

Abstract
The RREB1::MRTFB (former RREB1::MKL2) fusion characterizes ectomesenchymal chondromyxoid tumors of the tongue (EMCMT). Only five molecularly confirmed extra-glossal EMCMT cases have been reported recently; all occurring at head and neck or mediastinal sites. We herein describe five new cases including the first two extracranial/extrathoracic cases. The tumors occurred in three male and two female patients with an age ranging from 18 to 61 years (median, 28). Three tumors were located in the head and neck (jaw, parapharyngeal space and nasopharyngeal wall) and two in the soft tissue (inguinal and presacral). The tumor size ranged from 3.3 to 20 cm (median, 7). Treatment was surgical without adjuvant treatment in all cases. Two cases were disease-free at 5 and 17 months; other cases were lost to follow-up. Histologically, the soft tissue cases shared a predominant fibromyxoid appearance, but with variable cyto-architectural pattern (cellular perineurioma-like whorls and storiform pattern in one case and large polygonal granular cells embedded within a chondromyxoid stroma in the other). Two tumors (inguinal and parapharyngeal) showed spindled to ovoid and round cells with a moderately to highly cellular nondescript pattern. One sinonasal tumor closely mimicked nasal chondromesenchymal hamartoma (NCMH). Mitotic activity was low (0 to 5 mitoses/10 hpfs). Immunohistochemical findings were heterogeneous with variable expression of S100 (2/5), EMA (2/3), CD34 (1/4), desmin (1/4) and GFAP (1/3). Targeted RNA sequencing revealed the same RREB1::MRTFB fusion in all cases, with exon 8 of RREB1 being fused to exon 11 of MRTFB. This study expands the topographic spectrum of RREB1::MRTFB fusion-positive mesenchymal neoplasms, highlighting a significant morphological and phenotypic diversity. Overall, RREB1::MRTFB-rearranged neoplasms seem to fall into two subcategories: Tumors with lobulated, chondroid or myxochondroid epithelioid morphology (Case 2 and 3) and those with more undifferentiated hypercellular spindle cell phenotype (Case 1, 4, 5). Involvement of extracranial/extrathoracic sites and the NCMH-like pattern are novel. The biology of these likely indolent or benign tumors remains to be verified in the future.
Keywords: RREB1, MRTFB, MKL2, fibromyxoid, perineurioma, sarcoma, soft tissue
INTRODUCTION
Ectomesenchymal chondromyxoid tumor (EMCMT) is a rare, histologically distinctive, site-specific mesenchymal neoplasm of unknown histogenetic origin. Since its first description by Smith et al in 1995 [1], the histogenesis of this tumor has been the subject of ongoing debate. Likewise, its significant predilection (almost exclusive) for the anterior dorsal tongue remains obscure [1]. Smith et al have coined the term “ectomesenchymal chondromyxoid tumor”, based on the assumption that this enigmatic lesion originates from misplaced uncommitted neural crest-derived ectomesenchymal cells [1]. Alternatively, a myoepithelial origin (salivary gland-derived or soft tissue origin) has been proposed by some authors, but rejected by others [2, 3].
To date, no more than 100 cases of EMCMT have been reported in the literature (reviewed in ref. [4, 5, 6]). The neoplastic cells of EMCMT uniformly express vimentin, GFAP and pankeratin with frequent but variable reactivity with S100, smooth muscle actin, p63, and CD57, while desmin, myogenin, synaptophysin and calponin are less commonly expressed, usually with limited reactivity [1, 7, 8].
Recently, Dickson et al reported recurrent RREB1::MRTFB (formerly RREB1::MLK2) fusions in EMCMT, detected in 90% of cases using targeted RNA sequencing [8]. In contrast, rare cases harbored EWSR1 rearrangements [8, 9].
While the majority of tumors originate on the anterior dorsum of the tongue, few cases deviated from this and originated from the posterior part or other lateral borders of the tongue instead [1, 8]. Extra-glossal examples of EMCMT are exceedingly rare [10–16] and only five of them have been confirmed by genotyping (Table 1; [13–16]). We herein describe five new tumors carrying this gene fusion including the first two cases originating from extracranial/ extrathoracic soft tissue sites.
Table 1:
Clinicopathological features of previously reported and current RREB1::MRTFB fusion positive extra-glossal neoplasms.
| No | Case/ reference | Age/sex | Site | Size cm | Treatment | Outcome (duration) |
|---|---|---|---|---|---|---|
| 1 | Siegfried et al (2018) [13] | 53/M | Retro-/parapharyngeal | 3.5 | Surgery + CRT | NED (10 mo) |
| 2 | Makise et al (2020) [14] | 25/F | Neck/ superior mediastinum left | 5.6 | Surgery (piece-by-piece) | NED (27 mo) |
| 3 | Makise et al (2020) [14] | 73/F | Superior mediastinum left | 4.3 | Surgery (piece-by-piece) | NED (18 mo) |
| 4 | Bubola et al (2020) [15] | 37/F | Body of mandible right | 3 | Surgery | NA |
| 5 | Mechtersheimer et al (2021) [16] | 73/F | Posterior middle nasal turbinate right | 3.5 | Surgery | NA |
| 6 | Current Case 1 | 61/M | Inguinal/proximal thigh left | 8 | Surgery | NED (17 mo) |
| 7 | Current Case 2 | 36/F | Presacral region | 20 | Surgery | Lost to follow-up |
| 8 | Current Case 3 | 28/F | Jaw (follicular cyst?) | NA | Surgery | Lost to follow-up |
| 9 | Current Case 4 | 28/M | Parapharyngeal space | 6 | Surgery | NED (5 mo) |
| 10 | Current Case 5 | 18/M | Posterior nasopharyngeal wall | 3.3 | Surgery | Very recent case |
CRT=chemoradiotherapy; mo=months; NA=not available; NED=no evidence of disease.
MATERIALS AND METHODS
Case 1 was a routine case treated at the University Hospital, Erlangen, Germany. Cases 2 to 5 were identified in the consultation files of the authors (AA, CRA). The tissue specimens were fixed in formalin and processed routinely for histopathology. Due to the consultation nature of the cases, immunohistochemistry (IHC) was performed in different laboratories and the stains applied varied from case to case, based on tissue availability and the initial differential diagnostic considerations (details of the staining protocols and antibody sources are available upon request).
Next generation sequencing
For Cases 1 and 3 – 4, RNA was isolated from formalin-fixed paraffin embedded (FFPE) tissue sections using RNeasy FFPE Kit of Qiagen (Hilden, Germany) and quantified spectrophotometrically using NanoDrop-1000 (Waltham, United States). Molecular analysis was performed using the TruSight RNA Fusion Panel (Illumina, Inc., San Diego, CA, USA) with 500 ng RNA as input according to the manufacturer`s protocol. The library was sequenced on a MiSeq (Illumina, Inc., San Diego, CA, USA) with 3.4 million reads, and sequences were analyzed using the RNA-Seq Alignment workflow, version 2.0.1 (Illumina, Inc., San Diego, CA, USA). The Integrative Genomics Viewer (IGV), version 2.2.13 (Broad Institute, REF) was used for data visualization [17].
Cases 2 and 5 were subjected to targeted RNA sequencing (Archer FusionPlex Custom Solid Panel) to assess for gene fusions. The detailed procedure of Anchored Multiplex PCR RNA sequencing assay has been previously described [18, 19]. Unidirectional gene-specific primers were designed to target specific exons in 123 genes known to be involved in oncogenic fusions in solid tumors.
Fluorescence in-situ hybridization (FISH) on interphase nuclei from paraffin-embedded 4-micron sections was performed applying custom bacterial artificial chromosomes (BAC) probes covering and flanking genes of interest (RREB1 and MRTFB) as previously described [8]. DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution, as previously described [20]. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems, Newton, MA). A positive score was interpreted when at least 20% of the nuclei showed a split-apart signal in the break-apart assay. Nuclei with incomplete set of signals were omitted from the score.
Based on morphological similarity to Case 3, we tested two additional nasal chondromesenchymal hamartoma (NCMH)-like tumors for fusions using same panel (one case) and the Archer FusionPanel (for the other case). Moreover, all three NCMH-like tumors were tested for the presence of DICER1 mutations as described previously [21].
RESULTS
Clinical and demographic features
The tumors occurred in three male and two female patients whose age ranged from 18 to 61 years (median, 28) (Table 1). Three tumors were located in the head and neck (one case each in the maxilla, the parapharyngeal space and the posterior nasopharyngeal wall) and two in the soft tissue (one in the right inguinal area and one in the presacral region (representative imaging is shown in Fig. 1A–C)). The tumor size ranged from 3.3 to 20 cm (median, 7). The largest tumor (Case 2) originated in the presacral soft tissue mass abutting but not involving the bone that was resected. Treatment was radical surgical excision in Case 1 and 2, and variable local marginal or nonradical surgical modalities in Case 3 to 5. No adjuvant treatment was given to any of the patients. Case 1 and 4 were disease-free at 17 and 5 months follow-up, respectively; other cases were either recent or have been lost to follow-up.
FIGURE 1.
