

Abstract
Paracrine cell-cell communication is central to all developmental processes, ranging from cell diversification to patterning and morphogenesis. Precise calibration of signaling strength is essential for the fidelity of tissue formation during embryogenesis and tissue maintenance in adults. Membrane-tethered ubiquitin ligases can control the sensitivity of target cells to secreted ligands by regulating the abundance of signaling receptors at the cell surface. We discuss two examples of this emerging concept in signaling: (1) the ZNRF3/RNF43 transmembrane ubiquitin ligases that regulate responses to WNT and bone morphogenetic protein ligands and (2) the membrane-recruited ligase MGRN1 that controls hedgehog and melanocortin receptor signaling. We focus on the mechanistic logic of these systems, illustrated by structural and protein interaction models enabled by AlphaFold. We suggest that membrane-tethered ubiquitin ligases play a widespread role in remodeling the cell surface proteome to control responses to extracellular ligands in diverse biological contexts.
Keywords: Developmental Signaling, WNT signaling, Hedgehog signaling, melanocortin receptor signaling, morphogen signaling, ubiquitylation, ubiquitin ligases, signaling sensitivity, patterning, development, regeneration, stem cells
1. Introduction
The fates of signaling receptors and other membrane proteins are regulated by ubiquitylation during all stages of their life cycle: protein quality in the endoplasmic reticulum (ER), sorting, trafficking and expulsion into exosomes, endocytic clearance, and lysosomal degradation (Foot, Henshall, and Kumar 2017). The attachment of ubiquitin (Ub) and polyubiquitin chains to Lys residues on a target protein is carried out by the intricate interplay of three conserved families of enzymes whose structures and molecular mechanisms have been revealed by various approaches (Cappadocia and Lima 2018; Hershko, Ciechanover, and Varshavsky 2000). First, Ub is activated by its attachment to a ubiquitin activating enzyme (E1) through a thioester linkage. Second, the activated Ub is transferred to a Cys on a ubiquitin conjugating enzyme (E2). Finally, ubiquitin ligase enzymes (E3s) facilitate the transfer of Ub from the E2 to specific Lys residues on a substrate protein or to a previously conjugated Ub in a growing polyubiquitin chain. Given the presence of seven Lys residues that decorate the Ub surface, linear or branched chains containing various combinations of Ub linkages can be attached to the substrate, and this topographically diverse ‘Ub code’ can drive different outcomes (Kwon and Ciechanover 2017). E3s provide the crucial substrate specificity to the ubiquitylation reaction, and sometimes this recognition event requires the assembly of large multiprotein complexes (Harper and Schulman 2021; Morreale and Walden 2016; N. Zheng and Shabek 2017). The Really Interesting New Gene (RING) E3s comprise the largest family (~600 members), characterized by the presence of a compact RING domain nucleated by a pair of bound Zn2+ ions (Deshaies and Joazeiro 2009). RING domains recruit a Ub-charged E2 and position it optimally for transfer of Ub to a substrate that is captured by a separate recognition module (Metzger et al. 2014). HECT E3s (~29 members) and RBR (RING-between-RING) E3s (13 members) mediate Ub transfer through a two-step process involving a thioester intermediate between Ub and a catalytic Cys on the E3 itself prior to the transfer of Ub onto the substrate (N. Zheng and Shabek 2017). An additional level of regulation is afforded by ~100 deubiquitylating enzymes (DUBs) that remove Ub from proteins (Clague, Urbé, and Komander 2019).
In this chapter we focus on Ub modifications performed by RING E3s that have recognizable transmembrane (TM) helices. Approximately 50 of the ~600 annotated RING E3s fall into this class (W. Li et al. 2008; Fenech et al. 2020; Neutzner et al. 2011). However, the actual number of membrane-tethered E3s may be significantly larger, since cytoplasmic E3s can be recruited to the plasma membrane by stable association with a TM co-receptor, and such complexes are difficult to predict by sequence analysis alone. We use the term “membrane-tethered” to refer to both classes of E3s – those that are anchored to the membrane by an intrinsic TM domain and those that are recruited by non-covalent association with a TM protein. Much of the research in this area has been on TM E3s that function in the ER as part of the ER-associated degradation (ERAD) system and other protein quality control pathways (Fenech et al. 2020; Foot, Henshall, and Kumar 2017; Sardana and Emr 2021). For example, TM E3s such as the prototype yeast protein Hrd1 ubiquitylate misfolded ER proteins that are retro-translocated through a pore-like assembly to the cytoplasm, tagging them for proteasomal degradation (Phillips and Miller 2021). However, a growing number of structurally distinct membrane-tethered E3s have been shown to function outside of the ER to regulate the abundance of signaling receptors at the cell surface, and consequently the sensitivity of cells to signaling ligands. We will describe three such systems that function in developmental signaling pathways to control tissue patterning and morphogenesis, as well as in stem cell self-renewal, tissue homeostasis and regeneration. We anticipate that regulation of signaling strength in target cells – the cells exposed to signaling ligands – by membrane-tethered E3s will emerge as a general control mechanism in signaling pathways beyond those discussed in this chapter.
The recognition mechanisms that these membrane-tethered E3s employ to bind their targets and position their RING domains for effective Ub transfer to the cytoplasmic chains of substrate receptors remain largely unknown. We take advantage of the recent advances in the prediction of protein folds and protein-protein interactions by deep learning-based programs like AlphaFold and RoseTTaFold (Jumper et al. 2021; Tunyasuvunakool et al. 2021; Baek et al. 2021; Evans et al. 2021; Bryant, Pozzati, and Elofsson 2021) to create models of multimodular TM E3 complexes. These methods are useful to generate hypotheses for how E3s recognize substrates through extracellular, TM and intracellular contacts, and how they may themselves be regulated by ligands. We note that all the structures shown in the figures represent AlphaFold models unless indicated otherwise.
2. Classification of membrane-tethered E3s
Excluding the E3s involved in protein quality control pathways in the ER, membrane-tethered E3s fall into three broad architectural classes (Figure 1).
Figure 1.
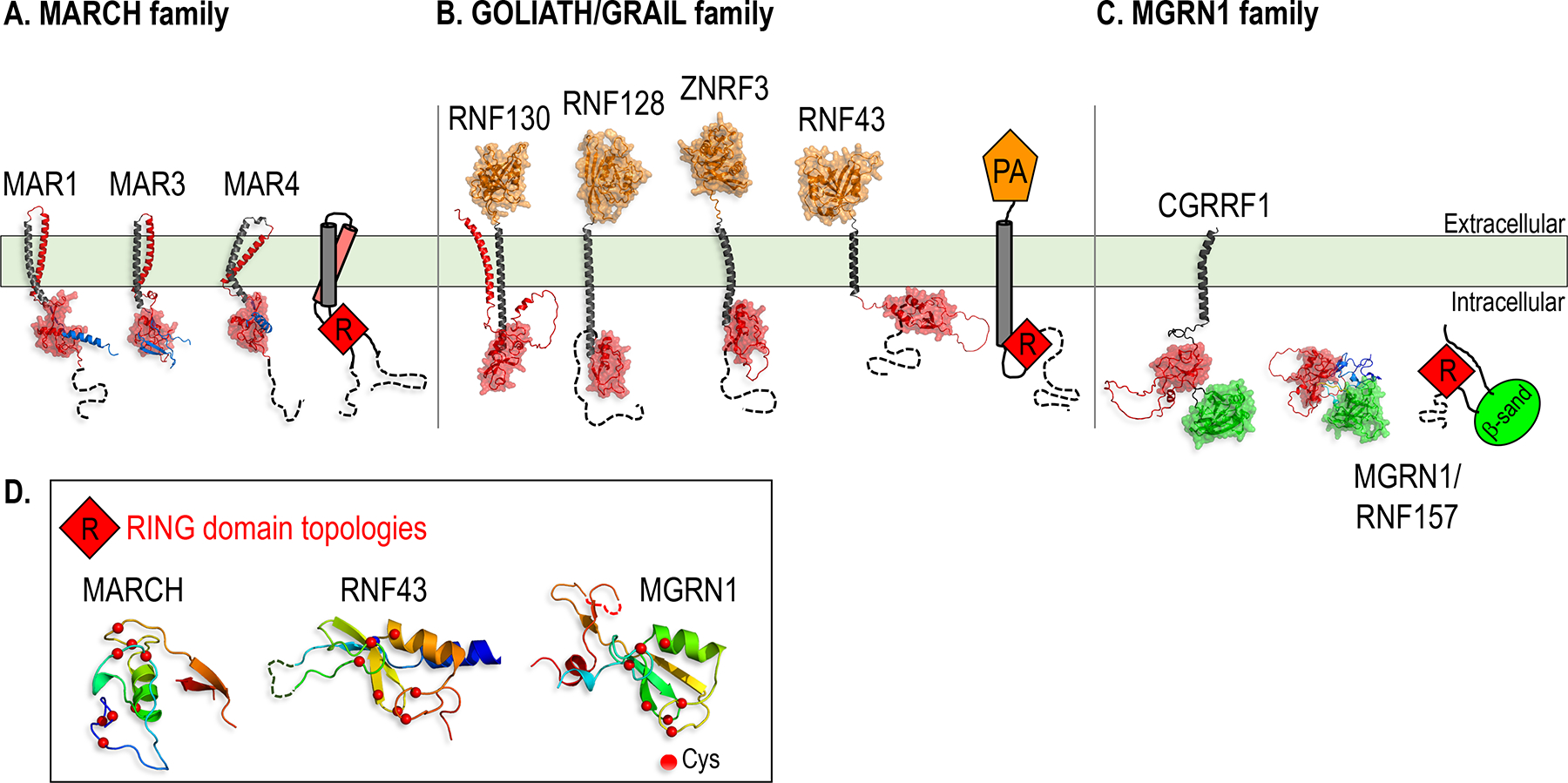
Structural models of the three classes of TM and membrane-associated E3s discussed in this chapter.
A-C. AlphaFold models of representative members of the MARCH (A), GOLIATH/GRAIL (B) and MGRN1 (C) E3 families, with cartoons used throughout the figures to represent each family. The RING domain, which recruits a Ub-charged E2 for Ub transfer to the substrate, is colored red and shown with a space filling surface in the structural models, and is shown as a red diamond labeled “R” in the cartoons in this and all subsequent figures. All the structures shown in the figures represent AlphaFold models unless indicated otherwise with a Protein Data Bank (PBD) ID shown in italics, and are drawn to the same scale within each figure, except for structures shown in boxes. Dotted lines denote unstructured segments of the proteins for which folds could not be predicted. Molecular graphics were generated with PyMOL (www.pymol.org).
A. In the MARCH family, substrate recognition is accomplished by two closely linked TM helices (gray and red) folded as a hairpin, and Ub transfer is catalyzed by a tightly associated RING domain.
B. Members of the GOLIATH/GRAIL family contain an extracellular PA domain that can bind to ligands and serve in substrate recognition.
C. The MGRN1 family is characterized by a RING domain juxtaposed to a putative substrate binding β-sandwich domain (β-sand, green). MGRN1 and RNF157 lack TM helices, but are recruited to the membrane by interactions with single pass TM proteins (see Figures 6 and 7), while CGRRF1 is tethered to the membrane by a single TM helix.
D. Topologies of the RING domains in one representative member of each of the three E3 families shown.
2.A. Membrane-associated RING-CH (MARCH) TM E3s
Homologs of the MARCH proteins were first identified as gene products that allow viruses to evade the host immune response by downregulating class I major histocompatibility complex (MHC-I) proteins (reviewed in (Bauer, Bakke, and Morth 2017)). MARCH proteins have been implicated in regulating the cell surface expression and trafficking of many single-pass TM proteins that play a role in T-cell activation: class I and II MHC proteins (antigen presentation), ICAM-1 (cell-cell adhesion), CD4 (T-cell co-receptor), CD86 (costimulatory signal), and cytokine receptors. Eleven MARCH family members have been recognized by the close similarity of their distinctive RING domains. Seven of these (MAR1-4, 8-9 and 11) contain a tight hairpin of two TM helices that follows an N-terminal RING module, two of them (MAR5-6) have more complex arrangements of multiple angled TM stretches, and two outliers (MAR7 and 10) have a single C-terminal TM helix (Figures 1A and 2A). The compact RING-TM-TM portion of the major group of MARCH TM E3s is predicted to form their only structured part, although their cytoplasmic chains, composed largely of long disordered segments at both the N- and C-termini, likely carry cryptic modification sites and short interaction motifs. This conserved, ~160 residue long RING-TM-TM module is capable of both recruiting a Ub-charged E2 via its juxtamembrane RING domain and recognizing the substrate to catalyze Ub transfer. Therefore, substrate recognition likely involves intra-membrane binding of one or multiple TM helices in the substrate to the MARCH TM hairpin motif.
Figure 2.
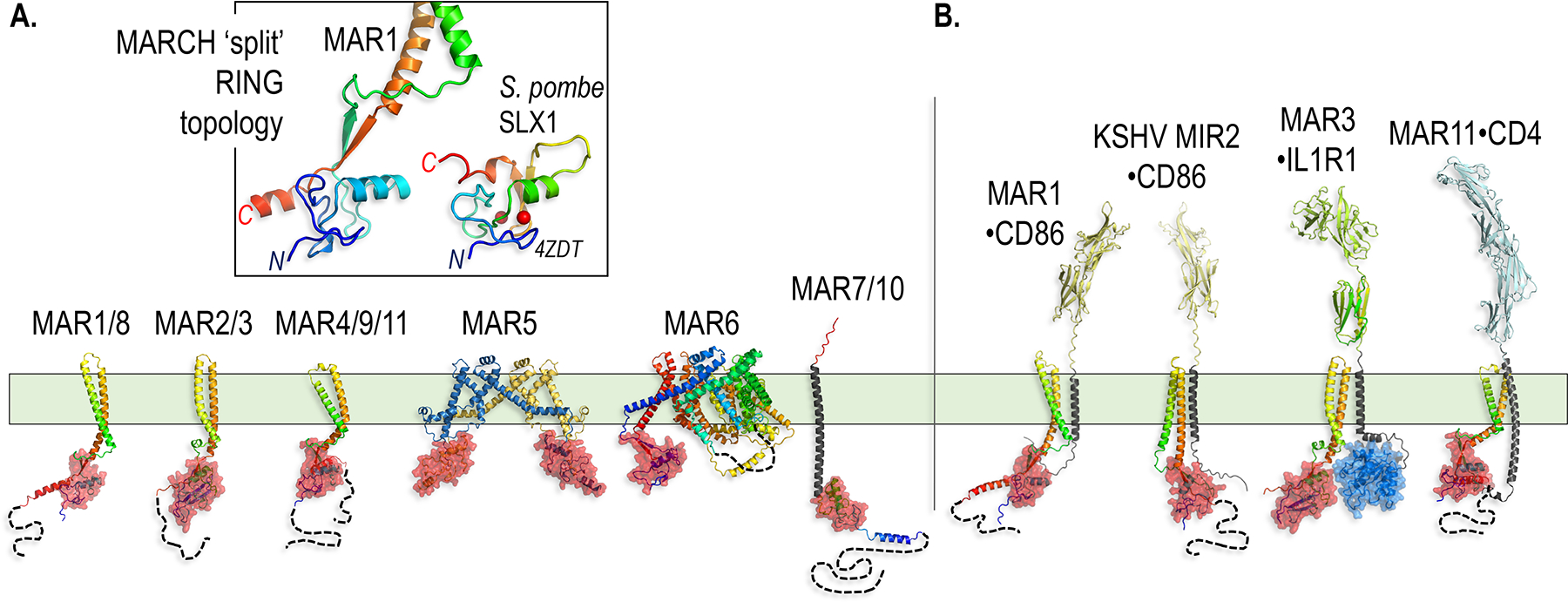
The MARCH family TM E3s and their substrate recognition mechanisms.
A. AlphaFold models of representatives of the eleven MARCH family members (MAR1/8, MAR2/3, MAR4/9/11 and MAR7/10 have similar structures, so only one of each group is shown in the figure). The unique ‘split’ RING topology is highlighted in the box (see main text for description). For comparison, the bipartite RING domain of MAR1 is shown next to the RING domain from the Saccharomyces pombe (S. pombe) protein SLX1 (PDB ID 4ZDT) (Lian, Xie, and Qian 2016).
B. Models of MARCH family members with their substrates highlight the importance of interactions between TM helices within the plane of the membrane. CD86 can be targeted by both MAR1 and the viral homolog MIR2 with slightly divergent folds and mechanisms.
Modeling of the shared RING-TM-TM module reveals that the MARCH RING domain is bipartite, built primarily by the canonical Zn2+-binding motif located just before the TM hairpin, but completed by a conserved β-strand that immediately follows the second TM helix (Figure 2A). As a result, the MARCH RING domain is closely juxtaposed to the hairpin TM structure at the level of the inner leaflet of the plasma membrane, and may be uniquely responsive to structural rearrangements within the TM hairpin motif upon substrate recognition inside the plasma membrane (Trenker et al. 2021). The more complex TM architectures of MARCH5 and 6 still display this bipartite RING domain structure, but with some variations. MAR5 adopts a predicted dimer fold that reconstitutes each of the two RING domains with a polypeptide chain from their respective partner subunits (Figure 2A). MAR6 reunites the N-terminal portion of the RING domain with a β-strand that follows the C-terminal TM helix, with an intervening 610 residue sequence that crosses the plasma membrane multiple times (Figure 2A).
The clearest indication that the distinctive MARCH hairpin-TM structure is responsible for substrate recognition comes from a comparative study describing the engagement and Ub modification of CD86, a protein that provides costimulatory signals to T cells, by two distinct MARCH-class E3 ligases: human MAR1 and Modulator of Immune Recognition 2 (MIR2), a viral MARCH homolog from Kaposi’s sarcoma herpesvirus (KSHV) (Figure 2B) (Trenker et al. 2021).
2.B. GOLIATH/GRAIL family TM E3s
The ZNRF3 and RNF43 ubiquitin ligases that regulate WNT and bone morphogenetic protein (BMP) signaling and are a major focus of this chapter, are related to the GOLIATH/GRAIL family of membrane-embedded E3s. Members of this family have a common domain architecture: an N-terminal extracellular Protease Associated (PA) domain connected by a linker of varying length to a single TM helix, closely followed by a cytoplasmic RING domain (Figure 1B). In the human proteome, we find twelve PA-TM-RING E3s (RNF13, 43, 128, 130, 133, 148-150, 167, 203-204, and 215) and two outlier members that lack the PA domain, RNF24 and RNF122. In some of these E3s, the RING domain is predicted to pack against the last two turns of an extended TM helix, restricting their conformational flexibility (Figure 3). One point of variability between the members of this family revealed by AlphaFold modeling is the seamless extension of the TM helix into an amphipathic cytoplasmic helix, which forms a rigid scaffold that positions the RING domain at different distances from the plasma membrane. The distance ranges from practically no extension of the TM helix (as in the case of RNF43, in which the RING domain is connected through a linker to a short cytoplasmic extension of the helix) to 5 helical turns (for RNF203 and ZNRF3) or even 8 helical turns (for RNF130, also known as GOLIATH). RNF130 has a second, C-terminally distal TM helix that packs against the canonical TM helix (Figure 3) in a manner reminiscent of some MARCH family E3s (Figure 2).
Figure 3.
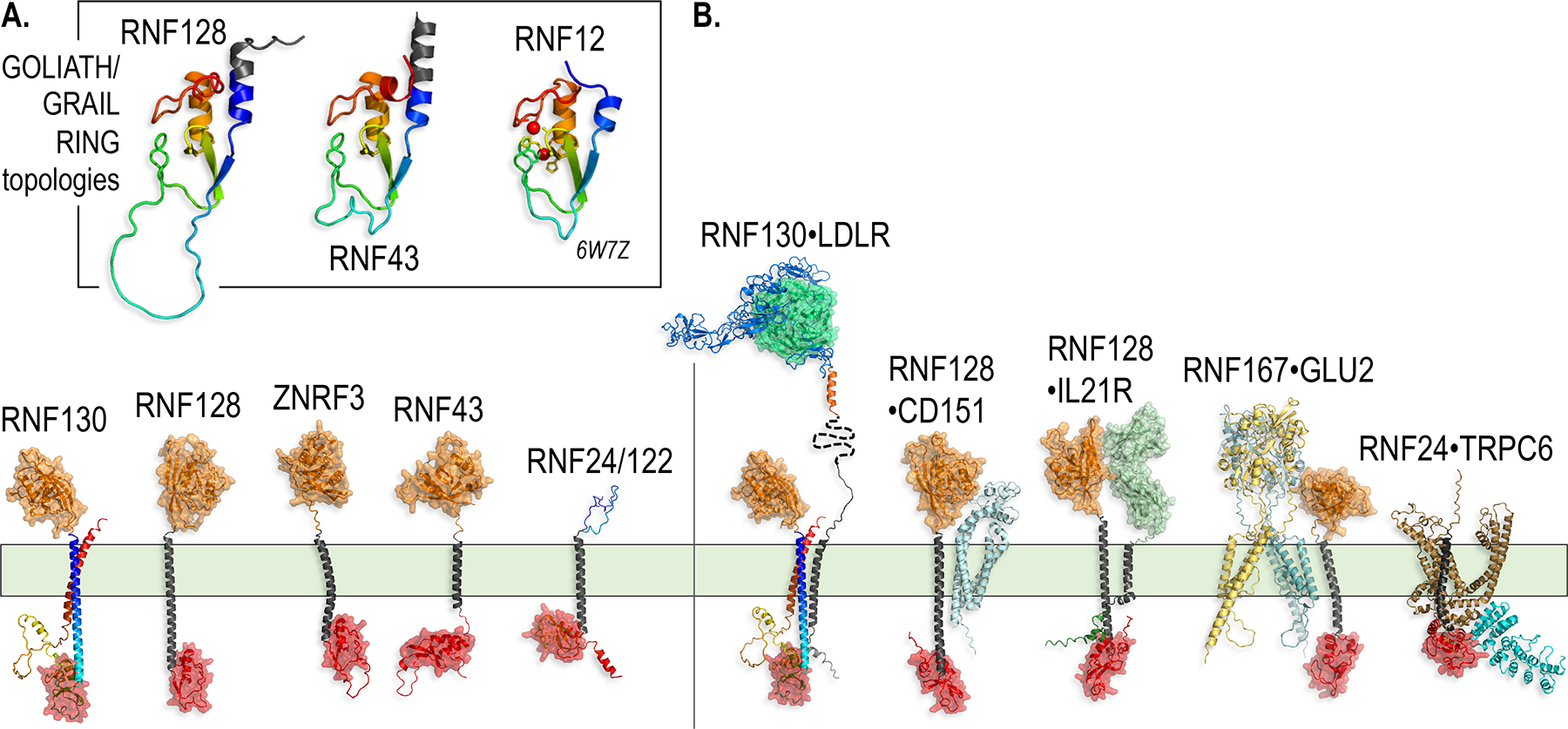
The GOLIATH/GRAIL family TM E3s and their substrate recognition mechanisms.
A. AlphaFold models of GOLIATH/GRAIL family members (RNF122/24 have similar structures, so only one of them is shown). The RING domain topologies for RNF128 and RNF43 are highlighted in the box. While no structures of the RING domain of GOLIATH/GRAIL family members have been solved, the RING domain most closely resembles that of the crystal structure of RNF12 (PDB ID 6W7Z) (Middleton, Zhu, and Day 2020), also shown for comparison.
B. AlphaFold models of GOLIATH/GRAIL family members interacting with their substrates suggest the importance of recognition events that span extracellular, TM and intracellular domains. The PA domain (orange) of RNF128 binds to the extracellular domains of substrates (Lineberry et al. 2008).
The best studied of the PA-TM-RING proteins is RNF128, also known as gene related to anergy in lymphocytes (GRAIL) (reviewed in (Whiting et al. 2011)). RNF128 suppresses T-cell responsiveness and cytokine transcription by ubiquitylating and downregulating multiple cell surface molecules involved in T-cell activation, including CD83, CD81, CD151 and CD40L (Su et al. 2009; Lineberry et al. 2008). The Drosophila GOLIATH family members have been shown to ubiquitylate the SNAP receptor (SNARE) protein VAMP3: loss-of-function mutations in GOLIATH or GODZILLA in flies result in the accumulation of membrane proteins in Rab5-positive giant endosomes due to defects in recycling endosome trafficking (Yamazaki et al. 2013). A relative of this family in C. elegans, a protein called PLR-1, regulates the density of multiple receptors for WNT ligands on the plasma membrane (Moffat et al. 2014). While the mechanism of substrate recognition by these PA-TM-RING proteins remains incompletely understood, studies of RNF128 suggest that the PA domain directly binds to the extracellular domains of substrate TM proteins, recruiting them for ubiquitylation by the cytoplasmic RING domain (Figure 3) (Lineberry et al. 2008). Thus, substrate recognition and ubiquitylation are segregated on opposite sides of the plasma membrane. However, ZNRF3 and RNF43, two TM E3s belonging to a distinct branch of the GOLIATH/GRAIL family, may require a cytoplasmic adaptor protein for substrate recognition, as discussed later (Jiang et al. 2015).
