

Abstract
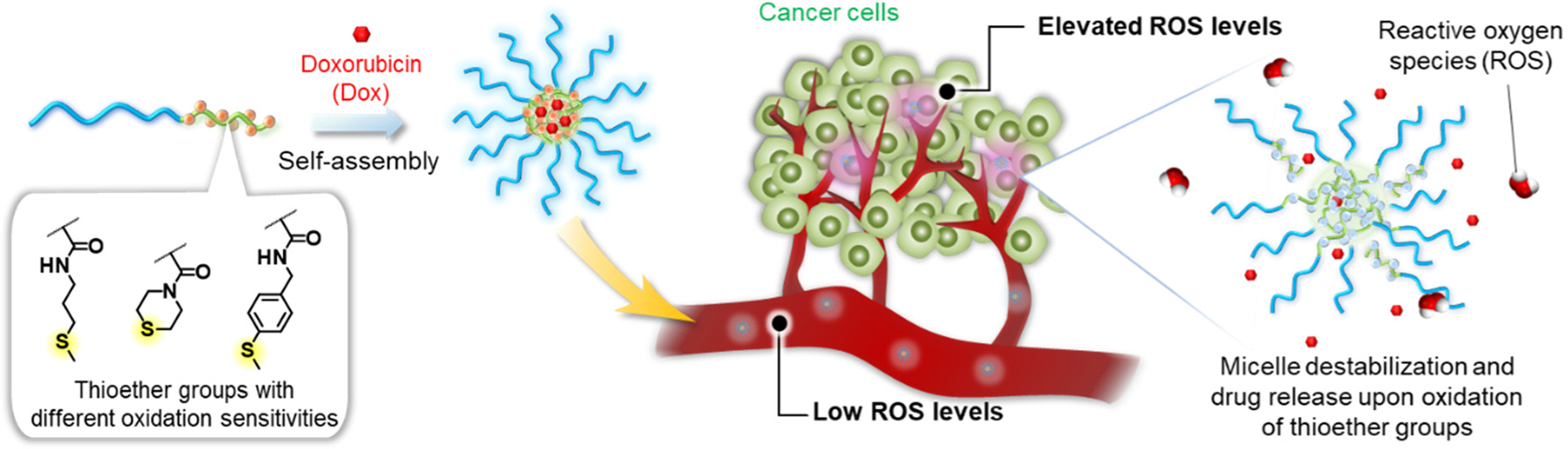
Oxidation-sensitive drug delivery systems (DDS) have attracted attention due to the potential to improve efficacy and safety of chemotherapeutics. These systems are designed to release the payload in response to oxidative stress conditions, which are associated with many types of cancer. Despite extensive research on the development of oxidation-sensitive DDS, the lack of selectivity towards cancer cells over healthy cells remains a challenge. Here, we report the design and characterization of polymeric micelles containing thioether groups with varying oxidation sensitivities within the micellar core, which become hydrophilic upon thioether oxidation leading to destabilization of the micellar structure. We first used the thioether model compounds, 3-methylthiopropylamide (TPAM), thiomorpholine amide (TMAM), and 4-(methylthio)benzylamide (TPhAM), to investigate the effect of the chemical structures of the thioethers on the oxidation by hydrogen peroxide . TPAM shows the fastest oxidation followed by TMAM and TPhAM, showing that the oxidation reaction of thioethers can be modulated by changing the substituent groups bound to the sulfur atom. We next prepared micelles containing these different thioether groups within the core (TP, TM and TPh micelles). The micelles containing the thioether groups with a higher oxidation sensitivity were destabilized by at lower concentration. Micelle destabilization was also tested in human liver cancer (HepG2) cells and human umbilical vein endothelial cells (HUVECs). The TP micelles having the highest oxidation sensitivity were destabilized in both HepG2 cells and HUVECs while the TPh micelles, which showed the lowest reactivity towards , were stable in those cell lines. The TM micelles possessing a moderate oxidation sensitivity were destabilized in HepG2 cells but were stable in HUVECs. Furthermore, the micelles were loaded with doxorubicin (Dox) to evaluate their potential in drug delivery applications. Among the micelles, the TM micelles loaded with Dox showed the enhanced relative toxicity in HepG2 cells over HUVECs. Therefore, our approach to fine-tune the oxidation sensitivity of the micelles has potential for improving therapeutic efficacy and safety of drugs in cancer treatment.
Keywords: Oxidation sensitive, polymeric micelles, thioether, cancer, drug delivery
Graphical Abstract
INTRODUCTION
Elevated levels of reactive oxygen species (ROS) such as hydrogen peroxide , superoxide anion radical (O2ꞏ−), singlet oxygen (1O2), and the hydroxyl radical (ꞏOH) are commonly observed in different types of cancer.1–2 These species are produced during mitochondrial electron transport in aerobic respiration as well as by the action of nicotinamide adenine dinucleotide phosphate oxidases to cause oxidative cellular damage including lipid peroxidation, DNA cleavage and protein modification.3 While ROS are maintained at basal levels in normal cells, many cancer cells exhibit increased rates of ROS production due to high metabolic activity and impaired antioxidant systems.4–6 This redox imbalance in cancer cells contributes to the characteristic tumor microenvironment, which stimulates tumor growth, invasion, and metastasis.
In the field of drug delivery, this oxidative tumor microenvironment has attracted attention as one of the hallmarks of cancer that provide a unique opportunity for cancer-specific drug delivery. Recently, many types of oxidation-sensitive drug carriers that release drugs in response to ROS have been developed with the goal to enhance drug accumulation in cancer tissues. These drug carriers, including polymeric micelles, polymersomes and other polymeric nanoparticles, are composed of amphiphilic polymers containing an oxidation-sensitive building block that alters its water solubility or degrades upon oxidation by ROS. Examples of such building blocks are polymers containing thioether,7–9 arylboronate ester10–11 and thioketal moieties12–13.
Thioether-containing polymers are among the most widely used building blocks to confer oxidation sensitivity to drug carriers. Thioethers are known to be oxidized by ROS, analogous to the oxidation of methionine residues of proteins under oxidative stress.14 Along with this oxidation reaction, thioethers, which have low dipolar moments, are converted to polar sulfoxides and, in part, sulfones. Therefore, thioether-containing polymers undergo a hydrophobic-hydrophilic phase transition upon oxidation. Hubbell and colleagues were the first to report oxidation-sensitive polymersomes and polymeric micelles prepared from amphiphilic block copolymers consisting of a hydrophobic poly(propylene sulfide) (PPS) block and a hydrophilic poly(ethylene glycol) (PEG) block. These nanostructures were destabilized in the presence of under physiological conditions8, 15 and improved delivery of a wide variety of drugs, such as siRNA,16 cyclosporin A17 and ovalbumin18 to cancer and dendritic cells. Following these reports, other types of thioether-containing polymers such as poly(methionine)19–20 and poly(N-acryloyl thiomorpholine)9 have been developed. Recently, poly(ester-thioether)s have been reported as biodegradable and oxidation-sensitive polymers.21 The oxidation of these polymers can be slowed down by increasing the hydrophobicity of the polymer backbone structures.
Despite the promising results of oxidation-sensitive drug carriers, their selectivity towards cancer remains questionable. Thus far, most studies have focused on improving the ROS sensitivity of drug carriers. For example, sulfur in thioether-containing polymers has been replaced with selenium or tellurium to increase their susceptibility to oxidation.22–23 However, drug carriers with a high ROS sensitivity may also respond to ROS at basal levels in healthy tissue (~1–10% of ROS levels in cancer)6 leading to non-specific drug release. Indeed, according to the report by Gupta et al., while drug release from PPS-based polymeric micelles was enhanced in murine macrophages that overproduce ROS in response to lipopolysaccharide activation, a significant amount of drug release was also observed for non-activated macrophages.24 This indicates that PPS-based drug carriers can also be destabilized in healthy tissues which have low levels of ROS. Therefore, fine-tuning of the ROS sensitivity is needed to develop drug carriers that are stable in healthy tissues, but release payloads in tumor tissues associated with elevated ROS production.6
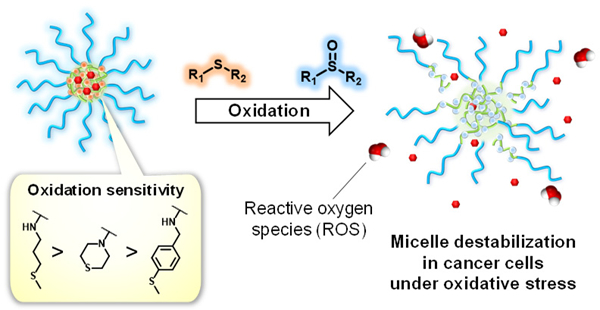
Here, we present thioether-containing polymeric micelles having different oxidation sensitivities (Figure 1). Since the oxidation of the thioether groups are expected to be dependent on the electronic and steric effects of the substituent groups attached to the sulfur atom, we hypothesize that the oxidation sensitivity of polymeric micelles can be controlled by designing amphiphilic block copolymers with different thioether pendant groups. We prepared polymeric micelles having different thioether groups and investigated their destabilization in the presence of as well as in cancer and normal cells. Furthermore, these micelles were loaded with doxorubicin (Dox) to evaluate cytotoxicity of these micelles to demonstrate the potential applications in chemotherapy.
Figure 1.
Oxidation-sensitive polymeric micelles based on thioether groups with fine-tuned oxidation sensitivities for site-specific drug release in cancer tissues that exhibit elevated reactive oxygen species (ROS) levels.
EXPERIMENTAL SECTION
Determination of the oxidation rate of the thioether model compounds by using the iodide oxidation assay.
The thioether model compound (1–3) were dissolved in 2.16 mL of 10% DMF/PBS (pH7.4) and mixed with 0.27 mL of in PBS (pH7.4) to give final concentrations of 10 mM of the model compound and 0.003 wt% . The solution was kept at 20°C and at the indicated time points, 0.2 mL of the solution was withdrawn and mixed with 0.2 mL of a 1000 mM NaI solution in 10% DMF/Phosphate buffered saline (PBS, pH7.4). After reacting for 15 min, the samples were transferred to a quartz cuvette and the absorbance at 350 nm due to the anion formed by oxidation of the anion by remaining was measured. The amount of unreacted was calculated as follows:
| (Eq. 1) |
After collecting the last data points, the thioether model compound/ solutions (2.7 mL) were lyophilized and the residue was extracted with CHCl3 (4 mL total). After removing solvent in a flow of air and drying under vacuum at 40°C, the residue was dissolved in deuterated chloroform to measure 1H NMR (Figure S1). 1H NMR spectra were in agreement with those of the oxidized model compounds (4-6).
Molecular Modeling.
Density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP).25 The generalized gradient approximation GGA-PBE26 functional was used to account for electron exchange-correlation effects. The projector augmented wave (PAW) method27 was used to represent the ionic cores. All the calculations were spin polarized. The energy cutoff for the plane wave function was 400 eV. The break condition for self-consistent interaction is 1 × 10−6. Ionic relaxation was stopped when the forces on all atoms were smaller than −0.02 eV/Å. The k-point was employed throughout entire DFT calculations.