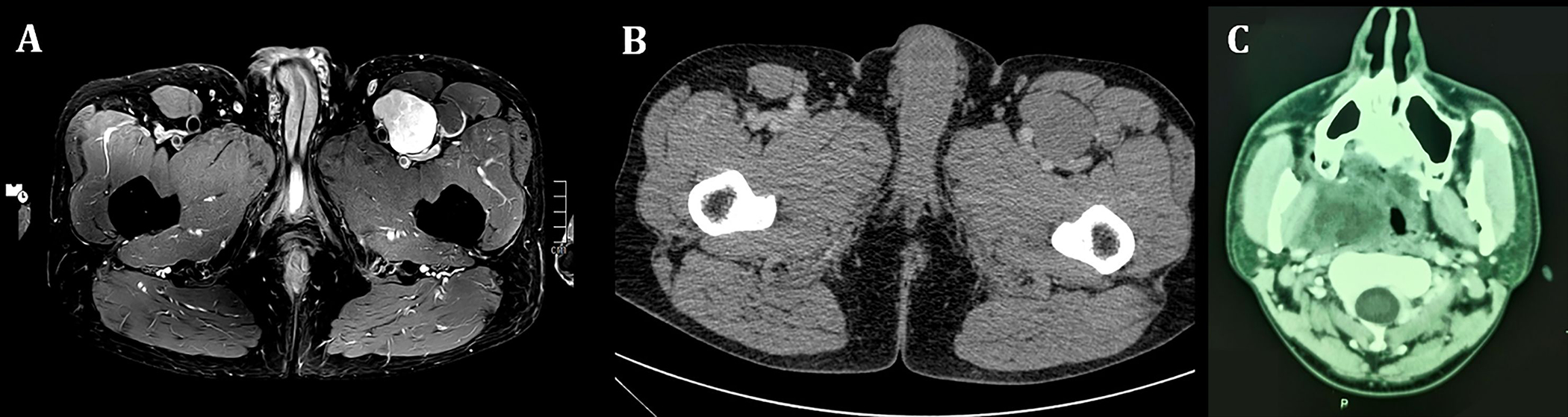
Representative radiological images of extra-glossal RREB1::MRTFB-positive neoplasms. A (magnetic resonance imaging of Case 1): A PDW TSE SPAIR axial image shows a well circumscribed, heterogeneously hyperintense mass in the left anterior thigh with contact to the femoral artery and vein (PDW=proton density-weighted; TSE=Turbo Spin Echo; SPAIR=SPectral Attenuated Inversion Recovery). B: Computed tomography (CT) of the pelvis (same Case) shows an isodense soft tissue mass with sharp margins and contact to the femoral artery and vein as well as to the sartorius and the rectus femoris. C: Axial post-contrast CT through the level of the maxillary sinus in Case 4. An ill-defined, inhomogeneous, partly cystic soft tissue mass in the right masticator space with lateral displacement of the medial pterygoid muscle, medial displacement of the pharynx, and contact to the carotid space.
Pathological findings
All tumors were well circumscribed but non-encapsulated. Their cut-surfaces were described as firm to soft with variable lobulation. Case 1 was diagnosed as unclassified low-grade fibromyxoid neoplasm on initial core needle biopsies (based on non-descript morphology and negative limited immunostains) and primary surgical resection recommended. Other cases were diagnosed initially as low-grade unclassified mesenchymal neoplasms using varying descriptive terms based on immunophenotypes (Cases 2, 4 and 5) and nasal chondromesenchymal hamartoma (Case 3).
Histologically, the soft tissue tumors (Case 1 and 2) shared a predominant fibromyxoid appearance, but with varying cyto-architectural patterns. Case 1 presented as a vaguely lobulated cellular neoplasm composed of slender to focally plump, bland spindle cells arranged in prominent whorls, short fascicles and a storiform pattern with frequent perivascular onionskin pattern (Fig. 2A, B). Focal myxoid stromal change, thickened vessels surrounded by concentric neoplastic cells (Fig. 2C, D), focal rhythmic arrangement of the nuclei alternating with paucicellular trabeculae (Fig. 2E, F), and large foci of ischemic type necrosis were present.
FIGURE 2.
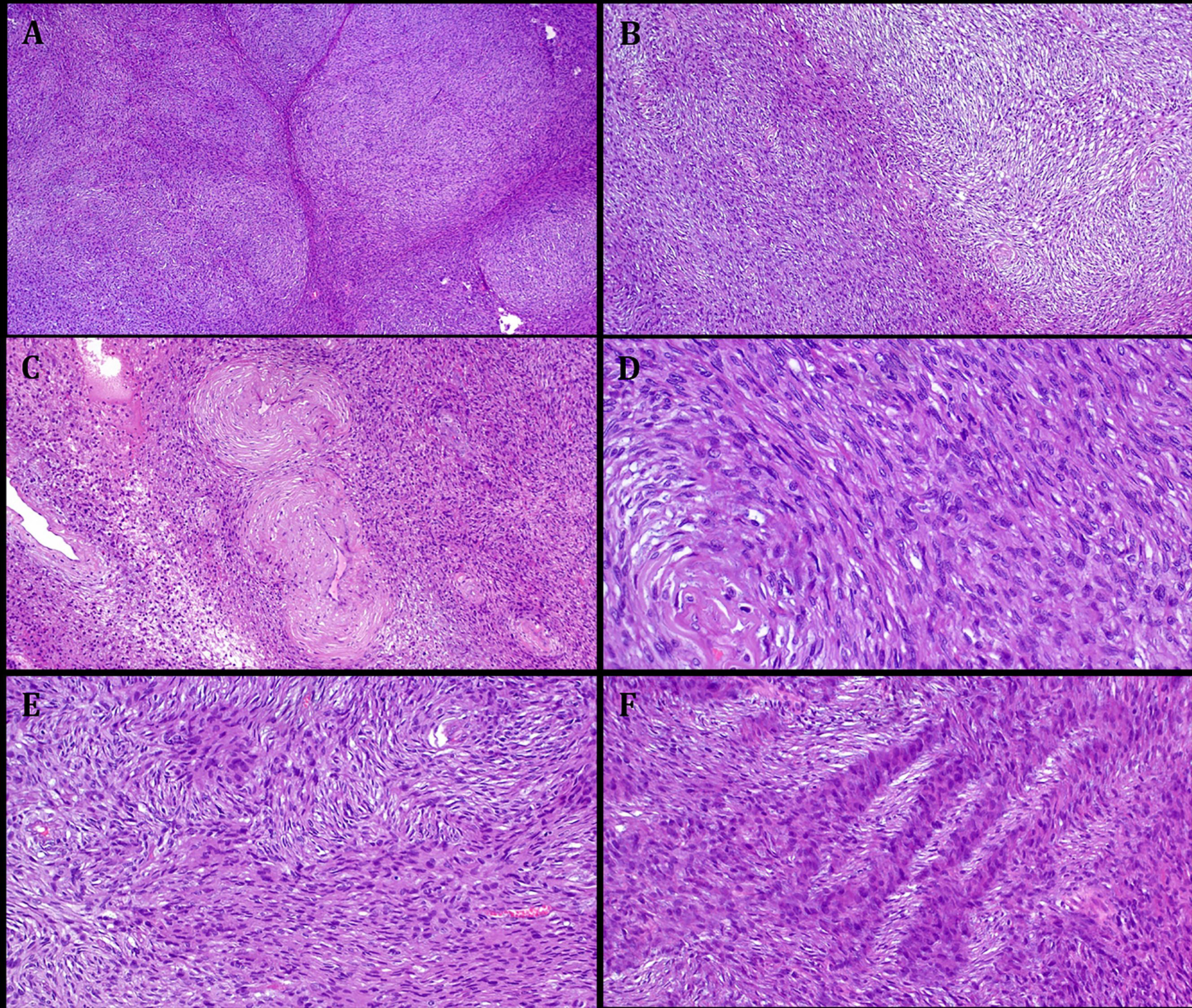
Representative images illustrating the histological features in Case 1. A: at low power, a cellular lobulated neoplasm is seen. B: abrupt transition from myxoid to non-myxoid cellular areas. C: thickened vessels surrounded by concentric neoplastic cells. D: high power shows elongated slender spindle cells, note perineurioma-like perivascular whorls. E: prominent storiform pattern with pale stained and moderately eosinophilic alternating fascicles cut transversally. F: distinct alternation of pale stained and moderately eosinophilic fascicles cut longitudinally.
At low power, Case 2 showed variable lobulation with several small lobules embedded within a chondromyxoid background stroma (Fig. 3A–C). The neoplastic cells were predominantly plump ovoid to medium-sized to large epithelioid cells with well demarcated, variably vacuolated (Fig. 3D) or pale granular cytoplasm (Fig. 3E). Numerous intranuclear pseudoinclusions were noted (Fig. 3E). A variable spindle cell component was seen, occasionally with prominent degenerative cytological atypia (Fig. 3F).
FIGURE 3.
