

Summary
Cytokines are key messengers by which immune cells communicate and they drive many physiological processes, including immune and inflammatory responses. Early discoveries demonstrated that cytokines, such as the interleukin family members and TNFα regulate synaptic scaling and plasticity. Still, we continue to learn more about how these traditional immune system cytokines affect neuronal structure and function. Different cytokines shape synaptic function on multiple levels ranging from fine-tuning neurotransmission, to regulating synapse number, to impacting global neuronal networks and complex behavior. These recent findings have cultivated an exciting and growing field centered on the importance of immune system cytokines for regulating synapse and neural network structure and function. Here, we highlight the latest findings related to cytokines in the central nervous system and their regulation of synapse structure and function. Moreover, we explore how these mechanisms are becoming increasingly important to consider in diseases – especially those with a large neuroinflammatory component.
eTOC
Immunity contributes to brain function in both health and disease. Here, Zipp, Bittner & Schafer review how immune cytokines shape synaptic function on multiple levels ranging from fine-tuning neurotransmission, to shaping synaptic function, to impacting global neuronal networks and behaviour.
Introduction
One neuron communicates with next at the synapse (see Box 1 for glossary of terms) - the site at which neurotransmitter is released to control neural circuit (see Box 1 for glossary of terms) function. However, it is now emerging that immune system cytokines, canonically involved in peripheral inflammatory processes, are playing important neuromodulatory roles to regulate neurons and their synapses. Indeed, immune mechanisms have now been shown to modulate synapse development, plasticity, and cognition in the central nervous system (CNS). Further, these immune-based mechanisms are now appreciated to have profound consequences on the normal function of the CNS (i.e. social behavior, cognition and balanced mental state), and for diseases involving inflammation of the CNS. Among the immune molecules that regulate neural circuits are immune cytokines, such as interferons, interleukins, and chemokines, and their cognate receptors. In the periphery, cytokines are typically expressed at low levels basally but their release is further stimulated upon an inflammatory stimulus (pathogen-associated molecular pattern molecules (PAMPS), damage-associated molecular pattern molecules (DAMPs), etc.) binding its cognate receptor on a cell membrane to stimulate a transcriptional increase in cytokines and their subsequent release. Intriguingly, numerous studies have now identified that many of these cytokines and their receptors are expressed by resident CNS cells, including neurons and glia, where they modulate baseline electrophysiological properties of synapses, synaptic remodeling, plasticity, and circuit-wide activity. To this end, cytokine signaling pathways such as via interferon gamma (IFNγ), TNF-α, IL-1, IL-4, IL-6 and IL-13 have been identified as key regulators of social behavior as well as learning and memory. Additionally, some immune cytokines are even present within the CNS during normal neurodevelopment in prenatal life and play multifaceted roles in brain circuit development.
Box 1. Glossary of terms.
Synapses: structures that allow neurons to transmit neurotransmitter from one neuron to the next. Generally, a synapse is comprised of a presynaptic terminal of one neuron, which then releases presynaptic vesicles containing neurotransmitter into the synaptic cleft to bind neurotransmitter receptors embedded in the postsynaptic membrane of a neighboring neuron.
Neural circuits: a population of neurons connected by synapses to carry out a specific function upon activation. The connection of neural circuits forms neuronal networks.
Glutamate: the major excitatory neurotransmitter.
γ-aminobutyric acid (GABA): the major inhibitory neurotransmitter.
Excitatory synaptic transmission: communication between neurons such that one neuron is stimulated to release more neurotransmitter, which is typically glutamate.
Excitatory postsynaptic currents (EPSCs): are electrophysiological measures of excitatory synaptic transmission.
Neural excitability: neural excitability depends on the membrane potential of the postsynaptic neuron that can be altered by neurotransmitters released at synapses. The membrane potential is set by permeable ion channels and active pumping mechanisms. During synaptic transmission, neurotransmitters depolarize the membrane potential until voltage-gated sodium channels start an action potential. Neurotransmitters have either excitatory (i.e., depolarizing such as glutamate) or inhibitory (i.e., hyperpolarizing such as GABA) effects.
Inhibitory tone: neurons that receive synaptic inputs from GABAergic neurons are prevented from firing and releasing neurotransmitter to neighboring neurons. This tone is maintained until either a sufficiently potent excitatory synaptic stimulus overcomes the inhibitor or until the inhibitory synaptic input is released.
Inhibitory postsynaptic currents (IPSCs): see EPSC. IPSCs are associated with inhibitory GABAergic transmission.
N-methyl-D-aspartate receptor (NMDAR): a ligand-gated ion channel on the postsynaptic membrane that binds the excitatory neurotransmitter glutamate. Upon neurotransmitter binding, this receptor allows calcium ions to flow into the postsynaptic neuron to generate mEPSPs or EPSPs.
α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR): another type of ligand-gated ion channel on the postsynaptic membrane that binds glutamate and facilitates excitatory neurotransmission. This receptor underlies most forms of synaptic plasticity.
GABAA receptor (GABAAR): a ligand-gated ion channel on the postsynaptic membrane that binds the inhibitory neurotransmitter GABA. Upon neurotransmitter binding, this receptor allows chloride ions to flow into the postsynaptic neuron to generate mIPSPs or IPSPs.
Miniature excitatory postsynaptic potential (mEPSP): a postsynaptic potential is a change in membrane potential due to neurotransmitter binding a ligand-gated ion channel on the postsynaptic neuron. This ligand binding makes a postsynaptic neuron more likely to generate electrical activity necessary for neurotransmitter signaling to a neighboring neuron. mEPSPs are recorded in the presence of the sodium channel blocker tetrodotoxin to block all spontaneous neuronal activity.
Miniature inhibitory postsynaptic potential (IPSP): see mEPSP. mIPSPs are associated with GABAergic transmission. They are inhibitory and generally decrease the potential of a neuron to communicate with the next.
Dendritic spine: small protrusions along excitatory neuron dendrites that contain postsynaptic machinery necessary for excitatory synaptic transmission.
Hippocampus: an area of the brain that plays a major role in learning and memory formation.
Cortex: the brains outermost layer that is subdivided into many regions that regulate a multitude of functions such as sensory processing, memory, learning, emotions, arousal, and consciousness.
Thalamus: a brain region that is a relay station for most sensory information coming into and out of the brain.
Synaptic plasticity: plasticity is the ability of the brain to change and adapt to new information. Synaptic plasticity controls how effectively neurons communicate with each other. This is closely linked to synaptic strength, i.e., the amount of current produced in a postsynaptic neuron by an action potential in the presynaptic neuron.
Synaptic pruning: a natural process in the brain of most mammals between early childhood and adulthood, wherein unneeded or redundant synapses are eliminated. This process is thought to maintain efficient brain function during learning and has been linked to neurodevelopmental disorders such as schizophrenia or autism spectrum disorder.
Contextual fear conditioning: a learning paradigm in which an animal learns to associate a novel context with an aversive mild foot shock.
While many different types of immune mechanisms have now been identified to regulate synaptic connectivity (Major Histocompatibility class I molecules, complement, etc.),(1, 2) this review will focus on the most recent findings that immune cytokines act as modulators of synapses in the CNS during homeostasis. We note that the term “cytokine” can be interpreted broadly to include molecules that also act as growth factors, such as brain-derived neurotrophic factor (BDNF). This review will focus on molecules that have more traditionally been associated with the peripheral immune system (such as the tumor necrosis factor family, the interferons, interleukins, and chemokines). We will also discuss how this complex interplay between cytokines and synapses has implications for neural circuit dysfunction in disease.
Cytokine regulation of basal excitatory synaptic transmission
Some of the most compelling evidence supporting a role for cytokines as regulators of synapses are early experiments showing that these molecules can directly impact basal excitatory and inhibitory synaptic function and structure (Figure 1). In vitro work is foundational and is now supported by investigation of the interplay between cytokines and synapses in vivo. Here, we briefly review this foundational work as a basis for understanding the mechanistic underpinnings by which these cytokines modulate synapse plasticity and function in vivo, which we discuss later in this review. In this first part, we focus on the regulation of excitatory synaptic transmission.
Figure 1: The effects of immune cytokines on baseline physiology of synapses.