TPAM (1), TMAM (2), and TPhAM (3) and their corresponding oxidized products (4–6) were placed in a large box of 25×25×25 Å3 for geometry optimization as shown in Figure S2. The vibrational frequencies were obtained based on the simple harmonic approximation of the optimized molecular structures. The S-O bond stretching modes were corroborated by FT-IR spectroscopy. Bader charge analysis28 of the S atom in Compounds 1–3 was also performed to evaluate their nucleophilicity.
Polymer synthesis Poly(pentafluorophenyl acrylate) homopolymer (PPFPA, 7).
PPFPA polymer (7) was synthesized by RAFT polymerization of pentafluorophenyl acrylate (PFPA) using azobisisobutyronitrile (AIBN) as the initiator and 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid as the chain transfer agent (CTA). PFPA (914.4 mg, 3.84 mmol), CTA (28.0 mg, 0.0768 mmol), and AIBN (1.26 mg, 0.00768 mmol) were dissolved in 1,4-dioxane (Total volume: 3.8 mL). The clear yellow solution was deoxygenated by five freeze-pump-thaw cycles under an inert atmosphere of argon and placed in an oil bath at 60°C. After 24 h, the reaction was stopped by cooling in liquid nitrogen and exposing to air. The clear yellow solution was added dropwise to 80 mL of EtOH to precipitate the polymer. The polymer was collected by filtration, washed with EtOH (3 x 25 mL) and dried under reduced pressure to yield a yellow solid (677.7 mg, 72%). The polymer was characterized by 1H NMR (Figures S3) and GPC. The degree of polymerization was calculated to be 52 by 1H NMR.
Poly(N-acryloyl morpholine)-PPFPA diblock copolymer (PAM-PPFPA, 8).
PAM-PPFPA polymer (8) was synthesized by RAFT polymerization of N-acryloyl morpholine (AM) using AIBN as the initiator and PPFPA polymer (7) as the macro CTA. AM (282.3 mg, 2 mmol), Polymer 7 (253.5 mg, 0.02 mmol) and AIBN (0.328 mg, 0.002 mmol) were dissolved in 1,4-dioxane (Total volume: 2 mL). The clear yellow solution was deoxygenated by five freeze-pump-thaw cycles under an inert atmosphere of argon and placed in an oil bath at 60°C. After 24 h, the reaction was stopped by cooling in liquid nitrogen and exposing to air. The clear yellow solution was added dropwise to 40 mL of Et2O to precipitate the polymer. The polymer was collected by filtration, washed with Et2O (3 x 15 mL) and dried under reduced pressure to yield a yellow solid (449.9 mg, 84%). The polymer was characterized by 1H NMR (Figure S4), 19F NMR (Figure 4) and GPC. The degree of polymerization was calculated to be 97 by 1H NMR.
Figure 4.
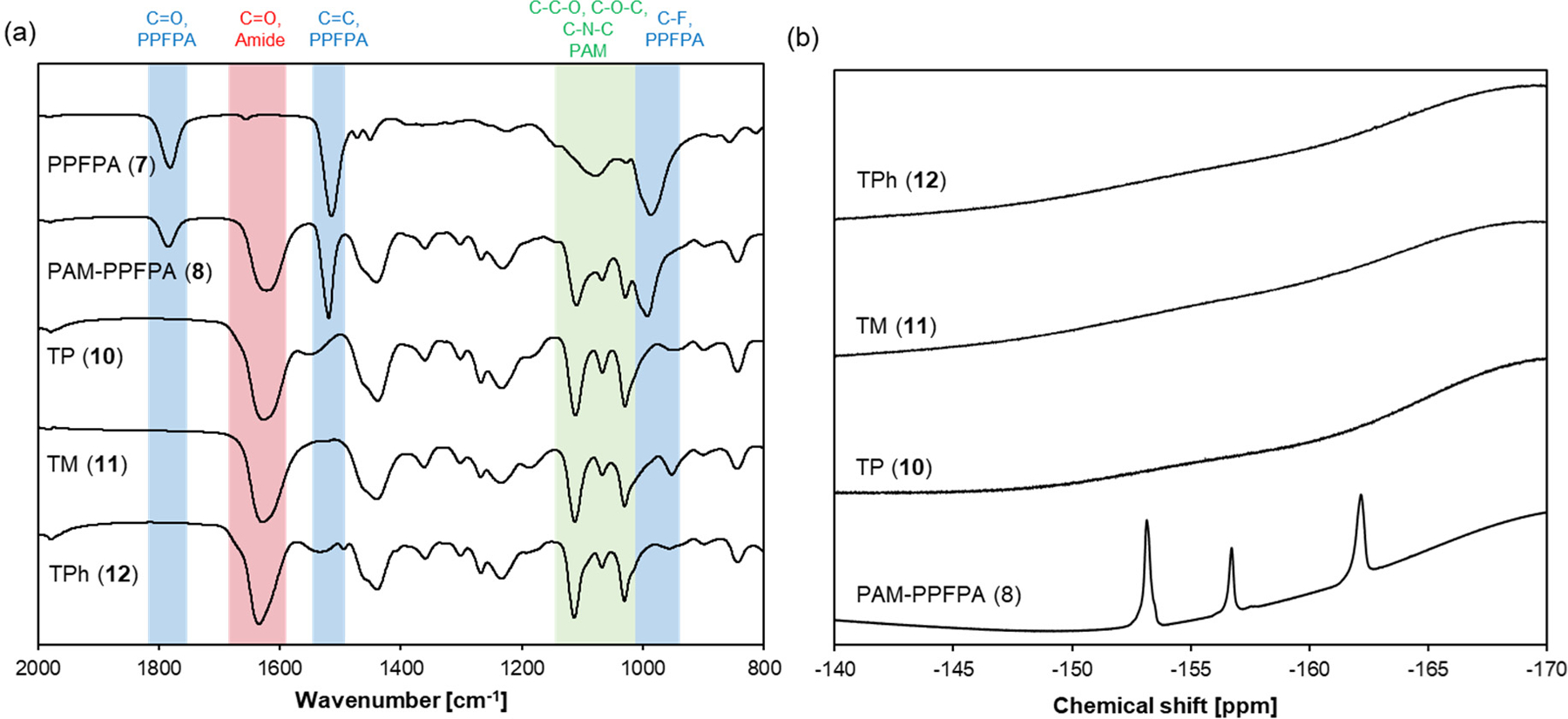
(a) FT-IR spectra and (b) 19F NMR of Polymers 7, 8, 10, 11 and 12.
CTA end group removal from PAM-PPFPA via radical-induced reduction (Polymer 9).
The CTA end group was removed by radical-induced reduction.29 Polymer 8 (393 mg, 0.0146 mmol), tris(trimethylsilyl)silane (18.1 mg, 22.3 μL, 0.0728 mmol) and AIBN (4.8 mg, 0.0291 mmol) were dissolved in 1.5 mL of 1,4-dioxane. The clear yellow solution was deoxygenated by five freeze-pump-thaw cycles under an inert atmosphere of argon and placed in an oil bath at 70°C. After 24 h, the reaction was stopped by cooling in liquid nitrogen and exposing to air. The colorless solution was added dropwise to 30 mL of Et2O to precipitate the polymer. The polymer was collected by filtration, washed with Et2O (3 x 10 mL) and dried under reduced pressure to yield a white solid (346.7 mg, 88%). The polymer was characterized by 1H NMR (Figure S5) and GPC. Polymers 10–12. A typical procedure to prepare TP polymer (10) is as follow: Polymer 9 (41.4 mg, 82.5 μmol of PFPA groups) was dried at 40 °C under vacuum for 5 h in a reaction vial with a septum cap and dissolved in 2 mL anhydrous DMF followed by the addition of TP-NH2 (43 mg, 46 μL, 412 μmol). After three evacuate-purge cycles under inert atmosphere of argon, the solution was placed at 50 °C for 24 h. The solution was cooled to room temperature and added dropwise to 40 mL of Et2O to precipitate the polymer. The polymer was collected by filtration, washed with Et2O (3 x 10 mL) and dried under reduced pressure. The crude product was dissolved in 1 mL THF and kept at −20°C for 24 h. The pentafluorophenol (PFP)-amine salt, which had precipitated out from the solution, was removed by filtering through a plug of glasswool. The clear solution was added dropwise to 10 mL Et2O to precipitate the polymer. The polymer was collected by filtration and dried under reduced pressure. For synthesis of TPh polymer (12), 1 eq. of TEA relative to TPhꞏHCl was added to deprotonate the amino group. The polymers were characterized by 1H NMR (Figures S6–8), 19F NMR (Figure 4) and FT-IR.
Polymers 13–15.
Polymer 9 was dried at 40 °C under vacuum for 24 h. Polymer 9 was dissolved in anhydrous DMF at 15 mg/mL, followed by the addition of FITC-NH2ꞏTFA (16) (0.05 eq. relative to PFPA group) and TEA (1 eq. relative to 16). After three evacuate-purge cycles under inert atmosphere of argon, the solution was placed at 50 °C. After 24 h, thioether amines (2 eq. relative to PFPA groups) were added to the reaction mixture under argon. For synthesis of TPh/FITC polymer (15), 1 eq. of TEA relative to TPhꞏHCl was added to deprotonate the amino group. After three evacuate-purge cycles under inert atmosphere of argon, the solution was placed at 50 °C for 24 h. The polymer was isolated from unreacted FITC-NH2, thioether amines and the PFP-amine salt by Sephadex LH-20 size exclusion column chromatography (column diameter: 13 mm, length: 250 mm) using DMF as the eluent. The absorbance at 280 nm of each fraction was measured. The fractions containing polymer were combined, diluted 2x with distilled water and dialyzed (RC membrane, MWCO 3.5kDa) against 1 L of distilled water for 2 d, with regularly replacing the water. Polymers were recovered by lyophilization.
Micelle preparation.
Polymers 10–15 were dissolved in NMP at 50 mg/mL and added dropwise to distilled water under stirring (Volume fraction (v/v) of NMP=10%. Final concentration of polymer: 5 mg/mL). After stirring for 1 d at room temperature, the solution was dialyzed (Dialysis cassette, MWCO 2kDa) against 1 L of distilled water for 2 d. The dialysis water was replaced regularly. The micelles were characterized by DLS, GPC-MALLS (Eluent: PBS, pH7.4) and cryoTEM. To determine the micelle concentrations after dialysis, 0.5 mL of the micelle solution was lyophilized and the amount of polymer was determined gravimetrically.
Change in size distribution of the micelles in the presence of .
The micelles 10-12 were mixed with in PBS and incubated at 37°C for 24 h. The size distribution of the micelles was measured by DLS.
Fluorescence recovery measurement of the micelles containing self-quenched fluorophores in the presence of .
