

Abstract
Background:
Elevated pulmonary vascular resistance (PVR), platelet adhesion, coagulation activation, and inflammation are prominent features of xenolung rejection. Here we evaluate the role of thromboxane and histamine on PVR, and their contribution to other lung xenograft injury mechanisms.
Methods:
GalTKO.hCD46 single pig lungs were perfused ex vivo with fresh heparinized human blood: lungs were either treated with 1-Benzylimidazole (1-BIA) combined with histamine receptor blocker famotidine (n=4) or diphenhydramine (n=6), 1-BIA alone (n=6) or were left untreated (n=9).
Results:
Six of the nine control experiments (GalTKO.hCD46 untreated), “survived” until elective termination at 4h. Without treatment, initial PVR elevation within the first 30min resolved partially over the following hour, and increased progressively during the final 2h of perfusion. In contrast, 1-BIA, alone or in addition to either antihistamine treatment, was associated with low stable PVR. Combined treatments significantly lowered the airway pressure when compared to untreated reference. Although platelet and neutrophil sequestration and coagulation cascade activation were not consistently altered by any intervention, increased terminal wet/dry weight ratio in untreated lungs was significantly blunted by combined treatments.
Conclusion:
Combined thromboxane and histamine pathway blockade prevents PVR elevation and significantly inhibits loss of vascular barrier function when GalTKO.hCD46 lungs are perfused with human blood. Platelet activation and platelet and neutrophil sequestration persist in all groups despite efficient complement regulation, and appear to occur independent of thromboxane and histamine antagonism. Our work identifies thromboxane and histamine as key mediators of xenolung injury and defines those pathways as therapeutic targets to achieve successful xenolung transplantation.
Keywords: lung, xenotransplantation, ex-vivo perfusion, pulmonary vascular resistance, thromboxane, histamine, GalTKO.hCD46
Introduction
Xenotransplantation is a promising future treatment option for end-stage organ failure patients, possibly solving the limited availability of human donor organs (1–6). Genetic engineering progress coupled with advances in understanding the “rejection” mechanisms leading to xenograft failure have contributed to increasing xenogeneic organ survival in various models (7–13). When organs with the galactosyl-transferase gene knocked out (GalTKO) also express one or more human complement pathway regulatory proteins (hCPRPs), xenograft failure is typically associated with coagulation cascade activation in the recipient, and an inflammatory response in the xenogeneic organ (14–17), particularly during human blood perfusion of pig pulmonary xenografts (15,18–19). Platelet sequestration as well as thromboxane and histamine elaboration are identified as early events during GalTKO.hCD46 (15) as well as wild-type (WT), GalTKO, and other hCPRP-expressing (20–21) porcine lung xenograft rejection, and have been implicated in elevated pulmonary vascular resistance (PVR), loss of lung vascular barrier function, and subsequent organ failure in these circumstances and in other lung models (22–28).
In this series of experiments we ask whether thromboxane, alone or in conjunction with histamine, mediates increased pulmonary vascular resistance and loss of vascular barrier function in GalTKO.hCD46 lungs evaluated in an established xenogeneic ex vivo human blood perfusion model.
Materials and Methods:
Animals
Genetically engineered pigs (BW 15–17kg) lacking the alpha-Gal epitope (GalTKO), expressing human membrane cofactor protein (hCD46), were supplied by Revivicor (Blacksburg, VA). Human CD46 expression was homozygous in all donor pigs. (GalTKO.hCD46 pigs used in previously published experiments were heterozygous for hCD46 expression (15,29).)
All procedures were approved by the IACUC at the University of Maryland School of Medicine and were conducted in compliance with NIH guidelines for the care and use of laboratory animals.
Lung Harvest
Induction of anesthesia and surgical organ dissection were performed as previously described (30–32). Prior to flushing the lungs, 1-benzylimidazole (1-BIA, 5mg/kg BW; Sigma-Aldrich), and synthetic prostaglandin I2 (0.06mg/kg, Remodulin (treprostinil), United Therapeutics) were administered intravenously and allowed to circulate for several minutes.
Lung Perfusion
The right and left lungs were separately perfused via the pulmonary artery using side-by-side circuits fashioned from silicon tubing and polyurethane connectors as previously described (32). Pulmonary artery flow was measured and recorded with a flowmeter (Transonic Systems Inc., Model TTFM 73–0146). The Digimed System Integrator (Micro-Med) continuously recorded PA- and airway pressures via transducers integrated into the perfusion (Ismatec MCP rollerpump, IDEX) and ventilator (Harvard apparatus respirator, model 613) circuits, respectively, using PowerLab 16/35 and LabChart 7 pro (AD Instruments). Lungs were ventilated at 15 breath/minute (tidal volume approximately 10mL/kg). Pulmonary vascular resistance (PVR) was estimated by using the equation: PVR = Pulmonary Artery Pressure/ Pulmonary Artery Flow. Lung survival time was defined as the interval after initiation of lung perfusion when the lung function reached one of three “lung failure” criterion: 1) PVR >600mmHg*min/L; 2) development of gross tracheal edema filling the endotracheal tube; or 3) intraparenchymal sequestration of >85% of the reservoir’s perfusate volume. If lungs did not meet a “survival” criterion, experiments were electively terminated after 4h of perfusion.