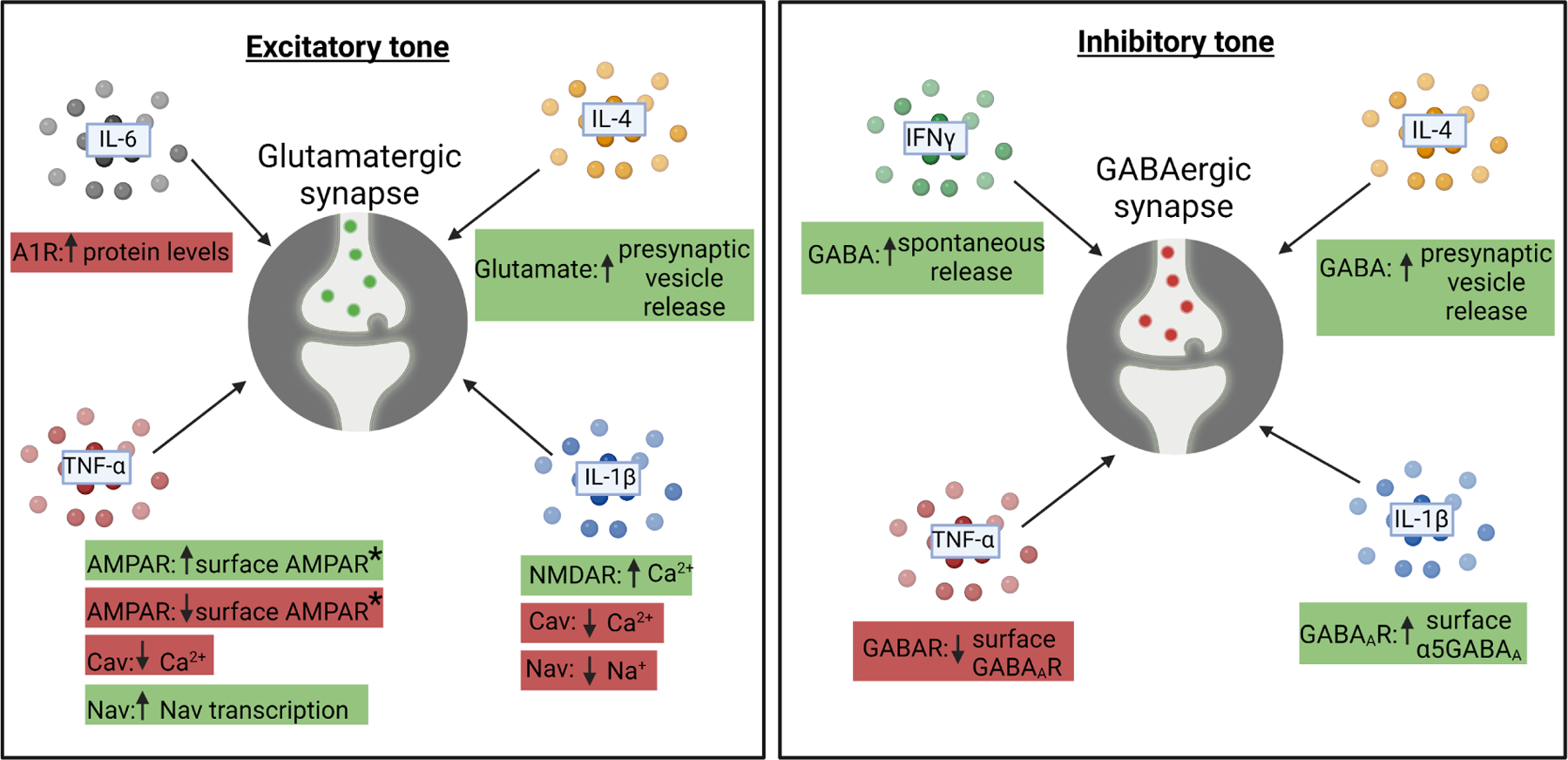
It is now appreciated that cytokines can impact the baseline function of excitatory (left) and inhibitory (right) synapses. They can regulate excitatory (left) and inhibitory (right) neurotransmission on the postsynaptic membrane by regulating the amount of ion channels or neurotransmitter receptors expressed or localized to the membrane. They can regulate the amounts of synaptic vesicles on the presynaptic side and they can regulate adrenergic signaling, which subsequently modulates synaptic activity. Red boxes denote an effect of decreasing excitatory (left) or inhibitory (right) transmission and green boxes denote an effect of increasing excitatory (left) or inhibitory (right) transmission. A1R=A1 adrenergic receptor, AMPAR=α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, Cav= voltage-gated calcium (Ca2+) channel, Nav=voltage-gated sodium (Na+) channel, GABA=γ-aminobutyric acid, GABAR= γ-aminobutyric acid receptor, GABAAR= γ-aminobutyric acid type A receptor.
The actions of IL-1β on excitatory synapses exemplifies the direct interplay between cytokines and synaptic function. IL-1β is part of the interleukin 1 family of cytokines. After being cleaved by caspase 1 into a mature form, it is then secreted and binds its cognate receptor IL-1R1. This receptor binding elicits the binding of the co-receptor IL-1RAcP to the IL-1β-IL1R1 complex and activation of downstream signaling, typically through MyD88. Early in vitro work, which was also supported in an in vivo seizure model, showed that IL-1R1 is expressed by excitatory glutamatergic neurons where it associates with the glutamate receptor N-methyl-D-aspartate receptor (NMDAR) (see Box 1 for glossary of terms) subunit NR2B.(3, 4) This work further showed that IL-1R1/NR2B association upon IL-1β binding facilitated NR2B phosphorylation and enhanced Ca2+ flux through the NMDAR upon glutamatergic synaptic transmission, which increased neuronal excitability. Conversely, a number of largely in vitro studies have suggested that IL-1β generally dampens neuronal excitability by decreasing the flow of ions through voltage-gated Na+ (Nav), Ca2+ (Cav) and K+ (Kv) channels.(5)
Besides IL-1β, TNF-α has emerged as a major driver of neuronal excitability. Unlike IL-1β-mediated regulation of NMDARs, TNF-α regulates another type of glutamate receptor in excitatory neurons called α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs) (see Box 1 for glossary of terms). Also, while IL-1β generally inhibits voltage-gated ion channels to limit excitability, TNF-α can increase the excitability of neurons by enhancing the transcription or Nav1.3 and Nav1.8 upon in vivo.(6) TNF-α belongs to the type II transmembrane family that can be expressed in a membrane-bound form (mTNF-α), which can be cleaved into a secreted form (sTNF-α). sTNF-α activates TNFR1, whereas mTNF-α typically acts through TNFR2. In neurons in the cortex (see Box 1 for glossary of terms), spinal cord, amygdala, and the periphery, sTNF-α binding to TNFR1 results in increased glutamate receptor 1 (GluR1)-containing AMPARs to increase neuronal excitability (discussed further in homeostatic scaling section) (reviewed in (5)). TNF-α can further enhance the excitability of cerebellar Purkinje neurons in vivo by activating phosphatases, which decreases small conductance Ca2+-activated K+ channels (SK channels).(7) The cellular sources of TNF-α, as well as IL-1β and other cytokines that regulate basal excitatory neurotransmission remains an open question. However, in vivo evidence supports microglia as a major source of TNF-α in the hippocampus (see Box 1 for glossary of terms), striatum (see Box 1 for glossary of terms), and cortex,(7–11) as well as astrocytes and Bergman glia in the cerebellum.(12, 13)
Interestingly, IL-6 has seemingly opposing effects to TNF-α and IL-1β. That is, IL-6 generally works to dampen basal neural activity. For example, early data in hippocampal cultures suggested that the IL-6 inhibits excitatory neurotransmission by promoting upregulation of adenosine A1 receptors, which will induce a decrease in neural activity.(14) IL-6 has also been shown to dampen basal neural activity in cultured neurons by decreasing Ca2+ flux through transcriptional regulation of Cav channels.(15, 16) Towards more in vivo relevance, mice were made to overexpress IL-6 in astrocytes (GFAP-IL6) and basal excitatory neurotransmission was decreased in cerebellar Purkinje neurons.(17) However, the endogenous cellular source(s) of IL-6, the downstream signaling critical to elicit these IL-6 dependent effects on basal excitatory neurotransmission, and the in vivo significance of IL-6 on basal neurotransmission in the absence of transgenic overexpression remain open questions. Still, an intriguing aspect of all these studies showing IL-1β, TNF-α, and IL-6-dependent regulation of basal excitatory neurotransmission is that these cytokines can have opposing effects, which could serve to o maintain circuit homeostasis normally and during inflammation.
Cytokine regulation of inhibitory tone
In addition to regulating excitability through glutamate receptors and voltage-gated ion channels, immune system cytokines have also been implicated in regulating inhibitory γ-aminobutyric acid (GABA) (see Box 1 for glossary of terms) tone in the CNS. One emerging key regulator of inhibitory tone (see Box 1 for glossary of terms) is the type 2 immune cytokine IL-4. Specifically, the IL-4 receptor alpha (IL-4Rα) was demonstrated to be expressed presynaptically in excitatory and inhibitory hippocampal neurons and deficiency in IL-4Rα in vivo in mice led to a decrease in synaptic vesicle pools, which contain neurotransmitter, in these neurons.(18) In mouse hippocampal slices, IL-4Rα deficiency decreased the basal activity of both excitatory and inhibitory neurons as indicated by electrophysiological recordings of miniature excitatory postsynaptic currents (mEPSCs) (see Box 1 for glossary of terms) and miniature inhibitory postsynaptic currents (mIPSCs) (see Box 1 for glossary of terms). This resulted in increased spontaneous postsynaptic currents (sPSCs) (see Box 1 for glossary of terms) reflecting the net result of excitatory and inhibitory currents at the neuron level. The altered presynaptic function eventually resulted in a higher excitatory drive, which is in line with predominant influence of the inhibitory plasticity on neuronal excitability.(19) The authors furthermore showed that acute IL-4 application increased mIPSC frequencies and this electrophysiological change at synapse was through activation of protein kinase C gamma (PKCγ) signaling downstream of IL-4Rα. The study indicates that decreased inhibitory synapse function in IL-4Rα-deficient neurons is the largest influence on elevated excitatory drive leading to increased cortical excitability in vivo.(18) This enhanced network excitability due to IL-4Rα deficiency translated into increased exploratory and locomotor behaviors as well as decreased anxiety levels. Concomitantly, fear learning was impaired, although less pronounced. Thus, IL-4 and IL-4Rα play a role in maintaining CNS synaptic function, neural excitability (see Box 1 for glossary of terms), and overall behavior through its actions on both excitatory and inhibitory neurons. IL-4-dependent effects on inhibitory transmission are further supported by other work showing that IL-4 is T cell derived and regulates learning and memory in vivo through its actions on GABAergic transmission (discussed later in Hebbian plasticity).(20) Here, the single nucleus transcriptome of dentate gyrus neurons activated during a behavioral task to induce memory formation (contextual fear conditioning, see Box 1 for glossary of terms) indicated that IL-4Rα regulated the expression of genes associated with synapse organization, synaptic plasticity (see Box 1 for glossary of terms) and regulation of synapse structure. One important consideration for all these studies related to IL-4/IL-4Rα signalling is IL-13. IL-13 also binds IL-4Rα and mice deficient in IL-13 have similar phenotypes with those deficient in IL-4.(21, 22) Thus, both cytokines, which are likely T cell derived, could be regulating excitatory neurotransmission, through IL-4Rα.
