

Abstract
Precision medicine uses genetic, environmental and lifestyle factors to more accurately diagnose and treat disease in specific groups of patients, and it is considered one of the most promising medical efforts of our time. The use of genetics is arguably the most data-rich and complex components of precision medicine. The grand challenge today is the successful assimilation of genetics into precision medicine that translates across different ancestries, diverse diseases and other distinct populations, which will require clever use of artificial intelligence (AI) and machine learning (ML) methods. Our goal here was to review and compare scientific objectives, methodologies, datasets, data sources, ethics and gaps of AI/ML approaches used in genomics and precision medicine. We selected high-quality literature published within the last 5 years that were indexed and available through PubMed Central. Our scope was narrowed to articles that reported application of AI/ML algorithms for statistical and predictive analyses using whole genome and/or whole exome sequencing for gene variants, and RNA-seq and microarrays for gene expression. We did not limit our search to specific diseases or data sources. Based on the scope of our review and comparative analysis criteria, we identified 32 different AI/ML approaches applied in variable genomics studies and report widely adapted AI/ML algorithms for predictive diagnostics across several diseases.
Keywords: artificial intelligence, machine learning, gene expression, gene variant, predictive analysis
Introduction
Genetic studies can reveal biomarkers that diagnose, determine risk and predict treatment outcomes for a wide variety of diseases [1]. Most genetic research investigates biological insights, disease mechanisms and disease risks by comparing healthy and diseased populations, which can overlook individual and subgroup variations [2]. DNA and RNA sequencing (RNA-seq) are the two most used methods in genetic research. Genetic variation, which encompasses DNA (gene) and RNA (gene expression) differences, is a fundamental element to understanding the genetics of diseases [3]. DNA sequencing can identify associations between genomic variants and diseases [4, 5], while RNA-seq can identify associations between RNA expression variations and diseases [6]. Combining multiple gene variants and/or gene expression differences into polygenic biomarkers can increase predictive power. Indeed, high and low polygenic scores from DNA can assess the probability of getting diseases [7]. While promising, the grand challenge here is analyzing the huge volume of known (and unknown) variants and using this information to diagnose, determine risk and predict treatment outcomes within diverse groups of humans [4].
This challenge is being met with precision medicine, which aims to translate this vast pool of genetic data to enhance disease outcomes accurately and safely [8]. However, the successful implementation of precision medicine remains difficult for heterogeneous ancestry groups and other distinct populations [8]. The convergence of genomics data and staggering developments in artificial intelligence (AI) and machine learning (ML) have the potential to address this issue [9]. Applied AI/ML approaches allow learning from a continuum of dataset sizes displaying heterogeneous levels of granularity [10]. AI/ML offers multiple supervised and unsupervised approaches to analyze genomics data which have been developed into multivariate statistical tools [9]. The successful implementation of these AI/ML approaches has the potential to support the development of enhanced systems-level understanding of diseases to decipher genomic regulatory networks. AI/ML approaches offer statistical analysis and classification of clinical genomics data for identifying best sources of information and predicting patients at high risk. Furthermore, AI/ML can be applied to capture genetic sequences for chronic diseases, phenotype categorization based on knowledge about human diseases and potential subtypes, and population sizing to create dimensions for common, rare and orphan diseases, and for understanding salutogenesis [9].
AI/ML approaches include Random Forest (RF) [11, 12]; Support Vector Machine (SVM) [13, 14]; Gradient Boosting [15, 16]; Extreme Gradient Boosting (XGBoost) [17, 18]; Elastic net regularized generalized linear model [19, 20]; Logistic Regression (LR) [21, 22]; Artificial Neural Network (ANN) [23–25]; Naïve Bayes (NB) [26, 27]; Bayesian Additive Regression Trees (BART) [28]; Bayesian Networks [29]; Greedy Thick Thinning algorithm [30]; K-Nearest Neighbor (K-NN) [31]; Decision Tree (DT) [32]; Linear Discriminant Analysis (LDA) [33]; Quadratic Discriminant Analysis (QDA) [34]; Gaussian Process Classification (GPC) [35]; AdaBoost (AB) [36]; Non-negative Matrix Factorization (NMF) [37, 38]; C4.5 [39, 40]; Formal Concept Analysis (FCA) [41]; Clustering [42, 43]; Multivariate Linear Regression (MLR) [44]; Genetic Algorithm (GA) [45]; Logit Boost [46]; Analysis of Variation for Association with Disease (AVA,Dx) [47]; OncoCast-MPM Machine-Learning Risk-Prediction Model (OncoCast-MPM) [48]; Combined Annotation Dependent Depletion (CADD) [49]; Very Efficient Substitution Transposition (VEST) [50]; Random Committee Ensemble Learning [51]; Deep Learning Neural Networks (DNNs) [52, 53]; MERGE [54]; Expectation–Maximization (EM) [55]; Generalized linear models (GLM) [56], J48 and Hidden Markov Model [57]. AI/ML approaches can be utilized to identify and assess genetic risk variants among individuals, especially those who are predisposed to having a disease. Successful implementation of AI/ML approaches has the potential to replace homogeneity with heterogeneity, caused by the existing genetic and statistical approaches [9]. However, the important questions here are, which AI/ML approach is appropriate for which data analytic problem? And, are results reproducible? During the process of choosing and implementing AI/ML algorithms, it is important to measure and avoid algorithmic bias [58].
In this study, our focus was to review, compare and report scientific objectives, methodology, development, performance evaluation, datasets, data sources, ethics and gaps of AI/ML approaches applied in the field of genomics (Table 1). We defined our criteria for the selection of high-quality literature published within the last 5 years (2017–22) that were indexed and available through PubMed Central. Our scope is limited and mainly includes those articles that report the application of AI/ML algorithms for statistical and predictive analyses using whole genome and/or whole exome sequencing (WGS/WES) identified gene variants, and RNA-seq and microarray gene expression differences. However, we are open to variable diseases among diverse populations, open data archives and annotation databases, and sequencing technologies used to produce raw WGS/WES and RNA-seq data. Our scope included reviewing and outlining the most relevant approaches to the goals of this study. Our findings report 32 different AI/ML algorithms [11–57] and approaches applied in 24 heterogeneous studies [47, 48, 51, 59–79] (Figure 1). Our literature search was performed using standalone and combinations of different keywords, including, but not limited to, ‘artificial intelligence’, ‘machine learning’, ‘algorithms’, ‘gene expression data’, ‘gene variant data’, ‘whole genome’, ‘whole exome’, ‘RNA-seq’, ‘sequence data’, ‘predictive analysis’, ‘precision medicine’, and various disorders. Further, itemized details are attached in Supplementary Material 1 available online at http://bib.oxfordjournals.org/, and literature selection workflow is added to Supplementary Material 2 available online at http://bib.oxfordjournals.org/.
Table 1.
Comparative analysis of AI/ML approaches using gene expression and gene variant data
| # | Year | PMIDs | Diseases | Study objectives | Machine learning and statistical algorithms | Statistical tools, packages and environments | Raw data | Processed data | Secondary, statistical, and downstream data analysis | Subjects | Single/multi-mics | Databases | Ethics |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2017 | PMID:28795970 | IBD | Classification and prioritization of genes to detect new genes connected to IBD | RF, SVM, Extreme gradient boosting (xgbTree) and Elastic net regularized generalized linear model (glmnet) | Benjamini–Hochberg, Bonferroni correction, one-sided Mann–Whitney U test, Fisher’s exact test | RNA-seq | Gene expression data | Gene Ontology (GO) Enrichment analysis (terms and pathway) | 513 (180 CD, 149 UC, 94 colorectal neoplasms, 90 control) | Single omics (transcriptomic) | GEO, GWAS, ClinVar database, MSigDB, KEGG, Pathway interaction database | N/A |
| 2 | 2019 | PMID: 31270349 | SLE | (1) Stratification of the subject as active and inactive SLE state with the help of raw data and test its potential to stratify, (2) identification of the best classifier/classifiers and (3) identification of the combinations of variables that make classification possible at best | GLM, k-nearest neighbors (K-NN), and RF | False discovery rate, Adjusted Rand Index, Rank–rank hypergeometric overlap | RNA-seq | Gene expression data | DEGS analysis- > Enrichment analysis(WGCNA), GO analysis and GSVA | N/A | Single omics (transcriptomic) | GEO | N/A |
| 3 | 2017 | PMID: 28269885 | CD | To estimate the performance of predictivity of three different techniques as classifiers to identity extra-intestinal manifestation in CD and compare with the existing method | NB, BART and Bayesian networks (BN) implemented using a Greedy Thick Thinning algorithm- learning, EM algorithm- learning conditional probabilities | Bayesian methods: NB, BART and Bayesian Networks, Greedy Thick Thinning algorithm; EM | WGS | Variant data (SNP) | Statistical analysis | 152 (75 Extra-intestinal manifestation, 77 control) | Single omics (genomic) | Self-generated, dbSNP | N/A |
| 4 | 2019 | PMID: 31564248 | CD | Disease prediction model by using previously unknown disease genes | AVA,Dx, SVM | VQSR, ANNOVAR | WGS | Variant data (Exonic) | Annotation using ANNOVAR, pathway enrichment analysis (ConsensusPath database) | 2855 (2793 CD, 62 control) | Single omics (genomic) | European Genome-Phenome Archive, Genotype-Tissue Expression Project, PopGen Biobank | Ethically approved |
| 5 | 2018 | PMID: 30204480 | Obesity | (1) Stratification of individuals into obese and non-obese and evaluation of obesity risk. (2) Comparison of predictive performance of various models | SVM, k-nearest neighbor (K-NN), and DT Feature selecting algorithms-stepwise MLR, DT and genetic algorithms | ANOVA | WGS | Variant data (SNP) | Genome-wide SNP and statistical analysis | 129 (74 obese, 65 control) | Single omics (genomic) | dbSNP | Ethically approved |
| 6 | 2019 | PMID: 31200905 | Colon cancer | Comparison of various machine learning algorithms for: (1) identification of differential genes of high risk using statistical tests, (2) prediction of cancer genes by using a ML strategy | LDA, QDA, NB, GPC, SVM, ANN, LR, DT, AB and RF | WCSRS, t test, Kruskal–Wallis (KW) and F-test | RNA-seq | Gene expression data | DEG analysis - > Statistical [WCSRS, t test, Kruskal–Wallis (KW) and F-test] | 62 (40 cancer, 22 control) | Single omics (transcriptomic) | Kent ridge biomedical data repository | N/A- No ethics approval is required for this dataset |
| 7 | 2016 | PMID: 27587275 | Breast cancer | Classification, characterization and prediction of breast cancer using mutation profiles | NMF Clustering, RF, NB, C4.5, SVM and K-NN | Wilcoxon rank-sum, Benjamini–Hochberg (FDR) | WES | Variant data (Somatic and non-synonymous SNV) | DEG analysis- > Enrichment analysis (Gene Set Enrichment Analysis (GSEA)), pathway analysis (Ingenuity Pathway Analysis) | 358 | Single omics (genomic) | TCGA | N/A |
| 8 | 2021 | PMID: 34332931 | Malignant pleural mesothelioma | Evaluation of risk scores and classification of the patients into low-risk and high-risk groups | OncoCast-MPM machine learning risk prediction model (elastic-net penalized Cox proportional hazard models) | χ2 test in R. lasso-penalized Cox regression. Kaplan–Meier survival curves with log-rank testing Concordance probability estimate | WGS | Variant data | Risk stratification and statistical analysis | 194 | Single omics (genomic) | MSK-IMPACT,TCGA | N/A |
| 9 | 2018 | PMID: 29298978 | Acute myeloid leukemia (AML) | Identification of molecular gene expression markers for precise treatment of acute myeloid leukemia | MERGE (mutation, expression hubs, known regulators, genomic CNV, and methylation) | Pearson’s, Spearman, ElasticNet | RNA-seq | Gene expression data | Covariate, association and prioritized subset analysis | 30 | Single omics (transcriptomic) | GEO | Ethically approved |
| 10 | 2020 | PMID: 32318621 | Alzheimer’s disease (AD) | Identification of genomic markers for precise therapy of AD (Phase 2a clinical study) | FCA - unsupervised | Integrated in the Knowledge Extraction and Management (KEM) environment | WES, RNA seq | Variant and Gene expression data | Association rules and linear mixed effect (LME) model analysis | 32 | Multi-omics (genomic & transcriptomic) | Self-generated | N/A |
| 11 | 2021 | PMID: 34220416 | Major depressive disorder (MDD) | Identification of potential peripheral blood transcriptome biomarkers and development of a MDD prediction model using peripheral blood transcriptomes | SVM, RF, K-Nearest Neighbors (K-NN) and NB | Positive predictive value, Matthews correlation coefficient | RNA-seq | Gene expression data | DEG analysis- > Statistical, KEGG pathway enrichment analysis | 302 (9 different datasets) | Single omics (transcriptomic) | GEO | N/A |
| 12 | 2020 | PMID: 33099536 | Ulcerative colitis | Identification of susceptibility genes and disease prediction development for ulcerative colitis disease prediction | RF and ANN | Benjamini–Hochberg | RNA-seq | Gene expression data | DEG- > GO enrichment analysis and KEGG enrichment analysis (Pathway) | 2 datasets | Single omics (transcriptomic) | GEO, GO, KEGG | N/A |
| 13 | 2021 | PMID: 33645908 | Major depressive disorder (MDD) | Stratification of MDD patients and control samples, and understanding pathophysiology | XGBoost (eXtreme Gradient Boosting implementation) | N/A | RNA-seq | Gene expression data | DEGs analysis- > GO enrichment analysis (GSA) | 390 (314 MD, 76 control) | Single omics (transcriptomic) | dbGaP, GEO | Ethically approved |
| 14 | 2019 | PMID: 29704323 | Schizophrenia (SCZ) | Prediction of high-risk individuals (5090 exomes) | eXtreme Gradient Boosting implementation (XGBoost), L1.logistic regression (lasso regularized), SVM and RF | ANNOVAR, Pearson correlation | WES | SNV(Variant data), DNM (Mutation-small insertions and deletions) | Annotated with ANNOVAR. KEGG enrichment analysis(Pathway) | 5090 (2545 SCZ, 2545 control) | Single omics (genomic) | dbGaP | Ethically approved |