Perfusate Preparation
Fresh whole blood was collected from two healthy human blood donors (~450ml each) into a blood collection bag containing 64ml of CPDA-1. Two units (~240ml each) of type-compatible thawed plasma were added for each unit of blood to obtain an initial perfusate volume of ~2L with a pre-perfusion hematocrit of ~16%. Heparin (3IU/ml blood, Heparin Sodium Injection, Sagent), calcium chloride (1.3 to 1.6mg/ml blood, American Regent, Inc.) to neutralize the CPDA chelating agent, and 8.4% sodium bicarbonate (up to 0.8mg/ml blood, target pH of 7.36 to 7.44, Hospira, Inc.) were added to the blood pool. All blood components were thoroughly mixed before circuit priming (1L per circuit). In our experience, no significant differences in anti-non-gal antibody levels are found in the pre-perfusion perfusate, probably as a consequence of pooling six donors (two bags of human blood plus four bags of human plasma).
Experimental groups
Treatment of the perfusate was with famotidine (Pepcid, a selective H2 receptor antagonist; 11.2mg/L, n=4) or diphenhydramine (Benadryl, an H1 receptor inverse agonist; 44.8mg/L, n=6), in combination with 1-benzylimidazole (a thromboxane synthase inhibitor, 1-BIA; 112mg/L), added as a single dose to the perfusate, >5 min prior to the start of each experiment. Six GalTKO.hCD46 single lungs were perfused with 1-BIA alone and 9 untreated GalTKO.hCD46 single lungs comprise additional reference groups.
Sampling regimen
Baseline blood samples were taken after blood preparation (“pre” sample), and after drug addition (where indicated) and circulating the blood in the perfusion circuit for at least 5min (time 0 sample). Further samples were collected from the pulmonary veins at 5, 15, 30, 60, 120 and 240min after lung perfusion was initiated. All blood samples were stored at −70°C.
Lung tissue samples (wedge biopsies) were collected pre-perfusion and 10, 30, 60, 120 and 240min of perfusion.
Hematologic Analysis
Blood cell counts were enumerated by standard automated techniques (Antech Diagnostics and Hemavet 950FS Hematology Analyzer, Drew Scientific in duplicate) in blood samples collected in ethylenediaminetetraacetic acid (EDTA).
Enzyme-Linked Immunosorbent Assays
Beta-thromboglobulin (βTG), prothrombin fragments 1+2 (F1+2), were measured by commercial enzyme-linked immunosorbent assay (ELISA) in plasma samples collected in CTAD tubes as previously described (31 EDTA plasma was used to measure histamine (ELISA kit Starfish) and C3a levels (Microvue Complement C3a Plus, Quidel) (29). Blood, collected in EDTA tubes containing 100μl of meclofenamate (10μg/ml, Sigma-Aldrich), was used for thromboxane (Thromboxane B2 EIA Kit).
Flow cytometry
Blood samples were stained by monoclonal antibodies specific for CD41 (as marker for platelets) (AbD Serotec) and CD62P (expressed by activated platelets) (BD Pharmingen) and acquired on a FACSCalibur (BD Biosciences). Platelets and platelet aggregates, identified by size and presence of CD41 staining, were analyzed for expression of CD62P by FACS analysis. Results were expressed as percentage of CD62P-positive cells among CD41-positive cells.
Wet/Dry Ratio
Tissue edema level was approximated by sampling a portion of the upper lower lung lobe at time of elective termination (4 hours). Samples from lungs failing before this time point (3 lungs in the control group) were excluded from the analysis. Tissue weight was measured immediately after collection, as well as after completely drying under a chemical fume hood (>2 weeks). The ratio of the weights is represented as wet-to-dry weight ratio (WDR).
Histology
Lung biopsies obtained during the perfusion and terminally samples were trisected, processed as previously described (31) and compared to specimens, taken prior to the start of the perfusion.
Statistical analysis
Unless otherwise noted, all data are presented as mean and standard deviation of the mean (SD) for all variables except for survival time, which is expressed as median. Continuous variables were checked for normality. Variables were assessed with a one-way analysis of variance (ANOVA) test. P-values <0.05 were considered statistically significant. All statistical analyses were performed on a personal computer with the statistical package SigmaPlot for Windows (Version 11.0) or GraphPad InStat (Version 3.01).
Results
Graft Survival
All, but three untreated GalTKO.hCD46 control grafts, “survived” until elective termination at 4h [Fig.1]. Grafts that failed showed a rise in PVR, meeting failure criteria 15, 45 and 200min after perfusion was initiated. 1-BIA treatment of the perfusate, with or without additional histamine blockade (H-blockade), was associated with significantly increased lung survival when compared to untreated controls (p=0.014).
Figure 1. Cumulative lung survival.

Survival of ex vivo perfused porcine lungs, based on pre-defined lung failure endpoint criteria (31), or attainment of an arbitrary 4 hour interval with preserved graft function, is illustrated for each experimental group. Except for one untreated lungs, all lungs survived to elective termination (p=0.01 for untreated vs. 1-BIA±H-blocker treatment).
Pulmonary Function Parameters
Untreated lungs displayed a physiologically significant if transient pulmonary vascular resistance (PVR) rise during the first hour of perfusion, whereas lungs treated with 1-BIA (+/−H-blockade) maintained very low and stable PVR throughout this interval (e.g. at 30min: untreated 217±149 vs. 1-BIA 32±4mmHg*min/L, p=0.01) [Fig.2A]. Between 90 and 150min, untreated lungs’ PVR dropped to between 100 and 140mmHg*min/L, but rose progressively over the final two hours of perfusion (at 240min: untreated 270±118 vs. 1-BIA 112±119mmHg*min/L, p=0.04). PVR rose during the last hour in 2 of 6 1-BIA monotherapy lungs. In contrast, PVR rose only slightly in the two dual treatment groups, with minimal intra-group variability. PVR values after 20min of perfusion were constantly significantly lower in all 1-BIA treated groups when compared to the untreated control.