The micelle solutions (13-15) were mixed with in PBS (100 mM, pH 7.4) containing 10 mM EDTA in a 96 well plate (Micelle concentration: 0.05 mg/mL). The plate was sealed with a plate sealer and incubated at 37°C in the dark. At the indicated time points, fluorescence intensities (FI) (, ) of the samples were measured using a microplate reader. The percent increase in FI of the micelles was calculated as follows:
| (Eq. 2) |
Cell Culture.
Human liver cancer HepG2 cells were cultured in MEM supplemented with 10% fetal bovine serum and 100 U/mL penicillin- 100 μg/mL streptomycin (P/S, 100 U/mL - 100 μg/mL) in a 5% CO2 incubator at 37 °C. Human umbilical vein endothelial cells (HUVECs) were cultured in Medium 200 supplemented with LSGS and P/S (10 U/mL penicillin - 10 μg/mL) in a 5 % CO2 incubator at 37 °C.
Fluorescence recovery of the micelles containing self-quenched fluorophores in cells.
HepG2 cells and HUVECs were seeded in quadruple well glass-bottom dishes (HepG2: 1.0 x 104 cells/well, HUVEC: 5.0 x 103 cells/well). For HUVEC, the glass bottom dish was coated with 5% GFR Matrigel/Dulbecco’s PBS for 30 min at 37°C and washed with PBS prior to cell seeding. After culturing for 1 d, the medium was replaced with 100 μL/well of fresh culture medium (phenol-red free) containing the micelles 13-15 at 0.5 mg/mL. After culturing for 1 d, the medium was replaced with 100 μL/well of fresh medium containing the Hoechst 33342 dye (HepG2: 8 μM, HUVEC: 0.8 μM). Cells were incubated for 5 min at room temperature and observed by CLSFM. The images were analyzed using Image J software to evaluate the increase of the fluorescence intensity within cells. The mean fluorescence intensity was calculated by subtracting the mean fluorescence intensity of the background (non-cell area).
Preparation of Dox-loaded micelles.
DoxꞏHCl was dissolved in DMSO at 10 mM and 100 μL of this solution was mixed with 10 μL of 100 mM TEA/DMSO to deprotonate Dox. This solution (38.5 μL) was mixed with 5 μL of 100 mg/mL Polymer 10-12 /NMP solution and 6.5 μL of DMSO, and added dropwise to 450 μL of distilled water under stirring. After stirring in the dark for 1 d at room temperature, the solution was transferred to a dialysis cassette (MWCO 2kDa) and dialyzed against 1 L of distilled water for 2 d. The dialysis water was replaced regularly. The solution was centrifuged to remove dark red aggregates. The supernatant was collected and stored at −20°C.
Determination of Dox concentration.
The Dox-loaded micelle solution (10 μL) was lyophilized and redissolved in 10 μL of DMSO. The absorbance at 500 nm was measured (n=3). The concentration of Dox in the micelle solutions was determined using a standard curve of Dox, which was prepared by dissolving deprotonated Dox (DoxꞏHCl mixed with 1 equivalent of TEA) in DMSO at different concentrations.
Cytotoxicity of Dox-loaded micelles.
HepG2 cells and HUVECs were seeded in a 96-well plate (HepG2: 5.0 x 103 cells/well, HUVEC: 3.0 x 103 cells/well) and cultured for 1 d. After the medium was replaced with 50 μL/well of fresh medium containing the Dox-loaded micelle solution (10 vol%) or DoxꞏHCl/DMSO (1 vol%), the cells were cultured for 3 d in a CO2 incubator. The medium was replaced with 100 μL of 0.5 mg/ml of MTT in medium. The cells were cultured for 5 h and the formazan crystals were dissolved in 100 μL/well of 100 mg/mL sodium dodecyl sulfate (SDS) solution in 0.01 M HCl (aq). The absorbance at 570 nm was measured using a microplate reader and metabolic activity was expressed as % of the absorbance of the nontreated samples. The half maximal inhibitory concentration was determined using a two-parameter logistic regression model:
| (Eq. 3) |
where is the Dox concentration and is the Hill coefficient.
RESULTS AND DISCUSSION
Effect of chemical structure on oxidation of thioether model compounds.
Thioethers are known to be oxidized by biologically relevant oxidants such as hydrogen peroxide . The proposed mechanism of thioether oxidation by involves the transfer of oxygen by nucleophilic attack of the sulfur atom onto .30 Therefore, it is expected that the oxidation sensitivity of thioethers depends on the nucleophilicity of the sulfur atom, which can be fine-tuned by changing the substituent groups.
To test this hypothesis, we prepared three different thioether amide compounds, 3-methylthiopropylamide (TPAM, 1), thiomorpholine amide (TMAM, 2), and 4-(methylthio)benzylamide (TPhAM, 3) (Scheme 1, see experimental details in the supporting information), and investigated their oxidation by . We first confirmed the formation of the corresponding oxidized product(s) after reacting the model compounds with for 28 h for 1 and 2 and 7 d for 3 at 25°C. As shown by 1H NMR (Figures 2a and d), 1 was oxidized to the sulfoxide and partially the sulfone (TPAM-SO, 4) by . Similarly, the formation of the sulfoxide and sulfone (TMAM-SO, 5) was observed for 2 (Figures 2b and e). In the case of 3, only the sulfoxide (TPhAM-SO, 6) was detected (Figures 2c and f). Furthermore, FT-IR analysis showed the sulfoxide S=O stretching vibration at 1000–1050 cm−1 for 4-6 as well as the sulfone S=O stretching vibration at 1100–1150 cm−1 for 4 and 5 indicating that the thioethers were oxidized to sulfoxides and, in part, sulfone (Figures 2g–i). According to DFT calculations, the vibrational frequencies for the S=O stretching in the sulfoxide species 4-6 are 1066 cm−1, 1049 cm−1, and 1072 cm−1, respectively. These values agree rather well with values determined experimentally by FT-IR, and thus confirming the formation of sulfoxides.
Scheme 1.
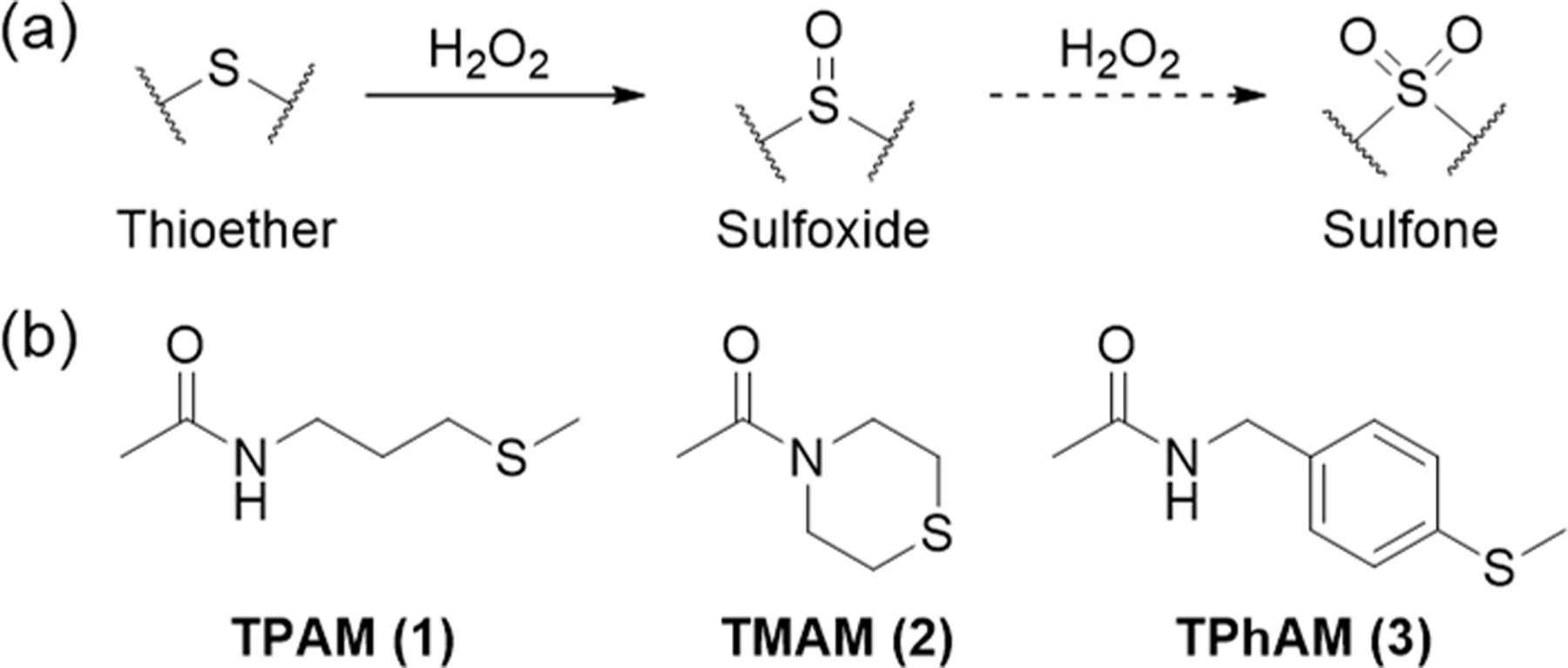
Thioether oxidation. (a) Oxidation of thioethers to sulfoxides and sulfones by . (b) Chemical structures of the thioether model compounds.
Figure 2.