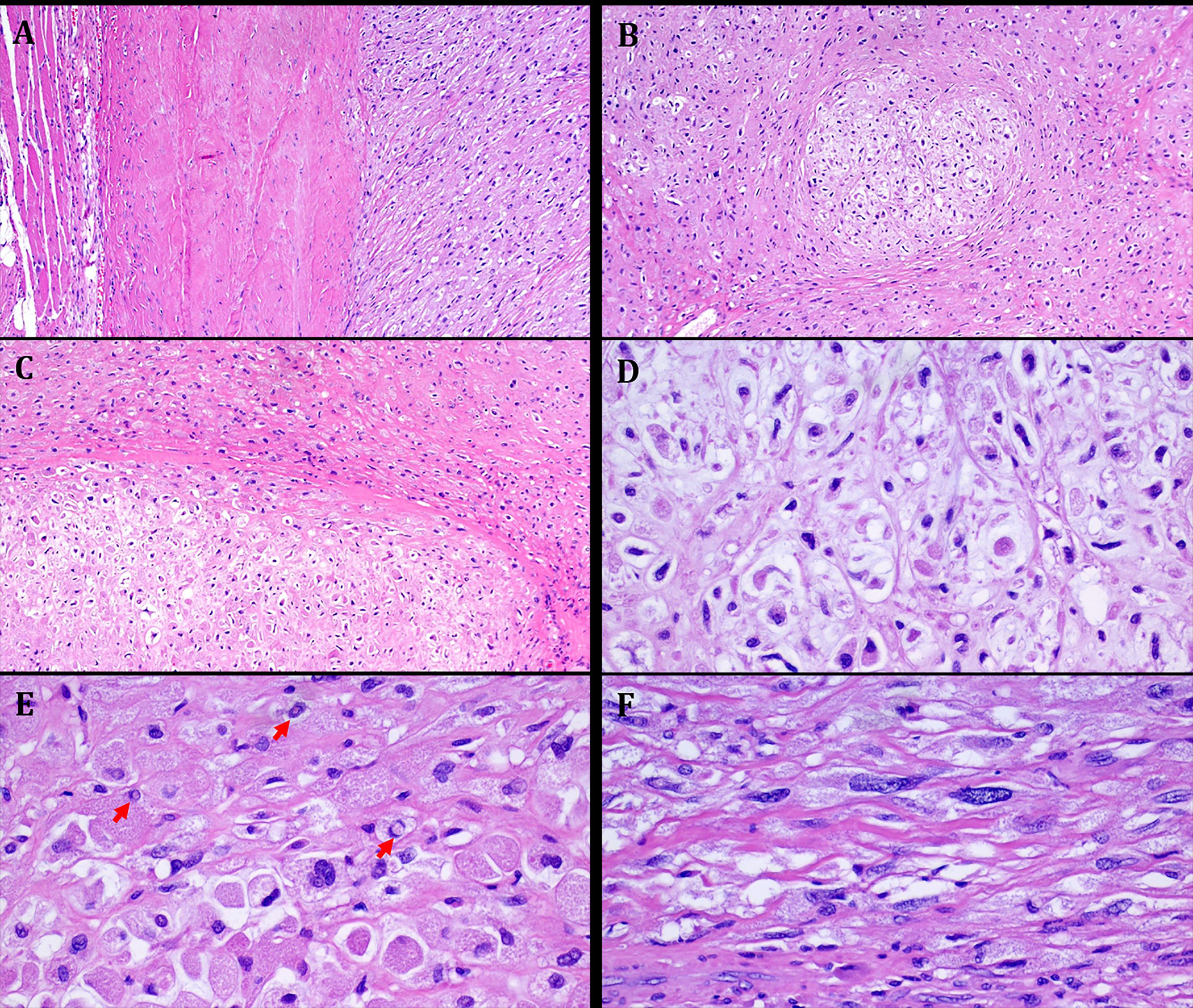
Representative images illustrating the histological features in Case 2. A. well-circumscribed tumor periphery surrounded by fascia and muscle on left. B: at low power, chondromyxoid lobules are seen embedded within hyaline variably sclerosed stroma. C: higher magnification showing abrupt transition between chondromyxoid and hyaline areas. D: admixture of large epithelioid variably vacuolated tumor cells and scattered spindle cells seen at high power. E: prominent eosinophilic and granular cytoplasmic quality, note numerous nuclear pseudoinclusions (arrows). F: scattered spindle cells with hyperchromatic atypical looking nuclei.
Case 3 was strikingly similar to NCMH with multiple smaller lobules of primitive chondromyxoid tissue entrapping mature bone trabeculae and scattered foci of fat with a frequent concentric arrangement of the myxoid nodules (Fig. 4). Case 4 was highly cellular composed of a nondescript arrangement of spindled to ovoid and round cells with variable fascicular to storiform patterns of growth, pericytoma-like vasculature and prominent cystic features (Fig. 5). Case 5 showed diffusely confluent nests and sheets of monomorphic ovoid and focally spindled to fusiform cells with variable pseudofollicular spaces containing pale stained oedematous vacuolated secretory material (Fig. 6). Focal gaping vessels were noted in the spindle cell areas. All cases lacked coagulative necrosis and significant atypia. Mitotic activity was low, ranging from 0 to 5 mitoses/10 hpfs.
FIGURE 4.
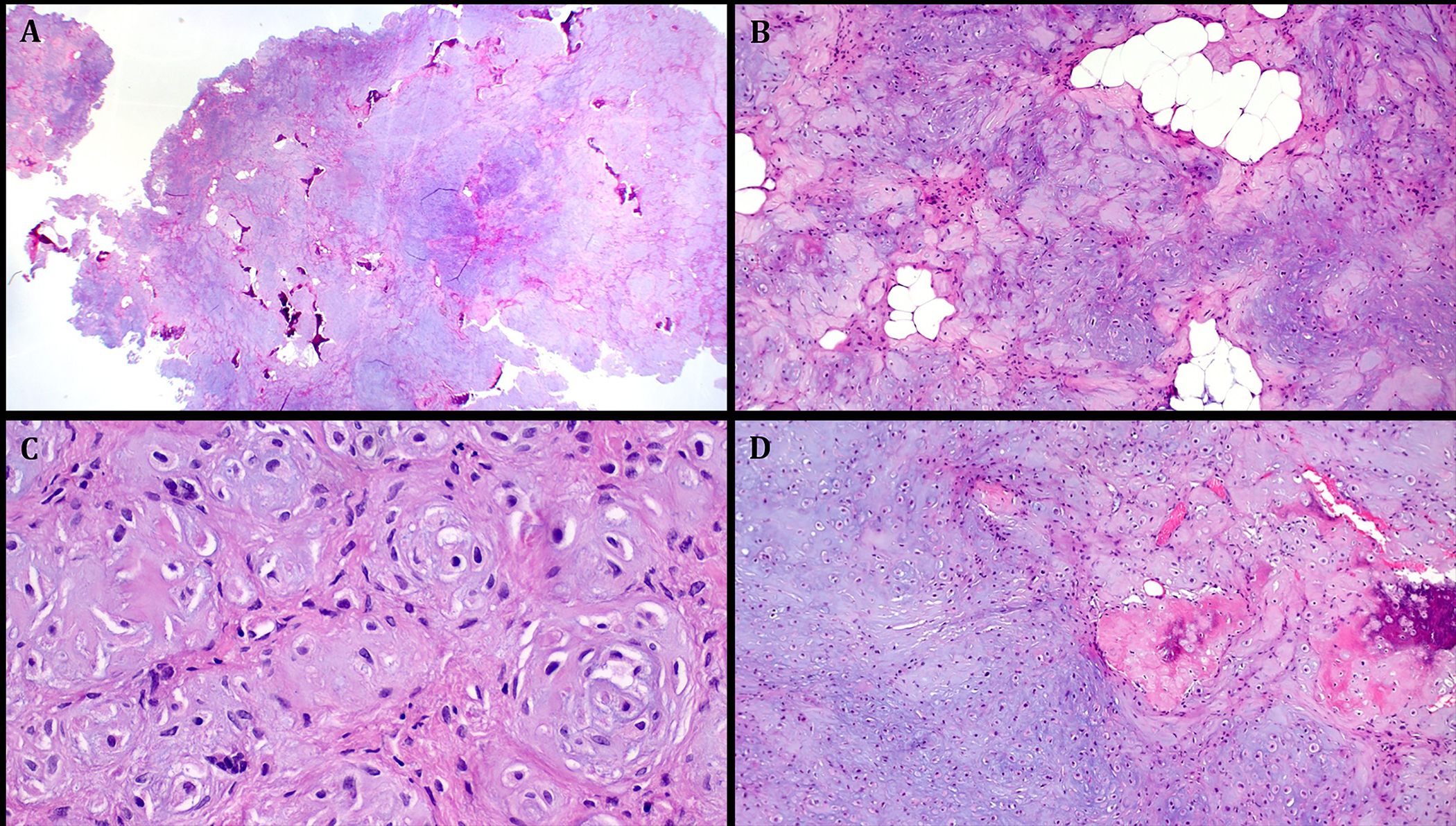
Representative images illustrating the histological features in Case 3. A: large partially polypoid chondromyxoid mass with scattered bone islands seen at low power. B: prominent microlobulation with islands of mature fat. C: higher magnification showing concentric arrangements of mcirolobules with intervening primitive fibroblastic-like spindled mesenchymal cells. D: Focus of bone merging with immature chondromyxoid mesenchymal tissue.
FIGURE 5.