| 15 | 2020 | PMID: 32111185 | Schizophrenia (SCZ) and Autism spectrum disorder (ASD) | Comparison of the architecture of the genomes of SCZ and ASD. (1) To identify if SCZ and ASD patients can be differentiated just based on supervised learning analysis from WES data. (2) Prioritization of genetic features by supervised learning algorithm and identification of central hub genes using unsupervised clustering | Regularized GBM (XGBoost implementation) and unsupervised hierarchical clustering | N/A | WES | Variant data | GO Enrichment analysis (Pathway) | 2392 | Single omics (genomic) | dbGaP, NDAR | N/A |
| 16 | 2021 | PMID: 34199109 | Ovarian failure (OF) | Identification of blood-based gene variant profiles for precise treatment | Clustering and RF | Shapiro–Wilk test, Wilcoxon and Fisher | WES | Variant data (non-synonymous rare variants) | Annotated with SnpEff software | 150 (118 OF, 32 controls) | Single omics (genomic) | IGSR, Self-generated, nomAD, dbSNP, Genecards, Uniprot, Gene Ontology | Ethically approved |
| 17 | 2020 | PMID: 33109206 | Premature ovarian failure (POF) | Identification of novel and candidate variants associated with premature ovarian failure | Bioinformatics analysis, Variant Effect Scoring Tool and CADD | CADD, VEST | WES | Variant data (SNV, InDel variants) | Annotated with MAF ExAC/ gnomAD/ 1000G/ KRGDB and pathogenicity score: CADD, VEST | 44 (34 POF, 10 controls) | Single omics (genomic) | DisGeNET, Monarch, MalaCards, NCBI, USCS Genome Browser, varsome, NCBI SRA, | Ethically approved. |
| 18 | 2016 | PMID: 27980626 | Hypertension | Disease model to predicting disease risk by genotypes, utilizing gene expression and rare variant data | Radial and linear SVM and LR | N/A | WGS and Microarray | Gene expression data and Variant data (rare variants) | Supervised machine learning methods (MLMs) analysis | N/A | Multi-omics (genomic & transcriptomic) | Genetic Analysis Workshop 19 (GAW19) | N/A |
| 19 | 2019 | PMID: 30462833 | Risk of Illness | Evaluation of microbial risk assessment (MRA) | RF, SVM, Neural Networks(NN), Gradient boosting (GBM), and Logit Boost (LB) | N/A | WGS | Variant data | Microbial risk and pathway analysis | 245 strains | Single omics (genomic) | WGS (Leekitcharoenphon, Nielsen, Kaas, Lund, & Aarestrup, 2014; Pielaat et al., 2013) and phenotypic data (Pielaat et al., 2013) | N/A |
| 20 | 2021 | PMID: 33999966 | Sepsis | Classification of individuals and comparison of different algorithms | DT, RF, SVM and DNNs | Benjamini–Hochberg | Microarray | Gene expression data | DE, resilience and prediction analysis | 1786 (1354 sepsis, 86 SIRS, 346 control) | Single omics (transcriptomic) | NCBI, GEO and EMBL-EBI ArrayExpress | Ethically approved |
| 21 | 2021 | PMID: 33570011 | Prostate cancer | Classification and detection of prostate cancer in medical diagnosis (normal or tumor cases) | Random committee ensemble learning | CFS method | Microarray | Gene expression data | Statistical analysis | N/A | Single omics (transcriptomic) | The European Nucleotide Archive (ENA)-PRJEB19256 | N/A |
| 22 | 2021 | PMID: 34054599 | Autism | Identification of subgroups of patients | RF classification and SVM, | Robust multi-array analysis, CPDB analysis | WGS | Gene expression data | DE analysis- > GO enrichment analysis and Pathway Analysis (KEGG, WikiPathways, BioCarta, and Reactome pathway database) and Transcriptome-Wide Association Analysis | 31 | Single omics (transcriptomic) | NCBI, GEO -(U48705, M87338, X51757, X69699), KEGG, WikiPathways, BioCarta, and Reactome pathway database. GO Biological Process database, ConsensusPathDB | Ethically approved |
| 23 | 2021 | PMID: 33681364 | Cancer | Identification of tumor tissue of origin | GBDT, K-Nearest neighbor (K-NN), DT, AB and SVM | N/A | RNA-seq | Gene expression data | DEG analysis- > GO enrichment analysis (pathway) | 9 datasets | Single omics (transcriptomic) | ICGC Data Portal, GEO, TCGA | N/A |
| 24 | 2021 | PMID: 34343245 | Ovarian Cancer | Identification of combinational therapies for ovarian cancer | RF, Gradient boosting (GB) and XGBoost | Wilcoxon Test, PRISM, Spearman correlation–Pearson correlation, mean-squared error and mean absolute error | DSS,RNA-seq, WGS, scRNA-seq | Gene expression data | Predictive analysis | 4 datasets | Single omics (transcriptomic) | HERCULES project | N/A |
Table includes number of studies, year published, PMID, disease, study objectives, machine learning algorithms applied, statistical tools and packages used, raw data used, processed data generated, secondary and downstream data analysis, subjects involved, single/multi-omics, databases and ethics.
Figure 1.

AI/ML approaches using gene variant and gene expression data for traditional bioinformatics and predictive analysis. Figure includes 24 AI/ML approaches, variable diseases [inflammatory bowel disease (IBD); systemic lupus erythematosus (SLE); colon cancer (CC); acute myeloid leukemia (AML); major depressive disorder (MDD); ulcerative colitis (UC); sepsis (Sep.); prostate cancer (PC.); Alzheimer’s disease (AD); hypertension (Hyp.); ovarian cancer (OC); Crohn’s disease (CD); obesity (Ob.); breast cancer (BC); malignant pleural mesothelioma (MPM); schizophrenia (SCZ); autism spectrum disorder (ASD); ovarian failure (OF); premature ovarian failure (POF); risk of illness (RI); autism (Au.)] and AI/ML algorithms [Generalized linear models (GLM); Genetic Algorithm (GA); Multivariate Linear Regression (MLR); Random Forest (RF); Bayesian Networks (BN); Support Vector Machine(SVM); Expectation–Maximization (EM); Bioinformatics Analysis (BI); Random committee ensemble learning (RC); Elastic net regularized generalized linear model (GLMNET); Linear discriminant analysis (LDA); Quadratic Discriminant Analysis (QDA); AdaBoost(AB); Formal Concept Analysis (FCA); Combined Annotation Dependent Depletion (CADD); Very Efficient Substitution Transposition (VEST); Deep Learning Neural Networks (DNNs); Decision Tree (DT); LogitBoost (LB); Gradient boosting (GB); Extreme gradient boosting (XGB); Gaussian Process Classification (GPC); Logistic Regression (LR); Artificial Neural Network (ANN); Greedy Thick Thinning algorithm (GTT); Neural Networks(NN); K-Nearest Neighbors (K-NN); Clustering (CU); Non-negative Matrix Factorization (NMF); Naïve Bayes (NB); MERGE (mutation, expression hubs, known regulators, genomic CNV and methylation); Bayesian Additive Regression Trees (BART)].
AI/ML approaches in genomics and precision medicine
Classifying analytical tasks based on available predictor variables is a key step in correctly addressing the problem of choosing appropriate AI/ML algorithm(s) for analysis in genomics. In this study, we report 24 different peer-reviewed and published scientific studies, applying variable supervised/unsupervised ML algorithms to genomic (WGS/WES- variant) and transcriptomic (RNA-seq and Microarray- gene expression) data for bioinformatics, statistics and predictive analyses (Figure 2, and Table 2). The studies covered extracted data and shared genomic data for a wide variety of diseases [83–102], e.g. inflammatory bowel disease (IBD) [80], systemic lupus erythematosus (SLE) [81], Crohn’s disease (CD) [82], obesity [83], colon cancer [84], breast cancer [85], acute myeloid leukemia (AML) [86], Alzheimer’s disease (AD) [87], major depressive disorder (MDD) [88], ulcerative colitis (UC) [89], schizophrenia (SCZ) [90], autism spectrum disorder (ASD) [91], premature ovarian failure (POF) [92], hypertension [93], autism [94], sepsis [95], prostate cancer [96], malignant pleural mesothelioma [97], ovarian cancer [98] and other cancer disorders [99] (Figure 1 and Supplementary Material 3 available online at http://bib.oxfordjournals.org/).
Figure 2.

Total number of machine learning algorithms applied for predictive analysis. Figure includes algorithms: Random Forest (RF), Support Vector Machine (SVM), Gradient Boosting, Extreme gradient boosting XGBoost, Elastic net regularized generalized linear model, Logistic regression (LR), Artificial neural network (ANN), Naïve Bayes (NB), Bayesian Additive Regression Trees, Bayesian Networks, Greedy Thick Thinning algorithm, k-nearest neighbor (K-NN), Decision tree (DT), Linear discriminant analysis (LDA), Quadratic discriminant analysis (QDA), Gaussian process classification (GPC), Adaboost (AB), Non-negative Matrix Factorization (NMF), C4.5, Formal concept analysis (FCA), Clustering (Unsupervised), Multivariate Linear Regression (MLR), Genetic Algorithm (GA), Logit Boost, AVA,Dx (Analysis of Variation for Association with Disease), OncoCast-MPM machine-learning risk-prediction model, Combined Annotation Dependent Depletion (CADD), Very Efficient Substitution Transposition (VEST), Random Committee Ensemble Learning (RCEL), Deep Learning Neural Networks (DNNs), MERGE, and Expectation–maximization (EM).
Table 2.
Total number of algorithms applied for predictive analysis
| AI/ML algorithms and approaches | Total count |
|---|---|
| SVM | 13 |
| RF | 12 |
| XGBoost | 5 |
| LR | 5 |
| NB | 5 |
| DT | 5 |
| ANN | 4 |
| k-nearest neighbor (K-NN) | 4 |
| AB | 3 |
| Gradient Boosting | 3 |
| LDA | 2 |
| QDA | 2 |
| GPC | 2 |
| Clustering (Unsupervised) | 2 |
| Elastic net regularized generalized linear model | 1 |
| BART | 1 |
| Bayesian Networks | 1 |
| Greedy Thick Thinning algorithm | 1 |
| NMF | 1 |
| C4.5 | 1 |
| FCA | 1 |
| MLR | 1 |
| GA | 1 |
| Logit Boost | 1 |
| AVA,Dx | 1 |
| OncoCast-MPM machine-learning risk-prediction model | 1 |
| CADD | 1 |
| Very Efficient Substitution Transposition (VEST) | 1 |
| Random committee ensemble learning | 1 |
| DNNs | 1 |
| MERGE | 1 |
| EM | 1 |
XGBoost, RF, elastic net regularized linear model and SVM-based identification of novel candidate risk genes for IBD [59]
Isakov et al. presented a supervised ML method to study the classification and prioritization of known genes to detect new genes connected to IBD [59]. ML algorithms were applied on gene expression data, mainly extracted from Gene Expression Omnibus (GEO). Five hundred and thirteen samples from readily available datasets were used to train and validate this model. This was broken down to 180 CD patients, 149 UC patients, 94 colorectal neoplasms patients and 90 healthy individuals. The authors also chose to focus on a single omics approach where they studied transcriptomic data and its potential in identifying IBD genes. Overall methodology involved implementation of XGBoost, elastic net regularized generalized linear model, RF and SVM with polynomial kernel. The original data set was split into two sets where 75% was used for training of the model and the remaining 25% was used for validation. A 5-fold cross validation technique was performed for 10 times to improve the performance of the model. A combined model was also constructed using all the above models that resulted in area under the ROC curve (AUC) = 0.852, sensitivity = 0.634, specificity = 0.914 and accuracy = 0.847 for the training data; and AUC = 0.829, sensitivity = 0.577, specificity = 0.880 and accuracy = 0.808 for the testing data. Every gene has 1027 features annotated and feature selection using regularized form of logistic regression identified terms associated with immunity and inflammation as features that can characterize IBD. The method effectively distinguished IBD risk genes from non-IBD genes and recognized unknown features that corresponded to IBD. A total of 347 genes had a high prediction score. Furthermore, the model found 67 novel genes (e.g. RELT, CCL18, TNFRSF10B, LILRB2, TNFRSF10D, etc.) that were previously not studied under other IBD publications. The main benefit of this research was the discovery of novel gene associations in the context of IBD. The authors recommended that these genes be studied more as new drug targets for IBD. Additionally, they can provide a better understanding of the pathophysiology of IBD.
GLM, K-NN and RF-based lupus disease prediction [60]
The scope of this article was focused on the beneficial aspects of transcriptomic data in predicting lupus disease [60]. Kegerreis et al. stratified subjects based on active and inactive SLE state and identified the best classifier/classifiers [60]. Authors filtered and implemented conventional bioinformatics methods, supervised and unsupervised ML techniques on three datasets of gene expression data, extracted from GEO. They performed and reported differential expression (DE) analysis [102] of active and inactive patient samples using the filtered genes. A total of 7848 genes were investigated, and it was observed that heterogeneity existed within the three studies (GSE39088, GSE45291 and GSE49454). Comparing these genes revealed that 5170 genes were unique to one study, 1234 occurred in two studies and 36 genes were present in all three studies. Unsupervised hierarchical clustering was not successful in distinguishing the patients into active and inactive status accurately. The absence of commonality questions the ability to properly distinguish the differentially expressed genes (DEGs) into active and inactive patients from the data set. The conventional bioinformatics methods do not bolster well enough, even though the gene expression data can distinguish the active SLE patients for accurate results. This signified the importance of the requirement for better practices. Gene set variation analysis [103] enrichment performed using 25 cell-specific gene modules on the dataset revealed that 12 of the 25 cell-specific modules had enrichment scores with significant Spearman correlations to SLE Disease Activity Index (SLEDAI; P < 0.05). Furthermore, 14 cell-specific gene modules scored with major differences between active and inactive patients (Welch’s t-test, P < 0.05) [60]. This was not enough to predict the disease status as no module managed to segregate the active and inactive SLE patients. Three ML techniques GLM, K-NN and RF were used as classifiers using two validation methods: 10-fold cross-validation (CV) and study-based CV. The validation method using the 10-fold CV along with the use of gene expression values yielded a better result for all three ML classifiers. Out of all the classifiers, RF had the highest performance with 83% accuracy and an area under the curve (AUC) value of 0.89 for the 10-fold CV. The accuracy of the validation method performed using study-based CV technique was close to 50% when using expression data. When applied on the module enrichment scores, they significantly improved the performance of RF test to 65% and K-NN test accuracy to about 70%. This study was limited to a small dataset used to train the classifiers. The authors state that although a large number of SLE data sets are available online, they are not annotated with SLEDAI [60, 101]. The authors predict that their approach has the potential to design a blood test that can predict SLE activity as well as to better understand organ involvement in SLE.
