

Abstract
Expression quantitative trait loci (eQTLs) have been touted as the missing piece that can bridge the gap between genetic variants and phenotypes. Over the past decade, we have witnessed a sharp rise of effort in the identification and application of eQTLs. The successful application of eQTLs relies heavily on their reproducibility. The current eQTL databases such as Genotype-Tissue Expression (GTEx) were populated primarily with eQTLs deriving from germline single nucleotide polymorphisms and normal tissue gene expression. The novel scenarios that employ eQTL models for prediction purposes often involve disease phenotypes characterized by altered gene expressions. To evaluate eQTL reproducibility across diverse data sources and the effect of disease-specific gene expression alteration on eQTL identification, we conducted an eQTL study using 5178 samples from The Cancer Genome Atlas (TCGA). We found that the reproducibility of eQTLs between normal and tumor tissues was low in terms of the number of shared eQTLs. However, among the shared eQTLs, the effect directions were generally concordant. This suggests that the source of the gene expression (normal or tumor tissue) has a strong effect on the detectable eQTLs and the effect direction of the eQTLs. Additional analyses demonstrated good directional concordance of eQTLs between GTEx and TCGA. Furthermore, we found that multi-tissue eQTLs may exert opposite effects across multiple tissue types. In summary, our results suggest that eQTL prediction models need to carefully address tissue and disease dependency of eQTLs. Tissue–disease-specific eQTL databases can afford more accurate prediction models for future studies.
Keywords: eQTL, SNP, tissue specificity, disease specificity
Introduction
Gene expression and single nucleotide polymorphisms (SNPs) are two of the most studied genomic features. High-throughput gene expression profiling has been commonly utilized to understand the human transcriptome and its connection with disease. As of October 2017, gene expression data from 2 234 695 samples of 4348 studies had been deposited into the Gene Expression Omnibus [1]. Genotyping technology has enabled mass screening for SNPs whose allele frequencies are statistically associated with disease susceptibility. The NHGRI-EBI Catalog of published Genome-Wide Association Studies (GWAS catalog) in October 2017 [2] has curated 58 993 SNP disease associations from 2724 GWAS studies.
One of the major criticisms of GWAS studies is that, thus far, the identified SNPs have yet to generate any clinical useful utility for treatment or prognosis. It is difficult to establish biological relevance for GWAS SNPs because the majority of the SNPs do not reside in protein-coding genes. For example, in the GWAS catalog, only ∼3.6% of the 40525 unique SNPs are located in protein-coding regions while ∼96.4% lie in noncoding regions (equally proportioned between the intergenic regions and intronic regions) [3, 4]. One theory that attempts to explain GWAS SNPs’ effect on disease risk is through long-range regulation of gene expression [5], also known as expression quantitative trait loci (eQTLs).
An eQTL is defined as the regulatory association between a genomic locus (such as an SNP) and expression of a gene. eQTLs are commonly divided into two categories according to the distance between an SNP and the coupled gene. A `cis-eQTL’ denotes an eQTL where the SNP resides within the gene or the flanking regions of the gene. `Trans-eQTLs’, on the other hand, are eQTLs with the SNP lying beyond the flanking boundaries (commonly 106 nt) of the gene.
Research on eQTLs has enjoyed increasing popularity over the past few years, with the Genotype-Tissue Expression (GTEx) project spearheading the efforts. GTEx collects and analyzes multiple human tissues from donors who are also densely genotyped to assess genome-wide genetic variations and transcriptome-wide gene expression. As a result, GTEx yields a comprehensive eQTL database consisting of 19 582 739 eQTLs deriving from 44 human tissue types [6]. Because of the enormous combinatorial complexity, GTEx as well as many genome-wide eQTL studies typically focuses on cis-eQTLs rather than trans-eQTLs. To date, the GTEx eQTL resource has been incorporated as the backbone of gene expression imputation models such as PrediXcan [7], which exploit the eQTL information to impute gene expression from SNP data and thereby prioritize genes implicated in the disease etiology [7]. Nevertheless, certain aspects of GTEx design remain open for discussion. For example, GTEx purposefully restricts itself to healthy human subjects, but the applications of GTEx eQTL data frequently extend to disease scenarios. An interesting question emerges as to how consistently eQTLs can be inferred from distinct tissue types, particularly, in normal samples versus in cancer samples.
The Cancer Genome Atlas (TCGA) is a completed consortium project that collected multiple layers of omics data from hundreds to thousands of patients of various cancer types. Unlike GTEx, which recruits exclusively healthy subjects, TCGA accrued both normal and tumor tissues from cancer patients. Given the availability of both SNP and gene expression data from TCGA, we carried out a study to answer three major questions unsolvable by GTEx. First, we thoroughly investigated the recurrence of eQTLs and the concordance of shared eQTLs among diverse combinations of genotyping and expression profiling sources. Secondly, we assessed the repeatability of GTEx eQTLs in TCGA data. Thirdly, we studied the degree of eQTLs’ tissue specificity across a dozen TCGA and 44 GTEx tissue types. Besides, we also investigated how the quantity and consistency of detectable eQTLs are influenced by sample size and statistical stringency, advising on practical ways to improve robustness in future eQTL detection. Our results help clarify important questions that preclude more confident and wider applications of GTEx eQTL data.
Methods
Pre-computed eQTLs associated with 44 tissue types were downloaded from the GTEx consortium. The TCGA SNP and gene expression data of 12 cancer types [breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), head and neck squamous cell carcinoma (HNSC), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), ovarian serous cystadenocarcinoma (OV), pancreatic adenocarcinoma (PAAD), prostate adenocarcinoma (PRAD), rectum adenocarcinoma (READ), skin cutaneous melanoma (SKCM) and stomach adenocarcinoma (STAD)] were downloaded from the Genomic Data Commons. The genotyping data went through rigorous quality control as described in our previous publication [8]. Eight tissue types match between TCGA and GTEx (breast, colon, liver, lung, ovary, pancreas, prostate and stomach). TCGA sample size varies by cancer type, and not all cancer types have normal samples paired with tumor samples. Generally, every subject has at least one tumor sample, while only ∼10% subjects have normal samples. Our tumor-normal comparative analyses required strictly paired normal and tumor samples, hence being restricted to certain subsets of TCGA samples. In total, our study incorporated TCGA data of 5178 samples (tumor and normal) from 4761 cancer subjects. Of the 12 cancer types, BRCA has the most samples making it the most ideal data set for in-depth study of certain questions.