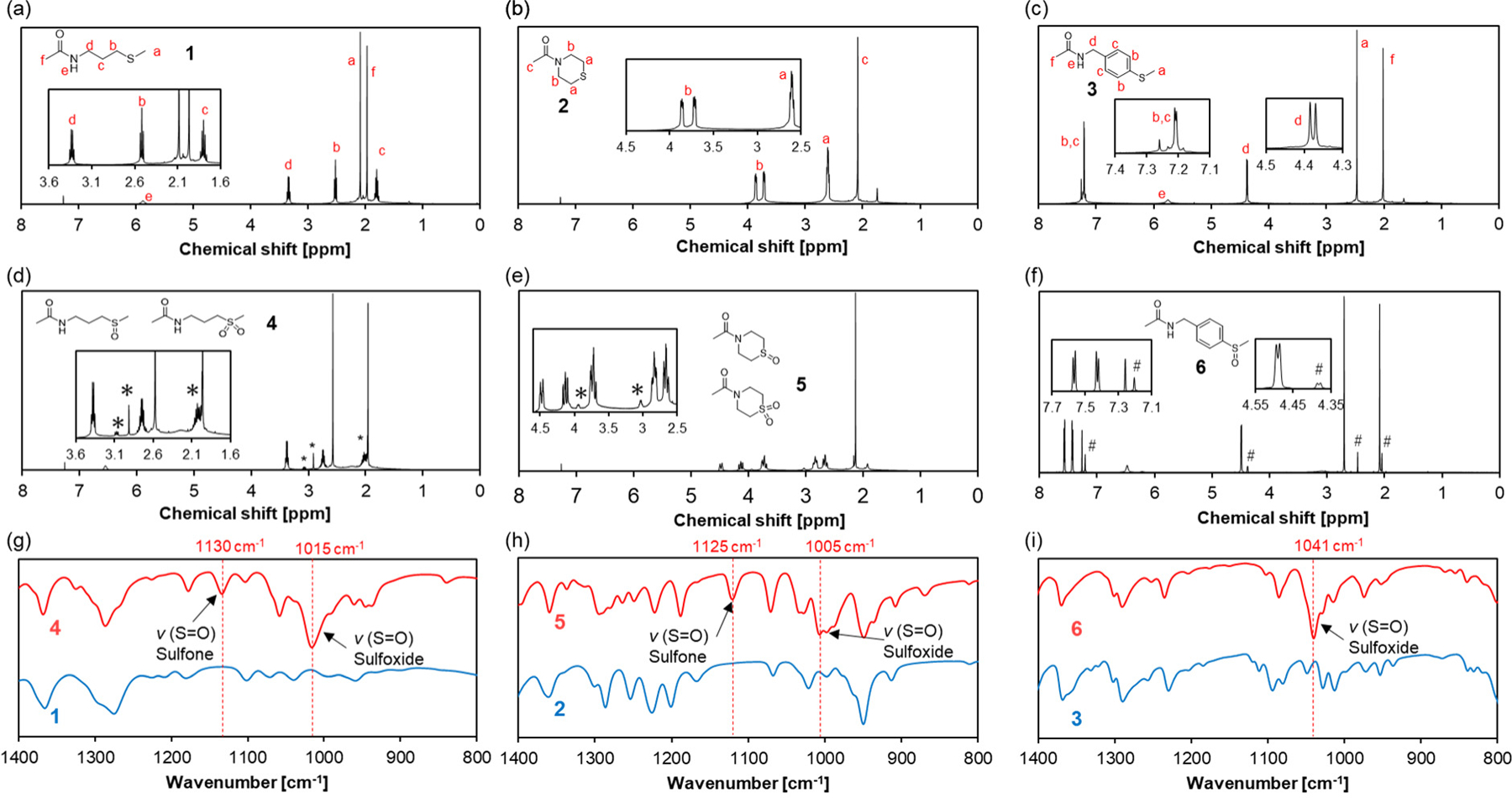
Characterization of the thioether model compounds and their oxidized product(s). The model compounds (1-3) were incubated with (1 equiv.) at 25°C for 28 h for TPAM and TMAM and 7 d for TPhAM to yield the corresponding oxidized species (4-6). 1H NMR spectra of (a) TPAM 1, (b)TMAM 2, (c) TPhAM 3, (d) TPAM-SO 4, (e) TMAM-SO 5, and (f) TPhAM-SO 6. *: signals due to the sulfone. #: signals from unreacted 3. FT-IR spectra of (g) 1 and 4, (h) 2 and 5, and (i) 3 and 6.
The oxidation reaction of 1-3 was followed by measuring the remaining at the indicated time points using the iodide oxidation assay.31 In the presence of 1, was consumed completely within 20 h (Figure 3). The oxidation of 3 was much slower and 40% of was remaining after 48 h. This might be due to the electron withdrawing effect of the phenyl group, which decreases electron density on the sulfur atom and lower its nucleophilicity thereby slowing down the oxidation by . On the other hand, 2 showed intermediate reactivity with 10% of remaining after 48 h. Since the nucleophilicity of the sulfur atom is expected to be similar for 1 and 2, the slower oxidation of 2 seems to be related to the steric effect of the ring structure. Furthermore, 1H NMR of the samples after 48 h showed the formation of the corresponding oxidized species under this experimental condition (Figure S1).
Figure 3.
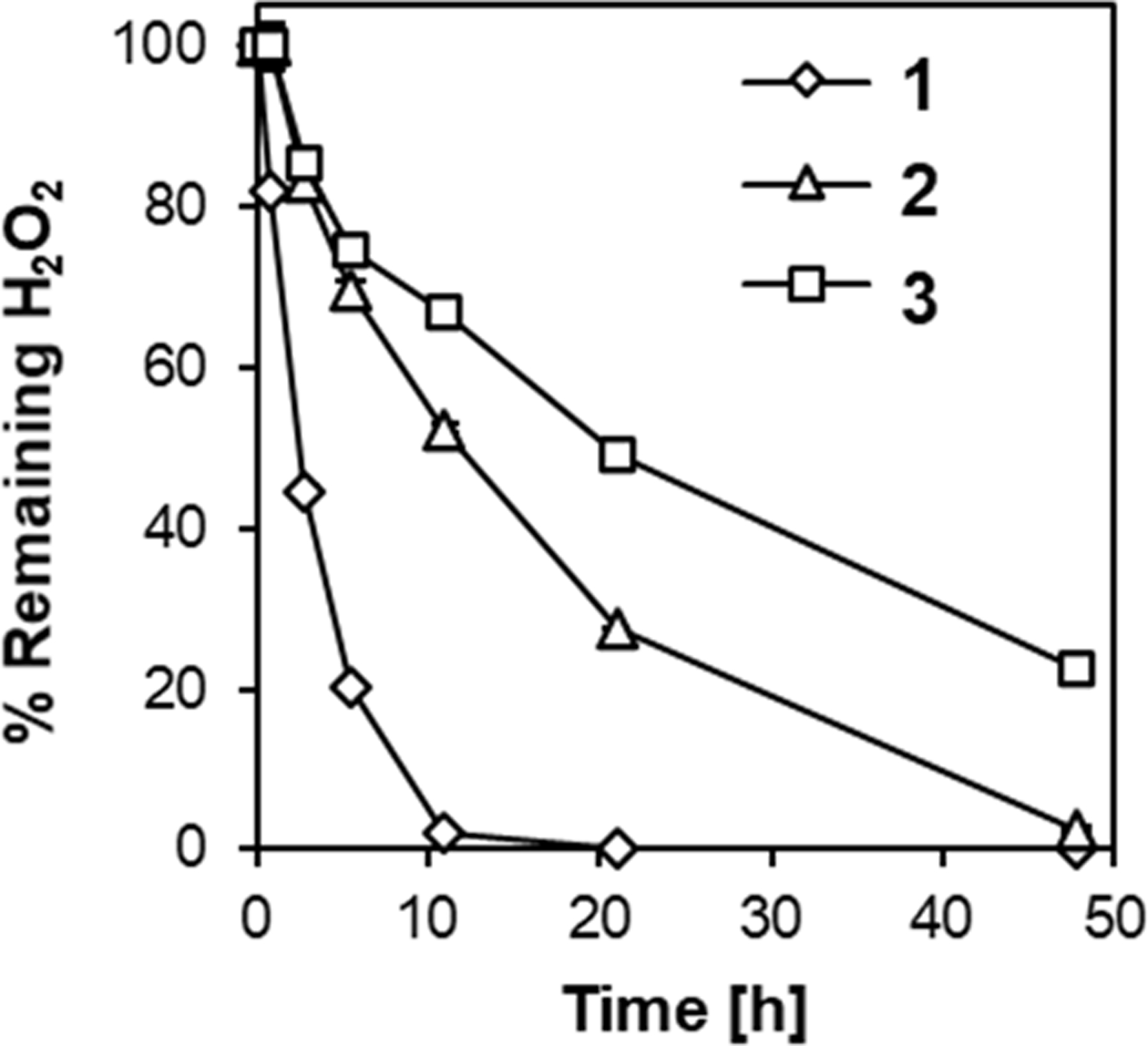
Oxidation rate of the thioether model compounds (1-3). consumption upon the addition of the model compounds was monitored in 100 mM PBS (pH 7.4) at 20°C by the iodide oxidation assay. Model compounds: 10 mM. wt%. n=3.
To evaluate the contribution of the nucleophilicity of thioethers to the oxidation sensitivity, we performed Bader charge analysis on the sulfur atom of these model compounds (Table 1). As for the linear thioether compounds (1 and 3), the thioether compound with a more negatively charged sulfur atom of 1 accelerates oxidation (Figure 3), which is in agreement with the mechanism proposed for thioether oxidation, where nucleophilic attack of the sulfur atom on an oxygen atom of leads to the formation of sulfoxide.30 On the other hand, while the charge on the sulfur atom in 2 was higher than that of 1, the oxidation of 2 was slower than that of 1. This unexpected result may relate to its cyclic structure. It has been reported that one of the key steps of thioether oxidation is the formation of a thioether- adduct, which is affected by both electronic and steric effects of the substituent groups.32 Considering the ring strain of 2, it is possible that the formation of this intermediate is less favored compared to 1, thereby slowing down its oxidation. In addition, accessibility of these hydrophobic thioethers in an aqueous environment may also play a role in the observed oxidation sensitivity. Estimated logP values, a measure of the hydrophobicity of a compound, for the model compounds supports this hypothesis.
Table 1.
Bader charge analysis on the sulfur atom and the estimated log P values of the model compounds.
| Charge on S atom [e] | LogP valuea | |
|---|---|---|
| 1 | − 0.010 | 0.273 |
| 2 | − 0.042 | 0.554 |
| 3 | + 0.074 | 1.436 |
Calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02
Synthesis of thioether-conjugated diblock copolymers.
Encouraged by the model compound results, we designed amphiphilic diblock copolymers consisting of a hydrophilic poly(N-acryloyl morpholine) (PAM) block and a hydrophobic block having different thioether groups as shown in Scheme 2. We first synthesized a poly(pentafluorophenyl acrylate) polymer (PPFPA, 7) by reversible addition-fragmentation chain transfer (RAFT) polymerization using 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid as the chain transfer agent (CTA). Polymer 7 was used as the macro CTA to synthesize the PAM-PPFPA diblock copolymer 8. The obtained polymers had narrow size distributions with Mw/Mn below 1.02 (Table 2, Figure S9). The trithiocarbonate end group of 8 was removed by radical-induced reduction to yield Polymer 9. Successful removal of the end group was confirmed by the absence of the peak due to the CTA group in the 1H NMR spectrum (Figure S5) and the absorbance at 310 nm in the GPC elution profile (Figure S9f). Polymer 9 was modified with amine-containing thioether compounds by substituting the pentafluorophenyl ester group to form amides,33–34 yielding TP (10), TM (11) and TPh (12) polymers. Successful conjugation of the thioether amines was confirmed by the absence of C-F stretching vibration (1000 cm−1), C=C stretching vibration of the aromatic ring (1510 cm−1), and C=O stretching vibration of activated ester group (1780 cm−1) in the FT-IR spectra and the absence of the pentafluorphenyl ester peaks in the 19F spectra (Figure 4). The degree of the thioether group conjugation was determined by 1H NMR (Figure S6–8).
Scheme 2.