Similar to IL-4 and IL-13, IFNγ derived from meningeal T cells from has also emerged as potent regulator of inhibitory neurotransmission. IFNγ is the sole member of the type II interferons. It is typically produced by T cells and natural killer cells and is a potent amplifier of peripheral inflammation. It was first shown in mice deficient in T cells that there were learning and memory defects.(23–25) Later work demonstrated that meningeal T cell derived IFNγ acted on GABAergic neurons expressing the IFNγ receptor IFNGR1 in the prefrontal cortex to increase GABAergic currents in projection neurons.(26) More recently, IFNγ as shown to act presynaptically, likely through nitric oxide, to increase the release of the inhibitory neurotransmitter GABA.(27, 28) The behavioral manifestations of IFNγ-dependent changes in GABAergic transmission includes learning and memory impairments and deficits in social behavior.(23–26)
As IL-4 and IFNγ have emerged as key regulators of GABAergic tone in the CNS, it is also important to consider other immune cytokines. For example, in vitro work has suggested that IL-1β increases tonic inhibitory tone by regulating the number and subunit content of GABAA receptors (see Box 1 for glossary of terms) on neurons through activation of P38-MAPK downstream of IL-1R1.(29) Towards a cellular source of IL-1β, a group studying the effects of ethanol exposure on GABAergic transmission demonstrated that IL-1β can be derived from neurons and microglia to promote GABA release in vivo.(30) In contrast, TNF-α decreases inhibitory tone in vitro and in vivo by inducing protein phosphatase 1-dependent endocytosis of surface GABAA receptors.(31, 32) The cellular source of TNF-α in this context remains an open question but it is highly enriched in microglia throughout the brain.(8–11, 33) Similar to excitatory neurotransmission, it is intriguing to consider that the opposing effects of all these cytokines on GABAergic transmission are important to maintain circuit homeostasis.
Cytokine regulation of synaptic structure
In addition to regulation of basal function, cytokines have emerged as important regulators of synapse structure, which ultimately impacts basal neurotransmission. Most of this evidence has been in the context of development where neurons must first grow their axonal projections to their postsynaptic targets. This is followed by a period of exuberant synaptogenesis and then a period of synaptic pruning (see Box 1 for glossary of terms), whereby a subset of these synapses that form in excess are eliminated while other synapses are strengthened and elaborated in the circuit.
Some of the first evidence suggesting a role for cytokines in the regulation of structural synaptic connectivity was in the context of microglia-mediated synaptic pruning. Microglia are a resident CNS macrophage that have emerged as key regulators of synaptic pruning by engulfing and removing a subset of synapses.(34) In these studies, the effects of cytokines on synapses are less direct and were mediated indirectly through cytokine effects on microglia, which subsequently engulf and remove structural synapses. For example, ablation of the fractalkine receptor (CX3CR1), a G-protein-coupled (Gαi) chemokine receptor highly enriched in microglia, resulted in a delay in synapse maturation in the hippocampus and barrel cortex(35, 36) and a delay in the removal of excess synapses by microglia in the hippocampus in vivo.(35) In more recent studies, it was shown that the fractalkine ligand CX3CL1 is expressed in neurons and promotes microglia to remove synapses via CX3CR1 in an activity-dependent manner.(37) Further, deficiency in CX3CR1 results in defects in functional connectivity, decreased social behavior, and enhanced repetitive behaviors.(38)
Related to the role of CX3CL1-CX3CR1 signaling in synapse removal by microglia, IL-33- IL1RL1 (also known as ST2) signaling was also identified to regulate microglial synaptic pruning in vivo.(39) IL-33 is an IL-1 family member that plays important roles in type-2 innate immunity with well-described roles as a cellular alarmin. In the developing spinal cord, it was shown that IL-33 produced by astrocytes stimulates microglia expressing IL1RL1 to engulf and remove excess synapses from α-motor neurons in the spinal cord.(39, 40) In contrast, in the adult hippocampus, astrocyte-derived IL-33-IL1RL1 signaling promotes homeostatic synapse formation in response to prolonged decreased neuronal activity and promotes spatial memory formation.(41) However, neurons in the adult hippocampus also seem to produce a distinct isoform of IL-33 called Il33b.(42) In contrast to astrocytes, conditional deletion of IL-33 in neurons resulted in a defect in extracellular matrix remodeling by microglia through IL1RL1 and a subsequent failure of new spine growth and decreased neurogenesis in the hippocampus.(42) These studies reveal that IL-33 is a critical regulator of synapse numbers and its effects can differ depending on the cellular source of IL-33, the IL-33 isoform, and the developmental stage.
Another aspect of synapse pruning and integration of new synapses into a circuit is stabilizing and strengthening synapses that are not eliminated. Interestingly, another cytokine of the TNF superfamily called cytokine tumor necrosis factor-like weak inducer of apoptosis or TWEAK (TNFSF12) and its receptor FN14 have been now implicated in synapse stabilization and maturation.(43, 44) In the developing visual thalamus (see Box 1 for glossary of terms), FN14 in neurons promoted mature bulbous spines. However, this FN14 effect was inhibited upon binding to TWEAK derived from microglia. Thus, FN14 normally promotes the formation and/or maintenance of synapses, and this effect is blocked near TWEAK-expressing microglia.(44) The signaling downstream of FN14 in neurons in this context remains an open question but another group has shown some insight into the downstream signaling in the adult hippocampus. This group showed recently in the adult hippocampus that TWEAK binding to Fn14 changes the phosphorylation state of several synaptic proteins and decreases basal synaptic transmission.(45) This change in synaptic protein phosphorylation and decrease in transmission could result in destabilization of synapses, which is supported by experiments in this same study in which blocking TWEAK-Fn14 signaling protected synaptic impairments in Alzheimer’s and stroke-relevant mouse models.
Cytokine regulation of functional plasticity
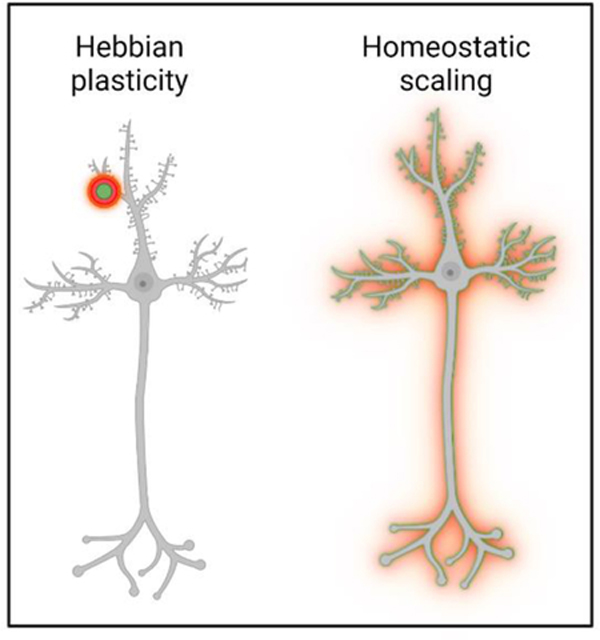
Through mechanisms that likely involve the modulation of baseline properties of neurons described above (Figure 1), cytokines have also been shown to regulate Hebbian and homeostatic forms of synaptic plasticity (Box 2). Hebbian mechanisms, including long-term potentiation (LTP) and long-term depression (LTD), are relatively rapid modifications of synaptic transmission at individual synapses. In contrast, homeostatic synaptic scaling stabilizes plasticity and maintains proper circuit function by scaling up or down the excitability of the entire neuron. Here, we review mounting evidence that immune cytokines are involved in both forms of plasticity.
Box 2– Hebbian and Homeostatic forms of synaptic plasticity.
Hebbian mechanisms such as long-term potentiation (LTP) and long-term depression (LTD) are fast and persisting input-specific modifications of synaptic transmission at individual synapses. This plasticity allows for a stable, but at the same time flexible, system. In contrast, homeostatic synaptic scaling stabilizes plasticity and maintains proper circuit function by scaling up or down the excitability of the entire neuron. Throughout our lifetime, the brain faces the challenge of keeping neuronal networks functional under varying conditions. This requires continuous fine-tuning of neuronal networks to not only dynamically adapt to new challenges, but also to return to stable set points. Processes ensuring this stability in the nervous system act by homeostatic alteration of synaptic strength, homeostatic synaptic scaling. During times of prolonged changes in neural activity, the neuron adjusts the strength of all its synapses to maintain a ‘set point’ level of activity. To achieve this, neurons must detect perturbation of activity via internal sensors and then translate the information in order to regulate synaptic neurotransmitter receptor expression and/or localization. Still, the distinction and/or connection between Hebbian plasticity and homeostatic scaling remains an open question, but it has been proposed that homeostatic mechanisms serve to keep LTP and LTD-based mechanisms in balance (67).