Figure 2. Pulmonary vascular resistance and airway pressure during perfusion.

PVR is expressed as a function of perfusion time, by experimental group (A). Time 0 represents measurements obtained during the first minute of lung perfusion. Organs in the 1-BIA treated groups maintained low and very stable PVR values, while untreated reference groups displayed bi-phasic PVR elevation, with a significant PVR rises at 30 minutes, and again during the last two hours of the 4-hour perfusion interval, a physiologic profile typical of thromboxane elaboration in other models (49). 1-BIA-treated groups show significantly lower PVR values between 20 and 240min of perfusion when compared to the untreated reference (* p<0.05). Airway pressures were significantly reduced throughout the perfusion when lungs were treated with 1-BIA alone or 1-BIA in combination with H1-blocker (B) (# p<0.05).
Airway pressures (AWP) showed a delayed rise and after 20min of perfusion significantly lower values in groups that were treated with 1-BIA when compared to untreated groups (e.g. at 60min: 1-BIA 5.9±0.9 vs. untreated 7.4±0.5cmH2O, p=0.002) [Fig.2B]. Similarly, the additional administration of H-blockade lowered AWPs further, especially when diphenhydramine was used (e.g. at 240 min: 1-BIA/diphenhydramine 6.6±1.2 vs. 1-BIA 8.6±1.0cmH2O, p=0.015)
Platelet Sequestration and Activation
In all groups, platelet sequestration of about 45–70% was detected at 5min [Fig. 3A]. There was no statistically significant difference between control, 1-BIA and 1-BIA/diphenhydramine treatment groups. Platelet sequestration in association with 1-BIA/Famotidine treatment appeared to show greater sequestration; due to the low number of experiments in this group, statistical differences could not be calculated. Substantial intra-group variability in platelet sequestration was observed.
Figure 3. Platelet activation and thromboxane elaboration.

Blood platelet counts measured by automated counting in pulmonary vein effluent samples collected serially over the course of ex vivo lung perfusion. Platelet sequestration was expressed as the percentage of platelets remaining in the perfusate at each time point (A). Activation of platelets was assessed by measuring the proportion of CD41+ platelets expressing CD62P by flow cytometry (B) and by measuring plasma levels of βTG released from platelet granules (C). (D) Plasma thromboxane B2 levels. Data is expressed as change from the baseline and shown as the mean ± SD of surviving experiments.
Platelet activation (P-Selectin [Fig 3B] and β-thromboglobulin, [Fig. 3C]) was increased in 1-BIA/famotidine experiments (n too low to calculate p-value). Other groups did not differ statistically during the perfusion. Only at the 4h time point, CD62P expression on platelets in the 1-BIA/diphenhydramine group was significantly lower than seen in the untreated control group (ΔCD62P 2.0±2.4 vs. 7.4±4.7%, p=0.03).
Thromboxane Plasma Levels
The rise in plasma thromboxane B2 levels at 1 and 4h in reference groups not treated with 1-BIA (e.g., ΔTXB2 at 4h: 51.9±19.8) was significantly inhibited by 1-BIA treatment (all 1-BIA treated groups p<0.001 vs. control), and was lowest when famotidine was combined with 1-BIA (1.4±1.0ng/mL) [Fig.3D]. Supp. Fig. 1A shows the absolute values measured in each group.
Complement and Coagulation Cascade Activation
All experimental groups showed similar kinetics of complement activation, measured by plasma C3a [Fig. 4A]. While all groups displayed similar thrombin generation within the first hour, lungs treated with 1-BIA and famotidine tended to be associated with lower thrombin formation at subsequent intervals when compared to their reference group (e.g. ΔF1+2 at 4h: 1-BIA/famotidine 15.7±8.8 vs. control 26.0±12.0nM; n too low for stat. analysis) [Fig. 4B].
Figure 4. Complement and coagulation cascade activation.

(A) Plasma levels of complement activation byproduct C3a, expressed as the amount of complement fragments produced above the pre-perfusion baseline. All groups showed statistically similar complement activation during perfusion. (B) Activation of the coagulation cascade was detected by the formation of thrombin measured by plasma levels of fragments F1+2. In groups that had received 1-BIA treatment, ΔF1+2 values tended to be lower throughout the perfusion, but differences did not achieve statistical significance.
Other parameters associated with lung xenograft failure
Neutrophil sequestration was observed in all experimental groups [Fig. 5]. After 4 hours of perfusion, only 8±5 (untreated) to 18±16% (1-BIA treated) of the initial PMN count were still detectable in the perfusate. During the first hour, PMN sequestration was delayed when 1-BIA was administered alone (at 30min: 69±46; vs. 35±14% for untreated [p=0.12]).
Figure 5. Neutrophil sequestration.

Neutrophil sequestration from the circulation during the ex vivo perfusion interval. Data is expressed as change from the baseline and shown as the mean ± SD of surviving experiments. Sequestration, during the first hour of perfusion, tended to be delayed in lungs where 1-BIA was administered alone (* p=0.1 vs. untreated GalTKO.hCD46). After 1 hour of perfusion, all groups showed similar results.