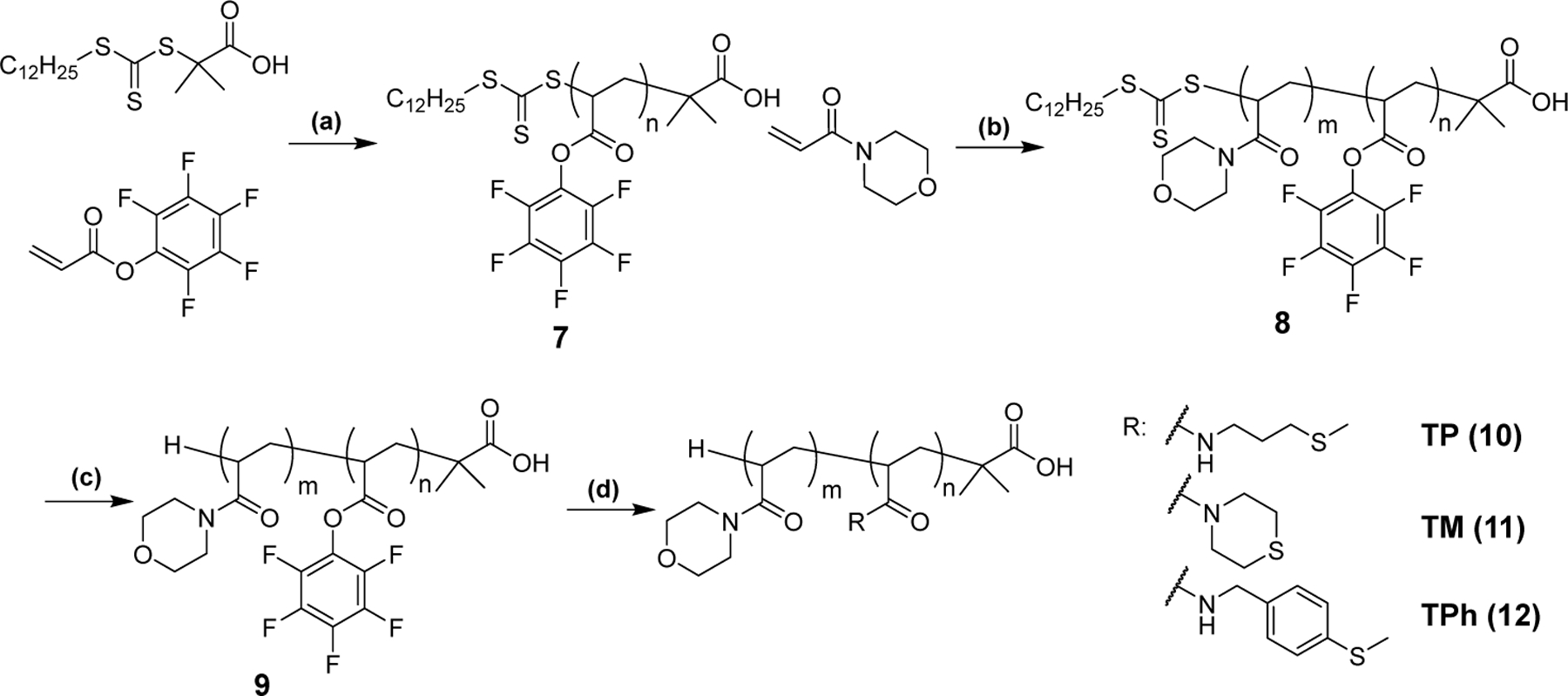
Synthesis scheme of the thioether-conjugated block copolymers. (a) AIBN, 1,4-dioxane, 60 °C, 24 h, (b) AIBN, 1,4-dioxane, 60 °C, 24 h, (c) tris(trimethylsilyl)silane, AIBN, 1,4-dioxane, 70 °C, 24 h, (d) DMF, thioether amine, 50 °C, 24 h.
Table 2.
Characterization of the thioether-containing block copolymers.
| Entry | n a | m a | Mn (NMR) [g/mol] |
Mn (GPC) b
[g/mol] |
Mw (GPC) b
[g/mol] |
Mw /Mn b |
|---|---|---|---|---|---|---|
| PPFPA (7) | 52 | - | 1.27 x 104 | 8.69 x 103 | 8.73 x 103 | 1.01 |
| PAM-PPFPA (8) | 52 | 97 | 2.64 x 104 | 2.34 x 104 | 2.38 x 104 | 1.02 |
Determined by 1H NMR.
Determined by GPC-MALLS (Eluent: THF)
Preparation of the thioether-containing micelles.
The micelles were prepared from the thioether-conjugated diblock copolymers (10-12) by self-assembly in water. The size distributions of the self-assembled structures were measured by DLS and GPC-MALLS (Table 3, Figure S10). TP (10) polymer formed two populations (about 10 and 100 nm) as shown by DLS. Contrary, TM and TPh polymers (11 and 12) formed monodisperse micelles with diameters (Dh) of 25 and 35 nm, respectively. GPC-MALLS analysis revealed that TM and TPh micelles are composed of 67 and 99 polymers. As for TP micelles, we observed two peaks at around 6 and 10 min as shown in Figure S10a. The weakly aggregating behavior of TP micelles may be related to the less hydrophobic nature of TP as suggested by the calculated logP values for the model compounds (Table 1). Due to the heterogeneous size distribution of TP micelles, we were unable to determine the molar mass by MALLS and estimated the molecular weight of the peak maxima (Mp) using the protein standard (Figure S10a and d). Furthermore, we examined the morphology of the micelles by vitrifying solutions prior to cryoTEM imaging, which confirmed the formation of monodisperse spherical structures from TM and TPh polymers (Figure 5b, c). TP polymers, on the other hand, show heterogeneous aggregates (Figure 5a).
Table 3.
Characterization of the micelles prepared from thioether-conjugated polymers.
| Entry | x a |
Dh
b [nm] |
PDI c | Molar mass e [g/mol of micelles] |
Nagg g |
|---|---|---|---|---|---|
| TP (10) | 38 | 10±4, 95±66 d | - | 7.6 x 104, 9.1 x 105 f | 4, 44 |
| TM (11) | 36 | 25 | 0.15 | 1.4 x 106 | 67 |
| TPh (12) | 37 | 35 | 0.14 | 2.2 x 106 | 99 |
x: number of thioether groups per polymer as determined by 1H NMR.
Dh: Hydrodynamic diameter as determined from the cumulant fit (DLS).
PDI: Polydispersity index as determined from the cumulant fit (DLS).
Sample contains two populations. The hydrodynamic diameters of these populations were determined using the CONTIN analysis.
Molar mass (Mn) of the micelles as determined by GPC-MALLS (Eluent: PBS, pH7.4)
Molar mass of the peak maxima (Mp) was estimated based on the BEH200 SEC Protein Standard (see Figure S10).
Nagg: Aggregation number = Molar mass of micelles/Molecular weight of polymer.
Figure 5.

Cryo-TEM image of the micelles. (a) TP, (b) TM, (c) TPh. Scale bars: 100 nm.
Destabilization of the micelles via thioether oxidation by .
We tested whether the oxidation of the thioether groups leads to destabilization of the micelles. The micelles were incubated with in PBS at 37°C for 1 d and measured by DLS (Figure 6). The intensity of the scattered light of TP micelles decreased when treated with at as low as 0.0003 wt%, showing dissociation of the micelles in the presence of . As for TM micelles, while no clear change was observed at 0.0003 wt% of , the scattered light intensity significantly decreased at > 0.03 wt% of . In addition to the decrease in the scattered light intensity, the formation of large structures of sub micrometer was observed, which seems to be due to the aggregation of the partially oxidized/destabilized micelles. TPh micelles were more stable than TM micelles and the effect of was only observed at 0.3 wt% or higher. Similar to TM micelles, TPh micelles also formed larger aggregates upon the addition of . Furthermore, to show that micelle dissociation was induced by the oxidation of the thioether groups, the presence of the corresponding sulfoxide/sufone was confirmed by FT-IR. As shown in Figure 7, the S=O vibrations of the oxidized species were observed for TP, TM, and TPh micelles treated with 3 wt% for 24 h.
Figure 6.
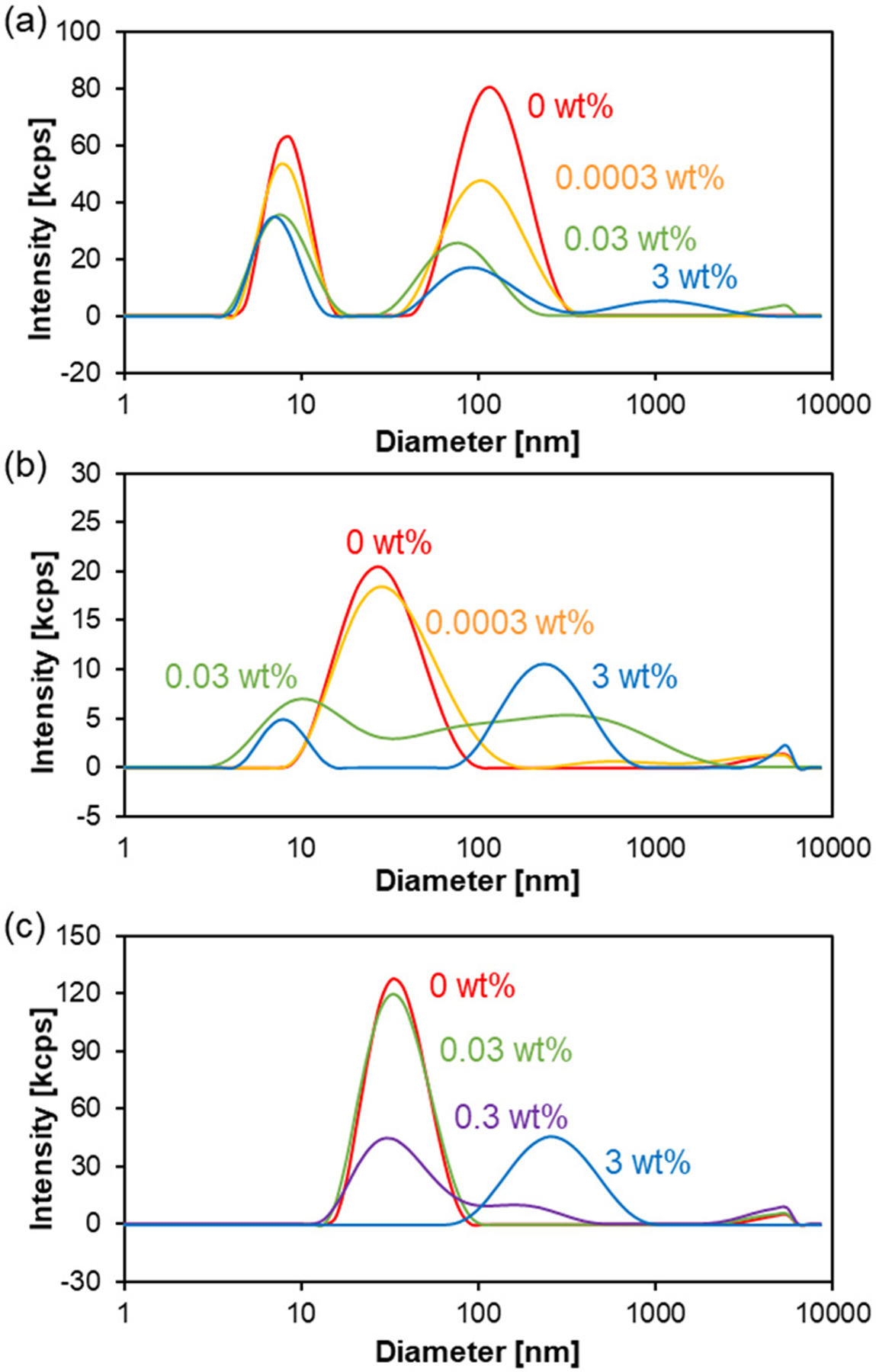
Change in size distribution of the thioether-containing micelles upon oxidation. The micelles were incubated with at different concentrations in PBS at 37°C for 24 h and the size distribution of the micelles was measured by DLS. (a) TP, (b) TM, (c) TPh micelles.