First, we discuss immune cytokine modulation of Hebbian mechanisms (Box 2). There is a large body of literature demonstrating that the addition of cytokines to brain slices ex vivo can modify Hebbian plasticity, including LTP and LTD (reviewed in (46, 47)). In much of this work, these effects are blocked by their cognate receptor antagonists. Here, we will focus on studies that provide genetic evidence that cytokines can modulate Hebbian forms of plasticity (LTP and LTD), which underlie learning and memory in vivo.
Some of the first genetic evidence that supported a role for cytokines in Hebbian plasticity in vivo involved manipulation of IL-1β signaling. In mice overexpressing a negative regulator of IL-1β (IL-1ra) or in mice deficient in the IL-1 receptor IL1R1, learning and memory and hippocampal LTP were impaired.(48–50) Large increases in IL-1β in vivo similarly impaired hippocampal-dependent memory, but smaller increases in IL-1β in the hippocampus improved memory.(48) Adding to the complexity, IL-1β appears to have a stronger effect on hippocampal-dependent learning in younger 2–4 month-old mice compared to older 6 month-old mice.(51) That is, young mice deficient in IL-1β or IL1RL1 had defects in spatial learning but these defects were no longer present in older mice. These results suggest more robust neuromodulatory effects of IL-1β on Hebbian plasticity in young circuits, which are concentration dependent. It is intriguing to consider that the decrease in IL-1β-dependent plasticity in older mice may be an underlying factor by which plasticity decreases during aging. The downstream signaling eliciting these IL-1β-dependent effects on Hebbian plasticity are far from being elucidated, but one possibility is through BDNF--a molecule that regulates LTP at the synapse and is regulated by IL-1β in other contexts.(52) Interestingly, TWEAK-FN14 signaling, which also alters basal neurotransmission (discussed above), dampens LTP in hippocampal slices similar to IL-1β. Unlike IL-1β, the concentration-dependent effects of TWEAK on this plasticity have not been investigated.(45)
IL-6 and TNF-α have also been described to regulate Hebbian plasticity. Similar to IL-1β, these effects are highly dependent on concentration.(53–55) One explanation for these concentration-dependent effects of CNS cytokines on LTP could be cell-type specificity. Cytokines can directly impinge on neurons expressing cytokine receptors to impact NMDARs or AMPARs (discussed in previous section) and plasticity. But, at higher concentrations, these cytokines may also signal to glial cells, which secondarily impact synaptic plasticity to have different effects. For example, neuroinflammatory levels of TNF-α can act through astrocytes expressing TNFR1 to enhance presynaptic glutamate release in the hippocampus and impair contextual memory in mice.(56) Still, the cellular source(s) of TNF-α, as well as IL-1β and IL-6, to regulate Hebbian plasticity remains an open question. At least in the context of homeostatic plasticity (discussed below), microglia are emerging as a major source of TNF-α.(8, 33)
Besides cytokines derived locally from resident CNS cells, cytokines derived from immune cells in the meninges can regulate Hebbian forms of plasticity. This includes IL-4. It was first shown that mice deficient in T cells or IL-4 had defects in hippocampal-dependent learning and memory and LTP.(20, 23, 24) Moreover, the behavioral effects on learning and memory were attenuated upon adoptive transfer with wild-type, but not IL-4−/−, T cells.(24) Since this initial work, it has now been shown that T cell-derived IL-4 is acting on inhibitory neurons, which express the receptor IL-4Rα.(18, 20) Although it is less clear what is downstream of IL-4Rα in inhibitory neurons to elicit changes in learning and memory, PKCγ has been shown to be downstream of IL-4Rα in neurons to elicit changes in network excitability.(18)
Molecules belonging to the chemokine family of cytokines have also been identified to regulate Hebbian plasticity. This has been most widely studied with respect to two different G-protein coupled chemokine receptors, CX3CR1 and C-C chemokine receptor 5 (CCR5). In ex vivo hippocampal slices, it was first shown that CX3CL1 depressed glutamatergic transmission and impaired LTP, which was blocked in CX3CR1−/− slices.(57–59) Similar depression of LTP by CX3CL1-CX3CR1 signaling was shown in vivo in the spinal cord.(60) In contrast, other work has shown a defect in hippocampal LTP in CX3CR1−/− slices and concomitant defects in behavioral learning and memory in vivo.(61) Like with other cytokines, conflicting data in these studies could arise from variations in experimental conditions such as concentration-dependent effects of CX3CL1 on LTP, differing ages of mice, sex of the mice, brain regions, or differences in LTP induction paradigms. These are all important considerations as one interprets results related to plasticity. For example, LTP or LTD will be more restricted in older mice and the induction of plasticity will differ in different brain regions depending on the composition of neurons and glial cells. LTP inductions can also vary widely across labs and can yield results that are seemingly contradictory. Thus, experimental methods should always be taken into strong consideration when evaluating and comparing these studies.
CCR5 is another G-protein coupled chemokine receptor discovered to inhibit Hebbian plasticity. CCR5 is most widely studied in the context of HIV infection where it normally promotes viral infection in macrophages.(62) CCR5 antagonists effectively block HIV in patients and improve cognitive impairment in patients.(63, 64) Strikingly, genetic ablation of CCR5 resulted in increased levels of MAPK and cAMP-responsive element-binding protein (CREB) in the hippocampus and enhanced hippocampal LTP in slices.(65) It was proposed that CCR5 is normally a break on MAPK and CREB-dependent signaling. Most recently, the same group showed that CCR5 and its ligand CCL5 dictate the temporal window for memory linking.(66) Briefly, memory linking occurs when two memories are acquired within a short time period such that the retrieval of one memory is more likely to trigger the retrieval of the other memory by activating the same ensemble of neurons. Neuronal CCR5 increased in expression following contextual fear conditioning and ablating this signaling in hippocampal neurons resulted in a blockade in neuronal ensemble formation and subsequent memory linking.(66) This work lays the foundation for further understanding the cellular sources of the ligands for CX3CR1 and CCR5 and determining how this chemokine signaling mechanistically impacts downstream signaling in neurons and glia to impact plasticity.
Besides Hebbian plasticity, another form of plasticity that modulates neuronal networks is homeostatic synaptic scaling (Box 2). During times of prolonged changes in neural activity, the neuron scales all of its synapses up or down to maintain a ‘set point’ level of activity.(67) Thus, synaptic scaling is thought to counteract Hebbian forms of plasticity to maintain circuit homeostasis (Box 2). Intriguingly, TNF-α has now emerged as a key regulator of synaptic scaling by regulating the expression and localization of synaptic neurotransmitter receptors. This began with seminal work showing that a chronic decline in hippocampal neuronal activity in vitro triggers glial TNF-α release, which induces an increase in surface AMPARs at the neuronal membrane.(68, 69) It was further shown that TNF-α rapidly increases surface levels of β3 integrin to mediate AMPAR accumulation.(70)
Since these initial findings, cell-specific inducible Cre lines or gene expression have been used to show that microglia are a major cellular source of TNF-α necessary for modulating synapse plasticity and homeostatic scaling.(8, 9, 33) Towards a structural correlate, it was recently shown by in vivo imaging in cortical neurons that dendritic spines (see Box 1 for glossary of terms) became larger during synaptic scaling.(71) Importantly, TNF-α can regulate synaptic scaling differently depending on the neurotransmitter receptor. For example, TNF-α induces an increase in GluR1 subunit-containing AMPA receptors in cultured hippocampal neurons through a phosphatidylinositol 3 kinase (PI3K)-dependent process.(31, 32) Remarkably, in the same neuronal cultures, it induces the endocytosis of GABAA receptors, resulting in fewer surface GABAA receptors and decreased inhibitory synaptic strength.(31, 32) TNF-α can also have opposing effects on receptor membrane insertion or removal depending on the neuron subtype.(8, 32) This work is reviewed in the following discussion on the role of immune system cytokines at synapses in disease.
Cytokine modulation of synapse and neuronal circuits - implications for disease
Considering the importance of cytokines for synaptic transmission, plasticity, structure and overall behavior, it is conceivable that cytokines are relevant for synaptic dysfunction in disease. While studies on cytokine involvement in neurological disease are numerous, we focus here specifically on studies that show a direct impact of cytokines on synapses in the in vivo disease context.
Some of the strongest links between cytokines and synaptic dysfunction were established in multiple sclerosis (MS) (Figure 2), where loss of synapses correlates with functional decline.(72–75) Much of this work has shown that cytokines can regulate a number of different MS pathologies, such as demyelination and axon degeneration, which impact synapses downstream. For example, IL-4 has been shown to protect axons and promote axonal outgrowth via modulation the F-actin cytoskeleton, which, in turn, likely protects synapses.(76)
Figure 2: Immune cytokine impact on network activity in pathology.