2.C. MGRN1 family membrane-recruited E3s
E3s lacking a TM helix can nevertheless be tightly tethered to the plasma membrane via direct, non-covalent association with an integral membrane protein. Mahogunin RING finger 1 (MGRN1, also known as RNF156) and its vertebrate-specific paralog RNF157 are the only examples of such ubiquitin ligases described to date (Figure 1C). These E3s are associated with two single-pass TM proteins to regulate Hedgehog and melanocortin receptor signaling (Kong et al. 2020; Lin He, Eldridge, et al. 2003). Interestingly, MGRN1 and RNF157 are related to CGRRF1 (also known as RNF197), an E3 that is anchored to the membrane by a single N-terminal TM segment (without an extracellular domain) and has been implicated in ERAD (Figure 1) (Glaeser et al. 2018). It is likely that other cytoplasmic E3s also associate with TM partners to ubiquitylate membrane proteins, but the cytoplasmic sequence motifs or cryptic structural modules in the co-receptors that drive complex formation have not been cataloged, with the exception of MGRN1.
In the following sections we describe in detail one example of membrane-embedded E3s – ZNRF3 and RNF43 – and one example of a membrane-recruited E3 – MGRN1 – within the context of the developmental signaling systems in which they have been best characterized.
3. The R-spondin-ZNRF3/RNF43 signaling system tunes WNT and BMP receptor levels by regulated ubiquitylation
The ZNRF3 and RNF43 PA-TM-RING E3s have been most extensively studied in the context of the R-spondin system, a signaling module that tunes the abundance of cell surface receptors in the WNT (Hoppler and Moon 2014) and BMP (Derynck and Miyazono 2017) signaling pathways by regulated ubiquitylation, endocytosis and lysosomal degradation (see review by (Niehrs 2012) for a timeline of the discovery and initial characterization of the R-spondin system). Recent work has also uncovered ZNRF3/RNF43-independent roles for R-spondins as WNT pathway agonists (Carmon et al. 2014) and antagonists (Reis and Sokol 2021), and in regulating other signaling pathways including TGFβ (Zhou et al. 2017), ERK/FGF (M. Zhang et al. 2017; Reis and Sokol 2020), EGFR (Yue et al. 2021; Stevens and Williams 2021), MAPK (C. Zheng et al. 2020) and estrogen receptor regulation via cAMP-PKA signaling (Geng et al. 2020). Since these systems do not use ZNRF3/RNF43, which is the focus of this chapter, we will not discuss them further.
The four members of the R-spondin family of secreted glycoproteins (RSPO1-4) were discovered in close succession and immediately linked to activation of WNT/β-catenin signaling or stabilization of β-catenin (Kamata et al. 2004; J.-Z. Chen et al. 2002; Kazanskaya et al. 2004; K.-A. Kim et al. 2005). The first report describing RSPO2 also suggested it may negatively regulate TGF-β signaling, but it was unclear if this was a secondary consequence of WNT signaling modulation or an independent effect (Kazanskaya et al. 2004). RSPOs were later also linked to regulation of β-catenin-independent WNT signaling, in particular the WNT/planar cell polarity (PCP) pathway (Ohkawara, Glinka, and Niehrs 2011). However, the precise mechanism of WNT/β-catenin signaling regulation by RSPOs remained unclear, and their receptors unknown.
Leucine-rich repeat (LRR) G protein-coupled receptor 5 (LGR5) was discovered as a common WNT target gene in normal intestinal crypts and in colon cancer, and was later shown to be an exquisite marker of many types of WNT-driven adult stem cells (reviewed in (W. de Lau et al. 2014; Barker, Tan, and Clevers 2013)). LGR5 and its close paralogs LGR4 and LGR6 (throughout the chapter, we refer jointly to these three members of the LGR family simply as ‘LGRs’) were independently identified as RSPO receptors by several groups (Carmon et al. 2011; W. de Lau et al. 2011; Glinka et al. 2011; Ruffner et al. 2012). Shortly thereafter, ZNRF3 and RNF43 were described as the effectors of RSPO signaling (Koo et al. 2012; Hao et al. 2012). ZNRF3 and RNF43 target WNT receptors for ubiquitylation and lysosomal degradation, and binding of RSPOs to both LGRs and ZNRF3/RNF43 prevents this process by promoting clearance of ZNRF3/RNF43 from the plasma membrane. Thus, the outcome of RSPO signaling through this mechanism is the accumulation of WNT receptors at the plasma membrane, which results in increased sensitivity of cells to WNT ligands. Additionally, the heparan sulfate proteoglycan (HSPG) syndecan 4 was also identified as an RSPO3 receptor involved in activation of WNT/PCP signaling (Ohkawara, Glinka, and Niehrs 2011).
Experiments in cells and mice lacking LGR4/5/6 then led to the unexpected discovery that RSPO2 and RSPO3 can signal independently of LGRs (Lebensohn and Rohatgi 2018; Szenker-Ravi et al. 2018). Similar findings were reported in 293T cells lacking LGR4 (Park et al. 2018). LGR-independent signaling was shown to be physiologically relevant, since mice lacking LGR4/5/6 did not exhibit many of the phenotypes observed in mice lacking RSPO2 or RSPO3, suggesting that RSPO2 and RSPO3 could still promote signaling in Lgr4/5/6 triple knock-out (KO) mice (Szenker-Ravi et al. 2018). In the absence of LGRs, RSPOs were shown to use HSPGs such as glypicans and syndecans as alternative receptors to promote potentiation of WNT/β-catenin signaling through a mechanism that still required interactions between RSPOs and ZNRF3/RNF43, as well as internalization of RNF43 (Lebensohn and Rohatgi 2018; Dubey et al. 2020).
More recently, RSPO2 and RSPO3 were shown to downregulate Type I BMP receptor levels through another LGR-independent mechanism (Lee et al. 2020). In this context, RSPO binding to ZNRF3/RNF43 and to the BMP receptor bone morphogenetic protein receptor type-1A (BMPR1A, also known as ALK3) promoted internalization and degradation of BMPR1A. This mechanism is very different from the way in which RSPOs regulate WNT receptor levels: binding of RSPOs to ZNRF3/RNF43 and BMPR1A directly downregulates BMPR1A levels, whereas binding of RSPOs to ZNRF3/RNF43, LGRs and/or HSPGs indirectly upregulates WNT receptors by preventing ZNRF3/RNF43 from inducing their ubiquitin-dependent internalization and lysosomal degradation.
In the following sections, we first describe the system architecture of the different RSPO-ZNRF3/RNF43 signaling modalities introduced above, including the protein components, their relevant domains and interactions, and some of the post-translational regulation relevant to their signaling properties. We then discuss the mechanisms for each of the three signaling modalities, considering similarities and differences between them. Finally we discuss some physiological and pathological contexts in which these divergent signaling modalities operate, and consider the prospect of leveraging the modular nature of the RSPO-ZNRF3/RNF43 signaling system for therapeutic targeting.
3.A. System architecture - components, domains and interactions
The RSPO-ZNRF3/RNF43 signaling system includes five main interacting components: ligands, engagement receptors, effector receptors, target receptors and adaptors. While some of these components have been previously referred to using these terms (i.e. LGR4/5/6 have been called ‘engagement receptors’ and ZNRF3/RNF43 ‘effector receptors’ for RSPOs (Xie et al. 2013; P.-H. Chen et al. 2013)), here we define them as follows. Ligands comprise the four members of the RSPO family that initiate the signaling cascade. Engagement receptors are TM or membrane-tethered cell surface proteins that engage RSPO ligands. They include LGR4/5/6, HSPGs such as glypicans and syndecans, and the type I BMP receptor BMPR1A. Effector receptors are the TM ubiquitin ligases ZNRF3 and RNF43, which also engage RSPO ligands and transduce the signal by directly or indirectly modulating the abundance of cell surface receptors. Target receptors are the WNT receptors frizzled (FZD) and low-density lipoprotein receptor-related protein 6 (LRP6), and the type I BMP receptor BMPR1A; the final outcome of RSPO signaling is to effect changes in the cell surface abundance of these receptors, and in so doing, tune the sensitivity of cells to WNT and BMP ligands. BMPR1A is unique in that it is both an engagement receptor and a target receptor, since it binds RSPOs directly and its abundance on the cell surface is regulated by ZNRF3/RNF43. Finally, adaptors are proteins that mediate the specificity of the ZNRF3/RNF43 ubiquitin ligases towards their target receptors. Dishevelled (DVL) is the only such adaptor described so far. In the following sections we describe the domain structure of these components and the interactions relevant to RSPO-ZNRF3/RNF43 signaling (Figures 4 and 5). We focus on the mammalian proteins, but descriptions of these components in other species can be found in the various reviews cited.
Figure 4.
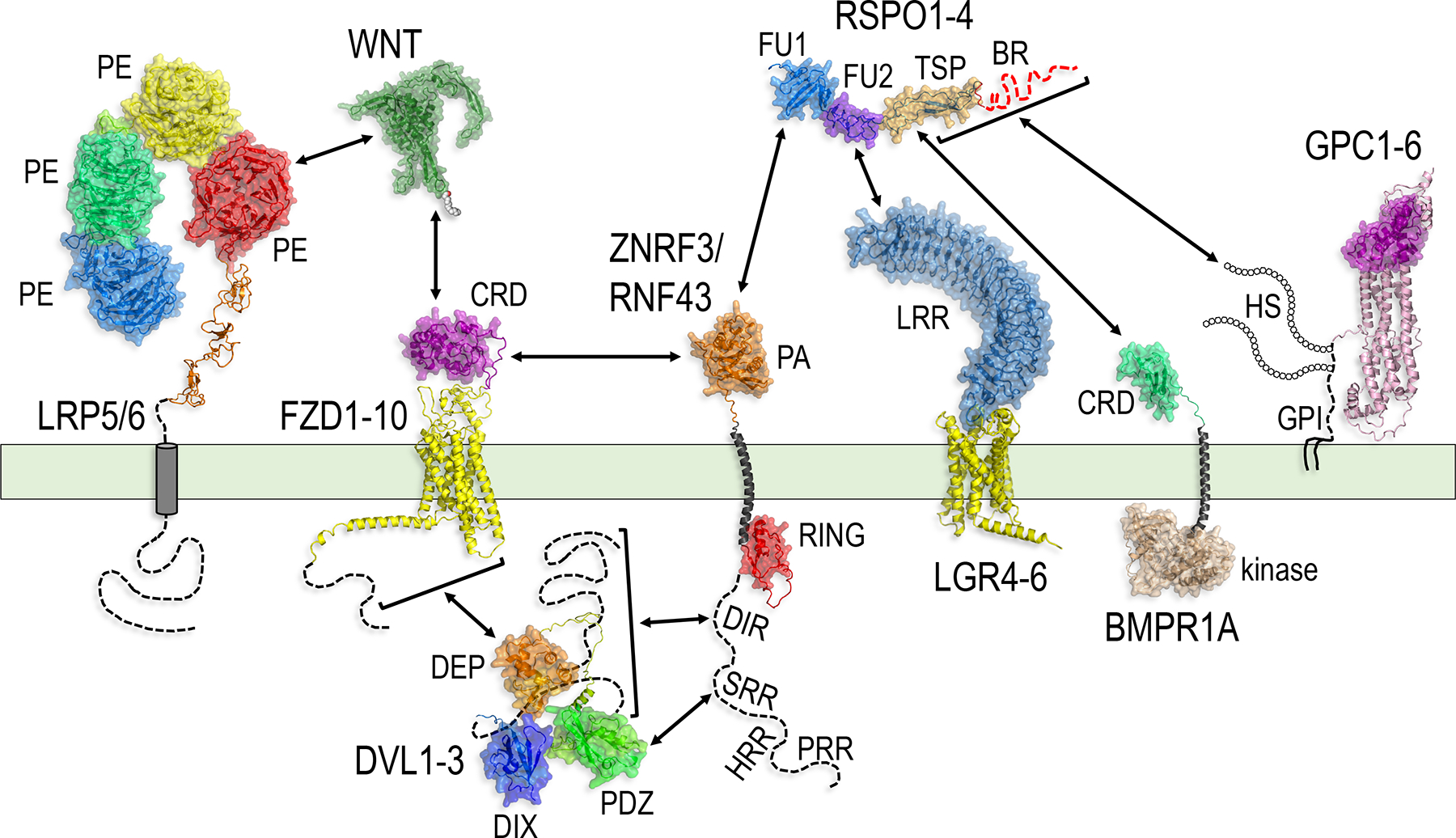
The RSPO-ZNRF3/RNF43 signaling system: components, domains and interactions.
AlphaFold models of the major components of the RSPO-ZNRF3/RNF43 system, indicating the domains and protein-protein interactions (double arrows) relevant for signal transduction. See main text for description. Dotted lines represent parts of the polypeptide chain for which the structure could not be predicted by AlphaFold. The HS chains and GPI anchor of GPC1–6 were drawn to represent their approximate sites of attachment to the polypeptide chain, but are not intended to depict their actual structures or dimensions.
Figure 5.
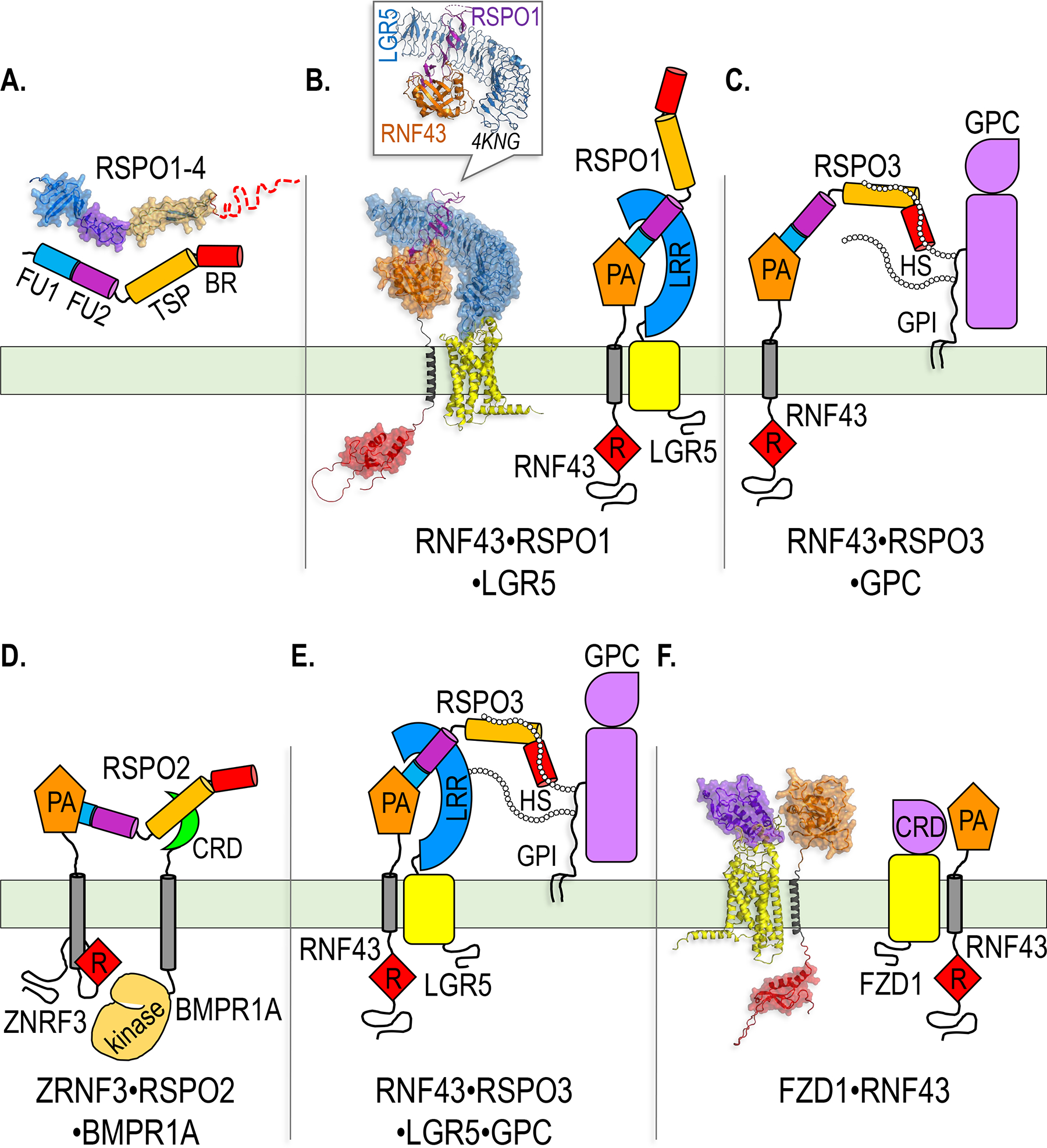
Protein complexes involved in ZNRF3/RNF43-mediated, LGR- and HSPG-dependent potentiation of WNT signaling, and BMPR1A-dependent inhibition of BMP signaling by RSPOs.
A. AlphaFold model and cartoon representation of RSPO1–4, showing the predicted modular architecture of the FU1, FU2, TSP and BR domains.
B. AlphaFold model and cartoon representation of the RNF43-RSPO1-LGR5 ternary complex that regulates WNT signaling by driving ZNRF3/RNF43 internalization and lysosomal degradation. In the model, a fragment of RSPO1 composed only of the FU1 and FU2 domains is shown, while in the cartoon representation full-length RSPO1 is shown to illustrate that the TSP/BR domains would extend into an open space not occupied by other polypeptides. The box shows the structure, solved by X-ray crystallography (PDB ID 4KNG), of the extracellular LRR domain of LGR5 and the PA domain of RNF43 bound to the RSPO1 FU1-FU2 fragment (P.-H. Chen et al. 2013). Note that the crystal structure is nearly superimposable with the AlphaFold model. In B-F, ZNRF3 or RNF43 are arbitrarily shown for illustrative purposes, but both E3s are thought to mediate all of these signaling modalities.
C and D. Cartoon representations of the ternary complexes that mediate HSPG-dependent potentiation of WNT signaling (C) and BMPR1A-dependent inhibition of BMP signaling (D) by RSPOs.
E. Cartoon representation of a hypothetical quaternary complex that could promote simultaneous LGR- and HSPG-dependent potentiation of WNT signaling by RSPOs. While the existence of such a complex has not been confirmed experimentally, it is compatible with the spatial arrangement of the relevant domains in RSPO based on solved crystal structures (B), and is consistent with the ability of the TSP/BR domains of RSPO3, as well as HSPGs, to potentiate WNT/β-catenin signaling beyond the levels promoted by the FU1-FU2 fragment and LGRs alone (Lebensohn and Rohatgi 2018; Dubey et al. 2020).
F. AlphaFold model and cartoon representation of a FZD1-RNF43 complex. The model suggests that the FZD1 CRD would interact with the PA domain of RNF43 and drive contacts between the TM helix of RNF43 and the 7TM of FZD1, potentially orienting the RING domain for ubiquitin transfer.
3.A.i. Ligands: RSPOs
RSPO1-4 are the four members of the R-spondin subfamily of thrombospondin type 1 (TSP) repeat-containing proteins. In addition to the eponymous TSP domain, all RSPOs contain two tandem Cys-rich furin-like repeats near the N-terminus, referred to as furin-like repeat 1 (FU1) and furin-like repeat 2 (FU2), connected by a flexible hinge, and a C-terminal region rich in basic amino acids (Lys and Arg), referred to as the basic region (BR) (Figures 4 and 5A). This domain architecture is highly conserved among the four RSPOs ((K.-A. Kim et al. 2008); reviewed in (W. B. M. de Lau, Snel, and Clevers 2012)), suggesting common functions. However, the length of the BR tail varies significantly between family members.
The FU1 domain of RSPOs interacts with the extracellular PA domain of ZNRF3 and RNF43 (Figures 4 and 5B–D). Conserved residues in the RSPO FU1 domain and the ZNRF3 or RNF43 extracellular domain (ECD) form an extensive interface comprising a mixture of hydrophobic and complementary charged interactions, as shown by a series of X-ray crystallographic structures (P.-H. Chen et al. 2013; Peng, de Lau, Madoori, et al. 2013; Zebisch et al. 2013; Zebisch and Jones 2015a). One distinctive feature of the FU1 domain, termed the ‘Met-finger’ because it contains a Met residue at the tip, inserts into a hydrophobic pocket in the ZNRF3/RNF43 ECD as a key determinant of the RSPO-ZNRF3/RNF43 interaction, and may account for the difference in the binding affinities between the four RSPO family members and ZNRF3/RNF43 (Zebisch et al. 2013). Point mutations in residues R66 and Q71 within the FU1 domain of RSPO1 (and corresponding residues in other RSPOs) abolish the interaction between RSPOs and ZNRF3/RNF43 (Xie et al. 2013; Zebisch et al. 2013), although there is some discrepancy between experiments about the extent to which these mutations impair potentiation of WNT signaling (Xie et al. 2013; Lebensohn and Rohatgi 2018).
The FU2 domain of RSPOs interacts with the large LRR array in the ECD of LGRs primarily through hydrophobic interactions, although charged interactions between residues in the FU1 domain and LGRs have also been described (Figures 4 and 5B) (P.-H. Chen et al. 2013; Peng, de Lau, Forneris, et al. 2013; D. Wang et al. 2013; Xu et al. 2013; Zebisch and Jones 2015a). Point mutations in residues F106 and/or F110 within the FU2 domain of RSPO1 (and corresponding residues in other RSPOs) abrogate binding of RSPOs to LGRs and eliminate potentiation of WNT signaling (Peng, de Lau, Forneris, et al. 2013; Xie et al. 2013). RSPO proteins containing point mutations in these FU2 domain residues are therefore useful reagents to study LGR-independent modes of RSPO signaling (Lebensohn and Rohatgi 2018; Dubey et al. 2020; Szenker-Ravi et al. 2018; Park et al. 2018).
The FU1 and FU2 domains used to be considered the ‘business end’ of the mature RSPO proteins (W. de Lau et al. 2014), since a fragment comprising these two domains is necessary and sufficient to potentiate WNT signaling (Kazanskaya et al. 2004; S.-J. Li et al. 2009; K.-A. Kim et al. 2008). For this reason, all of the structural studies described above were done with this minimal fragment. However, we now know that these two domains make only a partial contribution to the full repertoire of RSPO functions, since they are insufficient to signal through LGR-independent mechanisms (Lebensohn and Rohatgi 2018; Dubey et al. 2020; Lee et al. 2020) and are significantly less potent than the full-length proteins when signaling through LGRs (Lebensohn and Rohatgi 2018; Dubey et al. 2020).
The TSP and BR domains that extend from the FU1-FU2 domains interact with heparin – a glycosaminoglycan polymer that mimics heparan sulfate (HS) – and with the HS chains of HSPGs such as glypicans and syndecans (Figures 4 and 5C) (C.-F. Chang et al. 2016; Ohkawara, Glinka, and Niehrs 2011; Glinka et al. 2011; Ren et al. 2018; Nam et al. 2006; Bell et al. 2008). While the TSP and BR domains are often described separately, molecular modeling predicts that the positively charged surface of the TSP and BR domains forms a continuous binding interface for heparin (Ayadi 2008; Dubey et al. 2020). Indeed, the TSP and BR domains can individually mediate binding of RSPOs to heparin (Nam et al. 2006) and RSPO constructs containing either the TSP or BR domain can induce HSPG-dependent potentiation of WNT signaling (Lebensohn and Rohatgi 2018). The TSP domain is also required for binding of RSPO2 and RSPO3 to BMPR1A during downregulation of BMP signaling (Figures 4 and 5D) (Lee et al. 2020).
3.A.ii. Engagement receptors: LGRs, HSPGs, BMPR1A
LGR4/5/6 were identified and validated as RSPO receptors through various independent approaches (Carmon et al. 2011; W. de Lau et al. 2011; Glinka et al. 2011; Ruffner et al. 2012). They are classified as the three Class B members of the LGR subgroup of the rhodopsin family of G protein-coupled receptors (GPCRs) (reviewed in (W. de Lau et al. 2014; Barker, Tan, and Clevers 2013)) and mark stem cells in many embryonic and adult tissues ((Kinzel et al. 2014); reviewed in (Koo and Clevers 2014; Leung, Tan, and Barker 2018)). They contain a large ECD consisting of 16–17 LRRs followed by a hinge region and the distinctive 7TM domain of rhodopsin-like GPCRs (Figure 4). The concave face of the curved structure formed by the LRR array interacts with the FU2 domain of RSPOs (Figures 4 and 5B) (P.-H. Chen et al. 2013; Peng, de Lau, Forneris, et al. 2013; D. Wang et al. 2013; Xu et al. 2013; Zebisch and Jones 2015a). No G protein-coupled signaling activity triggered by binding to RSPO ligands has been reported (Carmon et al. 2011; W. de Lau et al. 2011), supporting the notion that LGRs transduce RSPO signals through other mechanisms discussed below.