Multi-ML algorithms to analyze interactions among genetic and clinical factors in IBD patients [61]
In a study involving CD, Menti et al., estimated the predictive performance of three classifiers and identified the extra-intestinal manifestation (EIM) in this gastrointestinal disease using a single omics (genomic) approach [61]. The classifiers, NB, BART and Bayesian networks were used to perform statistical analysis. The authors used the Greedy Thick Thinning algorithm and the EM algorithm on the variant data retrieved from the Single Nucleotide Polymorphism Database (dbSNP). The genetic polymorphisms of NOD2, CD14, TNFA, IL12B and IL1RN genes were considered as a variable along with the other variables like disease characteristics and risk factors. It was found that the role of CD14 is insignificant for predicting EIM. The analysis of this study showed that Bayesian networks performed better than NB and BART. Bayesian networks achieved an accuracy of 82% with clinical factors, and it went up to 89% when additional genetic information was included. Out of 152 patients, 75 had EIM. The predicted outcome showed the presence of IL and TNF contributed to a 10% increase in the accuracy validated by the disease phenotype [61]. This research highlighted the ability of Bayesian networks to accurately predict EIM in CD patients [61]. However, the major limitation in this study was the overfitting bias associated with the small sample size and the lack of external validation. The authors propose the use of Bayesian networks in predictive clinical settings due to its precision.
AVA, Dx–SVM pipeline for classifying CD signal from variome analysis [47]
Wang et al. proposed a novel ML pipeline, AVA,Dx that uses SVM to predict CD [47]. This model uses a single omics approach by employing genomic variant data consisting of a training dataset, testing dataset, a panel of data extracted from European Genome-Phenome Archive and the Genotype-Tissue Expression Project. This study had a total of 2855 samples were used to train and validate the different panels. Out of those samples, 2793 samples were from CD patients and 62 were from control patients. Additionally, the authors employed an ethnicity annotation for these samples. The functional effects of exome SNVs were combined and presented to generate gene scores which filtered 173 013 variants to 13 957 affected [47]. The predictive model was constructed based on external disease genes and gene sets retrieved from the computer feature selection. These genes were used in SVM model and trained in the leave-one-out CV technique. Along with the default gene score method, four other different gene scoring methods were used. It was observed that the default gene score performed better, thus signifying the usage of functional effects of variants in assessing the genetics of the disease. When the top Pascal-ranked CD genome-wide association study (GWAS) [104] genes were used in the construction of SVM model building using the training set. It was seen that these genes had a better performance ability than the random gene model with external genes set. The 175 Pascal top-ranked genes model attained a high ROC AUC of 0.70. The assessment of feature and computationally selected genes showed Disease Overfitted (DISO) sets having more than 100 genes not associated with CD in the training data of healthy individuals (HCs). Pascal GWAS175 was outperformed by other feature selection models like KS5max and DKMcost125. The feature selected genes effectively distinguished between CD and healthy individuals. Feature selection identified already known genes (LRRK2 and KIAA1109) and novel unreported genes: DKMcost125 genes NOD2, LSP1 and CCR6, and KS5max genes IL19 and ATF4. Three cutoff values were used in AVA,Dx for differentiating healthy versus Crohn’s-affected (14.3, 45 and 0). It was seen that higher scoring subjects had CD. These scores were not estimated, and the authors thought that it should not be further used for understanding the intensity of the disease. The evaluation of the predictions for the current model differentiated healthy individuals less accurately than the diseased patients but compared better than the baseline approach. AVA,Dx needed only 111 individuals to construct a model, which is much less than the individuals needed in previous studies that did not use this algorithm. The authors concluded that a larger panel and better gene scoring methods could improve the performance ability of this algorithm.
This research study was restricted to the AVA,Dx model. Firstly, the model’s predictive capabilities diminished when the test panel sequences did not share enough loci with the CD-train panel. Secondly, the AVA,Dx model could only recognize the genetic model of CD patients rather than healthy patients. Thus, it is unable to label unaffected patients as healthy [47]. Nevertheless, this model has many benefits that are not limited to the capacity to distinguish genes related to diseases without the need for large study panels. The strength of the AVA,Dx model lies in sequencing methods and panel differences. Hence, the authors recommend that this model be used for identifying disease-relevant variants not only for Crohn’s disease but also other complex diseases that have a genetic factor.
Multi-ML implementation for obesity risk evaluation [62]
A study on obesity evaluated the obesity risk aimed to stratify the individuals into obese and non-obese and compare the predictive performance of various models using single-nucleotide polymorphisms (SNPs) data retrieved from dbSNP [62]. Wang et al. used three algorithms—SVM, K-NN, DT with Stepwise MLR, DT and GA—as feature selection methods to the data. The ratio of training to testing was 1:4 and 5-fold CV was performed for 100 rounds. For models trained with 130 SNPs, the SVM model had a better performance ability when compared to both the K-NN model and the J48 model in sensitivity and specificity with an average accuracy of 0.67. Out of the 130 SNPs, 74 were obtained from obese patients and 65 were obtained from non-obese patients. Sex and age were other clinical factors that the authors also used in this study. Comparison of the feature selection algorithms to determine the informative SNPs suggested that the SVM and K-NN models, when trained with MLR, had the best performance with high accuracy. However, the J48 model, when trained with DT, showed the best performance. Using stepwise MLR, nine SNPs were associated with obesity (rs10501087, rs17700144, rs2287019, rs534870, rs660339, rs7081678, rs718314, rs9816226 and rs984222) along with gender selected as an informative feature. Four SNPs (rs10501087, rs534870, rs718314 and rs984222) were significantly associated with obesity risk (P < 0.05). The SVM model trained using selected nine SNPs attained the highest accuracy (0.71). All the nine SNPs were reported to have been associated with obesity. This single omics study concludes that the SNPs selected successfully identified obesity risk [62]. A major benefit of this research was that SVM initiated a predictive model that was better at analyzing genetic factors associated with obesity when compared to other ML algorithms.
Multi-ML algorithms for identification of high-risk, colon cancer differential genes by statistical tests [63]
This study involved comparing various ML algorithms for the identification of high-risk, differential genes in colon cancer by statistical tests [63]. In addition, Maniruzzaman et al. predicted cancer-associated genes using gene expression data extracted from Kent ridge biomedical data repository, USA. Transcriptomic data from 62 patients were utilized in the single omics approach by the authors. Out of the 62, 40 were cancer patients and 22 were healthy individuals. In addition to gene expression data, age and gender were other clinical data used in this study. This study follows a two-level analysis. In the first step, the feature selection identified the most important genes using four statistical methods like Wilcoxon sign rank sum (WCSRS), t-test, Kruskal–Wallis and F-test followed by a second step to select the best classifier among the 10 classifiers LDA, QDA, NB, GPC, SVM, ANN, LR, DT, AB and RF. Forty combinations in total with 4 statistical tests and 10 classifiers were designed. Kernel optimization was performed; Poly-2 kernel for GP-based classifier and RBF kernel for SVM were selected. As the P values decreased, the accuracy increased suggesting that the classifiers trained for the important genes, which have less noise. A graph plotted the number of significant genes against the cut-off point of P-values suggested that WCSRS is the most efficient test. Inter-comparison of the classifiers demonstrated that the highest classification accuracy was given by the RF classifier and the lowest classification accuracy was given by NB. Optimized ML system suggested that the best combination of statistical tests and classifiers was WCSRS test combined with RF-based classifier with a high classification accuracy of 99.81%. [63]. Although microarray technology is not in use now, the authors recommend using microarray gene expression data for future work and incorporating that into their model.
Multi-ML implementation for breast cancer model classification [64]
Vural et al. highlighted a new breast cancer model classification system for stratifying individuals into subgroups [64]. The authors implemented an unsupervised NMF clustering method, which identified the subgroups based on single omics system [64]. Other supervised ML algorithms like RF, SVM, C4.5, NB and K-NN were applied for classification on the variant data (somatic and non-synonymous SNV) downloaded from The Cancer Genome Atlas (TCGA). The NMF clustering grouped the whole data with 358 genes into three groups containing 169, 121 and 68 patients, respectively. The clusters were named as follows: Cluster 1 as early-stage-enriched cluster, Cluster 2 as a mixed cluster and Cluster 3 as late-stage-enriched cluster with AUC values 0.88, 0.8 and 0.95, respectively. It was observed that 358 genes had higher mean mutation scores in the late-stage-enriched cluster 3 than those in the early-stage-enriched cluster 1, which suggests that these genes may have amassed deleterious mutations which led to the advancement of the breast cancer disease condition. Out of the 358 genes, APC and BRCA2 were listed as tumor suppressor genes and MLL was listed as an oncogene. Information gain attribute evaluator was used as a feature selection approach. Ranker algorithms were implemented in Weka for feature evaluation and search. Five ML algorithms were used for testing to pick the best-suited algorithm. The RF classification algorithm was found to be the best as it achieved the best 10-fold CV accuracy of 70.86% whereas K-NN was the least effective [64]. An extensive benefit associated with this research is that somatic mutation profile data can be used to determine breast cancer subtypes. The authors conclude that their model can be used to predict and identify other tumor types.
OncoCast-MPM ML-driven risk prediction for malignant pleural mesothelioma [48]
A novel disease prediction model was built by Zauderer et al. for stratifying individuals into high-risk and low-risk groups [48]. A total of 194 samples were used in this single omics study. The authors also utilized clinical data such as age, gender, smoking history and age at diagnosis. The OncoCast-MPM ML risk-prediction model, an ensemble learning model was implemented on the Memorial Sloan Kettering Cancer Center-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) and TCGA cohort which generated risk scores for individuals with malignant pleural mesothelioma [48]. The MSK-IMPACT cohort received different ranges of the risk score than the TCGA cohort. The ratio of training to testing data was 2:1. Hierarchical clustering divided the patients of MSK-IMPACT into two groups: high-risk (27%) and low-risk (73%). It was also able to effectively stratify the TCGA cohort into low-risk and high-risk groups. Feature selection using univariate analysis identified CDKN2A and CDKN2B deletions along with the mutations of TP53, TERT, GNAS and DICER1 to be unfavorable, and epithelioid histology along with the presence of BAP1 or PBRM1 mutations to be favorable [48]. Feature selection using the multi-variate analysis identified features, including BAP1 and PBRM1 mutations, epithelioid histology, a smoking or tobacco history, and reported classic occupational asbestos exposure as favorable features, and male gender, CDKN2A and CDKN2B deletions, TP53 and TERT mutations, age, advanced stage disease and biphasic histology as unfavorable features. It was observed that the median overall survival for the low-risk group (30·8 months) for the MSK-IMPACT was higher than the high-risk group (13·9 months). The model was cross-validated with the train model for every 200 iterations and attained 0.67 as cross-validated estimate. To understand the survival in the high-risk group, treatment patterns with active agents were studied and these were the same for the low-risk and the high-risk groups [48]. The high-risk group consisted of 27 early-stage disease, 17 surgery, 29 epithelioid, 17 biphasic and 6 sarcomatoid histology. The TCGA cohort was used for validation and the results were just like the MSK-IMPACK cohort. The risk model found 33 high-risk individuals and 41 low-risk individuals with median overall survival higher in the low-risk (23·6 months) group than the high-risk group (13·6 months). The disease stages and the histology were same for the two cohorts. BAP1 alterations were more frequent in MSK-IMPACT than TCGA cohort. Comparison of genes enriched in OncoCast-MPM genes and stage stratification showed that six genes enriched in the former (high risk-TERT, NF2, TP53 and LATS2; and low risk- SETD2 and BAP1) and one in the latter (advanced stage disease—TP53 and early-stage disease—none) [48]. The model successfully classified the individuals and was more accurate than the existing stage and histology model. The limitation of this study was the OncoCast-MPM model not being prospectively validated. This is required to ensure that the analysis is not undermined by the external variables. Due to the growing field of molecular testing, OncoCast-MPM requires regular updates. However, OncoCast has shown to provide an accurate prognosis estimation for malignant pleural mesothelioma patients. Since this predictive model is available freely online, the authors recommend that it can be used for clinical and real datasets. This will allow the model to undergo prospective and independent validation.
Using a novel model, MERGE to identify gene markers for the targeted treatment for AML [65]
A new model called the MERGE (mutation, expression hubs, known regulators, genomic copy number variation and methylation) was developed by Lee et al. that identified gene markers for precise and targeted treatment for AML [65]. Transcriptomic data from a total of 30 AML patients were compiled for this study. Additionally, drug concentrations and sensitivities for 53 different drugs were utilized. Authors implemented MERGE on the gene expression data extracted from GEO. Unlike conventional statistical methods, MERGE uses the ‘MERGE’ score, which indicates gene–drug associations. Higher the MERGE score, more the number of associations with drugs. A graph plotting MERGE score against the weighted combination of driver features indicated that expression clustering is a significant feature and can be used to study tumor driver. Methylation was found to be the least significant feature. The results of this whole study indicated that SMARCA4 is a potential biological marker. High expression of SMARCA4 appeared to be sensitive to topoisomerase II inhibitors, mitoxantrone and etoposide in AML [65]. The main benefit of this research was that the authors organized genes based on their potential to drive cancer according to their multi-dimensional information. This original model can be further used to study the extent of gene–drug interactions.