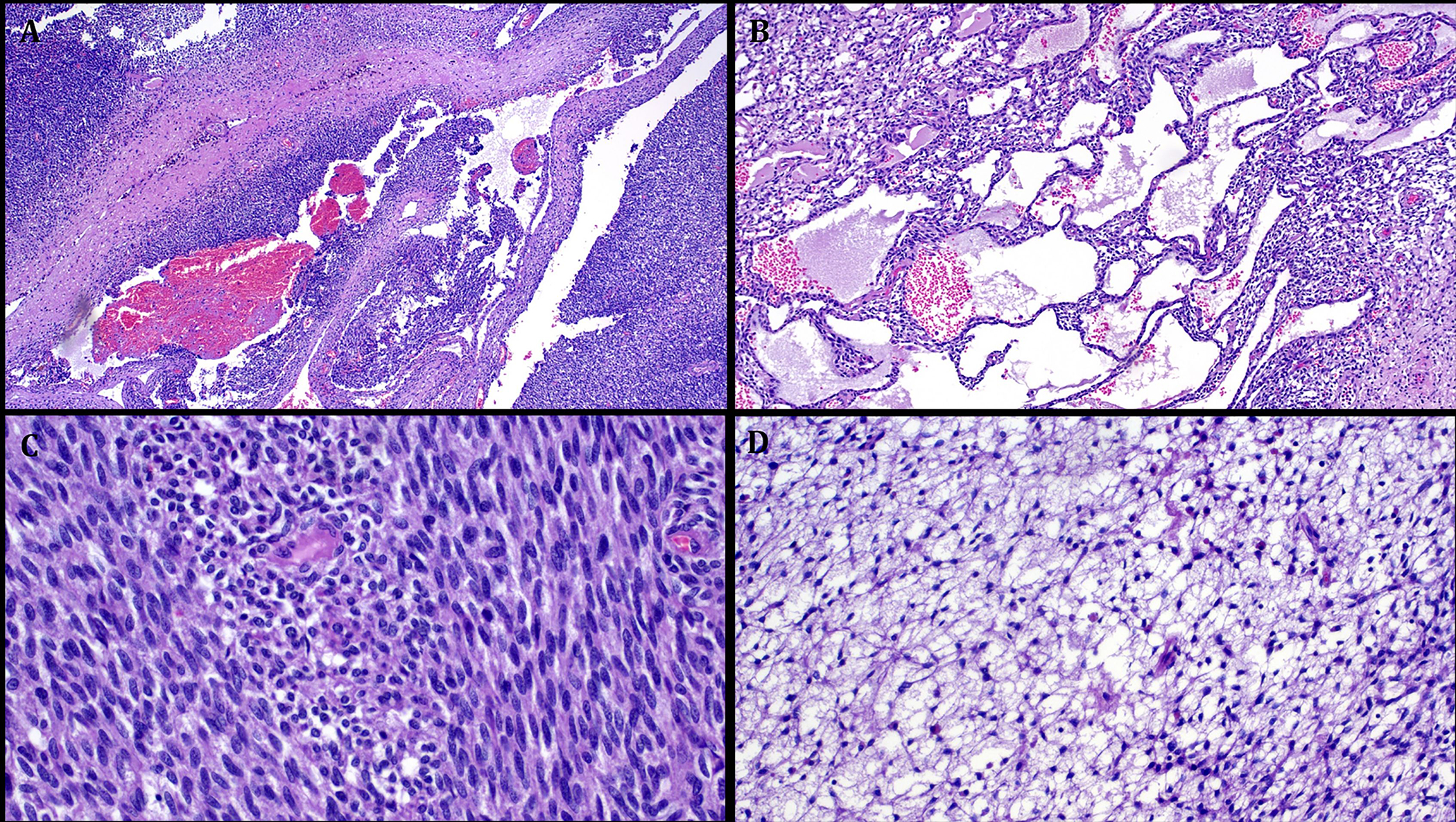
Representative images illustrating the histological features in Case 4. A: low power view showing highly cellular neoplasm with alternating solid and cystic areas. B: prominent macrocystic stromal degeneration. C: highly cellular fibrosarcoma-like areas (with little to no mitoses). D: prominent primitive reticular-myxoid stromal features reminiscent of EMCMT of tongue.
FIGURE 6.
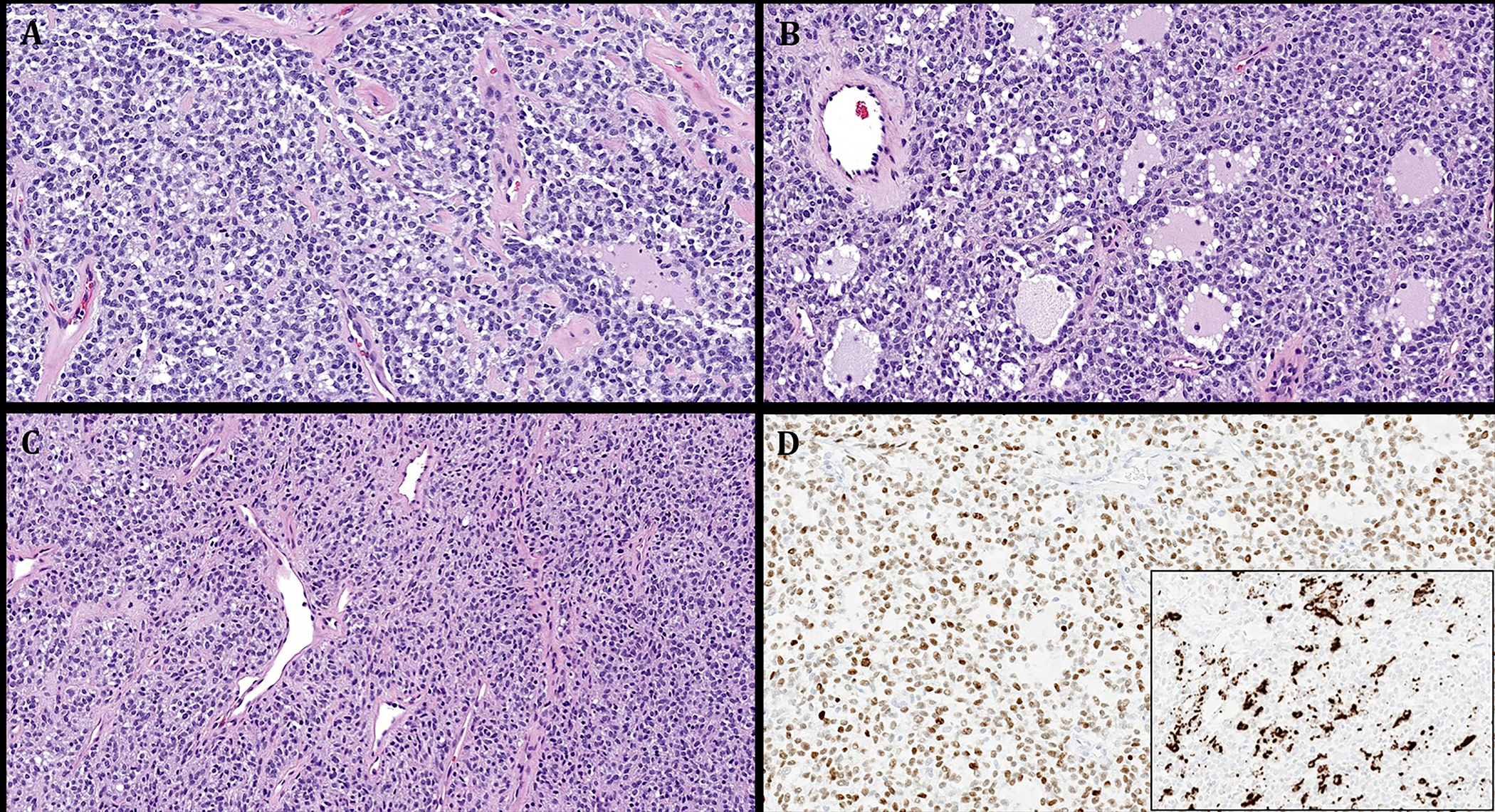
Representative images illustrating the histological features in Case 5. A: monotonous round to ovoid bland cells arranged into non-descript confluent sheets. B: prominent pseudofollicular spaces are seen. C: cellular spindle cell areas with thin-walled gaping vessels. D: main image: diffuse expression of MyoD1. Subimage: variable expression of desmin.
Immunohistochemical finings
Immunohistochemical findings were overall heterogeneous with variable expression of S100 (2/5), EMA (2/3), CD34 (1/4), synaptophysin (1/1), desmin (1/4), MyoD1 (1/2), myogenin (1/2) and GFAP (1/3).
Case 1 revealed prominent but variable expression of CD34 (diffuse in some lobules and negative or variable in others, Fig. 7A, B). EMA was expressed in <5% of the neoplastic cells (Fig. 7C). Very few cells expressed Claudin1. Case 2 showed diffuse strong expression of S100 (Fig. 7D) as well as CD68 and EMA (Fig. 7E) in rare cells. Moreover, Case 4 revealed strong and diffuse expression of synaptophysin (Fig. 7F). Case 3 strongly expressed S100 and weakly SATB2. Remarkably, the nasopharyngeal wall tumor (Case 5) revealed prominent expression of MyoD1 (Fig. 6D, main image) and focal reactivity for desmin (Fig. 6D, subimage) and myogenin, indicating partial rhabdomyoblastic differentiation. Other markers as listed in Table 2 (including SOX10) tested negative.
FIGURE 7.