The TCGA transcriptome data were normalized in the form of Reads Per Kilobase Million (RPKM) [9], containing 20 153 genes per sample. The TCGA SNP data were generated with the Affymetrix Genome-Wide Human SNP Array 6.0 that contains 934968 SNPs. Matrix eQTL [10] was employed to compute eQTLs from TCGA data. By default, Matrix eQTL uses P < 0.01 as the eQTL output threshold and also provides the false discovery rate (FDR) for each outputted eQTL. In GTEx, all reported eQTLs were selected by FDR < 0.05. Thus, we conducted our investigations at two thresholds: P < 0.01 and FDR < 0.05. To curtail influence from outlier SNPs, we excluded SNPs with <5% minor allele frequency (MAF). Because GTEx data set contains cis-eQTL only, we focused our analysis exclusively on cis-eQTLs, scrutinizing the 106 nt upstream and downstream from the gene.
Technically, an eQTL is composed of three elements: the SNP location, the effect allele and the affected gene. An effect allele is the allele that was used during the computation of eQTL. Switching the allele within an eQTL reverts the direction of association. We took special precaution to ensure that each pair of eQTLs in comparative analyses has the same effect allele.
Somatic mutations are thought to lie at the heart of early tumorigenesis, whereas altered gene expression plays a functional role in phenotypic presentation [11]. Both somatic mutations and gene expression alterations have been extensively observed in human cancer. We hypothesized that genotype alteration and gene expression dysregulation may translate to variation of eQTLs detected in the tumor samples than in the normal samples. This hypothesis was tested with TCGA data.
In TCGA, genomic data were collected from multiple sources (DNA: blood, normal tissue and tumor tissue; RNA: normal tissue and tumor tissue). This allows a number of combinations for eQTL computation. It is expected that the genotypes could have minor differences among these three sources due to somatic mutations and noise [12]. The detectable difference between SNPs from tumor and normal tissues was limited to homozygous versus heterozygous difference, because genotyping arrays were limited to the detection of two predefined alleles. The majority of the publicly available genotyping data were generated from blood. To circumvent the noise caused by somatic mutations in tumor tissues, we required that the pair of eQTLs in comparison must have the same two alleles in the testing populations. For completeness, six types of cis-eQTLs were computed from the TCGA data:
-
(i)
eQTL1: normal tissue SNP–normal tissue gene expression
-
(ii)
eQTL2: normal tissue SNP–tumor tissue gene expression
-
(iii)
eQTL3: tumor tissue SNP–tumor tissue gene expression
-
(vi)
eQTL4: germline blood SNP–normal tissue gene expression
-
(v)
eQTL5: germline blood SNP–tumor tissue gene expression
-
(vi)
eQTL6: tumor tissue SNP–normal tissue gene expression.
We used various combinations of these six types of eQTLs throughput the analyses depending on the goal and sample size requirement. eQTL6 is a scenario that is highly unlikely to happen in practical studies. Thus, it was only used in the tumor versus normal comparison for proof-of-concept purpose. During our comparative analysis of shared eQTLs between two data sets, we consider the two eQTLs to be consistent if the effects (beta) have same direction; otherwise, we consider the two eQTLs to be in conflict. The overlap percentage between any two eQTL types is defined as the number of eQTL detected by both eQTL types divided by the smaller set of eQTL detected by the these two types of eQTLs.
Results
Number of eQTL detected
Matrix eQTL identified tens to hundreds of thousands of eQTLs within each cancer type in TCGA (Table 1). The sample size was clearly positively correlated to the number of eQTL detected (Figure S1). Using P < 0.01 as the detection threshold, a Spearman correlation of 0.78 was observed between the number of detected eQTLs and the sample size; when using FDR < 0.05 as the threshold, a Spearman correlation of 0.89 was observed. No leveling-off effect can be observed for the number of eQTL detected. The total possible SNP-gene pairs in TCGA data is around 18.8 billion, which indicates that further increasing the sample size will likely continue to increase the number of eQTLs detected. To reach saturation of detectable eQTL, substantial larger data sets are required.
Table 1 .