Figure 7.
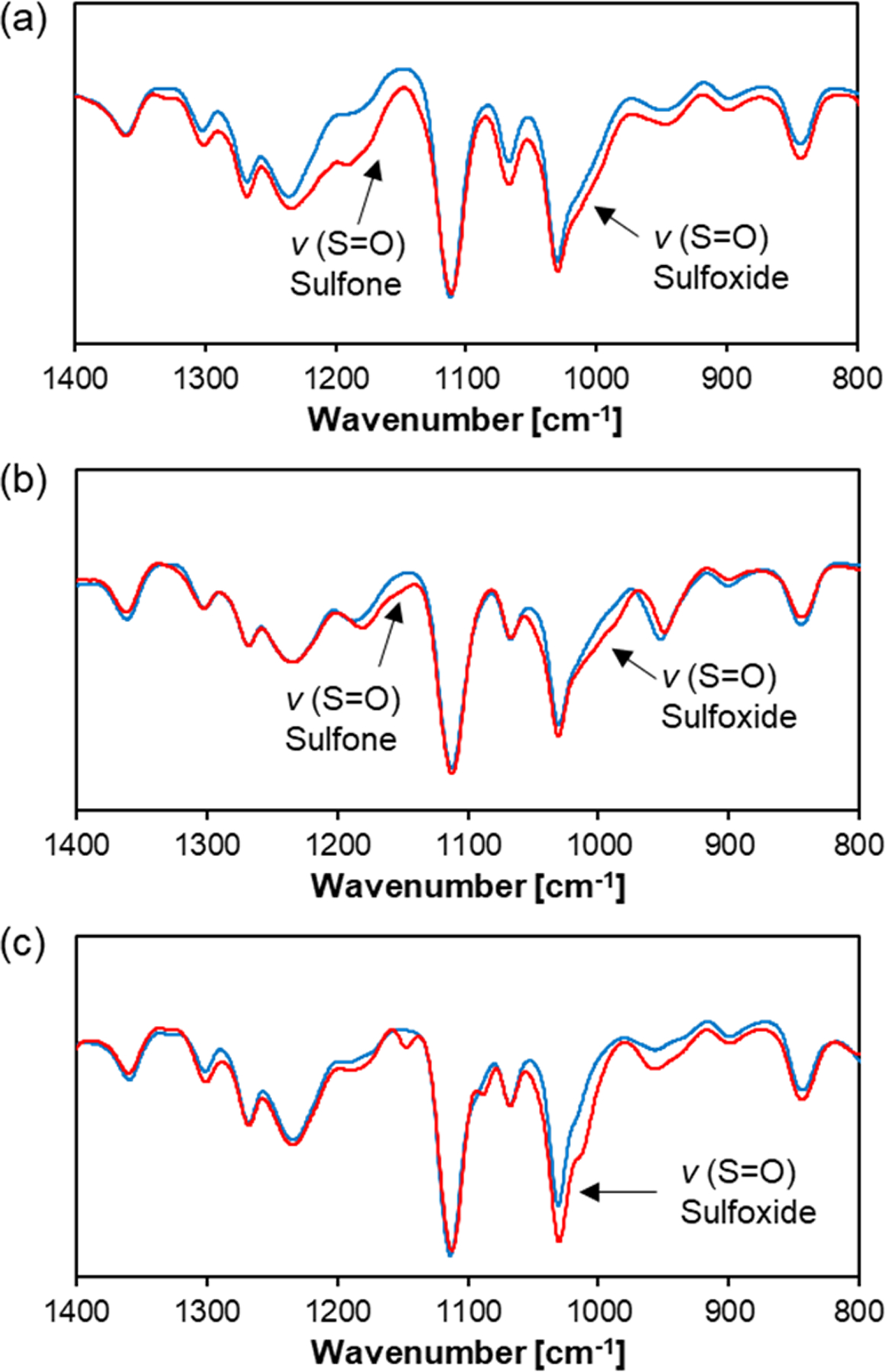
FT-IR spectra of thioether-containing micelles before (blue) and after treatment (red). The micelles were incubated with 3 wt% at 25°C for 24 h. (a) TP, (b) TM, and (c) TPh micelles.
Destabilization of thioether-containing micelles in cells.
To monitor the destabilization of the micelles under physiologically relevant conditions, we synthesized Polymer 13-15 (Figure 8a) and prepared micelles containing self-quenched fluorescein isothiocyanate (FITC) fluorophores in the micelle core. The intact micelles are expected to be non-fluorescent due to self-quenching of FITC, whereas dissociation of the micelles will cause a recovery of fluorescence.35
Figure 8.
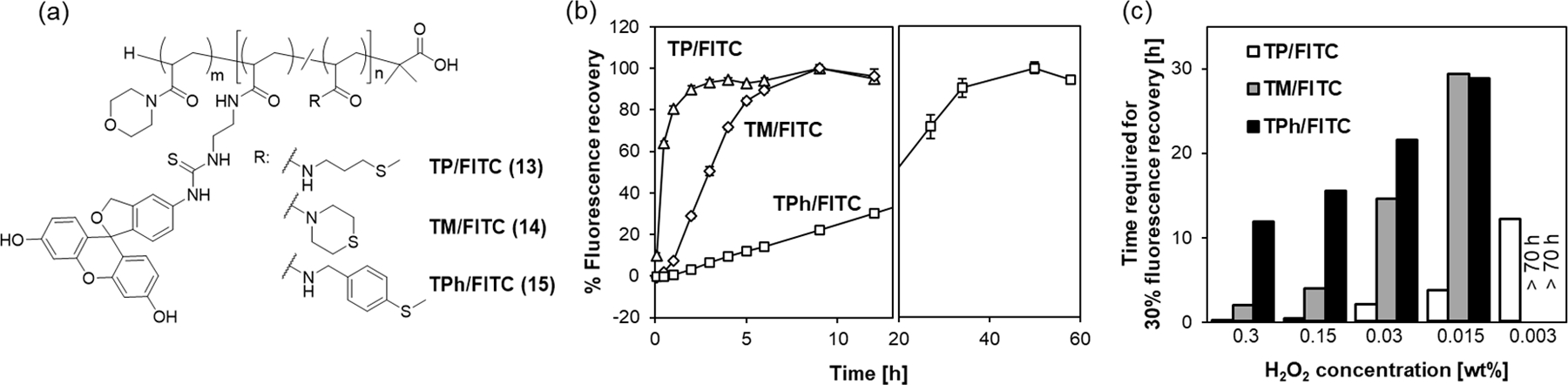
Destabilization of the micelles in the presence of as detected by the fluorescence recovery assay. (a) Chemical structure of the thioether/FITC-conjugated polymers 13-15. The micelles prepared from these polymers contain self-quenched FITC groups within the core. The fluorescence of the FITC groups is recovered upon the destabilization of the micellar structure. (b) Fluorescence recovery of self-quenched FITC fluorophores caused by the destabilization of the micellar structures. The TP/FITC, TM/FITC and TPh/FITC micelles were incubated with 0.3 wt% in PBS at 37°C and the fluorescence intensity (, ) was measured at different time points. TP/FITC: triangles, TM/FITC: diamonds, TPh/FITC: squares. n=3. (c) Time required for 30% fluorescence recovery of the micelles in the presence of at different concentrations.
The micelles were incubated in 0.3 wt% at 37°C, and the fluorescence recovery of the micelles was monitored (Figure 8b). A rapid increase of fluorescence intensity was observed for the TP/FITC micelles, which reached a plateau after 2 h. The TM/FITC micelles showed a slower fluorescence increase compared to the TP/FITC micelles and complete dissociation was observed after 9 h. Furthermore, much slower fluorescence recovery was observed for the TPh/FITC micelles, which required 50 h for complete dissociation. These results are in good agreement with the reactivity trend of the thioether model compounds towards oxidation (Figure 3). We also repeated the same experiment using at lower concentrations. As shown in Figures 8c and S11, the fluorescence recovery of the micelles was slowed down significantly by lowering the concentrations. At all concentrations, TP micelles showed the fastest fluorescence recovery followed by TM and TPh micelles.
We next evaluated the destabilization of the thioether/FITC-containing micelles in human liver cancer cells (HepG2) and human umbilical vein endothelial cells (HUVECs). It is known that the ROS production is upregulated in cancer cells compared to normal cells.38–41 Therefore, we hypothesized that the micelles would be destabilized in the presence of HepG2 cells but be more stable in the presence of HUVECs.
Cells were treated with the thioether/FITC-containing micelles and observed by CLSFM at different time points (Figure 9a–h). The fluorescence intensity within cells was quantified using Image J software (Figure 9i, j). The bright fluorescence spots due to the destabilized micelles were observed in both HepG2 cells and HUVECs treated with TP/FITC micelles. This result indicates that TP/FITC micelles are destabilized in both cancer and normal cells. On the other hand, TM/FITC micelles, fluorescence was clearly observed in HepG2 cells but not in HUVECs showing the cancer cell-specific destabilization of these micelles. Furthermore, the increase in fluorescence intensity was not observed in both cell lines treated with TPh/FITC micelles, which aligns with the low oxidation sensitivity of TPh micelles.
Figure 9.