Histamine Plasma Levels
Histamine was rapidly elaborated in the untreated reference group but thereafter stayed at relatively stable levels (ΔHistamine 160–190nM) between 2 and 4 hours of perfusion [Fig. 6A]. Histamine release in groups that were treated with either H1- or H2-receptor blocker in combination with 1-BIA was slightly lower at 15min but increased to the highest values of all groups at the 4h interval (ΔHistamine 852±504 for 1-BIA/famotidine [n too low for stat. analysis] and 540±231 for 1-BIA/diphenhydramine treatment vs. 190±79nM for untreated [p=0.006]). In the 1-BIA group, one outlier increased the average value at the 240min mark from 346 to 509 nM. Supp. Fig. 1B shows the absolute values measured in each group.
Figure 6. Histamine elaboration and wet-to-dry weight ratio.

(A) Histamine levels at 4 hours were significantly increased in groups treated with either histamine-receptor blocker. * p=0.005 for 1-BIA/diphenhydramine vs. untreated. (B) In 1-BIA/diphenhydramine treated lungs, wet-to-dry weight ratio (WDR) was significantly reduced when compared to the untreated reference (4.0±0.5 vs. 5.1±1.1, p<0.5). While the addition of 1-BIA/famotidine resulted in the lowest WDR (3.9±1.0), low experiment number did not allow p-value calculations.
Wet/Dry Ratio
Combined treatment with 1-BIA and histamine receptor blockade resulted in significantly lower terminal WDR than found in untreated lungs [Fig. 6B]. When 1-BIA was combined with diphenhydramine, WDR was 4.0±0.5 significantly lower than in untreated controls (5.1±1.1, p<0.05). 1-BIA administration alone did not significantly lower WDR when compared to the untreated group (4.2±0.8, p=0.14). The addition of 1-BIA/famotidine to the perfusate was associated with the lowest WDR (3.9±1.0) but due to the low number of experiments in this group, statistical analysis could not be performed.
Histology
Tissue samples collected before initiation of xenogeneic perfusion (“pre”sample) displayed normal microscopic lung anatomy with air-filled alveoli and thin inter-alveolar septae [Fig. 7A,D]. Biopsies taken at the time of elective termination, showed still relatively well-preserved pulmonary tissue with only little polymorphonuclear granulocyte infiltration [Fig. 7B,C,E, F]. All final tissue samples displayed interstitial hemorrhage and inter-alveolar wall thickening, which were subjectively the least prominent in tissue samples, collected from 1-BIA/famotidine treated experiments.
Figure 7. Representative lung histology.
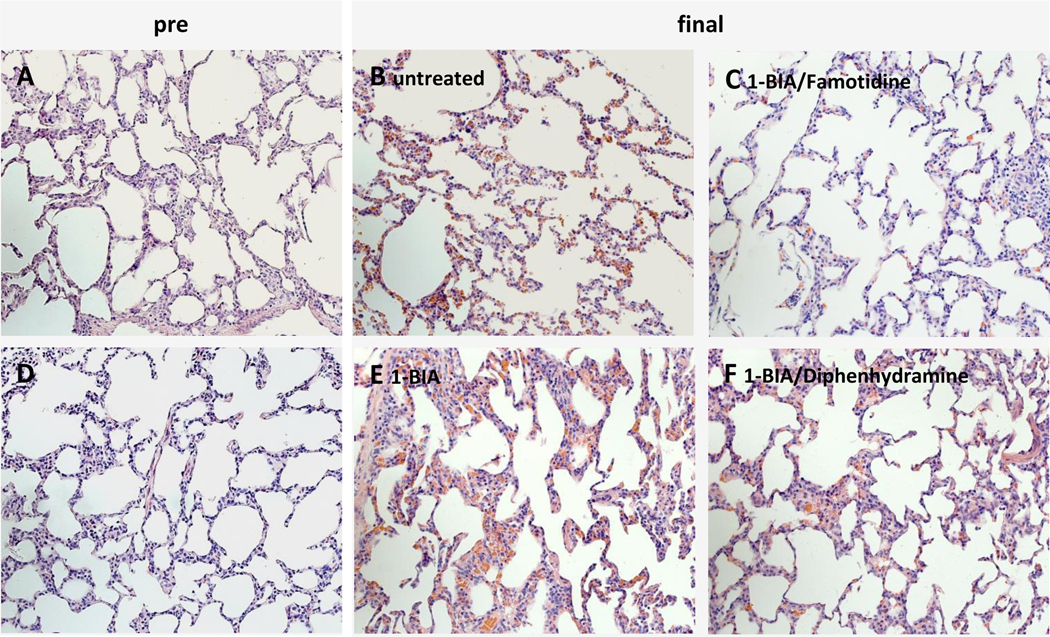
Lung biopsies, H&E, 20X. Pictures A and D show representative pre-perfusion biopsies. All terminal tissue biopsies (B, C, E, F) show relatively well-preserved pulmonary tissue with interstitial hemorrhage in varying degree, and only little polymorphonuclear granulocyte infiltration.
Discussion
Rejection of pulmonary xenografts is associated with activation of numerous pro-inflammatory pathways, some of which have been partly addressed by genetically modifying the donor pigs. Introduction of the α-galactosyl-transferase knockout (GalTKO pigs) eliminates the anti-Gal-antibody-mediated immune response (31,33–34), and addition of human complement-regulatory proteins (e.g. hCD46, hCD55, hCD59) reduces the complement driven organ injury (15,35–36). Other work by us and our collaborators has explored whether human genes that regulate the coagulation cascade (e.g. hTBM, hEPCR, hCD39) may diminish residual coagulation cascade dysregulation, thrombocytopenia, or neutrophil sequestration (37–39). Despite incremental progress reported to date with these modifications, PVR elevation, thromboxane and histamine elaboration and development of tissue edema remain prevalent in xenogenically perfused GalTKO.hCPRP lungs, often preceding organ injury and failure (15,35,39).