Unsupervised algorithm—FCA approach-based significant genomic biomarker discovery to study the drug response for Alzheimer’s disease therapy [66]
In this multi-omics study, Hampel et al. presented an unsupervised algorithm FCA integrated in the Knowledge Extraction and Management (KEM) environment using genomic (Variant and Gene expression), pharmacological and clinical data, with efficacy endpoints taken from ANAVEX2-73-002 and ANAVEX2-73-003 [66]. ANAVEX2-73-002 was a Blarcamesine Phase 2a clinical trial with 32 individuals with mild-to-moderate AD and which further continued as a 208-week extension study, ANAVEX2-73-003. The discussed model recognized the predictors of response by association and ranking them. During the reported analysis, the authors studied 3 145 630 associations among all the features. The applied strict filtration process significantly decreased the number of associations, as only 15 were linked to the clinical outcomes. These 15 associations had an average blood concentration of Blarcamesine above 4 ng/ml and this improved the Mini-Mental State Examination (MMSE) outcome and Alzheimer’s Disease Cooperative Study-Activities of Daily Living scale (ADCS-ADL) scores.
Authors identified and reported two DNA variants, SIGMAR1 p.Gln2Pro (rs1800866) and COMT p.Leu146fs (rs113895332/rs61143203) [66]. SIGMAR1 targeted by Blarcamesine is a significant drug target with the functions of maintaining cellular homeostasis, which delays or stops neurodegeneration and enhances the synaptic compensatory responses. The COMT gene is linked with memory and other neurological behavioral roles. Parameters including ANAVEX2-73 concentration levels, baseline MMSE score, SIGMAR1 p.Gln2Pro, and COMT p.Leu146fs variants, age, gender, APOEϵ4 genotype status, and Donepezil co-medication were used in the linear mixed effect models with change in MMSE (88%) and ADCS-ADL (78%). The differences are linked with a disproportion of APOEϵ4 genotype status in the arms. It was noticed from this analysis that the higher Blarcamesine mean concentration arm refined the therapeutic responses in the adjusted MMSE and ADCS-ADL compared to the low or the medium arm at 148 weeks. Besides time, APOEϵ4 status and Blarcamesine mean concentration were important predictors. Other important variables in this method were SIGMAR1 p.Gln2Pro, COMT p.Leu146fs and APOE4ϵ4 status interactions with time. Unadjusted values verified the results and individuals having improved therapeutic response biomarkers at week 57 had improved therapeutic response in ADCS-ADL at 148 weeks when compared to the individuals without these biomarkers or reference population given care. The overall approach of FCA is exemplary for the identification of biomarkers in early data. The authors concluded that these findings will be used to determine if patients have a better response to the drug Blarcamesine in other clinical trials.
SVM, RF, K-NN and NB approaches to identify potential MDD peripheral blood transcriptome biomarkers with ML [67]
Zhao et al. identified and used potential peripheral blood transcriptome biomarkers to develop an MDD prediction model [67]. The authors utilized nine different datasets, and each had a sample size of 128, 67, 18, 22, 45, 22, 12, 16 and 160 for algorithm implementation in this study. Clinical data such as age, gender and ethnicity were also recorded. They implemented SVM, RF, K-NN and NB on the gene expression data from GEO, where the RF was used to select the features. The meta-analysis of the genes showed 137 DEGs in which 66 are upregulated and 71 of them are downregulated. The genes that were significantly (false discovery rate <0.05) differentially expressed were TPST1, ARG1, KLRB1, WWC3, AKR1C3, and MAFG. These six genes were linked with the immune process, hormonal metabolism and inflammatory response and played an important role in the diagnosis of MDD by acting as a potential biomarker. Authors observed and reported that genes had a significant expression difference with a single-gene diagnostic method, which proves that single gene model is not efficient. Authors then used the transcriptome data from the discovery sets to perform the meta-analysis and it was seen that 114 DEGs were identified. Feature selection of these data resulted in feature set containing 108 genes that were implemented upon with four models. All models produced an average AUC value, whereas the SVM had the highest average AUC with values of average AUC of 0.82, an accuracy of 0.75, sensitivity of 0.78 and specificity of 0.74 in the train dataset. SVM had a better predictive performance when compared to the single-gene diagnostic model [67]. This study is limited by lack of knowledge regarding the amount of data needed to demonstrate the model’s predictive ability. Additionally, further independent validation is required to determine the accuracy of the model. The authors propose that this model could be used to discover transcriptional markers related to other mental illnesses.
Gene-based predictive RF and ANN-based model for the diagnosis of UC [68]
This study aimed to identify susceptibility genes using an RF algorithm and develop a new model for the prediction of UC using ANNs [68]. GSE109142 and GSE92415 were used as training and validation data sets, respectively. This model used a single omics approach consisting of transcriptomic gene expression data where the training dataset (GSE109142) had 781 upregulated and 127 downregulated DEGs. Two datasets were employed for this study that have been used in previous studies. These datasets contained both cases and controls. RF implementation identified the top five UC associated genes to be FAM65C, CSF3R, CSF3, POM121L9P and FER1L4 where CSF3R was found to be the best-characterized DEG among all the UC-associated genes [68]. UC prediction model attained ROC-AUC score and PR-AUC score of 0.9847 and 0.9444, respectively. Validation using GSE92415 yielded ROC-AUC and PR-AUC scores of 0.9506 and 0.9747, respectively. The major limitation of this study was the environmental factors associated with UC. This can cause the model to be restrained in terms of predictive power. It is the authors’ recommendation to perform external validation on their model. This model can be used in future work for early diagnosis and newer biochemical treatments for UC.
Supervised XGBoost implementation for depressive disorder stratification using for brain and blood mRNA profiles [69]
Qi et al. presented a model using supervised ML algorithm XGBoost implementation that effectively differentiated the MDD cases and controls using the gene expression data retrieved from GEO [69]. In this single omics study, a sample size of 390 individuals consisting of 314 MDD patients and 76 healthy individuals. 80% of the data were used for training and the remaining 20% of the data were used for testing. The model trained on brain mRNA gene expression data using a 10 CV method yielded an AUC value of 0.72 [69]. The use of the baseline model (same CV approach but with randomly permuted labels) on the same data yielded an average AUC of 0.55. Comparing these two models using the Wilcoxon signed rank revealed that the trained model had better performance ability than the baseline model. The final brain mRNA model had 62 genes in total and attained an AUC value of 0.76 on the test dataset. A model consisting only of covariates had an average 10-fold CV AUC value of 0.83, whereas a model with covariates and gene expression attained an average 10-fold CV AUC value of 0.71. This indicated the absence of a significant effect on the model performance due to the inclusion of the covariates. External validation by dorsolateral prefrontal cortex gene expression values attained an AUC of 0.62. The literature review of the genes predicted by ML (ENPEP, COX6A1) overlapped with the gene sets, revealing the association of these genes with depression. The model implementation on the blood mRNA data with the CV achieved an average AUC of 0.64 whereas the base-line model attained an AUC of 0.56 [69]. Comparison of these two models using the Wilcoxon signed rank revealed that the trained model had better performance ability than the baseline model. The final model had 1376 genes and an AUC of 0.61. The covariate analysis showed the relation between the performance and the smoking status of an individual. Other genes that were identified to be associated with depression are CX3CR1, TMEM245, COL4A1, PRAMEF1, TMEM52, A2M, DDC-AS1, GRP88, GALR3, VPS53 and CRYBA1 [69]. The main advantage of this study was highlighting the importance of implementing blood mRNA-based ML models as a tool for precision medicine. The predictive model employed in this research aids in early diagnosis and determining the different subtypes of MDD. The authors recommend that different types of data, such as transcriptome data, be analyzed using this model.
Multi-ML-based identification of subjects with high potential risk for SCZ [70]
WES data of 2545 subjects and 2545 unaffected subjects from the database of Genotypes and Phenotypes (dbGaP), study phs000473.v1.p1, were studied by Trakadis et al. [70]. The authors aimed to identify subjects with high potential risk for SCZ. The data were processed, and variant data [single nucleotide variants (SNVs) and small insertions and deletions] were annotated using ANNOVAR. Rare variants and DNMs of four neuropsychiatric disorders from 3555 trios and 36 studies were collected. Feature filtration was performed using Pearson correlation (having >90% Pearson correlation), which filtered features from 1155 to 17 138. Supervised ML algorithms RF, Lasso regularized (L1) logistic regression, XGBoost and SVM were applied to the data. XGBoost attained the highest accuracy, specificity, sensitivity, precision, recall and F1 score values of 85.7%, 86.6%, 84.9%, 86.9%, 84.9% and 85.9%, respectively. 70% of the data were used for training and 30% of the data were used for testing. The high value (0.95) of AUC implies that the patients can be differentiated from controls effectively and accurately. The analysis using the XGBoost algorithm on top 50 genes indicated the connection of SCZ with neuropsychiatric diseases and few of the potentially relevant genes included GRP, MYCBP2, CNDP1, ARL1, CAP1, GPRIN2, RASGRP3 and CHI3L1. The authors concluded that the current model can be improved by combining different levels of data like neuroimaging data, transcriptome data, genomic data and phenotypic information like speech during the training of the model. The limitations of this research stems from not analyzing the clinical characteristics data for patients with SCZ. This caused the data to not be generalized to larger groups of patients. Since SCZ has environmental aspects (in utero and neonatal), the authors propose that their model can be trained with different datasets, for instance transcriptome and genomic data, to increase its predictability.
XGBoost implementation and clustering for comparative analysis of SCZ and ASD [71]
This single omics study focused on comparing the genetic architecture of SCZ and ASD [71]. This study included a sample size of 2392 families with ASD. The criteria were not limited to age (patients had to be at least 36 months old), as it also included for the patients to not have extensive birth complications and not have a known genetic disorder. Sardaar et al. analyzed the WES data using regularized gradient boosted machines (GBMs) for the identification of the significant genetic features followed by a gene clustering approach for the identification of the mutated subsets of genes. 70% of the whole data were used for training and validation. The remaining 30% of the unseen data were used as test data. It was delineated that supervised algorithms are sufficient to differentiate SCZ and ASD. Boosted regression tree models of the Gradient Boosting algorithm showed an accuracy of 86% for the SNV-based model and 88% for the gene-based model. A 5-fold CV implemented for extra validation yielded 88% average validation accuracy for both gene-based model and SNV-based model. One hundred and fifty-one genes were found to be overlapping from both the SNV approach and the gene approach and the top 10 significant overlapped genes from both the approaches include SARM1, QRICH2, AKAP1, PCLO, TSPO2, ABCC3, KIF13A, FAN1, CCDC155 and PRPF31 using the boosted regression trees model. Clustering of these 151 genes identified 3 and 2 clustered gene groups in SCZ and ASD respectively 67 and 38 genes, respectively. An important benefit of this study was illustrated with the exome-based ML analysis. This type of analysis allowed for the investigation of distinct diseases that have a genetic similarity. The authors proposed focusing on common variants when it comes to ML analysis of mental illnesses due to the presence of this type of variant as a contributing factor for SCZ and ASD.
Clustering and RF application for the identification of variant profiles in ovarian failure [72]
This study identified blood-based gene variant profiles for the precise treatment of ovarian failure [72]. A total sample size of 150 individuals was utilized in this study. Out of these, 118 were patients suffering from OF and 32 were healthy individuals. Additionally, the authors had some other clinical criteria in place. The subjects had to be females, under 40 years of age and had no history of pelvic surgery, autoimmune diseases or chemotherapy. Henarejos-Castillo et al. used unsupervised clustering and the supervised ML algorithm, RF, on the variant data. WES data were collected from a study conducted between 2017 and 2019 [72]. A pipeline was used for the filtration of the genomic variants. To build the gene-targeted and non-targeted hypotheses, 161 209 variants were used. A total of 2395 and 63 928 variants were identified in the targeted approach and non-targeted approach, respectively. The authors did not pursue the targeted hypothesis due to a lower number of variants being associated with ovarian physiology when compared to the non-targeted hypothesis. The 63 928 variants demonstrated moderate to deleterious effects, with most changes being missense in the untranslated region or in structural interaction and frameshifts. Out of the 63 928 variants, 116 had significant differences (P < 0.01) in the distributions of allele frequencies between the cases and the controls. One of the variants was a missense variant (p.Ala301Gly) in the macrophage stimulating 1-like (MST1L) gene found in 14 controls and 4 cases. Sixty-two genes were affected by 66 variants of uncertain significance absent in controls with high case prevalence in >10% of cases. Three out of 66 variants were already known to be related to infertility (MSH3, GGT1 and AQP8). The 66 variants were clustered together giving rise to two subtypes of ovarian failure (A and B). Two genomic subtypes were differentiated. RF implemented on two models with a 10-fold CV (Model 1 and Model 2) yielded average accuracy, average sensitivity and average specificity values of 93.3%, 93.31% and 96.57%, respectively, in the Model 1 and yielded accuracy, average sensitivity and average specificity values of 97.2%, 97.2% and 99.2%, respectively, in Model 2. SPEP1 and GAB4 missense variants were responsible for the segregation of ovarian failure patients into 14.4% and 85.6%, which corresponds to types A and B, respectively [72]. The authors concluded that the identified variants are potential preventive biomarkers in fertility preservation programs.
VEST, CADD and bioinformatics analysis-based investigation of variants for POF [73]
Jin et al. studied the pathogenic variants responsible for POF by implementing bioinformatics analysis for variant filtration and ML algorithms VEST and CADD to identify the pathogenic variants [73]. WES analysis of 34 Korean individuals from Seoul CHA Hospital was performed along with 10 controls. A list of POF genes was downloaded from four public databases, DisGeNET, Monarch, MalaCards and NCBI; gene and variants were studied on VarSome. Nine heterozygous variants were identified by the screening of genes known to cause POF that were carried by 8 of the 34 individuals (24%). In four individuals, variants in Minichromosome maintenance-8 (MCM8) and MCM9 were identified. MCM8 and MCM9 are associated with homologous recombination and DNA repair [73]. In patient 15 (P15), a rare variant in the EIF2B3 gene and a variant in the PREPL gene were identified. The EIF2B3 variant is related to vanishing white matter (VWM) disease and leukodystrophy. Studies have proven that ovarian failure is generally seen in women with VWM disease. ERCC6 variant and an HFM1 variant were detected in two patients (P5 and P23). Moreover, SALL4 and TG variants were also detected. Seventy-two variants in 72 genes previously not known to be related to POF were identified. The authors also identified and reported new variants in the genes ADAMTSL1 and FER1L6, which may have a role in the development of reproductive organs and folliculogenesis [73]. The main advantage of this research was the confirmation that the WES model can be used for studying the genetic source of POF, which will in turn provide a better understanding of the disease pathogenesis. The authors recommend collecting data from a more diverse POF population to increase the prediction ability of the WES model.