The number of eQTLs identified
| Cancer | SNP source | RNA source | Sample size | P < 0.01 | FDR < 0.05 |
|---|---|---|---|---|---|
| BRCA | Blood | Normal tissue | 51 | 83 601 | 1206 |
| Normal tissue | Normal tissue | 92 | 95 274 | 3832 | |
| Normal tissue | Tumor tissue | 135 | 126 921 | 2361 | |
| Blood | Tumor tissue | 976 | 492 732 | 210 861 | |
| Tumor tissue | Tumor tissue | 1093 | 286 117 | 123 880 | |
| COAD | Blood | Normal tissue | 24 | 76 541 | 206 |
| Normal tissue | Normal tissue | 39 | 83 472 | 774 | |
| Normal tissue | Tumor tissue | 50 | 76 612 | 286 | |
| Blood | Tumor tissue | 248 | 149 898 | 9245 | |
| Tumor tissue | Tumor tissue | 285 | 156 076 | 14 614 | |
| HNSC | Blood | Normal tissue | 42 | 73 725 | 123 |
| Normal tissue | Normal tissue | 38 | 74 781 | 152 | |
| Normal tissue | Tumor tissue | 74 | 87 819 | 575 | |
| Blood | Tumor tissue | 485 | 135 550 | 14 261 | |
| Tumor tissue | Tumor tissue | 518 | 133 927 | 17 046 | |
| LIHC | Normal tissue | Normal tissue | 48 | 93 119 | 1305 |
| Normal tissue | Tumor tissue | 80 | 81 787 | 448 | |
| Blood | Tumor tissue | 304 | 157 571 | 8219 | |
| Tumor tissue | Tumor tissue | 369 | 185 761 | 24 751 | |
| LUAD | Blood | Normal tissue | 20 | 75 792 | 0 |
| Normal tissue | Normal tissue | 57 | 89 783 | 2811 | |
| Normal tissue | Tumor tissue | 175 | 118 930 | 2718 | |
| Blood | Tumor tissue | 398 | 127 974 | 11 097 | |
| Tumor tissue | Tumor tissue | 514 | 154 612 | 18 236 | |
| LUSC | Blood | Normal tissue | 29 | 80 480 | 525 |
| Normal tissue | Normal tissue | 50 | 88 379 | 1974 | |
| Normal tissue | Tumor tissue | 236 | 104 463 | 4593 | |
| Blood | Tumor tissue | 296 | 107 886 | 5737 | |
| Tumor tissue | Tumor tissue | 500 | 121 220 | 13 573 | |
| OV | Normal tissue | Tumor tissue | 59 | 83 720 | 298 |
| Blood | Tumor tissue | 235 | 124 805 | 4538 | |
| Tumor tissue | Tumor tissue | 301 | 122 106 | 26 408 | |
| PAAD | Normal tissue | Tumor tissue | 30 | 73 608 | 5 |
| Blood | Tumor tissue | 147 | 103 957 | 4397 | |
| Tumor tissue | Tumor tissue | 178 | 112 951 | 5406 | |
| PRAD | Blood | Normal tissue | 43 | 88 715 | 601 |
| Normal tissue | Normal tissue | 50 | 90 019 | 850 | |
| Normal tissue | Tumor tissue | 113 | 111 779 | 3267 | |
| Blood | Tumor tissue | 422 | 163 626 | 28 547 | |
| Tumor tissue | Tumor tissue | 494 | 189 650 | 38 982 | |
| READ | Normal tissue | Normal tissue | 10 | 52 087 | 0 |
| Blood | Tumor tissue | 86 | 87 203 | 1043 | |
| Tumor tissue | Tumor tissue | 94 | 99 735 | 1969 | |
| SKCM | Blood | Tumor tissue | 103 | 80 831 | 948 |
| Tumor tissue | Tumor tissue | 103 | 94 434 | 1636 | |
| STAD | Blood | Normal tissue | 23 | 128 906 | 22 |
| Normal tissue | Normal tissue | 33 | 206 675 | 188 | |
| Normal tissue | Tumor tissue | 86 | 114 554 | 358 | |
| Blood | Tumor tissue | 348 | 143 044 | 8428 | |
| Tumor tissue | Tumor tissue | 415 | 139 381 | 10 758 |
Comparative analysis: tumor versus normal
Across the 12 TCGA cancers, we identified ∼3% genotype difference between germline blood and tumor samples [8]. This hypothesis was tested by comparing the quantity and effect directions of distinct eQTL sets: eQTL1, eQTL2, eQTL3 and eQTL6 (see Methods for definitions). eQTL4 and eQTL5 were not used in the comparative analysis due to limited number of paired samples between germline blood SNP and normal tissue gene expression. Thresholds of both P < 0.01 and FDR < 0.05 were adopted for deriving finite eQTL sets.
The eQTL comparison results for paired tumor and normal tissues in eight types of cancers in TCGA were summarized in Figure 1 and Table S1. At P < 0.01 (Figure 1A), one observation that immediately stood out was that even though similar numbers of eQTLs were identified for all four definitions of eQTL using exactly the same samples, the overlap between them had a wide range depending on the source of SNP and gene expression used. Across the eight cancer types, between eQTL1 (normal tissue SNP—normal tissue gene expression) and eQTL2 (normal tissue SNP—tumor tissue gene expression), the average overlap is 4.54% (range: 2.47–7.39%); between eQTL1 and eQTL3 (tumor tissue SNP—tumor tissue gene expression), the average overlap is 4.26% (range: 2.34–6.97%); between eQTL2 and eQTL3, the average overlap is 66.90% (range: 58.68–79.74%); and between eQTL1 and eQTL6 (tumor tissue SNP—normal tissue gene expression), the average overlap is 72.63% (range: 64.33–82.21%). Clearly, the gene expression difference between the paired tumor and normal tissues played a larger role in the observed eQTLs than the SNP differences did. When the source of the gene expression differs, regardless of the source of the SNPs, the overlap between the two sets of eQTLs remained low. When the source of the gene expression was fixed, the overlap between the two sets of eQTLs was high, regardless of the sources of SNPs. The difference between eQTL2 versus eQTL3 and eQTL1 versus eQTL6 should be primarily contributed by the differences of genotypes between normal and tumor tissues. When using a more stringent threshold of FDR < 0.05 (Figure 1B), the proportion of overlap increased substantially (Table S1), except for certain cancer types with smaller sample size that identified no eQTLs with FDR < 0.05.
Figure 1 .