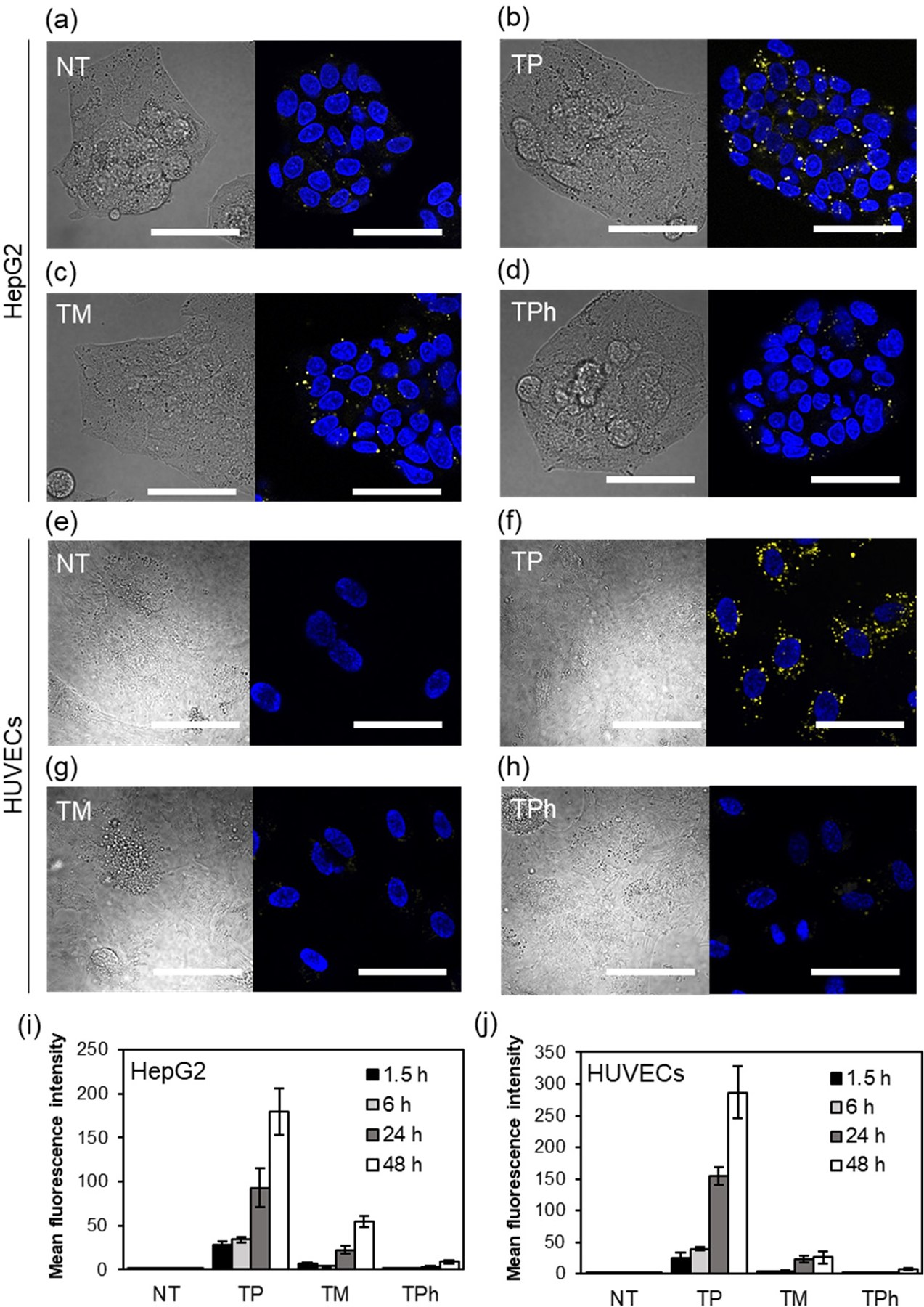
Destabilization of the thioether/FITC-containing micelles in (a-d) HepG2 cells and (e-h) HUVECs. Cells were treated with the micelles for 24 h and the increase in the fluorescence intensity of the micelles due to dissociation was observed by CLSFM. (a, e) No treatment, (b, f) TP/FITC, (c, g) TM/FITC, (d, h) TPh/FITC micelles Left panel: DIC, Right panel: Hoechst 33342 (nucleus, blue) and FITC (yellow), Scale bars: 50 μm. (i, j) Intracellular fluorescence intensity in (i) HepG2 cells and (j) HUVECs after 1.5, 6, 24 and 48 h. The mean fluorescence intensity inside the cells was determined using Image J software. n=5.
Cytotoxicity of the Dox loaded micelles.
To explore the potential application of the micelles for drug delivery applications, we encapsulated the anticancer drug doxorubicin (Dox) in the micelles to evaluate their functions as drug carriers. Dox and the Polymers 10-12 (1:5 by weight) were mixed in N-methyl pyrrolidone (NMP) and added dropwise to water to prepare Dox-loaded micelles. After unencapsulated Dox was removed by dialysis, the amount of Dox loaded in the micelles was quantified by UV-Vis spectroscopy. The weight fraction of Dox in the Dox-loaded micelles was 8.5 –10.8 wt% as shown in Table S1.
The cytotoxicity of the Dox-loaded micelles was evaluated in HepG2 cells and HUVECs by MTT assay and the values were determined (Figure 10 and Table 4). At the concentrations tested micelles alone did not affect cell viability as shown in Figure S12. To evaluate the relative cytotoxicity in HepG2 cells over HUVECs, the ratios , where a higher ratio means enhanced toxicity in HepG2 cells and/or reduced toxicity in HUVECs, were calculated. The Dox-loaded TP micelles showed a similar ratio with Dox alone, indicating that encapsulation of the Dox does not improve selectivity towards cancer cells. On the other hand, the ratio of the Dox-loaded TM micelles was much higher than that of Dox alone showing that TM micelles enhances toxicity of Dox in HepG2 cells relative to HUVECs. Furthermore, the Dox-loaded TPh micelles showed a low ratio, which might be due to the high stability of TPh micelles that prevents release of a sufficient amount of Dox to HepG2 cells. These results clearly show that TM micelles, which exhibit moderate ROS sensitivity, seems to be the most promising drug carrier to expand the therapeutic window of Dox.
Figure 10.
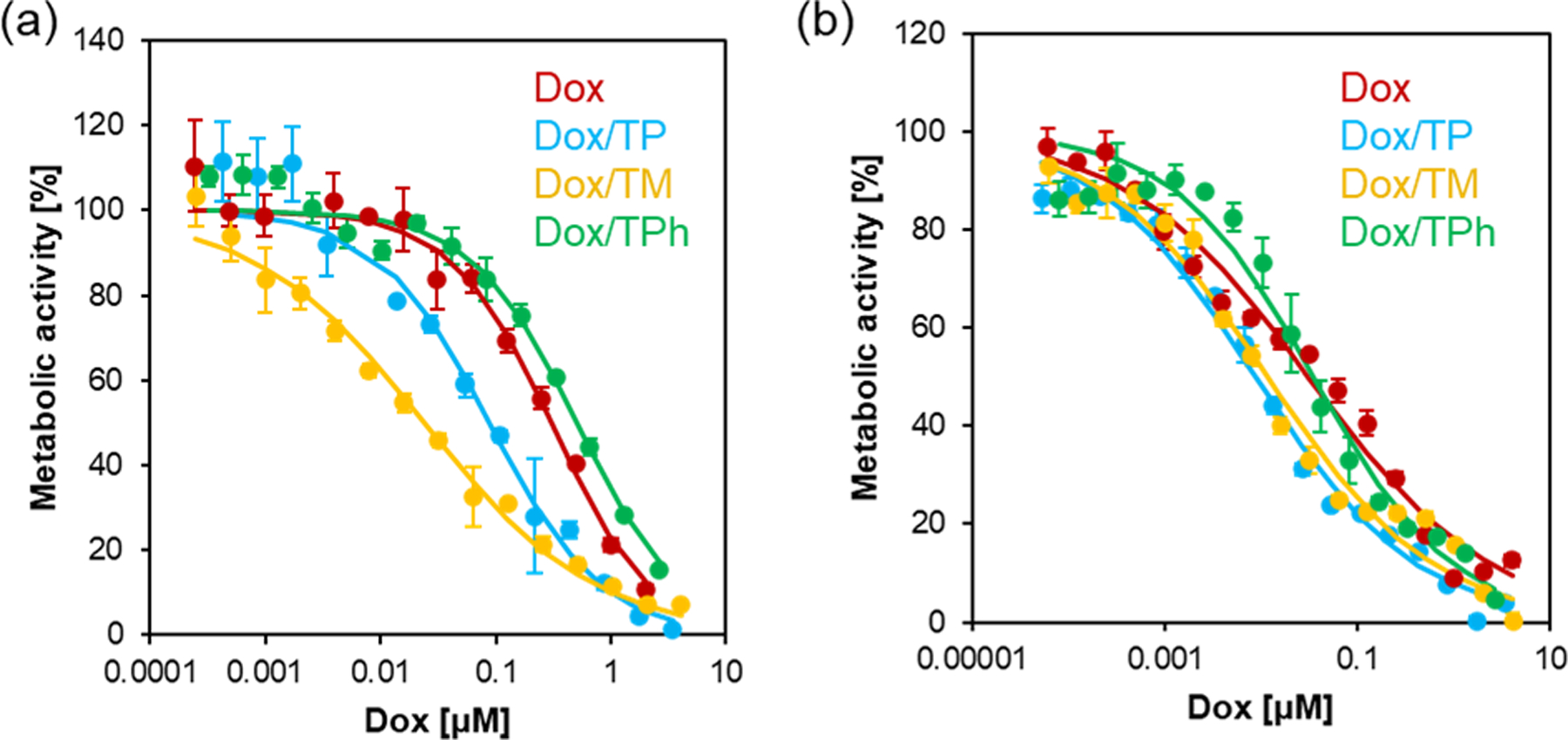
Cytotoxicity of the Dox-loaded micelles in (a) HepG2 cells and (b) HUVECs. Cells were cultured in the presence of Dox·HCl and Dox-loaded micelles for 3 d. Cell viability was measured by MTT assay. n=3.
Table 4.
values of Dox and the Dox-loaded micelles in HepG2 cells and HUVECs as determined by MTT Assay.
| Entry | |||
|---|---|---|---|
| HepG2 | HUVEC | ||
| Dox | 0.30 | 0.031 | 0.10 |
| Dox/TP | 0.087 | 0.0088 | 0.10 |
| Dox/TM | 0.024 | 0.012 | 0.49 |
| Dox/TPh | 0.51 | 0.035 | 0.069 |
CONCLUSION
We herein report thioether-containing polymeric micelles with different oxidation sensitivities. The effect of substituent groups on the oxidation of thioether was first investigated using different thioether model compounds, 3-methylthiopropylamide (TPAM), thiomorpholine amide (TMAM), and 4-(methylthio)benzylamide (TPhAM). TPAM showed much faster oxidation than TPhAM, which contains a less nucleophilic sulfur atom, according to Bader charge analysis. On the other hand, the cyclic thioether TMAM with a slightly higher charge density than TPAM exhibited slower oxidation than TPAM, but faster than TPhAM. These results indicate that both electronic and steric effects contribute to the oxidation rate of thioethers. A similar trend was observed for the polymeric micelles containing these thioether groups (TP, TM, and TPh micelles). The effect of the oxidation of thioether groups on the micellar structures was confirmed both in the presence of and human liver cancer HepG2 cells, which produce elevated levels of ROS. The destabilization rate in response to was fastest for TP micelles followed in order by TM and TPh micelles. Furthermore, cytotoxicity of the Dox-loaded thioether-containing micelles was investigated to explore the potential applications in chemotherapy. Among the micelles, TM micelles significantly increased toxicity of Dox in HepG2 cells but protected normal cells (HUVECs) from Dox cytotoxicity. These results clearly show that fine-tuning oxidation sensitivities of the thioether groups is a promising approach for rationally designing cancer-targeted drug delivery systems that release drugs in cancer tissues under oxidative stress but remain stable in healthy tissues.