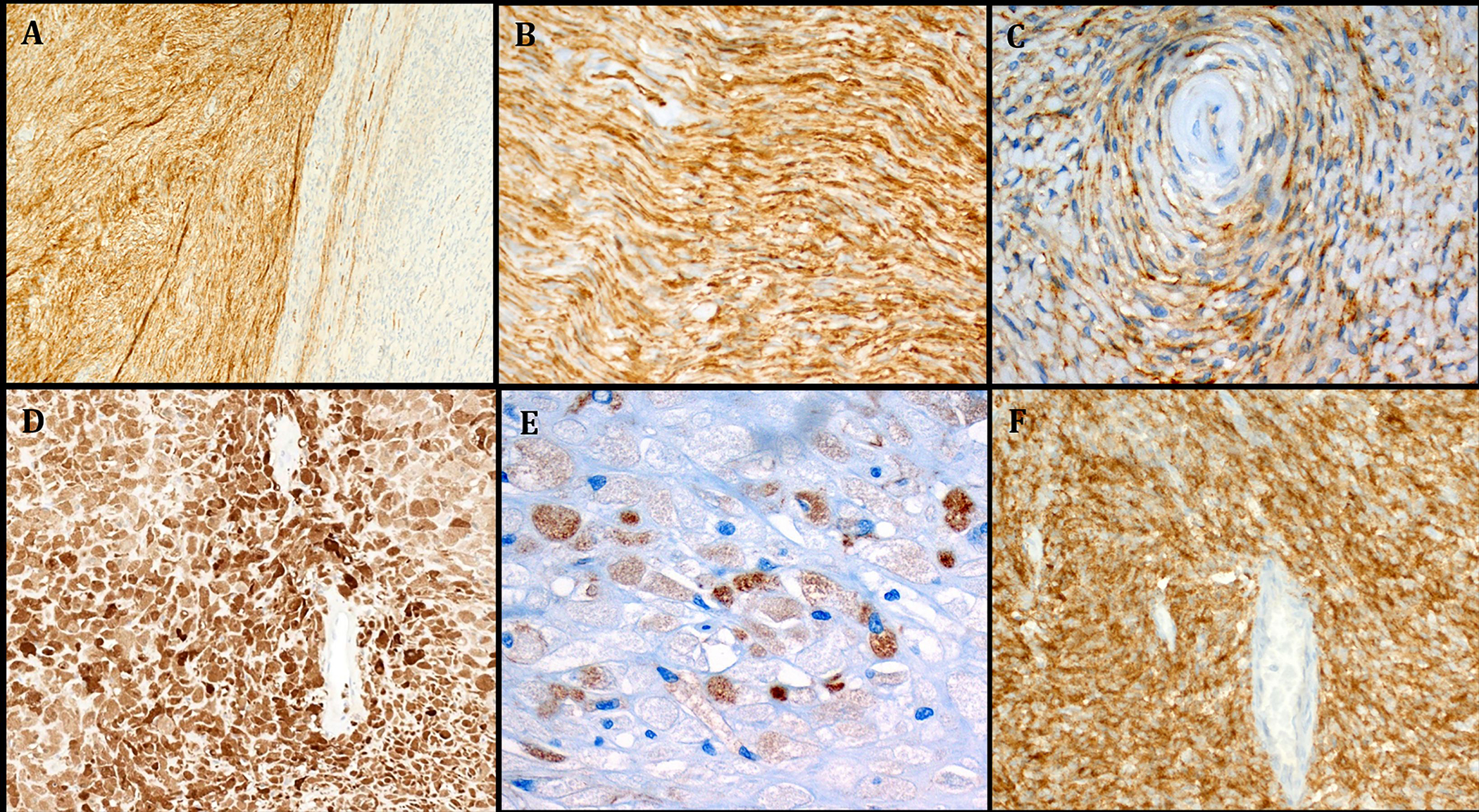
A: Immunohistochemistry showed strong expression of CD34 in some but not all lobules in Case 1. B: higher magnification of A highlighting the perineurioma-like CD34 pattern. C: focal EMA expression in the perivascular whorls (same case). D: Case 2 showed strong and diffuse S100 expression. E: scattered tumor cells stained for EMA (Case 2). F: Case 4 showed diffuse and strong expression of synaptophysin.
Table 2:
Immunohistochemical and molecular findings in RREB1::MRTFB fusion positive extra-glossal neoplasms
| No | Case/reference | Reported as | Histology patterns | Mitoses/10 hpfs | Positive IHC | Negative IHC | Fusion findings |
|---|---|---|---|---|---|---|---|
| 1 | Siegfried et al, 2018 [13] | Biphenotypic sarcoma | Monomorphic spindle cells fibrosarcoma-like | 4 | SMA, S100, desmin, myogenin | h-caldesmon, SOX10, EMA, p63, MUC4, AE1/AE3, CD34, STAT6, MDM2 | RREB1 exon 8/ MRTFB exon 11 |
| 2 | Makise et al, 2020 [14] | Unclassified/extra-glossal EMCMT | Round > > spindle | <1 | S100, GFAP, SMA, EMA, AE1/3, ER | Desmin, myogenin, SOX10, HMB45, MUC4, STAT6, CD34, BCOR, NKX2–2 | RREB1 exon 8/ MRTFB exon 11 |
| 3 | Makise et al, 2020 [14] | Unclassified/extra-glossal EMCMT | Spindle >> round > myxoid | <1 | S100, GFAP, SMA, EMA, AE1/3, pan-TRK, ER | AE1/AE3, desmin, myogenin, CD34, STAT-6, MUC4, SOX10, GLUT1, claudin-1, MDM2, DOG1, SSTR2a, ALK | RREB1 exon 8/ MRTFB exon 11 |
| 4 | Bubola et al (2020) [15] | EMCMT | EMCMT | 0 | SMA, S100, CD56, desmin, GFAP | SOX10, keratin (AE1/ AE3), chromogranin, calponin | RREB1 exon 8/ MRTFB exon 11 |
| 5 | Mechtersheimer et al, 2021 [16] | Biphenotypic sarcoma | Monomorphic spindle cells fibrosarcoma-like | 1 | SMA, S100, EMA, CD34 | Desmin, myogenin, GFAP, STAT6, AE1/AE3 | RREB1 exon 8/ MRTFB exon 11 |
| 6 | Current Case 1 | Unclassified fibromyxoid neoplasm | Fibromyxoid, perineurioma-like whorls | 5 | CD34, EMA (F+), claudin1 (F+) | Pankeratin, STAT6, SMA, desmin, GLUT1, S100, SOX10, GFAP, ERG, MUC4, INI1 retained | RREB1 exon 8/ MRTFB exon 11 |
| 7 | Current Case 2 | Unclassified fibromyxoid neoplasm | Fibromyxoid, chondromyxoid lobules | 0 | S100, CD68, EMA (F+) | Pankeratin, Brachyury, Claudin1, GFAP, MUC4, CD34, SOX10. INI1 retained | RREB1 exon 8/ MRTFB exon 11 |
| 8 | Current Case 3 | Chondromyxoid sinonasal hamartoma | Chondromyxoid hamartoma-like | 0 | S100, CD56 (focal), SATB2 (wk.) | Pankeratin, ERG, ALK, Brachyury, MUC4, Pan-Trk, SSTR2A, Desmin, MDM2 | RREB1 exon 8/ MRTFB exon 11 |
| 9 | Current Case 4 | Unclassified spindle and round cell neoplasm | Monomorphic spindle and round cell, highly cellular | 2 | Synaptophysin, GFAP, CD56, patchy CD99 and PAX8 | MUC4, NUT, Pan-TRK, STAT6, SS18 and ALK, EMA, p63, CK19, TLE1, S100, SMA, SOX10, CD34, CK34BetaE12, desmin, myogenin, Myo-D1, WT1, Chromogranin-A, Calcitonin, TTF1 | RREB1 exon 8/ MRTFB exon 11 |
| 10 | Current Case 5 | Low-grade mesenchymal neoplasm with rhabdomyoblastic differentiation and RREB::MRTFB fusion, not RMS | Monomorphic ovoid and round cells | 1 | MyoD1, desmin (focal), myogenin (focal) | GFAP, S100, betacatenin, CD34, CD117, CD3, CD19, CD45 | RREB1 exon 8/ MRTFB exon 11 |
EMCMT=ectomesenchymal chondromyxoid tumor; F=focal; IHC=immunohistochemistry; hpf=high power field; RMS=rhabdomyosarcoma.
Molecular results
All five tumors revealed a RREB1::MRTFB gene fusion (Table 2). In all cases, the fusion gene contained exons 1 to 8 of RREB1 and exons 11 to 17 of MRTFB (Fig. 8). FISH analysis confirmed the rearrangements of both genes (Fig. 9).
FIGURE 8.
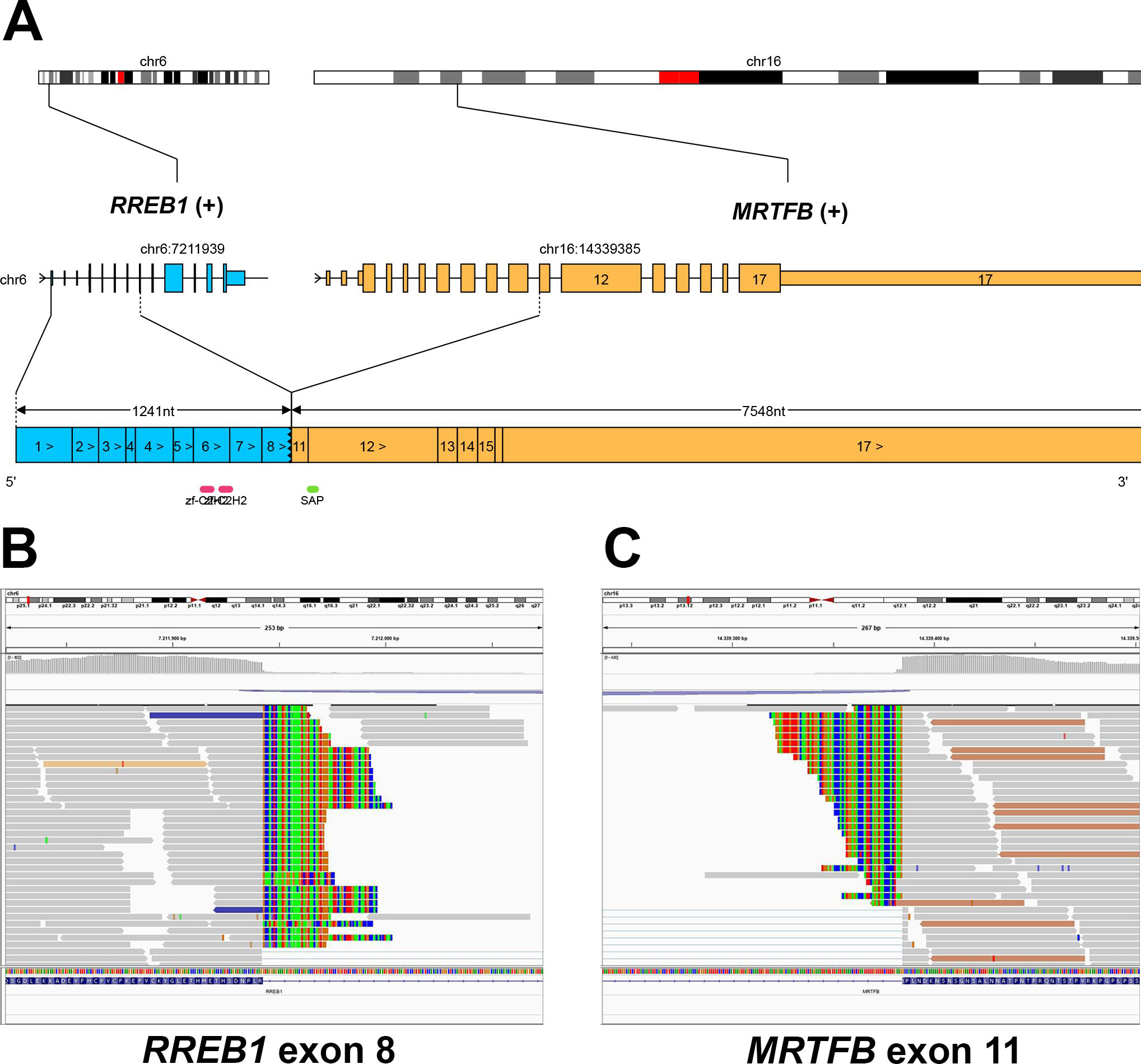
Graphical representation of the RREB1::MRTFB gene fusion (Case 1). The fusion gene comprised exons 1 to 8 of RREB1 and exons 11 to 17 of MRTFB (A). Representative IGV screenshots demonstrating split reads mapping to both 3`-region of RREB1 exon 8 (B) and to 5`-region of MRTFB exon 11 (C).