In prior work, we found that thromboxane was closely linked to the rise in PVR when WT porcine lungs were perfused with human blood (20–21). Because pretreatment of the pig lung donor with liposomal clodronate significantly attenuates thromboxane elaboration and PVR rise, we believe thromboxane is elaborated primarily from pulmonary intravascular macrophages, although platelets, lung endothelium, and other donor or recipient myeloid lineage cells may also contribute. To investigate the impact of thromboxane on GalTKO.hCPRP graft physiology and injury mode – which for convenience we refer to here as “rejection mechanisms” – in the current series of experiments we inhibited thromboxane synthesis with 1-BIA. Our results demonstrate that thromboxane elaboration is the pivotal mediator of PVR elevation in this model, since PVR remained very stable at low values throughout the perfusion time when thromboxane elaboration was substantially reduced in association with 1-BIA treatment, with or without added anti-histamine treatment. In contrast, the reference group without 1-BIA showed substantial thromboxane elaboration, quantitatively similar to that observed with GalTKO or GalTKO.hCD46 lungs in our prior report (15). PVR exhibited a significant biphasic rise, both within the first hour and again towards the end of perfusion, that is typical of thromboxane-mediated pulmonary vascular resistance elevation in endotoxin and other lung injury models. Although platelet and neutrophil adhesion and coagulation cascade activation were not significantly attenuated in any treatment group, and even dually treated lungs exhibited morphologic evidence of graft injury at elective experimental, vascular barrier function (as WDR) was substantially preserved when thromboxane inhibition was combined with either histamine receptor blocker. We conclude that these two innate immune pathways, thromboxane and histamine, contribute significantly to GalTKO.hCD46 lung xenograft injury rejection mechanisms. Targeting them together significantly attenuates PVR elevation and preserves lung vascular barrier function, two physiologically pivotal parameters of xenolung function. These significant improvements in regard to lung function and injury improvement, could also be found in an additional series of experiments, using the same drug treatments (1-BIA and H2-blockade) in transgenic lungs, additionally expressing human decay accelerating factor (hDAF/hCD55) [Supp. Fig. 2A–D].
Thromboxane synthase inhibition does not attenuate histamine release during pig lung perfusion with human blood, importantly demonstrating that other, thromboxane-independent pathways mediate activation of histamine-producing cells in this system. Histamine is elaborated by mast cells, basophils, and other myeloid lineage cells. We hypothesize that pulmonary intravascular macrophages (PIMs) constitute the principle histamine source in this model, based on our prior work (20); a contribution from other monocytes, macrophages, or other cell populations in the lung and human blood is possible. The upstream triggers for thromboxane and histamine elaboration also remain unknown, but may include complement or coagulation factor activation, cytokine elaboration, and platelet, neutrophil, and NK cell interactions that are either physiologic (driven by ‘normal’ activation pathways) or dysregulated due to cross-species molecular incompatibilities.
While anti-inflammatory effects have also been described (40–41), histamine is principally thought of as an important pro-inflammatory mediator. Particularly in anaphylactic, allergic and toxic reactions, histamine can cause hypotension, arrhythmia, asthma, urticaria, pruritus, edema, flushing and other significant symptoms; consequently, prophylaxis or treatment with antihistamines has become routine in current medical practice.
The role of histamine H1- and H2-receptors in the lung has previously been investigated. Various studies suggest that increased lung permeability and vasodilatation are mediated by H2 receptors, while the bronchoconstriction is ascribed primarily to an H1-receptor mechanism (42–47). However, H1-receptors have also been implicated in vasoconstriction in a lung circuit at higher histamine concentrations (47). In line with these effects, we have previously shown that histamine levels increase in the course of GalTKO and GalTKO.hCD46 lung perfusion, and histamine elaboration remained prolific despite thromboxane inhibition in WT pig lung perfusion. In addition, failure of autologously perfused pig lungs during perfusion ex vivo was associated with a significant histamine release (15,48).
Here we show for the first time that, in context of 1-BIA treatment, histamine receptor-driven mechanisms contribute to loss of vascular barrier function during ex vivo lung perfusion, since treatment with either histamine-type-receptor blocker famotidine (H2) or diphenhydramine (H1) attenuated WDR when combined with 1-BIA. While results with added histamine blockade were not significantly different from 1-BIA alone, an additive effect is mechanistically plausible, and appeared to be necessary to demonstrate a significant effect associated with combined treatment. Paradoxically, plasma histamine levels were significantly higher in the combined treatment groups after 4h of perfusion when compared to the untreated reference. This phenomenon could be attributable to the blocking of the histamine binding to the histamine-receptor, leaving more histamine free in the circulation; blockade of an autoregulatory “negative feedback” loop by both H1 and H2 histamine receptor antagonists seems less likely. One outlier in the 1-BIA group showed significantly higher histamine levels at the 240min mark than measured in the other experiments within this group. The average histamine level without the outlier was statistically similar to the control group, confirming that 1-BIA by itself does not increase the histamine level in the blood.
Study limitations: The number of experiments in which GalTKO.hCD46 lungs were treated with 1-BIA/famotidine is too low (n=4) to calculate statistical differences to the other study groups. In an attempt to confirm results obtained in this group, we also treated GalTKO.hCD46 lungs, expressing additional hCD55, with the same drug combination and compared this group with untreated GalTKO.hCD46.hCD55 and GalTKO.hCD46 ± 1-BIA/famotidine experiments. Main results for these experiments are displayed in Supp. Fig. 2A–D. In general, both, the GalTKO.hCD46.hCD55+1-BIA/famotidine treatment group and the untreated reference group show results similar to the ones found with GalTKO.hCD46 alone, supporting our conclusion that the effects of the treatment are biologically significant.