Supervised SVM- and LR-based model implementation for predicting hypertension [74]
Held et al. presented a supervised ML model for the prediction of hypertension disease using gene expression and rare variant data [74]. They implemented three algorithms, radial SVM, linear SVM and LR on WGS and next-generation sequencing (NGS) retrieved from Genetic Analysis Workshop 19 (GAW19). The three algorithms LR, linear SVM and radial SVM gave similar AUC values of 0.771, 0.777 and 0.771, respectively, when no genetic data but only covariates were included. The performance of Linear SVM was slightly better than the others. Linear SVM provided an AUC score of 54.9%, the highest value compared to other algorithms in majority of the cases, while radial SVM and LR had AUC scores of 19.7% and 25.4%, respectively. The addition of the causal and non-causal genes lowered the AUC for all the models. High noise and a greater number of simulations also decreased the predictive ability of the algorithms. The follow-up analysis performed conveyed that rise in the sensitivity of the hypertension variable increased the AUC for three algorithms with 0.03, 0.05 and 0.01 increase in the average AUC for radial SVM, linear SVM and LR, respectively. The predictive ability was decreased by the inclusion of gene expression data with the addition of genotype data at the same loci. Finally, the authors concluded the need for the development of better models for these kinds of studies [74]. To test the predictive ability of this model, the authors used gene expression data as well as genotype data at the same loci in one analysis, and genotype data only in the second analysis. The authors concluded that this did not the increase predictive ability of their model. Additionally, a small sample size was used for the AUC model. The authors recommend that larger datasets are used in the future to maximize the development of the model.
Multi-ML implementation for the risk prediction and variant analysis of bacterial illness [75]
A study on Listeria monocytogenes risk analysis was performed by Njage et al., using supervised ML methods on variant data obtained from 245 strains [75]. A comparison of algorithms RF, SVM (radial and linear), Gradient Boosting, neural networks (NN) and logic boost was performed using the 10-fold CV [75]. This revealed that NN, GBM and SVM (linear kernel) had the best performance with accuracy values of 0.89, 0.88 and 0.89, respectively. The authors used the SVM-linear kernel model for the final model construction. For the final model, 70% of the data were used for training and the remaining 30% were used for testing. The confusion matrix delineates at least 67% was predicted accurately by the model. The final SVM-linear model implemented using 10-fold CV, yielded an accuracy value of 89%. The variable importance measure model identified Inlk, Auto, GtcA, InlJ, IisY, IisD, IisX, IisH, IisB, Ami, GadA, ActA, InlF, lmo2026, FAM002725, FAM002729, FAM002728, FAM003296, FAM003297 and FAM003164 as the top 20 important genes. Virulence genes associated with highest frequencies of illness were FAM002725, FAM002728, FAM002729, InlF, InlJ, Inlk, IisY, IisD, IisX, IisH, IisB, lmo2026, and FAM003296. The study of the occurrence of these genes in various types of food matrices of origin of isolates revealed that the genes InlJ, Inlk and lmo2026 were highly present in all the sources of the isolates whereas lmo2026 was highly associated with ready-to-eat-food isolates. InlF highly occurred in both clinical isolates, and the dairy and composite food origin isolates. FAM002725, FAM002728 and FAM002729 genes were highly prevalent in the composite food isolates. The InlF gene appeared to be truncated in one of the subpopulations of L. monocytogenes, which justifies the less severity of the illness in the strains with these genes. The authors concluded that a major advantage of this approach is its ability to predict bacterial pathogenesis allowing for better reaction time. This will help increase food safety. In addition, the prediction of new strains based on this approach will help reduce and prevent outbreaks. The authors propose that this model be used on other types of bacterial pathogens.
DT, RF, SVM and DNN implementation for the classification of sepsis [76]
Gene expression data for sepsis from GEO and EMBL-EBI Array Express were classified by Schaack et al. [76]. In this transcriptome-focused study, a total of 1786 samples were involved in this study. Out of 1786, 1354 were sepsis patients, 86 were systemic inflammatory response syndrome (SIRS) patients and 346 were healthy individuals who were used as controls. A comparison of different algorithms, like DT, RF, SVM and DNNs, with the conventional DE analysis was performed. DE analysis of sepsis and non-sepsis demonstrated that 2361 genes were differentially expressed. Agglomerative hierarchical clustering identified three main subgroups of samples in which one of the clusters reordered samples from both sepsis and non-sepsis samples implying that this method was not feasible for use on the varied dataset. Thus, the authors performed ML-based methods for a better and more reliable diagnosis than the conventional differential analysis. For DNNs, 80% data were used for training and the remaining 20% were used for testing and validation (15%—testing, 5%—validation), whereas for the other ML algorithms −85% of the data were used for training and the remaining 15% data were used for testing. The performance of RF, SVM and DNNs on unaltered data resulted in mean values of AUC and probability of correct classification (PCC) of about 0.99 and 0.96, respectively. The DT algorithm showed poor results yet had high diagnostic performance (mean AUC of 0.946 and mean PCC of 0.924). Resilience testing was done in three stages, the DNN model was the least affected and the most reliable algorithm for classification. DEGs were removed and the residual 3571 genes (~60%) were processed. It was observed that DNN model results retain the same quality of classification. Whereas the other algorithms were affected. The study suggested that the DNN algorithm was reliable for the subtyping of diseases like sepsis, SIRS and trauma even using a small set of original gene expression data for training [76].
Random committee ensemble learning for the classification and detection of prostate cancer [51]
Gumaei et al. proposed a supervised ML method for the classification and detection of prostate cancer using transcriptomic approach [51]. The correlation feature selection (CFS) method was used as a feature selection method. Random committee (RC) ensemble learning algorithm using a 10-fold CV technique was implemented on the gene expression data retrieved from the European Nucleotide Archive. The authors chose the CFS method as it takes the correlation among the features in the feature selection and chose the RC algorithm as it solves the overfitting problems. Thirty-eight genes from 2135 genes were selected as important features. The confusion matrix accurately classified 49/50 normal tissues and 48/52 prostate tumors. The accuracy value and weighted average F1-score for the approach were 95.098% and 95.1%, respectively. The proposed approach gave the high recall metric value for normal tissues and for prostate tumors of 0.98 and 0.923, respectively, and the weighted average result of the recall metric was 0.951. The method received a low false positive (FP) rate for normal tissue and prostate tumor of 0.077 and 0.020, respectively, and the weighted average result of the FP rate was 0.048. To authenticate the accuracy of this proposed approach, the authors performed an experiment with all these features. Later, the accuracy of the result was compared with the accuracy value from the method performed with selected features. It was observed that higher accuracy was achieved when selected features were used than when all the features were used. By comparing and visualizing the accuracy results for the proposed approach and other related work, it was noted that the proposed approach performed better than the related work methods and techniques and received the highest accuracy [51]. The authors recommended performing further analyses using gene expression data to provide better ML-based diagnosis for prostate cancer.
Supervised RF and SVM implementation for the identification of ASD clusters [77]
Lin et al. presented a supervised ML algorithm for the identification of clusters for ASD [77]. A total of 31 children diagnosed with ASD were recruited for this single omics study. RF classification and SVM were applied to gene expression data downloaded from GEO. The transcriptome-wide association analysis reported 191 probes associated with SCQ scores with a P < 0.00001 in which 54 DEGs were selected with a fold-change >2. Two clusters were identified by the RF-partitioning around medoid (RF-PAM) analysis with a classification accuracy of 67.7% when the top 10 PCs were used, and a classification accuracy of 96.9% when all 191 probes were used for the generation of proximity matrix. The SVM approach implemented in 7:3 ratio of training data to testing data with top 10 PC scores gave a classification accuracy of 93.3% and a classification accuracy of 99.9% when all 191 probes were used in the analysis. The SVM clustering results presented that the first two principal components might classify support vectors with improved prediction confidence and equated with the results predicted by the individual probes. The authors concluded that both methods are effective and acceptable [77]. The authors propose that this method can be used for timely intervention of ASD which will allow appropriate interventions.
Gradient Boosting strategy for the identification of tumor tissue of origin [78]
In this study, Li et al. proposed a method to identify tumor tissue of origin in 20 types of solid tumors using the Gradient Boosting Decision Tree (GBDT) [78]. Expression data from GEO was used for this analysis. 20 501 genes in a total of 7713 samples from the TCGA data set were considered. Feature selection using GBDT determined the top 400 gene features. A 10-fold CV was implemented. GBDT was compared with other algorithms like K-NN, DT, AB and SVM. It was concluded that GBDT had the most accuracy of 96.1% for 20 cancer types and 83.5% for independent datasets. It was detected that the genes HOXB13, C19orf33, CRYAB, ACTG2, ACTA2, IGFBP2, CSRP1, RAB34, SMS, MAGOH, C21orf33, IDI1, TRIM27, ACTL6A and ILVBL had higher expression whereas the genes OR14A16, CRP and INS had lower expression [78]. The limitation of this research is limited to the small sample size as it can lead to incorrect predictions due to similar gene expression in the samples. The authors propose increasing the sample size to address this problem.
RF, Gradient Boosting and XGBoost for the identification of cancer-selective combinatorial therapies in ovarian cancer [79]
He et al. worked on combinational therapies for ovarian cancer. Supervised ML algorithms were implemented: RF, Gradient Boosting and XGBoost [79]. Binary variables were used as algorithm inputs for the prediction of 423 drug targets and 110 point mutations. WGS data from 4 HGSOC patients along with the expression levels of 698 cancer genes were studied. The genes consisted of ovarian and pan-cancer markers. The ovarian cancer markers were taken from the overexpressed genes seen in the differential gene expression of 76 HGSOC samples from the HERCULES study [79]. The pan-cancer markers were collected from AstraZeneca-Sanger Drug Combination Prediction DREAM Challenge. The pan-cancer genes are linked with cancer development, tumor suppressors and drug sensitivity or resistance. The whole data were split into two parts, training data (90%) and testing data (10%), and a 10-fold CV was used with a leave-drug-out CV setup. The study of the effect of using scRNA-seq from the EOC0939_pAsc sample on prediction indicated improvement in the prediction power of EOC0939 responses. There was no effect seen when scRNA-seq was considered for all samples, which suggests that scRNA-seq has importance. Model comparison identified that in four patient subpopulations [EOC0939_pAsc PAX8-, EOC1103_pOme1 PAX8+, EOC1103_pOme PAX8-, EOC1107_pAsc PAX8-], a single multi-patient model performed better than the patient-specific models. This indicated that using information from other patients enhanced predictions [79]. It was observed that patient-specific predictions were performed by the multi-patient models. The GB algorithm along with scRNA-seq yielded high accurate prediction. The model performed better with the low efficacy drugs. The drug combination predictions ranked the top combinations and one of the combinations was selected considering the robustness of the drug response, translational potential of the combination, previous success of the single agents of the combination in clinical trials and the known mechanistic interactions of the drug targets. Visualization by patientNet R/Shiny web application was done to understand the patient-customized co-vulnerability networks. A combination of Vistusertib (mTOR inhibitor) and A1155463 (BCL2L1 inhibitor) was selected for the sample EOC0939_pAsc, whereas for EOC1103pOme1 a combination of between Cobimetinib (MAP2K2 inhibitor) and BMS777607 (DDR1, MET and MERTK inhibitor) was selected. A combination of Verdinexor (XPO1 inhibitor) and AZD-8186 (PIK3C inhibitor) for EOC1103_pPer1 and for EOC1107_pAsc combination between AZD-5363 (AKT1/2 inhibitor) and Panobinostat (HDAC9 inhibitor) was selected [79]. A crucial benefit of this research is the predictive model that was created to consider the molecular diversity of cancer cells as well as the possible non-selectivity of drug interactions with target cells.
Discussion
To advance precision medicine, we need to apply intelligent prediction methods for finding disease-causal genetic variants with interactive AI/ML-based analysis, deep phenotyping and effective visualization [105]. We can accelerate our ability by using AI/ML algorithms to leverage and extend the information contained within the original data, and modelling patient-specific data (e.g. genomics, clinical and phenotypic) against publicly available annotation data (e.g. genes, variants, drugs and diseases) for understanding how coding and non-coding genomic variations connect to disease mechanisms. This will help us comprehend a wide variety of problems that we currently have trouble understanding and will have enormous impact in both basic and medical sciences. The current constraints in the implementation of AI/ML include, but are not limited to, recruitment of patients for research studies; translational integration of genomics and diversified public datasets; and development of AI/ML systems for data-intensive computational modelling to assist clinical decision-making. To enable a more widespread acceptance of AI/ML in clinical practice, the following grand challenges must be addressed: (i) handling inherent error rates in genome-wide and clinical data; (ii) generation, assessment and dissemination of continuous, longitudinal and AI/ML-ready datasets; (iii) dealing with the class imbalance problem [120, 121] and (iv) incorporating data standards, tools, and ethical and trustworthy AI/ML principles. Accurate and right evaluation methods are needed to assess the performance of ML models. We observed that small datasets often yield a greater classification accuracy, and this could be because of bias that is produced [122].