FIGURE 9.
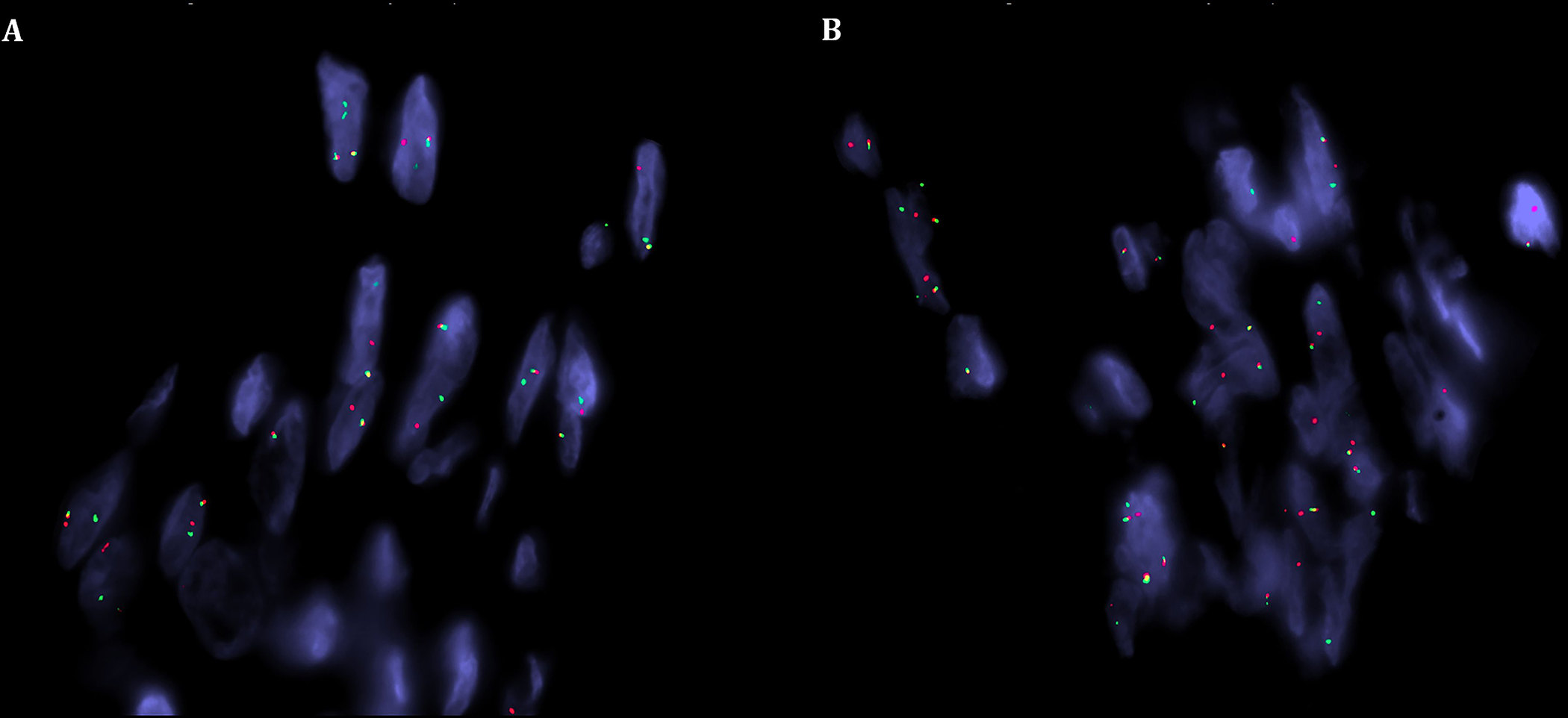
FISH analysis revealed a break-apart signal for both RREB1 (A) and MRTFB (B) (red, centromeric; green, telomeric) in keeping with balanced gene rearrangements.
Nasal chondromesenchymal hamartoma-like tumors
As one sinonasal tumor (Case 3) was strikingly similar to and initially diagnosed as nasal chondromesenchymal hamartoma (NCMH), we have tested two additional cases with similar histology to verify if these might belong to the same tumor spectrum. Case 3 occurred in a 28-year-old female in the jaw. The two additional NCMH-like tumors affected one female and one male aged 4 and 14 years, respectively. They originated in the nose and ethmoid/ maxillary sinus. Histologically, all three tumors displayed a stereotypical NCMH-like histology with a predominance of variably mature or primitive myxoid cartilage forming variably sized microlobules and blending with slightly cellular diffuse chondromyxoid stroma. Variable degree of concentric arrangement was seen in two cases and a fatty component in one. The lesion from the child showed more prominent and mature appearing bone formation. There was no epithelial component, entrapped glands, mitoses or atypia. Immunohistochemistry revealed consistent expression of S100 but no other lineage-specific or epithelial markers were expressed. All tumors were classified as NCMH, but they lacked pathogenic DICER1 mutations or other mutations in the 160 genes included in the panel used. Of these three DICER1-wild type cases, one revealed a RREB1::MRTFB fusion (Case 3 in table 1) and the other two had no detectable fusions. The clinicopathologic features of NCMH-like tumors are summarized in Table 3. Representative images of the fusion-positive case are depicted in Figure 4.
Table 3:
Clinicopathological features of nasal chondromesenchymal hamartoma-like tumors tested in this study
| No | Age (years)/sex | Site | Size cm | Positive IHC | Negative IHC | RNA sequencing | DICER1 status | Signs of DICER1 syndrome |
|---|---|---|---|---|---|---|---|---|
| 1 | 14/M | Ethmoid/maxillary left | 7 (in aggregate) | S100, CD34 | SOX10, ALK, betacatenin, ERG, SATB2, desmin, pankeratin, myogenin, EMA, GFAP, SMA | No fusion | No DICER1 mutations | No |
| 2* | 28/F | Jaw region 21–23 (follicular cyst?) | NA | S100, CD56 (focal), SATB2 (wk.) | Pankeratin, ERG, ALK, Brachyury, MUC4, Pan-Trk, SSTR2A, Desmin, MDM2 | RREB1::MTRFB | No DICER1 mutations | No |
| 3 | 04/F | Nasal left | 6 | S100, SATB2 (wk.) | - | No fusion | No DICER1 mutations | No |
DISCUSSION
The RREB1::MRTFB (former RREB1::MKL2) fusion has emerged recently as a defining feature in most if not all of ectomesenchymal chondromyxoid tumors (EMCMT) of the tongue. Dickson et al detected this fusion in 19 of 21 tumors (90%) [8]. A single tumor (5%) had an EWSR1::CREM fusion, and the remaining case lacked any known fusion gene by RNA Sequencing [8]. In addition, EWSR1 rearrangements (fusion partner unknown) have been detected by FISH in 27% of 11 cases in one study [9]. Notably the EWSR1::CREM fusion-positive case lacked all of the characteristic immunomarkers of EMCMT [8]. Whether these EWSR1-rearranged cases represent genetic variants of EMCMT or are more related to the EWSR1::CREM fusion family neoplasms remains currently controversial [8,9,22].
The MRTFB gene (AKA: MKL2 or Myocardin Like 2), mapped to 16p13.12, encodes for the Myocardin Related Transcription Factor B [23]. MRTFB is a transcription coactivator of serum response factor, involved in developmental processes such as smooth and skeletal muscle differentiation and in neuronal development [23–25]. In general, MRTFB (MKL2) fusions are uncommon. In the soft tissue, MRTFB (MKL2) is fused to C11orf95 in the majority of chondroid lipomas [26], a rare soft tissue neoplasm with subtle morphological similarities to EMCMT; both being characterized by prominent myxoid or chondromyxoid matrix.