Supplementary Material
ACKNOWLEDGMENT
We thank Ms. Arshiya Bhadu in Department of Materials Science and Engineering at The Pennsylvania State University for measuring dn/dc of the polymers. We thank Dr. Tapas K. Mal and Dr. Christy George of the nuclear magnetic resonance (NMR) facility at The Pennsylvania State University. We also thank Mr. Clint Barb and Mr. Christian Willcutt in Tim Taylor Department of Chemical Engineering at Kansas State University for performing preliminary experiments. This work was supported by Center for Molecular Analysis of Disease Pathways (CMADP), National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH), P20GM103638. MG and EDG acknowledge financial support from the U.S. National Science Foundation under award DMR 1905550.
Funding Sources
NIH NIGMS P20 GM103638.
NSF DMR 1905550.
ABBREVIATIONS
- PAM
poly(N-acryloyl morpholine)
- PPFPA
poly(pentafluorophenyl acrylate)
- TPAM
3-methylthiopropylamide
- TMAM
thiomorpholine amide
- TPhAM
4-(methylthio)benzylamide
- ROS
reactive oxygen species
Footnotes
ASSOCIATED CONTENT
Supporting Information. The following files are available free of charge at http://pubs.acs.org. Experimental details (Materials, Instrumentation, Synthetic procedure), 1H NMR spectra, GPC elution profiles, Fluorescence recovery assay results of FITC/thioether-containing micelles (PDF)
REFERENCES
- (1).Liou G-Y; Storz P Reactive oxygen species in cancer. Free Radical Research 2010, 44 (5), 479–496, DOI: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Pelicano H; Carney D; Huang P ROS stress in cancer cells and therapeutic implications. Drug Resistance Updates 2004, 7 (2), 97–110, DOI: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- (3).Chen Q; Vazquez EJ; Moghaddas S; Hoppel CL; Lesnefsky EJ Production of Reactive Oxygen Species by Mitochondria: CENTRAL ROLE OF COMPLEX III. Journal of Biological Chemistry 2003, 278 (38), 36027–36031, DOI: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- (4).Halliwell B; Clement MV; Long LH Hydrogen peroxide in the human body. FEBS Letters 2000, 486 (1), 10–13, DOI: 10.1016/S0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- (5).Valko M; Leibfritz D; Moncol J; Cronin MTD; Mazur M; Telser J Free radicals and antioxidants in normal physiological functions and human disease. The International Journal of Biochemistry & Cell Biology 2007, 39 (1), 44–84, DOI: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- (6).Sies H Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biology 2017, 11, 613–619, DOI: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Napoli A; Tirelli N; Kilcher G; Hubbell A New Synthetic Methodologies for Amphiphilic Multiblock Copolymers of Ethylene Glycol and Propylene Sulfide. Macromolecules 2001, 34 (26), 8913–8917, DOI: 10.1021/ma0108057. [DOI] [Google Scholar]
- (8).Napoli A; Valentini M; Tirelli N; Muller M; Hubbell JA Oxidation-responsive polymeric vesicles. Nat Mater 2004, 3 (3), 183–189, DOI: 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]
- (9).Sobotta FH; Hausig F; Harz DO; Hoeppener S; Schubert US; Brendel JC Oxidation-responsive micelles by a one-pot polymerization-induced self-assembly approach. Polymer Chemistry 2018, 9 (13), 1593–1602, DOI: 10.1039/C7PY01859B. [DOI] [Google Scholar]
- (10).Broaders KE; Grandhe S; Fréchet JMJ A Biocompatible Oxidation-Triggered Carrier Polymer with Potential in Therapeutics. Journal of the American Chemical Society 2011, 133 (4), 756–758, DOI: 10.1021/ja110468v. [DOI] [PubMed] [Google Scholar]
- (11).de Gracia Lux C; Joshi-Barr S; Nguyen T; Mahmoud E; Schopf E; Fomina N; Almutairi A Biocompatible Polymeric Nanoparticles Degrade and Release Cargo in Response to Biologically Relevant Levels of Hydrogen Peroxide. Journal of the American Chemical Society 2012, 134 (38), 15758–15764, DOI: 10.1021/ja303372u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wilson DS; Dalmasso G; Wang L; Sitaraman SV; Merlin D; Murthy N Orally delivered thioketal nanoparticles loaded with TNF-α–siRNA target inflammation and inhibit gene expression in the intestines. Nature Materials 2010, 9 (11), 923–928, DOI: 10.1038/nmat2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mahmoud EA; Sankaranarayanan J; Morachis JM; Kim G; Almutairi A Inflammation Responsive Logic Gate Nanoparticles for the Delivery of Proteins. Bioconjugate Chemistry 2011, 22 (7), 1416–1421, DOI: 10.1021/bc200141h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Oxidation of Methionine Residues of Proteins: Biological Consequences. Antioxidants & Redox Signaling 2003, 5 (5), 577–582, DOI: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
- (15).Cerritelli S; O’Neil CP; Velluto D; Fontana A; Adrian M; Dubochet J; Hubbell JA Aggregation Behavior of Poly(ethylene glycol-bl-propylene sulfide) Di- and Triblock Copolymers in Aqueous Solution. Langmuir 2009, 25 (19), 11328–11335, DOI: 10.1021/la900649m. [DOI] [PubMed] [Google Scholar]
- (16).Segura T; Hubbell JA Synthesis and in Vitro Characterization of an ABC Triblock Copolymer for siRNA Delivery. Bioconjugate Chemistry 2007, 18 (3), 736–745, DOI: 10.1021/bc060284y. [DOI] [PubMed] [Google Scholar]
- (17).Velluto D; Demurtas D; Hubbell JA PEG-b-PPS Diblock Copolymer Aggregates for Hydrophobic Drug Solubilization and Release: Cyclosporin A as an Example. Molecular Pharmaceutics 2008, 5 (4), 632–642, DOI: 10.1021/mp7001297. [DOI] [PubMed] [Google Scholar]
- (18).O’Neil CP; Suzuki T; Demurtas D; Finka A; Hubbell JA A Novel Method for the Encapsulation of Biomolecules into Polymersomes via Direct Hydration. Langmuir 2009, 25 (16), 9025–9029, DOI: 10.1021/la900779t. [DOI] [PubMed] [Google Scholar]
- (19).Yoo J; Sanoj Rejinold N; Lee D; Jon S; Kim Y-C Protease-activatable cell-penetrating peptide possessing ROS-triggered phase transition for enhanced cancer therapy. Journal of Controlled Release 2017, 264, 89–101, DOI: 10.1016/j.jconrel.2017.08.026. [DOI] [PubMed] [Google Scholar]
- (20).Rodriguez AR; Kramer JR; Deming TJ Enzyme-Triggered Cargo Release from Methionine Sulfoxide Containing Copolypeptide Vesicles. Biomacromolecules 2013, 14 (10), 3610–3614, DOI: 10.1021/bm400971p. [DOI] [PubMed] [Google Scholar]
- (21).Cheng F; Su T; Luo K; Pu Y; He B The polymerization kinetics, oxidation-responsiveness, and in vitro anticancer efficacy of poly(ester-thioether)s. Journal of Materials Chemistry B 2019, 7 (6), 1005–1016, DOI: 10.1039/C8TB02980F. [DOI] [PubMed] [Google Scholar]
- (22).Ma N; Li Y; Ren H; Xu H; Li Z; Zhang X Selenium-containing block copolymers and their oxidation-responsive aggregates. Polymer Chemistry 2010, 1 (10), 1609–1614, DOI: 10.1039/c0py00144a. [DOI] [Google Scholar]
- (23).Cao W; Gu Y; Li T; Xu H Ultra-sensitive ROS-responsive tellurium-containing polymers. Chemical Communications 2015, 51 (32), 7069–7071, DOI: 10.1039/C5CC01779C. [DOI] [PubMed] [Google Scholar]
- (24).Gupta MK; Meyer TA; Nelson CE; Duvall CL Poly(PS-b-DMA) micelles for reactive oxygen species triggered drug release. Journal of Controlled Release 2012, 162 (3), 591–598, DOI: 10.1016/j.jconrel.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kresse G; Furthmüller J Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B 1996, 54 (16), 11169–11186, DOI: 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- (26).Perdew JP; Burke K; Ernzerhof M Generalized Gradient Approximation Made Simple. Physical Review Letters 1996, 77 (18), 3865–3868, DOI: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- (27).Blöchl PE Projector augmented-wave method. Physical Review B 1994, 50 (24), 17953–17979, DOI: 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- (28).Yu M; Trinkle DR Accurate and efficient algorithm for Bader charge integration. The Journal of Chemical Physics 2011, 134 (6), 064111, DOI: 10.1063/1.3553716. [DOI] [PubMed] [Google Scholar]
- (29).Hasegawa U; Wang T; Chen JJY; Uyama H; van der Vlies AJ Furoxan-Bearing Micelles for Nitric Oxide Delivery. Macromolecular Bioscience 2016, 16 (7), 1009–1018, DOI: 10.1002/mabi.201500401. [DOI] [PubMed] [Google Scholar]
- (30).Chu J-W; Trout BL On the Mechanisms of Oxidation of Organic Sulfides by H2O2 in Aqueous Solutions. Journal of the American Chemical Society 2004, 126 (3), 900–908, DOI: 10.1021/ja036762m. [DOI] [PubMed] [Google Scholar]
- (31).Sanders Junglee LU, Huguette Sallanon, Félicie Lopez-Lauri. Optimized Assay for Hydrogen Peroxide Determination in Plant Tissue Using Potassium Iodide. American Journal of Analytical Chemistry 2014, 5 (11), 730–736, DOI: 10.4236/ajac.2014.511081 [DOI] [Google Scholar]
- (32).Srinivasan C; Kuthalingam P; Arumugam N Substituent and steric effects in the oxidation of alkyl aryl sulfides by peroxydisulfate. Canadian Journal of Chemistry 1978, 56 (24), 3043–3046, DOI: 10.1139/v78-497. [DOI] [Google Scholar]
- (33).Mohr N; Barz M; Forst R; Zentel R A Deeper Insight into the Postpolymerization Modification of Polypenta Fluorophenyl Methacrylates to Poly(N-(2-Hydroxypropyl) Methacrylamide). Macromolecular Rapid Communications 2014, 35 (17), 1522–1527, DOI: 10.1002/marc.201400249. [DOI] [PubMed] [Google Scholar]
- (34).Li Y; Duong HTT; Jones MW; Basuki JS; Hu J; Boyer C; Davis TP Selective Postmodification of Copolymer Backbones Bearing Different Activated Esters with Disparate Reactivities. ACS Macro Letters 2013, 2 (10), 912–917, DOI: 10.1021/mz4004375. [DOI] [PubMed] [Google Scholar]
- (35).Cho H; Indig GL; Weichert J; Shin H-C; Kwon GS In vivo cancer imaging by poly(ethylene glycol)-b-poly(ɛ-caprolactone) micelles containing a near-infrared probe. Nanomedicine: Nanotechnology, Biology and Medicine 2012, 8 (2), 228–236, DOI: 10.1016/j.nano.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.