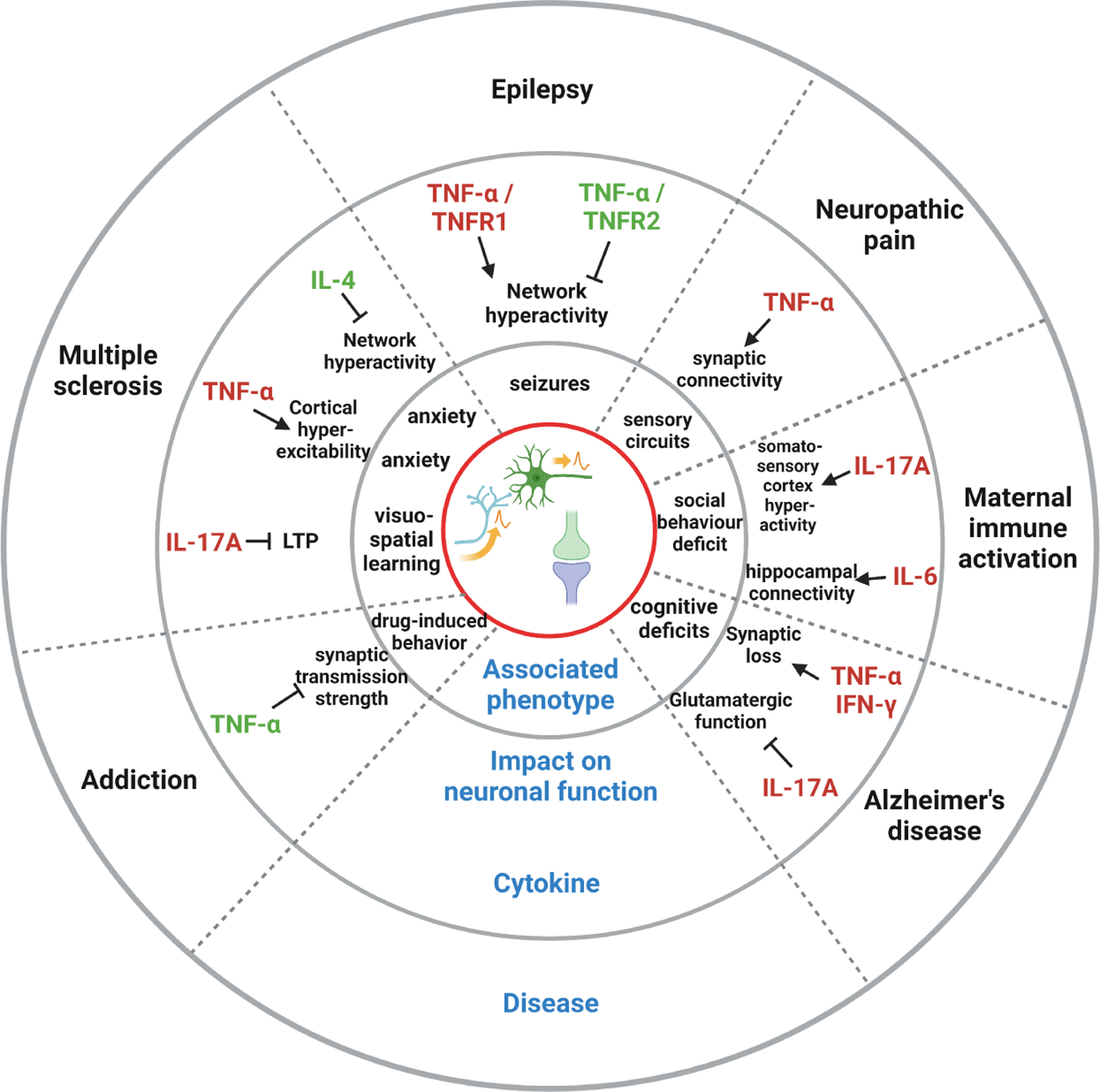
Emerging evidence reveals that immune cytokines play a role in different brain diseases by modifying neuronal function. Thereby, subsequent changes in synaptic transmission results in changes in network activity and brain plasticity and eventually leads to behavioral phenotypes.
An emerging potent regulator of MS, which has a more direct effect on synapses in disease, is TNF-α. In a mouse model relevant to MS (experimental autoimmune encephalomyelitis, EAE), peripherally-derived TNF-α was shown by in vivo two-photon live imaging to induce increased turnover of postsynaptic dendritic spines and presynaptic axonal boutons in the somatosensory cortex.(77) Further, stereotactic injection of IFNγ and TNF-α into rats that were previously immunized with myelin oligodendrocyte glycoprotein induced transient, focal cortical lesions and silencing of cortical neurons adjacent to the injury.(78) In contrast, in a mouse model of experimental relapsing-remitting neuroinflammation, TNF-α produced by neurons themselves caused an increased frequency of neuronal calcium, intensified spontaneous neuronal activity in the cortex, and heightened anxiety.(79) Additionally, both the increased cortical neuronal circuit activity and anxiety-like behavior in these mice were reduced by the local administration of the TNF-α inhibitor infliximab. This is further supported by data from humans in which overexcited cortical microcircuits and heightened anxiety are clinical features in many MS patients.(80) It is possible that TNF-α-dependent enhanced neuronal activity during MS may first serve a compensatory homeostatic scaling role following prolonged decreased activity during the relapsing phase of disease. Perhaps, later TNF-α becomes pathological leading to excitotoxicity and increased neuronal death.
In Alzheimer’s disease (AD) (Figure 2), elevated cytokines have been shown to be upstream of synapse loss. For example, type I interferon stimulates microglia-mediated synapse loss and memory impairment in AD-relevant neurodegeneration.(81, 82) In these AD-relevant mice, IFNγ receptor blockade or deletion of the IFN-I receptor (IFNAR) specifically from microglia reduced synapse loss, reactive microgliosis, and learning impairments. Towards a cellular source of IFNγ, another group has shown that IFNγ produced by T cells is critical to induce microglia-mediated synapse removal and learning impairments in flavivirus-induced neurodegeneration.(83)
Two other pro-inflammatory cytokines reported to play a role at synapses in AD are TNF-α and IL-17A. For example, in a rat model relevant to AD, TNF-α was shown to reduce inhibitory GABAergic neurotransmission and increase excitatory glutamatergic transmission prior to pathology.(84, 85) In addition, IL-17A producing γδ17 T cells were recently show to accumulate in the brain and the meninges of female, but not male transgenic AD mice, and were responsible for synaptic dysfunction and cognitive decline.(86) Towards a mechanism by which IL-17 is impacting synapse dysfunction and cognitive decline, IL-17A in other contexts is derived from meningeal γδ17 T cells, which can inhibit glutamatergic activity, short term memory and LTP.(87, 88)
Interestingly, some of the same immune system cytokines implicated in synaptic impairments in neurological disorders in adults, have also been implicated in neural circuit abnormalities in neurodevelopmental disorders. For example, offspring from mothers with a brain deficit in TNF-α exhibited low innate fear.(89) These data suggest that maternal-derived TNF-α can impact brain circuit development in utero to impact anxiety-like behaviors. In contrast, TNF-α signaling in the ventral hippocampus appears to sustain stress-induced synaptic changes and anxiety behavior in later life.(33) The later group further showed that TNF-α is elevated in microglia following stress and this induces an increase in AMPAR currents in the hippocampus (see also homeostatic scaling).
Besides this work on TNFα, some of the most compelling data supporting a role of cytokines on neural circuits in development are in models of maternal immune activation (MIA) (Figure 2). In mothers that have an infection (e.g., flu) during pregnancy, there is increased risk of offspring with autism spectrum disorders (ASD) and schizophrenia (SZ).(90) In rodent models of MIA, exposure of pregnant dams to a peripheral immune challenge, such as polyinosinic:polycytidylic acid (Poly(I:C)) a synthetic double stranded RNA that mimics a viral infection, can result in offspring that have behavioral deficits reminiscent of ASD or SZ.(90–92) Additionally, these Poly(I:C) offspring have cortical abnormalities in the primary somatosensory cortex and sociability defects.(93, 94) It was further shown that sociability defects and cortical abnormalities were largely due to IL-17A produced by maternal T helper 17 cells.(93) Interestingly, IL-17A has the opposite effect in adults through IL-17Ra expression in excitatory neurons in the somatosensory cortex.(94, 95) Direct injection of IL-17A into the primary adult somatosensory cortex or elevating IL-17A with lipopolysaccharide injection resulted in attenuation of sociability phenotypes in MIA offspring. A separate study further showed that meningeal γδ T cell-derived IL-17A induces baseline anxiety-like behavior in wildtype adult mice via the IL-17A-receptor expressed in cortical glutamatergic neurons.(96) It is likely that the cellular source of IL-17, the timing of its release, the brain region affected, and the concentration dictate distinct social or anxiety-like behavioral manifestations.