The RREB1 gene, mapped to 6p24.3, encodes for the Ras-Responsive Element-Binding Protein 1 [27]. This Kruppel-like protein from the zinc finger protein family has been originally isolated in a human medullary thyroid carcinoma cell line [27]. Via stimulation of the transcription of down-stream target genes, RREB1 influences vital processes such as cell proliferation, differentiation, tumorigenesis and metastasis [27]. Notably, genes associated with extracellular matrix proteases might be regulated via the Ras proteins, which are in turn under the influence of RREB1 [27]. Except for EMCMTs, RREB1 fusions are exceptionally rare in solid tumors. Complex RREB1::CMAHP and ELL::RREB1 fusions, concurrent with other translocations, have been described in a case of infantile acute myeloid leukemia [28].
EMCMTs usually are composed of uniform ovoid or fusiform cells forming multiple closely packed lobules with frequent multilobated nuclei and scattered atypical cells, embedded in a chondromyxoid background [1,7,8,15]. The cells of EMCMT stain diffusely with GFAP and pankeratin, but less frequently with S100 and SMA. Desmin expression is uncommon. Although the above features characterize the prototypical tumors, uncommon or unusual features have been encountered in a few cases, either focally or diffusely. These include hypercellularity, hyalinized stroma and necrosis [8].
To date, very few cases of extra-glossal EMCMT have been reported [10–16]. However, only five genuine extra-glossal EMCMTs have been verified by molecular testing (Table 1 and 2). These tumors originated predominantly in females (4:1) at a mean age of 53 years (range, 25 – 73) and involved exclusively head and neck (3 cases; parapharyngeal, sinonasal and mandibular [13,15,16]) and mediastinal (2 cases [14]) sites. Including our cases, a total of 10 molecularly verified extraglossal RREB1::MRTFB-rearranged neoplasms have been documented. Their ages ranged from 18 to 73 years (median, 36); 60% were females. Six of 10 cases were located in head and neck sites (two parapharyngeal, two in the jaw, one nasopharyngeal and one sinonasal). The remainder were either mediastinal (2) or have originated in extra-glossal, extra-mediastinal soft tissue sites (2); the latter two cases represent a novel location for these tumors. Following surgical treatment alone, all five patients with follow-up (range, 5 – 27 months; median, 17) were disease free indicating an indolent behavior. The immunophenotypes of these tumors varied greatly with variable expression of S100 (7/10), SMA (5/7), EMA (5/7), GFAP (4/7), desmin (3/9), myogenin (2/6), and AE1/AE3 (2/8) (Table 2). The two tumors with variable rhabdomyoblastic differentiation originated in the retro-/parapharyngeal space and the posterior nasopharyngeal wall. Notably, two tumors with RREB1::MRTFB fusion and myogenic differentiation were previously reported as biphenotypic sinonasal sarcoma [13].
These extra-glossal tumors showed variable resemblance to glossal EMCMTs but were dominated by variant histology including hypercellular nodules arranged into sheets and fascicles and hypocellular areas arranged into cords, reticular pattern, or haphazardly within a hyalinized stroma. Cellular storiform proliferation of bland spindle cells with perivascular hyalinization and occasional nuclear pseudoinclusions as well as myxoid areas containing round to polygonal cells and scattered binucleated cells were observed as well [13–16]. Overall, RREB1::MRTFB-rearranged neoplasms seem to fall into two morphological subcategories: Tumors with lobulated, chondroid or myxochondroid epithelioid phenotype (seen in 3 cases) and tumors with more undifferentiated hypercellular spindle cell morphology (seen in 7 cases; Table 2).
Currently, it remains controversial, whether these extra-glossal RREB1::MRTFB-rearranged neoplasms do represent genuine extra-glossal counterparts of EMCMT or they merely represent genetic variants in the spectrum of different site-specific independent entities. This controversy is reflected by the names used for them by different authors (e.g. BSS) and is mainly due to the variant histology and immunophenotype among reported cases.
Notably, several uncommon variant features reported in genuine EMCMT of the tongue were observed in most of our current cases. These uncommon features include the presence of cellular spindle cell areas showing a vaguely fascicular growth pattern and tumors with prominent eosinophilic hyaline stroma with superficial resemblance to hyaline cartilage. The first feature represents the predominant pattern seen in our Case 1 and 4, while the second was predominant in Case 2. This confirms at least some degree of analogy of the current cases with variant glossal EMCMT. However, the prominent granular cell pattern seen in Case 2 is unusual and potentially misleading. Moreover, the striking resemblance of one of our cases to perineurioma histologically and immunohistochemically (prominent expression of CD34 and limited focal reactivity for EMA and claudin1) is noteworthy. A perineurioma-like morphology with or without expression of perineurial markers has been reported in other translocation-associated sarcomas such as low-grade fibromyxoid sarcoma, representing a diagnostic pitfall [29]. Moreover, a recent study by Dickson et al identified a high frequency of VGLL3 gene fusions in tumors in the spectrum of hybrid schwannoma-perineurioma (detected in 14 of 18 cases) [30]. CHD7 (n=10), CHD9 (n=2), and MAMLD1 (n=2) were the partner genes encountered [30]. In addition, novel DST::BRAF and SQSTM1::CDX1 fusions were detected in two cases, while two tumors lacked identifiable fusions [30]. The authors suggested that hybrid schwannoma–perineurioma likely represents a distinct entity, unrelated to conventional schwannoma and perineurioma [30]. One of our cases illustrates the limited specificity of the perineurioma-like immunophenotype.
Two other findings in our series merit special attention. Case 3 was very similar to NCMH and indeed this was the original diagnosis. NCMH is a rare sinonasal lesion with strong association with the DICER1 syndrome [31,32]. The vast majority of affected patients are newborns or young children [31–34]. However, the cases we examined herein were diagnosed at an older age compared to the vast majority of NCMH. Moreover, the one harboring the RREB1::MRTFB fusion affected an adult. Although the morphology of these tumors is very similar to reported NCMH, it is possible that these tumors represent a distinctive chondromyxoid tumor unrelated to the DICER1 syndrome as evidenced by lack of DICER1 mutations in all three cases and instead presence of RREB1::MRTFB fusion in one of them. Analysis of more cases should shed light on the nosology and histogenesis of this distinctive lesion.
The two cases we reported from non-head-and-neck soft tissue sites are novel. Their morphology overlaps variably with glossal EMCMT and with variant extraglossal cases. Nevertheless, the morphological diversity of these tumors rather argues for potentially different entities unified by the presence of the RREB1::MRTFB fusion than for different faces of the same entity, but this remains a controversial issue to be addressed in future studies if more cases are reported. Their differential diagnosis includes a wide range of low-grade neoplasms showing fibromyxoid or chondromyxoid patterns as well as perineurial cell lesions. In particular, myoepithelial neoplasms, nerve sheath myxoma, extraskeletal myxoid chondrosarcoma and ossifying fibromyxoid tumor of soft parts need be ruled out by defined demographic, morphological, immunophenotypic, and genetic criteria.
Of interest, our Case 5 showed histological features potentially overlapping with BSS with rhabdomyoblastic differentiation [35–38]. Moreover, two tumors with identical RREB1::MRTFB fusion were reported previously as BSS [13,16]. However, there is no proof that these tumors represent genuine BSS as their immunophenotype is highly heterogeneous and may overlap with more than one entity including BSS. Notably, BSS, including also those cases with rhabdomyoblastic differentiation, is driven by PAX3 fusions (mostly fused to MAML3, less frequently to FOXO1, and rarely to NCOA2/1 and WWTR1) and they have not been reported to harbor the RREB1::MRTFB fusion [35–38].
In summary, we have described five RREB1::MRTFB-positive extraglossal mesenchymal neoplasms bringing up the total number of reported cases to 10. Our study for the first time documents the occurrence of these tumors in extraglossal, extra-mediastinal soft tissue sites. From the reported cases, it seems that these extraglossal RREB1::MRTFB fusion-driven neoplasms are biologically indolent with predilection for the head and neck soft tissue in adults with slight overrepresentation of females. These neoplasms do not fit exactly any defined tumor entity, but show some overlap with reported extra-glossal EMCMT. A potential relationship of the NCMH-like cases to genuine NCMH remains to be verified. Moreover, identification of more cases is mandatory to delineate the morphological spectrum and the biological behavior of this rare entity.
Funding:
Supported in part by: P50 CA217694 (CRA), P50 CA140146 (CRA), P30 CA008748 (CRA), Cycle for Survival (CRA).