Some common limitations in using AI/ML algorithms involved small sample size of the disease datasets, which leads to uncertainty of a model’s predictive ability [60, 74, 78]. The model’s predictive capabilities are reduced without a strong training dataset. Small sample size can lead to incorrect predictions due to similar gene expression in the samples. Larger datasets are needed to maximize the development of the mature model. Another constraint that was observed as a direct cause of a smaller dataset and lack of external validation was the overfitting bias that needs to be addressed in future work [61]. Other limitations focus on the individual models when compared to one another. The strength of the AVA,Dx model lies in sequencing methods and panel differences. The AVA,Dx model’s predictive capabilities diminished when the test panel sequences did not share enough loci with the disease-training panel. In addition, the AVA,Dx model could only recognize the genetic model of disease patients rather than healthy patients. Thus, it is unable to label unaffected patients as healthy [47]. OncoCast-MPM model requires regular updates [48]. The predictive ability of SVM was decreased by the inclusion of gene expression data with the addition of genotype data at the same loci. The DNN algorithm was reliable for the subtyping of diseases. The major benefits of utilizing an AI/ML model are the identification of novel gene–disease associations giving a better understanding of disease biology and providing early diagnosis or even preventing ailments altogether. They could be used to accurately diagnose a disease and present an initial prognosis that can aid medical personnel [47, 48, 61–71]. Other findings could include gene sets associated with chronic disorders that could be used as biomarkers for drug targets [47, 48, 59–71]. Some models accurately predicted drug responses to a disease. Feature selection also impacted the outcome of the model and can result in clustering. Clinical characteristics and environmental factors can also restrain the predictive power of the model.
The most widely used AI/ML algorithms are RF and SVM. The DNN algorithm was found to be reliable for subtyping of diseases even using a small set of original gene expression data for training. AVA,Dx could be used to construct a model even with a small sample size. Classifying tasks based on available predictor variables is a key step to correctly address the problem of choosing a suitable AI/ML algorithm. List of variables used in different AI/ML algorithms include abundances with transcripts per million, mean expressed transcript length, quantified gene, quality metrics, fragments per kilobase million, mapping quality, individual with variant, allele count, genetic loci-reference position, list of variants, high/moderate impact variants, genotype and filters (Table 3). It was also noted that these AI/ML algorithms, depending on the disease, may give variable prediction for disease diagnosis. Some beneficial factors were limited to the scope of the papers. For instance, authors identified the variants that can be used as a selective biomarker when it comes to fertility preservation programs as a substitute for other invasive procedures and tests [72]. While others were able to use their algorithms to diagnosis cancers of unknown primary origins using gradient boosting classifiers [78]. A major suggestion cited by the authors was that their models be validated in further studies in larger and diverse datasets such as transcriptome and microarray gene expression data [48, 51, 63, 69, 70, 73, 74] to allow for increased accuracy.
Table 3.
List of variables used in AI/ML algorithms using gene-expression and gene variant data
| Algorithm name | Variables: Gene expression | Variables: Gene variant |
|---|---|---|
| SVM | TPM; METL; QG; QM; FPKM | MQ; I/V; AC; GL; filters |
| RF | TPM; METL; QM; FPKM | MQ; GL; L/V; M/IV; filters |
| DT | METL; TPM; QG; FPKM | GL |
| LR | TPM; METL; QG | AC; genotype |
| ANN | TPM; METL; QG | Genotype |
| K-nearest neighbor (k-NN) | QG; METL | AC |
| Gradient Boosting (GB) | FPKM; METL | AC |
| NB | QG | I/V |
| AB | TPM; METL | N/A |
| XGBoost | TPM; METL | M/IV; filters |
| Elastic net regularized generalized linear model | TPM | N/A |
| BART | N/A | I/V |
| Bayesian networks | N/A | I/V |
| Greedy Thick Thinning algorithm | N/A | I/V |
| LDA | METL; QG | N/A |
| QDA | METL; QG | N/A |
| GPC | METL; QG | N/A |
| NMF | N/A | AC |
| C4.5 | N/A | AC |
| FCA | N/A | Genotype |
| Clustering (Unsupervised) | N/A | Filters; GL |
| MLR | N/A | I/V |
| GA | N/A | I/V |
| Logit Boost | N/A | GL |
| AVA,Dx | N/A | MQ |
| OncoCast-MPM machine-learning risk-prediction model | N/A | M/IV |
| CADD | N/A | M/IV; filters |
| Very Efficient Substitution Transposition (VEST) | N/A | M/IV; filters |
| Random committee ensemble learning | QG | N/A |
| DNNs | QM | N/A |
| MERGE | QG | N/A |
| EM | N/A | I/V |
Table includes following variables: abundances with transcripts per million (TPM); mean expressed transcript length (METL); quantified gene (QG); quality metrics (QM); fragments per kilobase million (FPKM); mapping quality (MQ); individual with variant (I/V); allele count (AC); genetic loci reference position (GL); list of variants (L/V); high/moderate impact variants (MI/V); genotype; and filters. N/A stands for not applicable.
To efficiently implement AI/ML in genomics, it is important to properly construct the prediction model. Critical steps involve (i) data collection, quality inspection, cleansing and AI/ML-ready generation; (ii) data modelling with the establishment of correct associations between predictive input variables and expected outcomes and (iii) training and validation of the model to evaluate the predictive performance [106]. During cases, when gene variant and gene expression data are high volume, it is significant to ensure the right balance between training and actual datasets to avoid overfitting. Knowledgebase of phenotypes and biomarkers is required to perform longitudinal population study for analyzing the effects of treatments and establishment of relevant scientific research [105]. We need to generate AI/ML-ready datasets, and create training data models to apply ML algorithms for predictive analysis. Currently, processed and analyzed gene expression and gene variant data through available genomic pipelines are not available in AI/ML-ready formats. With its availability as AI/ML input, it can be directly used for predictive analysis and deep phenotyping. There is an unmet need to develop cross-platform, user/researcher/physician-friendly and AI/ML-driven scientific software applications to facilitate automated, reproducible, and timely heterogeneous and high-volume gene variant and gene expression predictive data analysis in clinical settings.
The clinical interpretation of the significance of specific gene variants can be unique to a patient [105]. Therefore, it critical to understand how diseases are related to each other and the genetic basis of common diseases, which genes predispose one to a medical condition, and how rare genetic variants contribute to diseases [106]. We need to integrate clinical and genomics data for deep phenotyping and to reveal cases where absence of genotype–phenotype associations likely resulted in uncertainty in patient care. This will be established by assimilating past and current personal medical history with whole genome and transcriptome sequences to tailor therapy with the best response by analyzing an individuals’ genetic makeup. However, a key challenge in this realm is NGS interpretation with clinical relevance. Variability in interpretation for sequence variants is due, in part, to the lack of standard curated information to support clinical decision-making. With the rightful application of AI/ML approaches, we need to support researchers in identifying biomarkers for health assessment and supporting the process of genetic testing with the classification of susceptibility genes to detect changes of clinical significance necessary for customized therapy and personalized treatment.
In this study, our focus was primarily on WGS/WES-based gene variant and RNA-seq-driven gene expression data. However, genomics is not limited to these but include other NGS data types. Recently, scientists have carried out and reported research implementing different AI/ML approaches using single-cell RNA sequencing (scRNA-seq) [107–116] (e.g. clustering single-cell RNA-sequencing data [107]; discovery of minimum marker gene combinations for cell type [108]; valuating distribution and prognostic value of new tumor-infiltrating lymphocytes [109]; modelling cellular complex systems in Alzheimer’s disease [110]; systematic experiments on ab initio knowledge discovery with ML methods on single-cell RNA-seq data of early embryonic development [111]; investigating functionally significant interactions within and between cancer cells and T cells [112]; implementing ML for spatially resolved transcriptomics (SRT) data analysis [113]; Deconvolute gene expression data with deep learning [114]; functional classification of regulatory elements from single-cell and bulk ATAC-seq data with deep learning [115]; and predict regulatory networks and key regulatory genes with ML [116]) and Single-Cell ATAC-Seq (scATAC-Seq) [117–119] (e.g. BABEL generated single-cell expression for fine-grained classification of complex cell states [117]; using SCATE to estimate activities of individual CREs [118] and using deep learning to identify the islet cell type of action across genetic signals of type 2 diabetes predisposition [119]). Going forward, it is important to implement and investigate the potential of different AI approaches and ML algorithms at heterogeneous NGS data types for effective, predictive, genomics and personalized medicine.
Conclusion
SVM and RF are the most applied AI/ML algorithms, and GB, LR, ANN, NB, K-NN, dDT and AB are among the commonly used AI/ML algorithms in genomics for bioinformatics, statistics and predictive analyses of a wide variety of diseases. While SVM provides high accuracy for both regression and classification tasks, its computing ability is limited to smaller datasets. Additionally, SVM hyperparameters need to be adjusted to prevent overfitting and underfitting. It also tends to underperform when the target classes overlap or when the number of properties for each data point is greater than the number of training data specimens. Deep neural network algorithms like k-NN and ANN are usually preferred over SVM when it comes to larger datasets. RF is a type of ensemble learning model that employs decision trees to predict an outcome to solve regression and classification problems. RF is preferred over SVM when it comes to small datasets as it can provide predictions without the need for hyperparameter tuning. However, the drawbacks of RF are not limited to its slow computing ability with larger datasets. In addition to its being time-consuming to create so many trees for prediction, instances of covariate shift do not allow for the extrapolation of data. Thus, reducing the inerrability of the decision trees. XGBoost and deep neural network are two algorithms that are usually preferred to RF due to their high computing capability and accuracy. Furthermore, to support efficient implementation of AI/ML approaches, our intensive search showed that no prior research has been reported for the AI/ML-ready gene expression and variant data generation.
Key Points
The convergence of genomics and transcriptomics data, along with staggering developments in AI/ML, has the potential to elevate diagnostic and predictive analyses of major causes of mortality, modifiable risk factors and other clinically actionable information.
AI/ML approaches can utilize broad dataset sizes with heterogeneous levels of granularity and offer multiple supervised and unsupervised approaches to analyze gene variant and gene expression data with the potential for development of multivariate statistical tools.
To practice genetic and AI/ML-driven personalized medicine, we need to develop pipelines to generate and disseminate AI/ML-ready data, and address ethical issues, which involve protected health information associated with genomic datasets.
Supplementary Material
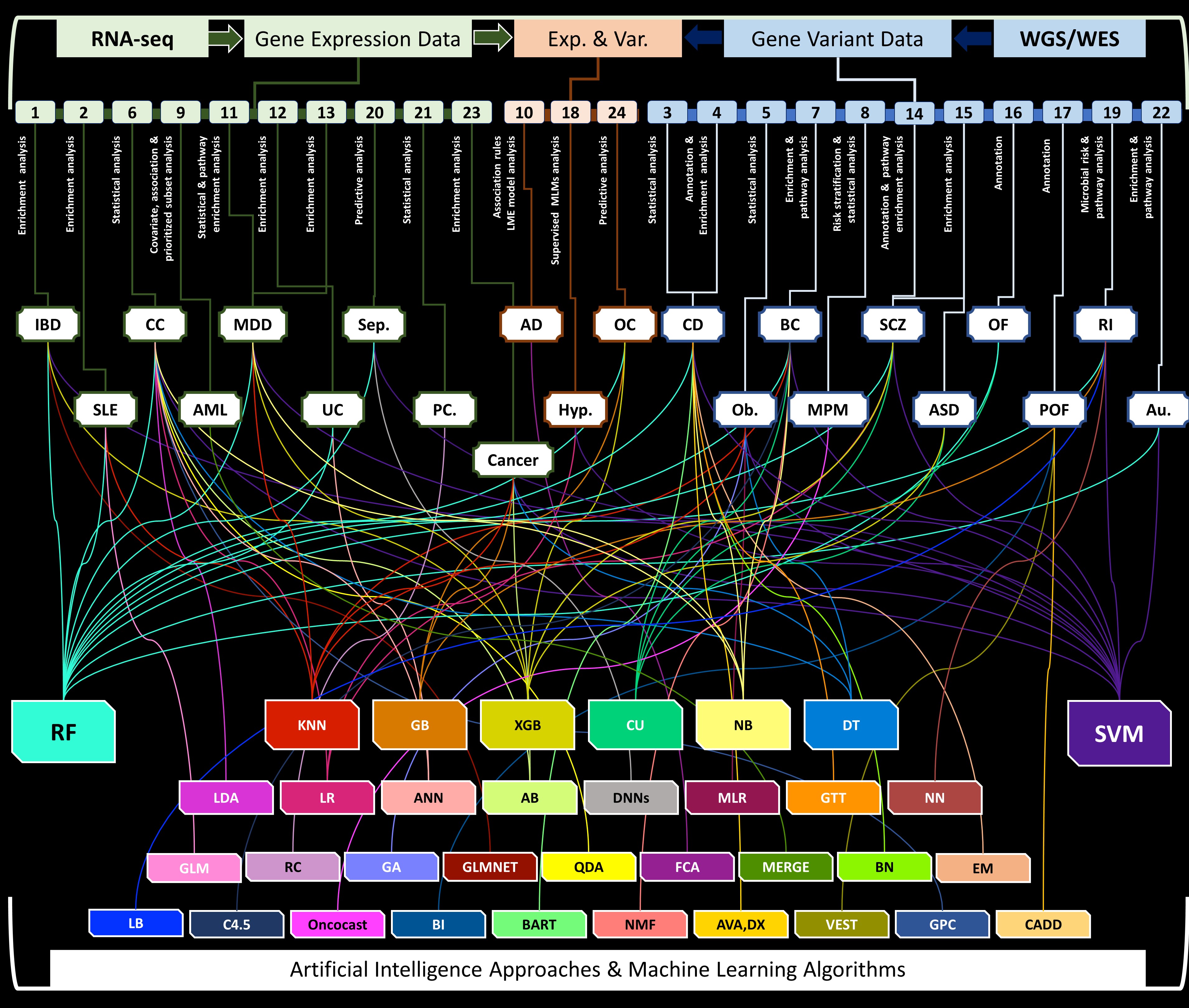{kind=link}
Acknowledgements
We appreciate great support by the Rutgers Institute for Health, Health Care Policy and Aging Research (IFH); Department of Medicine, Rutgers Robert Wood Johnson Medical School (RWJMS); and Rutgers Biomedical and Health Sciences (RBHS), at the Rutgers, The State University of New Jersey. We thank members and collaborators of Ahmed Lab at the Rutgers (IFH, RWJMS, RBHS) for their support, participation and contribution to this study. We would like to give special thanks to Dr Christopher Bonin and Dr Geneva Hargis for stylistic and native speaker corrections.
Author Biographies
Sreya Vadapalli is a research assistant at the Ahmed Lab, Rutgers Institute for Health, Health Care Policy and Aging Research, Rutgers University-New Brunswick, NJ.