We have not evaluated H1 or H2 receptor antagonists independently (without thromboxane synthase inhibition). Given the expense and complexity of this model, and the high probability that thromboxane effects would dominate in this model if antihistamines were used alone, we could not justify prioritizing study of antihistamine effects alone. The modest marginal effect on vascular permeability with either H1 or H2 antagonism coupled with the strong effects associated with 1-BIA, alone or in combination, guided this experimental choice.
We conclude that thromboxane and histamine play key roles in pulmonary vasoreactivity and vascular permeability during xenogeneic lung rejection. The current study confirms and extends prior observations using wild-type pigs (20), showing that not even the combined genetic modifications tested GalTKO.hCD46 (+/− hCD55 [supp. data]) was sufficient to attenuate thromboxane elaboration or prevent loss of lung vascular barrier function, and that addition of histamine receptor blockade (both H1 or H2) to thromboxane synthase inhibition demonstrated a synergic effect lead to the lowest tissue edema (WDR).
Pharmacologic thromboxane synthase inhibition coupled with histamine receptor blockade is associated with relatively preserved lung physiology, function, and morphology despite evidence that cell adhesive and coagulation pathway dysregulation mechanisms are not significantly inhibited. As practical consequence, we hypothesize that targeting both pathways, (potentially blocking the synthase of histamine [e.g. by alpha-fluoromethyl histidine] instead of the histamine receptor, to avoid high histamine blood levels) will also contribute to extend life-supporting graft survival in in vivo xenogeneic transplant models. Until upstream triggers for thromboxane and histamine release are identified and addressed, preferably by genetic modifications to the pig, pharmacologic targeting of these pathways will facilitate study of other lung xenograft injury mechanisms, and promote progress toward clinical application.
Supplementary Material
(A) Addition of 1-BIA significantly inhibits thromboxane elaboration when compared to the untreated control experiments (# p<0.01 for all 1-BIA groups vs. untreated at 1 and 4h). (B) When 1-BIA was combined with either histamine-receptor blocker, histamine elaboration was significantly higher at the 4h time point when compared to the untreated control (* p<0.01). The addition of 1-BIA alone did not reach significance (p=0.08).
Significant results, found with GalTKO.hCD46 transgenic pig lungs, were confirmed in lungs, additionally expressing hCD55. Treatment, as well as control groups show very similar curves for PVR (A), AWP (B), TXB2 (C) and Histamine (D) whether hCD55 was expressed or not.
Acknowledgements
Grant support / Funding:
NIH U19AI090959
United Therapeutics sponsored research agreement.
List of non-standardized abbreviations:
- AWP
airway pressure
- 1-BIA
1-benzylimidazole
- βTG
β-thromboglobulin
- BW
body weight
- CD62P
P-selectin
- CPDA-1
citrate phosphate dextrose adenine solution
- F1+2
Prothrombin fragments 1 + 2
- GalTKO
α1,3-galactosyl transferase knockout
- hCD39
human ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1)
- hCD46
human membrane cofactor protein, hMCP
- hCD55
human decay-accelerating factor, hDAF
- hCD59
human MAC-inhibitory protein, protectin
- hCPRP
human complement regulatory protein
- hDAF
human decay-accelerating factor, hCD55
- hEPCR
human endothelial protein C receptor, hCD201
- hTBM
human thrombomodulin, hCD141
- PA
pulmonary artery
- PMN
polymorphonuclear leukocyte
- PVR
pulmonary vascular resistance
- WDR
Wet/Dry Ratio
Footnotes
Disclosures:
RNP serves without compensation on Revivicor’s Scientific Advisory Board.
DLA is Executive VP and CSO and a full-time employee of Revivicor, Inc.
CJP is head of xenotransplantation research and development at Revivicor, Inc.
Revivicor, Inc. is a wholly owned subsidiary of United Therapeutics, Inc.