Footnotes
Conflict of interest: none
REFERENCES
- 1.Smith BC, Ellis GL, Meis-Kindblom JM, Williams SB. Ectomesenchymal chondromyxoid tumor of the anterior tongue. Nineteen cases of a new clinicopathologic entity. Am J Surg Pathol. 1995;19:519–30. [DOI] [PubMed] [Google Scholar]
- 2.Nikitakis NG, Argyris P, Sklavounou A, Papadimitriou JC. Oral myoepithelioma of soft tissue origin: report of a new case and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;110:e48–51. [DOI] [PubMed] [Google Scholar]
- 3.Goveas N, Ethunandan M, Cowlishaw D, Flood TR. Ectomesenchymal chondromyxoid tumour of the tongue: Unlikely to originate from myoepithelial cells. Oral Oncol. 2006;42:1026–8. [DOI] [PubMed] [Google Scholar]
- 4.Aldojain A, Jaradat J, Summersgill K, Bilodeau EA. Ectomesenchymal Chondromyxoid Tumor: A Series of Seven Cases and Review of the Literature. Head Neck Pathol. 2015;9:315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Truschnegg A, Acham S, Kqiku L, Jakse N, Beham A. Ectomesenchymal chondromyxoid tumor: a comprehensive updated review of the literature and case report. Int J Oral Sci. 2018;10(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naidoo S, Roode GJ, Bütow KW, Meer S. Ectomesenchymal Chondromyxoid Tumor: A Rare Association With an Asymmetrical Soft Palate Cleft. Cleft Palate Craniofac J. 2021. Aug 30:10556656211035029. [DOI] [PubMed] [Google Scholar]
- 7.Laco J, Mottl R, Höbling W, Ihrler S, Grossmann P, Skalova A, Ryska A. Cyclin D1 Expression in Ectomesenchymal Chondromyxoid Tumor of the Anterior Tongue. Int J Surg Pathol. 2016;24:586–94. [DOI] [PubMed] [Google Scholar]
- 8.Dickson BC, Antonescu CR, Argyris PP, Bilodeau EA, Bullock MJ, Freedman PD, Gnepp DR, Jordan RC, Koutlas IG, Lee CH, Leong I, Merzianu M, Purgina BM, Thompson LDR, Wehrli B, Wright JM, Swanson D, Zhang L, Bishop JA. Ectomesenchymal Chondromyxoid Tumor: A Neoplasm Characterized by Recurrent RREB1-MKL2 Fusions. Am J Surg Pathol. 2018;42:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Argyris PP, Bilodeau EA, Yancoskie AE, Trochesset D, Pambuccian SE, Wetzel SL, Shah SS, Edelman M, Freedman P, Dolan M, Koutlas IG. A subset of ectomesenchymal chondromyxoid tumours of the tongue show EWSR1 rearrangements and are genetically linked to soft tissue myoepithelial neoplasms: a study of 11 cases. Histopathology. 2016;69:607–13. [DOI] [PubMed] [Google Scholar]
- 10.Nigam S, Dhingra KK, Gulati A. Ectomesenchymal chondromyxoid tumor of the hard palate--a case report. J Oral Pathol Med. 2006;35:126–8. [DOI] [PubMed] [Google Scholar]
- 11.Gouvêa AF, Díaz KP, Léon JE, Vargas PA, de Almeida OP, Lopes MA. Nodular lesion in the anterior hard palate. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:154–9. [DOI] [PubMed] [Google Scholar]
- 12.Stecco A, Quagliozzi M, Pino M, Spina P, Pia F, Boldorini R, Carriero A. An unusual case of ectomesenchymal chondromyxoid tumour of the left tonsillar bed: imaging and histopathologic features. BJR Case Rep. 2016;2:20150183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siegfried A, Romary C, Escudié F, Nicaise Y, Grand D, Rochaix P, Barres B, Vergez S, Chevreau C, Coindre JM, Uro-Coste E, Le Guellec S. RREB1-MKL2 fusion in biphenotypic “oropharyngeal” sarcoma: New entity or part of the spectrum of biphenotypic sinonasal sarcomas? Genes Chromosomes Cancer. 2018;57:203–210. [DOI] [PubMed] [Google Scholar]
- 14.Makise N, Mori T, Kobayashi H, Nakagawa K, Ryo E, Nakajima J, Kohsaka S, Mano H, Aburatani H, Yoshida A, Ushiku T. Mesenchymal tumours with RREB1-MRTFB fusion involving the mediastinum: extra-glossal ectomesenchymal chondromyxoid tumours? Histopathology. 2020;76:1023–1031. [DOI] [PubMed] [Google Scholar]
- 15.Bubola J, Hagen K, Blanas N, Weinreb I, Dickson BC, Truong T. Expanding Awareness of the Distribution and Biologic Potential of Ectomesenchymal Chondromyxoid Tumor. Head Neck Pathol. 2021;15:319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mechtersheimer G, Andrulis M, Delank KW, Volckmar AL, Zhang L, von Winterfeld M, Stenzinger A, R Antonescu C. RREB1-MKL2 fusion in a spindle cell sinonasal sarcoma: biphenotypic sinonasal sarcoma or ectomesenchymal chondromyxoid tumor in an unusual site? Genes Chromosomes Cancer. 2021;60:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative Genomics Viewer. Nature Biotechnology 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, Chen J, Robinson HE, Shim HS, Chmielecki J, Pao W, Engelman JA, Iafrate AJ, Le LP. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84. [DOI] [PubMed] [Google Scholar]
- 19.Zhu G, Benayed R, Ho C, Mullaney K, Sukhadia P, Rios K, Berry R, Rubin BP, Nafa K, Wang L, Klimstra DS, Ladanyi M, Hameed MR. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol. 2019;32:609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antonescu CR, Zhang L, Chang NE, Pawel BR, Travis W, Katabi N, Edelman M, Rosenberg AE, Nielsen GP, Dal Cin P, Fletcher CD. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agaimy A, Witkowski L, Stoehr R, Cuenca JCC, González-Muller CA, Brütting A, Bährle M, Mantsopoulos K, Amin RMS, Hartmann A, Metzler M, Amr SS, Foulkes WD, Sobrinho-Simões M, Eloy C. Malignant teratoid tumor of the thyroid gland: an aggressive primitive multiphenotypic malignancy showing organotypical elements and frequent DICER1 alterations-is the term “thyroblastoma” more appropriate? Virchows Arch. 2020;477:787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida A, Wakai S, Ryo E, Miyata K, Miyazawa M, Yoshida KI, Motoi T, Ogawa C, Iwata S, Kobayashi E, Watanabe SI, Kawai A, Mori T. Expanding the Phenotypic Spectrum of Mesenchymal Tumors Harboring the EWSR1-CREM Fusion. Am J Surg Pathol. 2019;43:1622–1630. [DOI] [PubMed] [Google Scholar]
- 23.Selvaraj A, Prywes R. Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum response factor, is required for skeletal myogenic differentiation. J Biol Chem. 2003;278:41977–87. [DOI] [PubMed] [Google Scholar]
- 24.Cen B, Selvaraj A, Prywes R. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J Cell Biochem. 2004;93:74–82. [DOI] [PubMed] [Google Scholar]
- 25.Mokalled MH, Johnson A, Kim Y, Oh J, Olson EN. Myocardin-related transcription factors regulate the Cdk5/Pctaire1 kinase cascade to control neurite outgrowth, neuronal migration and brain development. Development. 2010;137:2365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang D, Sumegi J, Dal Cin P, Reith JD, Yasuda T, Nelson M, Muirhead D, Bridge JA. C11orf95-MKL2 is the resulting fusion oncogene of t(11;16)(q13;p13) in chondroid lipoma. Genes Chromosomes Cancer. 2010;49:810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thiagalingam A, De Bustros A, Borges M, Jasti R, Compton D, Diamond L, Mabry M, Ball DW, Baylin SB, Nelkin BD. RREB-1, a novel zinc finger protein, is involved in the differentiation response to Ras in human medullary thyroid carcinomas. Mol Cell Biol. 1996;16:5335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuborgh A, Meyer C, Marschalek R, Preiss B, Hasle H, Kjeldsen E. Complex three-way translocation involving MLL, ELL, RREB1, and CMAHP genes in an infant with acute myeloid leukemia and t(6;19;11)(p22.2;p13.1;q23.3). Cytogenet Genome Res. 2013;141:7–15. [DOI] [PubMed] [Google Scholar]
- 29.Thway K, Fisher C, Debiec-Rychter M, Calonje E. Claudin-1 is expressed in perineurioma-like low-grade fibromyxoid sarcoma. Hum Pathol. 2009;40:1586–90. [DOI] [PubMed] [Google Scholar]
- 30.Dickson BC, Antonescu CR, Demicco EG, Leong DI, Anderson ND, Swanson D, Zhang L, Fletcher CDM, Hornick JL. Hybrid schwannoma-perineurioma frequently harbors VGLL3 rearrangement. Mod Pathol. 2021;34:1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.González IA, Stewart DR, Schultz KAP, Field AP, Hill DA, Dehner LP. DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma. Mod Pathol 2022;35:4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart DR, Messinger Y, Williams GM, Yang J, Field A, Schultz KA, Harney LA, Doros LA, Dehner LP, Hill DA. Nasal chondromesenchymal hamartomas arise secondary to germline and somatic mutations of DICER1 in the pleuropulmonary blastoma tumor predisposition disorder. Hum Genet 2014;133:1443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vasta LM, Nichols A, Harney LA, Best AF, Carr AG, Harris AK, Miettinen M, Schultz KAP, Kim HJ, Stewart DR. Nasal chondromesenchymal hamartomas in a cohort with pathogenic germline variation in DICER1. Rhinol Online. 2020;3:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDermott MB, Ponder TB, Dehner LP. Nasal chondromesenchymal hamartoma: an upper respiratory tract analogue of the chest wall mesenchymal hamartoma. Am J Surg Pathol 1998;22:425–33. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Bledsoe KL, Graham RP, Asmann YW, Viswanatha DS, Lewis JE, Lewis JT, Chou MM, Yaszemski MJ, Jen J, Westendorf JJ, Oliveira AM. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat Genet. 2014;46:666–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong WJ, Lauria A, Hornick JL, Xiao S, Fletcher JA, Marino-Enriquez A. Alternate PAX3-FOXO1 oncogenic fusion in biphenotypic sinonasal sarcoma. Genes Chromosomes Cancer. 2016;55:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang SC, Ghossein RA, Bishop JA, Zhang L, Chen TC, Huang HY, Antonescu CR. Novel PAX3-NCOA1 Fusions in Biphenotypic Sinonasal Sarcoma With Focal Rhabdomyoblastic Differentiation. Am J Surg Pathol. 2016;40:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fritchie KJ, Jin L, Wang X, Graham RP, Torbenson MS, Lewis JE, Rivera M, Garcia JJ, Schembri-Wismayer DJ, Westendorf JJ, Chou MM, Dong J, Oliveira AM. Fusion gene profile of biphenotypic sinonasal sarcoma: an analysis of 44 cases. Histopathology. 2016;69:930–936. [DOI] [PubMed] [Google Scholar]