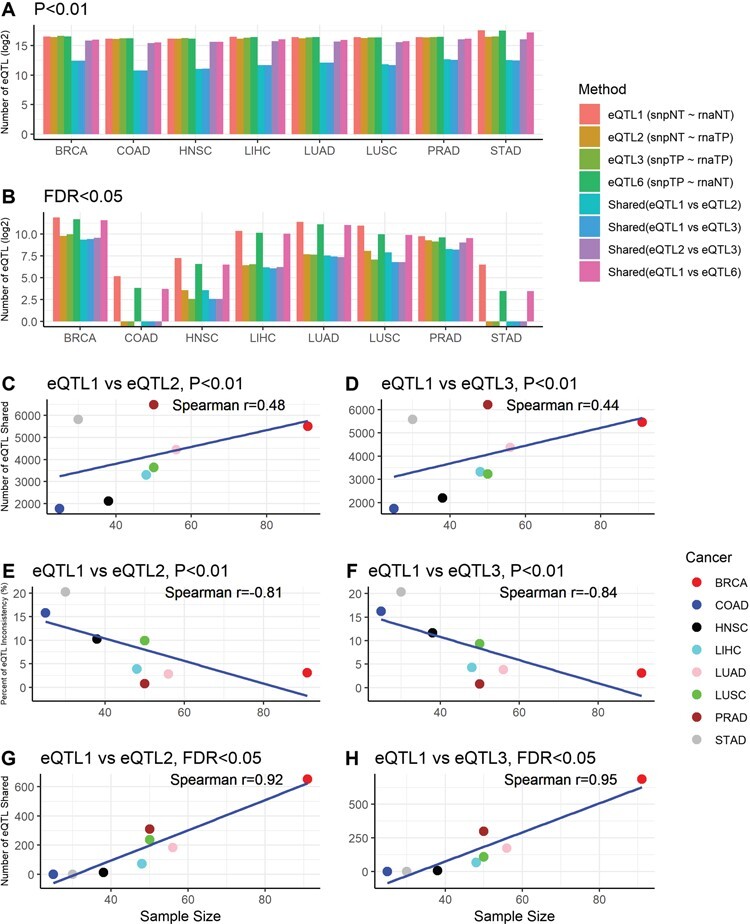
Results from eQTL analysis using paired normal and tumor samples from TCGA. A. Barplot that denotes the eQTLs detected in TCGA using paired normal and tumor samples using threshold P < 0.01. B. Barplot that denotes the eQTLs detected in TCGA using paired normal and tumor samples using threshold FDR < 0.05. The bar plots show that after applying a more stringent detection threshold, the proportion of shared eQTLs between normal and tumor tissues increased substantially. C and D. Scatter plots show positive Spearman correlation coefficients between sample size and number of shared eQTLs between eQTL pairs when using P < 0.01 as the eQTL detection threshold. E and F. Scatter plots show negative Spearman correlation coefficients between sample size and inconsistency rate between eQTL pairs when using P < 0.05 as the eQTL detection threshold. G and H. Scatter plots show positive Spearman correlation coefficients between sample size and number of shared eQTLs between eQTL pairs when using FDR < 0.01 as the eQTL detection threshold. The correlation became more significant after adopting a more stringent eQTL detection threshold. No scatter plots for sample size versus inconsistency rate under FDR < 005 were produced because all of the inconsistency rates were zeros.
Furthermore, we computed the inconsistency rate of eQTLs among the shared eQTLs (Table S1). The inconsistency between two identical eQTLs was defined by the inconsistency of their effect directions, not affected by the differences in the effect magnitude. Across the eight TCGA cancer types, when using P < 0.01, between eQTL1 and eQTL2, the average inconsistency rate was 8.53% (range: 0.75–20.2%); between eQTL1 and eQTL3, the average inconsistency is 8.71% (range: 0.79–20.37%); between eQTL2 and eQTL3, the average inconsistency is virtually zero for eQTL2 versus eQTL3 and eQTL1 versus eQTL6 across eight cancers types. Again, the source of gene expression played a more substantial role in eQTL inconsistency rate than source of the SNPs did. When the sources of the gene expression were the same, there was little to no inconsistency among the shared eQTLs. By using the stringent threshold of FDR < 0.05, we can virtually eliminate all of the inconsistent eQTLs. Although the number of data points were limited to eight, we were still able to observe positive correlations between the sample size and shared proportion of eQTLs, and negative correlations between sample size and the inconsistency rate (Figure 1C–H).
To complement the analyses resulting from only two distinct statistical thresholds, we investigated eQTL recurrence and concordance at five incremental P-value thresholds (0.01∼10−6). For all eight cancer types, the shared portion increased and the discordance decreased as more stringent P-values threshold were adopted (Figure S2). This informs that by imposing a sufficiently high statistical cut-off, housekeeping eQTLs may be detected even though gene expression was from different sources. Using the largest cohort in TCGA (BRCA), we conducted an eQTL reproducibility test by dividing the data set into five incrementing data sets. The smallest data set contained 200 subjects; the next data set was constructed by adding 200 subjects to the previous data set without altering the previous data set. This was to ensure that the smaller data set was always a subset of a larger data set. eQTL5 was selected for this analysis due to its large sample size and the likeliness that it mimics the potential future application setting of eQTLs. The number of detected eQTLs increased as the sample size increased and the percentage of shared eQTLs increased also as the number of the shared samples increased between any two sub-data sets in BRCA (Table 2). There were no inconsistent eQTLs between any pair of subsets.
Table 2 .
eQTL reproducibility test using BRCA subsets
| Set 1 sample size | Set 2 sample size | Set 1 eQTL | Set 2 eQTL | Set 1 versus set 2 overlap | Inconsistency rate |
|---|---|---|---|---|---|
| C200 | C400 | 3899 | 16 614 | 3350 | 0.0% |
| C200 | C600 | 3899 | 60 063 | 3460 | 0.0% |
| C200 | C800 | 3899 | 89 187 | 3445 | 0.0% |
| C400 | C600 | 16 614 | 60 063 | 15 002 | 0.0% |
| C400 | C800 | 16 614 | 89 187 | 14 655 | 0.0% |
| C600 | C800 | 60 063 | 89 187 | 47 673 | 0.0% |
Furthermore, we attempted to find the causes behind the eQTL difference between tumor and normal tissues. We hypothesized that eQTLs that are unique to tumor or normal tissues may be enriched in differentially expressed genes. We tested this by conducting an enrichment analysis on eQTL’s distribution. Using BRCA, the cohort with largest sample size as example, we found no significant enrichment of unique eQTLs in differentially expressed genes by Fisher’s exact tests (Figure S3, normal unique eQTL P = 0.28; tumor unique eQTL P = 0.12). eQTLs are computed based on linear relationship between SNP and gene expression. When tumor dysregulates gene expression, the alteration of the expression may happen in both directions, which might not be entirely reflected by differential expression analysis. However, such alterations can substantially affect the correlation between SNP and gene expression, thus resulting difference in eQTL detected.
Comparative analysis: TCGA versus GTEx
Except the impractical eQTL6, eQTLs 1–5 defined in TCGA were compared to GTEx in the eight matched tissue types. The complete results were summarized in Table S2. The numbers of shared eQTL were low. This may be partially due to the difference in SNP sets. When using P < 0.01, the average directional inconsistency rate between TCGA and GTEx in the shared eQTLs was 1.87% (range: 0.19–6.99%); when using the stringent threshold of FDR < 0.05, the average inconsistency rate dropped to 0.65% (range: 0.00–2.28%). These results suggest that reproducibility of eQTLs is high between independent data sets. Some of the inconsistencies were contributed by errors from genotyping or sequencing. The effect of these errors can be abated by imposing a stricter P-value threshold, which can result in higher reproducibility.