HSPGs have also been implicated as engagement receptors for RSPOs (Lebensohn and Rohatgi 2018; Dubey et al. 2020; Ohkawara, Glinka, and Niehrs 2011; Ren et al. 2018; Lebensohn et al. 2016). HSPGs are a diverse class of cell surface and extracellular matrix glycoproteins decorated with HS glycosaminoglycan polysaccharide chains (reviewed in (Sarrazin, Lamanna, and Esko 2011; Christianson and Belting 2014)). Abundant carboxyl and sulfate groups on the HS chains make them polyanionic, promoting interactions with polybasic domains on proteins. While HSPGs broadly include the cell surface-associated glypicans (GPCs) and syndecans (SDCs), the secreted extracellular matrix HSPGs (agrin, perlecan, type XVIII collagen) and the secretory vesicle proteoglycan serglycin, only GPCs and SDCs have been implicated as RSPO receptors. In mammals, there are six GPCs (GPCs1-6) and four SDCs (SDC1-4). GPCs are tethered to the plasma membrane through a glycosylphosphatidylinositol (GPI) anchor (Figure 4), while SDCs are single-pass TM proteins. HS chains are attached to the protein core of GPCs close to the plasma membrane, and to the protein core of SDCs at more peripheral sites. Ligands can bind either to the protein core or to the HS chains of HSPGs. RSPOs interact with the HS chains of HSPGs through the TSP and BR domains (Figures 4 and 5C) (C.-F. Chang et al. 2016; Nam et al. 2006; Bell et al. 2008) and this interaction is required for RSPOs to potentiate WNT/β-catenin signaling (Lebensohn and Rohatgi 2018; Dubey et al. 2020; Ren et al. 2018) as well as WNT/PCP signaling (Ohkawara, Glinka, and Niehrs 2011).
The cell surface abundance of the type I BMP receptor BMPR1A can also be regulated by RSPOs (Lee et al. 2020). In this case, BMPR1A appears to be both the engagement and target receptor, since its own internalization is triggered when RSPO2 or RSPO3 bind to it and cross-link it with the effector receptors ZNRF3/RNF43. BMPR1A is one of seven type I TGFβ receptors in humans. It contains a small extracellular Cys-rich domain (CRD), a TM domain, and an intracellular juxtamembrane domain rich in Gly and Ser residues (GS domain) followed by a Ser kinase domain (Figure 4) (reviewed in (Heldin and Moustakas 2016)). The BMPR1A ECD binds with high affinity to the TSP domains of RSPO2 and RSPO3 (Figures 4 and 5D) (Lee et al. 2020). The mechanism driving BMPR1A internalization following engagement of RSPOs is unknown (see discussion below).
3.A.iii. Effector receptors: ZNRF3/RNF43
The closely related ZNRF3 and RNF43 proteins are members of the GOLIATH/GRAIL family of PA-TM-RING E3s (Figure 3) (reviewed in (W. de Lau et al. 2014; Zebisch and Jones 2015b; Hao, Jiang, and Cong 2016)). They were discovered as WNT/β-catenin target genes whose expression was correlated with that of AXIN2 mRNA in primary tissue microarray data (Hao et al. 2012), or with LGR5-GFP abundance in LGR5+ small intestinal crypt stem cells (Koo et al. 2012). Both ZNRF3 and RNF43 contribute to a negative feedback mechanism which downregulates WNT receptor levels (Hao et al. 2012; Koo et al. 2012). A genome-wide, forward genetic screen in haploid human cells designed to find attenuators of WNT/β-catenin signaling – genes that when deleted enhanced signaling in the presence a low dose WNT ligand – later uncovered ZNRF3 as the top hit (Lebensohn et al. 2016). Since the HAP1 cells in which this screen was conducted do not express RNF43 mRNA, these unbiased screen results suggest that ZNRF3 is the most potent attenuator of WNT signaling in the genome, at least in haploid human cells.
The extracellular PA domain of ZNRF3 and RNF43 interacts with the FU1 domain of RSPOs (Figures 4 and 5B–E) as discussed above. Comparison of ZNRF3 ECD structures in isolation and in complex with RSPO ligands did not reveal major conformational differences (Zebisch et al. 2013), suggesting that signal transduction upon binding of RSPOs is unlikely to be an autonomous property of the ZNRF3/RNF43 proteins, instead requiring other components of the system. The PA domain of the GOLIATH/GRAIL family member RNF128 (Figure 3) interacts with transmembrane receptors such as CD40L and CD83 and targets them for ubiquitylation (Lineberry et al. 2008), suggesting that the PA domain of ZNRF3/RNF43 may do the same for the FZD family of WNT receptors. However, data regarding an interaction between the PA domain of ZNRF3/RNF43 and the ECD of FZD is conflicting, as we discuss later. The catalytic RING domain (Figure 4) is required for ubiquitylation of the WNT receptor FZD, which leads to its internalization and lysosomal degradation, resulting in decreased sensitivity to WNT ligands (Hao et al. 2012; Koo et al. 2012). The RING domain also appears to be required for membrane clearance of BMPR1A (Lee et al. 2020).
In addition to their defining PA, TM and RING domains, ZNRF3 and RNF43 have disordered cytoplasmic extensions containing a RING-proximal dishevelled-interaction region (DIR) followed by Ser-, His-, and Pro-rich regions (SRR, HRR and PRR, respectively) (Figure 4). The DIRs of ZNRF3 and RNF43 (comprising amino acids 346–528 and 325–454 of the human proteins, respectively) interact with the C-terminal two thirds of DVL (Figure 4), and this interaction is essential for ubiquitylation-dependent downregulation of FZD receptors (Jiang et al. 2015). Since DVL also interacts with FZD (Figure 4), it has been proposed that DVL promotes WNT receptor degradation by acting as a substrate adaptor that targets ZNRF3/RNF43 to FZD (Jiang et al. 2015). Another region of the RNF43 intracellular domain (ICD) located C-terminal to the DIR (comprising amino acids 478–596 of the human protein; Figure 4) has also been postulated to interact with DVL2, and this interaction is required for DVL-mediated inhibition of β-catenin-independent WNT signaling (Tsukiyama et al. 2015). RNF43-mediated FZD ubiquitylation (Tsukiyama et al. 2020), as well as ZNRF3/RNF43 internalization (L.-S. Chang et al. 2020; M. Kim, Reinhard, and Niehrs 2021), are also regulated by phosphorylation/dephosphorylation of the ICD.
3.A.iv. Target receptors: FZDs, LRP6, BMPR1A
FZD proteins (reviewed in (Niehrs 2012; MacDonald and He 2012; Y. Wang et al. 2016; Huang and Klein 2004) were the first WNT receptors to be identified (Bhanot et al. 1996). The 10 FZDs in humans (FZD1–10) are members of the Class F of the GPCR superfamily (reviewed in (Malbon 2004; Schulte and Bryja 2007; Schulte and Wright 2018). FZDs transduce both β-catenin-dependent and β-catenin-independent WNT signals, including those in the WNT/PCP, WNT/calcium (reviewed in (Niehrs 2012)) and WNT-dependent stabilization of proteins (WNT/STOP) pathways (Acebron et al. 2014). FZDs contain an extracellular CRD followed by a linker region, a 7TM domain, and an ICD of variable length (Figure 4). The FZD CRD interacts directly with WNT ligands (Figure 4) via contacts at two opposing faces of the globular CRD, with the principal interaction involving the palmitate group of WNT docking into a hydrophobic groove in the CRD (Janda et al. 2012). Replacement of several conserved Lys residues throughout the intracellular loops of the FZD 7TM and the ICD with Arg residues abrogated changes in FZD levels in response to ZNRF3/RNF43 over-expression or depletion (Koo et al. 2012; Hao et al. 2012), suggesting that these Lys residues may be ubiquitylated by ZNRF3/RNF43. However, the relative contributions of these potential ubiquitylation sites to regulation of FZD levels by ZNRF3/RNF43 have not been determined.
Low-density lipoprotein receptor (LDLR)-related proteins (LRPs) 5 and 6, and the Drosophila ortholog Arrow, are WNT co-receptors required for WNT/β-catenin signaling but not for β-catenin-independent WNT/PCP signaling (reviewed in (X. He et al. 2004; MacDonald and He 2012)). LRP5/6 are large (>1600 amino acids) single-pass TM proteins with an ECD formed by a closely packed set of four tandem β-propeller/epidermal growth factor repeats (PE domains), followed by three LDLR type A repeats (Figure 4). The LRP6 ECD interacts with WNT ligands in a manner that allows WNTs to simultaneously bind the FZD CRD (Figure 4), bridging them into a ternary receptor complex that triggers cytoplasmic WNT signaling (Tamai et al. 2000; Bourhis et al. 2010; Hirai et al. 2019; Chu et al. 2013). While LRP6 internalization and degradation is regulated by ZNRF3/RNF43 (Hao et al. 2012; L.-S. Chang et al. 2020; Giebel et al. 2021; M. Kim, Reinhard, and Niehrs 2021), the elements in LRP6 required for this regulation remain unknown.
The type I BMP receptor BMPR1A, discussed above, is unique among the targets of RSPO-ZNRF3/RNF43-dependent regulation in that it also engages RSPOs directly (Figures 4 and 5D) (Lee et al. 2020), and can therefore be considered both a target and an engagement receptor. However, unlike in the case of WNT receptors, it is unclear whether regulation of cell surface BMPR1A abundance by RSPO-ZNRF3/RNF43-mediated endocytosis and lysosomal degradation involves BMPR1A ubiquitylation.
3.A.v. Adaptors: DVL
The three DVL proteins (DVL1–3 in humans) are crucial intracellular components of both β-catenin-dependent and β-catenin-independent WNT signaling pathways (reviewed in (Gao and Chen 2010; Sharma et al. 2018; MacDonald and He 2012)). They bind the cytoplasmic segments of FZD receptors and route WNT signals to the WNT/β-catenin or WNT/PCP pathways by forming distinct signaling complexes (reviewed in (Mlodzik 2016; Gammons and Bienz 2018)). DVLs interact with a diverse array of proteins through three highly conserved modules connected by flexible linkers that mediate their molecular functions: an N-terminal Dishevelled, Axin (DIX) domain, a central Postsynaptic density 95, Discs large, Zona occludens-1 (PDZ) domain, and a C-terminal Dishevelled, Egl-10, Pleckstrin (DEP) domain (Figure 4). The DIX domain undergoes dynamic head-to-tail homo-polymerization (Schwarz-Romond et al. 2007; Kishida et al. 1999), which leads to formation of DVL assemblies (Schwarz-Romond, Metcalfe, and Bienz 2007), and can also undergo hetero-polymerization with the related DAX domain of AXIN (Fiedler et al. 2011; Kishida et al. 1999). The PDZ domain interacts with many proteins that mediate both WNT/β-catenin and WNT/PCP signaling, and may be involved in distinguishing between these two pathways (reviewed in (Sharma et al. 2018)). The interaction between the PDZ domain and a KTXXXW motif in the intracellular C-terminal tail of FZD recruits DVL to the WNT receptor complex and is crucial for transduction of WNT signals (Umbhauer et al. 2000; H.-C. Wong et al. 2003). The DEP domain also targets DVL to the plasma membrane (reviewed in (Consonni, Maurice, and Bos 2014)). It has a positively charged surface that likely interacts with phospholipids (H. C. Wong et al. 2000; Simons et al. 2009), and the DEP domain together with the C-terminal region of DVL interacts with a discontinuous motif in the FZD ICD (Tauriello et al. 2012).
DVL has been postulated as an adaptor required for recognition of FZD by ZNRF3/RNF43, a prerequisite step in promoting FZD degradation (Jiang et al. 2015). The three-way physical interaction between ZNRF3/RNF43, DVL and FZD is essential for the WNT/β-catenin inhibitory activity of ZNRF3/RNF43. This interaction is mediated by binding of the DVL DEP domain to FZD (Figure 4), and by contacts between segments in the C-terminal two thirds of DVL (notably excluding the DIX, PDZ, and DEP domains) and the DIR of ZNRF3/RNF43 (Figure 4). Accordingly, the DEP domain, but not the DIX or PDZ domains, are required for ZNRF3/RNF43-dependent FZD downregulation, and fusion of the DEP domain to ZNRF3/RNF43 eliminates the requirement of DVL to downregulate FZD levels (Jiang et al. 2015). An interaction between the PDZ domain of DVL and a region of the RNF43 ICD located C-terminal to the DIR is essential for inhibition of β-catenin-independent signaling through an undefined mechanism (Tsukiyama et al. 2015). This inhibition does not require the ubiquitin ligase activity of RNF43, or interactions between RNF43 and FZD, and does not result in downregulation of cell surface FZD.
3.B. Signaling mechanisms
In this section we describe three modalities of RSPO signaling mediated by ZNRF3/RNF43 that regulate cell surface receptors, we contrast their salient features, and we discuss their plausible underlying molecular mechanisms. Other mechanisms through which ZNRF3/RNF43 control WNT/β-catenin signaling that do not impinge on the regulation of cell surface receptor levels will not be addressed here, but we refer the reader to the primary literature (Spit et al. 2020; Loregger et al. 2015).
3.B.i. LGR-dependent, ZNRF3/RNF43-mediated potentiation of WNT/β-catenin signaling by RSPOs
The first full picture of a mechanism driving potentiation of WNT/β-catenin signaling by RSPOs emerged with the discovery that ZNRF3 and RNF43 promote ubiquitylation-dependent internalization and lysosomal degradation of the WNT receptors FZD and LRP6 (Hao et al. 2012; Koo et al. 2012). Following internalization, RNF43 and FZD co-localize in RAB5+ early endosomes, and the final fate of FZD is lysosomal rather than proteasomal degradation, as surmised from the fact that the process can be inhibited by the lysosomal inhibitor bafilomycin A, but not the proteasome inhibitor MG132 (Koo et al. 2012). Subsequent studies showed that at least three conditions contribute to ZNRF3/RNF43-mediated internalization and degradation of WNT receptors: 1. interaction of the adaptor protein DVL with both FZD and ZNRF3/RNF43 (Jiang et al. 2015), 2. phosphorylation of Ser residues in the SRR of ZNRF3/RNF43 (Tsukiyama et al. 2020) and 3. dephosphorylation of a 4Tyr motif in the DIR of ZNRF3 (L.-S. Chang et al. 2020; M. Kim, Reinhard, and Niehrs 2021). Therefore, in the absence of RSPOs, clearance of FZD and LRP6 from the plasma membrane results in decreased sensitivity of cells to WNT ligands.
In the presence of RSPO ligands, binding of the FU1 domain of RSPO to the PA domain of ZNRF3/RNF43 and of the FU2 domain of RSPO to the LRRs of LGR4/5/6 results in formation of a ternary complex (Figures 4 and 5B) (Xie et al. 2013; Moad and Pioszak 2013; P.-H. Chen et al. 2013; Zebisch and Jones 2015a). This molecular assembly triggers internalization of ZNRF3/RNF43, followed by lysosomal degradation, through a poorly understood process that requires the catalytic RING domain of ZNRF3/RNF43 (Hao et al. 2012) and can be counteracted through deubiquitylation of ZNRF3/RNF43 by the DUB USP42 (Giebel et al. 2021). As a consequence of ZNRF3/RNF43 clearance from the plasma membrane, ubiquitylation-dependent internalization and lysosomal degradation of FZD and LRP6 is diminished, leading to their accumulation on the cell surface (Hao et al. 2012). The outcome of this RSPO signaling modality is to increase the sensitivity of cells to WNT ligands.
Because ZNRF3/RNF43 and LGRs do not interact directly with each other, the secreted RSPOs must engage both of them simultaneously through the adjacent FU1 and FU2 domains, respectively, acting as molecular cross-linkers (Figure 5B) (Zebisch and Jones 2015b). The TSP and BR domains of RSPOs would appear to be dispensable for this mode of signaling, since they escape contact with either ZNRF3/RNF43 or LGRs. This is partially borne out by the fact that a fragment comprising only the FU1 and FU2 domains of RSPOs is sufficient to promote WNT/β-catenin signaling in cells and support the growth of small intestinal organoids (Kazanskaya et al. 2004; S.-J. Li et al. 2009; K.-A. Kim et al. 2008; Peng, de Lau, Forneris, et al. 2013). However, while this FU1-FU2 construct displays full signaling efficacy at sufficiently high concentrations, it is much less potent than the full-length protein containing the TSP/BR domains both in cells and in small intestinal organoids (K.-A. Kim et al. 2008; Lebensohn and Rohatgi 2018; Dubey et al. 2020), demonstrating that the TSP/BR domains contribute to signaling even in the presence of LGRs (Figure 5E).
3.B.ii. HSPG-dependent, ZNRF3/RNF43-mediated potentiation of WNT/β-catenin signaling by RSPOs
Unexpectedly, RSPO2 and RSPO3, but not RSPO1 or RSPO4, are capable of potentiating WNT/β-catenin signaling in cells and animals lacking LGRs, albeit with lower potency and efficacy than in cells containing LGRs (Lebensohn and Rohatgi 2018; Szenker-Ravi et al. 2018; Park et al. 2018). Furthermore, full length RSPO3 containing inactivating mutations in the LGR-interacting FU2 domain could still promote WNT/β-catenin signaling in haploid human cells, again with lower potency and efficacy than the wild type (WT) counterpart (Lebensohn and Rohatgi 2018). Similarly, RSPO2 and RSPO3 constructs lacking the BR domain and containing inactivating mutations in the LGR-interacting FU2 domain could also potentiate WNT/β-catenin signaling in HEK293 cells (Szenker-Ravi et al. 2018). These experiments, in which potentiation of WNT/β-catenin signaling was partially retained following perturbations of either the LGR receptors or the LGR-binding FU2 domain on the RSPO ligands themselves, conclusively demonstrated the capacity of RSPOs to signal independently of LGRs. This begged an urgent question: is there an alternative engagement receptor for RSPOs?
To answer that question, we mapped the domains on RSPO3 required for signaling in the absence of LGRs through mutagenesis, domain deletion and domain swapping experiments (Lebensohn and Rohatgi 2018). The ZNRF3/RNF43-interacting FU1 domain, and the HS-interacting TSP and/or BR domains of RSPO3 were required (constructs lacking either the TSP or the BR domain, but not both, could support signaling) (Figures 4 and 5C). Furthermore, replacing the TSP/BR domains of RSPO3 with those of RSPO1, which cannot signal without LGRs, did not impair signaling. These results suggested that LGR-independent signaling may be mediated by electrostatic interactions between the TSP and/or BR domains and the HS chains of HSPGs. Modeling of the TSP/BR domains of RSPO3 predicted two positively charged grooves lined by basic Lys and Arg residues that could potentially dock HS chains like those present in HSPGs (Dubey et al. 2020). Indeed, signaling by RSPO3 in cells lacking all LGRs was nearly completely abolished by three different manipulations that disrupted the interaction between the TSP/BR domains and the HS chains of HSPGs: 1. mutation of some Lys/Arg residues in the TSP/BR domains to charge-reversing Glu residues; 2. addition of heparin, which competes for binding to the HS chains of HSPGs; and 3. disruption of the gene encoding EXTL3, a glycosyltransferase specifically required for HSPG biosynthesis but dispensable for the synthesis of other glycosaminoglycans and proteoglycans (Lebensohn and Rohatgi 2018; Dubey et al. 2020).
The conclusive demonstration that interactions between the TSP and/or BR domains of RSPOs and the HS chains of HSPGs mediate LGR-independent signaling (Figure 5C) came from ligand engineering experiments (Dubey et al. 2020). A synthetic RSPO3 construct in which the entire TSP and BR domains were replaced with a single-chain variable fragment (scFv) that specifically binds to the HS chains of GPCs potentiated WNT/β-catenin signaling with the same potency and efficacy as WT RSPO3 in cells lacking LGRs. Experiments in which individual or entire families of HSPGs (including all GPCs or all SDCs) were eliminated in haploid human cells demonstrated that RSPO3 can signal in a redundant manner via either GPCs, SDCs or potentially another HSPG by engaging their HS chains rather than their protein cores (Dubey et al. 2020). Furthermore, genome-wide screens in haploid human cells lacking LGR4/5/6 did not reveal additional receptors required for potentiation of WNT signaling by RSPO3, making HSPGs the most likely engagement receptors for RSPOs in the absence of LGRs (Dubey et al. 2020).
The ZNRF3/RNF43-binding FU1 domain is also required for LGR-independent signaling (Figure 5C) (Lebensohn and Rohatgi 2018; Szenker-Ravi et al. 2018; Park et al. 2018). In fact, it is the FU1 domain rather than the HSPG-interacting TSP/BR domains that determines whether a given RSPO family protein can signal in the absence of LGRs. This was demonstrated by domain-swapping experiments in which the FU1 domain of RSPO3 conferred on RSPO1 the ability to signal without LGRs, and conversely an RSPO3 chimera containing the FU1 domain of RSPO1 lost its ability to signal without LGRs (Lebensohn and Rohatgi 2018). Because the affinities of the FU1 domains from RSPO3 (KD ~60 nM) and from RSPO1 (KD ~6.8 μM) towards ZNRF3 are markedly different (Zebisch et al. 2013), this difference may determine the requirement for LGRs (Lebensohn and Rohatgi 2018). We speculate that in the presence of a high-affinity interaction between the FU1 domain of RSPO2 (KD ~25 nM) or RSPO3 (KD ~60 nM) and ZNRF3 (Zebisch et al. 2013), the interaction between the FU2 domain and LGRs can be functionally replaced by the interaction between the TSP/BR domains and HSPGs. However, the lower-affinity interaction between the FU1 domain of RSPO1 (KD ~6.8 μM) or RSPO4 (KD ~300 μM) and ZNRF3 (Zebisch et al. 2013) would require the high-affinity interaction between the FU2 domain and LGRs (KD ~2–3 nM) (W. de Lau et al. 2011; Glinka et al. 2011; Zebisch et al. 2013) in order to signal.
While the TSP/BR domains are not required for signaling in the presence of LGRs, they substantially increase the potency of signaling by RSPOs in cells and small intestinal organoids (K.-A. Kim et al. 2008; Lebensohn and Rohatgi 2018; Dubey et al. 2020). In fact, at lower concentrations of RSPO3, the interaction of the FU2 domain with LGRs is not sufficient to drive efficient endocytosis of RNF43, and HSPG binding mediated by the TSP/BR domains is also required even in the presence of LGRs (Dubey et al. 2020). Furthermore, at limiting concentrations, RSPO3 was significantly more potent than RSPO1 in supporting the growth of intestinal organoids (Greicius et al. 2018), consistent with the ability of RSPO3 but not RSPO1 to signal through both LGR-dependent and LGR-independent mechanisms ((K.-A. Kim et al. 2008; Lebensohn and Rohatgi 2018; Dubey et al. 2020)). Although none of the structural studies discussed earlier included the TSP/BR domains of RSPOs, one of the structural models of the LGR5-RSPO1-RNF43 ternary complex suggested that the TSP/BR domains would extend into an open space not occupied by other polypeptides (Figure 5B) (P.-H. Chen et al. 2013), and would therefore be available to interact with other molecules such as HSPGs. This would allow RSPOs to bind two engagement receptors – LGRs and HSPGs – and an effector receptor – ZNRF3 or RNF43 – simultaneously (Figures 4 and 5E), consistent with the ability of HSPGs to potentiate LGR-dependent signaling (Dubey et al. 2020). Therefore, HSPGs may enhance the potency of RSPO signaling by trapping RSPOs near the cell surface, increasing their local concentration and promoting binding to LGRs. In support of this model, depletion of HS chains or removal of the TSP/BR domains reduces binding of RSPOs to the cell surface, while depletion of LGR4 does not (Ren et al. 2018).
We and others initially referred to the modality of RSPO signaling that takes place in the absence of LGRs as ‘LGR-independent’ (Lebensohn and Rohatgi 2018; Szenker-Ravi et al. 2018; Park et al. 2018), but it has since been shown to happen in more than one ‘variety’ (see next section). Therefore, in the context of the WNT/β-catenin pathway, where in the absence of LGRs RSPO signaling is mediated by HSPGs (Lebensohn and Rohatgi 2018; Dubey et al. 2020), we will henceforth refer to this modality as ‘HSPG-dependent’ RSPO signaling.
3.B.iii. BMPR1A-dependent, ZNRF3/RNF43-mediated inhibition of BMP signaling by RSPOs
A third mechanism of signaling by RSPOs, also independent of LGRs but mediated by ZNRF3/RNF43, has recently been described (Lee et al. 2020). In this case, RSPO2 and RSPO3, but not RSPO1 or RSPO4, antagonize BMP signaling in a process that is independent of WNT/β-catenin and WNT/PCP signaling. RSPO2 and RSPO3 interact directly with ZNRF3 and the type I BMP receptor BMPR1A (Figures 4 and 5D), triggering internalization and lysosomal degradation of BMPR1A. This results in decreased sensitivity of target cells to BMP ligands.