Similar to IL-17A, IL-6 has also been implicated in MIA-induced behavioral and synaptic changes. In a similar Poly(I:C) model of MIA or when pregnant dams were injected with IL-6 alone, there were behavioral defects reminiscent of ASD and SZ (prepulse inhibition and latent inhibition defects) in the offspring.(97) In the Poly(I:C) model, these effects were attenuated when pregnant dams were also injected with an antibody to block IL-6 or when Poly(I:C) was injected into IL-6−/− mice. Towards a synaptic mechanism, very recently it was demonstrated that injection of pregnant dams or their embryos with IL-6 elicited a synaptogenic transcriptional program after binding IL-6R-β-receptor subunit glycoprotein 130 receptor in neurons.(98) Ultimately, this transcriptional program elicited by IL-6-IL-6R signaling in neurons resulted in elevated structural and functional glutamatergic synapses in the hippocampus in the embryos and hyperconnectivity in adult offspring. It is possible that IL-6 and IL-17A are both inducing effects during MIA but on different neurons. This is likely due to neuron-specific expression of interleukin receptors. Moreover, the cellular sources of these cytokines remain to be fully deciphered. In the case of IL-17, this is appears to be largely T cell derived. However, the cellular source(s) of IL-6 is less clear.
Additional insights into how cytokines impact synapses in disease have been demonstrated in the context of epilepsy (Figure 2). For example, in mice deficient in interferon regulatory factor 8 (IRF8), a transcription factor necessary for microglia maturation, there was elevated TNF-α in microglia and increased susceptibility to lethal seizures.(9) If a blood-brain barrier (BBB) permeable TNF-α inhibitor, but not a BBB impermeable inhibitor, was administered, lethal seizures were attenuated in IRF8−/− mice. In contrast, TNF-α can also inhibit seizures when directly injected into the hippocampus.(99). It has been suggested that TNF-α can exert opposing effects on seizures depending on which receptor it binds. TNFR1 generally promotes seizure induction, while TNFR2 has the opposite effect.(100) In support of this idea, global deletion of TNFR1 (p55) results in a decrease in basal firing rate but no change in glutamate or KCl-induced neuronal activity, whereas global deletion of TNFR2 (p75) had the opposite effect.(101, 102)
In addition to TNF-α, elevated IL-1β and IL-18 promote seizures. IL-1β application to hippocampal slices induced hyperexcitability and blocking the IL-1β receptor IL1R1 blocked this hyperexcitability in vitro and attenuated seizures in mice in vivo.(103–105) This is likely through IL-1β effects on excitatory and inhibitory neurotransmission (Figure 1). In contrast, deficiency in a related cytokine IL-18 can exacerbate seizure activity by enhancing basal neurotransmission and increasing synapse numbers.(106) Interestingly, both cytokines are released upon NLRP3 inflammasome activation during neurodegeneration. It is possible that the two cytokines counteract each other in an attempt to maintain homeostasis.
An influence of TNF-α on synaptic function has also emerged in addiction (Figure 2). The original work on TNF-α in cortical neurons showed that it drove the insertion of AMPARs to regulate homeostatic scaling.(68, 69) Instead, in striatal neurons, TNF-α drives the internalization of synaptic AMPA receptors in the context of cocaine administration.(8) Repeated cocaine administration induced striatal microglia to release TNF-α, thereby depressing synaptic strength and limiting the development of behavioral sensitization. It was also shown that the increase in TNF-α production by microglia during cocaine administration can suppress cocaine-induced behavioral sensitization. Thus, microglial TNF-α has an adaptive role in the response to cocaine and it raises the possibility that TNF-α could be an effective treatment of drug-induced behavior. Similarly, during morphine withdrawal, there is a marked increase in TNF-α accompanied by diminished glutamatergic transmission in the lateral habenula.(107) As the lateral habenula normally encodes aversive stimuli and contributes to negative emotional states, the authors concluded that habenular TNF-α controls synaptic plasticity and behavioral adaptations to morphine.
Similar to addiction, TNF-α also appears to impact synapses and homeostatic plasticity during neuropathic pain (Figure 2). Pain-related information is transmitted through the spinal cord, as well as hippocampal and spinal neuronal circuits. It has been shown that this pain processing at the synaptic level is regulated by microglial TNF-α production.(108, 109) Moreover, the intracellular protease caspase-6 (CASP6) directly modulates spinal cord synaptic transmission and inflammatory pain by releasing TNF-α from microglia.(110)
Concluding Remarks
Immune system cytokines are emerging potent regulators of synapses. Many of these molecules are now appreciated to modify the overall excitatory and inhibitory tone of synaptic connectivity, which ultimately impacts functional and structural synaptic plasticity. Ultimately, this cytokine-dependent regulation of synapses impacts behavioral output, including learning and memory, anxiety, and social behaviors.
One intriguing aspect of cytokine function on synapses is that a single cytokine can have seemingly opposite effects (e.g., enhancing excitation and enhancing inhibition) depending on the concentration. Some cytokines can also have opposing effects to other cytokines on synapses. Therefore, it is difficult to surmise general principles by which cytokines modulate circuits. However, it is possible that the nervous system has evolved the counteracting, concentration-dependent effects of cytokines on synapses to help maintain circuits in homeostasis, particularly during times of inflammation. Technical advances are required to more precisely measure the local concentration of multiple cytokines simultaneously within neural circuits. For example, sensors to more accurately measure cytokine levels in vivo would be key to determine the local cytokine concentration at functional and dysfunctional synapses across different neuronal populations. Also, in many of the contexts described above, the cellular sources of the cytokine and cognate receptor are not fully elucidated. This is important to consider as same cytokine may have very different effects depending on the cell type and its receptor binding. Also, cytokines can be produced by resident CNS cells, but they can also originate from infiltrating immune cells or from immune cells within brain barriers (e.g., meninges). Therefore, mapping the cellular sources of cytokines, their cognate receptors, and downstream signaling will be important. Such a mapping strategy would better establish cytokine-dependent cell-to-cell interactions more concretely throughout the nervous system basally and during disease.
Another important aspect of cytokine-dependent modulation of synapses is the implications for disease. While previous work has emphasized cytokines as critical regulators of more generalized inflammation in the CNS, we are just beginning to unravel the impact of these molecules on synapse dysfunction in disease. For example, TNF-α is now well-described to affect the excitability of circuits, but these effects have not yet been systematically assessed in all brain pathologies. Also, determining how the entire “cytokine cocktail” impacts synapses in disease is complex and likely involves neurons and glial cells. These effects are concentration dependent and age dependent. It should be mentioned that additional work has been performed either reporting on peripheral immune challenge effects such as infection on circuits(111, 112) or indirectly pointing to the relevance of immune cytokine effects on neuronal function in behavioral disorders such as stress(11, 113) . Determining how to manipulate cytokine levels more locally and cell-specifically as well as unraveling the processes at the synapse itself will likely be necessary for developing effective therapeutics.
Finally, it is of utmost importance to confirm these crucial discoveries in the human system. Although this will partially consist of descriptive studies in humans, it is necessary to determine whether the exciting discoveries pertaining to the role of cytokines on synapses in animal models translate. One possibility is to test these cytokine-synapse interactions in human induced-pluripotent stem cell systems or in humanized mouse models. Together, these are key steps towards furthering our understanding of the broad impact of cytokines on synapses and for the development of new cytokine-based therapies for treating underlying synapse dysfunction in disease.