The inconsistency rate was compared (t-test) among tissue types, eQTL types and between the sources of RNA for the eQTLs with FDR < 0.05 (Figure 2). The type of cancer did not make significant difference in the consistency comparison between TCGA and GTEx (Figure 2A; Table S3). We found that inconsistency rates for eQTL1 and eQTL4 (Figure 2B), two types characterized with normal RNA source, were significantly lower than other types of eQTLs (eQTL1 P = 0.0027, eQTL4 P < 0.0001) (Figure 2C). The smaller inconsistency rate can be explained by the fact that eQTL1, eQTL4 and GTEx eQTLs are all derived from normal samples. Another related observation was that eQTL sets based on tumor RNA source (eQTL2, eQTL3 and eQTL5) had a greater inconsistency rate compared to the rest eQTL types based on normal RNA source (eQTL1 and eQTL4) (P < 0.0001). To further illustrate the confliction, we selected four example eQTLs that have strong (P < 10−8) but opposite effects between GTEx and TCGA (Figure S4). These four examples clearly demonstrated the existence of conflicting findings from independent data sets. Fortunately, such paradoxical cases account for only a minor portion of the shared eQTLs between TCGA and GTEx.
Figure 2 .
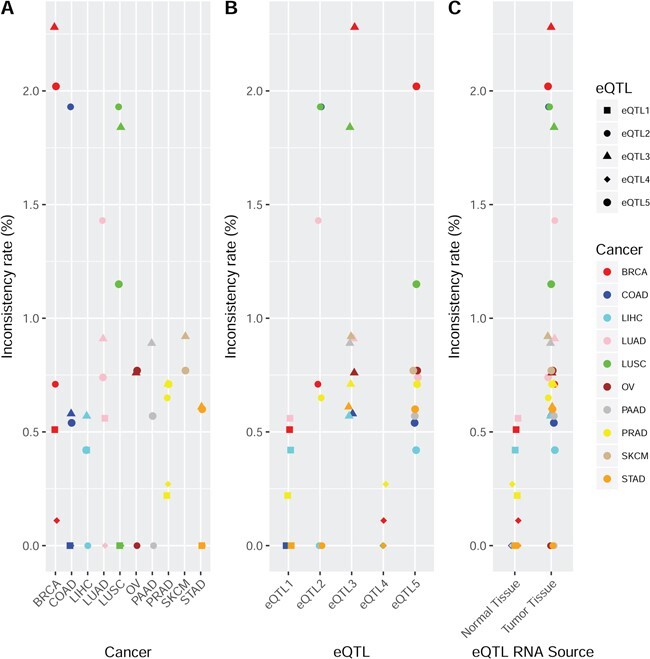
This figure presents the directional inconsistency rates among the shared eQTLs for TCGA versus GTEx. The detection threshold for eQTL in TCGA was FDR < 0.05. A. Directional inconsistency rate by cancer type in TCGA versus GTEx. No significantly different directional inconsistency rate was observed among tissue types. B. Directional inconsistency rate by eQTL type in TCGA versus GTEx. eQTL1 and eQTL4 were found to have significantly less directional inconsistency compared to other eQTL types. eQTL3 was found to have a significantly higher directional inconsistency rate compared to other eQTL types. These results reflect the fact that when the source of gene expression is the same as GTEx (normal tissue), the eQTLs are more likely to be concordant. When the source of gene expression was dramatically different (normal versus tumor tissues), the eQTLs are more likely to be in directional conflict. C. Directional inconsistency rate by source of the gene expression in TCGA versus GTEx. eQTLs produced from tumor tissues clearly had higher inconsistency rates than eQTLs produced from normal tissues compared to GTEx. This reiterates the point that the source of gene expression has more impact than the source of SNP for eQTLs.
Tissue specificity and inter-tissue concordance
It is commonly assumed that a portion of the eQTLs is tissue specific, while some might be more ubiquitous. To focus on the ubiquitous portion of the eQTLs, we performed multi-tissue analyses at significant thresholds using both P < 0.01 and FDR < 0.05 for TCGA data (Table 3). In GTEx, 18 593 eQTLs were observed in all 44 tissue types at FDR < 0.05. In TCGA, eQTL3 and eQTL5 were selected for this analysis due to their large sample size. When using P < 0.01, we found that 356 and 1098 eQTLs were presented in the 12 cancer types downloaded for eQTL3 and eQTL5, respectively; when using FDR < 0.05, we found that 79 and 204 eQTLs were presented in the 12 cancer types downloaded for eQTL3 and eQTL5, respectively. The majority of the eQTLs are tissue specific for both GTEx and TCGA. The overall distribution of multi-tissue eQTL can be observed in Figure 3A. Another trend we observed with this data is that the number of shared multi-tissue eQTLs is negatively associated with the number of tissues (Figure 3B).
Table 3 .
Shared multi-tissue eQTLs between GTEx and TCGA
| Number of tissues | Multi-tissue eQTLs in GTEx and TCGA | Shared multi-tissue eQTLs | ||||||
|---|---|---|---|---|---|---|---|---|
| GTEx | TCGA | GTEx eQTLs | TCGA eQTL3 | TCGA eQTL5 | GTEx eQTL3 | GTEx eQTL5 | eQTL3 eQTL5 | GTEx eQTL3 eQTL5 |
| 4 | 2 | 1 218 661 | 36 693 | 23 354 | 14 581 | 13 931 | 18 408 | 11 924 |
| 8 | 3 | 673 314 | 18 081 | 11 982 | 8128 | 7400 | 10 180 | 6679 |
| 12 | 4 | 449 072 | 10 884 | 7897 | 5242 | 4904 | 6767 | 4418 |
| 16 | 5 | 315 284 | 7189 | 5703 | 3605 | 3566 | 4824 | 3134 |
| 20 | 6 | 233 277 | 4864 | 4183 | 2521 | 2605 | 3339 | 2133 |
| 24 | 7 | 174 470 | 3234 | 3082 | 1675 | 1918 | 2298 | 1422 |
| 28 | 8 | 127 329 | 2003 | 2320 | 1057 | 1438 | 1479 | 887 |
| 32 | 9 | 91 346 | 1224 | 1798 | 550 | 1032 | 919 | 474 |
| 36 | 10 | 67 229 | 683 | 1015 | 264 | 502 | 478 | 218 |
| 40 | 11 | 45 594 | 289 | 599 | 88 | 340 | 193 | 80 |
| 44 | 12 | 18 593 | 79 | 204 | 19 | 63 | 60 | 19 |
Figure 3 .
