–153. DOI: 10.1126/science.aay3638 [DOI] [PubMed] [Google Scholar]
- 12.Willekens B; Presas-Rodríguez S; Mansilla MJ; Derdelinckx J; Lee W-P; Nijs G; De Laere M; Wens I; Cras P; Parizel P; Van Hecke W; Ribbens A; Billiet T; Adams G; Couttenye M-M; Navarro-Barriuso J; Teniente-Serra A; Quirant-Sánchez B; Lopez-Diaz de Cerio A; Inogés S; Prosper F; Kip A; Verheij H; Gross CC; Wiendl H; Van Ham MSM; Ten Brinke A; Barriocanal AM; Massuet-Vilamajó A; Hens N; Berneman Z; Martínez-Cáceres E; Cools N; Ramo-Tello C. Tolerogenic Dendritic Cell-Based Treatment for Multiple Sclerosis (MS): A Harmonised Study Protocol for Two Phase I Clinical Trials Comparing Intradermal and Intranodal Cell Administration. BMJ Open 2019, 9 (9). DOI: 10.1136/bmjopen-2019-030309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vries, I. J. M; Krooshoop Daniëlle JEB; Scharenborg NM; Lesterhuis WJ; Diepstra JHS; van Muijen GNP; Strijk SP; Ruers TJ; Boerman OC; Oyen WJG; Adema GJ; Punt CJA; Figdor CG. Effective Migration of Antigen-Pulsed Dendritic Cells to Lymph Nodes in Melanoma Patients Is Determined by Their Maturation State. Cancer Res. 2003, 63 (1), 12–17. [PubMed] [Google Scholar]
- 14.Phillips BE; Garciafigueroa Y; Trucco M; Giannoukakis N. Clinical Tolerogenic Dendritic Cells: Exploring Therapeutic Impact on Human Autoimmune Disease. Front. Immunol. 2017, 8. DOI: 10.3389/fimmu.2017.01279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willekens B; Cools N. Beyond the Magic Bullet: Current Progress of Therapeutic Vaccination in Multiple Sclerosis. CNS Drugs 2018, 32 (5), 401–410. DOI: 10.1007/s40263-018-0518-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou F; Ciric B; Zhang G-X; Rostami A. Immunotherapy Using Lipopolysaccharide-Stimulated Bone Marrow-Derived Dendritic Cells to Treat Experimental Autoimmune Encephalomyelitis. Clin. Exp. Immunol. 2014, 178 (3), 447–458. DOI: 10.1111/cei.12440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mansilla MJ; Contreras-Cardone R; Navarro-Barriuso J; Cools N; Berneman Z; Ramo-Tello C; Martínez-Cáceres EM Cryopreserved Vitamin D3-Tolerogenic Dendritic Cells Pulsed with Autoantigens as a Potential Therapy for Multiple Sclerosis Patients. J. Neuroinflammation 2016, 13 (1). DOI: 10.1186/s12974-016-0584-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verma V; Kim Y; Lee M-C; Lee J-T; Cho S; Park I-K; Min JJ; Lee JJ; Lee SE; Rhee JH Activated Dendritic Cells Delivered in Tissue Compatible Biomatrices Induce in-Situ Anti-Tumor CTL Responses Leading to Tumor Regression. Oncotarget 2016, 7 (26), 39894–39906. DOI: 10.18632/oncotarget.9529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver JD; Headen DM; Aquart J; Johnson CT; Shea LD; Shirwan H; García AJ Vasculogenic Hydrogel Enhances Islet Survival, Engraftment, and Function in Leading Extrahepatic Sites. Sci. Adv. 2017, 3 (6). DOI: 10.1126/sciadv.1700184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han WM; Anderson SE; Mohiuddin M; Barros D; Nakhai SA; Shin E; Amaral IF; Pêgo AP; García AJ; Jang YC Synthetic Matrix Enhances Transplanted Satellite Cell Engraftment in Dystrophic and Aged Skeletal Muscle with Comorbid Trauma. Sci. Adv. 2018, 4 (8). DOI: 10.1126/sciadv.aar4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pashuck ET; Stevens MM Designing Regenerative Biomaterial Therapies for the Clinic. Sci. Transl. Med. 2012, 4 (160). DOI: 10.1126/scitranslmed.3002717 [DOI] [PubMed] [Google Scholar]