Habiba Abdelhalim is a research assistant at the Ahmed Lab, Rutgers Institute for Health, Health Care Policy and Aging Research, Rutgers University-New Brunswick, NJ.
Saman Zeeshan is a bioinformatics research scientist and postdoctoral research associate at the Rutgers Cancer Institute of New Jersey, Rutgers University-New Brunswick, NJ.
Zeeshan Ahmed is an assistant professor of Medicine – Tenure Track and Core Member at the Rutgers Institute for Health, Health Care Policy and Aging Research; and Department of Medicine – Division of General Internal Medicine, Rutgers Robert Wood Johnson Medical School, Rutgers Biomedical and Health Sciences, Rutgers University-New Brunswick. Zeeshan Ahmed is an adjunct assistant professor at the Department of Genetics and Genome Sciences, UConn School of Medicine, UConn Health, CT, and a full academic member of the Rutgers Microbiology and Molecular Genetics; Center for Cancer Health Equity, Rutgers Cancer Institute of New Jersey; Rutgers Human Genetics Institute of New Jersey, NJ.
Contributor Information
Sreya Vadapalli, Rutgers Institute for Health, Health Care Policy and Aging Research, Rutgers University, 112 Paterson St, New Brunswick, NJ, USA.
Habiba Abdelhalim, Rutgers Institute for Health, Health Care Policy and Aging Research, Rutgers University, 112 Paterson St, New Brunswick, NJ, USA.
Saman Zeeshan, Rutgers Cancer Institute of New Jersey, Rutgers University, 195 Little Albany St, New Brunswick, NJ, USA.
Zeeshan Ahmed, Rutgers Institute for Health, Health Care Policy and Aging Research, Rutgers University, 112 Paterson St, New Brunswick, NJ, USA; Department of Medicine, Robert Wood Johnson Medical School, Rutgers Biomedical and Health Sciences, 125 Paterson St, New Brunswick, NJ, USA.
Author contributions
Z.A. proposed and led the study. S.V., H.A. and Z.A. conducted review and comparative research. S.Z. and Z.A. evaluated reported findings. Z.A. drafted the manuscript, and all authors participated in writing, revision and review and have approved it for publication.
Funding
Institute for Health, Health Care Policy and Aging Research (IFH); Rutgers Robert Wood Johnson Medical School, Rutgers Biomedical and Health Sciences at the Rutgers, The State University of New Jersey.
References
- 1. Zeeshan S, Xiong R, Liang BT, et al. 100 Years of evolving gene-disease complexities and scientific debutants. Brief Bioinform 2020;21(3):885–905. 10.1093/bib/bbz038. [DOI] [PubMed] [Google Scholar]
- 2. Ahmed Z, Zeeshan S, Mendhe D, et al. Human gene and disease associations for clinical-genomics and precision medicine research. Clin Transl Med 2020;10(1):297–318. 10.1002/ctm2.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martin AR, Kanai M, Kamatani Y, et al. Publisher correction: clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 2021;53(5):763. 10.1038/s41588-021-00797-z. [DOI] [PubMed] [Google Scholar]
- 4. Ahmed Z, Renart EG, Zeeshan S. Genomics pipelines to investigate susceptibility in whole genome and exome sequenced data for variant discovery, annotation, prediction and genotyping. PeerJ 2021;9:e11724. 10.7717/peerj.11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahmed Z, Renart EG, Mishra D, et al. JWES: a new pipeline for whole genome/exome sequence data processing, management, and gene-variant discovery, annotation, prediction, and genotyping. FEBS Open Bio 2021;11(9):2441–52. 10.1002/2211-5463.13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ahmed Z, Renart EG, Zeeshan S, et al. Advancing clinical genomics and precision medicine with GVViZ: FAIR bioinformatics platform for variable gene-disease annotation, visualization, and expression analysis. Hum Genomics 2021;15(1):37. 10.1186/s40246-021-00336-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med 2020;12(1):44. 10.1186/s13073-020-00742-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahmed Z. Practicing precision medicine with intelligently integrative clinical and multi-omics data analysis. Hum Genomics 2020;14(1):35. 10.1186/s40246-020-00287-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahmed Z, Mohamed K, Zeeshan S, et al. Artificial intelligence with multi-functional machine learning platform development for better healthcare and precision medicine. Database (Oxford) 2020;2020:baaa010. 10.1093/database/baaa010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahmed Z. Intelligent health system for the investigation of consenting COVID-19 patients and precision medicine. Pers Med 2021;18(6):573–82. 10.2217/pme-2021-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rigatti SJ. Random Forest. J Insur Med 2017;47(1):31–9. 10.17849/insm-47-01-31-39.1. [DOI] [PubMed] [Google Scholar]
- 12. Chen X, Ishwaran H. Random forests for genomic data analysis. Genomics 2012;99(6):323–9. 10.1016/j.ygeno.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Byvatov E, Schneider G. Support vector machine applications in bioinformatics. Appl Bioinforma 2003;2(2):67–77. [PubMed] [Google Scholar]
- 14. Huang S, Cai N, Pacheco PP, et al. Applications of support vector machine (SVM) learning in cancer genomics. Cancer Genomics Proteomics 2018;15(1):41–51. 10.21873/cgp.20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. González-Recio O, Jiménez-Montero JA, Alenda R. The gradient boosting algorithm and random boosting for genome-assisted evaluation in large data sets. J Dairy Sci 2013;96(1):614–24. 10.3168/jds.2012-5630. [DOI] [PubMed] [Google Scholar]
- 16. Ying J, Wang Q, Xu T, et al. Diagnostic potential of a gradient boosting-based model for detecting pediatric sepsis. Genomics 2021;113(1 Pt 2):874–83. 10.1016/j.ygeno.2020.10.018. [DOI] [PubMed] [Google Scholar]
- 17. Liu K, Chen W, Lin H. XG-PseU: an eXtreme Gradient Boosting based method for identifying pseudouridine sites. Mol Gen Genom 2020;295(1):13–21. 10.1007/s00438-019-01600-9. [DOI] [PubMed] [Google Scholar]
- 18. Parente DJ. PolyBoost: an enhanced genomic variant classifier using extreme gradient boosting. Proteomics Clin Appl 2021;15(2–3):e1900124. 10.1002/prca.201900124. [DOI] [PubMed] [Google Scholar]
- 19. Ogutu JO, Schulz-Streeck T, Piepho HP. Genomic selection using regularized linear regression models: ridge regression, lasso, elastic net and their extensions. BMC Proc 2012;6(Suppl 2):S10. 10.1186/1753-6561-6-S2-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Candia J, Tsang JS. eNetXplorer: an R package for the quantitative exploration of elastic net families for generalized linear models. BMC Bioinformatics 2019;20(1):189. 10.1186/s12859-019-2778-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nick TG, Campbell KM. Logistic regression. Methods Mol Biol 2007;404:273–301. 10.1007/978-1-59745-530-5_14. [DOI] [PubMed] [Google Scholar]
- 22. Sperandei S. Understanding logistic regression analysis. Biochem Med 2014;24(1):12–8. 10.11613/BM.2014.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zou J, Han Y, So SS. Overview of artificial neural networks. Methods Mol Biol 2008;458:15–23. 10.1007/978-1-60327-101-1_2. [DOI] [PubMed] [Google Scholar]
- 24. Zhang Z. A gentle introduction to artificial neural networks. Ann Transl Med 2016;4(19):370. 10.21037/atm.2016.06.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmidhuber J. Deep learning in neural networks: an overview. Neural Netw 2015;61:85–117. 10.1016/j.neunet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 26. Langarizadeh M, Moghbeli F. Applying naive Bayesian networks to disease prediction: a systematic review. Acta Informatica Medica 2016;24(5):364–9. 10.5455/aim.2016.24.364-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malovini A, Barbarini N, Bellazzi R, et al. Hierarchical naive Bayes for genetic association studies. BMC Bioinformatics 2012;13 Suppl 14(Suppl 14):S6. 10.1186/1471-2105-13-S14-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tan YV, Roy J. Bayesian additive regression trees and the general BART model. Stat Med 2019;38(25):5048–69. 10.1002/sim.8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Friedman N, Linial M, Nachman I, et al. Using Bayesian networks to analyze expression data. J Comput Biol 2000;7(3–4):601–20. 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 30. Liu Z, Malone B, Yuan C. Empirical evaluation of scoring functions for Bayesian network model selection. BMC Bioinformatics 2012;13(Suppl 15):S14. 10.1186/1471-2105-13-S15-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Z. Introduction to machine learning: k-nearest neighbors. Ann Transl Med 2016;4(11):218. 10.21037/atm.2016.03.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kingsford C, Salzberg SL. What are decision trees? Nat Biotechnol 2008;26(9):1011–3. 10.1038/nbt0908-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ricciardi C, Valente AS, Edmund K, et al. Linear discriminant analysis and principal component analysis to predict coronary artery disease. Health Informatics J 2020;26(3):2181–92. 10.1177/1460458219899210. [DOI] [PubMed] [Google Scholar]
- 34. Ryback RS, Eckardt MJ, Rawlings RR, et al. Quadratic discriminant analysis as an aid to interpretive reporting of clinical laboratory tests. JAMA 1982;248(18):2342–5. 10.1001/jama.1982.03330180088048. [PubMed] [Google Scholar]
- 35. Liu H, Ong YS, Yu Z, et al. Scalable Gaussian process classification with additive noise for non-Gaussian likelihoods. IEEE Trans Cybern 2021;1–13. 10.1109/TCYB.2020.3043355. [DOI] [PubMed] [Google Scholar]
- 36. Chen S, Shen B, Wang X, et al. A strong machine learning classifier and decision stumps based hybrid AdaBoost classification algorithm for cognitive radios. Sensors (Basel, Switzerland) 2019;19(23):5077. 10.3390/s19235077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Z, Michailidis G. A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinformatics (Oxford, England) 2016;32(1):btv544–8. 10.1093/bioinformatics/btv544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frigyesi A, Höglund M. Non-negative matrix factorization for the analysis of complex gene expression data: identification of clinically relevant tumor subtypes. Cancer Informat 2008;6:275–92. 10.4137/cin.s606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lamy JB, Ellini A, Ebrahiminia V, et al. Use of the C4.5 machine learning algorithm to test a clinical guideline-based decision support system. Stud Health Technol Inform 2008;136:223–8. [PMC free article] [PubMed] [Google Scholar]
- 40. Wiharto W, Kusnanto H, Herianto H. Interpretation of clinical data based on C4.5 algorithm for the diagnosis of coronary heart disease. Healthc Inform Res 2016;22(3):186–95. 10.4258/hir.2016.22.3.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keller BJ, Eichinger F, Kretzler M. Formal concept analysis of disease similarity. AMIA Joint Summits on Translational Science proceedings. AMIA Jt Summits Transl Sci 2012;2012:42–51. [PMC free article] [PubMed] [Google Scholar]
- 42. Frades I, Matthiesen R. Overview on techniques in cluster analysis. Methods Mol Biol 2010;593:81–107. 10.1007/978-1-60327-194-3_5. [DOI] [PubMed] [Google Scholar]
- 43. Rodriguez MZ, Comin CH, Casanova D, et al. Clustering algorithms: a comparative approach. PLoS One 2019;14(1):e0210236. 10.1371/journal.pone.0210236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eberly LE. Multiple linear regression. Methods Mol Biol 2007;404:165–87. 10.1007/978-1-59745-530-5_9. [DOI] [PubMed] [Google Scholar]
- 45. Katoch S, Chauhan SS, Kumar V. A review on genetic algorithm: past, present, and future. Multimed Tools Appl 2020;80(5):8091–126. 10.1007/s11042-020-10139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim K, Seo M, Kang H, et al. Application of LogitBoost classifier for traceability using SNP Chip data. PLoS One 2015;10(10):e0139685. 10.1371/journal.pone.0139685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Y, Miller M, Astrakhan Y, et al. Identifying Crohn's disease signal from variome analysis. Genome Med 2019;11(1):59. 10.1186/s13073-019-0670-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zauderer MG, Martin A, Egger J, et al. The use of a next-generation sequencing-derived machine-learning risk-prediction model (OncoCast-MPM) for malignant pleural mesothelioma: a retrospective study. Lancet Digital Health 2021;3(9):e565–76. 10.1016/S2589-7500(21)00104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rentzsch P, Witten D, Cooper GM, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47(D1):D886–94. 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Douville C, Masica DL, Stenson PD, et al. Assessing the pathogenicity of insertion and deletion variants with the Variant Effect Scoring Tool (VEST-Indel). Hum Mutat 2016;37(1):28–35. 10.1002/humu.22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gumaei A, Sammouda R, Al-Rakhami M, et al. Feature selection with ensemble learning for prostate cancer diagnosis from microarray gene expression. Health Informatics J 2021;27(1):1460458221989402. 10.1177/1460458221989402. [DOI] [PubMed] [Google Scholar]
- 52. Choi RY, Coyner AS, Kalpathy-Cramer J, et al. Introduction to machine learning, neural networks, and deep learning. Transl Vis Sci Technol 2020;9(2):14. 10.1167/tvst.9.2.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Georgevici AI, Terblanche M. Neural networks and deep learning: a brief introduction. Intensive Care Med 2019;45(5):712–4. 10.1007/s00134-019-05537-w. [DOI] [PubMed] [Google Scholar]
- 54. Attimonelli M, Lanave C, Liuni S, et al. MERGE: a software package for generating a single data-base starting from EMBL and GenBank collections. Nucleic Acids Res 1988;16(5):1681–2. 10.1093/nar/16.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Do CB, Batzoglou S. What is the expectation maximization algorithm? Nat Biotechnol 2008;26(8):897–9. 10.1038/nbt1406. [DOI] [PubMed] [Google Scholar]
- 56. Zheng B, Agresti A. Summarizing the predictive power of a generalized linear model. Stat Med 2000;19(13):1771–81. [DOI] [PubMed] [Google Scholar]
- 57. Eddy SR. Hidden Markov models. Curr Opin Struct Biol 1996;6(3):361–5. 10.1016/s0959-440x(96)80056-x. [DOI] [PubMed] [Google Scholar]