Acknowledgments and Funding:
Original work within this review was supported by the German Research Foundation (DFG; CRC-TR-128 to FZ and SB, CRC 1080 to FZ), and the Progressive MS Alliance (PA-1604–08492, BRAVEinMS to FZ). Additional work was supported by NIMH-R01MH113743 (DPS), NINDS-R01NS117533 (DPS), NIA-RF1AG068281 (DPS), and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (DPS). Schematic drawing was created with BioRender.com. The authors thank Dr. Cheryl Ernest for proofreading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schafer DP, Stevens B. Synapse elimination during development and disease: immune molecules take centre stage. Biochem Soc Trans 2010;38(2):476–81. [DOI] [PubMed] [Google Scholar]
- 2.Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron 2009;64(1):93–109. [DOI] [PubMed] [Google Scholar]
- 3.Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain : a journal of neurology 2008;131(Pt 12):3256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. The Journal of neuroscience : the official journal of the Society for Neuroscience 2003;23(25):8692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015;96(Pt A):70–82. [DOI] [PubMed] [Google Scholar]
- 6.He XH, Zang Y, Chen X, Pang RP, Xu JT, Zhou X, et al. TNF-alpha contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain 2010;151(2):266–79. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto M, Kim M, Imai H, Itakura Y, Ohtsuki G. Microglia-Triggered Plasticity of Intrinsic Excitability Modulates Psychomotor Behaviors in Acute Cerebellar Inflammation. Cell reports 2019;28(11):2923–38 e8. [DOI] [PubMed] [Google Scholar]
- 8.Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D. Microglial TNF-alpha Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron 2016;90(3):483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feinberg PA, Becker SC, Chung L, Ferrari L, Stellwagen D, Anaclet C, et al. Elevated TNF-α Leads to Neural Circuit Instability in the Absence of Interferon Regulatory Factor 8. J Neurosci 2022;42(32):6171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 2014;34(36):11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nie X, Kitaoka S, Tanaka K, Segi-Nishida E, Imoto Y, Ogawa A, et al. The Innate Immune Receptors TLR2/4 Mediate Repeated Social Defeat Stress-Induced Social Avoidance through Prefrontal Microglial Activation. Neuron 2018;99(3):464–79.e7. [DOI] [PubMed] [Google Scholar]
- 12.Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nature neuroscience 2001;4(7):702–10. [DOI] [PubMed] [Google Scholar]
- 13.Shim HG, Jang SS, Kim SH, Hwang EM, Min JO, Kim HY, et al. TNF-alpha increases the intrinsic excitability of cerebellar Purkinje cells through elevating glutamate release in Bergmann Glia. Sci Rep 2018;8(1):11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biber K, Pinto-Duarte A, Wittendorp MC, Dolga AM, Fernandes CC, Von Frijtag Drabbe Kunzel J, et al. Interleukin-6 upregulates neuronal adenosine A1 receptors: implications for neuromodulation and neuroprotection. Neuropsychopharmacology 2008;33(9):2237–50. [DOI] [PubMed] [Google Scholar]
- 15.Motagally MA, Lukewich MK, Chisholm SP, Neshat S, Lomax AE. Tumour necrosis factor alpha activates nuclear factor kappaB signalling to reduce N-type voltage-gated Ca2+ current in postganglionic sympathetic neurons. The Journal of physiology 2009;587(Pt 11):2623–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma SH, Li B, Huang HW, Peng YP, Qiu YH. Interleukin-6 inhibits L-type calcium channel activity of cultured cerebellar granule neurons. J Physiol Sci 2012;62(5):385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson TE, Ur CL, Gruol DL. Chronic interleukin-6 exposure alters electrophysiological properties and calcium signaling in developing cerebellar purkinje neurons in culture. Journal of neurophysiology 2002;88(1):475–86. [DOI] [PubMed] [Google Scholar]
- 18.Hanuscheck N, Thalman C, Domingues M, Schmaul S, Muthuraman M, Hetsch F, et al. Interleukin-4 receptor signaling modulates neuronal network activity. The Journal of experimental medicine 2022;219(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carvalho TP, Buonomano DV. Differential effects of excitatory and inhibitory plasticity on synaptically driven neuronal input-output functions. Neuron 2009;61(5):774–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herz J, Fu Z, Kim K, Dykstra T, Wall M, Li H, et al. GABAergic neuronal IL-4R mediates T cell effect on memory. Neuron 2021;109(22):3609–18 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brombacher TM, Nono JK, De Gouveia KS, Makena N, Darby M, Womersley J, et al. IL-13-Mediated Regulation of Learning and Memory. J Immunol 2017;198(7):2681–8. [DOI] [PubMed] [Google Scholar]
- 22.Li S, Olde Heuvel F, Rehman R, Aousji O, Froehlich A, Li Z, et al. Interleukin-13 and its receptor are synaptic proteins involved in plasticity and neuroprotection. Nature communications 2023;14(1):200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brynskikh A, Warren T, Zhu J, Kipnis J. Adaptive immunity affects learning behavior in mice. Brain, behavior, and immunity 2008;22(6):861–9. [DOI] [PubMed] [Google Scholar]
- 24.Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. The Journal of experimental medicine 2010;207(5):1067–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proceedings of the National Academy of Sciences of the United States of America 2004;101(21):8180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-gamma in regulating neuronal connectivity and social behaviour. Nature 2016;535(7612):425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flood L, Korol SV, Ekselius L, Birnir B, Jin Z. Interferon-gamma potentiates GABA(A) receptor-mediated inhibitory currents in rat hippocampal CA1 pyramidal neurons. Journal of neuroimmunology 2019;337:577050. [DOI] [PubMed] [Google Scholar]
- 28.Dohne N, Falck A, Janach GMS, Byvaltcev E, Strauss U. Interferon-gamma augments GABA release in the developing neocortex via nitric oxide synthase/soluble guanylate cyclase and constrains network activity. Frontiers in cellular neuroscience 2022;16:913299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang DS, Zurek AA, Lecker I, Yu J, Abramian AM, Avramescu S, et al. Memory deficits induced by inflammation are regulated by alpha5-subunit-containing GABAA receptors. Cell reports 2012;2(3):488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel RR, Khom S, Steinman MQ, Varodayan FP, Kiosses WB, Hedges DM, et al. IL-1beta expression is increased and regulates GABA transmission following chronic ethanol in mouse central amygdala. Brain, behavior, and immunity 2019;75:208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pribiag H, Stellwagen D. TNF-alpha downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013;33(40):15879–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 2005;25(12):3219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kemp GM, Altimimi HF, Nho Y, Heir R, Klyczek A, Stellwagen D. Sustained TNF signaling is required for the synaptic and anxiety-like behavioral response to acute stress. Molecular psychiatry 2022;27(11):4474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frost JL, Schafer DP. Microglia: Architects of the Developing Nervous System. Trends in cell biology 2016. [DOI] [PMC free article] [PubMed]
- 35.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011;333(6048):1456–8. [DOI] [PubMed] [Google Scholar]
- 36.Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E. Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 2012;32(43):15106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nature neuroscience 2019;22(7):1075–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nature neuroscience 2014;17(3):400–6. [DOI] [PubMed] [Google Scholar]
- 39.Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018;359(6381):1269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He D, Xu H, Zhang H, Tang R, Lan Y, Xing R, et al. Disruption of the IL-33-ST2-AKT signaling axis impairs neurodevelopment by inhibiting microglial metabolic adaptation and phagocytic function. Immunity 2022;55(1):159–73 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Fu WY, Cheung K, Hung KW, Chen C, Geng H, et al. Astrocyte-secreted IL-33 mediates homeostatic synaptic plasticity in the adult hippocampus. Proceedings of the National Academy of Sciences of the United States of America 2021;118(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen PT, Dorman LC, Pan S, Vainchtein ID, Han RT, Nakao-Inoue H, et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 2020;182(2):388–403 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheadle L, Tzeng CP, Kalish BT, Harmin DA, Rivera S, Ling E, et al. Visual Experience-Dependent Expression of Fn14 Is Required for Retinogeniculate Refinement. Neuron 2018;99(3):525–39 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheadle L, Rivera SA, Phelps JS, Ennis KA, Stevens B, Burkly LC, et al. Sensory Experience Engages Microglia to Shape Neural Connectivity through a Non-Phagocytic Mechanism. Neuron 2020;108(3):451–68 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagy D, Ennis KA, Wei R, Su SC, Hinckley CA, Gu RF, et al. Developmental synaptic regulator, TWEAK/Fn14 signaling, is a determinant of synaptic function in models of stroke and neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America 2021;118(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prieto GA, Cotman CW. Cytokines and cytokine networks target neurons to modulate long-term potentiation. Cytokine Growth Factor Rev 2017;34:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bourgognon JM, Cavanagh J. The role of cytokines in modulating learning and memory and brain plasticity. Brain Neurosci Adv 2020;4:2398212820979802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 2007;32(8–10):1106–15. [DOI] [PubMed] [Google Scholar]
- 49.Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter-Levin G, et al. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 2003;13(7):826–34. [DOI] [PubMed] [Google Scholar]
- 50.Spulber S, Mateos L, Oprica M, Cedazo-Minguez A, Bartfai T, Winblad B, et al. Impaired long term memory consolidation in transgenic mice overexpressing the human soluble form of IL-1ra in the brain. Journal of neuroimmunology 2009;208(1–2):46–53. [DOI] [PubMed] [Google Scholar]
- 51.Takemiya T, Fumizawa K, Yamagata K, Iwakura Y, Kawakami M. Brain Interleukin-1 Facilitates Learning of a Water Maze Spatial Memory Task in Young Mice. Frontiers in behavioral neuroscience 2017;11:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlos AJ, Tong L, Prieto GA, Cotman CW. IL-1beta impairs retrograde flow of BDNF signaling by attenuating endosome trafficking. Journal of neuroinflammation 2017;14(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pickering M, O’Connor JJ. Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog Brain Res 2007;163:339–54. [DOI] [PubMed] [Google Scholar]
- 54.Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neuroscience letters 1996;203(1):17–20. [DOI] [PubMed] [Google Scholar]
- 55.Bellinger FP, Madamba SG, Campbell IL, Siggins GR. Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neuroscience letters 1995;198(2):95–8. [DOI] [PubMed] [Google Scholar]
- 56.Habbas S, Santello M, Becker D, Stubbe H, Zappia G, Liaudet N, et al. Neuroinflammatory TNFalpha Impairs Memory via Astrocyte Signaling. Cell 2015;163(7):1730–41. [DOI] [PubMed] [Google Scholar]