References
- 1.LAMM V, HARA H, MAMMEN A, DHALIWAL D, COOPER DK. Corneal blindness and xenotransplantation. Xenotransplantation. 2014;21(2):99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.KIM MK, CHOI HJ, KWON I, et al. The International Xenotransplantation Association consensus statement on conditions for undertaking clinical trials of xenocorneal transplantation. Xenotransplantation. 2014;21(5):420–30. [DOI] [PubMed] [Google Scholar]
- 3.COOPER DK, SATYANANDA V, EKSER B, et al. Progress in pig-to-non-human primate transplantation models (1998–2013): a comprehensive review of the literature. Xenotransplantation. 2014;21(5):397–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.COOPER DK, HARA H, EZZELARAB M, ET AL. The potential of genetically-engineered pigs in providing an alternative source of organs and cells for transplantation. J Biomed Res. 2013;27(4):249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MATSUMOTO S, TAN P, BAKER J, et al. Clinical porcine islet xenotransplantation under comprehensive regulation. Transplant Proc. 2014;46(6):1992–5. [DOI] [PubMed] [Google Scholar]
- 6.SAMY KP, MARTIN BM, TURGEON NA, KIRK AD. Islet cell xenotransplantation: a serious look toward the clinic. Xenotransplantation. 2014;21(3):221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MOHIUDDIN MM, SINGH AK, CORCORAN PC, et al. One-year heterotopic cardiac xenograft survival in a pig to baboon model. Am J Transplant. 2014;14(2):488–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.COWAN PJ, COOPER DK, D’APICE AJ. Kidney xenotransplantation. Kidney Int. 2014;85(2):265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.BYRNE GW, MCGREGOR CG. Cardiac xenotransplantation: progress and challenges. Curr Opin Organ Transplant. 2012;17(2):148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.KIM K, SCHUETZ C, ELIAS N, et al. Up to 9-day survival and control of thrombocytopenia following alpha1,3-galactosyl transferase knockout swine liver xenotransplantation in baboons. Xenotransplantation. 2012;19(4):256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.YEH H, MACHAIDZE Z, WAMALA I, et al. Increased transfusion-free survival following auxiliary pig liver xenotransplantation. Xenotransplantation. 2014;21(5):454–64. [DOI] [PubMed] [Google Scholar]
- 12.HERING BJ, WALAWALKAR N. Pig-to-nonhuman primate islet xenotransplantation. Transpl Immunol. 2009;21(2):81–6. [DOI] [PubMed] [Google Scholar]
- 13.COWAN PJ, COOPER DK, D’APICE AJ. Kidney xenotransplantation. Kidney Int. 2014;85(2):265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.EZZELARAB MB, EKSER B, AZIMZADEH A, et al. Systemic inflammation in xenograft recipients precedes activation of coagulation. Xenotransplantation. 2014. Sep 11. doi: 10.1111/xen.12133. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.BURDORF L, STODDARD T, ZHANG T, et al. Expression of human CD46 modulates inflammation associated with GalTKO lung xenograft injury. Am J Transplant. 2014;14(5):1084–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MAEDA A, KAWAMURA T, UENO T, USUI N, EGUCHI H, MIYAGAWA S. The suppression of inflammatory macrophage-mediated cytotoxicity and proinflammatory cytokine production by transgenic expression of HLA-E. Transpl Immunol. 2013;29(1–4):76–81. [DOI] [PubMed] [Google Scholar]
- 17.LIN CC, EZZELARAB M, SHAPIRO R, et al. Recipient tissue factor expression is associated with consumptive coagulopathy in pig-to-primate kidney xenotransplantation. Am J Transplant. 2010;10(7):1556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.HARRIS DG, QUINN KJ, FRENCH BM, et al. Meta-analysis of the independent and cumulative effects of multiple genetic modifications on pig lung xenograft performance during ex vivo perfusion with human blood. Xenotransplantation. 2015. Mar-Apr;22(2):102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.HARRIS DG, QUINN KJ, DAHI S, BURDORF L, AZIMZADEH AM, PIERSON RN 3rd. Lung xenotransplantation: recent progress and current status. Xenotransplantation. 2014;21(6):496–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.COLLINS BJ, BLUM MG, PARKER RE, et al. Thromboxane mediates pulmonary hypertension and lung inflammation during hyperacute lung rejection. J Appl Physiol (1985). 2001;90(6):2257–68. [DOI] [PubMed] [Google Scholar]
- 21.PIERSON RN 3RD, PARKER RE. Thromboxane mediates pulmonary vasoconstriction and contributes to cytotoxicity in pig lungs perfused with fresh human blood. Transplant Proc. 1996;28(2):625. [PubMed] [Google Scholar]
- 22.SELIG WM, PATTERSON CE, HENRY DP, RHOADES RA. Role of histamine in acute oleic acid-induced lung injury. J Appl Physiol (1985). 1986;61(1):233–9. [DOI] [PubMed] [Google Scholar]
- 23.MEYRICK B, BRIGHAM KL. Increased permeability associated with dilatation of endothelial cell junctions caused by histamine in intimal explants from bovine pulmonary artery. Exp Lung Res. 1984;6(1):11–25. [DOI] [PubMed] [Google Scholar]
- 24.SIELAFF TD, SUGERMAN HJ, TATUM JL, BLOCHER CR. Successful treatment of adult respiratory distress syndrome by histamine and prostaglandin blockade in a porcine Pseudomonas model. Surgery. 1987;102(2):350–7. [PubMed] [Google Scholar]
- 25.MANIATIS NA, KOTANIDOU A, CATRAVAS JD, ORFANOS SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49(4–6):119–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.KIM TH, YOON HJ, LIM CM, KIM EK, KIM MJ, KOH Y. The role of endogenous histamine on the pathogenesis of the lipopolysaccharide (LPS)-induced, acute lung injury: a pilot study. Inflammation. 2005;29(2–3):72–80. [DOI] [PubMed] [Google Scholar]