- 58. de Hond A, Leeuwenberg AM, Hooft L, et al. Guidelines and quality criteria for artificial intelligence-based prediction models in healthcare: a scoping review. NPJ Digital Med 2022;5(1):2. 10.1038/s41746-021-00549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Isakov O, Dotan I, Ben-Shachar S. Machine learning-based gene prioritization identifies novel candidate risk genes for inflammatory bowel disease. Inflamm Bowel Dis 2017;23(9):1516–23. 10.1097/MIB.0000000000001222. [DOI] [PubMed] [Google Scholar]
- 60. Kegerreis B, Catalina MD, Bachali P, et al. Machine learning approaches to predict lupus disease activity from gene expression data. Sci Rep 2019;9(1):9617. 10.1038/s41598-019-45989-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Menti E, Lanera C, Lorenzoni G, et al. Bayesian machine learning techniques for revealing complex interactions among genetic and clinical factors in association with extra-intestinal manifestations in IBD patients. AMIA Annu Symp Proc 2017;2016:884–93. [PMC free article] [PubMed] [Google Scholar]
- 62. Wang HY, Chang SC, Lin WY, et al. Machine learning-based method for obesity risk evaluation using single-nucleotide polymorphisms derived from next-generation sequencing. J Comput Biol 2018;25(12):1347–60. 10.1089/cmb.2018.0002. [DOI] [PubMed] [Google Scholar]
- 63. Maniruzzaman M, Jahanur Rahman M, Ahammed B, et al. Statistical characterization and classification of colon microarray gene expression data using multiple machine learning paradigms. Comput Methods Prog Biomed 2019;176:173–93. 10.1016/j.cmpb.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 64. Vural S, Wang X, Guda C. Classification of breast cancer patients using somatic mutation profiles and machine learning approaches. BMC Syst Biol 2016;10(Suppl 3):62. 10.1186/s12918-016-0306-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee SI, Celik S, Logsdon BA, et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat Commun 2018;9(1):42. 10.1038/s41467-017-02465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hampel H, Williams C, Etcheto A, et al. A precision medicine framework using artificial intelligence for the identification and confirmation of genomic biomarkers of response to an Alzheimer's disease therapy: analysis of the Blarcamesine (ANAVEX2-73) phase 2a clinical study. Alzheimers Dement 2020;6(1):e12013. 10.1002/trc2.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhao S, Bao Z, Zhao X, et al. Identification of diagnostic markers for major depressive disorder using machine learning methods. Front Neurosci 2021;15:645998. 10.3389/fnins.2021.645998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li H, Lai L, Shen J. Development of a susceptibility gene based novel predictive model for the diagnosis of ulcerative colitis using random forest and artificial neural network. Aging 2020;12(20):20471–82. 10.18632/aging.103861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Qi B, Ramamurthy J, Bennani I, et al. Machine learning and bioinformatic analysis of brain and blood mRNA profiles in major depressive disorder: a case-control study. Am J Med Genetics B Neuropsychiatr Genetics 2021;186(2):101–12. 10.1002/ajmg.b.32839. [DOI] [PubMed] [Google Scholar]
- 70. Trakadis YJ, Sardaar S, Chen A, et al. Machine learning in schizophrenia genomics, a case-control study using 5,090 exomes. Am J Med Genet B Neuropsychiatr Genet 2019;180(2):103–12. 10.1002/ajmgb.32638. [DOI] [PubMed] [Google Scholar]
- 71. Sardaar S, Qi B, Dionne-Laporte A, et al. Machine learning analysis of exome trios to contrast the genomic architecture of autism and schizophrenia. BMC Psychiatry 2020;20(1):92. 10.1186/s12888-020-02503-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Henarejos-Castillo I, Aleman A, Martinez-Montoro B, et al. Machine learning-based approach highlights the use of a genomic variant profile for precision medicine in ovarian failure. J Pers Med 2021;11(7):609. 10.3390/jpm11070609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jin H, Ahn J, Park Y, et al. Identification of potential causal variants for premature ovarian failure by whole exome sequencing. BMC Med Genet 2020;13(1):159. 10.1186/s12920-020-00813-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Held E, Cape J, Tintle N. Comparing machine learning and logistic regression methods for predicting hypertension using a combination of gene expression and next-generation sequencing data. BMC Proc 2016;10(Suppl 7):141–5. 10.1186/s12919-016-0020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Njage P, Henri C, Leekitcharoenphon P, et al. Machine learning methods as a tool for predicting risk of illness applying next-generation sequencing data. Risk Anal 2019;39(6):1397–413. 10.1111/risa.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schaack D, Weigand MA, Uhle F. Comparison of machine-learning methodologies for accurate diagnosis of sepsis using microarray gene expression data. PLoS One 2021;16(5):e0251800. 10.1371/journal.pone.0251800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lin PI, Moni MA, Gau SS, et al. Identifying subgroups of patients with autism by gene expression profiles using machine learning algorithms. Front Psychol 2021;12:637022. 10.3389/fpsyt.2021.637022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li R, Liao B, Wang B, et al. Identification of tumor tissue of origin with RNA-Seq data and using gradient boosting strategy. Biomed Res Int 2021;2021:6653793. 10.1155/2021/6653793. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 79. He L, Bulanova D, Oikkonen J, et al. Network-guided identification of cancer-selective combinatorial therapies in ovarian cancer. Brief Bioinform 2021;22(6):bbab272. 10.1093/bib/bbab272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011;474(7351):307–17. 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers 2016;2(1):16039. 10.1038/nrdp.2016.39. [DOI] [PubMed] [Google Scholar]
- 82. Baumgart DC, Sandborn WJ. Crohn's disease. Lancet (London, England) 2012;380(9853):1590–605. 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 83. Oussaada SM, van Galen KA, Cooiman MI, et al. The pathogenesis of obesity. Metab Clin Exp 2019;92:26–36. 10.1016/j.metabol.2018.12.012. [DOI] [PubMed] [Google Scholar]
- 84. Cappell MS. Pathophysiology, clinical presentation, and management of colon cancer. Gastroenterol Clin N Am 2008;37(1):1–24. 10.1016/j.gtc.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 85. Pearce L. Breast cancer. Nurs Stand 2016;30(51):15. 10.7748/ns.30.51.15.s16. [DOI] [PubMed] [Google Scholar]
- 86. Khwaja A, Bjorkholm M, et al. Acute myeloid leukaemia. Nat Rev Dis Primers 2016;2(1):16010. 10.1038/nrdp.2016.10. [DOI] [PubMed] [Google Scholar]
- 87. Eratne D, Loi SM, Farrand S, et al. Alzheimer's disease: clinical update on epidemiology, pathophysiology and diagnosis. Australas Psychiatry 2018;26(4):347–57. 10.1177/1039856218762308. [DOI] [PubMed] [Google Scholar]
- 88. Verduijn J, Milaneschi Y, Schoevers RA, et al. Pathophysiology of major depressive disorder: mechanisms involved in etiology are not associated with clinical progression. Transl Psychiatry 2015;5(9):e649. 10.1038/tp.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Feuerstein JD, Moss AC, Farraye FA. Ulcerative colitis. Mayo Clin Proc 2019;94(7):1357–73. 10.1016/j.mayocp.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 90. Stevens JR. Pathophysiology of schizophrenia. Clin Neuropharmacol 1983;6(2):77–90. 10.1097/00002826-198306000-00002. [DOI] [PubMed] [Google Scholar]
- 91. Anderson G. Autism spectrum disorder: pathophysiology and treatment implications. Curr Pharm Des 2019;25(41):4319–20. 10.2174/138161282541191230102715. [DOI] [PubMed] [Google Scholar]
- 92. Shelling AN. Premature ovarian failure. Reproduction (Cambridge, England) 2010;140(5):633–41. 10.1530/REP-09-0567. [DOI] [PubMed] [Google Scholar]
- 93. Folkow B. Pathophysiology of hypertension: differences between young and elderly. J Hypertens 1993;11(4):S21–4. [PubMed] [Google Scholar]
- 94. Hodges H, Fealko C, Soares N. Autism spectrum disorder: definition, epidemiology, causes, and clinical evaluation. Transl Pediatr 2020;9(Suppl 1):S55–65. 10.21037/tp.2019.09.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ (Clin Res ed) 2016;353:i1585. 10.1136/bmj.i1585. [DOI] [PubMed] [Google Scholar]
- 96. Repetto L, Granetto C, Hall RR. Prostate cancer. Crit Rev Oncol Hematol 1998;27(2):145–6. 10.1016/s10408428(97)10024-5. [DOI] [PubMed] [Google Scholar]
- 97. Van Marck E. Pathology of malignant mesothelioma. Lung Cancer (Amsterdam, Netherlands) 2004;45(Suppl 1):S35–6. 10.1016/j.lungcan.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 98. Kroeger PT, Jr, Drapkin R. Pathogenesis and heterogeneity of ovarian cancer. Curr Opin Obstet Gynecol 2017;29(1):26–34. 10.1097/GCO.0000000000000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tsimberidou AM, Fountzilas E, Nikanjam M, et al. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev 2020;86:102019. 10.1016/j.ctrv.2020.102019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9(1):559. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Uribe AG, Vilá LM, McGwin G, Jr, et al. The systemic lupus activity measure-revised, the Mexican Systemic Lupus Erythematosus Disease Activity Index (SLEDAI), and a modified SLEDAI-2K are adequate instruments to measure disease activity in systemic lupus erythematosus. J Rheumatol 2004;31(10):1934–40. [PubMed] [Google Scholar]
- 102. Costa-Silva J, Domingues D, Lopes FM. RNA-Seq differential expression analysis: an extended review and a software tool. PLoS One 2017;12(12):e0190152. 10.1371/journal.pone.0190152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14(1):1–15. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet 2018;19(8):491–504. 10.1038/s41576-018-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ahmed Z. Precision medicine with multi-omics strategies, deep phenotyping, and predictive analysis. Prog Mol Biol Transl Sci 2022. 10.1016/bs.pmbts.2022.02.002. [DOI] [PubMed] [Google Scholar]
- 106. Ahmed Z. Multi-omics strategies for personalized and predictive medicine: past, current, and future translational opportunities. Emerg Topics Life Sci 2022;ETLS20210244. 10.1042/ETLS20210244. [DOI] [PubMed] [Google Scholar]
- 107. Petegrosso R, Li Z, Kuang R. Machine learning and statistical methods for clustering single-cell RNA-sequencing data. Brief Bioinform 2020;21(4):1209–23. 10.1093/bib/bbz063. [DOI] [PubMed] [Google Scholar]
- 108. Aevermann B, Zhang Y, Novotny M, et al. A machine learning method for the discovery of minimum marker gene combinations for cell type identification from single-cell RNA sequencing. Genome Res 2021;31(10):1767–80. 10.1101/gr.275569.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Li L, Shen L, Ma J, et al. Evaluating distribution and prognostic value of new tumor-infiltrating lymphocytes in HCC based on a scRNA-Seq study with CIBERSORTx. Front Med 2020;7:451. 10.3389/fmed.2020.00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vrahatis AG, Vlamos P, Avramouli A, et al. Emerging machine learning techniques for modelling cellular complex systems in Alzheimer's disease. Adv Exp Med Biol 2021;1338:199–208. 10.1007/978-3-030-78775-2_24. [DOI] [PubMed] [Google Scholar]
- 111. Shah N, Li J, Li F, et al. An experiment on ab initio discovery of biological knowledge from scRNA-Seq data using machine learning. Patterns (New York, NY) 2020;1(5):100071. 10.1016/j.patter.2020.100071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chen Z, Yang X, Bi G, et al. Ligand-receptor interaction atlas within and between tumor cells and T cells in lung adenocarcinoma. Int J Biol Sci 2020;16(12):2205–19. 10.7150/ijbs.42080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hu J, Schroeder A, Coleman K, et al. Statistical and machine learning methods for spatially resolved transcriptomics with histology. Comput Struct Biotechnol J 2021;19:3829–41. 10.1016/j.csbj.2021.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Torroja C, Sanchez-Cabo F. Digitaldlsorter: deep-learning on scRNA-Seq to deconvolute gene expression data. Front Genet 2019;10:978. 10.3389/fgene.2019.00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Thibodeau A, Khetan S, Eroglu A, et al. CoRE-ATAC: a deep learning model for the functional classification of regulatory elements from single cell and bulk ATAC-seq data. PLoS Comput Biol 2021;17(12):e1009670. 10.1371/journal.pcbi.1009670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Li S, Yan H, Lee J. Identification of gene regulatory networks from single-cell expression data. Methods Mol Biol (Clifton, NJ) 2021;2328:153–70. 10.1007/978-1-0716-1534-8_9. [DOI] [PubMed] [Google Scholar]
- 117. Wu KE, Yost KE, Chang HY, et al. BABEL enables cross-modality translation between multiomic profiles at single-cell resolution. Proc Natl Acad Sci U S A 2021;118(15):e2023070118. 10.1073/pnas.2023070118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ji Z, Zhou W, Hou W, et al. Single-cell ATAC-seq signal extraction and enhancement with SCATE. Genome Biol 2020;21(1):161. 10.1186/s13059-020-02075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Rai V, Quang DX, Erdos MR, et al. Single-cell ATAC-Seq in human pancreatic islets and deep learning upscaling of rare cells reveals cell-specific type 2 diabetes regulatory signatures. Mol Metab 2020;32:109–21. 10.1016/j.molmet.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Schubach M, Re M, Robinson PN, et al. Imbalance-aware machine learning for predicting rare and common disease-associated non-coding variants. Sci Rep 2017;7(1):2959. 10.1038/s41598-017-03011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bugnon LA, Yones C, Milone DH, et al. Deep neural architectures for highly imbalanced data in bioinformatics. IEEE Trans Neural Netw Learn Systems 2020;31(8):2857–67. 10.1109/TNNLS.2019.2914471. [DOI] [PubMed] [Google Scholar]
- 122. Vabalas A, Gowen E, Poliakoff E, et al. Machine learning algorithm validation with a limited sample size. PLoS One 2019;14(11):e0224365. 10.1371/journal.pone.0224365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.