Domain analysis revealed that the FU1 and TSP domains of RSPO2 are required to antagonize BMP signaling (Figure 5D) (Lee et al. 2020). RSPO2 interacts with the BMPR1A ECD with high affinity (KD ~4.8 nM), comparable to the FU2-mediated RSPO-LGR interaction. The TSP domain of RSPO2 and RSPO3, but not the FU1, FU2 or BR domains, is required for binding to the BMPR1A ECD. Furthermore, domain-swapping experiments revealed that the capacity to downregulate BMP receptor levels resides in the TSP domain: while WT RSPO1 did not antagonize BMP signaling, an RSPO1 chimera containing the TSP domain of RSPO2 bound to BMPR1A and antagonized BMP signaling. siRNA-mediated knock-down of ZNRF3/RNF43 or overexpression of a dominant negative ZNRF3 lacking the RING domain prevented RSPO2-induced destabilization of BMPR1A and inhibition of BMP signaling. On the other hand, siRNA-mediated knock-down of LGR4/5 did not affect inhibition of BMP signaling by RSPO2. These results suggest that BMP antagonism by RSPO2 requires ZNRF3/RNF43 but not LGRs. Consistent with these requirements, the ZNRF3/RNF43-binding FU1 domain of RSPO2, but not the LGR-binding FU2 domain, was required to antagonize BMP receptor signaling (Lee et al. 2020).
RSPO2 triggers BMP receptor clearance from the cell surface by acting as a cross-linking ligand between BMPR1A and ZNRF3 (Figure 5D) (Lee et al. 2020). In vitro binding assays and colocalization experiments demonstrated that ZNRF3 interacted with BMPR1A in the presence of RSPO2, and formation of a ZNRF3-RSPO2-BMPR1A ternary complex depended on the FU1 and TSP domains. In cells that produce RSPO2, BMPR1A was absent from the plasma membrane but colocalized with ZNRF3 in cytoplasmic vesicles, as well as with the early endosome marker EEA1 and the lysosome marker Lamp1. Knock-down of RSPO2 abolished endosomal and lysosomal localization, and resulted in accumulation of BMPR1A at the plasma membrane. Therefore, RSPO2 bridges ZNRF3 and BMPR1A, and routes the ternary complex for lysosomal degradation, antagonizing BMP signaling. The authors proposed that a similar mechanism applies to RSPO3, but not RSPO1 or RSPO4 (Lee et al. 2020).
3.B.iv. Comparing different modalities of RSPO-ZNRF3/RNF43 signaling
The three different modalities of ZNRF3/RNF43-mediated RSPO signaling described so far, LGR-dependent potentiation of WNT/β-catenin signaling, HSPG-dependent potentiation of WNT/β-catenin signaling, and BMPR1A-dependent antagonism of BMP signaling, illustrate the versatile modularity of the RSPO-ZNRF3/RNF43 signaling system (Table 1). These signaling modes are defined by a ‘combinatorial code’ in which the FU1, FU2, TSP and/or BR domains of RSPOs interact with different combinations of engagement, effector and target receptors to modulate the WNT/β-catenin or BMP pathways (Figures 4, 5 and Table 1). Furthermore, differences in the extent to which individual domains of distinct RSPO ligands interact with these receptors, presumably determined by their binding affinities, dictates the modalities through which each RSPO ligand can signal (Table 1). Finally, depending on whether RSPOs engage target receptors and directly promote their membrane clearance, as in the case of BMPR1A, or indirectly affect target receptor levels by modulating ZNRF3/RNF43 ubiquitin ligase activity, as in the case of the WNT receptors FZD and LRP6, the functional outcome is either down- or up-regulation of the signaling pathway, respectively (Table 1).
TABLE 1.
Summary of the various signaling modalities regulated by the RNF43/ZNRF3 E3s
| Signaling modality | RSPO ligands | Required domains in RSPO | Engagement receptors | Effector receptors | Adapter | Target receptors | Up/down-regulation of target receptor by RSPO | Direct/indirect effect of RSPO binding on target receptor levels | Ubiquitylation of target receptor? |
|---|---|---|---|---|---|---|---|---|---|
| LGR-dependent regulation of WNT/CTNNB1 signaling | RSPOl-4 | FUI, FU2 | LGR4–6 | ZNRF3/RNF43 | DVL | FZD, LRP6 | Up-regulation | Indirect | Yes |
| HSPG-dependent regulation of WNT/CTNNB1 signaling | RSP02/3 | FUI, TSP/BR | HSPGs (GPCs, SDCs) | ZNRF3/RNF43 | DVL? | FZD, LRP6? | Up-regulation | Indirect | Yes |
| BMPRIA-dependent regulation of BMP signaling | RSP02/3 | FUI, TSP | BMPR1A | ZNRF3/RNF43 | ? | BMPR1A | Down-regulation | Direct | ? |
In accordance with the opposite ways in which ZNRF3/RNF43 function during regulation of the WNT receptors FZD and LRP6 versus regulation of the BMP receptor BMPR1A (Table 1), the molecular mechanisms leading to membrane clearance and lysosomal degradation of target receptors are different between the two pathways. Furthermore, the molecular mechanisms of ZNRF3/RNF43 internalization and lysosomal degradation triggered by binding of RSPOs to ZNRF3/RNF43 and to the different engagement receptors have not been fully elucidated. In the following two sections we discuss potential mechanisms controlling ZNRF3/RNF43-dependent membrane clearance and degradation of target receptors in the WNT and BMP pathways, as well as those controlling RSPO-dependent internalization of ZNRF3/RNF43 and engagement receptors. In the case of BMPR1A, these mechanisms are one and the same.
3.B.v. Molecular mechanisms controlling membrane clearance of target receptors in the WNT pathway
In the context of WNT signaling, ZNRF3/RNF43-dependent ubiquitylation of the target receptor FZD on Lys residues within the cytoplasmic loops of the 7TM domain and/or the C-terminal tail targets FZD to RAB5+ early endosomes and CD63+ lysosomes (Koo et al. 2012; Hao et al. 2012). This results in FZD internalization and lysosomal degradation, leading to decreased sensitivity to WNT ligands. ZNRF3 and RNF43 are most likely co-internalized with FZD – RNF43 co-localized with FZD5 in internal vesicles (Koo et al. 2012) – and this endocytic process is regulated by phosphorylation/dephosphorylation of a conserved ‘4Tyr’ motif within the DIR of ZNRF3 (L.-S. Chang et al. 2020; M. Kim, Reinhard, and Niehrs 2021).
Several lines of evidence demonstrate that the ubiquitin ligase activity of ZNRF3/RNF43 is required for FZD ubiquitylation. Overexpression of WT ZNRF3 or RNF43 increased ubiquitylation of FZD, decreased cell surface FZD levels and reduced WNT-induced pathway activity (Hao et al. 2012; Koo et al. 2012). Conversely, overexpression of ZNRF3 or RNF43 mutants containing inactivating point mutations in or altogether lacking the catalytic RING domain suppressed ubiquitylation, increased the plasma membrane expression and extended the half-life of FZD, abolishing the inhibitory effect of ZNRF3/RNF43 on WNT signaling (Hao et al. 2012; Koo et al. 2012; Loregger et al. 2015). Inactivating mutations in or deletion of the RING domain of ZNRF3/RNF43 also enhanced WNT-induced pathway activity by acting in a dominant-negative fashion (Hao et al. 2012; Koo et al. 2012; Loregger et al. 2015). Furthermore, FZD variants in which all conserved cytoplasmic Lys residues were mutated to Arg were not internalized upon expression of RNF43 (Koo et al. 2012), and the membrane levels of these FZD mutants did not increase upon depletion of ZNRF3 (Hao et al. 2012). Ubiquitylation of FZD was reduced in cells lacking the WNT pathway scaffold protein DVL, which as discussed earlier may serve as an adaptor that targets ZNRF3/RNF43 to FZD (Jiang et al. 2015). Finally, ZNRF3 and RNF43 could be co-immunoprecipitated with FZD (Hao et al. 2012; Koo et al. 2012). This compilation of experiments strongly supports a model in which ZNRF3/RNF43 directly ubiquitylate FZD, but we note that FZD ubiquitylation by ZNRF3/RNF43 has not been reconstituted in vitro with purified components.
A recently described ‘phospho-switch’ also modulates the ability of ZNRF3/RNF43 to regulate WNT receptor levels (Tsukiyama et al. 2020). Phosphorylation by casein kinase 1 of three Ser residues located in the SRR of RNF43 (Ser474, Ser475 and Ser476, also conserved in ZNRF3) was required for downregulation of cell surface FZD and for suppression of WNT/β-catenin signaling. Phosphorylation of RNF43 at these residues promoted ubiquitylation of FZD, and in turn its endocytosis and lysosomal degradation. The precise mechanism underlying regulation of FZD ubiquitylation by this phospho-switch remains unknown, but does not appear to involve changes in the protein-protein interactions (including binding to the E2 UbcH5C), oligomerization state or subcellular localisation of RNF43 (Tsukiyama et al. 2020).
How do ZNRF3/RNF43 recognize FZD for ubiquitylation? As discussed earlier, it has been proposed that DVL, which binds both the DIR of ZNRF3 and the ICD of FZD (Figure 4), acts as a substrate adaptor that targets ZNRF3/RNF43 to FZD (Jiang et al. 2015). Furthermore, direct binding of the ZNRF3/RNF43 PA domain to the CRD of FZD (Figure 4) has also been proposed as a recognition mechanism (Tsukiyama et al. 2015), but this subject is still debated ((Radaszkiewicz and Bryja 2020) and reviewed by (Tsukiyama, Koo, and Hatakeyama 2021)). One study detected an interaction between the RNF43 PA domain and the FZD CRD (Tsukiyama et al. 2015), while others did not (Peng, de Lau, Madoori, et al. 2013; Jiang et al. 2015). Several studies showed that deletion or replacement of the PA domain prevented ZNRF3/RNF43 from promoting FZD internalization and suppressing WNT/β-catenin signaling (Tsukiyama et al. 2015; Koo et al. 2012; Spit et al. 2020; Moffat et al. 2014), while another study found that deletion of the PA domain had none of these effects (Radaszkiewicz and Bryja 2020). AlphaFold modeling suggests that the FZD1 CRD is well positioned to interact with the PA domain of RNF43, which could drive contacts between the TM helix of RNF43 and the 7TM of FZD, and orient the RING domain for ubiquitin transfer (Figure 5F). So while the question of how FZD is recognized as a substrate by ZNRF3/RNF43 is still unresolved, one possibility is that extracellular contacts between the ZNRF3/RNF43 PA domain and the FZD CRD, intramembrane packing of ZNRF3/RNF43 and FZD TM helices, and intracellular interactions mediated by DVL all play a role in substrate recognition.
LRP6 internalization and degradation is also regulated by ZNRF3/RNF43 (Hao et al. 2012; L.-S. Chang et al. 2020; Giebel et al. 2021; M. Kim, Reinhard, and Niehrs 2021) and while ZNRF3 could be co-immunoprecipitated with LRP6 (Hao et al. 2012), no single domain or motif in LRP6 has been identified as a target of ubiquitylation or regulation by ZNRF3/RNF43. Therefore the mechanism of LRP6 receptor regulation by the RSPO-ZNRF3/RNF43 system has not been determined. Some possibilities include direct ubiquitylation by ZNRF3/RNF43 – although this has not been demonstrated experimentally – or co-internalization with FZD, mediated by WNT ligands or other mutual binding partners.
3.B.vi. Molecular mechanisms controlling membrane clearance of ZNRF3/RNF43 and engagement receptors
Importantly, ubiquitylation and internalization of WNT receptors is not regulated directly by interactions between RSPOs and these target receptors, but is instead prevented indirectly as a result of RSPO binding to and downregulating ZNRF3/RNF43 through LGR-dependent and/or HSPG-dependent mechanisms (Table 1). On the other hand, downregulation of BMPR1A is the direct result of RSPOs interacting with and promoting the internalization of ZNRF3/RNF43 (Table 1). Therefore, the mechanisms controlling membrane clearance of ZNRF3/RNF43 are crucial to the regulation of target receptors in both the WNT and BMP pathways.
During LGR-dependent signaling, binding of RSPOs to both ZNRF3/RNF43 and LGRs is required for internalization of the ternary complex, since mutation of key residues in the ZNRF3/RNF43-interacting FU1 domain or the LGR-interacting FU2 domain of RSPOs abolishes potentiation of WNTβ-catenin signaling (Xie et al. 2013; Peng, de Lau, Forneris, et al. 2013; Zebisch et al. 2013). However, the precise molecular mechanism whereby formation of this ternary complex drives its internalization is not fully understood. One model is that RSPO acts as a cross-linking ligand that couples ZNRF3/RNF43 to LGRs, and since LGR5 undergoes constitutive clathrin-mediated endocytosis (Snyder et al. 2013, 2017), mere coupling could result in the co-internalization of ZNRF3/RNF43. This is consistent with the finding that RSPO-triggered WNT/β-catenin signaling requires clathrin-mediated endocytosis (Glinka et al. 2011). Further support of this model comes from the fact that synthetic RSPO ligands that cross-link ZNRF3/RNF43 to constitutively endocytosed receptors can trigger ZNRF3/RNF43 internalization and upregulate WNT signaling. Engineered ‘surrogate RSPO’ bispecific ligands comprising a ZNRF3 or RNF43-specific scFv fused to the immune cytokine IL-2, which binds to the constitutively internalized IL-2 receptor CD25, leads to co-internalization of ZNRF3 and stimulation of WNT signaling in CD25+ cells (Luca et al. 2020). Additionally, synthetic RSPO2 ligands retaining only the ability to bind ZNRF3/RNF43 through the FU1 domain and fused to scFvs targeting them to the liver-specific asialoglycoprotein receptor (ASGR), which is predominantly expressed on hepatocytes and undergoes rapid endocytosis, increased cell surface FZD and enhanced WNT signaling specifically in cells that express ASGRs (Zhengjian Zhang et al. 2020). Similar results were obtained when these synthetic RSPO2 ligands were fused to the ubiquitously expressed cell surface receptor transferrin receptor 1, which undergoes continuous endocytosis (Zhengjian Zhang et al. 2020). Finally, the need for RSPOs can be bypassed altogether as long as their cross-linking functionality is provided: appending DmrA and DmrC heterodimerization domains to the C-termini of ZNRF3 and LGR4, respectively, enabled the membrane clearance of ZNRF3 in response to addition of an A/C dimerizer (Hao et al. 2012).
These disparate systems demonstrate that cross-linking ZNRF3/RNF43 to a constitutively endocytosed cell surface receptor, whether it be through RSPOs themselves or other artificial cross-linkers, can clear ZNRF3/RNF43 from the plasma membrane and promote upregulation of WNT receptors. However, there is evidence that cross-linking of ZNRF3/RNF43 to the engagement receptors is not sufficient in all physiological contexts, and ubiquitylation/deubiquitylation of ZNRF3/RNF43, or potentially engagement receptors, is also involved in regulating their internalization. First, the RING domain of ZNRF3 and RNF43 is required for RSPO1 (or for the A/C dimerizer discussed above) to reduce the membrane level of ZNRF3 (Hao et al. 2012), suggesting that membrane clearance requires the ubiquitin ligase activity of ZNRF3/RNF43. Furthermore, the intracellular portion of ZNRF3 and the full-length protein purified by immunoprecipitation exhibit RING domain-dependent auto-ubiquitylation in in vitro ubiquitylation assays (Hao et al. 2012; L.-S. Chang et al. 2020). Therefore, one possibility is that auto-ubiquitylation of ZNRF3 is required for internalization, although this mechanism has not been directly demonstrated. Second, deubiquitylation of ZNRF3/RNF43 by the DUB USP42 stabilizes ZNRF3/RNF43 at the plasma membrane and ‘stalls’ the LGR4-RSPO-ZNRF3/RNF43 ternary complex, preventing its clearance from the cell surface (Giebel et al. 2021). In this way USP42 antagonizes RSPOs by protecting ZNRF3/RNF43 from RSPO- and ubiquitin-dependent internalization, thereby increasing the ubiquitylation and turnover of FZD and LRP6 receptors, and inhibiting WNT signaling. Since ubiquitylation of membrane proteins can drive their internalization (reviewed in (MacGurn, Hsu, and Emr 2012)), auto-ubiquitylation of ZNRF3/RNF43 in response to RSPOs may therefore be a second mechanism promoting membrane clearance of ZNRF3/RNF43.
Alternatively or in addition to auto-ubiquitylation, ubiquitylation of another substrate by ZNRF3/RNF43, for instance the engagement receptors themselves, may promote endocytosis of the receptors and associated ZNRF3/RNF43 molecules. This hypothesis is supported by the fact that bringing RNF43 in close proximity to transmembrane proteins, including a synthetic GFP-TM-NanoLuc construct as well as the endogenous immune checkpoint protein programmed death-ligand 1 (PD-L1), can promote their internalization and lysosomal degradation (Cotton et al. 2021). In the case of PD-L1, a synthetic bispecific IgG, or ‘abTAC’, that bound to the ECDs of both RNF43 and PD-L1 was used to recruit RNF43 to PD-L1. Since neither of these two proteins are internalized or degraded constitutively, this experiment showed that recruitment of ZNRF3 and a target TM protein in close proximity is sufficient to induce internalization and lysosomal degradation of the target protein, independently of RSPOs. Therefore, simultaneous binding of RSPO ligands to ZNRF3/RNF43 and engagement receptors, which would bring them in close proximity, may be sufficient to promote ubiquitylation of the engagement receptors and internalization of the ternary complex. However, whether RSPOs actively regulate the ubiquitin ligase activity of ZNRF3/RNF43, and therefore affect the endocytic efficiency of this process, remains an unanswered question.
In the case of HSPG-dependent signaling, we surmise that RSPO2/3-mediated cross-linking of ZNRF3/RNF43 and HSPGs (Figure 5C) promotes ternary complex co-internalization driven by endocytosis of HSPGs. HSPGs are autonomous endocytosis receptors that can mediate the internalization of growth factors and morphogens among other ligands (reviewed in (Christianson and Belting 2014). They can undergo constitutive or ligand-induced endocytosis, followed in some cases by lysosomal degradation ((Burbach et al. 2003; Ilia V. Fuki, Meyer, and Williams 2000; I. V. Fuki et al. 1997; Wittrup et al. 2009). During HSPG-dependent potentiation of WNT/β-catenin signaling by RSPO3, RNF43 is internalized in a process that requires the interaction of the TSP/BR domains with HSPGs (Dubey et al. 2020). Since GPCs are tethered to the plasma membrane through a GPI anchor (Figure 4) and do not have an cytoplasmic domain that can be ubiquitylated by ZNRF3/RNF43, ternary complex internalization cannot be driven by ubiquitylation of the engagement receptor.
In contrast to the indirect regulation of WNT receptor internalization by RSPOs, BMPR1A clearance from the plasma membrane is driven by direct binding of RSPOs to both BMPR1A and ZNRF3/RNF43 (Table 1 and Figure 5D), which promotes internalization and lysosomal degradation of the ternary complex (Lee et al. 2020). The molecular mechanism through which the ZNRF3/RNF43-RSPO2/3-BMPR1A complex is internalized has not been defined. In this case, internalization of BMPR1A is the step being regulated rather than being a constitutive process like the endocytosis of LGRs or HSPGs. Therefore, the mere cross-linking of BMPR1A and ZNRF3/RNF43 by RSPOs would not be sufficient to drive internalization of either receptor. We surmise that ubiquitylation of either ZNRF3/RNF43 or BMPR1A, induced by binding of RSPO2 or RSPO3, is likely the main mechanism driving internalization of the ternary complex.
In summary, we described two molecular mechanisms that could drive ZNRF3/RNF43 internalization and lysosomal degradation: 1. co-internalization of ZNRF3/RNF43 promoted by RSPO-mediated cross-linking to a constitutively endocytosed engagement receptor, and 2. endocytosis driven by ubiquitylation of ZNRF3/RNF43, engagement receptors or both, promoted by RSPO-mediated ternary complex formation. The latter could be driven by regulated auto-ubiquitylation of ZNRF3/RNF43 or trans-ubiquitylation of the engagement receptors.
3.C. Physiological, pathological and therapeutic implications of modular RSPO-ZNRF3/RNF43 signaling
The RSPO-ZNRF3/RNF43 system has important functions during embryonic development and in adult tissue homeostasis. Aberrant regulation caused by mutations in ZNRF3/RNF43 or by RSPO fusions that cause elevated expression can lead to cancer. We refer the reader to some excellent reviews on the physiology and pathology of the RSPO-ZNRF3/RNF43 system (W. B. M. de Lau, Snel, and Clevers 2012; Nagano 2019; Raslan and Yoon 2019; Bugter, Fenderico, and Maurice 2021; Hao, Jiang, and Cong 2016; Steege, ter Steege, and Bakker 2021; Jin and Yoon 2012). Here we describe some of the principal phenotypes caused by disruption of different components of the system, and discuss how the discovery of the three RSPO signaling modalities presented earlier compels us to re-interpret these phenotypes. We also posit that the modular nature of RSPO proteins presents a unique opportunity to manipulate the RSPO-ZNRF3/RNF43 system for therapeutic benefit.
To the best of our knowledge, the comprehensive phenotype of the ubiquitous Znrf3/Rnf43 double KO mouse has not been published, but would be predicted to result in early embryonic lethality. However, conditional Znrf3/Rnf43 KO in the intestinal epithelium (driven by Cyp1a1-cre or Villin-creERT2) resulted in marked expansion of the proliferative compartment (with hyperproliferative cells containing high levels of β-catenin), upregulation of WNT target genes, and increased numbers of intestinal stem and Paneth cells (Koo et al. 2012). Clonal deletion of Znrf3/Rnf43 in the intestinal epithelium or in intestinal stem cells (driven by Lgr5-creERT2) resulted in adenoma formation with continuous expansion of stem cells and generation of Paneth cells, but no other differentiated cell types (Koo et al. 2012). Intestinal organoids derived from Znrf3/Rnf43 KO compound mutant mice grew faster than controls and lost the dependence on RSPO1 supplementation, but not on secreted WNT3A, consistent with the role of ZNRF3/RNF43 in mediating RSPO-dependent potentiation of WNT signaling (Koo et al. 2012). The WNT/β-catenin pathway is also a major regulator of liver metabolic zonation, development and regeneration (reviewed in (Monga 2014; Hu and Monga 2021; Yang et al. 2014)). Inducible, systemic combined deletion of Znrf3 and Rnf43 (driven by Rosa26-creERT2) induced hepatocyte proliferation and extended metabolic zonation, measured as a marked increase in the expression, as well as zonal expansion, of the liver-specific WNT/β-catenin target proteins GS and CYP2E1 (Planas-Paz et al. 2016). Furthermore, deletion of Znrf3 and Rnf43 specifically in hepatocytes (driven by Ad5cre virus) led to the formation of multiple liver tumors, primarily classified as hepatocellular carcinomas (T. Sun et al. 2021). Thus, ZNRF3/RNF43 control the hepatic WNT/β-catenin signaling gradient and metabolic liver zonation, and prevent liver tumor formation. Simultaneous disruption of znrf3 and rnf43 by injection of TALENs into two-cell stage Xenopus tropicalis embryos resulted in development of ectopic limbs ranging from diplopodia (duplication of digits) to complete polymelia (presence of supernumerary limbs), including quadruplication of forelimbs in extreme cases (Szenker-Ravi et al. 2018). These three examples, and many others not discussed here, demonstrate that loss of ZNRF3 and/or RNF43 results in elevated WNT/β-catenin signaling in various tissues. However, given that ZNRF3/RNF43 are negative regulators of all three RSPO signaling modalities, the phenotypes caused by their disruption do not distinguish the specific physiological functions of LGR-dependent, HSPG-dependent and BMPR1A-dependent RSPO signaling.
The expression patterns of the four RSPOs in mice are distinct (Nam, Turcotte, and Yoon 2007) and not surprisingly so are the phenotypes associated with their disruption, illustrating the pleiotropic roles of RSPOs during embryogenesis (reviewed in (W. B. M. de Lau, Snel, and Clevers 2012; Nagano 2019; Jin and Yoon 2012). Mutations in RSPO1 cause a rare human syndrome characterized by XX male sex reversal, palmoplantar hyperkeratosis (abnormal thickening of the palms and soles) and predisposition to squamous cell carcinoma of the skin (Parma et al. 2006). Loss of Rspo1 in mice confirmed that the absence of RSPO1 at the gonadal differentiation stage causes partial sex reversal (Tomizuka et al. 2008). Human mutations in RSPO2 cause tetra-amelia with lung hypo/aplasia syndrome (TETAMS), a severe condition characterized by amelia (the complete absence of limbs), lung hypo/aplasia, cleft lip-palate, and labioscrotal fold aplasia (Szenker-Ravi et al. 2018). Loss of Rspo2 in mice causes severe malformations of laryngeal-tracheal cartilages, limb malformations or amelia, palate malformations, and lung hypoplasia (Bell et al. 2008; Yamada et al. 2009; Nam et al. 2007; Aoki et al. 2008). Loss of Rspo3 in mice resulted in severe placental vascular defects, causing death of the mutant mice around embryonic day (E)10 (Aoki et al. 2007; Kazanskaya et al. 2008). Mutations in human RSPO4 were found in individuals affected with anonychia, a rare autosomal recessive congenital syndrome characterized by partial or complete absence of fingernails and toenails (Bergmann et al. 2006; Blaydon et al. 2006; Brüchle et al. 2008; Ishii et al. 2008).