- 22.Singh A. Biomaterials Innovation for next Generation Ex Vivo Immune Tissue Engineering. Biomaterials 2017, 130, 104–110. DOI: 10.1016/j.biomaterials.2017.03.015 [DOI] [PubMed] [Google Scholar]
- 23.Danhier F; Ansorena E; Silva JM; Coco R; Le Breton A; Préat V. PLGA-Based Nanoparticles: An Overview of Biomedical Applications. J. Controlled Release 2012, 161 (2), 505–522. DOI: 10.1016/j.jconrel.2012.01.043 [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y; Chan HF; Leong KW Advanced Materials and Processing for Drug Delivery: The Past and the Future. Adv. Drug Delivery Rev. 2013, 65 (1), 104–120. DOI: 10.1016/j.addr.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoo J-W; Irvine DJ; Discher DE; Mitragotri S. Bio-Inspired, Bioengineered and Biomimetic Drug Delivery Carriers. Nat. Rev. Drug Discovery 2011, 10 (7), 521–535. DOI: 10.1038/nrd3499 [DOI] [PubMed] [Google Scholar]
- 26.Raïch-Regué D; Glancy M; Thomson AW Regulatory Dendritic Cell Therapy: From Rodents to Clinical Application. Immunol. Lett. 2014, 161 (2), 216–221. DOI: 10.1016/j.imlet.2013.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voigtländer C; Röner S; Cierpka E; Theiner G; Wiethe C; Menges M; Schuler G; Lutz MB Dendritic Cells Matured with TNF Can Be Further Activated in Vitro and after Subcutaneous Injection in Vivo Which Converts Their Tolerogenicity into Immunogenicity. J. Immunother. 2006, 29 (4), 407–415. DOI: 10.1097/01.cji.0000210081.60178.b4 [DOI] [PubMed] [Google Scholar]
- 28.Lewis JS; Dolgova NV; Chancellor TJ; Acharya AP; Karpiak JV; Lele TP; Keselowsky BG The Effect of Cyclic Mechanical Strain on Activation of Dendritic Cells Cultured on Adhesive Substrates. Biomaterials 2013, 34 (36), 9063–9070. DOI: 10.1016/j.biomaterials.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas AM; Dong Y; Beskid NM; García AJ; Adams AB; Babensee JE Brief Exposure to Hyperglycemia Activates Dendritic Cells in Vitro and in Vivo. J. Cell. Physiol. 2019, 235 (6), 5120–5129. DOI: 10.1002/jcp.29380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park J; Babensee JE Differential Functional Effects of Biomaterials on Dendritic Cell Maturation. Acta Biomater. 2012, 8 (10), 3606–3617. DOI: 10.1016/j.actbio.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phelps EA; Enemchukwu NO; Fiore VF; Sy JC; Murthy N; Sulchek TA; Barker TH; García AJ Maleimide Cross-Linked Bioactive Peg Hydrogel Exhibits Improved Reaction Kinetics and Cross-Linking for Cell Encapsulation and in Situ Delivery. Adv. Mater. 2011, 24 (1), 64–70. DOI: 10.1002/adma.201103574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darling NJ; Hung Y-S; Sharma S; Segura T. Controlling the Kinetics of Thiol-Maleimide Michael-Type Addition Gelation Kinetics for the Generation of Homogenous Poly(Ethylene Glycol) Hydrogels. Biomaterials 2016, 101, 199–206. DOI: 10.1016/j.biomaterials.2016.05.053 [DOI] [PubMed] [Google Scholar]
- 33.Patterson J; Hubbell JA Enhanced Proteolytic Degradation of Molecularly Engineered Peg Hydrogels in Response to MMP-1 and MMP-2. Biomaterials 2010, 31 (30), 7836–7845. DOI: 10.1016/j.biomaterials.2010.06.061 [DOI] [PubMed] [Google Scholar]