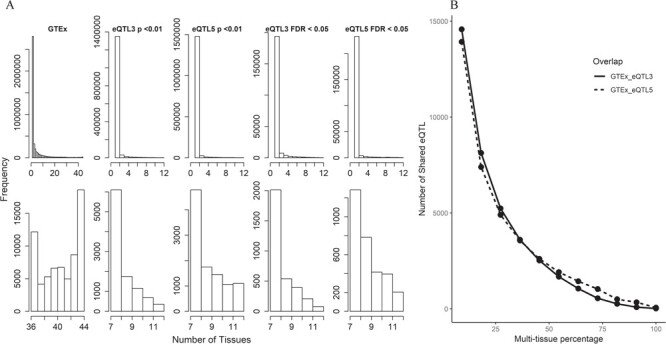
Analysis results for multi-tissue eQTLs in TCGA and GTEx. eQTL3 and eQTL5 from BRCA in TCGA were selected for this analysis due to their large sample size. Two eQTL detection thresholds were used (P < 0.01 and FDR < 0.05). A. Histograms display the distribution of multi-tissue eQTLs. The top panel of the histograms shows that the majority of the eQTLs are tissue specific or were associated in a low number of tissues. The lower panel of the histograms magnifies the right end tails of distributions in the upper panel. A smoother declining tails can be observed in TCGA data than in GTEx. This might be due to the fact that TCGA has much smaller number of tissue types. The GTEx also had smooth declining patterns until the right end of the tails. B. This figure depicts the negative relationship between shared multi-tissue eQTLs and percentage of all available tissue types. The analysis contains 44 tissue types from GTEx and 12 cancer types from TCGA. We roughly split the number of tissue and cancer types by ∼8–9% interval and computed the shared multi-tissue eQTLs.
Next we scrutinized the directional inconsistency for the multi-tissue eQTLs among tissue types (Figure 4A–F). The inconsistency rate was measured as the number of inconsistent tissue divided by the number of tissues that have found this eQTL. For example, if an eQTL was identified in 10 tissue types with eight positive effects and two negative effects, then the inconsistency rate would be 20%. We limited the minimum number of tissues required to define a multi-tissue eQTL to four. The results showed that multi-tissue eQTLs that are tissue specific tend to have a higher inconsistency rate, while eQTLs that are tissue independent tend to have a lower inconsistency rate. This observation is evidence that tissue-specific eQTLs are more likely to influence gene expression more specifically toward that tissue type, while the tissue-independent eQTLs exert similar directional effect on all tissue types.
Figure 4 .
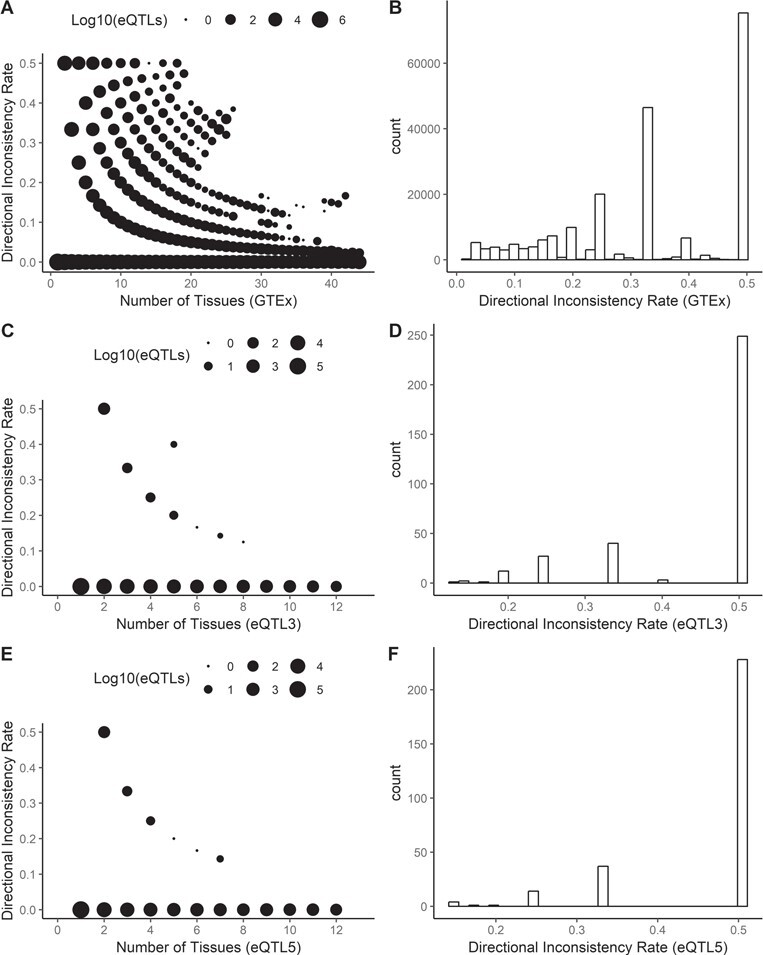
A multi-tissue eQTL may exert discordant effects across tissues. The inter-tissue discordance was measured as the number of tissues with the minority effect divided by the number of tissues that have captured this eQTL. For example, if an eQTL was identified in 10 tissue types with eight positive effects and two negative effects, the inter-tissue discordance would be 2/10 = 20%. A. C. E. Negative relationships between the number in multi-tissue and the inconsistency rate can be observed. This suggests that tissue-independent eQTLs are likely to have the same effect on all tissue types, and tissue-specific leaning eQTLs are more likely to exert a contrary effect based on tissue type. The number of dots drawn on the figure issignificantly higher than the number of dots visible due to overlapping of data points. B. D. F. These histograms depict the inconsistency rates for multi-tissue eQTLs. Multi-tissue eQTLs with zero inconsistency rates were not plotted.