While previously it may have been tempting to attribute these different phenotypes to the distinct expression patterns of RSPO1-4, an additional explanation must be considered in light of the multiple RSPO signaling modalities discussed in this chapter. Differences in RSPO KO phenotypes may also be explained by the capacity of distinct RSPOs to signal through LGR-dependent, HSPG-dependent and BMPR1A-dependent mechanisms. This possibility is supported by the finding that ubiquitous Lgr4/5/6 triple KO mice do not exhibit many of the phenotypes observed in Rspo2 KO or Rspo3 KO mice (Szenker-Ravi et al. 2018). Although the authors noted that some residual Lgr6 expression was retained in the Lgr4/5/6 triple KO mice, they suggested that it would have presumably originated from an alternative start codon downstream of the LGR6 signal peptide and would therefore result in non-functional protein. Lgr4/5/6 triple KO mice die around E14.5–18.5, but the embryos undergo normal development of the limbs and lungs, as well as normal placental vascularization, suggesting RSPO2 and RSPO3 signaling is largely unaffected. From these experiments it can be surmised that certain developmental processes governed by RSPO2 and RSPO3 occur independently of LGRs. However, other processes that are also regulated by RSPO2, such as palate and tongue development, rely on LGRs, since both Rspo2 KO and Lgr4/5/6 KO mice exhibit cleft palate and ankyloglossia (tongue-tie).
Comparison of the phenotypes caused by Lgr4/5/6 KO and by RSPO1-4 loss-of-function mutations in humans, mice and frogs can help distinguish between LGR-dependent and LGR-independent effects of RSPOs, but whether these effects are driven by potentiation of WNT signaling through HSPGs or by downregulation of BMP signaling through BMPR1A (or by yet other pathways regulated by RSPOs) is less clear. Little is known about the physiological contexts in which HSPG-dependent and BMPR1A-dependent RSPO signaling operate. During nephrogenesis, strong RSPO-dependent activation of WNT/β-catenin signaling is essential for nephron progenitors to differentiate and undergo mesenchymal to epithelial transition, and this process occurs largely in an LGR-independent manner (Vidal et al. 2020), suggesting the possibility that nephrogenesis is driven in part by HSPG-dependent RSPO signaling. In multiple myeloma cells, RSPO binds to SDC1 in a HS-dependent manner, and this event is required for optimal stimulation of WNT/β-catenin signaling (Ren et al. 2018). In Xenopus, RSPO2 cooperates with Spemann organizer effectors to inhibit BMP signaling during embryonic axis formation (Lee et al. 2020), and BMP signaling inhibition by RSPO2 maintains autocrine self-renewal in acute myeloid leukemia (AML) (R. Sun et al. 2021). Elucidating the complete repertoire of biological and pathological processes controlled by RSPOs through different signaling modalities will require a combination of approaches. Disrupting entire families of engagement receptors, as was done with the LGRs in mice (Szenker-Ravi et al. 2018), could provide additional insights, but this is a challenging prospect for HSPGs, since RSPO3 (and presumably RSPO2) can signal redundantly through GPCs, of which there are six members in mammals, and SDCs, of which there are an additional four (Dubey et al. 2020; Lebensohn and Rohatgi 2018).
Given the modular structure of RSPO proteins and the different domain requirements for distinct signaling modalities (Table 1), ‘modality-specific’ engineered RSPO ligands – ligands that can signal exclusively through a single signaling modality – could yield further insights. This concept was demonstrated by experiments with RSPO chimeras in which domains from RSPOs capable of signaling through distinct modalities were swapped, rendering them competent or incompetent, respectively, to signal through a different modality (Lebensohn and Rohatgi 2018; Lee et al. 2020). In other experiments, a domain required for one signaling modality was mutated, deleted or replaced by synthetic scFvs, leaving only the domains necessary to target RSPOs to engagement receptors that mediate a different signaling modality (Lebensohn and Rohatgi 2018; Dubey et al. 2020; Lee et al. 2020). Furthermore, the modularity of the RSPO-ZNRF3/RNF43 signaling system should enable the use of such modality-specific ligands for therapeutic or regenerative applications. The ability to target RSPOs to desired tissues through engagement of tissue-specific receptors has been demonstrated (Zhengjian Zhang et al. 2020; Luca et al. 2020) and could potentially be combined with modality-specific mutations to selectively target a single signaling pathway specifically in an affected tissue.
4. Regulation of Hedgehog and melanocortin receptor signaling by the membrane-recruited E3 MGRN1
MGRN1 and its paralog RNF157 belong to a unique class of E3s that do not contain a TM domain but are recruited to the plasma membrane through interactions with other single-pass TM proteins. The TM partners of MGRN1 regulate its substrate specificity, much like substrate adaptors do in multi-subunit RING class E3s (Metzger et al. 2014). We describe two such systems in which the association of MGRN1 with two related single-pass TM proteins directs it to ubiquitylate different GPCRs. We predict that the recruitment of cytoplasmic E3s to the membrane is a mechanism used more broadly to regulate the cell surface abundance of membrane receptors, and consequently to regulate signaling sensitivity.
4.A. The MGRN1-MEGF8-MOSMO complex, an attenuator of Hedgehog signaling strength
Mgrn1 encodes an eponymous RING family E3 (also known as RNF156, Figures 1C, 6, 7), and was identified as the mutated gene at the mahoganoid locus in mice (Phan et al. 2002; Lin He, Lu, et al. 2003). Mgrn1 has been studied because of its effects on coat color determination and spongiform neurodegeneration. However, ~25% of Mgrn1−/− embryos were also noted to die during gestation with heterotaxy and complex cardiac anomalies, suggesting a role for Mgrn1 during development (Cota et al. 2006).
Figure 6.
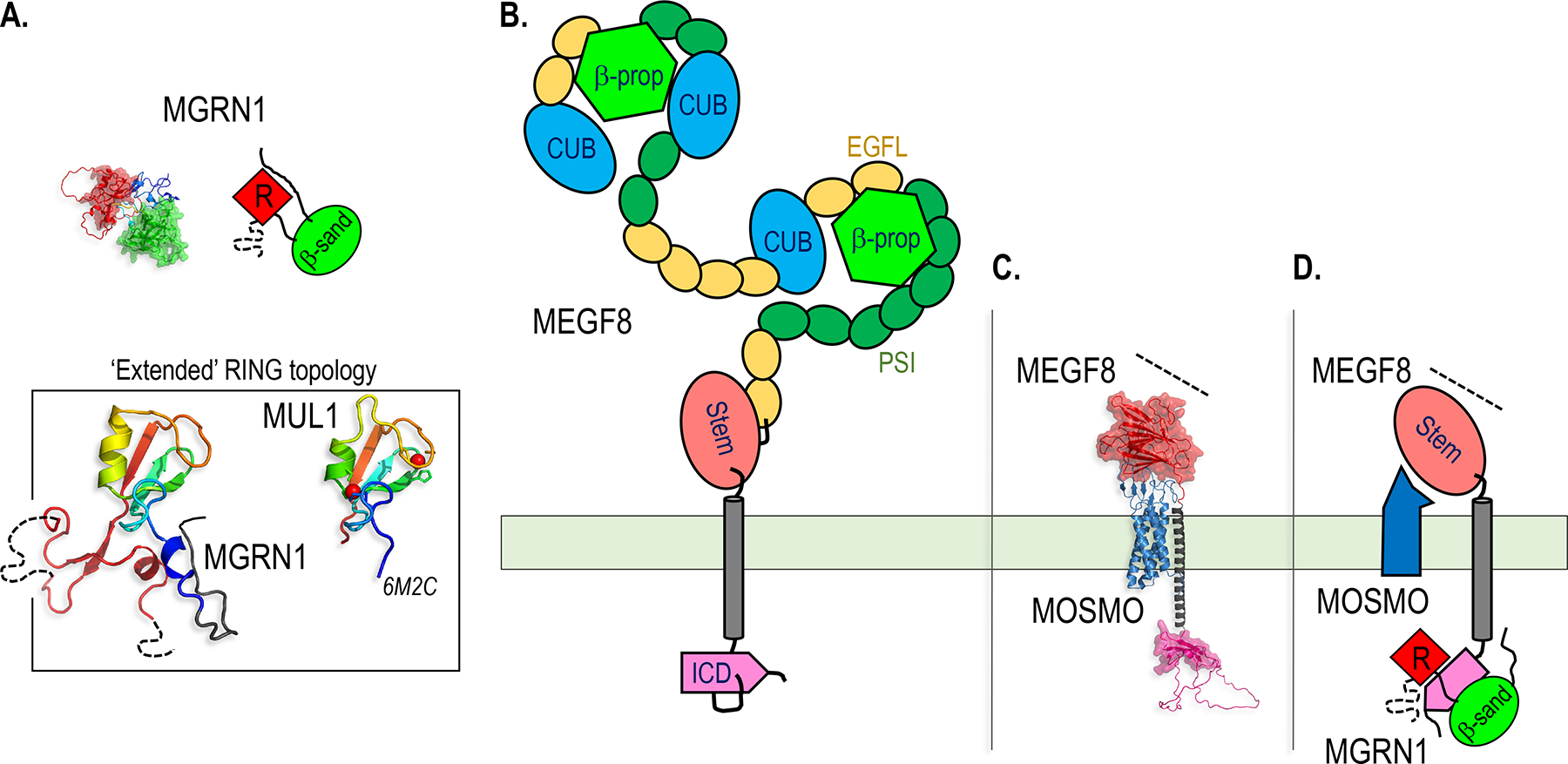
Architecture of the MMM complex, an attenuator of Hh signaling.
A. MGRN1 is a cytoplasmic E3 containing a RING domain and a β-sandwich domain. An AlphaFold structural model is shown on the left and a cartoon representation is shown on the right. The box shows the AlphaFold prediction of the MGRN1 ‘extended’ RING domain, which most closely resembles the structure of the RING domain from MUL1 (PDB ID 6M2C).
B. MEGF8 contains a massive ECD with a pseudo-repeat architecture. A central spine composed of multiple EGFL and PSI domains is decorated with two 6-blade β-propellers and three Complement C1r/C1s, Uegf, Bmp1 (CUB) domains. The extracellular domain is perched on a juxta-membrane, extracellular Stem domain followed by a TM helix that extends into the cytoplasm and connects to an ICD.
C. AlphaFold model of the 4-pass TM protein MOSMO (related to the Claudin family of 4TM proteins) complexed with a fragment of MEGF8 containing the Stem, TM and ICD. The Stem stacks on top of the extracellular β-sheet of MOSMO, promoting the ‘zippering’ of the 4TM bundle of MOSMO with the single TM helix of MEGF8.
D. Cartoon depicting the assembly of the MGRN1-MEGF8-MOSMO complex, excluding the large pseudo-repeat ECD of MEGF8 (shown in B).
Figure 7.
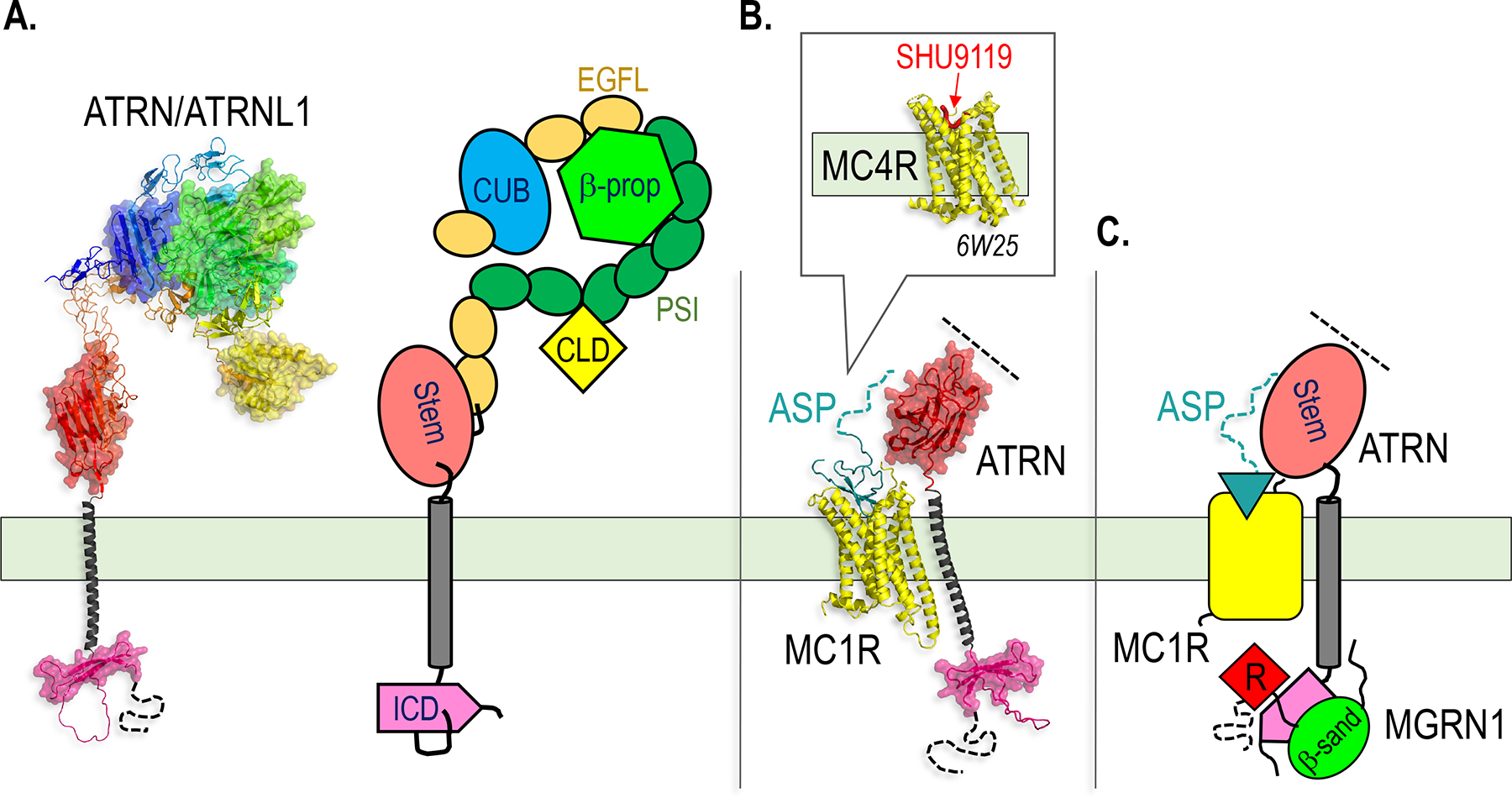
The MGRN1-ATRN complex, an attenuator of melanocortin receptor signaling.
A. AlphaFold model and cartoon representation of ATRN/ATRNL1 (ATRN and ATRNL1 are two closely related proteins, so only one of them is shown). Note the similarities between ATRN and MEGF8 (Figure 6B). The ECD of ATRN has only one of the two repeats present in MEGF8. ATRN has a cyclophilin-like domain (CLD) not found in MEGF8, but shares a 6-blade β-propeller and a CUB domain. The domain coloring in the cartoon on is matched to the structural model.
B and C. AlphaFold model (B) and cartoon representation (C) of how the ligand ASP could cross-link ATRN to MC1R. The N-terminus of ASP forms a β-hairpin that occupies a putative ligand binding site in MC1R. The box in (B) shows the solved structure of MC4R in complex with the antagonist SHU9119 (PDB ID 6W25) (Yu et al. 2020), which occupies the same site predicted to interact with ASP by AlphaFold. The C-terminus of ASP (dotted aqua line) is well positioned to interact with the Stem domain of ATRN.
MEGF8, the gene encoding multiple epidermal growth factor-like (EGFL) domains protein 8 (MEGF8, Figure 6) was amongst those compiled by a bioinformatics screen for genes that encode uncommonly large (>1000 amino acids) membrane-embedded proteins that contain multiple EGFL domains (Nakayama et al. 1998). This effort was motivated by the observation that these characteristics – large size, a TM domain and multiple EGFL domains – were seen in proteins that play important roles in cell-cell or cell-extracellular matrix interactions, such as AGRIN or receptors and ligands of the NOTCH family. MEGF8 was subsequently noted to have homology to a single-pass TM protein commonly referred to as Attractin (ATRN, Figures 6 and 7), which was identified by positional cloning of a mouse gene from the mahogany locus implicated in both body weight and coat color (discussed below) (Gunn et al. 1999; Nagle et al. 1999). Insights into the biological function of MEGF8 came from mouse embryos carrying loss-of-function mutations in Megf8, as well from human patients with a recessively inherited birth defect syndrome called Carpenter’s Syndrome (Engelhard et al. 2013; Z. Zhang et al. 2009; Twigg et al. 2012). In both cases, loss of MEGF8 resulted in heterotaxy (defects in left-right patterning of organs), severe congenital heart defects, pre-axial polydactyly, and skeletal and craniofacial defects. While Megf8 clearly plays a widespread role in the development of multiple tissues, the underlying mechanisms remained unclear. MEGF8 was proposed to be a modifier of BMP and nodal signaling due to its role in left-right patterning and peripheral axon guidance (Z. Zhang et al. 2009; Engelhard et al. 2013). In addition, a possible connection to Hedgehog (Hh) signaling was suggested by the observations that (1) Carpenter’s Syndrome can also be caused by mutations in RAB23, a negative regulator of Hh signaling and that (2) Carpenter’s Syndrome phenotypes resembled those of a related syndrome caused by mutations in the Hh transcription factor GLI3 (Twigg et al. 2012; Eggenschwiler, Espinoza, and Anderson 2001). However, the molecular mechanism of action of both MGRN1 and MEGF8 remained unknown despite the severe mouse and human developmental phenotypes observed when these proteins were lost.
MGRN1, MEGF8 and MOSMO, a completely uncharacterized 4-pass TM protein of the Claudin family, had never been linked to each other until they were all identified in a genome-wide, loss-of-function CRISPR screen designed to uncover attenuators of Hh signaling strength (Pusapati et al. 2018). This screen was performed by exposing cells to a sub-saturating concentration of the Hh ligand Sonic Hedgehog (SHH) and selecting cells carrying mutations that enhanced the activity of a Hh transcriptional reporter. Biochemical analyses demonstrated that all three proteins form a complex, which we named the MMM complex. MGRN1 stably associates with the C-terminal, cytoplasmic tail of MEGF8, and MOSMO associates with this MEGF8-MGRN1 subcomplex (Kong et al. 2020, 2021) (Figure 6). The MMM complex represents a new architecture for E3 complexes: a TM protein with a massive extracellular domain that stably associates with a cytoplasmic RING E3 through a short cytoplasmic tail (Figure 6).
4.A.i. Biochemical and cellular functions of the MMM complex
While there is little homology between the MMM proteins and ZNRF3/RNF43 (except for the presence of a RING domain in MGRN1), there are conceptual similarities in their mechanisms of action. The MMM complex regulates the abundance of the Hh signal transducer Smoothened (SMO) at the cell surface by ubiquitylation (Kong et al. 2020), similar to the way in which ZNRF3/RNF43 regulates the abundance of FZD receptors. SMO transmits the Hh signal across the membrane and is the closest relative of the FZD receptors in the Class F family of GPCRs. Loss of MGRN1, MEGF8 or MOSMO markedly reduced SMO endocytosis and degradation, leading to SMO accumulation on the cell surface and the membrane of the primary cilium (Pusapati et al. 2018; Kong et al. 2020, 2021). Primary cilia function as compartments for Hh signaling, and increased SMO accumulation in primary cilia enhances Hh signaling strength (Huangfu and Anderson 2005; Huangfu et al. 2003). Consequently, the concentration of SHH required to induce target genes is reduced by nearly an order of magnitude when any of the MMM proteins are lost (Pusapati et al. 2018). In summary, just as FZDs are target receptors for ZNRF3/RNF43, SMO is the target receptor for the MMM complex.
Both the integrity of the RING domain of MGRN1 and the physical interaction between MGRN1 and the MEGF8 ICD are required for ubiquitylation of SMO by the MMM complex (Kong et al. 2020). The MGRN1-MEGF8 interaction requires the amino acid sequence MASRPFA, a motif that is conserved in proteins of the MEGF8/ATRN family across evolution (Gunn et al. 1999; Nagle et al. 1999). Direct binding of SMO to MMM complex components has not been reported, so the mechanism of SMO recognition by the MMM complex remains to be elucidated. The large ECD of MEGF8 is dispensable for SMO recognition. A truncated protein containing only the TM domain (TMD) and ICD of MEGF8 (which cannot bind to MOSMO) is nonetheless sufficient to mediate SMO ubiquitylation, reduce its levels at the cell surface and dampen Hh signaling (Kong et al. 2020). However, it is not known whether this TMD-ICD segment of MEGF8 recognizes SMO directly or through an adaptor protein. Importantly, mere recruitment of MGRN1 to the plasma membrane is not sufficient to drive SMO ubiquitylation. The MEGF8 TMD (or the precise arrangement of the TMD relative to the ICD) is likely to be important because its replacement with the TMD of a different single-pass TM protein abolished SMO ubiquitylation despite the fact that MGRN1 was still recruited to the membrane and retained ubiquitin ligase activity (Kong et al. 2020). Therefore, MEGF8 likely functions as a substrate adaptor that recruits SMO, and perhaps other substrates, for ubiquitylation by MGRN1.
The function of the 4TM protein MOSMO remains to be fully elucidated. MOSMO is a Claudin family protein whose distinctive extracellular loop structure, which folds into a compact, disulfide-locked β-sheet, forms a cell surface interaction platform for a previously uncharacterized extracellular juxtamembrane domain in MEGF8 (Figure 6C) (Kong et al. 2021). Loss of MOSMO partially reduces MEGF8 levels at the cell surface, suggesting a role in trafficking (Kong et al. 2021). While MOSMO is not absolutely required for SMO ubiquitylation by the MEGF8-MGRN1 subcomplex in an overexpression system, the severe phenotypes of Mosmo−/− mice, which are similar to those of Megf8−/− mice, suggest that the association of MOSMO with MEGF8 and MGRN1 may in fact play a critical role in activation of the MMM complex.
A key unanswered question in MMM complex function is the role of the large MEGF8 ECD (Figure 6B). Just like ZNRF3/RNF43 are regulated by RSPO ligands, it is likely that the MMM complex is also regulated by interactions of the MEGF8 ECD with a soluble extracellular protein, a component of the extracellular matrix or another membrane protein on the same or adjacent cell. The unique architecture of the MMM complex suggests a regulatory mechanism whereby the TM topology of the MEGF8 substrate adaptor allows extracellular signals to directly regulate the selection and ubiquitylation of specific substrates by MGRN1 in the cytoplasm.
In both cultured cells and mouse embryos, loss of MMM components in target cells receiving Hh signals leads to elevated sensitivity to Hh ligands (Kong et al. 2020, 2021). The pattern of elevated Hh signaling in embryos lacking MMM components is distinct in a very specific way from that observed in embryos lacking Patched 1 (PTCH1) or Suppressor of Fused (SUFU), two negative regulators of Hh signaling. Loss of PTCH1 or SUFU leads to the ectopic activation of Hh signaling in multiple tissues, showing that these proteins suppress basal signaling activity even in the absence of Hh ligands (Goodrich et al. 1997; Cooper et al. 2005; Svard et al. 2006). In contrast, Hh signaling in MMM mutant mice remains dependent on Hh ligands: Hh target gene expression is localized correctly in the embryo, but the strength of signaling is elevated (Kong et al. 2021). Thus, like ZNRF3 and RNF43, the MMM proteins are best characterized as ‘attenuators’ of signaling rather than negative regulators, because their effects remain dependent on the presence of WNT or Hh ligands, respectively. In summary, the MMM proteins form a signaling module that calibrates how the Hh ligand gradient is decoded by target cells.
4.A.ii. Developmental roles of the MMM complex
A common function for the three proteins of the MMM complex, suggested by biochemical analyses, is also supported by mouse genetic studies (Kong et al. 2020, 2021; Cota et al. 2006; Z. Zhang et al. 2009). Mosmo−/− and Megf8−/− mouse embryos exhibit similar developmental defects, also shared by Carpenter’s Syndrome patients: heterotaxy, severe congenital heart defects, pre-axial digit duplication, skeletal defects, craniofacial defects and neurodevelopmental abnormalities. In addition, SMO abundance in the ciliary membrane is markedly elevated in nearly all Mosmo−/− and Megf8−/− embryonic tissues, consistent with observations in cultured cells (Kong et al. 2020, 2021). Some of these embryonic phenotypes were initially not observed in Mgrn1−/− embryos because MGRN1 is partially redundant with RNF157 (Cota et al. 2006; Kong et al. 2020). However, Mgrn1−/−;Rnf1571−/− embryos exhibit a constellation of defects very similar to those seen in Mosmo−/− and Megf8−/− embryos. Beyond common phenotypes, strong genetic interactions and gene dosage effects between mutant alleles of Megf8, Mgrn1 and Rnf157 support the conclusion that MGRN1/RNF157 and MEGF8 function together to regulate a common set of developmental processes (Kong et al. 2020).