- 57.Ragozzino D, Di Angelantonio S, Trettel F, Bertollini C, Maggi L, Gross C, et al. Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2006;26(41):10488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertollini C, Ragozzino D, Gross C, Limatola C, Eusebi F. Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacology 2006;51(4):816–21. [DOI] [PubMed] [Google Scholar]
- 59.Maggi L, Trettel F, Scianni M, Bertollini C, Eusebi F, Fredholm BB, et al. LTP impairment by fractalkine/CX3CL1 in mouse hippocampus is mediated through the activity of adenosine receptor type 3 (A3R). Journal of neuroimmunology 2009;215(1–2):36–42. [DOI] [PubMed] [Google Scholar]
- 60.Bian C, Zhao ZQ, Zhang YQ, Lu N. Involvement of CX3CL1/CX3CR1 signaling in spinal long term potentiation. PloS one 2015;10(3):e0118842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. The Journal of neuroscience : the official journal of the Society for Neuroscience 2011;31(45):16241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang M, Kamata M, Chen KN, Pariente N, An DS, Chen IS. Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J Gene Med 2010;12(3):255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hunt JS, Romanelli F. Maraviroc, a CCR5 coreceptor antagonist that blocks entry of human immunodeficiency virus type 1. Pharmacotherapy 2009;29(3):295–304. [DOI] [PubMed] [Google Scholar]
- 64.Ndhlovu LC, Umaki T, Chew GM, Chow DC, Agsalda M, Kallianpur KJ, et al. Treatment intensification with maraviroc (CCR5 antagonist) leads to declines in CD16-expressing monocytes in cART-suppressed chronic HIV-infected subjects and is associated with improvements in neurocognitive test performance: implications for HIV-associated neurocognitive disease (HAND). J Neurovirol 2014;20(6):571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou M, Greenhill S, Huang S, Silva TK, Sano Y, Wu S, et al. CCR5 is a suppressor for cortical plasticity and hippocampal learning and memory. eLife 2016;5. [DOI] [PMC free article] [PubMed]
- 66.Shen Y, Zhou M, Cai D, Filho DA, Fernandes G, Cai Y, et al. CCR5 closes the temporal window for memory linking. Nature 2022;606(7912):146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turrigiano G Homeostatic signaling: the positive side of negative feedback. Current opinion in neurobiology 2007;17(3):318–24. [DOI] [PubMed] [Google Scholar]
- 68.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science 2002;295(5563):2282–5. [DOI] [PubMed] [Google Scholar]
- 69.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature 2006;440(7087):1054–9. [DOI] [PubMed] [Google Scholar]
- 70.Cingolani LA, Thalhammer A, Yu LM, Catalano M, Ramos T, Colicos MA, et al. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron 2008;58(5):749–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barnes SJ, Franzoni E, Jacobsen RI, Erdelyi F, Szabo G, Clopath C, et al. Deprivation-Induced Homeostatic Spine Scaling In Vivo Is Localized to Dendritic Branches that Have Undergone Recent Spine Loss. Neuron 2017;96(4):871–82.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dutta R, Chang A, Doud MK, Kidd GJ, Ribaudo MV, Young EA, et al. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol 2011;69(3):445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Werneburg S, Jung J, Kunjamma RB, Ha SK, Luciano NJ, Willis CM, et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 2020;52(1):167–82 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jürgens T, Jafari M, Kreutzfeldt M, Bahn E, Brück W, Kerschensteiner M, et al. Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain 2016;139(Pt 1):39–46. [DOI] [PubMed] [Google Scholar]
- 75.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 1990;27(5):457–64. [DOI] [PubMed] [Google Scholar]
- 76.Vogelaar CF, Mandal S, Lerch S, Birkner K, Birkenstock J, Buhler U, et al. Fast direct neuronal signaling via the IL-4 receptor as therapeutic target in neuroinflammation. Sci Transl Med 2018;10(430). [DOI] [PubMed] [Google Scholar]
- 77.Yang G, Parkhurst CN, Hayes S, Gan WB. Peripheral elevation of TNF-alpha leads to early synaptic abnormalities in the mouse somatosensory cortex in experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America 2013;110(25):10306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jafari M, Schumacher AM, Snaidero N, Ullrich Gavilanes EM, Neziraj T, Kocsis-Jutka V, et al. Phagocyte-mediated synapse removal in cortical neuroinflammation is promoted by local calcium accumulation. Nat Neurosci 2021;24(3):355–67. [DOI] [PubMed] [Google Scholar]
- 79.Ellwardt E, Pramanik G, Luchtman D, Novkovic T, Jubal ER, Vogt J, et al. Maladaptive cortical hyperactivity upon recovery from experimental autoimmune encephalomyelitis. Nat Neurosci 2018;21(10):1392–403. [DOI] [PubMed] [Google Scholar]
- 80.Korostil M, Feinstein A. Anxiety disorders and their clinical correlates in multiple sclerosis patients. Mult Scler 2007;13(1):67–72. [DOI] [PubMed] [Google Scholar]
- 81.Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest 2020;130(4):1912–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roy ER, Chiu G, Li S, Propson NE, Kanchi R, Wang B, et al. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid beta plaques. Immunity 2022;55(5):879–94 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garber C, Soung A, Vollmer LL, Kanmogne M, Last A, Brown J, et al. T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nature neuroscience 2019;22(8):1276–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ren S, Breuillaud L, Yao W, Yin T, Norris KA, Zehntner SP, et al. TNF-alpha-mediated reduction in inhibitory neurotransmission precedes sporadic Alzheimer’s disease pathology in young Trem2(R47H) rats. The Journal of biological chemistry 2021;296:100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ren S, Yao W, Tambini MD, Yin T, Norris KA, D’Adamio L. Microglia TREM2(R47H) Alzheimer-linked variant enhances excitatory transmission and reduces LTP via increased TNF-alpha levels. eLife 2020;9. [DOI] [PMC free article] [PubMed]
- 86.Brigas HC, Ribeiro M, Coelho JE, Gomes R, Gomez-Murcia V, Carvalho K, et al. IL-17 triggers the onset of cognitive and synaptic deficits in early stages of Alzheimer’s disease. Cell Rep 2021;36(9):109574. [DOI] [PubMed] [Google Scholar]
- 87.Di Filippo M, Mancini A, Bellingacci L, Gaetani L, Mazzocchetti P, Zelante T, et al. Interleukin-17 affects synaptic plasticity and cognition in an experimental model of multiple sclerosis. Cell Rep 2021;37(10):110094. [DOI] [PubMed] [Google Scholar]
- 88.Ribeiro M, Brigas HC, Temido-Ferreira M, Pousinha PA, Regen T, Santa C, et al. Meningeal gammadelta T cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci Immunol 2019;4(40). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zupan B, Liu B, Taki F, Toth JG, Toth M. Maternal Brain TNF-α Programs Innate Fear in the Offspring. Curr Biol 2017;27(24):3859–63.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behavioural brain research 2009;204(2):313–21. [DOI] [PubMed] [Google Scholar]
- 91.Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. The Journal of neuroscience : the official journal of the Society for Neuroscience 2003;23(1):297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology 2003;28(10):1778–89. [DOI] [PubMed] [Google Scholar]
- 93.Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016;351(6276):933–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shin Yim Y, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, et al. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 2017;549(7673):482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature 2020;577(7789):249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal gammadelta T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol 2020;21(11):1421–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. The Journal of neuroscience : the official journal of the Society for Neuroscience 2007;27(40):10695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mirabella F, Desiato G, Mancinelli S, Fossati G, Rasile M, Morini R, et al. Prenatal interleukin 6 elevation increases glutamatergic synapse density and disrupts hippocampal connectivity in offspring. Immunity 2021;54(11):2611–31 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Balosso S, Ravizza T, Perego C, Peschon J, Campbell IL, De Simoni MG, et al. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann Neurol 2005;57(6):804–12. [DOI] [PubMed] [Google Scholar]
- 100.Weinberg MS, Blake BL, McCown TJ. Opposing actions of hippocampus TNFα receptors on limbic seizure susceptibility. Exp Neurol 2013;247:429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Balosso S, Ravizza T, Aronica E, Vezzani A. The dual role of TNF-alpha and its receptors in seizures. Exp Neurol 2013;247:267–71. [DOI] [PubMed] [Google Scholar]
- 102.Balosso S, Ravizza T, Pierucci M, Calcagno E, Invernizzi R, Di Giovanni G, et al. Molecular and functional interactions between tumor necrosis factor-alpha receptors and the glutamatergic system in the mouse hippocampus: implications for seizure susceptibility. Neuroscience 2009;161(1):293–300. [DOI] [PubMed] [Google Scholar]
- 103.Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A, et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proceedings of the National Academy of Sciences of the United States of America 2000;97(21):11534–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vezzani A, Moneta D, Richichi C, Aliprandi M, Burrows SJ, Ravizza T, et al. Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis . Epilepsia 2002;43 Suppl 5:30–5. [DOI] [PubMed] [Google Scholar]
- 105.Chiavegato A, Zurolo E, Losi G, Aronica E, Carmignoto G. The inflammatory molecules IL-1beta and HMGB1 can rapidly enhance focal seizure generation in a brain slice model of temporal lobe epilepsy. Frontiers in cellular neuroscience 2014;8:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tzeng TC, Hasegawa Y, Iguchi R, Cheung A, Caffrey DR, Thatcher EJ, et al. Inflammasome-derived cytokine IL18 suppresses amyloid-induced seizures in Alzheimer-prone mice. Proceedings of the National Academy of Sciences of the United States of America 2018;115(36):9002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Valentinova K, Tchenio A, Trusel M, Clerke JA, Lalive AL, Tzanoulinou S, et al. Morphine withdrawal recruits lateral habenula cytokine signaling to reduce synaptic excitation and sociability. Nat Neurosci 2019;22(7):1053–6. [DOI] [PubMed] [Google Scholar]
- 108.Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR. Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron 2018;100(6):1292–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, et al. TNF-α Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J Neurosci 2017;37(4):871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lü N, et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-α secretion. J Clin Invest 2014;124(3):1173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jiang J, Tang B, Wang L, Huo Q, Tan S, Misrani A, et al. Systemic LPS-induced microglial activation results in increased GABAergic tone: A mechanism of protection against neuroinflammation in the medial prefrontal cortex in mice. Brain Behav Immun 2022;99:53–69. [DOI] [PubMed] [Google Scholar]
- 112.Klawonn AM, Fritz M, Castany S, Pignatelli M, Canal C, Similä F, et al. Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity 2021;54(2):225–34.e6. [DOI] [PubMed] [Google Scholar]
- 113.McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, et al. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry 2018;23(6):1421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]