Furthermore, we compared the beta distributions between TCGA and GTEx (Figure S5), attempting to determine if there is any directional bias of eQTLs. No substantial directional bias was detected. We observed 19 highly ubiquitous multi-tissue eQTLs that were found in all 44 tissue types in GTEx and all 12 cancer types in TCGA (Table S4).
Discussion
The concept of eQTL was first introduced and tested in yeast in 2002 [13]. Accredited to the maturity of high-throughput sequencing technology, the past few years have seen a large effort in the curation and utilization of eQTLs in humans. The GTEx consortium project undoubtedly became the best-known resource for multi-tissue human eQTL resources. The application of eQTLs to predict gene expression in additional diseases rests heavily on the reproducibility of eQTLs. Further, potential limitations of the GTEx data include the use of postmortem tissue and the fact that all tissues in GTEx were considered normal. The magnitude of gene expression change in a diseased tissue is often ignored in the eQTL prediction model.
To survey the reproducibility between data sets of the same tissue type and between normal and diseased tissues, we carried out a thorough eQTL study using TCGA SNP and gene expression data. The comparisons between TCGA and GTEx were constrained by differences of SNP data collected. To make them comparable, we only examined the common portion of the eQTLs when comparing eQTL directional consistency between the two data sets. We found that sample size and MAF play the most pivotal roles in the power of eQTL detection [6]. Although TCGA contains large numbers of subjects, portion of our analyses was still limited by sample size due to requirement of pairing normal and tumor samples. Our analyses using TCGA data further validated the positive influence of sample size on eQTL detection. Moreover, the results suggest that increasing sample size or detection threshold likely leads to higher detection rate and more robust eQTLs.
The comparative analyses of eQTLs between paired normal and tumor in TCGA provided several important clues regarding eQTL robustness. The number and effect direction of eQTLs were much more sensitive to gene expression alteration than to genotype changes. Considering that the analyses were performed using the exact same set of SNPs and genes, the variation in the eQTLs can be almost entirely attributed to the difference in the source material (blood, normal tissue and tumor tissue). The shared proportion between normal and tumor eQTLs was low (∼5%), which suggests that eQTL sets inferred from tumor transcriptome are largely distinct from those inferred from normal transcriptome. For three cancer types (COAD, HNSC and STAD), the directional inconsistency was greater than 10% when P < 0.01 was used as eQTL detection threshold. These results cast some doubts on whether eQTLs computed from normal transcriptomes can be used to accurately predict gene expression in diseased tissues. The minor portion of shared eQTLs may concern genes less relevant to the studied phenotypes. Cancers are extreme, abnormal phenotypes that are subjected to more severe gene expression dysregulation. Diseases with etiology unrelated or marginally related with expression dysregulation may be more suitable application cases for eQTL models defined from normal tissue data. The effect direction discordance of eQTLs attenuated after applying FDR < 0.05. However, using more stringent threshold led to a drastic decline in the number of detected eQTLs, in some cases (e.g. COAD and STAD) resulting in null output, which also limits the application of eQTL models. In our study, the consistent eQTLs may be the result of genes not affected by cancer or genes whose expression scaled proportionally in the tumor tissues.
Current mainstream eQTL projects still have inferior sample size compared to traditional GWAS studies. One commonly proposed approach to increase power is to combine tissue types. Our multi-tissue analyses in GTEx and TCGA confirm that the majority of the eQTLs are tissue specific. The power of pooled-tissue eQTL analysis can be nullified by the multi-tissue eQTLs with contrary effects depending on tissue type. Pooled-tissue eQTL analysis may increase power only if directionally consistent effects were observed in all of the proposed tissue types independently.
Our analysis was limited by the paired sample size in TCGA and by the difference in the SNP sets between TCGA and GTEx. Furthermore, there have been arguments that cancer eQTL analysis needs to adjust for other possible confounding factors such as somatic copy number variation or methylation, etc. [14]. eQTLs are defined as `genomic regions that carry one or more DNA sequence variants that influence the expression level (typically mRNA abundance) of a given gene’ [15–17]. According to the definition, variation in expression levels of mRNAs is the final expected consequence, regardless the intermediate effects of other factors such as epigenetic variations. It is very common to only adjust factors that influence global gene expression of a sample, such as population structure, age, etc. but did not adjust epigenetic variations or other somatic alterations for each individual gene [18–27]. Many studies with both gene expression and DNA methylation data did not adjust methylation for eQTL identification [28–31]. Several studies using TCGA data to identify eQTLs did not adjust methylation nor somatic copy number alterations [32–35]. The latest publication of TCGA pan-cancer eQTL database [36] in 2018 also did not adjust for either copy number or methylation. Therefore, we used a perfectly accepted approach to identify eQTLs across different cancer types without adjusting methylation and somatic copy number alterations. Previous studies demonstrated the complicated mechanisms for regulating gene expression by eQTLs, including altering RNA sequence, RNA structure, transcription factor binding, miRNA binding, methylation and histone modification [17, 37]. However, this is beyond the scope for identification of eQTLs.
Based on the trends summarized from all of the presented analyses, we are confident to conclude that increasing sample size should increase the shared portion and reduce the directional inconsistency rate for eQTLs derived from distinct RNA sources or compiled from different projects (e.g. TCGA versus GTEx). Highly significant eQTLs were reproducible between normal and tumor tissues or across data sources, although they accounted for a small portion of detected eQTLs. Our results point out that it is challenging to predict the entire transcriptome of diseased phenotype with eQTL prediction model based purely on normal tissue. To correctly harvest the full potential of eQTLs, disease-specific eQTL databases should be assembled to provide more accurate prediction for future eQTL studies.
Supplementary Material
Quanhu Sheng is a research assistant professor at the Department of Biostatistics at Vanderbilt University. He is mostly interested in algorithm development, data analysis and software implementation in proteomics, glycomics and metabolomics.
David C. Samuels is an associate professor at the Department of Molecular Physiology and Biophysics at Vanderbilt University. His research interests include mitochondria, population genetics and computational model.
Hui Yu is a research fellow at the University of New Mexico, Comprehensive Cancer Center, her research areas include cancer genomics, genetics and computational methodology.