The widespread nature of the defects observed in mouse embryos carrying mutations in the MMM genes points to a central role of the MMM complex in the regulation of cell-cell communication. MMM complex components have been linked to nodal, BMP and Hh signaling, but a direct role in regulation of signaling components has only been established for SMO in the Hh pathway (Pusapati et al. 2018; Engelhard et al. 2013). The expression patterns of Megf8, Mgrn1 and Mosmo do not provide many clues to the developmental functions of the MMM complex: they are ubiquitously expressed in all three germ layers and in all major cardiac populations (Cota et al. 2006; W. Wang et al. 2020; Z. Zhang et al. 2009; Kong et al. 2021). Thus, control of MMM complex activity, perhaps by a ligand or by post-translational modifications, is likely to be the key regulated step that explains its tissue-specific roles.
An unresolved question is whether all of the embryonic defects seen in MMM mutant mice are caused by upregulation of SMO and elevated Hh signaling, or whether some are caused by regulation of other unidentified substrates that function in other pathways. The pre-axial digit duplication seen in MMM mutant embryos is likely to be driven by elevated Hh signaling, since this phenotype can be completely reversed by administration of vismodegib, a placenta-permeable small molecule inhibitor of SMO (Kong et al. 2020, 2021). However, vismodegib only partially rescues the congenital heart defects observed in MMM mutant embryos, which could be due to a suboptimal schedule of in utero vismodegib administration, or could also indicate that other pathways are regulated by the MMM complex. Further research will be necessary to comprehensively identify substrates of the MMM complex other than SMO and to test its role in other developmental signaling pathways.
Heterotaxy is a prominent feature of MMM mutant embryos and Carpenter’s Syndrome patients, and may be the root cause of the severe congenital heart defects observed in both (Kong et al. 2020, 2021; Cota et al. 2006; Z. Zhang et al. 2009; Twigg et al. 2012). The MMM complex regulates left-right patterning at an early stage in embryogenesis. In both Megf8−/− and Mgrn1−/− embryos, abnormal expression of all three canonical left-expressed genes (Nodal, Lefty, Pitx2) was observed (Z. Zhang et al. 2009; Cota et al. 2006). Conditional disruption of Megf8 in various cardiac lineages using a panel of Cre drivers (cTnt-Cre, Wt1-Cre, Tie2-Cre, Wnt1-Cre, Mesp1-Cre) did not reproduce the heart defects seen in the global Megf8 KO. Timed global deletion of Megf8 at E7.5 also did not reproduce the cardiac defects (W. Wang et al. 2020). These data suggest that Megf8 is required for cardiac development at a time point earlier than cardiac organogenesis and supports the hypothesis that the heart defects seen in MMM mutant mice are a consequence of disrupted left-right patterning early in development.
Current models suggest that Hh signaling plays a permissive role in left-right patterning of the mouse embryo: reduced Hh signaling caused by loss of SMO leads to a midline heart tube that fails to loop and an embryo that fails to turn (X. M. Zhang, Ramalho-Santos, and McMahon 2001; Levin et al. 1995; Tsiairis and McMahon 2009). While this model does not readily explain how the elevated Hh signaling seen in MMM mutant embryos leads to left-right patterning defects, loss-of-function mutations in the Hh negative regulator Sufu do cause abnormalities in embryo turning, heart looping and expression of the left-expressed gene Pitx2 (Cooper et al. 2005). Thus, normal left-right patterning may depend on a just-right, or ‘goldilocks,’ level of Hh signaling strength calibrated by the MMM complex. Alternatively, the MMM complex may regulate another pathway involved in left-right patterning, such as nodal or BMP signaling. Elucidating how the MMM complex regulates left-right patterning is critical to understanding its developmental roles and, consequently, the etiology of birth defects caused by mutations in MMM genes.
4.B. The MGRN1-ATRN system regulates melanocortin receptors
Regulation of Hh signaling by the MMM complex shares many themes with another E3 complex, formed by MGRN1 and the MEGF8-related protein ATRN, that functions in a paracrine fashion to regulate melanocortin receptor signaling (reviewed in (Lin He, Eldridge, et al. 2003)). The four melanocortin receptors, MC1R, MC2R, MC3R and MC4R, are GPCRs that bind to peptide agonists, including α-MSH and ACTH, and regulate diverse physiological processes in vertebrates. We focus here on the regulation of mouse coat color, which serves as a useful paradigm for paracrine cell-cell interactions that orchestrate both tissue patterning during development and tissue homeostasis in adults. Melanocortin receptor activity is controlled by agonists, like α-MSH, and inverse agonists. Agouti signaling protein (ASP) and agouti-related protein (AGRP) are both inverse agonists of the MC1R receptors in the skin and the MC4R receptors in the hypothalamus. They reduce basal and α-MSH-stimulated receptor activity (Lu et al. 1994).
Hair follicle melanocytes switch between producing the pigments eumelanin (dark) and pheomelanin (light). In mice, the presence of a subapical light band in an otherwise dark hair leads to the agouti coat color. This pheomelanic band in each hair is generated by a paracrine signaling interaction between dermal cells at the base of each hair follicle and neighboring melanocytes. The dark eumelanic hair pigmentation is driven by MC1R signaling in melanocytes. Transient secretion of ASP from the dermal cells inhibits MC1R signaling, causing a switch to pheomelanin synthesis and the generation of a light band on each hair. Ubiquitous and constitutive expression of ASP results in a light coat as well as hyperphagia and obesity, caused by inhibition of MC4R in the hypothalamus. This short-range dermal-melanocyte signaling circuit is similar to how RSPOs secreted by stromal cells influence WNT signaling in epithelial stem cells of the intestinal crypts (Greicius et al. 2018).
Mgrn1 and Atrn were identified as genes required for the inhibitory effect of ASP on melanocortin receptor signaling (Phan et al. 2002; Gunn et al. 1999; Lin He, Eldridge, et al. 2003; Nagle et al. 1999; Lin He, Lu, et al. 2003). The pigmentation changes caused by ectopic ASP expression can be suppressed by mutations in Mgrn1 and Atrn. Epistasis analyses have placed Mgrn1 and Atrn downstream of ASP but upstream of MC1R. Strikingly, genetic analyses show that Mgrn1 and Atrn are required for ASP signaling, despite the fact that purified ASP alone is a high-affinity antagonist of MC1R in biochemical assays (Willard et al. 1995). These genetic studies suggested that MGRN1 and ATRN are required for the inhibitory effects of ASP on MC1R in target melanocytes.
4.B.i. Cellular and biochemical models for the regulation of melanocortin receptors by ASP, MGRN1 and ATRN
ATRN is a single pass TM protein related to MEGF8. The massive extracellular domain of MEGF8 is composed of two tandem repeats (Figure 6B). ATRN is more compact and has only one of these repeats, but shares the single TM helix and short intracellular tail found in MEGF8 (Figure 7A). While the C-terminal domain of ASP binds to MC1R with high affinity (KD ~1 nM), the N-terminal chain of ASP binds to the ECD of ATRN with ~500-fold lower affinity (Figure 7B–C) (L. He et al. 2001; Willard et al. 1995). These genetic and biochemical studies are most consistent with a model in which the binding of ASP to ATRN transmits a signal to MGRN1 in the cytoplasm that ultimately leads to the downregulation of melanocortin receptors.
There are striking similarities between the ASP-MGRN1-ATRN module and the MMM complex: ATRN and MEGF8 are related proteins, MGRN1 is shared, and both systems regulate GPCRs. The simplest model that emerges from this comparison is that the MGRN1-ATRN complex ubiquitylates and downregulates MC1R and MC4R at the cell surface in response to binding of ASP. In this model, ASP acts as a ligand that cross-links melanocortin receptors to ATRN (Figure 7B–C), analogous to how RSPO2 and RSPO3 cross-link BMPR1A to ZNRF3/RNF43 (Figure 5). AlphaFold modeling suggests that ASP can simultaneously bind to MC1R through its N-terminus and to the STEM domain in ATRN through its C-terminus, thereby positioning the RING domain of ATRN for Ub transfer to the cytoplasmic surface of MC1R (Figure 7B–C).
There is some evidence to support this model. ASP promotes trafficking of MC4R to the lysosome and its subsequent degradation in a manner that depends on both MGRN1 and ATRN (B. Y. Kim et al. 2007; Overton and Leibel 2011). Loss of ATRN leads to elevated MC4R levels at the cell surface (Overton and Leibel 2011), analogous to how loss of MEGF8 leads to elevated SMO levels at the cell surface (Pusapati et al. 2018). However, a physical interaction between the C-terminal tail of ATRN and MGRN1 analogous to that between MEGF8 and MGRN1 has not been demonstrated, and neither has a role for such an interaction in melanocortin receptor downregulation. Notably, the MASRPFA motif in the MEGF8 cytoplasmic tail that is required for binding to MGRN1 is conserved in ATRN (Nagle et al. 1999; Kong et al. 2020), and this motif in the Drosophila ATRN ortholog is required for association with Drosophila MGRN1 (Nawaratne et al. 2021). Ubiquitylation of MC1R by an MGRN1-ATRN complex also remains to be established. MGRN1 has been shown to ubiquitylate MC2R, but it is not clear whether this reaction required ATRN (Cooray, Guasti, and Clark 2011). MGRN1 also ubiquitylates tumor suppressor gene 101 (TSG101), a component of the endosomal sorting complex required for transport-I (ESCORT-I) complex that mediates the trafficking of ubiquitylated cell surface receptors from the plasma membrane to the lysosome for degradation (Jiao et al. 2009; B. Y. Kim et al. 2007). Based on this finding, a different model has been proposed in which the effect of MGRN1 on melanocortin receptor trafficking is an indirect consequence of its regulation of TSG101. Further experiments will therefore be required to elucidate the biochemical function of the MGRN1-ATRN complex.
4.C. Evolutionary insights into MGRN1 and the MEGF8/ATRN protein family
MEGF8 and ATRN family proteins are conserved across metazoans and in their closest living relatives, the choanoflagellates (Pusapati et al. 2018). The Drosophila ortholog of Megf8 is also required for early embryonic development (Lloyd et al. 2018). However, MGRN1 is more widely distributed throughout evolution, found in all major eukaryotic lineages (Pusapati et al. 2018). The Arabidopsis thaliana ortholog of MGRN1, called LOG2, can be functionally replaced by human MGRN1 (D. D. Guerra et al. 2013). LOG2 is recruited to the plasma membrane by binding to the cytoplasmic tail of the single-pass TM protein glutamine dumper-1 (GDU1) (D. Guerra et al. 2017). The LOG2-GDU1 complex has been implicated in amino acid transport. These observations suggest that members of the MGRN1 family of RING E3s have evolved to associate with multiple single-pass TM proteins across eukaryotes to regulate the activity of diverse membrane receptors and transporters. In this scheme, MGRN1 provides the ubiquitin ligase function while the single-pass TM protein functions as a substrate adaptor to select targets for ubiquitylation.
This ancient membrane-tethered ubiquitylation system has likely been adapted to regulate signaling pathways at multiple times during evolution. While MGRN1 is found in all eukaryotes, Hh signaling is only present in a subset of the metazoan lineages where MEGF8 is found. The MGRN1-ATRN system seems to have been co-opted to regulate melanocortin receptor signaling much later in evolution, since ASP and melanocortin receptors are only found in vertebrates.
5. Conclusions
TM E3s can tune the sensitivity of cells to ligands by promoting the internalization and degradation of specific signaling receptors. They can target substrates constitutively, like ZNRF3/RNF43 target FZDs, or they can be regulated by secreted ligands that direct them to specific substrates, like RSPOs direct ZNRF3/RNF43 to BMPR1A or ASP directs MGRN1-ATRN to melanocortin receptors. Thus, TM E3s function like the intensively studied receptor kinases, enabling extracellular ligands to directly control the ubiquitylation of substrates in the cytoplasm. We propose that receptor regulation by TM E3s plays a widespread and understudied role in tuning cell sensitivity to paracrine signals that control embryonic development and tissue homeostasis, as exemplified by the conceptually analogous RSPO-ZNRF3/RNF43 and MMM modules that regulate WNT and BMP, or Hh signaling, respectively.
Compared to cytoplasmic or nuclear E3s, TM E3s face unique challenges in substrate recognition and modification because of a physical barrier, the plasma membrane, that creates three discrete zones for protein-protein interactions: the intracellular and extracellular spaces, and the plane of the membrane. These three regions likely create composite binding surfaces that promote the assembly of protein complexes in which the RING domain of the TM E3 is optimally positioned in the cytoplasm to transfer Ub to the substrate. In this respect, there are architectural and mechanistic parallels between the assembly of TM E3-substrate complexes and of cytokine-nucleated signaling receptor assemblies, which are driven by the ‘zippering’ together of the full complex, from binding of ligands to receptors, to ECD and TM contacts between receptors, and finally cytoplasmic domain interactions that may also recruit downstream signaling proteins (Spangler et al. 2015; Rosenbaum et al. 2020). These aggregate contacts contribute to the stability, lifetime and signaling strength of the receptor complex, and can be sites for therapeutic modulation or for engineering of tunable receptors.
For cytoplasmic and nuclear E3s, proteolysis-targeting chimeras (PROTACs) have emerged as a therapeutic modality that enables target degradation driven by small molecules (Schneider et al. 2021; Bond and Crews 2021). PROTACs function as bivalent linkers that direct E3s to specific substrates for ubiquitylation and degradation. Very recently, a strategy conceptually analogous to PROTACs has been applied to induce the degradation of PD-L1, a transmembrane immune checkpoint ligand, by RNF43 (Cotton et al. 2021). Targeting TM E3s to heterologous signaling receptors using small molecules, surrogate ligands or bivalent nanobody or antibody constructs is a promising strategy to modulate signaling strength for therapeutic purposes.
Molecular models and protein-protein interaction predictions enabled by AlphaFold and RoseTTaFold, with broad community access, will enable the rapid generation of hypotheses about how these TM E3s fold, assemble and recognize substrates. The ability of these new deep learning-based algorithms to predict protein folds and interactions relies in good part on their capacity to tease out the faintest signals of co-evolutionary linkage between amino acid positions on the same and on partner polypeptide chains by mining sequence databases. These algorithmic approaches should work well with the evolutionary pairings that underlie TM E3-substrate systems. We expect that the characterization of other TM E3-receptor systems will unravel new regulatory layers in many signaling pathways and enable new proteolysis-driven approaches to target TM and membrane-associated proteins.
Note Added in Proof
While the manuscript was being processed for publication, two groups reported on the regulation of additional cell surface proteins by ZNRF3/RNF43. Zhu and colleagues described the regulation of Hulula (Hwa), a determinant of the Spemann organizer and doral body axis formation in Xenopus laevis, by ZNRF3. ZNRF3 binds and ubiquitylates Hwa, thereby regulating its lysosomal trafficking and degradation (Zhu et al. 2021). Radaszkiewicz and colleagues described the regulation of β-catenin-independent, WNT5A-induced signaling by RNF43 in normal physiology and during melanoma invasion (Radaszkiewicz et al. 2021). RNF43 interacted with components of the WNT/PCP signaling pathway, including the receptors ROR1 and ROR2, and the signal transducers VANGL1 and VANGL2. RNF43 triggered VANGL2 ubiquitylation and proteasomal degradation, promoted ROR1 internalization, and inhibited ROR2 activation. We presume that some of the mechanisms described in this chapter may apply to the regulation of ZNRF3/RNF43 in these and other yet to be discovered contexts.
Acknowledgements
AML is supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. RR is supported by grants from the NIH (GM118082, HL157103, HD101980). We thank Feng Cong, Christof Niehrs and Sara Konopelski Snavely for feedback on the manuscript.
References
- Acebron Sergio P., Karaulanov Emil, Berger Birgit S., Huang Ya-Lin, and Niehrs Christof. 2014. “Mitotic Wnt Signaling Promotes Protein Stabilization and Regulates Cell Size.” Molecular Cell 54 (4): 663–74. [DOI] [PubMed] [Google Scholar]
- Aoki Motoko, Kiyonari Hiroshi, Nakamura Harukazu, and Okamoto Hitoshi. 2008. “R-spondin2 Expression in the Apical Ectodermal Ridge Is Essential for Outgrowth and Patterning in Mouse Limb Development.” Development, Growth & Differentiation 50 (2): 85–95. [DOI] [PubMed] [Google Scholar]
- Aoki Motoko, Mieda Michihiro, Ikeda Toshio, Hamada Yoshio, Nakamura Harukazu, and Okamoto Hitoshi. 2007. “R-spondin3 Is Required for Mouse Placental Development.” Developmental Biology 301 (1): 218–26. [DOI] [PubMed] [Google Scholar]
- Ayadi Leila. 2008. “Molecular Modelling of the TSR Domain of R-Spondin 4.” Bioinformation. 10.6026/97320630003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek Minkyung, Frank DiMaio Ivan Anishchenko, Dauparas Justas, Ovchinnikov Sergey, Gyu Rie Lee Jue Wang, et al. 2021. “Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network.” Science 373 (6557): 871–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker Nick, Tan Shawna, and Clevers Hans. 2013. “Lgr Proteins in Epithelial Stem Cell Biology.” Development 140 (12): 2484–94. [DOI] [PubMed] [Google Scholar]
- Bauer Johannes, Bakke Oddmund, and Morth J. Preben. 2017. “Overview of the Membrane-Associated RING-CH (MARCH) E3 Ligase Family.” New Biotechnology 38 (Pt A): 7–15. [DOI] [PubMed] [Google Scholar]
- Bell Sheila M., Schreiner Claire M., Wert Susan E., Mucenski Michael L., Scott William J., and Whitsett Jeffrey A.. 2008. “R-Spondin 2 Is Required for Normal Laryngeal-Tracheal, Lung and Limb Morphogenesis.” Development 135 (6): 1049–58. [DOI] [PubMed] [Google Scholar]
- Bergmann C, Senderek J, Anhuf D, Thiel CT, Ekici AB, Poblete-Gutierrez P, van Steensel M, et al. 2006. “Mutations in the Gene Encoding the Wnt-Signaling Component R-Spondin 4 (RSPO4) Cause Autosomal Recessive Anonychia.” American Journal of Human Genetics 79 (6): 1105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, and Nusse R. 1996. “A New Member of the Frizzled Family from Drosophila Functions as a Wingless Receptor.” Nature 382 (6588): 225–30. [DOI] [PubMed] [Google Scholar]
- Blaydon Diana C., Ishii Yoshiyuki, O’Toole Edel A., Unsworth Harriet C., Muy-Teck Teh, Rüschendorf Franz, Sinclair Claire, et al. 2006. “The Gene Encoding R-Spondin 4 (RSPO4), a Secreted Protein Implicated in Wnt Signaling, Is Mutated in Inherited Anonychia.” Nature Genetics 38 (11): 1245–47. [DOI] [PubMed] [Google Scholar]
- Bond Michael J., and Crews Craig M.. 2021. “Proteolysis Targeting Chimeras (PROTACs) Come of Age: Entering the Third Decade of Targeted Protein Degradation.” RSC Chemical Biology 2 (3): 725–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourhis E, Tam C, Franke Y, Bazan JF, and Ernst J. 2010. “Reconstitution of a Frizzled8· Wnt3a· LRP6 Signaling Complex Reveals Multiple Wnt and Dkk1 Binding Sites on LRP6.” Journal of Biological https://www.jbc.org/article/S0021-9258(20)87312-8/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüchle Nadina Ortiz, Frank Jorge, Frank Valeska, Senderek Jan, Akar Ahmet, Koc Erol, Rigopoulos Dimitris, Maurice van Steensel Klaus Zerres, and Bergmann Carsten. 2008. “RSPO4 Is the Major Gene in Autosomal-Recessive Anonychia and Mutations Cluster in the Furin-like Cysteine-Rich Domains of the Wnt Signaling Ligand R-Spondin 4.” The Journal of Investigative Dermatology 128 (4): 791–96. [DOI] [PubMed] [Google Scholar]
- Bryant P, Pozzati G, and Elofsson A. 2021. “Improved Prediction of Protein-Protein Interactions Using AlphaFold2.” bioRxiv. 10.1101/2021.09.15.460468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugter Jeroen M., Fenderico Nicola, and Maurice Madelon M.. 2021. “Mutations and Mechanisms of WNT Pathway Tumour Suppressors in Cancer.” Nature Reviews. Cancer 21 (1): 5–21. [DOI] [PubMed] [Google Scholar]
- Burbach Brandon J., Friedl Andreas, Mundhenke Christoph, and Rapraeger Alan C.. 2003. “Syndecan-1 Accumulates in Lysosomes of Poorly Differentiated Breast Carcinoma Cells.” Matrix Biology. 10.1016/s0945-053x(03)00009-x. [DOI] [PubMed] [Google Scholar]
- Cappadocia Laurent, and Lima Christopher D.. 2018. “Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism.” Chemical Reviews 118 (3): 889–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmon Kendra S., Gong Xing, Lin Qiushi, Thomas Anthony, and Liu Qingyun. 2011. “R-Spondins Function as Ligands of the Orphan Receptors LGR4 and LGR5 to Regulate Wnt/β-Catenin Signaling.” Proceedings of the National Academy of Sciences of the United States of America 108 (28): 11452–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmon Kendra S., Gong Xing, Yi Jing, Thomas Anthony, and Liu Qingyun. 2014. “RSPO-LGR4 Functions via IQGAP1 to Potentiate Wnt Signaling.” Proceedings of the National Academy of Sciences of the United States of America 111 (13): E1221–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Chiung-Fang, Hsu Li-Sung, Weng Chieh-Yu, Chen Chih-Kai, Wang Shu-Ying, Chou Yi-Hwa, Liu Yan-Yu, et al. 2016. “N-Glycosylation of Human R-Spondin 1 Is Required for Efficient Secretion and Stability but Not for Its Heparin Binding Ability.” International Journal of Molecular Sciences 17 (6). 10.3390/ijms17060937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Ling-Shih, Kim Minseong, Glinka Andrey, Reinhard Carmen, and Niehrs Christof. 2020. “The Tumor Suppressor PTPRK Promotes ZNRF3 Internalization and Is Required for Wnt Inhibition in the Spemann Organizer.” eLife 9 (January). 10.7554/eLife.51248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Jin-Zhong, Wang Shu, Tang Rong, Yang Quan-Sheng, Zhao Enpeng, Chao Yaoqiong, Ying Kang, Xie Yi, and Mao Yu-Min. 2002. “Cloning and Identification of a cDNA That Encodes a Novel Human Protein with Thrombospondin Type I Repeat Domain, hPWTSR.” Molecular Biology Reports 29 (3): 287–92. [DOI] [PubMed] [Google Scholar]