- 34.Turk BE; Huang LL; Piro ET; Cantley LC Determination of Protease Cleavage Site Motifs Using Mixture-Based Oriented Peptide Libraries. Nat. Biotechnol. 2001, 19 (7), 661–667. DOI: 10.1038/90273 [DOI] [PubMed] [Google Scholar]
- 35.Windsor WT; Syto R; Tsarbopoulos A; Zhang R; Durkin J; Baldwin S; Paliwal S; Mui PW; Pramanik B. Disulfide Bond Assignments and Secondary Structure Analysis of Human and Murine Interleukin 10. Biochemistry 1993, 32 (34), 8807–8815. DOI: 10.1021/bi00085a011 [DOI] [PubMed] [Google Scholar]
- 36.Klompus S; Solomon G; Gertler A. A Simple Novel Method for the Preparation of Noncovalent Homodimeric, Biologically Active Human Interleukin 10 in Escherichia Coli—Enhancing Protein Expression by Degenerate PCR of 5′ DNA in the Open Reading Frame. Protein Expression Purif. 2008, 62 (2), 199–205. DOI: 10.1016/j.pep.2008.07.013 [DOI] [PubMed] [Google Scholar]
- 37.Yang F; Wan Y; Liu J; Yang X; Wang H; Tao K; Han J; Shi J; Hu D. Expression and Purification of RHIL-10-RGD from Escherichia Coli as a Potential Wound Healing Agent. J. Microbiol. Methods 2016, 127, 62–67. DOI: 10.1016/j.mimet.2016.05.025 [DOI] [PubMed] [Google Scholar]
- 38.Rubinstein M. Polymer physics; Oxford University Press: Oxford, 2014. [Google Scholar]
- 39.Welzel PB; Prokoph S; Zieris A; Grimmer M; Zschoche S; Freudenberg U; Werner C. Modulating Biofunctional Starpeg Heparin Hydrogels by Varying Size and Ratio of the Constituents. Polymers 2011, 3 (1), 602–620. DOI: 10.3390/polym3010602 [DOI] [Google Scholar]
- 40.Stabenfeldt SE; Munglani G; García AJ; LaPlaca MC Biomimetic Microenvironment Modulates Neural Stem Cell Survival, Migration, and Differentiation. Tissue Eng., Part A 2010, 16 (12), 3747–3758. DOI: 10.1089/ten.tea.2009.0837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Enemchukwu NO; Cruz-Acuña R; Bongiorno T; Johnson CT; García JR; Sulchek T; García AJ Synthetic Matrices Reveal Contributions of ECM Biophysical and Biochemical Properties to Epithelial Morphogenesis. J. Cell Biol. 2015, 212 (1), 113–124. DOI: 10.1083/jcb.201506055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Apoorva FNU; Tian YF; Pierpont TM; Bassen DM; Cerchietti L; Butcher JT; Weiss RS; Singh A. Award Winner in the Young Investigator Category, 2017 Society for Biomaterials Annual Meeting and Exposition, Minneapolis, MN, April 05–08, 2017: Lymph Node Stiffness-Mimicking Hydrogels Regulate Human B-Cell Lymphoma Growth and Cell Surface Receptor Expr. J. Biomed. Mater. Res, Part A 2017, 105 (7), 1833–1844. DOI: 10.1002/jbm.a.36031 [DOI] [PubMed] [Google Scholar]
- 43.Acharya AP; Dolgova NV; Moore NM; Xia C-Q; Clare-Salzler MJ; Becker ML; Gallant ND; Keselowsky BG The Modulation of Dendritic Cell Integrin Binding and Activation by RGD-Peptide Density Gradient Substrates. Biomaterials 2010, 31 (29), 7444–7454. DOI: 10.1016/j.biomaterials.2010.06.025 [DOI] [PubMed] [Google Scholar]
- 44.Discher DE; Janmey P; Wang Y -li. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310 (5751), 1139–1143. DOI: 10.1126/science.1116995 [DOI] [PubMed] [Google Scholar]
- 45.Keselowsky BG; Collard DM; García AJ Surface Chemistry Modulates Fibronectin Conformation and Directs Integrin Binding and Specificity to Control Cell Adhesion. J. Biomed. Mater. Res, Part A 2003, 66A (2), 247–259. DOI: 10.1002/jbm.a.10537 [DOI] [PubMed] [Google Scholar]