- 27.HIROSE T, AOKI E, DOMAE M, ISHIBASHI M, IKEDA T, TANAKA K. Protective effect of a thromboxane synthetase inhibitor, OKY-1581, on increased lung vascular permeability in pulmonary microembolization in dogs. Prostaglandins Leukot Med. 1983;10(2):187–96. [DOI] [PubMed] [Google Scholar]
- 28.ISHITSUKA Y, MORIUCHIH, ISOHAMA Y, at al. A selective thromboxane A2 (TXA2) synthase inhibitor, ozagrel, attenuates lung injury and decreases monocyte chemoattractant protein-1 and interleukin-8 mRNA expression in oleic acid-induced lung injury in guinea pigs. J Pharmacol Sci. 2009;111(2):211–5. [DOI] [PubMed] [Google Scholar]
- 29.BURDORF, RINER A, RYBAK E, et al. Glycoproteins Ib and IIb/IIIa, and vWF modulate platelet sequestration and activation during GalTKO.hCD46 pig lung perfusion by human blood. Xenotransplantation. 2016. May;23(3):222–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.SCHROEDER C, ALLAN JS, NGUYEN BN, et al. Hyperacute rejection is attenuated in GalT knockout swine lungs perfused ex vivo with human blood. Transplant Proc. 2005;37(1):512–3. [DOI] [PubMed] [Google Scholar]
- 31.NGUYEN BN, AZIMZADEH AM, SCHROEDER C, et al. Absence of Gal epitope prolongs survival of swine lungs in an ex vivo model of hyperacute rejection. Xenotransplantation. 2011;18(2):94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.BURDORF L, AZIMZADEH AM, PIERSON RN 3RD. Xenogeneic lung transplantation models. Methods Mol Biol. 2012;885:169–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.HISASHI Y, YAMADA K, KUWAKI K, et al. Rejection of cardiac xenografts transplanted from alpha1,3-galactosyltransferase gene-knockout (GalT-KO) pigs to baboons. Am J Transplant. 2008;8(12):2516–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.COOPER DK, EZZELARAB M, HARA H, AYARES D. Recent advances in pig-to-human organ and cell transplantation. Expert Opin Biol Ther. 2008;8(1):1–4. [DOI] [PubMed] [Google Scholar]
- 35.WESTALL GP, LEVVEY BJ, SALVARIS E, et al. Sustained function of genetically modified porcine lungs in an ex vivo model of pulmonary xenotransplantation. J Heart Lung Transplant. 2013;32(11):1123–30. [DOI] [PubMed] [Google Scholar]
- 36.MCGREGOR CG, RICCI D, MIYAGI N, et al. Human CD55 expression blocks hyperacute rejection and restricts complement activation in Gal knockout cardiac xenografts. Transplantation. 2012;93(7):686–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MOHIUDDIN MM, SINGH AK, CORCORAN PC, et al. Genetically engineered pigs and target-specific immunomodulation provide significant graft survival and hope for clinical cardiac xenotransplantation. J Thorac Cardiovasc Surg. 2014;148(3):1106–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.IWASE H, EKSER B, HARA H, et al. Regulation of human platelet aggregation by genetically modified pig endothelial cells and thrombin inhibition. Xenotransplantation. 2014;21(1):72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.HARRIS DG, BENIPAL PK, GAO Z, et al. Activated Protein C decreases thrombosis on porcine endothelium transgenic for human endothelial protein C receptor - a novel mechanism to decrease porcine xenograft injury. Journal of Surgical Research 2014;186(2):579 [Google Scholar]
- 40.ØSTERUD B, OLSEN JO. Pro- and anti-inflammatory effects of histamine on tissue factor and TNFα expression in monocytes of human blood. Thromb Res. 2014;133(3):477–80. [DOI] [PubMed] [Google Scholar]
- 41.BHATTACHARYA SK, DAS N. Anti-inflammatory effect of intraventricularly administered histamine in rats. Agents Actions. 1985;17(2):150–2. [DOI] [PubMed] [Google Scholar]
- 42.SCHULMAN ES, LIU MC, PROUD D, MACGLASHAN DW JR, LICHTENSTEIN LM, PLAUT M. Human lung macrophages induce histamine release from basophils and mast cells. Am Rev Respir Dis. 1985;131(2):230–5. [DOI] [PubMed] [Google Scholar]
- 43.NOMURA H, SATO E, KOYAMA S, et al. Histamine stimulates alveolar macrophages to release neutrophil and monocyte chemotactic activity. J Lab Clin Med. 2001. Oct;138(4):226–35. [DOI] [PubMed] [Google Scholar]
- 44.BRAUDE S, ROYSTON D, COE C, BARNES PJ. Histamine increases lung permeability by an H2-receptor mechanism. Lancet. 1984;2(8399):372–4. [DOI] [PubMed] [Google Scholar]
- 45.BARNES PJ. Histamine receptors in the lung. Agents Actions Suppl. 1991;33:103–22. [DOI] [PubMed] [Google Scholar]
- 46.RUSSELL PC, WRIGHT CE, BARER GR, HOWARD P. Histamine induced pulmonary vasodilatation in the rat: site of action and changes in chronic hypoxia. Eur Respir J. 1994;7(6):1138–44. [PubMed] [Google Scholar]
- 47.BARER GR, EMERY CJ, MOHAMMED FH, MUNGALL IP. H1 and H2 histamine actions on lung vessels; their relevance to hypoxic vasoconstriction. Q J Exp Physiol Cogn Med Sci. 1978;63(2):157–69. [DOI] [PubMed] [Google Scholar]
- 48.AZIMZADEH A, ZORN GL 3RD, BLAIR KS, et al. Hyperacute lung rejection in the pig-to-human model. 2. Synergy between soluble and membrane complement inhibition. Xenotransplantation. 2003;10(2):120–31. [DOI] [PubMed] [Google Scholar]
- 49.BRIGHAM KL, OGLETREE M, SNAPPER J, HINSON J, PARKER RE. Prostaglandins and lung injury. Chest. 1983;83(5 Suppl):70S-72S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Addition of 1-BIA significantly inhibits thromboxane elaboration when compared to the untreated control experiments (# p<0.01 for all 1-BIA groups vs. untreated at 1 and 4h). (B) When 1-BIA was combined with either histamine-receptor blocker, histamine elaboration was significantly higher at the 4h time point when compared to the untreated control (* p<0.01). The addition of 1-BIA alone did not reach significance (p=0.08).
Significant results, found with GalTKO.hCD46 transgenic pig lungs, were confirmed in lungs, additionally expressing hCD55. Treatment, as well as control groups show very similar curves for PVR (A), AWP (B), TXB2 (C) and Histamine (D) whether hCD55 was expressed or not.