- Chen Po-Han, Chen Xiaoyan, Lin Zhenghong, Fang Deyu, and He Xiaolin. 2013. “The Structural Basis of R-Spondin Recognition by LGR5 and RNF43.” Genes & Development 27 (12): 1345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson Helena C., and Belting Mattias. 2014. “Heparan Sulfate Proteoglycan as a Cell-Surface Endocytosis Receptor.” Matrix Biology: Journal of the International Society for Matrix Biology 35 (April): 51–55. [DOI] [PubMed] [Google Scholar]
- Chu Matthew Ling-Hon, Ahn Victoria E., Choi Hee-Jung, Daniels Danette L., Nusse Roel, and Weis William I.. 2013. “Structural Studies of Wnts and Identification of an LRP6 Binding Site.” Structure 21 (7): 1235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague Michael J., Urbé Sylvie, and Komander David. 2019. “Breaking the Chains: Deubiquitylating Enzyme Specificity Begets Function.” Nature Reviews. Molecular Cell Biology 20 (6): 338–52. [DOI] [PubMed] [Google Scholar]
- Consonni Sarah V., Maurice Madelon M., and Bos Johannes L.. 2014. “DEP Domains: Structurally Similar but Functionally Different.” Nature Reviews. Molecular Cell Biology 15 (5): 357–62. [DOI] [PubMed] [Google Scholar]
- Cooper AF, Yu KP, Brueckner M, Brailey LL, Johnson L, McGrath JM, and Bale AE. 2005. “Cardiac and CNS Defects in a Mouse with Targeted Disruption of Suppressor of Fused.” Development 132 (19): 4407–17. [DOI] [PubMed] [Google Scholar]
- Cooray Sadani N., Guasti Leonardo, and Clark Adrian J. L.. 2011. “The E3 Ubiquitin Ligase Mahogunin Ubiquitinates the Melanocortin 2 Receptor.” Endocrinology 152 (11): 4224–31. [DOI] [PubMed] [Google Scholar]
- Cota Christina D., Bagher Pooneh, Pelc Piotr, Smith C. Owen, Bodner Christina R., and Gunn Teresa M.. 2006. “Mice with Mutations in Mahogunin Ring Finger-1 (Mgrn1) Exhibit Abnormal Patterning of the Left-Right Axis.” Developmental Dynamics: An Official Publication of the American Association of Anatomists 235 (12): 3438–47. [DOI] [PubMed] [Google Scholar]
- Cotton Adam D., Nguyen Duy P., Gramespacher Josef A., Seiple Ian B., and Wells James A.. 2021. “Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1.” Journal of the American Chemical Society 143 (2): 593–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck Rik, and Miyazono Kōhei. 2017. The Biology of the TGF-[Beta] Family. Cold Spring Harbor Laboratory Press. [Google Scholar]
- Deshaies Raymond J., and Joazeiro Claudio A. P.. 2009. “RING Domain E3 Ubiquitin Ligases.” Annual Review of Biochemistry 78: 399–434. [DOI] [PubMed] [Google Scholar]
- Dubey Ramin, Kerkhof Peter van, Jordens Ingrid, Malinauskas Tomas, Pusapati Ganesh V., McKenna Joseph K., Li Dan, et al. 2020. “R-Spondins Engage Heparan Sulfate Proteoglycans to Potentiate WNT Signaling.” eLife 9 (May). 10.7554/eLife.54469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler JT, Espinoza E, and Anderson KV. 2001. “Rab23 Is an Essential Negative Regulator of the Mouse Sonic Hedgehog Signalling Pathway.” Nature 412 (6843): 194–98. [DOI] [PubMed] [Google Scholar]
- Engelhard C, Sarsfield S, Merte J, Wang Q, Li P, Beppu H, Kolodkin AL, Sucov HM, and Ginty DD. 2013. “MEGF8 Is a Modifier of BMP Signaling in Trigeminal Sensory Neurons.” eLife 2: e01160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans Richard, O’Neill Michael, Pritzel Alexander, Antropova Natasha, Senior Andrew, Green Tim, Žídek Augustin, et al. 2021. “Protein Complex Prediction with AlphaFold-Multimer.” bioRxiv. 10.1101/2021.10.04.463034. [DOI] [Google Scholar]
- Fenech Emma J., Lari Federica, Charles Philip D., Fischer Roman, Marie Laétitia-Thézénas Katrin Bagola, Paton Adrienne W., et al. 2020. “Interaction Mapping of Endoplasmic Reticulum Ubiquitin Ligases Identifies Modulators of Innate Immune Signalling.” eLife 9 (July). 10.7554/eLife.57306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler Marc, Carolina Mendoza-Topaz Trevor J. Rutherford, Mieszczanek Juliusz, and Bienz Mariann. 2011. “Dishevelled Interacts with the DIX Domain Polymerization Interface of Axin to Interfere with Its Function in down-Regulating β-Catenin.” Proceedings of the National Academy of Sciences. 10.1073/pnas.1017063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foot Natalie, Henshall Tanya, and Kumar Sharad. 2017. “Ubiquitination and the Regulation of Membrane Proteins.” Physiological Reviews 97 (1): 253–81. [DOI] [PubMed] [Google Scholar]
- Fuki Ilia V., Meyer Marie E., and Williams Kevin Jon. 2000. “Transmembrane and Cytoplasmic Domains of Syndecan Mediate a Multi-Step Endocytic Pathway Involving Detergent-Insoluble Membrane Rafts.” Biochemical Journal. 10.1042/0264-6021:3510607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuki IV, Kuhn KM, Lomazov IR, Rothman VL, Tuszynski GP, Iozzo RV, Swenson TL, Fisher EA, and Williams KJ. 1997. “The Syndecan Family of Proteoglycans. Novel Receptors Mediating Internalization of Atherogenic Lipoproteins in Vitro.” Journal of Clinical Investigation. 10.1172/jci119685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammons Melissa, and Bienz Mariann. 2018. “Multiprotein Complexes Governing Wnt Signal Transduction.” Current Opinion in Cell Biology 51 (April): 42–49. [DOI] [PubMed] [Google Scholar]
- Gao Chan, and Chen Ye-Guang. 2010. “Dishevelled: The Hub of Wnt Signaling.” Cellular Signalling 22 (5): 717–27. [DOI] [PubMed] [Google Scholar]
- Geng Ajun, Wu Ting, Cai Cheguo, Song Wenqian, Wang Jiqiu, Yu Qing Cissy, and Yi Arial Zeng. 2020. “A Novel Function of R-spondin1 in Regulating Estrogen Receptor Expression Independent of Wnt/β-Catenin Signaling.” eLife 9 (August). 10.7554/eLife.56434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel Nicole, Jaime-Soguero Anchel de, Del Arco Ana García, Landry Jonathan J. M., Tietje Marlene, Villacorta Laura, Benes Vladimir, Fernández-Sáiz Vanesa, and Acebrón Sergio P.. 2021. “USP42 Protects ZNRF3/RNF43 from R-Spondin-Dependent Clearance and Inhibits Wnt Signalling.” EMBO Reports 22 (5): e51415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser Kathrin, Urban Manuela, Fenech Emma, Voloshanenko Oksana, Kranz Dominique, Lari Federica, Christianson John C., and Boutros Michael. 2018. “ERAD-Dependent Control of the Wnt Secretory Factor Evi.” The EMBO Journal 37 (4). 10.15252/embj.201797311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka Andrei, Dolde Christine, Kirsch Nadine, Huang Ya-Lin, Kazanskaya Olga, Ingelfinger Dierk, Boutros Michael, Cruciat Cristina-Maria, and Niehrs Christof. 2011. “LGR4 and LGR5 Are R-Spondin Receptors Mediating Wnt/β-Catenin and Wnt/PCP Signalling.” EMBO Reports 12 (10): 1055–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, and Scott MP. 1997. “Altered Neural Cell Fates and Medulloblastoma in Mouse Patched Mutants.” Science 277 (5329): 1109–13. [DOI] [PubMed] [Google Scholar]
- Greicius Gediminas, Kabiri Zahra, Sigmundsson Kristmundur, Liang Chao, Bunte Ralph, Singh Manvendra K., and Virshup David M.. 2018. “PDGFRα+ Pericryptal Stromal Cells Are the Critical Source of Wnts and RSPO3 for Murine Intestinal Stem Cells in Vivo.” Proceedings of the National Academy of Sciences of the United States of America 115 (14): E3173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra Damian, Chapiro Sonia M., Pratelli Réjane, Yu Shi, Jia Weitao, Leary Julie, Pilot Guillaume, and Callis Judy. 2017. “Control of Amino Acid Homeostasis by a Ubiquitin Ligase-Coactivator Protein Complex.” The Journal of Biological Chemistry 292 (9): 3827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra Damian D., Pratelli Réjane, Kraft Edward, Callis Judy, and Pilot Guillaume. 2013. “Functional Conservation between Mammalian MGRN1 and Plant LOG2 Ubiquitin Ligases.” FEBS Letters 587 (21): 3400–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn TM, Miller KA, He L, Hyman RW, Davis RW, Azarani A, Schlossman SF, Duke-Cohan JS, and Barsh GS. 1999. “The Mouse Mahogany Locus Encodes a Transmembrane Form of Human Attractin.” Nature 398 (6723): 152–56. [DOI] [PubMed] [Google Scholar]
- Hao Huai-Xiang, Jiang Xiaomo, and Cong Feng. 2016. “Control of Wnt Receptor Turnover by R-Spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer.” Cancers 8 (6). 10.3390/cancers8060054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Huai-Xiang, Xie Yang, Zhang Yue, Charlat Olga, Oster Emma, Avello Monika, Lei Hong, et al. 2012. “ZNRF3 Promotes Wnt Receptor Turnover in an R-Spondin-Sensitive Manner.” Nature 485 (7397): 195–200. [DOI] [PubMed] [Google Scholar]
- Harper J. Wade, and Schulman Brenda A.. 2021. “Cullin-RING Ubiquitin Ligase Regulatory Circuits: A Quarter Century Beyond the F-Box Hypothesis.” Annual Review of Biochemistry 90 (June): 403–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin Carl-Henrik, and Moustakas Aristidis. 2016. “Signaling Receptors for TGF-β Family Members.” Cold Spring Harbor Perspectives in Biology 8 (8). 10.1101/cshperspect.a022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Gunn TM, Bouley DM, Lu XY, Watson SJ, Schlossman SF, Duke-Cohan JS, and Barsh GS. 2001. “A Biochemical Function for Attractin in Agouti-Induced Pigmentation and Obesity.” Nature Genetics 27 (1): 40–47. [DOI] [PubMed] [Google Scholar]
- He Lin, Eldridge Adam G., Jackson Peter K., Gunn Teresa M., and Barsh Gregory S.. 2003. “Accessory Proteins for Melanocortin Signaling: Attractin and Mahogunin.” Annals of the New York Academy of Sciences 994 (June): 288–98. [DOI] [PubMed] [Google Scholar]
- He Lin, Lu Xin-Yun, Jolly Aaron F., Eldridge Adam G., Watson Stanley J., Jackson Peter K., Barsh Gregory S., and Gunn Teresa M.. 2003. “Spongiform Degeneration in Mahoganoid Mutant Mice.” Science 299 (5607): 710–12. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A, and Varshavsky A. 2000. “Basic Medical Research Award. The Ubiquitin System.” Nature Medicine 6 (10): 1073–81. [DOI] [PubMed] [Google Scholar]
- He Xi, Semenov Mikhail, Tamai Keiko, and Zeng Xin. 2004. “LDL Receptor-Related Proteins 5 and 6 in Wnt/beta-Catenin Signaling: Arrows Point the Way.” Development 131 (8): 1663–77. [DOI] [PubMed] [Google Scholar]
- Hirai Hidenori, Matoba Kyoko, Mihara Emiko, Arimori Takao, and Takagi Junichi. 2019. “Crystal Structure of a Mammalian Wnt–frizzled Complex.” Nature Structural & Molecular Biology 26 (5): 372–79. [DOI] [PubMed] [Google Scholar]
- Hoppler Stefan P., and Moon Randall T.. 2014. Wnt Signaling in Development and Disease: Molecular Mechanisms and Biological Functions. John Wiley & Sons. [Google Scholar]
- Huangfu D, and Anderson KV. 2005. “Cilia and Hedgehog Responsiveness in the Mouse.” Proceedings of the National Academy of Sciences of the United States of America 102 (32): 11325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, and Anderson KV. 2003. “Hedgehog Signalling in the Mouse Requires Intraflagellar Transport Proteins.” Nature 426 (6962): 83–87. [DOI] [PubMed] [Google Scholar]
- Huang Hui-Chuan, and Klein Peter S.. 2004. “The Frizzled Family: Receptors for Multiple Signal Transduction Pathways.” Genome Biology 5 (7): 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Shikai, and Monga Satdarshan P.. 2021. “Wnt/-Catenin Signaling and Liver Regeneration: Circuit, Biology, and Opportunities.” Gene Expression. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii Yoshiyuki, Wajid Muhammad, Bazzi Hisham, Fantauzzo Katherine A., Barber Alison G., Blaydon Diana C., Nam Ju-Suk, Yoon Jeong K., Kelsell David Peter, and Christiano Angela M.. 2008. “Mutations in R-Spondin 4 (RSPO4) Underlie Inherited Anonychia.” The Journal of Investigative Dermatology 128 (4): 867–70. [DOI] [PubMed] [Google Scholar]
- Janda Claudia Y., Waghray Deepa, Levin Aron M., Thomas Christoph, and Garcia K. Christopher. 2012. “Structural Basis of Wnt Recognition by Frizzled.” Science 337 (6090): 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Xiaomo, Charlat Olga, Zamponi Raffaella, Yang Yi, and Cong Feng. 2015. “Dishevelled Promotes Wnt Receptor Degradation through Recruitment of ZNRF3/RNF43 E3 Ubiquitin Ligases.” Molecular Cell 58 (3): 522–33. [DOI] [PubMed] [Google Scholar]
- Jiao Jian, Sun Kaihua, Walker Will P., Bagher Pooneh, Cota Christina D., and Gunn Teresa M.. 2009. “Abnormal Regulation of TSG101 in Mice with Spongiform Neurodegeneration.” Biochimica et Biophysica Acta 1792 (10): 1027–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Yong-Ri, and Yoon Jeong Kyo. 2012. “The R-Spondin Family of Proteins: Emerging Regulators of WNT Signaling.” The International Journal of Biochemistry & Cell Biology 44 (12): 2278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper John, Evans Richard, Pritzel Alexander, Green Tim, Figurnov Michael, Ronneberger Olaf, Tunyasuvunakool Kathryn, et al. 2021. “Highly Accurate Protein Structure Prediction with AlphaFold.” Nature 596 (7873): 583–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata Tomoyuki, Katsube Ken-Ichi, Michikawa Makoto, Yamada Masahito, Takada Shinji, and Mizusawa Hidehiro. 2004. “R-Spondin, a Novel Gene with Thrombospondin Type 1 Domain, Was Expressed in the Dorsal Neural Tube and Affected in Wnts Mutants.” Biochimica et Biophysica Acta 1676 (1): 51–62. [DOI] [PubMed] [Google Scholar]
- Kazanskaya Olga, Glinka Andrei, Barrantes Ivan del Barco, Stannek Peter, Niehrs Christof, and Wu Wei. 2004. “R-Spondin2 Is a Secreted Activator of Wnt/beta-Catenin Signaling and Is Required for Xenopus Myogenesis.” Developmental Cell 7 (4): 525–34. [DOI] [PubMed] [Google Scholar]
- Kazanskaya Olga, Ohkawara Bisei, Heroult Melanie, Wu Wei, Maltry Nicole, Augustin Hellmut G., and Niehrs Christof. 2008. “The Wnt Signaling Regulator R-Spondin 3 Promotes Angioblast and Vascular Development.” Development 135 (22): 3655–64. [DOI] [PubMed] [Google Scholar]
- Kim Bong Yoon, Olzmann James A., Barsh Gregory S., Chin Lih-Shen, and Li Lian. 2007. “Spongiform Neurodegeneration-Associated E3 Ligase Mahogunin Ubiquitylates TSG101 and Regulates Endosomal Trafficking.” Molecular Biology of the Cell 18 (4): 1129–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Kyung-Ah, Kakitani Makoto, Zhao Jingsong, Oshima Takeshi, Tang Tom, Binnerts Minke, Liu Yi, et al. 2005. “Mitogenic Influence of Human R-spondin1 on the Intestinal Epithelium.” Science 309 (5738): 1256–59. [DOI] [PubMed] [Google Scholar]
- Kim Kyung-Ah, Wagle Marie, Tran Karolyn, Zhan Xiaoming, Dixon Melissa A., Liu Shouchun, Gros Delphine, et al. 2008. “R-Spondin Family Members Regulate the Wnt Pathway by a Common Mechanism.” Molecular Biology of the Cell 19 (6): 2588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Minseong, Reinhard Carmen, and Niehrs Christof. 2021. “A MET-PTPRK Kinase-Phosphatase Rheostat Controls ZNRF3 and Wnt Signaling.” eLife 10 (September): e70885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzel Bernd, Pikiolek Monika, Orsini Vanessa, Sprunger Joëlle, Isken Andrea, Zietzling Svenja, Desplanches Magali, et al. 2014. “Functional Roles of Lgr4 and Lgr5 in Embryonic Gut, Kidney and Skin Development in Mice.” Developmental Biology 390 (2): 181–90. [DOI] [PubMed] [Google Scholar]
- Kishida Shosei, Yamamoto Hideki, Hino Shin-Ichiro, Ikeda Satoshi, Kishida Michiko, and Kikuchi Akira. 1999. “DIX Domains of Dvl and Axin Are Necessary for Protein Interactions and Their Ability To Regulate β-Catenin Stability.” Molecular and Cellular Biology. 10.1128/mcb.19.6.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Jennifer H., Young Cullen B., Pusapati Ganesh V., Espinoza F. Hernán, Patel Chandni B., Beckert Francis, Ho Sebastian, et al. 2021. “Gene-Teratogen Interactions Influence the Penetrance of Birth Defects by Altering Hedgehog Signaling Strength.” Development, September. 10.1242/dev.199867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Jennifer H., Young Cullen B., Pusapati Ganesh V., Patel Chandni B., Ho Sebastian, Krishnan Arunkumar, Jiuann-Huey Ivy Lin, et al. 2020. “A Membrane-Tethered Ubiquitination Pathway Regulates Hedgehog Signaling and Heart Development.” Developmental Cell 55 (4): 432–49.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo Bon-Kyoung, and Clevers Hans. 2014. “Stem Cells Marked by the R-Spondin Receptor LGR5.” Gastroenterology 147 (2): 289–302. [DOI] [PubMed] [Google Scholar]
- Koo Bon-Kyoung, Spit Maureen, Jordens Ingrid, Low Teck Y., Stange Daniel E., Wetering Marc van de, van Es Johan H., et al. 2012. “Tumour Suppressor RNF43 Is a Stem-Cell E3 Ligase That Induces Endocytosis of Wnt Receptors.” Nature 488 (7413): 665–69. [DOI] [PubMed] [Google Scholar]
- Kwon Yong Tae, and Ciechanover Aaron. 2017. “The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy.” Trends in Biochemical Sciences 42 (11): 873–86. [DOI] [PubMed] [Google Scholar]
- Lau Wim B. M. de, Snel Berend, and Clevers Hans C.. 2012. “The R-Spondin Protein Family.” Genome Biology 13 (3): 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau Wim de, Lau Wim de, Barker Nick, Low Teck Y., Koo Bon-Kyoung, Li Vivian S. W., Teunissen Hans, et al. 2011. “Lgr5 Homologues Associate with Wnt Receptors and Mediate R-Spondin Signalling.” Nature. 10.1038/nature10337. [DOI] [PubMed] [Google Scholar]
- Lau Wim de, Peng Weng Chuan, Gros Piet, and Clevers Hans. 2014. “The R-spondin/Lgr5/Rnf43 Module: Regulator of Wnt Signal Strength.” Genes & Development 28 (4): 305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensohn Andres M., Dubey Ramin, Neitzel Leif R., Ofelia Tacchelly-Benites Eungi Yang, Marceau Caleb D., Davis Eric M., et al. 2016. “Comparative Genetic Screens in Human Cells Reveal New Regulatory Mechanisms in WNT Signaling.” eLife 5 (December). 10.7554/eLife.21459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensohn Andres M., and Rohatgi Rajat. 2018. “R-Spondins Can Potentiate WNT Signaling without LGRs.” eLife 7 (February). 10.7554/eLife.33126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Hyeyoon, Seidl Carina, Sun Rui, Glinka Andrey, and Niehrs Christof. 2020. “R-Spondins Are BMP Receptor Antagonists in Xenopus Early Embryonic Development.” Nature Communications 11 (1): 5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung Carly, Tan Si Hui, and Barker Nick. 2018. “Recent Advances in Lgr5 Stem Cell Research.” Trends in Cell Biology. 10.1016/j.tcb.2018.01.010. [DOI] [PubMed] [Google Scholar]
- Levin M, Johnson RL, Stern CD, Kuehn M, and Tabin C. 1995. “A Molecular Pathway Determining Left-Right Asymmetry in Chick Embryogenesis.” Cell 82 (5): 803–14. [DOI] [PubMed] [Google Scholar]
- Lian Fu-Ming, Xie Si, and Qian Chengmin. 2016. “Crystal Structure and SUMO Binding of Slx1-Slx4 Complex.” Scientific Reports 6 (January): 19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lineberry Neil, Su Leon, Soares Luis, and Fathman C. Garrison. 2008. “The Single Subunit Transmembrane E3 Ligase Gene Related to Anergy in Lymphocytes (GRAIL) Captures and Then Ubiquitinates Transmembrane Proteins across the Cell Membrane.” The Journal of Biological Chemistry 283 (42): 28497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Sheng-Jian, Yen Ten-Yang, Endo Yoshimi, Klauzinska Malgorzata, Baljinnyam Bolormaa, Macher Bruce, Callahan Robert, and Rubin Jeffrey S.. 2009. “Loss-of-Function Point Mutations and Two-Furin Domain Derivatives Provide Insights about R-spondin2 Structure and Function.” Cellular Signalling 21 (6): 916–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Wei, Bengtson Mario H., Ulbrich Axel, Matsuda Akio, Reddy Venkateshwar A., Orth Anthony, Chanda Sumit K., Batalov Serge, and Joazeiro Claudio A. P.. 2008. “Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 That Regulates the Organelle’s Dynamics and Signaling.” PloS One 3 (1): e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd Deborah L., Toegel Markus, Fulga Tudor A., and Wilkie Andrew O. M.. 2018. “The Drosophila Homologue of MEGF8 Is Essential for Early Development.” Scientific Reports 8 (1): 8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loregger Anke, Grandl Martina, Mejías-Luque Raquel, Allgäuer Michael, Degenhart Kathrin, Haselmann Verena, Oikonomou Christina, et al. 2015. “The E3 Ligase RNF43 Inhibits Wnt Signaling Downstream of Mutated β-Catenin by Sequestering TCF4 to the Nuclear Membrane.” Science Signaling 8 (393): ra90. [DOI] [PubMed] [Google Scholar]
- Luca Vincent C., Miao Yi, Li Xingnan, Hollander Michael J., Kuo Calvin J., and Garcia K. Christopher. 2020. “Surrogate R-Spondins for Tissue-Specific Potentiation of Wnt Signaling.” PLOS ONE. 10.1371/journal.pone.0226928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, and Wilkison WO. 1994. “Agouti Protein Is an Antagonist of the Melanocyte-Stimulating-Hormone Receptor.” Nature 371 (6500): 799–802. [DOI] [PubMed] [Google Scholar]
- MacDonald Bryan T., and He Xi. 2012. “Frizzled and LRP5/6 Receptors for Wnt/β-Catenin Signaling.” Cold Spring Harbor Perspectives in Biology 4 (12). 10.1101/cshperspect.a007880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn Jason A., Hsu Pi-Chiang, and Emr Scott D.. 2012. “Ubiquitin and Membrane Protein Turnover: From Cradle to Grave.” Annual Review of Biochemistry. 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- Malbon Craig C. 2004. “Frizzleds: New Members of the Superfamily of G-Protein-Coupled Receptors.” Frontiers in Bioscience: A Journal and Virtual Library 9 (May): 1048–58. [DOI] [PubMed] [Google Scholar]
- Metzger Meredith B., Pruneda Jonathan N., Klevit Rachel E., and Weissman Allan M.. 2014. “RING-Type E3 Ligases: Master Manipulators of E2 Ubiquitin-Conjugating Enzymes and Ubiquitination.” Biochimica et Biophysica Acta 1843 (1): 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton AJ, Zhu J, and Day CL. 2020. “The RING Domain of RING Finger 12 Efficiently Builds Degradative Ubiquitin Chains.” Journal of Molecular Biology 432 (13): 3790–3801. [DOI] [PubMed] [Google Scholar]
- Mlodzik Marek. 2016. “The Dishevelled Protein Family: Still Rather a Mystery After Over 20 Years of Molecular Studies.” Current Topics in Developmental Biology 117 (January): 75–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moad Heather E., and Pioszak Augen A.. 2013. “Reconstitution of R-Spondin:LGR4:ZNRF3 Adult Stem Cell Growth Factor Signaling Complexes with Recombinant Proteins Produced in Escherichia Coli.” Biochemistry. 10.1021/bi401090h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat Laura L., Robinson Ryan E., Bakoulis Anastasia, and Clark Scott G.. 2014. “The Conserved Transmembrane RING Finger Protein PLR-1 Downregulates Wnt Signaling by Reducing Frizzled, Ror and Ryk Cell-Surface Levels in C. Elegans.” Development 141 (3): 617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monga Satdarshan (paul) Singh. 2014. “Role and Regulation of β-Catenin Signaling During Physiological Liver Growth.” Gene Expression. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morreale Francesca Ester, and Walden Helen. 2016. “Types of Ubiquitin Ligases.” Cell 165 (1): 248–248.e1. [DOI] [PubMed] [Google Scholar]