- 46.Phelps EA; Headen DM; Taylor WR; Thulé PM; García AJ Vasculogenic Bio-Synthetic Hydrogel for Enhancement of Pancreatic Islet Engraftment and Function in Type 1 Diabetes. Biomaterials 2013, 34 (19), 4602–4611. DOI: 10.1016/j.biomaterials.2013.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bott K; Upton Z; Schrobback K; Ehrbar M; Hubbell JA; Lutolf MP; Rizzi SC The Effect of Matrix Characteristics on Fibroblast Proliferation in 3 D Gels. Biomaterials 2010, 31 (32), 8454–8464. DOI: 10.1016/j.biomaterials.2010.07.046 [DOI] [PubMed] [Google Scholar]
- 48.Khetan S; Guvendiren M; Legant WR; Cohen DM; Chen CS; Burdick JA Degradation-Mediated Cellular Traction Directs Stem Cell Fate in Covalently Crosslinked Three-Dimensional Hydrogels. Nat. Mater. 2013, 12 (5), 458–465. DOI: 10.1038/nmat3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kouwenhoven M; Özenci V; Tjernlund A; Pashenkov M; Homman M; Press R; Link H. Monocyte-Derived Dendritic Cells Express and Secrete Matrix-Degrading Metalloproteinases and Their Inhibitors and Are Imbalanced in Multiple Sclerosis. J. Neuroimmunol. 2002, 126 (1–2), 161–171. DOI: 10.1016/s0165-5728(02)00054-1 [DOI] [PubMed] [Google Scholar]
- 50.Zdanov A. Structural Features of the Interleukin-10 Family of Cytokines. Curr. Pharm. Des. 2004, 10 (31), 3873–3884. DOI: 10.2174/1381612043382602 [DOI] [PubMed] [Google Scholar]
- 51.Walter MR The Molecular Basis of IL-10 Function: From Receptor Structure to the Onset of Signaling. Curr. Top. Microbiol. Immunol. 2014, 191–212. DOI: 10.1007/978-3-662-43492-5_9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shouval DS; Ouahed J; Biswas A; Goettel JA; Horwitz BH; Klein C; Muise AM; Snapper SB Interleukin 10 Receptor Signaling. Adv. Immunol. 2014, 177–210. DOI: 10.1016/b978-0-12-800267-4.00005-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soderquist RG; Milligan ED; Harrison JA; Chavez RA; Johnson KW; Watkins LR; Mahoney MJ Pegylation of Interleukin-10 for the Mitigation of Enhanced Pain States. J. Biomed. Mater. Res, Part A 2009, 9999A. DOI: 10.1002/jbm.a.32611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dendrou CA; Fugger L; Friese MA Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol. 2015, 15 (9), 545–558. DOI: 10.1038/nri3871 [DOI] [PubMed] [Google Scholar]
- 55.Becher B; Spath S; Goverman J. Cytokine Networks in Neuroinflammation. Nat. Rev. Immunol. 2016, 17 (1), 49–59. DOI: 10.1038/nri.2016.123 [DOI] [PubMed] [Google Scholar]
- 56.Thomas AM; Beskid NM; Blanchfield JL; Rosado AM; García AJ; Evavold BD; Babensee JE Localized Hydrogel Delivery of Dendritic Cells for Attenuation of Multiple Sclerosis in a Murine Model. J. Biomed. Mater. Res, Part A 2020, 109 (7), 1247–1255. DOI: 10.1002/jbm.a.37118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duval K; Grover H; Han L-H; Mou Y; Pegoraro AF; Fredberg J; Chen Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32 (4), 266–277. DOI: 10.1152/physiol.00036.2016 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 58.Müller G; Müller A; Tüting T; Steinbrink K; Saloga J; Szalma C; Knop J; Enk AH Interleukin-10-Treated Dendritic Cells Modulate Immune Responses of Naive and Sensitized T Cells in Vivo. J. Invest. Dermatol. 2002, 119 (4), 836–841. DOI: 10.1046/j.1523-1747.2002.00496.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.