Scott Ness is a Professor of Cancer Genomics and Director of the genomics and bioinformatics shared resource. Dr. Ness focuses on translational genetics and molecular medicine.
Ying-yong Zhao is a professor at the Northwest University China. His research is focused on genomics and genetics of chronic kidney disease.
Yan Guo is an associate professor in the Department of Internal Medicine, University of New Mexico. He is also the director of Bioinformatics Shared Resources of the University of New Mexico. Comprehensive Cancer Center.
Contributor Information
Quanhu Sheng, Department of Biostatistics, Vanderbilt University Medical Center, Nashville, TN, USA.
David C Samuels, Vanderbilt Genetics Institute, Dept. of Molecular Physiology and Biophysics, Vanderbilt University Medical School, Nashville, TN, USA.
Hui Yu, Department of Internal Medicine, University of New Mexico, Albuquerque, NM, USA.
Scott Ness, Department of Internal Medicine, University of New Mexico, Albuquerque, NM, USA.
Ying-yong Zhao, Key Laboratory of Resource Biology and Biotechnology in Western China, School of Life Sciences, Northwest University, Xi’an, Shaanxi, China.
Yan Guo, Department of Internal Medicine, University of New Mexico, Albuquerque, NM, USA.
Key Points
eQTLs are not only tissue specific, they are also disease specific.
Expression dysregulation can substantially affect the number and direction of eQTLs.
Multi-tissue eQTLs may exert inconsistent directional effect dependent on tissue type.
Funding
National Cancer Institute, (grant/award no: ‘P30CA118100’).
References
- 1. Barrett T, Edgar R. Mining microarray data at NCBI's Gene Expression Omnibus (GEO)*. Methods Mol Biol 2006;338:175–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009;106:9362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Freedman ML, Monteiro AN, Gayther SA, et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet 2011;43:513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blattler A, Yao L, Witt H, et al. Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol 2014;15:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen JQ, Tian WD. Explaining the disease phenotype of intergenic SNP through predicted long range regulation. Nucleic Acids Res 2016;44:8641–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lonsdale J, Thomas J, Salvatore M, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gamazon ER, Wheeler HE, Shah KP, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 2015;47:1091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo M, Yue W, Samuels DC, et al. Quality and concordance of genotyping array data of 12,064 samples from 5840 cancer patients. Genomics 2018:10.1016/j.ygeno.2018.06.001. [DOI] [PMC free article] [PubMed]
- 9. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012;28:1353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sager R. Expression genetics in cancer: shifting the focus from DNA to RNA. Proc Natl Acad Sci USA 1997;94:952–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo Y, Zhao SL, Sheng QH, et al. The discrepancy among single nucleotide variants detected by DNA and RNA high throughput sequencing data. BMC Genomics 2017;18(Suppl 6):690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brem RB, Yvert G, Clinton R, et al. Genetic dissection of transcriptional regulation in budding yeast. Science 2002;296:752–5. [DOI] [PubMed] [Google Scholar]
- 14. Li QY, Seo JH, Stranger B, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 2013;152:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rockman MV, Kruglyak L. Genetics of global gene expression. Nat Rev Genet 2006;7:862–72. [DOI] [PubMed] [Google Scholar]
- 16. Clyde D. Disease genomics: transitioning from association to causation with eQTLs. Nat Rev Genet 2017;18:271. [DOI] [PubMed] [Google Scholar]
- 17. Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet 2015;16:197–212. [DOI] [PubMed] [Google Scholar]
- 18. GTEx Consortium . Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang W, Gamazon ER, Zhang X, et al. SCAN database: facilitating integrative analyses of cytosine modification and expression QTL. Database (Oxford) 2015;2015: 10.1093/database/bav025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xia K, Shabalin AA, Huang S, et al. seeQTL: a searchable database for human eQTLs. Bioinformatics 2012;28:451–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang L, Morar N, Dixon AL, et al. A cross-platform analysis of 14,177 expression quantitative trait loci derived from lymphoblastoid cell lines. Genome Res 2013;23:716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu CH, Pal LR, Moult J. Consensus genome-wide expression quantitative trait loci and their relationship with human complex trait disease. OMICS 2016;20:400–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ongen H, Andersen CL, Bramsen JB, et al. Putative cis-regulatory drivers in colorectal cancer. Nature 2014;512:87–90. [DOI] [PubMed] [Google Scholar]
- 24. Brynedal B, Choi J, Raj T, et al. Large-scale trans-eQTLs affect hundreds of transcripts and mediate patterns of transcriptional co-regulation. Am J Hum Genet 2017;100:581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bryois J, Buil A, Evans DM, et al. Cis and trans effects of human genomic variants on gene expression. PLoS Genet 2014;10:e1004461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yao C, Joehanes R, Johnson AD, et al. Dynamic role of trans regulation of gene expression in relation to complex traits. Am J Hum Genet 2017;100:985–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stranger BE, Montgomery SB, Dimas AS, et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet 2012;8:e1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife 2013;2:e00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bell JT, Pai AA, Pickrell JK, et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol 2011;12:R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bonder MJ, Luijk R, Zhernakova DV, et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat Genet 2017;49:131–8. [DOI] [PubMed] [Google Scholar]
- 31. Wagner JR, Busche S, Ge B, et al. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol 2014;15:R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen QR, Hu Y, Yan C, et al. Systematic genetic analysis identifies Cis-eQTL target genes associated with glioblastoma patient survival. PLoS One 2014;9:e105393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whitington T, Gao P, Song W, et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat Genet 2016;48:387–97. [DOI] [PubMed] [Google Scholar]
- 34. Xie K, Liang C, Li Q, et al. Role of ATG10 expression quantitative trait loci in non-small cell lung cancer survival. Int J Cancer 2016;139:1564–73. [DOI] [PubMed] [Google Scholar]
- 35. Loo LWM, Lemire M, Le Marchand L. In silico pathway analysis and tissue specific cis-eQTL for colorectal cancer GWAS risk variants. BMC Genomics 2017;18:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gong J, Mei SF, Liu CJ, et al. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res 2018;46:D971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shastry BS. SNPs: impact on gene function and phenotype. Methods Mol Biol 2009;578:3–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.