- Nagano Kenichi. 2019. “R-Spondin Signaling as a Pivotal Regulator of Tissue Development and Homeostasis.” The Japanese Dental Science Review 55 (1): 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle DL, McGrail SH, Vitale J, Woolf EA, Dussault BJ Jr, DiRocco L, Holmgren L, et al. 1999. “The Mahogany Protein Is a Receptor Involved in Suppression of Obesity.” Nature 398 (6723): 148–52. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Nakajima D, Nagase T, Nomura N, Seki N, and Ohara O. 1998. “Identification of High-Molecular-Weight Proteins with Multiple EGF-like Motifs by Motif-Trap Screening.” Genomics 51 (1): 27–34. [DOI] [PubMed] [Google Scholar]
- Nam Ju-Suk, Park Emily, Turcotte Taryn J., Palencia Servando, Zhan Xiaoming, Lee Jackie, Yun Kyuson, Funk Walter D., and Yoon Jeong Kyo. 2007. “Mouse R-spondin2 Is Required for Apical Ectodermal Ridge Maintenance in the Hindlimb.” Developmental Biology 311 (1): 124–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Ju-Suk, Turcotte Taryn J., Smith Peter F., Choi Sangdun, and Jeong Kyo Yoon. 2006. “Mouse Cristin/R-Spondin Family Proteins Are Novel Ligands for the Frizzled 8 and LRP6 Receptors and Activate β-Catenin-Dependent Gene Expression.” Journal of Biological Chemistry. 10.1074/jbc.m508324200. [DOI] [PubMed] [Google Scholar]
- Nam Ju-Suk, Turcotte Taryn J., and Yoon Jeong Kyo. 2007. “Dynamic Expression of R-Spondin Family Genes in Mouse Development.” Gene Expression Patterns. 10.1016/j.modgep.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Nawaratne Vindhya, Kudumala Sirisha, Kakad Priyanka Prakash, and Godenschwege Tanja A.. 2021. “The Conserved MASRPF Motif in the Attractin Homolog, Distracted, Is Required for Association with Drosophila E3-Ligase Mgrn1.” microPublication Biology 2021 (July). 10.17912/micropub.biology.000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutzner Albert, Neutzner Melanie, Benischke Anne-Sophie, Ryu Seung-Wook, Frank Stephan, Youle Richard J., and Karbowski Mariusz. 2011. “A Systematic Search for Endoplasmic Reticulum (ER) Membrane-Associated RING Finger Proteins Identifies Nixin/ZNRF4 as a Regulator of Calnexin Stability and ER Homeostasis.” The Journal of Biological Chemistry 286 (10): 8633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs Christof. 2012. “The Complex World of WNT Receptor Signalling.” Nature Reviews. Molecular Cell Biology 13 (12): 767–79. [DOI] [PubMed] [Google Scholar]
- Ohkawara Bisei, Glinka Andrei, and Niehrs Christof. 2011. “Rspo3 Binds Syndecan 4 and Induces Wnt/PCP Signaling via Clathrin-Mediated Endocytosis to Promote Morphogenesis.” Developmental Cell 20 (3): 303–14. [DOI] [PubMed] [Google Scholar]
- Overton John D., and Leibel Rudolph L.. 2011. “Mahoganoid and Mahogany Mutations Rectify the Obesity of the Yellow Mouse by Effects on Endosomal Traffic of MC4R Protein.” The Journal of Biological Chemistry 286 (21): 18914–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Soohyun, Cui Jie, Yu Wangsheng, Wu Ling, Carmon Kendra S., and Liu Qingyun J.. 2018. “Differential Activities and Mechanisms of the Four R-Spondins in Potentiating Wnt/β-Catenin Signaling.” The Journal of Biological Chemistry 293 (25): 9759–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parma Pietro, Radi Orietta, Vidal Valerie, Marie Christine Chaboissier Elena Dellambra, Valentini Stella, Guerra Liliana, Schedl Andreas, and Camerino Giovanna. 2006. “R-spondin1 Is Essential in Sex Determination, Skin Differentiation and Malignancy.” Nature Genetics. 10.1038/ng1907. [DOI] [PubMed] [Google Scholar]
- Peng Weng Chuan, Wim de Lau Federico Forneris, Granneman Joke C. M., Huch Meritxell, Clevers Hans, and Gros Piet. 2013. “Structure of Stem Cell Growth Factor R-Spondin 1 in Complex with the Ectodomain of Its Receptor LGR5.” Cell Reports 3 (6): 1885–92. [DOI] [PubMed] [Google Scholar]
- Peng Weng Chuan, Wim de Lau Pramod K. Madoori, Forneris Federico, Granneman Joke C. M., Clevers Hans, and Gros Piet. 2013. “Structures of Wnt-Antagonist ZNRF3 and Its Complex with R-Spondin 1 and Implications for Signaling.” PloS One 8 (12): e83110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan Loan K., Lin Feng, LeDuc Charles A., Chung Wendy K., and Leibel Rudolph L.. 2002. “The Mouse Mahoganoid Coat Color Mutation Disrupts a Novel C3HC4 RING Domain Protein.” The Journal of Clinical Investigation 110 (10): 1449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips Ben P., and Miller Elizabeth A.. 2021. “Membrane Protein Folding and Quality Control.” Current Opinion in Structural Biology 69 (August): 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas-Paz Lara, Orsini Vanessa, Boulter Luke, Calabrese Diego, Pikiolek Monika, Nigsch Florian, Xie Yang, et al. 2016. “The RSPO–LGR4/5–ZNRF3/RNF43 Module Controls Liver Zonation and Size.” Nature Cell Biology. 10.1038/ncb3337. [DOI] [PubMed] [Google Scholar]
- Pusapati Ganesh V., Kong Jennifer H., Patel Bhaven B., Krishnan Arunkumar, Sagner Andreas, Kinnebrew Maia, Briscoe James, Aravind L, and Rohatgi Rajat. 2018. “CRISPR Screens Uncover Genes That Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog.” Developmental Cell 44 (1): 113–29.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radaszkiewicz Tomasz, and Bryja Vítězslav. 2020. “Protease Associated Domain of RNF43 Is Not Necessary for the Suppression of Wnt/β-Catenin Signaling in Human Cells.” Cell Communication and Signaling: CCS 18 (1): 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radaszkiewicz Tomasz, Michaela Nosková Kristína Gömöryová, Olga Vondálová Blanářová Katarzyna Anna Radaszkiewicz, Markéta Picková Ráchel Víchová, et al. 2021. “RNF43 Inhibits WNT5A-Driven Signaling and Suppresses Melanoma Invasion and Resistance to the Targeted Therapy.” eLife 10 (October). 10.7554/eLife.65759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raslan Ahmed A., and Jeong Kyo Yoon. 2019. “R-Spondins: Multi-Mode WNT Signaling Regulators in Adult Stem Cells.” The International Journal of Biochemistry & Cell Biology. 10.1016/j.biocel.2018.11.005. [DOI] [PubMed] [Google Scholar]
- Reis Alice H., and Sokol Sergei Y.. 2020. “Rspo2 Antagonizes FGF Signaling during Vertebrate Mesoderm Formation and Patterning.” Development 147 (10). 10.1242/dev.189324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2021. “Rspo2 Inhibits TCF3 Phosphorylation to Antagonize Wnt Signaling during Vertebrate Anteroposterior Axis Specification.” Scientific Reports 11 (1): 13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Zemin, Andel Harmen van, Lau Wim de, Hartholt Robin B., Maurice Madelon M., Clevers Hans, Kersten Marie José, Spaargaren Marcel, and Pals Steven T.. 2018. “Syndecan-1 Promotes Wnt/β-Catenin Signaling in Multiple Myeloma by Presenting Wnts and R-Spondins.” Blood 131 (9): 982–94. [DOI] [PubMed] [Google Scholar]
- Rosenbaum Mette Ishøy, Clemmensen Louise S., Bredt David S., Bettler Bernhard, and Strømgaard Kristian. 2020. “Targeting Receptor Complexes: A New Dimension in Drug Discovery.” Nature Reviews Drug Discovery. 10.1038/s41573-020-0086-4. [DOI] [PubMed] [Google Scholar]
- Ruffner Heinz, Sprunger Joëlle, Charlat Olga, Juliet Leighton-Davies Bianka Grosshans, Salathe Adrian, Zietzling Svenja, et al. 2012. “R-Spondin Potentiates Wnt/β-Catenin Signaling through Orphan Receptors LGR4 and LGR5.” PloS One 7 (7): e40976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardana Richa, and Emr Scott D.. 2021. “Membrane Protein Quality Control Mechanisms in the Endo-Lysosome System.” Trends in Cell Biology 31 (4): 269–83. [DOI] [PubMed] [Google Scholar]
- Sarrazin Stephane, Lamanna William C., and Esko Jeffrey D.. 2011. “Heparan Sulfate Proteoglycans.” Cold Spring Harbor Perspectives in Biology 3 (7). 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider Melanie, Radoux Chris J., Hercules Andrew, Ochoa David, Dunham Ian, Zalmas Lykourgos-Panagiotis, Hessler Gerhard, et al. 2021. “The PROTACtable Genome.” Nature Reviews. Drug Discovery 20 (10): 789–97. [DOI] [PubMed] [Google Scholar]
- Schulte Gunnar, and Bryja Vítezslav. 2007. “The Frizzled Family of Unconventional G-Protein-Coupled Receptors.” Trends in Pharmacological Sciences 28 (10): 518–25. [DOI] [PubMed] [Google Scholar]
- Schulte Gunnar, and Wright Shane C.. 2018. “Frizzleds as GPCRs – More Conventional than We Thought!” Trends in Pharmacological Sciences 39 (9): 828–42. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond Thomas, Fiedler Marc, Shibata Naoki, Butler P. Jonathan G., Kikuchi Akira, Higuchi Yoshiki, and Bienz Mariann. 2007. “The DIX Domain of Dishevelled Confers Wnt Signaling by Dynamic Polymerization.” Nature Structural & Molecular Biology. 10.1038/nsmb1247. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond Thomas, Metcalfe Ciara, and Bienz Mariann. 2007. “Dynamic Recruitment of Axin by Dishevelled Protein Assemblies.” Journal of Cell Science. 10.1242/jcs.002956. [DOI] [PubMed] [Google Scholar]
- Sharma Monica, Isabel Castro-Piedras Glenn E. Simmons Jr, and Pruitt Kevin. 2018. “Dishevelled: A Masterful Conductor of Complex Wnt Signals.” Cellular Signalling 47 (July): 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons Matias, Gault William J., Gotthardt Daniel, Rohatgi Rajeev, Klein Thomas J., Shao Youming, Lee Ho-Jin, et al. 2009. “Electrochemical Cues Regulate Assembly of the Frizzled/Dishevelled Complex at the Plasma Membrane during Planar Epithelial Polarization.” Nature Cell Biology 11 (3): 286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder Joshua C., Rochelle Lauren K., H. Kim Lyerly, Marc G. Caron, and Lawrence S. Barak. 2013. “Constitutive Internalization of the Leucine-Rich G Protein-Coupled Receptor-5 (LGR5) to the Trans-Golgi Network.” The Journal of Biological Chemistry 288 (15): 10286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder Joshua C., Rochelle Lauren K., Ray Caroline, Pack Thomas F., Bock Cheryl B., Lubkov Veronica, Lyerly H. Kim, Waggoner Alan S., Barak Larry S., and Caron Marc G.. 2017. “Inhibiting Clathrin-Mediated Endocytosis of the Leucine-Rich G Protein-Coupled Receptor-5 Diminishes Cell Fitness.” The Journal of Biological Chemistry 292 (17): 7208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler Jamie B., Moraga Ignacio, Mendoza Juan L., and Garcia K. Christopher. 2015. “Insights into Cytokine–Receptor Interactions from Cytokine Engineering.” Annual Review of Immunology. 10.1146/annurev-immunol-032713-120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spit Maureen, Fenderico Nicola, Jordens Ingrid, Radaszkiewicz Tomasz, Lindeboom Rik G. H., Bugter Jeroen M., Cristobal Alba, et al. 2020. “RNF 43 Truncations Trap CK 1 to Drive Niche-independent Self-renewal in Cancer.” The EMBO Journal 39 (18). 10.15252/embj.2019103932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steege Eline J. Ter, ter Steege Eline J., and Bakker Elvira R. M.. 2021. “The Role of R-Spondin Proteins in Cancer Biology.” Oncogene. 10.1038/s41388-021-02059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens Payton D., and Williams Bart O.. 2021. “LGR4: Not Just for Wnt Anymore?” Cancer Research. 10.1158/0008-5472.can-21-2266. [DOI] [PubMed] [Google Scholar]
- Su Leon L., Iwai Hideyuki, Lin Jack T., and Fathman C. Garrison. 2009. “The Transmembrane E3 Ligase GRAIL Ubiquitinates and Degrades CD83 on CD4 T Cells.” Journal of Immunology 183 (1): 438–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Rui, He Lixiazi, Lee Hyeyoon, Glinka Andrey, Andresen Carolin, Daniel Hübschmann Irmela Jeremias, Karin Müller-Decker Caroline Pabst, and Niehrs Christof. 2021. “RSPO2 Inhibits BMP Signaling to Promote Self-Renewal in Acute Myeloid Leukemia.” Cell Reports 36 (7): 109559. [DOI] [PubMed] [Google Scholar]
- Sun Tianliang, Annunziato Stefano, Bergling Sebastian, Sheng Caibin, Orsini Vanessa, Forcella Pascal, Pikiolek Monika, et al. 2021. “ZNRF3 and RNF43 Cooperate to Safeguard Metabolic Liver Zonation and Hepatocyte Proliferation.” Cell Stem Cell 28 (10): 1822–37.e10. [DOI] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, and Teglund S. 2006. “Genetic Elimination of Suppressor of Fused Reveals an Essential Repressor Function in the Mammalian Hedgehog Signaling Pathway.” Developmental Cell 10 (2): 187–97. [DOI] [PubMed] [Google Scholar]
- Szenker-Ravi Emmanuelle, Altunoglu Umut, Leushacke Marc, Célia Bosso-Lefèvre Muznah Khatoo, Hong Thi Tran Thomas Naert, et al. 2018. “RSPO2 Inhibition of RNF43 and ZNRF3 Governs Limb Development Independently of LGR4/5/6.” Nature 557 (7706): 564–69. [DOI] [PubMed] [Google Scholar]
- Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, and He X. 2000. “LDL-Receptor-Related Proteins in Wnt Signal Transduction.” Nature 407 (6803): 530–35. [DOI] [PubMed] [Google Scholar]
- Tauriello Daniele V. F., Jordens Ingrid, Kirchner Katharina, Slootstra Jerry W., Kruitwagen Tom, Bouwman Britta A. M., Noutsou Maria, et al. 2012. “Wnt/β-Catenin Signaling Requires Interaction of the Dishevelled DEP Domain and C Terminus with a Discontinuous Motif in Frizzled.” Proceedings of the National Academy of Sciences of the United States of America 109 (14): E812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizuka Kazuma, Horikoshi Kaori, Kitada Rina, Sugawara Yuriko, Iba Yumi, Kojima Ayako, Yoshitome Akiko, et al. 2008. “R-spondin1 Plays an Essential Role in Ovarian Development through Positively Regulating Wnt-4 Signaling.” Human Molecular Genetics 17 (9): 1278–91. [DOI] [PubMed] [Google Scholar]
- Trenker Raphael, Wu Xinyu, Nguyen Julie V., Wilcox Stephen, Rubin Alan F., Call Matthew E., and Call Melissa J.. 2021. “Human and Viral Membrane-Associated E3 Ubiquitin Ligases MARCH1 and MIR2 Recognize Different Features of CD86 to Downregulate Surface Expression.” The Journal of Biological Chemistry 297 (1): 100900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiairis Charisios D., and Andrew P. McMahon. 2009. “An Hh-Dependent Pathway in Lateral Plate Mesoderm Enables the Generation of Left/right Asymmetry.” Current Biology: CB 19 (22): 1912–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama Tadasuke, Fukui Akimasa, Terai Sayuri, Fujioka Yoichiro, Shinada Keisuke, Takahashi Hidehisa, Yamaguchi Terry P., Ohba Yusuke, and Hatakeyama Shigetsugu. 2015. “Molecular Role of RNF43 in Canonical and Noncanonical Wnt Signaling.” Molecular and Cellular Biology 35 (11): 2007–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama Tadasuke, Koo Bon-Kyoung, and Hatakeyama Shigetsugu. 2021. “Post-Translational Wnt Receptor Regulation: Is the Fog Slowly Clearing?: The Molecular Mechanism of RNF43/ZNRF3 Ubiquitin Ligases Is Not yet Fully Elucidated and Still Controversial.” BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology 43 (4): e2000297. [DOI] [PubMed] [Google Scholar]
- Tsukiyama Tadasuke, Zou Juqi, Kim Jihoon, Ogamino Shohei, Shino Yuki, Masuda Takamasa, Merenda Alessandra, et al. 2020. “A Phospho-Switch Controls RNF43-Mediated Degradation of Wnt Receptors to Suppress Tumorigenesis.” Nature Communications 11 (1): 4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunyasuvunakool Kathryn, Adler Jonas, Wu Zachary, Green Tim, Zielinski Michal, Augustin Žídek Alex Bridgland, et al. 2021. “Highly Accurate Protein Structure Prediction for the Human Proteome.” Nature 596 (7873): 590–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Lloyd D, Jenkins D, Elcioglu NE, Cooper CD, Al-Sannaa N, Annagur A, et al. 2012. “Mutations in Multidomain Protein MEGF8 Identify a Carpenter Syndrome Subtype Associated with Defective Lateralization.” American Journal of Human Genetics 91 (5): 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbhauer M, Djiane A, Goisset C, Penzo-Méndez A, Riou JF, Boucaut JC, and Shi DL. 2000. “The C-Terminal Cytoplasmic Lys-Thr-X-X-X-Trp Motif in Frizzled Receptors Mediates Wnt/beta-Catenin Signalling.” The EMBO Journal 19 (18): 4944–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal Valerie Pi, Fariba Jian-Motamedi Samah Rekima, Gregoire Elodie P., Emmanuelle Szenker-Ravi Marc Leushacke, Reversade Bruno, Chaboissier Marie-Christine, and Schedl Andreas. 2020. “R-Spondin Signalling Is Essential for the Maintenance and Differentiation of Mouse Nephron Progenitors.” eLife 9 (May). 10.7554/eLife.53895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Huang B, Zhang S, Yu X, Wu W, and Wang X. 2013. “Structural Basis for R-Spondin Recognition by LGR4/5/6 Receptors.” Genes & Development. 10.1101/gad.219360.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Wenfeng, Zheng Xiaoling, Song Hejie, Yang Junjie, Liu Xiangyang, Wang Ye, Zhang Min, and Zhang Zhen. 2020. “Spatial and Temporal Deletion Reveals a Latent Effect of Megf8 on the Left-Right Patterning and Heart Development.” Differentiation; Research in Biological Diversity 113 (May): 19–25. [DOI] [PubMed] [Google Scholar]
- Wang Yanshu, Chang Hao, Rattner Amir, and Nathans Jeremy. 2016. “Frizzled Receptors in Development and Disease.” Current Topics in Developmental Biology 117 (January): 113–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting Chan C., Su Leon L., Lin Jack T., and Fathman C. Garrison. 2011. “GRAIL: A Unique Mediator of CD4 T-Lymphocyte Unresponsiveness.” The FEBS Journal 278 (1): 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard DH, Bodnar W, Harris C, Kiefer L, Nichols JS, Blanchard S, Hoffman C, Moyer M, Burkhart W, and Weiel J. 1995. “Agouti Structure and Function: Characterization of a Potent Alpha-Melanocyte Stimulating Hormone Receptor Antagonist.” Biochemistry 34 (38): 12341–46. [DOI] [PubMed] [Google Scholar]
- Wittrup Anders, Zhang Si-He, Gerdy B. ten Dam, Toin H. van Kuppevelt, Per Bengtson, Maria Johansson, Johanna Welch, Matthias Mörgelin, and Mattias Belting. 2009. “ScFv Antibody-Induced Translocation of Cell-Surface Heparan Sulfate Proteoglycan to Endocytic Vesicles.” Journal of Biological Chemistry. 10.1074/jbc.m109.036129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HC, Mao J, Nguyen JT, Srinivas S, Zhang W, Liu B, Li L, Wu D, and Zheng J. 2000. “Structural Basis of the Recognition of the Dishevelled DEP Domain in the Wnt Signaling Pathway.” Nature Structural Biology 7 (12): 1178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Hing-C, Bourdelas Audrey, Krauss Anke, Lee Ho-Jin, Shao Youming, Wu Dianqing, Mlodzik Marek, Shi De-Li, and Zheng Jie. 2003. “Direct Binding of the PDZ Domain of Dishevelled to a Conserved Internal Sequence in the C-Terminal Region of Frizzled.” Molecular Cell. 10.1016/s1097-2765(03)00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Yang, Zamponi Raffaella, Charlat Olga, Ramones Melissa, Swalley Susanne, Jiang Xiaomo, Rivera Daniel, et al. 2013. “Interaction with Both ZNRF3 and LGR4 Is Required for the Signalling Activity of R-Spondin.” EMBO Reports 14 (12): 1120–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Kai, Xu Yan, Rajashankar Kanagalaghatta R., Robev Dorothea, and Nikolov Dimitar B.. 2013. “Crystal Structures of Lgr4 and Its Complex with R-spondin1.” Structure 21 (9): 1683–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Wakako, Nagao Kenji, Horikoshi Kaori, Fujikura Ayako, Ikeda Eiji, Inagaki Yoshimasa, Kakitani Makoto, et al. 2009. “Craniofacial Malformation in R-spondin2 Knockout Mice.” Biochemical and Biophysical Research Communications 381 (3): 453–58. [DOI] [PubMed] [Google Scholar]
- Yamazaki Yasuo, Christina Schönherr Gaurav K. Varshney, Dogru Murat, Hallberg Bengt, and Palmer Ruth H.. 2013. “Goliath Family E3 Ligases Regulate the Recycling Endosome Pathway via VAMP3 Ubiquitylation.” The EMBO Journal 32 (4): 524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Jing, Mowry Laura E., Kari Nichole Nejak-Bowen Hirohisa Okabe, Diegel Cassandra R., Lang Richard A., Williams Bart O., and Monga Satdarshan P.. 2014. “Beta-Catenin Signaling in Murine Liver Zonation and Regeneration: A Wnt-Wnt Situation!” Hepatology. 10.1002/hep.27082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Fei, Jiang Weiyu, Ku Amy T., Young Adelaide I. J., Zhang Weijie, Souto Eric P., Gao Yankun, et al. 2021. “A Wnt-Independent LGR4-EGFR Signaling Axis in Cancer Metastasis.” Cancer Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Jing, Gimenez Luis E., Hernandez Ciria C., Wu Yiran, Wein Ariel H., Gye Won Han Kyle McClary, et al. 2020. “Determination of the Melanocortin-4 Receptor Structure Identifies Ca2+ as a Cofactor for Ligand Binding.” Science 368 (6489): 428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebisch Matthias, and Jones E. Yvonne. 2015a. “Crystal Structure of R-Spondin 2 in Complex with the Ectodomains of Its Receptors LGR5 and ZNRF3.” Journal of Structural Biology 191 (2): 149–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2015b. “ZNRF3/RNF43--A Direct Linkage of Extracellular Recognition and E3 Ligase Activity to Modulate Cell Surface Signalling.” Progress in Biophysics and Molecular Biology 118 (3): 112–18. [DOI] [PubMed] [Google Scholar]
- Zebisch Matthias, Xu Yang, Krastev Christos, Bryan T. MacDonald, Chen Maorong, Gilbert Robert J. C., He Xi, and Jones E. Yvonne. 2013. “Structural and Molecular Basis of ZNRF3/RNF43 Transmembrane Ubiquitin Ligase Inhibition by the Wnt Agonist R-Spondin.” Nature Communications 4: 2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Min, Zhang Ping, Liu Yunsong, Lv Longwei, Zhang Xiao, Liu Hao, and Zhou Yongsheng. 2017. “RSPO3-LGR4 Regulates Osteogenic Differentiation Of Human Adipose-Derived Stem Cells Via ERK/FGF Signalling.” Scientific Reports 7 (February): 42841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, and McMahon AP. 2001. “Smoothened Mutants Reveal Redundant Roles for Shh and Ihh Signaling Including Regulation of L/R Symmetry by the Mouse Node.” Cell 106 (2): 781–92. [PubMed] [Google Scholar]
- Zhang Z, Alpert D, Francis R, Chatterjee B, Yu Q, Tansey T, Sabol SL, et al. 2009. “Massively Parallel Sequencing Identifies the Gene Megf8 with ENU-Induced Mutation Causing Heterotaxy.” Proceedings of the National Academy of Sciences of the United States of America 106 (9): 3219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Zhengjian, Broderick Caroline, Nishimoto Marni, Yamaguchi Teppei, Lee Sung-Jin, Zhang Haili, Chen Hui, et al. 2020. “Tissue-Targeted R-Spondin Mimetics for Liver Regeneration.” Scientific Reports 10 (1): 13951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Chang, Zhou Fan, Liang Liang Shi Gui Fang Xu, Zhang Bin, Wang Lei, Zhuge Yuzheng, Xiao Ping Zou, and Yi Wang. 2020. “R-spondin2 Suppresses the Progression of Hepatocellular Carcinoma via MAPK Signaling Pathway.” Molecular Cancer Research: MCR 18 (10): 1491–99. [DOI] [PubMed] [Google Scholar]
- Zheng Ning, and Shabek Nitzan. 2017. “Ubiquitin Ligases: Structure, Function, and Regulation.” Annual Review of Biochemistry 86 (June): 129–57. [DOI] [PubMed] [Google Scholar]
- Zhou Xiaolin, Geng Liying, Wang Degeng, Yi Haowei, Talmon Geoffrey, and Wang Jing. 2017. “R-Spondin1/LGR5 Activates TGFβ Signaling and Suppresses Colon Cancer Metastasis.” Cancer Research 77 (23): 6589–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Xuechen, Wang Pan, Wei Jiale, Li Yongyu, Zhai Jiayu, Zheng Tianrui, and Tao Qinghua. 2021. “Lysosomal Degradation of the Maternal Dorsal Determinant Hwa Safeguards Dorsal Body Axis Formation.” EMBO Reports, October, e53185. [DOI] [PMC free article] [PubMed] [Google Scholar]